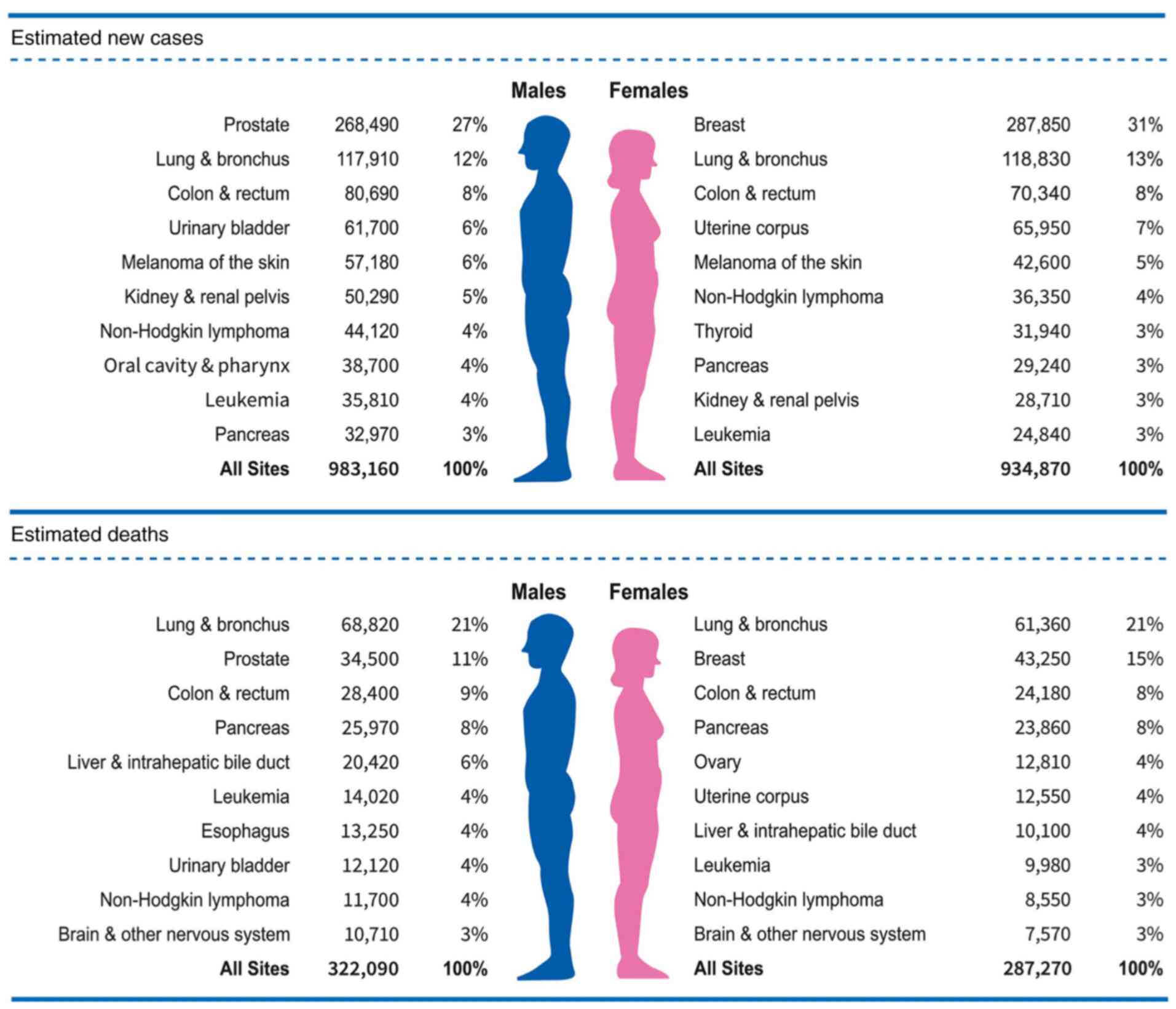
Breast cancer is the most commonly diagnosed cancer
(31% of the total cancer incidence) and the second highest cause of
cancer-associated death (15% of the total cancer-associated
mortality) in women worldwide (Fig.
1) (1). Among all breast
cancer subtypes, hormone receptor (HR)+ luminal subtype
breast cancer is the most common form of breast cancer, accounting
for 75% of the total breast cancer cases and 70% of metastatic
breast cancer (MBC) cases (2).
Endocrine therapy is the main treatment for HR+ luminal
subtype breast cancer, but its effectiveness is restricted by drug
resistance, which is almost inevitable in patients with advanced
breast cancer (ABC) (3-6). In recent years, the use of targeted
therapy combined with endocrine therapy to overcome the endocrine
therapy resistance of specific populations has provided a new
therapeutic prospect for patients with HR+ breast cancer
(7). One of the most basic
biological characteristics of malignant tumors is the malignant
transformation and uncontrolled proliferation of tumor cells caused
by the disorder of cell cycle regulation. CDK4/6 inhibitors restore
the cell cycle by selectively inhibiting cyclin-dependent kinases 4
and 6 (CDK4/6), and block cell proliferation in a variety of tumor
cells, including those of breast cancer. CDK4/6 inhibitors can
effectively improve the prognosis of patients with
HR+/human epidermal growth factor receptor 2
(HER2)− breast cancer, but different individuals have
different sensitivities to CDK4/6 inhibitors. The present review
aims to discuss the mechanism of CDK4/6 inhibitors, drug resistance
mechanisms and treatment strategies after resistance.
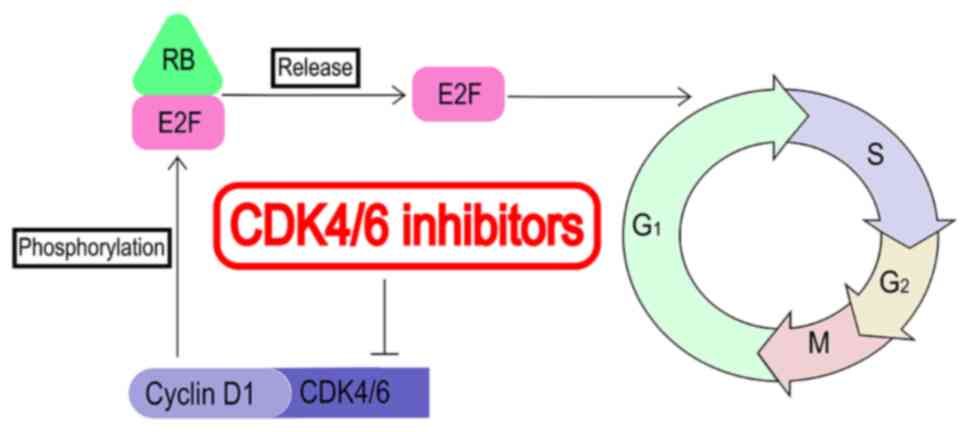
To ensure that each cell cycle is completed
accurately, complex regulatory mechanisms exist in the normal cell
cycle. CDK4/6 is a key regulator of the cell cycle, acting by
forming a complex with cyclin D (8). This complex can directly
phosphorylate retinoblastoma gene (RB), then release transcription
factor E2F and promote the transcription of cell cycle-related
genes, promoting the cell cycle from the G1 mitosis
phase to the S phase, leading to DNA replication (Fig. 2) (9). In estrogen receptor (ER)+
breast cancer, estrogen induces the activation of the ER signaling
pathway, which leads to the upregulation of the expression of
cyclin D and CDK4/6, and further leads to uncontrolled cell
proliferation (10,11). CDK4/6 is activated in
ER+ breast cancer; therefore, CDK4/6 inhibitors can
significantly inhibit the progression of ER+ breast
cancer.
Based on the data from three series of clinical
trials, namely PALOMA, MONALEESA and MONARCH, three CDK4/6
inhibitors have been approved by the USA Food and Drug
Administration (FDA) and European Medicines Agency for the
treatment of patients with HR+/HER2− ABC:
Palbociclib (Ibrance®; Pfizer, Inc.), ribociclib
(Kisqali®; Novartis International AG) and abemaciclib
(Verzenio®; Eli Lilly and Company) (12-17). In addition, a new CDK4/6
inhibitor, dalpiciclib, was reported in the 2021 ASCO meeting
(18). The DAWNA-1 study showed
that the dalpiciclib + fulvestrant group exhibited a significantly
improved median progression-free survival time (15.7 vs. 7.2
months; hazard ratio, 0.42; P<0.0001) compared with the placebo
+ fulvestrant group in patients with
HR+/HER2− ABC whose condition had relapsed or
advanced after endocrine therapy (Table I) (19). In addition, the safety of
dalpiciclib (SHR6390) was confirmed in clinical trial NCT03481998
(20).
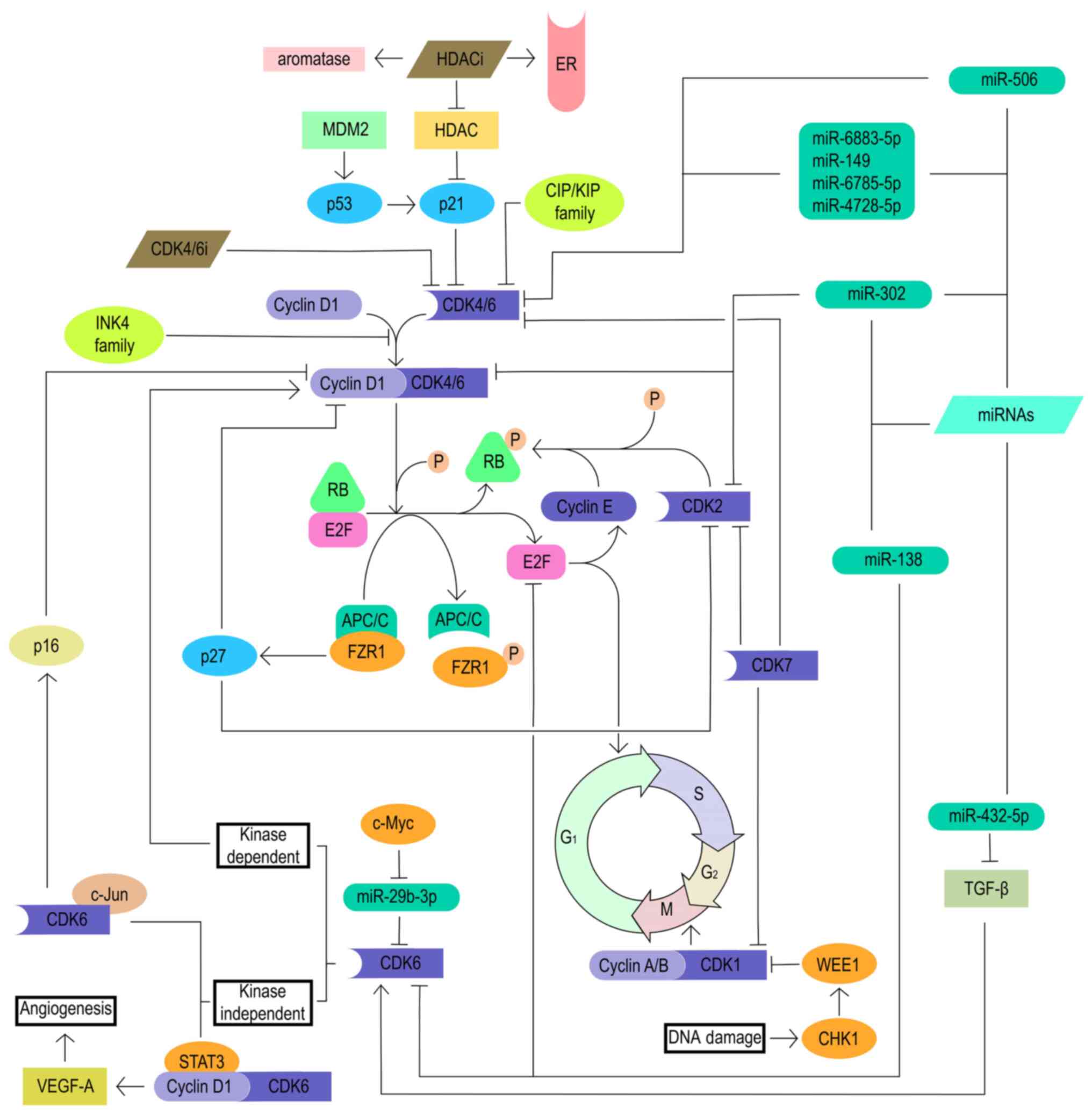
As a tumor suppressor, RB is the main target of the
cyclin D-CDK4/6 complex and controls the CDK4/6-RB1 pathway and the
cell cycle. Loss of RB is one of the most important reasons for the
development of resistance to CDK4/6 inhibitors (9). The main reason for the loss of RB is
the inactivion of the RB1 gene by a mutation. Despite the loss of
RB, the constitutive progression of the cell cycle continues
through the activation of other cell cycle mechanisms, including
the E2F and cyclin E-CDK2 axes, indicating that the progression of
the cell cycle from G1 to S phase has lost its
dependence on CDK4/6 (21). This
indicates that the combination of cyclin E-CDK2 axis inhibitors and
CDK4/6 inhibitors may reverse the resistance of CDK4/6 inhibitors
in patients with loss of RB (Fig.
3).
As aforementioned, CDK4/6 plays a vital role in the
progression of the cell cycle from the G1 phase to the S
phase. Various mechanisms such as gene amplification, mutation and
epigenetic changes can upregulate CDK4/6, thereby activating the
cyclin D-CDK4/6-RB pathway, which leads to a decreased blocking
effect of CDK4/6 inhibitors on cell cycle progression (28-30). In addition to kinase-dependent
functions, CDK6 also has some non-kinase-dependent functions. CDK6
upregulates the transcription of p16 in the presence of STAT3 and
cyclin D (31). In addition, CDK6
and c-Jun synergistically upregulate VEGF-A, induce tumor
angiogenesis, and usually promote cancer progression and drug
resistance (32,33). A previous study found that the
oncogene c-Myc could reduce the inhibitory effect of microRNA
(miRNA/miR)-29b-3p on CDK6 by downregulating miR-29b-3p, and that
activated CDK6 further induced breast cancer resistance to
palbociclib (34).
Cyclin E-CDK2 also plays an important role in the
progression of the cell cycle from the G1 phase to the S
phase. A previous study emphasized that after CDK4/6
inhibitor-resistant cells lost their dependence on cyclin D-CDK4/6
signaling, the inhibition of a variety of alternative signal
pathways, including the cyclin E-CDK2 pathway, could significantly
inhibit cell growth. CDK2 inhibitors effectively reduced the growth
of cells overexpressing cyclin E1 (35).
Moreover, CDK7 is a cell cycle regulator. CDK7 plays
the role of CDK-activating kinase (CAK) and participates in the
G1 and G2 phases by maintaining the activity
of CDK1/2/4/6 (36). It has been
reported that the increase in CDK7 expression confers resistance to
CDK4/6 inhibitors (Fig. 3)
(37). At the 2021 SABCS meeting,
a study reported the confirmed efficacy and safety of the first
oral selective CDK7 inhibitor samuraciclib (CT7001) + fulvestrant
in patients with HR+/HER2− ABC (38).
In addition, a study has found that histone
deacetylases (HDACs) have an inhibitory effect on the intrinsic CDK
inhibitor p21CIP1, and HDAC inhibitors can upregulate
the expression of p21CIP1. Whether in vitro or
in vivo, HDAC inhibitors can increase the expression of ER
and aromatase, and restore the sensitivity of breast cancer cells
to hormone blockade (41). Adding
entinostat or tucidinostat to exemestane can improve the mPFS time
(entinostat: 4.3 vs. 2.3 months; P=0.055; tucidinostat: 7.4 vs. 3.8
months; P=0.033), and significant improvement has been observed in
patients with non-steroidal aromatase inhibitor (NSAI) resistance
(42,43). This supports the hypothesis that
HDAC inhibition may enhance the activity of CDK4/6 inhibitors
through the CIP/KIP family, suggesting that the combination of HDAC
inhibitors and CDK4/6 inhibitors may be beneficial to patients with
CDK4/6 inhibitor resistance (Fig.
3).
WEE1 belongs to the serine/threonine protein kinase
family, which plays a key role in ensuring accurate DNA
replication, maintaining chromosome integrity, blocking abnormal
cell DNA replication and G2/M phase transition during
the cell cycle (44). WEE1 and
CDK1 synergistically inhibit DNA-damaged cells from entering
mitosis, whereas WEE1 inhibition promotes mitosis and propagates
genomic instability by forcing the cell through successive
replication cycles, ultimately resulting in apoptosis from mitotic
catastrophe. It was found that WEE1 inhibitors combined with
chemotherapy could synergistically strengthen the DNA damage to
tumor cells and block cell cycle transition (44). At the same time, a study has shown
that overexpression of WEE1 can induce tumor resistance to CDK4/6
inhibitors. Therefore, inhibiting WEE1 can increase the sensitivity
of CDK4/6 inhibitor-resistant cells to CDK4/6 inhibitors (Fig. 3) (45).
FZR1 forms the anaphase-promoting complex
(APC/C)-FZR1 complex by activating the ubiquitin ligase APC/C. The
APC/C-FZR1 complex interacts with RB during the G1 phase
of the cell cycle (46). In
addition, the APC/C-FZR1 complex further upregulates the natural
CDK inhibitor p27 by degrading S-phase kinase-associated protein 2,
leading to the decrease in the expression of CDK2, CDK4 and CDK6
(47). Therefore, the loss of
FZR1 leads to resistance to CDK4/6 inhibitors, the mechanism of
which needs further exploration (Fig.
3).
MDM2 is a protein that negatively regulates p53
activity. MDM2 inhibitors can activate p53 by disrupting the
MDM2-p53 complex (48). p53 can
activate the natural CDK4 inhibitor p21CIP1, leading to
cell cycle arrest (49).
NVP-CGM097 is one of the first new-generation inhibitors and has
entered phase I clinical trials (NCT01760525) (9). Although the mechanism of action of
MDM2 inhibitors still needs further research and exploration, MDM2
inhibition may be a new therapeutic target for the treatment of
resistance to CDK4/6 inhibitors (Fig.
3).
Numerous preclinical studies have discovered the
role of miRNAs in the progression of various tumors, including
glioblastoma, ovarian cancer and colon cancer. miR-302 induces cell
cycle G1/S arrest by inhibiting the cyclin D-CDK4/6 and
cyclin E-CDK2 pathways (50).
miR-138 induces cell cycle G1/S arrest by directly
targeting cell cycle genes such as CDK6, E2F2 and E2F3 (51). miR-506 directly targets CDK4 and
CDK6, and further inhibits CDK4/6-forkhead box M1 signaling to
inhibit cell proliferation (52).
A family of miRNAs containing miR-6883-5p, miR-149, miR-6785-5p and
miR-4728-5p inhibit cell proliferation by directly targeting the
untranslated regions of CDK4/6 mRNAs (53). miR-432-5p induces the
overexpression of CDK6 in ER+ breast cancer cells by
inhibiting the TGF-β pathway, further antagonizing the effect of
CDK4/6 inhibitors (54).
Therefore, targeted regulation of associated miRNAs provides a
therapeutic direction for patients with breast cancer who are
resistant to CDK4/6 inhibitors (Fig.
3).
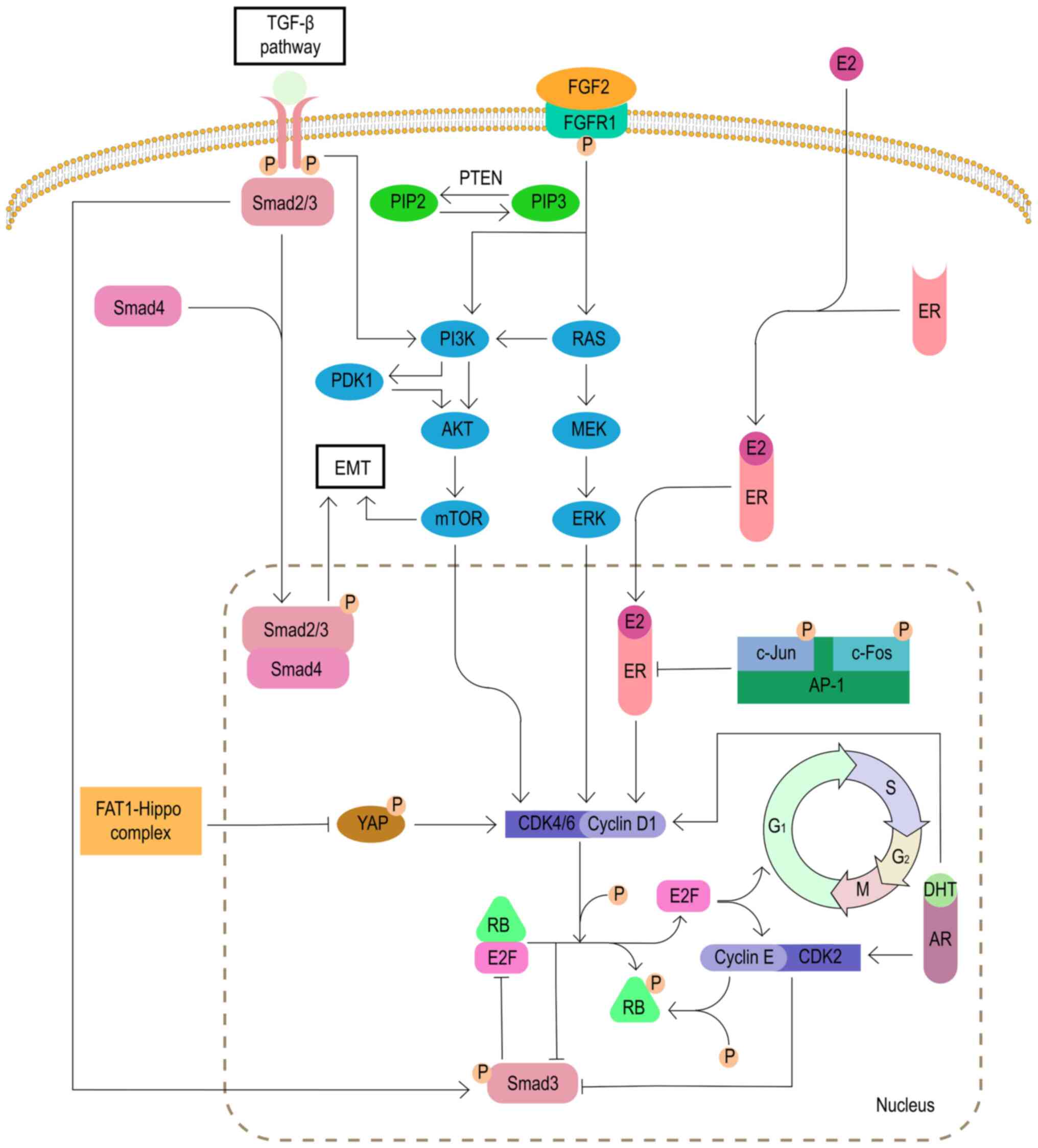
The FGFR1 signaling pathway is related to pivotal
biological processes, including proliferation, differentiation and
cell survival (55). FGFR1 plays
an important role in cancer progression. In endocrine-resistant
breast cancer cells, the expansion of FGFR1 activates the PI3K/AKT
and RAS/MEK/ERK signaling pathways, which is associated with CDK4/6
inhibitor resistance (56). In
the MONALEESA-2 trial, combined treatment with FGFR tyrosine kinase
inhibitor lucitanib eliminated the resistance to ribociclib, a
specific CDK4/6 inhibitor (57).
Therefore, inhibition of FGFR1 pathway may be a viable option to
overcome resistance to CDK4/6 inhibitors (Fig. 4).
It has been reported that the PIK3/AKT/mTOR
signaling pathway is associated with resistance to CDK4/6
inhibitors. A study has shown that direct inhibition of the
PI3K/AKT/mTOR pathway can reduce the expression of cyclin D and
cause cell cycle arrest, which may be the potential mechanism of
PI3K/AKT/mTOR pathway inhibitors (58).
In addition, as a negative regulator of the AKT/mTOR
signaling pathway, PTEN deletion causes tumor cells to develop
resistance to CDK4/6 inhibitors by activating AKT (59). It has also been found that the
deletion of PTEN induces the downregulation of the nuclear CDK
inhibitor protein p27 (coded by CDKN1B), which also leads to
resistance to CDK4/6 inhibitors (Fig.
4) (60).
The MAPK signaling pathway is one of the signaling
pathways downstream of FGFR1. The MAPK pathway is also known as the
RAS/RAF/MEK/ERK pathway based on the composition of its key
kinases. Therefore, MEK/ERK is a key target for the treatment of
patients with resistance to CDK4/6 inhibitors. A study has found
that the MEK1/2 inhibitor selumetinib combined with fulvestrant and
palbociclib can effectively inhibit the proliferation of breast
cancer cells resistant to CDK4/6 inhibitors (Fig. 4) (57).
The activity of breast cancer cyclin D-CDK4/6
complex depends on the activation of ER/PR induced by estrogen and
progesterone hormones (61). A
study has indicated that the resistance of CDK4/6 inhibitors is
associated with changes in ER/PR levels (30). Therefore, for patients with CDK4/6
inhibitor resistance due to the loss of ER/PR expression,
HR− breast cancer subtype relative treatments may be a
good choice.
As a transcription factor that regulates cyclin D1,
the AP-1 family consists of homodimers and heterodimers of the Jun,
Fos, activation transcription factor and transcription factor MAF
sub-families (62). A study has
found that 20-40% of human patients with breast cancer have high
levels of c-Jun activation, which inhibits the activity of ER and
activates the transcription of cyclin D1 to induce CDK4/6 inhibitor
resistance (63). Currently,
various AP-1 inhibitors are under active development, and a
selective c-Fos/AP-1 inhibitor (T-5224) has entered the second
phase of clinical trials (64).
The efficacy of AP-1 inhibitors for patients with breast cancer
resistant to CDK4/6 inhibitors needs further exploration, but the
results are worth looking forward to (Fig. 4).
It has been reported that the acquired resistance of
CDK4/6 inhibitors is associated with the activation of AR, which is
expressed in >70% of breast cancer cases (65). A study has found ER signal loss
and AR signal activation in the palbociclib-resistant breast cancer
cell line MCF-7pR. The non-aromatizable androgen
5-α-dihydrotestosterone-activated AR promotes G1/S
transition by activating cyclin D1, cyclin E1 and CDK2, thereby
inducing resistance to CDK4/6 inhibitors. As a new type of
selective AR inhibitor, the combination therapy of enzalutamide and
palbociclib can resensitize breast cancer cells to palbociclib and
reverse the resistance to the drug (66). Therefore, AR inhibitors may be one
of the treatment options for patients with breast cancer who are
resistant to CDK4/6 inhibitors (Fig.
4).
The tumor suppressor FAT atypical cadherin 1 (FAT1)
belongs to the cadherin superfamily and interacts with the Hippo
and β-catenin signaling pathways (25). In a genome analysis of 348 cases
of ER+ breast cancer after CDK4/6 inhibitor treatment,
loss of function mutations of FAT1 were detected in CDK4/6
inhibitor-resistant patients. Loss of FAT1 led to the inhibition of
the Hippo pathway and induced the nuclear localization of
Yes-related protein/tafazzin to induce CDK6 expression. At the same
time, the inactivation of neurofibrin 2, a component of the
hippocampus pathway, also increased the expression of CDK6. The two
approaches synergistically reduced the sensitivity to CDK4/6
inhibitors (Fig. 4) (67).
EMT refers to the transformation of cells from
epithelial cells to mesenchymal cells. The process plays an
important role in embryonic development and tissue reconstruction,
but EMT also confers the ability to invade and metastasize to
cancer cells. The inhibition of CDK4/6 can induce EMT by activating
the TGF-β-Smad and PI3K/AKT/mTOR signaling pathways (68-70). Phosphorylated TGF-β activates
Smad2 and Smad3, and then forms a complex with Smad4, resulting in
EMT by activation of the EMT transcription factor (70). However, it has been found that the
inhibition of Smad3 can also induce resistance to CDK4/6 inhibitors
(71). Inhibition of Smad3
induces the recovery of cell cycle arrest by releasing its blockage
of E2F from the Rb-E2F complex (72). In addition, the inhibition of
Smad3 is associated with the activation of the cyclin E-CDK2 axis
(73). As aforementioned,
miR-432-5p antagonizes the effects of CDK4/6 inhibitors by
inhibiting the TGF-β pathway. Therefore, the TGF-β pathway has a
very complicated association with CDK4/6 inhibitors, and more
research is needed (Fig. 4).
Treatment with endocrine therapy drugs combined with
CDK4/6 inhibitors has become the international recommended
treatment plan for HR+ ABC without visceral crisis, and
the level I recommended treatment plan of the 2020 CSCO guidelines
also agrees with this (74).
However, CDK4/6 inhibitors can significantly delay but not prevent
the emergence of acquired resistance of endocrine therapy. At
present, there is no uniform recommended treatment option for
patients who have progressed after treatment with endocrine therapy
drugs combined with CDK4/6 inhibitors. Generally, the various
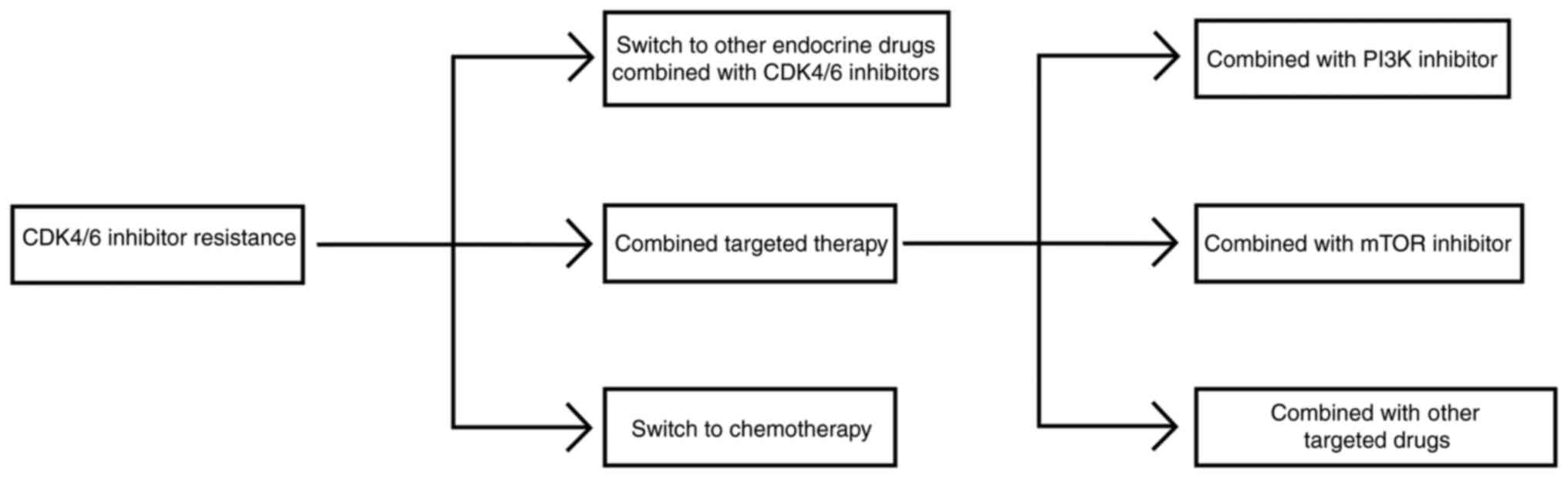
treatment options can be divided into three categories: Switching
to other endocrine therapy + CDK4/6i, combining targeted therapy
and switching to chemotherapy (Fig.
5).
The MonarchE and nextMONARCH studies have confirmed
the effectiveness of abemaciclib combined with endocrine therapy
(75,76). Another study from Harvard Medical
School suggested that for patients who had previously received the
CDK4/6 inhibitor palbociclib or ribociclib, switching to
abemaciclib monotherapy or a combination with endocrine therapy
after the disease progressed resulted in a mPFS time of 5.4 months
(77). As reported at the 2022
ASCO meeting, a phase II clinical study (MAINTAIN study) compared
the effect of receiving switch endocrine therapy ± ribociclib after
the progression on CDK4/6 inhibitors (78). The PFS time of the combination
group was prolonged by 2.5 months (5.29 vs. 2.76 months; P=0.006)
compared with the monotherapy group (Table II). A phase II clinical study
(PACE study) compared the post-progression effect of receiving
fulvestrant ± palbociclib on aromatase inhibitors (AIs) combined
with CDK4/6 inhibitors and is currently ongoing (79). The results of the EMERALD phase 3
study presented at the 2021 SABCS meeting showed that elacestrant
(an oral selective ER administration) was more effective than AI in
patients with ER+/HER2− MBC who had advanced
following prior endocrine therapy in combination with CDK4/6
inhibitors (80,81). Therefore, switching to other
endocrine drugs combined with CDK4/6 inhibitors is one of the
options after drug resistance.
PIK3CA is one of the most commonly mutated genes in
breast cancer. Approximately 40% of patients with HR+
and HER2− ABC have PIK3CA mutations. PIK3CA mutations
can promote endocrine resistance through the activation of the PI3K
pathway, which is associated with a poor prognosis (82).
As an α-selective PI3K inhibitor, alpelisib
demonstrated its efficacy for the first time in the SOLAR-1 study
(74). In the SOLAR-1 study,
alpelisib + fulvestrant was compared with placebo + fulvestrant for
patients with HR+/HER2− ABC with PIK3CA
mutations who progressed during or after treatment with AI. The PFS
time was prolonged by 5.3 months (11 vs. 5.7 months; P=0.00065)
(Table II). The 2020 ESMO
meeting reported the final results of this study. Compared with
that of the placebo + fulvestrant group, the median overall
survival (mOS) time of alpelisib + fulvestrant group was prolonged
by 7.9 months (39.3 vs. 31.4 months) [https://dailyreporter.esmo.org/esmo-congress-2020/articles/solar-1-trial-reports-overallsurvival-benefits-in-breast-cancer-patients-with-limited-options-of-treatment]
(83). Alpelisib has also been
approved by the FDA for marketing due to these study results.
However, <10% of the patients enrolled in the SOLAR-1 study
received CDK4/6 inhibitor treatment. The BYLieve study reported at
the 2020 ASCO meeting addresses the question of whether alpelisib
plays the same role in patients who progress after treatment with
CDK4/6 inhibitors (84). Patients
with PIK3CA mutations who progressed after first-line treatment
with AIs combined with CDK4/6 inhibitors were enrolled in cohort A
and received alpelisib combined with fulvestrant as the second-line
treatment. At the 2020 ASCO meeting, the mPFS time was reported as
7.3 months, and the 6-month PFS rate was 50.4% (85). This time was 11.0 months in the
SOLAR-1 study, suggesting that the first-line CDK4/6 inhibitor
treatment may not affect the efficacy of the follow-up PI3K
inhibitor (alpelisib) treatment (86). In addition, patients with PIK3CA
mutations who progressed after fulvestrant combined with CDK4/6
inhibitor first-line treatment were enrolled in cohort B in the
BYLieve study and received alpelisib combined with letrozole as the
second-line treatment. At the 2020 SABCS meeting, the mPFS time was
reported as 5.7 months, and the 6-month PFS rate was 46.1%
(Table II) (87). Therefore, for patients with PIK3CA
mutations who have progressed after endocrine therapy combined with
CDK4/6 inhibitor first-line treatment, the treatment of endocrine
therapy combined with PI3K inhibitors can be a choice for
patients.
The mTOR pathway is an important signaling pathway
downstream of PI3K, so mTOR inhibitors have received as much
attention as PI3K inhibitors. Everolimus is a representative of the
mTOR inhibitors, and its effect has been confirmed in the BOLERO-2
study. In the BOLERO-2 study, for patients who progressed after
NSAI treatment, the everolimus plus exemestane (SAI) group had
significantly longer mPFS and mOS times than the placebo plus
exemestane group (mPFS: 6.93 vs. 2.83 months; HR, 0.43;
P<0.0001; mOS: 31.0 vs. 26.6 months; HR, 0.89; P=0.14) (Table II). In patients who progressed
after treatment with CDK4/6 inhibitors, the study also showed a
good therapeutic effect of everolimus (88). The TRINITI clinical study reported
at the 2019 ASCO meeting enrolled patients who progressed after
treatment with CDK4/6 inhibitors (89). After receiving exemestane,
everolimus and ribociclib combination therapy, the clinical benefit
rate at 24 weeks reached 41%, which exceeds the pre-defined primary
endpoint threshold (>10%). The mPFS time of the overall
population reached 5.7 months (90). Therefore, mTOR inhibitors seem to
be a good option for patients who are resistant to CDK4/6
inhibitors.
Resistance to CDK4/6 inhibitors is a complex process
involving multiple different mechanisms, including CDK2 and CDK7
activation, and FGFR1 mutations, among others. Therefore, a series
of corresponding clinical studies are currently underway to explore
whether the addition of these new targeted drugs can overcome the
resistance of CDK4/6 inhibitors.
AKT acts as a bridge connecting the PI3K and mTOR
signaling pathways. The TAKTIC study evaluated the efficacy of
AKT-1 inhibitor ipatasertib + endocrine therapy ± CDK4/6 inhibitor
palbociclib in the treatment of patients with
HR+/HER2− ABC. The interim analysis of group
C (ipatasertib + fulvestrant + palbociclib) was reported at the
2020 ASCO meeting (91). The
results showed that for patients who had failed previous CDK4/6
inhibitor treatment, AKT inhibitor + CDK4/6 inhibitor + endocrine
therapy achieved good clinical effects in some patients (8/12) and
was well tolerated. The follow-up results of the TAKTIC study
remain to be seen. Therefore, AKT inhibitors may become one of the
treatment choices after CDK4/6 inhibitor resistance.
HDAC inhibitors are another of the targeted drugs
most focused upon. The ACE study showed that the mPFS time of the
chidamide combined with exemestane group was longer than that of
the placebo combined with exemestane group (7.4 vs. 3.8 months;
P=0.033) (Table II) (43). At the 2021 SABCS meeting, another
HADC inhibitor, entinostat, was shown to improve PFS time in
patients with HR+/HER2− ABC with AI
resistance. Therefore, HDAC inhibitors also provide new options for
patients who are resistant to CDK4/6 inhibitors.
B-cell lymphoma-2 (BCL2) is an estrogen-responsive
gene and anti-apoptotic protein overexpressed in ~80% of patients
with HR+ breast cancer (92). Venetoclax combined with tamoxifen
had a tolerable safety profile and significant activity in patients
with ER+/HER2− ABC that overexpresses BCL2 in
a phase I clinical trial (93).
In addition, an ongoing phase I clinical trial, PALVEN
(NCT03900884), is evaluating the safety and efficacy of AI + CDK4/6
inhibitor + venetoclax triple therapy as first-line treatment for
patients with ER+/HER2− ABC with BCL2
overexpression. Another ongoing phase II clinical trial, Veronica
(NCT03584009), is evaluating venetoclax + fulvestrant vs.
fulvestrant in patients who have progressed after CDK4/6 inhibitor
therapy. There was no significant difference in PFS time between
the combination and monotherapy groups in the preliminary results
of the Veronica trial presented at the ASCO 2021 meeting (2.69 vs.
1.94 months; P=0.7853) (Table
II) (94,95). However, with the support of
results from further clinical studies, BCL2 inhibitors are still
expected to be a second-line option after CDK4/6 inhibitor
resistance.
CDK4/6 inhibitors can effectively inhibit the
proliferation of regulatory T cells and enhance the function of
effector T cells in the tumor microenvironment, which provides a
theoretical basis for the combination of CDK4/6 inhibitors with
immune checkpoint inhibitors (96). An ongoing phase IB clinical trial,
JPCE (NCT02779751), is evaluating the efficacy and safety of
abemaciclib in combination with pembrolizumab (PD-1 inhibitor) in
the treatment of HR+/HER2− MBC. Another
ongoing phase II clinical trial, PACE (NCT03147287), is evaluating
the efficacy of fulvestrant, palbociclib and avelumab (PD-L1
inhibitor) in patients with ER+/HER2− breast
cancer who are resistant to palbociclib.
At present, treatment options after resistance to
CDK4/6 inhibitors are still limited and full of controversy. There
is no clear and systematic treatment strategy internationally. The
treatment options after CDK4/6 inhibitor resistance can be
basically divided into three categories: The first is to switch to
other endocrine drugs combined with CDK4/6 inhibitors, the second
is to used combined targeted therapy and the third is to switch to
cytotoxic drugs for chemotherapy. However, the choice of treatment
after CDK4/6 inhibitor resistance is mainly based on the number of
lines of endocrine therapy combined with CDK4/6 inhibitor treatment
used in previous treatment, the combination of drugs, the length of
treatment time, the current metastatic site, the size of the
metastatic load, whether there is a target for mutation in the
circulating tumor DNA and the economic situation of the patient.
After comprehensive consideration, patients should be provided with
an individualized treatment plan.
The treatment solution after CDK4/6 inhibitor
resistance is still the focus of research by scientists. At
present, delaying resistance is more important for patients who use
CDK4/6 inhibitors. For example, attempts should be made to use
CDK4/6 inhibitors as the second line treatment as much as possible.
In this way, the early occurrence of acquired drug resistance can
be avoided. Combined targeted therapy is also a common combination
method in clinical practice, which also helps delay the occurrence
of drug resistance. However, it also depends on the status of the
disease. If the disease progresses rapidly, it may be necessary to
speed up the use of CDK4/6 inhibitors to control the condition.
Therefore, in order to more accurately determine the resistance
mechanism of CDK4/6 inhibitors and find new targets to overcome the
resistance of CDK4/6 inhibitors, further exploratory research and
clinical verification are needed.
Not applicable.
JH investigated and wrote the original draft of the
manuscript. ZS and LZ reviewed and edited the manuscript. JL
proposed the theme of the review and helped in writing the
manuscript. All authors read and approved the final manuscript.
Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
This study was supported by the National Natural Science
Foundation of China (grant no. 82172917).
|
1
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2022. CA Cancer J Clin. 72:7–33. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dai X, Li T, Bai Z, Yang Y, Liu X, Zhan J
and Shi B: Breast cancer intrinsic subtype classification, clinical
use and future trends. Am J Cancer Res. 5:2929–2943.
2015.PubMed/NCBI
|
|
3
|
Cardoso F, Costa A, Senkus E, Aapro M,
André F, Barrios CH, Bergh J, Bhattacharyya G, Biganzoli L, Cardoso
MJ, et al: 3rd ESO-ESMO international consensus guidelines for
advanced breast cancer (ABC 3). Ann Oncol. 28:31112017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Milani A, Geuna E, Mittica G and Valabrega
G: Overcoming endocrine resistance in metastatic breast cancer:
Current evidence and future directions. World J Clin Oncol.
5:990–1001. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Clarke R, Tyson JJ and Dixon JM: Endocrine
resistance in breast cancer-an overview and update. Mol Cell
Endocrinol. 418:220–234. 2015. View Article : Google Scholar :
|
|
6
|
Brufsky AM and Dickler MN: Estrogen
receptor-positive breast cancer: Exploiting signaling pathways
implicated in endocrine resistance. Oncologist. 23:528–539. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu W and Xu B: Overcoming resistance to
endocrine therapy in hormone receptor-positive human epidermal
growth factor receptor 2-negative (HR+/HER2−)
advanced breast cancer: A meta-analysis and systemic review of
randomized clinical trials. Front Med. 15:208–220. 2021. View Article : Google Scholar
|
|
8
|
Nair BC and Vadlamudi RK: Regulation of
hormonal therapy resistance by cell cycle machinery. Gene Ther Mol
Biol. 12:3952008.PubMed/NCBI
|
|
9
|
Spring LM, Wander SA, Andre F, Moy B,
Turner NC and Bardia A: Cyclin-dependent kinase 4 and 6 inhibitors
for hormone receptor-positive breast cancer: Past, present, and
future. Lancet. 395:817–827. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Altucci L, Addeo R, Cicatiello L, Germano
D, Pacilio C, Battista T, Cancemi M, Petrizzi VB, Bresciani F and
Weisz A: Estrogen induces early and timed activation of
cyclin-dependent kinases 4, 5, and 6 and increases cyclin messenger
ribonucleic acid expression in rat uterus. Endocrinology.
138:978–984. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Geum D, Sun W, Paik SK, Lee CC and Kim K:
Estrogen-induced cyclin D1 and D3 gene expressions during mouse
uterine cell proliferation in vivo: Differential induction
mechanism of cyclin D1 and D3. Mol Reprod Dev. 46:450–458. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Finn RS, Crown JP, Lang I, Boer K,
Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, et
al: The cyclin-dependent kinase 4/6 inhibitor palbociclib in
combination with letrozole versus letrozole alone as first-line
treatment of oestrogen receptor-positive, HER2-negative, advanced
breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study.
Lancet Oncol. 16:25–35. 2015. View Article : Google Scholar
|
|
13
|
Finn RS, Boer K, Bondarenko I, Patel R,
Pinter T, Schmidt M, Shparyk YV, Thummala A, Voitko N, Bananis E,
et al: Overall survival results from the randomized phase 2 study
of palbociclib in combination with letrozole versus letrozole alone
for first-line treatment of ER+/HER2- advanced breast cancer
(PALOMA-1, TRIO-18). Breast Cancer Res Treat. 183:419–428. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Turner NC, Slamon DJ, Ro J, Bondarenko I,
Im SA, Masuda N, Colleoni M, DeMichele A, Loi S, Verma S, et al:
Overall survival with palbociclib and fulvestrant in advanced
breast cancer. N Engl J Med. 379:1926–1936. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hortobagyi GN, Stemmer SM, Burris HA, Yap
YS, Sonke GS, Paluch-Shimon S, Campone M, Petrakova K, Blackwell
KL, Winer EP, et al: Updated results from MONALEESA-2, a phase III
trial of first-line ribociclib plus letrozole versus placebo plus
letrozole in hormone receptor-positive, HER2-negative advanced
breast cancer. Ann Oncol. 30:18422019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Slamon DJ, Neven P, Chia S, Jerusalem G,
De Laurentiis M, Im S, Petrakova K, Valeria Bianchi G, Martín M,
Nusch A, et al: Ribociclib plus fulvestrant for postmenopausal
women with hormone receptor-positive, human epidermal growth factor
receptor 2-negative advanced breast cancer in the phase III
randomized MONALEESA-3 trial: Updated overall survival. Ann Oncol.
32:1015–1024. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sledge GW Jr, Toi M, Neven P, Sohn J,
Inoue K, Pivot X, Burdaeva O, Okera M, Masuda N, Kaufman PA, et al:
The effect of abemaciclib plus fulvestrant on overall survival in
hormone receptor-positive, ERBB2-negative breast cancer that
progressed on endocrine therapy-MONARCH 2: A randomized clinical
trial. JAMA Oncol. 6:116–124. 2020. View Article : Google Scholar
|
|
18
|
Xu B, Zhang Q, Zhang P, Hu X, Li W, Tong
Z, Sun T, Teng Y, Wu X, Ouyang Q, et al: Dalpiciclib versus placebo
plus fulvestrant in HR+/HER2- advanced breast cancer that relapsed
or progressed on previous endocrine therapy (DAWNA-1): A
multi-center, randomized, phase 3 study. J Clin Oncol. 39(15
Suppl): S1002. 2021. View Article : Google Scholar
|
|
19
|
Xu B, Zhang Q, Zhang P, Hu X, Li W, Tong
Z, Sun T, Teng Y, Wu X, Ouyang Q, et al: Dalpiciclib or placebo
plus fulvestrant in hormone receptor-positive and HER2-negative
advanced breast cancer: A randomized, phase 3 trial. Nat Med.
27:1904–1909. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang P, Xu B, Gui L, Wang W, Xiu M, Zhang
X, Sun G, Zhu X and Zou J: A phase 1 study of dalpiciclib, a
cyclin-dependent kinase 4/6 inhibitor in Chinese patients with
advanced breast cancer. Biomark Res. 9:242021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Malumbres M, Sotillo R, Santamaria D,
Galán J, Cerezo A, Ortega S, Dubus P and Barbacid M: Mammalian
cells cycle without the D-type cyclin-dependent kinases Cdk4 and
Cdk6. Cell. 118:493–504. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guarducci C, Bonechi M, Boccalini G,
Benelli M, Risi E, Di Leo A, Malorni L and Migliaccio I: Mechanisms
of resistance to CDK4/6 inhibitors in breast cancer and potential
biomarkers of response. Breast Care (Basel). 12:304–308. 2017.
View Article : Google Scholar
|
|
23
|
Siebert R, Willers CP and Opalka B: Role
of the cyclin-dependent kinase 4 and 6 inhibitor gene family p15,
p16, p18 and p19 in leukemia and lymphoma. Leuk Lymphoma.
23:505–520. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Green JL, Okerberg ES, Sejd J, Palafox M,
Monserrat L, Alemayehu S, Wu J, Sykes M, Aban A, Serra V and
Nomanbhoy T: Direct CDKN2 modulation of CDK4 alters target
engagement of CDK4 inhibitor drugs. Mol Cancer Ther. 18:771–779.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Finn RS, Crown JP, Ettl J, Schmidt M,
Bondarenko IM, Lang I, Pinter T, Boer K, Patel R, Randolph S, et
al: Efficacy and safety of palbociclib in combination with
letrozole as first-line treatment of ER-positive, HER2-negative,
advanced breast cancer: Expanded analyses of subgroups from the
randomized pivotal trial PALOMA-1/TRIO-18. Breast Cancer Res.
18:672016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Turner NC, Liu Y, Zhu Z, Loi S, Colleoni
M, Loibl S, DeMichele A, Harbeck N, André F, Bayar MA, et al:
Cyclin E1 expression and palbociclib efficacy in previously treated
hormone receptor-positive metastatic breast cancer. J Clin Oncol.
37:1169–1178. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Finn RS, Liu Y, Zhu Z, Martin M, Rugo HS,
Diéras V, Im SA, Gelmon KA, Harbeck N, Lu DR, et al: Biomarker
analyses of response to cyclin-dependent kinase 4/6 inhibition and
endocrine therapy in women with treatment-Naïve metastatic breast
cancer. Clin Cancer Res. 26:110–121. 2020. View Article : Google Scholar
|
|
28
|
Wu A, Wu B, Guo J, Luo W, Wu D, Yang H,
Zhen Y, Yu X, Wang H, Zhou Y, et al: Elevated expression of CDK4 in
lung cancer. J Transl Med. 9:382011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Olanich ME, Sun W, Hewitt SM, Abdullaev Z,
Pack SD and Barr FG: CDK4 amplification reduces sensitivity to
CDK4/6 inhibition in fusion-positive rhabdomyosarcoma. Clin Cancer
Res. 21:4947–4959. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang C, Li Z, Bhatt T, Dickler M, Giri D,
Scaltriti M, Baselga J, Rosen N and Chandarlapaty S: Acquired CDK6
amplification promotes breast cancer resistance to CDK4/6
inhibitors and loss of ER signaling and dependence. Oncogene.
36:2255–2264. 2017. View Article : Google Scholar :
|
|
31
|
Tigan AS, Bellutti F, Kollmann K, Tebb G
and Sexl V: CDK6-a review of the past and a glimpse into the
future: From cell-cycle control to transcriptional regulation.
Oncogene. 35:3083–3091. 2016. View Article : Google Scholar
|
|
32
|
Kollmann K, Heller G, Schneckenleithner C,
Warsch W, Scheicher R, Ott RG, Schäfer M, Fajmann S, Schlederer M,
Schiefer AI, et al: A kinase-independent function of CDK6 links the
cell cycle to tumor angiogenesis. Cancer Cell. 30:359–360. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gacche RN and Assaraf YG: Redundant
angiogenic signaling and tumor drug resistance. Drug Resist Updat.
36:47–76. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ji W, Zhang W, Wang X, Shi Y, Yang F, Xie
H, Zhou W, Wang S and Guan X: c-myc regulates the sensitivity of
breast cancer cells to palbociclib via c-myc/miR-29b-3p/CDK6 axis.
Cell Death Dis. 11:7602020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Etemadmoghadam D, Au-Yeung G, Wall M,
Mitchell C, Kansara M, Loehrer E, Batzios C, George J, Ftouni S,
Weir BA, et al: Resistance to CDK2 inhibitors is associated with
selection of polyploid cells in CCNE1-amplified ovarian cancer.
Clin Cancer Res. 19:5960–5971. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schachter MM, Merrick KA, Larochelle S,
Hirschi A, Zhang C, Shokat KM, Rubin SM and Fisher RP: A Cdk7-Cdk4
T-loop phosphorylation cascade promotes G1 progression. Mol Cell.
50:250–260. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Martin LA, Pancholi S, Ribas R, Gao Q,
Simigdala N, Nikitorowicz-Buniak J, Johnston SR and Dowsett M:
Abstract P3-03-09: Resistance to palbociclib depends on multiple
targetable mechanisms highlighting the potential of drug holidays
and drug switching to improve therapeutic outcome. Cancer Res. 77(4
Suppl): P3-03-092017. View Article : Google Scholar
|
|
38
|
Howell SJ, Krebs MG, Lord S, Kenny L, Bahl
A, Clack G, Ainscow E, Arkenau HT, Mansi JL, Palmieri C, et al:
265P Study of samuraciclib (CT7001), a first-in-class, oral,
selective inhibitor of CDK7, in combination with fulvestrant in
patients with advanced hormone receptor positive HER2 negative
breast cancer (HR+BC). Ann Oncol. 32(Suppl 5): S477–S478. 2021.
View Article : Google Scholar
|
|
39
|
Orlando S, Gallastegui E, Besson A, Abril
G, Aligué R, Pujol MJ and Bachs O: p27Kip1 and p21Cip1 collaborate
in the regulation of transcription by recruiting cyclin-Cdk
complexes on the promoters of target genes. Nucleic Acids Res.
43:6860–6873. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Álvarez-Fernández M and Malumbres M:
Mechanisms of sensitivity and resistance to CDK4/6 inhibition.
Cancer Cell. 37:514–529. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sabnis GJ, Goloubeva O, Chumsri S, Nguyen
N, Sukumar S and Brodie AM: Functional activation of the estrogen
receptor-α and aromatase by the HDAC inhibitor entinostat
sensitizes ER-negative tumors to letrozole. Cancer Res.
71:1893–1903. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yardley DA, Ismail-Khan RR, Melichar B,
Lichinitser M, Munster PN, Klein PM, Cruickshank S, Miller KD, Lee
MJ and Trepel JB: Randomized phase II, double-blind,
placebo-controlled study of exemestane with or without entinostat
in postmenopausal women with locally recurrent or metastatic
estrogen receptor-positive breast cancer progressing on treatment
with a nonsteroidal aromatase inhibitor. J Clin Oncol.
31:2128–2135. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jiang Z, Li W, Hu X, Zhang Q, Sun T, Cui
S, Wang S, Ouyang Q, Yin Y, Geng C, et al: Tucidinostat plus
exemestane for postmenopausal patients with advanced, hormone
receptor-positive breast cancer (ACE): A randomised, double-blind,
placebo-controlled, phase 3 trial. Lancet Oncol. 20:806–815. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Matheson CJ, Backos DS and Reigan P:
Targeting WEE1 kinase in cancer. Trends Pharmacol Sci. 37:872–881.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pandey K, An HJ, Kim SK, Lee SA, Kim S,
Lim SM, Kim GM, Sohn J and Moon YW: Molecular mechanisms of
resistance to CDK4/6 inhibitors in breast cancer: A review. Int J
Cancer. 145:1179–1188. 2019. View Article : Google Scholar :
|
|
46
|
Ramanujan A and Tiwari S: APC/C and
retinoblastoma interaction: Cross-talk of retinoblastoma protein
with the ubiquitin proteasome pathway. Biosci Rep. 36:e003772016.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fujita T, Liu W, Doihara H and Wan Y:
Regulation of Skp2-p27 axis by the Cdh1/anaphase-promoting complex
pathway in colorectal tumorigenesis. Am J Pathol. 173:217–228.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Laroche-Clary A, Chaire V, Algeo MP,
Derieppe MA, Loarer FL and Italiano A: Combined targeting of MDM2
and CDK4 is synergistic in dedifferentiated liposarcomas. J Hematol
Oncol. 10:1232017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cox LS: Multiple pathways control cell
growth and transformation: Overlapping and independent activities
of p53 and p21Cip1/WAF1/Sdi1. J Pathol. 183:134–140. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lin SL, Chang DC, Ying SY, Leu D and Wu
DT: MicroRNA miR-302 inhibits the tumorigenecity of human
pluripotent stem cells by coordinate suppression of the CDK2 and
CDK4/6 cell cycle pathways. Cancer Res. 70:9473–9482. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Qiu S, Huang D, Yin D, Li F, Li X, Kung HF
and Peng Y: Suppression of tumorigenicity by microRNA-138 through
inhibition of EZH2-CDK4/6-pRb-E2F1 signal loop in glioblastoma
multiforme. Biochim Biophys Acta. 1832:1697–1707. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liu G, Sun Y, Ji P, Li X, Cogdell D, Yang
D, Parker Kerrigan BC, Shmulevich I, Chen K, Sood AK, et al:
MiR-506 suppresses proliferation and induces senescence by directly
targeting the CDK4/6-FOXM1 axis in ovarian cancer. J Pathol.
233:308–318. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lulla AR, Slifker MJ, Zhou Y, Lev A,
Einarson MB, Dicker DT and El-Deiry WS: miR-6883 family miRNAs
target CDK4/6 to induce G1 phase cell-cycle arrest in
colon cancer cells. Cancer Res. 77:6902–6913. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Cornell L, Wander SA, Visal T, Wagle N and
Shapiro GI: MicroRNA-mediated suppression of the TGF-β pathway
confers transmissible and reversible CDK4/6 inhibitor resistance.
Cell Rep. 26:2667–2680.e7. 2019. View Article : Google Scholar
|
|
55
|
Turner N and Grose R: Fibroblast growth
factor signalling: From development to cancer. Nat Rev Cancer.
10:116–129. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Turner N, Pearson A, Sharpe R, Lambros M,
Geyer F, Lopez-Garcia MA, Natrajan R, Marchio C, Iorns E, Mackay A,
et al: FGFR1 amplification drives endocrine therapy resistance and
is a therapeutic target in breast cancer. Cancer Res. 70:2085–2094.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Formisano L, Lu Y, Servetto A, Hanker AB,
Jansen VM, Bauer JA, Sudhan DR, Guerrero-Zotano AL, Croessmann S,
Guo Y, et al: Aberrant FGFR signaling mediates resistance to CDK4/6
inhibitors in ER+ breast cancer. Nat Commun. 10:13732019.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kilker RL and Planas-Silva MD: Cyclin D1
is necessary for tamoxifen-induced cell cycle progression in human
breast cancer cells. Cancer Res. 66:11478–11484. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Costa C, Wang Y, Ly A, Hosono Y, Murchie
E, Walmsley CS, Huynh T, Healy C, Peterson R, Yanase S, et al: PTEN
loss mediates clinical cross-resistance to CDK4/6 and PI3Kα
inhibitors in breast cancer. Cancer Discov. 10:72–85. 2020.
View Article : Google Scholar
|
|
60
|
Bencivenga D, Caldarelli I, Stampone E,
Mancini FP, Balestrieri ML, Della Ragione F and Borriello A:
p27Kip1 and human cancers: A reappraisal of a still
enigmatic protein. Cancer Lett. 403:354–365. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Finn RS, Aleshin A and Slamon DJ:
Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen
receptor-positive breast cancers. Breast Cancer Res. 18:172016.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Shaulian E and Karin M: AP-1 in cell
proliferation and survival. Oncogene. 20:2390–2400. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Shen Q, Uray IP, Li Y, Zhang Y, Hill J, Xu
XC, Young MR, Gunther EJ, Hilsenbeck SG, Colburn NH, et al:
Targeting the activator protein 1 transcription factor for the
prevention of estrogen receptor-negative mammary tumors. Cancer
Prev Res (Phila). 1:45–55. 2008. View Article : Google Scholar
|
|
64
|
Tewari D, Nabavi SF, Nabavi SM, Sureda A,
Farooqi AA, Atanasov AG, Vacca RA, Sethi G and Bishayee A:
Targeting activator protein 1 signaling pathway by bioactive
natural agents: Possible therapeutic strategy for cancer prevention
and intervention. Pharmacol Res. 128:366–375. 2018. View Article : Google Scholar
|
|
65
|
McNamara KM, Yoda T, Takagi K, Miki Y,
Suzuki T and Sasano H: Androgen receptor in triple negative breast
cancer. J Steroid Biochem Mol Biol. 133:66–76. 2013. View Article : Google Scholar
|
|
66
|
Ji W, Shi Y, Wang X, He W, Tang L, Tian S,
Jiang H, Shu Y and Guan X: Combined androgen receptor blockade
overcomes the resistance of breast cancer cells to palbociclib. Int
J Biol Sci. 15:522–532. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Li Z, Razavi P, Li Q, Toy W, Liu B, Ping
C, Hsieh W, Sanchez-Vega F, Brown DN, Da Cruz Paula AF, et al: Loss
of the FAT1 tumor suppressor promotes resistance to CDK4/6
inhibitors via the Hippo pathway. Cancer Cell. 34:893–905.e8. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Liu F and Korc M: Cdk4/6 inhibition
induces epithelial-mesenchymal transition and enhances invasiveness
in pancreatic cancer cells. Mol Cancer Ther. 11:2138–2148. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Moustakas A and Heldin CH: Non-Smad
TGF-beta signals. J Cell Sci. 118:3573–3584. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Zelivianski S, Cooley A, Kall R and Jeruss
JS: Cyclin-dependent kinase 4-mediated phosphorylation inhibits
Smad3 activity in cyclin D-overexpressing breast cancer cells. Mol
Cancer Res. 8:1375–1387. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Yang J, Song K, Krebs TL, Jackson MW and
Danielpour D: Rb/E2F4 and Smad2/3 link survivin to TGF-beta-induced
apoptosis and tumor progression. Oncogene. 27:5326–5338. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Decker JT, Wan L, Shea LD and Jeruss JS:
Abstract P4-03-16: Cyclin E affects Smad3 pathway in trastuzumab
resistant HER2+ breast cancer. Cancer Res. 78(4 Suppl): pp.
P4-03-162018, View Article : Google Scholar
|
|
74
|
Jiang Z, Song E, Wang X, Wang H, Wang X,
Wu J, Yin Y, Zhang Q, Chen J, Che W, et al: Guidelines of Chinese
society of clinical oncology (CSCO) on diagnosis and treatment of
breast cancer (2020 version). 2020. Transl Breast Cancer Res.
1:272020. View Article : Google Scholar
|
|
75
|
Johnston SRD, Harbeck N, Hegg R, Toi M,
Martin M, Shao ZM, Zhang QY, Martinez Rodriguez JL, Campone M,
Hamilton E, et al: Abemaciclib combined with endocrine therapy for
the adjuvant treatment of HR+, HER2-, node-positive, high-risk,
early breast cancer (monarchE). J Clin Oncol. 38:3987–3998. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Hamilton E, Cortes J, Ozyilkan O, Chen SC,
Petrakova K, Manikhas A, Jerusalem G, Hegg R, Huober J, Chapman SC,
et al: nextMONARCH: Abemaciclib monotherapy or combined with
tamoxifen for metastatic breast cancer. Clin Breast Cancer.
21:181–190.e2. 2021. View Article : Google Scholar
|
|
77
|
Wander SA, Zangardi M, Niemierko A,
Kambadakone A, Kim LSL, Xi J, Pandey AK, Spring L, Stein C, Juric
D, et al: A multicenter analysis of abemaciclib after progression
on palbociclib in patients (pts) with hormone receptor-positive
(HR+)/HER2-metastatic breast cancer (MBC). J Clin Oncol. 37(15
Suppl): S10572019. View Article : Google Scholar
|
|
78
|
Kalinsky K, Accordino MK, Chiuzan C, Mundi
PS, Trivedi MS, Novik Y, Tiersten A, Raptis G, Baer LN, Oh SY, et
al: A randomized, phase II trial of fulvestrant or exemestane with
or without ribociclib after progression on anti-estrogen therapy
plus cyclin-dependent kinase 4/6 inhibition (CDK 4/6i) in patients
(pts) with unresectable or hormone receptor-positive (HR+),
HER2-negative metastatic breast cancer (MBC): MAINTAIN trial. J
Clin Oncol. 40(17 Suppl): LBA1004. 2022. View Article : Google Scholar
|
|
79
|
Mayer EL, Wander SA, Regan MM, DeMichele
A, Forero-Torres A, Rimawi MF, Ma CX, Cristofanilli M, Anders CK,
Bartlett CH, et al: Palbociclib after CDK and endocrine therapy
(PACE): A randomized phase II study of fulvestrant, palbociclib,
and avelumab for endocrine pre-treated ER+/HER2-metastatic breast
cancer. J Clin Oncol. 36(15 Suppl): TPS11042018. View Article : Google Scholar
|
|
80
|
Bardia A, Aftimos P, Bihani T,
Anderson-Villaluz AT, Jung J, Conlan MG and Kaklamani VG: EMERALD:
Phase III trial of elacestrant (RAD1901) vs endocrine therapy for
previously treated ER+ advanced breast cancer. Future Oncol.
15:3209–3218. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Bidard FC, Kaklamani VG, Neven P, Streich
G, Montero AJ, Forget F, Mouret-Reynier MA, Sohn JH, Taylor D,
Harnden KK, et al: Elacestrant (oral selective estrogen receptor
degrader) versus standard endocrine therapy for estrogen
receptor-positive, human epidermal growth factor receptor
2-negative advanced breast cancer: Results from the randomized
phase III EMERALD trial. J Clin Oncol. JCO22003382022.Epub ahead of
print. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Mosele F, Stefanovska B, Lusque A, Tran
Dien A, Garberis I, Droin N, Le Tourneau C, Sablin MP, Lacroix L,
Enrico D, et al: Outcome and molecular landscape of patients with
PIK3CA-mutated metastatic breast cancer. Ann Oncol. 31:377–386.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
André F, Ciruelos EM, Juric D, Loibl S,
Campone M, Mayer IA, Rubovszky G, Yamashita T, Kaufman B, Lu YS, et
al: Alpelisib plus fulvestrant for PIK3CA-mutated, hormone
receptor-positive, human epidermal growth factor
receptor-2-negative advanced breast cancer: Final overall survival
results from SOLAR-1. Ann Oncol. 32:208–217. 2021. View Article : Google Scholar
|
|
84
|
Rugo HS, Lerebours F, Ciruelos E,
Drullinsky P, Borrego MR, Neven P, Park YH, Prat A, Bachelot T,
Juric D, et al: Alpelisib (ALP) + fulvestrant (FUL) in patients
(pts) with PIK3CA-mutated (mut) hormone receptor-positive (HR+),
human epidermal growth factor receptor 2-negative (HER2-) advanced
breast cancer (ABC) previously treated with cyclin-dependent kinase
4/6 inhibitor (CDKi) + aromatase inhibitor (AI): BYLieve study
results. J Clin Oncol. 38(15 Suppl): S10062020. View Article : Google Scholar
|
|
85
|
Bartsch R: ASCO 2020: Highlights in breast
cancer. Memo. 14:58–61. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
André F, Ciruelos E, Rubovszky G, Campone
M, Loibl S, Rugo HS, Iwata H, Conte P, Mayer IA, Kaufman B, et al:
Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced
breast cancer. N Engl J Med. 380:1929–1940. 2019. View Article : Google Scholar
|
|
87
|
Suppan C: Post San Antonio update-my top
three abstracts! Memo-Mag Eur Med Oncol. 14:244–246. 2021.
|
|
88
|
Piccart M, Hortobagyi GN, Campone M,
Pritchard KI, Lebrun F, Ito Y, Noguchi S, Perez A, Rugo HS, Deleu
I, et al: Everolimus plus exemestane for hormone-receptor-positive,
human epidermal growth factor receptor-2-negative advanced breast
cancer: Overall survival results from BOLERO-2†. Ann Oncol.
25:2357–2362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Bardia A, Hurvitz SA, DeMichele A, Clark
AS, Zelnak AB, Yardley DA, Karuturi MS, Sanft TB, Blau S, Hart LL,
et al: Triplet therapy (continuous ribociclib, everolimus,
exemestane) in HR+/HER2-advanced breast cancer postprogression on a
CDK4/6 inhibitor (TRINITI-1): Efficacy, safety, and biomarker
results. J Clin Oncol. 37(15 Suppl): S10162019. View Article : Google Scholar
|
|
90
|
Bardia A, Hurvitz SA, DeMichele A, Clark
AS, Zelnak A, Yardley DA, Karuturi M, Sanft T, Blau S, Hart L, et
al: Phase I/II trial of exemestane, ribociclib, and everolimus in
women with HR+/HER2− advanced breast cancer
after progression on CDK4/6 inhibitors (TRINITI-1). Clin Cancer
Res. 27:4177–4185. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Wander SA, Juric D, Supko JG, Micalizzi
DS, Spring L, Vidula N, Beeler M, Habin KR, Viscosi E, Fitzgerald
DM, et al: Phase Ib trial to evaluate safety and anti-tumor
activity of the AKT inhibitor, ipatasertib, in combination with
endocrine therapy and a CDK4/6 inhibitor for patients with hormone
receptor positive (HR+)/HER2 negative metastatic breast cancer
(MBC) (TAKTIC). J Clin Oncol. 38(15 Suppl): S10662020. View Article : Google Scholar
|
|
92
|
Martin LA and Dowsett M: BCL-2: A new
therapeutic target in estrogen receptor-positive breast cancer?
Cancer Cell. 24:7–9. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Lok SW, Whittle JR, Vaillant F, The CE, Lo
LL, Policheni AN, Bergin ART, Desai J, Ftouni S, Gandolfo LC, et
al: A phase Ib dose-escalation and expansion study of the BCL2
inhibitor venetoclax combined with tamoxifen in ER and
BCL2-positive metastatic breast cancer. Cancer Discov. 9:354–369.
2019. View Article : Google Scholar
|
|
94
|
A phase II study comparing the efficacy of
venetoclax + fulvestrant vs fulvestrant in women with estrogen
receptor-positive, Her2-negative locally advanced or metastatic
breast cancer who experienced disease recurrence or progression
during or after CDK4/6 inhibitor therapy (Veronica).
|
|
95
|
Lindeman GJ, Bowen R, Jerzak KJ, Song X,
Decker T, Boyle FM, McCune SL, Armstrong A, Shannon CM, Bertelli G,
et al: Results from VERONICA: A randomized, phase II study of
second-/third-line venetoclax (VEN) + fulvestrant (F) versus F
alone in estrogen receptor (ER)-positive, HER2-negative, locally
advanced, or metastatic breast cancer (LA/MBC). J Clin Oncol. 39(15
Suppl): pp. S10042021, View Article : Google Scholar
|
|
96
|
Goel S, DeCristo MJ, Watt AC, BrinJones H,
Sceneay J, Li BB, Khan N, Ubellacker JM, Xie S, Metzger-Filho O, et
al: CDK4/6 inhibition triggers anti-tumour immunity. Nature.
548:471–475. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ogata R, Kishino E, Saitoh W, Koike Y and
Kurebayashi J: Resistance to cyclin-dependent kinase (CDK) 4/6
inhibitors confers cross-resistance to other CDK inhibitors but not
to chemotherapeutic agents in breast cancer cells. Breast Cancer.
28:206–215. 2021. View Article : Google Scholar
|
|
98
|
Rugo HS, Cristofanilli M, Loibl S, Harbeck
N, DeMichele A, Iwata H, Park YH, Brufsky A, Theall KP, Huang X, et
al: Prognostic factors for overall survival in patients with
hormone receptor-positive advanced breast cancer: Analyses from
PALOMA-3. Oncologist. 26:e1339–e1346. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Princic N, Aizer A, Tang DH, Smith DM,
Johnson W and Bardia A: Predictors of systemic therapy sequences
following a CDK 4/6 inhibitor-based regimen in post-menopausal
women with hormone receptor positive, HEGFR-2 negative metastatic
breast cancer. Curr Med Res Opin. 35:73–80. 2019. View Article : Google Scholar
|
|
100
|
Kolyadina IV, Bolotina L, Zhukova L,
Vladimirova LU, Sultanbaev A, Karabina E, Ganshina I, Ovchinnikova
E, Kolyadina IV, Antonova G, et al: The effectiveness and safety of
eribulin therapy in HR-positive HER2-negative metastatic breast
cancer post-CDK4/6 inhibitor therapy in Russian clinical practice.
J Clin Oncol. 39(15 Suppl): e130352021. View Article : Google Scholar
|