Introduction
Osteoarthritis (OA) is the most common degenerative
disease affecting the joints worldwide, and frequently affects the
elderly population. The pathological features of OA include the
erosion of articular cartilage, subchondral bone sclerosis and
synovitis (1,2). Although various studies have
reported that sex, age and obesity are the main risks factors for
OA, the pathophysiology of OA has not been fully elucidated to date
(3,4). It has been demonstrated that
inflammatory cytokines play a critical role in the initiation and
progression of OA, including interleukin (IL)-1β, tumor necrosis
factor (TNF), IL-6, etc. (5,6).
Among these, IL-1β is considered the most crucial one (7). The expression level of IL-1β has
been found to be increased in the synovial fluid and cartilage
tissue of patients with OA (8).
IL-1β affects the metabolic process of chondrocytes by increasing
the expression of MMPs, which contributes to cartilage degradation
(9). IL-1β also induces the
production of the inflammatory mediators, nitric oxide (NO) and
prostaglandin E2 (PGE2), which leads to bone resorption and
extracellular matrix (ECM) degradation (10). Therefore, novel effective agents
aiming to suppress IL-1β expression, thus inhibiting IL-1β-induced
MMPs and inflammatory mediators, may attenuate the progression of
OA (11-14). For example, wogonin, a
plant-derived small molecule, has been shown to play a potent
anti-inflammatory and cartilage protective role by activating the
reactive oxygen species (ROS)/ERK/nuclear factor erythroid
2-related factor 2 (NRF2) signaling pathway in chondrocytes of
patients with OA (11).
Stachydrine, a bioactive alkaloid, has been found to attenuate
IL-1β-induced inflammation through the NF-κB signaling pathway in
chondrocytes of patients with OA (14). The NF-κB pathway is known to be
involved in IL-1β-induced inflammation. Following stimulation with
IL-1β, p65 is translocated from the cytoplasm to the nucleus, where
it stimulates the expression of multiple inflammatory or catabolic
genes, including inducible NO synthase (iNOS), TNF-α and MMPs
(15). NF-κB signaling plays a
critical role in the pathogenesis of OA (164). It has been reported
that the activation of the NRF2/heme oxygenase-1 (HO-1) signaling
inhibits the IL-1β-induced activation of NF-κB in chondrocytes
(17). NRF2 is a transcription
factor that regulates the cellular response to oxidative stress and
affects the expression of superoxide dismutase and HO-1 (18). Previous studies have demonstrated
that NRF2 plays a pivotal role in cartilage integrity through
anti-oxidative and anti-apoptotic functions (19,20). In addition, an in vivo
study demonstrated that the deletion of NRF2 led to more severe
damage in cartilage (21).
Therefore, the activation of the NRF2 signaling pathway and the
inhibition of the NF-κB signaling pathway play major
chondroprotective roles in alleviating the symptoms of OA.
Caffeic acid phenethyl ester (CAPE), a natural
flavonoid compound, is one of the major bioactive ingredients of
propolis (22). It is also widely
found in fruits, vegetables and grains, and has been shown to
exhibit multiple health benefits (23). Previous studies have indicated
that CAPE has antioxidant, antitumor, anti-apoptotic,
anti-inflammatory and neuroprotective functions (24-27). CAPE has been reported to be a
potent inhibitor of NF-κB (28).
Liang et al (29) reported
that CAPE suppressed the proliferation and metastasis of
nasopharyngeal carcinoma cells by inhibiting the NF-κB signaling
pathway. Lim et al (30)
reported that CAPE suppressed skin inflammation by inhibiting NF-κB
activation. Furthermore, another study demonstrated that CAPE
decreased ROS levels and exerted a neuroprotective effect in a
mouse model of Alzheimer's disease via the activation of the
NRF2/HO-1 signaling pathway (31). In addition, as previously
demonstrated, CAPE suppressed the inflammation-triggered
myeloperoxidase activity and production of pro-inflammatory
cytokines in colitis (32). These
results confirm the effectiveness and safety of CAPE as an
anti-inflammatory agent. However, the role of CAPE in other
age-related and inflammation-related diseases, such as OA, remains
unclear. Elmali et al (33) reported that CAPE could alleviate
unilateral anterior cruciate ligament transection-induced cartilage
destruction. However, the chondroprotective effects of CAPE and its
underlying mechanisms remain elusive. Therefore, the present study
investigated the anti-inflammatory effects and the underlying
mechanisms of action of CAPE in the treatment of OA.
Materials and methods
Reagents
CAPE was purchased from Shanghai Aladdin Biochemical
Technology Co., Ltd. IL-1β was purchased from ProteinTech Group,
Inc. The Cell Counting Kit-8 (CCK-8) was purchased from Beyotime
Institute of Biotechnology. Griess Reagent Nitrite Measurement kit
(cat. no. 13547) was purchased from Cell Signaling Technology, Inc.
The ELISA kits for human PGE2 (cat. no. SEKH-0414), Toluidine Blue
solution and bovine serum albumin (BSA) were purchased from Beijing
Solarbio Science & Technology Co., Ltd.
Penicillin/streptomycin, fetal bovine serum (FBS) and DMEM were
purchased from Gibco (Thermo Fisher Scientific, Inc.).
Primary chondrocyte isolation and
culture
Human chondrocytes were isolated from human
articular cartilage tissues that were procured from patients with
femoral neck fracture. The collection of cartilage tissues from
patients was approved by the Ethics Committee of Shenzhen Second
People's Hospital (Shenzhen, China). Patients with an average age
of 64.8 years (aged 60-68 years, two males and three females) who
were subjected to total knee replacement surgery at the Shenzhen
Second People's Hospital from January to March, 2021 participated
in the study. All patients provided written informed consent. The
articular cartilage tissues were washed with PBS and cut into 1
mm3 pieces. The sections were then cultured with
collagenase II (2 g/l) for 4 h at 37°C (31). Upon centrifugation at 108 × g for
5 min at 24°C, the cells were cultured in DMEM with 10% (v/v) FBS
and 1% (v/v) antibiotics (penicillin/streptomycin) in an atmosphere
containing 5% CO2 at 37°C. The culture medium was
changed every other day. Only cells from the first three passages
were used in the present study. Chondrocyte morphology was examined
using toluidine blue staining. In brief, the culture medium was
aspirated and washed twice with PBS. Toluidine blue solution for 5
min at room temperature, and the same amount of distilled water was
added and mixed evenly. Images were acquired using an Olympus
microscope (Olympus Corporation) after standing for 15 min at room
temperature.
Animal models
Sprague-Dawley male wild-type rats (n=12; 8 weeks
old; weighing 300-350 g) were acquired from Cloud-Clone Corp. The
rats were maintained in a clean environment at 27°C under a 12-h
light/dark cycle with 50% humidity, and had free access to adequate
food and water. The animal protocols and the experimental
procedures were in agreement with the Animal Care and Use Committee
of Southwest Medical University (approval no. 20211124-043). The
model of OA was established by the destabilization of the medial
meniscus (DMM) through surgical treatment as previously described
(32). Briefly, the rats were
anesthetized with 2% (w/v) pentobarbital (40 mg/kg) via
intraperitoneal injection. The capsule of the right knee joint was
opened though the medial patellar tendon and the medial meniscus
tibial ligament was cut using microsurgical scissors. The medial
meniscus tibial ligament was not cut in the sham-operated control
group. In the present study, the rats were randomly divided into
three groups as follows: The sham-operated control group (sham),
the OA group (DMM) and the CAPE-treated OA group (DMM + CAPE). Rats
in the CAPE treatment group were administered 10 mg/kg/2 days CAPE,
for 8 weeks. At the end of the experiment, all animals were
sacrificed by cervical dislocation under anesthesia through an
intraperitoneal injection of pentobarbital sodium (50 mg/kg), and
respiratory arrest was used to confirm animal death. During the
course of the experiment, any rats that were unable to eat or
drink, had difficulty breathing and had lost 20% of their body
weight before the experiment, were regarded as having reached the
humane endpoint and were thus immediately euthanized. None of the
rats reached the aforementioned end points in this experiment and
thus none were euthanized before the end of the experiment.
Cell transfection with NRF2
small-interfering RNA (siRNA)
Negative control (NC) siRNA and NRF2 siRNA were
purchased from Shanghai GenePharma Co., Ltd. Chondrocytes were
plated and cultured in six-well plates for 24 h, and then
transfected with 80 nM NC and NRF2 siRNA for 48 h using
Lipofectamine® 2000 siRNA transfection reagent (Thermo
Fisher Scientific, Inc.). The sequences of the NRF2 and NC siRNAs
were as follows: 1#NRF2 siRNA: sense, 5′-GGG AGG AGC UAU UAU CCA
UTT-3′ and antisense, 5′-AUG GAU AAU AGC UCC UCC CTT-3′; and 2#NRF2
siRNA sense, 5′-GCC CAU UGA UGU UUC UGA UTT-3′ and antisense,
5′-AUC AGA AAC AUC AAU GGG CTT-3′; and 3#NRF2 siRNA sense, 5′-GCC
UGU AAG UCC UGG UCA UTT-3′ and antisense, 5′-AUG ACC AGG ACU UAC
AGG CTT-3′; and NC siRNA sense, 5′-UUC UCC GAA CGU GUC ACG UTT-3′
and antisense, 5′-ACG UGA CAC GUU CGG AGA ATT-3′; NC FAM-siRNA
sense, 5′-UUC UCC GAA CGU GUC AC GUT T-3′ and antisense, 5′-ACG UGA
CAC GUU CGG AGA ATT-3′.
CCK-8 assay
The cytotoxicity of CAPE on human chondrocytes was
evaluated using CCK-8 assay. In brief, chondrocytes were plated
into 96-well plates in serum-free medium for 24 h, and then treated
with various concentrations of CAPE (0, 1, 3, 5, 10, 20 and 40
µM) for 24 h. The chondrocytes were then washed with PBS,
and 100 µl DMEM containing 10 µl CCK-8 solution was
added to each well, followed by incubation at 37°C for 3 h. The
optical density value of the wells was measured at a wavelength of
450 nm using a microplate reader (Thermo Fisher Scientific,
Inc.).
Griess reaction and ELISA
Chondrocytes were plated and cultured in 6-well
plates, and pre-treated with CAPE (0, 10 or 20 µM). After 24
h, IL-1β (10 ng/ml) was added, followed by additional 24 h of
incubation at 37°C. The content of NO and PGE2 in each well was
detected using the Griess reaction and ELISA kits, respectively,
according to the manufacturer's protocols.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the chondrocytes using
TRIzol® reagent (Takara Bio, Inc.), and 1 µg
total RNA was used to synthesize cDNA using the PrimeScript™ RT
reagent kit with gDNA Eraser (Takara Bio, Inc.), and the conditions
were as follow: 15 min at 37°C, followed by 5 sec at 85°C. The
total volume of q-PCR was 10 µl, including 5 µl 2X
SYBR Master Mix (Beyotime Institute of Biotechnology), 0.25
µl each primer and 4.5 µl diluted cDNA. qPCR was
conducted in a CFX96Real-Time PCR System, and the thermocycling
conditions were as follows: 10 min at 95°C, followed by 40 cycles
of 15 sec at 95°C and 1 min at 60°C. GAPDH was used as the internal
control for normalization. The relative mRNA of the target genes
was calculated using the 2−ΔΔCq method (36). The primer sequences of the target
genes are presented in Table
I.
 | Table IPrimer sequences used in reverse
transcription-quantitative PCR. |
Table I
Primer sequences used in reverse
transcription-quantitative PCR.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| iNOS |
GACGAGACGGATAGGCAGAG′ |
CACATGCAAGGAAGGGAACT |
| COX-2 |
GAGAGATGTATCCTCCCACAGTCA |
GACCAGGCACCAGACCAAAG |
| MMP3 |
TGCGTGGCAGTTTGCTCAGCC |
GAATGTGAGTGGAGTCACCTC |
| MMP13 |
GGCTCCGAGAAATGCAGTCTTTCTT | ATCAAATGGGTAGAAG
TCGCCATGC |
| Adamts5 |
GGTCAAGGTCCCATGTGCAAC |
GAATGCGGCCATCTTGTCATC |
|
Aggrecan |
AGGATGGCTTCCACCAGTGT |
GGCATAAAAGACCTCACCCTCC |
| Collagen
II |
CTGTCCTTCGGTGTCAGGG |
CGGCTTCCACACATCCTTAT |
| GAPDH |
GGAGCGAGATCCCTCCAAAAT |
GGCTGTTGTCATACTTCTCATGG |
Western blot analysis
The cells were lysed by radio immunoprecipitation
assay (RIPA) buffer (Thermo Fisher Scientific, Inc.) containing 1
mM PMSF to extract total protein. The protein concentration was
detected with a BCA protein assay kit (Beyotime Institute of
Biotechnology). Subsequently, 40 µg denatured protein
solution were separated by 12% SDS-PAGE (Wuhan Servicebio
Technology Co., Ltd.) and transferred to a PVDF membrane (Merck
KGaA), which was incubated in methanol for 1 min at room
temperature. The methanol was then removed and the membrane was
equilibrated in transfer buffer until ready to use. Upon transfer
at 100 V for 1 h at 4°C, the membrane was blocked with 5% skimmed
milk for 1 h at room temperature and then incubated with primary
antibodies against GADPH (1:1,000; cat. no. AP0066; Bioworld
Technology, Inc.), MMP3 (1:5,000; cat. no. 66338-1-Ig; ProteinTech
Group, Inc.), MMP13 (1:1,000; cat. no. 18165-1-AP; ProteinTech
Group, Inc.), a disintegrin and metalloproteinase with
thrombospondin motif-5 (Adamts5; 1:500; cat. no. A2836; ABclonal
Biotech Co., Ltd.), iNOS (1:500; cat. no. 18985-1-AP; ProteinTech
Group, Inc.), cyclooxygenase-2 (COX-2; 1:500; cat. no. 12375-1-AP;
ProteinTech Group, Inc.), lamin B1 (1:1,000; cat. no. AF1408;
Beyotime Institute of Biotechnology), NRF2 (1:2,000; cat. No.
16396-1-AP; ProteinTech Group, Inc.), HO-1 (1:1,000; cat. no.
10701-1-AP; ProteinTech Group, Inc.), p65 (1:1,000; cat. no. 8242;
Cell Signaling Technology Inc.), phosphorylated (p)-p65 (1:1,000;
cat. no. 3033; Cell Signaling Technology Inc.), IκBα (1:1,000, cat.
no. 4814; Cell Signaling Technology Inc.), p-IκBα (1:1,000, cat.
no. 2859; Cell Signaling Technology Inc.) and collagen II (1:500;
cat. no. PA5-99159, Thermo Fisher Scientific, Inc.) overnight at
4°C. Subsequently, the membranes were incubated with a
HRP-conjugated goat anti-rabbit (1:1,000; cat. no. A0208; Beyotime
Institute of Biotechnology Inc.) or goat anti-mouse IgG (1:1,000;
cat. no. A0216; Beyotime Institute of Biotechnology Inc.) secondary
antibody for 2 h at room temperature. Finally, an ECL reagent (cat.
no. 34580; ThermoFisher Scientific, Inc.) was used to detect the
blots. The ECL signal was captured using Odyssey FC Imager (Gene
Company, Ltd.), and GAPDH and Lamin B1 were used as loading
controls for normalization. Quantitative analysis was performed
using ImageJ 1.53e software (National Institutes of Health).
Immunofluorescence
Chondrocytes were seeded in a glass plate, cultured
for 12 h, pre-treated with or without CAPE (20 µM) for 24 h,
and then co-incubated with or without IL-1β (10 ng/ml) for 24 h.
Subsequently, the cells were washed with PBS and fixed with 4%
paraformaldehyde for 15 min. The cells were then treated with 0.1%
TritonX-100 for 5 min and blocked with 5% BSA for 1 h at room
temperature. The cells were then incubated with primary antibodies
against collagen II (1:100; cat. no. PA5-99159, Thermo Fisher
Scientific, Inc.), MMP3 (1:100; cat. no. 66338-1-Ig; ProteinTech
Group, Inc.), p65 (1:400; cat. no. 8242; Cell Signaling Technology
Inc.) and NRF2 (1:500; cat. no. 16396-1-AP; ProteinTech Group,
Inc.) overnight at 4°C. The following day, the cells were incubated
with Alexa Fluor® 647-conjugated goat anti-rabbit IgG
(1:500; cat. no. A0468; Beyotime Institute of Biotechnology) and
Alexa Fluor® 647-conjugated goat anti-mouse IgG
secondary antibodies (1:500; cat. no. A0473; Beyotime Institute of
Biotechnology) for 1 h at room temperature in the dark and exposed
to DAPI (Beyotime Institute of Biotechnology) for 5 min at room
temperature. Finally, the cells were observed under a confocal
microscope (ZEISS GmbH) and analyzed using ImageJ 1.53e
software.
Molecular docking of CAPE with the
Kelch-like ECH-associated protein 1 (Keap1)-NRF2 complex
The structure data file (SDF) of CAPE (Pubchem ID:
5281787) was downloaded from the PubChem website (https://pubchem.ncbi.nlm.nih.gov/) and converted
to PDB format on OpenBabel 3.1.1. The structure of Keap1-NRF2
complex was downloaded from the RCSB Protein Data Bank (https://www.rcsb.org/). AutoDockTools1.5.7 software
was used to modify the receptor protein, such as water removal and
hydrogenation, and also to modify ligand small molecule, such as
hydrogenation. Next, the molecular docking analysis of receptor
protein and ligand small molecule was performed using
AutoDockTools. Eventually, ligand binding flexibility with the
binding pocket residues were drawn using PYMOL 2.5.2.
Histopathological analysis
The knee joint tissue was fixed with 4%
paraformaldehyde for 24 h at 4°C and then decalcified with 10% EDTA
solution for 2 weeks. The tissue was then dehydrated, cleared,
embedded in paraffin blocks and sliced to obtain frontal serial
sections at a thickness of 5 µm. The slides were then
stained with Safranin O/Fast Green (S/O) (cat. no. G1053; Beijing
Solarbio Science & Technology Co., Ltd.). In brief, the slides
were strained with Fast Green for 2 min, washed with water, and
soaked in 1% hydrochloric acid and alcohol for 10 sec at room
temperature. Then strained with Safranin O for 5 sec, rapid
dehydrated in absolute ethanol for 5 sec, 2 sec and 10 sec, and
sealed with neutral resin (Sinopharm Group Chemical Reagent Co.,
Ltd.) at room temperature. The extent of cartilage degeneration was
assessed using a Nikon E100 microscope (Nikon Corporation) and
evaluated using the Osteoarthritis Research Society International
(OARSI) scoring system as described previously (37).
Immunohistochemical assay
Immunohistochemical assay was performed as
previously described (38).
Briefly, the slides were deparaffinized and rehydrated. For antigen
repair, the slides were incubated with 0.4% pepsin (Sangon Biotech
Co., Ltd.) in 5 mM HCl at 37°C for 20 min, and then blocked with 5%
BSA for 30 min. The slides were then incubated with primary
antibodies against collagen Ⅱ (1:50; cat. no. PA5-99159, Thermo
Fisher Scientific, Inc.), MMP3 (1:500; cat. no. 66338-1-Ig;
ProteinTech Group, Inc.), MMP13 (1:500; cat. no. 18165-1-AP;
ProteinTech Group, Inc.) and NRF2 (1:500; cat. no. 16396-1-AP;
ProteinTech Group, Inc.) overnight at 4°C. Finally, the slides were
incubated with HRP-conjugated goat anti-rabbit (1:50; cat. no.
A0208; Beyotime Institute of Biotechnology, Inc.) or goat
anti-mouse IgG (1:50; cat. no. A0216; Beyotime Institute of
Biotechnology, Inc.) secondary antibody for 50 min at room
temperature, and images were captured using an Olympus BX51
microscope (Olympus Corporation).
Statistical analysis
Data are expressed as the mean ± SD. All experiments
were repeated at least three times. Statistical analyses were
performed using GraphPad Prism version 8.0 software (GraphPad
Software, Inc.). Comparisons between two groups were performed
using an unpaired two-tailed Student's t-test. Comparisons among
multiple groups were done using one-way ANOVA test followed by a
post-hoc Test (Bonferroni). A value of P<0.05 was considered to
indicate a statistically significant difference.
Results
Cytotoxic effects of CAPE on human
chondrocytes
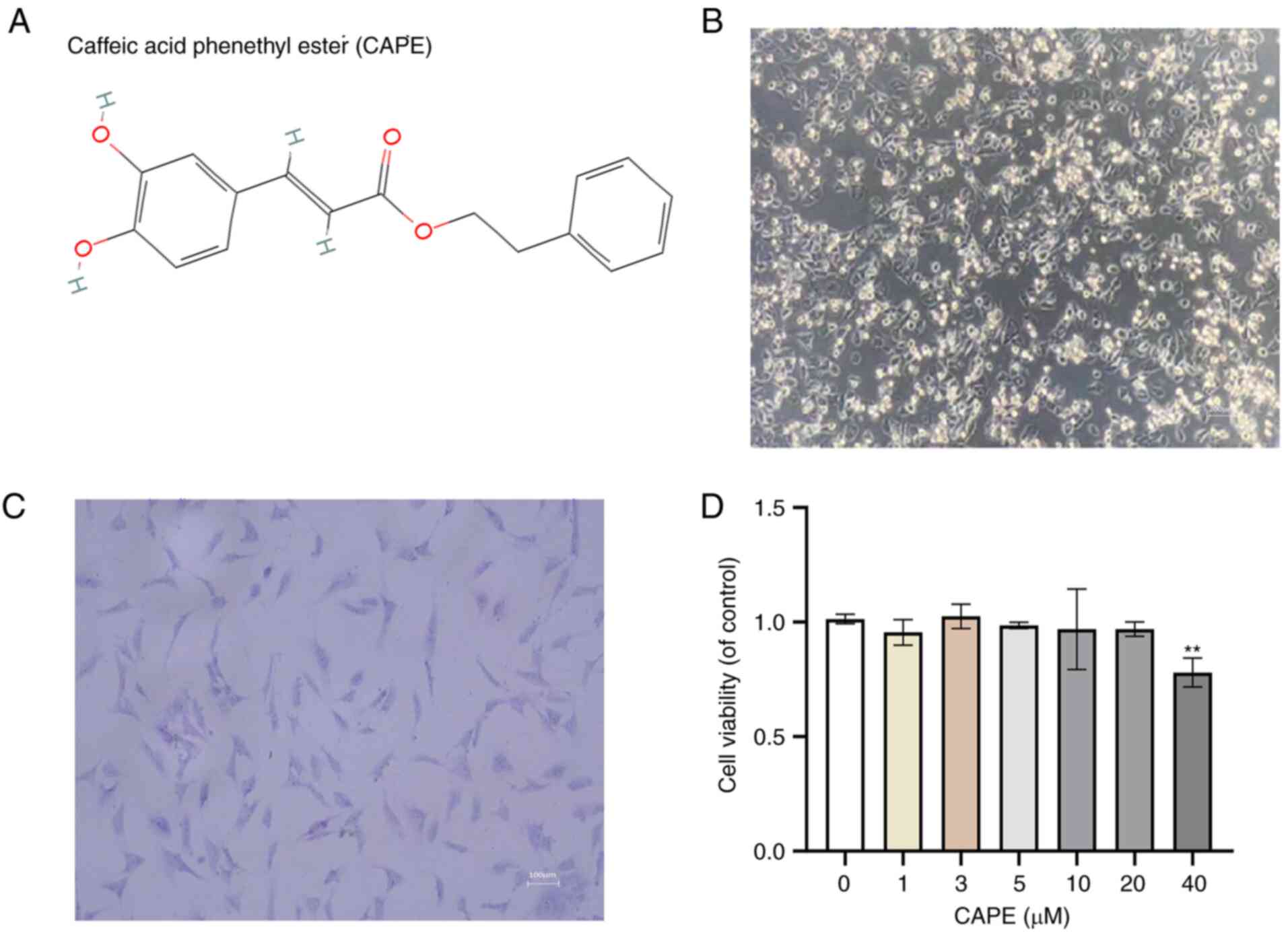
The molecular chemical structure of CAPE is
illustrated in Fig. 1A, and the
morphology of the human chondrocytes are illustrated in Fig. 1B and C, respectively. To measure
the cytotoxic effect of CAPE on human chondrocytes, the
chondrocytes were treated with various concentrations of CAPE (0,
1, 3, 5, 10, 20 and 40 µmol/l) for 24 h. CAPE cytotoxicity
was detected using CCK-8 assay. The results revealed that there was
no obvious cytotoxic effect of CAPE on human chondrocytes when the
cells were incubated with 1, 3, 5, 10 or 20 µM CAPE
(Fig. 1D). However, the viability
of the chondrocytes was significantly inhibited following treatment
with 40 µM CAPE (Fig. 1D).
Therefore, CAPE was used at 10 and 20 µM in the subsequent
experiments.
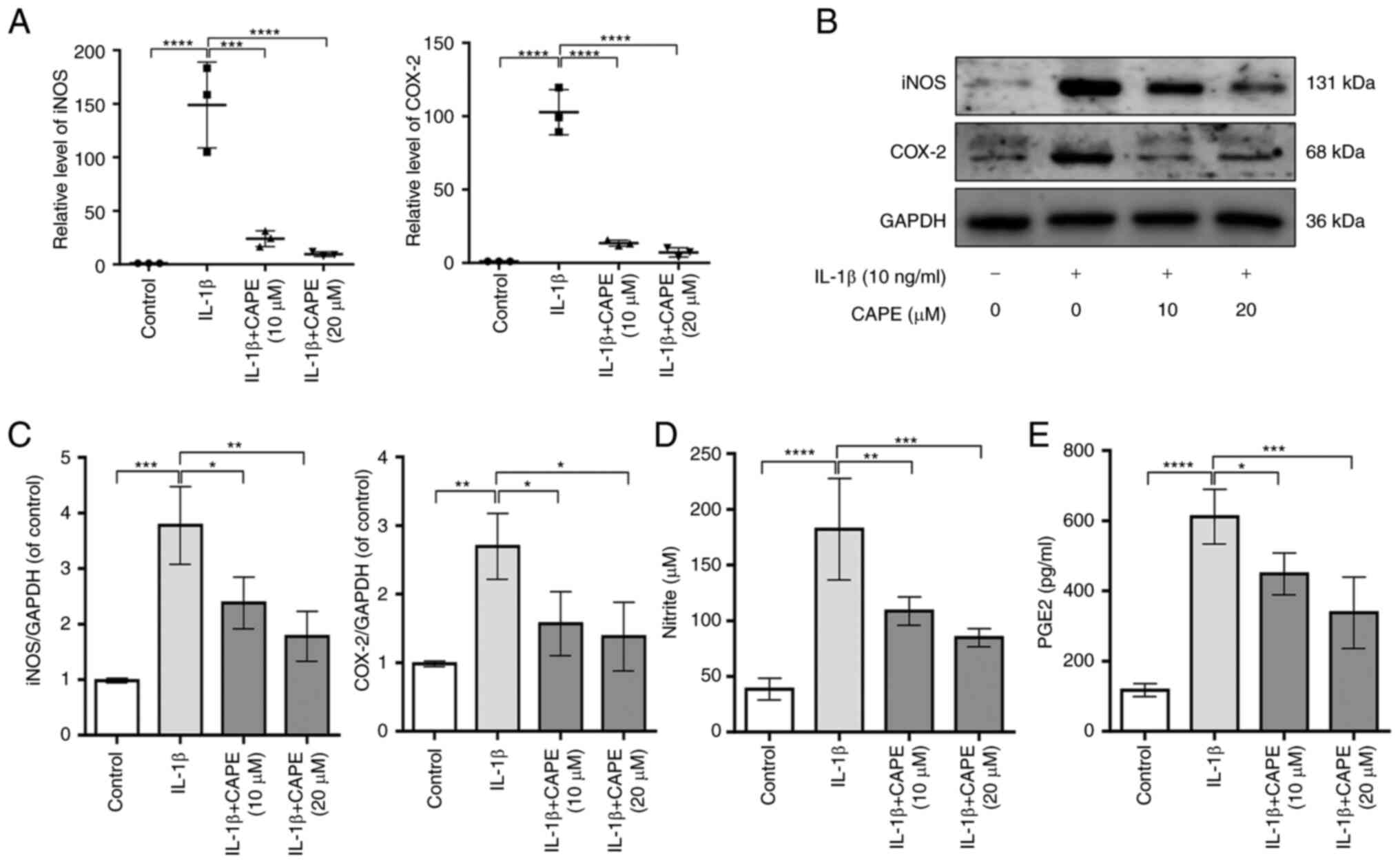
CAPE blocks the levels of IL-1β-induced
inflammatory mediators in human chondrocytes
To examine the effects of CAPE on the IL-1β-induced
inflammation of chondrocytes, the expression and production levels
of COX-2, iNOS, NO and PGE2 were measured using RT-qPCR, western
blot analysis and ELISA. The results revealed that CAPE at the
concentrations of 10 and 20 µM inhibited the expression of
COX-2 and iNOS stimulated by IL-1β at the protein and mRNA level
(Fig. 2A-C). In addition, the
results of Griess reaction and ELISA revealed that CAPE at
concentrations of 10 and 20 µM suppressed the production of
NO and PGE2 stimulated by IL-1β (Fig.
2D and E). Therefore, these results indicated that CAPE
inhibited the expression and production of IL-1β-induced
inflammatory mediators in human chondrocytes, particularly at the
concentrations of 10 and 20 µM.
CAPE attenuates the IL-1β-induced
degradation of the ECM in human chondrocytes
To determine the effects of CAPE on IL-1β-induced
ECM synthesis and degradation in chondrocytes, the effects of CAPE
on the expression of collagen II, aggrecan, MMP3, MMP13 and Adamts5
were examined using RT-qPCR, western blot analysis and
immunofluorescence. The results revealed that the mRNA expression
levels of collagen II and aggrecan were increased by CAPE at 10 and
20 µM under IL-1β stimulation, while the mRNA expression
levels of MMP3, MMP13 and Adamts5 were inhibited (Fig. 3A). In addition, the protein level
of collagen II was increased by CAPE at 10 and 20 µM
following IL-1β stimulation, while the protein levels of MMP3,
MMP13 and Adamts5 were inhibited (Fig. 3B). The immunofluorescence staining
of collagen II and MMP3 revealed that CAPE activated collagen II
expression and inhibited MMP3 expression following IL-1β
stimulation (Fig. 3C and D).
Therefore, these data indicated that CAPE attenuated the
IL-1β-induced ECM degradation in human chondrocytes by preventing
the degradation of collagen II and the production of ECM
degradative enzymes.
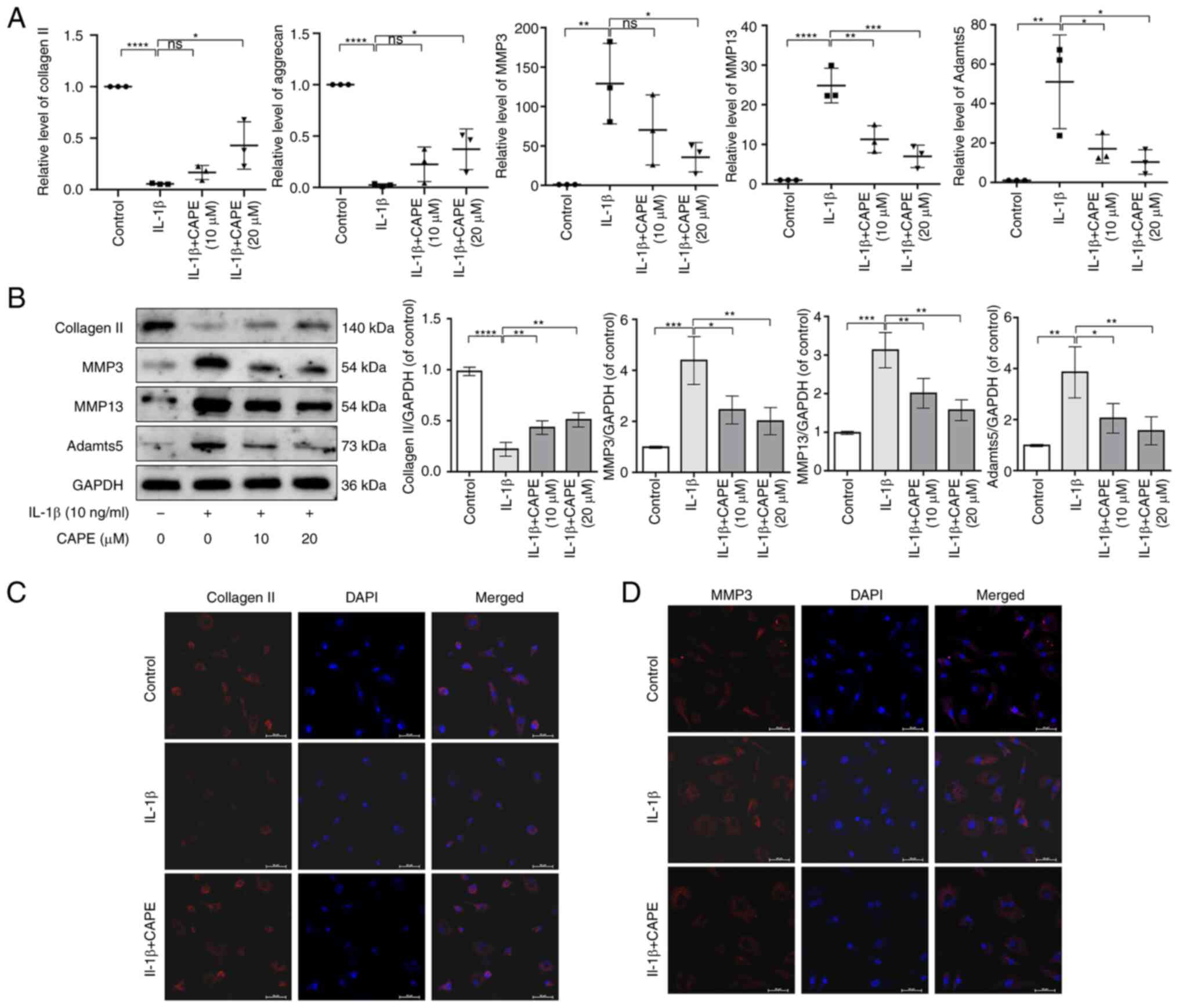 | Figure 3CAPE attenuates the IL-1β-induced
extracellular matrix degradation in human chondrocytes. (A) The
mRNA expression of collagen II, aggrecan, MMP3, MMP13 and Adamts5
was detected using reverse transcription-quantitative PCR. (B) The
protein expression of collagen II, MMP3, MMP13 and Adamts5 was
detected using western blot analysis and quantified. (C) Collagen
II and (D) MMP3 expression was detected by immunofluorescence.
Scale bar, 50 µm. *P<0.05,
**P<0.01, ***P<0.001 and
****P<0.0001. CAPE, caffeic acid phenethyl ester;
Adamts5, a disintegrin and metalloproteinase with thrombospondin
motif-5. |
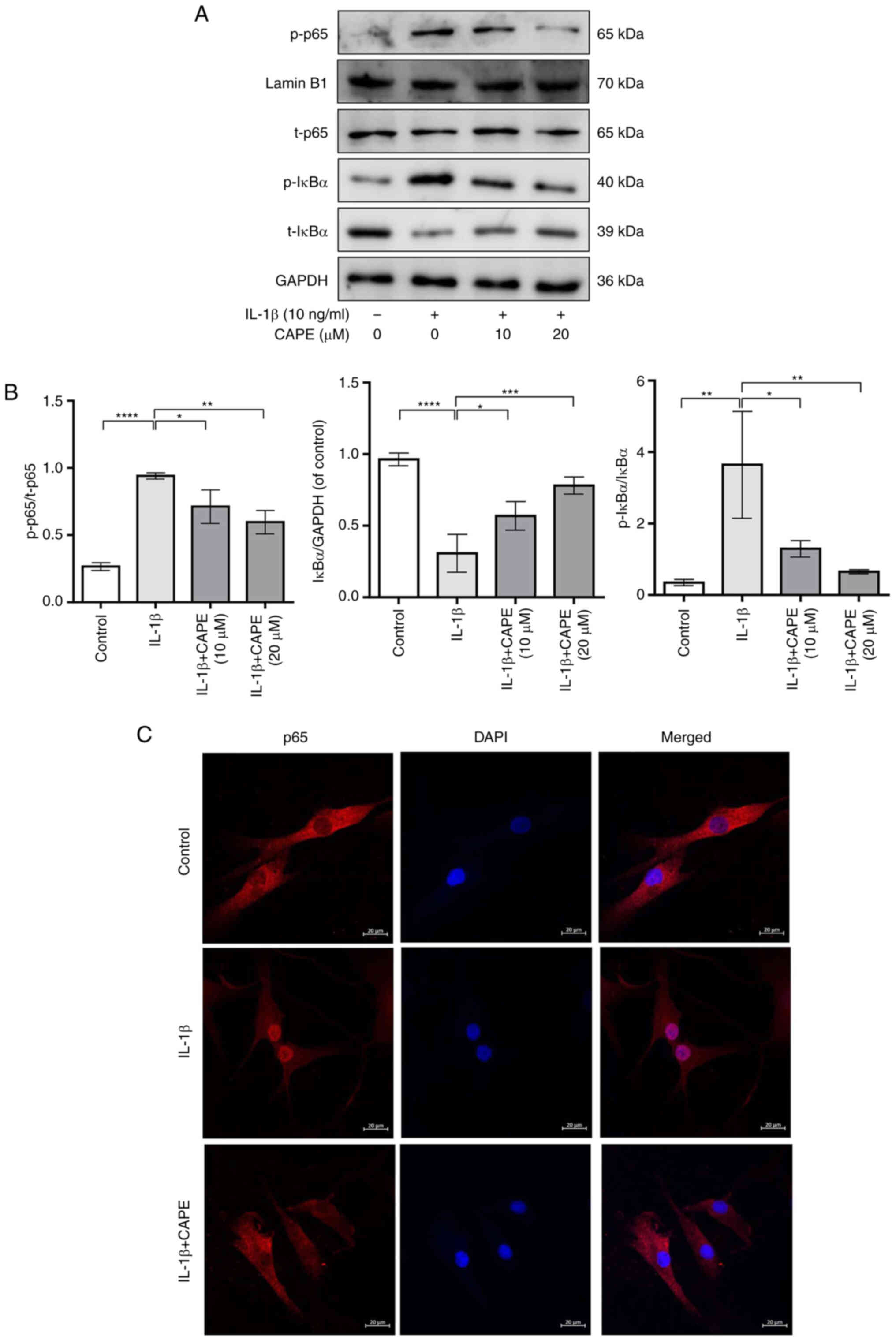
CAPE prevents IL-1β-induced NF-κB
activation in human chondrocytes
To explore the anti-inflammatory mechanisms of CAPE
in IL-1β-stimulated chondrocytes, the expression of p65, p-p65,
IκBα and p-IκBα was detected using western blot analysis and
immunofluorescence. The results revealed that the phosphorylation
of p65 was significantly increased by IL-1β stimulation, while it
was significantly inhibited by pre-treatment with CAPE (Fig. 4A and B). Additionally,
pre-treatment with CAPE inhibited the protein level of IkBα and the
phosphorylation level of IkBα in IL-1β-stimulated chondrocytes
(Fig. 4A and B). The
immunofluorescence staining of p65 revealed that pre-treatment with
CAPE decreased the expression of p65 in the nucleus of
IL-1β-stimulated chondrocytes (Fig.
4C). Therefore, these data suggested that CAPE prevented the
IL-1β-induced NF-κB activation by inhibiting the translocation of
p65 and the degradation of IκBα in human chondrocytes.
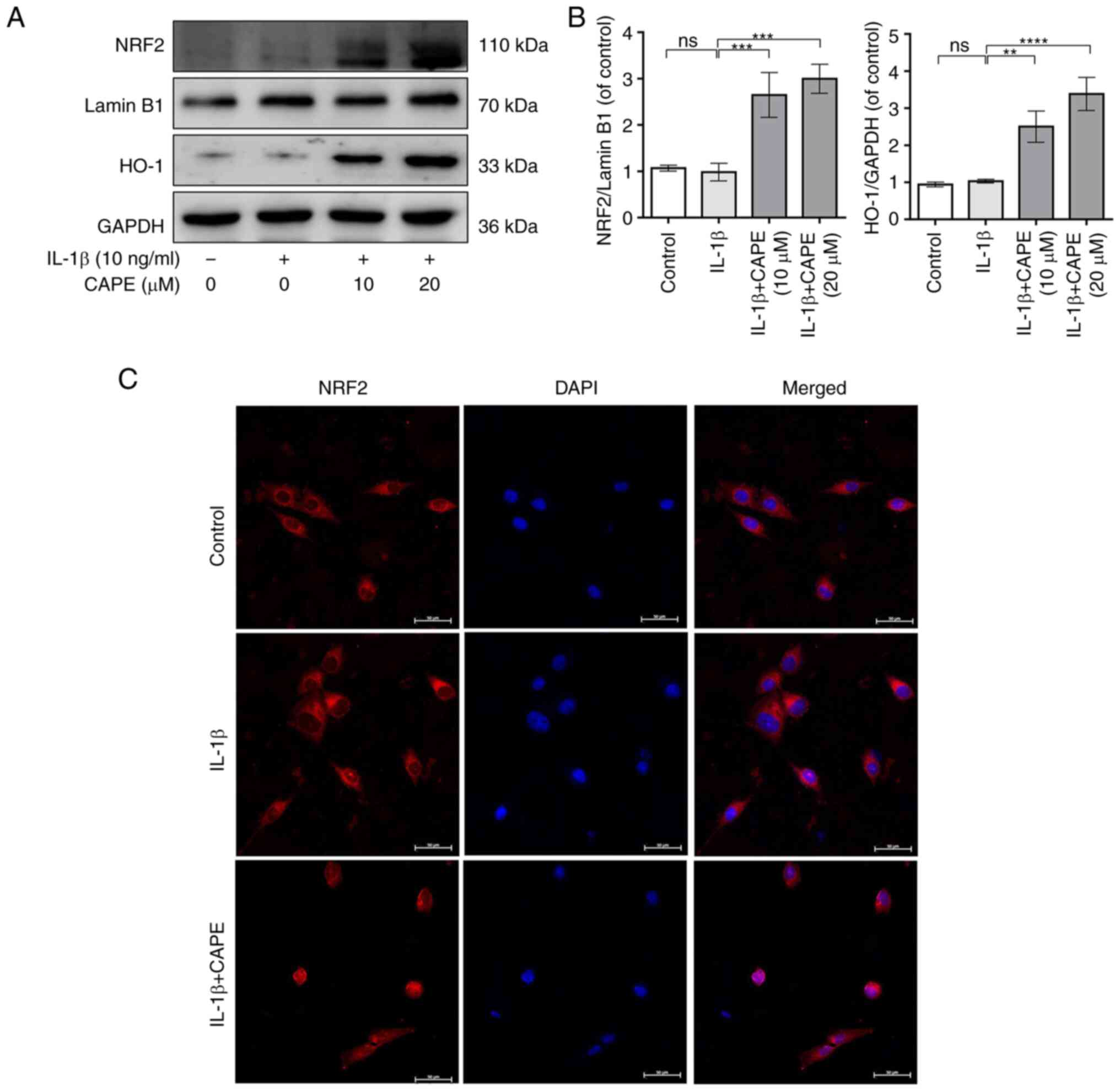
CAPE upregulates the NRF2/HO-1 signaling
pathway in IL-1β-stimulated human chondrocytes
To investigate the role of the NRF2/HO-1 signaling
pathway in the anti-inflammatory effects of CAPE, the expression of
NRF2 and HO-1 in IL-1β-stimulated human chondrocytes was detected
using western blot analysis and immunofluorescence. The results
revealed that CAPE increased the expression of NRF2 in a
concentration-dependent manner (Fig.
S1). Additionally, the results revealed that the protein levels
of NRF2 and HO-1 were increased by 10 and 20 µM CAPE under
IL-1β stimulation (Fig. 5A and
B). The immunofluorescence staining of NRF2 revealed that
pre-treatment with CAPE increased the expression of NRF2 in the
nuclei of IL-1β-stimulated chondrocytes (Fig. 5C). Therefore, these data suggested
that CAPE exerted anti-inflammatory effects in IL-1β-stimulated
chondrocytes through the activation of the NRF2/HO-1signaling
pathway.
To further confirm that the anti-inflammatory
effects of CAPE are mediated via the NRF2/HO-1 signaling pathway in
IL-1β-stimulated chondrocytes, NRF2 siRNA was transfected into
chondrocytes to silence the expression of NRF2 (Fig. S2). The results revealed that the
silencing of NRF2 inhibited the protein levels of NRF2 and HO-1
which had been increased by pre-treatment with CAPE in the
IL-1β-stimulated chondrocytes, which suggested that the
CAPE-induced activation of the NRF2/HO-1 signaling activation was
inhibited by the silencing of NRF2 (Fig. 6A and B). The silencing of NRF2
increased the phosphorylation levels of p65 in the nuclei of
IL-1β-stimulated chondrocytes which had been decreased by
pre-treatment with CAPE; this suggested that the CAPE-mediated
suppression of NF-κB signaling was abolished by the silencing of
NRF2 (Fig. 6A and B).
Furthermore, downregulation of NRF2 could inhibit the protein
levels of iNOS and COX-2 by pre-treatment with CAPE in
IL-1β-induced chondrocytes, which suggested that the CAPE-mediated
suppression of inflammation was abolished by downregulation of NRF2
(Fig 6A and B). In addition, the
silencing of NRF2 in IL-1β-stimulated chondrocytes increased the
protein levels of MMP3, MMP13 and Adamts5, whereas it decreased the
protein levels of collagen II upon pre-treatment with CAPE
(Fig. 6C and D). Therefore, these
data indicated that CAPE regulated NF-κB signaling and the
inflammatory response though the NRF2/HO-1 signaling pathway in
IL-1β-stimulated chondrocytes.
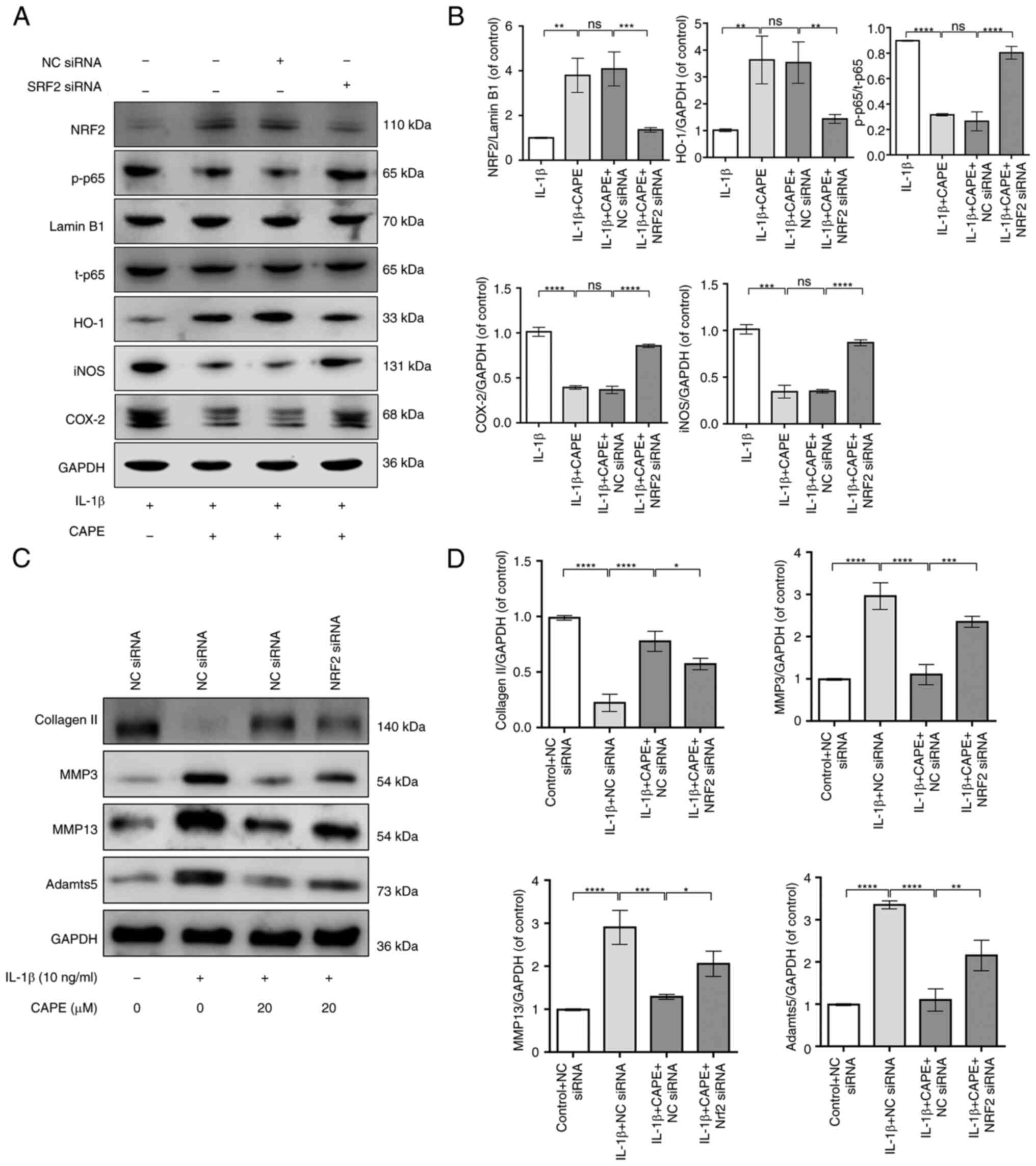 | Figure 6Downregulation of NRF2 inhibits the
anti-inflammatory effects of CAPE on human chondrocytes. The
protein expression of NRF2, HO-1, p-p65, iNOS and COX-2 was (A)
detected using western blot analysis and (B) quantified. (C) The
protein expression of collagen II, MMP3, MMP13 and Adamts5 was (C)
detected using western blotting and (D) quantified.
*P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. CAPE,
caffeic acid phenethyl ester; NRF2, nuclear factor erythroid
2-related factor 2; HO-1, heme oxygenase-1; iNOS, inducible nitric
oxide synthase; COX-2, cyclooxygenase 2. |
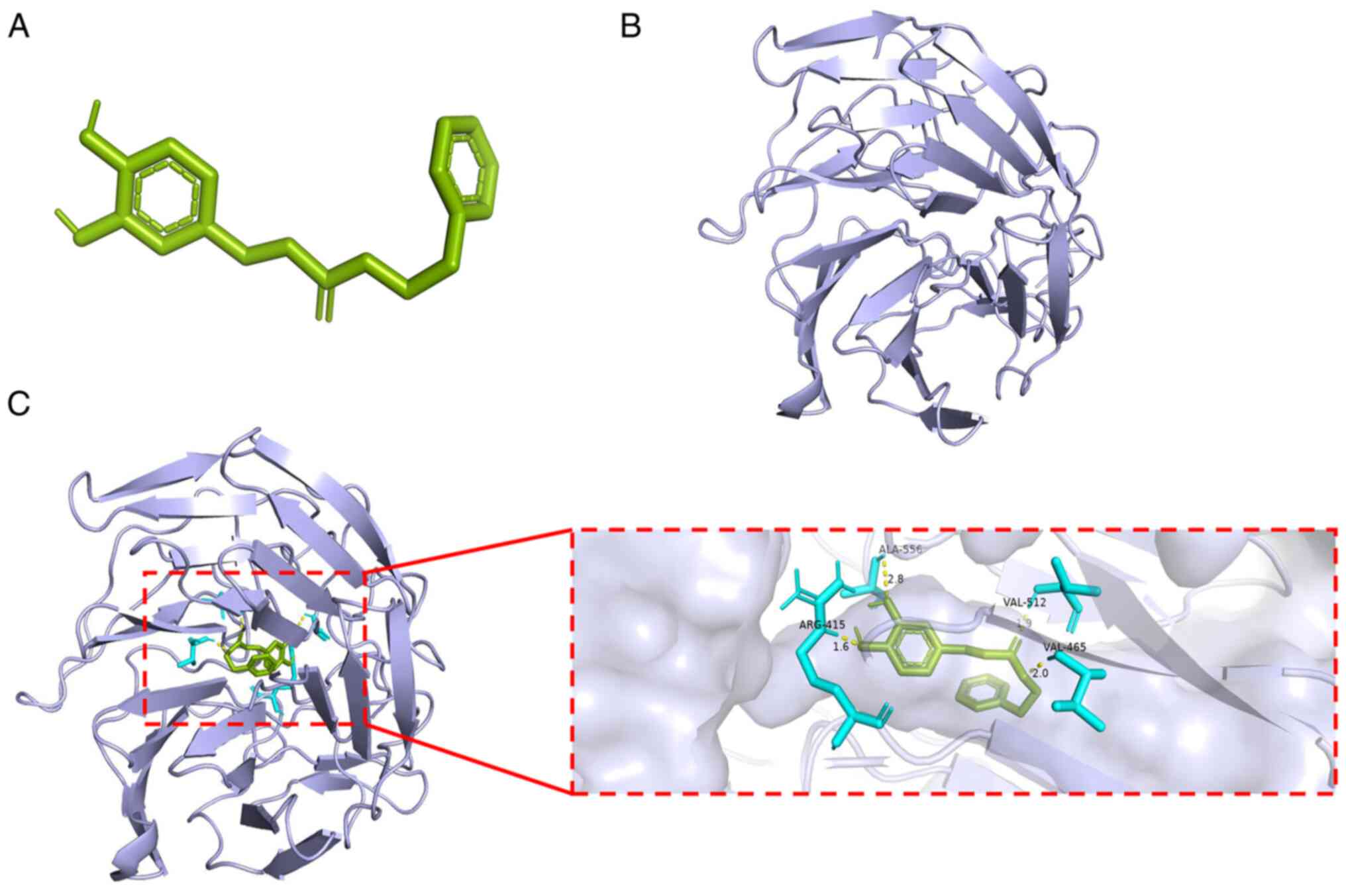
Molecular docking between CAPE and the
Keap1-NRF2 complex
In order to evaluate whether there is any affinity
between CAPE and Keap1-NRF2 complex, computational molecular
docking analysis was performed. The structures of CAPE and the
Keap1-NRF2 complex (PDB ID: 2FLU) were obtained and analyzed
(Fig. 7A and B). It was found
that CAPE interacted with and docked at the Keap1-NRF2 complex
binding site. High-affinity (-7.39 kcal/mol) hydrogen binding
events were observed between the residues of Arg415, Val465 and
Val512 in CAPE and Keap1-NRF2 complex (Fig. 7C). Therefore, these results
indicated that CAPE probably inhibited the development of OA by
interacting with the Keap1-NRF2 complex, inhibiting ubiquitination
and promoting nuclear translocation of NRF2.
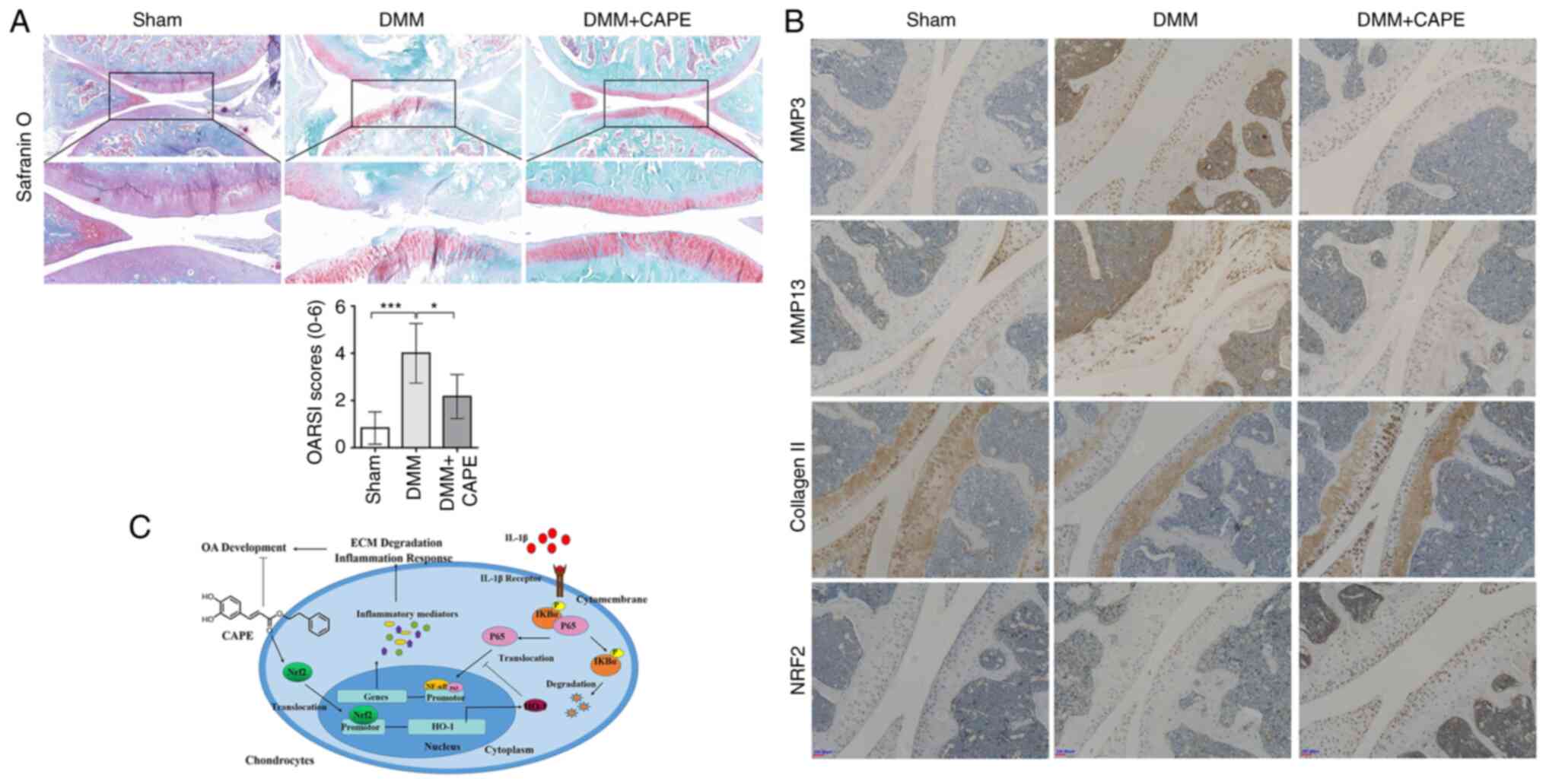
CAPE attenuates OA progression in a rat
model of DMM
To examine the effects of CAPE on OA progression
in vivo, a model of OA was established using rats, followed
by an intra-peritoneal injection of 10 mg/kg CAPE. S/O staining
revealed that the articular cartilage exhibited a normal, red-dyed
area and a smooth surface in the sham control group, whereas the
thickness of the articular cartilage was significantly reduced in
the DMM group (Fig. 8A). Compared
with that in the DMM group, the red-dyed area was thicker and the
surface of articular cartilage was smoother in the CAPE-treated
group (Fig. 8A), which suggested
that CAPE played a role in attenuating the degradation of cartilage
matrix. The immunohistochemistry of MMP3, MMP13, collagen Ⅱ and
NRF2 was also performed in the OA model. In the DMM group, the
expression of MMP3 and MMP13 was markedly increased compared with
that in the sham group, while the DMM + CAPE group exhibited a
decreased expression of MMP3 and MMP13 (Fig. 8B). In the DMM group, the
expression of collagen Ⅱ was markedly decreased compared with that
in the sham group, while the DMM + CAPE group exhibited a high
expression (Fig. 8B). In the DMM
group, the expression of NRF2 was not markedly different from that
in the sham group, while the DMM + CAPE group showed increased
expression in the cell nuclei (Fig.
8B).
Discussion
OA is a common degenerative disease affecting the
joints; however, there are currently no effective therapeutic drugs
for the clinical treatment of OA (39). Currently, OA is considered to be a
chronic and low-grade inflammatory disease. A better understanding
of the inflammatory pathophysiology of OA may lead to the
identification of novel potential therapeutic drugs (40). Natural products have already
exhibited potential for use in the treatment of OA by inhibiting
inflammatory processes (41).
CAPE, a natural polyphenolic product, is extracted from the bark of
conifer trees and propolis (22).
It has been reported that CAPE has antioxidant, antimicrobial,
anti-inflammatory, anti-cancer and immunomodulatory functions
(42-44). Numerous studies have recommended
the use of CAPE for the treatment of heart, kidney, liver and
neurological diseases, as well as for diabetes and cancer (42-44). A previous study demonstrated that
intra-articular injections of CAPE significantly decreased
cartilage destruction in vivo, which indicated that CAPE was
potentially useful as a therapeutic drug (33). However, the mechanisms through
which how CAPE protects cartilage and attenuates OA are currently
unknown. To the best of our knowledge, the present study identified
for the first time, the effects of CAPE on IL-1β-induced human
chondrocytes in vitro and explored the potential molecular
mechanisms involved in the prevention of OA.
Chondrocytes are the only cell type in cartilage.
Multiple pro-inflammatory cytokines, including IL-1β, TNF-α and
IL-6 can influence the functions of chondrocytes and may thus be
implicated in the pathogenesis process of OA (45). It is well-accepted that IL-1β can
be used to mimic OA by inducing an inflammatory response in
chondrocytes (7-9). In the present study, IL-1β was used
to induce OA in chondrocytes, and it was found that IL-1β
significantly increased the inflammatory responses of
chondrocytes.
CAPE has been reported to possess an
anti-inflammatory function (44).
CAPE has been found to attenuate amyloid-β oligomer-induced
neurodegeneration, neuroinflammation and memory impairment in mice
by inhibiting oxidative stress through the NRF2/HO-1 signaling
pathway (31). A previous study
also demonstrated that CAPE exerted protective effects against
Helicobacter pylori-induced gastritis in Mongolian gerbils,
which was attributed to its ability to suppress inflammatory
mediators, including TNF-α, IL-2, IL-6, IL-8 and iNOS through the
inhibition of NF-κB activation (46). In addition, CAPE has been shown to
prevent colitis-associated cancer by post-transcriptionally
inhibiting the NOD-, LRR- and pyrin domain-containing protein 3
inflammasome (47). The present
study found that CAPE inhibited the levels of IL-1β-induced
inflammatory mediators, including NO, PGE2, iNOS and COX-2 in human
chondrocytes. Therefore, the results of the present study suggested
that CAPE exerted its anti-inflammatory effects by suppressing the
levels of inflammatory mediators in IL-1β-stimulated
chondrocytes.
The ECM degradation of articular cartilage is one of
the main features in the process of OA development (48). During the development of OA,
chondrocytes produce and secrete ECM degradative enzymes, including
family members of MMPs and Adamts, in response to inflammatory
factors, which can promote the degradation of the ECM (49-51), while the generation of collagen II
and aggrecan is decreased (51).
The present study found that the levels of ECM degradative enzymes,
including MMP3, MMP13 and Adamts5, were increased in
IL-1β-stimulated chondrocytes, while the level of collagen II was
decreased under IL-1β stimulation. Pre-treatment with CAPE
inhibited the generation of ECM degradative enzymes and promoted
the generation of collagen II in IL-1β-induced chondrocytes, which
indicated that CAPE could prevent the ECM degradation of articular
cartilage. In addition, in the model of DMM OA, CAPE mitigated the
progression of OA, which was consistent with the results of the
in vitro experiments, suggesting that CAPE may be an
effective drug for the treatment of OA.
Previous studies have indicated that the NF-κB
transcription factor plays a central role in the pathogenesis of OA
(52,53). Notably, NF-κB is activated by
pro-inflammatory factors, excessive mechanical stresses associated
factors and ECM degradation products during the development of OA.
NF-κB then promotes the transcription of catabolic genes, including
the MMPs and Adamts families, and triggers the expression of
pivotal inflammatory factors of OA, including iNOS, COX-2 and PGE2
(16,54). Therefore, the inhibition of NF-κB
provides an effective strategy with which to protect cartilage from
damage in OA. The results of the present study demonstrated that
pre-treatment with CAPE inhibited the translocation of p65 from the
cytoplasm to the nucleus in IL-1β-stimulated chondrocytes,
indicating that CAPE exerted an anti-inflammatory effect by
inhibiting the activation of the NF-κB signaling pathway to protect
cartilage from destruction.
NRF2 is a crucial transcription factor that mediates
the response to ROS and inflammation (55). The activation of NRF2 has been
reported to repress the activity of NF-κB, leading to a decrease in
the production of inflammatory factors (56,57). Increasing evidence has indicated
that the NRF2/HO-1 signaling pathway plays a pivotal role in the
protection of joint cartilage during OA pathogenesis (58). The activation of the NRF2/HO-1
signaling pathway inhibits the production of MMPs and inflammatory
factors to reduce the degradation of ECM in OA chondrocytes
(19,59,60). A previous study reported that CAPE
exerted its anti-inflammatory effects by inducing the activation of
the NRF2/HO-1 signaling pathway; CAPE attenuated
2,4,6-trinitrobenzene sulfonic acid-induced colitis via the
activation of the NRF2 signaling pathway in the rat inflamed colon
(61). CAPE has also been shown
to protect the periodontal status, which is attributed to its
ability to suppress inflammation through activation of the
NRF2/HO-1 signaling pathway and the inhibition of the NF-κB
signaling pathway (62).
Additionally, CAPE has been found to prevent liver fibrosis via the
upregulation of NRF2 (63). In
the present study, CAPE reduced the inflammatory response and
attenuated the degradation of ECM via the upregulation of NRF2, the
activation of the NRF2/HO-1 signaling pathway and the inhibition of
the NF-κB signaling pathway. In the physiological state, NRF2 was
ubiquitinated by binding with Keap1 in the cytoplasm. Under
stimulation, NRF2 dissociates from Keap1 and enters the nucleus to
form a coactivator complex, which can improve the driving function
(64). Therefore, in the present
study, the interaction between CAPE and Keap1/NRF2 complex was
analyzed by computational molecular docking. The results of the
present study indicated that CAPE interacting with Keap1 led to the
release of NRF2 from the Keap1/NRF2 complex, which is essential for
NRF2 activation.
Although the effects of CAPE on OA have been
analyzed, there are relatively few studies available on its
mechanisms of action. In the study by Elmali et al (33), it was reported that CAPE
attenuated unilateral anterior cruciate ligament
transection-induced cartilage destruction in vivo. In the
study by Pichler et al (65), it was found that CAPE reduced the
mRNA levels of IL-1β and MMP-13 under treatment with the galectin
mixture, Gal-1/-3/-8. As CAPE has been previously described as an
effective inhibitor of the NF-κB-dependent expression of Gal-7 in
breast cancer cells, Pichler et al (65) hypothesized that the function of
CAPE in galectin-induced chondrocytes occurred via the NF-κB signal
pathway. However, this was not confirmed experimentally. In the
study by Wang et al (66),
the role of CAPE in the apoptosis of TNF-α induced chondrocytes and
in the expression of MMP-2 and MMP-9 in vitro was
investigated. However, they did not fully elucidate the role and
related mechanisms of CAPE in OA. Therefore, these studies only
simply described the role of CAPE in OA, but did not discuss the
mechanisms. The present study investigated the anti-inflammatory
effects of CAPE and the anti-degradation of the ECM in
IL-1β-stimulated chondrocytes in vitro and in vivo
for the first time (to the best of our knowledge), and confirmed
experimentally that CAPE participated in OA regulation as an NF-κB
inhibitor by regulating the NRF2/HO-1 signaling pathway. Therefore,
the findings of the present study are in agreement with those from
previous studies (33,65,66), and is comprehensive and novel to a
certain extent. However, there are several limitations to the
present study. Firstly, the binding between CAPE and the Keap1/NRF2
complex needs to be confirmed by co-immunoprecipitation. In
addition, in terms of morphology, the present study only observed
the morphology of normal chondrocytes and did not use OA
chondrocytes as a positive control. Furthermore, the effects of
CAPE need to be tested in NRF2-knockout mice. Therefore, further
studies are required to confirm and further elaborate on the
present findings.
In conclusion, the present study provided novel
insight into the potential protective effects of CAPE in OA. CAPE
attenuate IL-1β-induced inflammation and ECM degradation through
activation of the NRF2/HO-1 signaling pathway and the inhibition of
the NF-κB signaling pathway (Fig.
8C). In addition, the intraperitoneal injection of CAPE
ameliorated the degradation of cartilage matrix in vivo.
Therefore, CAPE may have potential for use in the treatment of
OA.
Supplementary Data
Availability of data and materials
The analyzed data sets generated during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
QY and WeiS conceived and designed the experiments.
YC, DH, LJ and QH performed the experiments. WeiS, JY and WX
conducted the literature search and the processing of the figures.
JY, WX, JX, QY and WeichaoS analyzed the data and wrote the
manuscript. QY and WeichaoS confirm the authenticity of all the raw
data. All authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
All individuals provided informed consent for the
use of human specimens in clinical experiments. The present study
was approved by the Ethics Committees of Shenzhen Second People's
Hospital (approval no. ethic NO.:20211215005-FS01). The animal
protocols and experimental procedures were in agreement with and
were approved by the Animal Care and Use Committee of Southwest
Medical University (approval no. 20211124-043).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by funds from the National
Natural Sciences Foundation of China (grant no. 82003126), the
Scientific Research Foundation of Southwest Medical University
(grant no. 2021ZKMS009), Luzhou Science and Technology Program
(grant no. 2021-JYJ-71), the Shenzhen Science and Technology
Projects (grant nos. JSGG20191129094218565, JCYJ20190807102601647
and JCYJ20210324103604013), and the Sichuan Science and Technology
Program (grant no. 2022NSFSC1368).
References
|
1
|
Martel-Pelletier J, Barr AJ, Cicuttini FM,
Conaghan PG, Cooper C, Goldring MB, Goldring SR, Jones G, Teichtahl
AJ and Pelletier JP: Osteoarthritis. Nat Rev Dis Primers.
2:160722016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Loeser RF, Goldring SR, Scanzello CR and
Goldring MB: Osteoarthritis: A disease of the joint as an organ.
Arthritis Rheum. 64:1697–1707. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bijlsma JW, Berenbaum F and Lafeber FP:
Osteoarthritis: An update with relevance for clinical practice.
Lancet. 377:2115–2126. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Glyn-Jones S, Palmer AJR, Agricola R,
Price AJ, Vincent TL, Weinans H and Carr AJ: Osteoarthritis.
Lancet. 386:376–387. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kapoor M, Martel-Pelletier J, Lajeunesse
D, Pelletier JP and Fahmi H: Role of proinflammatory cytokines in
the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 7:33–42.
2011. View Article : Google Scholar
|
|
6
|
Chow YY and Chin KY: The role of
inflammation in the pathogenesis of osteoarthritis. Mediators
Inflamm. 2020:82939212020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang B, Kang X, Xing Y, Dou C, Kang F, Li
J, Quan Y and Dong S: Effect of microRNA-145 on IL-1beta-induced
cartilage degradation in human chondrocytes. FEBS Lett.
588:2344–2352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Eymard F, Pigenet A, Citadelle D,
Flouzat-Lachaniette CH, Poignard A, Benelli C, Berenbaum F,
Chevalier X and Houard X: Induction of an inflammatory and
prodegradative phenotype in autologous fibroblast-like synoviocytes
by the infrapatellar fat pad from patients with knee
osteoarthritis. Arthritis Rheumatol. 66:2165–2174. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tu C, Huang X, Xiao Y, Song M, Ma Y, Yan
J, You H and Wu H: Schisandrin A inhibits the IL-1β-induced
inflammation and cartilage degradation via suppression of MAPK and
NF-κB signal pathways in rat chondrocytes. Front Pharmacol.
10:412019. View Article : Google Scholar
|
|
10
|
Chabane N, Zayed N, Afif H, Mfuna-Endam L,
Benderdour M, Boileau C, Martel-Pelletier J, Pelletier JP, Duval N
and Fahmi H: Histone deacetylase inhibitors suppress
interleukin-1beta-induced nitric oxide and prostaglandin E2
production in human chondrocytes. Osteoarthritis Cartilage.
16:1267–1274. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Khan NM, Haseeb A, Ansari MY, Devarapalli
P, Haynie S and Haqqi TM: Wogonin, a plant derived small molecule,
exerts potent anti-inflammatory and chondroprotective effects
through the activation of ROS/ERK/Nrf2 signaling pathways in human
Osteoarthritis chondrocytes. Free Radic Biol Med. 106:288–301.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shuai C, Liu G, Yang Y, Qi F, Peng S, Yang
W, He C, Wang G and Qian G: A strawberry-like Ag-decorated barium
titanate enhances piezoelectric and antibacterial activities of
polymer scaffold. Nano Energy. 74:1048252020. View Article : Google Scholar
|
|
13
|
Shuai C, Xu Y, Feng P, Wang G, Xiong S and
Peng S: Antibacterial polymer scaffold based on mesoporous
bioactive glass loaded with in situ grown silver. Chemical
Engineering J. 374:304–315. 2019. View Article : Google Scholar
|
|
14
|
Wu H, Zhang M, Li W, Zhu S and Zhang D:
Stachydrine attenuates IL-1beta-induced inflammatory response in
osteoarthritis chondrocytes through the NF-κB signaling pathway.
Chem Biol Interact. 326:1091362020. View Article : Google Scholar
|
|
15
|
Hu X, Li R, Sun M, Kong Y, Zhu H, Wang F
and Wan Q: Isovitexin depresses osteoarthritis progression via the
Nrf2/NF-kappaB pathway: An in vitro study. J Inflamm Res.
14:1403–1414. 2021. View Article : Google Scholar :
|
|
16
|
Rigoglou S and Papavassiliou AG: The NF-κB
signalling pathway in osteoarthritis. Int J Biochem Cell Biol.
45:2580–2584. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zeng J, Chen Y, Ding R, Feng L, Fu Z, Yang
S, Deng X, Xie Z and Zheng S: Isoliquiritigenin alleviates early
brain injury after experimental intracerebral hemorrhage via
suppressing ROS- and/or NF-κB-mediated NLRP3 inflammasome
activation by promoting Nrf2 antioxidant pathway. J
Neuroinflammation. 14:1192017. View Article : Google Scholar
|
|
18
|
Saha S, Buttari B, Panieri E, Profumo E
and Saso L: An overview of Nrf2 signaling pathway and its role in
inflammation. Molecules. 25:54742020. View Article : Google Scholar :
|
|
19
|
Khan NM, Ahmad I and Haqqi TM: Nrf2/ARE
pathway attenuates oxidative and apoptotic response in human
osteoarthritis chondrocytes by activating ERK1/2/ELK1-P70S6K-P90RSK
signaling axis. Free Radic Biol Med. 116:159–171. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shao Z, Pan Z, Lin J, Zhao Q, Wang Y, Ni
L, Feng S, Tian N, Wu Y, Sun L, et al: S-allyl cysteine reduces
osteoarthritis pathology in the tert-butyl hydroperoxide-treated
chondrocytes and the destabilization of the medial meniscus model
mice via the Nrf2 signaling pathway. Aging (Albany NY).
12:19254–19272. 2020. View Article : Google Scholar
|
|
21
|
Cai D, Yin S, Yang J, Jiang Q and Cao W:
Histone deacetylase inhibition activates Nrf2 and protects against
osteoarthritis. Arthritis Res Ther. 17:2692015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu J, Omene C, Karkoszka J, Bosland M,
Eckard J, Klein CB and Frenkel K: Caffeic acid phenethyl ester
(CAPE), derived from a honeybee product propolis, exhibits a
diversity of anti-tumor effects in pre-clinical models of human
breast cancer. Cancer Lett. 308:43–53. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Murtaza G, Karim S, Akram MR, Khan SA,
Azhar S, Mumtaz A and Asad MHH: Caffeic acid phenethyl ester and
therapeutic potentials. Biomed Res Int. 2014:1453422014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tolba MF, Omar HA, Azab SS, Khalifa AE,
Abdel-Naim AB and Abdel-Rahman SZ: Caffeic acid phenethyl ester: A
review of its antioxidant activity, protective effects against
ischemiareperfusion injury and drug adverse reactions. Crit Rev
Food Sci Nutr. 56:2183–2190. 2016. View Article : Google Scholar
|
|
25
|
Hao R, Song X, Li F, Tan X, Sun-Waterhouse
D and Li D: Caffeic acid phenethyl ester reversed cadmium-induced
cell death in hippocampus and cortex and subsequent cognitive
disorders in mice: Involvements of AMPK/SIRT1 pathway and
amyloid-tau-neuroinflammation axis. Food Chem Toxicol.
144:1116362020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu M, Li F, Huang Y, Zhou T, Chen S, Li
G, Shi J, Dong N and Xu K: Caffeic acid phenethyl ester ameliorates
calcification by inhibiting activation of the AKT/NF-kappaB/NLRP3
inflammasome pathway in human aortic valve interstitial cells.
Front Pharmacol. 11:8262020. View Article : Google Scholar
|
|
27
|
Lee HE, Yang G, Kim ND, Jeong S, Jung Y,
Choi JY, Park HH and Lee JY: Targeting ASC in NLRP3 inflammasome by
caffeic acid phenethyl ester: A novel strategy to treat acute gout.
Sci Rep. 6:386222016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Natarajan K, Singh S, Burke TR Jr,
Grunberger D and Aggarwal BB: Caffeic acid phenethyl ester is a
potent and specific inhibitor of activation of nuclear
transcription factor NF-kappa B. Proc Natl Acad Sci USA.
93:9090–9095. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liang Y, Feng G, Wu L, Zhong S, Gao X,
Tong Y, Cui W, Qin Y, Xu W, Xiao X, et al: Caffeic acid phenethyl
ester suppressed growth and metastasis of nasopharyngeal carcinoma
cells by inactivating the NF-κB pathway. Drug Des Devel Ther.
13:1335–1345. 2019. View Article : Google Scholar :
|
|
30
|
Lim KM, Bae S, Koo JE, Kim ES, Bae ON and
Lee JY: Suppression of skin inflammation in keratinocytes and
acute/chronic disease models by caffeic acid phenethyl ester. Arch
Dermatol Res. 307:219–227. 2015. View Article : Google Scholar
|
|
31
|
Morroni F, Sita G, Graziosi A, Turrini E,
Fimognari C, Tarozzi A and Hrelia P: Neuroprotective effect of
caffeic acid phenethyl ester in A mouse model of Alzheimer's
disease involves Nrf2/HO-1 pathway. Aging Dis. 9:605–622. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Khan MN, Lane ME, McCarron PA and
Tambuwala MM: Caffeic acid phenethyl ester is protective in
experimental ulcerative colitis via reduction in levels of
pro-inflammatory mediators and enhancement of epithelial barrier
function. Inflammopharmacology. 26:561–569. 2018. View Article : Google Scholar :
|
|
33
|
Elmali N, Ayan I, Türköz Y, Mizrak B,
Germen B and Bora A: Effect of caffeic acid phenethyl ester on
cartilage in experimental osteoarthritis. Rheumatol Int.
22:222–226. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lu H, Fu C, Kong S, Wang X, Sun L, Lin Z,
Luo P and Jin H: Maltol prevents the progression of osteoarthritis
by targeting PI3K/Akt/NF-κB pathway: In vitro and in vivo studies.
J Cell Mol Med. 25:499–509. 2021. View Article : Google Scholar
|
|
35
|
Glasson SS, Blanchet TJ and Morris EA: The
surgical destabilization of the medial meniscus (DMM) model of
osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage.
15:1061–1069. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
37
|
Glasson SS, Chambers MG, Van Den Berg WB
and Little CB: The OARSI histopathology initiative-recommendations
for histological assessments of osteoarthritis in the mouse.
Osteoarthritis Cartilage. 18(Suppl 3): pp. S17–S23. 2010,
View Article : Google Scholar
|
|
38
|
Gu M, Jin J, Ren C, Chen X, Gao W, Wang X,
Wu Y, Tian N, Pan Z, Wu A, et al: Akebia Saponin D suppresses
inflammation in chondrocytes via the NRF2/HO-1/NF-κB axis and
ameliorates osteoarthritis in mice. Food Funct. 11:10852–10863.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
da Costa BR, Reichenbach S, Keller N,
Nartey L, Wandel S, Jüni P and Trelle S: Effectiveness of
non-steroidal anti-inflammatory drugs for the treatment of pain in
knee and hip osteoarthritis: A network meta-analysis. Lancet.
390:e21–e33. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Berenbaum F: Osteoarthritis as an
inflammatory disease (osteoarthritis is not osteoarthrosis!).
Osteoarthritis Cartilage. 21:16–21. 2013. View Article : Google Scholar
|
|
41
|
Deligiannidou GE, Papadopoulos RE,
Kontogiorgis C, Detsi A, Bezirtzoglou E and Constantinides T:
Unraveling natural products' role in osteoarthritis management-an
overview. Antioxidants (Basel). 9:3482020. View Article : Google Scholar
|
|
42
|
Balaha M, Filippis BD, Cataldi A and di
Giacomo V: CAPE and neuroprotection: A review. Biomolecules.
11:1762021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Menezes da Silveira CCS, Luz DA, da Silva
CCS, Prediger RDS, Martins MD, Martins MAT, Fontes-Júnior EA and
Maia CSF: Propolis: A useful agent on psychiatric and neurological
disorders? A focus on CAPE and pinocembrin components. Med Res Rev.
41:1195–1215. 2021. View Article : Google Scholar
|
|
44
|
Murtaza G, Sajjad A, Mehmood Z, Shah SH
and Siddiqi AR: Possible molecular targets for therapeutic
applications of caffeic acid phenethyl ester in inflammation and
cancer. J Food Drug Anal. 23:11–18. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu-Bryan R and Terkeltaub R: Emerging
regulators of the inflammatory process in osteoarthritis. Nat Rev
Rheumatol. 11:35–44. 2015. View Article : Google Scholar :
|
|
46
|
Toyoda T, Tsukamoto T, Takasu S, Shi L,
Hirano N, Ban H, Kumagai T and Tatematsu M: Anti-inflammatory
effects of caffeic acid phenethyl ester (CAPE), a nuclear
factor-kappaB inhibitor, on Helicobacter pylori-induced gastritis
in Mongolian gerbils. Int J Cancer. 125:1786–1795. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dai G, Jiang Z, Sun B, Liu C, Meng Q, Ding
K, Jing W and Ju W: Caffeic acid phenethyl ester prevents
colitis-associated cancer by inhibiting NLRP3 inflammasome. Front
Oncol. 10:7212020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Guilak F, Nims RJ, Dicks A, Wu CL and
Meulenbelt I: Osteoarthritis as a disease of the cartilage
pericellular matrix. Matrix Biol. 71-72:40–50. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lu G, Li L, Wang B and Kuang L:
LINC00623/miR-101/HRAS axis modulates IL-1β-mediated ECM
degradation, apoptosis and senescence of osteoarthritis
chondrocytes. Aging (Albany NY). 12:3218–3237. 2020. View Article : Google Scholar
|
|
50
|
Boehme KA and Rolauffs B: Onset and
progression of human osteoarthritis-can growth factors,
inflammatory cytokines, or differential mirna expression
concomitantly induce proliferation, ECM degradation, and
inflammation in articular cartilage? Int J Mol Sci. 19:22822018.
View Article : Google Scholar
|
|
51
|
Rahmati M, Nalesso G, Mobasheri A and
Mozafari M: Aging and osteoarthritis: Central role of the
extracellular matrix. Ageing Res Rev. 40:20–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Choi MC, Jo J, Park J, Kang HK and Park Y:
NF-κB signaling pathways in osteoarthritic cartilage destruction.
Cells. 8:7342019. View Article : Google Scholar
|
|
53
|
Lepetsos P, Papavassiliou KA and
Papavassiliou AG: Redox and NF-κB signaling in osteoarthritis. Free
Radic Biol Med. 132:90–100. 2019. View Article : Google Scholar
|
|
54
|
Saito T and Tanaka S: Molecular mechanisms
underlying osteoarthritis development: Notch and NF-κB. Arthritis
Res Ther. 19:942017. View Article : Google Scholar
|
|
55
|
Tonelli C, Chio IIC and Tuveson DA:
Transcriptional regulation by Nrf2. Antioxid Redox Signal.
29:1727–1745. 2018. View Article : Google Scholar :
|
|
56
|
Sivandzade F, Prasad S, Bhalerao A and
Cucullo L: NRF2 and NF-B interplay in cerebrovascular and
neurodegenerative disorders: Molecular mechanisms and possible
therapeutic approaches. Redox Biol. 21:1010592019. View Article : Google Scholar
|
|
57
|
Lee Y, Shin DH, Kim JH, Hong S, Choi D,
Kim YJ, Kwak MK and Jung Y: Caffeic acid phenethyl ester-mediated
Nrf2 activation and IkappaB kinase inhibition are involved in
NFkappaB inhibitory effect: Structural analysis for NFkappaB
inhibition. Eur J Pharmacol. 643:21–28. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Alcaraz MJ and Ferrandiz ML: Relevance of
Nrf2 and heme oxygenase-1 in articular diseases. Free Radic Biol
Med. 157:83–93. 2020. View Article : Google Scholar
|
|
59
|
Chen X, Huang C, Sun H, Hong H, Jin J, Bei
C, Lu Z and Zhang X: Puerarin suppresses inflammation and ECM
degradation through Nrf2/HO-1 axis in chondrocytes and alleviates
pain symptom in osteoarthritic mice. Food Funct. 12:2075–2089.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Li JW, Wang RL, Xu J, Sun KY, Jiang HM,
Sun ZY, Lv ZY, Xu XQ, Wu R, Guo H, et al: Methylene blue prevents
osteoarthritis progression and relieves pain in rats via
upregulation of Nrf2/PRDX1. Acta Pharmacol Sin. 43:417–428. 2022.
View Article : Google Scholar :
|
|
61
|
Kim H, Kim W, Yum S, Hong S, Oh JE, Lee
JW, Kwak MK, Park EJ, Na DH and Jung Y: Caffeic acid phenethyl
ester activation of Nrf2 pathway is enhanced under oxidative state:
Structural analysis and potential as a pathologically targeted
therapeutic agent in treatment of colonic inflammation. Free Radic
Biol Med. 65:552–562. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Stahli A, Maheen CU, Strauss FJ, Eick S,
Sculean A and Gruber R: Caffeic acid phenethyl ester protects
against oxidative stress and dampens inflammation via heme
oxygenase 1. Int J Oral Sci. 11:62019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Li M, Wang XF, Shi JJ, Li YP, Yang N, Zhai
S and Dang SS: Caffeic acid phenethyl ester inhibits liver fibrosis
in rats. World J Gastroenterol. 21:3893–3903. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Bellezza I, Giambanco I, Minelli A and
Donato R: Nrf2-Keap1 signaling in oxidative and reductive stress.
Biochim Biophys Acta Mol Cell Res. 1865:721–733. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Pichler KM, Weinmann D, Schmidt S, Kubista
B, Lass R, Martelanz L, Alphonsus J, Windhager R, Gabius HJ and
Toegel S: The dysregulated galectin network activates nf-kappaB to
induce disease markers and matrix degeneration in 3D pellet
cultures of osteoarthritic chondrocytes. Calcif Tissue Int.
108:377–390. 2021. View Article : Google Scholar
|
|
66
|
Wang Y, Li DL, Zhang XB, Duan YH, Wu ZH,
Hao DS, Chen BS and Qiu GX: Increase of TNFalpha-stimulated
osteoarthritic chondrocytes apoptosis and decrease of matrix
metalloproteinases 9 by NF-κB inhibition. Biomed Environ Sci.
26:277–283. 2013.PubMed/NCBI
|