1. Mitochondrial physiology
Mitochondrial biogenesis
The biogenesis of mitochondria refers to the process
through which mitochondria grow in number and/or size. This is
mediated by physiological stimuli, including physical exercise,
dietary restrictions and temperature (1). Mitochondrial biogenesis (Fig. 1) is transcriptionally controlled
through the activation of peroxisome proliferator-activated
receptor-gamma coactivator 1α (PGC-1α) (2). PGC-1α has specific tissue
distribution and is mainly located in tissues with high energy
requirements or high oxidative activity, such as the heart,
skeletal muscle, liver and white or brown adipose tissue,
suggesting that it is closely related to the energy metabolism of
the body (3). The Ppargc1a
gene structure contains a well-conserved binding site for a cAMP
response element-binding protein (CREB) that allows activated CREB
to bind and promote PGC-1α expression (4,5).
Furthermore, PGC-1α can be activated by reduced adenosine
triphosphate (ATP)/adenosine monophosphate (AMP) levels mediated by
AMP-activated protein kinase (AMPK) that functions as a cellular
energy sensor (1). Thus,
activated PGC-1α leads to the consecutive stimulation of several
transcription factors, including nuclear respiratory factors
(NRFs)1 and 2, that promote the expression of nuclear genes that
are responsible for controlling transcriptionally the majority of
the subunits of mitochondrial complexes (6,7)
and peroxisome proliferator-activated receptors (PPARs) (8). Furthermore, PGC-1α can promote
oxidative phosphorylation (OXPHOS) gene expression, which encodes
proteins that constitute the electron transport chain (ETC) and are
responsible for ATP synthesis. Last but not least, PGC-1α
cooperates with PPARα to regulate the expression of mitochondrial
fatty acid oxidation (FAO) enzymes and transport proteins, enabling
increases in FAO pathway activity in coordination with
mitochondrial biogenesis (9). The
aforementioned findings indicate that PGC-1a masters cellular
mechanisms related to substrate utilization (fatty acids, glucose)
and their intra-mitochondrial oxidation to produce energy for
cellular demands (10,11).
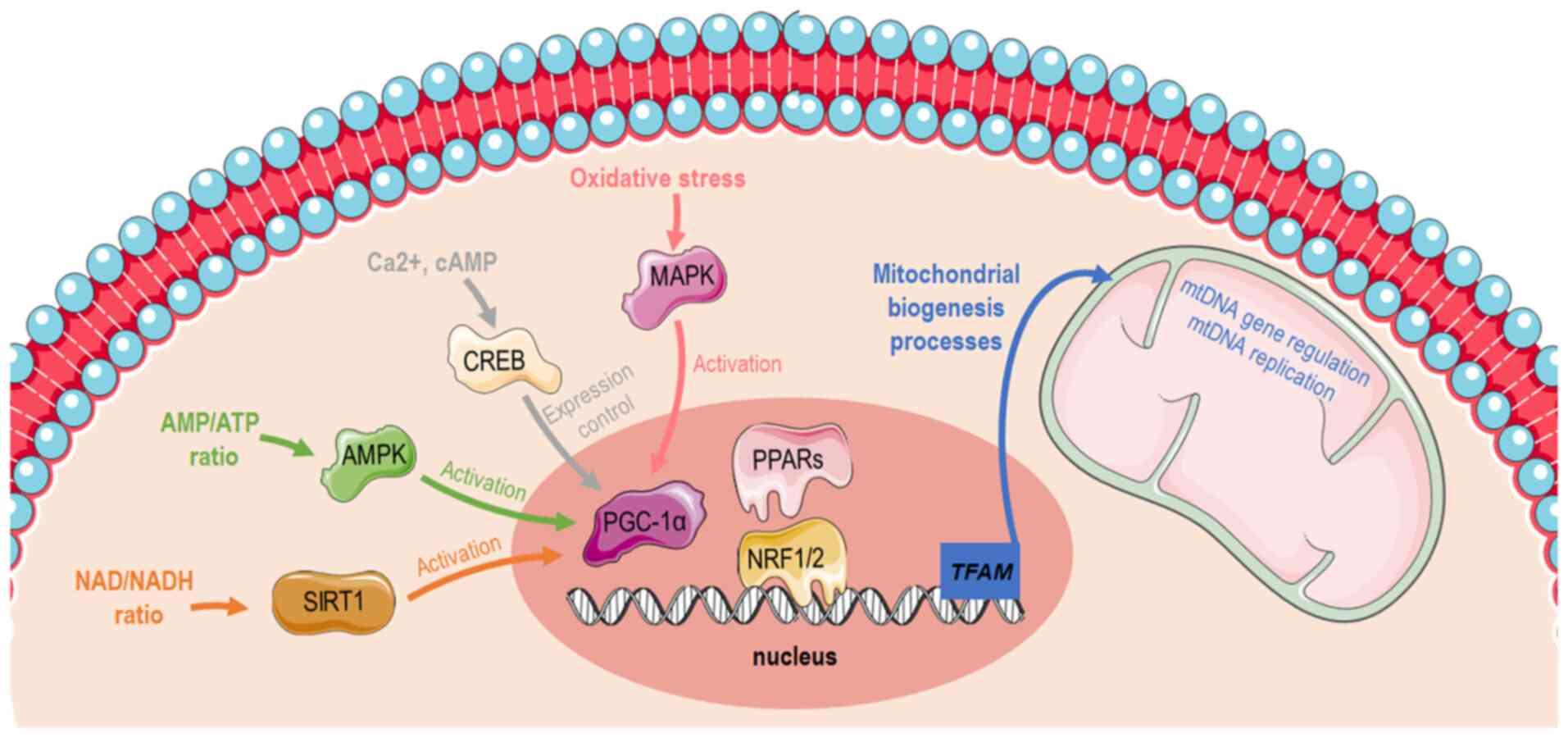 | Figure 1Graphical illustration depicting
distinct pathways that induces mitochondrial biogenesis via PGC-1α
activation. The image was created using the Smart servier medical
art website; smart.servier.com. PGC-1α,
peroxisome proliferator-activated receptor-gamma coactivator 1α;
AMP, adenosine monophosphate; ATP, adenosine triphosphate; SIRT1,
sirtuin 1; AMPK, AMP-activated protein kinase; CREB, cAMP response
element-binding protein; PPARs, peroxisome proliferator-activated
receptors; NRF, nuclear respiratory factor; TFAM, mitochondrial
transcription factor A; mtDNA, mitochondrial DNA. |
There is an ample amount of available literature on
the molecular pathways that induce mitochondrial biogenesis and
function during conditions of high energy demands, such as
exercise. One bout of acute exercise in the muscle is sufficient to
initiate transcriptional signaling toward mitochondrial biogenesis.
More elaborately, exercise increases intracellular calcium levels,
allowing the calcium/calmodulin-dependent protein kinase
IV-dependent phosphorylation and the subsequent activation of CREB
(12,13). As previously described, PGC-1α is
induced in the skeletal muscle by AMPK when ATP levels are low
(10,14). In the liver, the hormonal and
nutritional regulation of hepatic gluconeogenesis occurs mainly
through the modulation of the transcriptional coactivator PGC-1α.
During glucose deprivation, cells sensitize the need for additional
substrates and thus stimulate the gluconeogenesis program and
mitochondrial biogenesis through CREB and PGC-1a in order to supply
mitochondria with new glucose molecules and build more available
'factories' in order to generate sufficient amounts of energy.
Therefore, gluconeogenesis and mitochondrial biogenesis are tightly
coupled to allow hepatocytes to adapt in low available glucose
levels. Sirtuin (SIRT)1 protein expression is induced in the liver
through a nutrient-signaling response mediated by pyruvate kinase.
SIRT1 then activates forkhead box protein O1 (FOXO1) (15) and PGC-1α through their
deacetylation (16).
Subsequently, FOXO1 and PGC1a co-orchestrate the gluconeogenesis
program, enabling organism energetic stability through glucose
utilization (15).
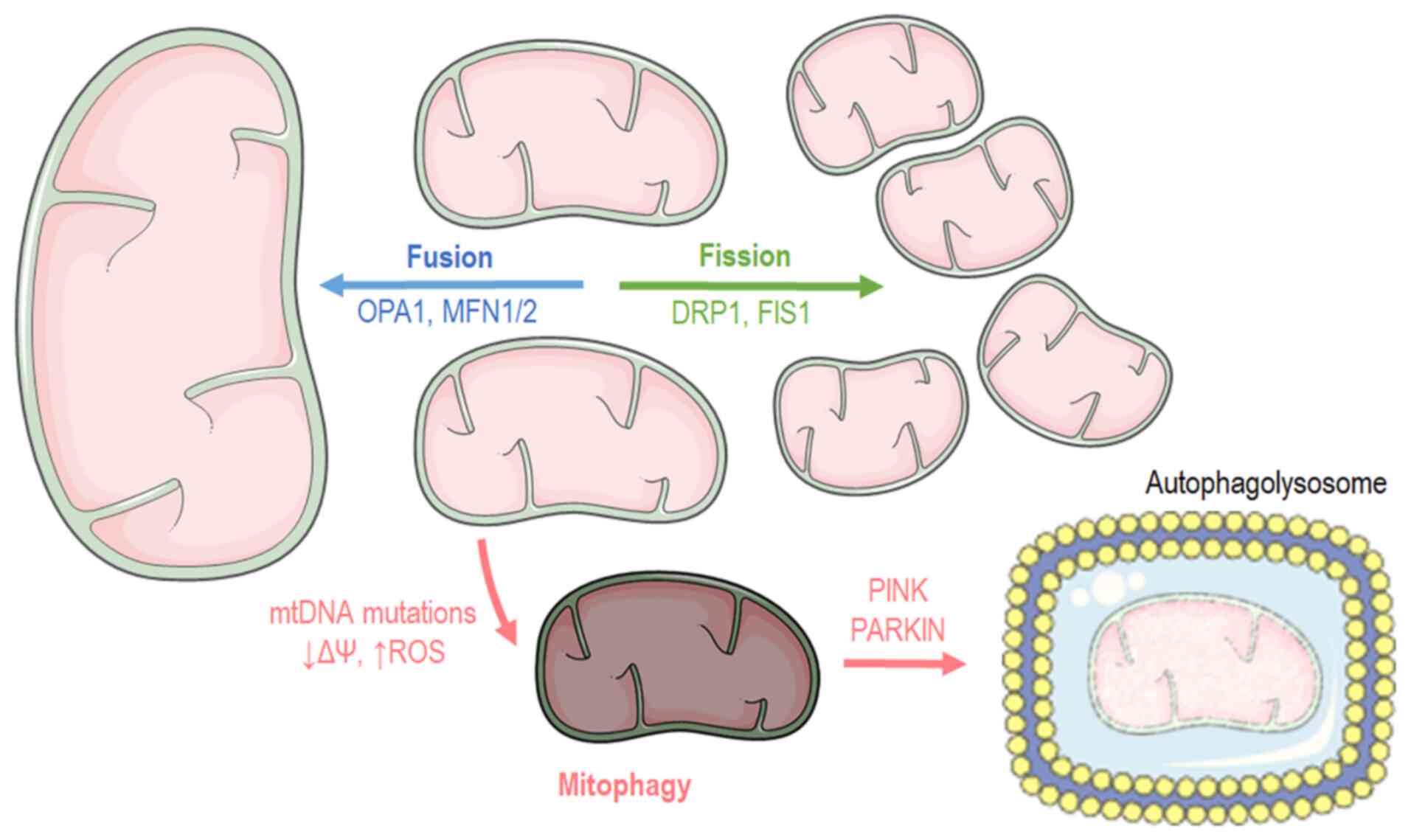
Mitochondrial dynamics:
Fusion-fission
Cells have distinct mechanisms depending on their
cycle status or their state to maintain mitochondrial morphology
and proper function. Mitochondria carry out two processes, fusion
and fission (Fig. 2), which are
of paramount importance for self-controlling organelle quality
(17,18). Mitochondrial fusion occurs when
nearby mitochondria merge, while mitochondrial fission takes place
when a mitochondrion needs to break apart into two separate
organelles. Mitochondrial fusion occurs during the early stages of
the S and G1 phases (19),
ensuring mainly three main functions: Respiration, ATP production
and intramitochondrial material exchange (20). At the G-S1 cellular phase,
mitochondria shape a giant, hyper-fused network characterized by
higher ATP production capacity (19). Additionally, it has been found
that fusion contributes to mitochondrial repair and elongation
while the substrates of the fused mitochondrial are optimally
exploited for respiration. At the structural level, mitochondrial
fusion is accomplished when both the inner mitochondrial membrane
(IMM) and outer mitochondrial membrane (OMM) of the fused
organelles merge, respectively (20), while at a molecular level, three
different proteins are responsible for the structural fusion: In
the OMM, both mitofusins 1 and 2 (MFN1 and MFN2), while in the IMM,
optic atrophy 1 (OPA1) (18).
In contrast to fusion, the mitochondrial fission
process usually occurs during the S, G2 and M phases due to the
need for the even separation of organelles that will be present in
offspring cells (21,22). Fission is beneficial when
mitochondria are dysfunctional. Mitochondria divide and stimulate
the authophagy pathway (mitophagy) (23). Briefly, mitochondrial fission
begins when the endoplasmic reticulum (ER) is recruited and
stimulates mtDNA replication. Fission is orchestrated by the
dynamin-related protein 1 (DRP1), which is a GTPase that is
recruited in the mitochondrial surface and anchored by complexes
that are constructed from different mitochondrial proteins, such as
mitochondrial fission 1 (FIS1), mitochondrial fission factor and
mitochondrial dynamics protein 49 and 51 (MiD49 and MiD51)
(18).
Mitophagy
Macroautophagy is a genetically programmed and
conserved catabolic process (24)
in which cytosol portions and/or complete organelles are engulfed
by double-membrane structures known as autophagosomes, that fuse
with lysosomes in order to form single membrane structures known as
autophagolysosomes (25).
Mitophagy is the macroautophagy process through which the
mitochondria are driven towards degradation (Fig. 2) (26,27). Mitophagy also constitutes a
mitochondrial quality control mechanism, preventing mitochondrial
dysfunction, a hallmark of cellular aging (28). Several autophagy-related genes
(ATGs) regulate autophagy or autophagy-related process,
transcriptional control that is evolutionary preserved since 30
ATGs have also been described in yeasts (29,30). The recognition of dysfunctional
mitochondria is mediated by the p62 and PARKIN proteins. PARKIN is
an ubiquitin ligase that translocates into the mitochondria and
tags them in order to become processed for degradation. Another
protein, the mitochondrial receptor NIX, binds the cytoskeleton
related protein, such as like gamma-aminobutyric acid type A
receptor-associated protein (GABARAP) and microtubule-associated
protein 1 light chain 3 (LC3, Atg8) (31), driving mitochondria towards
apoptosis since it regulates PARKIN translocation into mitochondria
(32). On the onset of
Parkin-mediated mitophagy, PARKIN interacts with PTEN-induced
kinase 1 (PINK1) in order to finalize dysfunctional mitochondria
labeling (33). Subsequently,
ATG9a and the ULK1/2 can depolarize mitochondria, a biochemical
manifestation that is responsible for the recruitment of additional
downstream autophagy-related proteins (ATG) apart from LC3.
Finally, LC3 recruitment drives mitochondria into the autophagosome
for decomposition (34). Another
molecular pathway that stimulates mitophagy includes the
transcriptional factor, FoxO3, which regulates autophagy through
Bcl-2 inter-acting protein 3 (BNIP3) (35). BNIP3 belongs to the BH3-only
proteins of the Bcl-2 family and can induce cellular apoptosis and
mitophagy (36). Mitophagy
pathways are closely aligned with those of mitochondrial dynamics.
As such, mitochondrial fission appears to be the first step and a
pre-requisite of the mitophagy process (37).
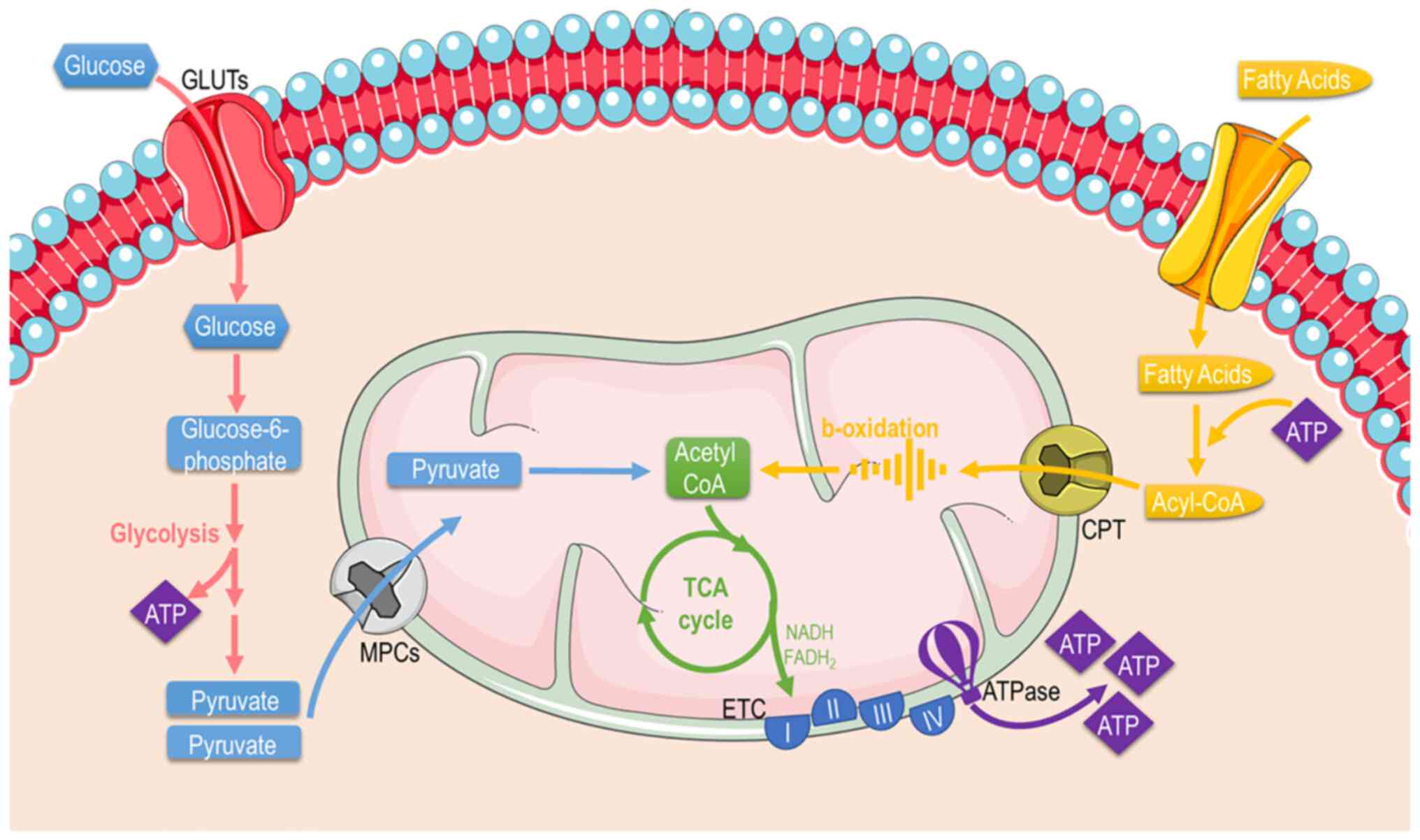
Mitochondrial function
Mitochondria are mainly considered the cellular
'powerplants' with their foremost function to supply cells with
energy through producing the energetic 'coin' ATP. Apart from
energy metabolism, the mitochondria contribute to different aspects
of cellular biology, such as signaling, differentiation, cell
cycle, growth and cellular death (38). Thereafter, mitochondrial and
cellular metabolism and are tightly coupled. Mitochondria possess
specific metabolic lines to process different substrates and
generate ATP (Fig. 3). These
include pathways that pyruvate (derived from glucose or lactate),
fatty acids, and amino acids are eventually oxidized to gear
protons onto NADH and FADH2. NADH and FADH2 carry these electrons
to the ETC, forming an electrochemical gradient that drives
ATP-synthetase to produce ATP through oxidative
phosphorylation.
Fuel metabolism
Glucose: Glucose is almost continuously
cellular available through facilitated diffusion conducted by
several isotypes of glucose transporters present in different cell
types (39). Glucose levels are
elevated in the circulation after feeding or through hepatic
gluconeogenesis (Fig. 4); hepatic
release during fasting, allows tissues to uptake glucose and
generate pyruvate through glycolysis, producing an initially low
number of ATP molecules. Subsequently, mitochondria can utilize
glucose through the form of pyruvate in order to produce ATP.
Depending on nutrient and oxygen availability, glucose is converted
into pyruvate or lactate through pyruvate and lactate
dehydrogenase, respectively. Under physiological conditions,
pyruvate is transported across the double-mitochondrial membrane
via mitochondrial pyruvate carrier (MPC) (40). Oxygen deprivation mitigates
oxidative phosphorylation, and net energy is produced through
anaerobic glycolysis and lactate generation as the end-product.
Lactate can be converted back to pyruvate and glucose in
hepatocytes through a different type of lactate dehydrogenase
during oxygen abundancy, resulting though a net negative balance of
ATP (41). Therefore, the Cori
cycle cannot be maintained relentlessly. On the other hand, the
inhibition of MPC proteins that block pyruvate shuttling inside the
mitochondria, forces the mitochondria into a metabolic reprogram
and to depend mainly on fatty acids and glutamine. The
glutaminolysis pathway allows glutamine to be oxidized in the
mitochondria and stimulates the Krebs cycle, producing
a-ketoglutarate or pyruvate via malic enzymes (42). Surprisingly, at high glucose
levels, cells lose their ability to utilize the excessive pyruvate
that is available, leading to cellular glucotoxicity through the
activation of polyol pathway, protein kinase C, increased advanced
glycation end-products, and hexosamine pathway flux (43). Thereafter, although the hypothesis
that excessive glucose facilitates higher energy production, the
mitochondria are actually vulnerable in toxic intermediates that
glucotoxicity is generating, resulting into mitophagy and cellular
death (44). This indicate that
glucose homeostasis is crucial for the proper functioning of the
mitochondria and cells.
Fatty acids (FAs): FAs are the main metabolic
substrates for the mitochondria of cardiac and skeletal muscle in
order to suffice their energy demands (45). FAs derive from the white adipose
tissue in the form of albumin-bound FAs or via the lipoprotein
lipase-dependent degradation of very-low-density lipoprotein
(46). FA intracellular uptake is
facilitated through different carriers and proteins, such as fatty
acid transporter protein 1, plasma membrane-associated fatty
acid-binding protein, and long-chain fatty acid transporter and
fatty acid translocase CD36 (FAT/CD36). Subsequently, FAs enter the
mitochondria or peroxisomes (they process long-chain fatty acids
and branched-chain fatty acids) through CPT1 and ABCD1 and are
catabolized (46) through β- and
α-oxidation, supplying the mitochondria with fuel substrates
(47). The β-oxidation rate and
the levels of its main product, acetyl-CoA, dictate the energy
cellular demand, since the lack of ATP to cover increased cellular
needs results in the enhanced tricarboxylic acid (TCA) cycle
activity OXPHOS. Similarly, NADH and acetyl-CoA decrease leads to
β-oxidation flux stimulation (48). On the other hand, the level of
CPT1 activity significantly determines the β-oxidation rate in
cardiac or skeletal muscle (49).
As aforementioned, the end-products of β-oxidation are acetyl-CoA,
which enters the TCA cycle, and NADH and FADH2, that are
required for the proper flow of electrons in the ETC, providing the
necessary gradient for F-ATPase to produce ATP energy molecular
coins (50).
2. Diets
High-fat diet (HFD)
A HFD was the mainstream diet followed as a dietary
habit in the 1980s, even though an introductory study that focused
on its effects on health was assessed back in 1958 from Ancel Keys
and is termed the Seven Countries Study (51), indicating that this dietary habit
had already been incriminated. HFD is referred to a diet in which
at least 30-35% of the amount of total calories are derived from
both unsaturated and saturated fats (52) (https://health.gov/our-work/food-nutrition/previous-dietary-guidelines/1980).
In addition to the popular processed foods, numerous other foods
have a high fat content, including but not limited to animal fat,
chocolate, butter and oily fish. Commonly higher in fat content,
the majority of processed foods are easier to obtain as they are
normally more economical considering socioeconomical factors, such
as a lower family income. A number of dishes among different
cultures and ethnicities, such as fried foods or 'soul food'
contain ingredients with a high fat content, such as oils, butters
and fats to increase flavor and appeal. A HFD is not a common
everyday diet for humans, but rather an experimental protocol with
which to create a disease model in animals and mimic the metabolic
adaptations that this creates in cellular physiology (53,54). More factors, such genetic and
environmental factors can contribute to obesity, generating a more
perplexed field that warrants further attention.
HFD leads to obesity and is responsible for the
induction of insulin resistance, which is one of the most critical
pathophysiological manifestations (55). This pathophysiology is emerging
since the capacity of non-adipose tissues for lipid storage is met,
and additionally, lipids cause lipotoxicity that affects cellular
function and cell fate. In white adipose tissue, excessive energy
intake causes tissue hypertrophy and the hyperplasia of adipocytes
(56). The latter manifestations
stimulate lipolysis in fat cells and ultimately lead to elevated
circulating levels of free fatty acids (57). As aforementioned, an increase in
FA oxidation in the mitochondria is responsible for elevating lipid
catabolism and energy production, through β-oxidation and Krebs
cycle respectively (58).
Adiponectin, leptin, acylation stimulating protein and resistin,
which are hormones that are secreted by adipocytes play a critical
role in regulating mitochondrial biogenesis and insulin sensitivity
(59-62). During HFD consumption, adiponectin
levels are decreased, leading to the diminished activation of the
cellular pathways that usually stimulate. More specifically,
adiponectin physiologically binds to its receptors, AdipoR1
(abundantly expressed in skeletal muscle and AdipoR2, activating
AMPK, which finally leads to the stimulation of glucose uptake and
FA oxidation (63). AMPK has also
been implicated in the regulation of PGC-1α, the master regulator
of mitochondrial biogenesis (62). More specifically, AMPK can
directly interact and phosphorylate PGC-1α, thus increasing its
transcriptional activity (64).
Mitochondrial biogenesis during HFD
Impaired mitochondrial biogenesis is one of the
well-described pathophysiological adaptations that a HFD promotes
(Table I). More specifically, in
a previous study, insulin-sensitive male mice that were fed a HFD
for 3 days exhibited decreased PGC-1a mRNA transcript levels in
skeletal muscle (65), indicating
a mechanism through which a HFD decreases the expression levels of
genes that are necessary for OXPHOS and mitochondrial biogenesis.
The prolonged downregulation of this molecular reprogramming can
lead to mitochondrial dysfunction that is found in prediabetic
conditions and insulin resistance, that eventually leads to
insulin-dependent type 2 diabetes (T2D). In line with the above
finding, in another study, C57Bl/6 mice treated with HFD for 3
weeks also exhibited reduced PGC1a mRNA and protein levels in
skeletal muscle (65).
Furthermore, as previously demonstrated 6-week-old male
Sprague-Dawley rats that were fed a HFD for 28 weeks (60 kcal% fat)
exhibited a reduced mtDNA copy number, suggesting impaired
mitochondrial biogenesis in the myocardium (66). A reduction in mitochondrial
abundance and mitochondrial dysfunction can eventually lead to both
systolic or diastolic heart failure (heart failure with reduced
ejection fraction and heart failure with preserved ejection
fraction) and both can cause mortality if they remain undiagnosed
and untreated. Following that, another study revealed that the gene
expression levels of PPARGC1α were diminished in the cardiac tissue
of 6-week-old Wistar rats fed a HFD (45 kcal% fat) for 10 weeks
(67). Another study also linked
mitochondrial abnormal function with diastolic dysfunction.
However, male and female 2-month-old Wistar rats that were fed a
HFD for 26 weeks had increased PGC-1α and mitochondrial
transcription factor A (TFAM) protein levels in the gastrocnemius
muscle (62). The same study
suggested that the effects of a HFD on mitochondrial biogenesis may
be sex-dependent, since male rats exhibited a greater increase in
PGC-1α and TFAM levels compared to females (62). Similar findings were demonstrated
in a study in which 8-week-old male C57BL/6 mice and male Wistar
rats exhibited increased PGC-1α protein levels in skeletal muscle
following 5 or 20 weeks of being fed a HFD (68). Furthermore, another study
suggested that augmented lipid availability and decreased muscle
mitochondrial fatty acid oxidative capacity due to the HFD could
not generate insulin resistance and elevated intramuscular lipid
abundance. Nonetheless, for a shorter period if time under a HFD (4
weeks), PGC-1α protein levels have been found to increase with no
concomitant elevation in PPARGC1a mRNA levels in the skeletal
muscle of male Wistar rats. This is related to the activation of
PPARδ, due to increased free fatty acids that mediate a
post-transcriptional increase in PGC-1α levels (69). These findings argue with the
hypothesis that an impaired mitochondrial content is responsible
for the development of insulin resistance.
 | Table ISummary table indicating the
mitochondrial manifestations and the molecular targets that are
affected by the high-fat diet. |
Table I
Summary table indicating the
mitochondrial manifestations and the molecular targets that are
affected by the high-fat diet.
High-fat diet
|
|---|
| Phenotype | Tissue (host) | Molecular
target | (Refs.) |
|---|
| Biogenesis | Skeletal
muscle
C57BL/6J mice | ↧PPARGC1a mRNA and
protein levels | (63) |
| Biogenesis | Cardiac
muscle
Sprague-Dawley rats | ↧mtDNA copy
number | (64) |
| Biogenesis | Cardiac
muscle
Wistar rats | ↧PGC-1a mRNA and
protein levels | (65) |
| Biogenesis | Skeletal
muscle
Wistar rats | ↥PGC-1α and TFAM
protein levels | (60) |
| Biogenesis | Skeletal
muscle
C57BL/6J mice and Wistar rats | ↥PGC-1α
protein | (66) |
| Biogenesis | Skeletal
muscle
Wistar rats | ↥PGC-1α
protein
↥PPRAGC1a mRNA levels | (67) |
| Function | C57Bl/6 mice | ↧ OXPHOS-related
mRNA transcripts
↧ cytochrome c protein levels | (63) |
| Function |
Cardiomyocytes
Sprague-Dawley rats | ↧ mitochondrial
respiration
↧ citrate synthase activities
↧ ATP
↥ AMP/ATP ratio | (58) |
| Function | Skeletal
muscle
C57BL/6 mice | ↥ protein
expression of all subunits from the respiratory chain | (66) |
| Function | Skeletal
muscle
Wistar rats | ↥COX1, COX4, UCP3
and Cyt b protein levels | (67) |
| Function |
Myocardium
Wistar rats | ↧ mRNA expression
levels of the OXPHOS genes
↧ protein expression of NDUFB5 | (65) |
| Dynamics |
Myocardium
Sprague-Dawley rats | ↥ FIS1 protein | (64) |
| Dynamics | Diaphragm
C57BL/6 mice | ↧ MFN2
protein
↥ DRP1protein | (70) |
| Dynamics | Skeletal
muscle
Sprague-Dawley rats | - MFN2
protein
↥ DRP1 and FIS1proteins | (71) |
| Dynamics | Skeletal
muscle
C57BL/6 mice | - MFN1
protein
↥ MFN2 and OPA1 protein | (72) |
| Mitophagy | Skeletal
muscle
non-obese sedentary male subjects
and endurance-trained male runners | - PINK1 | (73) |
| Mitophagy |
Cardiomyocytes
Tg-Mito-Keima transgenic mice | ↥ acidity
(mitochondria come into contact with the acidic milieu of lysosomes
during mitophagy) | (74) |
| Mitophagy | Cardiac
muscle
C57BL/6 mice | ↧ PARKIN
protein
- PARKIN mRNA | (75) |
Mitochondrial function during HFD
As aforementioned, a prolonged period on a HFD
affects the substrate availability that mitochondria can utilize,
and thus, mitochondria need to adapt their molecular machinery to
generate ATP efficiently in sufficient amounts, particularly in
high-energy consuming tissues, such as the brain and myocardium.
Numerous studies have tried to shed light on the effects of a HFD
for different durations upon mitochondrial function in various
tissues (Table I). Even though
differentiated levels of substrates enter the cellular environment
due to insulin resistance, master regulators of
mitochondrial-substrate utilization are highly affected. More
specifically, FOXO1 and PPARa transcriptionally control glucose and
fatty acid utilization (61,70). Recently, KLF5, another
transcriptional factor, completes a triangle that these
transcriptional factors form, in order to orchestrate substrate
utilization and mitochondrial function during diabetic
cardiomyopathy (71). Moreover,
C57Bl/6 mice fed a HFD have been shown to have OXPHOS-related mRNA
transcripts followed by decreased cytochrome c protein
levels (65), indicating that not
only reduced mitochondrial abundance, as aforementioned, but also
the decreased expression of genes related to mitochondrial function
can result in diastolic and/or systolic dysfunction in
insulin-resistant pathophysiological states. Similarly, a reduced
mitochondrial respiration, complex I-III and citrate synthase
activities have been detected in HFD-fed (60% kcal) rats (66), constituting important findings
that justify an impaired cardiac function. The previous findings
are also supported by ATP depletion, followed by an increase in the
AMP/ATP ratio, that has been observed in the mitochondria of
cardiomyocytes (66). By
contrast, a study in which 8-week-old male C57BL/6 mice were fed a
HFD (45% kcal) for 5 or 20 weeks, demonstrated elevated levels for
all subunits from the respiratory chain in the skeletal muscle
(68), most probably as a
compensatory mechanism of impaired efficiency. Additionally, HFD
administration for 4 weeks in male Wistar rats has been shown to
increase the levels of proteins involved in the OXPHOS, such as
cytochrome c oxidase (COX)I, COXIV, uncoupling protein (UCP)
3 and cytochrome b (69). In
another study, the mRNA expression levels of the OXPHOS genes were
shown to be significantly decreased in the myocardium of 6-week-old
Wistar rats. In addition, the protein expression of NDUFB5 (complex
I), one of the OXPHOS subunits in the mitochondria, was found to be
lower in the HFD group (67).
Even though some studies, as aforementioned, have demonstrated an
increase in the OXPHOS complex, possibly as a compensatory
mechanism in order to allow mitochondria to produce more energy,
the HFD appears to impair mitochondrial function, affecting
cellular energetics and ATP turnover.
Mitochondrial dynamics during the
HFD
Mitochondrial fusion and fission are cellular
processes that numerous studies have tried to shed light under the
HFD (Table I). As previously
noted, Chen et al (66)
examined the impact of the HFD on the mitochondria of rat
cardiomyocytes. Male Sprague-Dawley rats fed a HFD (60% kcal)
exhibited elevated levels of mitochondrial FIS1 (66). In another study, 4-week-old
healthy male C57BL/6 mice fed a HFD for 16 weeks exhibited a
reduced relative expression of mitochondrial fusion protein 2
(MFN2), while the expression of mitochondrial DRP1 was detected at
higher levels (72). Another
previous study demonstrated that in 8-week-old male Sprague-Dawley
rats fed a HFD for 6 weeks, changes in the expression levels of a
protein related to mitochondrial fusion and fission were observed.
More specifically, elevated levels of DRP1 and FIS1 were assessed
in the HFD-treated group, while MFN2 levels remained unaltered
(73). That study suggested that
the HFD and low-intensity endurance training modulated
mitochondrial dynamics, contributing to insulin sensitivity,
whereas they failed to generate an additive effect on mitochondrial
biogenesis. In line with the above, no change in the MFN1 protein
levels were detected in male C57BL/6 mice (4-week-old) fed a HFD
(74). However, MFN2 and OPA1
protein levels were assessed at a lower level in the HFD-fed mice
(74). These findings suggest
that moderate aerobic training can only mitigate insulin resistance
and mitochondrial dysfunction that are induced by obesity.
Mitophagy during HFD
The protein content of total PINK1 in skeletal
muscle has been detected at similar levels between groups before
and after a high-fat meal in healthy non-obese, sedentary males and
10 endurance-trained male runners (75), suggesting that mitophagy is not
necessary for metabolic flexibility in the healthy population.
Studies concerning the effects of the HFD on mitophagy markers have
also been conducted in transgenic mice that exclusively express
Mito-Keima in cardiomyocytes. Mito-Keima Red is a fluorescent
oligopeptide of which the excitation shifts to higher wavelength
when mitochondria are placed in an acidic environment, such as
lysosomes during mitophagy. Wild-type and transgenic Tg-Mito-Keima
mice fed a HFD (60 kcal% fat) for 2 months, have been shown to
exhibit mitophagy (elevated LC3II levels), followed by lower
mitochondrial abundance (mtDNA/nuDNA) (76). Additionally, ATG7 appears to play
critical role in mitophagy stimulation under a HFD since ATG7-KO
mice have been shown to exhibit an alleviated phenotype after the
respective treatment protocol (76). The aforementioned study suggests
that mitophagy in cardiomyocytes is a quality control process
during HFD consumption, and its activation can serve as a
therapeutic target against HFD-induced diabetic cardiomyopathy. In
another study, impaired mitophagy activity was observed in the
myocardium of C57BL/6 mice fed a HFD for 24 weeks, which was driven
through a reduction of PARKIN in the mitochondria that were
supposed to be recruited by PINK1. Moreover, the HFD failed to
alter Parkin mRNA levels, suggesting that possible
independent transcriptional mechanism(s) are involved (77).
Caloric restriction (CR)
CR that has been studied since the beginning of the
20th century (78), is a
nutritional approach that reduces calorie consumption without
leading to malnutrition. Additionally, it is worth mentioning that
during CR, adequate levels of vitamins and minerals should be
maintained. CR typically involves a chronic reduction (20-30%) in
energy intake from the standard calorie intake (79,80). In accordance with the current
scientific literature, the term CR is often used interchangeably
with dietary restriction (DR). However, CR is a partial example of
DR as DR protocols include CR (81). CR has been shown to improve health
and extend longevity in several studies in organisms with varying
levels of complexity, ranging from yeast to humans (79,80,82-85). In contrast to starvation, the CR
diet is able to lower glucose levels and increase ketone bodies
levels in the plasma within normal physiological ranges (86).
Mitochondrial biogenesis during caloric
restriction
Several studies have shed light on the effects of CR
on mitochondrial biogenesis (Table
II). Initially, CR has been studied in vitro or ex
vivo to examine the effects on the molecular level and the
distinctive pathways that are affected. Studies performed in both
skeletal muscle C2C12 cell line or murine primary skeletal
myoblasts treated under CR conditions have shown that the
NAD+/NADH ratio is increased, leading to subsequent
SIRT1 activation (87). That
study highlights that AMPK, NAMPT and SIRT1 are necessary molecules
of a functional pathway that permits cells from skeletal muscle
origin to sense and react to substrate deprivation and
availability. It is known that an increased SIRT1 activity
triggered by elevated NAD+ levels can increase the
transcriptional activity of PGC-1α (87) and FOXO1 (88), stimulating multiple pathways that
need to be activated due to energy depletion and refer mainly to
proliferative and metabolic processes. In a previous study,
following PGC-1a and FOXO1 activation, both NRF1 and NRF2βγ
transcript levels were found to be increased after CR for 24 h in
HeLa cells (89). However, the
same study revealed that CR treatment did not induce PPARα levels
(89). Therefore, the
mitochondria can adapt their bioenergetics under CR conditions and
at the same time are able to stimulate mechanisms to reduce
oxidative damage. Moreover, primary hepatocytes from 12-month-old
male Fischer 344 rats that have received CR (40% restriction) have
exhibited elevated expression levels of PGC-1α and PPARα (89). In line with the above, in
vivo studies have revealed that the CR (30% restriction) diet
in male C57BL/6 mice for 1, 9 and 18 months have increased PGC-1a
protein levels in the skeletal muscle (90). Following that, the mRNA of several
mitochondrial-relevant proteins, such as NRF1, Core 1, COXIV, Atps
and COX content, have also been found to be elevated (90). Moreover, the SIRT3 (mainly
expressed in the mitochondria) protein levels have been found to be
increased in the skeletal muscle of C57BL/6 male mice after 12
months on the CR diet (91).
SIRT3 activation is able to stimulate PGC1a through the CREB or
AMPK signaling pathway (91),
inducing the mitochondrial biogenesis molecular program. Of note,
the CR diet in F344BNF1 male rats has been shown to result in the
complete prevention of the age-related reduced levels of PGC-1α in
the liver (92). Thereafter CR
can be used as a therapeutic approach to alleviate age-dependent
mitochondrial confinement. In agreement with this finding, PGC-1α
activation has also been found in the liver of 12-24-month-old male
Fischer 344 rats that have been on a CR diet for 25 months
(93). Finally, the effects of CR
on mitochondrial biogenesis have also been assessed in humans. In
healthy and overweight participants on the CR diet for 6 months,
the expression levels of SIRT1 and PPARGC1A in skeletal muscle were
found to be elevated in non-obese young participants (94). The same study found that the
expression of the PGC-1a target TFAM was increased, a protein that
is a key activator of mitochondrial transcription and genome
replication which is also used as a determinant for mitochondrial
abundance (94).
| Table IISummary table indicating the
mitochondrial manifestations and the molecular targets that are
affected during the caloric restriction diet. |
Table II
Summary table indicating the
mitochondrial manifestations and the molecular targets that are
affected during the caloric restriction diet.
Caloric restriction
|
|---|
| Phenotype | Tissue (host) | Molecular
target | (Refs.) |
|---|
| Biogenesis | HeLa cells | ↥mRNA NRF1 and
NRF2
- PPARα protein | (87) |
| Biogenesis | Primary
hepatocytes
Fischer 344 rats | ↥ PGC-1α and PPARα
proteins | (87) |
|
Biogenesis/function | Skeletal
muscle
C57BL/6 mice | ↥ PGC-1a
protein
↥ mRNA Nrf1, Core 1, Cox IV cytochrome c oxidase | (88) |
|
Biogenesis/function | Skeletal
muscle
C57BL/6 mice | ↥ SIRT3
protein | (89) |
| Biogenesis | Liver
F344BNF1 rats | Prevents the
age-related reduced levels of PGC-1α | (90) |
| Biogenesis | Liver
Fischer-344 rats | ↧ PGC-1a
protein | (91) |
|
Biogenesis/function | Skeletal muscle
human | ↥ mRNA SIRT1 and
PPARGC1A | (92) |
| Function | Skeletal
muscle
B6C3F1 mice | ↧ Complex I, III,
and IV activity | (96) |
| Function | Skeletal and
cardiac muscle
F344BN rats | ↧ Complex IV
activity | (99) |
| Function | Cardiac
muscle
Wistar rats | ↧ Complex I and III
activity | (98) |
| Function | Skeletal
muscle
F344BN rats | ↥ High-affinity
binding sites of complex IV | (99) |
| Function | Skeletal
muscle
C57BL/6 mice | - Citrate synthase
activity
↥ mRNA levels of several mitochondrial associated proteins
(PPARGC-1α, NRF1, Core 1, COXIV, Atps) and cytochrome c oxidase
content | (88) |
| Function | Skeletal
muscle
Sprague-Dawley rats | ↥ mRNA transcripts
associated with mitochondrial ATP production (subunits of
cytochrome-c oxidase; COXI, II, III, IV, Va and VIII, and NADH
dehydrogenase | (100) |
| Function | Skeletal and
cardiac muscles
Fischer 344 rats | - ATP content | (101) |
| Function | Adipocytes
overweight
healthy men and women | ↧ Expression of
genes encoding ATP synthase subunits, cytochrome c oxidase, NADH
dehydrogenase | (102) |
| Function | Skeletal
muscle
obese men and women | ↥ Citrate synthase
activity | (103) |
| Function | Skeletal muscle of
overweight
male and female subjects | - Activity of
beta-hydroxyacyl-CoA dehydrogenase, citrate synthase, and
cytochrome c oxidase II | (92) |
| Function | Skeletal muscle of
non-obese
male and female subjects | - Activity of
citrate synthase, beta-hydroxyacyl-CoA dehydrogenase, and
cytochrome c oxidase II | (92) |
| Dynamics | Liver and skeletal
muscle
C57BL/6J mice | ↥ MFN2 levels in
liver
↥ MFN1 and NRF1 levels in the skeletal muscle | (108) |
| Dynamics | Skeletal
muscle
Male C57BL/6 mice | ↥ MFN2 and OPA1
protein levels
↥ DRP1 protein levels | (105) |
| Dynamics | Soleus and
gastrocnemius
male Sprague-Dawley rats | ↥ MFN2 protein
levels
↥ DRP1 protein levels
| (109) |
| Dynamics |
Gastrocnemius
male Sprague-Dawley rats | ↥ OPA1 and MFN1
proteins levels | (112) |
| Dynamics |
Hepatocytes
male C57BL/6 mice | No marked effects
on the expression levels (DRP1, MFN1, MFN2 and OPA1) | (110) |
| Dynamics |
Hepatocytes
male C57BL/6 mice | ↥ FIS1 and DRP1
proteins levels no change in MFN1, MFN2, and OPA1 protein
levels | (111) |
| Mitophagy | Skeletal
muscle
C57BL/6 mice | ↥ PINK1 and PARKIN
protein | (105) |
| Mitophagy | Kidney
Fischer 344 rats | ↥ LC3/ATG8 and
BNIP3 protein
↧ PINK1 protein | (106) |
| Mitophagy | Primary hepatocytes
C57BL/6 and GFP-LC3 transgenic male mice | ↧ Mitochondrial
membrane potential | (107) |
Mitochondrial function during caloric
restriction
The activation of AMPK and SIRTs followed by the
concomitant inhibition of mTOR downstream pathways are crucial
molecular events that the CR diet is able to promote in order for
cells to fine-tune oxidative metabolism, as well as the
mitochondrial biogenesis and turnover (Table II). It is well established that
AMPK and PGC-1α are key molecules that orchestrate these pathways
(95). CR is also known to lower
reactive oxygen species (ROS) production through enhanced
mitochondrial aerobic metabolism and to increase the activity of
antioxidant enzymes in the cardiovascular system and the skeletal
muscle (96,97). In the gastrocnemius derived from
10-month-old B6C3F1 female mice fed a CR diet (40%
restriction), the activities of complexes I, III, and IV were found
to be diminished (98). In line
with the that study, 8- to 10-month-old rats fed a CR diet for
4.5-6.5 months exhibited lower complex IV activity in the
mitochondria in both skeletal and cardiac muscle (99). That study suggested that even
though the CR diet can attenuate the decline in mitochondrial
function that is related to cellular aging, this beneficial effect
is insignificant compared with the impact that the CR diet has upon
mitochondrial biogenesis. In line with that previous study, in
another study, the CR diet (40% restriction) for 3 months in male
and female 5-month-old Wistar rats was shown to result in a
decrease at the activities of both complexes I and III (100). This adaptation to the CR diet
can justify the lower mitochondrial-derived superoxide formation
and the effect that the CR diet has upon age-related disorders. The
above has been also confirmed by the higher complex IV efficiency
in response to long-term CR administration that has been described
in the skeletal muscle of F344BN rats due to an increase in
high-affinity binding sites of complex IV (101). In another study, the CR (30%
restriction) diet in male C57BL/6 mice for 1, 9 and 18 months did
not result in any effect on citrate synthase activity (90). By contrast, the mRNA levels of
several mitochondrial-associated proteins (Ppargc-1α, NRF1, Core 1,
COXIV and Atps) and the COX content were increased in skeletal
muscle (90). Following that,
another study revealed that male Sprague-Dawley rats that fed the
CR diet for 36 weeks exhibited an increase in the expression levels
of genes associated with mitochondrial ATP production (six subunits
of COX; COXI, II, III, IV, Va, VIII and NADH dehydrogenase) in
skeletal muscle (102). The
CR-related increase that has been reported, particularly in genes
that encode proteins related to oxidative stress scavenging, may
contribute to the impact of CR on cellular longevity. On the other
hand, different levels of CR diets in male Fischer 344 rats
throughout their life (10% restriction; at 3.5 months, 25%
restriction; at 3.75 months, 40% restriction; at 4 months until
mortality) failed to enhance or alleviate the age-related decrease
in the ATP content of the mitochondria isolated from gastrocnemius
muscle (103). The effect of the
CR diet on mitochondrial function has also been assessed in humans.
Firstly, overweight healthy males and females on the CR diet (25%
restriction) for 6 months exhibited a downregulation in the levels
of essential genes encoding subunits of ATP synthase, COX and NADH
dehydrogenase in adipocytes (104). By contrast, in aged obese males
and females (60-75 years old) that followed a CR diet, an increase
in citrate synthase was observed in skeletal muscle (105). Another study revealed that
healthy male and female overweight participants on a CR diet (25%
restriction) for 6 months, did not exhibit an altered activity of
β-hydroxyacyl-CoA dehydrogenase, citrate synthase and COXII
(94). Finally, CR in overweight
non-obese participants did not lead to changes in the levels of
citrate synthase, β-hydroxyacyl-CoA dehydrogenase and COXII levels
(94). These results indicate
that the CR has a broad effect on the transcriptome in different
tissues, although it does not improve mitochondrial function in
overweight and obese participants.
Mitophagy during caloric restriction
The CR is one of the most potent non-genetic
triggers for initiating the mitophagy process (106) (Table II). Specifically, the highest
values of PINK1 and Parkin have been detected in the skeletal
muscle of male 10-week-old C57BL/6 mice fed the CR diet for 18
months (107). In line with that
previous study, in another study, the expression of the
autophagosome formation marker, LC3/Atg8, and BNIP3 was increased,
while that of PINK1 was markedly decreased in the kidneys of male
3-month-old Fischer 344 rats after 20 months of CR treatment
(108). Moreover, nutrient
deprivation induces mitophagy in primary hepatocytes from wild-type
and GFP-LC3 transgenic male mice (109). The aforementioned findings
suggest that the CR diet stimulates the mitophagy program in cells
in a possible attempt to discard energy consuming and not fully
functional mitochondria.
Mitochondrial dynamics during CR
As it is expected, a lack of energy intake is able
to affect mitochondrial dynamics, inducing the fusion phenotype in
mitochondria (Table II). A
previous in vivo study using male C57BL/6 mice fed a CR diet
(40% restriction) for 6 months revealed increased levels of MFN2
and OPA1 in skeletal muscle (107). A similar finding has also found
in mice fed a CR diet (40% restriction) with less calories, that
expressed increased levels of MFN1 and NRF1 (110). Additionally, the prolonged
duration of CR was found to have no effect on mitochondrial fusion,
proposing that initial energy deprivation led to a rather early
control of mitochondrial fusion (107). On the contrary, 18 months of CR
has been shown to result in higher expression levels of DRP1, which
as aforementioned, is able to stimulate mitochondrial fission.
Thereafter, distinct molecular signatures are required to
orchestrate and control mitochondrial fission and fusion dynamics
in mice fed a CR diet that also depend on the duration of the diet
per se (107). The CR diet is
also able to further induce mitochondrial fusion in the skeletal
muscle of aged mice (in which mitochondrial fusion has already been
induced) (111). On the
contrary, the CR diet can partially inhibit the induction of
mitochondrial dynamics in the glycolytic muscles of aged mice
(111). This discrepancy
suggests that the anti-aging effects of the CR diet in
mitochondrial dynamics may be restricted to highly oxidative
muscular fibers. The effects of the CR diet have also been studied
in the liver. C57BL/6 mice that fed a CR diet (40% restriction)
have been shown to exhibit no change in mitochondrial fusion
related proteins in hepatocytes (112,113). On the contrary, the study
showcased an elevation at DRP1 levels in mitochondria-enriched
fractions of hepatocytes (113),
indicating that a similar CR diet is able to affect mitochondrial
dynamics differently between tissues. Another study revealed that
the CR diet was not able to change the mitochondrial abundance in
hepatocytes, but stimulated MFN2 levels (110).
The effects of the CR diet have been also studied in
combination with exercise. As previously demonstrated, initially,
the CR diet was able to decrease the impact of resistance training
in muscle hypertrophy since rats that received both the CR diet and
resistance training exhibited elevated fusion-related proteins
levels (OPA1 and MFN1), while no effect was exerted on
fission-regulatory proteins (FIS1 and DRP1) (114). That study suggests that CR
promotes mitochondrial adaptation in the skeletal muscle, since it
attenuates the effect of resistance training upon muscle
hypertrophy. Finally, another study demonstrated that both CR and
exercise training in obese rats was able to alleviate reduced
mitochondrial fusion stimulated by HFD, since they induced MFN2
expression and reduced the phosphorylation of DPR1 protein at
Ser616 (115). In
this case, the CR diet appears to act synergistically with exercise
against the HFD-induced mitochondrial fusion.
Ketogenic diet (KD)
The KD was first applied to a patient with epilepsy
back in the 1920s (116). The KD
is a diet consisting of low carbohydrates, a high-fat content, and
moderate amounts of protein. Different types of the KD type have
been introduced in the scientific literature, including i) the
classic DK (CKD); ii) the less restrictive 'modified Atkins diet'
(mAD); and iii) the 'medium-chain triglyceride' KD (MCT-KD)
(117). The CKD refers to the
diet in which every 4 g of fat accounts for 1 g of protein plus
carbohydrates, reducing the carbohydrates (118). mAD has a different composition
since every 1 g of fat corresponds to 1 g of protein plus
carbohydrates (119). The ratio
of MCT-KD is similar to that of CKD, although the ketone bodies are
generated efficiently as the AMPA receptors are directly inhibited
by the medium-chain triglycerides that are used in MCT-KD.
Consequently, cellular energetics are shifted towards mitochondrial
biogenesis (120).
Generally, the KD can alter the organism's metabolic
state and shift its reliance from carbohydrates to FAs. This
adaptation also leads to an increase in FA oxidation,
gluconeogenesis and ketogenesis, with produced ketone bodies
entering the blood circulation (121). Acetoacetate, β-hydroxybutyrate
(β-HB) and acetone are the major forms of ketone bodies generated
during the KD (117).
Furthermore, the KD can stimulate numerous pathways, upregulating
proteins that participate in biochemical systems related to
cellular bioenergetics, such as the TCA cycle (citrate synthase,
malate dehydrogenase), the OXPHOS system (CI, CII, CIII, CIV, CV
and cytochrome c) and to FA oxidation [carnitine
palmitoyl-transferase, medium-chain acyl-CoA dehydrogenase (MCAD),
long-chain acyl-CoA dehydrogenase, very-long-chain acyl-CoA
dehydrogenase, β-hydroxyacyl-CoA dehydrogenase] and ketolysis
(β-hydroxybutyrate dehydrogenase) (122).
Mitochondrial function during the
ketogenic diet
Generally, the KD has been reported to stimulate
pathways that induce mitochondrial abundance, enhance mitochondrial
performance and regulate antioxidant mechanisms (123-126) (Table III). Low-level redox signaling
molecules, such electrophiles and H2O2 can
stimulate adaptive pathways such as the protective transcription
factor, NF E2-related factor 2 and as a result, lead to the
increased production of antioxidants (e.g., glutathione) and
detoxification enzymes (120).
Moreover, the KD has been found to increase cytosolic and
mitochondrial protein acetylation and alter protein succinylation
patterns (121). More
specifically, male mice fed the KD have been shown to exhibit
increased mitochondrial FA oxidation in the liver, while a
reduction of markers of hepatic de novo lipogenesis has been
detected in comparison to mice that fed a high fat/high sucrose
'western' diet (WD) (126).
Furthermore, another study revealed that the KD for 4 months
increased mitochondrial activity in younger animals (127). On the contrary, the prolonged
duration (14 months) of the KD was found to be required to increase
mitochondrial mass and function in elder animals in comparison with
mice fed the isocaloric diet (127). It was suggested that the
improvement of mitochondrial function due to the KD may contribute
to the improved longevity; hence, no direct link has been
determined between enhanced mitochondrial function with muscle
mass, strength, and muscular aging. In line with this previous
study, increased mitochondrial mass was also observed in the study
by Parry et al (128).
More specifically, increased citrate synthase activity that
corroborates with a higher mitochondria volume was found in the
liver and gastrocnemius of rats fed the KD compared to rats fed
standard chow. Additionally, the KD-fed rats had a higher median
lifespan (128). In another
study, rats fed the KD and exposed to resistance exercise training,
exhibited more efficient coupling of complex II substrates in the
skeletal muscle mitochondria compared with the control group that
underwent the same exercise regime but received the isocaloric
western diet (125). Those
authors suggested that their insights may reflect unknown
biochemical connections between axes involving ketone bodies or
medium-chain triglycerides that modulate mitochondrial function
(122). Another study revealed
that in the skeletal muscles of aged male C57BL/6 mice, an increase
in the levels of markers of mitochondrial content (citrate
synthase, complex I and complex IV activity) was observed following
long-term exposure to a KD (14 months) (129). On the contrary, in another
study, the long duration of the KD in 4-month-old male rats was
shown to result in lower gastrocnemius maximal citrate synthase
activity and caused damage to the respiratory control in the
gastrocnemius myotubes, while it did not alter mitochondial
abundance and quality in the brain and liver (130). Similarly, a decrease has been
documented in cytochrome c and complex IV levels in the
hepatocytes of C57Bl/6 male mice that fed the KD (124). The aforementioned results
suggest that the short-term use of the KD can promote mitochondrial
biogenesis and/or alter mitochondrial physiology in specific
tissues, with skeletal muscle being susceptible to KD-induced
alterations (127). Due to
contradictory findings, further investigations are required in the
future to shed more light and delineate the net impact of KD on
mitochondrial mass and function.
| Table IIISummary table indicating the
mitochondrial manifestations and the molecular targets that are
affected during the ketogenic diet. |
Table III
Summary table indicating the
mitochondrial manifestations and the molecular targets that are
affected during the ketogenic diet.
Ketogenic diet
|
|---|
| Phenotype | Tissue (host) | Molecular
target | (Refs). |
|---|
|
Biogenesis/function | Cardiac muscle
Sprague-Dawley rats Interscapular brown adipose tissue (IBAT)
C57BL/6 mice | ↥ Mitochondrial
number
↥ Electron transport chain proteins UCP1 protein
↥ AMPK, PPAR-γ, PGC-1α proteins | (137) |
| Biogenesis | Liver Wistar
rats | ↥ mRNA PPARGC1α and
TFAM | (139) |
| Biogenesis | Skeletal muscle,
liver, brain from Fisher Rats and mice | - PGC-1α protein
levels | (124,126) |
|
Biogenesis/dynamics | Skeletal muscle
C57BL/6 mice | ↥ PGC-1α
protein
↥ Mitochondrial fission/fusion related genes | (141) |
| Function | Liver male C57BL/6
mice | ↥ Mitochondrial
fatty acid oxidation
↥ de novo lipogenesis | (124) |
| Function | Skeletal muscle
C57BL/6 mice | ↧ Mitochondrial
activity (TFAM, SIRT1, SIRT3, PGC-1a) in younger animals
↥ Mitochondrial mass (level of OXPHOS) and function (citrate
synthase activity) in elder animals | (125) |
| Function | Liver and skeletal
muscle Fisher rats | ↥ Citrate synthase
activity
↥ Mitochondrial volume | (126) |
| Function | Skeletal muscle
Sprague-Dawley rats | ↥ Efficiency of
coupling of complex II substrates (respiratory control ratio of
isolated mitochondria) | (123) |
| Function | Liver C57Bl/6
mice | ↧ Hepatic
cytochrome c and complex IV levels | (122) |
| Function | Skeletal muscle
Fisher rats | ↧ citrate synthase
activity | (128) |
|
Function/dynamics | Cardiac muscle
type-2 diabetic db/db mice | ↧ Mitochondrial
fission (mitochondrial number, size)
↥ PI3K, p-Akt, Akt, mitochondrial respiratory control ratio, ATP
content | (134) |
| Function | Myocardium tissue
of male Sprague-Dawley rats | ↧ HDAC2 leading to
the transcriptional activation of SIRT7 | (129) |
| Function | Cybrid cell lines
from a patient with Kearns-Sayre syndrome (KSS) | ↥ Mitochondrial
volume | (130) |
| Function | Brain and muscle
tissue BTBR mice | ↥ Mitochondrial
morphological deformations | (131) |
| Function | Skeletal muscle
from healthy human subjects | ↥ Mitochondrial
respiratory control ratio (mitochondrial O2 consumption
and membrane potential index)
↥ ATP production
↥ ATP/H2O2 ratio | (132) |
| Function | Skeletal muscle
Fisher rats | ↧
H2O2 emission
↥ Mitochondrial respiration
↥↥ Cell viability | (133) |
| Function | Skeletal muscle
Twinkledupl transgenic mice | ↧ Amount of
cytochrome c oxidase negative muscle fibers | (136) |
| Function | Liver male C57BL/6
mice | ↥ mRNA expression
PPARGC1α | (140) |
| Dynamics | Liver C57Bl/6 and
BTBR mice | ↧ Mitochondrial
amount (decrease mtDNA)
↥ BNIP3 mRNA and protein | (143) |
| Dynamics | Skeletal muscle
C57BL/6 mice | ↥ Markers
mitochondrial content (citrate synthase, Complex I, and Complex IV
activity) | (127) |
| Dynamics | Cardiomyocytes of
rabbits with heart failure | ↧ Mitofusin 2
(MFN2) and dynamin-related protein 1 (DRP1) | (144) |
| Dynamics | SH-SY-5Y cells | ↧ Dynamin-related
protein1 (DRP1) | (145) |
| Mitophagy | Hepatocytes of male
and female Wistar rats | ↧ Markers of de
novo lipogenesis (FAS, ACC) | (139) |
| Mitophagy | Liver C57BL/6 and
BTBR mice | ↥ BNIP3
protein | (143) |
It is known that the KD is able to shift cellular
energy production reliance towards FA oxidation and not towards
glycolysis. This is a consequence of the elevated levels of ketone
bodies basically in the liver. Xu et al (131) reported the detrimental effect
that is caused due to extensive KD or frequent deep fasting. The
main manifestations reported were fibrotic lesions in the
myocardium of the rats (131).
This was followed by a reduction in mitochondrial biogenesis and
cellular respiration, while cardiomyocyte apoptosis was found to be
stimulated. At the molecular level, their study added that during
the increase in ketone body β-HB levels, the acetylation of the
histone Sirt7 promoter was maintained due to HDAC2
inhibition. The aforementioned molecular cascade was able to lead
to the transcriptional activation of Sirt7 (131) that inhibited the expression of
genes related to mitochondrial ribosome-encoding and mitochondrial
biogenesis, promoting cardiomyocyte apoptosis and myocardial
fibrosis (131). In a previous
study, β-HB supplementation in cybrid cell lines that contain a 1.9
kb partial deletion of mtDNA (the 'FLP' deletion) derived from a
patient with Kearns-Sayre syndrome (KSS) was found to increase
mitochondria volume (132),
while β-HB treatment improved mitochondrial morphological
imperfections in brain and muscle tissue at BTBR mice (133), which present hallmarks of
idiopathic autism. The authors of that study also reported that
β-HB supplementation increased the diminished mitochondrial size
that was induced in BTBR mice, signifying that larger fused
mitochondria exhibit a higher ATP production and lower ROS
levels.
Miller et al (134) investigated the changes in the
mitochondria of skeletal muscle in healthy individuals who followed
the KD combined with exercise training, focusing on periodized
resistance training, power training and high-intensity interval
training. They observed that the KD increased the mitochondrial
respiratory control ratio (mitochondrial O2 consumption
and membrane potential index), ATP production and the
ATP/H2O2 ratio (134). The latter depicts an increase in
the efficiency of energy production compared with the oxidative
burst. In another study, β-HB treatment in 5-month-old male Fisher
rats resulted an improvement of mitochondrial respiration followed
by less H2O2 generated by the mitochondria,
without affecting mitochondrial abundancy (135). A previous study conducted on
mice that had manifestations of T2D and fed the KD revealed that
the KD was able to reduce the mitochondrial respiratory control
ratio and the ATP content (136). Notably, it has been demonstrated
that homozygous mice with a missense mutation in Med30 that exhibit
cardiomyopathy die soon after weaning, while they are healthy at
the lactation stage (137). When
weaning mice are exposed to a KD, their viability is extended after
weaning, which is attributed to differences in genes involved in
oxidative phosphorylation and mitochondrial integrity (137). Thus, lethal mitochondrial
cardiomyopathy caused by a mutation in Med30 can be partially
reversed through dietary control (137). In a different pathophysiological
in vivo model, the KD was shown to reduce the progression
rate in a disease mouse model (transgenic mouse line) of late-onset
mitochondrial myopathy (138).
More specifically, the KD diminished the amount of COX-negative
muscle fibers, which is a hallmark of mitochondrial respiratory
chain deficiencies. However, the mtDNA quality or quantity remained
unaffected after the KD, while the study highlighted that
mitochondrial biogenesis was stimulated followed by liver lipid
restoration (138). The
aforementioned data suggest that minor mitochondrial dysfunction
can influence skeletal muscle energy metabolism by drifting towards
anaerobic glycolysis. Furthermore, if lipids can enter
mitochondria, then the cells reprogram and use FAs as energy
substrates, boosting β-oxidation. In another study, 8-week-old male
C57BL/6J mice fed D-β-hydroxybutyrate-(R)-1,3 butanediol
monoester [ketone ester (KE)], replacing the carbohydrate diet
content for 1 month, exhibited a higher number of mitochondria and
an increase in ETC proteins levels, UCP1 and mitochondrial
biogenesis-regulating proteins in the interscapular brown adipose
tissue (139). An increase in
mitochondrial UCP expression and activity was also observed by
Sullivan et al (140) in
juvenile mice that received a high-fat KD (HF-KD). In addition, ROS
levels after the HF-KD were found to be decreased, and this
suggested a potent neuroprotection activity of the KD (140). UCP activation is able to remove
protons from mitochondrial protein, decreasing the flow of ETC.
Therefore, UCP activation can indirectly diminish ROS mitochondrial
production, serving as an additional axis through which the KD can
function in order to promote its beneficial effect.
Mitochondrial biogenesis and mitophagy
during the KD
Male C57BL/6J mice and rats fed the KD have been
shown to have elevated PGC-1a and TFAM levels (141,142) in comparison with respective
animals fed a standard chow diet. On the contrary, other studies
have failed to document changes in the PGC-1a levels of sedentary
male rodents in the gastrocnemius, the liver or brain (126,128). In addition, an increase of
PPARGC1a mRNA levels has been observed in the gastrocnemius tissue
of male Sprague-Dawley rats who voluntarily exercised on wheels
regardless of the diet they were fed (WD or KD) (125). It can also be suggested that
exercise training and the KD can correct the high-fat induced
elevated mitochondrial content (122).
The majority of studies conducted on rodents have
used a low-protein KD. As such, recently, Huang et al
(143) investigated the possible
alterations of a normal-protein KD (NPKD) upon the substrate
oxidative capacity in the skeletal muscle of C57BL/6 male mice
which were also introduced to an exercise training regime. The
application of NPKD or exercise training alone could not produce
changes in the mitochondrial content, while their combined
application was found to increase both mitochondrial fission/fusion
markers and PGC-1α levels. Furthermore, the NPKD/exercise training
combination has been found to shift cellular metabolism towards
enhanced lipid utilization, since both mitochondrial and
peroxisomal lipid oxidation are stimulated (143). Even though that study suggested
a mechanistic adaptation of skeletal muscle due to KD, further
studies are warranted to fully elucidate the effects of a KD and/or
exercise on mitochondrial biogenesis and function.
As regards brain metabolism, data obtained from
experiments using male Sprague-Dawley rats have revealed that a
calorie-restricted KD can improve brain metabolism via an
anti-convulsant mechanism involving mitochondrial biogenesis. More
elaborately, increases in transcripts that encode mitochondrial
proteins, mitochondrial abundance and increases in the
phosphocreatine/creatine index followed by higher glutamate levels
have been found (144). It was
also indicated that the calorie-restricted KD was able to promote
brain metabolism and that the KD anti-convulsant mechanism affected
mitochondrial biogenesis with increased energy stores of
alternative substrates (144).
In another study performed on mice with T2D fed the KD,
mitochondrial fission was reduced, while the improvement of overall
mitochondrial function was documented. More specifically, the KD
was found to regulate the expression levels of mitochondrial
dynamics-related proteins (OPA1 and FIS1) (136). Furthermore, in mice fed the KD,
the expression levels of BNIP3 gene (mitophagy regulator gene) have
been found to be increased in the liver; therefore, it was
suggested that the KD may act as a mitophagy activator (145). On the contrary, in another
study, mitophagy was not induced in the hepatocytes of male and
female Wistar rats fed the KD (90, 5% fat) for 7 weeks (141), even though the KD increased the
levels of sequesterosome-1 (p62), a scaffold protein for LC3
(141). Furthermore, another
study that examined the effect of the KD (75% kcal fat) in male
C57Bl/6 and BTBR mice, revealed elevated BNIP3 gene and protein
expression levels in the livers of KD-fed mice, indicating the
potential activation of mitophagy (145).
Mitochondrial dynamics during the KD
In a previous study, in juvenile male C57Bl/6 and
BTBR mice fed the KD (75% fat) for 10-14 days, a decrease in the
levels of mitochondrial proteins in the liver was observed, despite
a concomitant increase in gene expression (145). Additionally, decreased levels of
mtDNA were documented in the liver, indicating a decrease in
mitochondrial abundancy. In the same study, BNIP3 gene and protein
expression levels were found to be increased in the liver, a
hallmark of activated mitophagy, while in the brain, BNIP gene
expression and protein levels remained unaltered (145). Since the KD is characterized by
increased β-HB levels, β-HB supplementation in myocytes from young
and aged mice β-HB has been documented to be beneficial for
mitochondrial repair, even though the expression of MFN2 and DRP1
is reduced by 50%, contributing to impaired fusion-fission process
(146). It has been suggested
that increased β-HB levels can repair mitochondria in the failing
aging myocardium, an effect that is alleviated by the impaired
MFN2-DRP1 axis and the reduced PARKIN levels (146). The mitochondrial translocation
of DRP1, a key regulator of mitochondrial fission, has also been
found to be suppressed in SH-SY-5Y cells, treated with β-HB. As a
result, mitochondrial fission is inhibited during ketone bodies
administration (147),
suggesting that KD restores mitochondrial integrity through the
mitochondrial translocation of DRP1and suppresses ER stress,
exerting neuroprotective effects.
Fasting
Fasting is defined as a voluntary abstinence from
food and drink for specified, recurring periods of time, ranging
from 12 h to 3 weeks in humans (148). Fasting can also be considered a
CR diet in periods of free access to food which are interrupted by
fasting (149). Several studies
that have been performed by other research groups and the authors,
as well as ours indicate that the overall improvement of health
through fasting involves the beneficial modulation of energy
substrates, adaptive cellular stress response, signaling pathways
that enhance mitochondrial health, DNA repair and autophagy
(150-153). Organisms respond to different
types of fasting by minimizing anabolic processes, enhancing stress
resistance, recycling damaged molecules, stimulating mitochondrial
biogenesis, and promoting cell survival, all of which support
improvements in health and disease resistance (150,154). The energy intake, as well as the
fasting duration between meals, can alter the NAD+/NADH,
AMP/ATP and acetyl-CoA/CoA ratios. More specifically, during
fasting, the AMP/ATP ratio is increased, leading to AMPK
activation, and promoting pathways related to cellular repair and
anabolic inhibition. Furthermore, acetyl coenzyme A (CoA) and
NAD+ serve as cofactors for epigenetic modifiers, such
as SIRTs (155). As
aforementioned, SIRTs deacetylate FOXOs and PGC-1α, rendering them
transcriptionally active (156).
Therefore, their activation results in the expression of genes
involved in stress resistance and mitochondrial biogenesis
(154,157).
Mitochondrial biogenesis during
fasting
Several studies have reported that fasting
stimulates mitochondrial biogenesis (Table IV). In fasted C2C12 myotubes,
both PGC-1a and SIRT1 deacetylation are elevated (158). Moreover, the fasting effect on
the muscles of adult male northern elephant seals revealed that the
phosphorylation levels of AMPK and SIRT1 mRNA ere increased, while
PGC-1a expression remained unaltered (159). Furthermore, a previous study
demonstrated that 6-week-old mice fasted for 14, 24 or 48 h
exhibited an induction of PGC-1α in the liver (160). Similarly, the hepatic expression
of PGC-1α at the mRNA level has been found to be elevated following
a 24-h fastin C57BL/6 mice (161). The studies suggest that cells
have a dual switch constituted by SIRT1 deacetylation and PGC-1a
activation that sense substrate fluctuation and availability. The
duration of fasting may have a differential effect on
differentiated transcriptional and post-translational metabolic
response that also can be affected by different tissue background
and physiological habits that evolutionary exist. The latter may
indicate the unaltered expression of PGC-1a in elephant seals that
may support the energetic demands associated with the prolonged
fasting in adult seals.
| Table IVSummary table indicating the
mitochondrial manifestations and the molecular targets that are
affected during fasting. |
Table IV
Summary table indicating the
mitochondrial manifestations and the molecular targets that are
affected during fasting.
Fasting
|
|---|
| Phenotype | Tissue (host) | Molecular
target | (Refs.) |
|---|
|
Biogenesis/function | C2C12 myοtubes | ↥ PGC-1α
protein
↥ SIRT1-dependent deacetylation | (88) |
| Biogenesis | Liver
HNF4α floxxed mice | ↥ PGC -1a
protein | (158) |
| Biogenesis | Liver
C57BL/6 mice | ↥ mRNA
PPARGC1α | (159) |
| Biogenesis | Skeletal muscle
male northern elephant seals | ↥ mRNA AMPK,
SIRT1
- mRNA expression PPARGC1a | (157) |
| Biogenesis | Hippocampal neurons
cultures from embryonic day 17 Sprague-Dawley rats | ↥ PGC-1α | (161) |
| Biogenesis | Skeletal muscle of
male northern elephant seals | ↥ AMPK
phosphorylation
↥ mRNA SIRT1
- PGC-1a protein | (157) |
| Biogenesis | Hippocampal neurons
cultures cells | ↥ mRNA
PPARGC1α | (161) |
|
Biogenesis/function | Muscle, liver and
blood C57BL/6J mice | ↥ Skeletal muscle
mRNA levels of NRF1, NRF2, TFAM
↥ Hepatic mRNA level of PGC-1α
↥ Plasmatic level of glucose-6-phosphate | (162) |
| Function | Liver, cardiac and
skeletal muscle Wistar rats | ↥ Hexokinases
activity in liver, cardiac and skeletal muscle
↥ FoF1 activity in skeletal muscle | (163) |
| Function | Skeletal muscle
OF-1 mice | ↥ Lipid
catabolism | (164) |
| Function | Liver
Ames dwarf mice | ↥ Stable or liver
OXPHOS
↧ Components | (165) |
| Function | Adipose tissue
ob/ob mice | ↥ UCP1 protein | (168) |
| Function | Adipose tissue
C57BL/6 mice | ↥ UCP1
protein
↧ Mitochondrial content in brown adipose tissue
↧ UCP1 protein mRNA levels | (169) |
| Function | Human skeletal
muscle | No difference on
markers of mitochondrial metabolism (PGC-1a, SIRT3, MFN2)
↧ ROS production | (147) |
| Function | Liver
male FVB mice | ↥ Expression of
enzymes of the tricarboxylic-acid (TCA) cycle and oxidative
phosphorylation | (172) |
| Function | Skeletal muscle of
ducklings | ↧ Activity of
succinate-cytochrome c reductase
↧ Oxidative phosphorylation activities | (171) |
| Function | Liver and skeletal
muscles of ducklings | ↧ Oxidative
phosphorylation activity
↥ Coupling efficiency | (170) |
| Function | Human PBMCs | ↥ mRNA of genes
related to fatty acid β-oxidation | (173) |
| Dynamics | Human peripheral
blood
mononuclear cells (PBMCs) | ↥ Mitochondrial
fission (electron microscopy images)
↧ Mitochondrial fusion (electron microscopy images) | (175) |
| Dynamics | Liver
C57Bl6J mice | ↥ Mitochondrial
size before feeding
↧ Mitochondrial size after feediing | (176) |
| Dynamics | Human adipose
tissue | ↧ Genes involved in
mitochondrial regulation (MRPS35, MRPL33, MRPL51, TOMM7, TOMM22,
NDUFA12, NDUFS5, ATP5F1E, ATP5PD) | (174) |
| Mitophagy | Cardiac
muscle
C57BL/6 mice | ↥ Mitophagy-related
targets | (178) |
| Mitophagy | Liver
C57BL/6 mice | ↥ BNIP3
protein | (179) |
| Mitophagy | Skeletal
muscle
Human subjects | - LC3BI and BNIP3
protein
- LC3II:I ratio and p62/SQSTM1 protein | (180) |
Importantly, mitophagy and mitochondrial function
have been implicated in the pathophysiology of depression (162). Therefore, they can serve as
novel therapeutic targets to alleviate the inflammation that
triggers depression. Furthermore, fasting is able to promote
mitochondrial biogenesis in hippocampal neuron cultures from
embryonic day 17, upregulating PGC-1α levels, similar to exercise
(163). This finding may provide
new therapeutic interventions against neurodegenerative disorders
that include synaptic degeneration since they are characterized by
reduced levels of PGC-1a. Recently, a study that compared fasting
with high-intensity intermittent exercise (HIIE) found increased
TFAM, NRF1 and NRF2 gene expression levels in skeletal muscle,
PPARGC1α levels in the liver, and elevated levels of
glucose-6-phosphate in the plasma of C57BL/6 male mice (164). These observations suggest that
fasting promotes mitochondrial biogenesis and enhances cellular
stress adaptation in skeletal muscle.
Mitochondrial function during
fasting
Real-Hohn et al (165) investigated the effects
intermittent fasting (IF; every other day) in combination with a
high-intensity intermittent exercise (IF/HIIE) for 8 weeks in
Wistar rats (165). Their study
revealed an increase in hexokinase activity in the liver, heart,
and skeletal muscle, followed by an elevated skeletal muscle FoF1
activity. Additionally, they documented a molecular reprogramming
in the white adipose tissue due to IF as regards UCP1,
monocarboxylate transporter 1 and GLUT4 expression. However, every
other day feeding alone can also promote changes in mitochondrial
function. More specifically, every other day feeding can increase
lipid catabolism in the muscle mitochondria of male OF-1 mice
(166), preventing lipid
peroxidation and muscular deterioration eventually. Furthermore,
every other day feeding has also been shown to lead to stable or
increased liver OXPHOS components in Ames dwarf mice compared to
wild-type mice (167). These
findings suggest that anabolic hormones contribute to the
differential effects of strict dietary regimens, such as fasting.
The ability of Ames swarf mice to adapt better via metabolism
reprogramming and oxidative damage handling, indicates that more
parameters should be taken into consideration before scientists
bring translational outcomes to humans. In addition, IF appears to
reverse some of the manifestations of the metabolic syndrome in
rodents, stimulating autophagy (168,169).
Fasting is also known to affect the function of the
mitochondria in adipocytes. More specifically, a 16-week isocaloric
fasting in leptin-deficient ob/ob mice was shown to increase UCP1
protein levels in subcutaneous white adipose tissue (170), suggesting that fasting affects
insulin and other adipokines, such as leptin in order to mitigate
distinct molecular programs in the adipose tissue. In line with
these findings, in another study, in 6-week-old male C57BL/6 mice
subjected to fasting every other day for 6 and 30 days, an increase
in UCP1 protein was also documented (171). However, in the same study, the
mitochondrial content of the brown adipose tissue was found to be
decreased with a concomitant decrease in UCP1 protein and mRNA
levels (171). That study
suggests that the effect of every other day fasting is
tissue-specific, since the activation of brown adipose tissue
relies mainly on β-adrenergic receptor (β-AR) signaling and that
the effect of fasting on white adipose tissue is β-AR-independent.
In another study, no difference was detected in markers of
mitochondrial metabolism (PGC-1a, SIRT3 and MFN2) at the skeletal
muscle of human subjects who fasted for 8 weeks (24-h fasting,
three times per week) (149).
However, a key finding of the same study was that mitochondrial ROS
production in the skeletal muscle diminished due to fasting
(149), an insight that has also
been documented in other types of diet, such as the CR, as
aforementioned.
Experiments using C2C12 myotubes mimicking fasting
and feeding conditions have shown enhanced mitochondrial oxidative
phosphorylation via an increase in the expression of genes
implicated in mitochondrial physiology (cytochrome c, COXVa)
and fatty acid utilization (MCAD, CPT-1b and PDK4) (158). In another study, 1 week of
fasting was applied to 5-week-old ducklings, resulting an
upregulation of the coupling efficiency in the mitochondria of the
liver and skeletal muscle, followed by a reduction in oxidative
phosphorylation (172). That
study suggested that energy preservation was maintained through
diminished mitochondrial activity, and this was counterbalanced
with an elevation in mitochondrial coupling efficiency, allowing
cells to conserve endogenous fuel stores during fasting periods.
The above suggestion was further confirmed through an additional
study in which a 6-day fasting period resulted a decrease in
oxidative phosphorylation activity without affecting the inner
membrane proton conductance at the mitochondria of the skeletal
muscle of ducklings (173). In
addition, the activity of succinate-cytochrome c reductase,
which involves complex III and co-enzyme Q pool, has been shown to
be significantly decreased (173). Moreover, in another study, a
24-h fasting period in 6-week-old male FVB mice resulted in an
induction of the expression of enzymes of the TCA cycle and
oxidative phosphorylation in the liver (174). Additionally, that study
demonstrated that a short-term fasting period affected the energy
production in hepatocytes, while prolonged fasting stimulated
glucose production from the liver (174). Fasting for 24 and 8 h in
human-derived peripheral blood mononuclear cells (PBMCs) has been
shown to result in a distinct gene expression pattern, with 74% of
the genes with an altered expression being similar in both time
points tested. More specifically, in both time points tested, an
increased expression was detected in genes related to fatty acid
β-oxidation, and a reduced expression was documented in genes
associated with the TCA cycle (175). That study suggests that these
changes may reflect the nutrition-related metabolic alterations
that fasting generates, possibly orchestrated by PPARα. Finally, a
downregulation of genes associated with mitochondrial regulation
has been assessed in adipose tissue derived from humans that had a
time-restricted eating protocol for 8 weeks. More specifically,
these genes are involved in mitochondrial biogenesis and oxidative
phosphorylation pathways (MRPS35, MRPL33 and MRPL51) mitochondrial
ribosome protein translocation (TOMM7 and TOMM22), and the ETC and
ATP synthase (NDUFA12, NDUFS5, ATP5F1E and ATP5PD) (176). These findings support the
hypothesis that time-restricted feeding can act as a preventive or
therapeutic regime to exert glycemic control in humans diagnosed
with T2D or pre-diabetes.
Mitochondrial dynamics and mitophagy
during fasting
In a previous study, in humans who fasted for 16 h,
it was found that during the fasting re-feeding transition, the
mitochondrial fission in human PBMCs was induced (177). The authors of that study
considered that these changes were observed due to diminished
interactions between the mitochondria and ER. Likewise, the
interaction of the mitochondria with the ER has been suggested to
sense glucose availability in the liver. More specifically, in
C57BL/6 mice that have been fasted overnight, an elevated
mitochondrial volume of their hepatocytes has been documented, a
hallmark of mitochondrial fusion (178). The same study also revealed that
feeding promoted mitochondrial fission, since cellular
'powerhouses' were found to be smaller (178). The disruption of the dynamic
adaptation of mitochondria-associated ER membranes has been
incriminated by an enhanced insulin resistance. Similarly, a 12-h
overnight fasting period in human subjects can promote
mitochondrial fission in the skeletal muscle, which has also been
associated with lipid oxidation (179). Additionally, this study
indicates that MFN2, not MFN1 or OPA1, contributes more to skeletal
muscle mitochondria fusion-fission processes and that lipid
droplets interact with mitochondria via MFN2 rather than MFN1
(179). A 2-day fasting period
for C57BL/6 mice has been shown to lead to an induction of
mitochondrial autophagy with the use of Mito-Keima (180). Furthermore, in male C57BL/6
mice, BNIP3 protein expression has been shown to be markedly
induced above basal levels after 6 h of fasting, with a further
induction by 24 h of fasting (180). By contrast, no induction of NIX
in the liver of fasted mice has been observed (181). Finally, non-significant
decreases in LC3BI and BNIP3 protein levels have been documented
during fasting; however, the preserved LC3II:I ratio and p62/SQSTM1
protein content suggests that autophagy/mitophagy has not been
robustly activated in the skeletal muscle of human subjects after 8
h of fasting (182). The last
contradictory results suggest that the induction of several
molecular pathways due to fasting are early events in the
adaptation of human tissues to a lack of substrates.
Other diet schemes
Mediterranean diet (MedD)
The first to define the MedD was Ancel Keys
(51), as the diet pattern which
included a low percentage of saturated fats and a high proportion
of vegetable oils. MedD was mainly noted during the 1960s in Greece
and northern Italy (183). MedD,
in its traditional form, originates from civilizations inhabiting
close to the Mediterranean Sea, and as a result, has a strong
association with the social behaviors and lifestyles of the
individuals inhabiting that region (184). Furthermore, UNESCO recognized
the MedD as 'an intangible cultural heritage that is deeply rooted
into its geographical origin and whose agricultural and dietary
practices have a responsible interaction with the environment'
(184). From a literature review
conducted by Davis et al (185), it was shown that that
descriptions concerning MedD are common among publications. More
specifically, the major components of a MedD pattern include olive
oil as the main culinary fat and the high consumption of
plant-derived foods (fruit, vegetables, legumes, nuts and seeds,
and whole grain cereals); the moderate intake of red wine; and the
moderate consumption of seafood and dairy products, such as yogurt
and cheese (185). However,
dairy products, such as whole milk, butter or cream are not
suggested. Furthermore, the MedD guidelines propose the moderate
intake of poultry and eggs, and the low consumption of sweet
desserts, red and processed meats (186). It can be drawn from the above
that the MedD does not exclude specific food sources or define a
caloric limit, but instead focuses more on the frequency at which
each food type should be consumed (187). However, there are no clear
guidelines for serving numbers or serving sizes.
The MedD has been found to reduce the progression,
and in a number of cases, to prevent the development of cancer,
cardiovascular disease, depression, obesity, diabetes, erectile
dysfunction, asthma and cognitive decline (187). In addition, the benefits of the
MedD include the reduced risk of metabolic syndrome and total
mortality. The MedD is one of the most studied diets s regards to
its association with cardiovascular disease (187). It is noteworthy that the Seven
Countries Study revealed that farmers inhabiting the Crete region
had the lowest cardiovascular mortality rate, even though they
consumed some of the largest amounts of fats (51).
Numerous in vitro and in vivo studies
have been conducted in order to demonstrate the effects of the MedD
on mitochondrial physiology (Table
V). More importantly, these studies report the effect of
isolated compounds that are present in the MedD and usually they
have been challenged in in vitro and in vivo models
that mimic several diseases or pathophysiological conditions.
| Table VSummary table indicating the
mitochondrial manifestations and the molecular targets that are
affected during Mediterranean, Nordic, and mixed scheme diets. |
Table V
Summary table indicating the
mitochondrial manifestations and the molecular targets that are
affected during Mediterranean, Nordic, and mixed scheme diets.
Mediterranean diet
|
|---|
| Phenotype | Diet and tissue
(host) | Molecular
target | (Refs.) |
|---|
|
Biogenesis/function | HUVECs
VEGF-induced mitochondrial dysfunction | ↥ NRF1,
TFAM
↧ mtDNA, complex IV activity, mitochondria respiration | (186) |
|
Biogenesis/function | C57BL/6 mice
treated with LPS | ↥ SIRT1,
PGC-1a
↥ Cox5b, Cox7a1, Cox8b, and complexes I, II, III, and IV | (187) |
|
Biogenesis/function | HUVECs ox-LDL
treated | ↥ SIRT1, AMPK,
PGC-1a
↧ mito-ROS | (188) |
|
Biogenesis/function | C57BL/6 mice | ↥ SIRT1,
PGC-1a | (189-191) |
|
Biogenesis/function |
Hepatocytes
Megalobrama amblycephala fish | ↥ ATP
turnover
↥ Mitochondrial abundance
↥ PGC-1a/b, NRF1, TFAM
↥ Citrate synthase activity | (192) |
| Function | C57BL/6 mice | ↥ Complex I and
II
↧ Complex V | (193) |
|
Biogenesis/function | HePG2
cells
C57BL/6 mice | ↥ mtDNA, ATP,
OXPHOS
↥ AMPK, PGC-1a
↥ Complex I and IV activity, mitochondria respiration | (194) |
| Function | Myocardium of
cardiac surgery
patients | ↥
β-oxidation
↥ PPARγ | (195) |
| Function | Muscle of
overweight/T2D patients | ↥ State 3
respiration
↥ Mitochondrial function
↧ Complex IV | (197) |
| Function | Whole blood at
resistance trained human subjects | ↥ ATP turnover↧
mitoROS | (198) |
| Dynamics | C57BL/6 mice | ↧ DRP1
↧ PARP | (193) |
|
Dynamics/biogenesis | PBMCs
C57BL/6 mice | ↥ PGC-1a,
NRF1
↧ Oxidative stress
↥ MFN1, MFN2, FIS1 and Beclin-1 gene expression | (199) |
|
Dynamics/biogenesis | HePG2
cells
C57BL/6 mice | ↥ Oxygen
consumption
↥ Citrate synthase activity
↧ Oxidative stress
↥ MFN1, MFN2, OPA1
↧DRP1 and FIS1 | (200) |
|
Nordic diet
|
| Phenotype | Diet and tissue
(host) | Molecular
target | (Refs.) |
|
| Function | PBMCs from human
subjects with metabolic syndrome | Regulation of genes
related to mitochondrial function, ETC and ROS production
↧ NRF1 and NRF2 expression | (201) |
Mixed schemes
|
| Phenotype | Diet and tissue
(host) | Molecular
target | (Refs.) |
|
| Function | High fat-keto diet
in hippocampi
C3HeB/FeJ juvenile mice | ↥ Mitochondrial
uncoupling protein (UCP) expression and activity
↥ ROS amounts | (138) |
|
Mixed schemes
|
| Phenotype | Diet and tissue
(host) | Molecular
target | (Refs.) |
|
| Biogenesis | Keto and Western
diet with. voluntarily exercise
Skeletal muscle Sprague-Dawley rats | ↥ mRNA
PPARGC1a | (123) |
|
Function/dynamics | Keto diet and
exercise training; Skeletal muscle of C57BL/6 mice | ↥ mitochondrial
fission/fusion markers
↥ PGC-1α levels | (141) |
| Dynamics | Skeletal
muscle
CR diet in female obese-insulin resistant (HFD) Wistar rats | Restore of
mitochondrial dynamics imbalance induced by HFD (↥ MFN2 levels, ↧
DRP1 phosphorylation) | (113) |
|
Biogenesis/function | Calorie-restricted
ketogenic diet; Brain of Sprague-Dawley rats | ↥ mRNA transcripts
that encode mitochondrial proteins
↥ Mitochondrial abundance
↥ Flutamate levels
↥ Phosphocreatine/creatine index | (142) |
As such, polyphenols are one of the main family of
constitutes that are present in large quantities in foods and
drinks of the MedD. More specifically, a flavonoid that is found in
large quantities in red wine, delphinidin, is able to increase the
mitochondrial activity through complex IV activity and the
mitochondrial abundance. At molecular levels, delphinidin has been
found to upregulate the expression of TFAM, TFB2M and NRF1 that are
implicated in the mitochondrial biogenesis molecular program
(188). In another study, in
which endothelial cells with VEGF-induced mitochondrial dysfunction
were treated with lycopene, an upregulation in the expression of
genes that encode proteins related to both mitochondrial biogenesis
and function, such as several COX members, PGC-1α and SIRT1 was
found (189). Similarly, SIRT1
and PGC-1α have been found to be stimulated by chlorogenic acid,
that is present in coffee beans and apples, in human umbilical vein
endothelial cells exposed to ox-LDL. Following increased
biogenesis, chlorogenic acid treatment has also been shown to
alleviate elevated mitochondrial ROS levels (190). One of the most known SIRT1
activators that is used in biomedical science is resveratrol, that
is found mainly in red wine and grapes. Resveratrol is able to
stimulate mitochondrial function through SIRT1 and PGC-1α
activation and prevent several pathophysiological conditions, such
as insulin resistance and cardiac failure (191-193). Another well-studied polyphenol
that is present in olive oil is hydroxytyrosol. It has been found
that it can increase mitochondrial abundance via the induction of
the AMPK signaling pathway, inducing PGC-1α, TFAM and NRF-1
(194). Furthermore,
hydroxytyrosol is able to induce mitochondrial activity, since it
has been found to elevate complex I, II and decrease complex V
expression (195). Ginger
extract that is also used extensively in the MedD and the Asian
traditional diet, can stimulate the AMPK/PGC-1α axis in both HepG2
cells and in skeletal muscle, liver, and brown adipose tissue in
mice. Moreover, mitochondrial abundance and the expression of
OXPHOS-related proteins are also increased in all tissues tested
(196).
Some studies have also been performed on human
subjects/patients. Initially, a study that was conducted on
patients that received a diet rich in polyunsaturated FAs for 2-3
weeks before they underwent cardiac surgery, revealed an
upregulation of PPARγ followed by an improvement of β-oxidation and
reduced a oxidative burst (197). In line with that study,
treatment with docosahexaenoic acid, that can be found in fish, was
also documented to decrease mitochondrial ROS and stimulate the
expression of genes that are implicated in fatty acid metabolism
(198). Resveratrol has also
been administrated to patients who are overweight and/or with T2DM
(199). It was revealed that
mitochondrial function was improved since state 3 respiration was
elevated, and the expression of complex IV was diminished (199). Finally, a study in which
healthy, resistance-trained, male subjects received a mixture of
ancient peat and apple extract demonstrated an elevation in ATP
turnover, increased mitochondrial function followed by a reduction
in mitochondrial ROS production (200).
In vitro studies have also aimed to document
the effects of the MedD on the other aspects of mitochondrial
physiology, such as mitophagy and fusion/fission. Hydroxytyrosol
supplementation as aforementioned, has been found to decrease DRP1
and PARP levels, indicating a reduction of mitochondrial fission
(195). In addition, ferulic
acid that is present in fruits and vegetables, such as tomatoes,
sweet corn and rice has been shown to improve the mitochondrial
biogenesis molecular program through the induction of PGC-1a,
PGC-1β and NRF1. It has also been found that it can increase all
axes of dynamics and mitophagy, since it stimulates the expression
of MFN1, MFN2, FIS1 and Beclin-1 (201). Finally, butyrate, that is a
short-chain fatty acid, has been shown to increase mitochondrial
fusion, since it is able to induce MFN1, MFN2 and OPA1 expression
followed by a concomitant decrease in DRP1 and FIS1, that control
mitochondrial fission (202).
Nordic diet
The Nordic diet is considered the new MedD and
consists of whole, fresh, seasonal, local foods of the Scandinavian
region. The Nordic diet also discourages the consumption of heavily
processed foods and is rich in whole grains, berries, fruits,
vegetables, fish and low-fat dairy products. Data available
concerning the effects of the Nordic diet and mitochondrial
function are extremely limited. A clinical trial study that was
performed on the healthy Nordic diet, revealed that it altered the
expression of genes related to mitochondrial function and thereby
reduced ROS production in PBMCs form patients with metabolic
syndrome. Apart from the involvement of genes that encode proteins
of the ETC, the Nordic diet has been shown to downregulated both
NRF1 and NRF2 levels, suggesting a reduction in the oxidative burst
(203) (Table V). Following the hypothesis that
the Nordic diet contains similar ingredients with the MedD, similar
effects are expected in future studies concerning other aspects of
mitochondrial physiology.
3. Clinical impact
The majority of researchers consider that the only
function of the mitochondria is to generate energy for cellular
demands. Hence, the mitochondria, though their functions are able
to contribute to cellular metabolism, redox signaling and ion
homeostasis. All these aspects are important for cellular survival
and well-being. The dysregulation of their function suggests their
involvement in the pathogenesis or/the progression of numerous
human diseases or pathophysiologies, including, diabetes, cancer,
neurodegenerative and cardiovascular diseases, sepsis, traumatic
injury, inflammation, aging and, frailty and loss of elasticity in
the skin. Despite the intense focus of the scientific community
concerning mitochondrial physiology, one study out of every 154
studies indexed in PubMed since 1998 has studied mitochondrial
function (204), the exact role
and mechanisms through which the mitochondria contribute to human
diseases remain elusive, opening new challenges for basic and
translational biomedical science. The up-to-date information
incriminates oxidative stress that is generated from the
mitochondria as the main causality or a secondary aggravating
effect that drive several tissue pathophysiologies.
Nutrition and dietary habits play a critical role
in mitochondrial physiology, since they provide mitochondria with
all necessary (or not), substrates for proper (or not) function.
For that, dietary habits, such as HFD, are able to aggravate
mitochondrial function and render the mitochondria a significant
determinant towards the development of metabolic diseases, such as
diabetes or metabolic syndrome. On the contrary, dietary
interventions, such as the KD, fasting or MedD, can modulate the
molecular environment of already dysfunctional mitochondria in
several diseases, including cardiac failure, Alzheimer's disease,
and cancer, alleviating several manifestations towards better
living. It has also been well-established that fasting and MedD can
contribute to longevity, since they have the ability to maintain
the mitochondria in a 'fit' state. Due to the fact that diet is a
daily habit, clinicians and other scientists are allowed to
optimize their interventions that modulate cellular 'powerhouses'
and impact human health.
4. Conclusions
The mitochondria have the flexibility to easily
adapt to exogenous parameters and preserve their proper
functionality in order to generate the adequate energy for cellular
demands. Differentiations in substrate availability due to
different dietary habits are being sensitized by the mitochondria
followed by their molecular rewiring. This orchestrates the
mitochondria to undergo changes in their function, the regulation
of biogenesis and mitophagy, and their 'self-recycling' through
fusion and fission (Fig. 5).
Conclusively, a HFD impairs mitochondrial function and fusion,
while it can induce mitochondrial fission. CR diets appear to
reduce mitochondrial biogenesis and function but stimulate
mitophagy. On the contrary, the KD appears to induce mitochondrial
function and biogenesis, while contradictory results exist for
mitochondrial dynamics and mitophagy. Finally, fasting and MedD
appear to induce mitochondrial biogenesis and fine-tune
mitochondrial function. On the contrary, fasting appears to induce
mitochondrial recycle and the clearance of problematic
mitochondrial units, while the MedD has the opposite effect on
mitochondrial dynamics and the autophagic process.
Availability of data and materials
Not applicable.
Authors' contributions
IDK, VNSG, DAS and DK contributed to the conception
and the design of the study. IDK, EV, MA, CA, ZS, FT, VNSG and DK
contributed to the drafting of the manuscript, including the
literature search, and to the revisions. All authors have read and
approved the final manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
DAS is the Editor-in-Chief for the journal, but had
no personal involvement in the reviewing process, or any influence
in terms of adjudicating on the final decision, for this article.
The other authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
No funding was received.
References
|
1
|
Jornayvaz FR and Shulman GI: Regulation of
mitochondrial biogenesis. Essays Biochem. 47:69–84. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dorn GW II, Vega RB and Kelly DP:
Mitochondrial biogenesis and dynamics in the developing and
diseased heart. Genes Dev. 29:1981–1991. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qiu Z, Wei Y, Song Q, Du B, Wang H, Chu Y
and Hu Y: The role of myocardial mitochondrial quality control in
heart failure. Front Pharmacol. 10:14042019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Herzig S, Long F, Jhala US, Hedrick S,
Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, et al:
CREB regulates hepatic gluconeogenesis through the coactivator
PGC-1. Nature. 413:179–183. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu Z, Huang X, Feng Y, Handschin C, Feng
Y, Gullicksen PS, Bare O, Labow M, Spiegelman B and Stevenson SC:
Transducer of regulated CREB-binding proteins (TORCs) induce
PGC-1alpha transcription and mitochondrial biogenesis in muscle
cells. Proc Natl Acad Sci USA. 103:14379–14384. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Au HC and Scheffler IE: Promoter analysis
of the human succinate dehydrogenase iron-protein gene-both nuclear
respiratory factors NRF-1 and NRF-2 are required. Eur J Biochem.
251:164–174. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Scarpulla RC: Nuclear activators and
coactivators in mammalian mitochondrial biogenesis. Biochim Biophys
Acta. 1576:1–14. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Goffart S and Wiesner RJ: Regulation and
co-ordination of nuclear gene expression during mitochondrial
biogenesis. Exp Physiol. 88:33–40. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vega RB, Huss JM and Kelly DP: The
coactivator PGC-1 cooperates with peroxisome proliferator-activated
receptor alpha in transcriptional control of nuclear genes encoding
mitochondrial fatty acid oxidation enzymes. Mol Cell Biol.
20:1868–1876. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Puigserver P, Wu Z, Park CW, Graves R,
Wright M and Spiegelman BM: A cold-inducible coactivator of nuclear
receptors linked to adaptive thermogenesis. Cell. 92:829–839. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu Z, Puigserver P, Andersson U, Zhang C,
Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC and
Spiegelman BM: Mechanisms controlling mitochondrial biogenesis and
respiration through the thermogenic coactivator PGC-1. Cell.
98:115–124. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Handschin C, Rhee J, Lin J, Tarr PT and
Spiegelman BM: An autoregulatory loop controls peroxisome
proliferator-activated receptor gamma coactivator 1alpha expression
in muscle. Proc Natl Acad Sci USA. 100:7111–7116. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Akimoto T, Sorg BS and Yan Z: Real-time
imaging of peroxisome proliferator-activated receptor-gamma
coactivator-1alpha promoter activity in skeletal muscles of living
mice. Am J Physiol Cell Physiol. 287:C790–C796. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zong H, Ren JM, Young LH, Pypaert M, Mu J,
Birnbaum MJ and Shulman GI: AMP kinase is required for
mitochondrial biogenesis in skeletal muscle in response to chronic
energy deprivation. Proc Natl Acad Sci USA. 99:15983–15987. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Daitoku H, Sakamaki J and Fukamizu A:
Regulation of FoxO transcription factors by acetylation and
protein-protein interactions. Biochim Biophys Acta. 1813:1954–1960.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rodgers JT, Lerin C, Haas W, Gygi SP,
Spiegelman BM and Puigserver P: Nutrient control of glucose
homeostasis through a complex of PGC-1alpha and SIRT1. Nature.
434:113–118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu YJ, McIntyre RL, Janssens GE and
Houtkooper RH: Mitochondrial fission and fusion: A dynamic role in
aging and potential target for age-related disease. Mech Ageing
Dev. 186:1112122020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tilokani L, Nagashima S, Paupe V and
Prudent J: Mitochondrial dynamics: Overview of molecular
mechanisms. Essays Biochem. 62:341–360. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mitra K, Wunder C, Roysam B, Lin G and
Lippincott-Schwartz J: A hyperfused mitochondrial state achieved at
G1-S regulates cyclin E buildup and entry into S phase. Proc Natl
Acad Sci USA. 106:11960–11965. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sharma A, Ahmad S, Ahmad T, Ali S and Syed
MA: Mitochondrial dynamics and mitophagy in lung disorders. Life
Sci. 284:1198762021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Margineantu DH, Gregory Cox W, Sundell L,
Sherwood SW, Beechem JM and Capaldi RA: Cell cycle dependent
morphology changes and associated mitochondrial DNA redistribution
in mitochondria of human cell lines. Mitochondrion. 1:425–435.
2002. View Article : Google Scholar
|
|
22
|
Mitra K: Mitochondrial fission-fusion as
an emerging key regulator of cell proliferation and
differentiation. Bioessays. 35:955–964. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Amiri M and Hollenbeck PJ: Mitochondrial
biogenesis in the axons of vertebrate peripheral neurons. Dev
Neurobiol. 68:1348–1361. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mizushima N: Autophagy: Process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bursch W, Ellinger A, Kienzl H, Török L,
Pandey S, Sikorska M, Walker R and Hermann RS: Active cell death
induced by the anti-estrogens tamoxifen and ICI 164 384 in human
mammary carcinoma cells (MCF-7) in culture: The role of autophagy.
Carcinogenesis. 17:1595–1607. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu L, Strandberg L and Lenardo MJ: The
selectivity of autophagy and its role in cell death and survival.
Autophagy. 4:567–573. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Petrovski G and Das DK: Does autophagy
take a front seat in lifespan extension? J Cell Mol Med.
14:2543–2551. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen G, Kroemer G and Kepp O: Mitophagy:
An emerging role in aging and age-associated diseases. Front Cell
Dev Biol. 8:2002020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Klionsky DJ, Cregg JM, Dunn WA Jr, Emr SD,
Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M
and Ohsumi Y: A unified nomenclature for yeast autophagy-related
genes. Dev Cell. 5:539–545. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nakatogawa H, Suzuki K, Kamada Y and
Ohsumi Y: Dynamics and diversity in autophagy mechanisms: Lessons
from yeast. Nat Rev Mol Cell Biol. 10:458–467. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rogov VV, Suzuki H, Marinković M, Lang V,
Kato R, Kawasaki M, Buljubašić M, Šprung M, Rogova N, Wakatsuki S,
et al: Phosphorylation of the mitochondrial autophagy receptor Nix
enhances its interaction with LC3 proteins. Sci Rep. 7:11312017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ding WX, Ni HM, Li M, Liao Y, Chen X,
Stolz DB, Dorn GW II and Yin XM: Nix is critical to two distinct
phases of mitophagy, reactive oxygen species-mediated autophagy
induction and Parkin-ubiquitin-p62-mediated mitochondrial priming.
J Biol Chem. 285:27879–27890. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Springer W and Kahle PJ: Regulation of
PINK1-Parkin-mediated mitophagy. Autophagy. 7:266–278. 2011.
View Article : Google Scholar
|
|
34
|
Itakura E, Kishi-Itakura C, Koyama-Honda I
and Mizushima N: Structures containing Atg9A and the ULK1 complex
independently target depolarized mitochondria at initial stages of
Parkin-mediated mitophagy. J Cell Sci. 125:1488–1499.
2012.PubMed/NCBI
|
|
35
|
Chaanine AH, Kohlbrenner E, Gamb SI,
Guenzel AJ, Klaus K, Fayyaz AU, Nair KS, Hajjar RJ and Redfield MM:
FOXO3a regulates BNIP3 and modulates mitochondrial calcium,
dynamics, and function in cardiac stress. Am J Physiol Heart Circ
Physiol. 311:H1540–H1559. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mammucari C, Milan G, Romanello V, Masiero
E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J,
et al: FoxO3 controls autophagy in skeletal muscle in vivo. Cell
Metab. 6:458–471. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Twig G, Elorza A, Molina AJ, Mohamed H,
Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al:
Fission and selective fusion govern mitochondrial segregation and
elimination by autophagy. EMBO J. 27:433–446. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tait SW and Green DR: Mitochondria and
cell signalling. J Cell Sci. 125:807–815. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Navale AM and Paranjape AN: Glucose
transporters: Physiological and pathological roles. Biophys Rev.
8:5–9. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
McCommis KS and Finck BN: Mitochondrial
pyruvate transport: A historical perspective and future research
directions. Biochem J. 466:443–454. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Glancy B, Kane DA, Kavazis AN, Goodwin ML,
Willis WT and Gladden LB: Mitochondrial lactate metabolism: History
and implications for exercise and disease. J Physiol. 599:863–888.
2021. View Article : Google Scholar
|
|
42
|
Yoo HC, Yu YC, Sung Y and Han JM:
Glutamine reliance in cell metabolism. Exp Mol Med. 52:1496–1516.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Stefano GB, Challenger S and Kream RM:
Hyperglycemia-associated alterations in cellular signaling and
dysregulated mitochondrial bioenergetics in human metabolic
disorders. Eur J Nutr. 55:2339–2345. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Melser S, Lavie J and Bénard G:
Mitochondrial degradation and energy metabolism. Biochim Biophys
Acta. 1853:2812–2821. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
King KL, Stanley WC, Rosca M, Kerner J,
Hoppel CL and Febbraio M: Fatty acid oxidation in cardiac and
skeletal muscle mitochondria is unaffected by deletion of CD36.
Arch Biochem Biophys. 467:234–238. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Duncan RE, Ahmadian M, Jaworski K,
Sarkadi-Nagy E and Sul HS: Regulation of lipolysis in adipocytes.
Annu Rev Nutr. 27:79–101. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lodhi IJ and Semenkovich CF: Peroxisomes:
A nexus for lipid metabolism and cellular signaling. Cell Metab.
19:380–392. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fillmore N and Lopaschuk GD: Targeting
mitochondrial oxidative metabolism as an approach to treat heart
failure. Biochim Biophys Acta. 1833:857–865. 2013. View Article : Google Scholar
|
|
49
|
Aon MA, Bhatt N and Cortassa SC:
Mitochondrial and cellular mechanisms for managing lipid excess.
Front Physiol. 5:2822014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Carley AN, Taegtmeyer H and Lewandowski
ED: Matrix revisited: Mechanisms linking energy substrate
metabolism to the function of the heart. Circ Res. 114:717–729.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Keys A, Menotti A, Karvonen MJ, Aravanis
C, Blackburn H, Buzina R, Djordjevic BS, Dontas AS, Fidanza F, Keys
MH, et al: The diet and 15-year death rate in the seven countries
study. Am J Epidemiol. 124:903–915. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Schwingshackl L and Hoffmann G: Comparison
of effects of long-term low-fat vs high-fat diets on blood lipid
levels in over-weight or obese patients: A systematic review and
meta-analysis. J Acad Nutr Diet. 113:1640–1661. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Casper RC, Sullivan EL and Tecott L:
Relevance of animal models to human eating disorders and obesity.
Psychopharmacology (Berl). 199:313–329. 2008. View Article : Google Scholar
|
|
54
|
Della Vedova MC, Muñoz MD, Santillan LD,
Plateo-Pignatari MG, Germanó MJ, Rinaldi Tosi ME, Garcia S, Gomez
NN, Fornes MW, Gomez Mejiba SE and Ramirez DC: A mouse model of
diet-induced obesity resembling most features of human metabolic
syndrome. Nutr Metab Insights. 9:93–102. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Schrauwen P, Schrauwen-Hinderling V, Hoeks
J and Hesselink MK: Mitochondrial dysfunction and lipotoxicity.
Biochim Biophys Acta. 1801:266–271. 2010. View Article : Google Scholar
|
|
56
|
Jo J, Gavrilova O, Pack S, Jou W, Mullen
S, Sumner AE, Cushman SW and Periwal V: Hypertrophy and/or
hyperplasia: Dynamics of adipose tissue growth. PLoS Comput Biol.
5:e10003242009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yang A and Mottillo EP: Adipocyte
lipolysis: From molecular mechanisms of regulation to disease and
therapeutics. Biochem J. 477:985–1008. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Carracedo A, Cantley LC and Pandolfi PP:
Cancer metabolism: Fatty acid oxidation in the limelight. Nat Rev
Cancer. 13:227–232. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Roy C, Paglialunga S, Fisette A, Schrauwen
P, Moonen-Kornips E, St-Onge J, Hesselink MK, Richard D, Joanisse
DR and Cianflone K: Shift in metabolic fuel in
acylation-stimulating protein-deficient mice following a high-fat
diet. Am J Physiol Endocrinol Metab. 294:E1051–E1059. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Chen Z, Tao S, Li X and Yao Q: Resistin
destroys mitochondrial biogenesis by inhibiting the
PGC-1α/NRF1/TFAM signaling pathway. Biochem Biophys Res Commun.
504:13–18. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Blanquer-Rosselló MM, Santandreu FM,
Oliver J, Roca P and Valle A: Leptin modulates mitochondrial
function, dynamics and biogenesis in MCF-7 cells. J Cell Biochem.
116:2039–2048. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yamauchi T and Kadowaki T: Physiological
and pathophysiological roles of adiponectin and adiponectin
receptors in the integrated regulation of metabolic and
cardiovascular diseases. Int J Obes (Lond). 32(Suppl 7): S13–S18.
2008. View Article : Google Scholar
|
|
63
|
Ouchi N, Parker JL, Lugus JJ and Walsh K:
Adipokines in inflammation and metabolic disease. Nat Rev Immunol.
11:85–97. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Cantó C and Auwerx J: PGC-1alpha, SIRT1
and AMPK, an energy sensing network that controls energy
expenditure. Curr Opin Lipidol. 20:98–105. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sparks LM, Xie H, Koza RA, Mynatt R,
Hulver MW, Bray GA and Smith SR: A high-fat diet coordinately
downregulates genes required for mitochondrial oxidative
phosphorylation in skeletal muscle. Diabetes. 54:1926–1933. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chen D, Li X, Zhang L, Zhu M and Gao L: A
high-fat diet impairs mitochondrial biogenesis, mitochondrial
dynamics, and the respiratory chain complex in rat myocardial
tissues. J Cell Biochem. 119:96022018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kang KW, Kim OS, Chin JY, Kim WH, Park SH,
Choi YJ, Shin JH, Jung KT, Lim DS and Lee SK: Diastolic dysfunction
induced by a high-fat diet is associated with mitochondrial
abnormality and adenosine triphosphate levels in rats. Endocrinol
Metab (Seoul). 30:557–568. 2015. View Article : Google Scholar
|
|
68
|
Turner N, Bruce CR, Beale SM, Hoehn KL, So
T, Rolph MS and Cooney GJ: Excess lipid availability increases
mitochondrial fatty acid oxidative capacity in muscle: Evidence
against a role for reduced fatty acid oxidation in lipid-induced
insulin resistance in rodents. Diabetes. 56:2085–2092. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hancock CR, Han DH, Chen M, Terada S,
Yasuda T, Wright DC and Holloszy JO: High-fat diets cause insulin
resistance despite an increase in muscle mitochondria. Proc Natl
Acad Sci USA. 105:7815–7820. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Battiprolu PK, Hojayev B, Jiang N, Wang
ZV, Luo X, Iglewski M, Shelton JM, Gerard RD, Rothermel BA,
Gillette TG, et al: Metabolic stress-induced activation of FoxO1
triggers diabetic cardiomyopathy in mice. J Clin Invest.
122:1109–1118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Kyriazis ID, Hoffman M, Gaignebet L,
Lucchese AM, Markopoulou E, Palioura D, Wang C, Bannister TD,
Christofidou-Solomidou M, Oka SI, et al: KLF5 is induced by FOXO1
and causes oxidative stress and diabetic cardiomyopathy. Circ Res.
128:335–357. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Li N, Li HP, Zhang BY, Zhang L, Shen JM
and Li QY: Effect of high-fat diet on respiratory function and
diaphragm fibers in mice and its mitochondrial mechanism. Zhonghua
Yi Xue Za Zhi. 101:2893–2899. 2021.In Chinese. PubMed/NCBI
|
|
73
|
Kang YS, Seong D, Kim JC and Kim SH:
Low-intensity exercise training additionally increases
mitochondrial dynamics caused by high-fat diet (HFD) but has no
additional effect on mitochondrial biogenesis in fast-twitch muscle
by HFD. Int J Environ Res Public Health. 17:54612020. View Article : Google Scholar :
|
|
74
|
Heo JW, No MH, Cho J, Choi Y, Cho EJ, Park
DH, Kim TW, Kim CJ, Seo DY, Han J, et al: Moderate aerobic exercise
training ameliorates impairment of mitochondrial function and
dynamics in skeletal muscle of high-fat diet-induced obese mice.
FASEB J. 35:e213402021. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Tarpey MD, Davy KP, McMillan RP, Bowser
SM, Halliday TM, Boutagy NE, Davy BM, Frisard MI and Hulver MW:
Skeletal muscle autophagy and mitophagy in endurance-trained
runners before and after a high-fat meal. Mol Metab. 6:1597–1609.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Tong M, Saito T, Zhai P, Oka SI, Mizushima
W, Nakamura M, Ikeda S, Shirakabe A and Sadoshima J: Mitophagy is
essential for maintaining cardiac function during high fat
diet-induced diabetic cardiomyopathy. Circ Res. 124:1360–1371.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Shao D, Kolwicz SC Jr, Wang P, Roe ND,
Villet O, Nishi K, Hsu YA, Flint GV, Caudal A, Wang W, et al:
Increasing fatty acid oxidation prevents high-fat diet-induced
cardiomyopathy through regulating parkin-mediated mitophagy.
Circulation. 142:983–997. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Rous P: The influence of diet on
transplanted and spontaneous mouse tumors. J Exp Med. 20:433–451.
1914. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Fontana L and Partridge L: Promoting
health and longevity through diet: From model organisms to humans.
Cell. 161:106–118. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Holloszy JO and Fontana L: Caloric
restriction in humans. Exp Gerontol. 42:709–712. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Pignatti C, D'Adamo S, Stefanelli C,
Flamigni F and Cetrullo S: Nutrients and pathways that regulate
health span and life span. Geriatrics (Basel). 5:952020. View Article : Google Scholar
|
|
82
|
Ruetenik A and Barrientos A: Dietary
restriction, mitochondrial function and aging: From yeast to
humans. Biochim Biophys Acta. 1847:1434–1447. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Comfort A: Effect of delayed and resumed
growth on the longevity of a fish (lebistes reticulatus, peters) in
captivity. Gerontologia. 49:150–155. 1963. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Romey-Glüsing R, Li Y, Hoffmann J, von
Frieling J, Knop M, Pfefferkorn R, Bruchhaus I, Fink C and Roeder
T: Nutritional regimens with periodically recurring phases of
dietary restriction extend lifespan in Drosophila. FASEB J.
32:1993–2003. 2018. View Article : Google Scholar
|
|
85
|
Colman RJ, Beasley TM, Kemnitz JW, Johnson
SC, Weindruch R and Anderson RM: Caloric restriction reduces
age-related and all-cause mortality in rhesus monkeys. Nat Commun.
5:35572014. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Seyfried TN: Ketone strong: Emerging
evidence for a therapeutic role of ketone bodies in neurological
and neurodegenerative diseases. J Lipid Res. 55:1815–1817. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Fulco M, Cen Y, Zhao P, Hoffman EP,
McBurney MW, Sauve AA and Sartorelli V: Glucose restriction
inhibits skeletal myoblast differentiation by activating SIRT1
through AMPK-mediated regulation of Nampt. Dev Cell. 14:661–673.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Xiong S, Salazar G, Patrushev N and
Alexander RW: FoxO1 mediates an autofeedback loop regulating SIRT1
expression. J Biol Chem. 286:5289–5299. 2011. View Article : Google Scholar :
|
|
89
|
López-Lluch G, Hunt N, Jones B, Zhu M,
Jamieson H, Hilmer S, Cascajo MV, Allard J, Ingram DK, Navas P and
de Cabo R: Calorie restriction induces mitochondrial biogenesis and
bioenergetic efficiency. Proc Natl Acad Sci USA. 103:1768–1773.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Hempenstall S, Page MM, Wallen KR and
Selman C: Dietary restriction increases skeletal muscle
mitochondrial respiration but not mitochondrial content in C57BL/6
mice. Mech Ageing Dev. 133:37–45. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Palacios OM, Carmona JJ, Michan S, Chen
KY, Manabe Y, Ward JL III, Goodyear LJ and Tong Q: Diet and
exercise signals regulate SIRT3 and activate AMPK and PGC-1alpha in
skeletal muscle. Aging (Albany NY). 1:771–783. 2009. View Article : Google Scholar
|
|
92
|
Picca A, Pesce V, Fracasso F, Joseph AM,
Leeuwenburgh C and Lezza AM: Aging and calorie restriction
oppositely affect mitochondrial biogenesis through TFAM binding at
both origins of mitochondrial DNA replication in rat liver. PLoS
One. 8:e746442013. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zhu M, Miura J, Lu LX, Bernier M, DeCabo
R, Lane MA, Roth GS and Ingram DK: Circulating adiponectin levels
increase in rats on caloric restriction: The potential for insulin
sensitization. Exp Gerontol. 39:1049–1059. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Civitarese AE, Carling S, Heilbronn LK,
Hulver MH, Ukropcova B, Deutsch WA, Smith SR and Ravussin E;
CALERIE Pennington Team: Calorie restriction increases muscle
mitochondrial biogenesis in healthy humans. PLoS Med. 4:e762007.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Herzig S and Shaw RJ: AMPK: Guardian of
metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol.
19:121–135. 2018. View Article : Google Scholar :
|
|
96
|
López-Lluch G and Navas P: Calorie
restriction as an intervention in ageing. J Physiol. 594:2043–2060.
2016. View Article : Google Scholar :
|
|
97
|
Feldman JL, Dittenhafer-Reed KE and Denu
JM: Sirtuin catalysis and regulation. J Biol Chem. 287:42419–42427.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Desai VG, Weindruch R, Hart RW and Feuers
RJ: Influences of age and dietary restriction on gastrocnemius
electron transport system activities in mice. Arch Biochem Biophys.
333:145–151. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Hepple RT, Baker DJ, McConkey M, Murynka T
and Norris R: Caloric restriction protects mitochondrial function
with aging in skeletal and cardiac muscles. Rejuvenation Res.
9:219–222. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Colom B, Oliver J, Roca P and
Garcia-Palmer FJ: Caloric restriction and gender modulate cardiac
muscle mitochondrial H2O2 production and oxidative damage.
Cardiovasc Res. 74:456–465. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Hepple RT, Baker DJ, Kaczor JJ and Krause
DJ: Long-term caloric restriction abrogates the age-related decline
in skeletal muscle aerobic function. FASEB J. 19:1320–1322. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Sreekumar R, Unnikrishnan J, Fu A, Nygren
J, Short KR, Schimke J, Barazzoni R and Nair KS: Effects of caloric
restriction on mitochondrial function and gene transcripts in rat
muscle. Am J Physiol Endocrinol Metab. 283:E38–E43. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Drew B, Phaneuf S, Dirks A, Selman C,
Gredilla R, Lezza A, Barja G and Leeuwenburgh C: Effects of aging
and caloric restriction on mitochondrial energy production in
gastrocnemius muscle and heart. Am J Physiol Regul Integr Comp
Physiol. 284:R474–R480. 2003. View Article : Google Scholar
|
|
104
|
Lam YY, Ghosh S, Civitarese AE and
Ravussin E: Six-month calorie restriction in overweight individuals
elicits transcriptomic response in subcutaneous adipose tissue that
is distinct from effects of energy deficit. J Gerontol A Biol Sci
Med Sci. 71:1258–1265. 2016. View Article : Google Scholar
|
|
105
|
Menshikova EV, Ritov VB, Dube JJ, Amati F,
Stefanovic-Racic M, Toledo FGS, Coen PM and Goodpaster BH: Calorie
restriction-induced weight loss and exercise have differential
effects on skeletal muscle mitochondria despite similar effects on
insulin sensitivity. J Gerontol A Biol Sci Med Sci. 73:81–87. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Bagherniya M, Butler AE, Barreto GE and
Sahebkar A: The effect of fasting or calorie restriction on
autophagy induction: A review of the literature. Ageing Res Rev.
47:183–197. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Gutiérrez-Casado E, Khraiwesh H,
López-Domínguez JA, Montero-Guisado J, López-Lluch G, Navas P, de
Cabo R, Ramsey JJ, González-Reyes JA and Villalba JM: The impact of
aging, calorie restriction and dietary fat on autophagy markers and
mitochondrial ultrastructure and dynamics in mouse skeletal muscle.
J Gerontol A Biol Sci Med Sci. 74:760–769. 2019. View Article : Google Scholar :
|
|
108
|
Cui J, Shi S, Sun X, Cai G, Cui S, Hong Q,
Chen X and Bai XY: Mitochondrial autophagy involving renal injury
and aging is modulated by caloric intake in aged rat kidneys. PLoS
One. 8:e697202013. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Kim I and Lemasters JJ: Mitochondrial
degradation by autophagy (mitophagy) in GFP-LC3 transgenic
hepatocytes during nutrient deprivation. Am J Physiol Cell Physiol.
300:C308–C317. 2011. View Article : Google Scholar :
|
|
110
|
Rodríguez-López S, López-Bellón S,
González-Reyes JA, Burón MI, de Cabo R and Villalba JM:
Mitochondrial adaptations in liver and skeletal muscle to
pro-longevity nutritional and genetic interventions: The crosstalk
between calorie restriction and CYB5R3 overexpression in transgenic
mice. Geroscience. 42:977–994. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Faitg J, Leduc-Gaudet JP, Reynaud O,
Ferland G, Gaudreau P and Gouspillou G: Effects of aging and
caloric restriction on fiber type composition, mitochondrial
morphology and dynamics in rat oxidative and glycolytic muscles.
Front Physiol. 10:4202019. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Khraiwesh H, López-Domínguez JA, Fernández
del Río L, Gutierrez-Casado E, López-Lluch G, Navas P, de Cabo R,
Ramsey JJ, Burón MI, Villalba JM and González-Reyes JA:
Mitochondrial ultrastructure and markers of dynamics in hepatocytes
from aged, calorie restricted mice fed with different dietary fats.
Exp Gerontol. 56:77–88. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Khraiwesh H, López-Domínguez JA,
López-Lluch G, Navas P, de Cabo R, Ramsey JJ, Villalba JM and
González-Reyes JA: Alterations of ultrastructural and
fission/fusion markers in hepatocyte mitochondria from mice
following calorie restriction with different dietary fats. J
Gerontol A Biol Sci Med Sci. 68:1023–1034. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Kitaoka Y, Nakazato K and Ogasawara R:
Combined effects of resistance training and calorie restriction on
mitochondrial fusion and fission proteins in rat skeletal muscle. J
Appl Physiol (1985). 121:806–810. 2016. View Article : Google Scholar
|
|
115
|
Pattanakuhar S, Sutham W, Sripetchwandee
J, Minta W, Mantor D, Palee S, Pratchayasakul W, Chattipakorn N and
Chattipakorn SC: Combined exercise and calorie restriction
therapies restore contractile and mitochondrial functions in
skeletal muscle of obese-insulin resistant rats. Nutrition.
62:74–84. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Kang HC, Lee YM, Kim HD, Lee JS and Slama
A: Safe and effective use of the ketogenic diet in children with
epilepsy and mitochondrial respiratory chain complex defects.
Epilepsia. 48:82–88. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Qu C, Keijer J, Adjobo-Hermans MJW, van de
Wal M, Schirris T, van Karnebeek C, Pan Y and Koopman WJH: The
ketogenic diet as a therapeutic intervention strategy in
mitochondrial disease. Int J Biochem Cell Biol. 138:1060502021.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Dhamija R, Eckert S and Wirrell E:
Ketogenic diet. Can J Neurol Sci. 40:158–167. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Olgac A, İnci A, Okur İ, Biberoğlu G, Oğuz
D, Ezgü FS, Kasapkara ÇS, Aktaş E and Tümer L: Beneficial effects
of modified atkins diet in glycogen storage disease type IIIa. Ann
Nutr Metab. 76:233–241. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Augustin K, Khabbush A, Williams S, Eaton
S, Orford M, Cross JH, Heales SJR, Walker MC and Williams RSB:
Mechanisms of action for the medium-chain triglyceride ketogenic
diet in neurological and metabolic disorders. Lancet Neurol.
17:84–93. 2018. View Article : Google Scholar
|
|
121
|
Morris G, Maes M, Berk M, Carvalho AF and
Puri BK: Nutritional ketosis as an intervention to relieve
astrogliosis: Possible therapeutic applications in the treatment of
neurodegenerative and neuroprogressive disorders. Eur Psychiatry.
63:e82020. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Miller VJ, Villamena FA and Volek JS:
Nutritional ketosis and mitohormesis: Potential implications for
mitochondrial function and human health. J Nutr Metab.
2018:51576452018. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Milder J and Patel M: Modulation of
oxidative stress and mitochondrial function by the ketogenic diet.
Epilepsy Res. 100:295–303. 2012. View Article : Google Scholar :
|
|
124
|
Hutfles LJ, Wilkins HM, Koppel SJ,
Weidling IW, Selfridge JE, Tan E, Thyfault JP, Slawson C, Fenton
AW, Zhu H and Swerdlow RH: A bioenergetics systems evaluation of
ketogenic diet liver effects. Appl Physiol Nutr Metab. 42:955–962.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Hyatt HW, Kephart WC, Holland AM, Mumford
P, Mobley CB, Lowery RP, Roberts MD, Wilson JM and Kavazis AN: A
ketogenic diet in rodents elicits improved mitochondrial
adaptations in response to resistance exercise training compared to
an isocaloric western diet. Front Physiol. 7:5332016. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Kennedy AR, Pissios P, Out H, Roberson R,
Xue B, Asakura K, Furukawa N, Marino FE, Liu FF, Kahn BB, et al: A
high-fat, ketogenic diet induces a unique metabolic state in mice.
Am J Physiol Endocrinol Metab. 292:E1724–E1739. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Wallace MA, Aguirre NW, Marcotte GR,
Marshall AG, Baehr LM, Hughes DC, Hamilton KL, Roberts MN,
Lopez-Dominguez JA, Miller BF, et al: The ketogenic diet preserves
skeletal muscle with aging in mice. Aging Cell. 20:e133222021.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Parry HA, Kephart WC, Mumford PW, Romero
MA, Mobley CB, Zhang Y, Roberts MD and Kavazis AN: Ketogenic diet
increases mitochondria volume in the liver and skeletal muscle
without altering oxidative stress markers in rats. Heliyon.
4:e009752018. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Zhou Z, Hagopian K, López-Domínguez JA,
Kim K, Jasoliya M, Roberts MN, Cortopassi GA, Showalter MR, Roberts
BS, González-Reyes JA, et al: A ketogenic diet impacts markers of
mitochondrial mass in a tissue specific manner in aged mice. Aging
(Albany NY). 13:7914–7930. 2021. View Article : Google Scholar
|
|
130
|
Kephart WC, Mumford PW, Mao X, Romero MA,
Hyatt HW, Zhang Y, Mobley CB, Quindry JC, Young KC, Beck DT, et al:
The 1-week and 8-month effects of a ketogenic diet or ketone salt
supplementation on multi-organ markers of oxidative stress and
mitochondrial function in rats. Nutrients. 9:10192017. View Article : Google Scholar :
|
|
131
|
Xu S, Tao H, Cao W, Cao L, Lin Y, Zhao SM,
Xu W, Cao J and Zhao JY: Ketogenic diets inhibit mitochondrial
biogenesis and induce cardiac fibrosis. Signal Transduct Target
Ther. 6:542021. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Santra S, Gilkerson RW, Davidson M and
Schon EA: Ketogenic treatment reduces deleted mitochondrial DNAs in
cultured human cells. Ann Neurol. 56:662–669. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Ahn Y, Sabouny R, Villa BR, Yee NC,
Mychasiuk R, Uddin GM, Rho JM and Shutt TE: Aberrant mitochondrial
morphology and function in the BTBR mouse model of autism is
improved by two weeks of ketogenic diet. Int J Mol Sci.
21:32662020. View Article : Google Scholar :
|
|
134
|
Miller VJ, LaFountain RA, Barnhart E,
Sapper TS, Short J, Arnold WD, Hyde PN, Crabtree CD, Kackley ML,
Kraemer WJ, et al: A ketogenic diet combined with exercise alters
mitochondrial function in human skeletal muscle while improving
metabolic health. Am J Physiol Endocrinol Metab. 319:E995–E1007.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Parker BA, Walton CM, Carr ST, Andrus JL,
Cheung ECK, Duplisea MJ, Wilson EK, Draney C, Lathen DR, Kenner KB,
et al: β-Hydroxybutyrate elicits favorable mitochondrial changes in
skeletal muscle. Int J Mol Sci. 19:22472018. View Article : Google Scholar
|
|
136
|
Guo Y, Zhang C, Shang FF, Luo M, You Y,
Zhai Q, Xia Y and Suxin L: Ketogenic diet ameliorates cardiac
dysfunction via balancing mitochondrial dynamics and inhibiting
apoptosis in type 2 diabetic mice. Aging Dis. 11:229–240. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Krebs P, Fan W, Chen YH, Tobita K, Downes
MR, Wood MR, Sun L, Li X, Xia Y, Ding N, et al: Lethal
mitochondrial cardiomyopathy in a hypomorphic Med30 mouse mutant is
ameliorated by ketogenic diet. Proc Natl Acad Sci USA.
108:19678–19682. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Ahola-Erkkilä S, Carroll CJ,
Peltola-Mjösund K, Tulkki V, Mattila I, Seppänen-Laakso T, Oresic
M, Tyynismaa H and Suomalainen A: Ketogenic diet slows down
mitochondrial myopathy progression in mice. Hum Mol Genet.
19:1974–1984. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Srivastava S, Kashiwaya Y, King MT, Baxa
U, Tam J, Niu G, Chen X, Clarke K and Veech RL: Mitochondrial
biogenesis and increased uncoupling protein 1 in brown adipose
tissue of mice fed a ketone ester diet. FASEB J. 26:2351–2362.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Sullivan PG, Rippy NA, Dorenbos K,
Concepcion RC, Agarwal AK and Rho JM: The ketogenic diet increases
mitochondrial uncoupling protein levels and activity. Ann Neurol.
55:576–580. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Moore MP, Cunningham RP, Kelty TJ,
Boccardi LR, Nguyen NY, Booth FW and Rector RS: Ketogenic diet in
combination with voluntary exercise impacts markers of hepatic
metabolism and oxidative stress in male and female Wistar rats.
Appl Physiol Nutr Metab. 45:35–44. 2020. View Article : Google Scholar
|
|
142
|
Jornayvaz FR, Jurczak MJ, Lee HY,
Birkenfeld AL, Frederick DW, Zhang D, Zhang XM, Samuel VT and
Shulman GI: A high-fat, ketogenic diet causes hepatic insulin
resistance in mice, despite increasing energy expenditure and
preventing weight gain. Am J Physiol Endocrinol Metab.
299:E808–E815. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Huang TY, Linden MA, Fuller SE, Goldsmith
FR, Simon J, Batdorf HM, Scott MC, Essajee NM, Brown JM and Noland
RC: Combined effects of a ketogenic diet and exercise training
alter mitochondrial and peroxisomal substrate oxidative capacity in
skeletal muscle. Am J Physiol Endocrinol Metab. 320:E1053–E1067.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Bough KJ, Wetherington J, Hassel B, Pare
JF, Gawryluk JW, Greene JG, Shaw R, Smith Y, Geiger JD and
Dingledine RJ: Mitochondrial biogenesis in the anticonvulsant
mechanism of the ketogenic diet. Ann Neurol. 60:223–235. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Newell C, Shutt TE, Ahn Y, Hittel DS, Khan
A, Rho JM and Shearer J: Tissue specific impacts of a ketogenic
diet on mitochondrial dynamics in the BTBRT+tf/j mouse.
Front Physiol. 7:6542016. View Article : Google Scholar
|
|
146
|
Thai PN, Seidlmayer LK, Miller C, Ferrero
M, Dorn GW II, Schaefer S, Bers DM and Dedkova EN: Mitochondrial
quality control in aging and heart failure: Influence of ketone
bodies and mitofusin-stabilizing peptides. Front Physiol.
10:3822019. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Guo M, Wang X, Zhao Y, Yang Q, Ding H,
Dong Q, Chen X and Cui M: Ketogenic diet improves brain ischemic
tolerance and inhibits NLRP3 inflammasome activation by preventing
Drp1-mediated mitochondrial fission and endoplasmic reticulum
stress. Front Mol Neurosci. 11:862018. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Anton SD, Moehl K, Donahoo WT, Marosi K,
Lee SA, Mainous AG III, Leeuwenburgh C and Mattson MP: Flipping the
metabolic switch: Understanding and applying the health benefits of
fasting. Obesity (Silver Spring). 26:254–268. 2018. View Article : Google Scholar
|
|
149
|
Liu B, Hutchison AT, Thompson CH, Lange K,
Wittert GA and Heilbronn LK: Effects of intermittent fasting or
calorie restriction on markers of lipid metabolism in human
skeletal muscle. J Clin Endocrinol Metab. 106:e1389–e1399. 2021.
View Article : Google Scholar
|
|
150
|
Mattson MP, Longo VD and Harvie M: Impact
of intermittent fasting on health and disease processes. Ageing Res
Rev. 39:46–58. 2017. View Article : Google Scholar :
|
|
151
|
Ravanidis S, Grundler F, de Toledo FW,
Dimitriou E, Tekos F, Skaperda Z, Kouretas D and Doxakis E:
Fasting-mediated metabolic and toxicity reprogramming impacts
circulating microRNA levels in humans. Food Chem Toxicol.
152:1121872021. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Grundler F, Mesnage R, Goutzourelas N,
Tekos F, Makri S, Brack M, Kouretas D and Wilhelmi de Toledo F:
Interplay between oxidative damage, the redox status, and metabolic
biomarkers during long-term fasting. Food Chem Toxicol.
145:1117012020. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Wilhelmi de Toledo F, Grundler F,
Goutzourelas N, Tekos F, Vassi E, Mesnage R and Kouretas D:
Influence of long-term fasting on blood redox status in humans.
Antioxidants (Basel). 9:4962020. View Article : Google Scholar
|
|
154
|
de Cabo R and Mattson MP: Effects of
intermittent fasting on health, aging, and disease. N Engl J Med.
381:2541–2551. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Klimova N, Long A, Scafidi S and Kristian
T: Interplay between NAD+ and acetyl-CoA metabolism in
ischemia-induced mitochondrial pathophysiology. Biochim Biophys
Acta Mol Basis Dis. 1865:2060–2067. 2019. View Article : Google Scholar
|
|
156
|
Ren Z, He H, Zuo Z, Xu Z, Wei Z and Deng
J: The role of different SIRT1-mediated signaling pathways in toxic
injury. Cell Mol Biol Lett. 24:362019. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Antunes F, Erustes AG, Costa AJ,
Nascimento AC, Bincoletto C, Ureshino RP, Pereira GJS and Smaili
SS: Autophagy and intermittent fasting: The connection for cancer
therapy? Clinics (Sao Paulo). 73(Suppl 1): e814s2018. View Article : Google Scholar
|
|
158
|
Gerhart-Hines Z, Rodgers JT, Bare O, Lerin
C, Kim SH, Mostoslavsky R, Alt FW, Wu Z and Puigserver P: Metabolic
control of muscle mitochondrial function and fatty acid oxidation
through SIRT1/PGC-1alpha. EMBO J. 26:1913–1923. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Lee D, Martinez B, Crocker DE and Ortiz
RM: Fasting increases the phosphorylation of AMPK and expression of
sirtuin1 in muscle of adult male northern elephant seals (Mirounga
angustirostris). Physiol Rep. 5:e131142017. View Article : Google Scholar :
|
|
160
|
Rhee J, Inoue Y, Yoon JC, Puigserver P,
Fan M, Gonzalez FJ and Spiegelman BM: Regulation of hepatic fasting
response by PPARgamma coactivator-1alpha (PGC-1): Requirement for
hepatocyte nuclear factor 4alpha in gluconeogenesis. Proc Natl Acad
Sci USA. 100:4012–4017. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Zhang YK, Wu KC and Klaassen CD: Genetic
activation of Nrf2 protects against fasting-induced oxidative
stress in livers of mice. PLoS One. 8:e591222013. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Tripathi A, Scaini G, Barichello T,
Quevedo J and Pillai A: Mitophagy in depression: Pathophysiology
and treatment targets. Mitochondrion. 61:1–10. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Cheng A, Wan R, Yang JL, Kamimura N, Son
TG, Ouyang X, Luo Y, Okun E and Mattson MP: Involvement of PGC-1α
in the formation and maintenance of neuronal dendritic spines. Nat
Commun. 3:12502012. View Article : Google Scholar
|
|
164
|
Marosi K, Moehl K, Navas-Enamorado I,
Mitchell SJ, Zhang Y, Lehrmann E, Aon MA, Cortassa S, Becker KG and
Mattson MP: Metabolic and molecular framework for the enhancement
of endurance by intermittent food deprivation. FASEB J.
32:3844–3858. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Real-Hohn A, Navegantes C, Ramos K,
Ramos-Filho D, Cahuê F, Galina A and Salerno VP: The synergism of
high-intensity intermittent exercise and every-other-day
intermittent fasting regimen on energy metabolism adaptations
includes hexokinase activity and mitochondrial efficiency. PLoS
One. 13:e02027842018. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Rodríguez-Bies E, Santa-Cruz Calvo S,
Fontán-Lozano A, Peña Amaro J, Berral de la Rosa FJ, Carrión AM,
Navas P and López-Lluch G: Muscle physiology changes induced by
every other day feeding and endurance exercise in mice: Effects on
physical performance. PLoS One. 5:e139002010. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Brown-Borg HM and Rakoczy S: Metabolic
adaptations to short-term every-other-day feeding in long-living
Ames dwarf mice. Exp Gerontol. 48:905–919. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Singh R, Kaushik S, Wang Y, Xiang Y, Novak
I, Komatsu M, Tanaka K, Cuervo AM and Czaja MJ: Autophagy regulates
lipid metabolism. Nature. 458:1131–1135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Wan R, Ahmet I, Brown M, Cheng A, Kamimura
N, Talan M and Mattson MP: Cardioprotective effect of intermittent
fasting is associated with an elevation of adiponectin levels in
rats. J Nutr Biochem. 21:413–417. 2010. View Article : Google Scholar :
|
|
170
|
Kim YH, Lee JH, Yeung JL, Das E, Kim RY,
Jiang Y, Moon JH, Jeong H, Thakkar N, Son JE, et al:
Thermogenesis-independent metabolic benefits conferred by
isocaloric intermittent fasting in ob/ob mice. Sci Rep. 9:24792019.
View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Li G, Xie C, Lu S, Nichols RG, Tian Y, Li
L, Patel D, Ma Y, Brocker CN, Yan T, et al: Intermittent fasting
promotes white adipose browning and decreases obesity by shaping
the gut microbiota. Cell Metab. 26:672–685.e4. 2017. View Article : Google Scholar :
|
|
172
|
Monternier PA, Teulier L, Drai J,
Bourguignon A, Collin-Chavagnac D, Hervant F, Rouanet JL and
Roussel D: Mitochondrial oxidative phosphorylation efficiency is
upregulated during fasting in two major oxidative tissues of
ducklings. Comp Biochem Physiol A Mol Integr Physiol. 212:1–8.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Roussel D, Boël M and Romestaing C:
Fasting enhances mitochondrial efficiency in duckling skeletal
muscle by acting on the substrate oxidation system. J Exp Biol.
221:jeb1722132018.PubMed/NCBI
|
|
174
|
Sokolović M, Sokolović A, Wehkamp D, Ver
Loren van Themaat E, de Waart DR, Gilhuijs-Pederson LA, Nikolsky Y,
van Kampen AH, Hakvoort TB and Lamers WH: The transcriptomic
signature of fasting murine liver. BMC Genomics. 9:5282008.
View Article : Google Scholar
|
|
175
|
Bouwens M, Afman LA and Müller M: Fasting
induces changes in peripheral blood mononuclear cell gene
expression profiles related to increases in fatty acid
beta-oxidation: Functional role of peroxisome proliferator
activated receptor alpha in human peripheral blood mononuclear
cells. Am J Clin Nutr. 86:1515–1523. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Zhao L, Hutchison AT, Liu B, Yates CL,
Teong XT, Wittert GA, Thompson CH, Nguyen L, Au J, Manoogian ENC,
et al: Time-restricted eating improves glycemic control and dampens
energy-consuming pathways in human adipose tissue. Nutrition.
96:1115832022. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Castro-Sepúlveda M, Morio B, Tuñón-Suárez
M, Jannas-Vela S, Díaz-Castro F, Rieusset J and Zbinden-Foncea H:
The fasting-feeding metabolic transition regulates mitochondrial
dynamics. FASEB J. 35:e218912021. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Theurey P, Tubbs E, Vial G, Jacquemetton
J, Bendridi N, Chauvin MA, Alam MR, Le Romancer M, Vidal H and
Rieusset J: Mitochondria-associated endoplasmic reticulum membranes
allow adaptation of mitochondrial metabolism to glucose
availability in the liver. J Mol Cell Biol. 8:129–143. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Castro-Sepulveda M, Jannas-Vela S,
Fernández-Verdejo R, Ávalos-Allele D, Tapia G, Villagrán C, Quezada
N and Zbinden-Foncea H: Relative lipid oxidation associates
directly with mitochondrial fusion phenotype and
mitochondria-sarcoplasmic reticulum interactions in human skeletal
muscle. Am J Physiol Endocrinol Metab. 318:E848–E855. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Shirakabe A, Fritzky L, Saito T, Zhai P,
Miyamoto S, Gustafsson ÅB, Kitsis RN and Sadoshima J: Evaluating
mitochondrial autophagy in the mouse heart. J Mol Cell Cardiol.
92:134–139. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Glick D, Zhang W, Beaton M, Marsboom G,
Gruber M, Simon MC, Hart J, Dorn GW II, Brady MJ and Macleod KF:
BNip3 regulates mitochondrial function and lipid metabolism in the
liver. Mol Cell Biol. 32:2570–2584. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Islam H, Amato A, Bonafiglia JT, Rahman
FA, Preobrazenski N, Ma A, Simpson CA, Quadrilatero J and Gurd BJ:
Increasing whole-body energetic stress does not augment
fasting-induced changes in human skeletal muscle. Pflugers Arch.
473:241–252. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Martínez-González MA and Sánchez-Villegas
A: The emerging role of Mediterranean diets in cardiovascular
epidemiology: Monounsaturated fats, olive oil, red wine or the
whole pattern? Eur J Epidemiol. 19:9–13. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Guasch-Ferré M and Willett WC: The
Mediterranean diet and health: A comprehensive overview. J Intern
Med. 290:549–566. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Davis C, Bryan J, Hodgson J and Murphy K:
Definition of the Mediterranean diet; a literature review.
Nutrients. 7:9139–9153. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Martínez-González MA, Salas-Salvadó J,
Estruch R, Corella D, Fitó M and Ros E; PREDIMED INVESTIGATORS:
Benefits of the Mediterranean diet: Insights from the PREDIMED
study. Prog Cardiovasc Dis. 58:50–60. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Widmer RJ, Flammer AJ, Lerman LO and
Lerman A: The Mediterranean diet, its components, and
cardiovascular disease. Am J Med. 128:229–238. 2015. View Article : Google Scholar
|
|
188
|
Duluc L, Jacques C, Soleti R,
Andriantsitohaina R and Simard G: Delphinidin inhibits VEGF
induced-mitochondrial biogenesis and Akt activation in endothelial
cells. Int J Biochem Cell Biol. 53:9–14. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Wang J, Zou Q, Suo Y, Tan X, Yuan T, Liu Z
and Liu X: Lycopene ameliorates systemic inflammation-induced
synaptic dysfunction via improving insulin resistance and
mitochondrial dysfunction in the liver-brain axis. Food Funct.
10:2125–2137. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Tsai KL, Hung CH, Chan SH, Hsieh PL, Ou
HC, Cheng YH and Chu PM: Chlorogenic acid protects against
oxLDL-induced oxidative damage and mitochondrial dysfunction by
modulating SIRT1 in endothelial cells. Mol Nutr Food Res.
62:e17009282018. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Lagouge M, Argmann C, Gerhart-Hines Z,
Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P,
Elliott P, et al: Resveratrol improves mitochondrial function and
protects against metabolic disease by activating SIRT1 and
PGC-1alpha. Cell. 127:1109–1122. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Kalliora C, Kyriazis ID, Oka SI, Lieu MJ,
Yue Y, Area-Gomez E, Pol CJ, Tian Y, Mizushima W, Chin A, et al:
Dual peroxis ome-proliferator-activated-receptor-α/γ activation
inhibits SIRT1-PGC1 α axis and causes cardiac dysfunction. JCI
Insight. 5:e1295562019. View Article : Google Scholar
|
|
193
|
Baur JA, Pearson KJ, Price NL, Jamieson
HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K,
et al: Resveratrol improves health and survival of mice on a
high-calorie diet. Nature. 444:337–342. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Dong YZ, Li L, Espe M, Lu KL and
Rahimnejad S: Hydroxytyrosol attenuates hepatic fat accumulation
via activating mitochondrial biogenesis and autophagy through the
AMPK pathway. J Agric Food Chem. 68:9377–9386. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Cao K, Xu J, Zou X, Li Y, Chen C, Zheng A,
Li H, Li H, Szeto IM, Shi Y, et al: Hydroxytyrosol prevents
diet-induced metabolic syndrome and attenuates mitochondrial
abnormalities in obese mice. Free Radic Biol Med. 67:396–407. 2014.
View Article : Google Scholar
|
|
196
|
Deng X, Zhang S, Wu J, Sun X, Shen Z, Dong
J and Huang J: Promotion of mitochondrial biogenesis via activation
of AMPK-PGC1α signaling pathway by ginger (Zingiber officinale
Roscoe) extract, and its major active component 6-gingerol. J Food
Sci. 84:2101–2111. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Anderson EJ, Thayne KA, Harris M, Shaikh
SR, Darden TM, Lark DS, Williams JM, Chitwood WR, Kypson AP and
Rodriguez E: Do fish oil omega-3 fatty acids enhance antioxidant
capacity and mitochondrial fatty acid oxidation in human atrial
myocardium via PPARγ activation? Antioxid Redox Signal.
21:1156–1163. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Yoshino J, Smith GI, Kelly SC, Julliand S,
Reeds DN and Mittendorfer B: Effect of dietary n-3 PUFA
supplementation on the muscle transcriptome in older adults.
Physiol Rep. 4:e127852016. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
de Ligt M, Bruls YMH, Hansen J, Habets MF,
Havekes B, Nascimento EBM, Moonen-Kornips E, Schaart G,
Schrauwen-Hinderling VB, van Marken Lichtenbelt W and Schrauwen P:
Resveratrol improves ex vivo mitochondrial function but does not
affect insulin sensitivity or brown adipose tissue in first degree
relatives of patients with type 2 diabetes. Mol Metab. 12:39–47.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Joy JM, Vogel RM, Moon JR, Falcone PH,
Mosman MM, Pietrzkowski Z, Reyes T and Kim MP: Ancient peat and
apple extracts supplementation may improve strength and power
adaptations in resistance trained men. BMC Complement Altern Med.
16:2242016. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Perez-Ternero C, Werner CM, Nickel AG,
Herrera MD, Motilva MJ, Böhm M, Alvarez de Sotomayor M and Laufs U:
Ferulic acid, a bioactive component of rice bran, improves
oxidative stress and mitochondrial biogenesis and dynamics in mice
and in human mononuclear cells. J Nutr Biochem. 48:51–61. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Mollica MP, Mattace Raso G, Cavaliere G,
Trinchese G, De Filippo C, Aceto S, Prisco M, Pirozzi C, Di Guida
F, Lama A, et al: Butyrate regulates liver mitochondrial function,
efficiency, and dynamics in insulin-resistant obese mice. Diabetes.
66:1405–1418. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Myhrstad MCW, de Mello VD, Dahlman I,
Kolehmainen M, Paananen J, Rundblad A, Carlberg C, Olstad OK,
Pihlajamäki J, Holven KB, et al: Healthy nordic diet modulates the
expression of genes related to mitochondrial function and immune
response in peripheral blood mononuclear cells from subjects with
metabolic syndrome-A SYSDIET Sub-study. Mol Nutr Food Res.
e18014052019.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Brand MD, Orr AL, Perevoshchikova IV and
Quinlan CL: The role of mitochondrial function and cellular
bioenergetics in ageing and disease. Br J Dermatol. 169(Suppl 2):
S1–S8. 2013. View Article : Google Scholar
|