Introduction
Acute respiratory distress syndrome (ARDS) is
categorized as a minor disease, according to the Berlin definition
(1). However, ARDS and acute lung
injury (ALI) lead to major severe respiratory failure worldwide,
and are associated with a high morbidity and mortality due to
increased vascular permeability, alveolar-capillary membrane
dysfunction, the flooding of protein-rich fluid, alveolar
hemorrhage and fibrin deposition (2,3).
Following exposure to exogenous irritants, such as
lipopolysaccharide (LPS), the excessive accumulations and responses
of immune cells, including macrophages and neutrophils in the
infected area, are the key factors involved in the pathogenesis of
ALI/ARDS (4,5).
LPS is a major component of the outer membrane of
Gram-negative bacteria that binds to Toll like receptor-4 (TLR-4),
thus leading to the secretion of pro-inflammatory cytokines via
various signaling pathways (6,7).
Previous studies have demonstrated that LPS may directly affect
T-cell activity through the TLR ligand (8,9),
alter the ratio of CD4+/CD8+ T-cells, and
recruit neutrophils and macrophages into the inflamed area
(10). Based on their function
and location, macrophages present in the lungs are mainly
recognized as alveolar macrophages (AMs) and interstitial
macrophages (IMs) (11).
Importantly, during the process of ALI, AMs exert a host-protective
effect by killing microorganisms, secreting large amounts of
reactive oxygen radicals and tumor necrosis factor-a (TNF-a)
(12); IMs have more efficient
functions by releasing immunoregulatory cytokines, such as
interleukin (IL)-1β, keratinocyte-derived chemokine (CXCL1/KC) and
macrophage inflammatory protein 2 (MIP-2), inducing a specific
immune reaction in the pathogenesis of ALI/ARDS (13).
There is also evidence to indicate that activated
CD4+ T-cells are involved in ALI and regulate cytotoxic
T-lymphocyte antigen 4 (CTLA4) following exposure to LPS (14). In addition to CD4+
T-cells, transcription factors, such as retinoic acid
receptor-related orphan nuclear receptor γt (RORγt) trigger the
clonal expansion of pro-inflammatory Th17 cells (15). Existing evidence suggests that the
activation of the Th17 immune response enhances the inflammatory
response in various inflammatory and autoimmune disorders (16). Previous studies have documented
that the number of CD4+/CD8+ cells is
markedly increased in the lower respiratory tract area of lung
injury (17,18). The role of excessive immune
activation and the cytokine storm in lung injury induced by
COVID-19 has become a consensus (19,20). However, to date, the interaction
between T-cells and lung injury-mediated dysfunction has not yet
been fully elucidated. Therefore, further in-depth, systematic and
time-dependent studies on T-cell subsets
(CD4+/CD8+ ratio) and cytokine production, as
well as the related molecular mechanisms are warranted.
Nuclear factor E2-related factor 2 (Nrf2) and the
nucleotide-binding oligomerization domain (NOD)-like receptor
containing pyrin domain 3 (NLRP3) inflammasome are both regulated
by reactive oxygen species (ROS) under inflammatory conditions
(21). Previous studies have
demonstrated that Nrf2 plays a positive role in acute and chronic
inflammatory diseases (22,23). Under conditions of cellular
stress, Nrf2 is released from Kelch-like ECH-associated protein 1
(Keap1) through the proteasomal pathway, and subsequently, Nrf2
translocated to the nucleus to initiate the gene transcription,
such as heme oxygenase 1 (HO-1), NAD(P)H dehydrogenase [quinone] 1
(NQO1) and glutathione S-transferase-α. (24). The inflammasome is a
multifunctional protein complex composed of inactive NLRP3, adaptor
protein apoptosis-associated speck-like protein containing a CARD
(ASC) and caspase-1. Subsequently, activated caspase-1 leads to
changes in the form of IL-1β and IL-18 from a premature to a mature
one. Notably, these are the important indications of pyroptosis
(25,26). It has also been observed that Nrf2
is critical for NLRP3 inflammasome activation by ASC
oligomerization (27). Previous
studies have suggested that Nrf2 deficiency contributes to the
alleviation NLRP3 expression in acute and chronic inflammatory
diseases, and these results demonstrate an unexpected
pro-inflammatory effect of Nrf2 (22,23). In addition, the time-dependent,
pro-inflammatory role of Nrf2 remains a contradictory and undefined
issue. To date, the immunomodulatory and pro-inflammatory potential
of LPS in the NLRP3/Nrf2 pathway has not yet been extensively
investigated in a time-dependent manner in an in vivo model,
at least to the best of our knowledge.
Moreover, caspase-8 is a critical regulator that
activates the cleavage of gasdermin D (GSDMD)-dependent cell death
(28,29). Cleaved-GSDMD promotes membrane
pores, thus leading to the release of cytokine such as IL-1β and
others in the inflamed area (30). Therefore, understanding these cell
death processes is essential for the development of drugs for the
treatment of ALI.
The present study established acute and sub-chronic
models of ALI via the intratracheal instillation of 4 mg/kg LPS.
Taken together, the present study demonstrated the changes of the
potential inflammatory molecules and immune-related pathways in the
lung injury model at different time points. Moreover, the
inflammatory cell profiles in bronchoalveolar lavage fluid (BALF),
and multiple immune cell distribution and the redox status in lung
tissue, as well as the activation of the Nrf2/NLRP3 signaling
pathway and pyroptosis in the lungs of mice with LPS-induced ALI
were also determined.
Materials and methods
Mouse model of intratracheal injection of
LPS and experimental design
In the present study, 30 male C57/B6 mice, 6-7 weeks
old and weighing 20-25 g, were purchased from SLAC Laboratory
Animal (certificate no: SCXK2017-0016). All animal experiments were
approved by the Animal Care and Ethics Committee of Zhejiang
University, Hangzhou, China. All efforts were made to minimize
animal suffering. The mice were kept under standard day/night (12 h
light/12 h dark cycle, 45-55% relative humidity and a temperature
of 23-25°C) and pathogen-free conditions. The mice were randomly
divided into six groups of 5 mice in each (the control, LPS 4 h,
LPS 24 h, LPS 48 h, LPS 96 h and LPS 144 h groups).
First, the mice were anaesthetized with Avertin
(2,2,2-tribromoethanol; 0.2 ml/10 g; MilliporeSigma at 240 mg/kg by
an intraperitoneal (i.p.) injection according to body weight. After
5 min, the mice were place on the surgical tray in an appropriate
position. The neck skin was then sterilized with 75% ethanol, the
skin was cut using forceps and scissors, the neck skin was opened,
and the tracheal area was isolated very carefully to make the
airway visible. Subsequently, LPS (4 mg/kg body weight,
Escherichia coli, 0111: B4, MilliporeSigma) was injected
intratracheally into the mice using a microsyringe. LPS (4 mg/kg)
was used as previously described (31,32). In the control group, the same
volumes of 0.9% sodium chloride (NaCl) were administrated
intratracheally. At the different time points, the mice were deeply
anaesthetized and sacrificed for further analysis. Of note, no mice
died during the modelling or treatment process in the present
study.
Sample collection
The mice were sacrificed at 4, 24, 48, 96 and 144 h
after the LPS administration. For euthanasia, the mice (control
group and LPS groups) were deeply anesthetized with Avertin
(2,2,2-tribromoethanol; 480 mg/kg by i.p. injection. A few minutes
later, the heart rate of the mice disappeared due to an anesthesia
overdose. Blood was then collected by enucleating the mouse eyeball
and this was preserved at 4°C for 16-18 h. After 16-18 h, the blood
samples were centrifuged at 2,000 × g, 4°C for 10 min, and the
supernatant (upper layer) was collected as serum samples and
preserved at -80°C for determining cytokine levels. Subsequently, a
tracheal cannula was used to collect 0.5 ml BALF by injecting
ice-cold 1X PBS (Beijing Solarbio Science & Technology Co.,
Ltd.) into the lungs of mice (twice). The BALF was then centrifuged
at 2,000 × g for 10 min at 4°C. The BALF supernatant and blood
serum were then collected and kept at −80°C for the measurement of
cytokine levels. The third part of the right lung was used to
perform histopathological examinations; the first lobe of the left
lung was used for flow cytometric analysis and the remaining lung
lobes were frozen at −80°C for use in western blot analysis,
reverse transcription-quantitative PCR (RT-qPCR) and ROS generation
analysis.
Histopathological examination
For histological analysis, the lung tissue was fixed
in 10% neutral formalin for 48-72 h at room temperature, embedded
in paraffin, and sectioned at a thickness of 4 µm. Following
deparaffinization and rehydration using a series of laboratory
graded alcohol at different percentages (75%; 85%; 95%-I; 95%-II;
95% alcohol-III, dimethyl benzene-I and dimethyl benzene-II).
Alcohol and dimethyl benzene were obtained from Sinopharm Chemical
Reagent Co., Ltd. and Haoke Biological Technology Co., Ltd.,
respectively and the sections were stained with hematoxylin for 5
min and eosin solution for 10-12 sec at room temperature (Nanjing
Jiancheng Technology Co. Ltd.), and the tissue sections were rinsed
under running water. Finally, lung structures were observed under a
full slide scanning microscope (VS200 digital slide scanner BX51,
Olympus Corporation). The histopathological findings were then
determined based on neutrophil infiltration in the lung tissue. The
histopathological status of lung injury was scored according to the
Official American Thoracic Society Workshop Report (33). The parameters included the
following: a) Neutrophils in the alveolar space; b) neutrophils in
the interstitial space; c) hyaline membranes; d) proteinaceous
debris filling the airspaces; and e) alveolar septal thickening.
The total score was calculated using the following formula:
Score=[(20 × a) + (14 × b) + (7 × c) + (7 × d) + (2 × e)]/(number
of fields ×100).
Determination of cytokine levels in BALF
and serum using enzyme-linked immunosorbent assay (ELISA)
The collected BALF and blood were centrifuged at
1,000 × g for 8 min at 4°C, and the supernatant of BALF and serum
was used to evaluate the production of inflammatory cytokines using
ELISA kits, such as IL-1β (cat. no. DY401), CXCL1/KC (cat. no.
DY453), MIP-2 (cat. no. DY452), TNF-α (cat. no. DY410) (all from
R&D Systems, Inc.) following the manufacturer's protocol. The
optical density was detected at 450 nm using a microplate reader
(BioTek Instruments, Inc.). The result sample value was subtracted
from the bank value. The levels of IL-1β, CXCL1/KC, MIP-2 and TNF-α
were finally expressed in pg/ml.
Flow cytometric analysis
Single-cell suspensions were prepared from the left
lung tissues of the mice by cutting these into small sections and
digesting these with type I collagenase (3 mg/ml, MilliporeSigma)
and DNase I (30 µg/ml) for 45 min at 37°C in RPMI-1640
medium (cat. no. 10040, Corning, Inc.). The digested lungs were
mechanically disrupted using the flat portion of a plunger from a
3-ml syringe, then through a sterile filter (100 µm, Falcon,
BD Biosciences) followed by an additional 40-µm strainer
(Falcon, BD Biosciences). Red blood cells were lysed with 150 mM
NH4Cl, 10 mM KHCO3 and 0.1 mM EDTA. Following
two washes with phosphate-buffered saline (PBS), (Beijing Solarbio
Science & Technology Co., Ltd.), the cells were stained with
the Zombie Aqua™ Fixable Viability kit (cat. no. 423101; BioLegend,
Inc.) in the dark for 15-30 min at room temperature, followed by
washing once with cell staining buffer. Following the isolation of
live cells, BUV395 rat anti-mouse CD45 (1:80; cat. no. 564279),
PerCP-Cy™5.5 hamster anti-mouse CD3e (1:80; cat. no. 551163), FITC
rat anti-mouse CD4 (1:80; cat. no. 557307), APC-Cy™7 rat anti-mouse
CD8a (1:80; cat. no. 557654), PE-Cy™7 rat anti-mouse Ly-6G (1:80;
cat. no. 560601), PE rat anti-mouse F4/80 (1:80; cat. no. 565410),
BV650 rat anti-CD11b (1:80; cat. no. 563402) antibodies were used
with incubation at 4°C for 30 min. All antibodies were purchased
from BD Biosciences. Samples were evaluated using a CytoFLEX LX
flow cytometry analyzer (Beckman Coulter, Inc.). The results were
analyzed using CytExpert Version 2.4 software (Beckman Coulter,
Inc.).
Total RNA isolation and RT-qPCR
RNA was isolated from lung tissues using RNAiso plus
(Takara Bio, Inc.) according to the manufacturer's instructions and
reverse transcribed into cDNA using the PrimeScript™ RT reagent kit
from Takara Bio, Inc. qPCR was performed on the Bio-Rad C1000
real-time PCR system using SYBR-Green Master Mix reagent (Bio-Rad
Laboratories, Inc.). The PCR amplification reaction was as follows:
95°C for 30 sec, and subjected to 40 cycles of 95°C for 3 sec and
62°C for 30 sec. Primers were designed using the primer bank
website (https://pga.mgh.harvard.edu/primerbank/). The data
were evaluated using the 2−ΔΔCq formula for relative
quantitation and normalized to glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) (34). The
sequences of the primers for mouse gene expression are listed in
Table I (forward and
reverse).
 | Table ISequences of the primers used in the
present study. |
Table I
Sequences of the primers used in the
present study.
| Number | Primer name | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| I | TLR-4 |
CGCTTTCACCTCTGCCTTCACTACAG |
ACACTACCACAATAACCTTCCGGCTC |
| II | Nrf2 |
TCTTGGAGTAAGTCGAGAAGTGT |
GTTGAAACTGAGCGAAAAAGGC |
| III | GAPDH |
CATCACTGCCACCCAGAAGACTG |
ATGCCAGTGAGCTTCCCGTTCAG |
Western blot analysis
Total protein was extracted from the lung tissue
using cold radio immunoprecipitation assay (RIPA) lysis buffer
(including 1% protease inhibitor cocktail (Roche Diagnostics), 2%
PMSF (MilliporeSigma) and 1X PhosSTOP (Roche Diagnostics) and
centrifuged at 10,000 × g at 4°C for 10 min. The protein
concentration was evaluated using quick start Bradford 1X dye
reagent (Bio-Rad Laboratories, Inc.). Subsequently, 5X loading
buffer (Beyotime Institute of Biotechnology, Inc.) was added to the
protein sample and then denatured at 100°C for 5 min. Subsequently,
12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) was used to separate the proteins. The equivalent
loading of the gel was determined by quantitation of the protein
and by re-probing the membranes for GAPDH detection. Isolated
proteins were electroblotted onto nitrocellulose (NC) membranes
(BioTrace NT membranes, Gelman Laboratory) for 70 to 90 min at 300
mA (Mini-protein II System, Bio-Rad Laboratories, Inc.) and blocked
for h at room temperature with Tris-buffered saline containing 5%
BSA. The membranes were then incubated with the following primary
antibodies: TLR-4 (1:1,000; cat. no. ab13867, Abcam), anti-NLRP3
(1:1,000; cat. no. AG-20B-0014, Adipogen Life Sciences);
anti-procaspase-1/cleaved caspase-1 (1:1,000; cat. no. ab179515,
Abcam), p-Nrf2 (1:1,000; cat. no. db523, Diagbio), (http://www.diagbio.com/), anti-Nrf2 antibody (1:1,000;
cat. no. ab137550, Abcam); anti-Keap1 antibody (1:1,000; cat. no.
ab119403, Abcam), aAnti-GSDMD antibody (1:1,000; cat. no. ab155233,
Abcam), anti-caspase-8 (1:1,000; cat. no. ab227430, Abcam),
anti-ASC (1:1,000; cat. no. sc-514559, Santa Cruz Biotechnology,
Inc.) and anti-GAPDH (1:5,000; cat. no. db106, Diagbio) at room
temperature overnight. After washing with 1X TBST, the membranes
were incubated with HRP-conjugated secondary antibodies (1:5,000;
IRDye 800CW goat anti-rabbit; IRDye 680CW goat anti-mouse; LI-COR
Biosciences) for 1.5 h at room temperature. The membranes were
washed three times with 1X TBST, 5 min for each time. Images were
captured using an Oddessy CLx infrared laser dual color imaging
analysis system (Image Studio Ver 5.2, LI-COR Biosciences). The
density of each protein band on the membrane is reported as the
densitometric ratio between the protein of interest and GAPDH.
Measurement of ROS generation
ROS production in lung tissues was estimated using
the method described in the study by Socci et al (35), with minor modifications.
Homogenates of tissue samples were prepared using ice-cold 40 mM
Tris-HCl buffer (pH 7.4). Following sonication, tissue samples were
diluted with 0.25% with ice-cold Tris buffer. Subsequently, 100
µl sample solution was used to determine the protein
concentration. The remaining 0.25% pf the solution was divided into
two equal portions; one portion was mixed with 5 µM
dichlorodihydrofluorescein diacetate (DCFH-DA; MilliporeSigma) in
methanol, and the other portion was used as a blank (100 µl
methanol). All samples were then incubated in a water bath (37°C)
for 45 min. The DCF fluorescence intensities of the samples were
detected using a Varioskan Flash microplate reader (Thermo Fisher
Scientific, Inc.) at excitation and emission wavelengths of 485 and
525 nm, respectively.
Statistical analysis
Statistical analysis was performed using Graph prism
7 software (Graphpad Software, Inc.). The data are presented as the
mean ± SEM (n=4-5 mice per group). Differences between groups were
determined using one-way analysis of variance (ANOVA) followed by
the Bonferroni correction for the comparison of selected column
pairs, and the Kruskal-Wallis test followed by Dunn's post hoc
test. A value of P<0.05 was considered to indicate a
statistically significant difference.
Results
LPS induces lung injury in a
time-dependent manner
The most crucial pathological feature of LPS-induced
ALI is the accumulation and activation of neutrophils in lung
tissues. As shown in Fig. 1A, LPS
induced pathological changes in the lung tissues of the mice at 4,
24, 48, 96 and 144 h when compared with the control group, and the
highest lung injury scores were recorded from the LPS-4 h to LPS-96
h groups; the scores then exhibited a decreasing trend in the
LPS-144 h group (Fig. 1B).
Additionally, LPS exposure also resulted in the highest number of
white blood cells (WBCs) compared with the control group, with a
significant difference up to 96 h of LPS administration, with an
increasing trend from 4 h (Fig.
1C). From 4 to 96 h after LPS administration, the features of
lung injury were more evident. Notably, no detectable histological
damage was observed in the control group.
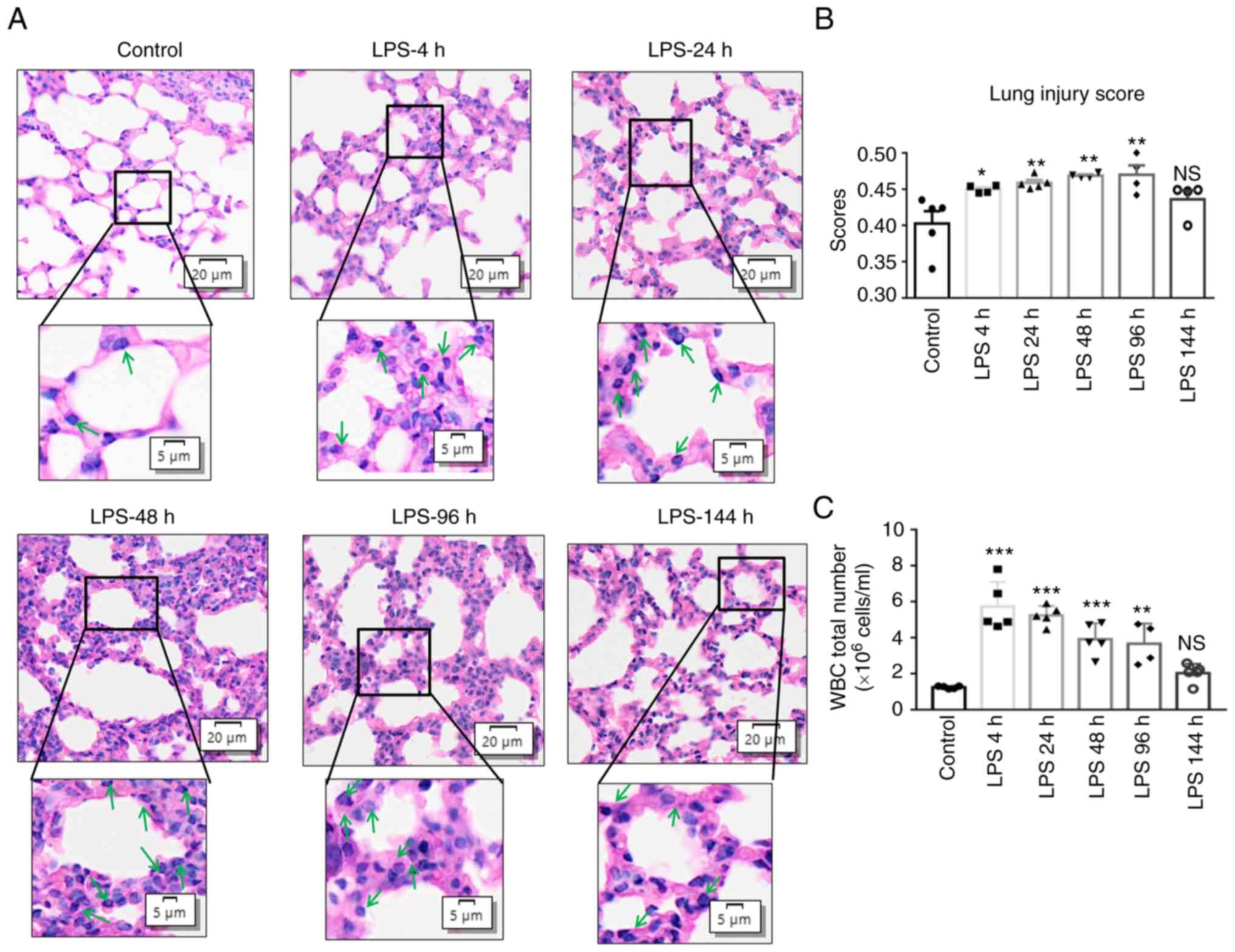 | Figure 1Time-dependent pathological effects
in the model of LPS-induced acute lung injury. C57BL/6 mice were
exposed to 4 mg/kg LPS via intratracheal installation and 0.9% NaCl
was administered to the control mice. Lung tissues and BALF were
collected, after 4, 24, 48, 96 and 144 h of LPS exposure and from
the control mice. (A) Histological structures of right lung lobes
stained with H&E for the LPS groups at different time points
and the control group (scale bars: Upper panels, 20 µM;
lower panels, 5 µM). Representative results from 5 mice are
shown for each group. H&E staining represents the structure of
neutrophils (green arrows). (B) Lung injury scores in lung tissue
from mice exposed to LPS. (C) Number of WBCs in BALF. Values are
presented as the mean ± SEM, n=4-5 mice per group. (C) One-way
ANOVA followed by the Bonferroni test and (B) the Kruskal-Wallis
test followed by Dunn's post hoc test were used to analyze
significant differences between the different time points of the
LPS groups vs. the control. *P<0.05,
**P<0.01 and ***P<0.001, vs. control.
NS, not significant; LPS, lipopolysaccharide; BALF, bronchoalveolar
lavage fluid; H&E, hematoxylin and eosin; WBC, white blood
cell. |
LPS induces the time-dependent activation
of T-cell subsets in lung tissue
T-cell subsets are critical mediators of immune
responses. A previous study demonstrated that the function of
T-helper cells was associated with the polarization, recruitment
and activation of immune cells during inflammation (36). The present study compared the
subpopulation, composition and activation/differentiation of
T-cells between the LPS-exposed groups and the control group, in
which cells were stained with CD4 FITCA (a marker of
CD4+ T-cells) CD8a APC-A750-A (a marker of
CD8+ T-cells). As shown in the flow cytometry images
(Fig. 2A and B) following
exposure to LPS, the numbers of CD4+ T-cells increased
to 70.68, 85.52, 40.58, 88.27 and 94.55% in the LPS-4 h, LPS-24 h,
LPS-96 h and LPS-144 h groups, respectively, although there were no
significant differences relative to the control group (Fig. 2C); of note, in lung tissue from
mice exposed to LPS, the numbers of CD8+ T-cells were
significantly inhibited at the different time points [LPS-4 h
(9.85%); LPS-24 h (7.90%); LPS-48 h (5.48%); LPS-96 h (11.71%) and
LPS-144 h (25.98%)] (Fig. 2D). In
addition, all LPS-exposed groups had significantly higher
CD4+ to CD8+ ratios than the normal control
group, and the highest peak of CD4+/CD8+
T-cell ratios was identified in the LPS-24 h group (Fig. 2E). These results thus indicated
that the abnormal differentiation of T-cells in responses to LPS
was associated with aberrant cytokine production in downstream
signaling pathways in ALI.
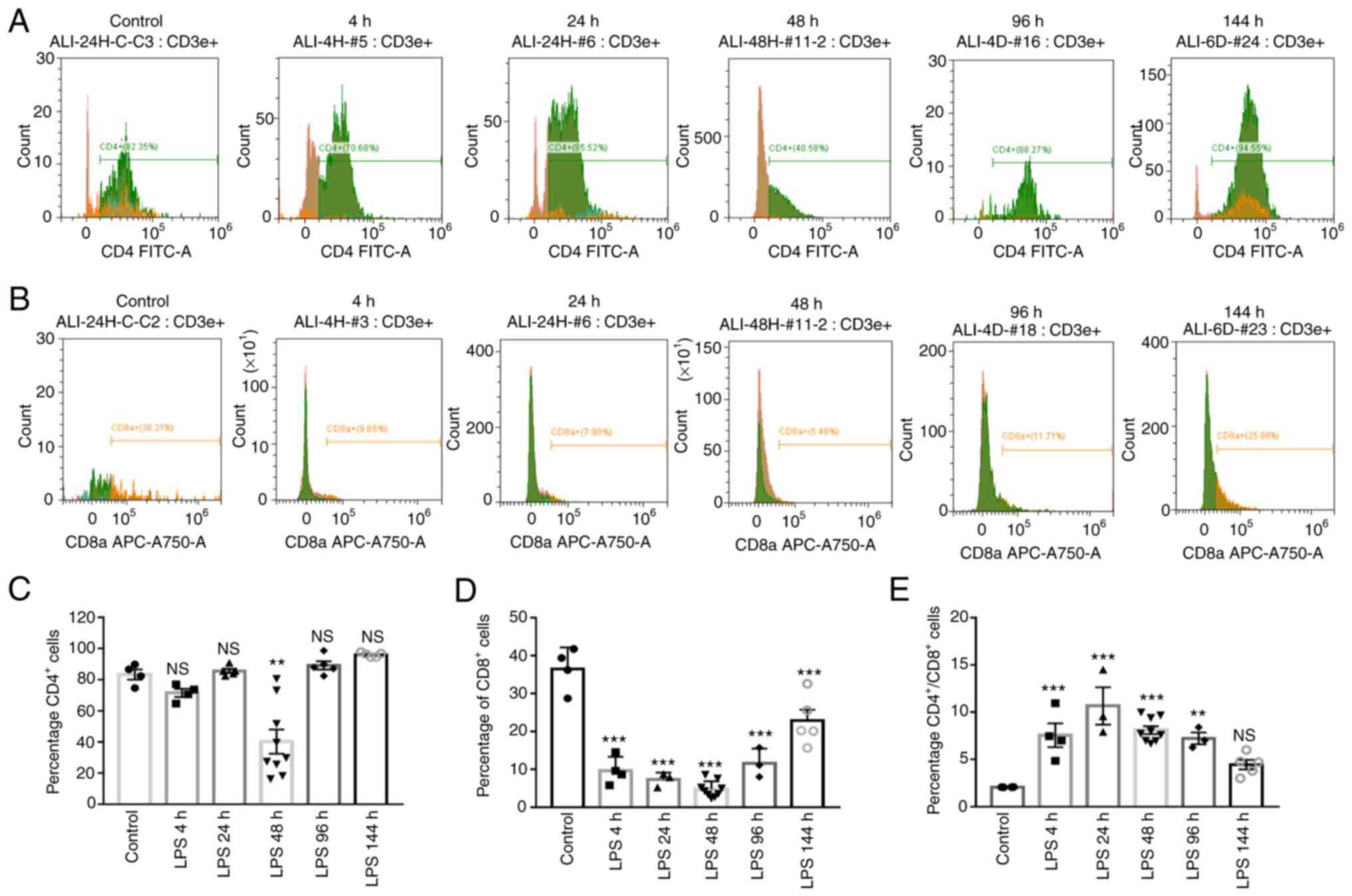 | Figure 2Regulation of CD4+,
CD8+ cells and the ratio of CD4+ to
CD8+ responses by LPS. C57BL/6 mice were exposed to 4
mg/kg LPS via intratracheal installation for 4, 24, 48, 96 and 144
h, and 0.9% NaCl was administered to the control mice orally. Lung
tissues were digested and stained with CD4 FIT C A (marker of
CD4+ T-cells) CD8a APC-A750-A (marker of CD8+
T-cells). (A and B) The percentages of CD4+ and
CD8+ T cells in lung tissue were determined using flow
cytometry. (C-E) CD4+, CD8+ T-cells, and the
CD4+/CD8+ ratios are presented in bar graphs.
The results are expressed as the mean ± SEM (n=4-5 animals in each
group). One-way ANOVA followed by the Bonferroni test were used to
analyze significant differences among the different time points of
the LPS group vs. the control. **P<0.01 and
***P<0.001, vs. control. NS, not significant; LPS,
lipopolysaccharide. |
Time-dependent activation of neutrophil
and alveolar/interstitial macrophages in lung tissue
The recruitment of macrophages and neutrophils plays
a crucial role in the process of ALI. Following LPS stimulation,
the whole left lung lobes from mice were digested and stained;
there was a significant increase in the staining for F4/80 (a
marker of macrophages) in the LPS-144 h group by 25.1% compared to
the control group (Fig. 3B and
E). Double immunostaining of the lung tissues confirmed that
the F4/80 and CD11b+ macrophages were confined to the
interstitial compartment, and the numbers of IMs were significantly
increased at 4 to 144 h following exposure to LPS (Fig. 3C and G). Additionally, the
intratracheal instillation of LPS induced neutrophil recruitment
into the lung tissue; however, the actual time point and mechanisms
involved remain unclear. AMs have previously been reported to be
involved in the accumulation of neutrophils in lung injury
(37). In the present study, as
shown by flow cytometry, the percentage of AMs (stained with
F4/80+ CD11b−) decreased from 4 to 144 h
following exposure to LPS (Fig. 3C
and F); however, the number of neutrophils (stained with LY6G
PE-CY7A) significantly increased during this time period (LPS-4 h,
LPS-24 h and LPS-48 h) compared to the control group (Fig. 3A and D).
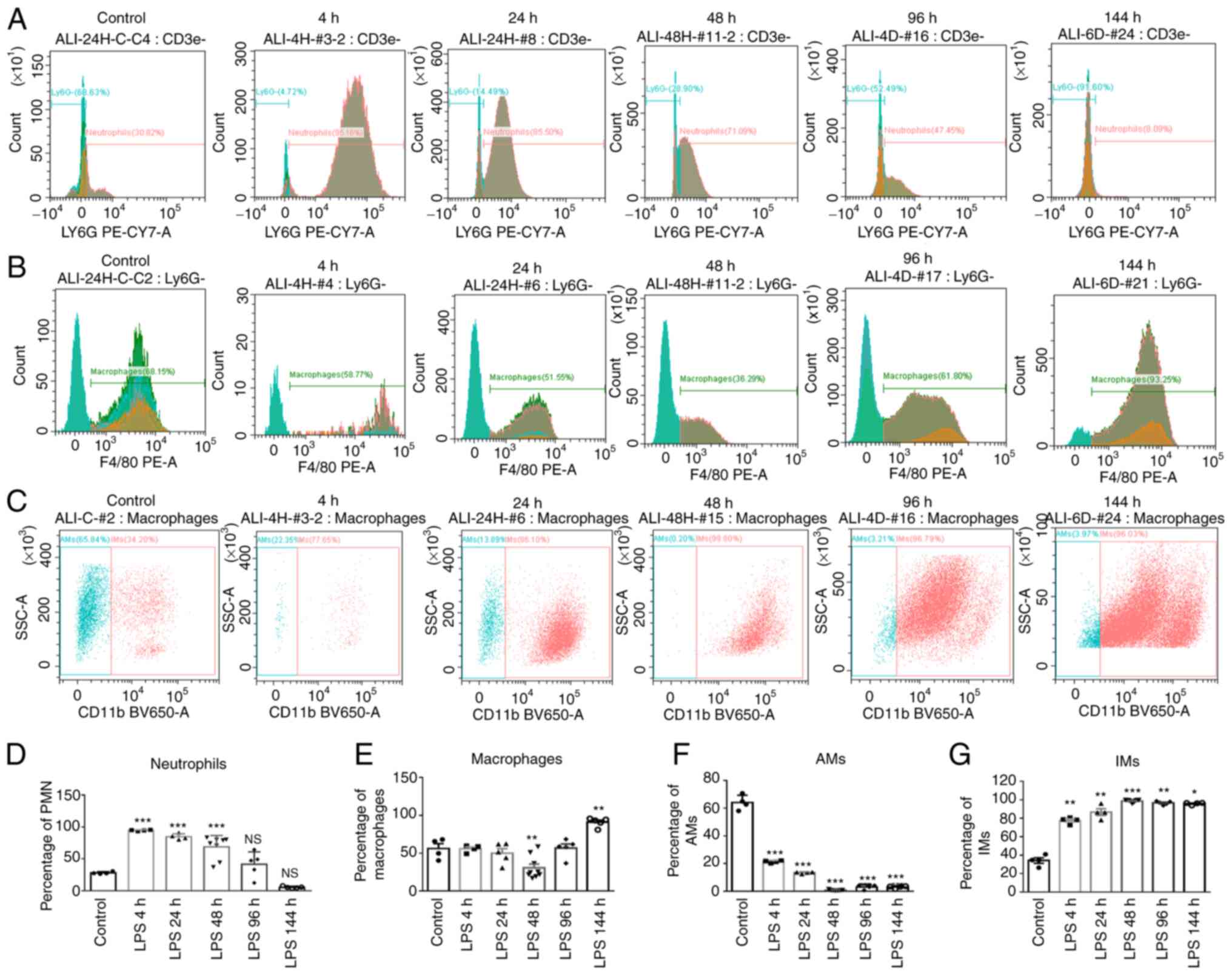 | Figure 3Neutrophils, macrophages, AMs and IMs
respond differentially to LPS in the model of ALI. C57BL/6 mice
were exposed to 4 mg/kg LPS via intratracheal installation for 4,
24, 48, 96 and 144 h, and 0.9% NaCl was administered to the control
mice orally. Lung cells isolated at indicated time points and
stained for F4/80 (marker of macrophages),
F4/80+CD11b− (marker of AMs), LY6G PE-CY7A
(stained for neutrophils), and F4/80+CD11b+
(marker of IMs). (A-C) Flow cytometric analysis of neutrophils,
macrophages, AMs and IMs. (D-G) The percentages of indicated cell
populations are presented in bar graphs. The results are
representative of five independent mice in each group (mean ± SEM,
n=5). One-way ANOVA followed by the Bonferroni test were used to
analyze significant differences among the different time points of
the LPS group vs. the control. *P<0.05,
**P<0.01 and ***P<0.001, vs. control.
NS, not significant; LPS, lipopolysaccharide; AMs, alveolar
macrophages; IMs, interstitial macrophages. |
Time-dependent effects of LPS on
pro-inflammatory cytokines BALF and serum
Accumulating evidence suggests that macrophages and
neutrophils are generally considered to be the major sources of
IL-1β, CXCL1/KC, MIP-2, TNF-α (38-40). In the present study, to evaluate
the time-dependent effects of LPS on the secretion of
pro-inflammatory cytokines in BALF and serum, samples were
collected at different time points from the mice in the LPS groups
(4, 24, 48, 96, 144 h) and the control group. As shown in Fig. 4, the secretion of IL-1β, CXCL1/KC,
MIP-2 and TNF-α in BALF and serum increased, and the maximal peak
of cytokine secretion following LPS exposure was reached at 4 h;
the levels then exhibited a decreasing trend in the from 96 to 144
h following the LPS instillation. These results demonstrated that
LPS induced time-dependent changes in the secretion of the
pro-inflammatory mediators, IL-1β, CXCL1/KC, MIP-2 and TNF-α, in
the lungs and serum of C57BL/6 mice. Of note, in the control group,
the cytokines were not constitutively expressed. Similar to the
present study, Bosnar et al (41) found that the levels of CXCL1,
TNF-α and IL-1β increased from 4 h following LPS stimulation in
inflammatory cells and lung tissue.
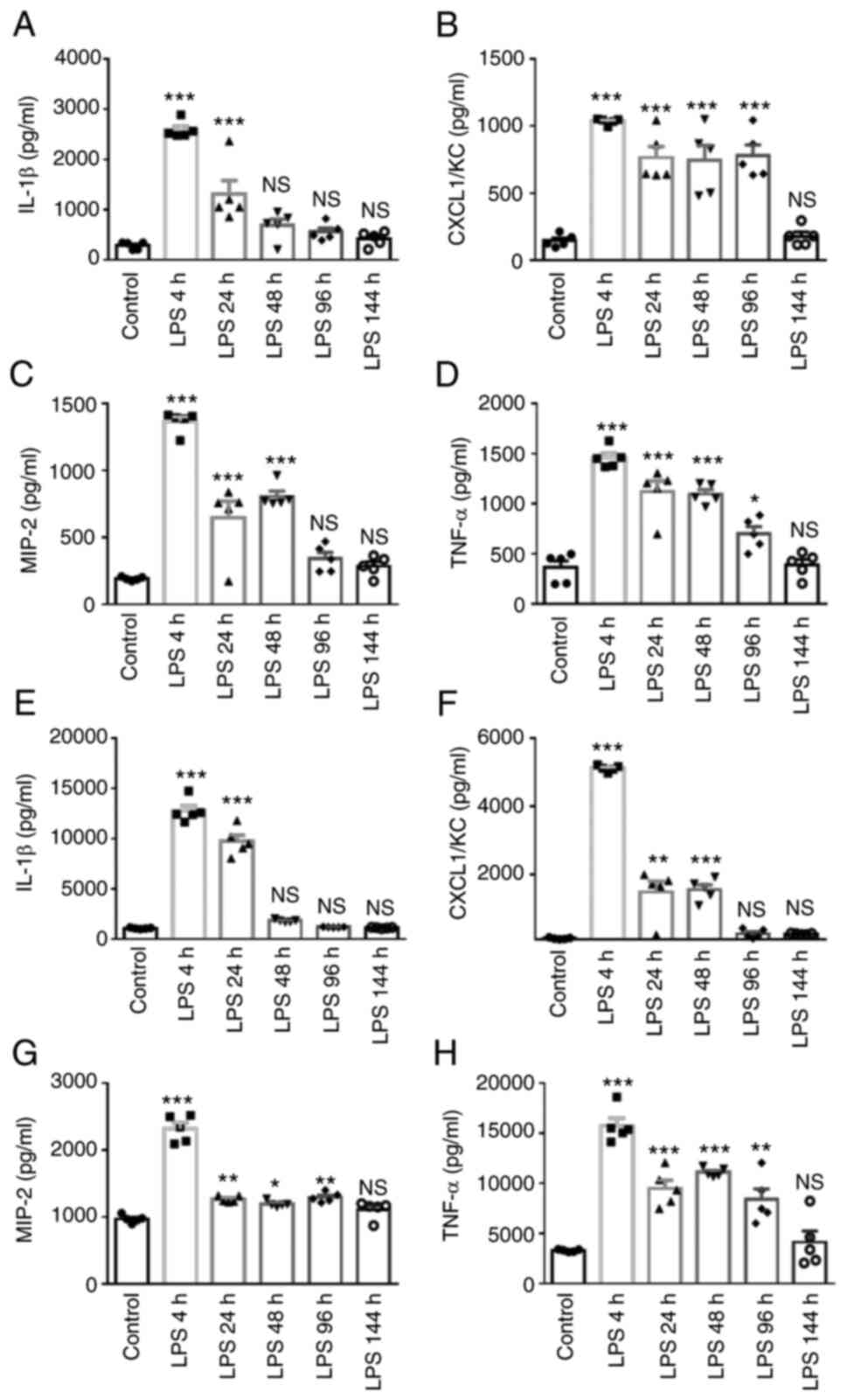 | Figure 4Time-dependent effects on
pro-inflammatory cytokines in BALF and serum from mice with
LPS-induced lung injury. C57BL/6 mice were exposed to with 4 mg/kg
LPS via intratracheal installation for 4, 24, 48, 96 and 144 h, and
0.9% NaCl was administered to the control mice orally. BALF and
serum were collected after for 4, 24, 48, 96 and 144 h of LPS
exposure and from the control mice. The cytokine levels in (A-D)
BALF and (E-H) serum were measured using ELISA and are shown as (A
and E) IL-1β, (B and F) CXCL1/KC; (C and G) MIP-2; (D and H) TNF-α
accordingly. The results are expressed as the mean ± SEM (n=5
animals in each group). One-way ANOVA followed by the Bonferroni
test were used to analyze significant differences among the
different time points of the LPS group vs. the control.
*P<0.05, **P<0.01 and
***P<0.001, vs. control. NS, not significant; LPS,
lipopolysaccharide; BALF, bronchoalveolar lavage fluid; MIP-2,
macrophage inflammatory protein 2. |
Time-dependent effects of LPS on ROS
production in lung tissue
The present study then examined the redox balance as
an indicator of oxidative stress at different time points in
LPS-induced lung tissue using an H2DCFDA fluorescence
probe. As shown in Fig. 5, the
level of fluorescence intensity in the lung tissue significantly
increased following LPS exposure at 4 h (36.39%), 24 h (48.33%), 48
h (55.40%), 96 h (39.04%) and 144 h (20.56%), whereas the highest
level was recorded in the LPS-48 h group, with a decreasing trend
observed at 144 h. These data suggested that ROS play a crucial
role in hindering normal structure in a time-dependent manner after
LPS induction.
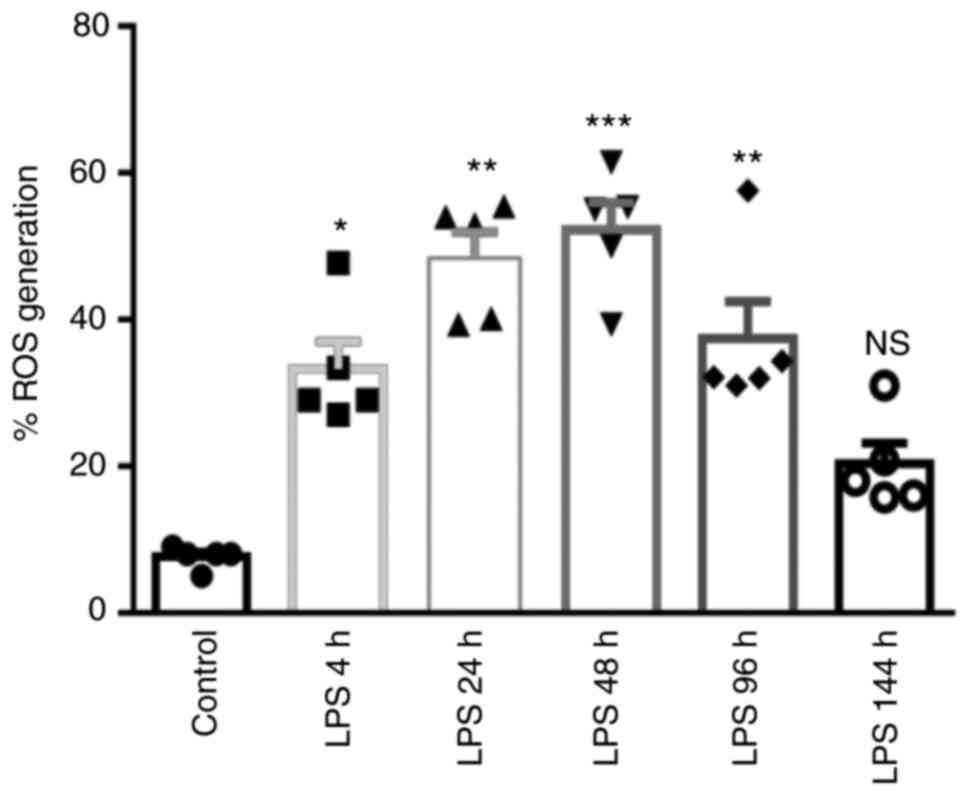 | Figure 5Time-dependent effects of LPS on ROS
production in lung tissue. C57BL/6 mice were exposed to with 4
mg/kg LPS via intratracheal installation for 4, 24, 48, 96 and 144
h, and 0.9% NaCl was administered to the control mice orally. Lung
tissues were collected after 4, 24, 48, 96 and 144 h of LPS
exposure and from the control mice. ROS production was determined
in lung tissue at different time points in the LPS groups and
control group. Data are presented as the mean ± SEM (n=5 animals in
each group). One-way ANOVA followed by the Bonferroni test were
used to analyze significant differences among the different time
points of the LPS group vs. the control. *P<0.05,
**P<0.01 and ***P<0.001, vs. control.
NS, not significant; LPS, lipopolysaccharide; ROS, reactive oxygen
species. |
Time-dependent changes in mRNA levels of
TLR-4 and Nrf2 in lung tissue induced by LPS
As shown in Fig. 6A
and B, the mRNA levels of TLR-4 significantly increased from 4
to 144 h following exposure to LPS compared with the control group,
and the maximal mRNA expression levels were detected after 24 h
(TLR-4) of LPS stimulation. The mRNA level of Nrf2 was
significantly increased from 4 to 48 h following exposure to LPS.
Notably, these data suggested that LPS affected the TLR-4 and Nrf2
mRNA levels in the early stages of lung injury.
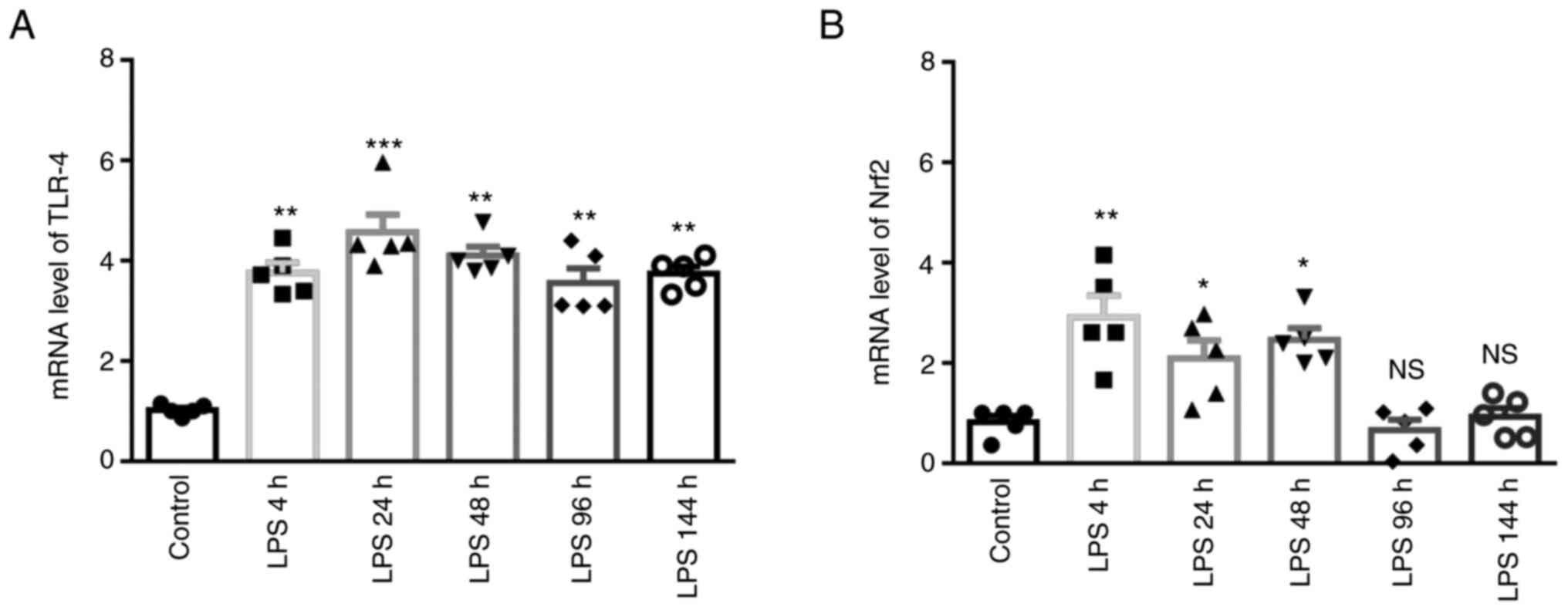 | Figure 6Time-dependent changes in the mRNA
levels of TLR-4 and Nrf2 in lung tissue from mice with LPS-induced
lung injury. C57BL/6 mice were exposed to with 4 mg/kg LPS via
intratracheal installation for 4, 24, 48, 96 and 144 h, and 0.9%
NaCl was administered to the control mice orally. Lung tissues were
collected after 4, 24, 48, 96 and 144 h of LPS exposure and from
the control mice. The mRNA levels of (A) TLR-4 and (B) Nrf2 were
determined using reverse transcription-quantitative PCR. The
results are expressed as the mean ± SEM (n=5 animals in each
group). One-way ANOVA followed by the Bonferroni test were used to
analyze significant differences among the different time points of
the LPS group vs. the control. *P<0.05,
**P<0.01 and ***P<0.001, vs. control.
NS, not significant; LPS, lipopolysaccharide; TLR-4, Toll like
receptor-4; Nrf2, nuclear factor E2-related factor 2. |
Time-dependent responses of NLRP3
inflammasome activation in lung tissue
The present study then evaluated the effects of LPS
on the activation of NLRP3 and cleaved caspase-1 in lung tissue in
a time-dependent manner. The expression levels of NLRP3 and cleaved
caspase-1 were markedly increased from 4 to 144 h following
exposure to LPS; the densitometric analysis of the western blots
revealed that the expression of NLRP3 at 24 h and that of cleaved
caspase-1 at 48 h were higher when compared to those of the control
group (Fig. 7). These data
suggested that the NLRP3 inflammasome and cleaved caspase-1
activity were altered in a time-dependent manner following exposure
to LPS.
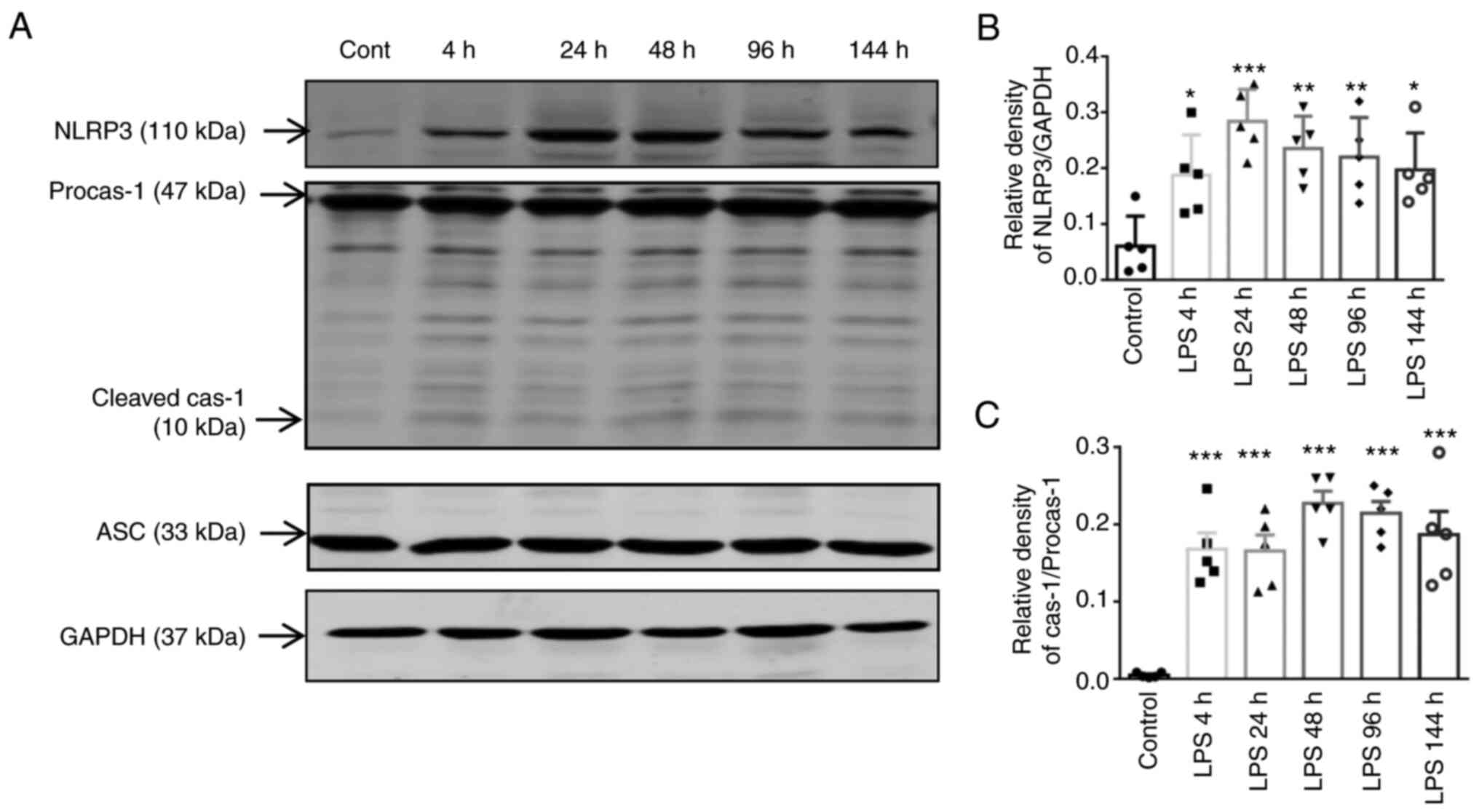 | Figure 7Time-dependent responses of NLRP3
inflammasome activation in lung tissue. C57BL/6 mice were exposed
to with 4 mg/kg LPS via intratracheal installation for 4, 24, 48,
96 and 144 h, and 0.9% NaCl was administered to the control mice
orally. Lung tissues were collected after 4, 24, 48, 96 and 144 h
of LPS exposure and from the control mice. (A) Protein expression
levels of NLRP3, ASC, procaspase-1/cleaved caspase-1 and GAPDH
(internal control) were determined using western blot analysis with
corresponding antibodies. (B and C) Relative expression levels of
the proteins were determined using densitometric analysis. Data are
presented as the mean ± SEM (n=5 animals in each group). One-way
ANOVA followed by the Bonferroni test were used to analyze
significant differences among the different time points of the LPS
group vs. the control. *P<0.05,
**P<0.01 and ***P<0.001, vs. control.
NLRP3, nucleotide-binding oligomerization domain (NOD)-like
receptor containing pyrin domain 3; LPS, lipopolysaccharide; ASC,
apoptosis-associated speck-like protein containing a CARD;
Procas-1, procaspase-1; cas-1, caspase-1. |
Time-dependent responses of TLR-4,
Keap-1, Nrf2 and HO-1 expression levels in lung tissue
Nrf2 serves as an important mediator regulating
ROS-dependent inflammasome activation in ALI (27). In the present study, to
investigate the possible mechanisms relevant to the Nrf2-related
signaling pathway, the expression of the pNrf2, tNrf2, keap-1 and
TLR-4 was first assayed using western blot analysis (Fig. 8A). LPS induced an increase in
TLR-4 and Keap-1 expression in a time-dependent manner, with
significant induction at 4, 24, 48 and 96 h (Fig. 8B and C). However, LPS induced the
phosphorylation of Nrf2 according to Keap-1, suggesting that p-Nrf2
upregulation was independent of Keap-1 expression. As shown in
Fig. 8D, the p-Nrf2 level was
significantly increased in the LPS-4 h, LPS-24 h and LPS-48 h
groups, respectively, compared with the t-Nrf2 level, as shown by
densitometric analysis. In addition, HO-1 expression was
significantly increased following LPS exposure from 4 to 96 h
(Fig. 8E). Taken together, these
results prompted us to further investigate the role of TLR-4
signaling components in regulating the initiation and expansion of
Nrf2/HO-1 signaling in LPS-induced lung injury.
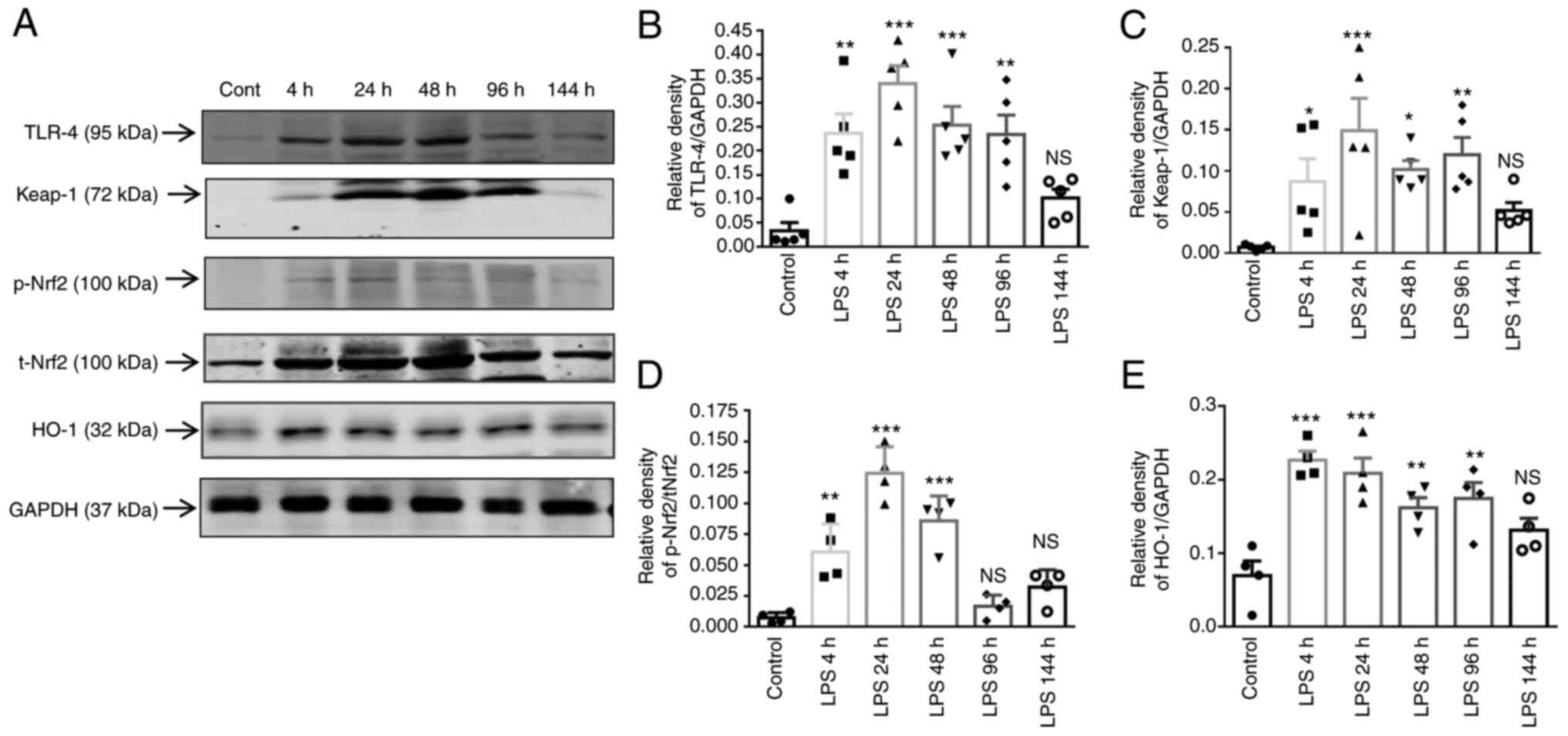 | Figure 8Time-dependent responses of TLR-4,
Keap-1, Nrf2 and HO-1 expression in lung tissue. C57BL/6 mice were
exposed to with 4 mg/kg LPS via intratracheal installation for 4,
24, 48, 96 and 144 h, and 0.9% NaCl was administered to the control
mice orally. (A) Protein expression levels of TLR-4, Keap-1 and
p-Nrf2, t-Nrf2, HO-1 and GAPDH (internal control) in the lungs of
mice were assessed using western blot analysis; (B-E) Relative
protein expression levels were determined using densitometric
analysis. Data are presented as the mean ± SEM (n=4 animals in each
group). One-way ANOVA followed by the Bonferroni test were used to
analyze significant differences among the different time points of
the LPS group vs. the control. *P<0.05,
**P<0.01 and ***P<0.001, vs. control.
*P<0.05, **P<0.01 and
***P<0.001, vs. control. NS, not significant; TLR-4,
Toll like receptor-4; LPS, lipopolysaccharide; Nrf2, nuclear factor
E2-related factor 2; p-, phosphorylated; t-, total. |
Time-dependent changes in the levels of
cleaved caspase-8 and cleaved GSDMD in lung tissue
In pyroptosis, IL-1β secretion and caspase-1
activation are prominent characteristics. Caspase-8 has been
reported to promote the cleavage of GSDMD and gasdermin E (GSDME)
in murine macrophages, leading to pyroptotic cell death (28). In the present study, the levels of
caspase-8, cleaved caspase-8, and the pyroptosis markers, GSDMD and
cleaved GSDMD were measured at different time points (4, 24, 48, 96
and 144 h) following exposure to LPS (Fig. 9A). The protein expression levels
of cleaved caspase-8 and cleaved GSDMD were higher at 48 and 4 h
following exposure to LPS (Fig. 9B
and C), while from 4 to 48 h following exposure to LPS, when
compared to the control group, the levels of these proteins
exhibited a significant difference. These results indicated that
LPS plays a critical role in pyroptosis by upregulating the cleaved
GSDMD levels in parallel with the activation of cleaved caspase-8
in ALI.
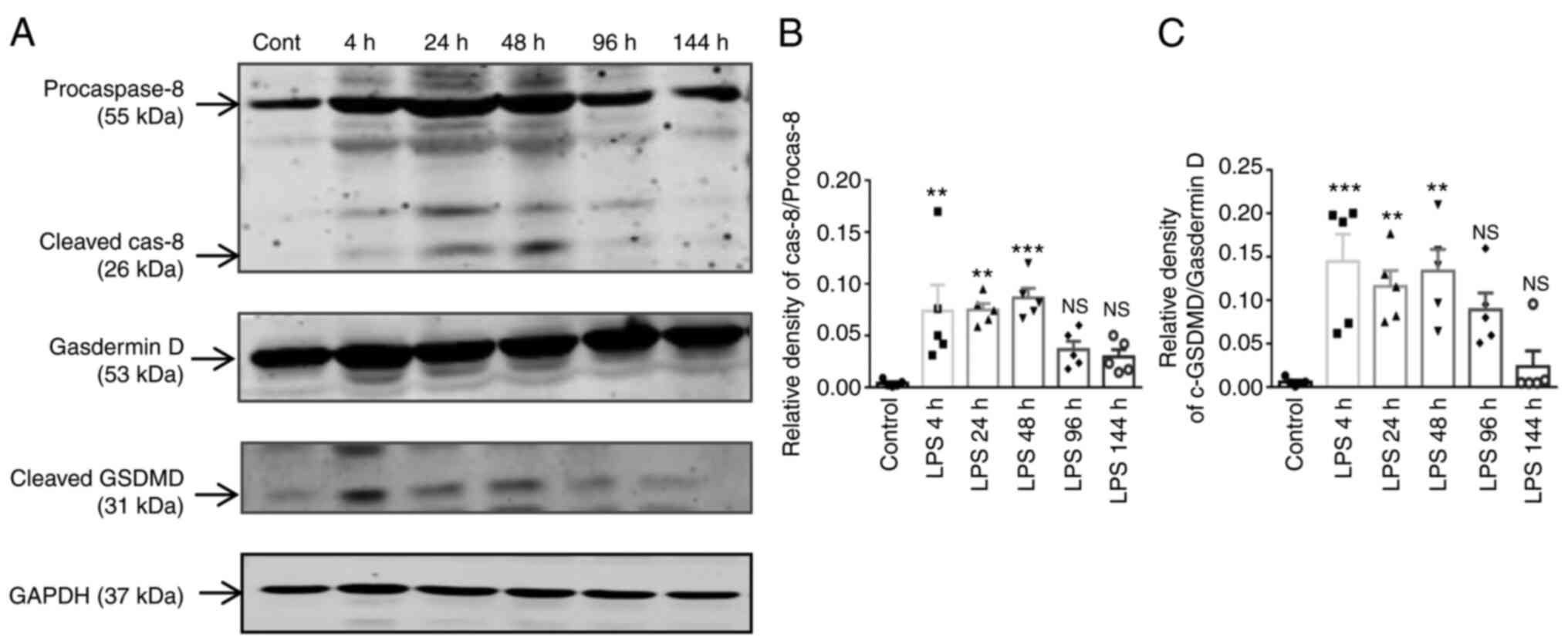 | Figure 9Time-dependent changes of cleaved
caspase-8 and cleaved GSDMD in lung tissue. C57BL/6 mice were
exposed to with 4 mg/kg LPS via intratracheal installation for 4,
24, 48, 96 and 144 h, and 0.9% NaCl was administered to the control
mice orally. Lung tissues were collected, after 4, 24, 48, 96 and
144 h of LPS exposure and from the control mice. (A) The protein
levels of procaspase-8, cleaved caspase-8, GSDMD and cleaved GSDMD
in lung tissue were evaluated using western blot analysis; (B and
C) the protein levels were quantified using densitometry. GAPDH was
used as the loading control. The results are expressed as the mean
± SEM (n=5 animals in each group). One-way ANOVA followed by the
Bonferroni test were used to analyze significant differences among
the different time points of the LPS group vs. the control.
**P<0.01 and ***P<0.001, vs. control.
NS, not significant; GSDMD, gasdermin D; LPS,
lipopolysaccharide. |
Discussion
The present study demonstrated the changing trends
in macrophages and neutrophils along with subsets of T-cells in the
lung tissues of mice with LPS-induced lung injury. Following the
intratracheal administration of LPS, a significant increase in the
activation of not only T-cells, but also macrophages and
neutrophils was observed during the ALI process in a time-dependent
manner. Furthermore, NLRP3/HO-1 activation, the subsequent release
of pro-inflammatory cytokines and pyroptosis of lung tissue were
found to be possible consequences of Nrf2/NLRP3 signaling.
LPS has been reported to induce neutrophil
accumulation by stimulating macrophages in the infected area, which
subsequently express distinct patterns of cytokines, chemokines and
surface markers in ALI (42). The
activation of macrophages and their subtypes (AMs and IMs) are
dynamic processes that release pro-inflammatory cytokines during
inflammation (12,43). Therefore, the present study
analyzed neutrophils, macrophages and their types (AMs and IMs) in
a mouse model of LPS-induced lung injury. It was found that LPS led
to the recruitment of macrophages during the sub-chronic phase of
lung injury. The suppression of AMs from LPS-4 to 144 h was also
demonstrated, which had a positive effect on lung injury and the
increase in IMs. In the model of ALI, the number of neutrophils
accumulated from 4 to 144 h of LPS treatment and was accompanied by
the production of inflammatory mediators. Due to certain
limitations, the present study we did not separate M1 and M2
macrophages. However, it was reported that the M2 phenotype was the
main form of these resident AMs. In the acute phase of ALI/ARDS,
resident alveolar macrophages, typically expressing the
alternatively activated phenotype (M2), shift into the classically
activated phenotype (M1) and release various potent
pro-inflammatory mediators (44).
The differentiation of CD4+ and
CD8+ T-cells has gained considerable attention in
assessing the immune system (45). On the other hand, previous studies
have suggested that LPS-induced TNF-α secretion in macrophages is
mitigated when CD4+ T-cells are depleted (46,47). Previously, challenge with LPS has
been shown to increase the number of CD4+ T-lymphocytes
in the inflammatory infiltration during ALI (48,49). Furthermore, CD4+
T-cells have been reported to be involved in the phenotypic
transformation of macrophages and neutrophils (36). At the onset of injury, the
activation of CD8+ T-cells protects the injured area by
laying the foundation for fibroblasts to enter and deposit the
collagen scar, as well as to begin to resolve neutrophil-mediated
inflammation (50,51). den Haan et al (52) suggested that the activation of
TLRs led to the suppression of CD8+ T-cell responses
after priming with OVA plus LPS or poly(I:C). Therefore, the
elucidation of the role of T-cells in promoting neutrophil and
macrophage migration at different stages of ALI is of utmost
urgency, as it may aid in diminishing inflammation in ALI. Herein,
the data illustrated that the numbers of neutrophils and macrophage
were increased along with the suppression of CD8+
T-cells following exposure to LPS. However, LPS had no significant
effect on CD4+ T-cell activation. In addition, the
present study evaluated the ratio of
CD4+:CD8+ cells; the flow cytometry data
revealed that the LPS installation markedly increased the
percentage of CD4+/CD8+ cells in lung
tissues. Similarly, the CD4+/CD8+ T-cell
ratio has been found to be significantly elevated in virus-induced
lower respiratory tract infection (17). Similar to the present study,
Hussein et al (53)
suggested that the higher CD4/CD8 ratio may be the result of
recruited CD4+ cells with decreased CD8+
T-cell numbers, and/or the effects of the disease with the release
of cytokines such as IL-1β, IL-6, TNF-α. The regulation of Th1,
Th2, Th17 and Tregs, plays a critical role in immune mechanisms by
interacting with other cells, and is associated with several
inflammatory immune-mediated disorders, mainly sub-chronic and
chronic inflammatory disorders (54). Moreover, the stimulation of these
cells can trigger TGF-β, IL-6, IL-1β and MIP-2 and may also
activate their transcriptional factor transcription factors, T-bet,
GATA-3, RORγt and Forkhead box protein P3) (55,56). A previous study demonstrated that
the stimulation of TNF-α triggered the expression of
neutrophil-attracting chemokines, such as CXCL1 and CXCL2 (57). The present study investigated the
mechanisms of regulatory T-cells and the secretion of the
pro-inflammatory cytokines, TNF-α, CXCL1/KC, IL-1β and MIP-2.
However, the results indicated that all pro-inflammatory cytokines
aggravated lung inflammation from 4 h (acute phase) to 96 h
(subacute phase) following the LPS administration. Moreover,
compared with the control group, the protein expression and mRNA
levels of TLR-4 were notably increased. Overall, a sustained LPS
exposure may exhaust the immune regulation between T-cell subsets,
as well as the recruitment of macrophages, neutrophils and the
secretion of pro-inflammatory cytokines through downstream TLR-4
signaling in the area of inflammation. Taken together, these data
may aid in the future investigations of therapeutic approaches for
immune abnormalities in models of LPS-induced lung injury.
There is increasing evidence to suggest that
inflammasome complexes are activated by various danger signals,
both endogenous and exogenous. Oxidative stress is known to play a
critical role in ROS production and inflammasome activation
(58). Even though researchers
have suggested two different pathways through which NLRP3 senses
changes in ROS, this remains an undefined issue.
Thioredoxin-interacting protein binds to thioredoxin under
homeostatic conditions, is liberated by ROS and can then interact
with NLRP3, resulting in inflammasome inactivation (59,60). Another important pathway is NADPH
oxidase and mitochondria dysfunction, and their aberrant actions
subsequently activate the inflammasome (61). Of note, Nrf2 is a pivotal
transcription factor that regulates intracellular redox balance by
activating antioxidant genes (62). Therefore, it was primarily
hypothesized that Nrf2 deletion promotes inflammasome formation by
regulating a large battery of genes, such as HO-1, that reduces
intracellular redox homeostasis. However, there is also an urgency
to develop novel strategies with which to reduce inflammation in
the early and late stages. The present study demonstrated that the
intratracheal administration of LPS in mice led to the upregulation
of Nrf2 activation (4 to 48 h) possibly involved in NLRP3 and
caspase-1 expression through the aggravation of ROS levels from 4
to 96 h; IL-1β production and the recruitment of neutrophils and
other cytokines and chemokines were also observed in the model of
lung injury. Therefore, it is suggested that the positive feedback
of the NLRP3/Nrf2/HO-1 pathway may be related to LPS-induced injury
in lung tissue in the early stages of lung injury via the
activation of immune cells and the release of various cytokines and
chemokines. It has been suggested that bone marrow-derived
macrophages (BMDMs) release mature IL-1β via Nrf2 signaling within
2 to 8 h following the activation of the NLRP3/caspase-1 complex;
Nrf2 activation has also been reported to contribute to the LPS-
and nigericin-induced ASC speck formation in BMDMs (27). It is therefore conceivable that
Nrf2-signaling can promote IL-1β responses by sensing the loss of
organelle integrity, similar to that shown by the NLRP3
inflammasome (61,63). Moreover, previous studies have
indicated that HO-1 overexpression can lead to deleterious effects
(64,65). However, the potential implications
of the Nrf2/HO-1 pathway in the crosstalk regulation of imbalanced
immune responses remain to be elucidated.
In addition to induction via death receptor
ligation, caspase-8-mediated pyroptosis may also be involved in
inflammasome components (66).
Furthermore, the presence of caspase-1 and -11 inflammasomes may be
involved in pyroptosis in macrophages (67), while the absence of cleaved
caspase-1 promotes apoptosis (68). However, macrophages and
neutrophils are considered as important participators in this
process (69). In addition,
neutrophil extracellular traps may contribute to ALI by promoting
the pyroptosis of alveolar macrophages and systemic inflammation
(70). Recently, a study
indicated that the TLR4-mediated activation of caspase-8 led to the
cleavage of GSDMD and the release IL-1β, resulting in pyroptosis
(71). It has also been reported
that caspase-1 cleaves GSDMD, and its N-terminal fragment
translocates to the outer membrane, where macropores are formed,
resulting in a lytic form of cell death (pyroptosis) (72,73). However, this process remains
unclear in the model of LPS-induced ALI. Herein, it was found that
the LPS-triggered caspase-8 and cleaved GSDMD activations were
markedly enhanced in the presence of caspase-1 expression at
similar time points from 4 to 48 h. This suggested that the
time-dependent recruitment of caspase-8 is sensitive to the
pyroptotic cascade, such as cleaved GSDMD expression in the model
of ALI.
In conclusion, the present study demonstrated that
acute and sub-chronic intratracheal administration of LPS enhanced
multiple inflammatory responses, macrophages, AMs, IMs and
neutrophils, as well as T-cell subsets from 4 to 96 h. The results
of the present study suggest that the aberrant function of
CD4+/CD8+ T-cell can exert profound effects
on neutrophils and macrophage activation, and is associated with
intervention in lung injury by modulating pro-inflammatory
cytokines in both acute and sub-chronic stages. Moreover, the
disruption of the redox balance and Nrf2/NLRP3 signaling pathway
may also be involved in LPS-induced immune responses by activating
pyroptosis. Therefore, the epigenetic program of T-cells and their
functions, and the inhibition of Nrf2/NLRP3 target activation may
lead to the development of novel therapeutic strategies for acute
and sub-chronic lung disease.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RD, NL and LZ participated in the western blot
analysis, RT-qPCR, ELISA, histopathological analysis, ROS analysis
and flow cytometry experiments. RD interpreted the results and
wrote the manuscript. LN and LZ interpreted the key flow cytometry
data. NL, YL and MNR contributed to the preparation of the animal
model, and to the collection of the animal tissue specimens and
interpreted the data. XC, ZH, XW, XZ contributed to the harvesting
of the animal tissues. XX and HT conceived all the experiments and
revised the manuscript RD, NL and LZ confirm the authenticity of
all the raw data. All authors confirmed and commented on previous
versions of the manuscript along with have read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Zhejiang University (Hangzhou, China), and the
experiments were performed in accordance with the National
Institutes of Health Guidelines for the Use of Laboratory
Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
Acknowledgments
The authors are grateful to the Core Facilities of
Zhejiang University School of Medicine (Hangzhou, China), for
technical assistance with flow cytometry and for processing
analysis.
Funding
The present study was supported by the Zhejiang Provincial
Natural Science Foundation (grant nos. LBY21H0100001, LYY18H310007
and LY18H310002), the Science Technology Department of Zhejiang
Province Project (grant no. 2017C37132), the Medical Health Science
and Technology Project of Zhejiang Provincial Health Commission
(grant nos. 2018246654 and 2022KY928), and the Pre-research project
of the National Natural Science Foundation of (grant no.
2018ZG12).
References
|
1
|
Ranieri VM, Rubenfeld GD, Thompson BT,
Ferguson ND, Caldwell E, Fan E, Camporota L and Slutsky AS: Acute
respiratory distress syndrome: The Berlin definition. JAMA.
307:2526–2533. 2012.PubMed/NCBI
|
|
2
|
Kushimoto S, Endo T, Yamanouchi S,
Sakamoto T, Ishikura H, Kitazawa Y, Taira Y, Okuchi K, Tagami T,
Watanabe A, et al: Relationship between extravascular lung water
and severity categories of acute respiratory distress syndrome by
the Berlin definition. Crit Care. 17:R1322013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Abdulnour REE and Levy BD: Acute lung
injury and the acute respiratory distress syndrome. Med Manag Surg
Patient A Textb Perioper Med Fifth Ed. 154–171. 2010.
|
|
4
|
Johnston LK, Rims CR, Gill SE, McGuire JK
and Manicone AM: Pulmonary macrophage subpopulations in the
induction and resolution of acute lung injury. Am J Respir Cell Mol
Biol. 47:417–426. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang A, Pan W, Lv J and Wu H: Protective
effect of amygdalin on LPS-induced acute lung injury by inhibiting
NF-κB and NLRP3 signaling pathways. Inflammation. 40:745–751. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tianzhu Z and Shumin W: Esculin inhibits
the inflammation of LPS-induced acute lung injury in mice via
regulation of TLR/NF-κB pathways. Inflammation. 38:1529–1536. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Park EJ, Kim YM, Kim HJ and Chang KC:
Luteolin activates ERK1/2- and Ca2+-dependent HO-1 induction that
reduces LPS-induced HMGB1, iNOS/NO, and COX-2 expression in
RAW264.7 cells and mitigates acute lung injury of endotoxin mice.
Inflamm Res. 67:445–453. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Marsland BJ, Nembrini C, Grün K, Reissmann
R, Kurrer M, Leipner C and Kopf M: TLR ligands act directly upon T
cells to restore proliferation in the absence of protein kinase
C-theta signaling and promote autoimmune myocarditis. J Immunol.
178:3466–3473. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mandraju R, Murray S, Forman J and Pasare
C: Differential ability of surface and endosomal TLRs to induce CD8
T cell responses in vivo. J Immunol. 192:4303–4315. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Imam F, Al-Harbi NO, Al-Harbi MM, Ansari
MA, Zoheir KMA, Iqbal M, Anwer MK, Hoshani ARA, Attia SM and Ahmad
SF: Diosmin downregulates the expression of T cell receptors,
pro-inflammatory cytokines and NF-κB activation against LPS-induced
acute lung injury in mice. Pharmacol Res. 102:1–11. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Boorsma CE, Draijer C and Melgert BN:
Macrophage heterogeneity in respiratory diseases. Mediators
Inflamm. 2013:7692142013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Byrne AJ, Mathie SA, Gregory LG and Lloyd
CM: Pulmonary macrophages: Key players in the innate defence of the
airways. Thorax. 70:1189–1196. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee JW, Chun W, Lee HJ, Min JH, Kim SM,
Seo JY, Ahn KS and Oh SR: The role of macrophages in the
development of acute and chronic inflammatory lung diseases. Cells.
10:8972021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nakajima T, Suarez CJ, Lin KW, Jen KY,
Schnitzer JE, Makani SS, Parker N, Perkins DL and Finn PW: T cell
pathways involving CTLA4 contribute to a model of acute lung
injury. J Immunol. 184:5835–5841. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Eberl G: RORγt, a multitask nuclear
receptor at mucosal surfaces. Mucosal Immunol. 10:27–34. 2017.
View Article : Google Scholar
|
|
16
|
Noack M and Miossec P: Th17 and regulatory
T cell balance in autoimmune and inflammatory diseases. Autoimmun
Rev. 13:668–677. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Connors TJ, Ravindranath TM, Bickham KL,
Bickham KL, Gordon CL, Zhang F, Levin B, Baird JS and Farber DL:
Airway CD8+ T cells are associated with lung injury during infant
viral respiratory tract infection. Am J Respir Cell Mol Biol.
54:822–830. 2016. View Article : Google Scholar :
|
|
18
|
Wong JJM, Leong JY, Lee JH, Albani S and
Yeo JG: Insights into the immuno-pathogenesis of acute respiratory
distress syndrome. Ann Transl Med. 7:5042019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kumar V: Toll-like receptors in
sepsis-associated cytokine storm and their endogenous negative
regulators as future immunomodulatory targets. Int Immunopharmacol.
89:1070872020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Soy M, Keser G, Atagündüz P, Tabak F,
Atagündüz I and Kayhan S: Cytokine storm in COVID-19: Pathogenesis
and overview of anti-inflammatory agents used in treatment. Clin
Rheumatol. 39:2085–2094. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hennig P, Garstkiewicz M, Grossi S,
Filippo MD, French LE and Beer HD: The crosstalk between Nrf2 and
Inflammasomes. Int J Mol Sci. 19:5622018. View Article : Google Scholar :
|
|
22
|
Garstkiewicz M, Strittmatter GE, Grossi S,
Sand J, Fenini G, Werner S, French LE and Beer HD: Opposing effects
of Nrf2 and Nrf2-activating compounds on the NLRP3 inflammasome
independent of Nrf2-mediated gene expression. Eur J Immunol.
47:806–817. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maier NK, Leppla SH and Moayeri M: The
cyclopentenone prostaglandin 15d-PGJ 2 inhibits the NLRP1 and NLRP3
inflammasomes. J Immunol. 194:2776–2785. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tonelli C, Chio IIC and Tuveson DA:
Transcriptional regulation by Nrf2. Antioxid Redox Signal.
29:1727–1745. 2018. View Article : Google Scholar :
|
|
25
|
Kelley N, Jeltema D, Duan Y and He Y: The
NLRP3 inflammasome: An overview of mechanisms of activation and
regulation. Int J Mol Sci. 20:33282019. View Article : Google Scholar :
|
|
26
|
Possomato-Vieira, José S and Khalil RAK:
Mechanism and regulation of NLRP3 inflammasome activation. Physiol
Behav. 176:139–148. 2016.
|
|
27
|
Zhao C, Gillette DD, Li X, Zhang Z and Wen
H: Nuclear factor E2-related factor-2 (Nrf2) is required for NLRP3
and AIM2 inflammasome activation. J Biol Chem. 289:17020–17029.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sarhan J, Liu BC, Muendlein HI, Li P,
Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR and
Poltorak A: Caspase-8 induces cleavage of gasdermin D to elicit
pyroptosis during Yersinia infection. Proc Natl Acad Sci USA.
115:E10888–E10897. 2018. View Article : Google Scholar :
|
|
29
|
Muendlein HI, Jetton D, Connolly WM,
Eidell KP, Magri Z, Smirnova I and Poltorak A: CFLIPL protects
macrophages from LPS-induced pyroptosis via inhibition of complex
II formation. Science. 367:1379–1384. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xia S, Hollingsworth LR and Wu H:
Mechanism and regulation of gasdermin-mediated cell death. Cold
Spring Harb Perspect Biol. 12:1–14. 2020. View Article : Google Scholar
|
|
31
|
Cen M, Ouyang W, Zhang W, Yang L, Lin X,
Dai M, Hu H, Tang H, Liu H, Xia J and Xu F: MitoQ protects against
hyper-permeability of endothelium barrier in acute lung injury via
a Nrf2-dependent mechanism. Redox Biol. 41:1019362021. View Article : Google Scholar
|
|
32
|
Tseng TL, Chen MF, Tsai MJ, Hsu YH, Chen
CP and Lee TJF: Oroxylin-a rescues LPS-induced acute lung injury
via regulation of NF-κB signaling pathway in rodents. PLoS One.
7:e474032012. View Article : Google Scholar
|
|
33
|
Matute-Bello G, Downey G, Moore BB,
Groshong SD, Matthay MA, Slutsky AS and Kuebler WM: An official
american thoracic society workshop report: Features and
measurements of experimental acute lung injury in animals. Am J
Respir Cell Mol Biol. 44:725–738. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
35
|
Socci DJ, Bjugstad KB, Jones HC, Pattisapu
JV and Arendash GW: Evidence that oxidative stress is associated
with the pathophysiology of inherited hydrocephalus in the H-Tx rat
model. Exp Neurol. 155:109–117. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Roberts CA, Dickinson AK and Taams LS: The
interplay between monocytes/macrophages and CD4+ T cell subsets in
rheumatoid arthritis. Front Immunol. 6:5712015. View Article : Google Scholar :
|
|
37
|
Dagvadorj J, Shimada K, Chen S, Jones HD,
Tumurkhuu G, Zhang W, Wawrowsky KA, Crother TR and Arditi M:
Lipopolysaccharide induces alveolar macrophage necrosis via CD14
and the P2x7 receptor leading to Interleukin-1α release. Immunity.
42:640–653. 2016. View Article : Google Scholar
|
|
38
|
Altemeier WA, Zhu X, Berrington WR, Harlan
JM and Liles WC: Fas (CD95) induces macrophage proinflammatory
chemokine production via a MyD88-dependent, caspase-independent
pathway. J Leukoc Biol. 82:721–728. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hatanaka E, Monteagudo PT, Marrocos MSM
and Campa A: Neutrophils and monocytes as potentially important
sources of proinflammatory cytokines in diabetes. Clin Exp Immunol.
146:443–447. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
An SJ, Pae HO, Oh GS, Choi BM, Jeong S,
Jang SI, Oh H, Kwon TO, Song CE and Chung HT: Inhibition of TNF-α,
IL-1β, and IL-6 productions and NF-κB activation in
lipopolysaccharide-activated RAW 264.7 macrophages by catalposide,
an iri glycoside isolated from Catalpa ovata G. Don (Bignoniaceae).
Int Immunopharmacol. 2:1173–1181. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bosnar M, Bošnjak B, Čužić S, Hrvacic B,
Marjanovic N, Glojnaric I, Culic O, Parnham MJ and Haber VE:
Azithromycin and clarithromycin inhibit lipopolysaccharide-induced
murine pulmonary neutrophilia mainly through effects on
macrophage-derived granulocyte-macrophage colony-stimulating factor
and interleukin-1β. J Pharmacol Exp Ther. 331:104–113. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Risso K, Kumar G, Ticchioni M, Sanfiorenzo
C, Dellamonica J, Guillouet-de Salvador F, Bernardin G, Marquette
CH and Roger PM: Early infectious acute respiratory distress
syndrome is characterized by activation and proliferation of
alveolar T-cells. Eur J Clin Microbiol Infect Dis. 34:1111–1118.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Janssen WJ, Barthel L, Muldrow A,
Oberley-Deegan RE, Kearns MT, Jakubzick C and Henson PM: Fas
determines differential fates of resident and recruited macrophages
during resolution of acute lung injury. Am J Respir Crit Care Med.
184:547–560. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen X, Tang J, Shuai W, Meng J, Feng J
and Han Z: Macrophage polarization and its role in the pathogenesis
of acute lung injury/acute respiratory distress syndrome. Inflamm
Res. 69:883–895. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Williams MA, Rangasamy T, Bauer SM,
Killedar S, Karp M, Kensler TW, Yamamoto M, Breysse P, Biswal S and
Georas SN: Disruption of the transcription factor Nrf2 promotes
pro-oxidative dendritic cells that stimulate Th2-like
immunoresponsiveness upon activation by ambient particulate matter.
J Immunol. 181:4545–4559. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
D'Souza NB, Mandujano FJ, Nelson S, Summer
WR and Shellito JE: CD4+ T lymphocyte depletion attenuates
lipopolysaccharide-induced tumor necrosis factor secretion by
alveolar macrophages in the mouse. Lymphokine Cytokine Res.
13:359–366. 1994.PubMed/NCBI
|
|
47
|
Crowe CR, Chen K, Pociask DA, Alcorn JF,
Krivich C, Enelow RI, Ross TM, Witztum JL and Kolls JK: Critical
role of IL-17RA in immunopathology of influenza infection. J
Immunol. 183:5301–5310. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chai YS, Chen YQ, Lin SH, Xie K, Wang CJ,
Yang YZ and Xu F: Curcumin regulates the differentiation of naïve
CD4+T cells and activates IL-10 immune modulation against acute
lung injury in mice. Biomed Pharmacother. 125:1099462020.
View Article : Google Scholar
|
|
49
|
Philippakis GE, Lazaris AC, Papathomas TG,
Zissis C, Agrogiannis G, Thomopoulou G, Nonni A, Xiromeritis K,
Nikolopoulou-Stamati P, Bramis J, et al: Adrenaline attenuates the
acute lung injury after intratracheal lipopolysaccharide
instillation: An experimental study. Inhal Toxicol. 20:445–453.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Haring JS, Badovinac VP and Harty JT:
Inflaming the CD8+ T cell response. Immunity. 25:19–29. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ilatovskaya DV, Pitts C, Clayton J,
Domondon M, Troncoso M, Pippin S and DeLeon-Pennell KY: CD8+
T-cells negatively regulate inflammation post-myocardial
infarction. Am J Physiol Hear Circ Physiol. 317:H581–H596. 2019.
View Article : Google Scholar
|
|
52
|
den Haan JMM, Kraal G and Bevan MJ:
Cutting edge: Lipopolysaccharide induces IL-10-producing regulatory
CD4 + T cells that suppress the CD8 + T cell response. J Immunol.
178:5429–5433. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hussein MR, Hassan HI, Hofny ERM, Elkholy
M, Fatehy NA, Elmoniem AEA, El-Din AME, Afifi OA and Rashed HG:
Alterations of mononuclear inflammatory cells, CD4/CD8+ T cells,
interleukin 1β, and tumour necrosis factor α in the bronchoalveolar
lavage fluid, peripheral blood, and skin of patients with systemic
sclerosis. J Clin Pathol. 58:178–184. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Cabrera-Perez J, Condotta SA, Badovinac VP
and Griffith TS: Impact of sepsis on CD4 T cell immunity. J Leukoc
Biol. 96:767–777. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hori S, Nomura T and Sakaguchi S: Control
of regulatory T cell development by the transcription factor Foxp3.
J Immunol. 198:981–985. 2017.PubMed/NCBI
|
|
56
|
Ivanov II, McKenzie BS, Zhou L, Tadokoro
CE, Lepelley A, Lafaille JJ, Cua DJ and Littman DR: The orphan
nuclear receptor RORγt directs the differentiation program of
proinflammatory IL-17+ T helper cells. Cell. 126:1121–1133. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Griffin GK, Newton G, Tarrio ML, Bu DX,
Maganto-Garcia E, Azcutia V, Alcaide P, Grabie N, Luscinskas FW,
Croce KJ and Lichtman AH: IL-17 and TNFα sustain neutrophil
recruitment during inflammation through synergistic effects on
endothelial activation1. J Immunol. 188:6287–6299. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wen H, Miao EA and Ting JPY: Mechanisms of
NOD-like receptor-associated inflammasome activation. Immunity.
39:432–441. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Abais JM, Xia M, Zhang Y, Boini KM and Li
PL: Redox regulation of NLRP3 inflammasomes: ROS as trigger or
effector? Antioxidants Redox Signal. 22:1111–1129. 2015. View Article : Google Scholar
|
|
60
|
Zhou R, Tardivel A, Thorens B, Choi I and
Tschopp J: Thioredoxin-interacting protein links oxidative stress
to inflammasome activation. Nat Immunol. 11:136–140. 2010.
View Article : Google Scholar
|
|
61
|
Zhou R, Yazdi AS, Menu P and Tschopp J: A
role for mitochondria in NLRP3 inflammasome activation. Nature.
469:221–226. 2011. View Article : Google Scholar
|
|
62
|
Vomund S, Schäfer A, Parnham MJ, Brüne B
and Von Knethen A: Nrf2, the master regulator of anti-oxidative
responses. Int J Mol Sci. 18:27722017. View Article : Google Scholar
|
|
63
|
Hornung V, Bauernfeind F, Halle A, Samstad
EO, Kono H, Rock KL, Fitzgerald KA and Latz E: Silica crystals and
aluminum salts activate the NALP3 inflammasome through phagosomal
destabilization. Nat Immunol. 9:847–856. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Jais A, Einwallner E, Sharif O, Gossens K,
Lu TTH, Soyal SM, Medgyesi D, Neureiter D, Paier-Pourani J,
Dalgaard K, et al: Heme oxygenase-1 drives metaflammation and
insulin resistance in mouse and man. Cell. 158:25–40. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Tronel C, Rochefort GY, Arlicot N, Bodard
S, Chalon S and Antier D: Oxidative stress is related to the
deleterious effects of heme oxygenase-1 in an in vivo
neuroinflammatory rat model. Oxid Med Cell Longev. 2013:2649352013.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Raghawan AK, Sripada A, Gopinath G,
Pushpanjali P, Kumar Y, Radha V and Swarup G: A disease-associated
mutant of NLRC4 shows enhanced interaction with SUG1 leading to
constitutive fadddependent caspase-8 activation and cell death. J
Biol Chem. 292:1218–1230. 2017. View Article : Google Scholar
|
|
67
|
Pierini R, Juruj C, Perret M, Jones CL,
Mangeot P, Weiss DS and Henry T: AIM2/ASC triggers
caspase-8-dependent apoptosis in francisella-infected
caspase-1-deficient macrophages. Cell Death Differ. 19:1709–1721.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Antonopoulos C, Russo HM, El Sanadi C,
Martin BN, Li X, Kaiser WJ, Mocarski ES and Dubyak GR: Caspase-8 as
an effector and regulator of NLRP3 inflammasome signaling. J Biol
Chem. 290:20167–20184. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Robinson N, Ganesan R, Hegedűs C, Kovács
K, Kufer TA and Virág L: Programmed necrotic cell death of
macrophages: Focus on pyroptosis, necroptosis, and parthanatos.
Redox Biol. 26:1012392019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Haitao Lee PP: Neutrophil extracellular
traps promoted alveolar macrophages pyroptosis in LPS induced
ALI/ARDS. Eur Respir J. 52:PA42842018.
|
|
71
|
Orning P, Weng D, Starheim K, Ratner D,
Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, et al: Pathogen
blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin
D and cell death. Science. 362:1064–1069. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Shi J, Zhao Y, Wang K, Shi X, Wang Y,
Huang H, Zhuang Y, Cai T, Wang F and Shao F: Cleavage of GSDMD by
inflammatory caspases determines pyroptotic cell death. Nature.
526:660–665. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Kayagaki N, Stowe IB, Lee BL, O'Rourke K,
Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT,
et al: Caspase-11 cleaves gasdermin D for non-canonical
inflammasome signalling. Nature. 526:666–671. 2015. View Article : Google Scholar : PubMed/NCBI
|














