Introduction
It has been reported that inflammation induced by
ischemia/reperfusion (I/R) injury mainly occurs in non-cardiac
cells, such as leukocytes, fibroblasts and endothelial cells
(1). When I/R injury occurs,
cardiac microvascular dysfunction results in reduced oxygen supply
to cardiac cells (2). Current
treatments for myocardial infarction (MI) are based on two
principles: Short-term revascularization and long-term angiogenesis
(3,4). Short-term revascularization, such as
coronary artery bypass grafting or percutaneous coronary
intervention, are widely used for treatment (3). However, the beneficial effects of
revascularization are limited owing to low myocardial microvascular
density and poor local perfusion at the infarct margin (5,6).
Moreover, it has been demonstrated that changes in capillary bed
structure after I/R injury may lead to reduced microcirculation
flow (7).
Cardiac microvascular endothelial cells (CMECs) are
the most common cells in the heart and are the basic components of
myocardial microcirculation; under normal conditions, CMECs can
secrete cytokines related to cardiac growth, contractile
performance and rhythm (8). In
addition, a previous study highlighted the role of CMEC dysfunction
in driving I/R injury in cardiomyocytes (9). It was also reported that the
sustained viability, the reduced apoptosis and the increase in
nitric oxide (NO) generation in CMECs after I/R injury could
alleviate myocardial I/R injury (10,11). Therefore, attenuating CMEC injury
may be able to protect against myocardial I/R-induced injury. A
recent study reported that growth arrest and DNA damage-inducible α
(GADD45A) expression was increased in ischemic myocardial cells and
could be targeted by microRNA (miR)-1283 to reduce
hypoxia/reoxygenation (H/R)-induced apoptosis of myocardial cells
(12). Nevertheless, its
expression in CMECs remains unknown.
GADD45A was found to be distributed in endothelial
cells of myocardial tissue in the Human Protein Atlas database
(13), but its expression in
endothelial cells of H/R-treated myocardial tissue is unknown.
Thus, we hypothesized that GADD45A may be involved in the
pathogenesis of MI by affecting the apoptosis and angiogenesis of
CMECs. Therefore, the present study aimed to explore whether
GADD45A participated in the apoptosis and dysfunction of CMECs
following myocardial I/R injury.
Materials and methods
Animal model
Male Sprague-Dawley rats (SPF grade; age, 6 weeks;
weight, 180-220 g; n=30) were fed under standard laboratory
conditions with a temperature of 27°C, 40-60% humidity and 12-h
light/dark cycle. The rats were acclimated to these conditions for
7 days and provided with free access to water and food. The rats
were then anesthetized with 1% pentobarbital sodium (30 mg/kg) by
intraperitoneal (i.p.) injection, and the myocardial I/R model was
established following the surgical protocol of a previous study
(10). A total of 21 rats were
successfully induced in the I/R model with a survival rate of 91.3%
(21/23), which was similar to previous studies (14,15); ischemia time may be the cause of
death of two rats. All rats were randomly divided into 4 groups
(n=7 rats/group): i) Control group (Sham operation), in which open
heart surgery was performed but the anterior descending branch of
the coronary artery was not ligated; ii) I/R group; iii) I/R +
lentiviral short hairpin-RNA-NC (Lv-sh-NC) group; and iv) I/R +
Lv-sh-GADD45A group. Lentiviral vectors containing shRNA targeting
GADD45A (Lv-sh-GADD45A, 5′-CGC AGA GCA GAA GAT CGA AAG-3′) and the
Lv-sh-NC (5'-TTC TCC GAA CGT GTC ACG T-3') were constructed by
Hanbio Biotechnology Co., Ltd. During ligation, 20 µM
lentivirus vectors (4×109 IFU/ml) were injected into the
pericardial tissue of rat hearts at four different places around
the infarcted area. No obvious side effects were found following
I/R surgery and administration of si-GADD45A.
At 7 days post-I/R surgery, the rats were
anesthetized with 1% pentobarbital sodium (30 mg/kg; i.p.) and
rapid thoracotomy was performed. Blood (10 ml) was collected from
the apex of the heart and serum was separated by first letting the
collected blood stand at 37°C for 30 min, and then centrifuged at
1,006 × g at 4°C for 15 min to detect the levels of creatine kinase
(CK), lactate dehydrogenase (LDH) and NO (described below). Next,
the rats were euthanized by the i.p. injection of 1% pentobarbital
sodium (150 mg/kg) and pre-cooled saline was injected into the apex
of the heart through an intravenous infusion device for irrigation.
Cessation of breathing for 3 min was used to verify death. After
full irrigation, the quickly separated heart was washed in saline,
placed on ice and dried with absorbent paper. The myocardial tissue
was collected for 2,3,5-triphenyltetrazolium chloride (TTC)
staining and other experiments, as described below.
Animal experiments were approval of the animal care
and ethics committee of Yan'an Hospital Affiliated to Kunming
Medical University (Kunming, China) and performed following the
ARRIVE guidelines (https://www.nc3rs.org.uk/arrive-guidelines). The
humane endpoints considered in this experiment were as follows: i)
The animals showed mental depression accompanied by hypothermia
(<37°C) in the absence of anesthesia; ii) the experiments were
terminated before the earliest indicator if an animal experienced
severe pain. Any animals reaching these endpoints were to be
euthanized with 1% pentobarbital sodium (150 mg/kg; i.p.).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from myocardial tissues or
CMECs using TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instruction. cDNA
was synthesized from the total RNA using the PrimeScript™ RT
Reagent kit (cat. no. RR037A; Takara Bio, Inc.) according to the
manufacturer's instructions. qPCR was conducted using the
SYBR® Premix EX Taq™ kit (Takara Bio, Inc.) and the
following thermocycling conditions: Initial denaturation for 5 min
at 94°C; followed by 40 cycles of denaturation for 20 sec at 94°C,
annealing for 20 sec at 65°C and elongation for 30 sec at 70°C. The
following primer sequences were used: Rat GADD45A forward, 5′-TAA
GCA AGA AGC CGG CAA GA-3′ and reverse, 5′-GGG TCT ACG TTG AGC AGC
TT-3′; human GADD45A forward, 5′-CGA AAG GAT GGA TAA GGT G-3′ and
reverse, 5′-GGA TCA GGG TGA AGT GGA-3′; GAPDH (rat) forward, 5′-TGT
GAA CGG ATT TGG CCG TA-3′ and reverse, 5′-GAT GGT GAT GGG TTT CCC
GT-3′; and GAPDH (human) forward, 5′-GCA CCG TCA AGG CTG AGA AC-3′
and reverse, 5′-GGA TCT CGC TCC TGG AAG ATG-3′. mRNA expression
levels were quantified using the 2−ΔΔCq method and
normalized to the internal reference gene GAPDH (16).
Western blotting
Total proteins were isolated from rat myocardial
tissue and cell samples using RIPA (Beyotime Institute of
Biotechnology) and semi-quantified using a BCA kit (Beyotime
Institute of Biotechnology). Proteins (20 µg/lane) were
separated on 12% gels by SDS-PAGE and transferred to PVDF membranes
(cat. no. FFP24; Beyotime Institute of Biotechnology). PVDF
membranes were blocked with 5% skimmed milk at room temperature for
2 h and then incubated at 4°C overnight with primary antibodies
against: GADD45A [1:1,000; cat. no. 4632; Cell Signaling
Technology, Inc.(CST)], CD31 (1:1,000; cat. no. ab281583; Abcam),
phosphorylated (p)-eNOS (1:1,000; cat. no. bs-3589R; BIOSS), eNOS
(1:1,000; cat. no. ab300071; Abcam), entothelin-1 (ET-1; 1:1,000;
cat. no. ab2786; Abcam), p-p38 MAPK (1:1,000; cat. no. bs-5476R;
BIOSS), p38 MAPK (1:1,000; cat. no. bs-0637R; BIOSS), p-JNK
(1:1,000; cat. no. 4668; CST), JNK (1:1,000; cat. no. 9252; CST),
early growth response 1 (EGR1; 1:1,000; cat. no. 4154; CST),
p-STAT3 (1:1,000; cat. no. ab32143; Abcam), STAT3 (1:1,000; cat.
no. ab68153; Abcam), VEGF (1:1,000; cat. no. 19003-1-AP;
Proteintech Group, Inc.), BCL2 (1:1,000; cat. no. ab196495; Abcam),
Bax (1:1,000; cat. no. ab32503; Abcam), cleaved caspase 3 (1:1,000;
cat. no. 9661; CST) and GAPDH (1:10,000; cat. no. ab181602; Abcam).
Subsequently, the membranes were incubated with HRP-conjugated goat
anti-rabbit (1:2,000; cat. no. ab6721; Abcam) or goat anti-mouse
(1:2,000; cat. no. ab6789; Abcam) secondary antibodies at 4°C for 1
h. BeyoECL Plus (Beyotime Institute of Biotechnology) was used to
visualized the protein bands, and densitometric analysis was
conducted using ImageJ 1.8.0 software (National Institutes of
Health).
TTC staining
The heart slices (1.5 mm) were firstly placed in TTC
solution (Sigma-Aldrich; Merck KGaA) with pH of 7.4 at 37°C for 15
min, and then fixed in 4% paraformaldehyde for 24 h at room
temperature. The color of the normal myocardium became red, and
that of the ischemic myocardium was gray. The MI area was
calculated using an EOS 90D digital camera (Canon, Inc.) to capture
images and the staining was quantified by ImageJ2x software
(National Institutes of Health).
Measurement of CK, LDH and NO levels
Serum (100 µl) was used to determine the
contents of LDH (Beyotime Institute of Biotechnology) and CK
(Sigma-Aldrich; Merck KGaA) in rats according the manufacturer's
protocols. NO level in the serum was detected using an NO kit (cat.
no. S0021S; Beyotime Institute of Biotechnology) in line with the
kit instructions. CK, LDH and NO levels were examined using a
CHEMIX-180 automatic biochemistry analyzer (Sysmex
Corporation).
H&E staining
The myocardial tissues were fixed in 4%
paraformaldehyde at room temperature. After 12 h, the tissues were
dehydrated in an ascending gradient of ethanol and then made
transparent with xylene. Next the tissues were embedded in paraffin
wax and sliced into 4-µm-thick sections. Finally, the
sections were stained with hematoxylin for 5 min at 4°C and then
with eosin for 3 min at 4°C. H&E staining was observed under a
BX43 light microscope (Olympus Corporation).
Immunofluorescence staining
Cardiac tissues and CMECs were fixed with 4%
paraformaldehyde at room temperature for 12 h and for 20 min,
respectively, and then permeabilized with 0.5% Triton X-100 at room
temperature for 5 min. The 5-µm-thick sections and CMECs
were blocked with 5% normal goat serum (Beijing Solarbio Science
& Technology Co., Ltd.) for 1 h at room temperature, incubated
at 4°C overnight with primary antibodies against CD31 (1:1,000; cat
no. 28083-1-AP; Proteintech Group, Inc.) and subsequently incubated
with goat anti-rabbit Alexa Fluor® 488 IgG secondary
antibody (1:100; cat. no. ab150077; Abcam) or goat anti-rabbit
Alexa Fluor® 555 IgG secondary antibody (1:200; cat. no.
ab150078; Abcam) at 37°C for 1.5 h in the dark. At least five
randomly selected fields were examined using an IX73 inverted
fluorescence microscope (Olympus Corporation).
TUNEL assay
Rat hearts were fixed at room temperature for 24 h
in 4% paraformaldehyde and dehydrated with 30% sucrose at room
temperature for 24 h. Subsequently, the hearts were embedded in
paraffin and then sliced into 4-5 µm sections. Apoptotic
cells in the heart tissues were stained using a One-step TUNEL
Apoptosis Detection kit (cat. no. C1086; Beyotime Institute of
Biotechnology) in accordance with manufacturer's protocol and
observed under an IX73 inverted fluorescence microscope (Olympus
Corporation).
Cell culture
Human CMECs were purchased from BLUEFBIO, and HUVECs
were from American Type Culture Collection; both were incubated in
Dulbecco's Modified Eagle Medium supplemented with 10% FBS (both
from HyClone; Cytiva) in an incubator containing 5% CO2
at 37°C.
H/R cell model and transfection
Plasmids inducing GADD45A silencing (siRNA-GADD45A-1
and -2; siB08722144103-1-5 and siB08722144126-1-5, respectively; 80
nM; Guangzhou RiboBio Co., Ltd.) or an siRNA-NC (siN0000002-1-5; 80
nM) were transfected into human CMECs using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) for 48 h at 37°C following the standard
procedures of the manufacturer. After 48 h, the transfected cells
were incubated in a hypoxic incubator (95% N2 and 5%
CO2) at 37°C for 45 min and then under normoxic
conditions (21% O2, 5% CO2 and 74%
N2) at 37°C for 90 min, then used for subsequent
experimentation.
In rescue experiments, prior to H/R induction, CMECs
were treated with the JNK and p38 MAPK activator, anisomycin (5
µM), or with an inhibitor of STAT3, AG490 (50
µmol/l), for 2 h at 37°C.
Cell Counting Kit-8 (CCK-8) viability
assay
CMEC viability in each group was detected using the
CCK-8 kit (Nanjing Jiancheng Bioengineering Institute) according to
the manufacturer's protocol. A total of 2×104 cells/well
were seeded into 96-well plates for 24 h at 37°C. Then, 10
µl CCK-8 solution was added to each well and the cells were
incubated for further 2 h at 37°C. Finally, a microplate reader was
used to assess the absorbance at λ=450 nm.
Flow cytometry
CMECs were cultured in 6-well plates
(1×106 cells/well) for 24 h at 37°C. The apoptotic level
of CMECs in each group was assessed by Annexin V/FITC-PI Apoptosis
Detection kit (Beyotime Institute of Biotechnology) according to
the manufacturer's protocol. Total apoptotic rates (equal to
early-plus late-stage apoptosis) were analyzed using a CytoFLEX
flow cytometer with FlowJo v10 software (FlowJo; BD
Biosciences).
Tube formation
Matrigel (50 µl) was spread into 96-well
plates at 37°C for 30 min. After H/R treatment for 30 min at 37°C,
100 µl HUVECs (2×105 cell/ml) were added to each
well. To confirm the role of JAK2-STAT3 pathway in angiogenesis,
JAK2-STAT3 pathway inhibitor AG490 was added. Angiogenesis was
observed after 6 h incubation at 37°C under a BX43 light microscope
(Olympus Corporation).
Statistical analysis
All experiments were repeated at least three times.
Data are presented as the mean ± SD and analyzed with GraphPad
Prism v7.0 (GraphPad Software; Dotmatics). One-way ANOVA was used
for comparisons among groups, followed by Tukey's post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Result
GADD45A silencing alleviates pathological
injury of myocardial I/R
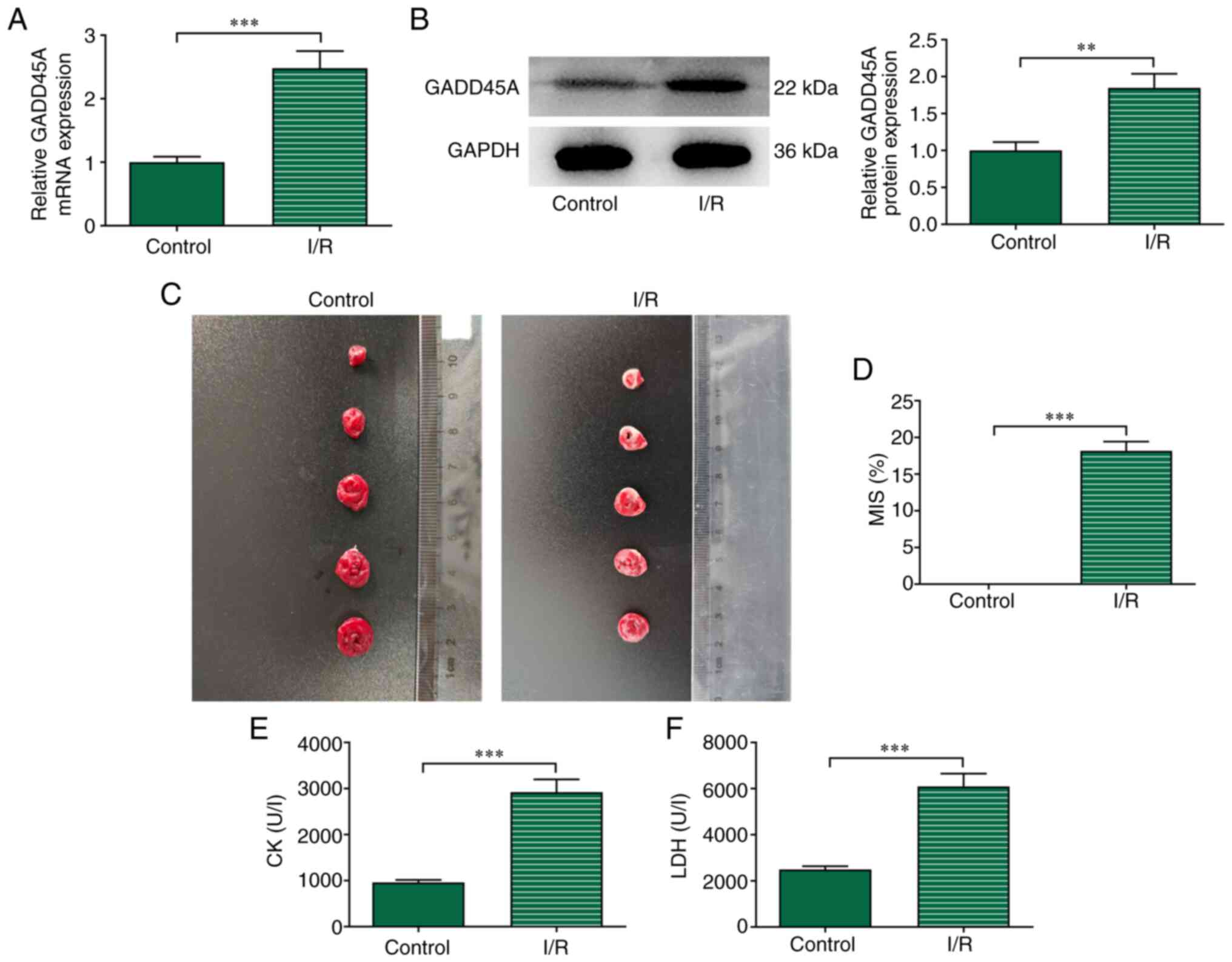
Results obtained from RT-qPCR and western blotting
revealed that GADD45A mRNA and protein expression levels,
respectively, of the I/R group were significantly elevated compared
with those in the Control group (Fig.
1A and B). TTC staining results showed that I/R induction
significantly increased the size of the ischemic area in rats
compared with the Control group (Fig.
1C and D). In addition, the levels of CK and LDH were
significantly higher in the I/R group compared with those in the
Control group (Fig. 1E and
F).
To assess the role of GADD45A in I/R induction,
GADD45A silencing was induced by intramyocardial injection of
Lv-si-GADD45A. GADD45A level was successfully decreased following
Lv-si-GADD45A induction in I/R model rats compared with that in I/R
+ Lv-si-NC group (Fig. 2A and B).
The sizes of the infarcted area were significantly decreased by
GADD45A silencing compared with those in the I/R + Lv-si-NC group
(Fig. 2C and D). In the Control
group rats, no changes in myocardial tissue were observed, whereas
myocardial fibers were disordered with nuclear splitting, edema and
enlarged myocardial spaces after I/R induction (Fig. 2E). Myocardial fiber arrangement
disorder and interstitial edema were improved in I/R +
Lv-si-GADD45A group rats compared with those in I/R + Lv-si-NC
group rats (Fig. 2E). In
addition, the levels of CK and LDH in the serum were significantly
decreased in I/R + Lv-si-GADD45A rats compared with those in the
I/R + Lv-si-NC group (Fig. 2F and
G).
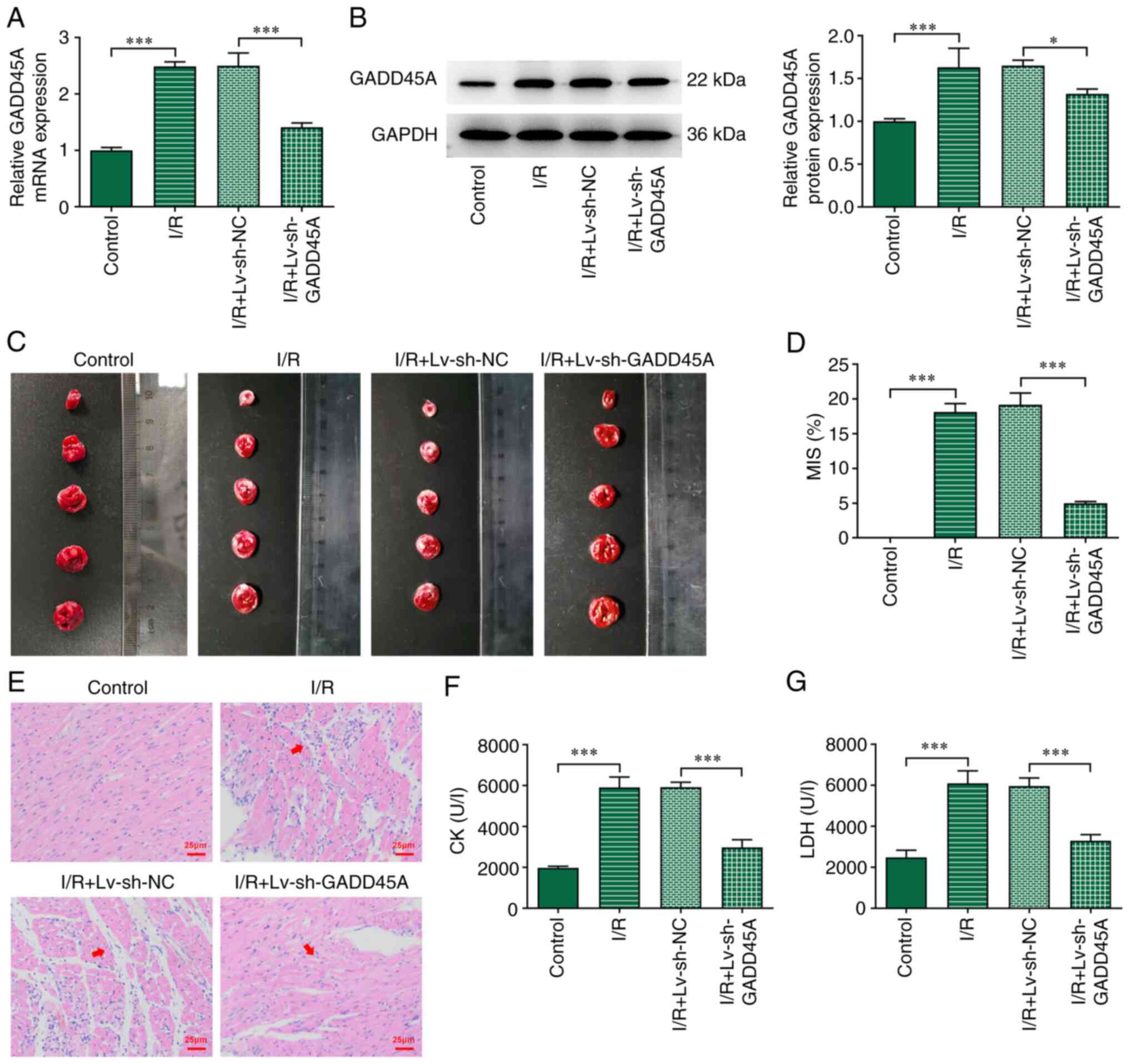 | Figure 2Effects of GADD45A silencing on
myocardial pathology. (A) mRNA and (B) protein expression levels of
GADD45A in heart tissues showing the transfection efficacy of
Lv-si-GADD45A on I/R-induced rats. (C) TTC staining of rat hearts.
(D) Myocardial infarct size. (E) H&E staining. Levels of (F) CK
and (G) LDH in serum. n=7; *P<0.05,
***P<0.001. CK, creatine kinase; GADD45A, growth
arrest and DNA damage-inducible α; I/R ischemia/reperfusion; Lv,
lentivirus; LDH, lactate dehydrogenase; MIS, myocardial infarct
size; NC, negative control; si, small interfering RNA; TTC,
2,3,5-triphenyltetrazolium chloride. |
GADD45A silencing alleviates loss of
vascular endothelial cells in myocardial I/R
To assess the effect of GADD45A on the loss of
vascular endothelial cells, the cells were stained with CD31 and
TUNEL. The data showed that I/R induction contributed to increased
apoptotic level and decreased CD31 staining compared with the
Control group (Fig. 3A); these
effects were reversed by GADD45A silencing. The protein expression
levels of CD31 and p-eNOS were significantly downregulated, whereas
that of ET-1 was upregulated in the I/R group compared with those
in the Control group (Fig. 3B);
however, GADD45A silencing significantly reversed this effect. In
addition, the significantly decreased expression of NO following
I/R was partially reversed in I/R model rats treated with
si-GADD45A (Fig. 3C). These
results demonstrated that GADD45A silencing may ameliorate the
injury induced by myocardial I/R.
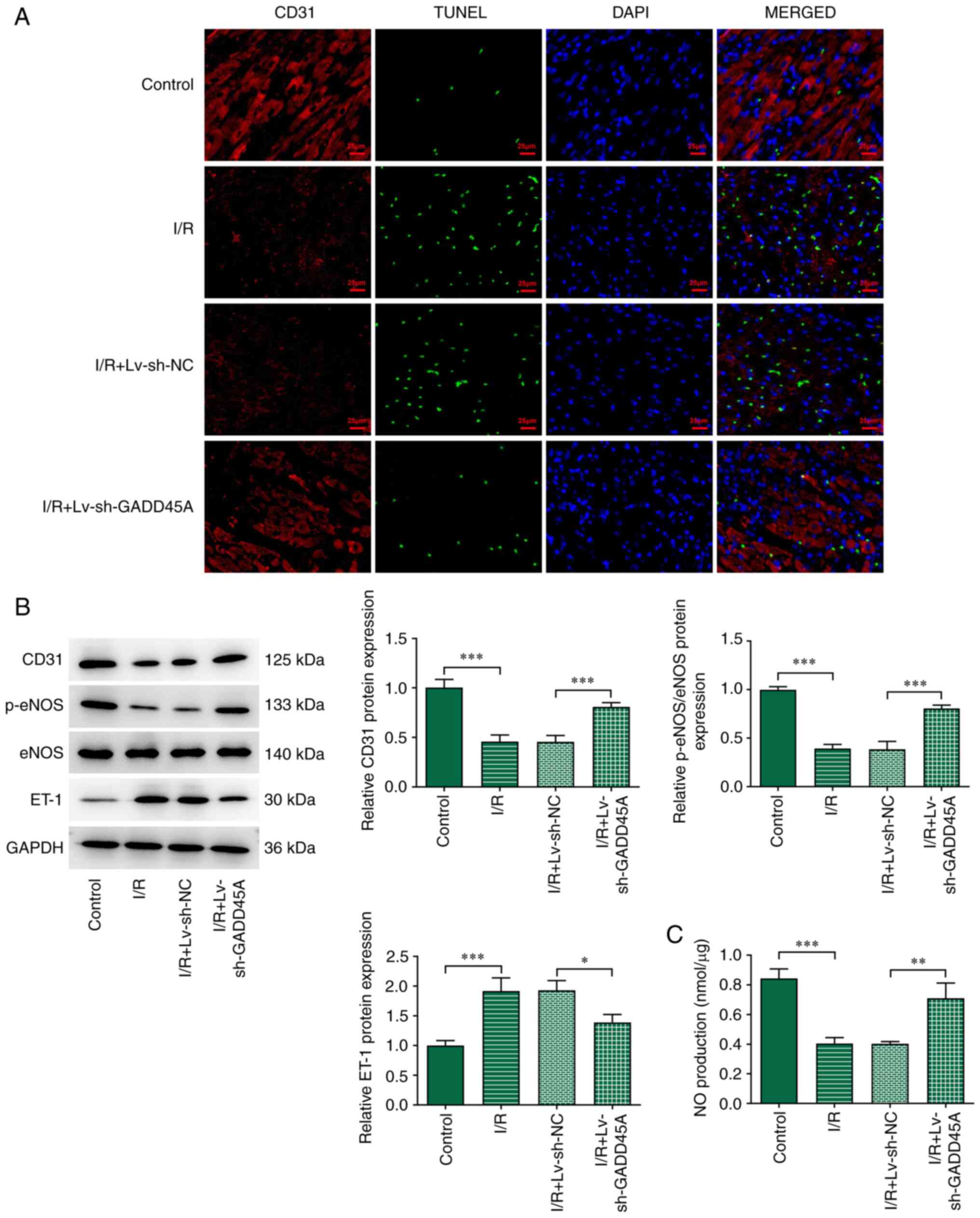 | Figure 3Effects of GADD45A on vascular
endothelial cell loss. (A) Dual-staining of CD31 and TUNEL in I/R
model rats with or without Lv-si-GADD45A treatment; DAPI was used
to stain the nuclei. (B) CD31, p-eNOS, eNOS and ET-1 expression
levels were examined by western blotting. (C) NO serum levels were
detected using an NO kit. Each experiment was repeated at least
three times. *P<0.05, ***P<0.001. eNOS,
endothelial NO synthase; ET-1, endothelin-1; GADD45A, growth arrest
and DNA damage-inducible α; I/R ischemia/reperfusion; Lv,
lentivirus; NC, negative control; NO, nitric oxide; p-,
phosphorylated; si, small interfering RNA. |
GADD45A silencing inactivates JNK/p38
MAPK signaling and activates STAT3/VEGF signaling in myocardial I/R
injured tissues
To assess the regulatory role of GADD45A in I/R, the
protein expression levels of p-p38 MAPK, p38 MAPK, p-JNK, JNK,
EGR1, p-STAT3, STAT3 and VEGF were determined. The levels of p-p38
MAPK, p-JNK and EGR1 were significantly increased by I/R induction
compared with those in the Control group, and these were
subsequently decreased following GADD45A silencing (Fig. 4A). Furthermore, the expression
levels of p-STAT3 and VEGF were significantly decreased in the I/R
group compared with the Control group (Fig. 4B); these effects were
significantly reversed in the I/R + Lv-si-GADD45A group compared
with rats in the I/R + Lv-si-NC group.
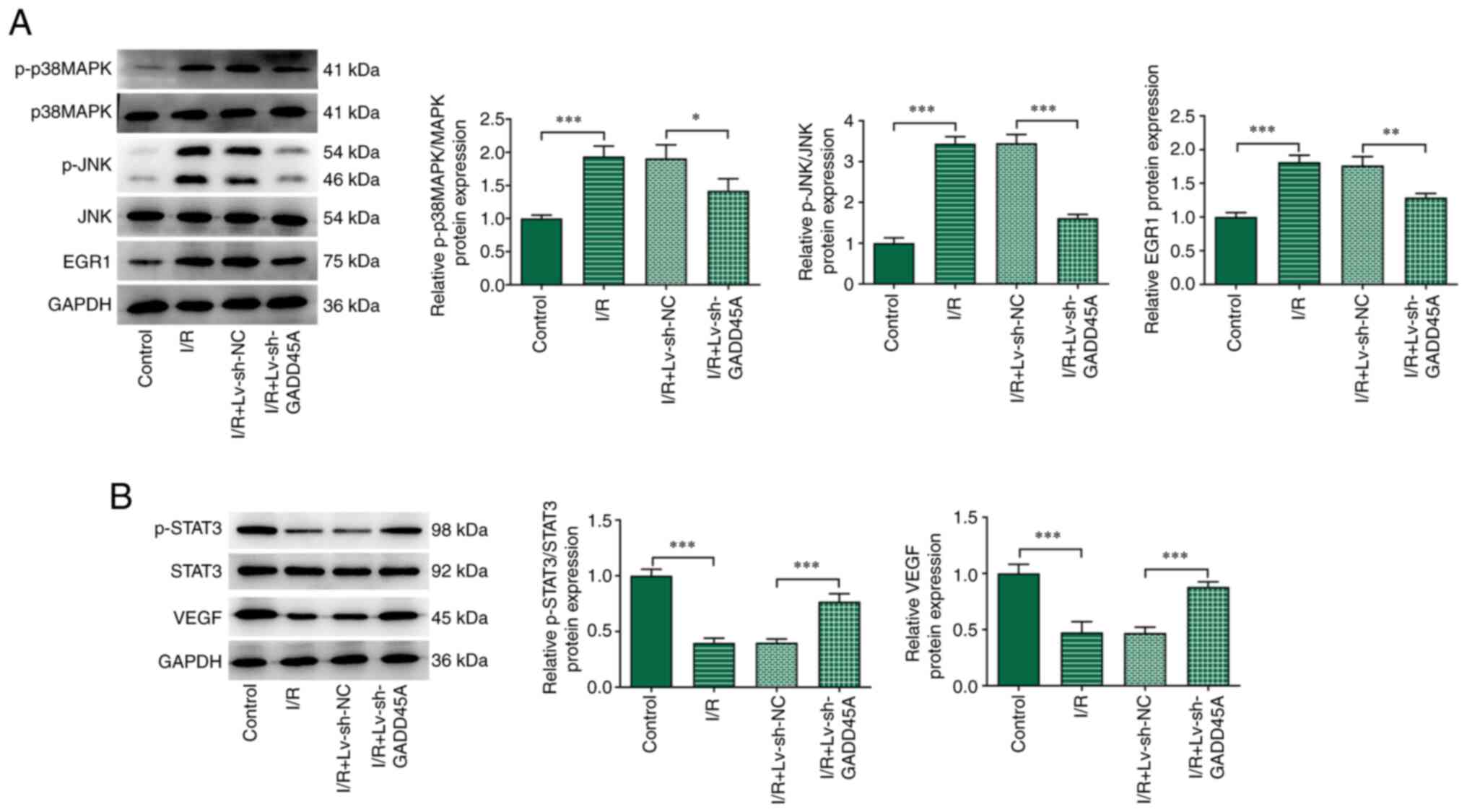 | Figure 4Effects of GADD45A on p38 MAPK/JNK
and STAT3/VEGF in vivo. (A) p-p38 MAPK, p-JNK, JNK, MAPK and
EGR1 protein expression levels were examined by western blotting in
I/R model rats with or without Lv-si-GADD45A treatment. (B)
p-STAT3, STAT3 and VEGF protein expression levels as determined by
western blotting. Each experiment was repeated at least three
times. *P<0.05, **P<0.01,
***P<0.001. EGR1, early growth response 1; GADD45A,
growth arrest and DNA damage-inducible α; I/R ischemia/reperfusion;
Lv, lentivirus; NC, negative control; p-, phosphorylated; si, small
interfering RNA. |
GADD45A silencing inhibits p38 MAPK/JNK
signaling and activates STAT3/VEGF signaling in H/R-induced
CMECs
To examine the mechanism by which GADD45A is
involved in H/R-induced CMEC injury in vitro, the cells were
subjected to H/R induction. H/R treatment significantly increased
GADD45A mRNA and protein expression levels compared with those in
the untreated Control group (Fig. 5A
and B, respectively). Next, the effects of GADD45A silencing on
CMECs were examined. RT-qPCR and western blotting analysis
confirmed that GADD45A mRNA and protein expression levels,
respectively, were successfully reduced following transfection with
siRNA-GADD45A-1 and siRNA-GADD45A-2 compared with siRNA-NC
transfection (Fig. 5C and D).
Based on the transfection efficiency, siRNA-GADD45A-1 was selected
for use in subsequent experiments. H/R stimulation activated p38
MAPK/JNK signaling and suppressed STAT3/VEGF signaling, as shown by
the increased levels of p-p38 MAPK and p-JNK (Fig. 5E), and the decreased levels of
p-STAT3 and VEGF in H/R group (Fig.
5F). In addition, EGR1 expression was increased compared with
the Control group (Fig. 5E).
GADD45A silencing reversed the effects of H/R induction on these
protein expression levels (Fig. 5E
and F).
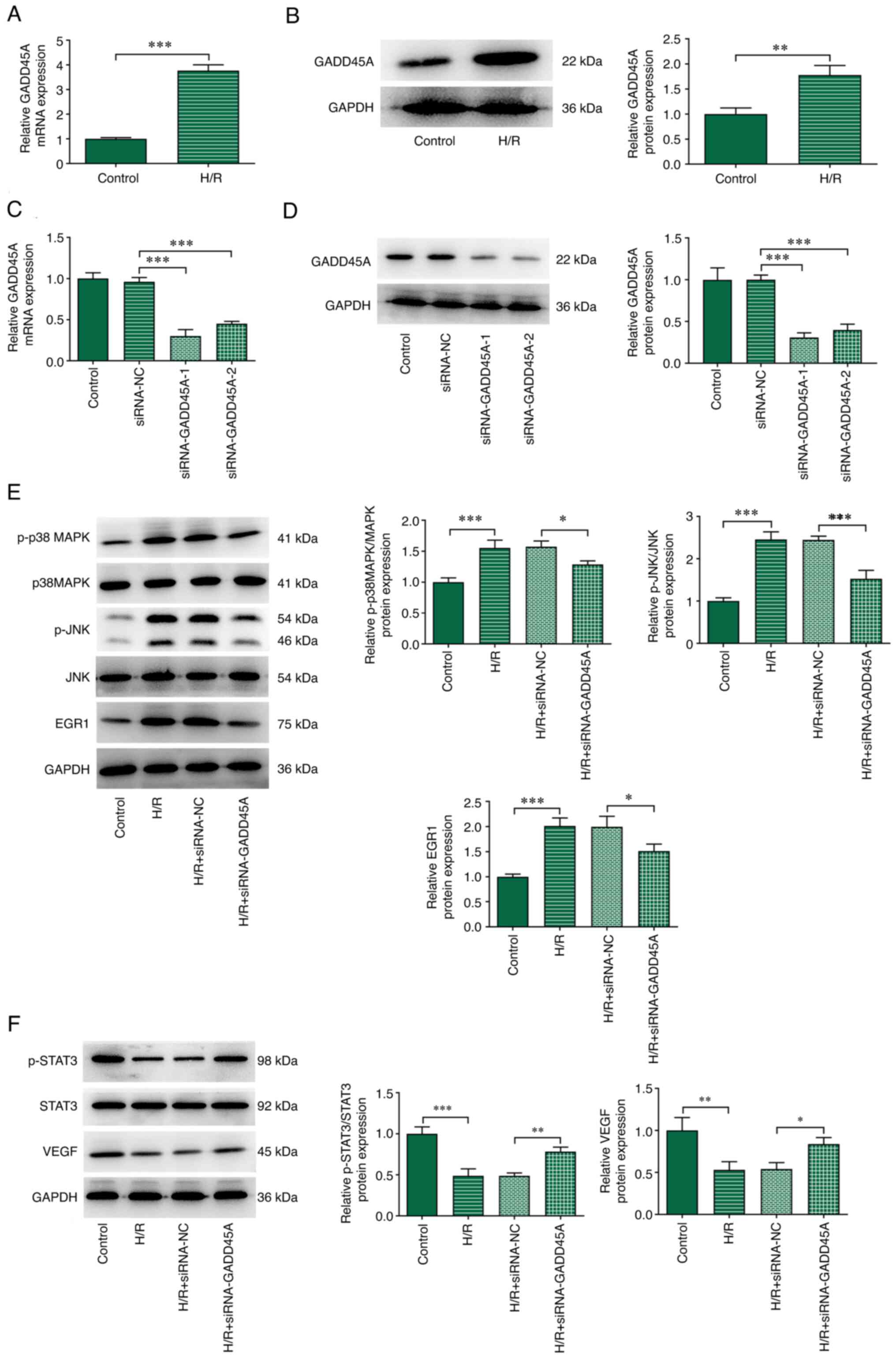 | Figure 5Effects of GADD45A on p38 MAPK/JNK
and STAT3/VEGF in vitro. CMECs were transfected
siRNA-GADD45A-1, siRNA-GADD45A-2 or siRNA-NC for 24 h and then
subjected to H/R treatment. (A) mRNA and (B) protein expression
levels of GADD45A expression in H/R-induced CMECs. (C) mRNA and (D)
protein expression levels of GADD45A expression in CMECs following
transfection with siRNA-GADD45A. (E) p-p38 MAPK, p38 MAPK, p-JNK,
JNK and EGR1 protein expression levels were determined by western
blotting. (F) p-STAT3, STAT3 and VEGF expression levels were
determined by western blotting. Each experiment was repeated at
least three times. *P<0.05, **P<0.01,
***P<0.001. CMEC, cardiac microvascular endothelial
cells; EGR1, early growth response 1; GADD45A, growth arrest and
DNA damage-inducible α; H/R, hypoxia/reoxygenation; NC, negative
control; p-, phosphorylated; siRNA, small interfering RNA. |
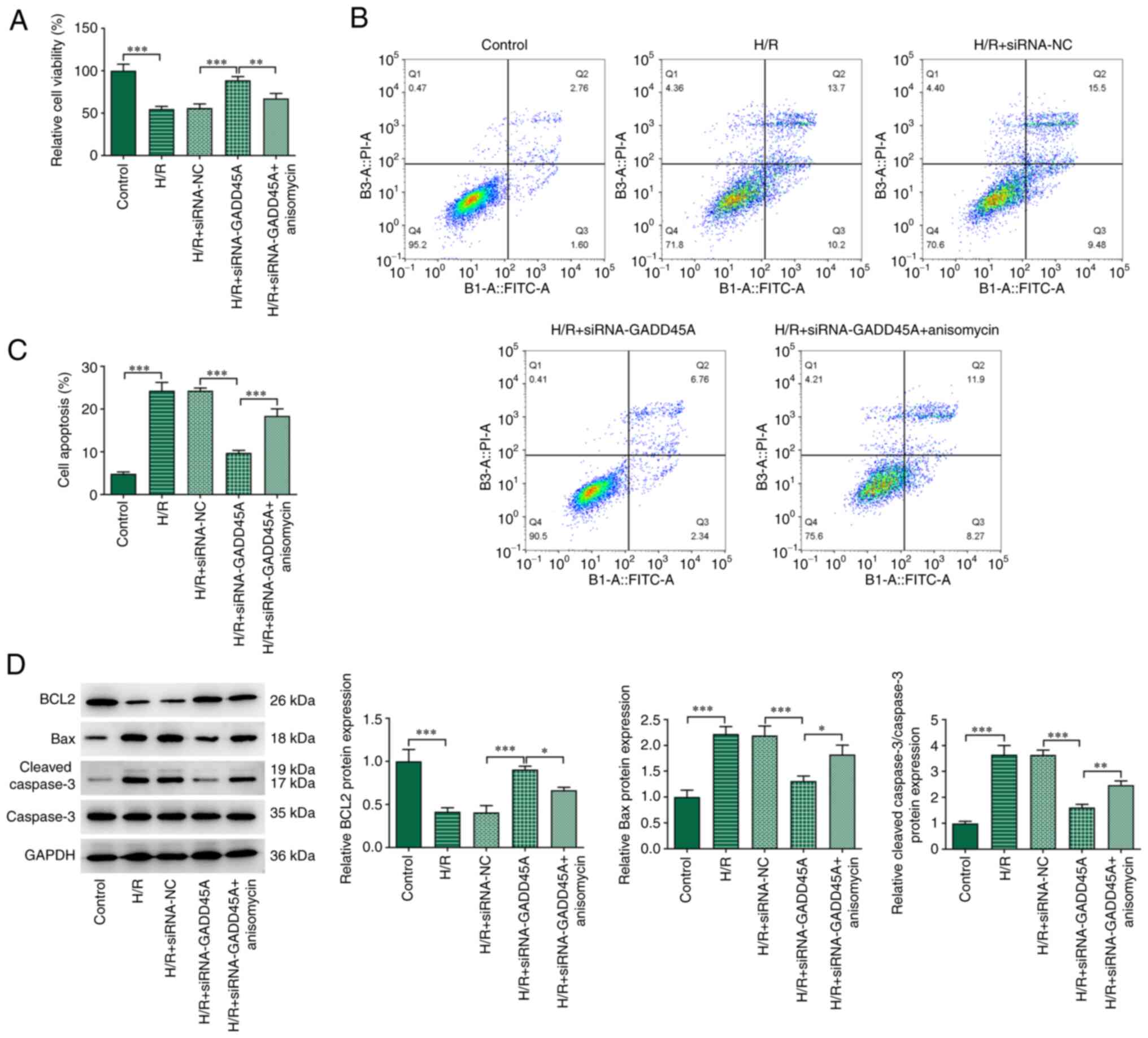
GADD45A silencing increases viability and
reduces apoptosis in H/R-induced CMECs through MAPK and ameliorates
angiogenesis through STAT3/VEGF
The mechanisms by which GADD45A may affect the
injury and apoptosis of CMECs induced by H/R were investigated. H/R
induction diminished cell viability and promoted apoptosis
(Fig. 6A-C). The knockdown of
GADD45A subsequently increased the viability and decreased the
apoptosis of H/R-induced CMECs. To verify that GADD45A modulated
viability and apoptosis in H/R-treated CMECs via J JNK/p38 MAPK
signaling, NK and p38 activator anisomycin was used here. Treatment
with anisomycin significantly reduced these effects. The declined
BCL2 expression and the elevated Bax and Cleaved caspase 3/caspase
3 expressions in the H/R group were all reversed by GADD45A
silencing (Fig. 6D). Furthermore,
anisomycin treatment also reduced BCL2 protein expression but
raised the expression levels of Bax and cleaved caspase 3 in CMECs
subjected to H/R + siRNA-GADD45A.
AG490 is an inhibitor of STAT3 phosphorylation,
which may enhance myocardial cell apoptosis and eliminate the
protective effect of ischemic pre-conditioning and ischemic
post-conditioning in the heart (17). The decreased CD31 expression
caused by H/R induction was elevated following GADD45A silencing
(Fig. 7A and B); however, AG490
treatment partially reversed this effect. The decreased protein
expression levels of CD31 and p-eNOS (Fig. 7B), as well as the reduction in NO
(Fig. 7C), in H/R-induced CMECs
were significantly increased in H/R + siRNA-GADD45A cells, which
were then reduced by AG490 treatment; ET-1 expression exhibited the
opposite trend. In the angiogenesis assay, fewer tubes were
observed in the H/R group compared with the Control group, whereas
the number of tubes was significantly increased in H/R cells
transfected with siRNA-GADD45A (Fig.
7D and E); H/R + siRNA-GADD45A cells treated with AG490 formed
fewer tubes compared with the untreated H/R + siRNA-GADD45A
group.
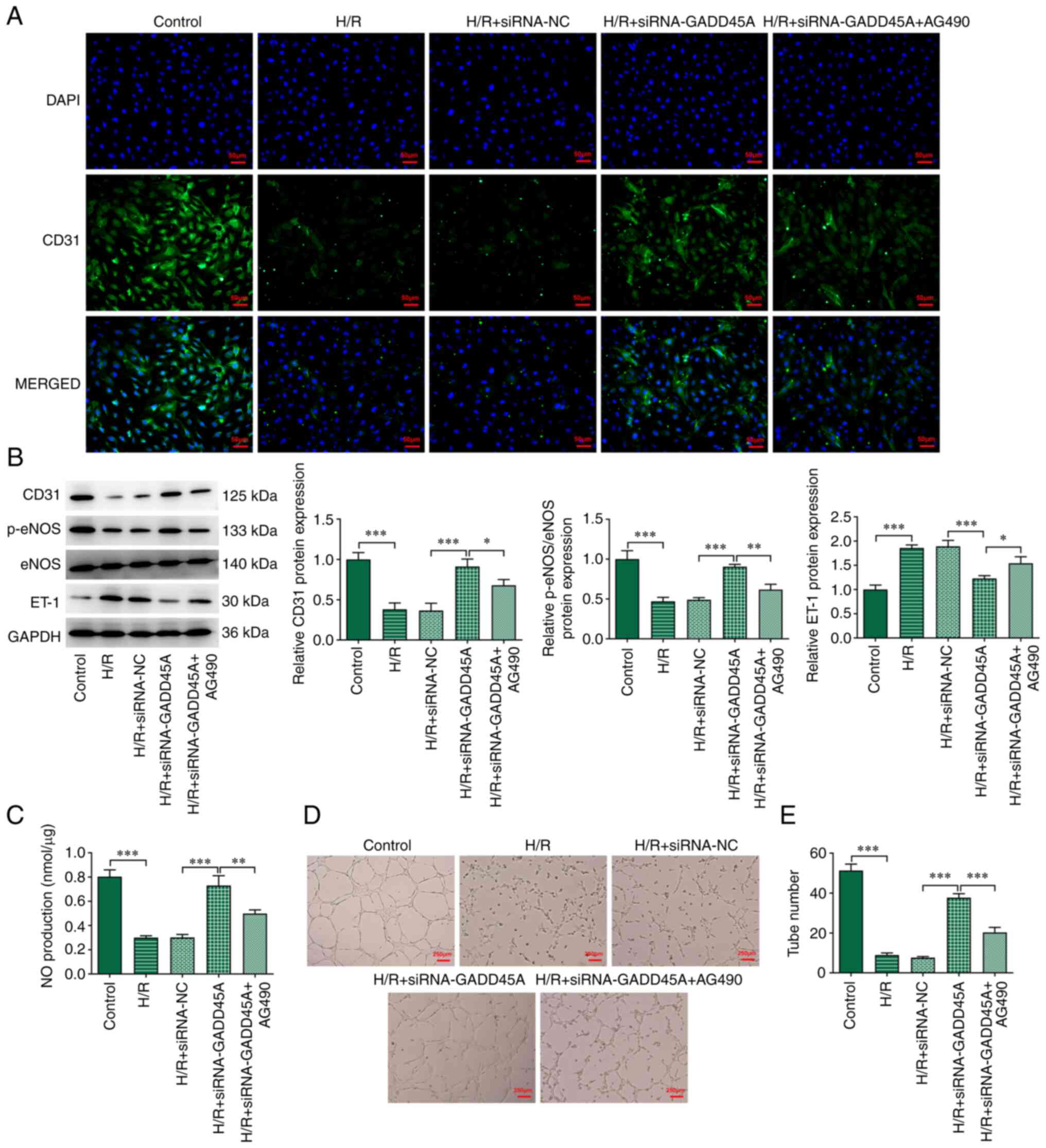 | Figure 7Effects of ATF6 on angiogenesis of
CMECs via the STAT3/VEGF signaling pathway. (A) Immunofluorescence
staining for CD31 in CMECs; DAPI was used to stain the nucleus. (B)
Protein expression levels of CD31, p-eNOS, eNOS and ET-1 were
determined by western blotting analysis. (C) NO serum levels were
determined using an NO kit. (D and E) Angiogenesis tube formation
assay. Each experiment was repeated at least three times.
*P<0.05, **P<0.01,
***P<0.001. CMEC, cardiac microvascular endothelial
cell; eNOS, endothelial NO synthase; ET-1, endothelin-1; GADD45A,
growth arrest and DNA damage-inducible α; H/R,
hypoxia/reoxygenation; NC, negative control; NO, nitric oxide; p-,
phosphorylated; siRNA, small interfering RNA. |
Discussion
As a member of the GADD45 family, which is a group
of stress sensors, GADD45A serves a crucial regulatory role in
various cellular functions, such as DNA repair, cell cycle
regulation and senescence, and genotoxic stress response (18). Importantly, GADD45A has been
reported to be upregulated in myocardial infarction, as shown in
I/R injury rat models (12). In
addition, GADD45A was able to inhibit proliferation and promote
apoptosis in H/R-induced cardiomyocyte injury (12). In the present study, it was shown
that GADD45A silencing decreased the size of the myocardial
infarcted area, improved myocardial pathological injury and
decreased the loss of vascular endothelial cells, demonstrating
that the targeting of GADD45A may serve a protective role against
I/R-induced injury. Expression of the angiogenesis-related protein
CD31 was significantly decreased in the I/R model rats, indicating
that a severe infarction occurred to the blood vessels. However,
this effect was significantly reduced by GADD45A silencing, which
suggested a protective role of GADD45A knockdown against the
infarction of blood vessels. Additionally, GADD45A silencing
increased CD31 expression and eNOS phosphorylation, as well as
decreasing the expression of vascular constriction factor ET-1.
Endothelium-generated eNOS is known to be involved in microvascular
relaxation and contraction (19).
The ratio of eNOS and ET-1 also participates in luminal stenosis
and vascular wall edema induced by I/R (20). Thus, I/R induction resulted in
myocardial injury by GADD45A, possibly by regulating the eNOS and
ET-1.
The present study provided evidence supporting the
newly recognized role of GADD45A in ameliorating I/R-induced injury
by regulating the MAPK and STAT3/VEGF signaling pathways. Previous
studies have shown that GADD45A could activate downstream JNK and
p38 signaling proteins, and its silencing suppressed the expression
of JNK and p38 (12,21,22). In addition, data from a previous
study suggested that I/R could upregulate the levels of JNK and p38
MAPK phosphorylation in CMECs (23), and the activation of JNK and MAPK
signals was involved in endothelial cell apoptosis (23). In addition, results from the
present study demonstrated that ERK1/2, JNK and p38 inhibitors
downregulated EGR1 expression in H/R-induced CMECs at various
levels. However, MAPK activators have the opposite effect on EGR1
expression, suggesting that ERK1/2, JNK and p38 are upstream
signaling proteins that induce EGR1 (24). The transcription factor EGR1 is
known to serve an important role in the pathophysiological damage
of a variety of cardiovascular diseases, including atherosclerosis
and cardiac hypertrophy (25).
Previous studies established that upregulation of EGR1 in the heart
induced inflammation after I/R injury (26), and the subsequent use of EGR1
targeting DNAzymes reduced infarct size and inflammatory marker
production in rodent and pig models (27,28). One study reported that GADD45A
could inhibit the expression of STAT3 signaling protein (29), and STAT3 played an important role
in cell survival (29). STAT3 is
required for the growth of myocardial capillaries after ischemic
injury, and loss of STAT3 in myocardial cells leads to reduced
capillarization in the left ventricle (30). By contrast, heart-specific STAT3
activation promotes cardiac vascular formation, and JAK/STAT3 and
ERK pathways are activated in angiogenesis and NO accumulation in
human umbilical vein endothelial cells (30,31).
The present study showed that I/R promoted GADD45A
expression, which may subsequently activate the p38 MAPK/JNK
pathway to inhibit cell viability and promote apoptosis, as well as
to suppress the STAT3/VEGF pathway to affect cell function in
CMECs. In summary, this research may provide novel insights into
the mechanism and therapy of ischemic cardiomyopathy.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YL designed the study. YW, HG and XC performed the
research. YW, ZL, YK and YJ analyzed the data. YW wrote the
manuscript, which was revised by YL. All authors contributed to
editorial changes in the manuscript. All authors read and approved
the final manuscript. YL and YW confirm the authenticity of all the
raw data.
Ethics approval and consent to
participate
Animal experiments were approved by the Animal Care
and Ethics Committee of Yan'an Hospital Affiliated to Kunming
Medical University (Kunming, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The study was supported by The Clinical Medicine Center for
Cardiovascular Disease of Yunnan Province (grant no.
ZX2019-08-01).
References
|
1
|
Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q,
Jin Q, Cao F, Tian F and Chen Y: Melatonin protects cardiac
microvasculature against ischemia/reperfusion injury via
suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis.
J Pineal Res. 63:e124132017. View Article : Google Scholar
|
|
2
|
Mezzaroma E, Toldo S, Farkas D, Seropian
IM, Van Tassell BW, Salloum FN, Kannan HR, Menna AC, Voelkel NF and
Abbate A: The inflammasome promotes adverse cardiac remodeling
following acute myocardial infarction in the mouse. Proc Natl Acad
Sci USA. 108:19725–19730. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Perrier S, Kindo M, Gerelli S and
Mazzucotelli JP: Coronary artery bypass grafting or percutaneous
revascularization in acute myocardial infarction? Interact
Cardiovasc Thorac Surg. 17:1015–1019. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wu X, Reboll MR, Korf-Klingebiel M and
Wollert KC: Angiogenesis after acute myocardial infarction.
Cardiovasc Res. 117:1257–1273. 2021. View Article : Google Scholar
|
|
5
|
Erbs S, Linke A, Schachinger V, Assmus B,
Thiele H, Diederich KW, Hoffmann C, Dimmeler S, Tonn T, Hambrecht
R, et al: Restoration of microvascular function in the
infarct-related artery by intracoronary transplantation of bone
marrow progenitor cells in patients with acute myocardial
infarction: The doppler substudy of the reinfusion of enriched
progenitor cells and infarct remodeling in acute myocardial
infarction (REPAIR-AMI) trial. Circulation. 116:366–374. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Olivetti G, Ricci R, Beghi C, Guideri G
and Anversa P: Response of the border zone to myocardial infarction
in rats. Am J Pathol. 125:476–483. 1986.PubMed/NCBI
|
|
7
|
Molyneux CA, Glyn MC and Ward BJ:
Oxidative stress and cardiac microvascular structure in ischemia
and reperfusion: The protective effect of antioxidant vitamins.
Microvasc Res. 64:265–277. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cui H, Li X, Li N, Qi K, Li Q, Jin C,
Zhang Q, Jiang L and Yang Y: Induction of autophagy by Tongxinluo
through the MEK/ERK pathway protects human cardiac microvascular
endothelial cells from hypoxia/reoxygenation injury. J Cardiovasc
Pharmacol. 64:180–190. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Leucker TM, Bienengraeber M, Muravyeva M,
Baotic I, Weihrauch D, Brzezinska AK, Warltier DC, Kersten JR and
Pratt PF Jr: Endothelial-cardiomyocyte crosstalk enhances
pharmacological cardioprotection. J Mol Cell Cardiol. 51:803–811.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhong J, Ouyang H, Sun M, Lu J, Zhong Y,
Tan Y and Hu Y: Tanshinone IIA attenuates cardiac microvascular
ischemia-reperfusion injury via regulating the
SIRT1-PGC1α-mitochondrial apoptosis pathway. Cell Stress
Chaperones. 24:991–1003. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bai J, Wang Q, Qi J, Yu H, Wang C, Wang X,
Ren Y and Yang F: Promoting effect of baicalin on nitric oxide
production in CMECs via activating the PI3K-AKT-eNOS pathway
attenuates myocardial ischemia-reperfusion injury. Phytomedicine.
63:1530352019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu C, Liu H, Sun Q and Zhang P: MicroRNA
1283 alleviates cardiomyocyte damage caused by
hypoxia/reoxygenation via targeting GADD45A and inactivating the
JNK and p38 MAPK signaling pathways. Kardiol Pol. 79:147–155. 2021.
View Article : Google Scholar
|
|
13
|
Pontén F, Jirström K and Uhlen M: The
human protein atlas-a tool for pathology. J Pathol. 216:387–393.
2008. View Article : Google Scholar
|
|
14
|
Hung YC, Kuo YJ, Huang SS and Huang TF:
Trimucrin, an Arg-Gly-Asp containing disintegrin, attenuates
myocardial ischemia-reperfusion injury in murine by inhibiting
platelet function. Eur J Pharmacol. 813:24–32. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qiu Y, Wu Y, Meng M, Luo M, Zhao H, Sun H
and Gao S: GYY4137 protects against myocardial ischemia/reperfusion
injury via activation of the PHLPP-1/Akt/Nrf2 signaling pathway in
diabetic mice. J Surg Res. 225:29–39. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
17
|
Lei S, Su W, Xia ZY, Wang Y, Zhou L, Qiao
S, Zhao B, Xia Z and Irwin MG: Hyperglycemia-induced oxidative
stress abrogates remifentanil preconditioning-mediated
cardioprotection in diabetic rats by impairing caveolin-3-modulated
PI3K/Akt and JAK2/STAT3 signaling. Oxid Med Cell Longev.
2019:98363022019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Salvador JM, Brown-Clay JD and Fornace AJ
Jr: Gadd45 in stress signaling, cell cycle control, and apoptosis.
Adv Exp Med Biol. 793:1–19. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang J, Toan S and Zhou H: New insights
into the role of mitochondria in cardiac microvascular
ischemia/reperfusion injury. Angiogenesis. 23:299–314. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li C, Ma Q, Toan S, Wang J, Zhou H and
Liang J: SERCA overexpression reduces reperfusion-mediated cardiac
microvascular damage through inhibition of the
calcium/MCU/mPTP/necroptosis signaling pathways. Redox Biol.
36:1016592020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li FH, Han N, Wang Y and Xu Q: Gadd45a
knockdown alleviates oxidative stress through suppressing the p38
MAPK signaling pathway in the pathogenesis of preeclampsia.
Placenta. 65:20–28. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tront JS, Hoffman B and Liebermann DA:
Gadd45a suppresses Ras-driven mammary tumorigenesis by activation
of c-Jun NH2-terminal kinase and p38 stress signaling resulting in
apoptosis and senescence. Cancer Res. 66:8448–8454. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu F, Liu Y and Xu J: Pro-BDNF contributes
to hypoxia/reoxygenation injury in myocardial microvascular
endothelial cells: Roles of receptors p75NTR and
sortilin and activation of JNK and caspase 3. Oxid Med Cell Longev.
2018:30914242018.
|
|
24
|
Lu S, Zhang Y, Zhong S, Gao F, Chen Y, Li
W, Zheng F and Shi G: N-n-butyl haloperidol iodide protects against
hypoxia/reoxygenation injury in cardiac microvascular endothelial
cells by regulating the ROS/MAPK/Egr-1 pathway. Front Pharmacol.
7:5202017. View Article : Google Scholar :
|
|
25
|
Khachigian LM: Early growth response-1 in
cardiovascular pathobiology. Circ Res. 98:186–191. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bhindi R, Khachigian LM and Lowe HC:
DNAzymes targeting the transcription factor Egr-1 reduce myocardial
infarct size following ischemia-reperfusion in rats. J Thromb
Haemost. 4:1479–1483. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bhindi R, Fahmy RG, McMahon AC, Khachigian
LM and Lowe HC: Intracoronary delivery of DNAzymes targeting human
EGR-1 reduces infarct size following myocardial ischaemia
reperfusion. J Pathol. 227:157–164. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rayner BS, Figtree GA, Sabaretnam T, Shang
P, Mazhar J, Weaver JC, Lay WN, Witting PK, Hunyor SN, Grieve SM,
et al: Selective inhibition of the master regulator transcription
factor Egr-1 with catalytic oligonucleotides reduces myocardial
injury and improves left ventricular systolic function in a
preclinical model of myocardial infarction. J Am Heart Assoc.
2:e0000232013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang F, Zhang W, Li D and Zhan Q: Gadd45a
suppresses tumor angiogenesis via inhibition of the mTOR/STAT3
protein pathway. J Biol Chem. 288:6552–6560. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hilfiker-Kleiner D, Hilfiker A, Fuchs M,
Kaminski K, Schaefer A, Schieffer B, Hillmer A, Schmiedl A, Ding Z,
Podewski E, et al: Signal transducer and activator of transcription
3 is required for myocardial capillary growth, control of
interstitial matrix deposition, and heart protection from ischemic
injury. Circ Res. 95:187–195. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Osugi T, Oshima Y, Fujio Y, Funamoto M,
Yamashita A, Negoro S, Kunisada K, Izumi M, Nakaoka Y, Hirota H, et
al: Cardiac-specific activation of signal transducer and activator
of transcription 3 promotes vascular formation in the heart. J Biol
Chem. 277:6676–6681. 2002. View Article : Google Scholar
|