Introduction
Hypertension (HP) is a common condition that
accounts for ~12% of primary care consultations globally and is one
of the prevailing causes of fatal cardiovascular diseases such as
stroke, arrhythmia and ischemic heart disease (1,2).
Poor long-term blood pressure management can result in cardiac
remodeling, which typically involves cardiomyocyte and endothelial
cell apoptosis (3-5). Autophagy has been identified as a
pertinent mechanism underlying hypertensive (HP) cardiac diseases
(6). Thus, autophagy likely plays
a role in cardiac HP, particularly in myocardial microvascular
endothelial cells, and may thus be a major contributor to
myocardial injury (7-10).
A previous study by the authors demonstrated that
the cardiac expression levels of autophagy proteins, including
microtubule-associated proteins 1A/1B light chain 3B (LC3),
Beclin-1 and autophagy-related 7 (Atg7), were decreased, whereas
the expression of p62 was increased in a diabetic mouse model
(11). By pharmaceutically
restoring the expression of these proteins, cardiac function
improved in these mice, further indicating that autophagy is
involved in diabetic cardiomyopathy (11). However, the interplay between
autophagy and HP myocardial injury, and the underlying mechanisms
remain to be fully elucidated.
One candidate protein potentially involved in the
interaction between autophagy and HP myocardial injury is mammalian
ste20-like kinase1 (Mst1). Mst1 is a protein kinase that can be
activated during cardiomyocyte apoptosis (12,13). Exposure to high glucose has been
shown to phosphorylate and activate Mst1 in microvascular
endothelial cells, leading to apoptosis (12). It has also been shown that
decreased autophagy is associated with apoptosis, which is a
critical step in the development of diabetic cardiomyopathy
(12). The persistent activation
of Mst1 has also been observed in dilated cardiomyopathy (DCM)
(14). In the present study, it
was hypothesized that the inhibition of Mst1 may induce autophagy
and confer protective effects against HP-induced myocardial injury.
The results obtained using animal and cell HP models suggest that
the inhibition of Mst1 can exert protective effects against
HP-related cardiomyocyte injuries.
Materials and methods
Mouse model
Mst1 knockout mice (Mst1−/− mice) were
purchased from Shanghai Model Organisms. The animals were housed in
the SPF animal laboratory of the Animal Experiment Center of
Xinjiang Medical University at a constant temperature of 22-24°C,
constant humidity (55±5%), artificial light and dark for 12 h, and
free access to water and food. The present study used male
Mst1−/− mice and C57BL/6 wild-type (WT) mice (8-10 weeks
old, weighing 20-25 g) to establish a mouse model of HP. All
experimental mice were randomly allocated into four groups (the
control, HP, HP + Ad-KD-Mst1 and HP + Ad-KD-NC groups) with 24 mice
in each group. Briefly, Mst1−/− and WT mice in the
control groups were subcutaneously infused with 0.9% NaCl using an
osmotic bump (ALZET Osmotic Pumps Model 2006, Durect Corp.) for 42
days, and mice in the HP groups were subcutaneously infused with
angiotensin II [Ang II; 2,000 ng/kg/min, A604098-0100, Sangon
Biotech (Shanghai) Co., Ltd.] for 42 days, as previously described
(15,16). In total, 96 mice were used in the
experiment. Systolic blood pressure was measured at baseline and
during Ang II infusion using a tail-cuff method. A body weight loss
>10% was set as the criteria for humane endpoints to remove mice
from the experiments. At the end of the experiments, the
exsanguination of 100 µl blood from the heart under general
anesthesia with 1% pentobarbital sodium (45 mg/kg; P3761,
MilliporeSigma) following the collection of the heart was used to
euthanize the mice. The body weight of the mice was 30-32 g at the
time of sacrifice. The present study was reviewed and approved by
Ethics Committee of The First Affiliated Hospital, Xinjiang Medical
University (approval no. IACUC20201116-13). All procedures
performed in experiments involving animals were in accordance with
the ethical standards of the institution or practice at which the
studies were conducted.
Cell model
Cardiac microvascular endothelial cells (CMECs;
C-12285, PromoCell) were cultured in endothelial cell growth medium
MV (C-22020, PromeCell) supplemented with 10% fetal bovine serum
(FBS), 100 U/ml penicillin and 100 µg/ml streptomycin at
37°C in a 5% CO2 humidified incubator. Ang II (100
µM, 24 h) was used to induce the in vitro HP cell
model as previously described (17,18). Mst1 silencing was achieved by
transfecting Ad-sh-Mst1 [MOI: 200; Hanheng Biotechnology (Shanghai)
Co., Ltd.] into the cells. The cultured CMECs were treated with 50
nM bafilomycin A1 (MiilliporeSigma) for 2 h to evaluate the
autophagic flux.
Adenovirus construction
The pHBAd-U6-MCS-CMV-GFP-Δloxp was recombined with
backbone pBHGlox(delta)E1, 3Cre in bacteria. The adenovirus was
packaged using LipofiterTM for 6 h at 37°C in 293A cells [Hanheng
Biotechnology (Shanghai) Co., Ltd.] cultured in Dulbecco's modified
Eagle's medium (DMEM) plus 10% FBS, as previously described
(19). The nucleotide shRNA of
Mst1 was cloned using sequence, 5′-GAA GAC TAT CGA GGC ACA ACC AAT
A-3′ and the negative control shRNA sequence was 5′-TTC TCC GAA CGT
GTC ACG TAA-3′.
Adenovirus transduction
Ad-sh-Mst1 or control Ad-sh-green fluorescent
protein (GFP) was designed and provided by Hanheng Biotechnology
(Shanghai) Co., Ltd. The titer of the adenoviruses used in the
present study was ~1.58×1010 PFU/ml. Prior to adenoviral
transfection, CMECs (1×106 ̸ml, 3rd generation) were
seeded into six-well plates until they reached a confluency of 80%.
The cells were then incubated with Ad-sh-Mst1 at an MOI of 200 for
48 h at 37°C. Fluorescence microscopy was used to observe GFP
expression. The mRNA level of Mst1 was further measured to evaluate
the effectiveness of adenoviral transfection.
Echocardiography
Transthoracic echocardiography was performed to
evaluate mouse cardiac structure and function using a Vevo 2100
ultrasound imaging system (VisualSonics). Briefly, the mice were
anesthetized with 1% pentobarbital sodium (45 mg/kg; P3761,
MilliporeSigma) and placed on a warmed platform. Ultrasound gel
(Ultrasonic coupling agent; TM-100, Tianjin Jinya Electronics Co.
Ltd.) was applied to the chest skin after preparation (mice were
depilated using the Vetin depilatory cream from the chest to the
subxiphoid process up to the left axilla, and the skin was
prepared). Subsequently, two-dimensional images and M-mode images
of the parasternal short-axis view were recorded using a diagnostic
ultrasound machine (Philips HD11 XE). The left ventricular
end-diastolic diameter (LVEDD), left ventricular end-systolic
diameter (LVESD), left ventricular ejection fraction (LVEF) and
left ventricular fractional shortening (LVFS) were then
calculated.
Histological analysis
The mouse hearts were harvested and fixed with 10%
formalin for 24 h. The hearts were then embedded in paraffin and
cut into 5-µm-thick sections. Myocardial morphology and
fibrosis were assessed by staining the sections with hematoxylin
and eosin (H&E) for 4 min at room temperature, and Masson's
trichrome for about 13-15 min at room temperature (D026-1-2;
Nanjing Jiancheng Biological Engineering Research Institute Co.
Ltd.), respectively, as previously described (20). Images were captured using a light
microscope (E200, Nikon Corporation). The cardiomyocyte size and
area of fibrosis were measured using ImageJ software (version
1.50i, National Institutes of Health).
Biochemical assays
Mouse heart samples were homogenized, and
supernatants were collected and used for biochemical measurements.
The concentrations of interleukin-6 (IL-6; EK206/3-48, Lianke
Biotechnology Co., Ltd.), tumor necrosis factor-α (TNF-α;
EK282/3-48, EK206/3-48, Lianke Biotechnology), malondialdehyde
(MDA; A003-1-1, Nanjing Jiancheng Biological Engineering Research
Institute Co. Ltd.), superoxide dismutase (SOD; A001-3-1, Nanjing
Jiancheng Biological Engineering Research Institute) and
glutathione peroxidase (GSH-PX, A005, Nanjing Jiancheng Biological
Engineering Research Institute) were measured using enzyme-linked
immunosorbent assays (ELISAs) using commercially available kits (as
indicated above) according to the manufacturer's instructions.
Measurement of apoptosis
Apoptotic cells were detected using a terminal
deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay
kit (MK1015, Boster Biological Technology Co. Ltd.). Briefly, heart
paraffin sections were prepared, dewaxed in xylene and hydrated in
ethanol. The sections were then incubated for 30 min at 37°C with
TUNEL reaction mixture according to the manufacturer's
instructions. Images were captured using a fluorescence microscope
(E200, Nikon Corporation). TUNEL-positive cells in 10 randomly
selected areas were quantified. In total, five visual fields under
the microscope were selected randomly to observe the proportion of
apoptotic cells in each visual field with the naked eye, and an
average apoptotic rate was then calculated using SPSS 19.0 software
(IBM Corp. USA).
Microvascular corrosion casting
Microvascular corrosion casting was conducted as
previously described (21) using
scanning electron microscopy (SEM). Briefly, after being euthanized
and perfused, the mice were infused with Mercox CL-2B (Dainippon
Ink and Chemicals, Inc.) diluted with monomeric methylmethacrylate
(Ladd Research Industries) at a flow rate of 41 ml/h. After the
hardening of the injected resin, the heart was washed and submerged
in 2% hydrochloric acid. The specimens were then washed, and
ice-embedded casts were freeze-dried. The mounted specimens were
either evaporated with carbon and gold or sputter-coated with gold
and examined using the XL-30 SEM (FEI, Eindhoven) at an
accelerating voltage of 10 kV.
Mitochondrial assays
The detection of mitochondrial membrane potential
was performed as described previously (22). In brief, the cells were detached
using Trypsin (0.25%, #25200-056, Gibco; Thermo Fisher Scientific,
Inc.), washed with PBS precooled to 4°C and incubated with JC-1
staining solution for 30 min at 37°C (C2006, Beyotime Institute of
Biotechnology). After 30 min, the cells were washed and detected
using fluorescence-activated cell sorting (BD LSRFortessa™ System,
BD Biosciences). The ratio of red to green fluorescence intensity
at an excitation wavelength of 490 nm indicated the mitochondrial
membrane potential.
Transmission electron microscopy (TEM)
detection of autophagosomes
Autophagosomes were detected using a transmission
electron microscope (TEM1230, JEOL, Ltd.) according to a previously
published study (23). In brief,
cultured CMECs were digested and washed, followed by 2.5%
glutaraldehyde fixation at 4°C, and the cells were then examined
using an electron microscope. Groups consisting of 10 fields were
selected for analysis.
Western blot analyses
The preparation of cell lysates (RIPA buffer,
AR0105, Boster Biological Technology) from in vitro and
in vivo samples was performed as previously described
(11). Equal amounts (50
µg for tissue lysates or 25 µg for cell lysates) of
protein were separated in 10% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis and then transferred to polyvinylidene fluoride
membranes. The membranes were incubated with antibodies against
MST1 (1:1,000, cat. no. 3682S), phosphorylated (p-)Mst1 (1:1,000,
cat. no. 49332S), Beclin-1 (1:1,000, cat. no. 3738S), p62 (1:1,000,
cat. no. 23214S), LC3B (1:1,000, cat. no. 2775S), cleaved caspase-3
(1:1,000), cleaved caspase-9 (1:1,000), BAX (1:1,000), Bcl-2
(1:1,000), PARP-1 (1:1,000) overnight at 4°C. All primary
antibodies were purchased from Cell Signaling Technology, Inc.
Following incubation with goat anti-rabbit IgG H&L (conjugated
with horseradish peroxidase, 1:5,000, cat. no. ab205718, Abcam) or
goat anti-mouse IgG H&L (conjugated with horseradish
peroxidase, 1:5,000, cat. no. ab205719, Abcam) for 2 h at room
temperature, the bands were detected with enhanced
chemiluminescence (SuperSignal West Pico PLUS Chemiluminescent
Substrate; cat. no. 34580; Thermo Fisher Scientific, Inc.), and the
density and size of the bands were quantified using ImageJ 1.50i
software (National Institutes of Health) by normalizing to β-actin
[1:1,000, Sangon Biotech (Shanghai) Co., Ltd.] and GAPDH (1:5,000;
cat. no. 5174, Cell Signaling Technology, Inc.).
Immunofluorescence
To visualize autophagy markers, the cells were
washed with cold phosphate-buffered saline (PBS) and fixed with 4%
formaldehyde solution for 20 min. This was followed by
permeabilization with 0.5% Triton X-100 for 20 min and incubation
with blocking buffer containing 1% bovine serum albumin (BSA) for
30 min at room temperature. Samples were incubated with primary
antibodies against LC3B (2 µg/ml, cat. no. 2775S) and p62 (2
µg/ml, cat. no. 23214) (both from Cell Signaling Technology,
Inc.) at 37°C for 2 h and secondary antibodies (2 µg/ml,
goat anti-rabbit IgG H&L labeled with Alexa Fluor488, cat. no.
ab150081, Abcam) at 37°C for 1 h. For myocardial tissue sections,
the samples were fixed with acetone at 4°C for 10 min, followed by
blocking with 5% BSA for 30 min at room temperature. Incubation
with primary antibodies was performed at 4°C overnight and with the
secondary antibodies at room temperature for 1 h. Staining without
primary antibodies was used as the negative control. Images were
obtained using a LSM 700 laser scanning confocal microscope (Carl
Zeiss AG).
Statistical analysis
Statistical analyses were conducted using SPSS 19.0
software (IBM Corp.). Data are expressed as the mean ± standard
deviation (SD) for the indicated number of experiments or mice. The
differences in means between two groups and multiple groups were
evaluated using an unpaired t-test and one-way analysis of
variance, respectively. Post-test comparisons for multiple analyses
were performed using Tukey's test. P-values were 2-sided and a
P-value <0.05 was considered to indicate a statistically
significant difference.
Results
Mst1 knockout ameliorates cardiac
structure and function in mice with HP
To evaluate the role of Mst1 in the hearts of mice
with HP, mouse model of HP was first constructed as described in
the 'Materials and methods'. The knockout of Mst1 significantly
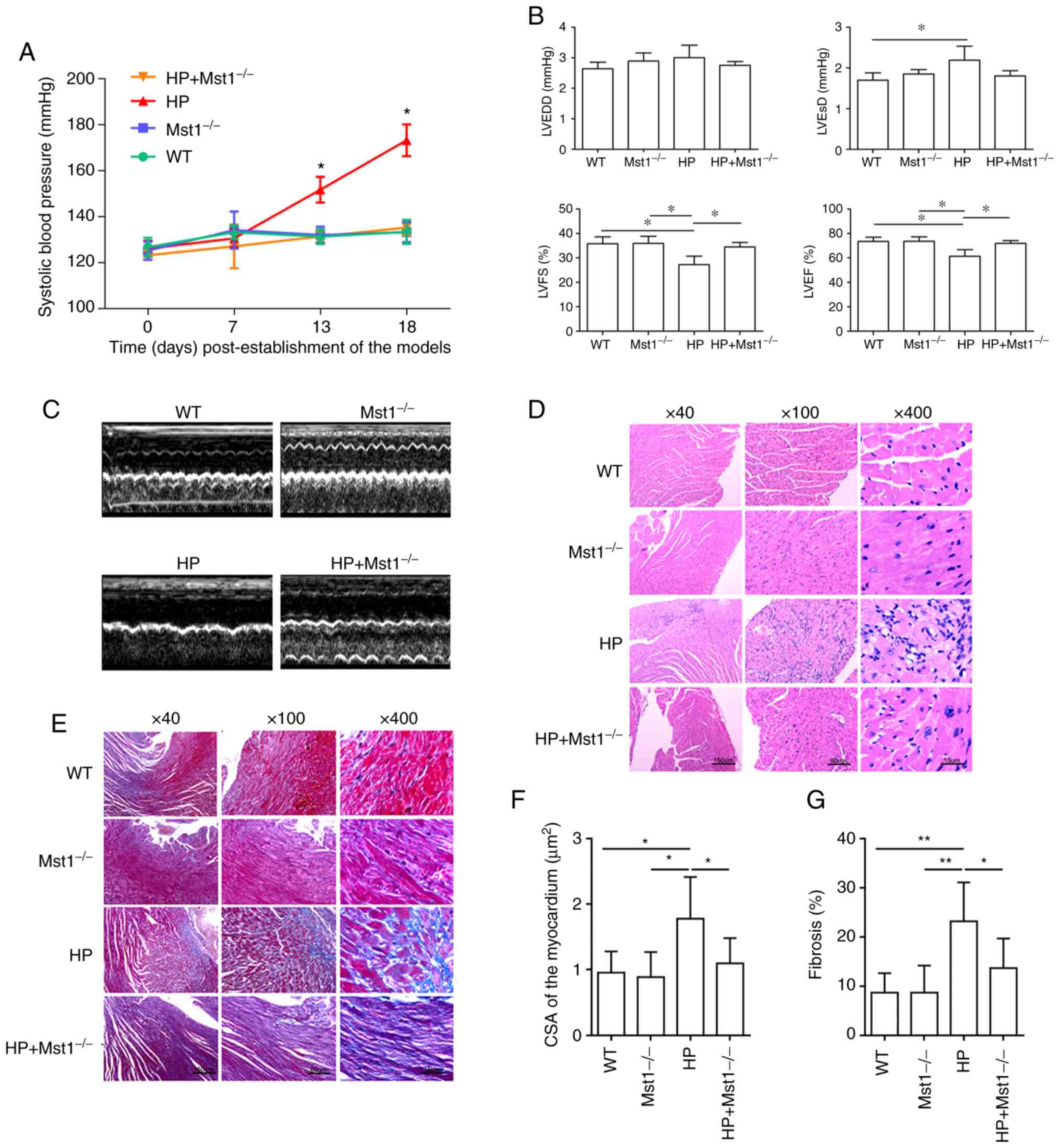
attenuated Ang II-induced HP (P<0.05, Fig. 1A). Echocardiography revealed an
impaired heart function in the HP group compared to the control
group, as indicated by both morphological changes and heart
function markers, including LVEDD, LVESD, LVEF and LVFS (all
P<0.05, Fig. 1B and C). Mst1
knockout in the HP group resulted in improved cardiac function (all
P<0.05, Fig. 1B and C),
suggesting a key role of Mst1 in HP. H&E staining (Fig. 1D) and Masson's trichrome staining
(Fig. 1E) revealed cardiomyocyte
hypertrophy and fibrosis (both P<0.05, Fig. 1F and G) in the HP group,
respectively. Mst1 knockout in the HP group significantly reduced
both cardiomyocyte hypertrophy and fibrosis (both P<0.05,
Fig. 1D-G), demonstrating the
potential benefits of the silencing of Mst1 to improve cardiac
function.
Mst1 knockout reduces HP-induced
inflammation and oxidative stress
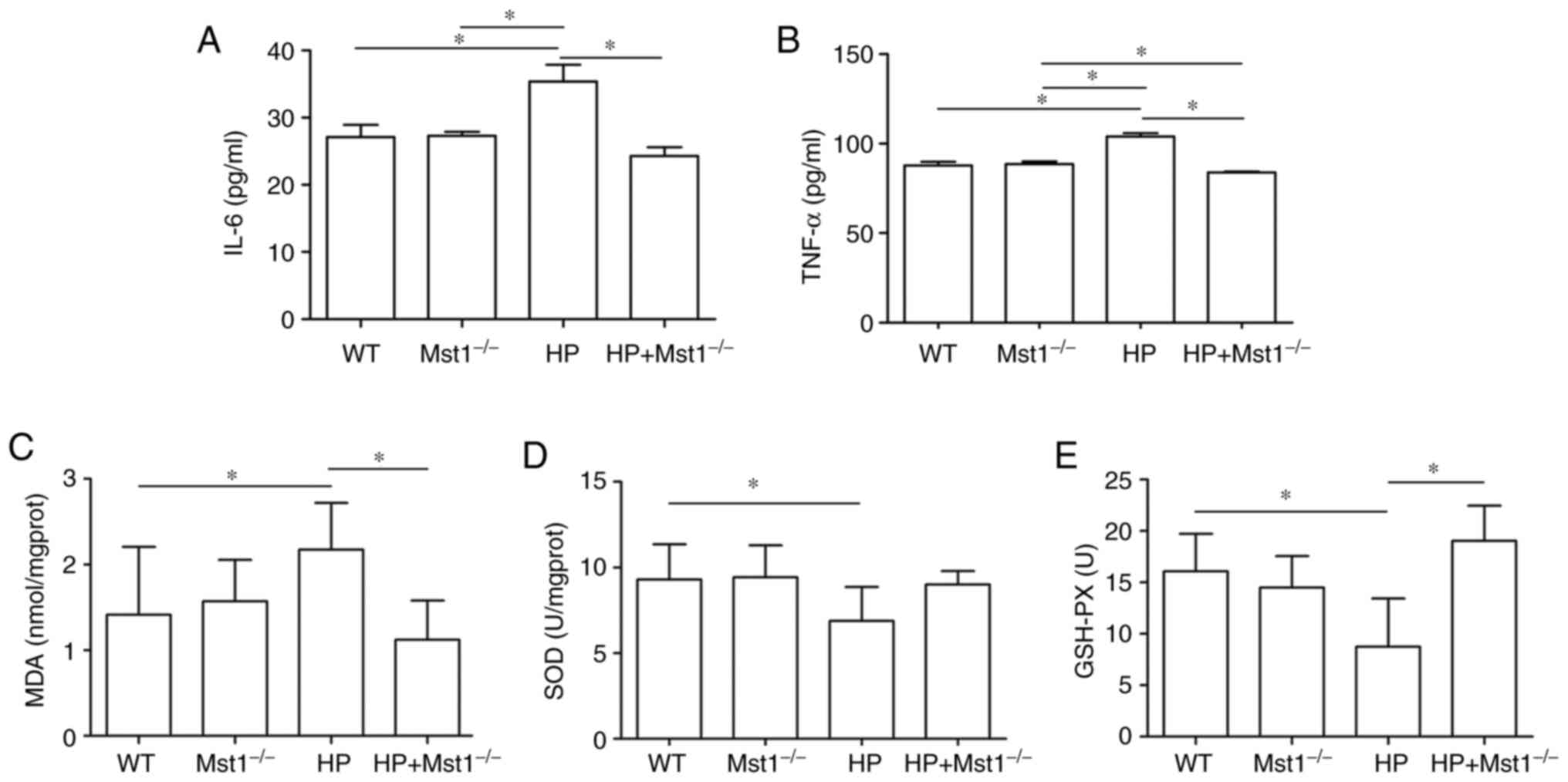
HP is often associated with the increased expression
of inflammatory factors in the heart, such as IL-6 and TNF-α
(24). Indeed, the present study
observed an increase in IL-6 (P<0.05) and TNF-α (P<0.05)
expression in the HP group. Both IL-6 and TNF-α expression levels
were decreased in Mst1−/− mice in the HP group (both
P<0.05, Fig. 2A and B),
indicating that the silencing of Mst1 expression can protect
against inflammation during HP myocardial injury. In addition, an
increase was found in oxidative stress, as evidenced by changes in
MDA, SOD and GSH-PX levels (all P<0.05, Fig. 2C-E), similar to the findings of a
previous study (25). Of note, a
significant increase in MDA, and a decrease in SOD and GSH-PX
levels were observed in the HP group (all P<0.05, Fig. 2C-E). Mst1 knockout in the HP group
markedly abrogated the HP-mediated induction of these oxidative
stress markers (all P<0.05, Fig.
2C-E).
Mst1 knockout decreases HP-induced
myocardial apoptosis
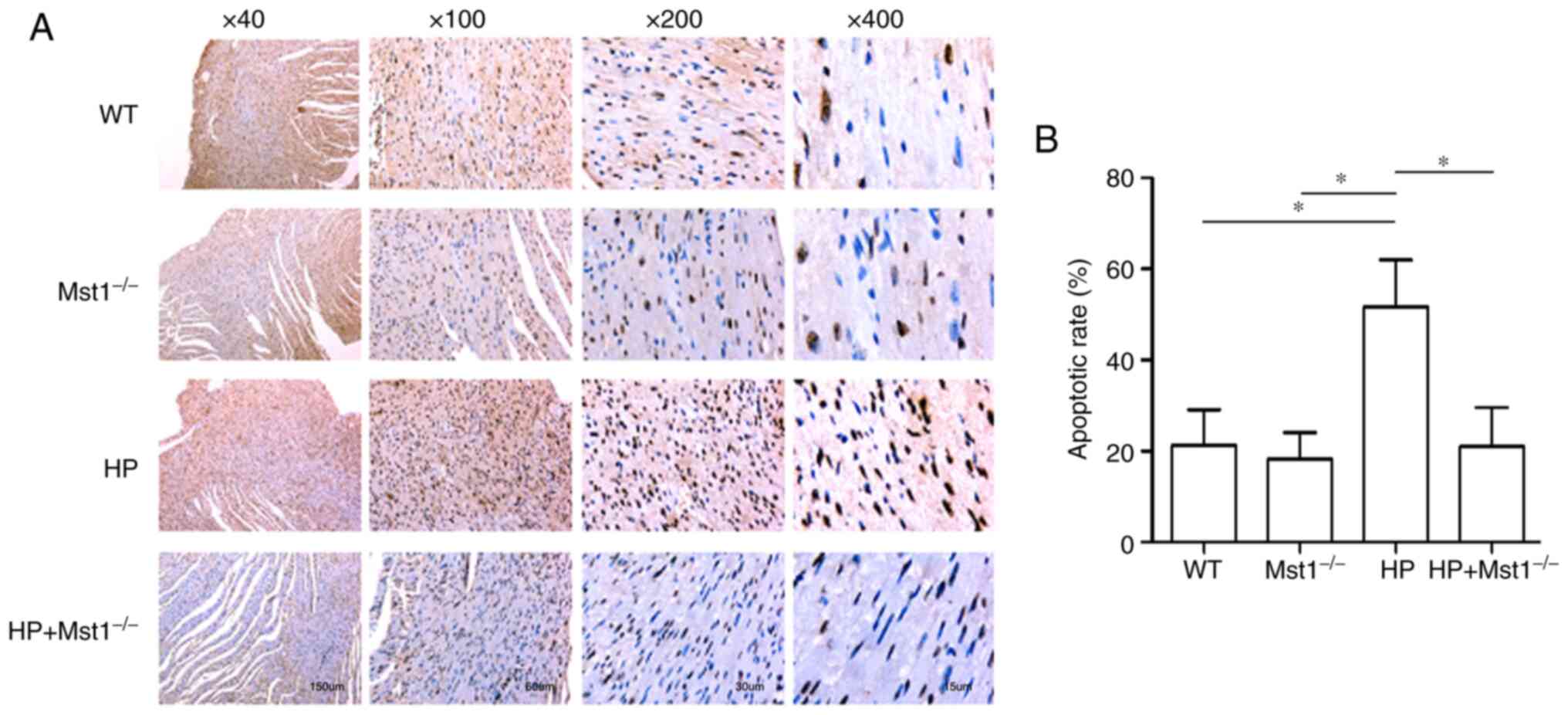
A key event in cardiac HP is the apoptosis of
cardiomyocytes and CMECs (10,12,26). In the present study, TUNEL
staining was used to compare apoptosis in myocardial sections from
the different groups. There was a marked increase in the apoptotic
ratio in the myocardial tissues in the HP group compared to the
non-HP groups (both P<0.05, Fig.
3). As was expected, Mst1 knockout abolished the HP-induced
increase in apoptosis in the HP group (both P<0.05, Fig. 3), further confirming the
beneficial roles of inhibiting Mst1 in cardiac HP. In addition, the
protein expression levels of apoptosis-related proteins were
measured. The expression of Bcl-2 was decreased, while the
expression levels of BAX, PARP-1, cleaved caspase-3 and cleaved
caspase-9 were increased in the HP group (all P<0.01, Fig 4). However, these changes induced by
HP were significantly attenuated in the Mst1−/− mice
(all P<0.01, Fig 4).
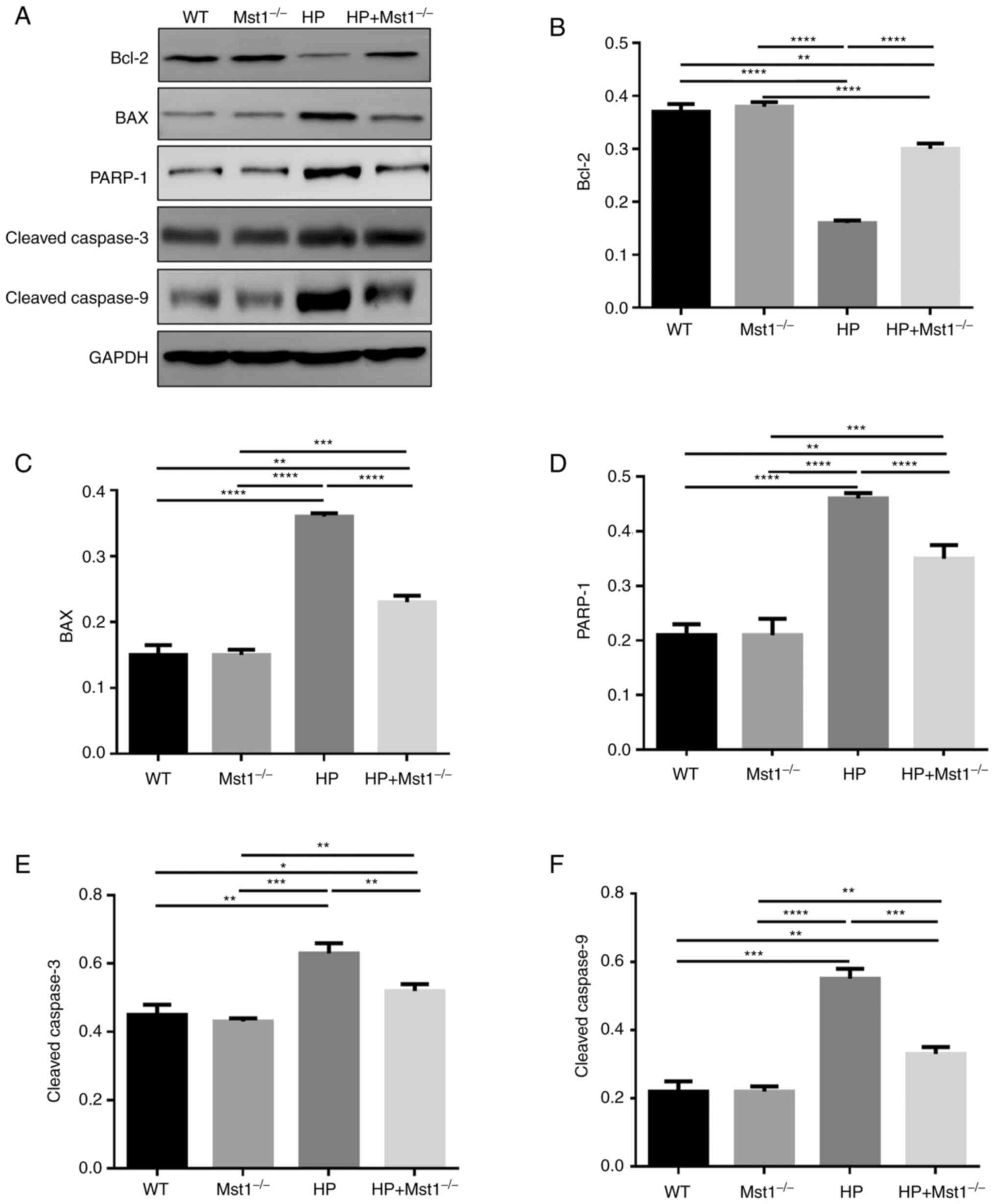 | Figure 4Expression of apoptosis-related
proteins. (A) representative western blots. Quantification of the
expression level of (B) Bcl-2, (C) BAX, (D) PARP-1, (E) cleaved
caspase-3, and (F) cleaved caspase-9 in WT, Mst1−/−, HP
and HP Mst1−/− mice. *P<0.05,
**P<0.01, ***P<0.001 and
****P<0.0001. WT, wild-type; Mst1, mammalian
ste20-like kinase 1; HP, hypertensive/hypertension. |
Mst1-deficient microvascular endothelial
cells exhibit an improved integrity and mitochondrial membrane
potential in HP models
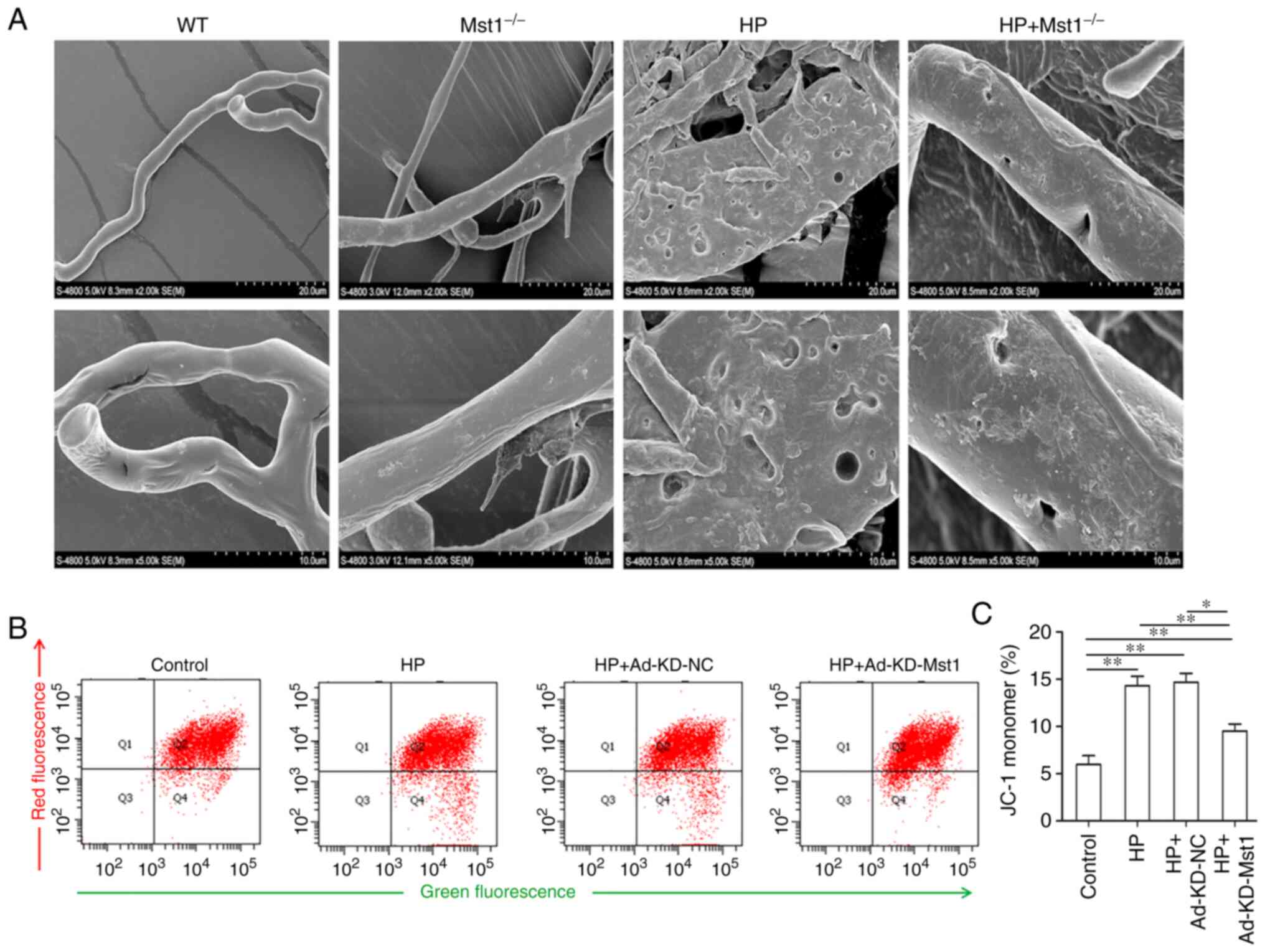
As microvascular endothelial cell apoptosis is
critical for endothelial integrity, and in turn for cardiac
microcirculation (26), the
present study hypothesized that the inhibition of Mst1 may improve
myocardial endothelial integrity in HP mice. The microvascular
corrosion casting results demonstrated that the microvascular
endothelial integrity was severely damaged in the HP group compared
to the non-HP groups, and Mst1 knockout efficiently ameliorated the
altered endothelial cell integrity (Fig. 5A), consistent with the function of
Mst1 in apoptosis. It was then hypothesized that Mst1 could also
improve mitochondrial function in HP-compromised myocardial
microvascular endothelial cells. It was found that Mst1 knockdown
attenuated the disproportion of mitochondrial membrane potential in
the HP endothelial cell model (P<0.05, Fig. 5B and C). To summarize, the
silencing of Mst1 improved microvascular endothelial cell functions
in the heart in HP.
Mst1 deficiency increases autophagy in HP
hearts and endothelial cells
The dysregulation of autophagy is frequently
associated with cardiac disease, and the balance between autophagy
and apoptosis has become a therapeutic target (8,9).
Using TEM, the present study found a decrease in the number of
autophagosomes in the HP CMECs compared to the control CMECs, and
Mst1 knockdown recovered the number of autophagosomes (Fig. 6A). Fluorescence microscopy
detected the expression of the key autophagy markers, p62 and LC3B,
in both in vivo myocardial tissue sections and in
vitro CMECs, providing additional evidence that Mst1 silencing
can reverse the inhibition of autophagy in cardiac HP in
vivo (all P<0.05, Fig.
6B-E). These findings suggest that modulating autophagy by
inhibiting Mst1 is a critical factor in treating HP injury.
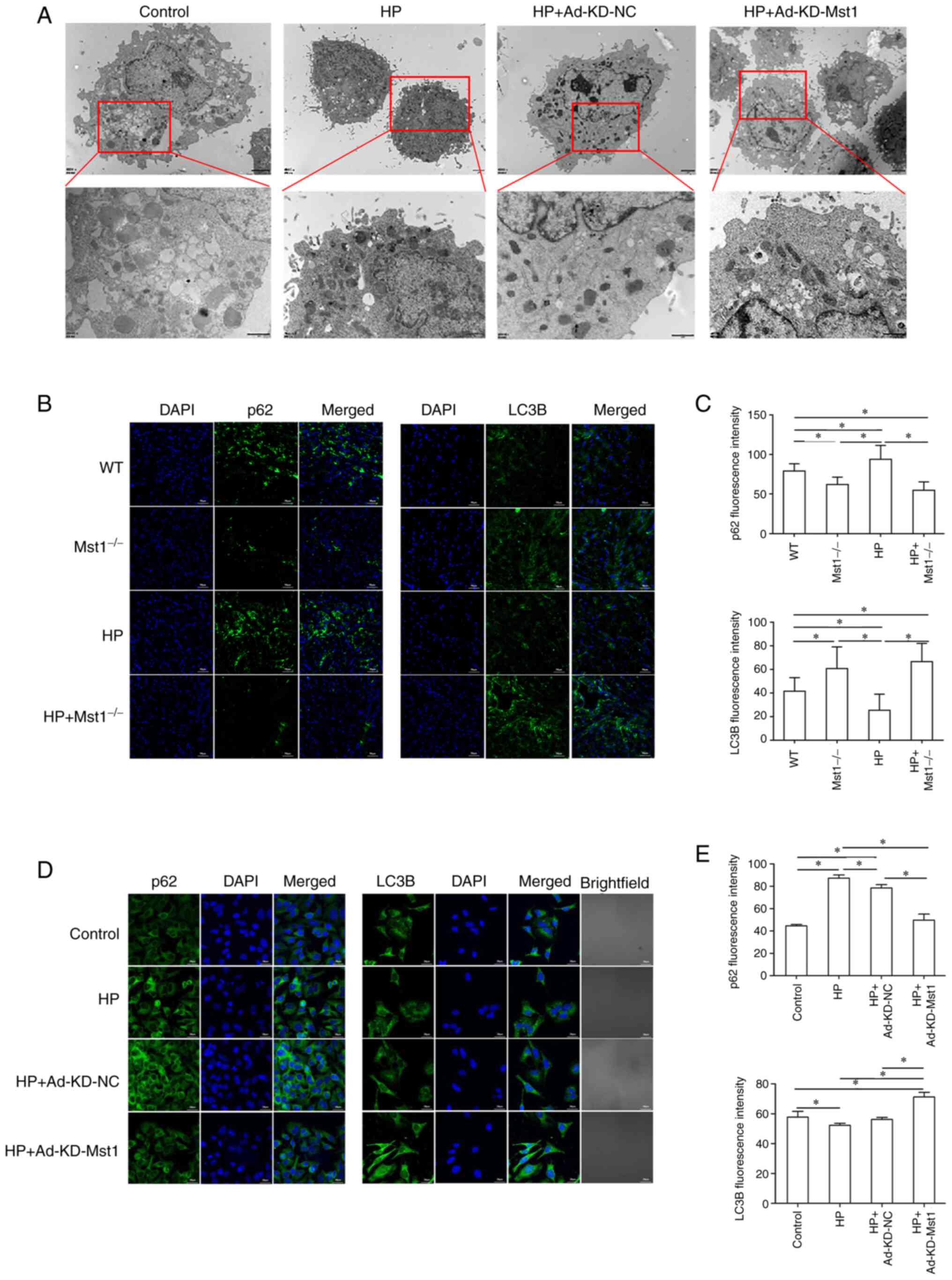 | Figure 6Increased autophagy in Mst1-deficient
endothelial cells. (A) TEM images of CMECs. Scale bar, 2 µm.
(B) Representative immunofluorescence images of p62 and LC3B in
mouse myocardium; magnification, ×200. (C) Quantitative assessment
of immunofluorescence images; n=3. (D) Representative
immunofluorescence images of p62 and LC3B in CMECs; magnification,
×200. (E) Quantitative assessment of immunofluorescence images;
n=3. *P<0.05. WT, wild-type; Mst1, mammalian
ste20-like kinase 1; HP, hypertensive/hypertension; LC3B,
microtubule-associated proteins 1A/1B light chain 3B; CMECs,
cardiac microvascular endothelial cells; KD, knockdown; NC,
negative control. |
Mst1 plays a critical role in modulating
autophagy in the HP heart
To verify the role of Mst1 in the HP heart, the
present study then examined the expression and activation of Mst1
and the autophagy markers, Beclin-1, LC3B and p62, in CMECs and
cardiac tissues using western blot analysis (all P<0.05,
Fig. 7). In both the in
vivo and in vitro HP models, it was observed that Mst1
deficiency, along with blocking its activation, corresponded with
the activation of autophagy, reflected by the increased expression
of Beclin-1, increased LC3B lipidation and decreased p62
accumulation (all P<0.05, Fig.
7). Together, these results suggest that Mst1 may be a master
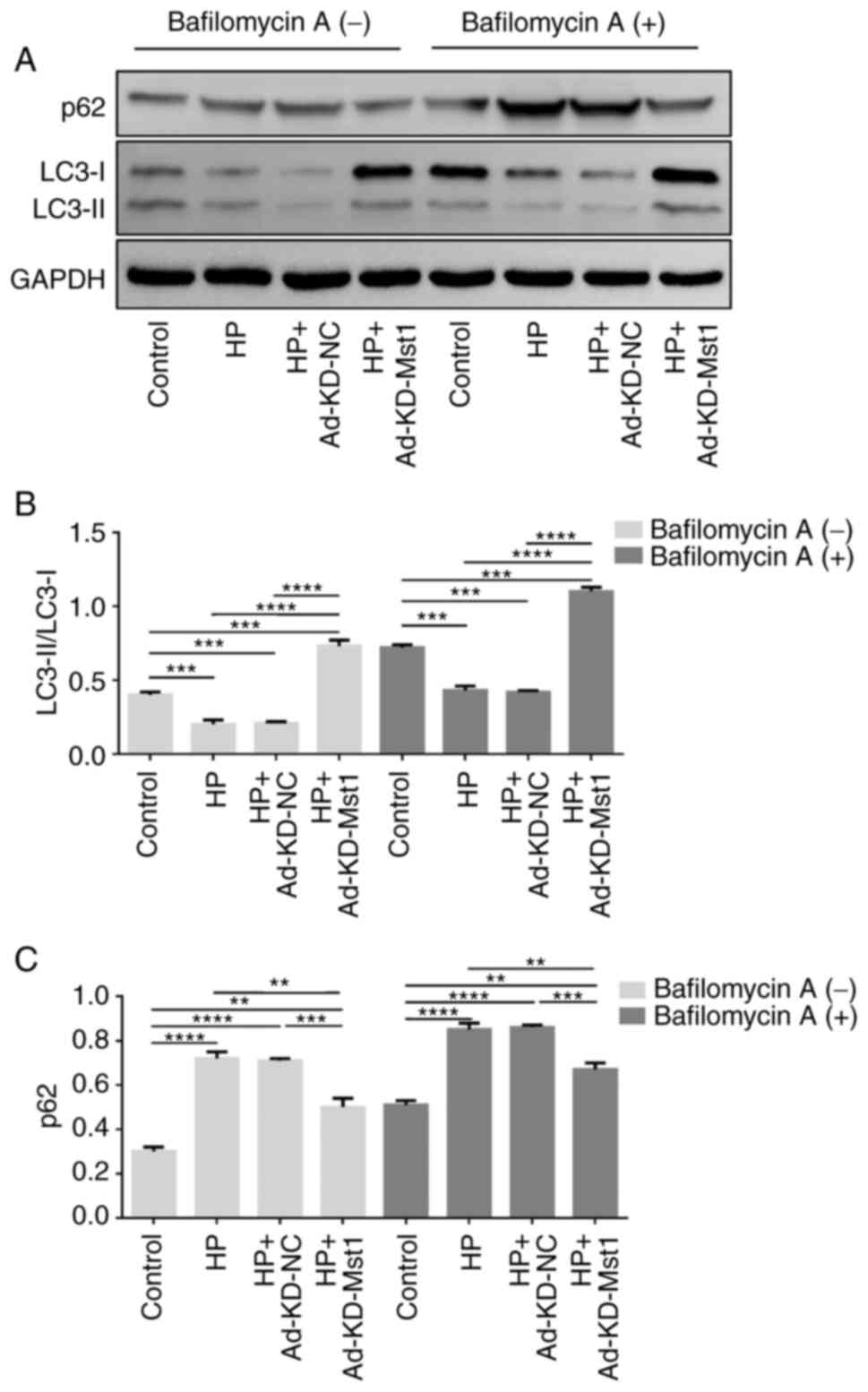
regulator of autophagy in mouse hearts in HP. Subsequently,
Cultured CMECs were treated with 50 nM bafilomycin A1 for 2 h to
determine the activity of the autophagic flux. Consistently, the
enhanced autophagic flux exerted by Mst1 deficiency was
demonstrated by an increased LC3 II/LC3 I ratio and a decreased p62
expression in the presence of bafilomycin A1 (Fig. 8).
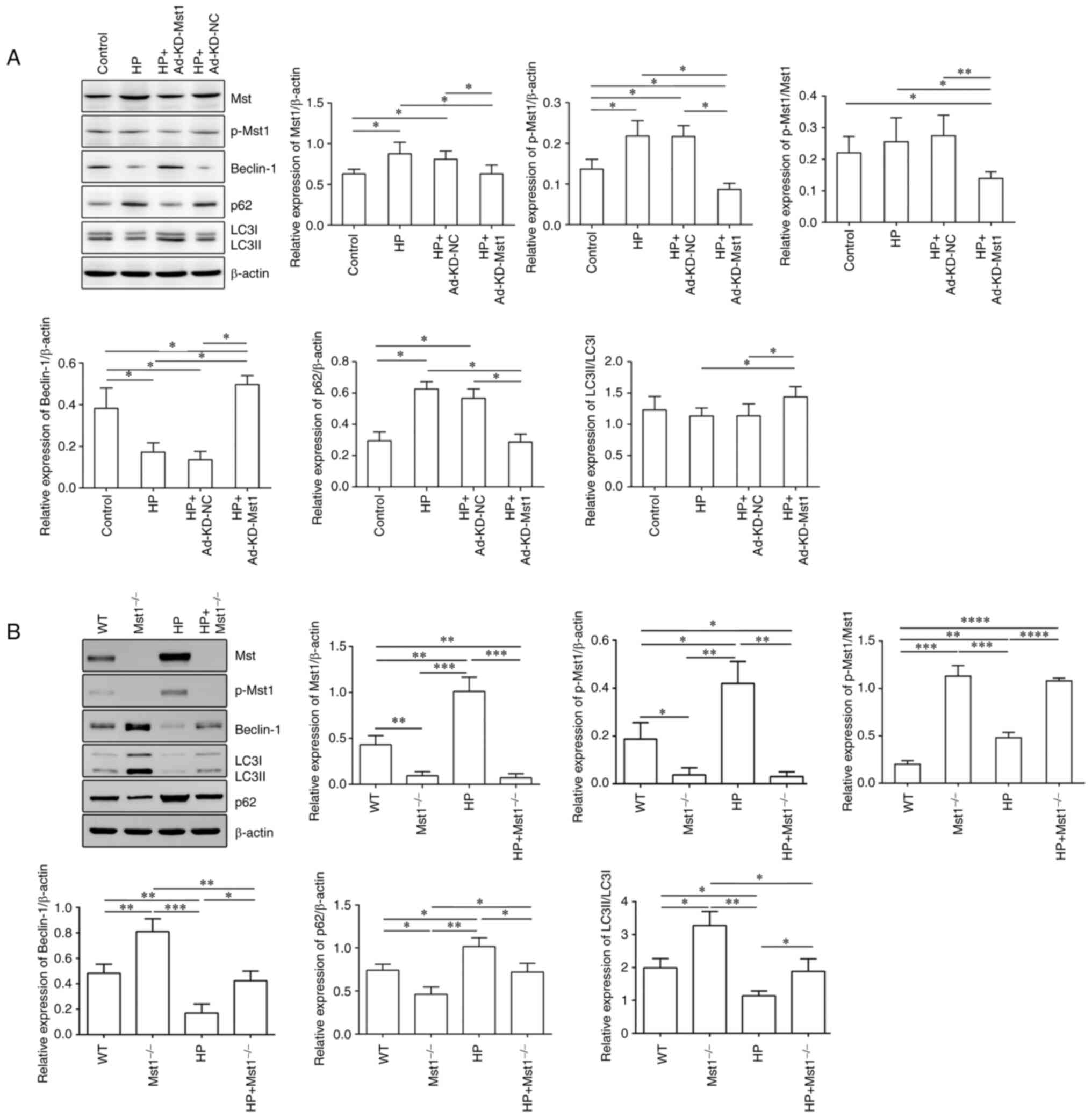 | Figure 7Modulation of core autophagy protein
expression by Mst1. (A) Representative western blots of Mst1,
p-Mst1, Beclin-1, p62, LC3B and β-actin proteins in CMECs and the
quantitative assessment of the western blots; n=4. (B)
Representative western blots of Mst1, p-Mst1, Beclin-1, p62, LC3B
and β-actin proteins in mouse myocardium and the quantitative
assessment of the western blots; n=3. *P<0.05,
**P<0.01, ***P<0.001 and
****P<0.0001. Mst1, mammalian ste20-like kinase 1;
LC3B, microtubule-associated proteins 1A/1B light chain 3B; CMECs,
cardiac microvascular endothelial cells; WT, wild-type; HP,
hypertensive/hypertension; KD, knockdown; NC, negative control. |
Discussion
Myocardial remodeling is a compensatory response to
HP myocardial injury and is indicated by cardiac hypertrophy,
fibrosis, inflammation, mitochondria injury, impaired autophagy and
an enhanced apoptosis (3). Severe
myocardial remodeling can even lead to arrhythmia and heart
failure. The present study demonstrated that Mst1, a protein kinase
activated during HP, is a key regulator of HP myocardial injury.
The silencing of Mst1 in vivo markedly ameliorated the
changes in cardiac structure and function, and reduced
inflammation, oxidative stress and the apoptosis of CMECs in the HP
mouse model. In addition, Mst1 knockdown in vitro restored
endothelial integrity, improved mitochondrial membrane potential
and activated autophagy, conferring protective effects in the HP
cell culture model.
Mst1 has been implicated as a key factor that
regulates the balance between apoptosis and autophagy in the
cardiovascular system (12,13,16,27,28). Generally, autophagy inhibits the
induction of apoptosis, and apoptosis blocks the autophagic process
(29). Impaired autophagy and
enhanced apoptosis are associated with HP heart disease (6). Therefore, Mst1 may play a role in HP
myocardial injury by regulating the balance between autophagy and
apoptosis. It has been reported that Mst1 plays a critical role in
regulating autophagy by phosphorylating LC3, a marker of autophagy
(30-32). In physiological settings, Mst1 is
a critical factor in the development of the cardiovascular system,
specifically as regards the modulation of cardiomyocyte growth and
apoptosis (27). Under
pathophysiological conditions, multiple stresses, such as oxidative
stress, ischemia and hypoxia, can activate Mst1. The persistent
activation of Mst1 has been shown to be associated with a variety
of cardiovascular diseases, including DCM, atherosclerosis and
ischemia/reperfusion injury (14,33,34). However, whether Mst1 plays a role
in myocardial injury induced by HP remains largely unknown.
In the present study, to investigate the role of
Mst1 in HP cardiomyopathy, a HP mouse model was constructed using
WT and Mst1−/− mice. The echocardiography results
revealed that Mst1 knockout attenuated the HP-induced impairment of
cardiac function as indicated by changes in LVFS and LVEF. H&E
and Masson's trichrome staining also revealed reduced cardiomyocyte
hypertrophy and fibrosis in the Mst1−/− mice compared
with the WT mice, indicating that the inhibition of Mst1 improves
cardiac function and remodeling in vivo. There is evidence
to indicate that Ang II-induced pathological cardiac hypertrophy is
associated with decreased autophagy (35-38). The findings in the present study
suggest that Mst1 may regulate autophagy and attenuate Ang
II-induced pathological cardiac hypertrophy in mice.
HP is often associated with the increased expression
of inflammatory factors and elevated oxidative stress in the heart
(26). The present study
demonstrated that in the HP mouse models, Mst1 knockout reduced the
expression of IL-6 and TNF-α compared with the WT group, suggesting
a protective effect of Mst1 knockout. In addition, less myocardial
oxidative stress was observed in the Mst1−/− mice, as
indicated by a reduction in MDA and GSH-PX compared with the WT
mice.
To further evaluate the function of Mst1 in HP,
myocardial apoptosis was measured in the Mst1−/− mice
and WT mice using TUNEL assay. As was expected, cardiomyocyte
apoptosis increased in the HP group, which was abolished by Mst1
knockout, supporting the protective role of Mst1 in HP-induced
cardiomyocyte apoptosis.
The present study also found that Mst1 knockout
significantly improved myocardial microvascular endothelial
integrity in HP mice, suggesting a role for Mst1 in endothelial
cell function, consistent with the findings of a previous study
(39). One factor that
contributes to HP is the apoptosis of microvascular endothelial
cells, as this directly leads to microcirculation reduction and
peripheral resistance increase, both of which result in elevated
blood pressure (10,40). Microcirculation impairment can
also cause organ injury due to a decrease in nutrient and oxygen
supply. Therefore, the perturbation of the microcirculation can
lead to a range of cardiovascular diseases, such as diabetic
cardiomyopathy, atherosclerosis, angina pectoris and
ischemia/reperfusion injury (26,41). A previous study found that high
glucose can phosphorylate and activate Mst1 in myocardial
microvascular endothelial cells, promote apoptosis, and inhibit
autophagy, resulting in diabetic cardiomyopathy (12).
Mitochondrial membrane potential reflects
mitochondrial function, and its dysregulation can initiate
apoptosis (42). The present
study assessed mitochondrial membrane potential in CMECs using JC-1
staining and observed that the knockdown of Mst1 in vitro
markedly reversed the Ang II-induced increase in membrane
potential, demonstrating a beneficial effect of inhibiting Mst1 on
mitochondrial health.
Physiological autophagy is a key mechanism in the
quality control of proteins and organelles and therefore, in cell
homeostasis (7,8). The dysregulation of autophagy under
conditions of stress is frequently implicated in heart diseases
(43). Using TEM, the present
study observed a marked increase in the number of autophagosomes in
the Mst1-deficient mouse myocardial microvascular endothelial cells
compared with the WT cells following the induction of HP.
Fluorescence microscopy and western blot analysis of the autophagy
markers, p62 and LC3B, further confirmed that the silencing of Mst1
increased autophagy in both the in vivo and in vitro
HP models.
In the heart, constitutive autophagy is not only a
homeostatic mechanism for maintaining cardiac structure and
function, but is also of utmost importance for the maintenance of
cardiac cellular integrity. However, the disruption of autophagy
may lead to heart failure (44).
The findings of the present study suggest that Mst1 plays roles in
the regulation of autophagy, apoptosis, oxidative stress and
inflammation in HP hearts. A possible mechanism involved is that
the loss of Mst1 primarily induces protective autophagy, which
subsequently inhibits apoptosis, oxidative stress and inflammatory
responses, leading to suppressed cardiomyocyte injury in HP. This
hypothesis warrants investigation in future studies. In summary,
Mst1 inhibition can considerably ameliorate HP-induced myocardial
injury which is associated with enhanced microvascular endothelial
cell autophagy and decreased apoptosis. Mst1−/− mice
were used in the present study, while further studies are required
in the future to determine whether the inhibition or downregulation
of Mst1 using pharmacological approaches prevents or improves
myocardial injury induced by HP. The promising beneficial effects
of Mst1 silencing may provide novel strategies for the treatment of
HP heart diseases.
Availability of data and materials
The datasets generated and analyzed during the
present study are available from the corresponding author upon
reasonable request.
Authors' contributions
LPW was involved in the conceptualization of the
study, and in the provision of data analysis and funding support.
RMH, BW and XX were involved in data curation and in the study
methodology. MYL, YHD, WW and CH were involved in the experiment
operation and data analysis. JL was involved in the
conceptualization of the study, as well as in data curation, formal
analysis, funding acquisition, the study methodology, project
administration, the provision of funding support, in the writing of
the original draft, and in the writing, reviewing and editing of
the manuscript. JL and LPW confirmed the authenticity of all the
raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was reviewed and approved by the
Ethics Committee of The First Affiliated Hospital, Xinjiang Medical
University (approval no. IACUC20201116-13). All procedures
performed involving animals were in accordance with the ethical
standards of the institution or practice at which the studies were
conducted.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the grants from the National
Natural Science Foundation of China (no. 81860048), the Tianshan
Youth Program from the Department of Science and Technology of
Xinjiang Uygur Autonomous Region (no. 2020Q038), and the Science
and Technology Support Projects from the Department of Science and
Technology of Xinjiang Uygur Autonomous Region (no. 2017E0268).
Abbreviations:
|
Ang II
|
angiotensin II
|
|
CMEC
|
cardiac microvascular endothelial
cell
|
|
DCM
|
dilated cardiomyopathy
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
|
GSH-PX
|
glutathione peroxidase
|
|
H&E
|
hematoxylin and eosin
|
|
HP
|
hypertensive/hypertension
|
|
IL-6
|
interleukin-6
|
|
LC3B
|
microtubule-associated proteins 1A/1B
light chain 3B
|
|
LVEDD
|
left ventricular end-diastolic
diameter
|
|
LVESD
|
left ventricular end-systolic
diameter
|
|
LVEF
|
left ventricular ejection fraction
|
|
LVFS
|
left ventricular fractional
shortening
|
|
MDA
|
malondialdehyde
|
|
Mst1
|
mammalian ste20-like kinase 1
|
|
SEM
|
scanning electron microscopy
|
|
SOD
|
superoxide dismutase
|
|
TEM
|
transmission electron microscopy
|
|
TNF-α
|
tumor necrosis factor-α
|
|
TUNEL
|
terminal deoxynucleotidyl transferase
dUTP nick-end labeling
|
References
|
1
|
Oparil S, Acelajado MC, Bakris GL,
Berlowitz DR, Cífková R, Dominiczak AF, Grassi G, Jordan J, Poulter
NR, Rodgers A and Whelton PK: Hypertension. Nat Rev Dis Primers.
4:180142018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McCormack T, Krause T and O'Flynn N:
Management of hypertension in adults in primary care: NICE
guideline. Br J Gen Pract. 62:163–164. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gonzalez A, Ravassa S, Lopez B, Moreno MU,
Beaumont J, José GS, Querejeta R, Bayés-Genís A and Díez J:
Myocardial remodeling in hypertension. Hypertension. 72:549–558.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gobé G, Browning J, Howard T, Hogg N,
Winterford C and Cross R: Apoptosis occurs in endothelial cells
during hypertension-induced microvascular rarefaction. J Struct
Biol. 118:63–72. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
White K, Dempsie Y, Caruso P, Wallace E,
McDonald RA, Stevens H, Hatley ME, Rooij EV, Morrell NW, MacLean MR
and Baker AH: Endothelial apoptosis in pulmonary hypertension is
controlled by a microRNA/programmed cell death 4/caspase-3 axis.
Hypertension. 64:185–194. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang ZV, Rothermel BA and Hill JA:
Autophagy in hypertensive heart disease. J Biol Chem.
285:8509–8514. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sciarretta S, Maejima Y, Zablocki D and
Sadoshima J: The role of autophagy in the heart. Annu Rev Physiol.
80:1–26. 2018. View Article : Google Scholar
|
|
8
|
Nemchenko A, Chiong M, Turer A, Lavandero
S and Hill JA: Autophagy as a therapeutic target in cardiovascular
disease. J Mol Cell Cardiol. 51:584–593. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang F, Jia J and Rodrigues B: Autophagy,
metabolic disease, and pathogenesis of heart dysfunction. Can J
Cardiol. 33:850–859. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Savoia C, Battistoni A, Calvez V, Cesario
V, Montefusco G and Filippini A: Microvascular alterations in
hypertension and vascular aging. Curr Hypertens Rev. 13:16–23.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu B, Lin J, Luo J, Han D, Fan M, Guo T,
Tao L, Yuan M and Yi F: Dihydromyricetin protects against diabetic
cardiomyopathy in streptozotocin-induced diabetic mice. Biomed Res
Int. 2017:37643702017.PubMed/NCBI
|
|
12
|
Zhang M, Zhang L, Hu J, Lin J, Wang T,
Duan Y, Man W, Feng J, Sun L, Jia H, et al: MST1 coordinately
regulates autophagy and apoptosis in diabetic cardiomyopathy in
mice. Diabetologia. 59:2435–2447. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maejima Y, Kyoi S, Zhai P, Liu T, Li H,
Ivessa A, Sciarretta S, Re DPD, Zablocki DK, Hsu CP, et al: Mst1
inhibits autophagy by promoting the interaction between Beclin1 and
Bcl-2. Nat Med. 19:1478–1488. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yamamoto S, Yang G, Zablocki D, Liu J,
Hong C, Kim SJ, Soler S, Odashima M, Thaisz J, Yehia G, et al:
Activation of Mst1 causes dilated cardiomyopathy by stimulating
apoptosis without compensatory ventricular myocyte hypertrophy. J
Clin Invest. 111:1463–1474. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Foulquier S, Namsolleck P, Van Hagen BT,
Milanova I, Post MJ, Blankesteijn WM, Rutten BP, Prickaerts J, Van
Oostenbrugge RJ and Unger T: Hypertension-induced cognitive
impairment: Insights from prolonged angiotensin II infusion in
mice. Hypertens Res. 41:817–827. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cheng Z, Zhang M, Hu J, Lin J, Feng X,
Wang S, Wang T, Gao E, Wang H and Sun D: Mst1 knockout enhances
cardiomyocyte autophagic flux to alleviate angiotensin II-induced
cardiac injury independent of angiotensin II receptors. J Mol Cell
Cardiol. 125:117–128. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Froese N, Kattih B, Breitbart A, Grund A,
Geffers R, Molkentin JD, Kispert A, Wollert KC, Drexler H and
Heineke J: GATA6 promotes angiogenic function and survival in
endothelial cells by suppression of autocrine transforming growth
factor beta/activin receptor-like kinase 5 signaling. J Biol Chem.
286:5680–5690. 2011. View Article : Google Scholar
|
|
18
|
Lu Y, Wang RH, Guo BB and Jia YP:
Quercetin inhibits angiotensin II induced apoptosis via
mitochondrial pathway in human umbilical vein endothelial cells.
Eur Rev Med Pharmacol Sci. 20:1609–1616. 2016.PubMed/NCBI
|
|
19
|
He TC, Zhou S, da Costa LT, Yu J, Kinzler
KW and Vogelstein B: A simplified system for generating recombinant
adenoviruses. Proc Natl Acad Sci USA. 95:2509–2514. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Van De Vlekkert D, Machado E and d'Azzo A:
Analysis of generalized fibrosis in mouse tissue sections with
Masson 's Trichrome staining. Bio Protoc. 10:e36292020. View Article : Google Scholar
|
|
21
|
Van Steenkiste C, Trachet B, Casteleyn C,
van Loo D, Hoorebeke LV, Segers P, Geerts A, Vlierberghe HV and
Colle I: Vascular corrosion casting: Analyzing wall shear stress in
the portal vein and vascular abnormalities in portal hypertensive
and cirrhotic rodents. Lab Invest. 90:1558–1572. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
De Biasi S, Gibellini L and Cossarizza A:
Uncompensated polychromatic analysis of mitochondrial membrane
potential using JC-1 and multilaser excitation. Curr Protoc Cytom.
72:7.32.1–7.32.11. 2015.
|
|
23
|
Arai R and Waguri S: Improved electron
microscopy fixation methods for tracking autophagy-associated
membranes in cultured mammalian cells. Methods Mol Biol.
1880:211–221. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tomiyama H, Shiina K, Matsumoto-Nakano C,
Ninomiya T, Komatsu S, Kimura K, Chikamori T and Yamashina A: The
contribution of inflammation to the development of hypertension
mediated by increased arterial stiffness. J Am Heart Assoc.
6:e0057292017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kuyumcu F and Aycan A: Evaluation of
oxidative stress levels and antioxidant enzyme activities in burst
fractures. Med Sci Monit. 24:225–234. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Suematsu M, Suzuki H, Delano FA and
Schmid-Schonbein GW: The inflammatory aspect of the
microcirculation in hypertension: Oxidative stress,
leukocytes/endothelial interaction, apoptosis. Microcirculation.
9:259–276. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang Y, Wang H, Ma Z, Hu W and Sun D:
Understanding the role of mammalian sterile 20-like kinase 1 (MST1)
in cardiovascular disorders. J Mol Cell Cardiol. 114:141–149. 2018.
View Article : Google Scholar
|
|
28
|
Feng X, Wang S, Yang X, Lin J, Man W, Dong
Y, Zhang Y, Zhao Z, Wang H and Sun D: Mst1 knockout alleviates
mitochondrial fission and mitigates left ventricular remodeling in
the development of diabetic cardiomyopathy. Front Cell Dev Biol.
8:6288422020. View Article : Google Scholar
|
|
29
|
Marino G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wilkinson DS, Jariwala JS, Anderson E,
Mitra K, Meisenhelder J, Chang JT, Ideker T, Hunter T, Nizet V,
Dillin A and Hansen M: Phosphorylation of LC3 by the Hippo kinases
STK3/STK4 is essential for autophagy. Mol Cell. 57:55–68. 2015.
View Article : Google Scholar :
|
|
31
|
Nieto-Torres JL, Shanahan SL, Chassefeyre
R, Chaiamarit T, Zaretski S, Landeras-Bueno S, Verhelle A, Encalada
SE and Hansen M: LC3B phosphorylation regulates FYCO1 binding and
directional transport of autophagosomes. Curr Biol. 31:3440–3449
e3447. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nieto-Torres JL, Encalada SE and Hansen M:
LC3B phosphorylation: Autophagosome's ticket for a ride toward the
cell nucleus. Autophagy. 17:3266–3268. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu W, Xu M, Zhang T, Zhang Q and Zou C:
Mst1 promotes cardiac ischemia-reperfusion injury by inhibiting the
ERK-CREB pathway and repressing FUNDC1-mediated mitophagy. J
Physiol Sci. 69:113–127. 2019. View Article : Google Scholar
|
|
34
|
Wang T, Zhang L, Hu J, Duan Y, Zhang M,
Lin J, Man W, Pan X, Jiang Z, Zhang G, et al: Mst1 participates in
the atherosclerosis progression through macrophage autophagy
inhibition and macrophage apoptosis enhancement. J Mol Cell
Cardiol. 98:108–116. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mehta PK and Griendling KK: Angiotensin II
cell signaling: Physiological and pathological effects in the
cardiovascular system. Am J Physiol Cell Physiol. 292:C82–C97.
2007. View Article : Google Scholar
|
|
36
|
Li AL, Lv JB and Gao L: MiR-181a mediates
Ang II-induced myocardial hypertrophy by mediating autophagy. Eur
Rev Med Pharmacol Sci. 21:5462–5470. 2017.PubMed/NCBI
|
|
37
|
Kishore R, Krishnamurthy P, Garikipati VN,
Benedict C, Nickoloff E, Khan M, Johnson J, Gumpert AM, Koch WJ and
Verma SK: Interleukin-10 inhibits chronic angiotensin II-induced
pathological autophagy. J Mol Cell Cardiol. 89:203–213. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou L, Ma B and Han X: The role of
autophagy in angiotensin II-induced pathological cardiac
hypertrophy. J Mol Endocrinol. 57:R143–R152. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Qin R, Lin D, Zhang L, Xiao F and Guo L:
Mst1 deletion reduces hyperglycemia-mediated vascular dysfunction
via attenuating mitochondrial fission and modulating the JNK
signaling pathway. J Cell Physiol. 235:294–303. 2020. View Article : Google Scholar
|
|
40
|
Dong Q, Xing W, Su F, Liang X, Tian F, Gao
F, Wang S and Zhang H: Tetrahydroxystilbene glycoside improves
microvascular endothelial dysfunction and ameliorates
obesity-associated hypertension in obese ZDF rats via inhibition of
endothelial autophagy. Cell Physiol Biochem. 43:293–307. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang B, Li BW, Li HW, Li AL, Yuan XC, Wang
Q and Xiu RJ: Enhanced matrix metalloproteinases-2 activates aortic
endothelial hypermeability, apoptosis and vascular rarefaction in
spontaneously hypertensive rat. Clin Hemorheol Microcirc.
57:325–338. 2014. View Article : Google Scholar
|
|
42
|
Zorova LD, Popkov VA, Plotnikov EY,
Silachev DN, Pevzner IB, Jankauskas SS, Babenko VA, Zorov SD,
Balakireva AV, Juhaszova M, et al: Mitochondrial membrane
potential. Anal Biochem. 552:50–59. 2018. View Article : Google Scholar :
|
|
43
|
Yan L, Vatner DE, Kim SJ, Ge H, Masurekar
M, Massover WH, Yang G, Matsui Y, Sadoshima J and Vatner SF:
Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci
USA. 102:13807–13812. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nah J, Fernandez AF, Kitsis RN, Levine B
and Sadoshima J: Does autophagy mediate cardiac myocyte death
during stress? Circ Res. 119:893–895. 2016. View Article : Google Scholar : PubMed/NCBI
|