Introduction
The C-X-C chemokine receptor type 4 (CXCR4) is a
Gαi protein-coupled receptor, and its only identified
ligand is the CXC chemokine stromal-derived factor-1 (SDF-1). The
SDF-1/CXCR4 axis is essential for organogenesis during embryonic
development, including hematopoiesis, cardiac septum formation, and
neuronal development; genetic deletion of CXCR4 or SDF-1 leads to
almost identical developmental defects and embryonic lethality
(1-3). In the adult animals, previous
evidence has shown that SDF-1/CXCR4 axis plays an indispensable
role in the maintenance of stem/progenitor cells in the bone marrow
niche, recruitment of stem/progenitor cells to sites of injury,
ischemic neovascularization and tissue repair (4-10).
These findings have led to the notion of targeting SDF-1/CXCR4 axis
for therapy of ischemic heart disease (10-14).
The role of CXCR4 signaling in cardiomyocyte
biology, however, is not well understood. Recent evidence suggested
that in the developing heart, SDF-1/CXCR4 signaling is crucial not
only for epicardia-guided coronary vessel formation but also for
the expansion of the second heart field (6,15).
In the adult, CXCR4 is expressed in cardiomyocytes across mice,
rats and humans (16). Notably,
SDF-1 treatment of isolated mouse papillary muscles or rat
cardiomyocytes attenuates β-adrenergic agonist Isoproterenol
(Iso)-induced calcium mobilization and contractility (17), and cardiac CXCR4 knockout mice
develop progressive cardiomyopathy (18). Importantly, CXCR4 is expressed at
high levels in the cardiomyocytes of patients with end-stage heart
failure (19), the first cuase of
death in the developed countries.
Heart failure is featured by reduced cardiac pump
function unable to maintain the systemic demand of blood supply,
for which the impaired β-adrenergic receptor (β-AR) signaling plays
a central role (20). β-ARs,
predominantly β1 (80%) and β2 (20%) ARs in a
normal mammalian heart, primarily couple Gαs, although
β2 has been shown to also couple Gαi. Upon
binding to ligands (catecholamine hormone epinephrine and
neurotransmitter norepinephrine) or agonist (isoproterenol), β-ARs
activate Gαs GTPase, leading to adenylyl cyclase
activation, cAMP production, and signaling events including protein
kinase A (PKA) activation and phosphorylation (activation) of
proteins involved in cellular calcium mobilization and contraction
(21). While acute activation of
β-ARs increase heart rate (chronotrophy) and contractility
(inotrophy) to augment cardiac performance, over or chronic
activation results in cardiomyocyte death, adverse cardiac
remodeling, and progressive deterioration of function (22-26). Thus, β blockers are one of most
widely used treatment and have been shown to increase the long-term
survival of patients with chronic heart failure (27,28). Furthermore, evidence over last
three decades suggested that β1 AR not β2 AR
plays a causal role in cardiomyocyte apoptosis and heart failure
(26,29-31). Mice lacking β1 and
β2 ARs or overexpressing β2 ARs have
essentially normal basal cardiac function (32,33), whereas mice overexpressing
β1 AR, Gαs, or active-PKA display a similar cardiac
phenotype, including initial increased responsiveness to
catecholamines and hypertrophy followed by cardiomyocyte death,
fibrosis, cardiac dilation and failure (34-36).
In the present study, the role of cardiomyocyte
CXCR4 in Iso-induced heart failure in cardiomyocyte-specific CXCR4
knockout (CMKO) mice was investigated. It was found that
these mice display normal heart structure and function in the basal
state but are more susceptibility to Iso-induced cardiomyocyte
apoptosis, fibrosis and heart failure, and that SDF-1/CXCR4
signaling attenuates Iso-induced cardiomyocyte death in a
Gαi dependent manner.
Materials and methods
Mice
Male, 8-10 weeks old mice on C57BL/6J background
with body weights between 25 and 27 g were used.
CXCR4fl/fl mice and α-MHC-Cre mice were obtained from
Jax lab and bred to generate cardiomyocyte-specific CXCR4 knockout
(MHC-Cre+; CXCR4f/f or CMKO) mice.
All animal experiments in the present study were approved (IACUC
approval no. 3005) by the Animal Care and Use Committee of Huazhong
University of Science and Technology (Wuhan, China) and performed
in compliance with the 'Guide for the Care and Use of Laboratory
Animals' (NIH publication) and relevant ethical regulations for
animal testing and research. Animal numbers for the experiments
were pre-calculated based on the reported similar studies from the
authors and other laboratories with 80% power (37,38). The basic characteristics of our
model animals were reported, and physiological and biochemical
measurements were performed in double-blinded fashion. After Iso or
Saline minipump implantation, the animal health and behaviour were
monitored daily for the first 3 days, then every other day until
they were euthanized at day 14. Over the course of the study, no
experimental animal experienced conditions needed for euthanasia
before the end of experiments. For euthanasia, the mice were placed
in a designated chamber with continuous introduction of 100%
CO2 at a fill rate of 50% of the chamber volume per min
and for a duration of 15 min; the death was verified by loss of
consciousness, head sinking, and disappearance of muscle tone. Then
the mice were subjected to a secondary method, either vital organ
removal (for 30 mice used in histological analyses) or cervical
dislocation (for 30 mice used in echocardiographic analyses), to
ensure death. For colony maintenance, mice were fed ad
libitum and maintained at 22-24°C ambient temperature and
40-60% humidity under a 12:12-h light: dark cycle.
Chemicals and bioagents
All chemicals and bioagents used in the present
study, including Iso, AMD3100, Gi inhibitor Pertussis toxin (all
from Sigma-Aldrich; Merck KGaA), SDF-1 (R&D Systems, Inc.) were
purchased from commercial sources and authenticated by the
providers. Their concentrations, Iso (100 µM), SDF-1 (500
ng/ml), and AMD3100 (100 ng/ml), used on H9C2 cells were based on
pilot studies by the authors and consistent with a similar study
previously reported (39).
Continuous Iso infusion model
CMKO and Cre mice received Iso (45
mg/kg/day) or saline (control) continuously via Alzet mini-osmotic
pumps (model 2004; Durect Corporation) for up to 28 days. Pumps
were prepared and subcutaneously implanted in the mice by following
the manufacturer's instructions. The surgery was performed under
anesthesia by intraperitoneal injection of sterile pentobarbital
sodium (50 mg/kg body weight, Sigma-Aldrich; Merck KGaA) and
post-operative care was performed by following our approved animal
study protocol. For pain management, Metacam was subcutaneously
injected (1 mg/kg) at the end of surgery and continued twice daily
for 2 days. The heart function was monitored via echocardiography
at day 0, 3, 7, and 14. Then the mice were euthanized, and the
cardiac tissues were harvested for histological analyses.
Echocardiography
Cardiac function was recorded and analyzed using
Vevo 770 high-resolution ultrasound system (VisualSonics, Inc.) as
previously described (10). Mice
were lightly anesthetized via 2% isoflurane inhalation and secured
with surgical tapes to the platform in supine position. Heart rates
were recorded. Data were collected after three days of training.
Parasternal long-axis view, short-axis view at the papillary muscle
level and 2-D guided M-mode images were recorded. Left ventricular
ejection fraction, internal diameter and anterior wall thickness
during systole and diastole were measured, and the measurements
were performed by an individual who was blinded to the treatment
assignments.
Histology
Both formalin-fixed paraffin-embedded (FFPE) and
fixed frozen (FF) sections were used in the present study. For FFPE
sections, cardiac tissues were fixed in 10% formalin overnight,
embedded in paraffin, and cut at 5 µm in thickness as
previously described (40). For
FF sections, tissues were fixed in 4% paraformaldehyde for 4 h,
dehydrated in 30% sucrose and embedded in OCT compound followed by
cryosectioning into 8 µm in thickness. A total of 3 sections
per heart and 6 fields per section were examined. The H&E
staining (Thermo Fisher Scientific, Inc.) was performed by
following standard procedures. The Sirius Red/Fast Green (Chondrex)
staining were performed by following manufacturer's instructions.
For TUNEL staining, FF sections or cells growing on slides were
fixed in 4% paraformaldehyde for 30 min, then permeabilized with
proteinase K solution (20 µg/ml; for tissue) or 0.2% Triton
X-100 (for cells) for 5 min at room temperature. After wash with
PBS, the sections were stained with TUNEL reagent (Sigma-Aldrich;
Merck KGaA) for 60 min at 37°C. After wash, the nuclei were counter
stained with DAPI for 7 min at room temperature. For each section,
4-6 fields were examined under fluorescent microscope. To analyze
tissue CXCR4 expression, anti-CXCR4 antibody (Abcam, cat. no.
ab124824; 1:200) was used in the histochemical staining, as
previously reported by the authors (9). The histological assessments and
analyses were performed by an individual blinded to treatment
assignments.
Cells
H9C2, a well characterized and widely used
cardiomyocyte cell line that was originally derived from rat
embryonic heart tissue, was used as the in vitro cardiomyocyte
model (41). The cells were
purchased from the American Type Culture Collection (cat. no.
CRL-1446), maintained in DMEM with 10% FBS, and used until passage
8.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl
tetrazolium bromide (MTT) assay
The MTT assay was performed to evaluate the
viability of H9C2 cells after treatment with test reagents in
96-well plates by using CellTiter 96 Nonradioactive Cell
Proliferation Assay kit (Promega Corporation). First, 15 µl
of dye solution was added to each well, and the plate was returned
to incubator for 4 h at 37°C. Then, 100 µl of
Solubilization/Stop Solution was added to each well. The colored
formazan product is stable at 4°C, and absorbance was recorded at
570 nm using a 96-well plate reader.
Lenti-CXCR4 shRNA
The lentiviral vector that carries CXCR4 shRNA or
non-targeting (NT) shRNA was obtained from Sigma-Aldrich; Merck
KGaA with the knockdown specificity and efficiency authenticated by
the company. The CXCR4-shRNA base sequence was 5′-GGA TCA GCA TCG
ATT CCT TCA-3′ and the NT-shRNA base sequence was 5′-TTC TCC GAA
CGT GTC ACG T-3′. The vectors were packaged following the
manufacturers' instructions. The multiplicity of infection was 2
for application on H9C2 cells, and the transduced cells were
selected in puromycin for 10 days before evaluation for CXCR4
expression and myocyte functions.
Reverse transcription-quantitative (RT-q)
PCR
RT-qPCR was performed via standard techniques as
previously described by the authors (9). Briefly, total RNA was extracted with
RNA Stat-60 (Tel-Test, Inc.), and RNA was reverse transcribed with
the Taqman Multiscribe RT kit (Applied Biosystems; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
qPCR was performed in duplicate with cDNA from 10 ng of RNA by
using the Lightcycler hybridization Probes Master Mix [Roche
Diagnostics (Shanghai) Co., Ltd.]; a negative control (lacking a
template) was included for each probe set. Relative gene expression
was calculated using the 2−ΔΔCq method (42) and normalized to GAPDH. The primer
sequences were as follows: mouse CXCR4 forward, 5′-CCT CGC CTT CTT
CCA CTG TT-3′ and reverse, 5′-CTG GGC AGA GCT TTT GAA CTT G-3′; and
mouse GAPDH forward, 5′-GGG TCC CAG CTT AGG TTC ATC-3′ and reverse,
5′-TAC GGC CAA ATC CGT TCA CA-3′.
Western blot analysis
Western blotting was performed via standard
techniques as previously described by the authors (43), using primary antibodies against
phosphorylated (p)-Akt, total Akt, β-actin and relevant horseradish
peroxidase (HRP)-linked secondary antibodies (all from Cell
Signaling Technology, Inc.). Briefly, the membrane was first
blotted with anti-p-Akt (cat. no. 9271; 1:1,000) at 4°C for
overnight, then with anti-rabbit IgG (cat. no. 7074; 1:5,000)
before application of chemiluminescence detection reagents and
image taking. Next, the membrane was placed in the stripping
solution [25 mM glycine-HCl, pH 2, 1% (w/v) SDS] with agitation for
30 min at room temperature, followed by the second round of
immunoblotting with the use of anti-total-Akt (cat. no. 2920;
1:1,000) and anti-Mouse IgG (cat. no. 7076; 1:5,000). β-actin was
immunoblotted in some experiments as loading control by using
anti-β-actin (cat. no. 4967; 1:1,000) and then anti-rabbit IgG
(cat. no. 7074; 1:5,000).
Statistical analysis
All values are presented as the mean ± SEM. Unpaired
two-tailed Student's t-test was used for comparison between two
means. One-way or two-way analysis of variance (ANOVA), followed by
Bonferroni post-hoc tests, were used in multiple (>2) group
comparisons with one or two independent variables, respectively.
GraphPad Prism 8 software (GraphPad Software, Inc.) was used for
statistical analysis. P<0.05 was considered to indicate a
statistically significant difference.
Results
CXCR4 is expressed in the adult
cardiomyocytes
The present investigation was initiated by assessing
the expression of CXCR4 in wild-type mouse hearts.
Immunohistological staining revealed that CXCR4 protein is
expressed in the cardiomyocytes (Fig. S1) in the pattern consistent with
plasmalemma localization as previously reported (16). This result is consistent with
previous studies and confirmed that CXCR4 is expressed in
cardiomyocytes.
Loss of CXCR4 in cardiomyocyte worsens
Iso-induced heart failure
To investigate the role of CXCR4 in cardiomyocyte, a
cardiomyocyte-specific CXCR4 knockout (CMKO) mouse line
was generated, αMHC-Cre;CXCR4f/f, by crossing
CXCR4f/f mice (44)
with αMHC-Cre mice. The resultant CMKO mice were born at
the expected Mendelian ratio and displayed no overt defect in gross
appearance or cardiac structure for at least up to 4 months old,
suggesting that CXCR4 expression specifically in the cardiomyocyte
lineage is not essential for heart development. To induce heart
failure, CMKO mice and control αMHC-Cre (Cre)
littermates (8-10 weeks old) were subjected to a 14-day mini-pump
delivery of Iso, a well characterized adrenergic agonist that
induces heart failure primarily as the result of activated
β1-adrenergic signaling and Gs stress. Serial echocardiography
revealed that Iso treatment induced heart failure in both groups of
mice; however, in CMKO mice, the left ventricular
contractility was worse, chamber was more dilated, and end-systolic
left-ventricular anterior wall thickness was thinner than in Cre
control littermates (Fig. 1). In
addition, it was observed that CMKO mice exhibit a
higher heart rate than control Cre littermates under the same
anesthesia regimen (Fig. S2).
These worsened parameters in CMKO mice suggested that
CXCR4 expression in cardiomyocytes plays a protective role in Gs
stress-induced heart failure.
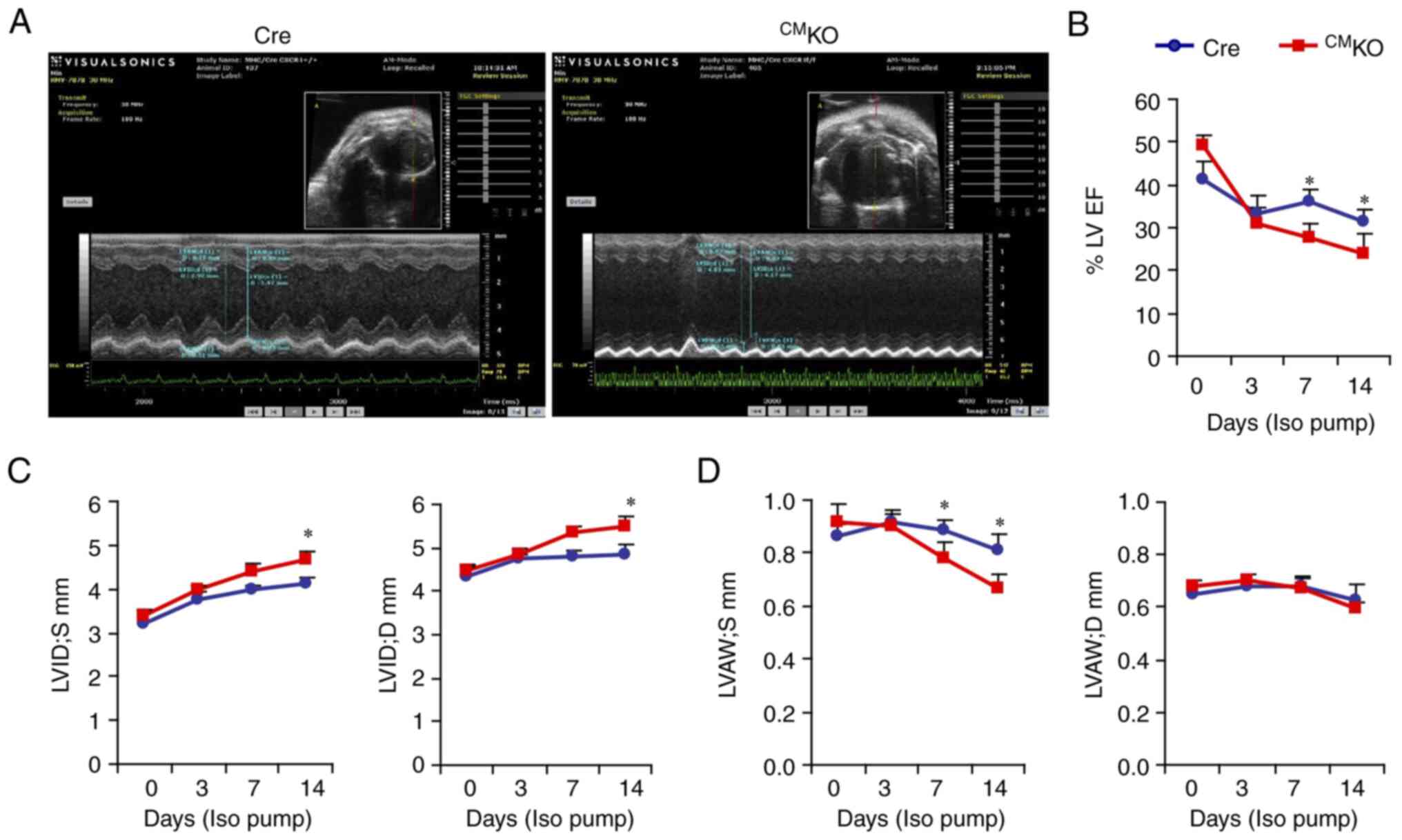 | Figure 1Loss of CXCR4 in cardiomyocytes
worsens Iso-induced cardiac dysfunction. CMKO (αMHC-Cre;
CXCR4f/f) mice and control αMHC-Cre (Cre) littermates
were treated with Iso (45 mg/kg BW/day) or saline via a
subcutaneously-implanted mini-osmotic pump for 14 days, then
cardiac functions were serially evaluated via echocardiography at
day 0 (baseline), 3, 7, and 14. (A) Representative image of
echocardiography. (B) LVEF. (C) LVID at end systole (S, left panel)
and end diastole (D, right panel). (D) Thickness of LVAW at end
systole (S, left panel) and end diastole (D, right panel). n=15 per
group. *P<0.05 (B-D: One-way ANOVA). Iso,
isoproterenol; CMKO, CXCR4 knockout; LVEF, left
ventricular ejection fraction; LVID, left ventricular internal
diameter; LVAW, left ventricular anterior wall; Iso,
isoproterenol. |
Loss of CXCR4 in cardiomyocytes worsens
Iso-induced cardiac adverse remodeling and apoptosis
In another set of experiments, mice were
administered Iso or saline via subcutaneous mini-pumps and
euthanized at day 14, and the hearts were excised. The heart size
and heart-weight (H.W.) to body-weight (B.W.) ratio appeared normal
and comparable between CMKO and Cre mice with Saline
treatment (Fig. 2A and B).
However, with Iso treatment, these measures were significantly
elevated in both groups and significantly deteriorated in
CMKO mice compared with Cre mice (Fig. 2A and B). Histological assessments
of cardiac tissue revealed that Iso induced a significantly
increased interstitial fibrosis (Sirius Red/Fast Green staining;
Fig. 2C and D) and apoptotic
cardiomyocytes (TUNEL staining; Fig.
2E and F) in CMKO mice. Thus, it was identified that
loss of CXCR4 in cardiomyocytes increases Iso-induced cardiac cell
death and adverse remodeling.
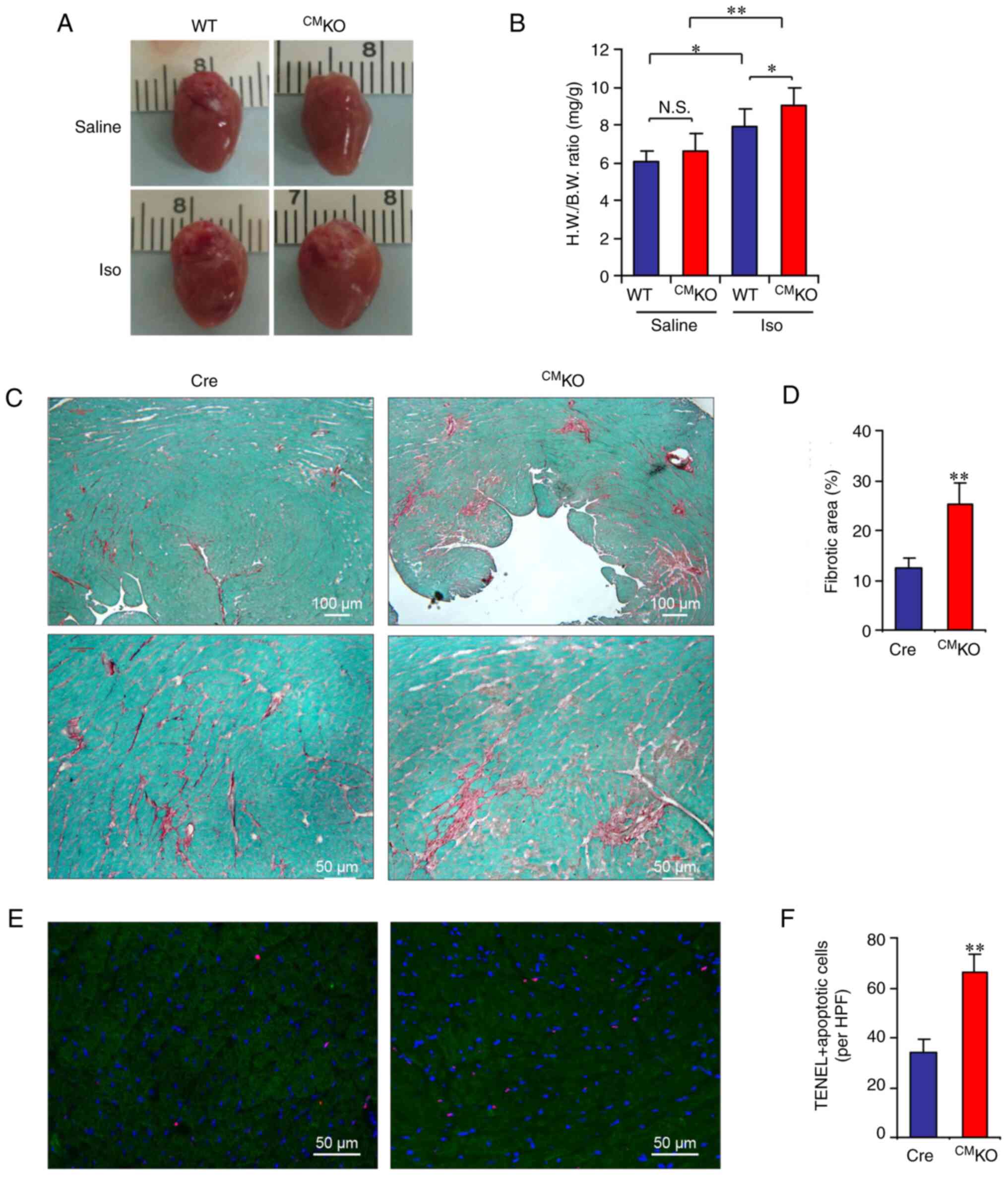 | Figure 2Loss of CXCR4 in cardiomyocytes
worsens Iso-induced cardiac adverse remodeling and apoptosis. Mice
were administered Iso or Saline via mini-pump implantation and at
day 14, euthanized. (A) Representative gross appearance of the
mouse hearts. (B) H.W. to B.W. ratio. (C and D) Representative
images of Sirius Red/Fast Green staining [C; Original
magnifications, ×40 for upper panels, ×100 for lower panels) and
(D) quantification of the interstitial fibrosis areas]. (E and F)
Representative images of (E) TUNEL (red) and DAPI (blue) double
staining and (F) quantification of the TUNEL+ apoptotic
cardiomyocytes. Original magnifications, ×100. HPF, high-powered
field. n=6 per group. *P<0.05 and
**P<0.01 (B: Two-way ANOVA; D and F: unpaired
two-tailed t-test). Iso, isoproterenol; H.W., heart weight; B.W.,
body weight; CMKO, CXCR4 knockout; WT, wild-type; N.S.,
not significant. |
SDF-1 treatment abrogates Iso-induced
apoptosis in cultured cardiomyocytes
Since CXCR4 is a Gi protein-coupled receptor, and Gi
protein-coupled receptor signaling is inhibitory to Gs effects, it
was hypothesized that activation of CXCR4 by SDF-1 counters
β-AR/Gαs signaling to improve cardiomyocyte survival. In cultured
H9C2 cardiomyocytes, treatment with Iso for 24 h significantly
reduced cell number and viability (Fig. 3A-C) and increased cell apoptosis
(Fig. 3D and E). While treatment
with SDF-1 or AMD3100 (CXCR4 antagonist) alone did not show a
significant effect, co-treatment with SDF-1 almost completely
prevented Iso-induced reduction in cell number and viability
(Fig. 3A-C) and significantly
attenuated Iso-induced apoptosis (Fig. 3D and E). Consistently, the level
of p-Akt, a major downstream effector of Gi signaling-activated
PI-3 kinase-β and cell survival, was diminished by Iso treatment
(Fig. 3F) however elevated by
SDF-1 treatment (Fig. 3G);
importantly, co-treatment with SDF-1 attenuated Iso-induced
reduction in p-Akt (Fig. 3H).
Collectively, these results suggested that CXCR4 protection of
Iso-induced cell death is mediated by SDF-1, and that SDF-1/CXCR4
signaling promotes cell survival.
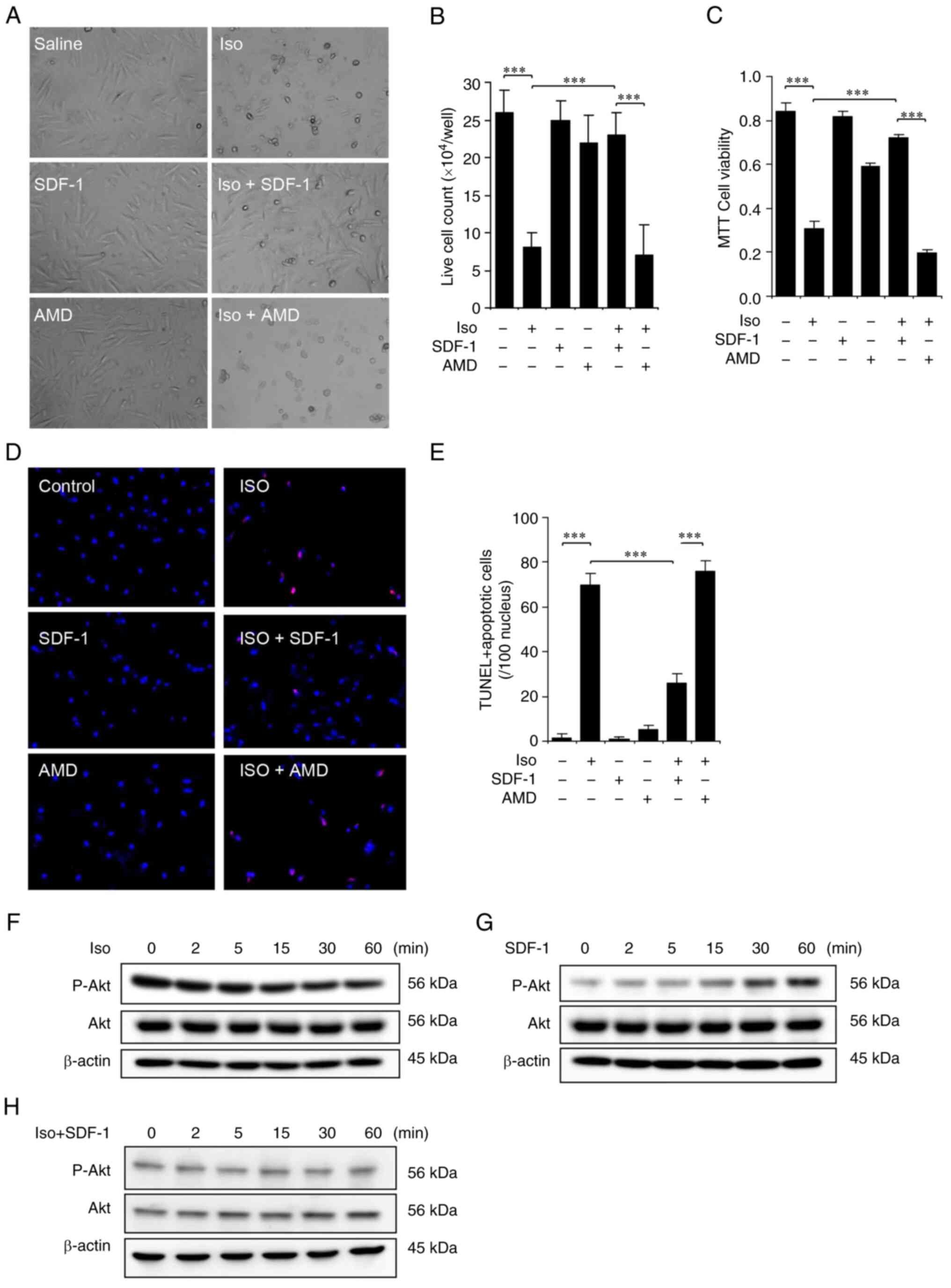 | Figure 3SDF-1/CXCR4 signaling attenuates
Iso-induced apoptosis in cardiomyocytes. H9C2 cardiomyocytes were
treated for 24 h with Saline, Iso (100 µM), SDF-1 (500
ng/ml), AMD3100 (100 ng/ml), Iso + SDF-1, or Iso + AMD3100, then
analyzed. (A) Microphotograph of the treated cells. Original
magnification, ×400. (B) Manual counts of live cells. (C) MTT
assays of viable cells. (D) TUNEL (red) and DAPI (blue) double
staining (original magnification, ×400). (E) quantification of the
TUNEL+ apoptotic cells. (F-H) Western blot analyses of
p-Akt and total Akt in cells treated with (F) Iso, (G) SDF-1 and
(H) Iso + SDF-1. n=6 per group. ***P<0.001 (B, C and
E: One-way ANOVA). Iso, isoproterenol; p-, phosphorylated. |
SDF-1/CXCR4 axis promotes cardiomyocyte
survival through Gai protein signaling
To understand the role of CXCR4 more definitively,
H9C2 cardiomyocytes were transduced with Lenti-CXCR4-shRNA, which
knocked down CXCR4 mRNA expression by ~90% (Fig. 4A). While knockdown of CXCR4 alone
did not affect cell number and viability, it worsened Iso-induced
reduction of cell number and viability and more importantly,
abolished SDF-1 mediated protection from Iso-induced reduction in
cell number and viability (Fig. 4B
and C). Consistently, knockdown of CXCR4 also diminished the
ability of SDF-1 to ameliorate Iso-induced p-Akt reduction
(Fig. 4D). These results
confirmed that SDF-1 mediated cell survival is dependent on CXCR4
expression. To further determine the role of Gi signaling in
SDF-1-mediated protection, H9C2 cells were treated with Iso/SDF-1
or Saline (-) in the presence or absence of a Gi-specific
inhibitor. Clearly, SDF-1 attenuated the Iso-induced cell number
and viability (Fig. 4E and F),
and the protection was lost in the presence of Gi inhibitor but not
vehicle control, confirming that SDF-1 protection from Iso-induced
cell death is mediated by Gi signaling.
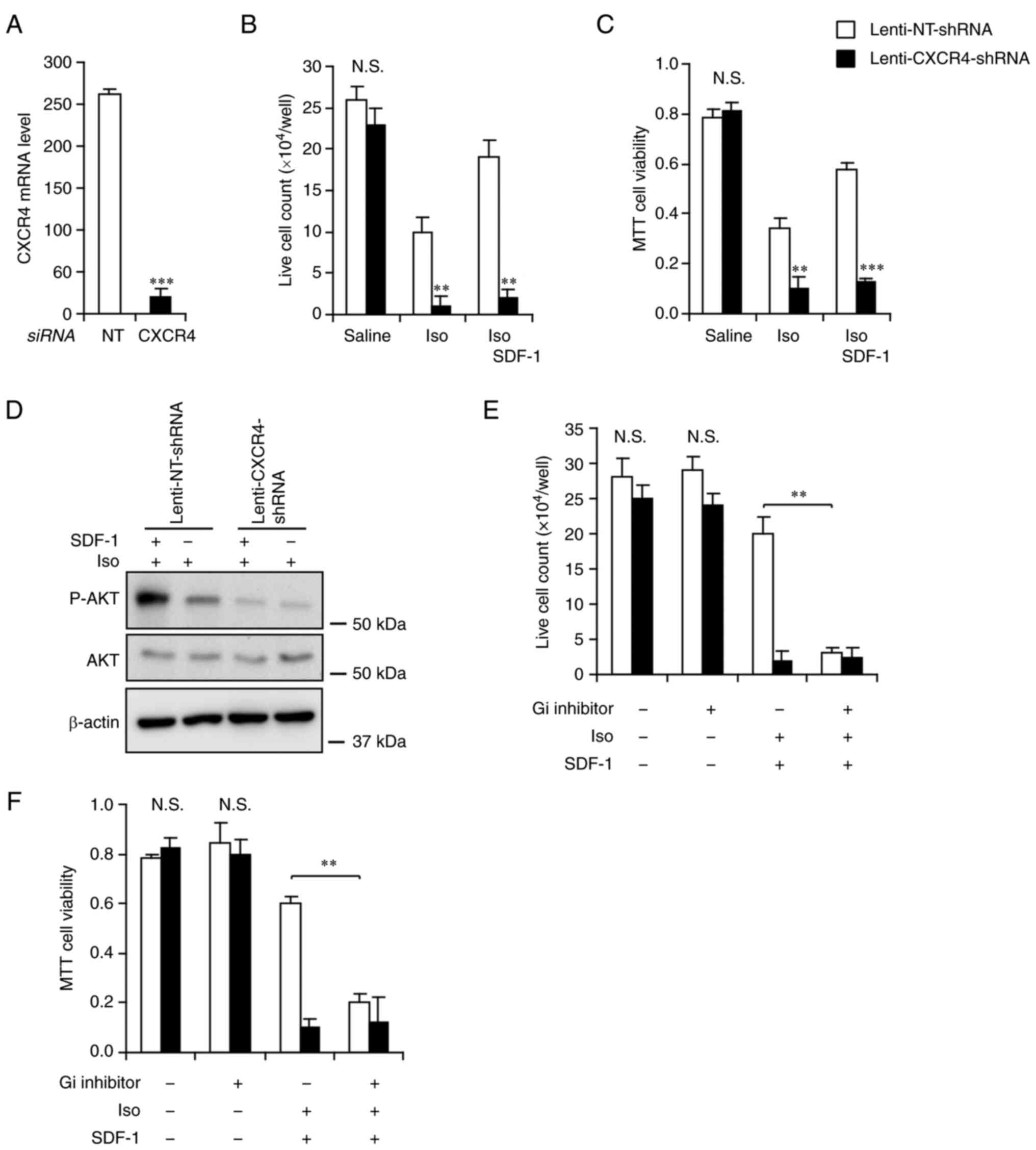 | Figure 4SDF-1/CXCR4 mediates cardiomyocyte
survival via Gi signaling. H9C2 cardiomyocytes were transduced with
Lenti-CXCR4-shRNA or Lenti-NT-shRNA and subjected to the following
experiments: (A) Reverse transcription-quantitative PCR analysis of
CXCR4 mRNA levels. (B and C) Treatment with Saline, Iso (100
µM), or Iso + SDF-1 (500 ng/ml) for 24 h, followed by (B)
manual counting of cell numbers and (C) MTT viability assays. n=6
per group. (D) Treatment with Iso (+) plus SDF-1 (+) or vehicle
(-), followed by western blotting of p-Akt and total Akt. (E and F)
Treatment with Saline, Iso, or Iso + SDF1 for 24 h in the presence
of Gi inhibitor Pertussis toxin (200 ng/ml) (+) or vehicle (-),
followed by (E) manual cell counts and (F) MTT viability assays.
n=6 per group. **P<0.01 and ***P<0.001.
A, unpaired two-tailed t-test; B and C, E and F, Two-way ANOVA.
shRNA, short hairpin RNA; NT, non-targeting; p-, phosphorylated;
N.S., not significant. |
Discussion
In the present study, it was identified that loss of
CXCR4 in cardiomyocytes renders the mice more vulnerable to
Iso-induced heart failure-characterized by worsened LV
contractility, greater LV dilation and thinner ventricular wall-
and that SDF-1 treatment potently protects cardiomyocytes from
Iso-induced cell death in a CXCR4 and Gi-dependent manner. These
data suggested that SDF-1/CXCR4 signaling plays a protective role
in β-adrenergic stress-induced cell death, cardiac adverse
remodeling and progression to dilation, and heart failure.
The results of cardiac functional analysis in CXCR4
CMKO mice are consistent with a previous study (45); however, in the present study
direct evidence was provided of Iso-induced cardiomyocyte apoptosis
in vivo and in vitro in the absence or downregulation
of CXCR4, respectively. Furthermore, it was demonstrated that this
effect is mediated by SDF-1 action and Gi protein signaling. The
extent of SDF-1 protection of Iso-induced cardiomyocyte death is
particularly striking. Previous studies suggested that
SDF-1/CXCR4-mediated cell survival from ischemic stress is linked
to Gβγ mediated activation of phosphoinositide 3-kinase (likely
PI3Kβ) and Akt (11,13,46). Indeed, it was found that SDF-1
induces a significantly increased p-Akt in cardiomyocytes and also
attenuates Iso-induced downregulation of p-Akt. Nevertheless, since
sustained activation of Gαs-coupled β-ARs results in apoptotic loss
of cardiomyocytes via a cAMP-dependent mechanism (30), SDF-1/CXCR4 signaling, through Gαi
activation, may also enhance cell survival by directly
downregulating adenylyl cyclase activity. Further experiments would
be needed to confirm this assertion. Furthermore, it was observed
that during progression of heart failure, Gai protein expression is
increased (20). This increase,
previously considered as a compensatory effect from altered β-AR
signaling, may in fact play an important protective role by
substantiating the effect of CXCR4 and potentially other Gai
coupled receptors.
Iso is the prototypical β-AR agonist with well
documented profile in positive inotropic/chronotropic effects and
induction of apoptosis (47), and
continuous infusion is commonly used in rodents to induce
phenotypic features of myocardial infarction and heart failure
(37). Our in vivo
analyses were particularly focused on the initial 14 days following
Iso treatment, which is considered the best time window for
detecting direct effects of adrenergic stress on cardiomyocytes in
this model (37). Nevertheless,
the detrimental effects are primarily mediated via β1-AR
and Gαs signaling, thus investigation of CXCR4, a
Gαi-coupled receptor, with Iso stress has a limitation,
because heart failure is a complex disease and involves mechanism
beyond β-AR and Gαs stress. Thus, examining the role of
SDF-1/CXCR4 with additional heart failure model, such as pressure
overload, is warranted in future studies.
Notably, it was observed that CXCR4 CMKO
mice display an elevated heart rate associated with progression to
heart failure. This is likely due to the dysregulated adrenergic
signaling, although detailed electrophysiologic features and
underlying mechanisms are unclear at present and remain a subject
of our ongoing investigations. Notably, upregulation of CXCR4
expression was observed in patients with atrial fibrillation (AF) a
decade ago (48). Recently, a
flurry of bioinformatics analyses of clinical high-throughput data
sets revealed that CXCR4 is a potent hub gene strongly associated
with the pathogenesis of AF (49-52). Previous studies with a mouse model
of AF suggested that CXCR4 antagonist AMD3100 can treat AF by
ameliorating calcium mishandling and tissue inflammation (50,52). Given that CXCR4 signaling can
mediate both cardiomyocyte survival and tissue inflammation, which
appear to have contrasting effects on AF, strategies that target
CXCR4 for treatment of cardiac arrhythmia would need to be
carefully evaluated before clinical application.
One of the limitations to the present study is the
limited scope of mechanistic study. Gain-of-function experiments
were performed only in cultured cardiomyocytes, but not in
vivo in animals. While the conclusion-that ablation of CXCR4
expression in cardiomyocytes exacerbates isoproterenol-induced cell
death and heart failure-is fully supported by the data, the
translational value of the present finding or if CXCR4 signaling
can be used for treatment of cardiac adverse remodeling and heart
failure remains to be rigorously investigated by using relevant
CXCR4 activator/agonists particularly in large animal models with
longer term follow-ups.
Lastly, CXCR4-null embryos exhibit dysplasia of the
ventricular septum (3). However,
a structural or functional defect in our CXCR4 CMKO
mouse line was not observed for at least 4 months after birth,
consistent with a previous study from Agarwal et al
(53). The findings of the
present study suggested that CXCR4 is required for septum formation
probably through a non-cardiomyocyte lineage or cardiomyocyte
progenitor cells prior to expressing α-MHC, which remains a subject
of future research by the authors.
In conclusion, the present results suggested that
SDF-1/CXCR4 signaling promotes cardiomyocyte survival and
attenuates heart failure induced by β-AR overactivation.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MC conceptualized the study, interpreted the data,
and wrote the manuscript. CC, KY, and XL performed the experiments
and analyzed the data. ND, QZ and FZ made intellectual
contributions and assisted in data interpretation. FZ edited the
manuscript. MC and CC confirm the authenticity of all the raw data.
All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
All animal experiments in the present study were
approved (IACUC approval no. 3005) by the Animal Care and Use
Committee of Huazhong University of Science and Technology (Wuhan,
China) and performed in compliance with the 'Guide for the Care and
Use of Laboratory Animals' (NIH publication) and all relevant
ethical regulations for animal testing and research.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This study was supported by NSFC (grant nos. 82070318 and
81570257) and Wuhan Science and Technology program (grant no.
2019020701011455).
References
|
1
|
Tachibana K, Hirota S, Iizasa H, Yoshida
H, Kawabata K, Kataoka Y, Kitamura Y, Matsushima K, Yoshida N,
Nishikawa S, et al: The chemokine receptor CXCR4 is essential for
vascularization of the gastrointestinal tract. Nature. 393:591–594.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nagasawa T, Hirota S, Tachibana K,
Takakura N, Nishikawa S, Kitamura Y, Yoshida N, Kikutani H and
Kishimoto T: Defects of B-cell lymphopoiesis and bone-marrow
myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature.
382:635–638. 1996. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zou YR, Kottmann AH, Kuroda M, Taniuchi I
and Littman DR: Function of the chemokine receptor CXCR4 in
haematopoiesis and in cerebellar development. Nature. 393:595–599.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Askari AT, Unzek S, Popovic ZB, Goldman
CK, Forudi F, Kiedrowski M, Rovner A, Ellis SG, Thomas JD,
DiCorleto PE, et al: Effect of stromal-cell-derived factor 1 on
stem-cell homing and tissue regeneration in ischaemic
cardiomyopathy. Lancet. 362:697–703. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jujo K, Ii M, Sekiguchi H, Hamada H,
Thorne T, Klyachko E, Clarke T, Ito A, Misener S, Renault MA and
Losordo DW: Bone marrow MMP-9 expression mediates therapeutic
effect of selective CXCR4 antagonist AMD3100 on myocardial
infarction. Circulation. 118:S536–S537. 2008.
|
|
6
|
Marin-Juez R, El-Sammak H, Helker CSM,
Kamezaki A, Mullapuli ST, Bibli SI, Foglia MJ, Fleming I, Poss KD
and Stainier DYR: Coronary revascularization during heart
regeneration is regulated by epicardial and endocardial cues and
forms a scaffold for cardiomyocyte repopulation. Dev Cell.
51:503–515.e504. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang FY, Xia TL, Li JL, Li CM, Zhao ZG,
Lei WH, Chen L, Liao YB, Xiao D, Peng Y, et al: The bifunctional
SDF-1-AnxA5 fusion protein protects cardiac function after
myocardial infarction. J Cell Mol Med. 23:7673–7684. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Abbott JD, Huang Y, Liu D, Hickey R,
Krause DS and Giordano FJ: Stromal cell-derived factor-1alpha plays
a critical role in stem cell recruitment to the heart after
myocardial infarction but is not sufficient to induce homing in the
absence of injury. Circulation. 110:3300–3305. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cheng M, Zhou J, Wu M, Boriboun C, Thorne
T, Liu T, Xiang Z, Zeng Q, Tanaka T, Tang YL, et al: CXCR4-mediated
bone marrow progenitor cell maintenance and mobilization are
modulated by c-kit activity. Circ Res. 107:1083–1093. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tang YL, Zhu W, Cheng M, Chen L, Zhang J,
Sun T, Kishore R, Phillips MI, Losordo DW and Qin G: Hypoxic
preconditioning enhances the benefit of cardiac progenitor cell
therapy for treatment of myocardial infarction by inducing CXCR4
expression. Circ Res. 104:1209–1216. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Saxena A, Fish JE, White MD, Yu S, Smyth
JWP, Shaw RM, DiMaio JM and Srivastava D: Stromal cell-derived
factor-1alpha is cardioprotective after myocardial infarction.
Circulation. 117:2224–2231. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Segers VF, Tokunou T, Higgins LJ,
MacGillivray C, Gannon J and Lee RT: Local delivery of
protease-resistant stromal cell derived factor-1 for stem cell
recruitment after myocardial infarction. Circulation.
116:1683–1692. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hu X, Dai S, Wu WJ, Tan W, Zhu X, Mu J,
Guo Y, Bolli R and Rokosh G: Stromal cell derived factor-1 alpha
confers protection against myocardial ischemia/reperfusion injury:
Role of the cardiac stromal cell derived factor-1 alpha CXCR4 axis.
Circulation. 116:654–663. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bromage DI, Taferner S, He Z, Ziff OJ,
Yellon DM and Davidson SM: Stromal cell-derived factor-1alpha
signals via the endothelium to protect the heart against
ischaemia-reperfusion injury. J Mol Cell Cardiol. 128:187–197.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xiong H, Luo Y, Yue Y, Zhang J, Ai S, Li
X, Wang X, Zhang YL, Wei Y, Li HH, et al: Single-cell
transcriptomics reveals chemo-taxis-mediated intraorgan crosstalk
during cardiogenesis. Circ Res. 125:398–410. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Segret A, Rucker-Martin C, Pavoine C,
Flavigny J, Deroubaix E, Châtel MA, Lombet A and Renaud JF:
Structural localization and expression of CXCL12 and CXCR4 in rat
heart and isolated cardiac myocytes. J Histochem Cytochem.
55:141–150. 2007. View Article : Google Scholar
|
|
17
|
Pyo RT, Sui J, Dhume A, Palomeque J,
Blaxall BC, Diaz G, Tunstead J, Logothetis DE, Hajjar RJ and
Schecter AD: CXCR4 modulates contractility in adult cardiac
myocytes. J Mol Cell Cardiol. 41:834–844. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
LaRocca TJ, Altman P, Jarrah AA, Gordon R,
Wang E, Hadri L, Burke MW, Haddad GE, Hajjar RJ and Tarzami ST:
CXCR4 cardiac specific knockout mice develop a progressive
cardiomyopathy. Int J Mol Sci. 20:22672019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Damas JK, Eiken HG, Oie E, Bjerkeli V,
Yndestad A, Ueland T, Tonnessen T, Geiran OR, Aass H, Simonsen S,
et al: Myocardial expression of CC- and CXC-chemokines and their
receptors in human end-stage heart failure. Cardiovasc Res.
47:778–787. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang J, Gareri C and Rockman HA:
G-protein-coupled receptors in heart disease. Circ Res.
123:716–735. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
de Lucia C, Eguchi A and Koch WJ: New
insights in cardiac β-adrenergic signaling during heart failure and
aging. Front Pharmacol. 9:9042018. View Article : Google Scholar
|
|
22
|
Packer M, Carver JR, Rodeheffer RJ,
Ivanhoe RJ, DiBianco R, Zeldis SM, Hendrix GH, Bommer WJ, Elkayam U
and Kukin ML: Effect of oral milrinone on mortality in severe
chronic heart failure. The PROMISE study research group. N Engl J
Med. 325:1468–1475. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shizukuda Y, Buttrick PM, Geenen DL,
Borczuk AC, Kitsis RN and Sonnenblick EH: Beta-adrenergic
stimulation causes cardiocyte apoptosis: Influence of tachycardia
and hypertrophy. Am J Physiol. 275:H961–H968. 1998.PubMed/NCBI
|
|
24
|
Asai K, Yang GP, Geng YJ, Takagi G, Bishop
S, Ishikawa Y, Shannon RP, Wagner TE, Vatner DE, Homcy CJ and
Vatner SF: Beta-adrenergic receptor blockade arrests myocyte damage
and preserves cardiac function in the transgenic G(salpha) mouse. J
Clin Invest. 104:551–558. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhu WZ, Wang SQ, Chakir K, Yang D, Zhang
T, Brown JH, Devic E, Kobilka BK, Cheng H and Xiao RP: Linkage of
beta1-adrenergic stimulation to apoptotic heart cell death through
protein kinase A-independent activation of Ca2+/calmodulin kinase
II. J Clin Invest. 111:617–625. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Whelan RS, Konstantinidis K, Xiao RP and
Kitsis RN: Cardiomyocyte life-death decisions in response to
chronic beta-adrenergic signaling. Circ Res. 112:408–410. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Packer M, Bristow MR, Cohn JN, Colucci WS,
Fowler MB, Gilbert EM and Shusterman NH: The effect of carvedilol
on morbidity and mortality in patients with chronic heart failure.
U.S. Carvedilol heart failure study group. N Engl J Med.
334:1349–1355. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu
Q, Makarewich C, Ai X, Li Y, Tang A, et al: Cardiotoxic and
cardioprotective features of chronic β-adrenergic signaling. Circ
Res. 112:498–509. 2013. View Article : Google Scholar
|
|
29
|
Singh K, Xiao L, Remondino A, Sawyer DB
and Colucci WS: Adrenergic regulation of cardiac myocyte apoptosis.
J Cell Physiol. 189:257–265. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Adams JW and Brown JH: G-proteins in
growth and apoptosis: Lessons from the heart. Oncogene.
20:1626–1634. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Steinberg SF: Beta1-adrenergic receptor
regulation revisited. Circ Res. 123:1199–1201. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rohrer DK, Desai KH, Jasper JR, Stevens
ME, Regula DP Jr, Barsh GS, Bernstein D and Kobilka BK: Targeted
disruption of the mouse beta1-adrenergic receptor gene:
Developmental and cardiovascular effects. Proc Natl Acad Sci USA.
93:7375–7380. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Milano CA, Allen LF, Rockman HA, Dolber
PC, McMinn TR, Chien KR, Johnson TD, Bond RA and Lefkowitz RJ:
Enhanced myocardial function in transgenic mice overexpressing the
beta-2-adrenergic receptor. Science. 264:582–586. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Engelhardt S, Hein L, Wiesmann F and Lohse
MJ: Progressive hypertrophy and heart failure in beta1-adrenergic
receptor transgenic mice. Proc Natl Acad Sci USA. 96:7059–7064.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Iwase M, Bishop SP, Uechi M, Vatner DE,
Shannon RP, Kudej RK, Wight DC, Wagner TE, Ishikawa Y, Homcy CK and
Vatner SF: Adverse effects of chronic endogenous sympathetic drive
induced by cardiac GS alpha overexpression. Circ Res. 78:517–524.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Antos CL, Frey N, Marx SO, Reiken S,
Gaburjakova M, Richardson JA, Marks AR and Olson EN: Dilated
cardiomyopathy and sudden death resulting from constitutive
activation of protein kinase a. Circ Res. 89:997–1004. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nichtova Z, Novotova M, Kralova E and
Stankovicova T: Morphological and functional characteristics of
models of experimental myocardial injury induced by isoproterenol.
Gen Physiol Biophys. 31:141–151. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chang SC, Ren S, Rau CD and Wang JJ:
Isoproterenol-induced heart failure mouse model using osmotic pump
implantation. Methods Mol Biol. 1816:207–220. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tan Y, Li Y, Xiao J, Shao H, Ding C,
Arteel GE, Webster KA, Yan J, Yu H, Cai L and Li X: A novel CXCR4
antagonist derived from human SDF-1beta enhances angiogenesis in
ischaemic mice. Cardiovasc Res. 82:513–521. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wu M, Zhou J, Cheng M, Boriboun C,
Biyashev D, Wang H, Mackie A, Thorne T, Chou J, Wu Y, et al: E2F1
suppresses cardiac neovascularization by down-regulating VEGF and
PlGF expression. Cardiovasc Res. 104:412–422. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kuznetsov AV, Javadov S, Sickinger S,
Frotschnig S and Grimm M: H9c2 and HL-1 cells demonstrate distinct
features of energy metabolism, mitochondrial function and
sensitivity to hypoxia-reoxygenation. Biochim Biophys Acta.
1853:276–284. 2015. View Article : Google Scholar :
|
|
42
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta DeltaC(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
43
|
Cheng M, Yang J, Zhao X, Zhang E, Zeng Q,
Yu Y, Yang L, Wu B, Yi G, Mao X, et al: Circulating myocardial
microRNAs from infarcted hearts are carried in exosomes and
mobilise bone marrow progenitor cells. Nat Commun. 10:9592019.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nie Y, Han YC and Zou YR: CXCR4 is
required for the quiescence of primitive hematopoietic cells. J Exp
Med. 205:777–783. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang ER, Jarrah AA, Benard L, Chen J,
Schwarzkopf M, Hadri L and Tarzami ST: Deletion of CXCR4 in
cardiomyocytes exacerbates cardiac dysfunction following
isoproterenol administration. Gene Ther. 21:496–506. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bresnick AR and Backer JM: PI3Kβ-A
versatile transducer for GPCR, RTK, and small GTPase signaling.
Endocrinology. 160:536–555. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhu WZ, Zheng M, Koch WJ, Lefkowitz RJ,
Kobilka BK and Xiao RP: Dual modulation of cell survival and cell
death by beta(2)-adrenergic signaling in adult mouse cardiac
myocytes. Proc Natl Acad Sci USA. 98:1607–1612. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang XX, Zhang FR, Zhu JH, Xie XD and Chen
JZ: Up-regulation of CXC chemokine receptor 4 expression in chronic
atrial fibrillation patients with mitral valve disease may be
attenuated by renin-angiotensin system blockers. J Int Med Res.
37:1145–1151. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu X, Zeng Y, Liu Z, Li W, Wang L and Wu
M: Bioinformatics analysis of the circRNA-miRNA-mRNA network for
atrial fibrillation. Medicine (Baltimore). 101:e302212022.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang YF, Meng LB, Hao ML, Li XY and Zou
T: CXCR4 and TYROBP mediate the development of atrial fibrillation
via inflammation. J Cell Mol Med. 26:3557–3567. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang J, Huang X, Wang X, Gao Y, Liu L, Li
Z, Chen X, Zeng J, Ye Z and Li G: Identification of potential
crucial genes in atrial fibrillation: A bioinformatic analysis. BMC
Med Genomics. 13:1042020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liu P, Sun H, Zhou X, Wang Q, Gao F, Fu Y,
Li T, Wang Y, Li Y, Fan B, et al: CXCL12/CXCR4 axis as a key
mediator in atrial fibrillation via bioinformatics analysis and
functional identification. Cell Death Dis. 12:8132021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Agarwal U, Ghalayini W, Dong F, Weber K,
Zou YR, Rabbany SY, Rafii S and Penn MS: Role of cardiac myocyte
CXCR4 expression in development and left ventricular remodeling
after acute myocardial infarction. Circ Res. 107:667–676. 2010.
View Article : Google Scholar : PubMed/NCBI
|














