Introduction
13-cis-retinoic acid (13CRA), a member of the
family of vitamin A compounds (1), plays a role in several cellular
processes, such as cell proliferation, differentiation and
apoptosis (2). At present, 13CRA,
a Food and Drug Administration-approved drug for severe acne, is a
readily available and well-tolerated agent that has been reported
to exhibit antitumor potential both in vitro and in
vivo in various types of cancer (3-8).
Previous preclinical studies have demonstrated that 13CRA inhibits
the proliferation of cancer cells, including small-cell lung,
gastric and breast cancer cells (4-6).
In addition, 13CRA induces apoptosis and exerts a suppressive
effect on the metastasis of melanoma and colon cancer (7,8).
13CRA has been investigated for its anticancer activity for several
years, and its potential mechanisms of action have been explored in
various cancer types; however, to the best of our knowledge, there
is no report available to date on the anticancer activity and
potential use of 13CRA in cholangiocarcinoma (CCA). Thus,
information on the roles of 13CRA in CCA cells may provide
critical, useful evidence for further studies.
CCA is an aggressive tumor with a poor prognosis due
to its late clinical presentation and lack of effective
non-surgical therapies (9).
Several risk factors for CCA have been identified, including
primary sclerosing cholangitis, hepatobiliary parasites,
hepatolithiasis, Caroli disease, choledochal cysts and Thorotrast
(10). In Asia, CCA is associated
with chronic infection with the liver flukes Opisthorchis
viverrini and Clonorchis sinensis, which is a main risk
factor for CCA development (11).
The majority of patients with CCA are diagnosed at a late state; at
this stage, the metastasis of CCA has already occurred. Surgical
treatment is not suitable for these patients, and only palliative
treatment is possible (9). For
patients with advanced and metastatic CCA, the therapeutic option
is adjuvant chemotherapy, and the standard regimen with
gemcitabine/cisplatin combination therapy leads to an overall
survival time of only ~1 year (12). Thus, there is a critical need for
the identification of novel and effective therapies for patients
with CCA at advanced and metastatic phases of the disease. Among
numerous efforts conducted to date aimed at improving the efficacy
of CCA treatment, targeting hallmarks of CCA carcinogenesis, such
as sustaining proliferative ability, epithelial-mesenchymal
transition (EMT), stemness and plasticity properties have
particularly attracted attention.
Cell proliferation is a key aspect of cancer
development and progression, and the self-renewal capacity of
cancer cells has been linked to cancer recurrence (13). Sustained proliferative signaling
via c-Myc, an oncogenic molecule, has been shown to result in the
altered expression or activity of cell cycle-related genes or
proteins, which can stimulate growth abnormality and cancer
progression (13,14). Previous studies have suggested the
anti-proliferative effects of 13CRA on cancer cells, including
breast and gastric cancer cells (5,6).
Furthermore, 13CRA has been shown to induce the cell cycle arrest
of the immortalized sebocyte cell line, SEB-1, at the G1
phase via the upregulation of p21 and the downregulation of cyclin
D1 proteins (15).
EMT is a cell plasticity-promoting phenomenon that
enables cancer cells (of epithelial origin) to acquire mesenchymal
features with invasive properties, which leads to metastatic
colonization (16,17). In CCA, the expression of
EMT-related transcription factors, such as Snail, a zinc finger
E-box-binding homeobox and Twist, is associated with a poor
prognosis (18). CCA exhibits
several mesenchymal phenotypic features, known to be associated
with an increased motility and invasiveness; these features are
also associated with mechanisms involving the downregulation of the
epithelial phenotypic molecules, E-cadherin (E-cad) and β-catenin,
and the upregulation of the EMT-related transcription factors,
Snail1, Twist and S100 calcium binding protein A4 (19). The downregulation of E-cad in CCA
cells also increases the expression of vimentin, a mesenchymal
marker, which subsequently leads to an increased migration
(20). Upon detachment from the
primary tumor through EMT, cancer cells leave their primary site
and initiate the metastatic process of cancer progression (16). The metastasis cascade is generally
a complex process involving five key steps of metastasis:
Migration/invasion, intravasation, survival/circulation,
extravasation and colonization/proliferation (21). The process of metastasis requires
multiple steps, such as the degradation of the extracellular matrix
by matrix metalloproteinases (MMPs), particularly MMP-1, MMP-2,
MMP-3 and MMP-9, which are abundantly released in malignant
cholangiocytes (9). Furthermore,
cell surface adhesion molecules, such as intercellular adhesion
molecule-1 (ICAM-1) also participate in the binding, recognition
and adhesion of cells, which are involved in the metastatic process
(16,22). The expression of the inflammatory
molecule, cyclooxygenase-2 (COX-2), is induced in response to
various stimuli, including inflammatory cytokines and MMPs, which
are associated with cancer metastasis (23-25). Previously, CCA has been reported
to exhibit an aggressive behavior, and to be associated with MMP-9,
ICAM-1 and COX-2, suggesting the suppression of these molecules as
targets for CCA treatment (26,27).
The present study aimed to evaluate the effects of
13CRA on the proliferation, migration, adhesion and invasion of CCA
cells. In addition, the potential mechanisms underlying the
cancer-suppressive effects of 13CRA on CCA cells were
investigated.
Materials and methods
CCA cell lines and culture
For the present study, two human CCA cell lines,
KKU-100 and KKU-213B, were kindly provided by Professor Banchob
Sripa (Department of Pathology, Faculty of Medicine, Khon Kaen
University, Khon Kaen, Thailand). Cell line authentication was
performed by short tandem repeat (STR) analysis. The DNA markers of
23 STR loci and the sex marker, amelogenin, were analyzed using the
AmpFLSTR Identifiler PCR Amplification kit (Applied Biosystems;
Thermo Fisher Scientific, Inc.). DNA STR analysis of the KKU-213B
and KKU-100 cells has been previously described (28-30) and the profile was similar to the
STR profile of KKU-213 cells deposited in JCRB Cell Bank, Japan.
The KKU-213B cells are tumor cell variants similar to the KKU-213
cells; Sripa et al (29)
demonstrated that the KKU-213B cells exhibited a greater motility
than the KKU-213 cells, as evidenced by Boyden chamber assays. The
cells were routinely cultured in Ham's F12 medium (Gibco; Thermo
Fisher Scientific, Inc.; pH 7.4) supplemented with 1 mM sodium
bicarbonate, 10 mM HEPES, 100 U/ml penicillin G, 50 µg/ml
gentamicin sulfate and 10% (v/v) fetal bovine serum (FBS) (Gibco;
Thermo Fisher Scientific, Inc.), and were maintained under 5%
CO2 in a humidified incubator at 37°C. Cells were
sub-cultured every 2-3 days at 80% confluency using 0.25% (v/v)
trypsin-EDTA (Gibco; Thermo Fisher Scientific, Inc.).
Sulforhodamine B (SRB) assay
The CCA cells were seeded into 96-wells at
7.5×103 cells/well, incubated overnight and then treated
with 0.00, 1.25, 2.50, 5.00, 10.00 and 20.00 µM 13CRA
(R6256; MilliporeSigma) in serum-free medium (Ham's F12 medium
without 10% FBS) for 12, 24 and 48 h under 5% CO2 in a
humidified incubator at 37°C. Following treatment, the cells were
incubated with ice-cold 10% trichloroacetic acid (T6399;
MilliporeSigma) at 4°C for 1 h, followed by washing with deionized
(DI) water and staining with 0.4% SRB in 1% acetic acid at room
temperature for 30 min. The SRB dye inside the cells was
solubilized with 10 mM Tris-base (pH 10.5) solution, and the
absorbance was then measured at 540 nm using a microplate reader
(Sunrise™ Elisa Plate Reader; Tecan Group, Ltd.). The experiments
were independently performed ≥3 times. Cell viability was expressed
as a percentage of cell cytotoxicity relative to the untreated
control, which was considered as no having cytotoxicity. The
half-maximal inhibitory concentration (IC50) was
calculated by fitting the dose-response curve using SigmaPlot v12
Software (SigmaPlot for Windows; Systat Software, Inc.).
Clonogenic survival assay
The CCA cells were seeded into a six-well plate at a
density of 2.5×105 cells/well overnight and treated with
0.00, 0.312, 0.625, 1.25, 2.50 and 5.00 µM 13CRA in
serum-free medium for 48 h. Following treatment, the CCA cells were
trypsinized, and the viable-treated cells were seeded into a new
six-well plate at a density of 600 cells/well in serum-containing
medium. The cells were incubated in 5% CO2 in a
humidified incubator at 37°C for an additional 8 days, and the
culture medium was freshly renewed every 3 days. Finally, the
colonies were fixed with absolute methanol for 30 min at 4°C and
stained with 0.25% crystal violet (61135, Sigma-Aldrich; Merck
KGaA) in 2% ethanol at room temperature for 30 min. Images of the
colonies were captured using the ChemiDoc™ MP Imaging system
(Bio-Rad, Laboratories, Inc.) and the number of colonies was then
counted using Image-Pro Plus v4.5.0.29 software (Media
Cybernetics). The colony forming ability was calculated as a
percentage by comparison with the untreated control cells. The
experiments were performed independently three times.
Cell cycle distribution analysis by
propidium iodide (PI) staining
The CCA cells were seeded in a six-well plate at a
density of 2.5×105 cells/well overnight. Th following
day, the cells were treated with 0.00, 1.25 and 2.50 µM
13CRA in serum-free medium for 48 h. At the end of treatment, the
cells were collected, washed twice with PBS and fixed with 70%
ethanol at −80°C for 4 h. The cell suspension was then maintained
at −20°C for 3 h before being subjected to staining with a PI
solution containing 0.5% Triton-X in PBS, 0.02 mg/ml PI and 0.02
mg/ml RNase A. The cell suspension was incubated for 1 h at 4°C in
the dark. The percentage of cells in the different phases of the
cell cycle (cell cycle distribution) was analyzed using a BD FACS
Canto™ II flow cytometer and FACSDiva™ software version V6.1.3
(both from BD Biosciences). Three independent experiments were
performed.
Cell adhesion assay
The CCA cells were seeded in a six-well plate at a
density of 2×105 cells/well overnight and treated with
0.00, 1.25 and 2.50 µM 13CRA in serum-free medium for 48 h.
Following treatment, the CCA cells were harvested and re-plated
into a fibronectin-coated 96-well plate at a density of
2×104 cells/well. Following 30 min of incubation at
37°C, the non-adherent cells were removed. The adhered cells were
washed gently with PBS, fixed with ice-cold absolute methanol and
washed with DI water prior to staining with 0.25% crystal violet at
room temperature for 30 min. The stained cells were captured under
an Eclipse TS100 microscope (Nikon Corporation). Finally, the
crystal violet was solubilized with 10% methanol in 5% glacial
acetic acid solution, and the absorbance was measured at 540 nm
using a microplate reader (Sunrise™ Elisa Plate Reader; Tecan
Group, Ltd.). The percentage of cell adhesion was calculated by
comparison with the untreated control cells.
Transwell migration assay
The CCA cells were seeded into the upper chamber of
a Transwell plate (8.0 µm Pore Polycarbonate Membrane
Insert, 3422, Corning, Inc.) at a density of 2×104
cells/well in serum-free medium and were allowed to attach to the
membrane overnight. On the following day, the cells were treated
with 0.00, 1.25 and 2.50 µM 13CRA in serum-free medium,
while the bottom chamber was filled with serum-containing medium
without 13CRA. Following 24 h of incubation at 37°C, the cells were
fixed with absolute methanol and stained with 0.25% crystal violet
in 2% ethanol at room temperature for 30 min. The non-migrated
cells were gently removed using a cotton swab, and the number of
migrated cells on the bottom side of the membrane of the Transwell
plate was quantified. The image of migrated cells was captured
under an Eclipse TS100 microscope (Nikon Corporation), the number
of cells was then counted using Image-Pro Plus v4.5.0.29 program
(Media Cybernetics, Inc.) and the percentage of migrated cells was
calculated by comparison with the untreated control cells. A total
of three independent experiments were performed.
Matrigel invasion assay
The CCA cells were seeded into the upper chamber of
a Transwell plate coated with Matrigel at a density of
2×104 cells/well in serum-free medium, and were allowed
to attach to the thin layer of Matrigel overnight. The cells were
treated with 0.00, 1.25 and 2.50 µM 13CRA in serum-free
medium, while the lower part of the chamber was filled with
serum-containing medium without 13CRA. Following incubation for 48
h at 37°C, the invaded cells were fixed with absolute methanol and
stained with 0.5% crystal violet in 2% ethanol at room temperature
for 30 min, while the non-invaded cells and the thin layer of
Matrigel on the upper Transwell chamber were gently removed using a
cotton swab. The number of invaded cells on the bottom side of the
Transwell plate was quantified under a light microscope. The number
of invaded cells was counted, and the percentage of invaded cells
was calculated by comparison with the untreated control cells. A
total of three independent experiments were performed.
Reverse transcription-quantitative PCR
(RT-qPCR)
The CCA cells were seeded in a six-well plate at a
density of 2.5×105 cells/well overnight and then treated
with 0.00, 1.25 and 2.50 µM 13CRA in serum-free medium for
24 h. Following treatment, total RNA was isolated using TRIzol™
reagent (15596026, Thermo Fisher Scientific, Inc.) following the
manufacturer's instructions. cDNA synthesis was performed in a
C1000™ thermal cycler (Bio-Rad Laboratories, Inc.) using 5X
iScript™ Reverse Transcription Supermix (Bio-Rad Laboratories,
Inc.) following the manufacturer's instructions. cDNA served as a
template for qPCR amplification. RT-qPCR was performed using Light
Cycler® 480II/384 (Roche Applied Science). The
thermocycling conditions were as follows: 95°C for 3 min, followed
by 40 cycles at 95°C for 15 sec and 60°C for 31 sec, 1 cycle of
melting curve (95°C for 5 sec, 72°C for 5 sec, and 97°C
continuous), and a cooling cycle (40°C for 10 min). To verify the
purity of the products, a melting curve analysis was performed
after each run. The specific primers used are listed in Table SI. The expression of target genes
was calculated and represented as a ratio to the expression of the
housekeeping reference gene, β-actin. The experiment was
performed independently three times.
Western blot analysis
The CCA cells were seeded in a six-well plate at a
density of 2.5×105 cells/well overnight and treated with
0.00, 1.25 and 2.50 µM 13CRA in serum-free medium. Following
treatment, the cells were lysed with RIPA cell lysis buffer
containing 1X Halt™ Protease Inhibitor Cocktail (87786; Thermo
Fisher Scientific, Inc.). The samples were then centrifuged at
13,000 × g, 4°C for 30 min and supernatants were collected and
stored at -80°C until use. The protein concentration of the cell
lysate solutions was determined using Bradford protein assay
(Bio-Rad Laboratories, Inc.) following the manufacturer's
instructions. A total of 30 µg of protein from whole cell
lysates was loaded onto 10% SDS-PAGE and separated using SE 260
mini-vertical gel electrophoresis (Hoefer, Inc.). For western
blotting, proteins in the gel were transferred onto PVDF membranes
using Owl™ HEP-1 Semidry Electroblotter (HEP-1; Thermo Fisher
Scientific, Inc.). The membranes were incubated with the primary
antibodies. Following incubation with the corresponding horseradish
peroxidase-conjugated secondary antibodies (all primary and
secondary antibodies, and conditions are presented in Table SII), the protein bands were
developed and detected using Luminata™ Forte Western HRP Substrate
(MilliporeSigma), and imaged using the ChemiDoc™ MP Imaging system
(Bio-Rad, Laboratories, Inc.). The intensity of the protein bands
was analyzed using Image Lab software version 6.0 (Bio-Rad
Laboratories, Inc.) and expressed as a ratio to the housekeeping
reference protein, β-actin. The experiments were independently
performed ≥3 times.
Statistical analysis
Data are expressed as the mean ± standard deviation
of ≥3 independent experiments. Comparisons between the untreated
control and the treatment groups were performed using one-way ANOVA
followed by Tukey's post hoc test using SigmaStat v2.0 software
(Systat Software Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
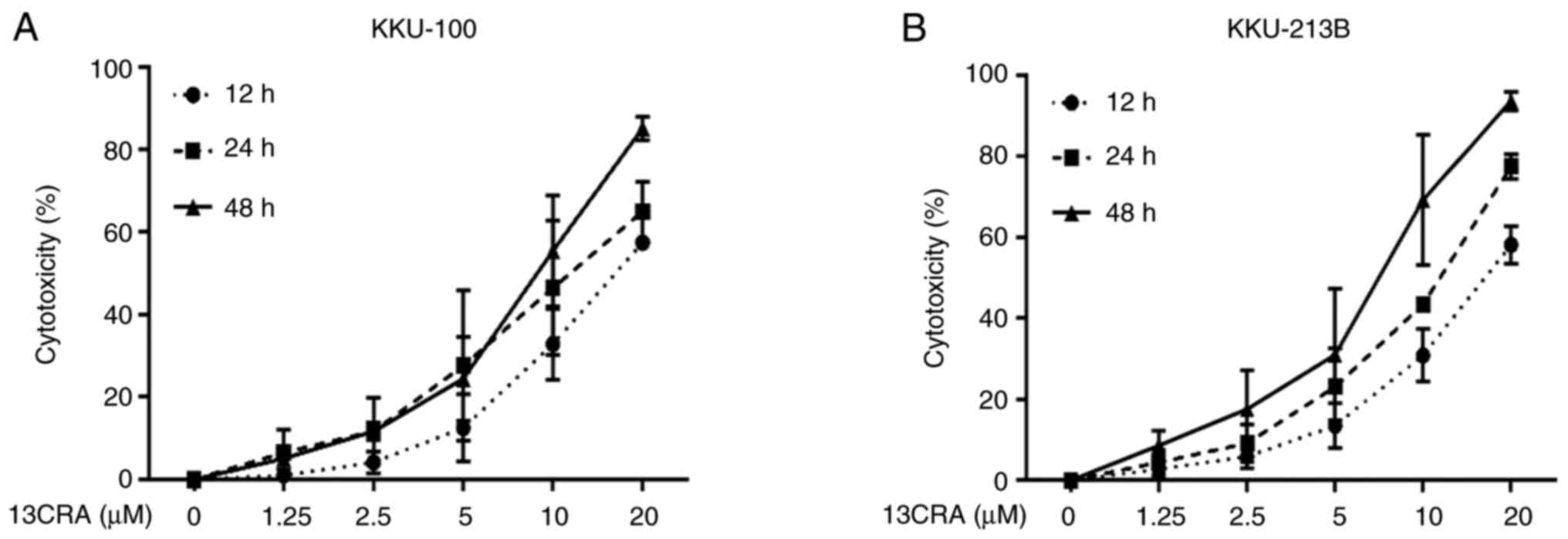
Effects of 13CRA on the viability of CCA
cells
In the present study, two CCA cell lines (KKU-100
and KKU-213B) were treated with increasing concentrations of 13CRA
ranging from 1.25 to 20 µM for 12, 24 and 48 h. The results
revealed that 13CRA suppressed the viability of both CCA cell lines
in a concentration- and time-dependent manner (Fig. 1). The half-maximal inhibitory
concentrations (IC50) of 13CRA for the viability of the
KKU-100 cells at 12, 24 and 48 h were 20.48±8.27, 12.35±3.60 and
9.33±7.53 µM, respectively. The IC50 values of
13CRA for the viability of the KKU-213B cells at 12, 24 and 48 h
were 20.27±16.34, 11.89±9.20 and 6.66±5.11 µM, respectively.
13CRA is a well-tolerated agent for severe acne; however, the
absence of experiments on non-cancerous cells used as a negative
control is a limitation of the present study. To explore the
tumor-suppressive activity of 13CRA at concentrations lower than
the IC50 values, concentrations of 1.25 and 2.5
µM 13CRA were used in the subsequent experiments.
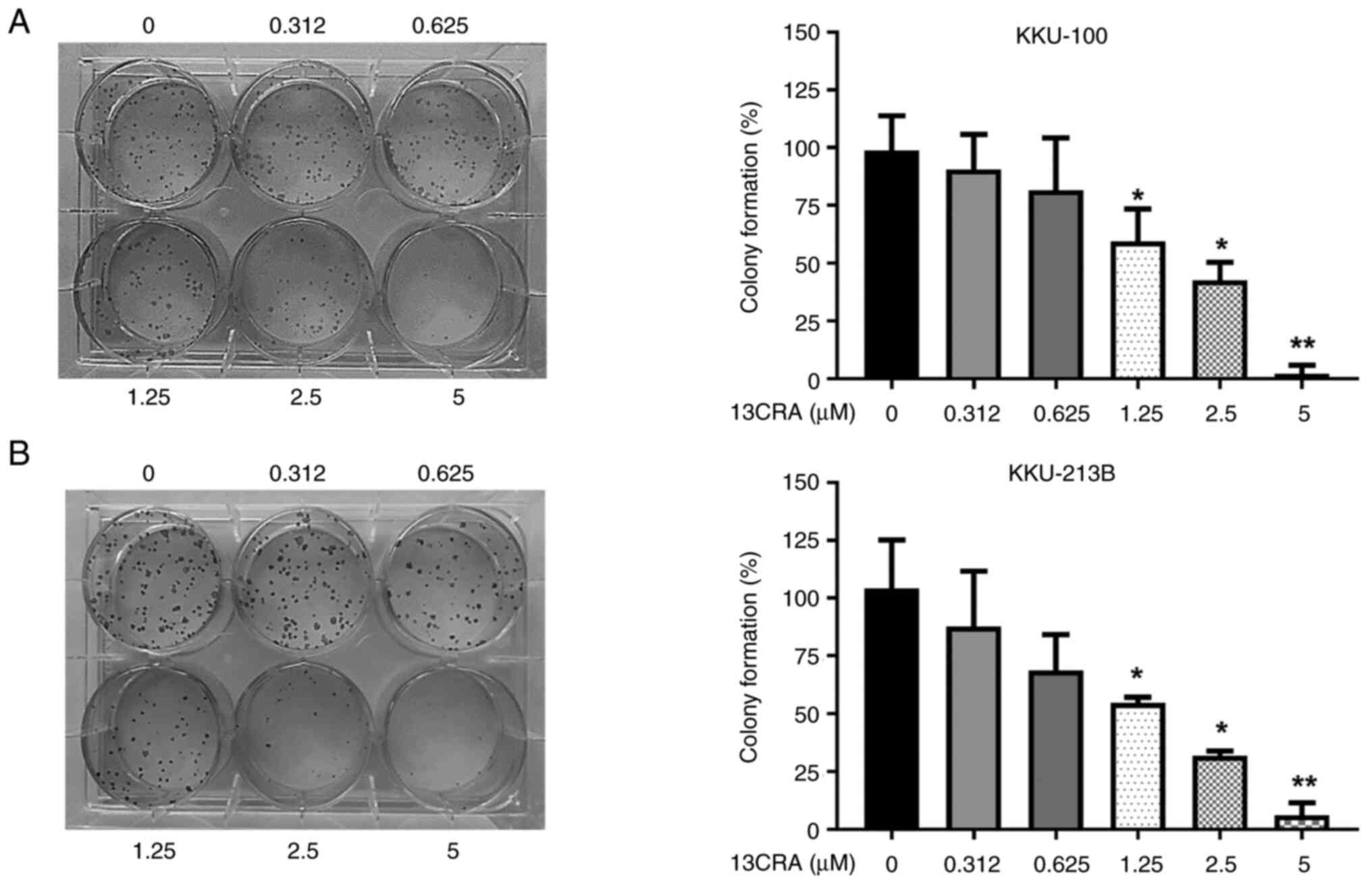
Effects of 13CRA on the clonogenic
self-renewal ability of CCA cells
To identify and quantify the effects of 13CRA on the
self-renewal ability of CCA cells, a clonogenic survival assay was
performed. Following treatment with 13CRA for 48 h, only the viable
13CRA-treated cells were cultured for an additional 8 days to allow
colony formation. The results revealed that 13CRA significantly
reduced the colony formation ability of both CCA cell lines in a
concentration-dependent manner. At a concentration of 2.5
µM, which induced minimal cytotoxicity (10-15%) in the
KKU-100 and KKU-213B cells, 13CRA markedly suppressed colony
formation compared with that of the untreated control cells
(>55%; Fig. 2). These results
indicated that 13CRA exerted a potent inhibitory effect on the
self-renewal ability of CCA cells.
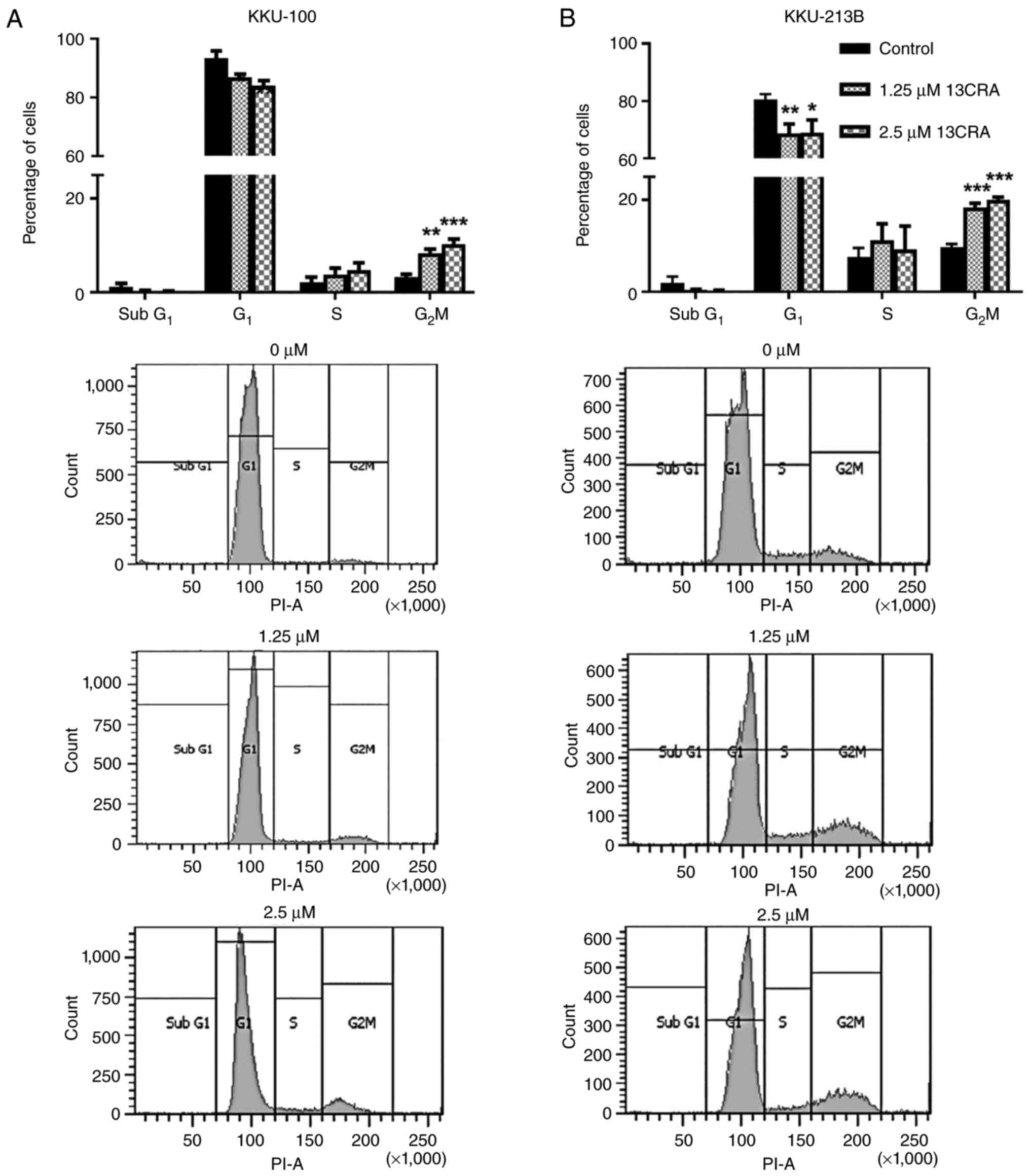
Effect of 13CRA on the cell cycle
progression of CCA cells
Since the present study demonstrated that 13CRA
exerted an anti-proliferative effect on CCA cells by inhibiting
their self-renewal ability, it was further investigated whether
this effect of 13CRA was caused by alterations of the cell cycle.
Thus, the cell cycle of 13CRA-treated and untreated CCA cells was
analyzed using flow cytometry. The results revealed that 1.25 and
2.5 µM 13CRA induced significant cell cycle arrest at the
G2/M phase, and decreased the number of cells at the
G1 phase in both CCA cell lines (Fig. 3) compared with the untreated
control cells. These findings suggested that the 13CRA-induced cell
cycle arrest at the G2/M phase resulted in the
inhibition of the proliferation of CCA cells.
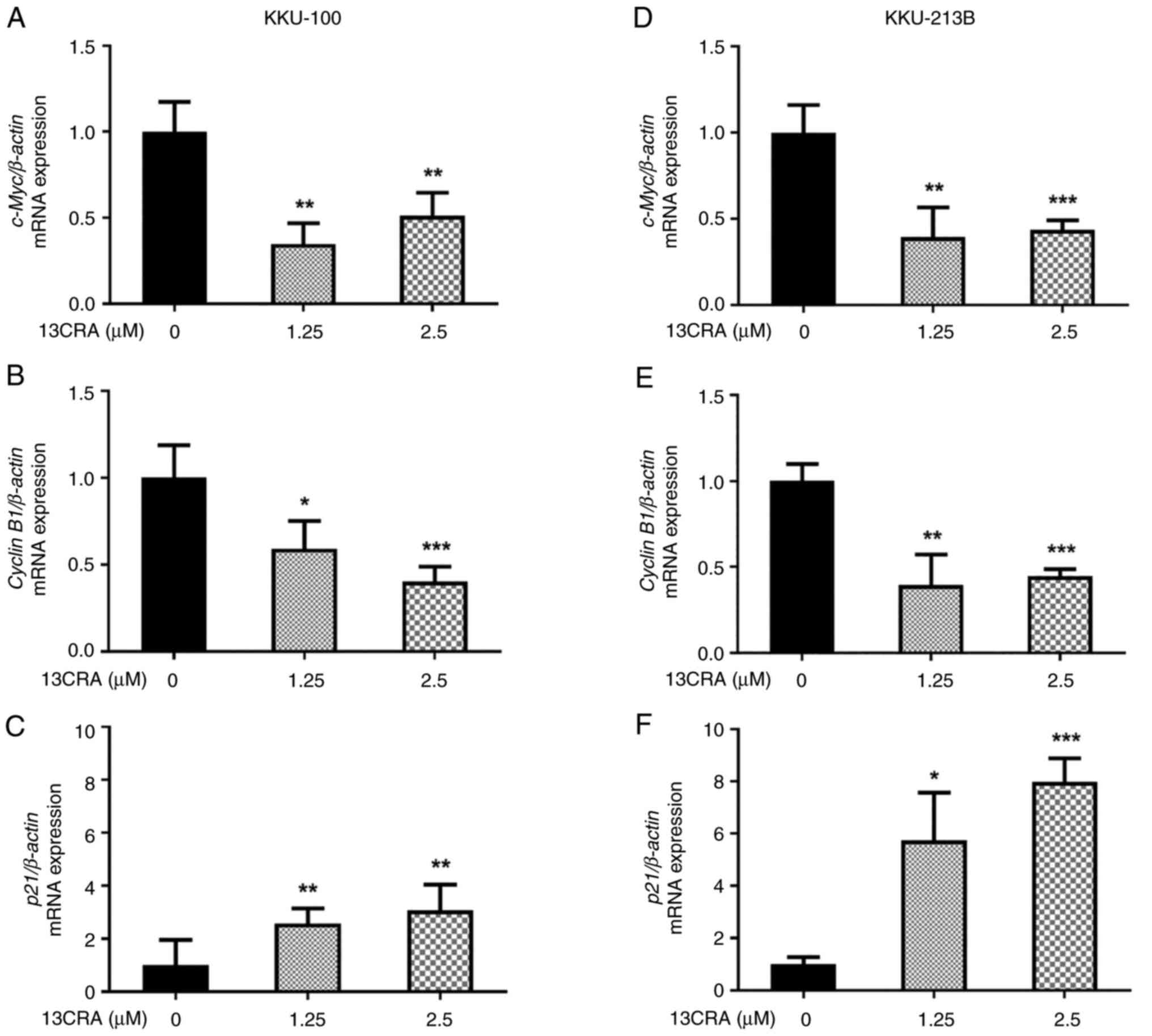
Effects of 13CRA on the expression of
cell cycle-regulatory genes in CCA cells
Since the present study demonstrated the inhibitory
effects of 13CRA on the cell cycle progression of CCA cells, the
effects of 13CRA on the mRNA expression of cell cycle-regulatory
genes, including p21, c-Myc and cyclin B1,
were further investigated using RT-qPCR. The results demonstrated
that, following treatment with 13CRA for 48 h, the expression of
p21, a cyclin-dependent kinase inhibitor, was upregulated,
while the expression of the cell cycle activator genes c-Myc
and cyclin B1 was downregulated (P<0.05; Fig. 4). These results suggested that
cell cycle arrest upon incubation with 13CRA was partly mediated by
the altered expression of cell-cycle regulatory genes.
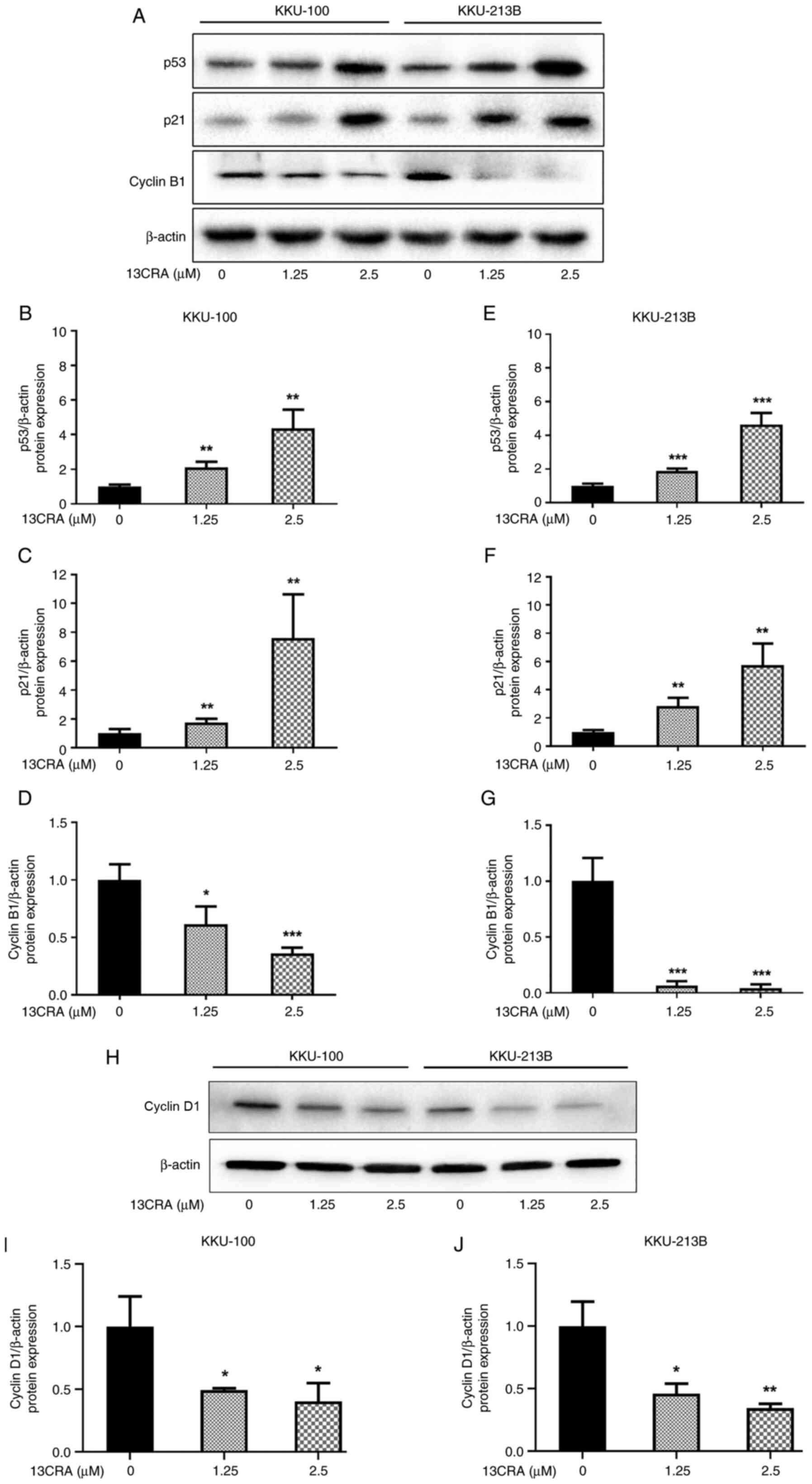
Effects of 13CRA on the expression of
cell cycle-regulatory proteins in CCA cells
To further confirm the effect of 13CRA on cell cycle
regulation, the expression of the cell cycle-related proteins,
including p53, p21, cyclin B1 and cyclin D1, was evaluated using
western blot analysis. The levels of p53 (the upstream regulator of
the cell cycle) and p21 (the cell cycle inhibitor) were
significantly increased (P<0.05) in both the KKU-100 (Fig. 5A-C) and KKU-213B (Fig. 5A, E and F) cells. Conversely, the
levels of cyclin B1 (the G2/M cell cycle driver) were
significantly decreased compared with those of the untreated
control cells (Fig. 5A, D and G).
Thus, in addition to the effects of 13CRA on the expression of the
p21, c-Myc and cyclin B1 genes, 13CRA also
induced an increase in the expression of the cell cycle inhibitors,
p53 and p21, as well as a decrease in the G2/M driver
cyclin B1, which eventually led to cell cycle arrest in the
G2/M phase. Since 13CRA induced a decrease in the number
of cells at the G1 phase of the cell cycle, the present
study investigated the expression of cyclin D1, which is the
regulatory protein of the G1 phase of the cell cycle.
Following treatment with 13CRA for 48 h, the protein level of
cyclin D1 significantly decreased in both CCA cell lines
(P<0.05) compared with that of the untreated control cells
(Fig. 5H-J).
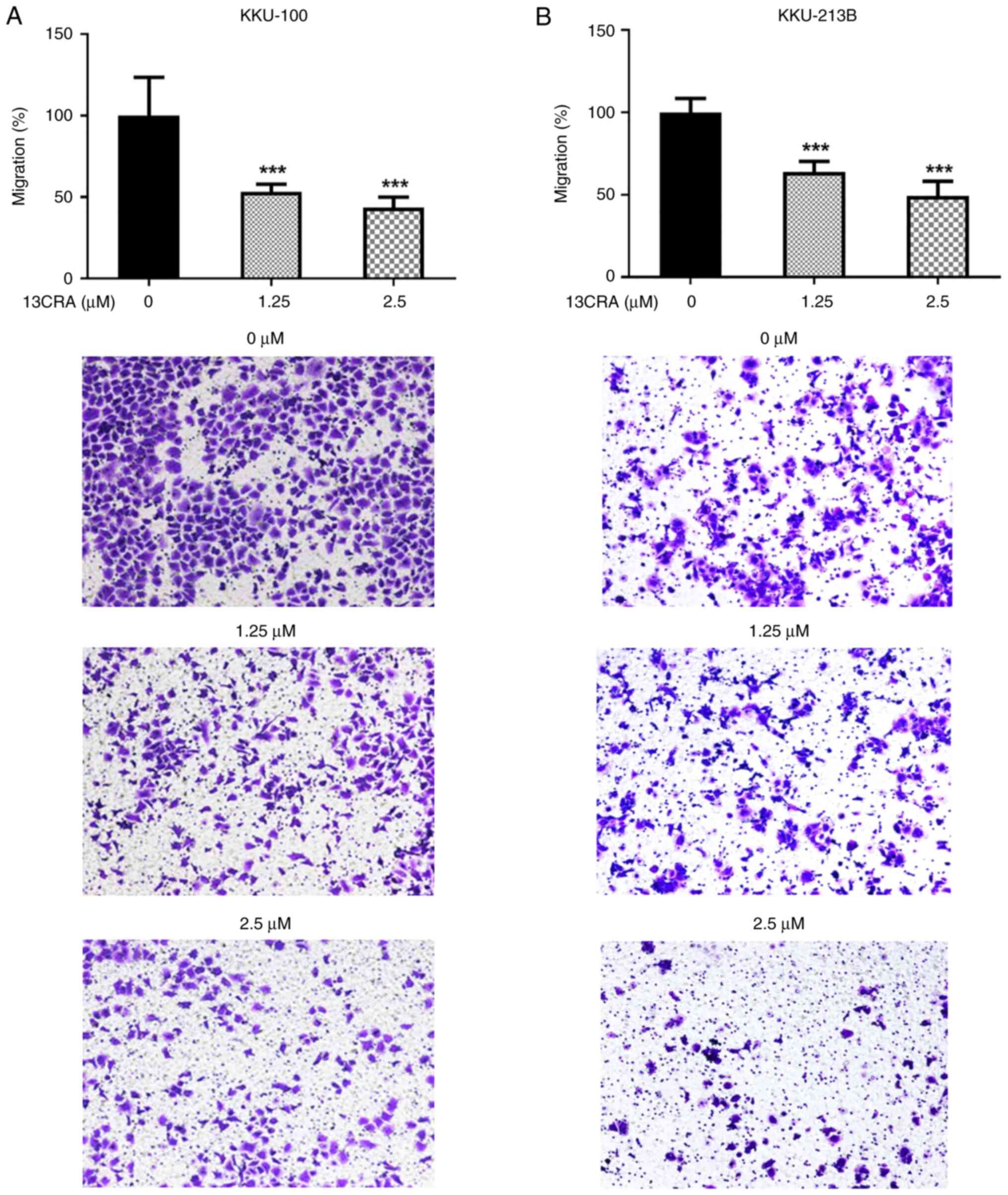
Effects of 13CRA on the migration of CCA
cells
The present study then evaluated the effects of
13CRA on the migratory ability of CCA cells using Transwell
migration assay. The results revealed that 13CRA significantly
suppressed the migration of both KKU-100 and KKU-213B CCA cells
compared with that of the untreated control cells (Fig. 6). At a concentration of 1.25
µM, which exerted markedly low cytotoxicity at 48 h of
treatment, 13CRA inhibited almost 50% of the cells migrating
through the Transwell membrane. These results indicated that 13CRA
potently suppressed the migratory ability of CCA cells.
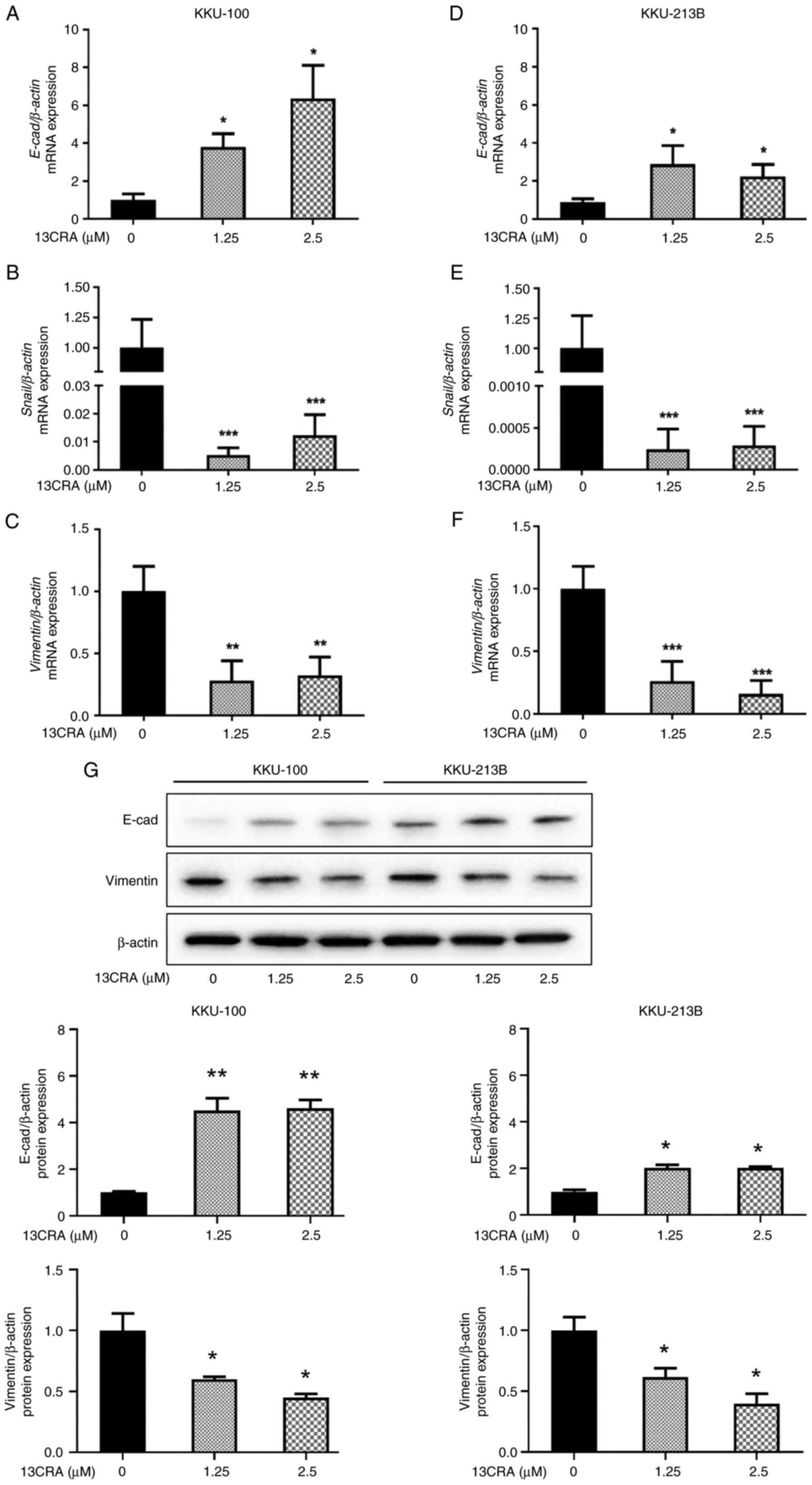
Effects of 13CRA on the expression of
EMT-related genes and proteins in CCA cells
EMT is a cell plasticity-promoting phenomenon that
allows cancer cells to migrate (16,17). Since the present study
demonstrated the inhibitory effects of 13CRA on the migratory
ability of CCA cells, it then investigated the effects of 13CRA on
the mRNA expression of EMT-related genes, including the epithelial
marker, E-cad, and the mesenchymal markers, Snail and
vimentin, using RT-qPCR. Treatment with 13CRA significantly
upregulated the expression of the epithelial marker gene,
E-cad, while it downregulated the expression of the
mesenchymal maker genes, Snail and vimentin, in both
the KKU-100 (Fig. 7A-C) and
KKU-213B (Fig. 7D-F) cells
(P<0.05). Furthermore, the effects of 13CRA on the expression of
two major proteins regulating EMT transition (E-cad and vimentin)
were investigated using western blot analysis. The results revealed
that 13CRA increased the expression of the epithelial marker
protein, E-cad, and decreased the expression of the mesenchymal
maker protein, vimentin, in both CCA cell lines (Fig. 7G). These results indicated that
13CRA modulated the expression of EMT-related genes and proteins in
CCA cells.
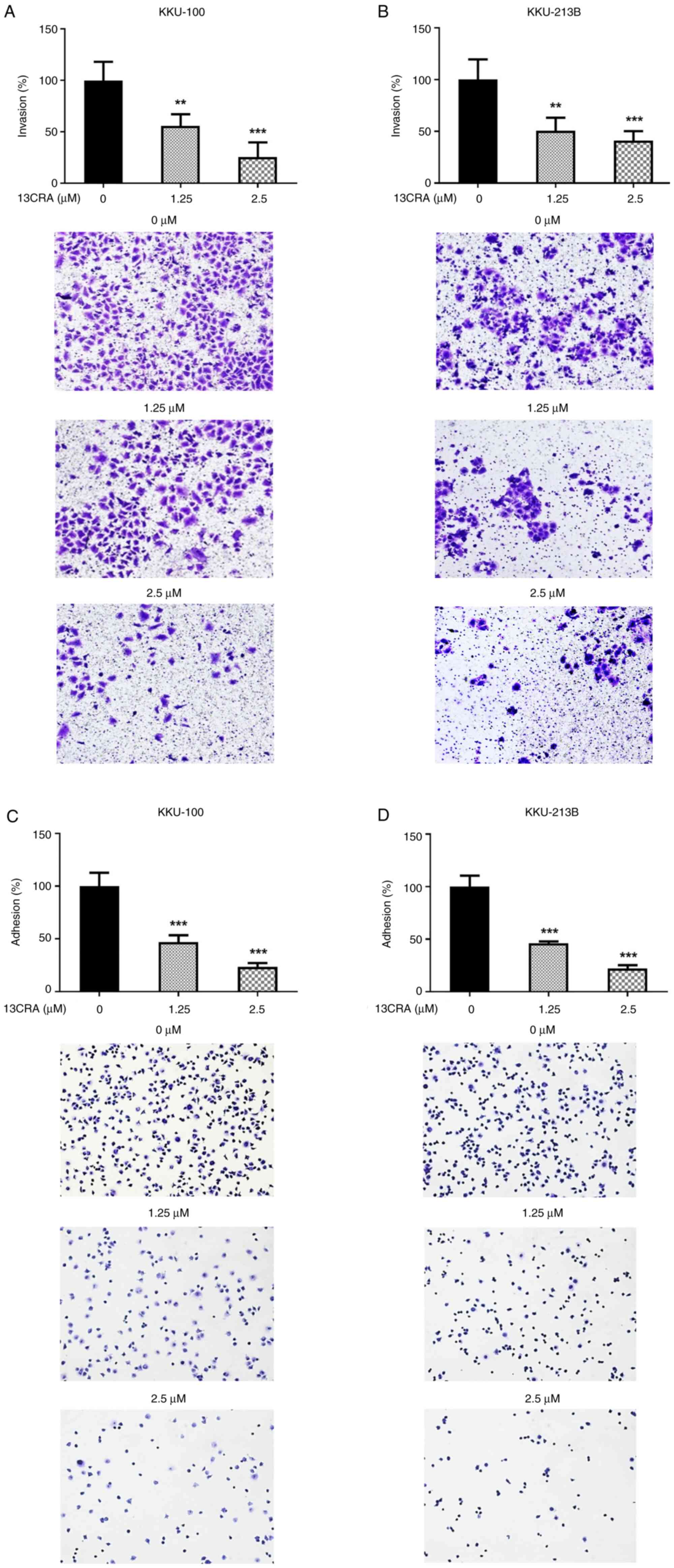
Effects of 13CRA on the invasion and
adhesion of CCA cells
Since cell invasion and adhesion are critical steps
contributing to the metastatic phenotype (16), the present study further examined
the effects of 13CRA on these two properties in CCA cells using
Matrigel invasion and cell adhesion assays. When both CCA cell
lines (KKU-100 and KKU-213B) were treated with 1.25 and 2.5
µM 13CRA for 48 h, the number of invaded cells through the
Transwell membrane coated with Matrigel was significantly
(P<0.05) reduced compared with that of the untreated controls
(Fig. 8A and B). Similarly, 13CRA
treatment induced a significant decrease in the percentage of
adherent cells on the surface of the culture plate in both CCA cell
lines compared with that of the untreated control cells (Fig. 8C and D).
Effects of 13CRA on the expression of
invasion- and adhesion-related genes and proteins in CCA cells
Since 13CRA treatment induced a significant
inhibition of the invasion and adhesion properties of CCA cells,
RT-qPCR was performed to explore the effects of 13CRA on the mRNA
expression of adhesion- and invasion-related genes, including
ICAM-1, COX-2, MMP-2 and MMP-9. Following
treatment for 48 h, treatment with 1.25 and 2.5 µM 13CRA
significantly downregulated the mRNA expression of ICAM-1,
COX-2 and MMP-2 in KKU-100 (Fig. 9A-C) and KKU-213B (Fig. 9E-G) cells (P<0.05).
Furthermore, there was a significant decrease in MMP-9 mRNA
expression in the 13CRA-treated KKU-213B cells (Fig. 9H) compared with that of the
untreated control cells.
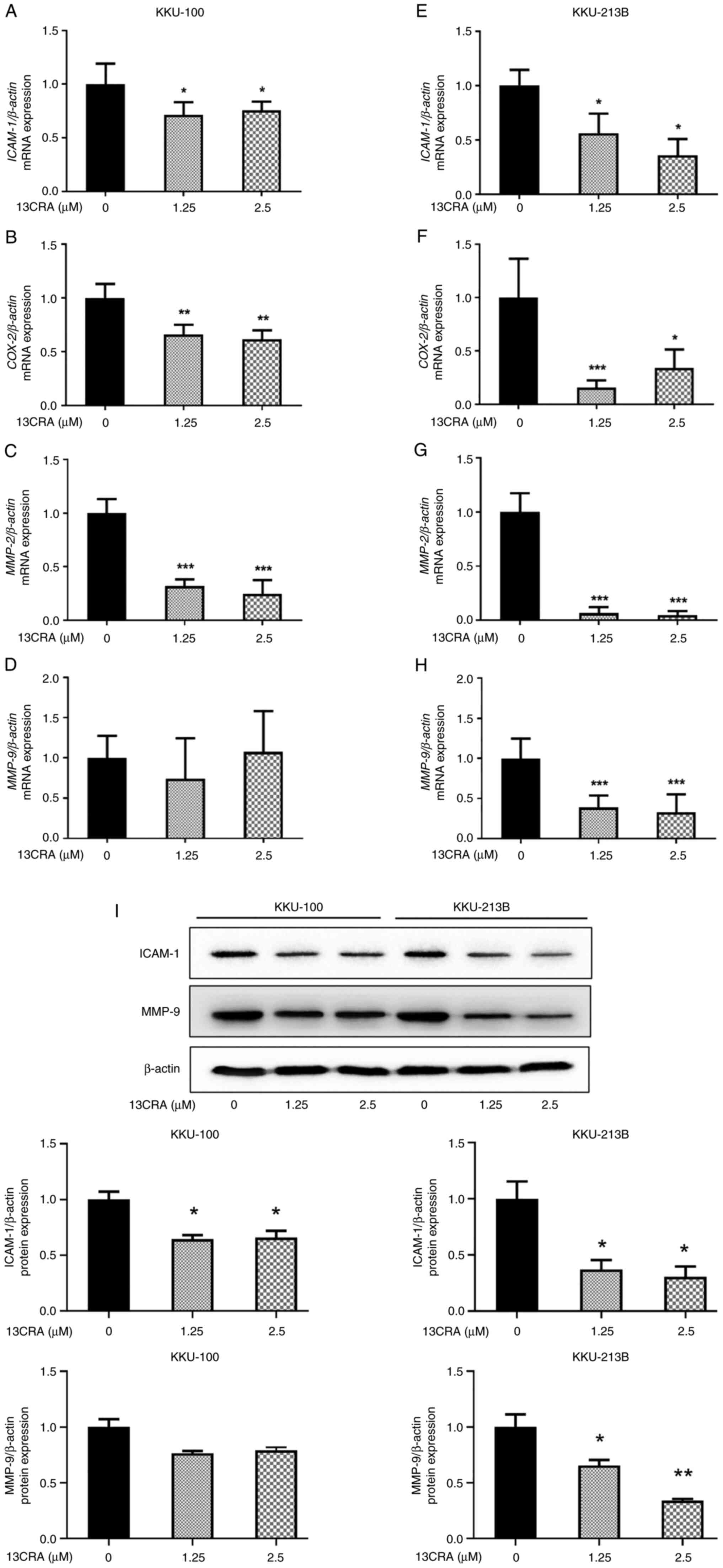 | Figure 913CRA downregulates the expression of
adhesion- and invasion-related genes and proteins in
cholangiocarcinoma cells. (A-D) KKU-100 and (E-H) KKU-213B cells
were treated with 1.25 or 2.5 µM 13CRA for 48 h. The
expression levels of ICAM-1, COX-2, MMP-2 and
MMP-9 genes were determined using reverse
transcription-quantitative PCR, and the quantification of the mRNA
levels of ICAM-1, COX-2, MMP-2 and
MMP-9 was performed via normalization to β-actin. (I)
The expression levels of ICAM-1 and MMP9 proteins were assessed
using western blot analysis and the intensity of the ICAM-1 and
MMP9 bands was quantified by normalization to β-actin. Data are
presented as the mean ± SD from three independent experiments.
*P<0.05, **P<0.01 and
***P<0.001 vs. the untreated control. 13CRA,
13-cis-retinoic acid; MMP, matrix metalloproteinase;
ICAM-1, intercellular adhesion molecule-1; COX-2,
cyclooxygenase-2. |
In addition, the present study further investigated
the effects of 13CRA on the expression of adhesion- and
invasion-related proteins using western blot analysis. The results
revealed that 13CRA decreased the expression of the
adhesion-related protein, ICAM-1, in both CCA cell lines (Fig. 9I). 13CRA decreased the expression
of the invasion-related protein, MMP-9, in KKU-213B cells, and a
decreasing trend in MMP-9 protein expression was also observed in
the KKU-100 cells treated with 13CRA (Fig. 9I). This suppressive effect of
13CRA on the expression of these major adhesion- and
invasion-related genes and proteins in CCA cells supported the
anti-invasion and anti-adhesion activities of 13CRA in CCA
cells.
Discussion
The findings of the present study revealed the
inhibitory effects of 13CRA on the self-renewal, migration,
invasion and adhesion of CCA cells. 13CRA suppressed cell
proliferation by inducing cell cycle arrest at the G2/M
phase and decreased the number of cells in the G1 phase.
Protein analyses using western blotting demonstrated that treatment
of the CCA cells with 13CRA induced a significant increase in the
expression of the cell cycle inhibitor proteins, p53 and p21, and
decreased the expression of cyclin B1 (a protein that regulates the
G2/M transition of the cell cycle) and cyclin D1 (a
regulatory protein of the G1 transition the cell cycle).
RT-qPCR analyses of mRNA expression revealed that 13CRA induced the
concurrent upregulation of p21 expression, and the
downregulation of c-Myc and cyclin B1 expression. In
addition, 13CRA markedly inhibited CCA cell migration by
controlling the expression of EMT-related genes (E-cad,
Snail and vimentin) and proteins (E-cad and
vimentin). Furthermore, 13CRA significantly reduced the invasion
and adhesion of CCA cells, and decreased the expression of
adhesion- and invasion-related genes (ICAM-1, COX-2,
MMP-2 and MMP-9) and proteins (ICAM-1 and MMP-9).
However, even though the anticancer activity of 13CRA in CCA cells
has been demonstrated in the present study, the absence of results
using cholangiocytes (cells used as a control) is a limitation of
the study.
13CRA has been extensively investigated for its
potential use in cancer treatment and prevention. Previous
preclinical studies have demonstrated that 13CRA inhibits the
proliferation, migration and invasion of several cancer types
(4-6,31,32). Previously,
all-trans-retinoic acid (ATRA), a retinoid used for the
treatment of acne and acute promyelocytic leukemia, has been shown
to exert apoptosis-inducing effects on CCA cells (30,33); the present study demonstrated the
cytotoxicity of 13CRA against KKU-100 and KKU-213B cells. Thus,
these results suggest the cytotoxicity of retinoid drugs towards
CCA cells. Considering the IC50 values, the magnitude of
the sensitivity to 13CRA between CCA cells (KKU-100 and KKU-213B)
used herein and breast cancer cells (triple-negative MDA-MB-231
cells) previously used was in the same micromolar range (6). Moreover, the blood levels in the
micromolar range of 13CRA in humans can be achieved by the oral
administration of 80 mg 13CRA twice daily (34). The present study demonstrated the
inhibitory effects of 13CRA on CCA; however, the absence of
experiments on non-cancerous cells used as a negative control is a
limitation of the study.
In the present study, 13CRA exerted a direct
cytotoxic effect and inhibited the self-renewal ability of two CCA
cell lines. It has previously been reported that 13CRA inhibits the
proliferation of breast and gastric cancer cells (5,6).
Considering the concentrations of 13CRA required for an
anti-proliferative effect, the CCA cells were more sensitive to
13CRA treatment than breast and gastric cancer cells (5,6).
Thus, to elucidate the possible underlying mechanisms of the
anti-proliferative effects of 13CRA, the present study analyzed the
cell cycle distribution patterns of 13CRA-treated cells using flow
cytometry. At the concentrations which slightly inhibited cell
proliferation, i.e., those lower than the IC50 value of
13CRA at 48 h, 13CRA inhibited cell cycle progression at the
G2/M phase and decreased the number of cells in the
G1 phase. Retinoid medications are known to induce cell
cycle arrest via the altered expression of genes and proteins of
cell cycle regulators in several cancer types, such as gastric
cancer, melanoma and leukemia cells (35-38). Previously, 13CRA was shown to
induce cell cycle arrest by decreasing DNA synthesis, increasing
p21 protein levels and decreasing cyclin D1 expression in human
sebocytes (15). In the present
study, 13CRA induced cell cycle arrest at the G2/M
phase, and altered the expression of the cell cycle regulatory
molecules, p21 and cyclin B1, at the mRNA and protein levels. In
addition, 13CRA altered the expression of upstream molecules
involved in cell cycle progression by upregulating p53 protein
expression. 13CRA also decreased the expression of cyclin D1, the
regulatory protein of the G1 transition of the cell
cycle. Similarly, ATRA was previously reported to induce cell cycle
arrest at the G0/G1 phase in a human
monoblastic cell line, which was associated with the marked
downregulation of c-Myc and cyclin E levels, and increased p21
expression (39). In the present
study, 13CRA inhibited the self-renewal ability of CCA cells
through G2/M arrest via the alteration of the expression
of cell cycle regulatory genes and proteins.
Targeting the processes indicative of hallmarks of
cholangiocarcinogenesis, such as EMT, stemness and plasticity
properties, has attracted particular attention for chemotherapy
(16). It has been previously
suggested that ATRA reverses the EMT process by upregulating the
levels of epithelial markers and downregulating those of
mesenchymal markers of colorectal cancer and hepatocellular
carcinoma cells (40,41). A previous study demonstrated that
13CRA inhibited vascular endothelial cell migration through the
suppression of NF-κB, a transcription factor that plays a major
role in mediating cancer metastasis (31). The present study revealed the
effects of 13CRA on EMT-related genes and proteins. Treatment of
the CCA cells with 13CRA suppressed vertical cell migration in
association with the reversal of EMT-related markers, i.e., the
upregulation of E-cad (an epithelial marker) and the
downregulation of Snail and vimentin (mesenchymal
markers), and it also increased the protein expression level of
E-cad and decreased the protein expression level of vimentin.
Notably, a previous study reported the markedly low expression of
E-cad in KKU-100 cells, while treatment with 13CRA led to the
upregulation of E-cad expression at both the mRNA and protein
levels (41).
Furthermore, the results of the present study
demonstrated the potent inhibitory effects of 13CRA on CCA cell
invasion and adhesion. At a concentration of 1.25 µM, which
exerted only minimal cytotoxicity, 13CRA inhibited >50% of cell
invasion and adhesion. Previous studies have also reported that
13CRA inhibits the adhesion of squamous cell carcinoma and reduces
the invasion of colon cancer cells; at 1 µM, 13CRA was shown
to exert ~50% inhibition (7,42).
Previous studies have revealed that 13CRA inhibits the invasion of
colon cancer by suppressing the expression of MMP-7 and COX-2
(7,43). The present study demonstrated that
the inhibitory effects of 13CRA on the invasion and adhesion of CCA
cells were possibly associated with the decreased expression of
adhesion- and invasion-related genes and proteins. Notably, the
lack of a change in MMP-9 mRNA expression in KKU-100 cells may be
caused by differences in genetic alterations and backgrounds
between the KKU-100 and KKU-213B CCA cells (28-30,44). Nevertheless, the expression of
MMP-9 exhibited a decreasing trend in KKU-100 cells, which
suggested that 13CRA may modulate other molecules associated with
the post-translational modification of MMP-9 that affect the
stability and activity of the MMP family (45). The inhibitory effects of 13CRA on
the proliferation, migration, invasion and adhesion of CCA cells
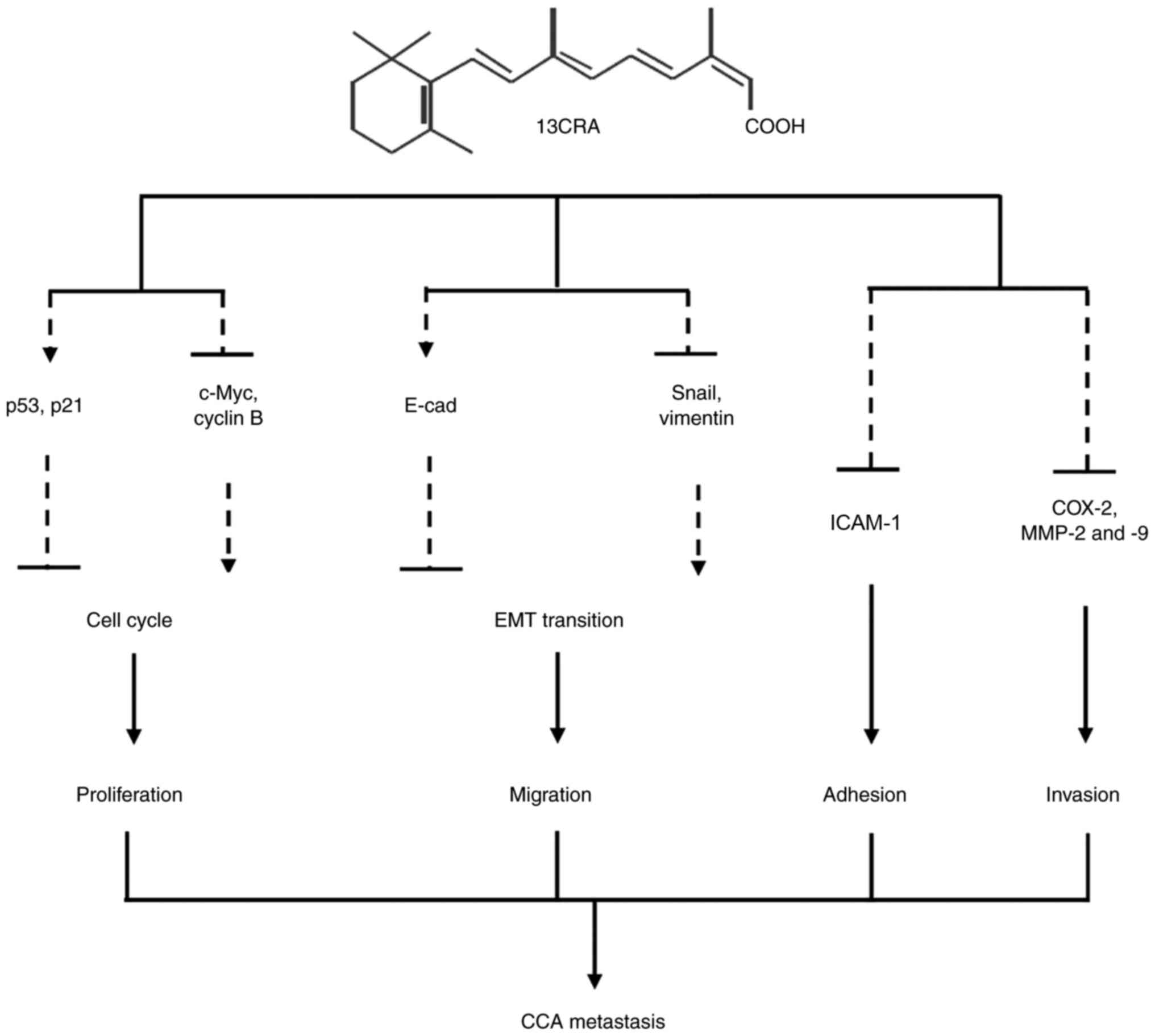
and the possible underlying mechanisms are summarized in Fig. 10. 13CRA can regulate the
expression of several key genes and proteins that control cancer
cell behavior including cell cycle progression, EMT and cell
invasion and adhesion. 13CRA modulates the expression of p53, p21,
c-Myc and cyclin B1, leading to the inhibition of the self-renewal
ability of CCA cells. 13CRA also controls the expression of E-cad,
Snail and vimentin, which subsequently alter EMT and cell
migration. In addition, 13CRA can modify the expression of ICAM-1,
COX-2, MMP-2 and MMP-9, impairing the invasion and adhesion of CCA
cells.
In conclusion, the present study demonstrated that
13CRA reduced cell proliferation by leading to cell cycle arrest at
the G2/M phase. 13CRA suppressed cell migration possibly
though the reversal of the expression of EMT-related genes and
proteins. 13CRA also inhibited cell invasion and adhesion by
suppressing the expression of adhesion- and invasion-related genes
and proteins. Thus, these results indicated that 13CRA may be
useful for CCA therapy; however, the anticancer activity of 13CRA
both in vitro and in vivo models warrants further
investigations to demonstrate the beneficial effects of 13CRA for
the treatment of CCA.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SB participated in performing experiments, analyzing
the results and writing the manuscript. VK, LS and SK collaborated
in analyzing the data and providing critical comments. AP
participated in designing the study, planning the experiments,
analyzing the results and writing the manuscript. SB and AP confirm
the authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Professor Yukifumi
Nawa for editing the MS through Publication Clinic KKU,
Thailand.
Funding
The present study was supported by the Cholangiocarcinoma
Research Institute, Khon Kaen University (grant no. 61003305). SB
was supported by a scholarship for Postgraduate Study Support of
the Faculty of Medicine, Khon Kaen University, Program Year
2016.
References
|
1
|
Meyskens FL Jr, Goodman GE and Alberts DS:
13-Cis-retinoic acid: Pharmacology, toxicology, and clinical
applications for the prevention and treatment of human cancer. Crit
Rev Oncol Hematol. 3:75–101. 1985. View Article : Google Scholar
|
|
2
|
Blaner WS: Cellular metabolism and actions
of 13-cis-retinoic acid. J Am Acad Dermatol. 45:S129–S135. 2001.
View Article : Google Scholar
|
|
3
|
Layton A: The use of isotretinoin in acne.
Dermatoendocrinol. 1:162–169. 2009. View Article : Google Scholar
|
|
4
|
Avis I, Mathias A, Unsworth EJ, Miller MJ,
Cuttitta F, Mulshine JL and Jakowlew SB: Analysis of small cell
lung cancer cell growth inhibition by 13-cis-retinoic acid:
Importance of bioavailability. Cell Growth Differ. 6:485–492.
1995.PubMed/NCBI
|
|
5
|
Jiang SY, Shyu RY, Chen HY, Lee MM, Wu KL
and Yeh MY: In vitro and in vivo growth inhibition of SC-M1 gastric
cancer cells by retinoic acid. Oncology. 53:334–340. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Toma S, Isnardi L, Raffo P, Dastoli G, De
Francisci E, Riccardi L, Palumbo R and Bollag W: Effects of
all-trans-retinoic acid and 13-cis-retinoic acid on breast-cancer
cell lines: Growth inhibition and apoptosis induction. Int J
Cancer. 70:619–627. 1997. View Article : Google Scholar
|
|
7
|
Adachi Y, Itoh F, Yamamoto H, Iku S,
Matsuno K, Arimura Y and Imai K: Retinoic acids reduce matrilysin
(matrix metalloproteinase 7) and inhibit tumor cell invasion in
human colon cancer. Tumour Biol. 22:247–253. 2001. View Article : Google Scholar
|
|
8
|
Guruvayoorappan C, Pradeep CR and Kuttan
G: 13-cis-retinoic acid induces apoptosis by modulating caspase-3,
bcl-2, and p53 gene expression and regulates the activation of
transcription factors in B16F-10 melanoma cells. J Environ Pathol
Toxicol Oncol. 27:197–207. 2008. View Article : Google Scholar
|
|
9
|
Banales JM, Marin JJG, Lamarca A,
Rodrigues PM, Khan SA, Roberts LR, Cardinale V, Carpino G, Andersen
JB, Braconi C, et al: Cholangiocarcinoma 2020: The next horizon in
mechanisms and management. Nat Rev Gastroenterol Hepatol.
17:557–588. 2020. View Article : Google Scholar :
|
|
10
|
Blechacz B: Cholangiocarcinoma: Current
knowledge and new developments. Gut Liver. 11:13–26. 2017.
View Article : Google Scholar :
|
|
11
|
Shin HR, Oh JK, Masuyer E, Curado MP,
Bouvard V, Fang YY, Wiangnon S, Sripa B and Hong ST: Epidemiology
of cholangio-carcinoma: An update focusing on risk factors. Cancer
Sci. 101:579–585. 2010. View Article : Google Scholar
|
|
12
|
Valle JW, Furuse J, Jitlal M, Beare S,
Mizuno N, Wasan H, Bridgewater J and Okusaka T: Cisplatin and
gemcitabine for advanced biliary tract cancer: A meta-analysis of
two randomised trials. Ann Oncol. 25:391–398. 2014. View Article : Google Scholar
|
|
13
|
Feitelson MA, Arzumanyan A, Kulathinal RJ,
Blain SW, Holcombe RF, Mahajna J, Marino M, Martinez-Chantar ML,
Nawroth R, Sanchez-Garcia I, et al: Sustained proliferation in
cancer: Mechanisms and novel therapeutic targets. Semin Cancer
Biol. 35(Suppl): S25–S54. 2015. View Article : Google Scholar
|
|
14
|
Miller DM, Thomas SD, Islam A, Muench D
and Sedoris K: c-Myc and cancer metabolism. Clin Cancer Res.
18:5546–5553. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nelson AM, Gilliland KL, Cong Z and
Thiboutot DM: 13-cis Retinoic acid induces apoptosis and cell cycle
arrest in human SEB-1 sebocytes. J Invest Dermatol. 126:2178–2189.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guan X: Cancer metastases: Challenges and
opportunities. Acta Pharm Sin B. 5:402–418. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tiwari N, Gheldof A, Tatari M and
Christofori G: EMT as the ultimate survival mechanism of cancer
cells. Semin Cancer Biol. 22:194–207. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vaquero J, Guedj N, Clapéron A, Nguyen
Ho-Bouldoires TH, Paradis V and Fouassier L: Epithelial-mesenchymal
transition in cholangiocarcinoma: From clinical evidence to
regulatory networks. J Hepatol. 66:424–441. 2017. View Article : Google Scholar
|
|
19
|
Cadamuro M, Nardo G, Indraccolo S,
Dall'olmo L, Sambado L, Moserle L, Franceschet I, Colledan M,
Massani M, Stecca T, et al: Platelet-derived growth factor-D and
Rho GTPases regulate recruitment of cancer-associated fibroblasts
in cholangiocarcinoma. Hepatology. 58:1042–1053. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Techasen A, Loilome W, Namwat N, Khuntikeo
N, Puapairoj A, Jearanaikoon P, Saya H and Yongvanit P: Loss of
E-cadherin promotes migration and invasion of cholangiocarcinoma
cells and serves as a potential marker of metastasis. Tumour Biol.
35:8645–8652. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fares J, Fares MY, Khachfe HH, Salhab HA
and Fares Y: Molecular principles of metastasis: A hallmark of
cancer revisited. Signal Transduct Target Ther. 5:282020.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dustin ML, Rothlein R, Bhan AK, Dinarello
CA and Springer TA: Induction by IL 1 and interferon-γ: Tissue
distribution, biochemistry, and function of a natural adherence
molecule (ICAM-1). J Immunol. 1986.137:245–254. View Article : Google Scholar
J Immunol. 186:5024–5033. 2011.
|
|
23
|
Fosslien E: Review: Molecular pathology of
cyclooxygenase-2 in cancer-induced angiogenesis. Ann Clin Lab Sci.
31:325–348. 2001.PubMed/NCBI
|
|
24
|
Wagenaar-Miller RA, Hanley G,
Shattuck-Brandt R, DuBois RN, Bell RL, Matrisian LM and Morgan DW:
Cooperative effects of matrix metalloproteinase and
cyclooxygenase-2 inhibition on intestinal adenoma reduction. Br J
Cancer. 88:1445–1452. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Itatsu K, Sasaki M, Yamaguchi J, Ohira S,
Ishikawa A, Ikeda H, Sato Y, Harada K, Zen Y, Sato H, et al:
Cyclooxygenase-2 is involved in the up-regulation of matrix
metalloproteinase-9 in cholangiocarcinoma induced by tumor necrosis
factor-alpha. Am J Pathol. 174:829–841. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tuponchai P, Kukongviriyapan V, Prawan A,
Kongpetch S and Senggunprai L: Myricetin ameliorates
cytokine-induced migration and invasion of cholangiocarcinoma cells
via suppression of STAT3 pathway. J Cancer Res Ther. 15:157–163.
2019.
|
|
27
|
Kaewmeesri P, Kukongviriyapan V, Prawan A,
Kongpetch S and Senggunprai L: Cucurbitacin B diminishes metastatic
behavior of cholangiocarcinoma cells by suppressing focal adhesion
kinase. Asian Pac J Cancer Prev. 22:219–225. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sripa B, Leungwattanawanit S, Nitta T,
Wongkham C, Bhudhisawasdi V, Puapairoj A, Sripa C and Miwa M:
Establishment and characterization of an opisthorchiasis-associated
cholangiocarcinoma cell line (KKU-100). World J Gastroenterol.
11:3392–3397. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sripa B, Seubwai W, Vaeteewoottacharn K,
Sawanyawisuth K, Silsirivanit A, Kaewkong W, Muisuk K, Dana P,
Phoomak C, Lert-Itthiporn W, et al: Functional and genetic
characterization of three cell lines derived from a single tumor of
an Opisthorchis viverrini-associated cholangiocarcinoma patient.
Hum Cell. 33:695–708. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Butsri S, Kukongviriyapan V, Senggunprai
L, Kongpetch S and Prawan A: All-trans-retinoic acid induces
RARB-dependent apoptosis via ROS induction and enhances cisplatin
sensitivity by NRF2 downregulation in cholangiocarcinoma cells.
Oncol Lett. 23:1792022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Seyfried TN and Huysentruyt LC: On the
origin of cancer metastasis. Crit Rev Oncog. 18:43–73. 2013.
View Article : Google Scholar :
|
|
32
|
Guruvayoorappan C and Kuttan G: 13
cis-retinoic acid regulates cytokine production and inhibits
angiogenesis by disrupting endothelial cell migration and tube
formation. J Exp Ther Oncol. 7:173–182. 2008.PubMed/NCBI
|
|
33
|
Chung KD, Jeong YI, Chung CW, Kim DH and
Kang DH: Anti-tumor activity of all-trans retinoic
acid-incorporated glycol chitosan nanoparticles against HuCC-T1
human cholangiocarcinoma cells. Int J Pharm. 422:454–461. 2012.
View Article : Google Scholar
|
|
34
|
Veal GJ, Cole M, Errington J, Pearson AD,
Foot AB, Whyman G and Boddy AV; UKCCSG Pharmacology Working Group:
Pharmacokinetics and metabolism of 13-cis-retinoic acid
(isotretinoin) in children with high-risk neuroblastoma-a study of
the United Kingdom children's cancer study group. Br J Cancer.
96:424–431. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nguyen PH, Giraud J, Staedel C,
Chambonnier L, Dubus P, Chevret E, Boeuf H, Gauthereau X, Rousseau
B, Fevre M, et al: All-trans retinoic acid targets gastric cancer
stem cells and inhibits patient-derived gastric carcinoma tumor
growth. Oncogene. 35:5619–5628. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ozeki M and Shively JE: Differential cell
fates induced by all-trans retinoic acid-treated HL-60 human
leukemia cells. J Leukoc Biol. 84:769–779. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang H, Satyamoorthy K, Herlyn M and
Rosdahl I: All-trans retinoic acid (atRA) differentially induces
apoptosis in matched primary and metastatic melanoma cells-a
speculation on damage effect of atRA via mitochondrial dysfunction
and cell cycle redistribution. Carcinogenesis. 24:185–191. 2003.
View Article : Google Scholar
|
|
38
|
Zhang H and Rosdahl I: Expression of p27
and MAPK proteins involved in all-trans retinoic acid-induced
apoptosis and cell cycle arrest in matched primary and metastatic
melanoma cells. Int J Oncol. 25:1241–1248. 2004.
|
|
39
|
Dimberg A, Bahram F, Karlberg I, Larsson
LG, Nilsson K and Oberg F: Retinoic acid-induced cell cycle arrest
of human myeloid cell lines is associated with sequential
down-regulation of c-Myc and cyclin E and posttranscriptional
up-regulation of p27(Kip1). Blood. 99:2199–2206. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cui J, Gong M, He Y, Li Q, He T and Bi Y:
All-trans retinoic acid inhibits proliferation, migration, invasion
and induces differentiation of hepa1-6 cells through reversing EMT
in vitro. Int J Oncol. 48:349–357. 2016. View Article : Google Scholar
|
|
41
|
Shi G, Zheng X, Wu X, Wang S, Wang Y and
Xing F: All-trans retinoic acid reverses epithelial-mesenchymal
transition in paclitaxel-resistant cells by inhibiting nuclear
factor kappa B and upregulating gap junctions. Cancer Sci.
110:379–388. 2019. View Article : Google Scholar
|
|
42
|
Speyer MT, Jackson JA and Burkey BB: The
effects of 13-cis retinoic acid on squamous cell carcinoma
proliferation and adhesion to extracellular matrix proteins.
Laryngoscope. 107:44–48. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jiang C, Wang Q, Xu Z, Li WS, Chen C, Yao
XQ and Liu FK: Cyclooxygenase-2 knockdown using retinoic acid
chalcone (RAC), a promising therapeutic strategy for colon cancer.
Am J Cancer Res. 5:2012–2021. 2015.PubMed/NCBI
|
|
44
|
Saensa-Ard S, Leuangwattanawanit S,
Senggunprai L, Namwat N, Kongpetch S, Chamgramol Y, Loilome W,
Khansaard W, Jusakul A, Prawan A, et al: Establishment of
cholangiocarcinoma cell lines from patients in the endemic area of
liver fluke infection in Thailand. Tumour Biol.
39:10104283177259252017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Madzharova E, Kastl P, Sabino F and Auf
dem Keller U: Post-translational modification-dependent activity of
matrix metalloproteinases. Int J Mol Sci. 20:30772019. View Article : Google Scholar : PubMed/NCBI
|