Introduction
Liver fibrosis is the pathological basis of most
liver diseases. Activation of static hepatic stellate cells (HSCs)
and disordered proliferation are the main developmental steps of
liver fibrosis (1-3). Drug toxicity, viral infection or
alcohol may cause static HSCs to lose intracellular lipid droplets,
release large amounts of extracellular matrix components, including
α-SMA and COL1A1, and transform from a non-split phenotype to a
myofibroblast-like cell phenotype (4,5).
In other words, HSCs switch from an inactive state to an active
state. Further exploration of the potential mechanism of HSCs
development is required to prevent liver fibrosis.
Previous studies have reported the pivotal role of
noncoding RNAs [including microRNAs (miRNAs/miRs), circular RNAs
and long noncoding RNAs] in HSCs (6,7).
miRNAs are a highly conserved form of noncoding single-stranded
RNA, which consist of 20-26 bases, and regulate a variety of
physiological and pathological processes in the body (8,9).
miR-29b-3p has been reported to serve a key role in a wide range of
pathological processes, such as myocardial injury, ion channel
opening and insulin resistance (10,11). Although its antifibrotic function
has been reported previously, the specific downstream mechanism has
not been completely clarified.
VEGFA is secreted by hepatocytes and HSCs, and is
the core factor for maintaining fenestration of hepatic sinusoidal
endothelial cells; however, the specific crosstalk of VEGFA in the
progression of hepatic fibrosis is unclear. VEGF has been reported
to induce the transformation of HSCs into fibroblasts that
proliferate and synthesize type I collagen in a rat model of liver
fibrosis (12). Moreover, VEGF
expression in HSCs has been shown to be significantly increased in
hypoxic environments (13).
Treatment of HSCs with recombinant VEGF and recombinant
angiopoietin-1 (Ang-1) can produce similar effects that transform
HSCs into fibroblasts, and contribute to the proliferation and
synthesis of type I collagen (12).
Autophagy is a ubiquitous phenomenon in eukaryotic
cells that has an important role in numerous aspects of life.
Autophagy is mainly mediated by the PI3K/AKT/mTOR, sirtuin (SIRT)
and AMP-activated protein kinase (AMPK) pathways. The PI3K/AKT/mTOR
pathway can negatively regulate autophagy-related protein activity,
and PI3K agonists effectively block autophagy (1-3).
Heat shock protein 60 (HSP60) is mainly distributed in the
cytoplasm and mitochondria, and is encoded by the nuclear genome.
Notably, HSP60 is important in the transport, activation and
folding of mitochondrial proteins; therefore, imbalance of the
HSP60 protein may induce mitochondrial destruction and apoptosis
via the mitochondrial pathway (14).
Dihydroartemisinin (DHA) is a natural and safe
antimalarial drug (15). Our
previous studies confirmed that DHA protected rats from liver
fibrosis induced by intra-peritoneal injection of carbon
tetrachloride (CCl4) or bile duct ligation by
influencing HSCs migration, contraction and senescence (16,17). However, to the best of our
knowledge, the potential relationship between DHA and miR-29b-3p,
and their specific downstream signals, have not been determined;
the present study aimed to address this.
Materials and methods
Reagents and antibodies
DHA (cat. no. D7439), CCl4 (cat. no.
488488) and dimethyl sulfoxide (cat. no. D2650) were purchased from
MilliporeSigma. 740Y-P (cat. no. P0175) was purchased from
MedChemExpress. Dulbecco's modified Eagle's medium (DMEM; cat. no.
11995-065), fetal bovine serum (FBS), phosphate-buffered saline
(PBS), penicillin-streptomycin solution (100X), mitomycin and
trypsin EDTA were purchased from Gibco, Thermo Fisher Scientific,
Inc. Small interfering RNA (siRNA) targeting VEGFA, VEGFR1 and
VEGFR2, and negative control (NC) siRNA were obtained by Nanjing
KeyGen Biotech. Co., Ltd. Wild-type (WT) 3′UTR-psiCHECK2-VEGFA and
mutant (MUT) 3′UTR-psiCHECK2-VEGFA plasmids were obtained by
Hedgehogbio Co., Ltd. P3*Flag-CMV-14-HSP60 and its NC
(empty plasmid) were purchased from Eproll. Co., Ltd.
AAV8-GP-3-rno-miR-29b-3p mimic, AAV8-GP-3-rno-miR-29b-3p mimic NC,
miR-29b-3p mimic, mir-29b-3p inhibitor and their NC vectors were
obtained by Jima Biological Company. The corresponding siRNA and
plasmid sequences are listed in Table SI.
Animal experiment
The animal experiment was approved by Institutional
and Local Committee of Nanjing University of Chinese Medicine
(approval no. A20402; Nanjing, China). The present study was
carried out based on the guidelines of the Care and Use of Animals
of Nanjing University of Chinese Medicine. A total of 36 male
Sprague Dawley rats (weight, 220-260 g; age, 6 weeks) were
purchased from Shanghai SLAC Laboratory Animal Co., Ltd. Rats were
maintained under a standard 12-h light/dark cycle, at an indoor
temperature of 20±2°C and humidity of 50±10%, and their bedding was
autoclaved. Rats received standard food (5 g/100 g/day) and
drinking water (11 ml/100 g/day). The health and behavior of the
animals were monitored once a day to ensure reliability of the
experiments. A liver fibrosis model was constructed via
intraperitoneal (IP) injection of CCl4 and olive oil
[1:9 (v/v); 0.5 ml/100 g body weight]. A total of 36 rats were
randomly divided into the following six groups (6 rats/group): i)
Control group, ii) CCl4 group, iii) CCl4 +
AAV8-miR NC group, iv) CCl4 + DHA group, v)
CCl4 + AAV8-miR group and vi) CCl4 + AAV8-miR
+ DHA group. The dosage of DHA and administration route were
selected based on our previous study (13). CCl4 and olive oil were
injected into rats in all of the groups, with the exception of the
control group, every other day from week 1 to week 8. Rats in the
control group received olive oil without CCl4. At the
beginning of week 4, rats in the CCl4 + AAV8-miR NC
group were administered AAV8-GP-3-rno-miR-29b-3p mimics NC
(6.02×1011 V.G/ml; 32 µl/100 g body weight),
whereas rats in the CCl4 + AAV8-miR and the
CCl4 + AAV8-miR + DHA group were administered
AAV8-GP-3-rno-miR-29b-3p mimics (4.16×1012 V.G/ml; 160
µl/100 g body weight) by caudal vein. At the beginning of
week 2, rats in the CCl4 + DHA group and the
CCl4 + AAV8-miR + DHA group were administered DHA by IP
injection (3 mg/ml; 2 g/100 g body weight). DHA was suspended in
olive oil and given once a day. The duration of the experiment was
2 months, starting from the first IP injection of CCl4
until the end of the 8th week. After the experiment, the mice were
weighed and were anesthetized with pentobarbital sodium (50 mg/kg).
Blood was collected from the orbital vein of rats at a volume of
0.5 ml quickly without delay and immediately underwent biochemical
index detection. Subsequently, the rats were euthanized by
intraperitoneal injection of an overdose of pentobarbital sodium
(100-150 mg/kg). Euthanasia was confirmed through the observation
of respiratory, heartbeat, pupil and nerve reflexes, and other
indications. Blood and liver samples were collected from rats in
each group for serum biochemical analysis, tissue
immunofluorescence, reverse transcription-quantitative PCR
(RT-qPCR) and western blot analysis.
Hematoxylin and eosin (H&E), Masson's
trichrome and Sirius Red staining
The rat liver tissues were fixed with 4%
paraformaldehyde at 37°C for 0.5 h. The liver tissues were then
dehydrated with various concentrations of alcohol, embedded in
paraffin and sectioned (5 µm). Subsequently, the sections
were stained with hematoxylin (cat. no. BP-DL019; Nanjing SenBeiJia
Biological Technology Co., Ltd.) for 5 min, washed with 1X PBS and
differentiated with 1% hydrochloric alcohol for a few seconds at
room temperature. Finally, the sections were stained with eosin
(cat. no. BP-DL010; Nanjing SenBeiJia Biological Technology Co.,
Ltd.) for 3 min at room temperature. Masson's trichrome and Sirius
red staining were used for the evaluation of collagen expression.
The Masson's trichrome kit (cat. no. BP-DL371; Nanjing SenBeiJia
Biological Technology Co., Ltd) and the Sirius red staining kit
(cat. no. BP-DL030; Nanjing SenBeiJia Biological Technology Co.,
Ltd.) were carried out according to the manufacturer's instructions
at room temperature. The representative images were obtained under
an inverted light microscope (Zeiss GmbH).
Cell culture
HSCs-LX-2 cells (BNCC337957) were purchased from
BeNa Culture Collection; Beijing Beina Chuanglian Institute of
Biotechnology. The cells were cultured in DMEM containing 10% FBS
and 1% penicillin-streptomycin solution (100X) at 37°C in a
humidified incubator with 5% CO2 and 95% air.
Drug intervention
After the HSCs were cultured until they adhered to
the wells, DHA was added at 10, 20 and 40 µM for 24 h at
37°C. In addition, cells were treated with 740Y-P (15 µM)
for 24 h at 37°C, and with the VEGFR1-specific inhibitor GNQWFI or
the VEGFR2-specific inhibitor SU5416 (0.02, 0.2, 2 and 10
µM) for 24 h at 37°C.
Cell transfection
According to the instructions, 100 nM siRNA-VEGFA,
miR-29b-3p mimic, miR-29b-3p inhibitor, siRNA-VEGFR1 and
siRNA-VEGFR2, and 500 ng/µl CMV-14-HSP60, as well as their
NCs (with the same concentration) were transfected into LX-2 cells
(3×105 cells/well) in 6-well microplates with 3
µl Lipofectamine® 2000 (cat. no. 11668027; Thermo
Fisher Scientific, Inc.). The ratio of plasmid or siRNA with
Lipofectamine 2000 was 1:1. The transfected cells were cultured in
normal conditions at 37°C for 12 h. RT-qPCR was then performed to
verify the transfection efficiency and subsequent experiments were
performed. Subsequent experimentation was performed after 12 h of
transfection.
RT-qPCR
The LX-2 cells were lysed with TRIzol®
(Invitrogen; Thermo Fisher Scientific, Inc.), total RNA was
collected with isopropanol (Shanghai Aladdin Biochemical Technology
Co., Ltd.) and RNA content was determined using an enzyme labeling
instrument. In addition, rat liver tissues were cut into pieces and
rapidly frozen with liquid nitrogen prior to extraction of total
RNA using TRIzol. RNA was then reverse transcribed into cDNA with
Hifair® II 1st Strand cDNA Synthesis kit (Shanghai
Yeasen Biotechnology Co., Ltd.) according to the manufacturer's
protocol. cDNA was amplified with Hieff Qreal-time PCR SYBR Green
main mixture (low Rox +) (Shanghai Yeasen Biotechnology Co., Ltd.)
and detected using an Applied Biosystems 7500 system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. qPCR was performed under the following
conditions: Initial denaturation at 95°C for 5 min, followed by 40
cycles at 95°C for 15 sec, 60°C for 60 sec and 72°C for 40 sec, and
a final step at 72°C for 5 min. The expression levels of mRNA and
miRNA were normalized to β-actin and U6 expression, respectively,
and the 2−ΔΔCq method was used to calculate the
expression levels (13). The
primer sequences used for RT-qPCR are listed in Table SI.
Western blot analysis
The LX-2 cells were washed with 1X PBS for three
times and then total proteins were extracted using RIPA lysis
buffer (cat. no. P0013C; Beyotime Institute of Biotechnology). The
rat liver tissues was cut into pieces and rapidly frozen with
liquid nitrogen prior to total protein extraction using RIPA lysis
buffer. Total protein concentration was determined using the BCA
protein analysis kit (cat. no. 23225; Thermo Fisher Scientific,
Inc.). The same concentration of proteins (30 µg/sample)
were separated by SDS-PAGE and the SDS gel percentage was chosen
according to the molecular level of the target protein (10, 12 or
15%). Proteins were then transferred to PVDF membranes
(microporous; cat. no. IPVH00010; MilliporeSigma) and blocked with
5% BSA (cat. no. ST025; Beyotime Institute of Biotechnology) at
37°C for 1 h in TBS + 0.1% Tween-20. The PVDF membranes were then
incubated with primary antibodies (1:1,000) at 4°C overnight and
with the secondary antibody (1:10,000) at 37°C for 2 h. Finally,
the membranes were incubated with ECL (cat. no. 32109; Thermo
Fisher Scientific, Inc.) and band intensity was semi-quantified by
Image Lab software v3.0 (Bio-Rad Laboratories, Inc.). The primary
antibodies β-actin (cat. no. AF7018) were purchased from Affinity
Biosciences Ltd. The primary antibodies against α-SMA (cat. no.
14395-1-AP), COL1A1 (cat. no. 67288-1-Ig), VEGFA (cat. no.
19003-1-AP) and VEGFR2 (cat. no. 26415-1-AP) were purchased from
ProteinTech Group, Inc. The primary antibody against ubiquitin
(cat. no. R26024) was purchased from Chengdu Zen Bioscience Co.,
Ltd. The primary antibodies against VEGFR1 (cat. no. A19132), AKT
(cat. no. A20799), phosphorylated (p)-AKT (cat. no. AP1259), mTOR
(cat. no. A2445), p-mTOR (cat. no. AP0115), ULK1 (cat. no. A8529),
p-ULK1 (cat. no. AP0736), caspase-9 (cat. no. A18676), cytochrome
c (cat. no. A4912), AIF (cat. no. A19536), LC3B (cat. no.
A19665), Beclin-1 (cat. no. A7353) and P62 (cat. no. A19700) were
purchased from ABclonal Biotech Co., Ltd. The HRP Goat Anti-Rabbit
IgG (H+L) secondary antibody (cat. no. AS014) and HRP Goat
Anti-Mouse IgG (H+L) secondary antibody (cat. no. AS003) were
purchased from ABclonal Biotech Co., Ltd.
Immunofluorescence staining
The protein expression levels of LX-2 cells were
measured by immunofluorescence staining. LX-2 cells
(1×105 cells/well) were seeded into a 24-well
microplate. After intervention, the cells were fixed with 4%
paraformaldehyde at 37°C for 0.5 h and blocked with 1% BSA at 37°C
for 2 h. The cells were incubated with primary antibodies against
α-SMA (cat. no. 14395-1-AP), COL1A1 (cat. no. 67288-1-Ig) and VEGFA
(cat. no. 19003-1-AP) (all from ProteinTech Group, Inc.) at 4°C
overnight (1:200) and with FITC-conjugated anti-rabbit or
anti-mouse secondary antibodies (1:200; cat. nos. ab6717 and
ab6785; Abcam) at 37°C for 2 h. All images were captured under an
inverted fluorescence microscope (Zeiss GmbH). Liver tissues (5
µm) were also assessed by immunofluorescence staining using
the same technique as aforementioned.
Oil red O staining
LX-2 cells (1×105 cells/well) were seeded
into 24-well microplates and stained with 200 µl oil red O
(Nanjing Jiancheng Bioengineering Institute) at room temperature
for 8 min according to the manufacturer's instructions. The
representative images were obtained under an inverted light
microscope (Zeiss GmbH).
Nile red staining
LX-2 cells (1×105 cells/well) were seeded
into 24-well microplates and fixed with 4% paraformaldehyde at 37°C
for 0.5 h. Subsequently, they were stained with 500 µl Nile
Red (Nanjing Jiancheng Bioengineering Institute) at 37°C for 10 min
according to the manufacturer's instructions. The representative
images were obtained under an inverted light microscope (Zeiss
GmbH).
Determination of triglyceride (TG)
levels
The TG levels were measured using the TG detection
kit (cat. no. SY6206; Yita Biological Co., Ltd.) according to the
manufacturer's instructions. After transfection with miR-29b-3p
mimic NC, miR-29b-3p mimic, miR-29b-3p inhibitor NC and miR-29b-3p
inhibitor, the optical density of cells from the four
aforementioned groups was determined at 570 nm using a plate reader
(Thermo Fisher Scientific, Inc.).
Determination of cholesterol levels
The total cholesterol (TC) levels were measured
using the TC detection kit (cat. no. SY6226; Yita Biological Co.,
Ltd.) according to the manufacturer's instructions. After
transfection with miR-29b-3p mimic NC, miR-29b-3p mimic, miR-29b-3p
inhibitor NC and miR-29b-3p inhibitor, the optical density of cells
from the four aforementioned groups was determined at 570 nm using
a plate reader (Thermo Fisher Scientific, Inc.).
IL-6 detection
IL-6 levels were measured using an IL-6 ELISA kit
(cat. no. RK00004; ABclonal Biotech Co., Ltd.) according to the
manufacturer's instructions. LX-2 cells (3×104
cells/well) were seeded into 96-well microplates and incubated at
37°C for 24 h in 5% CO2. After transfection with VEGFA
siRNA, VEGFR1 siRNA and VEGFR2 siRNA, the optical density of the
corresponding samples was determined at 570 nm using a plate reader
(Thermo Fisher Scientific, Inc.).
Luciferase assay
LX-2 cells (3×105 cells/well) were seeded
into a 6-well microplate. Subsequently, 100 nM miR-29b-3p mimic NC
or 100 nM miR-29b-3p mimic was transfected into LX-2 cells, and
then 200 ng WT 3′UTR-psiCHECK2-VEGFA vector or MUT
3′UTR-psiCHECK2-VEGFA vector (cat. no. HH-LUC-036, HedgehoBio
Science and Technology Co., Ltd.) was transfected into cells with
Lipofectamine 2000 at 37°C for 12 h in an atmosphere containing 5%
CO2. Subsequently, cells (1.0×105 cells/ml)
were lysed with the lysis buffer in the Luciferase Reporter Gene
Assay Kit (cat. no. 11401ES60; Shanghai Yeasen Biotechnology Co.,
Ltd.), collected and treated with luciferase test reagent (cat. no.
11401-B; Shanghai Yeasen Biotechnology Co., Ltd.). Luciferase
activity was measured using a Multiskan FC photometer (Thermo
Fisher Scientific, Inc.). Luciferase activity was normalized to a
blank control group.
Mitochondrial membrane potential
detection
LX-2 cells (1×105 cells/well) were seeded
into 24-well microplates and incubated for 24 h at 37°C in 5%
CO2. After intervention, the LX-2 cells were treated
with 10 µM JC-1 reagent (cat. no. SY0910; Yita Biological
Co., Ltd.) at 37°C for 15 min. Subsequently, JC-1 aggregate
fluorescence (excitation wavelength, 530 nm; emission wavelength,
583 nm) and JC-1 monomer fluorescence (excitation wavelength, 488
nm; emission wavelength, 525 nm) were measured using a Multiskan FC
photometer (Thermo Fisher Scientific, Inc.). Representative images
were captured using an inverted fluorescence microscope (Zeiss
GmbH).
Annexin V-FITC/PI double staining
Annexin V-FITC/PI double staining assay was
conducted with the Annexin V-FITC Apoptosis Detection Kit (Beyotime
Institute of Biotechnology). LX-2 cells were collected and
dissolved in Annexin V-FITC binding buffer at a density of
1.0×106 cells/ml. Subsequently, 100 µl sample
solution was treated with 5 µl FITC-conjugated Annexin V
reagent and 5 µl PI reagent, and incubated at room
temperature for 15 min in the dark. The percentages of cells within
each quadrant (Q1, Q3, Q3 and Q4) were determined using a Gallios
flow cytometer (Beckman Coulter, Inc.). The results were analyzed
by FlowJo software v10.6.2 (FlowJo, LLC).
Immunoprecipitation assay
LX-2 cells were washed with 1X PBS and total
proteins were extracted using 500 µl RIPA lysis buffer
(Beyotime Institute of Biotechnology). The homogenates were
centrifuged at 1,300 × g for 30 min at 4°C. Subsequently, 20% of
the lysate supernatant was used as the input for western blot
analysis and the remaining lysate was used to perform the
immunoprecipitation assay. The binding/detergent solution (20 mM
Na2HPO4, 0.15 M NaCl; pH 7.0) was prepared
for later use. The HSP60 antibody (cat. no. A0564) was purchased
from ABclonal Biotech Co., Ltd.; 5 µl HSP60 antibody was
added to the remaining lysate, and the proteins were shaken
overnight at 4°C. Subsequently, 50 µl protein A/G plus
agarose beads (Santa Cruz Biotechnology, Inc.) were added to the
previously prepared antigen-antibody binding complex at 4°C
overnight to make them couple with each other. Subsequently, the
bead-antibody-antigen complex was centrifuged at 1,300 × g for 3
min at 4°C. The supernatant was carefully extracted, and the
agarose beads were washed with 1 ml binding/detergent solution
three times. After washing, the magnetic bead-antibody-antigen
complex was added to 30 µl 2X SDS-PAGE loading buffer, mixed
and heated in a 95°C water bath for 15 min. The supernatant was
then taken for western blot analysis.
Cell Counting Kit-8 (CCK-8) assay
LX-2 cells (3×104 cells/well) were seeded
into 96-well microplates and incubated at 37°C for 24 h in 5%
CO2. After intervention, cells were treated with 10
µl CCK-8 reagent (Shanghai Yeasen Biotechnology Co., Ltd.)
and incubated at 37°C for 4 h. The optical density was determined
at 570 nm using a plate reader (Thermo Fisher Scientific,
Inc.).
Trypan blue staining
LX-2 cells (1×105 cells/well) were seeded
into 24-well microplates and incubated at 37°C for 24 h in 5%
CO2. After intervention, the cells were treated with
0.4% trypan blue solution (MilliporeSigma) at a ratio of 9:1 at
room temperature for 3 min. The representative images were obtained
under an inverted light microscope (Zeiss GmbH).
Adhesion assay
Rat tail tendon collagen type I (cat. no. C8062;
Beijing Solarbio Science & Technology Co., Ltd.) was used to
coat a 96-well microplate at the concentration of 2 mg/ml, which
was dissolved in 0.006 mol/l acetic acid. NaOH (0.1 mol/l) was then
added to promote gelation at 37°C for 2 h. The uncoated well was
used as the negative control. Each well was washed three times with
1X PBS, and the LX-2 cells (5×104 cells/well) were
seeded into the coated and uncoated wells, before being incubated
for 0.5, 1 and 2 h at 37°C in 5% CO2. Subsequently, each
well was washed with 1X PBS to remove the unattached cells. The
viability of adherent cells was measured using an MTT assay.
Dimethyl sulfoxide was used to dissolve the purple formazan and the
absorbance values at 490 nm were measured using a Synergy2
Microplate reader (BioTek Corporation). The representative images
at 2 h were obtained under an inverted light microscope (Zeiss
GmbH).
Wound healing assay
LX-2 cells (3×105 cells/well) were seeded
into a 6-well microplate until they formed a 90% confluent
monolayer. The tip of a 200-µl sterile pipette was used to
generate an artificial and uniform wound, and the unattached cells
were carefully removed with 1X PBS. Cells were firstly treated with
mitomycin at the concentration of 1 µg/ml at 37°C for 1 h in
5% CO2 to inhibit cell division and then incubated in
serum-free medium at 37°C for 12 h in 5% CO2. The
representative images at 0 and 12 h were captured under an inverted
light microscope (Zeiss GmbH). The horizontal distance of the wound
was calculated with ImageJ v1.8.0 (National Institutes of
Health).
Molecular docking prediction
AutoDock Vina molecular docking software was used to
simulate the combination of DHA and VEGFA proteins (http://autodock.scripps.edu/resources/raccoon)
(18).
Serum biochemistry
Blood was collected into clot activating tubes from
rat orbital veins and was then centrifuged at 300 × g for 15 min at
room temperature to collect the supernatant. An automatic
biochemical analyzer (Hitachi, Ltd.) was used to detect the
concentration of liver injury-related indicators alanine
aminotransferase (ALT), aspartate aminotransferase (AST) and
alkaline phosphatase (ALP), as well as the liver fibrosis-related
indicators hyaluronic acid (HA), laminin (LN), type III procollagen
(PC-III) and type IV collagen (IV-C) in rat serum.
Datasets
The enrichment analysis of differentially expressed
genes was conducted using bioinformatics analysis and Kyoto
Encyclopedia of Genes and Genomes (KEGG). The liver
fibrosis-related miRNAs were analyzed using the following
databases: GSE33857 (19),
GSE49012 (20), GSE59492
(21), GSE74872 (22) from Gene Expression Omnibus (GEO)
(http://www.ncbi.nih.gov/geo) (23), miRWalk (http://mirwalk.umm.uni-heidelberg.de/) (24), miR2Disease (http://www.mir2disease.org/) (25) and HMDD (http://www.cuilab.cn/hmdd) (26). The potential interactions between
VEGFA and HSP60 protein were predicted using the following
database: GeneMania (http://www.genemania.org/) (27). The target genes of miR-29b-3p were
predicted using the following databases: miRWalk (http://mirwalk.umm.uni-heidelberg.de/),
miRND (http://mirdb.org/) and TargetScan (https://www.targetscan.org/) (28). The thresholds in the GEO database
were set as follows: P<0.05, fold change of all and gene rank of
all. The KEGG enrichment analysis of differentially expressed genes
between patients with different stages of liver fibrosis in the
GSE33258 GEO dataset was performed using the ASSISTANT for Clinical
Bioinformatics (https://www.aclbi.com/) (29). For KEGG analysis, P<0.05 or FDR
<0.05 was considered to be a meaningful pathway [enrichment
score with -log10(P) >1.3].
Statistical analysis
The present data are presented as the mean ±
standard deviation. GraphPad Prism 7.0 was used to analyze the
significance of the results (GraphPad Software, Inc.). Statistical
significance was determined using an unpaired Student's t-test for
comparisons between two groups, or one-way ANOVA followed by
Tukey's post hoc test for comparisons among more than two groups.
P<0.05 was considered to indicate a statistically significant
difference. Each experiment was repeated at least three times.
Results
miR-29b-3p upregulation inhibits HSC
development in vitro
Our previous study revealed that lncRNA H19 can
prevent liver fibrosis in vivo and in vitro (30). The present study aimed to screen
liver fibrosis-related miRNAs and explore their underlying
mechanisms. Between normal and hepatic fibrosis samples, five
significantly differentially expressed miRNAs were identified in
four GEO datasets (GSE33857, GSE49012, GSE59492 and GSE74827), and
two differentially expressed miRNAs were identified in three public
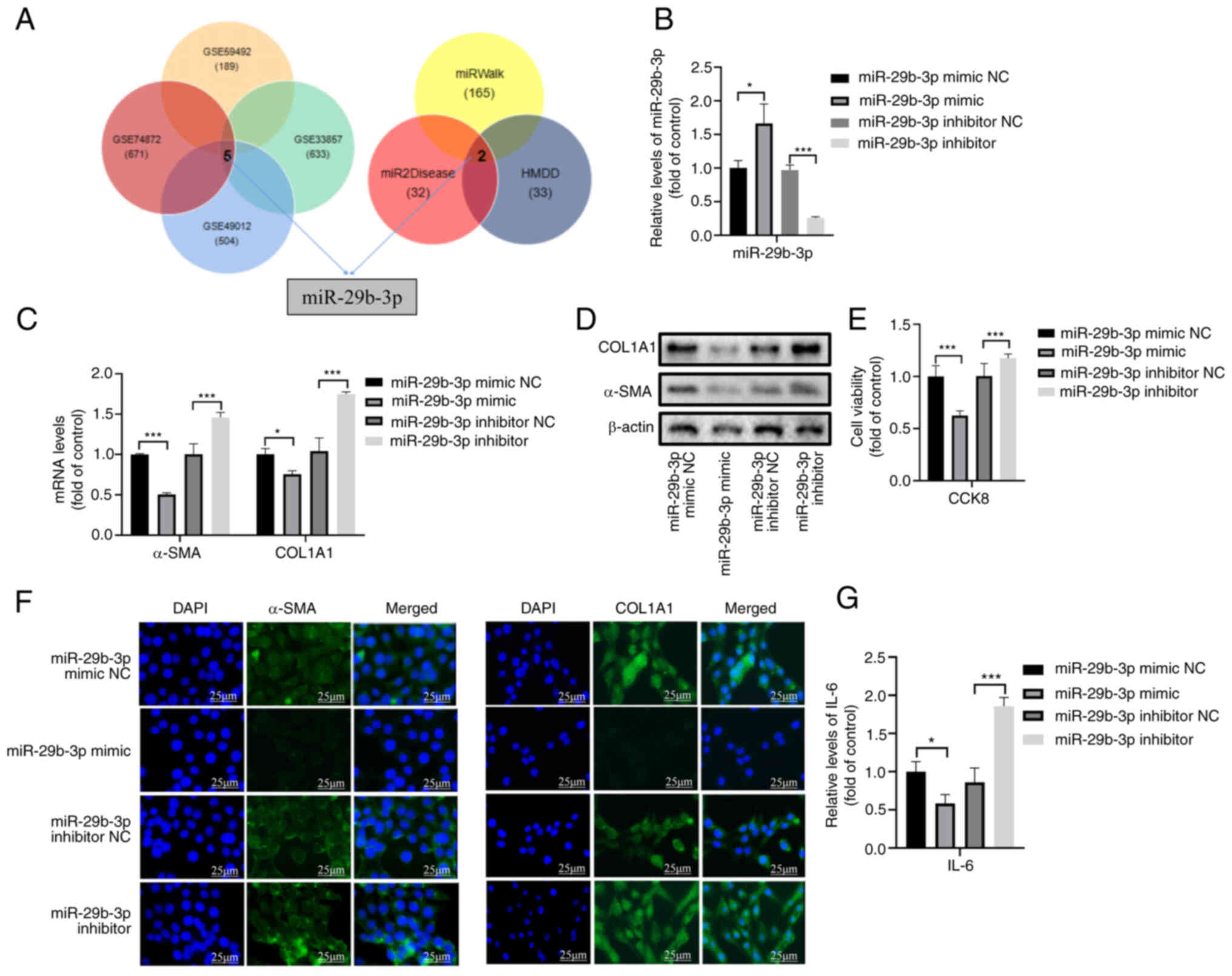
databases (miRWalk, miR2Disease and HMDD) (Fig. 1A). Only miR-29b was revealed to be
differentially expressed in all databases. Since miR-29b has two
subtypes miR-29b-1 and miR-29b-2, and their 3p-ends are the same
but their 5p-ends are different, miR-29b-3p was selected for
further analysis (Fig. 1A).
The transfection efficacy of the miR-29b-3p mimic
and miR-29b-3p inhibitor was confirmed by RT-qPCR (Fig. 1B). Notably, transfection with the
miR-29b-3p mimic decreased the mRNA and protein expression levels
of the fibrotic markers α-SMA and COL1A1 in LX-2 cells, whereas the
inhibitor increased them (Figs. 1C,
D and S1A). A previous study
confirmed that COL1A1 and COL3A1 are targets of miR-29b-3p in mouse
embryonic liver fibroblasts (31). In addition, the miR-29b-3p mimic
decreased HSC viability, whereas the inhibitor increased it
(Fig. 1E). Immunofluorescence
staining results also revealed that the expression levels of α-SMA
and COL1A1 were downregulated by the miR-29b-3p mimic but were
upregulated by the inhibitor (Fig.
1F). Furthermore, ELISA analysis revealed that the miR-29b-3p
mimic decreased the levels of IL-6, whereas the inhibitor increased
these levels (Fig. 1G).
miR-29b-3p reverses HSCs activation by
regulating lipid metabolism
To explore the main mechanism of miR-29b-3p in liver
fibrosis, KEGG analysis of differentially expressed genes between
patients with early (I, II) and late (III, IV) liver fibrosis in
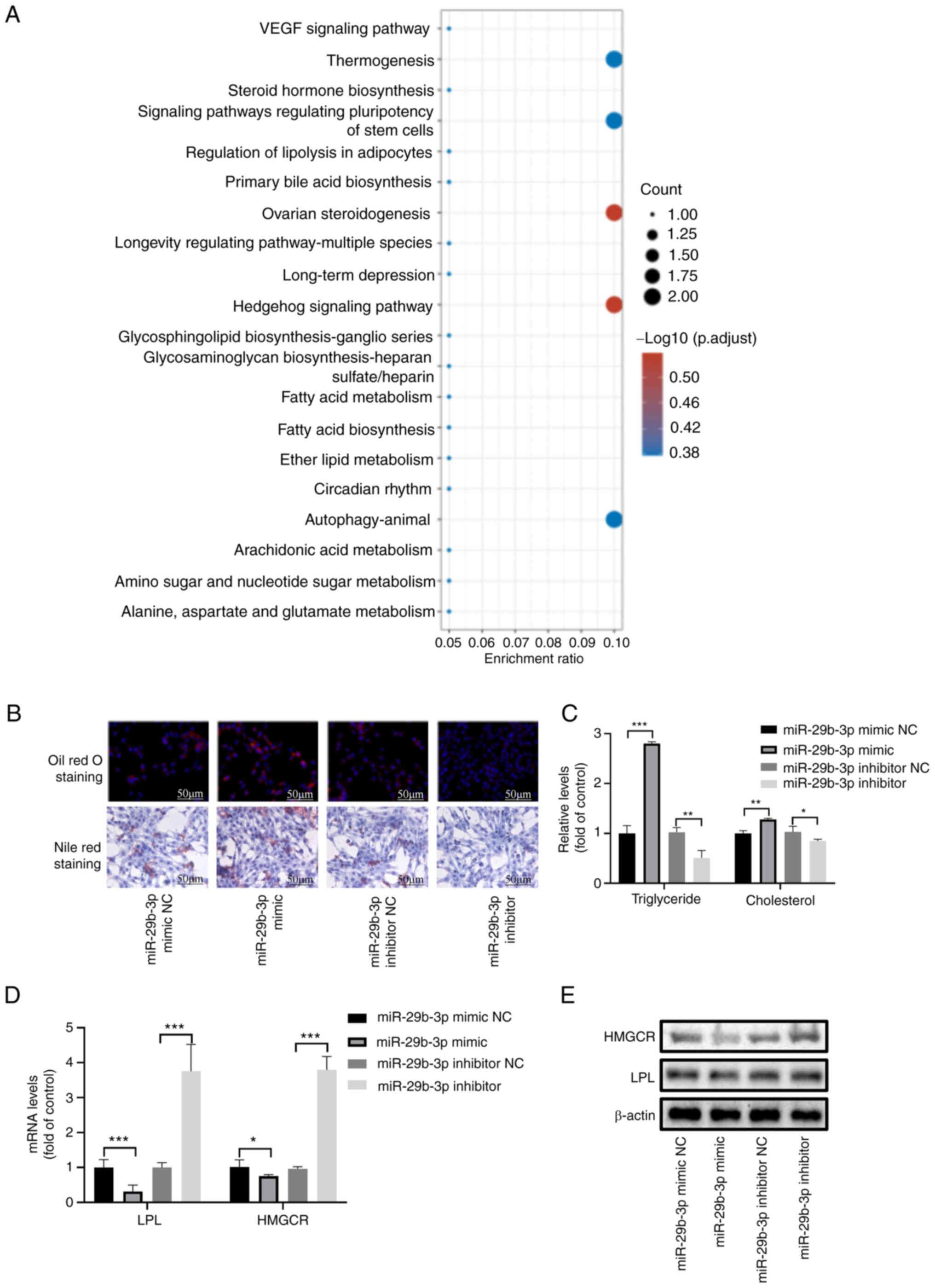
the GSE33258 dataset were assessed (Fig. 2A). In addition, KEGG analysis of
the target genes of hsa-miR-29b-3p was performed (Fig. S1B). 'VEGF signaling pathway' and
'fatty acid metabolism' were enriched in both. Moreover,
'Apoptosis-multiple species' and 'Autophagy-animal' were also
involved.
The miR-29b-3p mimic restored the lipid droplet
content in activated HSCs, as determined by oil red O and Nile red
staining; however, the miR-29b-3p inhibitor had no apparent effect
(Fig. 2B). The TG and TC assays
also confirmed that the miR-29b-3p mimic significantly increased
the levels of TG and TC, whereas the miR-29b-3p inhibitor decreased
these levels (Fig. 2C). It has
previously been reported that lipoprotein lipase (LPL) and HMGCR,
as key enzymes in TG and cholesterol metabolism, are target genes
of miR-29b-3p (32). The present
study demonstrated that the mRNA and protein expression of LPL and
HMGCR were inhibited by the miR-29b-3p mimic and activated by the
miR-29b-3p inhibitor (Figs. 2D, E
and S1C). Notably, the results
revealed that the miR-29b-3p mimic increased TC levels and
inhibited the expression levels of HMGCR, which is a rate-limiting
enzyme of cholesterol synthesis.
miR-29b-3p inhibits the proliferation of
activated HSCs through the VEGF pathway
Although lipid droplets were partially recovered in
cells transfected with the miR-29b-3p mimic, trypan blue staining
revealed that the miR-29b-3p mimic induced cell death (Fig. 3A). As shown in Figs. 2A and S1, the VEGF pathway may serve an
important role in the development of liver fibrosis.
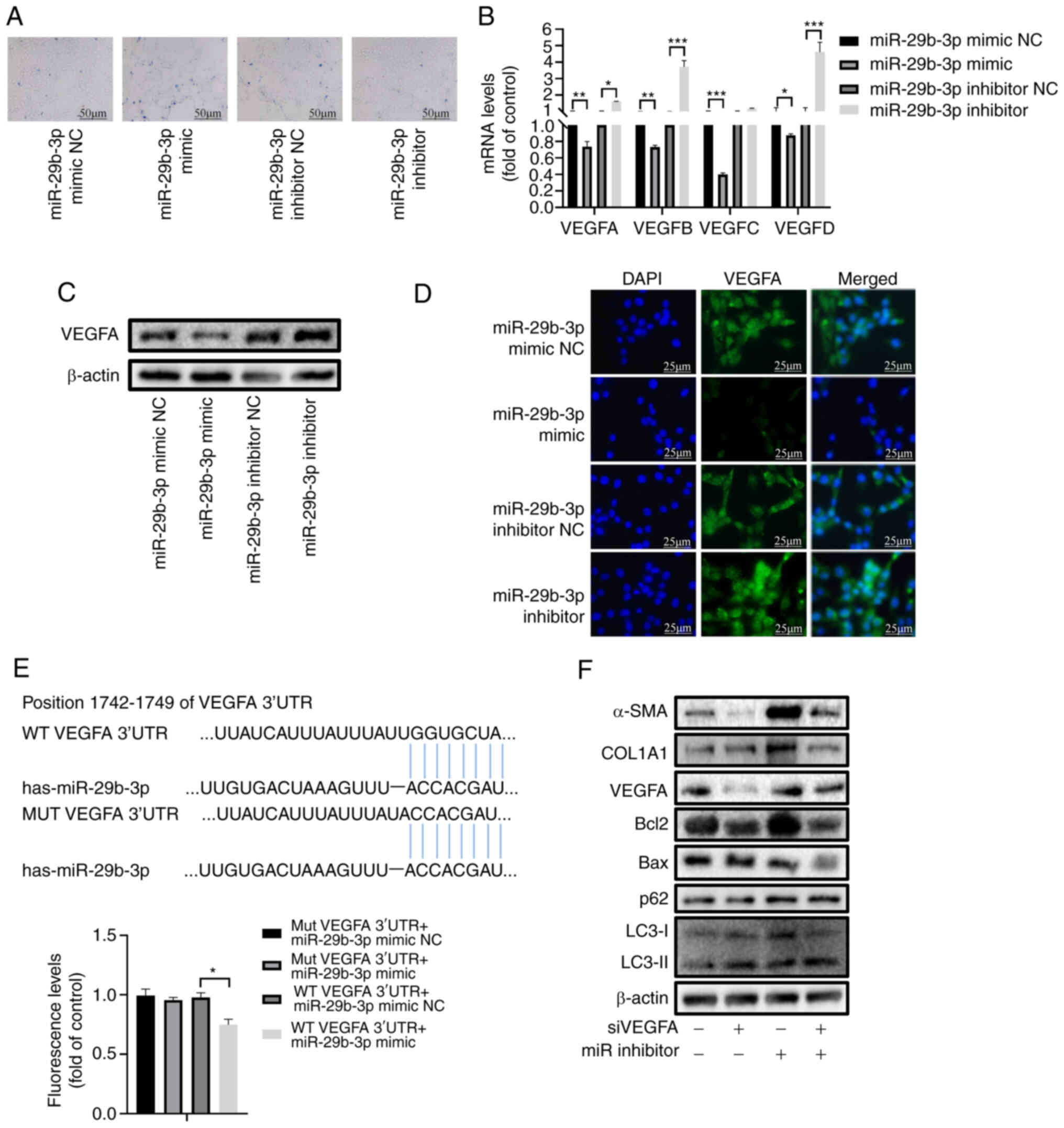 | Figure 3miR-29b-3p inhibits the proliferation
of activated hepatic stellate cells through the VEGF pathway. (A)
Trypan blue staining of LX-2 cells transfected with miR-29b-3p
mimic or inhibitor. (B) Reverse transcription-quantitative PCR
analysis of VEGFA, VEGFB, VEGFC and VEGFD. (C) Western blot
analysis of VEGFA, in LX-2 cells transfected with miR-29b-3p mimic
or inhibitor. (D) Immunofluorescence staining of VEGFA in LX-2
cells transfected with miR-29b-3p mimic or inhibitor. (E)
Luciferase assay testing VEGFA binding with miR-29b-3p. (F) Western
blot analysis of α-SMA, COL1A1, VEGFA, Bax/Bcl-2, p62 and
LC3-II/LC3-I in LX-2 cells transfected with miR-29b-3p mimic or
inhibitor. Data are expressed as the mean ± SD (n=3).
*P<0.05, **P<0.01,
***P<0.001. LPL, lipoprotein lipase; miR/miRNA,
microRNA; MUT, mutant; NC, negative control; WT, wild-type. |
The VEGF family has four members: VEGFA, VEGFB,
VEGFC and VEGFD. Except for VEGFC, the mRNA expression levels of
other VEGF genes were inhibited by the miR-29b-3p mimic and
activated by the miR-29b-3p inhibitor (Fig. 3B). VEGFA had the highest
expression among them (33). In
addition, VEGFA was predicted to be a target of miR-29b-3p through
TargetScan, miRDB and miRWalk analyses (Fig. S2A). The miR-29b-3p mimic
decreased the protein expression levels of VEGFA, whereas the
miR-29b-3p inhibitor had the opposite effect (Figs. 3C and S2B). Immunofluorescence assays revealed
that the miR-29b-3p mimic downregulated the expression levels of
VEGFA, whereas the inhibitor upregulated them (Fig. 3D). Notably, VEGFA was confirmed as
a direct target gene of miR-29b-3p via the luciferase assay
(Fig. 3E).
The present study further verified the function of
the miR-29b-3p/VEGFA axis. RT-qPCR confirmed successful
transfection of the cells with the VEGFA siRNA (Fig. S2C). Compared with in the control
group, VEGFA siRNA inhibited the protein expression levels of the
fibrotic biomarkers COL1A1 and α-SMA, whereas the miR-29b-3p
inhibitor increased the expression levels of these proteins. In
addition, VEGFA siRNA increased the expression levels of Bax/Bcl-2
and LC3-II/LC3-I while decreasing p62, whereas the miR-29b-3p
inhibitor had the opposite effects (Figs. 3F and S2D).
VEGFR1 and VEGFR2 serve different roles
in HSC activation
VEGFA has two main receptors: VEGFR1 and VEGFR2.
RT-qPCR and western blot analysis demonstrated that VEGFR1 and
VEGFR2 siRNAs were successfully transfected into LX-2 cells
(Fig. S3A-C). Wound healing and
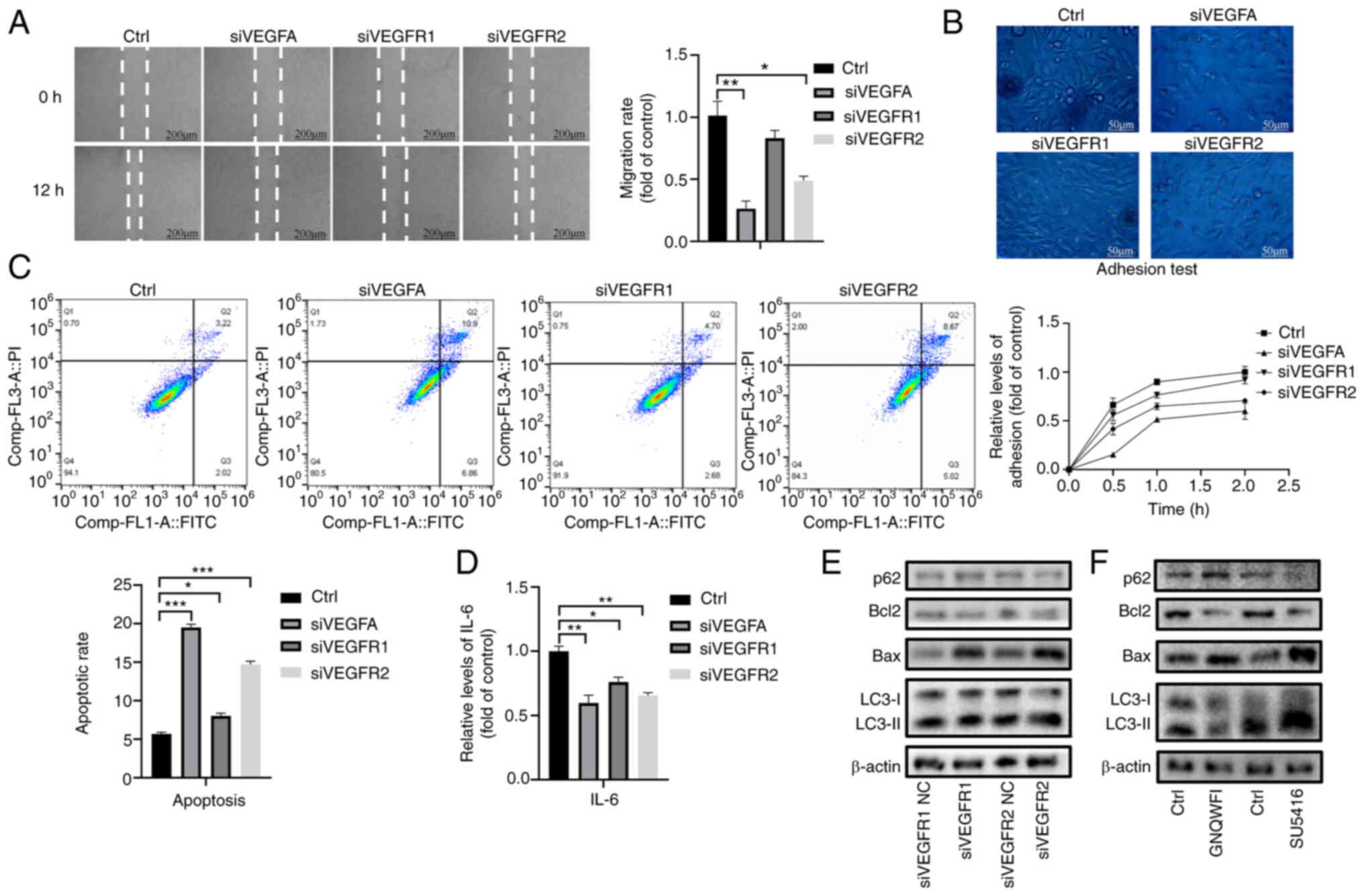
adhesion assays demonstrated that VEGFR2 knockdown significantly
inhibited the migratory and adhesive ability of LX-2 cells, whereas
VEGFR1 knockdown had little effect (Fig. 4A and B). In addition, Annexin
V-FITC/PI double staining and ELISA showed that VEGFR1 and VEGFR2
knockdown both induced the apoptosis and inhibited the inflammatory
reaction via decreasing IL-6 expression of LX-2 cells (Fig. 4C and D).
Notably, western blot analysis was performed to
detect the expression levels of autophagy-related proteins and it
was revealed that VEGFR1 knockdown inhibited the expression levels
of LC3-II/LC3-I and increased the expression levels of p62 in
activated HSCs, whereas VEGFR2 knockdown increased the expression
levels of LC3-II/LC3-I and inhibited the expression levels of p62
in activated HSCs (Figs. 4E and
S3D). LX-2 cells were further
treated with the VEGFR1-specific inhibitor GNQWFI and the
VEGFR2-specific inhibitor SU5416 at concentrations of 0.02, 0.2, 2
and 10 µM (Fig. S3E). The
results revealed that 2 µM GNQWFI and 0.2 µM SU5416
significantly inhibited the expression levels of VEGFR1 and VEGFR2,
respectively (Fig. S3C). Western
blot analysis demonstrated that the autophagy-related protein
LC3-II/LC3-I was decreased after GNQWFI treatment but increased
after SU5416 treatment (Figs. 4F
and S3F). In addition, both
knockdown of VEGFR1 and VEGFR2 increased the expression levels of
Bax/Bcl-2 (Figs. 4E, F and
S3D).
VEGFR2 knockdown induces autophagy via
PI3K/AKT/mTOR/ULK1, promotes ubiquitination of HSP60 and induces
protein degradation
Numerous studies have reported that the PI3K/AKT
pathway is the main downstream signal of VEGFR2 and is essential in
inducing autophagy (17,34,35). Western blot analysis revealed that
VEGFR2 siRNA significantly reduced the phosphorylation levels of
PI3K and AKT, and these changes were reversed by the PI3K-specific
agonist 740Y-P (Figs. 5A and
S4A). Western blot analysis also
revealed that VEGFR2 knockdown inhibited the phosphorylation of
mTOR and ULK1, whereas 740Y-P had the opposite effect (Fig. 5A).
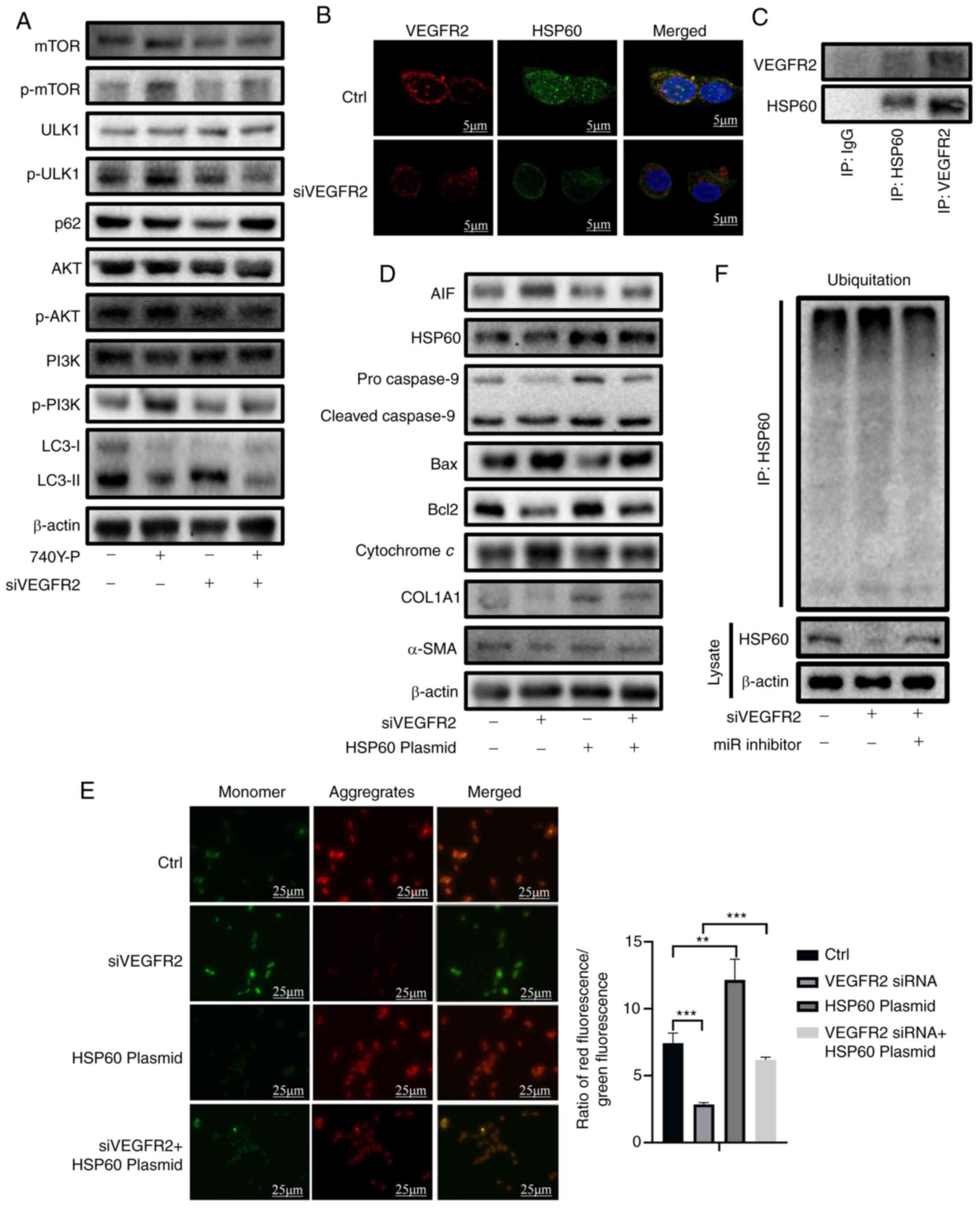 | Figure 5VEGFR2 knockdown induces autophagy
via the PI3K/AKT/mTOR/ULK1 axis and induces apoptosis by promoting
ubiquitination of HSP60. (A) Western blot analysis of PI3K, p-PI3K,
AKT, p-AKT, mTOR, p-mTOR, ULK1, p-ULK1, p62 and LC3-II/LC3-I in
LX-2 cells transfected with VEGFR2 siRNA and treated with 740Y-P.
(B) Colocalization of VEGFR2 and HSP60 as determined by
immunofluorescence. (C) Immunoprecipitation analysis of VEGFR2 and
HSP60 in LX-2 cells. (D) Western blot analysis of α-SMA, COL1A1,
AIF, HSP60, cleaved caspase-9/pro-caspase-9, Bax/Bcl-2 and
cytochrome c in LX-2 cells transfected with VEGFR2 siRNA and
HSP60 plasmid. (E) JC-1 assay of LX-2 cells transfected with VEGFR2
siRNA and HSP60 plasmid. Data are expressed as the mean ± SD (n=3).
(F) Western blot analysis of ubiquitination of HSP60 in LX-2 cells
transfected with VEGFR2 siRNA and miR-29b-3p inhibitor.
**P<0.01, ***P<0.001. HSP60, heat shock
protein 60; miR, microRNA; p-, phosphorylated; siRNA/si, small
interfering RNA. |
In addition to inducing autophagy, VEGFR2 knockdown
simultaneously promoted cell apoptosis (Fig. 4C). Analysis using GeneMania, a
protein interaction prediction website, revealed a potential
interaction between VEGFR2 and the mitochondrial stabilizing
protein HSP60 (Fig. S4B). A
fluorescence colocalization assay revealed that VEGFR2 and HSP60
were colocalized in the LX-2 cytoplasm under normal conditions, and
the colocalization level was markedly weakened after transfection
with VEGFR2 siRNA (Fig. 5B).
Immunoprecipitation assays showed that the VEGFR2 protein could
bind to the HSP60 protein (Fig.
5C). RT-qPCR and western blot analysis revealed that the
CMV-14-HSP60 plasmid was successfully transfected into LX-2 cells
(Figs. S4C, 4D and 5D). Western blot analysis also revealed
that the expression levels of AIF, cytochrome c and cleaved
caspase-9/pro-caspase-9 were markedly increased after VEGFR2
knockdown; these effects were reversed by HSP60 overexpression
(Figs. 5D and S4D). VEGFR2 knockdown also induced an
imbalance in mitochondrial membrane potential, but HSP60
overexpression reversed this imbalance (Fig. 5E).
The present results revealed that VEGFR2 knockdown
decreased the protein expression levels of HSP60, whereas the
miR-29b-3p inhibitor reversed this condition (Fig. 5F). Immunoprecipitation assays
showed that the ubiquitination of HSP60 increased after VEGFR2
knockdown, and miR-29b-3p intervention partly reduced this increase
(Figs. 5F and S4E). These findings indicated that
VEGFR2 may inhibit apoptosis by binding with HSP60 to prevent its
ubiquitination and degradation, and mitochondrial homeostasis
(36,37).
DHA prevents liver fibrosis by increasing
the expression of miR-29b-3p
A previous study screened the small molecule agonist
DHA from natural products and revealed that it could prevent
fibrosis; therefore, the present study aimed to confirm the
relationship between miR-29b-3p and DHA in fibrosis treatment
(30).
The CCK-8 assay showed that DHA decreased the
viability of LX-2 cells in a dose-dependent manner (Fig. 6A). Furthermore, miR-29b-3p
expression was significantly higher in HSCs treated with DHA
compared with that in the control group (Fig. 6B). The mRNA and protein expression
levels of LPL and VEGFA in HSCs were significantly decreased
following treatment with DHA in a dose-dependent manner (Figs. 6C, D and S5A). DHA also increased intracellular
TG and TC levels (Fig. 6E), and
inhibited HSP60 protein expression (Fig. 6D) in a dose-dependent manner.
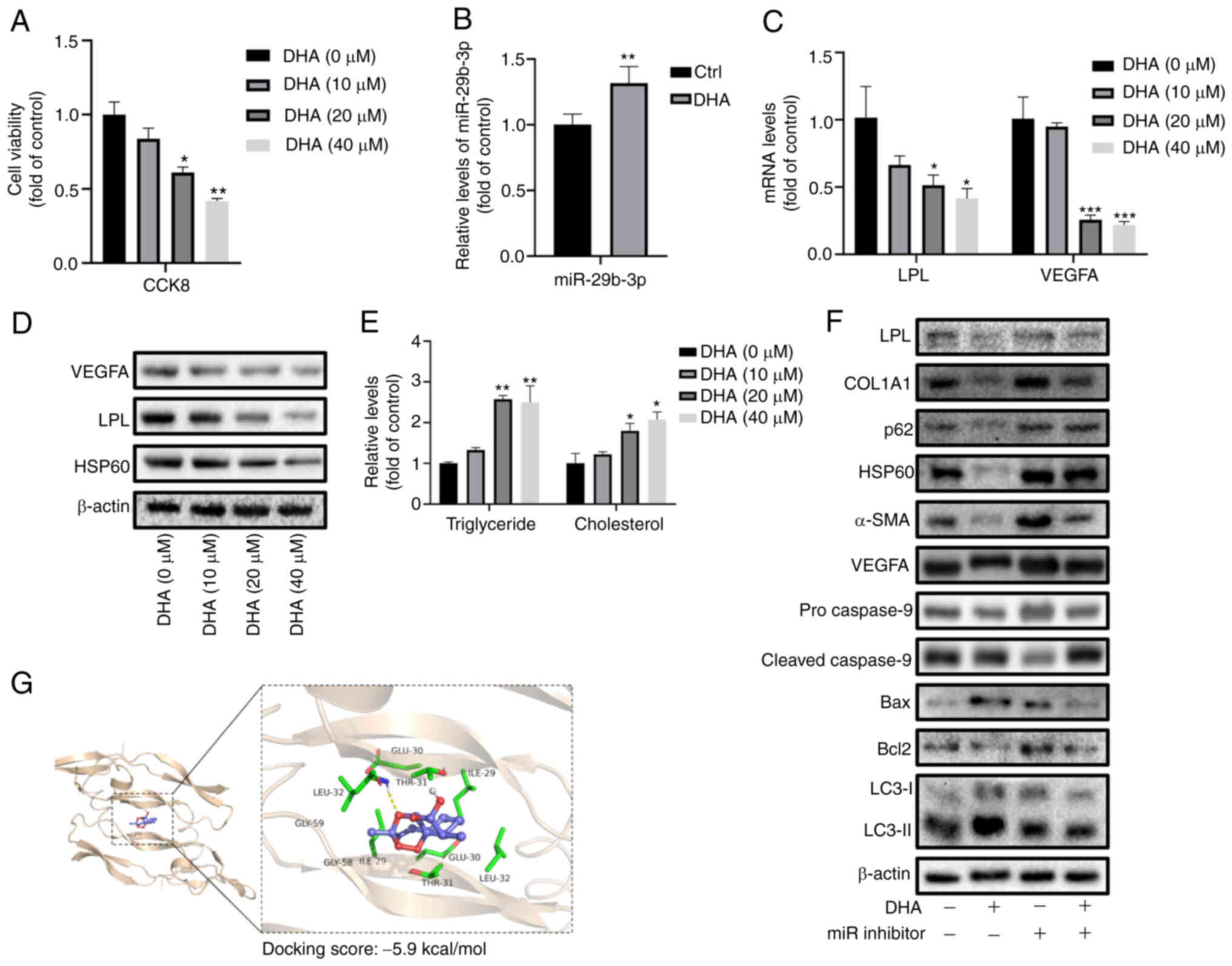 | Figure 6DHA prevents liver fibrosis by
increasing the expression of miR-29b-3p. (A) Viability of LX-2
cells treated with DHA at different concentrations.
*P<0.05, **P<0.01 vs. 0 µM DHA.
(B) RT-qPCR analysis of miR-29b-3p in LX-2 cells treated with
DHA.**P<0.01 vs. Ctrl. (C) RT-qPCR analysis of LPL
and VEGFA in LX-2 cells treated with DHA at different
concentrations. (D) Western blot analysis of LPL, VEGFA and HSP60
in LX-2 cells treated with DHA at different concentrations. (E)
Detection of triglyceride and cholesterol levels in LX-2 cells
treated with DHA at different concentrations.
*P<0.05, **P<0.01,
***P<0.001 vs. 0 µM DHA. (F) Western blot
analysis of Bax/Bcl-2, Beclin-1, LC3-II/LC3-I, cleaved
caspase-9/pro-caspase-9, VEGFA, α-SMA and COL1A1 in LX-2 cells
treated with DHA and transfected with miR-29b-3p inhibitor. (G)
Prediction of interaction between DHA and VEGFA protein via micro
thermal surge instrument. Data are expressed as the mean ± SD
(n=3). DHA, dihydroartemisinin; LPL, lipoprotein lipase; miR,
microRNA; RT-qPCR, reverse transcription-quantitative PCR. |
Furthermore, DHA treatment enhanced the protein
expression levels of Bax/Bcl-2, LC3-II/LC3-I and cleaved
caspase-9/pro-caspase-9, and decreased the expression levels of
p62, VEGFA, α-SMA and COL1A1 in LX-2 cells, whereas miR-29b-3p
inhibition reversed these effects (Figs. 6F and S5B). In addition, molecular docking
indicated a strong direct interaction of DHA with VEGFA protein
(docking score, -5.9 kcal/mol), thus indicating that DHA not only
regulated VEGFA mRNA via miR-29b-3p but also by directly acting on
the VEGFA protein (Fig. 6G).
DHA treatment prevents liver fibrosis and
hepatic injury in CCl4-treated rats via miR-29b-3p
To further confirm the specific mechanism
underlying the effects of DHA treatment on liver fibrosis, a rat
model of liver fibrosis was established via intraperitoneal
injection of CCl4. An AAV8 miR-29b-3p mimic was used to
increase miR-29b-3p expression via caudal vein injection. RT-qPCR
confirmed the successful intervention of AAV8 miR-29b-3p mimic in
the liver of rats (Fig. S5C).
Liver morphology evaluations confirmed the successful construction
of the liver fibrosis model, which exhibited obvious fibrotic
lesions compared with in the control group. Treatment with the
miR-29b-3p mimic and DHA markedly reduced fibrotic lesions in the
liver (Fig. 7A). In addition,
H&E staining was used to investigate the effects of the AAV8
miR-29b-3p mimic and DHA on hepatic injury and inflammation.
Intraperitoneal injection of CCl4 induced inflammatory
infiltration, hepatic fibrous septum formation and hepatocyte
disorder; however, these effects were attenuated by the AAV8
miR-29b-3p mimic and DHA. The results of Masson and Sirius Red
staining also revealed that large amounts of collagen and matrix
were deposited around the hepatic fibrous scar area in the model
group, but were effectively reduced by the AAV8 miR-29b-3p mimic
and DHA (Fig. 7A). Serological
examination demonstrated that hepatic injury biomarkers (ALT, AST,
ALP, HA111, LN, PC-III and IV-C) were higher in the model group
than in the AAV8 miR-29b-3p mimic and DHA treatment groups, which
is consistent with our previous conclusion that miR-29b-3p and DHA
inhibited HSC activity and reversed activated HSCs into a static
condition (Fig. 7B).
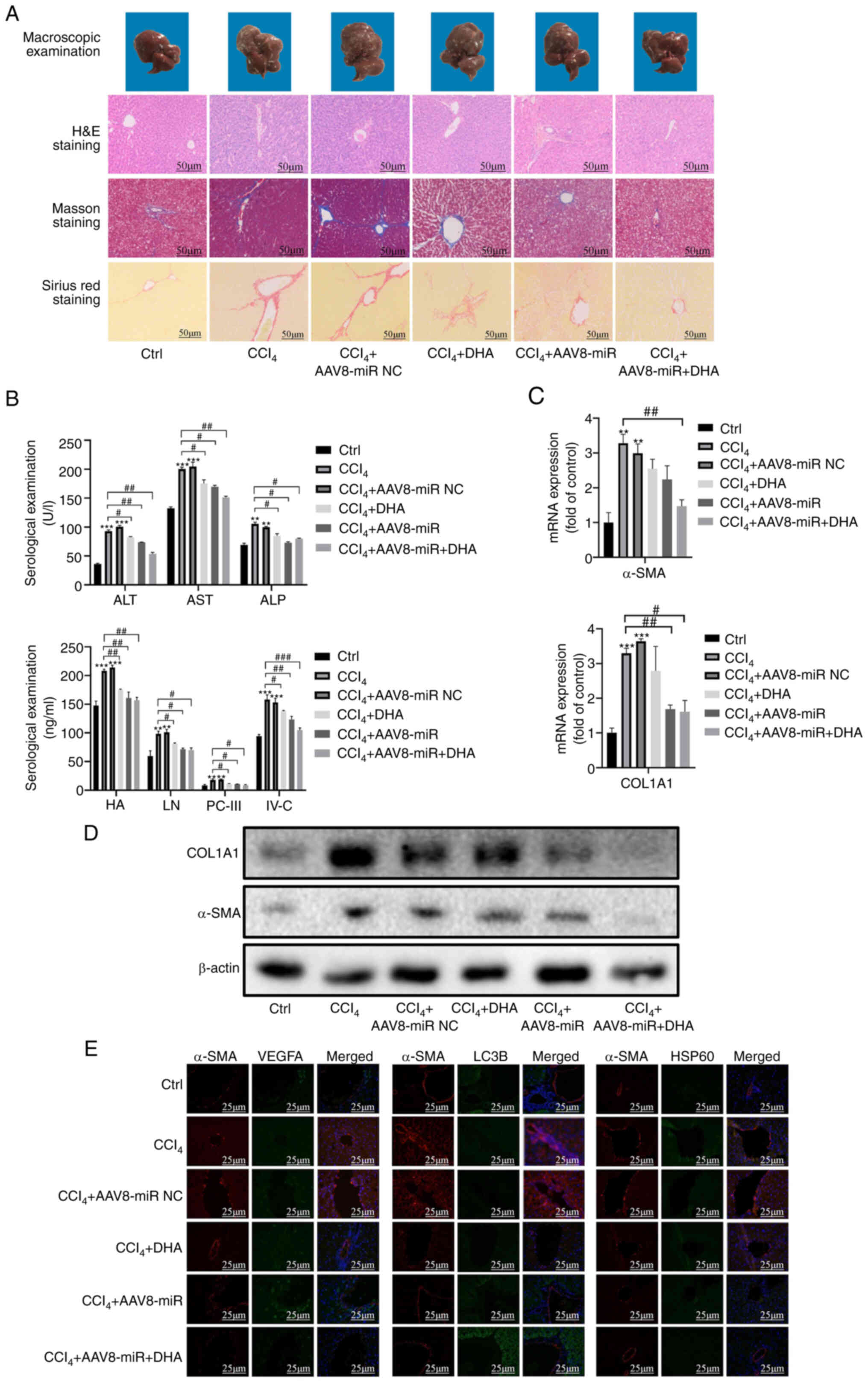 | Figure 7DHA treatment prevents liver fibrosis
and hepatic injury in CCl4-treated rats via miR-29b-3p.
(A) Liver pathological changes were observed by macroscopic
examination, and liver sections were stained with H&E, Masson
and Sirius Red. (B) Serological examination of injury biomarkers
(ALT, AST, ALP, HA111, LN, PC-III and IV-C). (C) Reverse
transcription-quantitative PCR of α-SMA and COL1A1 in liver tissue.
(D) Western blot analysis of α-SMA and COL1A1 in liver tissue. (E)
Immunofluorescence staining of VEGFA, HSP60 and LC3 in rat liver
tissue. α-SMA was used to stain hepatic stellate cells. Data are
expressed as the mean ± SD (n=3). **P<0.01,
***P<0.001 vs. Ctrl; #P<0.05,
##P<0.01 vs. CCl4. CCl4, carbon
tetrachloride; DHA, dihydroartemisinin; H&E, hematoxylin and
eosin; miR, microRNA; NC, negative control. |
The mRNA and protein expression levels of α-SMA and
COL1A1 were significantly upregulated in the model group, whereas
they were downregulated by the AAV8 miR-29b-3p mimic and DHA
(Figs. 7C, D and S5D). Immunofluorescence double staining
revealed that the expression of VEGFA and HSP60 in fibrotic tissue
was inhibited by the AAV8 miR-29b-3p mimic or DHA, whereas LC3B
expression was increased. Fibrotic liver tissue was stained with
α-SMA (Fig. 7E).
Discussion
The pathogenesis of liver fibrosis is complex and
difficult to elucidate. Liver fibrosis is a common pathological
process associated with most types of acute or chronic liver
disease, which can ultimately progress to liver cirrhosis and liver
cancer without timely treatment. Although the etiology of these
diseases differs, they are all characterized by hepatic fibrosis,
and early and long-term anti-hepatic fibrosis treatment are
recommended for these conditions (38,39); however, there is still no
effective method to cure liver fibrosis. Natural products have
attracted great interest from researchers because of their unique
chemical structures and diverse biological activity. In addition,
medicinal plants have been widely used to prevent and treat various
diseases. DHA is a derivative of artemisinin, which is a commonly
used antimalarial drug; however, DHA has also been reported to
exert anti-inflammatory, antitumor, antibacterial and antiviral
activities (40,41). We previously showed that DHA could
reduce CCl4-induced liver fibrosis in vivo, and
induce activation of HSCs aging and HSCs apoptosis in vitro
(15,16). The present study aimed to clarify
the mechanism by which DHA could inhibit HSCs activation and induce
cell death via miR-29b-3p.
Increasing evidence has shown that miRNAs have an
important role in cell survival and development, and great progress
has been achieved in miRNA-targeted therapy. A miR-34 mimic
encapsulated in lipid nanoparticles that inhibits tumor cell growth
has been evaluated in phase II clinical trials (NCT01829971)
(42), and locked nucleic acid
therapy inhibiting miR-122 for the treatment of hepatitis has also
entered clinical trials (43,44). The present results were consistent
with the prediction that miR-29b-3p has a key role in recovering
HSC lipid droplets and inducing cell death. Moreover, we aim to
collect liver samples from patients with liver fibrosis of
different stages, and to further compare the differential
expression of miR-29b-3p in a future study.
The main components of lipid droplets are retinol,
TG and cholesterol. Lipid droplet loss can induce static HSCs
activation. In the present study, miR-29b-3p promoted TG and TC
levels in HSCs, and reversed HSC activation. LPL is the
rate-limiting enzyme for the degradation of TGs into glycerol and
free fatty acids. Due to the special cell structure of lipid
droplets in HSCs, LPL may have an important role in metabolism of
lipid droplets. Previous studies have reported a strong correlation
between LPL and HSCs activation and liver fibrosis (45,46). The present study verified the
hypothesis that miR-29b-3p may participate in intracellular lipid
metabolism by regulating LPL and HMGCR via western blot analysis
and RT-qPCR analysis. The results revealed that LPL was regulated
by miR-29b-3p and influenced by DHA in a dose-dependent manner. LPL
regulates lipid metabolism and maintains an energy balance, and is
primarily located on the capillary endothelial cell surface. A
previous study reported that LPL was enhanced in HSCs, not
hepatocytes, in a model of nonalcoholic fatty liver disease
(6). Enhanced LPL has been
reported to induce the accumulation of free cholesterol and to
decrease TG levels in HSCs via TLR4- and Bambi-related signals,
increasing the susceptibility of HSCs to TGF-β-induced HSC
activation. The present study also confirmed that LPL inhibition in
HSCs contributed to the increase in TGs and recovery of lipid
droplets; with the decrease in LPL expression level detected
following intervention with the miR-29b-3p mimic, TG began to
accumulate in HSCs and lipid droplets gradually recovered. However,
the decrease in HMGCR conflicts with the increase in cholesterol;
HMGCR is a key enzyme in the cholesterol synthesis pathway.
Generally, increased HMGCR could can induce the synthesis of
cholesterol. Kurtz et al (32) reported a similar phenomenon. This
previous study found that among the genes upregulated by
miR-29b-3p, the anti-lipogenic deacetylase SIRT1 and aryl
hydrocarbon receptor exhibited the greatest differences. These two
genes may reverse the effect of HMGCR on cholesterol. Moreover,
other cholesterol enzymes, such as lecithin-cholesterol
acyltransferase and acyl-coenzyme A-cholesterol acyltransferase,
may also contribute to cholesterol upregulation. It was
hypothesized that this may explain the contradictory phenomenon
that HMGCR expression was decreased whereas cholesterol was
increased after miR-29b-3p mimic intervention.
Previous studies have shown that VEGF signaling in
sinusoidal endothelial cells negatively influences liver fibrosis
development (47,48). The present study aimed to explore
the function of the VEGF pathway in HSCs. VEGFA was identified as a
direct target of miR-29b-3p, which simultaneously regulated
multiple functions of HSCs, including adhesion, migration, survival
and the inflammatory response. Notably, VEGFR1 and VEGFR2, as
receptors of VEGFA, had different roles. Although knockdown of both
inhibited fibrosis, and caused apoptosis and inhibited
inflammation, VEGFR1 knockdown had no significant effect on cell
migration and adhesion. By contrast, VEGFR2 knockdown significantly
inhibited cell migration and adhesion. Notably, autophagy was
decreased after VEGFR1 silencing but was increased after VEGFR2
silencing. It was hypothesized that VEGFR1 and VEGFR2 competitively
bind to VEGFA, and that VEGFR2 plays a greater role after VEGFR1
silencing and protects cells from autophagy. By contrast, when
VEGFR2 was silenced, autophagy was increased. It has previously
been suggested that VEGFR2 is the main receptor of VEGF, whereas
VEGFR1 is considered a decoy receptor that weakens the binding
between VEGFA and VEGFR2 (48). A
previous study reported that the ULK1 complex may act as a bridge
connecting the upstream trophic receptor mTOR or the energy
receptor AMPK and downstream autophagy in vivo (15). Notably, p-mTOR can phosphorylate
ULK1 to disrupt the binding between ATG3 and ULK1, thus
destabilizing it and ultimately inhibiting the initiation of
autophagy (34,35). In the present study, it was
revealed that VEGFA/VEGFR2 regulated autophagy via the
PI3K/AKT/mTOR/ULK1 pathway. It was hypothesized that when VEGFR1
was silenced, VEGFR2 worked more and inhibited HSCs autophagy,
whereas when VEGFR2 was silenced, the autophagy of HSCs was
activated. However, autophagy is a complex cellular event involving
multigene regulation. The present study only preliminarily explored
the function of two VEGFRs through siRNA and specific inhibitors.
At present, the dynamic process of autophagy over time is not clear
and further research is required (15,35,37).
To the best of our knowledge, the present study is
the first to show that HSP60 is an intermediate protein in
DHA-induced mitochondrial apoptosis, and its ubiquitination and
degradation may be a key event leading to the imbalance of
mitochondrial homeostasis after VEGFR2 silencing. It has previously
been reported that after VEGF intervention, phosphorylation and
ubiquitination of zonula cluster-1 occur, further influencing
vascular permeability (36).
ATP7A has been identified as an interacting protein of VEGFR2 that
assists VEGFR2 entry into the cytoplasm. Where ATP7 is
ubiquitinated, autophagy is induced (37). In recent years, epigenetics has
emerged as an important research topic. An increasing number of
studies have shown that several epigenetic modifications, such as
methylation, acetylation and ubiquitination, participate in cell
survival. Furthermore, HSP60 has been reported to be important in
liver disease. Yang et al reported that the expression of
HSP60 was significantly upregulated in the liver tissue of mice
with liver fibrosis and that this upregulation was reversed by a
miR-29a mimic (49). Mazo et
al also revealed that the expression of HSP60 mRNA was
significantly increased in a rat model of nonalcoholic
steatohepatitis, whereas S-nitroso-N-acetylcysteine decreased the
expression of HSP60 and COL1A1 and alleviated liver fibrosis
(50). HSP60 is mainly involved
in protein transport and folding under physiological conditions.
When the cell is injured, HSP60 acts as a stress protein to resist
various damaging effects. In addition, HSP60 can repair
mitochondrial cytochrome c in combination with HSP10, and
can protect mitochondria (51).
HSP60 overexpression has been reported to increase the expression
levels of the Bcl-XL gene and reduce the expression of Bax,
stabilizing mitochondrial membrane potential and inhibiting
activation of the caspase-9-related pathway (52). The HSP60 amino acid sequence
contains numerous potential phosphorylation sites and other
post-translational modifications (PTMs), such as ubiquitination,
oxidation, S-nitrosylation and acetylation, can also occur in HSP60
protein. PTMs are essential for maintaining HSP60 function in a
number of processes, such as mitochondrial dysfunction, tumor
invasiveness, and delay or facilitation of apoptosis (53). The present study revealed that
VEGFR2 could protect HSP60 from ubiquitination and degradation.
Notably, VEGFR2 may also be involved in the
ubiquitination of several autophagy genes. The present study only
examined the mechanism by which VEGFR2 regulates autophagy through
the classical PI3K/AKT signaling pathway; however, the differences
in autophagy activity may be the result of a variety of epigenetic
modifications. Therefore, it is important to further explore the
interactions among chaperone proteins, VEGFR2, apoptosis and
autophagy, especially the regulatory crosstalk between different
epigenetic modifications.
There are some limitations in the present study.
Firstly, the LX-2 cell line was selected as the research subject,
whereas primary HSCs were not assessed. Secondly, this study did
not verify the correlation between HSP60, the PI3K/AKT pathway and
the miR-29b-3p/VEGFA axis. Thirdly, despite post-transcriptional
interactions between VEGFR2 and HSP60 proteins being detected in
LX-2 cells, protein-mRNA interactions during transcription may also
play key roles.
In conclusion, DHA may inhibit the activation of
HSCs and induce the programmed death of HSCs via the
miR-29b-3p/VEGFA axis. Furthermore, VEGFR2 was revealed to be
important in regulating autophagy and apoptosis via the
PI3K/AKT/mTOR/ULK1 pathway, and by binding to HSP60 to prevent its
ubiquitination and degradation (Fig.
S6). The present study provided a new perspective and target
for drug development in the treatment of liver fibrosis.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
SH, SZ and GY contributed to the conception of the
study. SH, SMS and GY contributed to the funding acquisition. SH,
YZ and GY performed the experiments. SH and SMS contributed to data
collection and systematic analysis. SLS contributed to the ethics
approval application and animal experiments. SH, JD and YJ
contributed to the design of methodology and use of software. GZ
and YZ contributed to analysis and manuscript preparation. SH, YJ
and GY provided resources, supervised the study and wrote the
original draft. SH and GY confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The authors confirm that all methods were carried
out in accordance with the relevant guidelines and regulations. The
study protocol and animal experiments were approved by the
Institutional and Local Committee of Nanjing University of Chinese
Medicine (approval no. A20402).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work was supported in part by the Natural Science
Foundation of Nanjing University of Traditional Chinese Medicine
(grant no. XZR2020072), Graduate Research and Innovation Projects
of Jiangsu Province (grant no. KYCX21_1637) and Graduate Research
and Innovation Projects of Jiangsu Province (grant no.
KYCX21_1732).
References
|
1
|
Kisseleva T and Brenner D: Molecular and
cellular mechanisms of liver fibrosis and its regression. Nat Rev
Gastroenterol Hepatol. 18:151–166. 2021. View Article : Google Scholar
|
|
2
|
Hernandez-Gea V and Friedman SL:
Pathogenesis of liver fibrosis. Annu Rev Pathol. 6:425–456. 2011.
View Article : Google Scholar
|
|
3
|
Parola M and Pinzani M: Liver fibrosis:
Pathophysiology, pathogenetic targets and clinical issues. Mol
Aspects Med. 65:37–55. 2019. View Article : Google Scholar
|
|
4
|
Kang N, Gores GJ and Shah VH: Hepatic
stellate cells: Partners in crime for liver metastases? Hepatology.
54:707–713. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Trivedi P, Wang S and Friedman SL: The
power of plasticitymetabolic regulation of hepatic stellate cells.
Cell Metab. 33:242–257. 2021. View Article : Google Scholar
|
|
6
|
Rupaimoole R and Slack FJ: MicroRNA
therapeutics: Towards a new era for the management of cancer and
other diseases. Nat Rev Drug Discov. 16:203–222. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Krol J, Loedige I and Filipowicz W: The
widespread regulation of microRNA biogenesis, function and decay.
Nat Rev Genet. 11:597–610. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dong H, Lei J, Ding L, Wen Y, Ju H and
Zhang X: MicroRNA: Function, detection, and bioanalysis. Chem Rev.
113:6207–6233. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim VN: MicroRNA biogenesis: Coordinated
cropping and dicing. Nat Rev Mol Cell Biol. 6:376–385. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Su T, Xiao Y, Xiao Y, Guo Q, Li C, Huang
Y, Deng Q, Wen J, Zhou F and Luo XH: Bone marrow mesenchymal stem
cells-derived exosomal MiR-29b-3p regulates aging-associated
insulin resistance. ACS Nano. 13:2450–2462. 2019.PubMed/NCBI
|
|
11
|
Xue Y, Fan X, Yang R, Jiao Y and Li Y:
miR-29b-3p inhibits post-infarct cardiac fibrosis by targeting FOS.
Biosci Rep. 40:BSR202012272020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Novo E, Cannito S, Zamara E, Valfrè di
Bonzo L, Caligiuri A, Cravanzola C, Compagnone A, Colombatto S,
Marra F, Pinzani M and Parola M: Proangiogenic cytokines as
hypoxia-dependent factors stimulating migration of human hepatic
stellate cells. Am J Pathol. 170:1942–1953. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sun J, Shi L, Xiao T, Xue J, Li J, Wang P,
Wu L, Dai X, Ni X and Liu Q: microRNA-21, via the HIF-1α/VEGF
signaling pathway, is involved in arsenite-induced hepatic fibrosis
through aberrant cross-talk of hepatocytes and hepatic stellate
cells. Chemosphere. 266:1291772021. View Article : Google Scholar
|
|
14
|
Huang YH and Yeh CT: Functional
Compartmentalization of HSP60-Survivin interaction between
mitochondria and cytosol in cancer cells. Cells. 9:232019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Z, Yao Z, Zhao S, Shao J, Chen A,
Zhang F and Zheng S: Interaction between autophagy and senescence
is required for dihydroartemisinin to alleviate liver fibrosis.
Cell Death Dis. 8:e28862017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen Q, Chen L, Kong D, Shao J, Wu L and
Zheng S: Dihydroartemisinin alleviates bile duct ligation-induced
liver fibrosis and hepatic stellate cell activation by interfering
with the PDGF-βR/ERK signaling pathway. Int Immunopharmacol.
34:250–258. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen Q, Chen L, Wu X, Zhang F, Jin H, Lu
C, Shao J, Kong D, Wu L and Zheng S: Dihydroartemisinin prevents
liver fibrosis in bile duct ligated rats by inducing hepatic
stellate cell apoptosis through modulating the PI3K/Akt pathway.
IUBMB Life. 68:220–231. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Seeliger D and de Groot BL: Ligand docking
and binding site analysis with PyMOL and Autodock/Vina. J Comput
Aided Mol Des. 24:417–422. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Murakami Y, Toyoda H, Tanahashi T, Tanaka
J, Kumada T, Yoshioka Y, Kosaka N, Ochiya T and Taguchi YH:
Comprehensive miRNA expression analysis in peripheral blood can
diagnose liver disease. PLoS One. 7:e483662012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vuppalanchi R, Liang T, Goswami CP,
Nalamasu R, Li L, Jones D, Wei R, Liu W, Sarasani V, Janga SC and
Chalasani N: Relationship between differential hepatic microRNA
expression and decreased hepatic cytochrome P450 3A activity in
cirrhosis. PLoS One. 8:e744712013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Blaya D, Coll M, Rodrigo-Torres D,
Vila-Casadesús M, Altamirano J, Llopis M, Graupera I, Perea L,
Aguilar-Bravo B, Díaz A, et al: Integrative microRNA profiling in
alcoholic hepatitis reveals a role for microRNA-182 in liver injury
and inflammation. Gut. 65:1535–1545. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Matsuura K, De Giorgi V, Schechterly C,
Wang RY, Farci P, Tanaka Y and Alter HJ: Circulating let-7 levels
in plasma and extracellular vesicles correlate with hepatic
fibrosis progression in chronic hepatitis C. Hepatology.
64:732–745. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang J, Huang J, Liu W, Ding L, Cheng D
and Xiao H: Identification of common oncogenic genes and pathways
both in osteosarcoma and Ewing's sarcoma using bioinformatics
analysis. J Immunol Res. 2022:36559082022.PubMed/NCBI
|
|
24
|
Wang Z, Zhang J, Feng T, Zhang D, Pan Y,
Liu X, Xu J, Qiao X, Cui W and Dong L: Construction of
lncRNA-Mediated competing endogenous RNA networks correlated With
T2 asthma. Front Genet. 13:8724992022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhong T, Li Z, You ZH, Nie R and Zhao H:
Predicting miRNA-disease associations based on graph random
propagation network and attention network. Brief Bioinform.
23:bbab5892022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li G, Sun J, Zhang J, Lv Y, Liu D, Zhu X,
Qi L, Chen Z, Ye Z, Su X and Li L: Identification of
Inflammation-related biomarkers in diabetes of the exocrine
pancreas with the use of weighted gene Co-Expression network
analysis. Front Endocrinol (Lausanne). 13:8398652022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mostafavi S and Morris Q: Combining many
interaction networks to predict gene function and analyze gene
lists. Proteomics. 12:1687–1696. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang S, Shen L and Luo H: Identification
and Validation of Key miRNAs and a microRNA-mRNA Regulatory Network
Associated with Ulcerative Colitis. DNA Cell Biol. 40:147–156.
2021. View Article : Google Scholar
|
|
29
|
Matsuura K, De Giorgi V, Schechterly C,
Wang RY, Farci P, Tanaka Y and Alter HJ: Circulating let-7 levels
in plasma and extracellular vesicles correlate with hepatic
fibrosis progression in chronic hepatitis C. Hepatology.
64:732–745. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xia S, Wang Z, Chen L, Zhou Y, Li Y, Wang
S, Chen A, Xu X, Shao J, Zhang Z, et al: Dihydroartemisinin
regulates lipid droplet metabolism in hepatic stellate cells by
inhibiting lncRNA-H19-induced AMPK signal. Biochem Pharmacol.
192:1147302021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cutroneo KR, White SL, Phan SH and Ehrlich
HP: Therapies for bleomycin induced lung fibrosis through
regulation of TGF-beta1 induced collagen gene expression. J Cell
Physiol. 211:585–589. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kurtz CL, Fannin EE, Toth CL, Pearson DS,
Vickers KC and Sethupathy P: Inhibition of miR-29 has a significant
lipid-lowering benefit through suppression of lipogenic programs in
liver. Sci Rep. 5:129112015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Apte RS, Chen DS and Ferrara N: VEGF in
signaling and disease: Beyond discovery and development. Cell.
176:1248–1264. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Aoki M and Fujishita T: Oncogenic Roles of
the PI3K/AKT/mTOR Axis. Curr Top Microbiol Immunol. 407:153–189.
2017.PubMed/NCBI
|
|
35
|
Song YM, Lee YH, Kim JW, Ham DS, Kang ES,
Cha BS, Lee HC and Lee BW: Metformin alleviates hepatosteatosis by
restoring SIRT1-mediated autophagy induction via an AMP-activated
protein kinase-independent pathway. Autophagy. 11:46–59. 2015.
View Article : Google Scholar :
|
|
36
|
Muthusamy A, Lin CM, Shanmugam S, Lindner
HM, Abcouwer SF and Antonetti DA: Ischemia-reperfusion injury
induces occludin phosphorylation/ubiquitination and retinal
vascular permeability in a VEGFR-2-dependent manner. J Cereb Blood
Flow Metab. 34:522–531. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ash D, Sudhahar V, Youn SW, Okur MN, Das
A, O'Bryan JP, McMenamin M, Hou Y, Kaplan JH, Fukai T and
Ushio-Fukai M: The P-type ATPase transporter ATP7A promotes
angiogenesis by limiting autophagic degradation of VEGFR2. Nat
Commun. 12:30912021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hunt NJ, Kang SWS, Lockwood GP, Le Couteur
DG and Cogger VC: Hallmarks of Aging in the Liver. Comput Struct
Biotechnol J. 17:1151–1161. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Brenner C, Galluzzi L, Kepp O and Kroemer
G: Decoding cell death signals in liver inflammation. J Hepatol.
59:583–594. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Efferth T: From ancient herb to modern
drug: Artemisia annua and artemisinin for cancer therapy. Semin
Cancer Biol. 46:65–83. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li Q, Ma Q, Cheng J, Zhou X, Pu W, Zhong X
and Guo X: Dihydroartemisinin as a sensitizing agent in cancer
therapies. Onco Targets Ther. 14:2563–2573. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hong DS, Kang YK, Borad M, Sachdev J,
Ejadi S, Lim HY, Brenner AJ, Park K, Lee JL, Kim TY, et al: Phase 1
study of MRX34, a liposomal miR-34a mimic, in patients with
advanced solid tumours. Br J Cancer. 122:1630–1637. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang L, Liao Y and Tang L: MicroRNA-34
family: A potential tumor suppressor and therapeutic candidate in
cancer. J Exp Clin Cancer Res. 38:532019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
van Rooij E and Kauppinen S: Development
of microRNA therapeutics is coming of age. EMBO Mol Med. 6:851–864.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Teratani T, Tomita K, Furuhashi H,
Sugihara N, Higashiyama M, Nishikawa M, Irie R, Takajo T, Wada A,
Horiuchi K, et al: Lipoprotein Lipase Up-regulation in hepatic
stellate cells exacerbates liver fibrosis in nonalcoholic
steatohepatitis in mice. Hepatol Commun. 3:1098–1112. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Duan X, Meng Q, Wang C, Liu Z, Liu Q, Sun
H, Sun P, Yang X, Huo X, Peng J and Liu K: Calycosin attenuates
triglyceride accumulation and hepatic fibrosis in murine model of
non-alcoholic steatohepatitis via activating farnesoid X receptor.
Phytomedicine. 25:83–92. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hu C and Jiang X: Role of NRP-1 in
VEGF-VEGFR2-Independent Tumorigenesis. Target Oncol. 11:501–505.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Simons M, Gordon E and Claesson-Welsh L:
Mechanisms and regulation of endothelial VEGF receptor signalling.
Nat Rev Mol Cell Biol. 17:611–625. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yang YL, Wang FS, Lin HY and Huang YH:
Exogenous therapeutics of Microrna-29a attenuates development of
hepatic fibrosis in cholestatic animal model through regulation of
phosphoinositide 3-Kinase p85 Alpha. Int J Mol Sci. 21:36362020.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mazo DF, de Oliveira MG, Pereira IV,
Cogliati B, Stefano JT, de Souza GF, Rabelo F, Lima FR, Ferreira
Alves VA, Carrilho FJ and de Oliveira CP:
S-nitroso-N-acetylcysteine attenuates liver fibrosis in
experimental nonalcoholic steatohepatitis. Drug Des Devel Ther.
7:553–563. 2013.PubMed/NCBI
|
|
51
|
Samali A, Cai J, Zhivotovsky B, Jones DP
and Orrenius S: Presence of a pre-apoptotic complex of
pro-caspase-3, HSP60 and HSP10 in the mitochondrial fraction of
Jurkat cells. EMBO. 18:2040–2048. 1999. View Article : Google Scholar
|
|
52
|
Caruso Bavisotto C, Alberti G, Vitale AM,
Paladino L, Campanella C, Rappa F, Gorska M, Conway de Macario E,
Cappello F, Macario AJL and Marino Gammazza A: HSP60
Post-translational modifications: Functional and pathological
consequences. Front Mol Biosci. 7:952020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Macario AJ and de Macario EC: Molecular
mechanisms in chaperonopathies: Clues to understanding the
histopathological abnormalities and developing novel therapies. J
Pathol. 250:9–18. 2020. View Article : Google Scholar
|