Introduction
Acute kidney injury (AKI) refers to a clinical
syndrome characterized by a sudden and severe decline in kidney
function, resulting from various factors. Due to its high incidence
and mortality rates, AKI has become a global clinical problem
(1,2). Despite certain advancements in the
prevention and treatment of this disease, a significant proportion
of patients who recover from AKI exhibit reduced kidney function
and progress to develop chronic kidney disease (CKD), including
end-stage renal disease (3-5).
Follow-up studies of hospitalized patients with AKI have shown that
15-30% of them progress to CKD (6-8).
Tubulointerstitial fibrosis (TIF) is a typical pathological feature
of CKD, and its extent serves as a marker for predicting CKD
progression (9). The occurrence
and development of TIF involves complex processes with the
participation of multiple cell types, with a particular focus on
fibroblasts that produce a large quantity of extracellular matrix
(ECM) components upon activation. However, increasing evidence has
demonstrated the significance of the contribution of renal tubular
cells in TIF. Severe AKI injuries lead to abnormal repair of
tubular cells, resulting in the production and secretion of various
cytokines, growth factors, pro-inflammatory molecules, and
pro-fibrotic molecules (10). The
increased bioactive molecules play a role in both autocrine
function, which is necessary for the de-differentiation, migration,
and proliferation of tubular cells during regeneration (10,11), and in normal paracrine function,
for recruiting leukocytes from circulation and activating
fibroblasts and other interstitial cells (11,12). The characteristics of activated
fibroblasts include elevated expression of contractile proteins,
increased cell motility, induction of pro-inflammatory genes, and
high-level deposition of collagen and other ECM proteins (13). TGF-β and the phosphorylated form
of SMAD2/3, which acts downstream in the signaling pathway, are
crucial intracellular signals in fibroblast activation, including
renal myofibroblast activation (14-16). However, the molecular mechanisms
underlying the dysregulated paracrine secretion of fibrotic factors
by injured tubular cells remain incompletely understood. Therefore,
a thorough investigation of the mechanisms leading to kidney
fibrosis following AKI and the identification of potential
intervention targets are crucial for preventing the chronic
progression of AKI.
In the current study, the transcriptomic dataset of
kidney tissues following ischemia-reperfusion injury [IRI; gene
expression omnibus (GEO) dataset GSE98622] was analyzed (17). The data indicated dynamic changes
in the expression of the flavin-containing monooxygenase (FMO)
family between 2 h and 14 days post-ischemia reperfusion. A recent
study has identified that FMO2 possesses a previously
uncharacterized enzyme-independent antifibrotic activity via the
cytochrome P450 2J3-SMAD ubiquitination regulatory factor 2
(SMURF2) axis (18).
Particularly, FMO1 and FMO2, which are abundantly expressed in the
kidney, are of significant interest. FMOs are a class of enzymes
that utilize flavin coenzymes to introduce a single oxygen atom
into chemical reactions. These enzymes interact with oxygen
molecules, transferring the oxygen atom to substrates and
undergoing oxidation-reduction reactions. They typically function
in conjunction with reducing agents, such as coenzymes NADH or
flavin adenine dinucleotide, to provide the necessary electrons for
the reactions (19). FMOs play
various roles in biological systems. FMO2 is primarily expressed in
human lungs, followed by the kidneys, and exhibits high activity
during infancy. It participates in the metabolism of exogenous
compounds, including certain drugs and environmental toxins, such
as sulfur-containing compounds (20). IRI triggers an inflammatory
response and members of the FMO family may be involved in
regulating the inflammation processes induced by IRI. These enzymes
may impact the intensity and duration of the inflammatory response
by modulating the synthesis and metabolism processes of
inflammatory mediators, such as leukocyte activation, cytokine
production, and cell adhesion molecule expression (21,22). It has been shown in a previous
study conducted by our research group that renal expression of FMO2
mitigates post-AKI functional impairment and immune cell
infiltration into the kidneys. Furthermore, members of the FMO
family may participate in tubular cell-mediated paracrine secretion
of pro-fibrotic factors, thereby influencing the fibrotic process.
It is important to note that FMO2 overexpression inhibits the
effects of TGF-β and the phosphorylation of SMAD2/3. FMO2
expression negatively regulates the TGF-β signaling pathway through
the induction of SMURF2 expression and its nuclear translocation.
SMURF2 serves as an intrinsic negative regulator of SMAD2/3
phosphorylation and of the TGF-β1 signaling pathway. Therefore, the
present study demonstrated that FMO2 acted as an inhibitor of the
TGF-β/SMAD2/3 signaling pathway, exerting a conservative
ameliorative effect on renal fibrosis and pathological remodeling.
However, the specific mechanisms and effects may vary depending on
the type of fibrosis and tissue specificity. Further investigations
will contribute to a deeper understanding of the specific roles of
the FMO family in the process of renal fibrosis, thereby providing
new targets for the treatment of this disease.
Materials and methods
Transcriptomic profiling and differential
expression analysis of a murine renal bilateral
ischemia-reperfusion mode
The GSE98622 (17)
mRNA expression profile dataset was downloaded from GEO (https://www.ncbi.nlm.nih.gov/geo/). The 'limma'
software package helped identify differentially expressed genes.
The 'heatmap' software packages of R software were used to draw
heat maps.
Experimental animals for renal
ischemia-reperfusion injury
Male C57BL/6 mice weighing 20-25 g were purchased
from the Experimental Animal Center of China Three Gorges
University (Yichang, China) and maintained under specific
pathogen-free conditions. All animal experiments were conducted in
accordance with the regulations of the relevant laws and approved
by the Ethics Committee of the Children's Hospital Affiliated to
Zhengzhou University (approval no. 2023-K-074).
Unilateral renal ischemia/reperfusion
model
Unilateral ischemic acute kidney injury (AKI) or
chronic kidney disease (CKD) mouse models were established by
clamping the left renal artery. Unilateral renal
ischemia-reperfusion was performed as recently described (7,11).
Briefly, mice were anesthetized by intraperitoneal injection of
pentobarbital (60 mg/kg) prior to surgery. The left renal artery
was isolated, and a microaneurysm clip was used to clamp the artery
for 28 min, inducing renal ischemia-reperfusion injury. After 28
min, blood flow was restored by releasing the clamp, and the layers
of muscle and skin were sutured sequentially. Throughout the entire
surgical procedure, the mice were maintained at a body temperature
of 36-37.2°C. One day before euthanasia, the non-clamped right
kidney was removed. Following a reperfusion period of either 24 h
or 14 days, mice were euthanized through cervical dislocation under
inhalation anesthesia using 3% isoflurane, and serum and kidney
tissue samples were collected. The control group underwent a sham
operation without clamping the renal artery.
Cell culture
Boston University mouse proximal tubule cells
(BUMPT; obtained from Dr Lieberthal and Dr Shwartz at Boston
University) were cultured in DMEM (Dulbecco's modified Eagle's
medium) (Thermo Fisher Scientific, Inc.) with 10% fetal bovine
serum, penicillin (100 U/ml) and streptomycin (100 μg/ml) in
a humidified atmosphere of 5% CO2 at 37°C. NRK-49F rat
kidney interstitial fibroblast cells were obtained from the
American Type Culture Collection. Cells were cultured in Dulbecco's
modified Eagle's medium/nutrient mixture F-12 (DMEM/F-12) medium
supplemented with 10% fetal bovine serum (Invitrogen; Thermo Fisher
Scientific, Inc.).
Gene delivery in vivo
In the animal study, 10 days prior to renal
ischemia, the recombinant lentiviral vector PGLV carrying FMO2
(Shanghai GenePharma, Co., Ltd.) was introduced into the mouse left
kidney as previously described (7,23).
Briefly, mice were anesthetized by intraperitoneal injection of
pentobarbital (60 mg/kg) prior to surgery (24). A total of 100 μl lentivirus
solution [1×105 transduction units (TU)/μl] was
injected into anesthetized mice with a 30 G syringe. The needle was
inserted from the lower pole to the upper pole of the kidney, and
the lentivirus solution was slowly injected while withdrawing the
needle.
Treatment of renal tubule cells with
TGF-β
BUMPT cells reached ~60% confluence, they were
exposed to serum-free DMEM supplemented with 10 ng/ml recombinant
human TGF-β for 48 h. Control cells were cultured in serum-free
medium without TGF-β. At specified time points, cells were
collected for analysis of mRNA and protein levels to detect
fibrogenic factors such as Vimentin, actin α 2, smooth muscle
(Acta2), platelet-derived growth factor subunit B (Pdgfb) and
cellular communication network factor 2 (Ccn2).
Collection of renal tubule conditioned
media and intervention on renal fibroblasts (NRK-49F)
For renal tubule cells, cells were seeded in 60 mm
culture dishes at a density of 1×106 cells/dish for
BUMPT cells. When cells reached ~60% confluence, they were treated
with 10 ng/ml TGF-β in serum-free DMEM for 48 h, while control
cells were maintained in serum-free medium without TGF-β for 48 h.
Subsequently, the medium for both TGF-β-treated cells and control
cells was replaced with complete medium without TGF-β, and further
incubated for 24 h to collect renal tubule cells-conditioned media.
The tubular cell-conditioned media were concentrated using
Amicon® Ultra-4 centrifugal filter devices (cat. no.
UFC8003; MilliporeSigma) with a molecular weight cutoff of 3 kDa. A
total of 4 ml of conditioned media were collected from each 60 mm
culture dish, and the supernatant was transferred to the
Milli-Q® water pre-rinsed centrifugal filter device. It
was then centrifuged at 7,500 × g for 1 h at 4°C using a
fixed-angle rotor to recover ~100 μl concentrated
conditioned media.
To treat fibroblasts with renal tubule
cell-conditioned media, NRK-49F fibroblasts were seeded in 12-well
plates at a density of 0.15×106 cells/well and reached
~80% confluence on the following day. After overnight serum
starvation in serum-free DMEM, NRK-49F fibroblasts were incubated
with renal tubule cell-conditioned media (diluted 100 μl in
1 ml serum-free DMEM) for 48 h. Cell morphology was monitored, and
cells were counted using the Bio-Rad TC20 automated cell counter
(Bio-Rad Laboratories, Inc.) after trypsin digestion. Subsequently,
cells were lysed for measurement of cellular protein and immunoblot
analysis. In experiments testing the effects of fibroblast growth
factor 2 (FGF2) and antibodies, antibodies or mouse IgG were
pre-incubated with renal tubule cell-conditioned media at room
temperature for 1.5 h before adding them to NRK-49F
fibroblasts.
Histology, immunohistochemistry,
immunofluorescence and TUNEL staining
Tissues were transferred to 4% paraformaldehyde and
fixed by leaving tissues at 4°C overnight, then embedded in
paraffin and cross-sectioned (4 μm) for histology
examination. H&E and Masson's trichrome were performed
according to manufacturer's instructions (Beijing Solarbio).
Immunohistochemistry and immunofluorescence techniques were
employed using 5 μm sections of mouse kidney tissues.
Antigen retrieval was performed using 10 mM sodium citrate, pH 6.0,
0.05% Tween (0.1 mmol sodium citrate in 5 ml + dH2O 45
ml + Tween X 100 25 μl; 100 mM sodium citrate stored at
4°C). Samples were boiled in the above buffer in a steamer for 60
min and then allowed to cool at room temperature for 20 min. The
slides were washed with PBS and blocked with blocking buffer [1%
bovine serum albumin (BSA), 1% goat serum, 0.3% Triton X-100 in
PBS] for 20-40 min at room temperature. After antigen retrieval and
blocking, primary antibodies against FMO2 (cat. no. NBP1-85952;
Novus Biologicals, LLC; 1:500 dilution), Kim-1 (cat. no. AF1750;
R&D Systems, Inc.; 1:200 dilution), macrophage (F4/80) (cat.
no. ab56297; Abcam; 1:50 dilution), myeloperoxidase (MPO; cat. no.
MAK283; MilliporeSigma; 1:200 dilution), α-SMA (cat. no. ab5694;
Abcam; 1:200 dilution) and Vimentin (cat. no. ab92547; Abcam; 1:200
dilution) were used to detect specific markers incubating at 4°C
overnight. For immunohistochemistry, sections were initially washed
thrice with PBS before 1-h incubation at 37°C with an HRP-labeled
secondary antibody. Diaminobenzidine tetrahydrochloride served as
the enzyme substrate for visualization, followed by hematoxylin
counterstaining. Images were then acquired using cellSens software
(V3.2; cellSens) at ×200 magnification. For tissue
immunofluorescence, samples were incubated for 1 h at 25°C with
1:2,000 diluted Alexa Fluor 488 and 555-conjugated secondary
antibodies (Cell Signaling Technology, Inc.). Nuclei were labeled
with DAPI from Beyotime Institute of Biotechnology, and
visualization was carried out at ×400 magnification using an
Olympus fluorescence microscope at ×200 magnification.
Tubular injury was assessed based on
histopathological changes, including cell lysis, loss of brush
border, and cast formation in renal tubules. A blinded examination
was performed, and the percentage of injured tubules was scored as
follows: 0 (no damage), 1 (<25%), 2 (25-50%), 3 (50-75%) and 4
(>75%). A total of 10 randomly selected fields were evaluated
per mouse for quantification, and the mean score was calculated as
the tubular injury score. The ImageJ 1.45 software (National
Institutes of Health) was used to calculate the collagen volume
fraction, that is, the percentage of the blue area (collagen) to
the total area of each field. Randomly selected 10 fields of view
per mouse kidney at a magnification of ×200 were scored for
quantification using an Olympus microscope and quantized with
cellSens software (V3.2; cellSens) or an LSM880 laser scanning
confocal microscope (Zeiss AG) system with a Plan-Apochromat
63x/1.4 objective and ZEN 2.3 software (Zeiss AG).
ImageJ 1.45 software (National Institutes of Health)
was used to obtain the area (%) values for α-SMA staining (the
percentage of the α-SMA positive green signal as a percentage of
the total area of each field).
To detect cell apoptosis, a TUNEL apoptosis
detection system (Promega Corporation) was utilized. The cellular
nuclei were stained with DAPI for a duration of 3-5 min at ambient
temperature, followed by inspection of the prepared sections under
a fluorescence microscope at a magnification of ×400. The number of
TUNEL-positive cells was counted from 10 randomly selected fields
of view per specimen in the outer medulla and kidney cortex regions
(25).
Western blot analysis
Western blot assays were used to identify protein
expression in kidney tissues and cells. Briefly, lysates containing
whole cells, nuclei, or cytosolic extracts (with 10-50 mg of
protein) were heated at 95°C for 5 min in Laemmli sample buffer
(Bio-Rad Laboratories, Inc.). Proteins were then separated by
polyacrylamide gel electrophoresis in acrylamide gels (8-15%) and
transferred using a Bio-Rad western system to polyvinylidene
difluoride membranes (Bio-Rad Laboratories, Inc.), which were
immediately placed in 5% non-fat milk in Tris-buffered saline (TBS,
50 mM Tris, pH 7.6, 150 mM NaCl)-Tween (0.1% Tween20) buffer for
blocking (1 h at 25°C). Membranes were incubated overnight with
antibodies against flavin-containing monooxygenase 1 (FMO1; cat.
no. PA5-95285; Invitrogen; Thermo Fisher Scientific, Inc.; 1:1,000
dilution), FMO2 (cat. no. NBP1-85952; Novus Biologicals, LLC;
1:500), SMURF2 (cat. no. Ab53316; Abcam; 1:1,000), Histone3 (cat.
no. Ab1791; Abcam; 1:1,000), phosphorylated SMAD3 (423/425) (cat.
no. 9520; Cell Signaling Technology, Inc.; 1:1,000), SMAD3 (cat.
no. 9523; Cell Signaling Technology, Inc.; 1:1,000), phosphorylated
SMAD2 (465/467) (cat. no. 18338; Cell Signaling Technology, Inc.;
1:1,000), SMAD2 (cat. no. 8685; Cell Signaling Technology, Inc.;
1:1,000), Vimentin (cat. no. ab92547; Abcam; 1:1,000), FGF2 (cat.
no. ab222932; Abcam; 1:1,000), PDGF (cat. no. ab178409; Abcam;
1:1,000), connective tissue growth factor (CTGF; cat. no. ab209780;
Abcam; 1:1,000), α-SMA (cat. no. ab5694; Abcam; 1:5,000) and GAPDH
(cat. no. CL594-60004; ProteinTech Group, Inc.; 1:20,000) at 4°C.
Membranes were then washed 3 times for 10 min in TBS-Tween buffer
and incubated with a horseradish peroxidase-conjugated anti-mouse
antibody (Servicebio; cat. no. GB23301; 1:10,000 dilution) or
anti-rabbit antibody (Servicebio; cat. no. GB23303; 1:10,000
dilution) at 25°C for 1 h. Films were scanned and signals detected
using a Bio-Rad calibrated densitometer (Bio-Rad Laboratories,
Inc.). ImageJ 1.45 software (National Institutes of Health) was
used for densitometry analysis. Statistical analysis was performed
using 3-5 biological replicates from 2-3 technical replicates.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted from whole kidney tissues or
cells using TRIzol® reagent (Ambion; cat. no. 368711),
according to the manufacturer's protocol. RNA reverse transcription
reagents (Vazyme; cat. no. r323-01) were used to generate cDNA from
1 μg total RNA using an amplicon (Pcrsurecycler8800)
according to the manufacturer's protocol. Real-time quantitative
TaqMan PCR reagents (Vazyme; cat. no. q712-02) were employed, along
with the TaqMan primers (Applied Biosystems; Thermo Fisher
Scientific, Inc.) listed below. qPCR was performed on a real-time
PCR instrument (Mx3005p), and data were analyzed using the
2−ΔΔCq method for quantification (26). The sequences of the primers were
as follows: Monocyte chemotactic protein 1 forward, 5′-CTG AGT TGA
CTC CTA CTG TGG A-3′ and reverse, 5′-TCT TCC CAG GGT CGA TAA
AGT-3′; IL-6 forward, 5′-CTG CAA GAG ACT TCC ATC CAG-3′ and
reverse, 5′-AGT GGT ATA GAC AGG TCT GTT GG-3′; Fgf2 forward, 5′-GCG
ACC CAC ACG TCA AAC TA-3′ and reverse, 5′-CCG TCC ATC TTC CTT CAT
AGC-3′; Ccn2 forward, 5′-GGC CTC TTC TGC GAT TTC G-3′ and reverse,
5′GCA GCT TGA CCC TTC TCG G-3′; transforming growth factor β1
(TGF-β1) forward, 5′-CCA CCT GCA AGA CCA TCG AC-3′ and reverse,
5′-CTG GCG AGC CTT AGT TTG GAC-3′; Pdgfb forward, 5′-TGC TGC ACA
GAG ACT CCG TA-3′ and reverse, 5′-GAT GAG CTT TCC AAC TCG ACT C-3′;
and GAPDH forward, 5′-AAG TTC AAC GGC ACA GTC AA-3′ and reverse,
5′-TCT CGC TCC TGG AAG ATG G-3′.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism version 9.0.0 (Dotmatics), and the data are presented as the
mean ± SEM. Normal distribution and homogeneity of variance were
assessed using a Shapiro-Wilk test and Bartlett's test,
respectively. For data that passed both normality and equality of
variance, comparisons between two groups were performed using an
unpaired 2-tailed Student's t-test, and multiple comparisons were
analyzed using an ANOVA followed by a Tukey's post-hoc test. If
not, the non-parametric Kruskal-Wallis test was used, followed by a
Dunn's test. P<0.05 was considered to indicate a statistically
significant difference.
Results
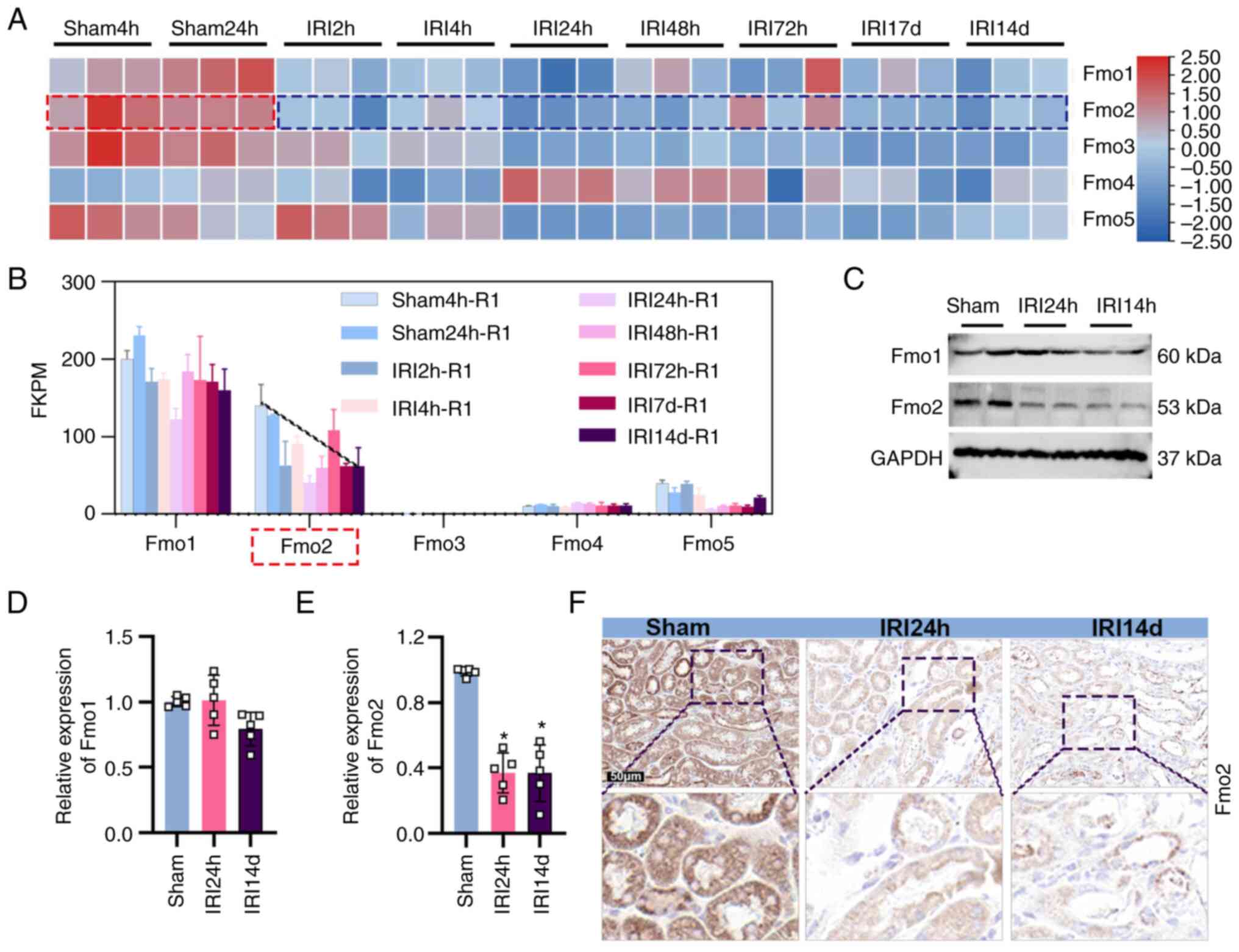
Dynamic expression of FMO2 in AKI-CKD
transformation
To determine the alterations in the expression
levels of the FMO family of enzymes during the process of AKI-CKD,
the transcriptomic data of mouse kidney tissues were analyzed from
the sham and IRI groups obtained from a public RNA sequencing
database (GEO dataset GSE98622) (17). The FMO family consists of five
subtypes (FMO1-5). Among them, FMO1 and FMO2 exhibited the highest
abundance values in the kidneys (Fig.
1A and B). It is important to note that only FMO2 indicated a
significant downregulation at the protein level in mouse kidney
tissues following IRI (Fig. 1C).
Consistent with the protein level changes, the mRNA levels of FMO2
were also lower in the IRI 24 h and IRI 14 day groups compared with
those of the sham group (Fig.
1E). However, no significant differences were noted at the
protein and mRNA levels of FMO1, which exhibited the highest
abundance in the kidneys among the FMO subtypes (Fig. 1C and D). Immunostaining analysis
was based on mouse kidney slices and the results further confirmed
the significant downregulation of FMO2 expression in the IRI 24 h
and IRI 14 day groups compared with the sham group. Therefore, the
alteration of FMO2 expression may be dynamically associated with
the occurrence of renal ischemic injury and fibrotic
remodeling.
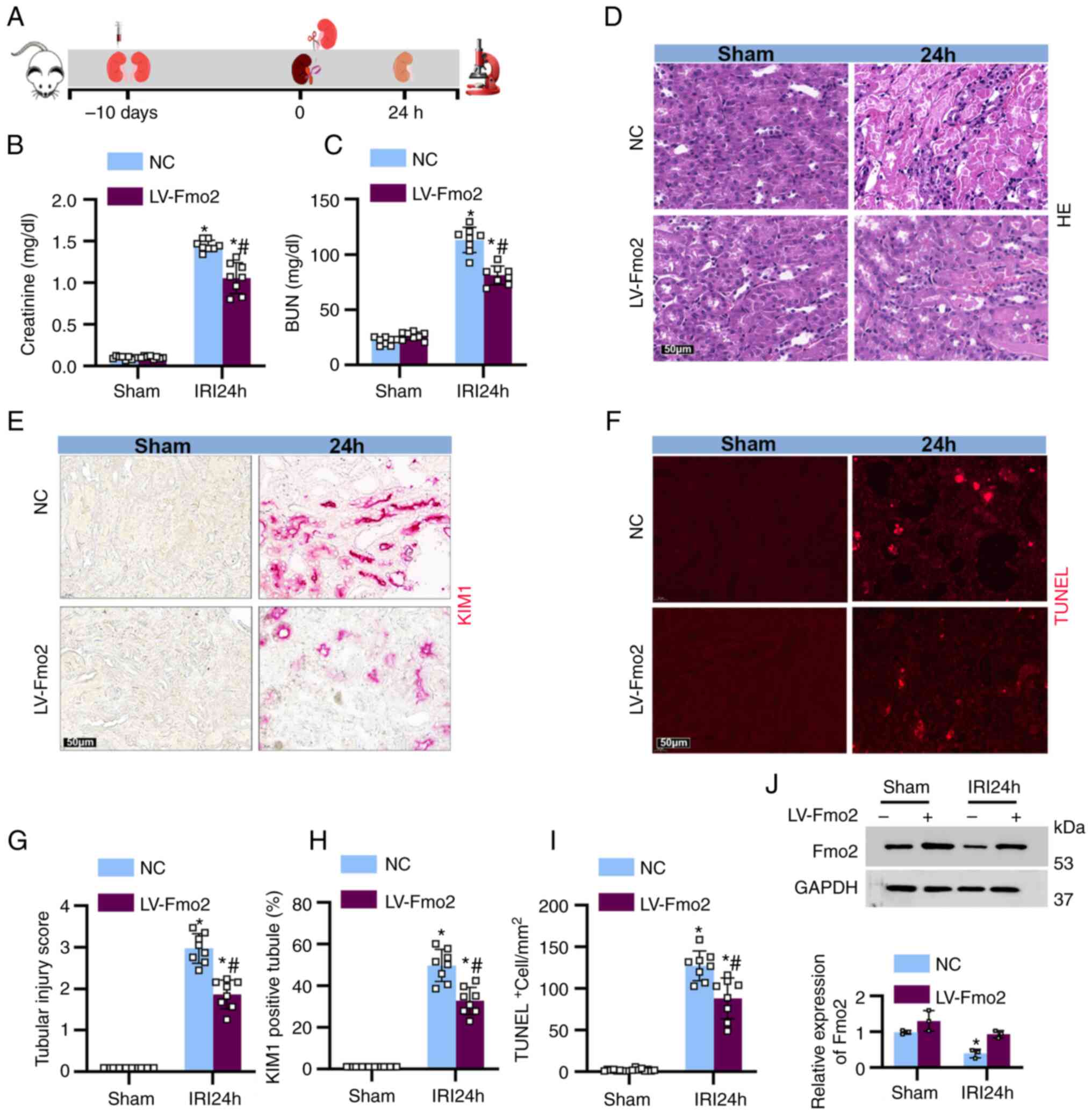
FMO2 alleviates ischemia-induced AKI
To investigate the role of FMO2 in renal AKI, mice
were injected with lentivirus (LV)-FMO2 or a negative control (NC)
10 days prior to unilateral renal ischemia. Following 28 min of
continuous ischemia and 24 h of reperfusion, the mice were
euthanized (Fig. 2A). The mice
treated with LV-FMO2 indicated a significant decrease in blood urea
nitrogen and serum creatinine levels following IRI stimulation
compared with the corresponding levels noted in the NC group
(Fig. 2B and C). The extent of
renal tissue damage was further assessed and examined using
H&E, kidney injury molecule (KIM)-1, and TUNEL staining
(Fig. 2D-F). It is important to
note that the renal tubular injury score reached 2.9 following 28
min of ischemia and 24 h of reperfusion, while mice treated with
LV-FMO2 exhibited less severe renal tubular injury with a score of
1.9 (Fig. 2G). Following IRI, the
percentage of KIM-1-positive tubular cells in the LV-FMO2-treated
group (57.6%) was significantly lower than that noted in the NC
group (38.4%, Fig. 2H). The
LV-FMO2-treated mice had a significantly lower number of apoptotic
cells (130/mm2) compared with that of the NC group
(98/mm2, Fig. 2I).
Following immunoblotting analysis, the levels of FMO2 were
significantly higher in the LV-FMO2-treated mice compared with
those of the LV-NC-treated mice (Fig.
2J). These results suggested that FMO2 may exert a protective
effect during the injury phase of ischemic AKI.
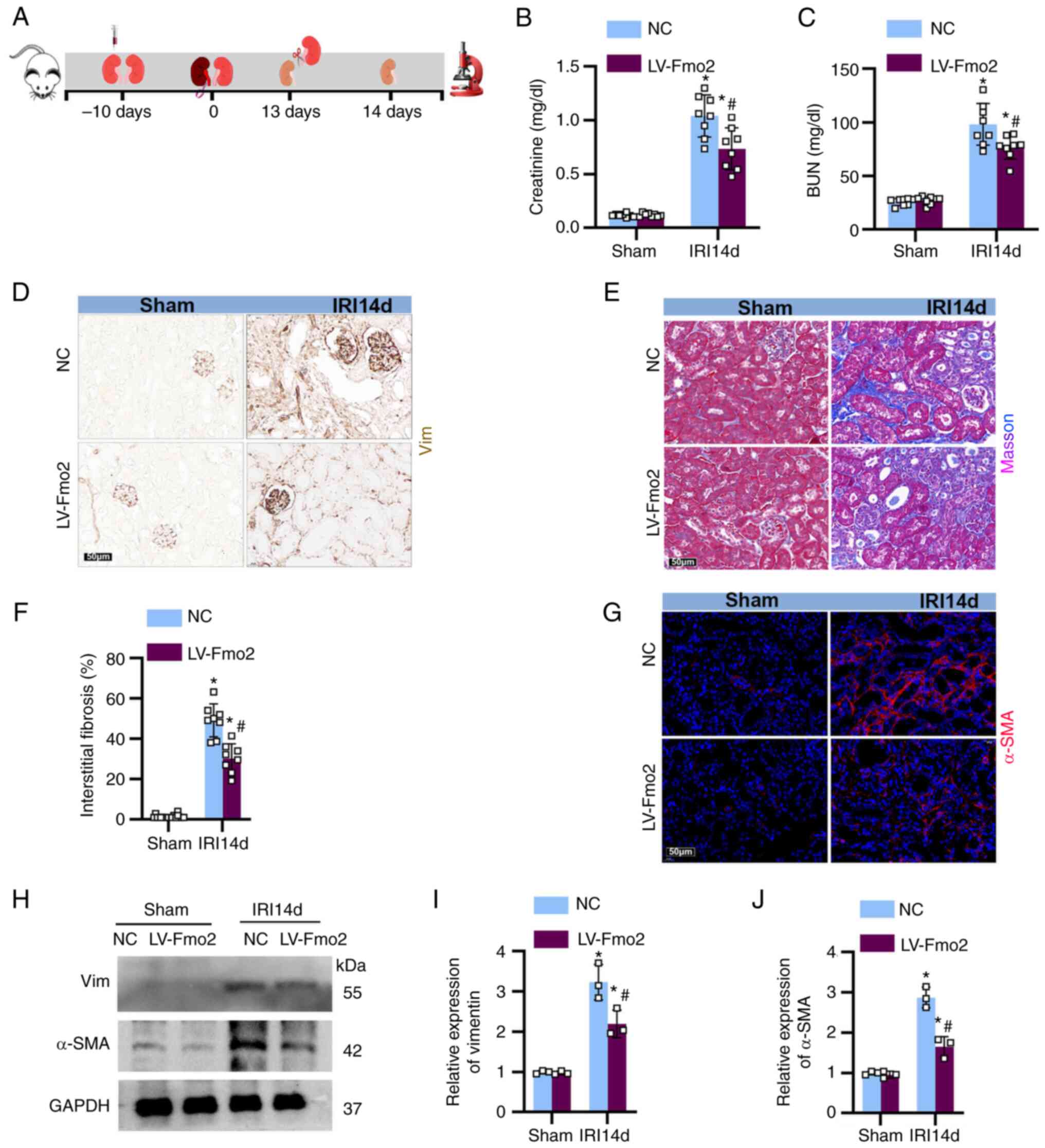
Expression of FMO2 in the kidney prevents
the development of fibrosis
To examine the impact of FMO2 on renal fibrosis
following ischemia-reperfusion, the mice were injected with LV
vectors containing LV-FMO2 or an NC into the kidney 10 days prior
to unilateral left renal ischemia. The mice underwent 28 min of
continuous ischemia, followed by unilateral right nephrectomy on
the 13th day following reperfusion, and were euthanized on the 14th
day (Fig. 3A). Following the
removal of the right kidney, the levels of blood urea nitrogen and
serum creatinine increased (Fig. 3B
and C). Staining for vimentin (Fig. 3D), Masson's trichrome (Fig. 3E) and α-SMA (Fig. 3G) revealed significant ECM
deposition in the renal tubulointerstitium on the 14th day
following ischemia-reperfusion treatment (Fig. 3F). Furthermore, western blot
analysis of and expression in the mouse kidneys (Fig. 3H-J) confirmed that overexpression
of FMO2 attenuated renal dysfunction and fibrosis induced by
IRI.
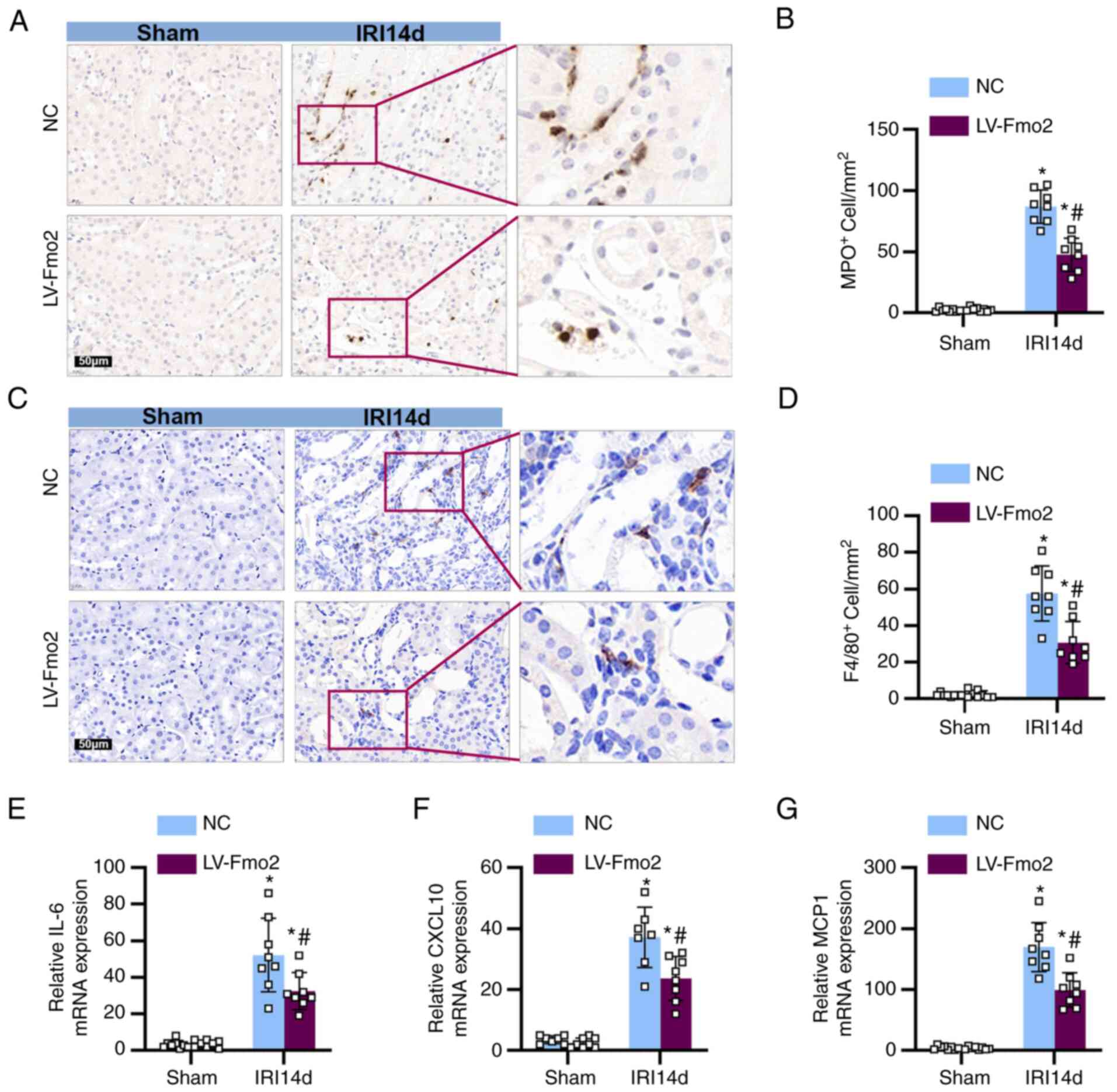
Expression of FMO2 reduces the
infiltration of immune cells into the kidneys
Subsequent evaluation of the impact of FMO2
expression on immune cell infiltration was conducted.
Immunohistochemical analysis using MPO staining indicated the
presence of neutrophils, while F4/80 staining indicated the
presence of macrophages. The expression of FMO2 limited the
inflammatory infiltration of neutrophils (Fig. 4A and B) and macrophages (Fig. 4C and D) compared with that noted
in the IRI 14-day NC group. In addition, qPCR analysis demonstrated
that the expression of FMO2 suppressed the expression levels of
pro-inflammatory cytokines, such as IL-6, chemokine ligand 1 and
monocyte chemoattractant protein 1 (Fig. 4E and G).
In vitro, decreased expression of FMO2 in
BUMPT cells diminishes the effect of TGF-β
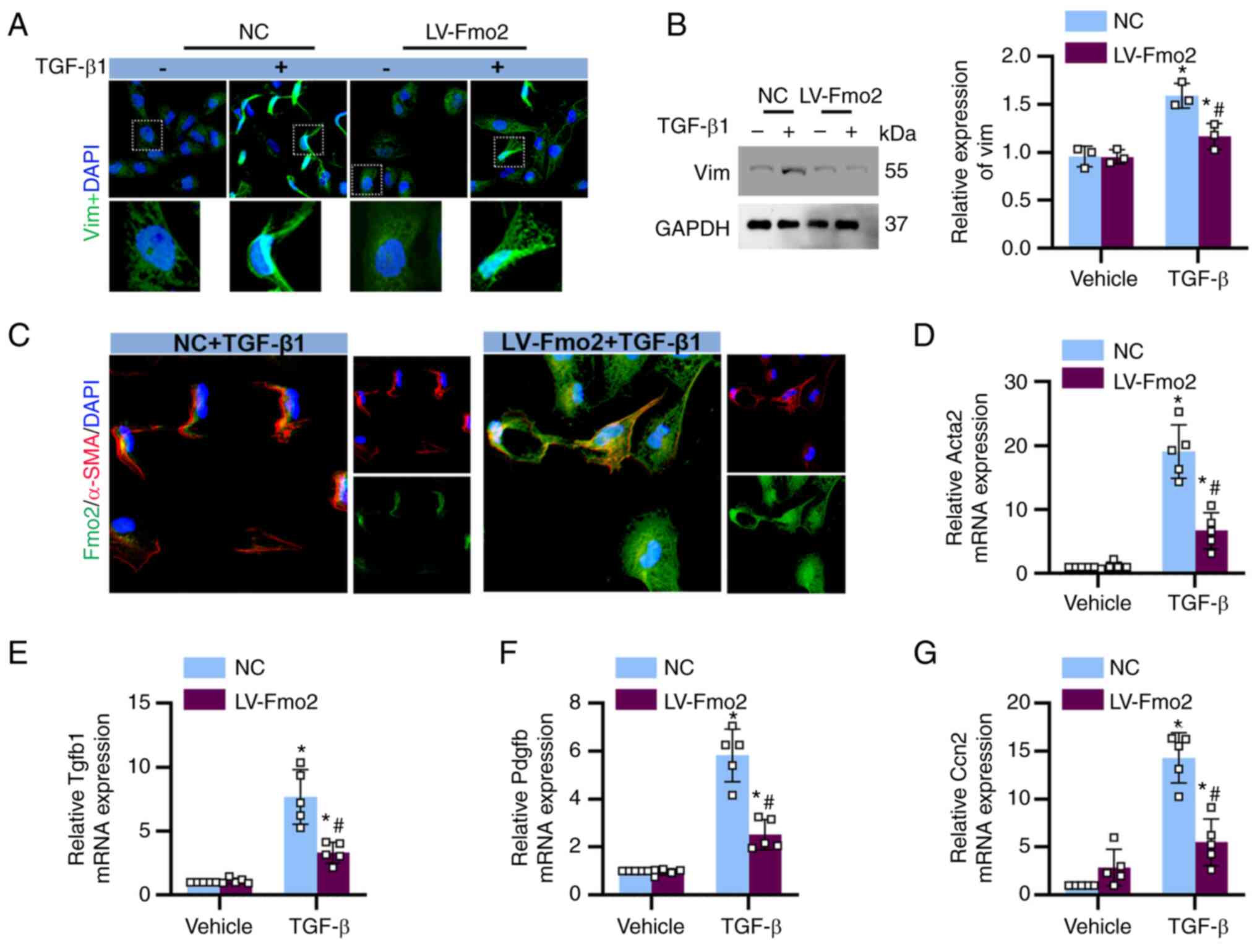
The in vitro data of the present study
suggested that the expression of FMO2 in tubular cells exerted
anti-fibrotic effects. In BUMPT cells cultured under basal
conditions without TGF-β stimulation, minimal expression of
vimentin and α-SMA was observed. However, significant expression of
vimentin and α-SMA was observed in BUMPT cells following TGF-β
stimulation. The expression of FMO2 effectively prevented the
responsiveness of BUMPT cells to TGF-β stimulation (Fig. 5A and B). Furthermore, fluorescence
co-staining of FMO2 and α-SMA revealed that TGF-β stimulation led
to a decrease in FMO2 expression and an increase in α-SMA
expression (Fig. 5C). qPCR
results demonstrated that the expression of FMO2 suppressed the
expression levels of fibrotic factors induced by TGF-β, including
α-SMA, TGF-β1, PDGF-β, and cellular communication network factor 2
(Fig. 5D-G).
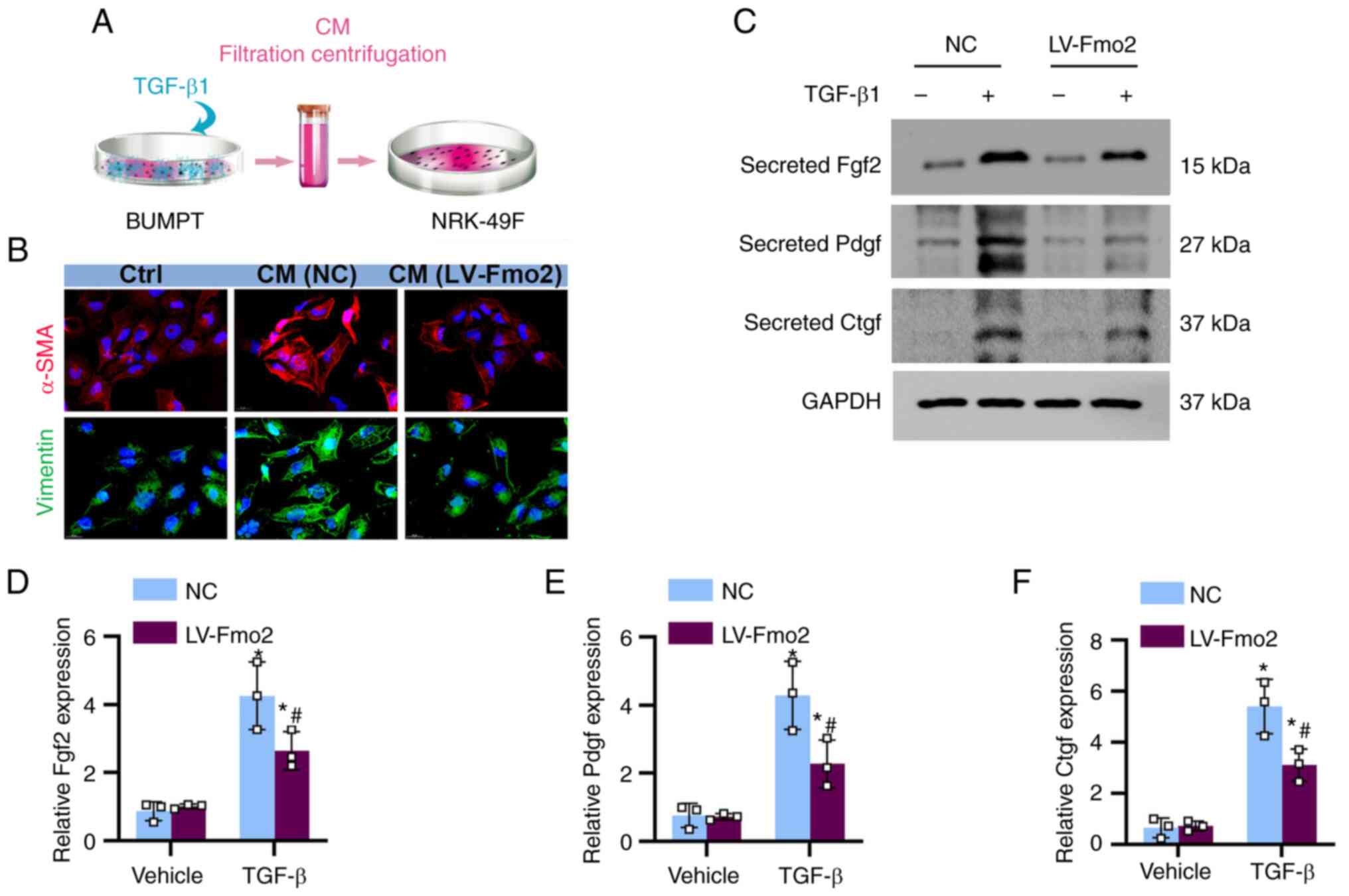
Expression of FMO2 reduces the paracrine
secretion of pro-fibrotic factors from BUMPT cells, inhibiting the
activation of fibroblasts
The results indicated that the conditioned medium
from TGF-β-induced BUMPT cells exerted an activating effect on
fibroblasts. The medium exposed to BUMPT cells containing TGF-β
exhibited a significant increase in the levels of paracrine
fibrotic factors, such as FGF2, PDGF, and CTGF. The expression of
FMO2 partially alleviated the paracrine secretion of FGF2 and other
factors (Fig. 6A). Conditioned
medium from TGF-β-treated BUMPT cells was collected, centrifuged,
and subsequently used to culture NRK-49F cells (Fig. 6B). Immunofluorescence staining of
NRK-49F cells revealed that the conditioned medium from
LV-FMO2-treated BUMPT cells exhibited a noticeable reduction in the
expression levels of α-SMA and vimentin (Fig. 6C).
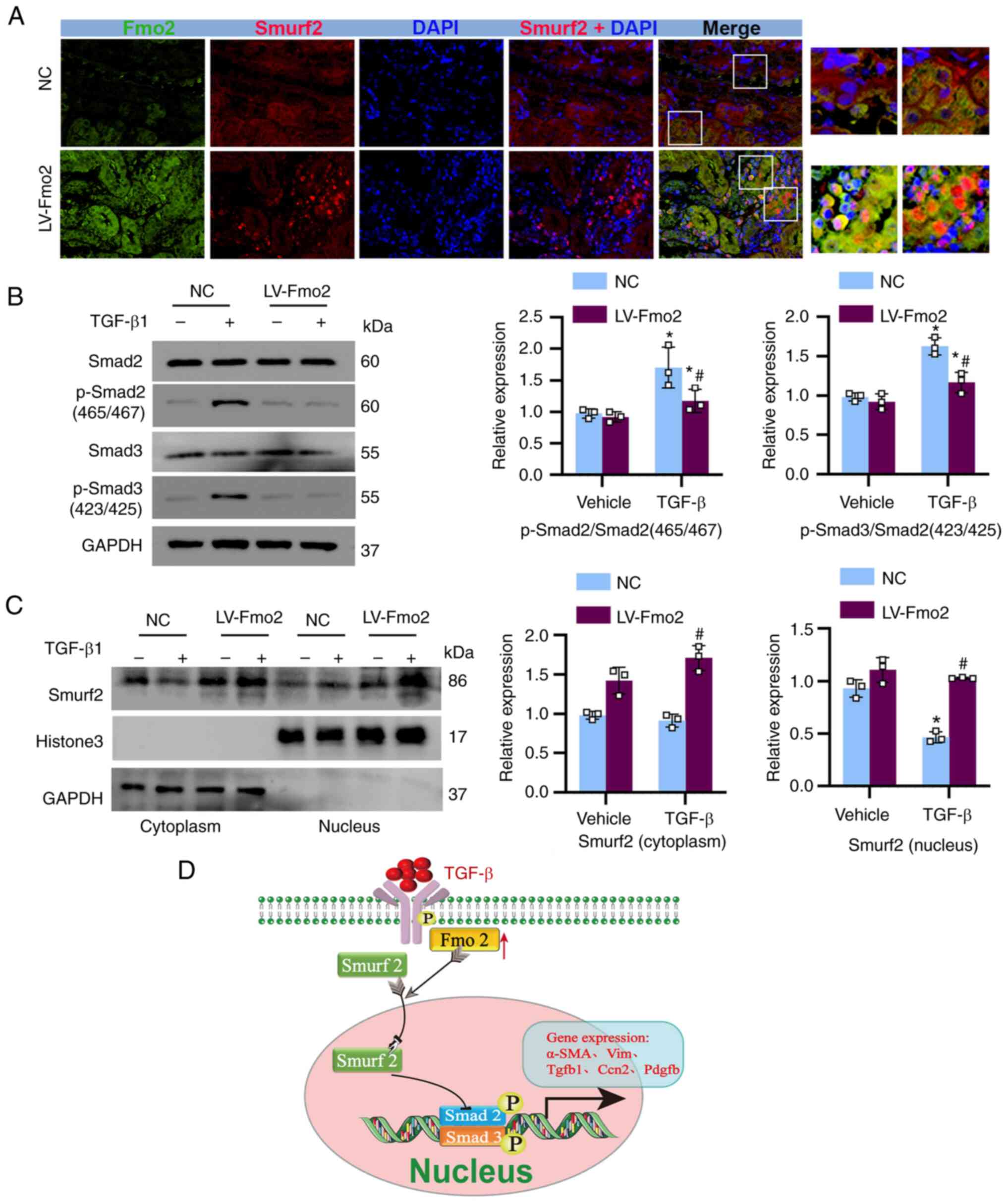
FMO2 inhibits the phosphorylation of
SMAD2/3 by promoting the translocation of SMURF2 to the
nucleus
Due to the cytoplasmic localization of FMO2, this
enzyme is unlikely to directly participate in transcriptional gene
regulation. Therefore, the transcriptional post-effects mediated by
FMO2 and its subsequent anti-fibrotic actions mediated via
inhibition of SMAD2/3 phosphorylation were examined. SMURF2, an E3
ubiquitin ligase, exhibits specific activity towards phosphorylated
SMAD2/3 in the nucleus (27) and
plays a crucial role in the negative regulation of the TGF-β
signaling pathway. Initially, co-immunofluorescence staining of
FMO2 (green) and SMURF2 (red) was performed in the kidney tissues
of IRI 14 day mice, revealing nuclear translocation of SMURF2 under
conditions of high FMO2 expression (Fig. 7A). The elevated expression of FMO2
in BUMPT cells significantly reduced the levels of phosphorylated
SMAD2/3, as determined by its phosphorylation state (Fig. 7B). Furthermore, by using nuclear
protein extraction and immunoblotting, it was confirmed that the
expression of FMO2 promoted the translocation of SMURF2 from the
cytoplasm to the nucleus (Fig.
7C). These data suggest that FMO2 ultimately facilitates the
nuclear translocation of SMURF2, by regulating its expression and
leads to inhibition of TGF-β signaling (Fig. 7D).
Discussion
The present study provides three primary
conclusions. First, the expression levels of FMO2 were evaluated,
which is a member of the FMO gene family, that are abundantly
expressed in the kidney, and dynamically associated with renal
ischemic injury and fibrotic remodeling. Overexpression of FMO2
alleviated renal ischemia-reperfusion-induced AKI, renal fibrosis,
and immune cell infiltration into the kidney. Second, FMO2
overexpression not only effectively blocked TGF-β-induced tubular
cell fibrosis but also inhibited the activation of aberrant tubular
cell-derived paracrine fibrogenic factors on fibroblasts. Thirdly,
FMO2 negatively regulated TGF-β-induced SMAD2/3 activation by
promoting the expression and nuclear translocation of SMURF2.
Therefore, FMO2 emerges as a previously unidentified modulator of
TGF-β signaling, playing a conserved key role in fibroblast
activation and fibrotic remodeling following injury.
The enzymes of the FMO family are primarily present
in animals and microorganisms. The canonical function of most FMO
gene family members involves enzymatic activity-dependent cellular
detoxification (19).
Unexpectedly, however, the FMO2-mediated suppression of TGF-β
activity is independent of its enzymatic activity. While the
nonenzymatic FMO2 isoform has been reported in mammals (27,28), its biological role has never been
clearly elucidated. They participate in the metabolism of various
substrates, including drugs and toxins, and are involved in
specific biological processes (29). The members of the FMO family are
predominantly expressed in tissues, such as the liver and kidneys,
exhibiting structural and functional diversity. The FMO family
consists of five subtypes (FMO1-5), each potentially differing in
tissue distribution, catalytic properties, and substrate
specificity; these enzymes play important roles in drug metabolism
(27). By using analysis of
transcriptomic data following renal IRI, it was discovered that the
expression of one member of the FMO gene family, FMO2, was highly
enriched in tubular cells and dynamically associated with renal
injury and fibrotic remodeling. However, previous studies have
shown that FMO2-mediated inhibition of TGF-β activity is
independent of its enzymatic activity (18). Although non-enzymatic isoforms of
FMO2 have been reported in humans and rats (30-32) and have been shown to influence the
lifespan of the nematode Caenorhabditis elegans (33), their biological roles remain
unclear. It is important to note that the downregulation of FMO2
expression in renal tubular cells is a conserved molecular
characteristic in response to injury and TGF-β stimulation, while
FMO2 inactivation is sufficient to promote the induction of
fibrosis and renal dysfunction. By contrast, overexpression of FMO2
alleviated renal ischemia-reperfusion-induced AKI, renal fibrosis,
and immune cell infiltration into the kidney. Therefore, FMO2
emerges as a previously unidentified modulator in the progression
from AKI to CKD, playing a crucial role in tubular cell fibroblast
transition, fibroblast activation, and interstitial fibrotic
remodeling following renal injury.
Impaired adaptive repair of the kidney following AKI
is a fundamental pathophysiological mechanism leading to renal
fibrosis and progression to CKD. Defective repair of proximal
tubular epithelial cells is a key factor in the progression from
AKI to CKD. In the early stages of injury, surviving tubular cells
mount an adaptive response, undergoing dedifferentiation and
proliferation to repair the damaged renal tubules (34). However, severe AKI injury can
result in aberrant repair of tubular cells, leading to the
production and secretion of various cytokines, growth factors,
pro-inflammatory molecules, and pro-fibrotic molecules (10). The increased levels of these
bioactive molecules play a role in both autocrine functions, which
are necessary for tubular cell regeneration via dedifferentiation,
migration, and proliferation, and paracrine functions, recruiting
circulating leukocytes and activating interstitial cells, such as
fibroblasts (35). During the
AKI-CKD process, aberrant paracrine secretion of fibrotic factors
by tubular cells, such as CTGF, PDGF and FGF2, stimulates adjacent
myofibroblasts to produce ECM components, which in turn activates
the fibroblasts (36). However,
the molecular mechanisms involved in the regulation of impaired
tubular cell paracrine secretion of fibrotic factors are not fully
understood. Previous studies have focused on the abnormal metabolic
adaptation of tubular cells in AKI, proposing that disrupted
adaptive energy metabolism has a crucial role in promoting CKD
progression (37,38).
In the present study, a unilateral
ischemia-reperfusion model was used to characterize the role of
FMO2 in AKI-CKD, and validation experiments were conducted in
immortalized cell lines. While data from these models consistently
demonstrate the beneficial effects of FMO2 in treating renal
fibrosis and CKD, they are not without limitations. The main
limitation is the lack of verification in FMO2 knockout mice.
Therefore, although the data from this model suggest the
involvement of FMO2 in the development and progression of renal
fibrosis, they do not exclude the contribution of potential other
detrimental factors in the absence of FMO2; moreover, they do not
specify the specific renal segment in which FMO2 inhibits fibrotic
reactions. Future studies are required to elucidate the role of
FMO2 in the AKI-to-CKD transition. Another limitation is that FMO2
is not solely expressed by tubular cells. It is also expressed in
other tubular segments and certain non-tubular cells. Future
research should determine the role of FMO2 in other cell
populations within the kidney. In the present study, conditioned
media were collected from TGF-β-stimulated renal tubular cells,
which were used to stimulate fibroblasts to secrete ECM. However,
the secretion of ECM was significantly reduced when renal tubular
cells were transfected with LV-expressing FMO2, suggesting the
presence of other active substances in the conditioned media
(following FMO2 overexpression) that play important roles (Fig. 6). Therefore, the findings of the
present study aid the confirmation of the specific effects of the
FMO2 gene on renal tubular cells. Although the in vivo and
in vitro models in the present study have certain
limitations, when considered together, the data from these models
suggest specific roles of FMO2 in AKI-CKD. Furthermore, a novel
function of FMO2 was discovered in the kidney as a signaling
regulator and not as a metabolic enzyme; the FMO2-SMURF2-mediated
negative regulatory circuitry modulates the phosphorylation and
activation of SMAD2/3 in response to TGF-β signaling in fibroblasts
(27,39).
In conclusion, the present study demonstrated that
FMO2 can regulate maladaptive repair and renal fibrosis following
AKI through the expression and nuclear translocation of SMURF2.
FMO2 not only effectively inhibited TGF-β-induced tubular cell
fibrosis but also suppressed the abnormal paracrine secretion of
fibrogenic factors by tubular cells. The regulation of fibrosis
mediated by FMO2 holds significant therapeutic implications by
inhibiting the transition from AKI to CKD.
During the transition from AKI to CKD, FMO2
modulates tubular cell fibrogenesis and paracrine secretion via
SMURF2, thereby impacting the outcome of this process.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
LW, HZ, JH and LS confirm the authenticity of all
the raw data. LS contributed to the conception of the study. LW, HZ
and JH performed the experiments. HZ, LW and JH performed the
analysis. HZ and LW drafted the manuscript. LS reviewed the
manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal experiments were performed in compliance
with the Animal Management Regulations of the Ministry of Health of
the P.R. China and approved by the Ethics Committee of the
Children's Hospital Affiliated to Zhengzhou University (approval
no. 2023-K-074).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work was supported by the National Natural Science
Foundation of China (grant no. NSFC82100340) and the Open Project
of Henan International Joint Laboratory of Prevention and Treatment
of Pediatric Diseases (grant no. EKB202203).
References
|
1
|
Hoste EAJ, Kellum JA, Selby NM, Zarbock A,
Palevsky PM, Bagshaw SM, Goldstein SL, Cerdá J and Chawla LS:
Global epidemiology and outcomes of acute kidney injury. Nat Rev
Nephrol. 14:607–625. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
James MT, Bhatt M, Pannu N and Tonelli M:
Long-term outcomes of acute kidney injury and strategies for
improved care. Nat Rev Nephrol. 16:193–205. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Turgut F, Awad AS and Abdel-Rahman EM:
Acute kidney injury: Medical causes and pathogenesis. J Clin Med.
12:3752023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhao ZB, Marschner JA, Iwakura T, Li C,
Motrapu M, Kuang M, Popper B, Linkermann A, Klocke J, Enghard P, et
al: Tubular epithelial cell HMGB1 promotes AKI-CKD transition by
sensitizing cycling tubular cells to oxidative stress: A rationale
for targeting HMGB1 during AKI recovery. J Am Soc Nephrol.
34:394–411. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kurata Y and Nangaku M: Use of antibiotics
as a therapeutic approach to prevent AKI-to-CKD progression. Kidney
Int. 104:418–420. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Patidar KR, Naved MA, Grama A, Adibuzzaman
M, Aziz Ali A, Slaven JE, Desai AP, Ghabril MS, Nephew L, Chalasani
N and Orman ES: Acute kidney disease is common and associated with
poor outcomes in patients with cirrhosis and acute kidney injury. J
Hepatol. 77:108–115. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shi L, Song Z, Li Y, Huang J, Zhao F, Luo
Y, Wang J, Deng F, Shadekejiang H, Zhang M, et al: MiR-20a-5p
alleviates kidney ischemia/reperfusion injury by targeting
ACSL4-dependent ferroptosis. Am J Transplant. 23:11–25. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mansour SG, Bhatraju PK, Coca SG, Obeid W,
Wilson FP, Stanaway IB, Jia Y, Thiessen-Philbrook H, Go AS, Ikizler
TA, et al: Angiopoietins as prognostic markers for future kidney
disease and heart failure events after acute kidney injury. J Am
Soc Nephrol. 33:613–627. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gewin LS: Renal fibrosis: Primacy of the
proximal tubule. Matrix Biol. 68-69:248–262. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sheng L and Zhuang S: New insights into
the role and mechanism of partial epithelial-mesenchymal transition
in kidney fibrosis. Front Physiol. 11:5693222020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu C, Hong Q, Zhuang K, Ren X, Cui S, Dong
Z, Wang Q, Bai X and Chen X: Regulation of pericyte metabolic
reprogramming restricts the AKI to CKD transition. Metabolism.
145:1555922023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lewis MP, Fine LG and Norman JT: Pexicrine
effects of basement membrane components on paracrine signaling by
renal tubular cells. Kidney Int. 49:48–58. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kuppe C, Ibrahim MM, Kranz J, Zhang X,
Ziegler S, Perales-Paton J, Jansen J, Reimer KC, Smith JR, Dobie R,
et al: Decoding myofibroblast origins in human kidney fibrosis.
Nature. 589:281–286. 2021. View Article : Google Scholar :
|
|
14
|
Gong H, Zheng C, Lyu X, Dong L, Tan S and
Zhang X: Inhibition of Sirt2 alleviates fibroblasts activation and
pulmonary fibrosis via Smad2/3 pathway. Front Pharmacol.
12:7561312021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Khalil H, Kanisicak O, Prasad V, Correll
RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, et al:
Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac
fibrosis. J Clin Invest. 127:3770–3783. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang Q, Ren GL, Wei B, Jin J, Huang XR,
Shao W, Li J, Meng XM and Lan HY: Conditional knockout of
TGF-βRII/Smad2 signals protects against acute renal injury by
alleviating cell necroptosis, apoptosis and inflammation.
Theranostics. 9:8277–8293. 2019. View Article : Google Scholar :
|
|
17
|
Liu J, Kumar S, Dolzhenko E, Alvarado GF,
Guo J, Lu C, Chen Y, Li M, Dessing MC, Parvez RK, et al: Molecular
characterization of the transition from acute to chronic kidney
injury following ischemia/reperfusion. JCI Insight. 2:e947162017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ni C, Chen Y, Xu Y, Zhao J, Li Q, Xiao C,
Wu Y, Wang J, Wang Y, Zhong Z, et al: Flavin containing
monooxygenase 2 prevents cardiac fibrosis via CYP2J3-SMURF2 axis.
Circ Res. July 5–2022.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Krueger SK, Williams DE, Yueh MF, Martin
SR, Hines RN, Raucy JL, Raucy JL, Dolphin CT, Shephard EA and
Phillips IR: Genetic polymorphisms of flavin-containing
monooxygenase (FMO). Drug Metab Rev. 34:523–532. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Siddens LK, Henderson MC, Vandyke JE,
Williams DE and Krueger SK: Characterization of mouse
flavin-containing monooxygenase transcript levels in lung and
liver, and activity of expressed isoforms. Biochem Pharmacol.
75:570–579. 2008. View Article : Google Scholar
|
|
21
|
Hsu DZ, Chu PY, Li YH, Chandrasekaran VR
and Liu MY: Role of flavin-containing-monooxygenase-dependent
neutrophil activation in thioacetamide-induced hepatic inflammation
in rats. Toxicology. 298:52–58. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang J, Chaluvadi MR, Reddy R, Motika MS,
Richardson TA, Cashman JR and Morgan ET: Hepatic flavin-containing
monooxygenase gene regulation in different mouse inflammation
models. Drug Metab Dispos. 37:462–468. 2009. View Article : Google Scholar :
|
|
23
|
Ding H, Li J, Li Y, Yang M, Nie S, Zhou M,
Yang X, Liu Y and Hou FF: MicroRNA-10 negatively regulates
inflammation in diabetic kidney via targeting activation of the
NLRP3 inflammasome. Mol Ther. 29:2308–2320. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shi L, Song Z, Li C, Deng F, Xia Y, Huang
J, Wu X and Zhu J: HDAC6 inhibition alleviates ischemia- and
Cisplatin-induced acute kidney injury by promoting autophagy.
Cells. 11:39512022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Moore CL, Savenka AV and Basnakian AG:
TUNEL assay: A powerful tool for kidney injury evaluation. Int J
Mol Sci. 22:4122021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
27
|
Lin X, Liang M and Feng XH: Smurf2 is a
ubiquitin E3 ligase mediating proteasome-dependent degradation of
Smad2 in transforming growth factor-beta signaling. J Biol Chem.
275:36818–36822. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Choi HS, Bhat A, Howington MB, Schaller
ML, Cox RL, Huang S, Beydoun S, Miller HA, Tuckowski AM, Mecano J,
et al: FMO rewires metabolism to promote longevity through
tryptophan and one carbon metabolism in C. elegans. Nat Commun.
14:5622023. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Krueger SK, Vandyke JE, Williams DE and
Hines RN: The role of flavin-containing monooxygenase (FMO) in the
metabolism of tamoxifen and other tertiary amines. Drug Metab Rev.
38:139–147. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bailleul G, Yang G, Nicoll CR, Mattevi A,
Fraaije MW and Mascotti ML: Evolution of enzyme functionality in
the flavin-containing monooxygenases. Nat Commun. 14:10422023.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Whetstine JR, Yueh MF, McCarver DG,
Williams DE, Park CS, Kang JH, Cha YN, Dolphin CT, Shephard EA,
Phillips IR and Hines RN: Ethnic differences in human
flavin-containing monooxygenase 2 (FMO2) polymorphisms: Detection
of expressed protein in African-Americans. Toxicol Appl Pharmacol.
168:216–224. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hugonnard M, Benoit E, Longin-Sauvageon C
and Lattard V: Identification and characterization of the FMO2 gene
in Rattus norvegicus: A good model to study metabolic and
toxicological consequences of the FMO2 polymorphism.
Pharmacogenetics. 14:647–655. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Leiser SF, Miller H, Rossner R, Fletcher
M, Leonard A, Primitivo M, Rintala N, Ramos FJ, Miller DL and
Kaeberlein M: Cell nonautonomous activation of flavin-containing
monooxygenase promotes longevity and health span. Science.
350:1375–1378. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu BC, Tang TT, Lv LL and Lan HY: Renal
tubule injury: A driving force toward chronic kidney disease.
Kidney Int. 93:568–579. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Garimella PS, Katz R, Waikar SS,
Srivastava A, Schmidt I, Hoofnagle A, Palsson R, Rennke HG,
Stillman IE, Wang K, et al: Kidney tubulointerstitial fibrosis and
tubular secretion. Am J Kidney Dis. 79:709–716. 2022. View Article : Google Scholar :
|
|
36
|
Livingston MJ, Shu S, Fan Y, Li Z, Jiao Q,
Yin XM, Venkatachalam MA and Dong Z: Tubular cells produce FGF2 via
autophagy after acute kidney injury leading to fibroblast
activation and renal fibrosis. Autophagy. 19:256–277. 2023.
View Article : Google Scholar :
|
|
37
|
Li Z, Lu S and Li X: The role of metabolic
reprogramming in tubular epithelial cells during the progression of
acute kidney injury. Cell Mol Life Sci. 78:5731–5741. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xu P, Chen C, Zhang Y, Dzieciatkowska M,
Brown BC, Zhang W, Xie T, Abdulmalik O, Song A, Tong C, et al:
Erythrocyte transglutaminase-2 combats hypoxia and chronic kidney
disease by promoting oxygen delivery and carnitine homeostasis.
Cell Metab. 34:299–316.e6. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang Z, Fan Y, Xie F, Zhou H, Jin K, Shao
L, Shi W, Fang P, Yang B, van Dam H, et al: Breast cancer
metastasis suppressor OTUD1 deubiquitinates SMAD7. Nat Commun.
8:21162017. View Article : Google Scholar : PubMed/NCBI
|