Introduction
Sepsis is defined as a syndrome of the systemic
inflammatory response caused primarily by infection. Sepsis-induced
cardiomyopathy (SIC) caused by sepsis is one of the manifestations
of multiple organ failure in sepsis. The primary manifestations are
myocardial inflammation, ventricular dilation, reduced
contractility and impaired ventricular response (1). Myocardial injury due to sepsis is
recognized as a serious global health problem, and it is a
significant cause of morbidity and mortality (2). Potential mechanisms of SIC include
myocardial cell inflammation, programmed cell death such as
apoptosis, pyroptosis (3),
ferroptosis (4), metabolic
changes, autophagy disturbances, and mitochondrial dysfunction
(5). Several recent papers have
shown that sepsis can induce ferroptosis in cardiomyocytes
(6-8), and thus causes cardiomyopathy and
dysfunction. Therefore, identifying a therapeutic agent to improve
myocardial injury in patients with sepsis may provide an important
reference to reduce the mortality of patients.
Ferroptosis is an iron-dependent means of programmed
cell death. It is a type of regulated cell death that is induced by
a combination of iron toxicity, lipid peroxidation, and plasma
membrane damage. Ferrous iron or lipoxygenase catalyzes liposome
peroxidation of highly expressed unsaturated fatty acids present at
the cell membrane to induce cell death (9); its primary feature is decreased
levels of glutathione (GSH) and glutathione peroxidase 4 (GPX4)
(10). Two GSH molecules serve
as electron donors to diminish phospholipid peroxide (PL-OOH) to
the corresponding alcohol under the action of GPX4 and generate
oxidized glutathione (GSSG) at the same time; GSSG can be reduced
by glutathione. The enzyme GSR utilizes NADPH to reduce GSH,
forming a cycle. Without deoxidation of peroxidative toxicity by
reduction to the corresponding alcohol via GPX4 action,
di/trioxycephalin [OO(O)-AA/AdA-PE) accumulates and propagates
lipid peroxidation to other phospholipids, eventually leading to
impaired membrane integrity and iron death. On the one hand, cell
ferroptosis is often accompanied by GSH depletion, decreased GPX4
activity, lipid peroxides that cannot be metabolized by the
GPX4-catalyzed reduction reaction, and lipids are oxidized by
ferrous iron in a Fenton reaction, producing ROS and damaging the
mitochondria, thereby further promoting the occurrence of
ferroptosis and inflammation (11). On the other hand, ferroptosis can
affect inflammation through its immunogenicity by release of DAMPs
or promoted COX and lipoxygenase activity through the release of
oxidized lipid mediators to trigger inflammation (9). Thus, identifying special pathogenic
mechanisms may contribute to the development of a therapeutic
strategy for myocardial recovery in patients with sepsis.
A previous study showed that the treatment of rat
cardiomyocyte cell line H9C2 with 1 μg/ml LPS for 24 h as a
model of sepsis in vitro can induce ferroptosis in H9C2
cells (7). In addition, there is
evidence that in H9C2 cells treated with LPS, the SIRT1/p53/SLC7A11
pathway is inhibited by LPS and causes inflammation and ferroptosis
(12). The SIRT1/p53/SLC7A11
pathway is an important regulatory pathway for ferroptosis
(13), and studies have shown
that activating this pathway by expressing the USP22 protein can
slow ferroptosis in H9C2 cells (12). SIRT1 has also been reported to
stimulate antioxidant-related protein expression, repair cells
damaged by oxidative stress, and prevent cell dysfunction. The
reduced SIRT 1 level leads to mitochondrial dysfunction by
increasing ROS levels, lipid peroxidation, and DNA damage (14). Therefore, the involvement of
SIRT1 and its downstream signaling pathways ensured its unique
roles in the progression of SIC.
Quercetin (QUE) is a dietary flavonoid widely
present in the plant kingdom with oxidative stress-regulating and
anti-inflammatory functions (15). In LPS-induced kidney injury, QUE
can exert anti-inflammatory and anti-oxidant effects by increasing
SIRT1 expression and inhibiting NF-κB signaling (16). In diabetic encephalopathy, QUE
can exert anti-inflammatory effects by increasing the expression of
SIRT1 and inhibiting the expression of NLRP3 (17). In suppressing cellular
ferroptosis, QUE can alleviate the ferroptosis of hepatocytes by
protecting mitochondria and reducing ROS (18). Similarly, QUE can inhibit
oxidative stress and ROS production in acute kidney injury, and
reduce inflammation and ferroptosis in rat renal tubular duct
epithelial cells (19). It has
also been shown that in rat cardiomyocytes H9C2, QUE protects
cardiomyocytes from hypoxia/reoxygenation-induced apoptosis by
modulating the ERK pathway, reducing oxidative stress and
mitochondrial damage (20).
Therefore, in the present study, the effects of QUE
and its underlying mechanisms in SIC were assessed to provide a
potential therapeutic option for the management of SIC.
Materials and methods
Patients
The present study was approved by the Ethics
Committee of the Affiliated Hospital of Shandong University of
Traditional Chinese Medicine and was performed in accordance with
the Declaration of Helsinki. A total of 20 healthy donors (male,
11; female, 9) with a mean age of 48.7±15.3 and 20 SIC patients
(male, 10; female, 10) aged 46.2±15.9 were enrolled in this study
and written informed consent was obtained. No significance was
observed between the healthy donors and the SIC patients concerning
sex and age. The inclusion criteria were: Patients were diagnosed
with sepsis according to the American College of Chest
Physicians/Society of Critical Care Medicine (ACCP/SCCM) (21). Individuals with a history of any
heart diseases and cardiac surgery, autoimmune diseases, malignant
tumors, or exposure to toxic drugs were excluded. From patients, 5
ml elbow venous blood was collected, and the serum was obtained via
centrifugation at 1,000 × g for 10 min at room temperature. The
serum levels of GPX4 (cat. no. ab282257), SIRT1 (cat. no.
ab171573), CK-MB (cat. no. ab193696), cTnI (cat. no. ab200016),
TNF-α (cat. no. ab181421), and IL-6 (cat. no. ab281515) (all from
Abcam) levels were measured using commercial ELISA kits and a
VersaMax microplate reader.
Cell culture
H9C2 myofibroblasts were purchased (Procell) and
cultured in DMEM containing 10% FBS and 1% antibiotics
penicillin/streptomycin (Procell Life Science & Technology Co.,
Ltd.) in an incubator with 5% CO2 and 95% air (v/v) at
37°C. To establish the SIC cell model, the H9C2 cells were treated
with LPS (0.5, 1.0, 1.5, and 2.0 μg/ml; Beijing Solarbio
Science & Technology Co., Ltd.) for 24 h. To determine the
appropriate concentration of QUE, different concentrations of QUE
(20, 40, 80, and 160 μM) (MedChemExpress) and 1.0
μg/ml LPS were applied to the cell culture for 24 h. The
levels of GSH, MDA, NADPH, and intracellular Fe2+ were
measured using commercial kits (Beyotime Institute of
Biotechnology) and a VersaMax reader.
Cell transfection
H9C2 cells were plated into six-well plates with the
density of 1×105. When cells reached 60-80% confluence,
the cells were transiently transfected with 50 nM siRNA-negative
control (NC) or siRNA-SIRT1 (siSIRT1) (synthesized by Shanghai
GenePharma Co.) by Lipofectamine 2000 (Invitrogen) according to the
manufacturer's protocol. The cells were cultivated in an incubator
at 37°C with 5% CO2 for 18-48 h. The transfection
efficiency was verified at 24 h after transfection at mRNA and
protein levels. After 48 h transfection, the transfected cells were
used for the subsequent experiments. The sequences of siRNA against
SIRT1 and the siRNA-negative control were as follows:
siSIRT1, sense: 5′-CCA GUA GCA CUA AUU CCA ATT-3′,
antisense: 5′-UUG GAA UUA GUG CCA CUG GTT-3′)
Negative NC, sense: 5′-UUC UCC GAA CGU GUC ACG
UTT-3′, antisense: 5′-ACG UGA CAC GUU CGG AGA ATT-3′).
MTT assay
H9C2 cells (1×104 cells/well) were
maintained in 96-well plates. Then, 20 μl MTT (Shanghai
Yeasen Biotechnology Co., Ltd.) was added and maintained for 4 h.
Then the formazan crystals were dissolved using 150 μl DMSO
(Shanghai Yeasen Biotechnology Co., Ltd.). The optical density (OD)
value was measured at 490 nm using a VersaMax microplate
reader.
Rats
This study was approved by the Laboratory Animal
Ethics Committee and followed the Guide for the Care and Use of
Laboratory Animals. A total of 32 adult Sprague-Dawley rats
(Beijing Fuhao Experimental Animal Breeding Center) were maintained
under a 12-h light/dark cycle with access to water and standard
chow. The rats were grouped as follows (n=8 per group): Normal,
model, model + 20 mg/kg QUE, model + 40 mg/kg QUE. The rats in the
normal group were injected with 150 and 150 μl DMSO was also
injected into the rats in the model group 6 h before SIC modeling.
The rats in the model + 20 mg/kg QUE and model + 40 mg/kg QUE
groups were injected with 150 μl DMSO + 20 or 40 mg/kg QUE
dehydrates 6 h before establishing the SIC model. To establish the
SIC model, the rats were anesthetized using 3% halothane. After
shaving and disinfecting, cecal ligation and puncture (CLP) was
performed using a median incision. CLP is referred to as the 'gold
standard' rodent model for abdominal sepsis, it creates a
continuously leaking, polymicrobial infectious focus in the
abdomen. This was performed following the protocol generated by
Drechsler and Osuchowski (22).
The abdominal cavity of rats was opened, and after exposing the
cecum, 1.5 cm of the cecum was ligated with a 3-0 silk thread and
punctured twice with an 18-gauge needle, the abdomen was sealed
with 5-0 thread. Bacteremia appeared 6 h after CLP and within 12-24
h, the developed clinical signs of a systemic bacterial infection
including fever, chills, erect hair, general weakness, and
decreased activity were observed in rats after CLP indicating
successful establishment of the model. Two investigators monitored
the health and behavior of rats every 4 h after CLP. Of the 32
adult Sprague-Dawley rats one died as a result of severe sepsis
after CLP, the 31 living rats were used for the subsequent
experiments. The serum levels of GPX4 (cat. no. ab243674), SIRT1
(cat. no. ab242725), CK-MB (cat. no. ab285275), and cTnI (cat. no.
ab24460) (all from Abcam) were measured 24 h after CLP using
commercial kits and a VersaMax microplate reader. A total of 48 h
after CLP, according to the results of the murine sepsis score
(MSS) scoring protocol the live rats were sacrificed using
CO2 at a volume displacement of 30-70% vol/min in
accordance with the AVMA Guidelines for the Euthanasia of Animals.
Then the cardiac tissues of the rats were collected for subsequent
experiments. The entire animal experiment lasted ~5 months
including animal ordering and purchase, feeding, SIC model
establishment, and the subsequent detection.
Histology
Heart tissue was fixed with 4% formaldehyde for
18-24 h at room temperature, embedded in paraffin, and sectioned
into 5 μm thickness. To detect inflammatory cell
infiltration and iron in the tissues, deparaffinized tissue
sections were hydrated using ethanol. Subsequently, sections were
processed with a hematoxylin and eosin staining kit according to
the manufacturer's protocol (Beyotime Institute of Biotechnology)
and a Prussian Blue Iron Staining kit (Shanghai Yeasen
Biotechnology Co., Ltd.) and observed with a microscope (Olympus
BX51) under the brightfield with the magnification of ×100 and
×400.
Reverse transcription-quantitative
PCR
RNA extraction was performed using an RNApure kit
(Bioteke Corporation). RT was performed using a Super M-MLV kit
(Shanghai Yeasen Biotechnology Co., Ltd.). qPCR was performed using
a QuantStudio 7 Pro (Applied Biosystems; Thermo Fisher Scientific,
Inc.) using a SYBR MasterMix (Beijing Solarbio Science &
Technology Co., Ltd.) and a 2× Power Taq PCR MasterMix (Bioteke
Corporation) and the relative changes in gene expression were
analyzed using the 2-ΔΔCq method (23). The sequences of primers were as
follows: SIRT1 forward, 5′-GAG TGT GCT GGA GGA TCT G-3′ and
reverse, 5′-TGC TCT GAT TTG TCT GGT GT-3′; and β-actin forward,
5′-ACC CGC GAG TAC AAC CTT CT-3′ and reverse, 5′-ATG GCT ACG TAC
ATG GCT GG-3′.
Western blotting
Proteins were extracted with RIPA Lysis Buffer
(Solarbio) and quantified using BCA kit (Beijing Solarbio Science
& Technology Co., Ltd.). 20 μg protein was loaded per
lane and separated with 10% SDS-PAGE (GenScript) and transferred to
PVDF membranes (MilliporeSigma), the primary antibodies used were:
Anti-GPX4 (1:1,000, cat. no. ab125066, 22 kDa), anti-PTGS2
(1:1,000, cat. no. ab179800, 69 kDa), anti-ferritin (1:1,000, cat.
no. ab75973, 21 kDa), anti-SIRT1 (1:1,000, cat. no. ab110304, 110
kDa), anti-acetyl-p53 (K382) (1:1,000, cat. no. ab75754, 53 kDa),
anti-SLC7A11 (1:1,000, cat. no. ab175186, 55 kDa), anti-TOM20
(1:1,000, cat. no. ab186735, 16 kDa), cytoplasmic cytochrome C
(1:1,000, cat. no. ab133504, 11 kDa), and β-actin (1:5,000, cat.
no., ab6276, 42 kDa) (all from Abcam), and blots were incubated
with primary antibody for 4°C for 12 h, after which membranes were
incubated the secondary HRP-conjugated antibody (1:2,000, cat. no.
ab6789) for 1 h at room temperature. Signals were visualized using
a DAB kit (Beijing Solarbio Science & Technology Co., Ltd.) and
processed using ImageJ 1.46R version (National Institutes of
Health).
Flow cytometry
A total of 1×105 H9C2 cells were seeded
in 6-well plates, treated with QUE for 24 h, digested and
resuspended in 150 μl binding buffer, and stained in the
dark with Annexin V-FITC/PI staining kit (BD Biosciences)
containing 5 μl FITC-conjugated Annexin V and 5 μl PI
for 15 min at room temperature. The apoptosis rate of H9C2 cells
was obtained using a FACSCalibur flow cytometer (BD Biosciences)
and analyzed by FlowJo 10.6.2 (Becton Dickinson & Company).
Lipid peroxidation assay
For this assay, an Image-iT lipid peroxidation assay
kit (Thermo Fisher Scientific, Inc.) was used. The cells were
treated with 25 μl Image-iT lipid peroxidation sensor and
incubated for 0.5 h at 37°C. The media was removed, and the cells
were washed with PBS (Beijing Solarbio Science & Technology
Co., Ltd.) and imaged by a fluorescence microscope (Olympus CKX53,
Olympus Corporation) with the magnification of ×200. The
fluorescence was read at two separate wavelengths.
Excitation/emission of 581/591 nm for the reduced dye (red), and
the other at excitation/emission of 488/510 nm for the oxidized dye
(green).
Immunofluorescence analysis
A total of 1 ml precooled 0.9% NaCl buffer was
thoroughly homogenized using 50 mg tissue, and after centrifugation
with 1,000 × g for 10 min at 4°C by a low speed freezing
centrifuge, 200 μl supernatant was mixed with 2 μl
dihydroethidium probe (Beijing Biolab Technology Co., Ltd.) and
incubated for 0.5 h at 37°C. The images were obtained using a
fluorescence microscope (Ex/Em=535/610 nm; Olympus CKX53, Olympus
Corporation).
Statistical analysis
The experiments were performed in triplicate and
repeated three times. Data are presented as the mean ± SEM.
GraphPad Prism version 9.3.3 (GraphPad Software, Inc.) was used for
analysis. Pearson's correlation test was used for correlation
analysis. Statistical significance was analyzed using an unpaired
Student's t-test between two groups or a one-way ANOVA with
Bonferroni post hoc analysis for comparisons between multiple
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
GPX4 and SIRT1 and inflammatory oxidative
stress marker levels are increased in the serum of SIC
patients
To assess the properties of the serum samples of 20
healthy donors and 20 SIC patients, blood samples were collected,
and serum was obtained. Next, the serum levels of GPX4, SIRT1,
markers of myocardial injury (CK-MB and cTnI), and markers of
inflammatory oxidative stress (TNF-α and IL-6) were measured. The
results indicated that the serum levels of GPX4 and SIRT1 were
lower whereas the levels of myocardial injury and inflammatory
oxidative stress markers were higher in the serum of SIC patients
compared with the serum of healthy donors (Fig. 1A-C). Using a Pearson's
correlation test, serum levels of GPX4 and SIRT1 in 20 SIC patients
were negatively correlated with the serum levels of CK-MB and cTnI
(Fig. 1D). This indicated that
cell ferroptosis was closely associated with SIC.
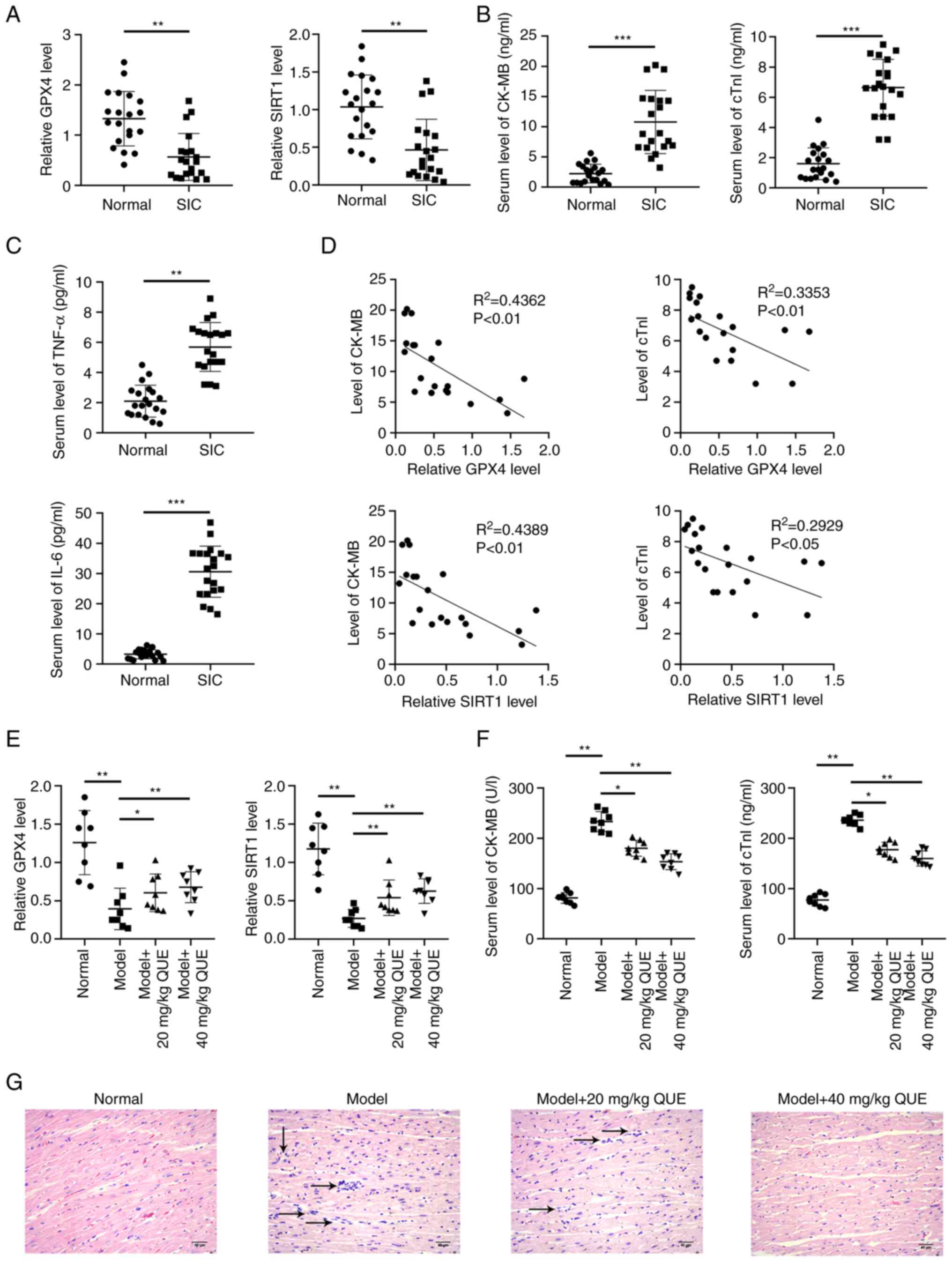 | Figure 1Measurement of the serum levels of
GPX4, SIRT1, markers of cardiac injury, and markers of the
oxidative stress responses in the clinical samples and rat SIC
models. Serum levels of (A) GPX-4 and SIRT1, (B) CK-MB and cTnI and
(C) TNF-α and IL-6 in SIC patients and healthy donors. (D)
Correlation between GPX4, SIRT1, CK-MB, and cTnI in the 20 SIC
patients. Serum levels of (E) GPX-4 and SIRT1, and (F) of cardiac
injury markers in the rat model of SIC. (G) Inflammatory cell
infiltration in the rat cardiac tissues was assessed using
hematoxylin and eosin staining. *P<0.05,
**P<0.01, ***P<0.001. GPX4, glutathione
peroxidase 4; SIC, Sepsis-induced cardiomyopathy. |
QUE increases the levels of GPX4 and
SIRT1 and decreases the levels of markers of myocardial injury in a
dose-dependent manner in vivo
To determine the potential protective effects of QUE
against SIC in vivo, the rats were divided into four groups
(normal, model, model +20 mg/kg QUE and model +40 mg/kg QUE) and
the serum levels of GPX4, SIRT1, and markers of myocardial injury
markers were tested. The SIC model group had lower levels of GPX4
and SIRT1 but higher levels of CK-MB and cTnI compared with the
normal group. The injection of QUE upregulated the levels of GPX4
and SIRT1 and it showed beneficial effects against cardiac injury
caused by sepsis based on the reduction in the levels of CK-MB and
cTnI, and this effect was dose-dependent manner, thus reducing SIC
(Fig. 1E and F). To investigate
the pathology of sepsis in inducing cardiac injury and the
mechanism of action of QUE, the rat cardiac tissues were stained
using hematoxylin and eosin and it was shown that the number of
inflammatory cells increased in the model group and injection of
QUE reduced inflammatory cell infiltration in cardiac tissues. When
40 mg/kg QUE was used, a better anti-inflammatory effect was
observed than 20 mg/kg QUE (Fig.
1G). These results showed that QUE upregulated the levels of
GPX4 and SIRT1 and reduced the levels of markers of myocardial
injury in a dose-dependent manner in vivo.
QUE inhibits LPS-induced ferroptosis of
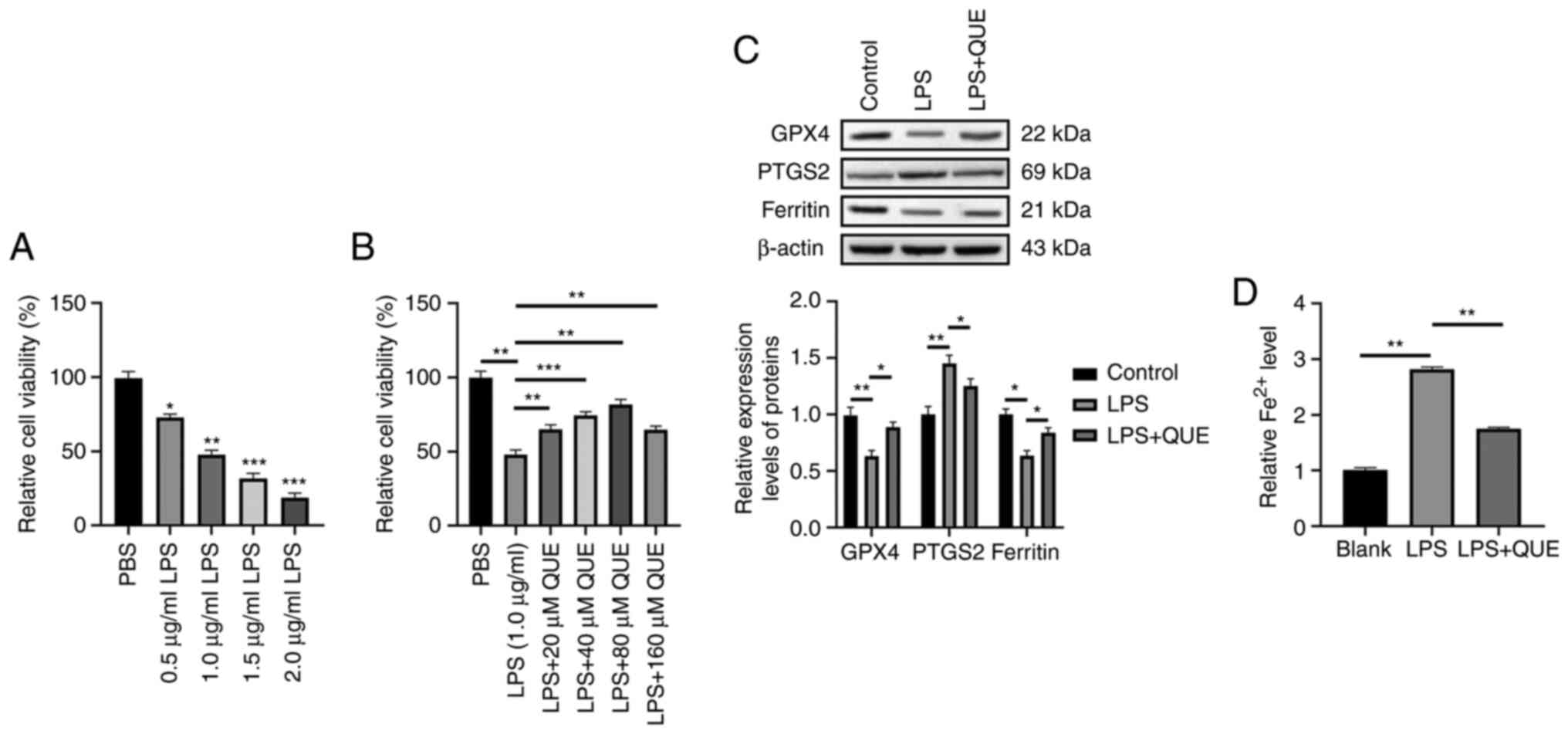
H9C2 cells in vitro
To explore the roles and molecular mechanism of QUE
in treating SIC, the H9C2 cells were treated with different
concentrations of LPS (0.5, 1.0, 1.5, or 2.0 μg/ml) for 24 h
and the results of the MTT assay showed that the viability of H9C2
cells decreased in a dose-dependent manner and the viability of
cells treated with 1.0 μg/ml LPS was ~50% of the viability
of cells treated with PBS (Fig.
2A). Thus, 1.0 μg/ml LPS was selected for the
establishment of the in vitro model of SIC. Next, different
concentrations of QUE (20, 40, 80, and 160 μM) and 1.0
μg/ml LPS to treat the H9C2 cells for 24 h. It was found
that the cell viability was elevated when the concentration of QUE
increased, but the viability was reduced when the concentration of
QUE was 160 μM. In the LPS + 80 μM QUE group, the
cell viability was ~80% of the viability of cells in the PBS group
(Fig. 2B). Therefore, 80
μM QUE was used for the subsequent experiments.
The anti-ferroptotic effect of QUE was examined
using western blotting and commercial kits, and it was found that
1.0 μg/ml LPS treatment increased the levels of PTGS2 and
intracellular Fe2+ and reduced the expression of GPX4
and ferritin. QUE decreased the levels of PTGS2 and intracellular
Fe2+ and increased the expression of GPX4 and ferritin
(Fig. 2C and D). These results
illustrated that QUE ameliorated ferroptosis of H9C2 cells induced
by 1.0 μg/ml LPS.
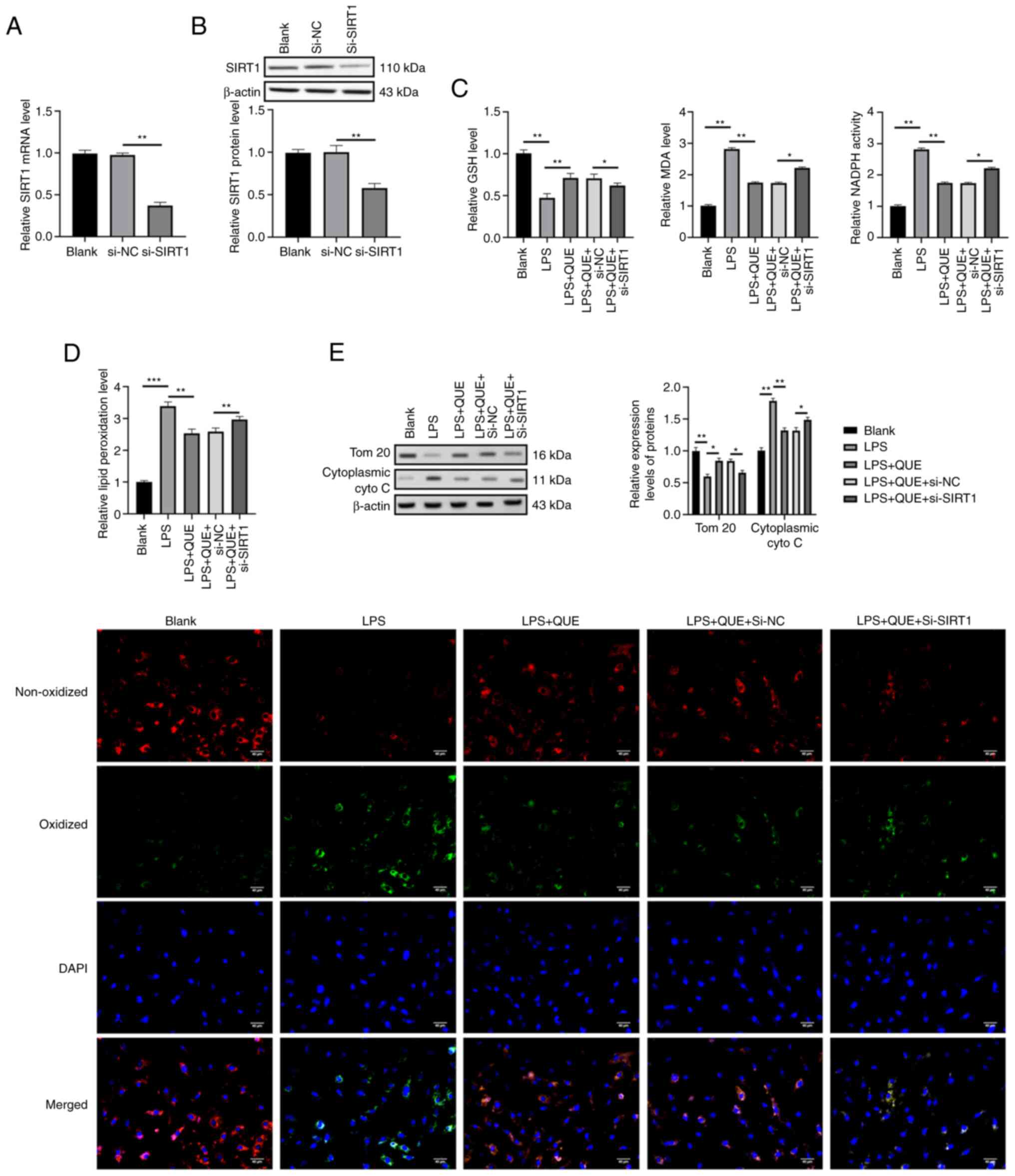
QUE suppresses LPS-induced oxidative
stress by mediating SIRT1 expression in H9C2 cells in vitro
Since it was found that QUE could increase the
expression levels of SIRT1 in vivo in Fig. 1E and both QUE (24) and SIRT1 (14) have been reported to exert
anti-oxidative roles; thus, whether QUE played anti-oxidative roles
via modulating SIRT1 expression in the in vitro SIC cell
model was investigated. siRNA against SIRT1 was transfected into
H9C2 cells to knock down endogenous SIRT1 expression and the
transfection efficiency was confirmed (Fig. 3A and B). Next, the levels of
oxidative stress response markers (GSH, MDA, NADPH, and lipid
peroxidation) in H9C2 cells were measured using ELISA and
immunofluorescent microscopy. The results showed that the LPS group
exhibited higher levels of MDA, NADPH, and lipid peroxidation but a
lower level of GSH compared with the blank group. QUE reduced the
oxidative stress response of cells by increasing the GSH levels and
decreasing the MDA, NADPH, and ROS levels. Additionally, knockdown
of SIRT1 partially counteracted the anti-oxidative effects of QUE
by increasing the levels of MDA, NADPH and lipid peroxidation and
reducing the levels of GSH (Fig. 3C
and D). Given the highly activated oxidative stress response
was associated with mitochondrial injury, the expression levels of
mitochondrial proteins were evaluated by western blotting. The
cells in the LPS group exhibited a higher level of cytoplasmic
cytochrome C and a lower level of TOM 20 compared with the blank
group, which indicated that LPS treatment induced mitochondrial
damage. The damaging effects of LPS were neutralized by QUE via
increasing the expression of TOM 20 and downregulating cytoplasmic
cytochrome C levels. Compared with the LPS + QUE +si-NC group, the
protective roles of QUE on mitochondria were then reversed by
si-SIRT1 in the LPS+QUE+si-SIRT1 group (Fig. 3E). This showed that LPS damaged
the mitochondria of H9C2 cells and QUE exerted a protective effect
against LPS-induced oxidative-related damages, but the antioxidant
impact of QUE was neutralized by the knockdown of SIRT1.
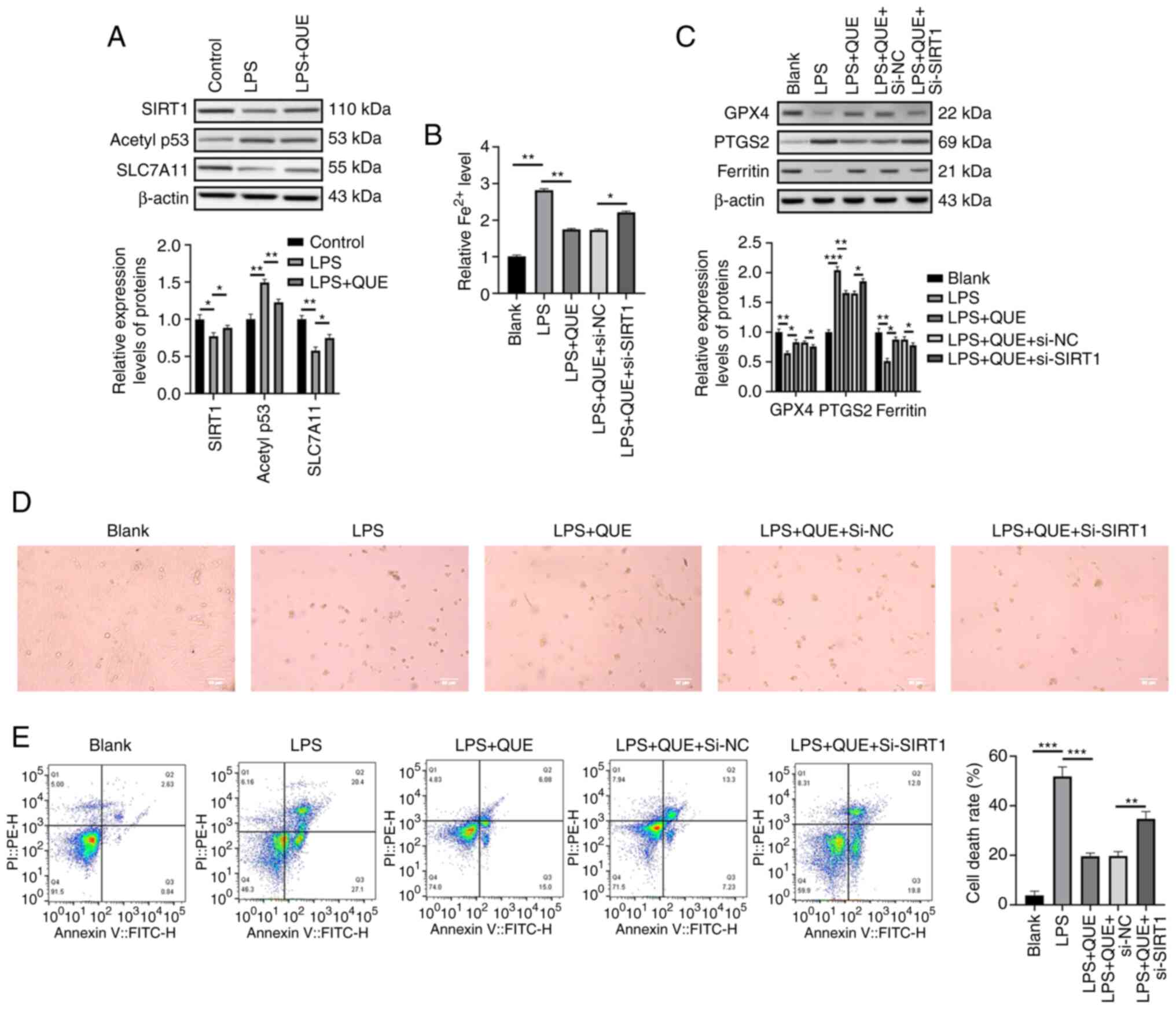
QUE activates the SIRT1/p53/SLC7A11
signaling pathway to inhibit ferroptosis of H9C2 cells in
vitro
As it was found that QUE exhibited anti-ferroptotic
and anti-oxidative effects on LPS-treated H9C2 cells, to explore
its underlying mechanism of action and the involvement of
downstream signaling pathways, western blotting was used and it was
found that LPS downregulated the expression of SIRT1 and SCL7A11
but upregulated the acetylated levels of p53. QUE suppressed the
effect of LPS (Fig. 4A). Next,
the ferroptosis of H9C2 cells was determined based on the levels of
ferroptosis markers via ELISA and western blotting to verify the
suppressive mechanism of QUE against cell ferroptosis. QUE exerted
an anti-ferroptotic effect by increasing the intracellular
Fe2+ levels as well as the expression levels of GPX4 and
ferritin and suppressing the expression levels of PTGS2 in the
LPS-induced ferroptosis of H9C2 cells. However, its influence was
partially reversed by the knockdown of SIRT1 in the
LPS+QUE+si-SIRT1 group compared to the LPS +QUE +si-NC group
(Fig. 4B and C). Subsequently,
the death of H9C2 cells was observed by microscopy and analyzed via
flow cytometry. It was found that the cells in the LPS group showed
a higher rate of apoptosis, and the administration of QUE reduced
this cell death. Knockdown of SIRT1 in the LPS+ QUE+si-SIRT1 group
increased the apoptosis of cells in contrast to the LPS + QUE +
si-NC group (Fig. 4D and E).
These results revealed that QUE inhibited LPS-induced ferroptosis
of H9C2 cells by activating the SIRT1/p53/SLC7A11 signaling in
vitro.
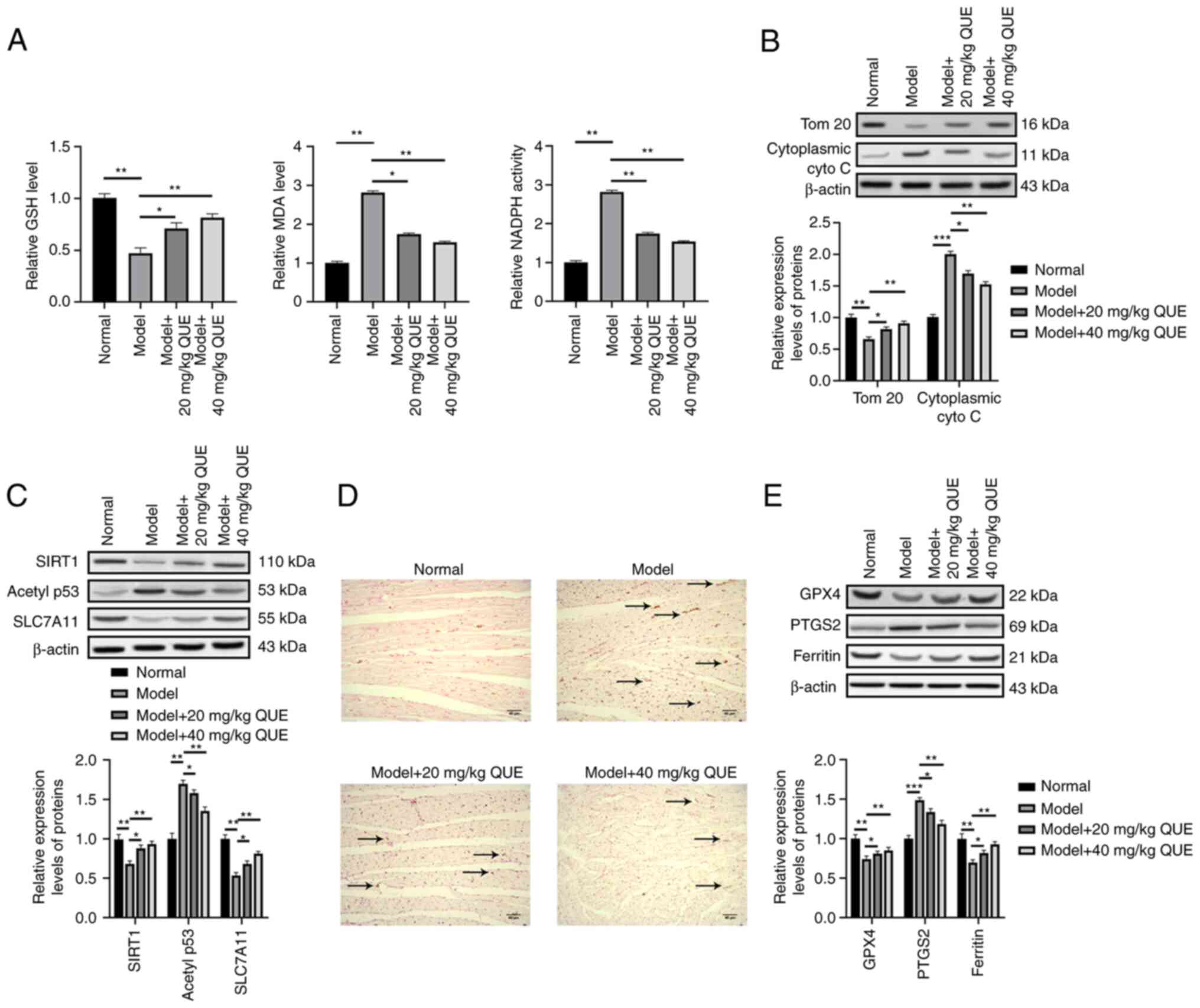
QUE activates the SIRT1/p53/SLC7A11
signaling pathway to inhibit ferroptosis in vivo
Since the protective effects of QUE in rat models
against SIC were determined, next, the molecular mechanism of
action was subsequently investigated in vivo. First, the
levels of oxidative stress response markers (GSH, MDA, and NADPH)
in the rat cardiac tissues were measured using commercial ELISA
kits to investigate the anti-oxidative effects of QUE. The results
showed that the model group exhibited higher levels of MDA and
NADPH but lower levels of GSH compared with the normal group. The
application of QUE reduced the oxidative stress response in a
dose-dependent manner by increasing GSH levels and reducing MDA and
NADPH levels (Fig. 5A). Next, to
explore the impact of QUE on mitochondria, which are the primary
cell organelles responsible for oxidative stress responses, the
expression levels of mitochondrial proteins in cardiac tissues were
evaluated due to the close relationships between oxidative stress
response and mitochondrial injury. The cardiac tissues in the model
group expressed higher levels of cytoplasmic cytochrome C and a
lower level of TOM 20 compared with the blank group, suggesting the
occurrence of mitochondrial damage; and this mitochondrial injury
was alleviated by QUE in a dose-dependent manner by increasing the
expression of TOM 20 and downregulating cytoplasmic cytochrome C
expression (Fig. 5B). This
showed that SIC damaged the mitochondria and QUE exerted a
mitochondrial protective and anti-oxidative effect against
sepsis.
Next, the expression levels of SIRT1, acetyl-p53
(K382), and SLC7A11 in the rat cardiac tissues were detected to
investigate the mechanism of action of QUE. The results showed that
the rats in the model group exhibited a lower level of SIRT1 and
SCL7A11 and a higher level of acetyl-p53. The injection of
QUE-activated SIRT1/p53/SLC7A11 signaling by upregulating the
expression of SIRT1 and SCL7A11 and decreasing acetyl-p53 levels,
and 40 mg/kg QUE exhibited a more potent effect than 20 mg/kg QUE
(Fig. 5C). Finally, the levels
of ferroptosis markers in the cardiac tissues were assessed using
Prussian blue staining and western blotting to verify the
anti-ferroptotic roles of QUE in vivo. The Prussian blue
staining showed that LPS increased the levels of ferric iron
deposition, while its effects were mitigated by QUE in a
dose-dependent manner, suggesting that the Fenton reaction could be
suppressed by QUE (Fig. 5D). The
results of western blotting also confirmed that the model group
exhibited a higher level of PTGS2 but lower levels of GSH and
ferritin. QUE ameliorated the ferroptosis by reducing PTGS2 and
increasing GSH and ferritin expression (Fig. 5E). These results showed that QUE
inhibited oxidative stress responses and also weakened cell
ferroptosis by activating the SIRT1/p53/SLC7A11 signaling pathway
to ameliorate SIC in rats in vivo.
Discussion
In the present study, the roles of QUE in SIC were
investigated both in vitro and in vivo. QUE exerted
anti-ferroptotic roles via activation of the SIRT1/p53/SLC7A11
pathway to protect mitochondria, reduce oxidative stress response,
and relieve SIC. These outcomes corroborated that QUE exhibited
potential therapeutic effects in SIC.
Clinically, sepsis-induced a complex myocardial
inflammatory response that resulted in myocardial dysfunction.
Sepsis can develop into septic shock or multiple organ dysfunction
if it is not controlled in a timely and effective manner (25). Despite advances in the
understanding of myocardial inflammatory responses, there are no
reliable targets and drugs to treat SIC (26). In the present study,
administration of LPS to H9C2 cells could induce SIC, stimulate
oxidative stress responses, and also induce cell ferroptosis which
was consistent with a study by Li et al (7). The execution of CLP in rats may
lead to tissue inflammation of rats to mimic SIC in vivo,
which was similar to a previous study (27).
Studies have shown that H9C2 cells undergo
ferroptosis due to various reasons such as ischemia/reperfusion
injury (28), chemotherapeutic
agents (29), and sepsis
(30). Ferroptosis is a type of
iron-related programmed cell death that differs from apoptosis,
necrosis, autophagy, and pyroptosis, and pharmacologically, this
cell death process can be inhibited by iron chelators and lipid
peroxidation inhibitors (31).
Following LPS-triggered ferroptosis of H9C2 cells, the cell
viability was decreased, the mitochondria were damaged, and the
oxidative stress response was activated. Additionally, the levels
of GPX4, which protects cells from ferroptosis, were downregulated.
Conditions that lead to GSH exhaustion directly affect the activity
and stability of GPX4, thereby making cells more susceptible to
ferroptosis (30). Additionally,
ferric iron deposition and inflammatory cell infiltration in the
cardiac tissues of the experimental sepsis rat model were observed
similar to that observed by Li et al (7).
Activation of the SIRT1/p53/SLC7A11 signaling
pathway is associated with ferroptosis. SIRT1 is an NAD+-dependent
deacetylase that directly deacetylates p53 and mediates its
pro-apoptotic function (32).
SIRT1 functions in catalyzing histone deacetylation and has broad
effects on several processes such as anti-inflammation, aging,
anti-oxidation, mitochondrial biogenesis, cellular senescence,
apoptosis, and circadian rhythms (33). It has been reported that
depletion of SIRT1 exacerbates I/R injury (34) and reducing SIRT1 expression
protected mice from alcohol-induced liver damage by reducing
ferroptosis of hepatocytes (35). In addition, p53 positively
regulated cell ferroptosis by inhibiting the expression of SLC7A11,
thereby promoting the production of reactive oxygen species
(13). Upregulated SLC7A11
expression may inhibit ROS-induced ferroptosis (36). Therefore, these findings suggest
that p53/SLC7A11 potentially mediates ferroptosis in cardiomyocytes
downstream of SIRT1. To show that QUE reduced cell ferroptosis by
increasing SIRT1 expression, in vitro experiments were
performed by knocking down SIRT1 after the application of QUE, and
the results showed that QUE could activate SIRT1/p53/SLC7A11 axis
to ameliorate ferroptosis of H9C2 cells and relieve SIC, consistent
a study by Ma et al (12). However, whether SIRT1 regulated
the ferroptosis of cardiomyocytes by deacetylation of P53 remains
to be determined. Additionally, to further confirm the results of
the present study, the dose of LPS (0.5, 1.0, 1.5, or 2) for the
establishment of the in vitro model should be increased
using a broader range (using concentration in orders of magnitudes)
as performed previously (37,38). Additionally, a ferroptosis
inducer group should also be included as a positive control of LPS
in cell modeling to improve the rigor of the design process. These
are the limitations of the present which will be taken into
consideration in future studies on the study of QUE, specifically
when determining the mechanism of myocardial injury induced by
sepsis.
In conclusion, QUE had protective effects on
mitochondria and oxidative stress responses and an anti-ferroptotic
effect on rat H9C2 cells in vitro and a rat model of SIC
in vivo by stimulating the SIRT1/p53/SLC7A11 pathway. Due to
the proinflammatory effect of SIC and the close relationship
between ferroptosis and inflammation, the roles of QUE on
LPS-induced pyroptosis in SIC using H9C2 cells and a rat model
highlighted its potential therapeutic value and its potential
mechanism, of action in the management of SIC.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
XL, YW and HZ conceived and designed the study. XZ,
QC, and XW prepared the materials, and acquired and analyzed the
data. XL and YW drafted the manuscript. All authors revised the
manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was performed in line with the principles
of the Declaration of Helsinki. The present study was approved by
the Ethics Committee of the Affiliated Hospital of Shandong
University of Traditional Chinese Medicine (approval no.
AF/SC-08/02.0). The animal experiments were reviewed and approved
by The Institutional Animal Care and Use Committee of the
Affiliated Hospital of Shandong University of Traditional Chinese
Medicine (approval no. MDL2022-06-15-01). Written informed consent
was obtained from all the patients.
Patient consent for publication
Informed consent was obtained for publication of the
patient data and images.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
No funding was received.
References
|
1
|
Walley KR: Sepsis-induced myocardial
dysfunction. Curr Opin Crit Care. 24:292–299. 2018.
|
|
2
|
Angus DC, Linde-Zwirble WT, Lidicker J,
Clermont G, Carcillo J and Pinsky MR: Epidemiology of severe sepsis
in the United States: Analysis of incidence, outcome, and
associated costs of care. Crit Care Med. 29:1303–1310. 2001.
|
|
3
|
Li N, Zhou H, Wu H, Wu Q, Duan M, Deng W
and Tang Q: STING-IRF3 contributes to lipopolysaccharide-induced
cardiac dysfunction, inflammation, apoptosis and pyroptosis by
activating NLRP3. Redox Biol. 24:1012152019.
|
|
4
|
Chen Z, Cao Z, Gui F, Zhang M, Wu X, Peng
H, Yu B, Li W, Ai F and Zhang J: TMEM43 protects against
sepsis-induced cardiac injury via inhibiting ferroptosis in mice.
Cells. 11:29922022.
|
|
5
|
Flierl MA, Rittirsch D, Huber-Lang MS,
Sarma JV and Ward PA: Molecular events in the cardiomyopathy of
sepsis. Mol Med. 14:327–336. 2008.
|
|
6
|
Kong C, Ni X, Wang Y, Zhang A, Zhang Y,
Lin F, Li S, Lv Y, Zhu J, Yao X, et al: ICA69 aggravates
ferroptosis causing septic cardiac dysfunction via STING
trafficking. Cell Death Discov. 8:1872022.
|
|
7
|
Li N, Wang W, Zhou H, Wu Q, Duan M, Liu C,
Wu H, Deng W, Shen D and Tang Q: Ferritinophagy-mediated
ferroptosis is involved in sepsis-induced cardiac injury. Free
Radic Biol Med. 160:303–318. 2020.
|
|
8
|
Zhou B, Zhang J, Chen Y, Liu Y, Tang X,
Xia P, Yu P and Yu S: Puerarin protects against sepsis-induced
myocardial injury through AMPK-mediated ferroptosis signaling.
Aging. 14:3617–3632. 2022.
|
|
9
|
Sun Y, Chen P, Zhai B, Zhang M, Xiang Y,
Fang J, Xu S, Gao Y, Chen X, Sui X and Li G: The emerging role of
ferroptosis in inflammation. Biomed Pharmacother.
127:1101082020.
|
|
10
|
Zhao X and Chen F: Propofol induces the
ferroptosis of colorectal cancer cells by downregulating STAT3
expression. Oncol Lett. 22:7672021.
|
|
11
|
Ursini F and Maiorino M: Lipid
peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic
Biol Med. 152:175–185. 2020.
|
|
12
|
Ma S, Sun L, Wu W, Wu J, Sun Z and Ren J:
USP22 protects against myocardial ischemia-reperfusion injury via
the SIRT1-p53/SLC7A11-dependent inhibition of ferroptosis-induced
cardiomyocyte death. Front Physiol. 11:5513182020.
|
|
13
|
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun
X, Kang R and Tang D: Ferroptosis: Process and function. Cell Death
Differ. 23:369–379. 2016.
|
|
14
|
Alam F, Syed H, Amjad S, Baig M, Khan TA
and Rehman R: Interplay between oxidative stress, SIRT1,
reproductive and metabolic functions. Curr Res Physiol. 4:119–124.
2021.
|
|
15
|
Karimi A, Naeini F, Azar VA, Hasanzadeh M,
Ostadrahimi A, Niazkar HR, Mobasseri M and Tutunchi H: A
comprehensive systematic review of the therapeutic effects and
mechanisms of action of quercetin in sepsis. Phytomedicine.
86:1535672021.
|
|
16
|
Lu S, Zhou S, Chen J, Zheng J, Ren J, Qi
P, Zhu Z and Li Z: Quercetin nanoparticle ameliorates
lipopolysaccharide-triggered renal inflammatory impairment by
regulation of Sirt1/NF-KB pathway. J Biomed Nanotechnol.
17:230–241. 2021.
|
|
17
|
Hu T, Lu XY, Shi JJ, Liu XQ, Chen QB, Wang
Q, Chen YB and Zhang SJ: Quercetin protects against diabetic
encephalopathy via SIRT1/NLRP3 pathway in db/db mice. J Cell Mol
Med. 24:3449–3459. 2020.
|
|
18
|
Jiang JJ, Zhang GF, Zheng JY, Sun JH and
Ding SB: Targeting mitochondrial ROS-mediated ferroptosis by
quercetin alleviates high-fat diet-induced hepatic lipotoxicity.
Front Pharmacol. 13:8765502022.
|
|
19
|
Wang Y, Quan F, Cao Q, Lin Y, Yue C, Bi R,
Cui X, Yang H, Yang Y, Birnbaumer L, et al: Quercetin alleviates
acute kidney injury by inhibiting ferroptosis. J Adv Res.
28:231–243. 2021.
|
|
20
|
Li F, Li D, Tang S, Liu J, Yan J, Chen H
and Yan X: Quercetin protects H9c2 cardiomyocytes against
oxygen-glucose deprivation/reoxygenation-induced oxidative stress
and mitochondrial apoptosis by regulating the ERK1/2/DRP1 signaling
pathway. Evid Based Complement Alternat Med. 2021:75221752021.
|
|
21
|
Li Y, Lu B, Yu M, Zhai J, Yao Y and Chai
Y: Diagnostic value and significance of serum miR-132 combined with
miR-223 for sepsis-induced cardiomyopathy. Exp Ther Med.
22:13962021.
|
|
22
|
Drechsler S and Osuchowski M: Cecal
ligation and puncture. Methods Mol Biol. 2321:1–8. 2021.
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
|
|
24
|
Sato S and Mukai Y: Modulation of chronic
inflammation by quercetin: The beneficial effects on obesity. J
Inflamm Res. 13:421–431. 2020.
|
|
25
|
Angus DC and van der Poll T: Severe sepsis
and septic shock. N Engl J Med. 369:840–851. 2013.
|
|
26
|
Hollenberg SM and Singer M:
Pathophysiology of sepsis-induced cardiomyopathy. Nat Rev Cardiol.
18:424–434. 2021.
|
|
27
|
Huang X, Zhang MZ, Liu B, Ma SY, Yin X and
Guo LH: Astragaloside IV attenuates polymicrobial sepsis-induced
cardiac dysfunction in rats via IKK/NF-κB pathway. Chin J Integr
Med. 27:825–831. 2021.
|
|
28
|
Zhang Y, Ren X, Wang Y, Chen D, Jiang L,
Li X, Li T, Huo M and Li Q: Targeting ferroptosis by polydopamine
nanoparticles protects heart against ischemia/reperfusion injury.
ACS Appl Mater Interfaces. 13:53671–53682. 2021.
|
|
29
|
Hou K, Shen J, Yan J, Zhai C, Zhang J, Pan
JA, Zhang Y, Jiang Y, Wang Y, Lin RZ, et al: Loss of TRIM21
alleviates cardiotoxicity by suppressing ferroptosis induced by the
chemotherapeutic agent doxorubicin. EBioMedicine.
69:1034562021.
|
|
30
|
Park TJ, Park JH, Lee GS, Lee JY, Shin JH,
Kim MW, Kim YS, Kim JY, Oh KJ, Han BS, et al: Quantitative
proteomic analyses reveal that GPX4 downregulation during
myocardial infarction contributes to ferroptosis in cardiomyocytes.
Cell Death Dis. 10:8352019.
|
|
31
|
Chen D, Eyupoglu IY and Savaskan N:
Ferroptosis and cell death analysis by flow cytometry. Methods Mol
Biol. 1601:71–77. 2017.
|
|
32
|
Chen H, Lin X, Yi X, Liu X, Yu R, Fan W,
Ling Y, Liu Y and Xie W: SIRT1-mediated p53 deacetylation inhibits
ferroptosis and alleviates heat stress-induced lung epithelial
cells injury. Int J Hyperthermia. 39:977–986. 2022.
|
|
33
|
Liu X, Liu J, Xiao W, Zeng Q, Bo H, Zhu Y,
Gong L, He D, Xing X, Li R, et al: SIRT1 Regulates N(6)
-methyladenosine RNA modification in hepatocarcinogenesis by
inducing RANBP2-dependent FTO SUMOylation. Hepatology.
72:2029–2050. 2020.
|
|
34
|
Chun SK, Lee S, Flores-Toro J, U RY, Yang
MJ, Go KL, Biel TG, Miney CE, Louis SP, Law BK, et al: Loss of
sirtuin 1 and mitofusin 2 contributes to enhanced
ischemia/reperfusion injury in aged livers. Aging Cell.
17:e127612018.
|
|
35
|
Zhou Z, Ye TJ, DeCaro E, Buehler B, Stahl
Z, Bonavita G, Daniels M and You M: Intestinal SIRT1 deficiency
protects mice from ethanol-induced liver injury by mitigating
ferroptosis. Am J Pathol. 190:82–92. 2020.
|
|
36
|
Jiang L, Kon N, Li T, Wang SJ, Su T,
Hibshoosh H, Baer R and Gu W: Ferroptosis as a p53-mediated
activity during tumour suppression. Nature. 520:57–62. 2015.
|
|
37
|
Meyer D, Telele S, Zelená A, Gillen AJ,
Antonucci A, Neubert E, Nißler R, Mann FA, Erpenbeck L, Boghossian
AA, et al: Transport and programmed release of nanoscale cargo from
cells by using NETosis. Nanoscale. 12:9104–9115. 2020.
|
|
38
|
Willis C, Morris JM, Danis V and Gallery
EDM: Cytokine production by peripheral blood monocytes during the
normal human ovulatory menstrual cycle. Hum Reprod. 18:1173–1178.
2003.
|