Introduction
Traumatic brain injury (TBI) is caused by blunt,
penetrating and accelerating or decelerating forces. Symptoms of
TBI include decreased levels of consciousness, memory loss or
amnesia, and other neurological or neuropsychological
abnormalities. TBI, which occurs mostly in young individuals, is
one of the major causes of disability, and in some cases, can lead
to death (1). Indeed, TBI is the
third most common cause of mortality worldwide. The annual global
incidence of TBI is >294 per 100,000 individuals (2,3).
TBI severely affects the quality of life of affected individuals
and is associated with a wide range of disabilities, including
sensory, motor and cognitive impairments, as well as affective
disorders (4). In addition to
primary mechanical injury occurring immediately following trauma,
secondary brain injuries, such as delayed intracranial hemorrhage,
oxidative stress, local inflammatory response and blood-brain
barrier injury, play a major role in neuronal death (1,5).
Following brain cell injury, the Ca2+ homeostasis of the
endoplasmic reticulum (ER) of neurons is destroyed, which can cause
ER stress; thus, ER stress is involved in the pathophysiology of
TBI (6,7).
The ER is an important cellular organelle
responsible for protein modification and processing, as well as the
folding, assembly and transportation of new peptide chains
(8). The unfolded protein
response (UPR) is the classic pathway that regulates the ER stress
response. Following the abnormal over-accumulation of proteins in
the ER, protein kinase R-like ER kinase (PERK), activating
transcription factor 6 (ATF6) and inositol-requiring protein 1
(IRE1) exhibit signs of stress (9). As a result, they signal the nucleus
and trigger a cellular response that leads to a decrease in protein
synthesis and an increase in ER capacity, initiating the ER stress
mechanism (10,11). If the ER stress cannot be
resolved through the aforementioned three pathways, the UPR can
also maintain the stability of the intracellular environment by
affecting cell signals mediated by multiple mitogen-activated
protein kinase (MAPK) signaling pathways, such as extracellular
regulated protein kinases (ERKs), p38 MAPK and C-Jun N-terminal
kinase (JNK) (12). The UPR is
involved in the biosynthetic stress of the ER, and the pathways
involved serve to maintain cellular homeostasis. By contrast,
stress-activated MAPK pathways are regulated by a diverse array of
intracellular and extracellular stresses and regulate cell death
and aging by integrating signals from the UPR, other cell stress
responses and other cellular signaling pathways (13,14).
In the context of ER stress, a number of pathologies
are characterized by changes in the cellular microenvironment,
including changes in the composition of hyaluronic acid (HA) in the
extracellular matrix (ECM). HA, a key component of the ECM,
participates in critical tasks, such as receptor protein attachment
and intercellular communication (15,16). In a recent study, transmembrane
protein 2 (TMEM2), a cell surface hyaluronidase, was shown to
convert high-molecular-weight hyaluronan (HMW-HA) into
low-molecular-weight hyaluronan (LMW-HA) to regulate ER stress, and
this process was likely mediated via the CD44-p38/ERK signaling
pathway (17).
TMEM2 is a recently discovered specific cell surface
hyaluronidase, which has been proven to be an effective regulator
of ER stress. TMEM2 participates in the regulation of ER stress
through the decomposition of HA in the ECM (2). CD44, a cell adhesion molecule, is a
transmembrane glycoprotein encoded by a single gene, which is
commonly expressed on the surface of various cells and tissues
(18,19). CD44 can affect apoptosis by
participating in ER stress (20). Recent studies have demonstrated
that TMEM2 can decompose HMW-HA (molecular weight >1,000 kDa)
into LMW-HA (molecular weight of ~20 kDa). These decomposed HMW-HAs
can enter cells through CD44 and affect p38/ERK signal
transduction, improve ER stress resistance and protect cells from
damage induced by ER stress (17). Notably, TMEM2 has been shown to
prolong the lifespan and improve the immunity of nematode worms,
and this pathway is independent of the classic UPR pathway after ER
stress (17). The present study
investigated the association between TMEM2 and neuronal apoptosis
following TBI. The authors aimed to clarify the neuroprotective
role of TMEM2 in TBI and determine whether it regulates ER stress
and inhibits the activation of the p38/ERK signaling pathway
through CD44 to alleviate brain edema and nerve cell death.
Materials and methods
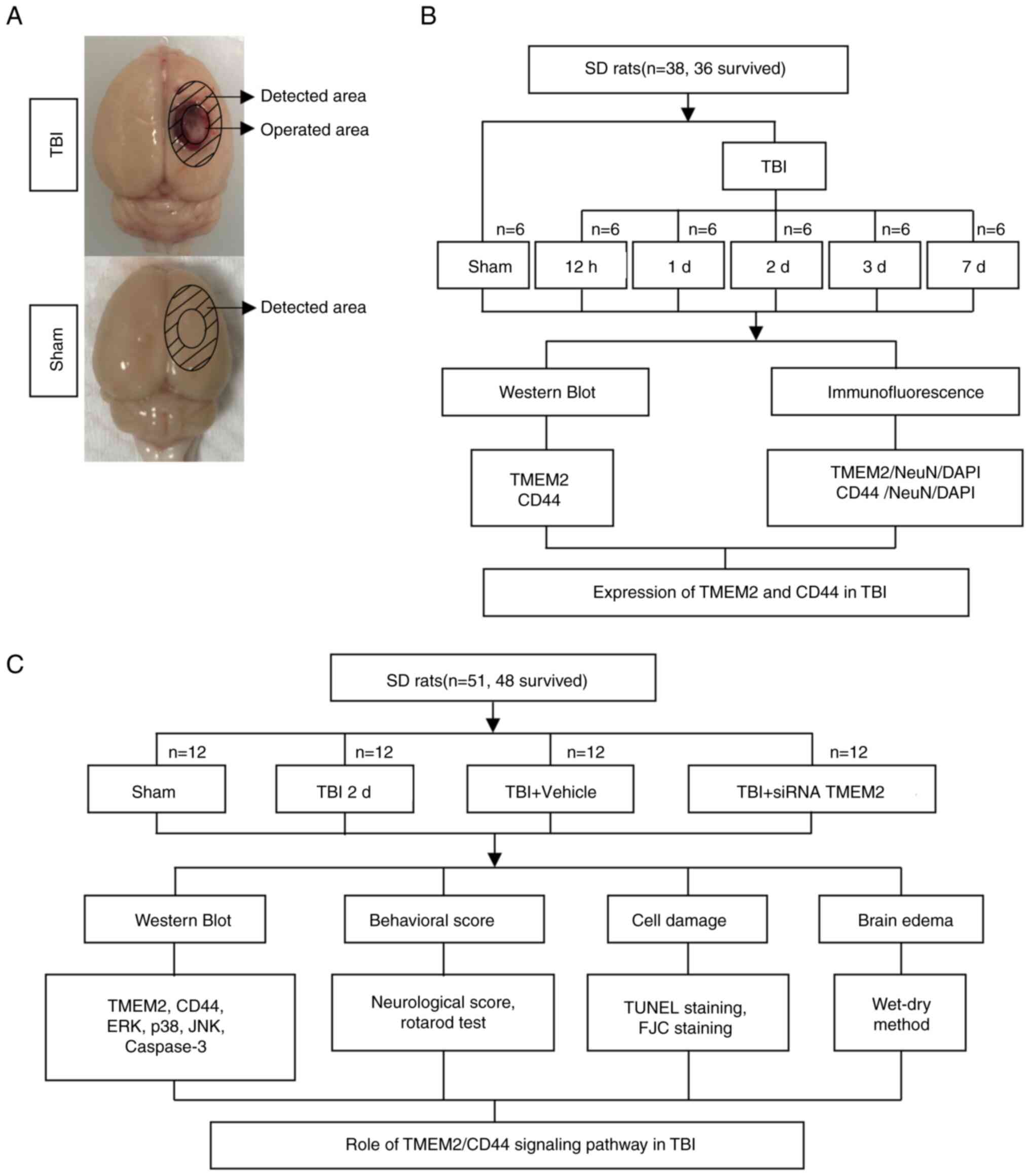
Study design and grouping
A rat model TBI of was employed in the present
study, and two blind experiments were performed, as illustrated in
Fig. 1. The rats exhibited no
obvious differences in body weight, feed intake and motor
function.
Experiment 1 was conducted to determine the time
course of TMEM2 and CD44 post-TBI (Fig. 1B). A total of 36 rats (36
surviving out of an initial cohort of 38) were randomly divided
into the sham-operated (sham), TBI 12-h, TBI 1-day, TBI 2-day, TBI
3-day and TBI 7-day groups (6 rats per group). Following the
induction of TBI (as described below), the brain tissue surrounding
the damaged area was collected and divided (Fig. 1A). Western blot (WB) analysis was
performed on the tissues collected from the front of the damaged
area, while tissues collected from the rear were subjected to
double immunofluorescence analysis.
Experiment 2 was conducted to establish the role of
the TMEM2/CD44 signaling pathway in TBI (Fig. 1C). A total of 48 rats (48
surviving out of a group of 51) were randomly assigned to the sham,
TBI 2-day, TBI + siRNA TMEM2 and TBI + vehicle groups (12 rats per
group). At 2 days post-TBI, which was based on Experiment 1, the
animals were euthanized and injured brain tissues were collected.
Tissues from 6 rats in each group were assessed using WB analysis,
terminal deoxynucleotidyl transferase-mediated dUTP nick-end
labeling (TUNEL) staining and Fluoro-Jade C (FJC) staining. An
additional 6 rats from each group were evaluated for brain edema,
and 6 rats per group were randomly selected for neurological score
assessment prior to euthanasia (2% sodium pentobarbital, 150 mg/kg,
i.p.) (Fig. 1C).
Animals
A total of 89 male Sprague-Dawley rats (weighing
280-320 g; 8 weeks old) were provided by Zhaoyan New Drug Research
Center (Suzhou, China), of which 84 were analyzed as two rats from
Experiment 1 and 3 rats from Experiment 2 died (the rats did not
recover from breathing arrest during modeling) during anesthesia or
modeling. The humane endpoints used in the present study were
cyanosis, dyspnea, mental depression and severe hypothermia
(<37°C). The rats were provided food and water ad
libitum. The rats were kept in an environment with a constant
temperature (25°C) and humidity (50%), with a 12-h light/dark
cycle. The experimental protocols were approved by the Animal
Ethics and Welfare Committee (AEWC) of Zhangjiagang TCM Hospital
Affiliated to Nanjing University of Chinese Medicine (Zhangjiagang,
China; protocol code 2022-4-1), and conformed to the guidelines on
the care and use of animals outlined by the National Institutes of
Health.
Establishment of the rat model of
TBI
Experimental TBI was established in the rats, as
described in a previous study (21). The rats underwent intraperitoneal
anesthesia with 1% sodium pentobarbital at 40 mg/kg and fixation
onto a stereotaxic apparatus (Shanghai Yuyan Instruments Co.,
Ltd.). A 5-mm parietal window to the right of the midline and
behind the coronal suture was created with a bone drill, and the
dura remained intact. A copper weight (4 mm in diameter and 5 mm in
height) was then placed in the bone window, and a 40-g steel rod
was dropped from a height of 25 cm onto the copper weight to induce
trauma. A short pause in the rat heart rate and breathing indicated
successful modeling. Following disinfection and suturing, the rats
were allowed to recover in a warm room. The rats in the sham group
were subjected to the aforementioned procedure, but without the
steel rod step.
TMEM2 siRNA injection
At 24 h prior to the induction of TBI, the TMEM2
siRNA and control siRNA groups received an intracerebroventricular
injection (500 pmol; Thermo Fisher, Inc.) after the rats were
anesthetized (1% sodium pentobarbital, 40 mg/kg, i.p.), mixed with
in vivo RNA transfection reagent [Engreen Biosystem Ltd.
(China)], according to a previously described method (22). TMEM2 siRNA/control siRNA was
added into 2 μl nuclease-free water to 500 pmol, and then
mixed with 1 μl transfection reagent, to a total solution of
3 μl. The TMEM2 siRNA was purchased from Thermo Fisher
Scientific, Inc. A non-targeted siRNA control (Thermo Fisher
Scientific, Inc.) was used as the control siRNA in the TBI +
vehicle group.
Tissue collection and sectioning
1% sodium pentobarbital (40 mg/kg, i.p.) anesthesia
was performed at the indicated time points post-injury. The rats
were perfused transcardially with 200 ml 0.9% normal saline, and
cortical specimens around the injury area (3 mm from the edge of
the injury site in the TBI group or the same location in Sham
animals) were obtained and placed on ice (Fig. 1A). A portion of the specimens
underwent snap freezing and storage at -80°C for use in WB
analysis. The remaining samples were fixed in 4% formalin
overnight, embedded in paraffin and sectioned at a thickness of 5
μm with a paraffin slicer (SLEE medical GmbH) for
immunofluorescence, TUNEL assay and FJC staining. The tissue
samples were extracted and selected by two pathologists who were
blinded to the grouping.
WB analysis
WB analysis was conducted using a previously
published method (23). Brain
tissues were mixed with a tissue protein extraction reagent (CWBio)
containing protease inhibitors before being homogenized on ice for
20 min. The homogenates were isolated by centrifugation at 12,000 ×
g and 4°C. A Pierce™ BCA Protein Assay kit (Thermo Fisher
Scientific, Inc.) was used for protein quantification. Equal
amounts of total protein (30 μg) were resolved by SDS-PAGE
and electro-transferred onto a PVDF membrane (MilliporeSigma).
Following membrane blocking with QuickBlock™ Blocking Buffer
(Beyotime Institute of Biotechnology) at an ambient temperature
(25°C) for 1 h, primary antibodies were added followed by
incubation overnight at 4°C. Subsequently, goat anti-rabbit
(1:5,000; cat. no. 65-6120, Invitrogen; Thermo Fisher Scientific,
Inc.) or anti-mouse IgG-HRP (1:5,000; cat. no. 62-6520, Invitrogen;
Thermo Fisher Scientific, Inc.) were added followed by incubation
at an ambient temperature (25°C) for 1 h. Finally, immunoblots were
detected using the Immobilon™ Western Chemiluminescent HRP
Substrate (Millipore), visualized with an imaging system (Bio-Rad
Laboratories, Inc.), and quantified using ImageJ 1.8.0 software
(National Institutes of Health). The primary antibodies used in
this experiment were as follows: Rabbit anti-TMEM2 (1:1,000; cat.
no. ab98348), rabbit anti-CD44 (1:2,000; cat. no. ab157107), rabbit
anti-p38 (1:1,000; cat. no. ab31828), rabbit anti-phosphorylated
(p-)p38 (1:1,000; cat. no. ab4822), rabbit anti-ERK (1:1,000; cat.
no. ab184699), rabbit anti-p-ERK (1:1,000; cat. no. ab201015),
rabbit anti-JNK (1:1,000; cat. no. ab179461), rabbit anti-p-JNK
(1:2,000; cat. no. ab307802) and rabbit anti-caspase-3 (1:2,000;
cat. no. ab184787) (all from Abcam), with rabbit anti-GAPDH
(1:1,0000, MilliporeSigma) used as the loading control.
Immunofluorescence staining
Double immunofluorescence staining was conducted as
described in a previous study (24). After dewaxing, the
paraffin-embedded sections were treated with an immunostaining
permeable solution (Beyotime Institute of Biotechnology) to rupture
the cell membranes. The cells were washed with phosphate-buffered
physiological saline (PBS) three times. Subsequently, the sections
were blocked with immunostaining block solution (Beyotime Institute
of Biotechnology) at an ambient temperature (25°C) for 30 min and
incubated at 4°C overnight with the following primary antibodies:
Rabbit anti-TMEM2 (1:100, cat. no. ab98348, Abcam), rabbit
anti-CD44 (1:100, cat. no. ab157107, Abcam) and mouse anti-NeuN
(1:300, cat. no. MAB377, Millipore, Merck). Following incubation,
the sections were washed three times with PBS and incubated with
the secondary antibodies, Alexa Fluor 488 donkey anti-rabbit IgG
antibody (1:800, cat. no. A-21206, Invitrogen; Thermo Fisher
Scientific, Inc.) and Alexa Fluor 555 donkey anti-mouse IgG
antibody (1:800, cat. no. A32773, Invitrogen; Thermo Fisher
Scientific, Inc.), for 1 h at room temperature. Finally,
4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) was used for
counterstaining at room temperature (25°C) for 10 min, and the
sections were imaged with a fluorescence microscope (Olympus
Corporation).
Neurological assessment
Neurological evaluation was performed 2 days
post-TBI, as previously described (25). The scoring system consisted of
the following seven components: i) Spontaneous activity; ii) axial
sensation; iii) vibrissae proprioception; iv) symmetry of limb
movement; v) lateral turning; vi) forelimb outstretching; and vii)
climbing. Each subtest was scored from 0 to 3, with a maximum score
of 21, where higher scores indicated less nerve damage.
Rotarod test
The rats were trained for 3 days prior to TBI by
placing them on an accelerated rotation cylinder with multiple
runways. After turning on the machine, the speed was slowly
increased from 4 to 40 r/min for 5 min, during which time the rats
walked on it to prevent falling. The trial ended if the animal fell
off the pedal. The duration the rats spent on the device was
recorded.
TUNEL staining
TUNEL assay was conducted to measure apoptosis, as
per the manufacturer's instructions (Beyotime Institute of
Biotechnology). Following dewaxing in xylene, the paraffin-embedded
sections were treated for 20 min with DNase-free proteinase K (20
μg/ml) at 37°C, followed by a 1-h incubation with TUNEL
working solution at 37°C in the dark. Subsequently, DAPI
Fluoromount-G™ [Yeasen Biotechnology (Shanghai) Co., Ltd.] was used
for counterstaining at room temperature (25°C) for 10 min prior to
observation under a fluorescent microscope (Olympus Corporation).
The apoptotic index was calculated as follows: (TUNEL-positive
cells)/(total cells) ×100%.
FJC staining
Degeneration was measured using FJC staining, as per
the manufacturer's instructions (Biosensis). After dewaxing in
xylene, the paraffin-embedded sections were transferred to solution
B [potassium permanganate and distilled water (1:9, v/v)] and
incubated for 10 min. The sections were then incubated with
solution C [FJC solution and distilled water (1:9, v/v)] for 30 min
in the dark and washed with distilled water. After drying at 60°C
for 10 min, the specimens were soaked in xylene for 5 min and
sealed with a Neutral Balsam [Yeasen Biotechnology (Shanghai) Co.,
Ltd.] prior to observation under a fluorescent microscope (Olympus
Corporation).
Brain edema
The assessment of brain edema was conducted using
the wet-dry method, as previously described (26). After separating the rat brains,
the brain sections were split into ipsilateral and contralateral
sides, and the wet weight was immediately determined with an
electronic balance (ME104/02, Mettler Toledo). Next, the brain
tissues were dried at 100°C for 24 h, and the dry weights were
obtained. The brain water content was quantified as follows: [(wet
weight-dry weight)/wet weight] ×100%.
Statistical analyses
Data are expressed as the mean ± standard deviation,
and GraphPad Prism 8.0 (GraphPad Prism, Inc.) was used for
analysis. A one-way analysis of variance (ANOVA) with post-hoc
Dunnett's multiple test was performed to compare the experimental
groups with the Sham group in Fig.
2. A Student's t-test was performed for the analysis of two
groups (Fig. 3). A one-way ANOVA
with Tukey's multiple comparisons post hoc test was performed to
compare multiple groups (Figs.
4-7; apart from Fig. 7E). A two-way ANOVA with Tukey's
multiple comparisons post hoc test was conducted to analyze brain
edema (Fig. 7E). A value of
P<0.05 was considered to indicate a statistically significant
difference.
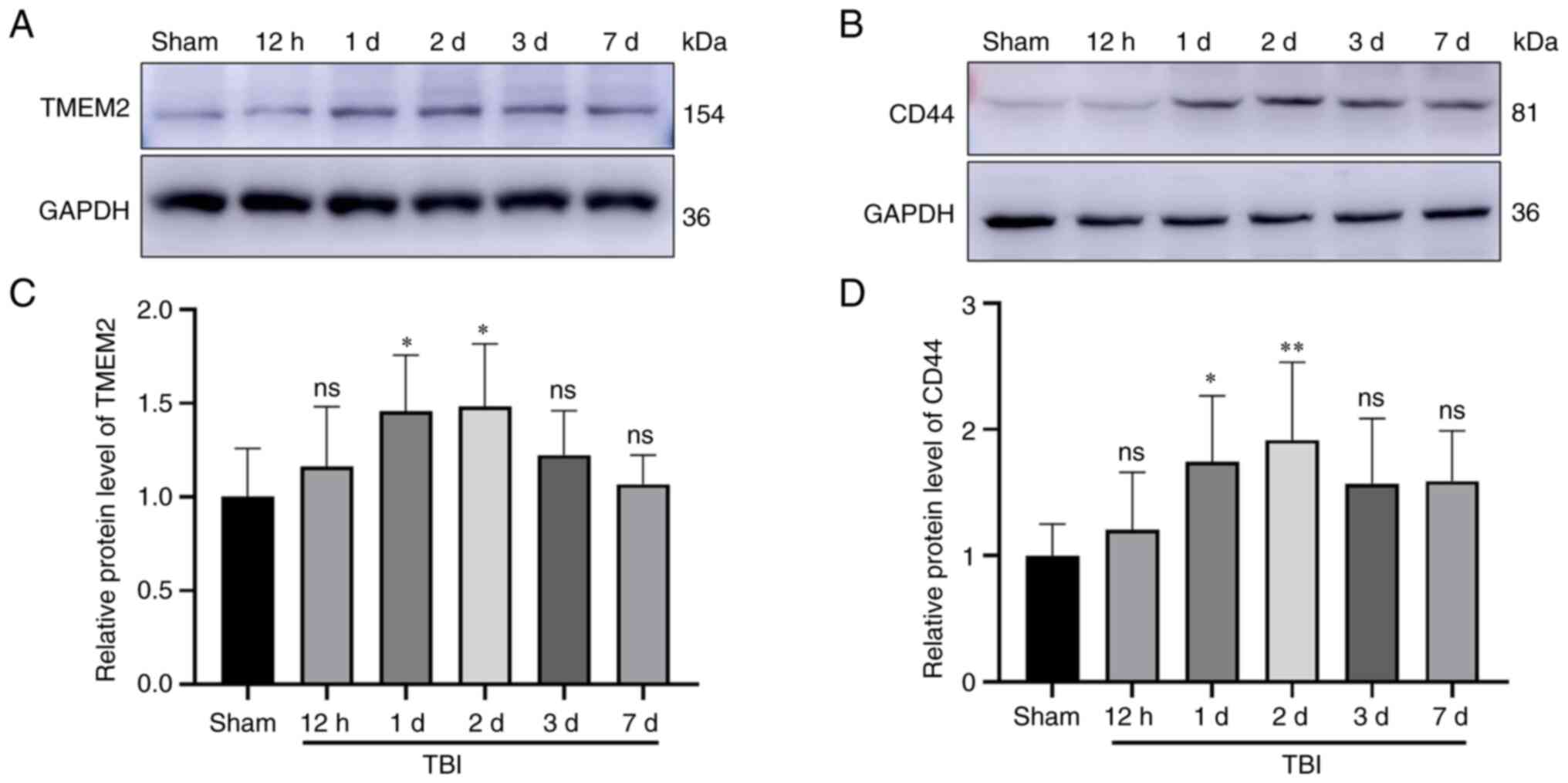 | Figure 2Expression of TMEM2 and CD44 in the
injured cortex following TBI. (A and C) TMEM2 and (B and D) CD44
expression levels in the TBI and sham groups at 12 h, and 1, 2, 3
and 7 days, as determined using western blot analysis. The relative
densities of each protein were normalized to those of the sham
group. Quantification was performed using ImageJ software, and the
mean value of the sham group was normalized to 1. Statistical
analysis was performed using a one-way analysis of variance
(ANOVA), followed by the Dunnett's post-hoc test; n=6 rats in each
group. *P<0.05 and **P<0.01, compared
to the Sham group; ns, not significant (P>0.05). TBI, traumatic
brain injury; TMEM2, transmembrane protein 2; sham, sham-operated;
d, days. |
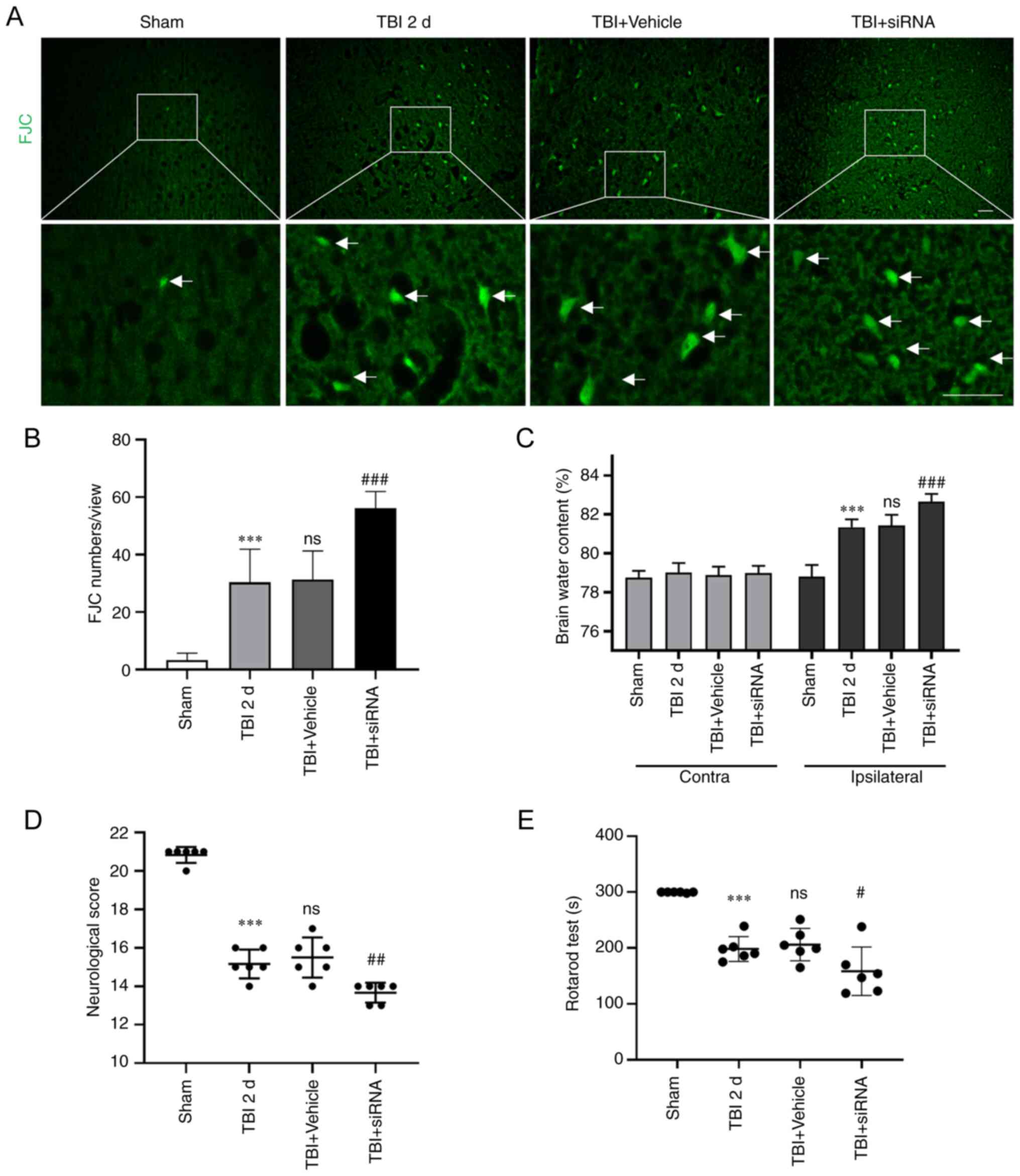 | Figure 7Degeneration, brain edema and
behavioral scores following intervention with transmembrane protein
2 siRNA post-TBI. (A and B) FJC (green) staining was performed to
detect degeneration. (C) Brain edema was measured using the wet-dry
method. (D) Neurological scores and (E) the rotarod test were used
to reflect behavioral scores. Statistical analyses were performed
using a one-way ANOVA, followed by the Tukey's post-hoc test
(except brain edema), and brain edema analysis was performed using
a two-way ANOVA followed by the Tukey's post-hoc test; n=6 rats in
each group. ***P<0.001, compared to the sham group;
#P<0.05, ##P<0.01 and
###P<0.001, compared to the TBI + vehicle group. ns,
not significant (P>0.05), compared to the TBI 2-day group. TBI,
traumatic brain injury; sham, sham-operated; d, days. |
Results
TMEM2 and CD44 protein levels in the rat
brain following TBI
The amount of TMEM2 and CD44 proteins was determined
at 12 h, and 1, 2, 3 and 7 days post-TBI using WB analysis. The
TMEM2 expression levels were elevated at 12 h post-TBI and peaked
at 2 days (Fig. 2A and C). The
CD44 expression levels were consistent with those of TMEM2 and were
increased at 12 h post-TBI, and peaked at 2 days (Fig. 2B and D). Both the TBI 1-day and
2-day groups differed significantly from the sham group.
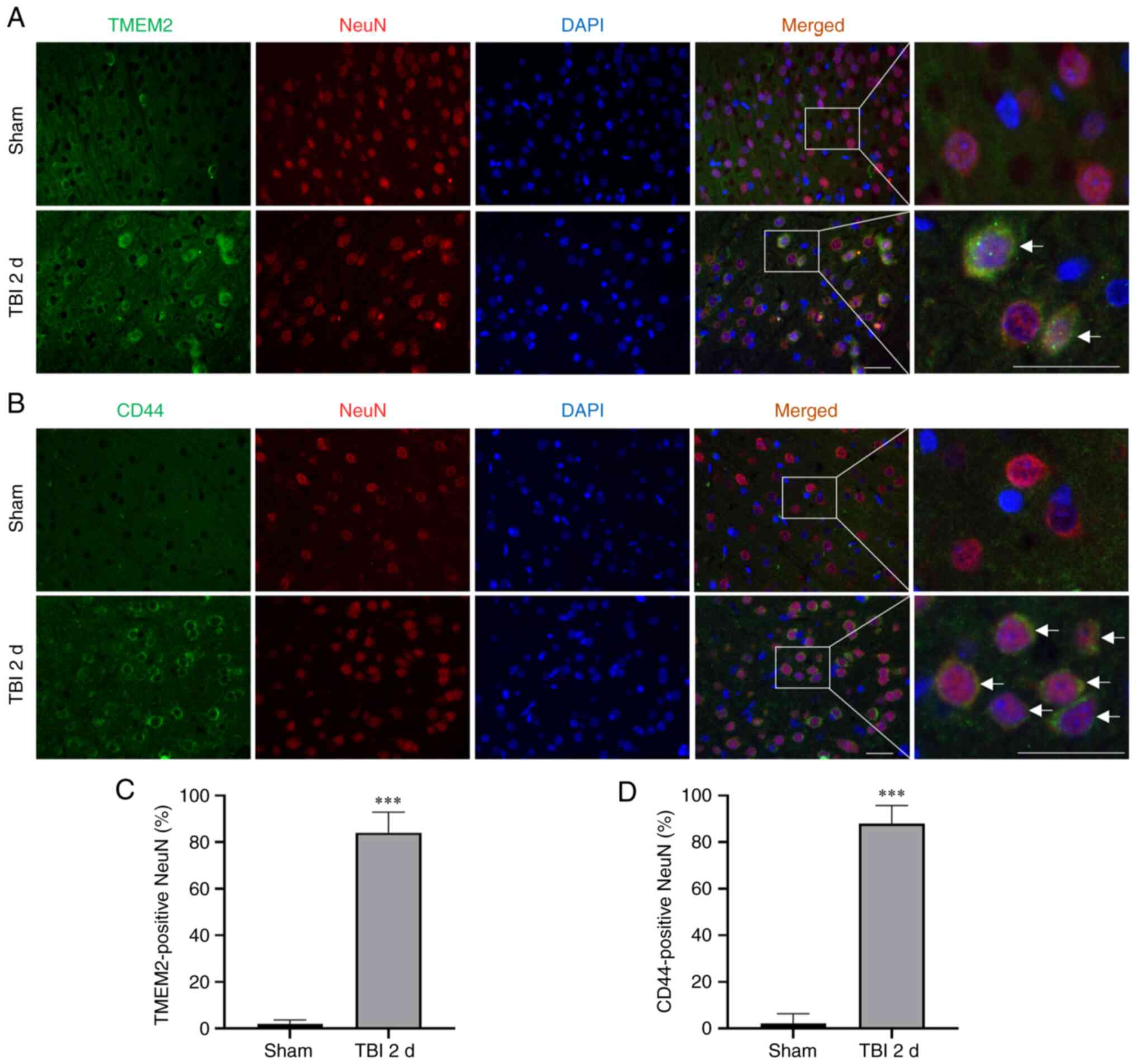
TMEM2 and CD44 expression in neurons
following TBI
Double immunofluorescence staining for NeuN was used
to determine the expression of TMEM2 and CD44. At 2 days post-TBI,
the number of TMEM2-positive neurons (Fig. 3A and C) and CD44-positive neurons
(Fig. 3B and D) increased in the
TBI group compared to the sham group.
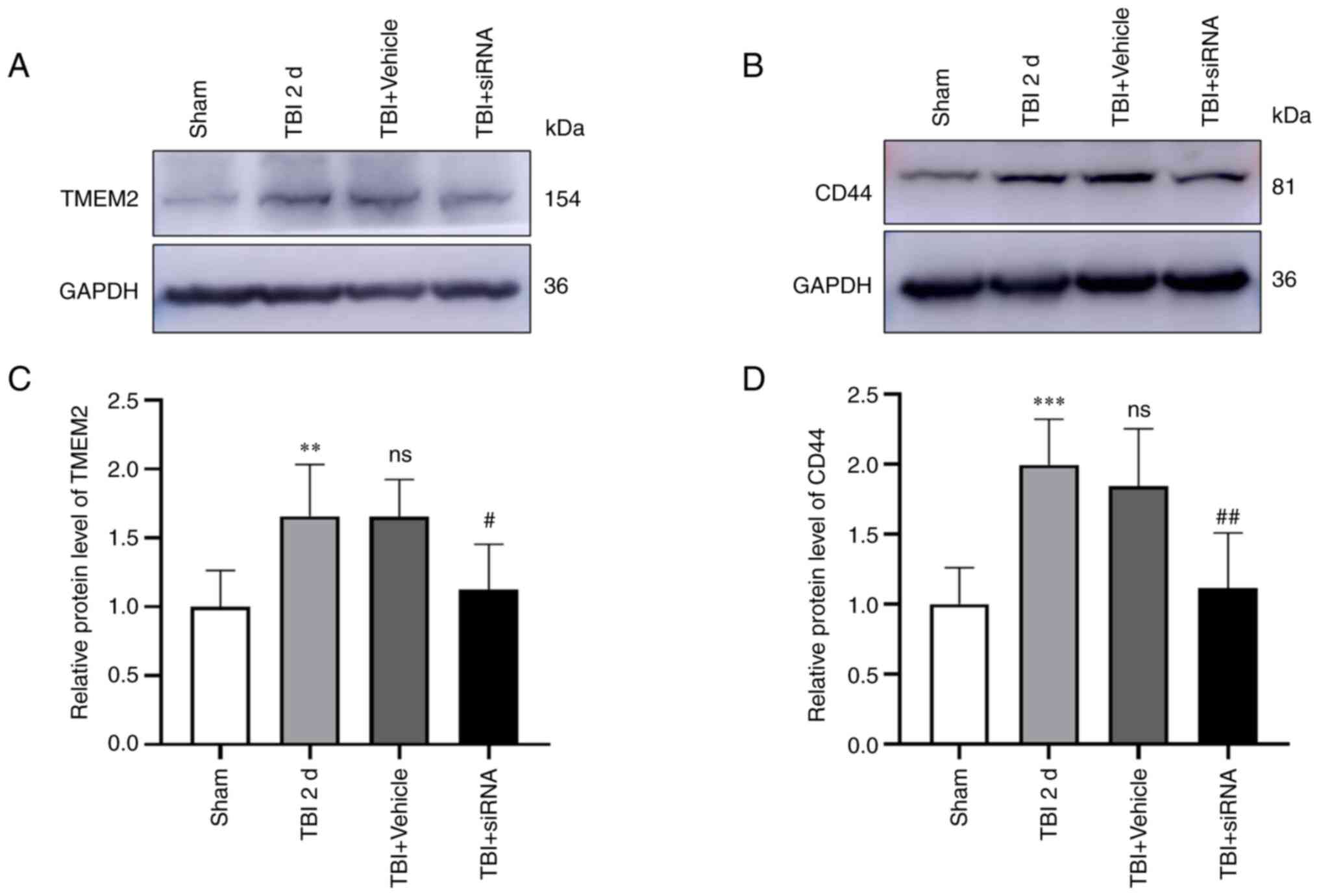
Effects of TMEM2 siRNA on TMEM2 and CD44
expression following TBI
Compared to the sham group, the expression of TMEM2
(Fig. 4A and C) and CD44
(Fig. 4B and D) increased
significantly in the TBI group, and the expression in the TBI and
TBI + vehicle groups was comparable. Additionally, the TMEM2 and
CD44 expression levels were markedly reduced in the TBI + siRNA
group compared to the levels in the TBI + vehicle group.
Effects of TMEM2 siRNA on the MAPK
signaling pathway following TBI
Compared to the sham group, the expression of p-p38
(Fig. 5A and B) and p-ERK
(Fig. 5C and D) was
significantly increased in the TBI group, with comparable amounts
in the TBI and TBI + vehicle groups. Additionally, the expression
of p-p38 and p-ERK in the TBI + siRNA group was markedly increased
compared to that in the TBI + vehicle group. However, the
expression of p-JNK did not differ significantly among the four
groups (Fig. 5E and F).
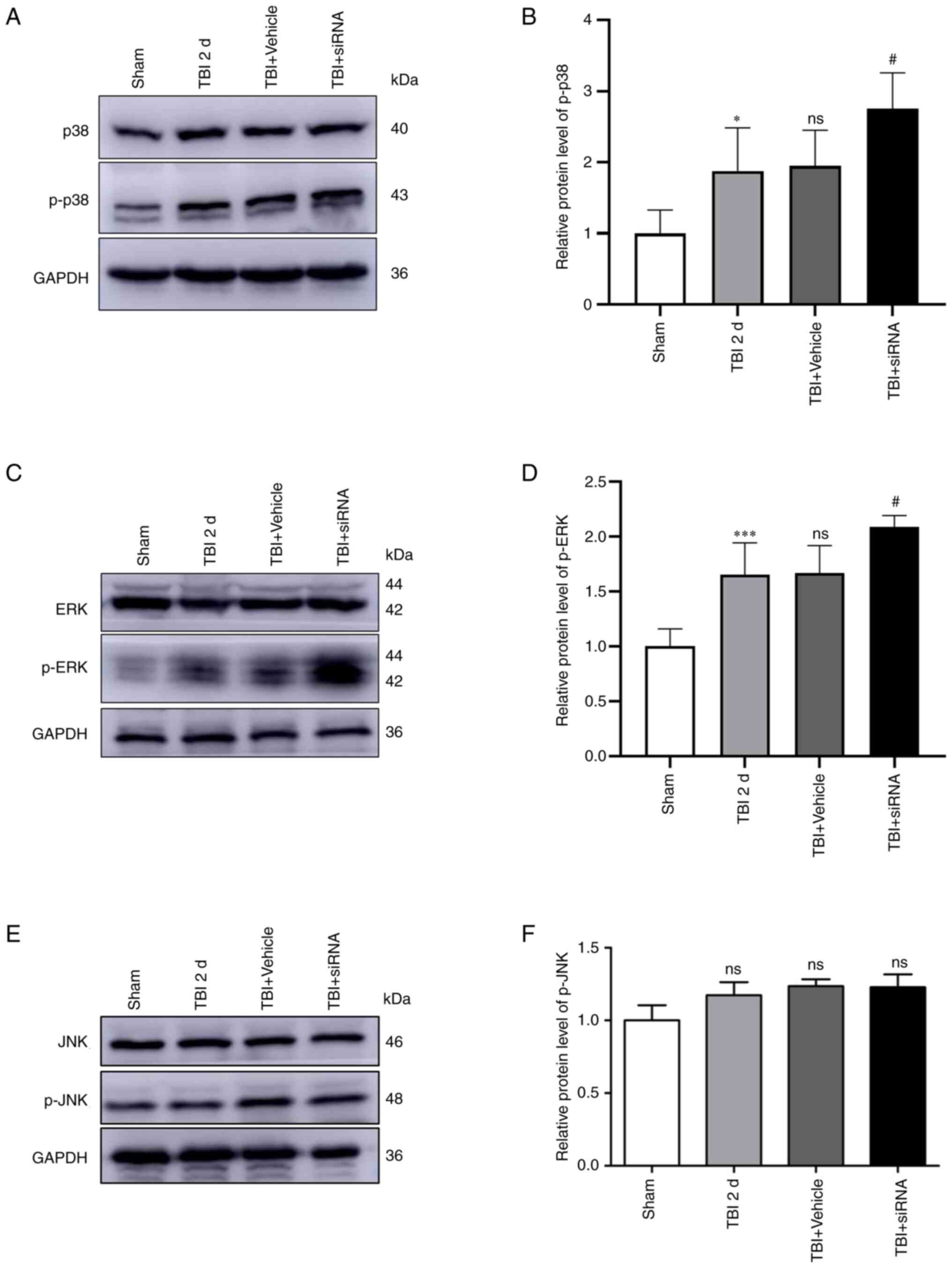 | Figure 5Expression of MAPK signaling
following intervention with TMEM2 siRNA post-TBI. Protein
expression levels of (A and B) p38/p-p38, (C and D) ERK/p-ERK, and
(E and F) JNK/p-JNK at 2 days post-TBI. Statistical analyses were
performed using a one-way ANOVA, followed by Tukey's post hoc test;
n=6 rats in each group. *P<0.05 and
***P<0.001, compared to the sham group;
#P<0.05, compared to the TBI + vehicle group; ns, not
significant (P>0.05), compared to the TBI 2 d group. TBI,
traumatic brain injury; sham, sham-operated; d, days. |
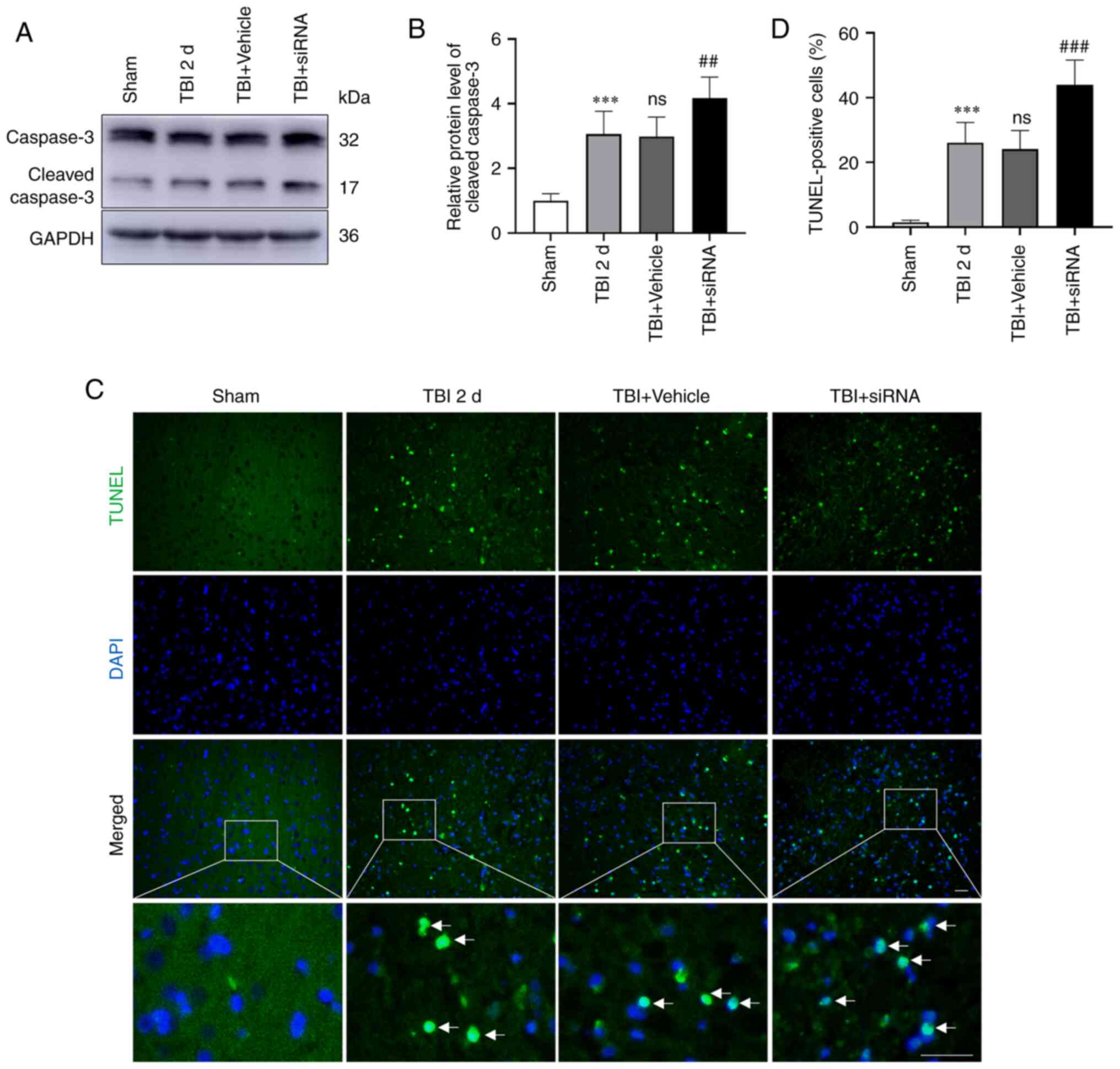
Neuronal apoptosis and degeneration in
rats with TBI following intervention with TMEM2 siRNA
The expression of caspase-3 (Fig. 6A and B) was evaluated, and TUNEL
staining (Fig. 6C and D) was
performed to determine the extent of neuronal apoptosis, while FJC
staining (Fig. 7A and B) was
performed to reflect degeneration. The results revealed that
neuronal apoptosis and degeneration were elevated in the TBI and
TBI + vehicle groups compared to the sham group. Moreover, the TBI
and TBI + vehicle groups had similar rates of neuronal degeneration
and apoptosis. Following TMEM2 siRNA intervention, neuronal
degeneration and apoptosis increased in the TBI + siRNA group
compared to those in the TBI + vehicle group.
Brain edema and behavioral scores
following intervention with TMEM2 siRNA
Compared to the sham group, the brain water content
in the injured hemispheres was significantly elevated in the TBI
group. The TBI and TBI + vehicle groups had comparable brain water
content. Additionally, post-TBI, cerebral edema was significantly
enhanced following intervention with TMEM2 siRNA (Fig. 7C). Additionally, the neurological
score (Fig. 7D) and rotarod test
duration (Fig. 7E) were reduced
in the TBI groups compared to those in the sham group. Moreover,
the scores in the TBI + siRNA group were substantially lower than
those in the TBI + vehicle group.
Discussion
TMEM2 is a type II transmembrane protein expressed
in numerous organs and systems, including the heart, brain, spinal
cord, eyes, heart, lungs, liver, kidneys, spleen, skeletal muscle,
articular cartilage, lymph node, bone marrow, thymus, bladder and
synovium (27,28). However, the expression and
function of TMEM2 are limited under various pathophysiological
conditions. The present study investigated the neuroprotective
effects of TMEM2/CD44 in rats with TBI and the possible underlying
mechanisms. Post-TBI, the expression of TMEM2 was increase in the
rats and peaked after 2 days (Fig.
2A and C). Double-immunofluorescence staining revealed that
TMEM2 was expressed on the surface of neuron cells, as shown in
Fig. 3A. Previous studies have
demonstrated that TMEM2 is located on the plasma membrane, and
represents a newly discovered cell surface HA-degrading enzyme
(29). Additionally, TMEM2 has
been shown to be involved in maintaining ER homeostasis (17) and to promote angiogenesis
(30).
When ER stress is induced by cell injury, the
sensors of UPR (PERK, IRE1 and ATF6) are activated to restore ER
homeostasis by blocking the synthesis of some proteins, as well as
by enhancing ER-specific molecular chaperones, protein degradation
pathways and autophagy (31).
When irreparable damage occurs, the UPR participates in the
terminal response, leading to apoptotic clearance (32), which has been reported in the TBI
model (33). In addition, the
MAPK signaling pathway (including p38, ERK and JNK pathways) is
also known to regulate apoptosis following ER stress. If ER stress
is not alleviated following injury, it will trigger cell death or
aging by influencing the MAPK signaling; this mechanism enables
cells to have certain flexibility during ER stress and can regulate
cell fate according to internal and external signals (13,34). Attenuating the phosphorylation of
p38 and ERK can improve the neurological score following TBI
(35). Alleviating ER stress and
inhibiting the phosphorylation of MAPK signaling can improve
apoptosis (36). TMEM2 responds
to ER stress through the MAPK pathway, independent of UPR signals
and maintains ER homeostasis. This sequence of events was
discovered by Schinzel et al (17), who found that TMEM2
overexpression protected wild-type human fibroblasts from ER stress
by regulating CD44. It has been proven that TMEM2/p38/ERK improves
ER folding capacity or limits the damage in response to ER stress
(37). However, to the best of
our knowledge, no previous study to date has investigated whether
TMEM2 regulates TBI-induced ER stress through the MAPK pathway. In
the present study, the expression of TMEM2 was found to increase in
the surrounding brain tissue following TBI (Fig. 2A). When TMEM2 was silenced using
siRNA, the CD44 expression levels were decreased (Fig. 4B), the expression of p-p38 and
p-ERK was increased (Fig. 5A and
C) and secondary brain injury following TBI was aggravated
(Figs. 6 and 7). These results indicate that TMEM2
exerts a protective effect on rats with TBI through the p38/ERK
pathway.
With the activation of the UPR, a number of
pathological manifestations in neurodegenerative diseases and some
malignant tumors exhibit significant changes in the cellular
microenvironment. Changes in the ER stress response and
glycosaminoglycan and HA composition in the ECM have been observed
(38,39). TMEM2 degrades HA in the ECM in a
Ca2+-dependent manner, resulting in HA internalization
and complete degradation in the lysosome (29,40). HA is a large macromolecule and
one of the main components of the ECM (41). Genetic and chemical inhibition of
typical UPR components suggests that HA decomposition increases ER
stress resistance. Low-molecular-weight fragments of HA appear to
be involved in regulating the MAPK pathway components, p38 and ERK,
by activating CD44 receptors and maintaining cell viability under
conditions of stress (42). The
hyaluronidase activity of TMEM2 and its products provide protection
against ER stress and mediate ER stress resistance through the
CD44/MAPK pathway. Therefore, the present study investigated the
effects of TMEM2 on secondary brain injury in rats with TBI through
MAPK signaling. The results revealed that when TMEM2 was silenced
using TMEM2 siRNA following TBI, the expression of TMEM2 and CD44
decreased (Fig. 4), and the
expression of p-p38 and p-ERK increased (Fig. 5A and C), which aggravated
apoptosis (Figs. 5E and 6B) and degeneration (Fig. 5A). However, the silencing of
TMEM2 had no effect on the expression of p-JNK (Fig. 5E). Previous research has also
found that TMEM2 mediates ER stress through the CD44 and MAPK
signaling pathways (ERK and p38 signaling) and protects cells from
ER stress (17). Although the
JNK pathway can also mediate apoptosis (43), as also demonstrated in the
present study, TMEM2/CD44 did not affect apoptosis through the JNK
pathway, and the specific mechanisms remain to be explored.
As a hyaluronidase, TMEM2 can decompose HA by
dissolution mechanisms, as well as decompose HA from HMW-HA
(>1,000 kDa) to LMW-HA (~20 kDa) (29,30). Specifically, TMEM2 is responsible
for degrading the extracellular HMW-HA into
moderate-molecular-weight HA (MMW-HA, 200-1,000 kDa). The
intracellular lysosomal hyaluronidase and glucosidase further
process the MMW-HA into smaller fragments (27). HA is involved in numerous
processes, including receptor protein attachment and intercellular
communications (44). HMW-HA
exerts anti-inflammatory and anti-angiogenic effects, while LMW-HA
promotes inflammatory and angiogenic responses (45,46). However, it does exert a
neuroprotective effect on the nervous system (47,48).
The present study had certain limitations, which
should be mentioned. The present study did not investigate the
changes and effects of HA following TBI. Moreover, it was not
verified whether TMEM2 decomposes HMW-HA into LMW-HA, or whether
LMW-HA enters cells to play a neuroprotective role. Therefore, in
future studies, the authors aim to verify the effects of HA on
secondary brain injury following TBI in vivo and in
vitro. Previous studies have also shown that HA regulates cell
adhesion, migration and proliferation through various interactions
with specific receptors on cell surfaces, such as CD44 (44,45). However, the possibility of the
interaction between HA and CD44 was did not explore herein;
therefore, the authors aim to evaluate the association between HA
and CD44 on the cell surface by co-immunoprecipitation in future
studies.
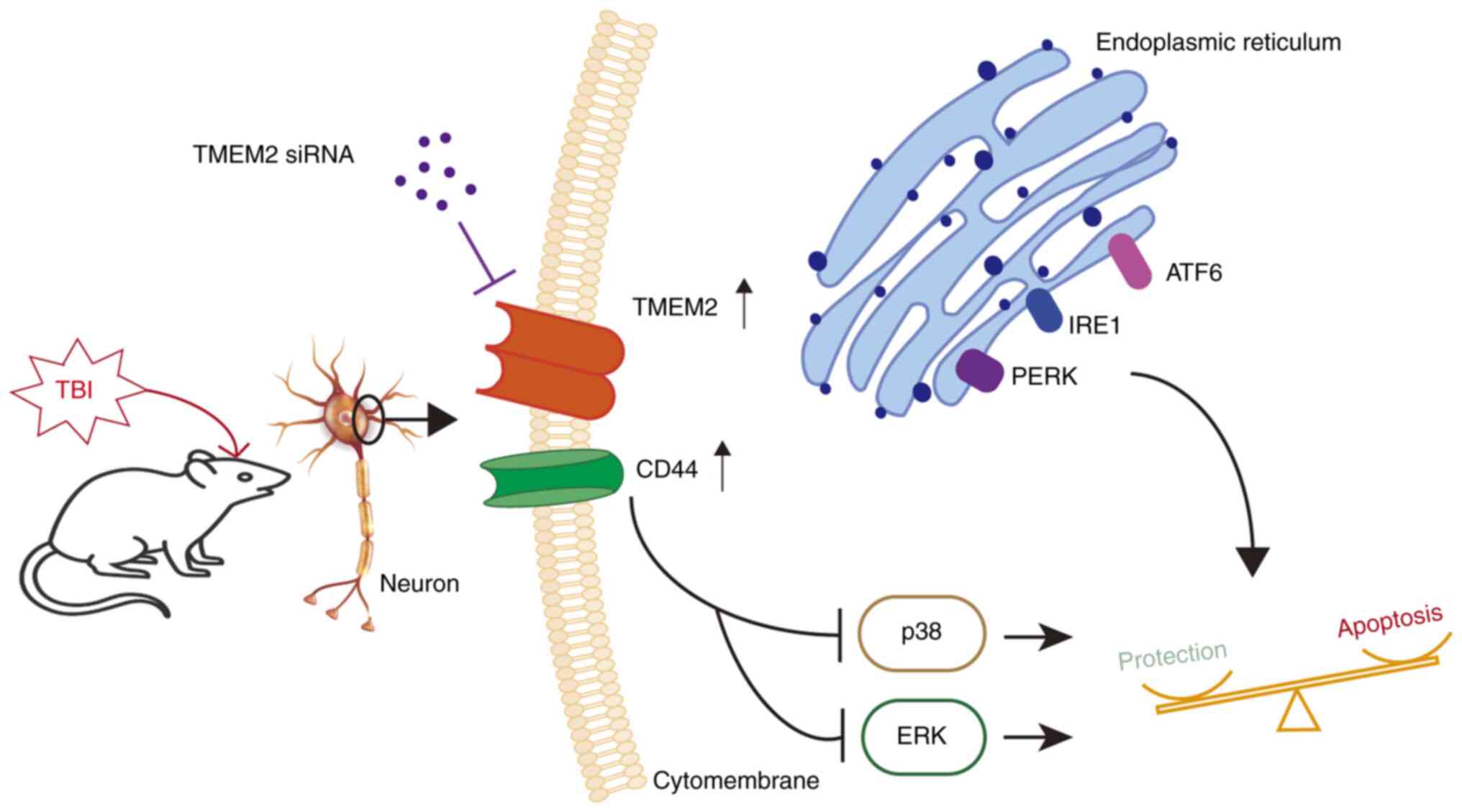
In conclusion, the present study investigated the
role of TMEM2 in a rat model of TBI. The results revealed that
TMEM2 was activated and activated CD44 on the surface of neurons,
which induced brain edema and apoptosis by inhibiting the p38 and
ERK pathways of MAPK, and alleviated the degree of secondary brain
injury (Fig. 8). Moreover, when
TMEM2 was silenced, cerebral edema and nerve injury were aggravated
by the upregulation of ERK and p38 signals. Therefore, TMEM2/CD44
represents a potential therapeutic target for TBI prevention and
control.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
MW, RG and BD contributed to the conception or
design of the study. MW, CW, YG, YH, LJ and MZ contributed to the
acquisition of data. MW and CW drafted the manuscript; YG and RG
processed the graphs and performed data analysis. MW and BD revised
the manuscript. All authors have read and approved the final
manuscript. YG, RG and BD confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
The present study was conducted in accordance with
the Declaration of Helsinki and was approved by the Animal Ethics
and Welfare Committee (AEWC) of Zhangjiagang TCM Hospital
Affiliated with Nanjing University of Chinese Medicine (protocol
code 2022-4-1).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the Enterprise School
Cooperative Education Project of the Ministry of Education (grant
no. 202102242005), the Zhangjiagang Key Health Personnel Training
Program (grant no. ZJGWSRC202003), the Suzhou Youth Science and
Technology Project (grant nos. KJXW2020062 and KJXW2021063), the
Suzhou Science and Technology Development Project (grant nos.
SKJY2021001 and SKJY2021003), the Suzhou Livelihood Science and
Technology Project (grant no. SYS2020054) and the Gusu Health
Personnel Training Project (grant no. GSWS2020104).
References
|
1
|
Dang B, Chen W, He W and Chen G:
Rehabilitation treatment and progress of traumatic brain injury
dysfunction. Neural Plast. 2017:15821822017.
|
|
2
|
Nguyen R, Fiest KM, McChesney J, Kwon CS,
Jette N, Frolkis AD, Atta C, Mah S, Dhaliwal H, Reid A, et al: The
International incidence of traumatic brain injury: A systematic
review and Meta-Analysis. Can J Neurol Sci. 43:774–785. 2016.
|
|
3
|
Dehghanian F, Soltani Z and Khaksari M:
Can mesenchymal stem cells act multipotential in traumatic brain
injury? J Mol Neurosci. 70:677–688. 2020.
|
|
4
|
Eastman CL, D'Ambrosio R and Ganesh T:
Modulating neuroinflammation and oxidative stress to prevent
epilepsy and improve outcomes after traumatic brain injury.
Neuropharmacology. 172:1079072020.
|
|
5
|
Li D, Ni H, Rui Q, Gao R and Chen G:
Deletion of Mst1 attenuates neuronal loss and improves neurological
impairment in a rat model of traumatic brain injury. Brain Res.
1688:15–21. 2018.
|
|
6
|
Wu J, He J, Tian X, Zhong J, Li H and Sun
X: Activation of the hedgehog pathway promotes recovery of
neurological function after traumatic brain injury by protecting
the neurovascular unit. Transl Stroke Res. 11:720–733. 2020.
|
|
7
|
Ni H, Rui Q, Xu Y, Zhu J, Gao F, Dang B,
Li D, Gao R and Chen G: RACK1 upregulation induces neuroprotection
by activating the IRE1-XBP1 signaling pathway following traumatic
brain injury in rats. Exp Neurol. 304:102–113. 2018.
|
|
8
|
Oakes SA and Papa FR: The role of
endoplasmic reticulum stress in human pathology. Annu Rev Pathol.
10:173–194. 2015.
|
|
9
|
Wang M, Law ME, Castellano RK and Law BK:
The unfolded protein response as a target for anticancer
therapeutics. Crit Rev Oncol Hematol. 127:66–79. 2018.
|
|
10
|
Choi SI, Lee E, Jeong JB, Akuzum B, Maeng
YS, Kim TI and Kim EK: 4-Phenylbutyric acid reduces mutant-TGFBIp
levels and ER stress through activation of ERAD pathway in corneal
fibroblasts of granular corneal dystrophy type 2. Biochem Biophys
Res Commun. 477:841–846. 2016.
|
|
11
|
Katayama T, Imaizumi K, Honda A, Yoneda T,
Kudo T, Takeda M, Mori K, Rozmahel R, Fraser P, George-Hyslop PS
and Tohyama M: Disturbed activation of endoplasmic reticulum stress
transducers by familial Alzheimer's disease-linked presenilin-1
mutations. J Biol Chem. 276:43446–43454. 2001.
|
|
12
|
Liu Y, Guyton KZ, Gorospe M, Xu Q, Lee JC
and Holbrook NJ: Differential activation of ERK, JNK/SAPK and
P38/CSBP/RK map kinase family members during the cellular response
to arsenite. Free Radic Biol Med. 21:771–781. 1996.
|
|
13
|
Hotamisligil GS and Davis RJ: Cell
signaling and stress responses. Cold Spring Harb Perspect Biol.
8:a0060722016.
|
|
14
|
Li W, Zhu J, Dou J, She H, Tao K, Xu H,
Yang Q and Mao Z: Phosphorylation of LAMP2A by p38 MAPK couples ER
stress to chaperone-mediated autophagy. Nat Commun. 8:17632017.
|
|
15
|
Vasvani S, Kulkarni P and Rawtani D:
Hyaluronic acid: A review on its biology, aspects of drug delivery,
route of administrations and a special emphasis on its approved
marketed products and recent clinical studies. Int J Biol Macromol.
151:1012–1029. 2020.
|
|
16
|
Garantziotis S and Savani RC: Hyaluronan
biology: A complex balancing act of structure, function, location
and context. Matrix Biol. 78-79:1–10. 2019.
|
|
17
|
Schinzel RT, Higuchi-Sanabria R, Shalem O,
Moehle EA, Webster BM, Joe L, Bar-Ziv R, Frankino PA, Durieux J,
Pender C, et al: The hyaluronidase, TMEM2, promotes ER homeostasis
and longevity independent of the UPR(ER). Cell. 179:1306–1318.e18.
2019.
|
|
18
|
He X, Shi X, Puthiyakunnon S, Zhang L,
Zeng Q, Li Y, Boddu S, Qiu J, Lai Z, Ma C, et al: CD44-mediated
monocyte transmigration across Cryptococcus neoformans-infected
brain microvascular endothelial cells is enhanced by HIV-1 gp41-I90
ectodomain. J Biomed Sci. 23:282016.
|
|
19
|
Thorne RF, Legg JW and Isacke CM: The role
of the CD44 transmembrane and cytoplasmic domains in co-ordinating
adhesive and signalling events. J Cell Sci. 117:373–380. 2004.
|
|
20
|
Dalal S, Zha Q, Daniels CR, Steagall RJ,
Joyner WL, Gadeau AP, Singh M and Singh K: Osteopontin stimulates
apoptosis in adult cardiac myocytes via the involvement of CD44
receptors, mitochondrial death pathway, and endoplasmic reticulum
stress. Am J Physiol Heart Circ Physiol. 306:H1182–H1191. 2014.
|
|
21
|
Gao F, Li D, Rui Q, Ni H, Liu H, Jiang F,
Tao L, Gao R and Dang B: Annexin A7 levels increase in rats with
traumatic brain injury and promote secondary brain injury. Front
Neurosci. 12:3572018.
|
|
22
|
Shi M, Gong Y, Wu M, Gu H, Yu J, Gao F,
Ren Z, Qian M, Dang B and Chen G: Downregulation of TREM2/NF-кB
signaling may damage the blood-brain barrier and aggravate neuronal
apoptosis in experimental rats with surgically injured brain. Brain
Res Bull. 183:116–126. 2022.
|
|
23
|
Gong Y, Wu M, Shen J, Tang J, Li J, Xu J,
Dang B and Chen G: Inhibition of the NKCC1/NF-κB signaling pathway
decreases inflammation and improves brain edema and nerve cell
apoptosis in an SBI rat model. Front Mol Neurosci.
14:6419932021.
|
|
24
|
Wu MY, Gao F, Tang JF, Shen JC, Gao R,
Dang BQ and Chen G: Possible mechanisms of the PERK pathway on
neuronal apoptosis in a rat model of surgical brain injury. Am J
Transl Res. 13:732–742. 2021.
|
|
25
|
Feng D, Wang B, Wang L, Abraham N, Tao K,
Huang L, Shi W, Dong Y and Qu Y: Pre-ischemia melatonin treatment
alleviated acute neuronal injury after ischemic stroke by
inhibiting endoplasmic reticulum stress-dependent autophagy via
PERK and IRE1 signalings. J Pineal Res. 62:2017. View Article : Google Scholar
|
|
26
|
Gong Y, Wu M, Gao F, Shi M, Gu H, Gao R,
Dang BQ and Chen G: Inhibition of the pSPAK/pNKCC1 signaling
pathway protects the bloodbrain barrier and reduces neuronal
apoptosis in a rat model of surgical brain injury. Mol Med Rep.
24:7172021.
|
|
27
|
Yamaguchi Y, Yamamoto H, Tobisawa Y and
Irie F: TMEM2: A missing link in hyaluronan catabolism identified?
Matrix Biol. 78-79:139–146. 2019.
|
|
28
|
Scott DA, Drury S, Sundstrom RA, Bishop J,
Swiderski RE, Carmi R, Ramesh A, Elbedour K, Srikumari
Srisailapathy CR, Keats BJ, et al: Refining the DFNB7-DFNB11
deafness locus using intragenic polymorphisms in a novel gene,
TMEM2. Gene. 246:265–274. 2000.
|
|
29
|
Yamamoto H, Tobisawa Y, Inubushi T, Irie
F, Ohyama C and Yamaguchi Y: A mammalian homolog of the zebrafish
transmembrane protein 2 (TMEM2) is the long-sought-after
cell-surface hyaluronidase. J Biol Chem. 292:7304–7313. 2017.
|
|
30
|
De Angelis JE, Lagendijk AK, Chen H, Tromp
A, Bower NI, Tunny KA, Brooks AJ, Bakkers J, Francois M, Yap AS, et
al: Tmem2 regulates embryonic vegf signaling by controlling
hyaluronic acid turnover. Dev Cell. 40:4212017.
|
|
31
|
Hetz C: The unfolded protein response:
Controlling cell fate decisions under ER stress and beyond. Nat Rev
Mol Cell Biol. 13:89–102. 2012.
|
|
32
|
Korennykh A and Walter P: Structural basis
of the unfolded protein response. Annu Rev Cell Dev Biol.
28:251–277. 2012.
|
|
33
|
Sun G, Zhao Z, Lang J, Sun B and Zhao Q:
Nrf2 loss of function exacerbates endoplasmic reticulum
stress-induced apoptosis in TBI mice. Neurosci Lett.
770:1364002022.
|
|
34
|
Darling NJ and Cook SJ: The role of MAPK
signalling pathways in the response to endoplasmic reticulum
stress. Biochim Biophys Acta. 1843:2150–2163. 2014.
|
|
35
|
Zhang J, Yi T, Cheng S and Zhang S:
Glucagon-like peptide-1 receptor agonist Exendin-4 improves
neurological outcomes by attenuating TBI- induced inflammatory
responses and MAPK activation in rats. Int Immunopharmacology.
86:1067152020.
|
|
36
|
Chen J, Chen J, Cheng Y, Fu Y, Zhao H,
Tang M, Zhao H, Lin N, Shi X, Lei Y, et al: Mesenchymal stem
cell-derived exosomes protect beta cells against hypoxia-induced
apoptosis via miR-21 by alleviating ER stress and inhibiting p38
MAPK phosphorylation. Stem Cell Res Ther. 11:972020.
|
|
37
|
Goncalves RLS and Hotamisligil GS: TMEM2
modulates ER stress in a Non-canonical manner. Cell Metab.
30:999–1001. 2019.
|
|
38
|
Brown MK and Naidoo N: The endoplasmic
reticulum stress response in aging and age-related diseases. Front
Physiol. 3:2632012.
|
|
39
|
Chanmee T, Ontong P and Itano N:
Hyaluronan: A modulator of the tumor microenvironment. Cancer Lett.
375:20–30. 2016.
|
|
40
|
Tammi MI, Oikari S, Pasonen-Seppanen S,
Rilla K, Auvinen P and Tammi RH: Activated hyaluronan metabolism in
the tumor matrix-Causes and consequences. Matrix Biol.
78-79:147–164. 2019.
|
|
41
|
Kudo Y, Sato N, Adachi Y, Amaike T, Koga
A, Kohi S, Noguchi H, Nakayama T and Hirata K: Overexpression of
transmembrane protein 2 (TMEM2), a novel hyaluronidase, predicts
poor prognosis in pancreatic ductal adenocarcinoma. Pancreatology.
20:1479–1485. 2020.
|
|
42
|
Mascaro M, Pibuel MA, Lompardia SL, Diaz
M, Zotta E, Bianconi MI, Lago N, Otero S, Jankilevich G, Alvarez E
and Hajos SE: Low molecular weight hyaluronan induces migration of
human choriocarcinoma JEG-3 cells mediated by RHAMM as well as by
PI3K and MAPK pathways. Histochem Cell Biol. 148:173–187. 2017.
|
|
43
|
Bai G, Wang H and Cui N: mTOR pathway
mediates endoplasmic reticulum stress-induced CD4+ T cell apoptosis
in septic mice. Apoptosis. 27:740–750. 2022.
|
|
44
|
Laurent TC, Laurent UB and Fraser JR: The
structure and function of hyaluronan: An overview. Immunol Cell
Biol. 74:A1–A7. 1996.
|
|
45
|
West DC, Hampson IN, Arnold F and Kumar S:
Angiogenesis induced by degradation products of hyaluronic acid.
Science. 228:1324–1326. 1985.
|
|
46
|
Vigetti D, Karousou E, Viola M, Deleonibus
S, De Luca G and Passi A: Hyaluronan: Biosynthesis and signaling.
Biochim Biophys Acta. 1840:2452–2459. 2014.
|
|
47
|
Wang J, Wang X, Wei J and Wang M:
Hyaluronan tetrasaccharide exerts neuroprotective effect and
promotes functional recovery after acute spinal cord injury in
rats. Neurochem Res. 40:98–108. 2015.
|
|
48
|
Wakao N, Imagama S, Zhang H, Tauchi R,
Muramoto A, Natori T, Takeshita S, Ishiguro N, Matsuyama Y and
Kadomatsu K: Hyaluronan oligosaccharides promote functional
recovery after spinal cord injury in rats. Neurosci Lett.
488:299–304. 2011.
|