Introduction
Myocardial ischemia (MI) is the leading cause of
death globally, accounting for ~16% of total deaths (1). Nearly half of MI cases die as a
result of sudden cardiac death (SCD) and lethal ventricular
arrhythmia (LVA) is the most common cause of SCD (2). Although progress has been made in
supportive care, efforts to prevent MI-induced SCD have failed;
cases are increasing and becoming a major public health problem
worldwide (3). Clinically, SCD
induced by MI commonly occurs within the first hour after a heart
attack and the incidence decreases exponentially thereafter
(4,5). Therefore, the early period is the
most dangerous period for LVA-SCD and is a key time to prevent SCD.
Nevertheless, the underlying mechanism of LVA-SCD during this
period is not fully understood.
As the primary source of reactive oxygen species
(ROS) during MI, mitochondrial (m)ROS predispose individuals to LVA
(6). Studies have uncovered
crosstalk between mROS and Ca2+ signaling via the
mitochondria-associated sarcoplasmic reticulum membrane (MAM)
(7,8). Ca2+/calmodulin-dependent
protein kinases (CaMKs), which are localized within the MAM, are
activated upon oxidation at methionine 282 (9). In turn, oxidized (ox-)CaMKII
phosphorylates ryanodine receptor 2 (RyR2) at serine 2814 via the
MAM, leading to diastolic Ca2+ leak from the
sarcoplasmic reticulum and triggering malignant arrhythmia
(10). CaMKII-M282 oxidation and
RyR2-S2814 phosphorylation are increased in patients with atrial
fibrillation and ventricular tachycardia (11,12), suggesting that both the mROS and
relevant Ca2+ leak can lead to LVA. To the best of our
knowledge, however, it has not been determined whether
mROS-Ca2+ crosstalk serves a pivotal role in the
development of LVA-SCD in early MI. The present study aimed to
explore the underlying mechanism of LVA-SCD within the early stage
of MI (up to 30 min post-MI), focusing on the mROS-Ca2+
crosstalk.
Materials and methods
Experimental mice
The present study was approved by the Medical Animal
Care and Welfare Committee at Shantou University Medical College
(Shantou, China; approval no. SUMC2020-035). Animal studies were
performed according to the guidelines from the National Institutes
of Health (NIH) Guide for the Care and Use of Laboratory Animals
(13).
A point mutation of serine (S) to alanine (A) at
position 2814 of the RyR2 (hereafter referred to as 'S2814A') was
introduced in C57/BL6 mice using CRISPR/Cas gene-editing technology
(Data S1) by Cyagen Biosciences
to address the role of RyR2-S2814 phosphorylation in
Ca2+ balance. They were bred and their offspring were
raised, mated, and bred at the Laboratory Animals Center of Shantou
University Medical College (Shantou, China) under 22-24°C, 60-65%
humidity and a 12/12-h light/dark cycle. Since males are more
likely to have coronary artery disease than females (14), the present study used only male
offspring of the fourth to five generation (n=32, age, ~8 weeks,
25-30 g). Animals had free access to rodent chow and clean drinking
water. Specific pathogen-free grade C57/BL6 male mice (n=130, age,
~8 weeks, 25-30 g) were obtained from Charles River Laboratories
(Beijing, China), and they were kept in the same housing
conditions.
Acute MI mouse model and MitoTEMPO
treatment
S2814A homozygous mutant (confirmed by DNA
sequencing; Supplementary Materials and methods) male mice and
their wild-type male littermates (n=152, age, ~8 weeks, 25-30 g)
were subjected to left coronary artery ligation (CAL). Briefly,
mice were anesthetized using 30 mg/kg body weight 1% pentobarbital
sodium in saline (Sigma-Aldrich; Merck KGaA) through
intraperitoneal injection. Then, lead II electrocardiogram (ECG)
was monitored using BL-420 Biological-Functional Experimental
System (Chengdu Taimeng Co., Ltd.). When the animal was deeply
anesthetized (no response to pinching of the toes or fingers),
artificial ventilation was established with a tidal volume of 2
ml/kg, an inspiratory/expiratory ratio of 1:2 and a respiratory
rate of 115 breaths/min. The thoracic cavity was opened, the
pericardium was cut and the main left coronary artery was ligated.
Following CAL, the elevated T waves indicated the success of the
ligation. Some mice developed LVA-SCD (MI-SCD group, n=21). The
remaining mice, who maintained a relatively normal ECG and survived
≥70 min after CAL, were defined as stable group (MI-S group, n=41).
S2814A mice were similarly subjected to MI, which was defined as
transgenic group (TG-MI group, n=32). To characterize the
protective mechanism of the point mutation, only mice that survived
early MI were used for experiments.
A group of wild-type mice (n=26) were administered
MitoTEMPO (2.0 mg/kg in saline; Sigma-Aldrich; Merck KGaA; cat. no.
SML0737) via tail vein injection 15 min before CAL operation, which
as defined as the MT-MI group, and we only recruited the surviving
mice (n=21) to determine the specific protective mechanism of
MitoTEMPO. Sham-operated mice (SO group, n=32) were anesthetized
and subjected to thoracotomy as aforementioned, excluding those
with abnormal ECG after operation. Surviving mice were euthanized
by over-anesthesia with sodium pentobarbital (90 mg/kg) (15). Mice who experienced massive
hemorrhage during the operation were excluded. Left ventricles were
immediately harvested after death and stored at -80°C for further
experiments.
ECG analysis
The corrected QT interval (QTc) was calculated with
Bazett's formula (QTc=QT interval/√RR interval (RRI)) and RRI was
normalized by heart rates (16).
For T wave variation analysis, one typical case was selected from
each experimental group and 10 complete ECG data (P wave, QRS
complex, and T wave) were read every 5 min for the first 20 min
after MI. Amplitudes of T waves were collected every 8 msec and
integrated to produce a polyline chart to determine T wave
variation (17).
Hypoxic model of H9c2 and dynamic
detection of mROS
A hypoxia model of H9c2 cells (purchased from the
Cell Bank of Chinese Academy of Sciences, Shanghai, China) was
generated to explore changes in mROS after hypoxia. H9c2 cells were
inoculated into 3.5-cm dishes (total cells:3×105) and
grown (37°C, 5% CO2) to 60-80% confluence in
high-glucose DMEM (Gibco) supplemented with 10% fetal bovine serum.
Penicillin/streptomycin (100 U/100 μg/ml) was used. The
cells were incubated at 37°C in Baker Ruskinin's InvivO2
(400) hypoxia workstation (I&L Biosystems GmbH) in 1%
O2, 5% CO2 and 94% N2 for 15, 30,
45 or 70 min, respectively. A group of cells undergoing 15 min
hypoxia was also pretreated with MitoTEMPO (0.5 μM) at 37°C
for 30 min. Cells were incubated at 37°C with MitoSOX™ probe
(Thermo Fisher Scientific, Inc.; 5 μM) for 15 min, and
fluorescence images (×200 magnification) were obtained in a Carl
Zeiss LSM 880 confocal microscope (Carl Zeiss GmbH) to evaluate
mROS levels. The excitation wavelength (λEx) and emission
wavelength (λEm) were set according to the manufacturer's
instructions. The fluorescence intensity was quantified using Image
J software (Image J 1.52i; NIH).
Detection of ROS and Ca2+ in
cytoplasm and mitochondria
ROS and Ca2+ were directly measured by
relative fluorescent probes. Briefly, hearts were immediately
retrieved after mouse death and pre-cooled in a frozen sectioning
machine for 20 min. Then, myocardial cryosections (5 μM)
were prepared immediately, followed by incubation at 37°C with
dihydroethidium (DHE; 2 μM; 30 min), MitoSOX (5 μM;
15 min), Fura-2 AM (1 μM; 30 min; all Thermo Scientific) or
Rhod-2 AM (2 μM; 15 min; MedChemExpress) to detect levels of
ROS and Ca2+ in the cytoplasm and mitochondria,
respectively. DAPI (50 μl, Beyotime) staining (10 min at
room temperature) was used to visualize nuclei of cardiomyocytes.
Fluorescence (×200 magnification) was measured using a Carl Zeiss
LSM 800 confocal microscope (Carl Zeiss) and the excitation
wavelength was set according to the manufacturer's instructions.
The fluorescence intensity was quantified using Image J
software.
Measurement of mitochondrial membrane
potential (MMP)
MMP was assessed by JC-1 staining according to
manufacturer's instructions. First, myocardial mitochondria from
the myocardium of the experimental mice were isolated with Tissue
Mitochondria Isolation kit (Beyotime Institute of Biotechnology).
MMP (ΔΨm) assay kit (Beyotime Institute of Biotechnology) with the
JC-1 probe was used to detect MMP Fluorescence images (×100
magnification) were obtained with an Olympus fluorescence
microscope to detect JC-1 monomers (green fluorescence, low
potential) and aggregates (red fluorescence, high potential) were
detected. The fluorescence intensity was quantified using Image J
software.
Measurement of reduced
glutathione/oxidized glutathione (GSH/GSSG) ratio
Oxidized and total glutathione levels in the left
ventricular tissue were measured with GSH and GSSG Assay kit
(Beyotime), according to the manufacturer's protocols. GSH levels
were calculated as follows: GSH=(total glutathione-GSSG) ×2. Data
are presented as the ratio of GSH to GSSG.
Western blot assay
The protein samples from ventricular tissue were
separated by T-PER™ (Thermo Fisher Scientific) and concentration
was measured by the bicinchoninic acid (BCA) assay kit (Beyotime).
Next, an equal amount of protein (25 μg/well) from each
sample was separated by SDS-polyacrylamide gels (10%) and
transferred to a PVDF membrane. After blocking with 2% BSA
(Sigma-Aldrich, 1 h at room temperature) the membrane was incubated
overnight at 4°C with primary antibodies against anti-ox-CaMKII
(1:1,000, Millipore Sigma; cat. no. 07-1387), anti-CaMKII (1:000,
Abcam; cat. no. ab181052), anti-phosphorylated (p-) RyR2S-2814
(1:500, Badrilla; cat. no. A010-31AP), anti-RyR2 (1:1,000, Thermo
Fisher Scientific, Inc.; cat. no. C3-33) and anti-GAPDH (1:10,000,
Abcam; cat. no. ab181602), respectively. Then the membranes were
incubated with corresponding secondary antibodies horseradish
peroxidase-linked anti-rabbit Ig G (1:10,000, Abcam; cat. no.
ab6721) or anti-mouse IgG (1:5,000, Abcam; cat. no. ab205719) for
one hour at room temperature. Immunoblotting was imaged with a
ChemiDoc MP (Bio-Rad Laboratories, Inc.) with ECL Western Blotting
Substrate (Solarbio), and the blot densitometry was quantified
using the Image Lab™ (v3.0) software (Bio-Rad). Expression of
CaMKII-M282 oxidation and RyR2-S2814 phosphorylation was normalized
to total levels of their respective proteins.
Phosphoproteome analysis
The phosphoproteome was analyzed according to
previous studies (18,19). A total of 1 mg protein from each
left ventricular myocardium was treated with the filter-aided
sample preparation (FASP) method (18), followed by digestion with trypsin
(1:50, w/w; Promega Corporation) overnight at 37°C. The peptides
were then dried by vacuum centrifugation (1,000 g, −25°C, 3 h),
followed by phosphorylated peptide enrichment using the
High-Select™ TiO2 Phosphopeptide Enrichment kit (Thermo
Fisher Scientific, Inc.), according to the manufacturer's
instructions.
Tandem Mass Tag (TMT) six plex™ Label Reagent Set
(Thermo Fisher Scientific, Inc.) was used to label the peptides.
The mixture was subsequently desalted, concentrated, dried and
lyophilized using a desalting column (C18 Stage Tips, Thermo). To
increase phosphopeptide, fractionation of labeled peptides using
Pierce™ High pH Reversed-Phase Peptide Fractionation kit (Thermo
Fisher Scientific, Inc.) was performed before LC-tandem mass
spectrometry (LC-MS/MS) analysis. Acquisition was performed on a
Thermo Scientific™ Orbitrap Elite mass spectrometer (Thermo Fisher
Scientific, Inc.), coupled to an EASY-nLC™ 1000 nano flow liquid
chromatography (Thermo Fisher Scientific, Inc.) equipped with a 75
μm × 105 mm PicoCHIP nano spray column packed with
Reprosil-PUR C18-AQ, 3 μm, 120 Å (New Objective, Inc.). A
total of 2 μg desalted peptide from each fraction were
loaded onto PicoCHIP analytical column (New Objective) and the
peptides were separated using a 120-min elution gradient at 300
nl/min [5-9% mobile phase B (acetonitrile, 0.1% formic acid) for 2
min, 9-27% for 90 min, 27-40% for 13 min, 40-90% for 5 min and
90-100% mobile phase B for 10 min]. Three replicates were performed
for each group.
Electron Spray Ionization (ESI) voltage was set to
2.0 kV, capillary temperature was 280°C, nebuliser pressure (psi)
was set to 100-300 bar, and cation mode was selected for
acquisition. MS data (scan range 350-1,800 m/z) were acquired from
Fourier transform mass spectrometry (FTMS) with a resolution of
60,000 at 200 m/z. Automatic gain control target was set to
3×106, and maximum injection time was 200 msec. MS2 data
were acquired in FTMS mode with a resolution of 15,000 at 200 m/z.
The 15 strongest ions with charges 2-5 were continuously separated
in data dependent acquisition (DDA) mode for high-energy
collision-induced dissociation at 40% normalized collision energy
with a dynamic exclusion time set to 60 sec, excluding ions with
single and unidentified charge states. All measurements were
internally calibrated using the mass option. The data were analyzed
(Data S1) using MaxQuant
software (Max-Planck-Institute of Biochemistry, version 2.1.4.0,
maxquant.org/) (20) and MSstats R package (version
2.2.7) to perform quality inspection and statistical analysis
(21).
According to the criteria of P<0.05, fold-change
(FC) >1.2 for up- and <0.83 for downregulation, the
differentially expressed phosphorylated proteins (DEPPs) were
screened. Functional enrichment analysis was performed using
Metascape (https://metascape.org/, v3.5.20230501)
and the Database for Annotation, Visualization and Integrated
Discovery (DAVID, https://david.ncifcrf.gov/, Dec 2021) to determine
enriched Kyoto Encyclopedia of Genes and Genomes (KEGG, https://www.genome.jp/kegg/, Nov 2022) pathways, Gene
ontology (GO, geneontology.org/), biological process (BP), cellular
component (CC) and molecular function (MF) (22).
Statistical analysis
Data were analyzed by SPSS (IBM Corp.; version 24.0)
and are expressed as the mean ± SD. Data were analyzed using
one-way ANOVA followed by Dunnett's multiple comparisons post hoc
test. Survival was assessed by Kaplan-Meier curves and log-rank
test. Figures were constructed using GraphPad Prism (version 8.0;
GraphPad Software Inc.; Dotmatics) and Visio software (version
2013, Microsoft Corporation). P<0.05 was considered to indicate
a statistically significant difference.
Results
High incidence of LVA-SCD occurs in early
MI
Mice with successful CAL displayed T wave elevation,
verifying induction of MI. Wild-type mice subjected to MI without
MitoTEMPO treatment or S2814A mutation had a high incidence
(~33.9%) of LVA-SCD within 30 min post-MI. SCD mice displayed
notable T wave variation and prolonged QT, which culminated in
lethal ventricular bradycardia. The surviving mice also showed
prolonged QT but to a lesser extent (Fig. 1B). MitoTEMPO treatment prevented
the ECG phenotype relevant to LVA and decreased SCD incidence
(14.8%). S2814A mutation was successfully constructed in C57/BL6
mice and confirmed by DNA sequencing (Fig. S1). Mutation reduced SCD
incidence as well (15.6%; Fig.
1; Table SI).
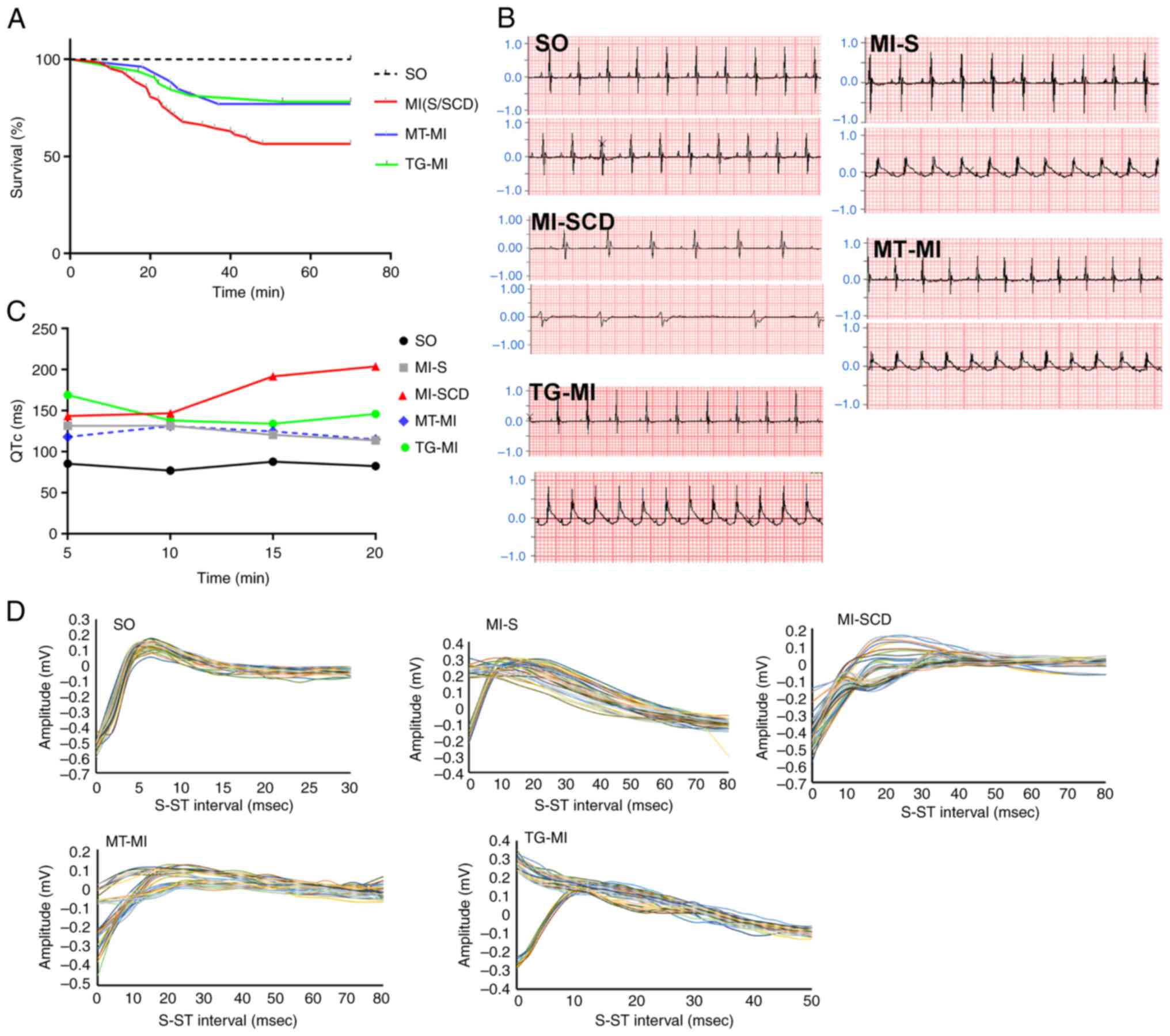 | Figure 1High incidence of LVA-SCD in early MI
is prevented by MitoTEMPO treatment or S2814A mutation. (A)
Kaplan-Meier curve (number of SCD cases to that of total cases:
SO=0/32, MI-S/SCD=21/62, MT-MI=4/26, TG-MI=5/32), illustrating a
high incidence of LVA-SCD in early MI and MitoTEMPO and S2814A
mutation could reduce propensity for LVA-SCD. (B) Lead II ECG.
Upper and lower traces show the ECGs before and after MI,
respectively. (C) QTc. (D) T wave variability. LVA, lethal
ventricular arrhythmia; SCD, sudden cardiac death; MI, myocardial
ischemia; SO, sham operation; MT, MitoTEMPO; TG, transgenic; ECG,
electrocardiogram; QTc, corrected QT. |
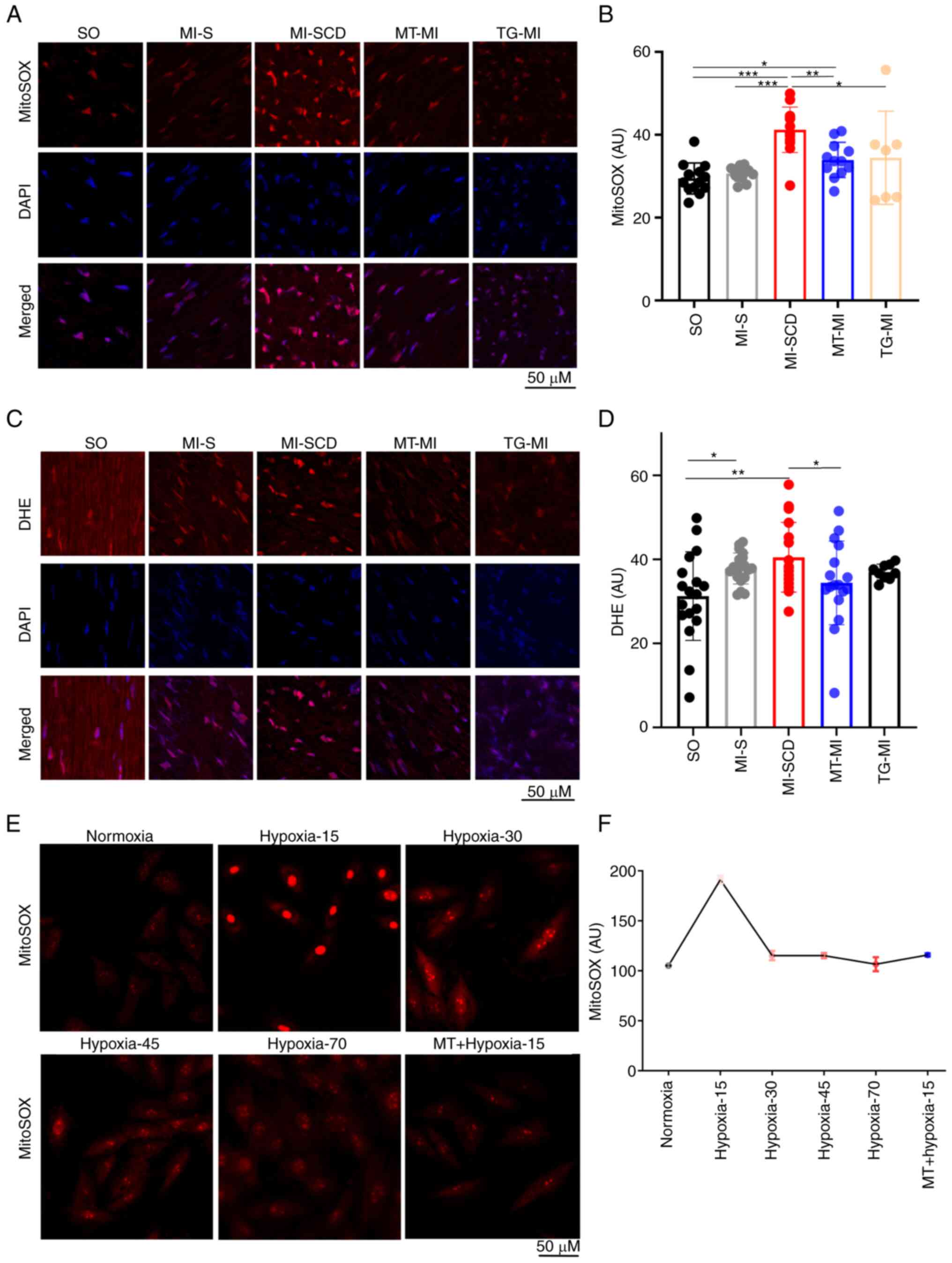
mROS burst serves a key role in the
development of LVA-SCD
LVA-SCD mice exhibited a notable increase in
myocardial mROS, producing more mROS than those undergoing longer
ischemia (Fig. 2A and B),
suggestive of an early burst of mROS following ischemia. To
validate this burst, mROS levels were measured in H9c2 cells
subjected to hypoxia (1% O2); hypoxia triggered mROS
burst at ~15 min post-hypoxia; this was suppressed by MitoTEMPO
(Fig. 2E and F). Therefore, the
time of the high SCD incidence overlapped with the peak mROS
emission after MI.
Consistently, SCD mice also demonstrated higher
levels of cytosolic ROS (cytoROS), as reflected by DHE intensity
(Fig. 2C and D). These mice had
elevated CaMKII-M282 oxidation and RyR2-S2814 phosphorylation
(Figs. 3A-C and S2; Tables SII and SIII), overloads of
cytoCa2+ and mitochondrial Ca2+
(mitoCa2+) and lower MMP and GSH/GSSG ratio in the
myocardium (Fig. 3D-I).
Consistent with in vivo mROS results, these changes were
greater in SCD mice than mice survived after MI, indicating that
the SCD-induced alterations occurred in a short time.
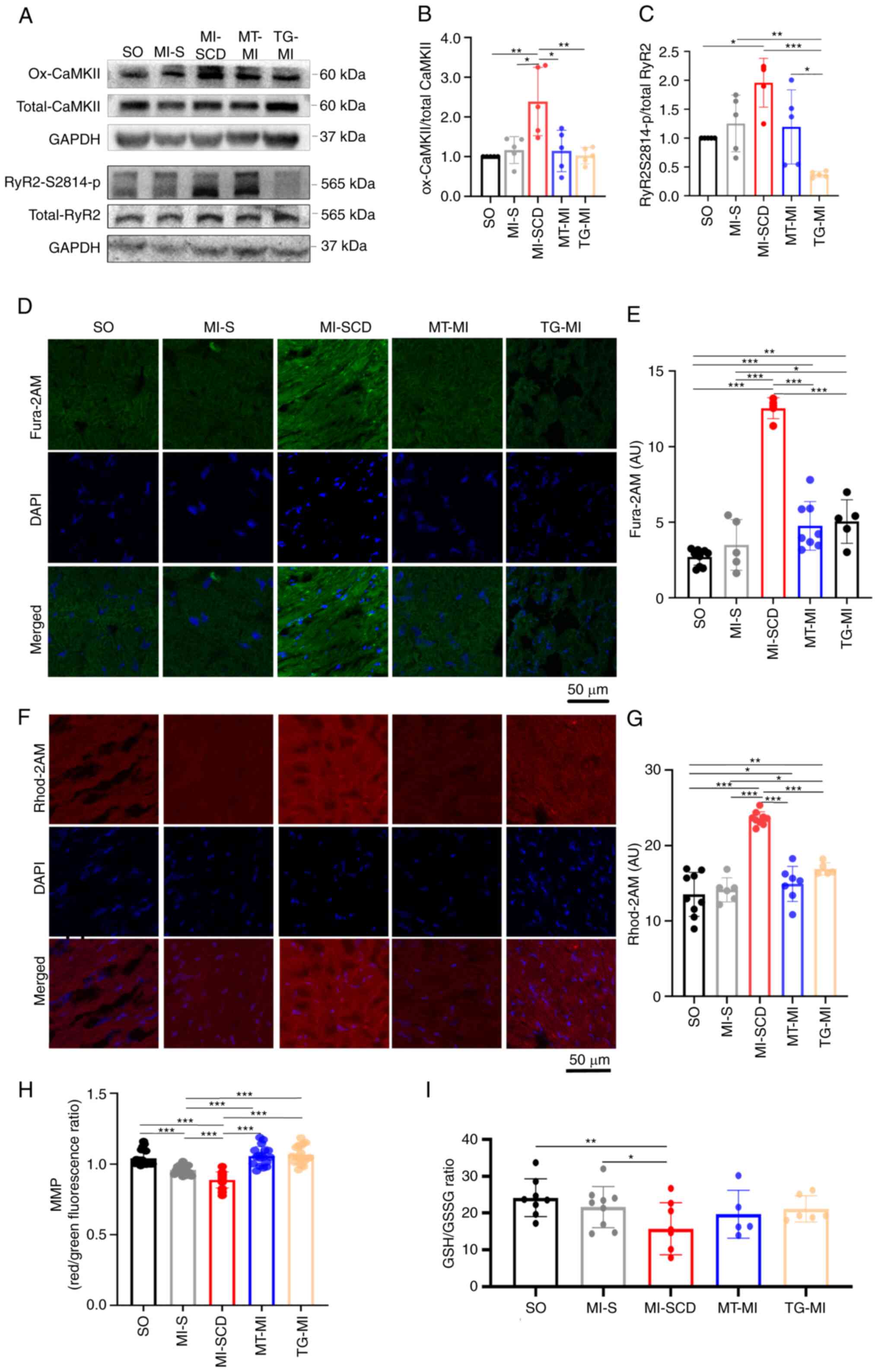 | Figure 3Increased CaMKII-M282 oxidation and
RyR2-S2814 phosphorylation cause cytoCa2+ and
mitoCa2+ overload in SCD mice. (A) Representative
western blot; (B) densitometry ratio of CaMKII-M282 oxidation to
total CaMKII and (C) densitometry ratio of RyR2-S2814
phosphorylation to total RyR2 (n=4-6). (D) Representative images of
Fura 2-AM in frozen heart sections; (E) Fura 2-AM intensity in
different groups (n=5-8). (F) Representative images of Rhod 2-AM in
frozen heart sections; (G) Rhod 2-AM intensity in different groups
(n=5-9); (H) MMP (14-18 fluorescence images from eight hearts in
each group; (I) GSH/GSSG ratio (n=5-9). *P<0.05,
**P<0.01, ***P<0.001. Ox-CaMKII,
oxidized Ca2+/calmodulin-dependent protein kinase;
p-RyR2-S2814, phosphorylation of ryanodine receptor 2 at Ser2814;
cyto, cytoplasm; mito, mitochondria; MMP, mitochondrial membrane
potential; GSH, reduced glutathione; GSSG, oxidized glutathione;
SCD, sudden cardiac death; MI, myocardial ischemia; SO, sham
operation; MT, MitoTEMPO; TG, transgenic; AU, arbitrary unit. |
MitoTEMPO and S2814A mutation decrease
the incidence of LVA-SCD in early MI
Following pretreatment with MitoTEMPO, most mice
survived 30 min post-MI (85.2%; Fig.
1A). MitoTEMPO inhibited the mROS burst and controlled the
cytoROS production (Fig. 2C and
D). As a consequence, MitoTEMPO reduced CaMKII-M282 oxidation
and subsequently decreased RyR2-S2814 phosphorylation (Figs. 3A-C and S2; Tables SII and SIII), preventing
overloads of myocardial cytoCa2+ and mitoCa2+
(Fig. 3D-G). Moreover, MitoTEMPO
restored MMP, GSH/GSSG ratio and ECG alterations such as prolonged
QTc and T wave variation (Figs.
1B-D and 3H and I).
MitoTEMPO effectively suppressed mROS emission, mitigated
Ca2+ imbalance and suppressed LVA-SCD in early MI.
Similarly, most S2814A mice also survived early MI
(84.4%; Fig. 1A), implying that
they were protected against LVA-SCD. Of note, S2814A mutation
prevented RyR2-S2814 phosphorylation despite upregulation of its
upstream kinase ox-CaMKII in early MI (Fig. 3A-C). As a result, the mutation
alleviated Ca2+ overload both in the cytoplasm and
mitochondria (Fig. 3D-G).
Subsequently, S2814A mutation decreased mROS release and
CaMKII-M282 oxidation (Figs.
2A-D and 3A-C). As seen with
MitoTEMPO treatment, S2814A mutation partly ameliorated
mitochondrial function and restored antioxidative capacity
(Figs. 1D and 3H and I). These data revealed that
S2814A mutation effectively corrected Ca2+ imbalance,
which decreased mROS production, thereby inhibiting LVA-SCD.
Myocardial phosphoproteome remodeling in
SCD is prevented by MitoTEMPO and S2814A mutation
Changes in the myocardial phosphoproteome were
assessed; 4,108 phosphorylated peptides were identified in 1,028
proteins. Phosphorylation of 583 proteins was significantly changed
in SCD mice; among these, phosphorylation was up- and downregulated
in 175 and 408 proteins, respectively, compared with the SO
controls. MI-S, MT-MI, and TG-MI groups) exhibited fewer changes
than SCD mice (Fig. 4B; Table SIV and V).
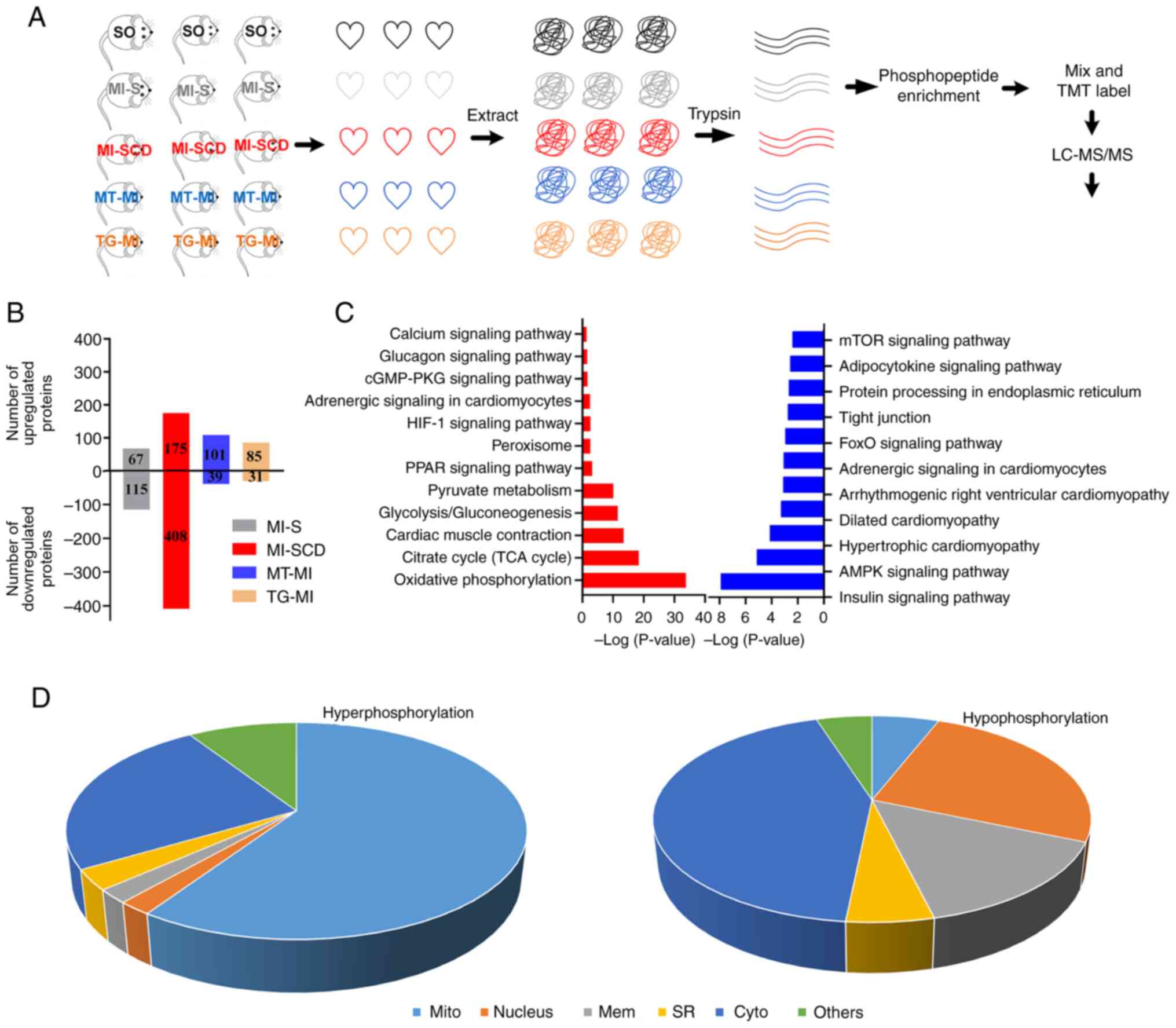 | Figure 4Myocardial phosphoproteome
alterations in SCD and surviving mice. (A) Design of
phosphoproteome experiments. (B) SCD mice exhibited more
alterations in protein phosphorylation than the surviving mice. (C)
Representative enriched Kyoto Encyclopedia of Genes and Genomes
pathways in the SCD mice. Red, upregulation; blue, downregulation.
(D) Cellular components of hyperphosphorylated proteins in SCD mice
were primarily enriched in the mitochondria, whereas those of
hypophosphorylated proteins were primarily enriched in the
cytoplasm. SCD, sudden cardiac death; SO, sham operation; MI,
myocardial ischemia; S, stable; MT, MitoTEMPO; TG, transgenic; TMT,
Tandem Mass Tag; LC-MS, Liquid Chromatography-Mass Spectrometry;
mito, mitochondria; mem, membrane; SR, sarcoplasmic reticulum;
cyto, cytoplasm. |
KEGG enrichment analysis of hyperphosphorylated
proteins in LVA-SCD revealed enrichment in 'HIF-1 signaling
pathway', citrate cycle (TCA cycle), 'Oxidative phosphorylation',
'Calcium signaling pathway', 'cGMP-PKG signaling pathway' and
'Glycolysis/gluconeogenesis' (Fig.
4C; Table SVI). Similarly,
BPs were primarily enriched in 'mitochondrial ATP synthesis coupled
proton transport', 'canonical glycolysis, 'NADH metabolic process',
and mitochondrial electron transport', 'ubiquinol to cytochrome c'
(Table SVII). Consistently, CCs
were primarily enriched in the mitochondria (Fig. 4D; Table SVIII). These results suggested
that hyperphosphorylation changes primarily occurred in the
mitochondria. MitoTEMPO and S2814A prevented most
hyperphosphorylation (Tables SIV
and SV), implying that changes in the mitochondrial
phosphoproteome are primarily mROS- and Ca2+-dependent
and may be a key influence on the formation of the
mROS-Ca2+ loop.
Enrichment analysis of hypophosphorylated proteins
revealed enrichment of signaling pathways, which primarily included
'insulin signaling pathway', 'spliceosome', 'AMPK signaling
pathway', 'FoxO signaling pathway', 'tight junction', 'protein
processing in endoplasmic reticulum', 'adipocytokine signaling
pathway' and 'mTOR signaling pathway' (Fig. 4C; Tables SIX and SX). CCs were mainly
enriched in the cytoplasm, different from those of
hyperphosphorylated proteins (Fig.
4D; Table SXI).
Discussion
The most important finding of the present study was
the high incidence of LVA-SCD in early MI. Since mice that had a
massive hemorrhage, respiratory failure and anesthesia allergy were
excluded, deaths occurring in the early MI resulted from lethal
bradycardia (SCD). From the electrophysiological perspective, the
SCD mice were characterized by lethal bradycardia and increased T
wave variation, which is associated with increased likelihood of
lethal arrhythmias and SCD (23). Collectively, the present study
confirmed that the early period of MI was the most common period
for LVA-SCD, similar to a previous study (4).
The present study used hypoxic H9c2 cells to mimic
ischemia. H9c2 cells are rat embryonic cardiomyocytes that share
many features with primary mouse cardiomyocytes, especially in
terms of energy metabolism patterns (such as cellular ATP levels,
bioenergetics and mitochondrial function). H9c2 cells are more
sensitive to hypoxic injury (24). Thus, using H9c2 cells were used
to confirm hypoxia-related mROS dynamic changes. In response to
hypoxia, H9c2 cells experienced an early mROS burst. Such an mROS
burst has been previously demonstrated (25). Hypoxia leads to mitochondrial
electron transfer chain (ETC) dysfunction, with the release of a
large amount of superoxide (O2·-) from
complexes III and I in the ETC (26). This accumulation sensitizes the
mitochondrial inner membrane anion channel (IMAC), promoting
release of O2·- into the cytoplasm in the
form of hydrogen peroxide (27).
This accounts for elevated cytoROS levels in SCD mice. Higher
cytoROS levels further activate the IMAC in adjacent mitochondria,
which leads to ROS-induced ROS release (RIRR) in mitochondria
(28), resulting in mROS burst.
Consistently, the SCD mice also demonstrated poor antioxidative
capacity and mitochondrial dysfunction, as reflected by lowered
GSH/GSSG ratio and MMP, both of which may facilitate the mROS burst
(29,30). Another explanation for the mROS
burst is that the compensatory mechanism, such as increasing
expression of antioxidant proteins, has not been set up to cope
with extensive oxidative stress upon early MI, hence allowing RIRR
(31). However, with increased
duration of ischemia, myocytes establish a compensatory mechanism
to reduce excessive mROS (32),
as indicated by lowered GSH/GSSG ratio and restoration of MMP
almost to normal levels in surviving mice.
Expectedly, MitoTEMPO, a combination of the
antioxidant TEMPO (2,2,6,6-tetramethylpiperidinyl-1-oxyl) and
lipophilic cation triphenyl phosphonium that is capable of targeted
removal of mROS (33), corrected
SCD-associated alterations induced by mROS, demonstrating the
effects of mROS burst on LVA-SCD development. Therefore, the effect
of mROS-Ca2+ interaction in LVA-SCD was assessed using
S2814A mice. Apart from the expected findings that the transgenic
mice exhibited decreased RyR2(Ser2814) phosphorylation and
Ca2+ content in the cytoplasm and mitochondria, the
mutation decreased the levels of mROS, which indicated that the
early mROS burst was Ca2+-dependent. MitoCa2+
overload activates protein kinase C (PKC) and subsequently increase
mROS (34). PKC enhances
phosphorylation of ETC proteins, which facilitates mROS production
(35). On the other hand,
excessive mROS lead to diastolic Ca2+ leak and
Ca2+ overload via the mROS-oxidized
CaMKII(M281)/phosphorylated RyR2(S2814) pathway. ROS directly
oxidize RyR2 and cause Ca2+ leakage. However, under
oxidative stress, increased RyR2 phosphorylation occurs earlier
than RyR2 oxidation (36).
Therefore, the activation of the above pathway is a primary
mechanism leading to Ca2+ imbalance. Altogether,
Ca2+ imbalance and mROS may form an mROS-Ca2+
loop that promotes LVA-SCD in early MI (Fig. 5). MitoTEMPO and S2814A mutation
block this loop and effectively curb LVA-SCD.
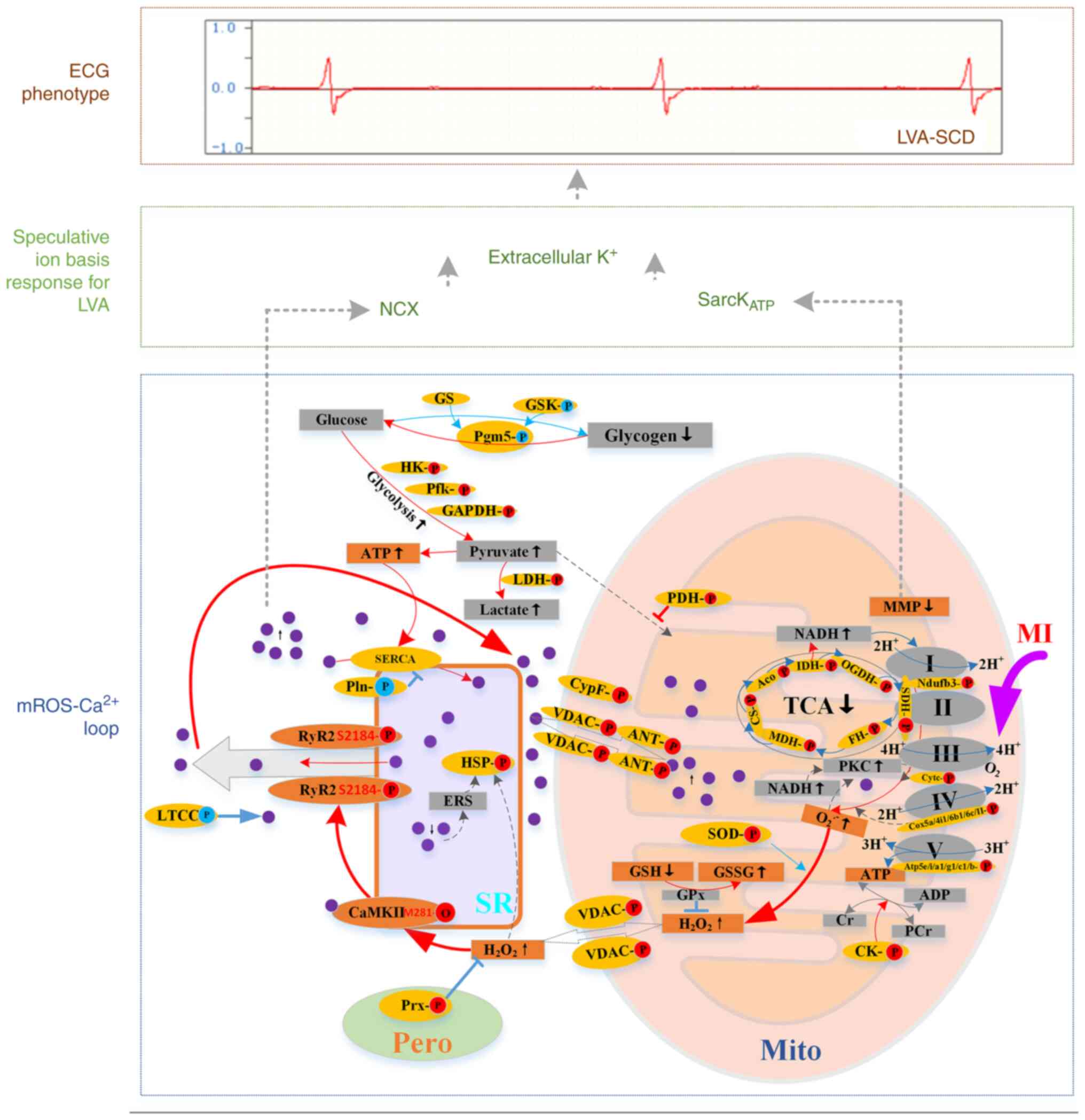 | Figure 5Scheme depicting the crosstalk of
mROS/ROS and Ca2+ signaling pathways and proposed
relevant ion bases that promote LVA-SCD. Red, upregulation; blue,
downregulation; purple circle, calcium ion. Mito, mitochondria; SR,
sarcoplasmic reticulum; I-V, Complex I-V; Cyt c, cytochrome C; COX,
cytochrome oxidase; Atp5, ATP synthase; SOD, superoxide dismutase;
VDAC, voltage-dependent anion channel; Cyp F, cyclophilin
F/peptidylprolyl isomerase F; ANT, adenine nucleotide translocase;
MDH, malate dehydrogenase; CS, citrate synthase; IDH, isocitrate
dehydrogenase; SDH, succinate dehydrogenase; OGDH, oxoglutarate
dehydrogenase; FH, fumarate hydratase; ERS, endoplasmic reticulum
stress; HSP, heat shock protein; HK, hexokinase; Pfk,
phosphofructokinase; PDH, pyruvate dehydrogenase; PKC, protein
kinase C; SERCA, sarco/endoplasmic reticulum Ca2+
ATPase; Pln, phospholamban; LTCC, L-type calcium channel; NCX,
Na+/Ca2+ exchanger; sarcKATP,
sarcolemmal ATP-sensitive K+ channel; ECG,
electrocardiogram; mROS, mitochondrial ROS; MI, myocardial
ischemia; TCA, tricarboxylic acid; Aco, aconitase; pero,
peroxisome; Mito, mitochondria; PCr, Phosphocreatine; CK, creatine
kinase; LVA-SCD, lethal ventricular arrhythmia-sudden cardia
death. |
Phosphorylation is a key post-translational
mechanism that participates in rapid pathophysiological processes
(37). Following a short period
of MI, the SCD mice exhibited notable phosphoproteome changes.
Surviving mice that experienced longer ischemia had fewer changes
in phosphoprotein levels. Notably, most hyperphosphorylated
proteins were localized in the mitochondria and primarily involved
in redox homeostasis, ion transport, mitochondrial electron
transport and energy balance. Elevated Ca2+ and ROS
activate PKC, which may be responsible for hyperphosphorylation
(34). ETC-localized proteins
were hyperphosphorylated in SCD, which decreases ETC function,
thereby promoting O2- burst (38,39). Meanwhile, enzymes involved in the
TCA cycle were hyperphosphorylated, which has been shown to inhibit
the TCA cycle (40).
Furthermore, voltage-dependent anion channel (VDAC), peptidylprolyl
isomerase F (cyclophilin F) and adenine nucleotide translocase A4/5
(ANT) were hyperphosphorylated in LVA-SCD. VDAC
hyperphosphorylation favors its closure, increasing Ca2+
influx into mitochondria (41).
Phosphorylation of cyclophilin F and ANT facilitate mPTP opening
(41,42). These findings explain
MitoCa2+ concomitant with cytoCa2+ overload
in mice that underwent SCD. In addition, superoxide mutase (SOD)
was hyperphosphorylated, which inactivates its function, thereby
allowing local O2-. Accumulation (43). Therefore, hyperphosphorylation of
mitochondrial protein was mainly related to the aggravation of
mitochondrial dysfunction and subsequent induction of the
mROS/Ca2+ cycle. Additionally glycolysis-associated
enzymes were hyperphosphorylated in LVA-SCD, which may activate
these enzymes and enhance glycolysis (44,45). SCD mice exhibited higher levels
of mROS as well as significant mitochondrial protein
phosphorylation alteration; these phosphorylation alterations were
prevented by MitoTEMPO, and S2814A mutation, which decreased mROS
levels in MI mice, could also normalize the phosphorylated
disturbance; therefore, it was hypothesized that most
mitochondria-associated phosphorylated alterations were associated
with the mROS/Ca2+ loop.
Upon formation of the mROS/Ca2+ loop,
several factors contribute to the occurrence of lethal ventricular
bradycardia. First, excessive mROS cause mitochondrial metabolic
sink, a state that cardiomyocytes are rendered inexcitable because
of the large background K+ (46). Second, cytoplasmic
Ca2+ overload leads to cytoplasmic Na+
overload by activating the Na+/Ca2+
exchanger, which further facilitates net intracellular net
K+ loss because of the need to maintain
electroneutrality and osmotic balance (47). Additionally, enhanced glycolysis
increases intracellular H+, which activates
Na+/H+ exchanger (48). All these alterations result in
extracellular K+ accumulation and decrease electrical
conduction within the ventricle, as displayed in ECG in
LVA-SCD.
A limitation of the present study is that
Ca2+ concentration was not detected dynamically. Here,
frozen sections were made immediately after death when notable cell
membrane damage did not occur. Meanwhile, some free Ca2+
may be released to the extracellular compartment during slice
preparation, which may cause signals from extracellular areas.
Since contraction requires excitation, the myocardium was
maintained in diastole after death because the excitation stopped.
Therefore, myocardial Ca2+ content represents
Ca2+ content in diastole.
In conclusion, early post-MI is the most common
stage for SCD. The formation of an mROS/Ca2+ loop plays
a critical role in promoting LVA-SCD. Mitochondrial proteomic
hyperphosphorylation may facilitate formation of this loop. Both
MitoTEMPO treatment and S2814A mutation prevent SCD-associated
changes and LVA-SCD. The present data imply an association between
mROS and Ca2+ imbalance, which warns that we need to
prevent the formation of the mROS/Ca2+ loop to
effectively control early SCD post-MI.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
DZ, YeZ, MZhu, XiaojuaZ, XiaojunZ, WT, QW, YL, LT,
ZZ, JL, YaZ and DW performed the experiments. DW designed the
study. DW and DZ wrote the manuscript. DZ, YeZ, MZhu, ML, DW and
MZha analyzed the data. All authors have read and approved the
final manuscript. DW and DZ confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
The study was approved by the Medical Animal Care
and Welfare Committee at Shantou University Medical College
(approval no. SUMC2020-035).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
CAL
|
coronary artery ligation
|
|
ETC
|
electron transport chain
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
LVA
|
lethal ventricular arrhythmia
|
|
MAM
|
mitochondria-associated sarcoplasmic
reticulum membrane
|
|
MI
|
myocardial ischemia
|
|
mROS
|
mitochondrial reactive oxygen
species
|
|
MMP
|
mitochondrial membrane potential
|
|
ox-CaMKII
|
oxidized
Ca2+/calmodulin-dependent protein kinase
|
|
RIRR
|
ROS-induced ROS release
|
|
RyR2
|
ryanodine receptor 2
|
|
SCD
|
sudden cardiac death
|
|
TCA
|
tricarboxylic acid cycle
|
Acknowledgments
The authors would like to thank Dr Ricardo Carnicer
Hijazo (University of Oxford, Oxford, UK) and Professor Stanley Lin
(SUMC, Shantou, China) for editing the manuscript.
Funding
The present study was supported by the Guangdong Province
General University Characteristic Innovation Project (grant no.
2021KTSCX032), Guangdong Natural Science Foundation (grant no.
2022A1515011119) and Innovative Team Research Program for
Universities of Guangdong Province (grant no. 2022KCXTD009).
References
|
1
|
World Health Organization (WHO): The top
10 causes of death. WHO; Geneva: 2020, https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death.
|
|
2
|
Chugh SS, Reinier K, Teodorescu C, Evanado
A, Kehr E, Al Samara M, Mariani R, Gunson K and Jui J: Epidemiology
of sudden cardiac death: Clinical and research implications. Prog
Cardiovasc Dis. 51:213–228. 2008.
|
|
3
|
Al-Khatib SM, Stevenson WG, Ackerman MJ,
Bryant WJ, Callans DJ, Curtis AB, Deal BJ, Dickfeld T, Field ME,
Fonarow GC, et al: 2017 AHA/ACC/HRS Guideline for Management of
Patients With Ventricular Arrhythmias and the Prevention of Sudden
Cardiac Death: Executive Summary: A Report of the American College
of Cardiology/American Heart Association Task Force on Clinical
Practice Guidelines and the Heart Rhythm Society. Circulation.
138:e210–e271. 2018.
|
|
4
|
Zaman S and Kovoor P: Sudden cardiac death
early after myocardial infarction: Pathogenesis, risk
stratification, and primary prevention. Circulation. 129:2426–2435.
2014.
|
|
5
|
Reed GW, Rossi JE and Cannon CP: Acute
myocardial infarction. Lancet. 389:197–210. 2017.
|
|
6
|
Jeong EM, Liu M, Sturdy M, Gao G, Varghese
ST, Sovari AA and Dudley SC Jr: Metabolic stress, reactive oxygen
species, and arrhythmia. J Mol Cell Cardiol. 52:454–463. 2012.
|
|
7
|
Csordas G, Weaver D and Hajnoczky G:
Endoplasmic Reticulum-Mitochondrial Contactology: Structure and
signaling functions. Trends Cell Biol. 28:523–540. 2018.
|
|
8
|
Wang JJ, Park KS, Dhimal N, Shen S, Tang
X, Qu J and Zhang SX: Proteomic analysis of retinal
mitochondria-associated ER membranes identified novel proteins of
retinal degeneration in long-term diabetes. Cells. 11:28192022.
|
|
9
|
Liu X, Wang S, Guo X, Li Y, Ogurlu R, Lu
F, Prondzynski M, de la Serna Buzon S, Ma Q, Zhang D, et al:
Increased Reactive Oxygen Species-Mediated
Ca(2+)/calmodulin-dependent protein kinase II activation
contributes to calcium handling abnormalities and impaired
contraction in barth syndrome. Circulation. 143:1894–1911.
2021.
|
|
10
|
Bertero E and Maack C: Calcium signaling
and reactive oxygen species in mitochondria. Circ Res.
122:1460–1478. 2018.
|
|
11
|
Purohit A, Rokita AG, Guan X, Chen B,
Koval OM, Voigt N, Neef S, Sowa T, Gao Z, Luczak ED, et al:
Oxidized Ca(2+)/calmodulin-dependent protein kinase II triggers
atrial fibrillation. Circulation. 128:1748–1757. 2013.
|
|
12
|
van Oort RJ, McCauley MD, Dixit SS,
Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG,
Anderson ME, et al: Ryanodine receptor phosphorylation by
calcium/calmodulin-dependent protein kinase II promotes
life-threatening ventricular arrhythmias in mice with heart
failure. Circulation. 122:2669–2679. 2010.
|
|
13
|
National Research Council of The National
Academies: Guide for the Care and Use of Laboratory Animals. 8th.
National Academy Press; Washington, DC: 2011
|
|
14
|
Albert CM, McGovern BA, Newell JB and
Ruskin JN: Sex differences in cardiac arrest survivors.
Circulation. 93:1170–1176. 1996.
|
|
15
|
Jasmin M, Ahn EH, Voutilainen MH, Fombonne
J, Guix C, Viljakainen T, Kang SS, Yu LY, Saarma M, Mehlen P and Ye
K: Netrin-1 and its receptor DCC modulate survival and death of
dopamine neurons and Parkinson's disease features. EMBO J.
40:e1055372021.
|
|
16
|
Koshy AN, Gow PJ, Testro A, Th AW, Ko J,
Lim HS, Han HC, Weinberg L, VanWagner LB and Farouque O:
Relationship between QT interval prolongation and structural
abnormalities in cirrhotic cardiomyopathy: A change in the current
paradigm. Am J Transplant. 21:2240–2245. 2021.
|
|
17
|
Dey S, DeMazumder D, Sidor A, Foster DB
and O'Rourke B: Mitochondrial ROS drive sudden cardiac death and
chronic proteome remodeling in heart failure. Circ Res.
123:356–371. 2018.
|
|
18
|
Xie D, Wu J, Wu Q, Zhang X, Zhou D, Dai W,
Zhu M and Wang D: Integrating proteomic, lipidomic and metabolomic
data to construct a global metabolic network of lethal ventricular
tachyarrhythmias (LVTA) induced by aconitine. J Proteomics.
232:1040432021.
|
|
19
|
Friedrich C, Schallenberg S, Kirchner M,
Ziehm M, Niquet S, Haji M, Beier C, Neudecker J, Klauschen F and
Mertins P: Comprehensive micro-scaled proteome and phosphoproteome
characterization of archived retrospective cancer repositories. Nat
Commun. 12:35762021.
|
|
20
|
Cox J and Mann M: MaxQuant enables high
peptide identification rates, individualized p.p.b.-range mass
accuracies and proteome-wide protein quantification. Nat
Biotechnol. 26:1367–1372. 2008.
|
|
21
|
Choi M, Chang CY, Clough T, Broudy D,
Killeen T, MacLean B and Vitek O: MSstats: An R package for
statistical analysis of quantitative mass spectrometry-based
proteomic experiments. Bioinformatics. 30:2524–2526. 2014.
|
|
22
|
Shang L, Wang Y, Li J, Zhou F, Xiao K, Liu
Y, Zhang M, Wang S and Yang S: Mechanism of Sijunzi Decoction in
the treatment of colorectal cancer based on network pharmacology
and experimental validation. J Ethnopharmacol. 302(Pt A):
1158762023.
|
|
23
|
Ramirez J, Orini M, Minchole A, Monasterio
V, Cygankiewicz I, Bayés de Luna A, Martínez JP, Pueyo E and Laguna
P: T-Wave morphology restitution predicts sudden cardiac death in
patients with chronic heart failure. J Am Heart Assoc.
6:e0053102017.
|
|
24
|
Kuznetsov AV, Javadov S, Sickinger S,
Frotschnig S and Grimm M: H9c2 and HL-1 cells demonstrate distinct
features of energy metabolism, mitochondrial function and
sensitivity to hypoxia-reoxygenation. Biochim Biophys Acta.
1853:276–284. 2015.
|
|
25
|
Hernansanz-Agustin P, Choya-Foces C,
Carregal-Romero S, Ramos E, Oliva T, Villa-Piña T, Moreno L,
Izquierdo-Álvarez A, Cabrera-García JD, Cortés A, et al: Na(+)
controls hypoxic signalling by the mitochondrial respiratory chain.
Nature. 586:287–291. 2020.
|
|
26
|
Yang KC, Kyle JW, Makielski JC and Dudley
SC Jr: Mechanisms of sudden cardiac death: Oxidants and metabolism.
Circ Res. 116:1937–1955. 2015.
|
|
27
|
Akar FG, Aon MA, Tomaselli GF and O'Rourke
B: The mitochondrial origin of postischemic arrhythmias. J Clin
Invest. 115:3527–3535. 2005.
|
|
28
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014.
|
|
29
|
Schafer FQ and Buettner GR: Redox
environment of the cell as viewed through the redox state of the
glutathione disulfide/glutathione couple. Free Radic Biol Med.
30:1191–1212. 2001.
|
|
30
|
Bagkos G, Koufopoulos K and Piperi C: A
new model for mitochondrial membrane potential production and
storage. Med Hypotheses. 83:175–181. 2014.
|
|
31
|
Crewe C, Funcke JB, Li S, Joffin N,
Gliniak CM, Ghaben AL, An YA, Sadek HA, Gordillo R, Akgul Y, et al:
Extracellular vesicle-based interorgan transport of mitochondria
from energetically stressed adipocytes. Cell Metab. 33:1853–1868
e11. 2021.
|
|
32
|
Ferko M, Andelova N, Szeiffova Bacova B
and Jasova M: Myocardial adaptation in pseudohypoxia: Signaling and
Regulation of mPTP via mitochondrial connexin 43 and cardiolipin.
Cells. 8:14492019.
|
|
33
|
Shetty S, Kumar R and Bharati S:
Mito-TEMPO, a mitochondria-targeted antioxidant, prevents
N-nitrosodiethylamine-induced hepatocarcinogenesis in mice. Free
Radic Biol Med. 136:76–86. 2019.
|
|
34
|
Hegyi B, Borst JM, Bailey LRJ, Shen EY,
Lucena AJ, Navedo MF, Bossuyt J and Bers DM: Hyperglycemia
regulates cardiac K(+) channels via O-GlcNAc-CaMKII and
NOX2-ROS-PKC pathways. Basic Res Cardiol. 115:712020.
|
|
35
|
Rebollo-Hernanz M, Zhang Q, Aguilera Y,
Martin-Cabrejas MA and Gonzalez de Mejia E: Relationship of the
phytochemicals from coffee and cocoa by-products with their
potential to modulate biomarkers of metabolic syndrome in vitro.
Antioxidants (Basel). 8:2792019.
|
|
36
|
Belevych AE, Terentyev D, Terentyeva R,
Nishijima Y, Sridhar A, Hamlin RL, Carnes CA and Györke S: The
relationship between arrhythmogenesis and impaired contractility in
heart failure: Role of altered ryanodine receptor function.
Cardiovasc Res. 90:493–502. 2011.
|
|
37
|
Humphrey SJ, James DE and Mann M: Protein
Phosphorylation: A major switch mechanism for metabolic regulation.
Trends Endocrinol Metab. 26:676–687. 2015.
|
|
38
|
Kalpage HA, Wan J, Morse PT, Zurek MP,
Turner AA, Khobeir A, Yazdi N, Hakim L, Liu J, Vaishnav A, et al:
Cytochrome c phosphorylation: Control of mitochondrial electron
transport chain flux and apoptosis. Int J Biochem Cell Biol.
121:1057042020.
|
|
39
|
Kane LA, Youngman MJ, Jensen RE and Van
Eyk JE: Phosphorylation of the F(1)F(o) ATP synthase beta subunit:
Functional and structural consequences assessed in a model system.
Circ Res. 106:504–513. 2010.
|
|
40
|
Guo X, Niemi NM, Hutchins PD, Condon SGF,
Jochem A, Ulbrich A, Higbee AJ, Russell JD, Senes A, Coon JJ and
Pagliarini DJ: Ptc7p dephosphorylates select mitochondrial proteins
to enhance metabolic function. Cell Rep. 18:307–313. 2017.
|
|
41
|
Wang Z, Ge Y, Bao H, Dworkin L, Peng A and
Gong R: Redox-sensitive glycogen synthase kinase 3β-directed
control of mitochondrial permeability transition: Rheostatic
regulation of acute kidney injury. Free Radic Biol Med. 65:849–858.
2013.
|
|
42
|
Feng J, Zhu M, Schaub MC, Gehrig P,
Roschitzki B, Lucchinetti E and Zaugg M: Phosphoproteome analysis
of isoflurane-protected heart mitochondria: Phosphorylation of
adenine nucleotide translocator-1 on Tyr194 regulates mitochondrial
function. Cardiovasc Res. 80:20–29. 2008.
|
|
43
|
Banks CJ and Andersen JL: Mechanisms of
SOD1 regulation by post-translational modifications. Redox Biol.
26:1012702019.
|
|
44
|
Dieni CA and Storey KB: Regulation of
hexokinase by reversible phosphorylation in skeletal muscle of a
freeze-tolerant frog. Comp Biochem Physiol B Biochem Mol Biol.
159:236–243. 2011.
|
|
45
|
Lee JH, Liu R, Li J, Wang Y, Tan L, Li XJ,
Qian X, Zhang C, Xia Y, Xu D, et al: EGFR-Phosphorylated platelet
isoform of phosphofructokinase 1 promotes PI3K activation. Mol
Cell. 70:197–210 e7. 2018.
|
|
46
|
Akar FG and O'Rourke B: Mitochondria are
sources of metabolic sink and arrhythmias. Pharmacol Ther.
131:287–294. 2011.
|
|
47
|
Landstrom AP, Dobrev D and Wehrens XHT:
Calcium signaling and cardiac arrhythmias. Circ Res. 120:1969–1993.
2017.
|
|
48
|
Barth AS and Tomaselli GF: Cardiac
metabolism and arrhythmias. Circ Arrhythm Electrophysiol.
2:327–335. 2009.
|