Introduction
Heart failure (HF) is an end-stage cardiovascular
disease, which has become a notable public health burden due to its
high morbidity and mortality rates (1,2).
Pathological cardiac hypertrophy is an early characteristic of HF,
which manifests as the enlargement of cardiomyocytes, increased
protein synthesis, reactivation of fetal genes and cardiac fibrosis
(3). Notably, there is a lack of
effective treatments for the management of pathological cardiac
hypertrophy in clinical practice (4). Investigating the molecular
mechanisms involved in the progression of cardiac hypertrophy may
provide novel therapeutic targets for treating HF.
Tripartite motif (Trim) proteins are a group of
proteins belonging to the E3 ubiquitin ligase family; these
proteins are involved in the post-translational modification of
proteins by catalyzing the binding of ubiquitin to its substrate
(5). Trim38 is a prototypical
Trim protein that is widely expressed in various cells, which
contains a RING domain, two B-box domains, a coiled-coil domain and
a PRY-SPRY domain, and can be activated by type I IFNs and viral
infection (6). Previous studies
have shown that Trim38 serves critical regulatory roles in innate
immune and inflammatory pathways, such as the Toll-like receptor
signaling pathway, GAS/STING pathway, and MDA5- and RIG-I mediated
antiviral response (7,8). Recently, it has been reported that
Trim38 can attenuate cardiac fibrosis after myocardial infarction
by suppressing TAK1 activation (9). Trim38 has also been shown to
mitigate the development of non-alcoholic fatty liver disease
(NAFLD) by promoting TAB2 degradation and inhibiting the MAPK
signaling pathway (10).
However, the role of Trim38 in pathological cardiac hypertrophy has
not yet been fully elucidated.
The present study assessed the expression levels of
Trim38 in a model of pressure overload-induced cardiac hypertrophy
and examined its role in this pathological process.
Materials and methods
Trim38 knockout (Trim38-KO) mice
Trim38-KO mice were generated using the clustered
regularly interspaced short palindromic repeats (CRISPR)/CRISPR
associated protein 9 (Cas9) system. The guide RNA (gRNA) was
designed using an online tool (http://chopchop.cbu.uib.no/). The target site was
located ~150 base pairs downstream of the start codon ATG on exon
1; the sequence of this region (5′-GCAATGTCAGCCCAAAAACA-3′)
disrupts the entire protein domain. The Trim38-specific gRNA was
cloned into the pUC57-sgRNA vector (cat. no. 51132; Addgene, Inc.).
The Cas9 expression vector pST1374-Cas9 (cat. no. 44758; Addgene,
Inc.) and the in vitro transcribed Trim38-sgRNA were
combined. This mixture was microinjected into single-cell C57BL/6
mouse embryos using the FemtoJet 5247 microinjection system
(Eppendorf SE). The injected embryos were then transferred into
three pseudopregnant female ICR mice (weight, 29-32 g; age, 8-9
weeks; Beijing Vital River Laboratory Animal Technology Co., Ltd.).
All mice were housed in a specific pathogen-free environment where
temperature (22±2°C), humidity (50-55%) and light cycles (12-h
light/dark cycle) were strictly regulated. In addition, the mice
had ad libitum access to both food and water. After a
gestation period of 19-21 days, the F0 generation mice were born. A
total of 2 weeks postpartum, genomic DNA was extracted from tail
snips to genotype the mice using the following primers:
Trim38-check forward (F)1, 5′-TGG GCT CAG ACT TTA GCA CG-3′;
Trim38-check reverse (R)1, 5′-TCT TCC CAA TAA CAG CGC CA-3′.
Animal model of cardiac hypertrophy
The male wild-type (WT) C57BL/6 mice (age, 8 weeks;
weight, 27-30 g) used were purchased from Beijing Vital River
Laboratory Animal Technology Co., Ltd. The WT mice and Trim38-KO
mice were randomly assigned to four experimental groups, with
initial sample sizes determined to account for potential attrition
during the modeling phase. The group allocations were as follows:
WT Sham (15 mice), WT TAC (15 mice), Trim38-KO Sham (15 mice) and
Trim38-KO TAC (20 mice). Animals were excluded if they either died
during surgery or failed to meet the predefined model success
criteria. After exclusions, a total of 10 mice per group were
included in the final dataset. Both WT and Trim38-KO mice underwent
either sham surgery or TAC surgery to induce pressure overload. The
mice were bred in a specific pathogen-free grade animal laboratory.
Housing conditions included no more than three mice per cage, with
free access to water and food, a 12-h light cycle, room temperature
maintained at 22±2°C and humidity controlled at 50-55%. During the
modeling process, anesthesia was induced via intraperitoneal
injection of a 3% sodium pentobarbital solution at a dose of 50
mg/kg. Transverse aortic constriction (TAC) was adopted to induce
cardiac hypertrophy (11). After
anesthesia, a 27G needle was placed parallel to the aorta. Next, a
needle holder was utilized to pass a 7-0 suture through the aorta
and ligate it together with the needle around the aorta. Once the
ligation was complete, the needle was rapidly removed to establish
aortic stenosis. The sham group underwent thoracotomy without
aortic ligation.
Histological analysis
Upon completion of the experimental period, mouse
hearts were harvested for analysis. The 8-week-old male C57BL/6
mice were euthanized via intraperitoneal injection of pentobarbital
sodium (150 mg/kg body weight) to ensure a rapid and painless
death. To confirm irreversible cessation of vital functions, the
following criteria were monitored: i) Cessation of cardiac
activity, verified by both thoracic palpation and direct visual
inspection post-thoracotomy; ii) bilateral pupillary dilation under
bright light examination; iii) absence of nociceptive reflexes,
demonstrated by unresponsiveness to a standardized toe-pinch
stimulus. Cardiac tissues were harvested only after confirmation of
death. The heart tissues were fixed with 4% paraformaldehyde for 24
h and subsequently dehydrated through a graded series of ethanol
prior to paraffin embedding. Specimens were sectioned at a
thickness of 5 µm, and hematoxylin and eosin (H&E)
staining was employed to assess cardiomyocyte size. For H&E
staining, heart sections were incubated in hematoxylin working
solution for 5 min followed by eosin solution for 3 min. In
addition, picro-sirius red (PSR) staining was utilized to evaluate
the extent of ventricular fibrosis. PSR staining involved applying
a 1% PSR solution to the sections and staining them in a humidified
chamber for 90 min. All procedures were conducted at room
temperature (20-25°C). H&E-stained sections were observed under
a standard light microscope, whereas PSR-stained sections were
examined under a polarized light microscope to facilitate collagen
fiber visualization. Images were captured at ×400 magnification and
data were analyzed using the imaging analysis system Image-Pro Plus
6.0 (Media Cybernetics, Inc.).
Cell culture
Following surface sterilization through immersion in
a 75% ethanol solution, 30 neonatal Sprague-Dawley rats (age, 1-3
days; SPF Biotechnology Co., Ltd.), were euthanized via
decapitation using large, sterile straight scissors. Neonatal rat
cardiomyocytes (NRCMs) were isolated from neonatal rats according
to previously established methods (12). Specifically, the hearts of
neonatal rats were excised, followed by removal of the atria while
retaining the ventricles. NRCMs were subsequently harvested after
treatment with 0.03% pancreatin and 0.04% type II collagenase. The
digestion process with pancreatin and collagenase was conducted at
37°C to maintain enzymatic activity. The initial digestion cycle
lasted for 15 min, followed by subsequent digestion cycles each
lasting 10 min; three cycles were performed until complete tissue
dissociation was achieved. Fibroblasts were eliminated using
differential adhesion. The AC16 human cardiomyocyte cell line (cat.
no. CC-Y1760) was obtained from Shanghai EK-Bioscience
Biotechnology Co., Ltd. Both NRCMs and AC16 cells were seeded into
6-well plates at a density of 2×105 cells/well and
cultured in DMEM/F12 (cat. no. 10565018) supplemented with 10%
fetal bovine serum (cat. no. 10099141C) and 100 U/ml
penicillin/streptomycin (cat. no. 15140122) (all from Gibco; Thermo
Fisher Scientific, Inc.) for 48 h under standard conditions (37°C,
5% CO2).
Recombinant vector construction
The Trim38-knockdown adenovirus was generated by
constructing a replication-deficient adenovirus vector carrying
short hairpin RNA (shRNA) targeting Trim38 (AdshTrim38), with the
AdshRNA adenovirus containing a non-specific shRNA used as the
control. The non-specific shRNA does not target any endogenous
genes, thereby eliminating the potential for non-specific gene
knockdown to interfere with experimental outcomes. Additionally, a
rat adenovirus overexpressing Flag-Trim38 (AdTrim38) and an
adenovirus containing dominant-negative TAK1 (Ad-dnTAK1) were
constructed, with an empty vector (AdVector) used as the control.
The dnTAK1 mutant effectively inhibits the normal activation of
endogenous WT TAK1. The plasmid backbone used for these constructs
was pAd-CMV (Thermo Fisher Scientific, Inc.) and 293 cells
(American Type Culture Collection) were used as the packaging cell
line for adenoviral production. Cells were transfected with 10
µg adenoviral plasmid using polyethyleneimine (PEI; cat. no.
24765-100; Polysciences, Inc.) at a DNA:PEI mass ratio of 1:3. The
DNA-PEI complex was formed in serum-free medium (~22°C, 15 min) and
added to cells. After 6 h, the medium was replaced with fresh
complete medium. Cells were cultured (37°C, 5% CO2) and
observations began on day 2 post-infection at a consistent time
point. Cytopathic effect (CPE) was assessed daily using an inverted
microscope, with cell rounding, detachment and vacuolation recorded
as key markers. CPE typically appeared within 5-7 days. For primary
virus (P1) harvest, when 50-60% of cells exhibited CPE, both the
cells and supernatant were collected. The mixture was centrifuged
at 800 × g for 10 min at 4°C, after which the supernatant was
resuspended. The cell suspension was then subjected to three
freeze-thaw cycles (liquid nitrogen/37°C water bath) to release
viral particles. The primary viral stock (P1 generation) harvested
from the initial transfection of 293 cells was used to infect fresh
293 cells. Subsequent passages (P2-P4) were generated by repeating
the harvest protocol under identical CPE progression criteria (50%
cellular pathology). All viral generations were processed through
identical centrifugation and freeze-thaw cycles as described for
P1. Following three freeze-thaw cycles to lyse the cells, the
lysate was clarified by centrifugation at 800 × g for 10 min at
4°C, and the resulting supernatant was layered onto a discontinuous
CsCl gradient (1.1, 1.3 and 1.4 g/ml) and ultracentrifuged (115,700
× g, 4°C, 2 h; TH641 rotor; Thermo Fisher Scientific, Inc.). The
viral band (between the 1.3 and 1.4 g/ml CsCl layers) was
collected, dialyzed (4°C, PBS + 5% glycerol), filtered (0.22
µm) and stored at −80°C.
Adenoviruses were used to infect NRCMs at a
multiplicity of infection (MOI) of 50 particles/cells for 24 h at
37°C followed by detection and identification. The shRNA sequence
targeting Trim38 was: 5′-GCA CAA AGG TCA TAC CTT ATT-3′. The
sequences used for the construction of the aforementioned
adenoviral vectors were as follows: AdshTrim38-rat-F, CCG GTA GCT
CTG TCT AAA GGTGGAACT CGA GTT CCA CCT TTA GAC AGA GCT ATT TTTG,
AdshTrim38-rat-R, AAT TCA AAA AGC ACA AAG GTC ATACCT TAT TCT CGA
GAA TAA GGT ATG ACC TTT GTGC; AdTrim38-rat-F, AGG CTA GCG ATA TCG
GAT CCA TGG GCT CAG ACT TTA GCA CAG TG, AdTrim38-rat-R, TCG TCC TTG
TAA TCA CTA GTCT TCT TTC CAT GAT TAT TTA TGG CAG GAG GGA;
Ad-dnTAK1-mouse-F, GAT GTC GCT ATT AAC CAG ATA GAA AGT G;
Ad-dnTAK1-mouse-R, CAC TTT CTA TCT GGT TAA TAG CGA CAT C.
Plasmid construction
The Trim38 gene was amplified utilizing PCR.
Subsequently, it was cloned into the GL107 vector [pSLen
ti-EF1-EGFP-P2A-Puro-CMV-MCS-3xFLAG-WPRE; OBiO Technology
(Shanghai) Corp., Ltd.] using Seamless Cloning Kit [cat. no.
SCNP-20/50; OBiO Technology (Shanghai) Corp., Ltd.]. This process
generated a Trim38 overexpression plasmid designed to induce the
stable overexpression of Trim38 in the AC16 cardiomyocyte cell
line. The forward sequencing primer (CMV-F) was 5′-CGC AAA TGG GCG
GTA GGC GTG-3′ and the reverse sequencing primer (101-R) was 5′-AGA
GAC AGC AAC CAG GAT-3′.
Transduction and transfection
NRCMs were infected with the corresponding
adenoviruses as aforementioned. AC16 cardiomyocytes were
transfected with Trim38 overexpression plasmid to overexpress
Trim38 using Fugene transfection reagent (cat. no. E2311; Promega
Corporation), whereas the control group was transfected with an
empty plasmid. Briefly, 2 µg plasmid DNA was used per well
in a 6-well plate. The complex (plasmid DNA-Fugene HD mixture) was
formulated at ~22°C and transfection was performed at 37°C. The
complex was incubated with the cells for 48 h under standard
conditions (37°C, 5% CO2) to facilitate transfection and
DNA expression. Subsequently, the medium was replaced with
serum-free DMEM/F12 for 12 h. Thereafter, phenylephrine (PE; 50
µM; cat. no. P6126; MilliporeSigma) was used to establish a
cardiomyocyte hypertrophy model at 37°C for 48 h. The control group
was treated with an equivalent volume of PBS under the same
conditions.
Immunofluorescence
NRCMs employed in the experiment were first fixed
with 3.7% formaldehyde at room temperature for 15 min and
permeabilized with 0.1% Triton X-100 for 20 min. After washing with
PBS, the cells were blocked with 8% BSA (cat. no. A1933;
MilliporeSigma) at room temperature for 60 min. A primary antibody
against α-actinin (1:100; cat. no. 05-384; MilliporeSigma) was then
applied and incubated overnight at 4°C. After removing the primary
antibody, a Alexa Fluor 488-conjugated secondary antibody [donkey
anti-mouse IgG (H+L); 1:200; cat. no. A21202; Invitrogen; Thermo
Fisher Scientific, Inc.] was added and incubated in the dark at
37°C for 60 min. The cells were counterstained with SlowFade Gold
antifade reagent containing DAPI for nuclear staining, and cured at
room temperature in the dark for 24 h. Alternatively, the cell
samples were prepared as aforementioned and stained overnight at
4°C with Actin-Tracker Red (1:100; cat. no. C2205S; Beyotime
Institute of Biotechnology). The following day, the samples were
directly mounted with DAPI and observed. Fluorescence images were
acquired using a confocal laser scanning microscope (TCS SP8; Leica
Microsystems GmbH). Image-Pro Plus 6.0 software was employed to
measure the cell surface area.
Reverse transcription-quantitative PCR
(RT-qPCR)
RNA was isolated from mouse heart tissue or NRCMs
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and cDNA synthesis was carried out through RT of
the RNA according to the manufacturer's protocol (cat. no.
04896866001; Roche Diagnostics). qPCR was performed to measure the
expression levels of the selected genes, utilizing a specific qPCR
kit (cat. no. 4913850001; Roche Diagnostics), with GAPDH used as an
internal control. Primer details are provided in Table SI. The thermal cycling
conditions were as follows: Pre-incubation at 95°C for 10 min;
followed by 40 cycles of amplification at 95°C for 10 sec, 60° for
30 sec, 72°C for 20 sec; melt curve at 95°C for 5 sec, 60° for 1
min, 95°C for 15 sec; and cooling at 40°C for 10 sec. Based on the
obtained crossing point values, relative quantification analysis
was conducted using the 2−ΔΔCq method (13).
Western blot analysis
Total protein was extracted from heart tissues or
cardiomyocytes using RIPA lysis buffer (100 mg SDS, 29.2 mg EDTA,
788.2 mg Tris-HCl, 200 mg sodium deoxycholate, 1 ml NP-40 and 876.6
mg NaCl dissolved in double-distilled water; pH 7.5; final volume,
100 ml). Protein concentration was determined using a BCA assay
kit. Subsequently, proteins (50 µg) were separated by
SDS-PAGE on a 10% gel and were transferred to PVDF membranes. After
blocking with 5% non-fat milk at room temperature for 1 h, the
membranes were incubated with primary antibodies at 4°C overnight.
The following day, appropriate secondary antibodies were added and
incubated for 1 h at room temperature in the dark. Immunoblots were
visualized using the Odyssey Imaging System (LI-COR Biosciences)
and the results were normalized to GAPDH levels as the internal
control. Protein expression was semi-quantified using Image Lab 5.1
software (Bio-Rad Laboratories, Inc.). Detailed information
regarding the antibodies is provided in Table SII.
Ubiquitination assays
AC16 cells transfected with the Trim38
overexpression plasmid and empty plasmid were lysed using a lysis
buffer added at four times the volume (8 M urea, 1% protease
inhibitor, 50 µM PR-619). The cells were sonicated in a 4°C
water bath for a total duration of 3 min using a high-intensity
ultrasonic processor (Scientz) with the following parameters: 3-sec
pulses followed by a 5-sec pause; power output, 220 W; frequency
range, 20-25 kHz. The supernatant was collected and the protein
concentration was determined using a BCA kit (cat. no. P0011;
Beyotime Institute of Biotechnology) according to the
manufacturer's instructions. Subsequently, the lysate was
precipitated with 20% TCA, centrifuged at 4,500 × g for 5 min at
4°C and washed with acetone. After air-drying, TEAB (200 µM)
was added to the precipitate and dispersed by ultrasound. Trypsin
was added at a ratio of 1:50 (enzyme to protein, w/w) and the
mixture was digested overnight. The next day, trypsin was added at
a ratio of 1:100 for a second digestion lasting 4 h. The solution
was reduced with dithiothreitol (5 mM) and alkylated with
iodoacetamide (11 mM) in the dark. To enrich the modified peptides,
the peptides dissolved in NETN buffer (100 mM NaCl, 1 mM EDTA, 50
mM Tris-HCl, 0.5% NP-40, pH 8.0). Each sample was incubated with 40
µl pre-washed anti-diglycyl-lysine antibody conjugated
agarose beads (cat. no. PTM1104; PTM Biolabs Inc.) at 4°C
overnight. Subsequently, the beads were washed four times with
immunoprecipitation buffer solution (PTM Biolabs Inc.) and twice
with deionized water. Bound peptides were subsequently eluted
through two consecutive washes with 0.1% trifluoroacetic acid (100
µl per wash; pH ~2) while subjected to vigorous vortexing.
The eluates were then combined and vacuum-dried. The resulting
peptides were desalted with C18 ZipTips (MilliporeSigma).
Liquid chromatography-tandem mass
spectrometry (LC-MS/MS) analysis
The peptides were dissolved in solvent A (0.1%
formic acid and 2% acetonitrile in water) and loaded onto a
homemade reversed-phase analytical column (length, 25 cm; internal
diameter, 100 µm). The mobile phase consisted of solvent A
and solvent B (0.1% formic acid in acetonitrile). Peptides were
separated using following gradient; 0-40 min, 6-22% solvent B;
40-52 min, 22-30% B; 52-56 min, 30-80% solvent B; 56-60 min, 80%
solvent B. All procedures were conducted at a constant flow rate of
450 nl/min on a NanoElute UHPLC system (Bruker Daltonics; Bruker
Corporation).
Subsequently, the peptides were electrosprayed
through a capillary source, generating charged droplets that
underwent desolvation via solvent evaporation to yield gaseous
ions. These ions were then analyzed in the timsTOF Pro2 mass
spectrometry (Bruker Daltonics; Bruker Corporation). The
electrospray voltage was set to 1.5 kV. Precursors and fragments
were analyzed at the TOF detector, with a MS/MS scan range from
100-1,700 m/z. The timsTOF Pro2 was operated in parallel
accumulation serial fragmentation (PASEF) mode. Precursors with
charge states 0-5 were selected for fragmentation, and 10
PASEF-MS/MS scans were acquired per cycle. The dynamic exclusion
was set to 24 sec. The resulting MS/MS data were processed using
MaxQuant search engine (v.1.6.15.0; https://www.maxquant.org/maxquant/) (14). Tandem mass spectra were searched
against the human SwissProt database (https://www.uniprot.org/) (15) concatenated with reverse decoy and
contaminants database. Trypsin/P was specified as cleavage enzyme
allowing up to 4 missing cleavages. Minimum peptide length was set
at 7 and maximum number of modifications per peptide was set at 5.
The mass tolerance for precursor ions was set at 20 ppm in the
first search and 20 ppm in the main search, and the mass tolerance
for fragment ions was set at 20 ppm. Due to sample pre-treatment
with reduction and alkylation, carbamidomethyl modification on
cysteine residues was set as a fixed modification; ubiquitylation
on lysine residues was specified as a variable modification to
identify ubiquitination sites. The false discovery rate of protein,
peptide and peptide-spectrum match was adjusted to <1%.
Functional enrichment-based cluster
analysis
Hierarchical clustering was conducted to classify
differentially expressed proteins in the Trim38 overexpression
group compared with the control group based on their function.
Based on their expression levels, differentially expressed proteins
were divided into four groups, namely Q1 to Q4, with Q4
representing the most significantly altered proteins. Gene Ontology
(GO) classification and Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathway enrichment analyses were both conducted on each of
the Q groups. GO annotation was accomplished by analyzing the
identified proteins with eggnog-mapper software v2.0 (https://github.com/eggnogdb/eggnog-mapper) (16) and the KEGG database (https://www.genome.jp/kegg/) (17) was used to conduct KEGG pathway
enrichment analysis. The Fisher's exact test was employed to
analyze the significance of KEGG pathway enrichment of
differentially expressed proteins. P<0.05 was considered to
indicate a statistically significant difference.
Statistical analysis
All values are presented as the mean ± standard
error of the mean. Normality was assessed using the Shapiro-Wilk
test. For normally distributed data, comparisons between two groups
were conducted using an unpaired two-tailed Student's t-test and
one-way ANOVA was employed to evaluate differences among multiple
groups. Post-hoc analysis was performed using Bonferroni correction
for data with equal variances and Tamhane's T2 test for data with
unequal variances. For data sets with skewed distributions,
non-parametric statistical methods were applied: The Mann-Whitney U
test for comparisons between two groups and the Kruskal-Wallis test
followed by Dunn's post-hoc test for multiple comparisons.
P<0.05 was considered to indicate a statistically significant
difference.
Results
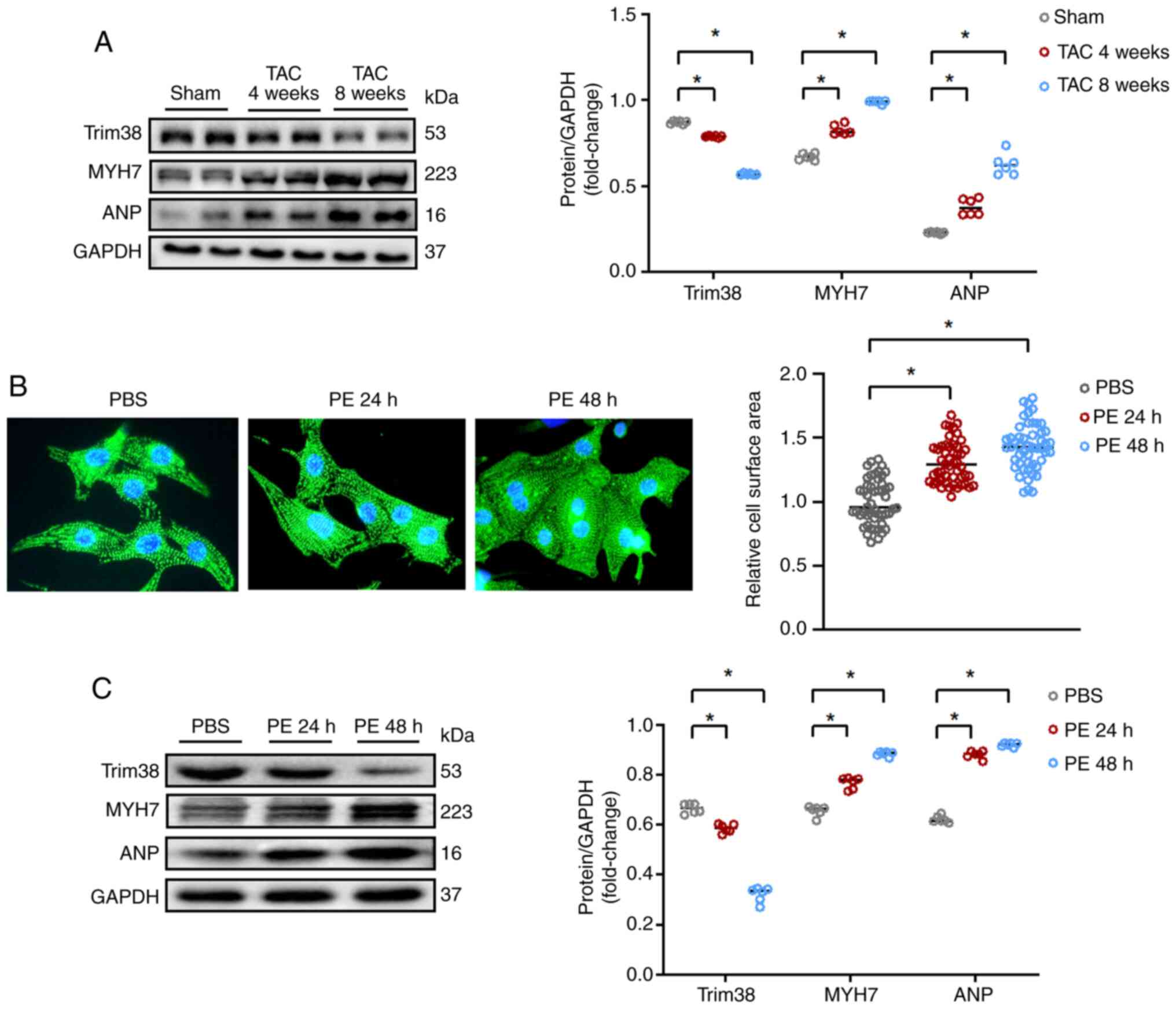
Trim38 expression is decreased in
hypertrophic myocardial tissues and cardiomyocytes
A TAC-induced mouse model of cardiac hypertrophy was
first established and the expression of Trim38 was detected using
western blotting. As shown in Fig.
1A, the expression levels of Trim38 were gradually decreased
after TAC, while the expression levels of biomarkers of cardiac
hypertrophy, such as myosin heavy chain 7 (MYH7) and atrial
natriuretic peptide (ANP), were increased. In addition, in
vitro experiments confirmed the reduced expression of Trim38 in
cardiomyocytes stimulated with PE. After 24 h of treatment with PE,
NRCMs displayed an altered morphology, indicating hypertrophy,
which was more pronounced after 48 h of PE treatment (Fig. 1B). Western blot analysis
(Fig. 1C) also demonstrated a
decrease in Trim38 expression in NRCMs treated with PE, alongside
increased levels of MYH7 and ANP, further confirming the
development of hypertrophy in the cells. Therefore, Trim38 may be
considered a potential target for cardiac hypertrophy.
Loss of Trim38 aggravates cardiac
hypertrophy
Trim38-KO mice were produced and Trim38 expression
was validated using western blotting to investigate whether Trim38
contributes to cardiac hypertrophy (Fig. 2A). At the baseline, there were no
significant differences in heart weight, myocardial hypertrophy and
myocardial fibrosis levels between WT sham mice and Trim38-KO sham
mice (Figs. 2B-F, and S1A and B). These findings suggested
that Trim38-KO had no significant impact on myocardial hypertrophy
and myocardial fibrosis under physiological conditions. However,
compared with in WT mice, heart weight, heart weight/body weight
ratio and heart weight/tibia length ratio were increased in
Trim38-KO mice 4 weeks after TAC (Fig. 2B). In addition, histological
analysis of cardiac tissue sections demonstrated a marked increase
in cardiomyocyte size upon H&E staining in the Trim38-KO TAC
group compared with in the WT TAC group (Fig. 2C). Furthermore, PSR staining
revealed that Trim38-KO significantly aggravated myocardial
fibrosis (Fig. 2D). Molecular
analyses, including qPCR and western blotting, confirmed that
Trim38-KO led to elevated mRNA and protein expression levels of
ANP, brain natriuretic peptide (BNP) and MYH7 in the heart
(Fig. 2E and G). Additionally,
Trim38-KO upregulated the expression of fibrosis markers, including
collagen type I α1 (collagen Ia), collagen III and connective
tissue growth factor (CTGF) following TAC (Fig. 2F and H). Taken together, these
findings suggested that Trim38-KO may contribute to the development
of cardiac hypertrophy.
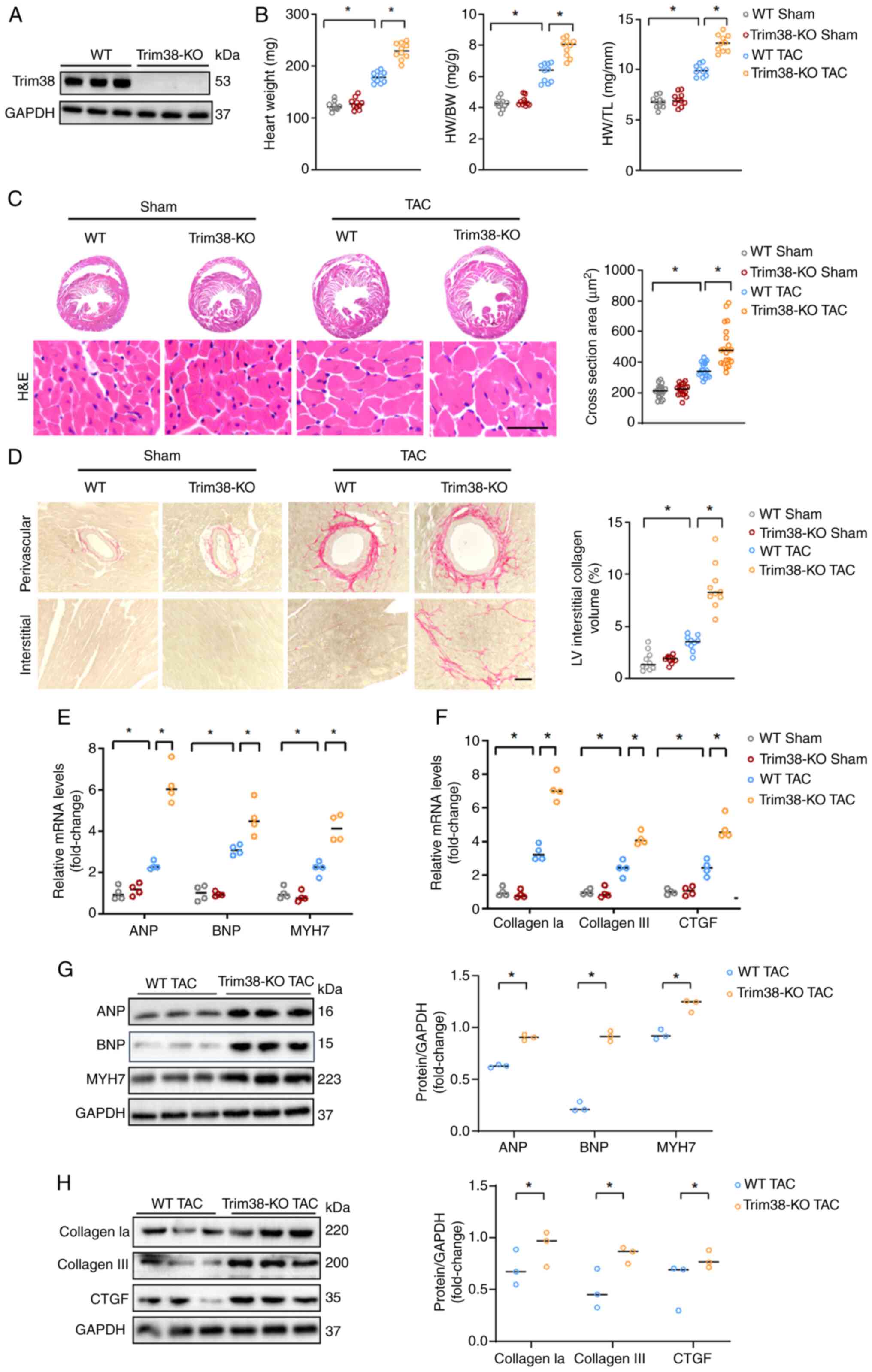 | Figure 2Trim38-KO exacerbates cardiac
hypertrophy. (A) Trim38 expression levels in Trim38-KO mice were
detected using western blot analysis. (B) HW, HW/BW ratio and HW/TL
ratio of WT and Trim38-KO mice subjected to TAC (n=10). (C) H&E
staining of heart tissue sections from WT mice and Trim38-KO mice
(scale bars, 25 µm). Cell section area was quantified in
each group (n>50 cells/group). (D) Picro-sirius red staining
showing fibrosis of heart tissue sections from WT and Trim38-KO
mice (scale bars, 50 µm). Collagen volume was quantified in
each group (n>10 files/group). mRNA expression levels of (E)
hypertrophic markers ANP, BNP and MYH7, and (F) collagen Ia,
collagen III and CTGF in each group (n=4 independent experiments).
Western blot analyses of the expression levels of (G) ANP, BNP and
MYH7, and (H) collagen Ia, collagen III and CTGF in WT mice and
Trim38-KO mice after TAC. Blots represent one of three replicated
experiments. *P<0.05. ANP, atrial natriuretic
peptide; BNP, brain natriuretic peptide; BW, brain weight; collagen
Ia, collagen type I α1; CTGF, connective tissue growth factor;
H&E, hematoxylin and eosin; HW, heart weight; KO, knockout; LV,
left ventricle; MYH7, myosin heavy chain 7; TAC, transverse aortic
constriction; TL, tibia length; Trim38, tripartite motif 38; WT,
wild-type. |
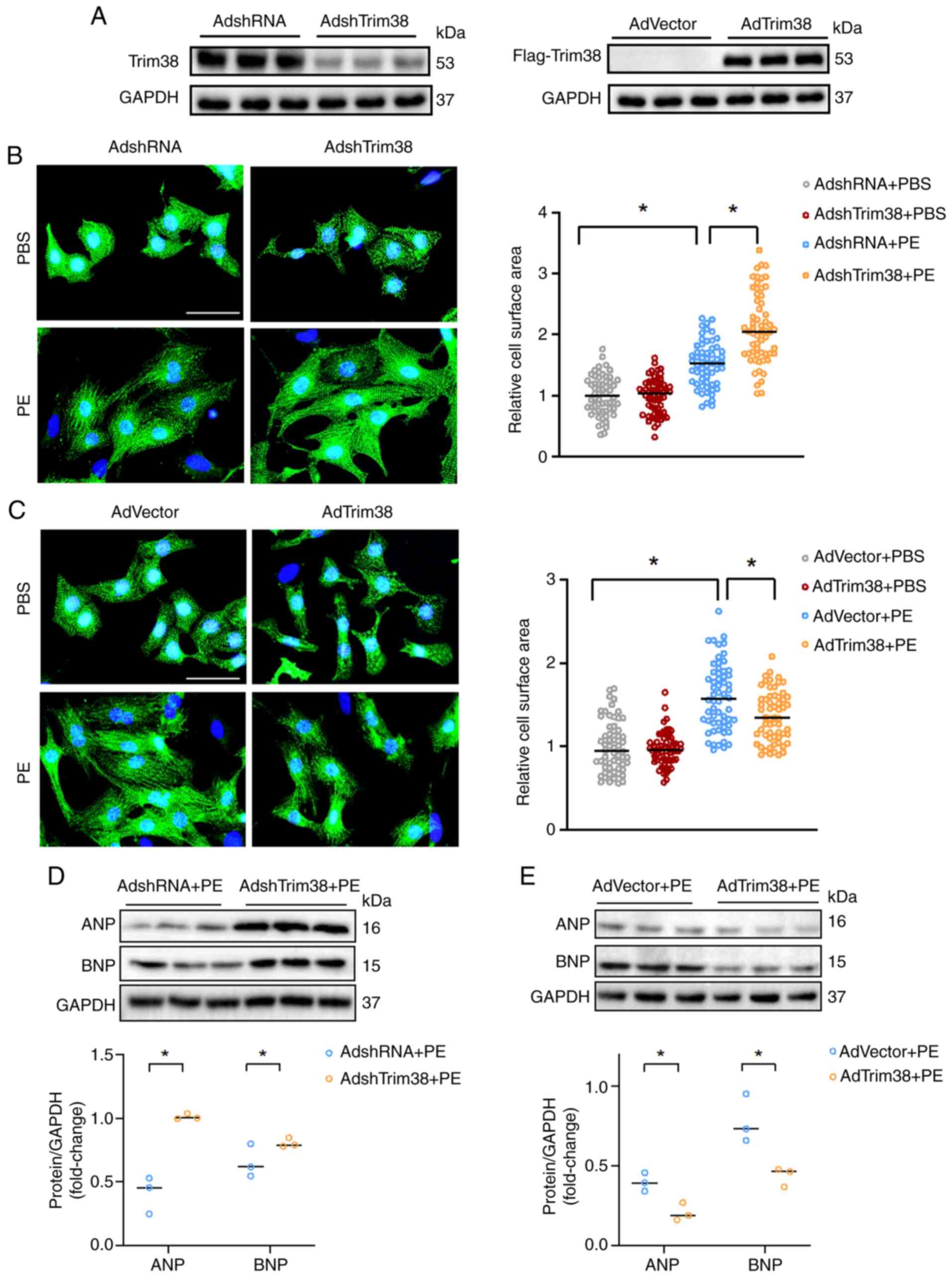
Trim38 ameliorates PE-induced
cardiomyocyte hypertrophy
The present study also investigated the role of
Trim38 in cardiomyocytes in vitro. AdshTrim38 infection was
used to downregulate Trim38 expression in NRCMs, while
AdFlag-Trim38 infection was used to overexpress Trim38 (Fig. 3A). After treatment with PE for 48
h, cells with Trim38 knockdown were of a larger size, and exhibited
enhanced expression levels of ANP and BNP, compared with in cells
infected with AdshRNA (Figs. 3B and
D, and S1C). By contrast,
Trim38 overexpression inhibited cardiomyocyte hypertrophy, and
downregulated PE-induced expression levels of ANP and BNP (Figs. 3C and E, and S1D). Thus, these findings demonstrated
that Trim38 could protect against cardiomyocyte hypertrophy in
vitro.
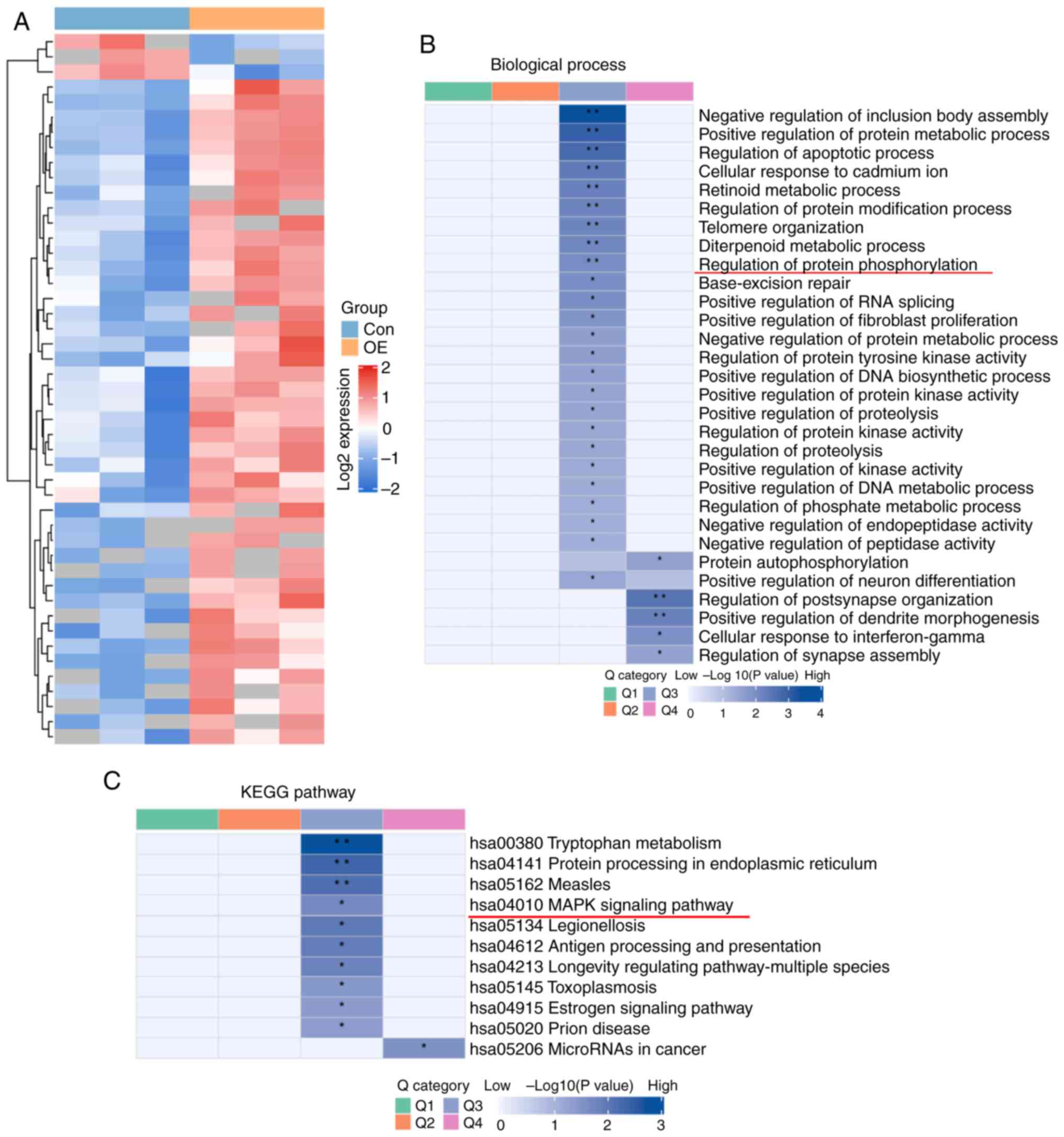
Trim38 protects against cardiac hypertrophy via
the TAK1/JNK/P38 signaling pathway. Given that Trim38 is an E3
ubiquitin ligase, ubiquitinomics analysis was conducted on
cardiomyocytes overexpressing Trim38 treated with PE to explore how
Trim38 regulates cardiac hypertrophy. A heat map demonstrating
differences in protein expression between the Trim38 overexpression
group and the control group is shown in Fig. 4A. Using cluster analysis,
differentially modified proteins were divided into four groups (Q1,
Q2, Q3 and Q4). GO enrichment analyses indicated that they were
involved in 'regulation of protein phosphorylation' (Fig. 4B). KEGG analysis indicated that
differentially modified proteins in the Q3 group (with fold changes
ranging from 1.5 to 2) were enriched in the 'MAPK signaling
pathway' (Fig. 4C). Therefore,
further studies placed a particular emphasis on phosphorylation of
the MAPK signaling pathway. Western blotting was conducted to
identify potential proteins involved in the MAPK signaling pathway,
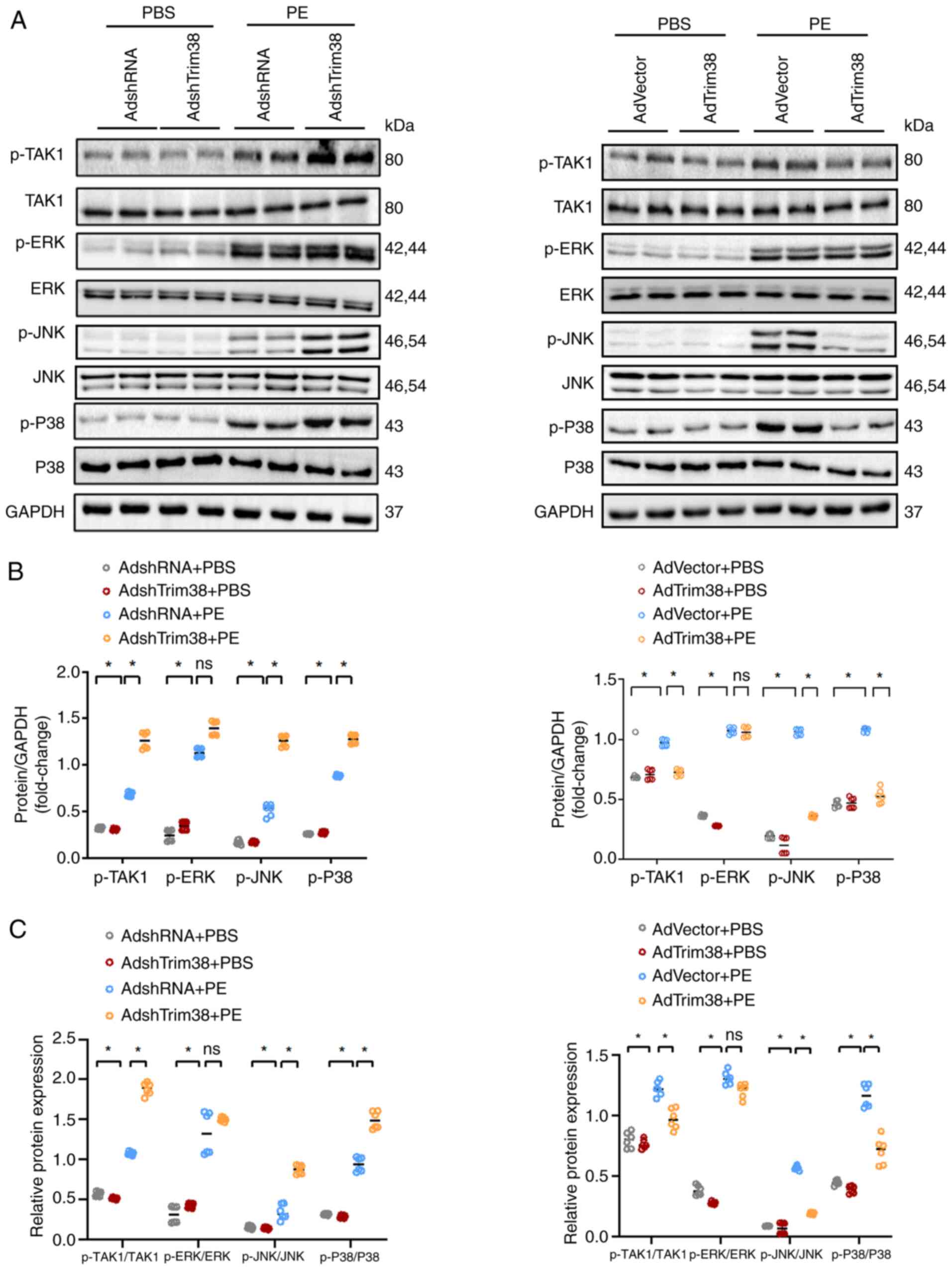
and it was unveiled that after TAC, TAK1 phosphorylation was
markedly upregulated in Trim38-KO mice compared with in the WT TAC
group (Fig. 5A-C). In addition,
Trim38-KO TAC mice exhibited a significant increase in the
phosphorylation of JNK and P38 during cardiac hypertrophy compared
with WT TAC mice (Fig. 5D-F).
Subsequently, in vitro experiments were conducted to confirm
the underlying mechanism. Western blotting showed that Trim38
knockdown enhanced the phosphorylation of TAK1, JNK and P38,
whereas Trim38 overexpression inhibited the phosphorylation of
TAK1, JNK and P38 in NRCMs treated with PE (Fig. 6A-C). Overall, these results
preliminarily indicated that Trim38 inhibited activation of the
MAPK pathway in cardiac hypertrophy.
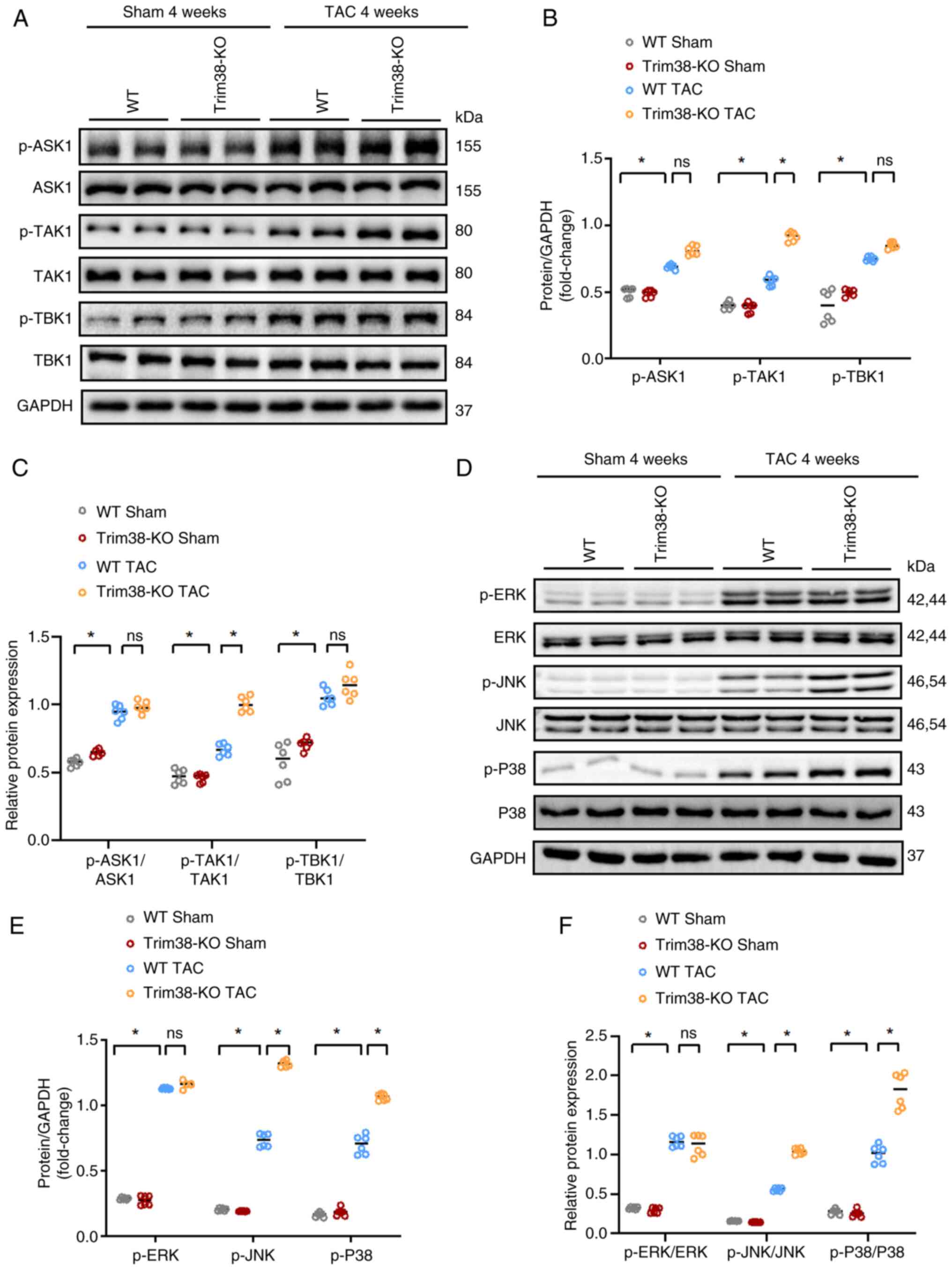 | Figure 5Trim38-KO leads to further activation
of the MAPK signaling pathway in mice after TAC. (A) Western blot
analysis comparing the total and p-levels of ASK1, TAK1 and TBK1
between Trim38-KO mice and WT mice. (B) Semi-quantification
analysis of phosphorylation levels of ASK1, TAK1 and TBK1 in each
group. (C) Ratios of p-/total protein levels for ASK1, TAK1 and
TBK1 in each group. (D) Western blot analysis of the total and
p-levels of ERK, JNK and P38 in Trim38-KO mice and WT mice. (E)
Semi-quantification of the phosphorylation levels of ERK, JNK and
P38 in hearts from Trim38-KO mice and WT mice. (F) p-ERK/total ERK,
p-JNK/total JNK and p-P38/total P38 ratios in each group. Blots
represent one of six replicated experiments. *P<0.05.
ASK1, apoptosis signal-regulating kinase 1; KO, knockout; p-,
phosphorylated; TAC, transverse aortic constriction; Trim38,
tripartite motif 38; WT, wild-type. |
Blocking the activity of TAK1 ameliorates
Trim38 knockdown-induced cardiomyocyte hypertrophy
Ad-dnTAK1 was generated, which lacks kinase
activity, to function as a TAK1 inhibitor. This allowed for
investigation as to whether Trim38 modulates cardiac hypertrophy
via the TAK1/JNK/P38 signaling pathway. The dnTAK1 harbors a
dominant-negative mutation that abolishes its kinase activity and
effectively neutralizes the function of WT TAK1. Upon infection
with Ad-dnTAK1, this mutant form effectively neutralizes the
activity of WT TAK1 while maintaining its own kinase-inactive
state.
Ad-dnTAK1 was co-transfected into NRCMs with either
AdshRNA or AdshTrim38, and the following observations were made.
Under PBS conditions, compared with in cells infected with AdshRNA,
the expression levels of TAK1 were increased in cells
co-transfected with Ad-dnTAK1 and AdshRNA (Ad-dnTAK1 + AdshRNA),
whereas the expression levels of p-TAK1 remained unchanged
(Fig. S2A). Similarly, the cell
area was not significantly altered between these two groups
(Fig. S2B). These findings
suggested that, despite the increased expression levels of TAK1,
its biological activity remained unaltered at baseline due to the
dominant-negative effect of Ad-dnTAK1; this prevents the
phosphorylation of TAK1 and any resultant phenotypic alterations in
the cells.
Upon treatment with PE, consistent with the
aforementioned findings, Trim38 knockdown in NRCMs resulted in
increased TAK1 phosphorylation (Fig.
7A) accompanied by an enlargement of cell area (Fig. 7B), and an upregulation of the
hypertrophy markers ANP and BNP compared with in NRCMs infected
with AdshRNA (Figs. 7C and
S1F). Notably, co-infection
with Ad-dnTAK1 and AdshRNA significantly suppressed TAK1
phosphorylation relative to AdshRNA alone. A parallel phenomenon
was observed in AdshTrim38-infected cells: Co-infection with
AdshTrim38 and Ad-dnTAK1 led to a significant reduction in TAK1
phosphorylation compared with infection with AdshTrim38 alone
(Figs. 7A and S1E). Crucially, comparing the NRCMs
co-infected with Ad-dnTAK1 and AdshTrim38 to those infected with
Ad-shTrim38 alone revealed that blockade of TAK1 activity via
Ad-dnTAK1 abrogated the pro-hypertrophic effects of Trim38
deficiency in cardiomyocytes (Figs.
7A-C and S1F).
Collectively, these findings indicated that Trim38 alleviated
cardiomyocyte hypertrophy in a manner that is dependent on TAK1
activity.
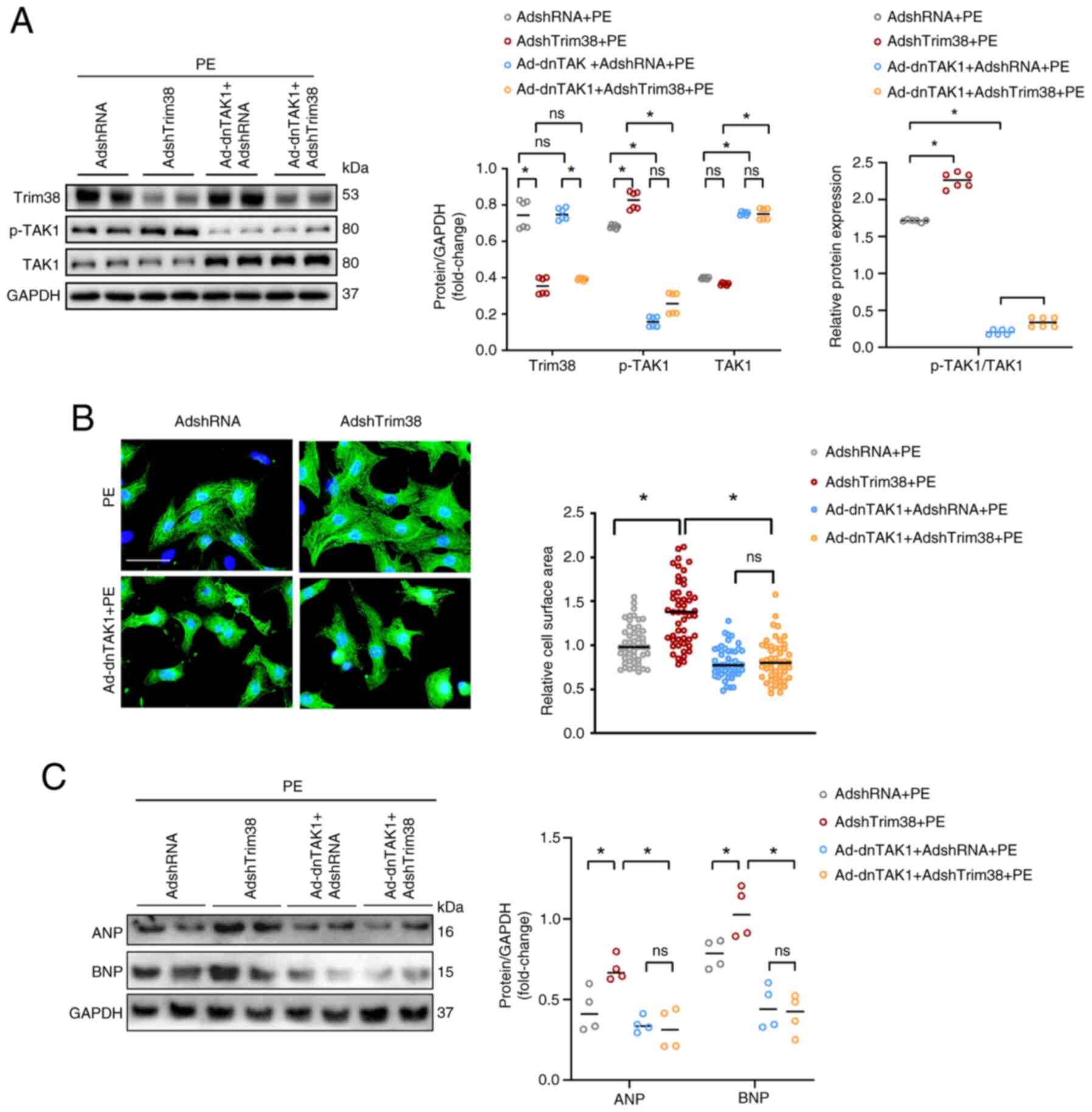 | Figure 7TAK1 is essential for Trim38-mediated
improvement of cardiac hypertrophy. (A) Western blot analysis and
semi-quantification of Trim38, TAK1 and p-TAK1 in neonatal rat
cardiomyocytes infected with the indicated Ads and treated with PE.
The p-TAK1/TAK1 ratio is also displayed. Blots represent one of six
replicated experiments. (B) Representative images of α-actinin
staining and quantification of cell cross-sectional area for each
group (n>50 cells/group) (scale bar, 20 µm). (C) Western
blot analysis and semi-quantitation of the expression levels of ANP
and BNP in each group. Blots represent one of four replicated
experiments. *P<0.05. Ad, adenovirus; ANP, atrial
natriuretic peptide; BNP, brain natriuretic peptide; dnTAK1,
dominant-negative TAK1; p-, phosphorylated; PE, phenylephrine; sh,
short hairpin; Trim38, tripartite motif 38. |
Discussion
A previous study revealed that Trim38 expression is
significantly downregulated within the cardiac tissues of mice
subjected to LAD coronary artery permanent ligation, a
well-characterized model of acute myocardial infarction (AMI), and
is also reduced in coronary blood samples from patients with AMI.
Furthermore, this previous study demonstrated that suppression of
Trim38 can enhance the proliferative and secretory activities of
cardiac fibroblasts (CFs), thereby exacerbating cardiac fibrosis
(9). Conversely, upregulation of
Trim38 expression has been observed to mitigate these fibrotic
responses, highlighting its protective role against cardiac
fibrosis under ischemic conditions (9). The present study revealed that
Trim38 confers protection against pathological myocardial
hypertrophy in response to pressure overload. Initial observations
demonstrated downregulated Trim38 expression in hypertrophic
cardiomyocytes and cardiac tissues. To further investigate this
phenomenon, Trim38-KO mice were generated. Under physiological
conditions, these mice exhibited no significant differences in
cardiac structure or function compared with their WT counterparts;
however, under conditions of pressure overload, Trim38-KO
exacerbated pathological hypertrophy, as evidenced by increased
heart weight, enlarged cardiomyocyte area in cardiac tissue
sections and elevated expression of hypertrophy markers, such as
MYH7, ANP and BNP, in Trim38-KO cardiac tissues. Consistent with
these in vivo findings, parallel in vitro experiments
demonstrated that Trim38 knockdown led to increased cardiomyocyte
size and upregulation of hypertrophy markers, whereas Trim38
overexpression resulted in reduced cell size and downregulation of
these markers under PE stimulation. Collectively, these findings
confirmed that Trim38 may have a crucial role in modulating
pathological hypertrophy. Specifically, Trim38 could attenuate
cardiac hypertrophy through inhibition of the JNK/P38 signaling
pathway. Mechanistic insights further revealed that regulation of
TAK1 phosphorylation by Trim38 is a critical factor in this
process. To the best of our knowledge, the present study is the
first to provide evidence that Trim38 alleviates pressure
overload-induced pathological hypertrophy by inhibiting the
TAK1/JNK/P38 signaling pathway.
The development of cardiac hypertrophy is
accompanied by adaptive changes in protein expression, which are
regulated by various factors, including gene transcription, mRNA
translation and post-translational modifications (PTMs) (18). PTMs increase the complexity and
diversity of proteins by adding or introducing new functional
groups (19). Ubiquitination is
a common form of PTM and a reversible process in which ubiquitin
binds to the amino acid side, or C-terminal or N-terminal of
proteins. Through diverse modifications, specifically the
structural variability of ubiquitin chains formed by distinct
lysine linkages (e.g., K48, K63) or linear M1 linkages, and
specific recognition mechanisms, ubiquitination can rapidly and
flexibly regulate signaling pathways without relying on changes in
gene expression or waiting for transcription and translation
processes. The dynamic nature of ubiquitination may make it more
efficient than transcriptional regulation, which requires the
activation of gene expression and the synthesis of new proteins
(20). Ubiquitination serves a
crucial role in determining protein structure, localization and
function, playing a complex and precise regulatory role in cardiac
hypertrophy (21). Trim family
proteins are involved in various processes associated with cardiac
hypertrophy through their ubiquitin ligase activity. These proteins
facilitate the ubiquitination and degradation of specific
substrates, which directly impacts the signaling pathways and
protein turnover associated with hypertrophic responses. Notably,
Trim65 has been demonstrated to prevent pathological cardiac
hypertrophy by promoting mitochondrial autophagy through the
Jak1/Stat1 signaling pathway (22). Moreover, Trim44 has been reported
to promote cardiac hypertrophy by upregulating the expression of
NOX4 and enhancing ferroptosis (23). The present study observed a
decreased expression of Trim38 in cardiac hypertrophy in
vivo and in vitro. After TAC, Trim38-KO increased cell
size, aggravated fibrosis, and upregulated the expression of
hypertrophic and fibrosis markers. Consequently, these findings
suggested that Trim38 serves a crucial role in alleviating pressure
overload-induced cardiac hypertrophy.
Ubiquitination experiments were conducted to
elucidate the underlying mechanisms of Trim38 in cardiac
hypertrophy. Using KEGG and GO enrichment analyses, several key
pathways were identified. Notably, the MAPK pathway emerged as a
classical pathway associated with the progression of cardiac
hypertrophy. It has previously been shown that Trim38 suppresses
cardiac fibrosis by attenuating the TAK1/MAPK signaling pathway in
CFs (9). Consequently, the
present study focused on the MAPK signaling pathway and discovered
that Trim38 can modulate phosphorylation within this pathway.
Multiple PTMs have been reported to maintain cellular homeostasis
(24), and the cooperation of
ubiquitination with other forms of PTMs is crucial for the
orchestration of biological activities. The interplay between
ubiquitination and phosphorylation regulates cellular processes and
has been extensively explored in previous studies (25-27). Skp1/cullin/F box proteins (SCF)
have been well identified as ubiquitin ligases responsible for
phosphorylation-mediated ubiquitination. The F-box domain of SCF
recognizes and binds to phosphodegron motifs, leading to substrate
polyubiquitination, which typically results in proteasomal
degradation (28). In addition
to direct interaction with phosphorylated substrates, Trim16 has
been demonstrated to ubiquitinate both TAK1 and p-TAK1, and promote
the proteasomal degradation of p-TAK1 in nonalcoholic
steatohepatitis through K-48 ubiquitination (29).
The MAPK signaling pathway is a highly conserved
pathway that facilitates the activation of numerous transcription
factors. It is activated in the hypertrophic myocardial tissue of
mice induced by PE or pressure overload and in diseased human
hearts (30). TAK1 functions as
an 'initiation switch' that activates the MAPK signaling pathway,
and upregulation of TAK1 phosphorylation appears to be an adaptive
mechanism after TAC. Cardiac-specific ablation of TAK1 in mice has
been shown to predispose them to cardiac hypertrophy and HF
(31). Furthermore, the results
of the present study revealed that overexpression of Trim38
inhibited the phosphorylation of TAK1, while Trim38 knockdown
upregulated p-TAK1 expression and activated the JNK/P38 signaling
pathway. To determine whether Trim38 protects against cardiac
hypertrophy via p-TAK1, dnTak1 was used to block TAK1 binding to
TAB1, thereby inhibiting TAK1 activation (32). p-TAK1 inactivation reversed cell
enlargement and inhibited the expression of hypertrophic markers
induced by Trim38 knockdown in vitro. Taken together, these
findings suggested that Trim38 may ameliorate cardiac hypertrophy
by inhibiting TAK1 phosphorylation.
The interaction between Trim38 and TAK1 has been
investigated in several studies. Mechanistically, TAK1 activation
necessitates the binding of TAB1, TAB2 and TAB3 to form the
TAK1-TAB complex (33). TAB1
triggers the phosphorylation of TAK1, whereas TAB2 links TAK1 to
the upstream proteins. Moreover, TAB2 and TAB3 serve a crucial role
in maintaining the activation of TAK1 (34). Notably, emerging evidence has
highlighted Trim38 as a key modulator of the TAK1-TAB complex
across diverse pathological contexts, suggesting its potential role
in fine-tuning TAK1-mediated signaling cascades. Previous studies
have indicated that Trim38 mediates lysosome-dependent degradation
of TAB2, thereby inhibiting TAK1 and the NFκB signaling pathway
in vitro (35,36). In CFs treated with PE, Trim38
overexpression has been shown to inhibit total ubiquitination and
K63-linked ubiquitination of TAK1, which seems to be inconsistent
with the fact that Trim38 acts as an E3 ubiquitin ligase that can
promote ubiquitination. Subsequent analysis in the same study
unveiled that Trim38 can promote TAB2 degradation, resulting in
reduced TAK1 ubiquitination and phosphorylation (9). A similar observation has been made
in NAFLD; Trim38 overexpression has been shown to downregulate TAB2
expression, thereby inhibiting the phosphorylation of TAK1 and MAPK
signaling pathways (10). The
present study confirmed previous findings and contributed
additional evidence that Trim38 can protect against cardiac
hypertrophy by suppressing TAK1 phosphorylation and activating the
JNK/P38 signaling pathway. Based on previous findings, it was
hypothesized that the reduction in TAK1 phosphorylation may be
attributed to increased Trim38-mediated degradation of TAB2. This
phenomenon could subsequently disrupt the binding of TAK1 to its
upstream adaptors. However, further studies are needed to fully
understand the underlying mechanisms.
There are several limitations in the present study
that warrant acknowledgment. Firstly, a notable limitation of the
study is the lack of cardiac ultrasound data, which prevented a
direct assessment of ventricular wall thickness and overall cardiac
function. While histological and molecular analyses provide
supportive evidence for cardiac hypertrophy, these methods cannot
fully replicate the distinct advantages of cardiac ultrasound in
evaluating cardiac structure and function. Future studies should
incorporate cardiac ultrasound to offer a more comprehensive
evaluation of cardiac function and myocardial hypertrophy-related
pathological changes. Additionally, the current study did not
provide conclusive evidence on the decrease of Trim38 in the blood
of patients with HF. Based on RNA-sequencing data from the Human
Protein Atlas (https://www.proteinatlas.org/ENSG00000111666-TRIM38)
(37) and GeneCards (https://www.genecards.org/cgi-bin/carddisp.pl?gene=TRIM38)
(38), Trim38 demonstrates low
expression levels in whole blood (TPM <1). This suggests
potentially low concentrations of TRIM38 in circulation, thereby
presenting challenges for its detection. Moreover, further
investigation is necessary to explore the specific mechanism
underlying the interaction between Trim38 and TAK1 in cardiac
hypertrophy. Finally, clinical studies are required to validate the
association between Trim38 and pressure overload-induced cardiac
hypertrophy.
In conclusion, the present study demonstrated that
Trim38 serves a protective role against pressure overload-induced
pathological cardiac hypertrophy by suppressing the TAK1/JNK/p38
signaling axis. By contrast, Trim38 deficiency exacerbated
myocardial hypertrophy and upregulated hypertrophic biomarkers
(ANP/BNP). Mechanistically, Trim38 deficiency may activate TAK1
phosphorylation and the downstream MAPK pathway. Conversely, its
overexpression has been shown to inhibit TAK1 phosphorylation and
downstream MAPK pathway activation. Crucially, the detrimental
effects of Trim38 knockdown were revealed to be mitigated by TAK1
inhibition, identifying TAK1 as a central mediator of
Trim38-mediated cardioprotection. To the best of our knowledge, the
present study is the first to highlight Trim38 as a novel negative
regulator of pathological cardiac remodeling, positioning it as a
promising therapeutic target for combating heart failure.
Supplementary Data
Availability of data and materials
The mass spectrometry proteomics data have been
deposited to the ProteomeXchange Consortium via the PRIDE partner
repository with the dataset identifier PXD060763 or at the
following URL: https://www.ebi.ac.uk/pride/archive/projects/PXD060763.
The other data generated in the present study may be requested from
the corresponding author.
Authors' contributions
LJ and LH conceived the study and provided financial
support. YP, LW, JX, XX and CG performed the experiments, collected
the data and prepared the figures. YP was a major contributor in
writing the manuscript. YP and LW confirm the authenticity of all
the raw data. All authors contributed to the article, and read and
approved the final version of the manuscript.
Ethics approval and consent for
participation
The animal study was reviewed and approved by
Shanghai Tongren Hospital Ethics Committee (approval no.
A2023-086-01; Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
HF
|
heart failure
|
|
Trim
|
tripartite motif
|
|
NAFLD
|
nonalcoholic fatty liver disease
|
|
MYH7
|
myosin heavy chain 7
|
|
ANP
|
atrial natriuretic peptide
|
|
TAC
|
transverse aortic constriction
|
|
PE
|
phenylephrine
|
|
KO
|
knockout
|
|
WT
|
wild-type
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
GO
|
Gene Ontology
|
|
PTMs
|
post-translational modifications
|
|
SCF
|
Skp1/cullin/F box proteins
|
|
H&E
|
hematoxylin and eosin
|
|
PSR
|
picro-sirius red
|
Acknowledgments
Not applicable.
Funding
No funding was received.
References
|
1
|
McDonagh TA, Metra M, Adamo M, Gardner RS,
Baumbach A, Böhm M, Burri H, Butler J, Čelutkienė J, Chioncel O, et
al: 2021 ESC guidelines for the diagnosis and treatment of acute
and chronic heart failure. Eur Heart J. 42:3599–3726. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Levy D, Larson MG, Vasan RS, Kannel WB and
Ho KK: The progression from hypertension to congestive heart
failure. JAMA. 275:1557–1562. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Martin TG, Juarros MA and Leinwand LA:
Regression of cardiac hypertrophy in health and disease: Mechanisms
and therapeutic potential. Nat Rev Cardiol. 20:347–363. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Katz AM and Rolett EL: Heart failure: When
form fails to follow function. Eur Heart J. 37:449–454. 2016.
View Article : Google Scholar
|
|
5
|
Dikic I: Proteasomal and autophagic
degradation systems. Annu Rev Biochem. 86:193–224. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hu MM and Shu HB: Multifaceted roles of
TRIM38 in innate immune and inflammatory responses. Cell Mol
Immunol. 14:331–338. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xue Q, Zhou Z, Lei X, Liu X, He B, Wang J
and Hung T: TRIM38 negatively regulates TLR3-mediated IFN-β
signaling by targeting TRIF for degradation. PLoS One.
7:e468252012. View Article : Google Scholar
|
|
8
|
Hu MM, Yang Q, Xie XQ, Liao CY, Lin H, Liu
TT, Yin L and Shu HB: Sumoylation promotes the stability of the DNA
sensor cGAS and the adaptor STING to regulate the kinetics of
response to DNA virus. Immunity. 45:555–569. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu Z, Hao C, Qian H, Zhao Y, Bo X, Yao Y,
Ma G and Chen L: Tripartite motif 38 attenuates cardiac fibrosis
after myocardial infarction by suppressing TAK1 activation via
TAB2/3 degradation. iScience. 25:1047802022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yao X, Dong R, Hu S, Liu Z, Cui J, Hu F,
Cheng X, Wang X, Ma T, Tian S, et al: Tripartite motif 38
alleviates the pathological process of NAFLD-NASH by promoting TAB2
degradation. J Lipid Res. 64:1003822023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pang Y, Ma M, Wang D, Li X and Jiang L:
TANK promotes pressure overload induced cardiac hypertrophy via
activating AKT signaling pathway. Front Cardiovasc Med.
8:6875402021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ehler E, Moore-Morris T and Lange S:
Isolation and culture of neonatal mouse cardiomyocytes. J Vis Exp.
6:501542013.
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
14
|
Cox J and Mann M: MaxQuant enables high
peptide identification rates, individualized p.p.b.-range mass
accuracies and proteome-wide protein quantification. Nat
Biotechnol. 26:1367–1372. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
UniProt Consortium: Uniprot: The universal
protein knowledgebase in 2023. Nucleic Acids Res. 51:D523–D531.
2023. View Article : Google Scholar :
|
|
16
|
Cantalapiedra CP, Hernández-Plaza A,
Letunic I, Bork P and Huerta-Cepas J: eggNOG-mapper v2: Functional
annotation, orthology assignments, and domain prediction at the
metagenomic scale. Mol Biol Evol. 38:5825–5829. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kanehisa M, Furumichi M, Tanabe M, Sato Y
and Morishima K: KEGG: New perspectives on genomes, pathways,
diseases and drugs. Nucleic Acids Res. 45:D353–D361. 2017.
View Article : Google Scholar :
|
|
18
|
Oka T, Akazawa H, Naito AT and Komuro I:
Angiogenesis and cardiac hypertrophy: Maintenance of cardiac
function and causative roles in heart failure. Circ Res.
114:565–571. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu X, Xu M, Geng M, Chen S, Little PJ, Xu
S and Weng J: Targeting protein modifications in metabolic
diseases: Molecular mechanisms and targeted therapies. Signal
Transduct Target Ther. 8:2202023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cockram PE, Kist M, Prakash S, Chen SH,
Wertz IE and Vucic D: Ubiquitination in the regulation of
inflammatory cell death and cancer. Cell Death Differ. 28:591–605.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Qiu M, Chen J, Li X and Zhuang J:
Intersection of the ubiquitin-proteasome system with oxidative
stress in cardiovascular disease. Int J Mol Sci. 23:121972022.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu H, Zhou Z, Deng H, Tian Z, Wu Z, Liu
X, Ren Z and Jiang Z: Trim65 attenuates isoproterenol-induced
cardiac hypertrophy by promoting autophagy and ameliorating
mitochondrial dysfunction via the Jak1/Stat1 signaling pathway. Eur
J Pharmacol. 949:1757352023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu L, Jia M, Xiao L, Wang Z, Yao R, Zhang
Y and Gao L: TRIM-containing 44 aggravates cardiac hypertrophy via
TLR4/NOX4-induced ferroptosis. J Mol Med (Berl). 101:685–697. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee JM, Hammarén HM, Savitski MM and Baek
SH: Control of protein stability by post-translational
modifications. Nat Commun. 14:2012023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Song L and Luo ZQ: Post-translational
regulation of ubiquitin signaling. J Cell Biol. 218:1776–1786.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu P, Cong X, Liao S, Jia X, Wang X, Dai
W, Zhai L, Zhao L, Ji J, Ni D, et al: Global identification of
phospho-dependent SCF substrates reveals a FBXO22 phosphodegron and
an ERK-FBXO22-BAG3 axis in tumorigenesis. Cell Death Differ.
29:1–13. 2022. View Article : Google Scholar :
|
|
27
|
Zhu GW, Chen H, Liu SY, Lin PH, Lin CL and
Ye JX: PPM1B degradation mediated by TRIM25 ubiquitination
modulates cell cycle and promotes gastric cancer growth. Sci Rep.
15:61602025. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Barbour H, Nkwe NS, Estavoyer B, Messmer
C, Gushul-Leclaire M, Villot R, Uriarte M, Boulay K, Hlayhel S,
Farhat B, et al: An inventory of crosstalk between ubiquitination
and other post-translational modifications in orchestrating
cellular processes. iScience. 26:1062762023. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang L, Zhang X, Lin ZB, Yang PJ, Xu H,
Duan JL, Ruan B, Song P, Liu JJ, Yue ZS, et al: Tripartite motif 16
ameliorates nonalcoholic steatohepatitis by promoting the
degradation of phospho-TAK1. Cell Metab. 33:1372–1388.e7. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dorn GN II and Force T: Protein kinase
cascades in the regulation of cardiac hypertrophy. J Clin Invest.
115:527–537. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li L, Chen Y, Doan J, Murray J, Molkentin
JD and Liu Q: Transforming growth factor β-activated kinase 1
signaling pathway critically regulates myocardial survival and
remodeling. Circulation. 130:2162–2172. 2024. View Article : Google Scholar
|
|
32
|
Xie M, Zhang D, Dyck JR, Li Y, Zhang H,
Morishima M, Mann DL, Taffet GE, Baldini A, Khoury DS and Schneider
MD: A pivotal role for endogenous TGF-beta-activated kinase-1 in
the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc
Natl Acad Sci USA. 103:17378–17383. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hirata Y, Takahashi M, Morishita T,
Noguchi T and Matsuzawa A: Post-translational modifications of the
TAK1-TAB complex. Int J Mol Sci. 18:2052017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xu YR and Lei CQ: TAK1-TABs complex: A
central signalosome in inflammatory responses. Front Immunol.
11:6089762021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hu MM, Yang Q, Zhang J, Liu SM, Zhang Y,
Lin H, Huang ZF, Wang YY, Zhang XD, Zhong B and Shu HB: TRIM38
inhibits TNFα- and IL-1β-triggered NF-κB activation by mediating
lysosome-dependent degradation of TAB2/3. Proc Natl Acad Sci USA.
111:1509–1514. 2014. View Article : Google Scholar
|
|
36
|
Kim K, Kim JH, Kim I, Seong S and Kim N:
TRIM38 regulates NF-κB activation through TAB2 degradation in
osteoclast and osteoblast differentiation. Bone. 113:17–28. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Uhlén M, Fagerberg L, Hallström BM,
Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C,
Sjöstedt E, Asplund A, et al: Proteomics. Tissue-based map of the
human proteome. Science. 347:12604192015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Stelzer G, Rosen N, Plaschkes I, Zimmerman
S, Twik M, Fishilevich S, Stein TI, Nudel R, Lieder I, Mazor Y, et
al: The genecards suite: From gene data mining to disease genome
sequence analyses. Curr Protoc Bioinformatics. 54:1–30. 2016.
|