Endoplasmic reticulum (ER) stress (ERS) is a
pathological state in which the normal function of the ER is
impaired, leading to the accumulation of unfolded or misfolded
proteins and disruption of Ca2+ homeostasis. To manage
ERS, cells activate an adaptive mechanism known as the unfolded
protein response (UPR) to restore metabolic balance and protein
folding capacity within the ER (1). On the other hand,
epithelial-mesenchymal transition (EMT) is a process in which
epithelial cells, stimulated by specific cytokines or
microenvironmental signals, lose their polarity and cell-cell
adhesion properties, gradually transforming into mesenchymal cells
with migratory and invasive capabilities. This transformation
contributes considerably to pathological processes such as tissue
remodelling, fibrosis and tumour metastasis (2). As two core processes in the field
of cell biology, ERS and EMT substantially influence the
initiation, progression and metastasis of fibrotic diseases and
tumours (3,4). However, the specific mechanisms by
which ERS directly or indirectly regulates EMT have not yet been
fully elucidated and related research remains limited. Therefore,
the aim of this study was to systematically explore the research
progress and targeted therapeutic strategies of ERS and EMT in
fibrotic diseases and tumours, and to elucidate the molecular
connections and regulatory mechanisms between the two processes.
Another objective was to provide a theoretical foundation to
further reveal the interactions between these complex processes in
the future, as well as to offer new insights and directions for the
innovation of clinical treatment strategies.
A heat shock protein called glucose-regulated
protein 78 (GRP78)/immunoglobulin heavy chain-binding protein (BiP)
dissociates from the transmembrane receptor, binds and processes
the unfolded proteins and activates the UPR, which is the cell's
protective pathway (5). Any
factor that disrupts protein function, such as hypoxia, nutrient
deprivation or oxidative stress, can cause the accumulation of
misfolded/unfolded proteins in the ER, a crucial organelle involved
in Ca2+ storage and release in the lumen of the ER
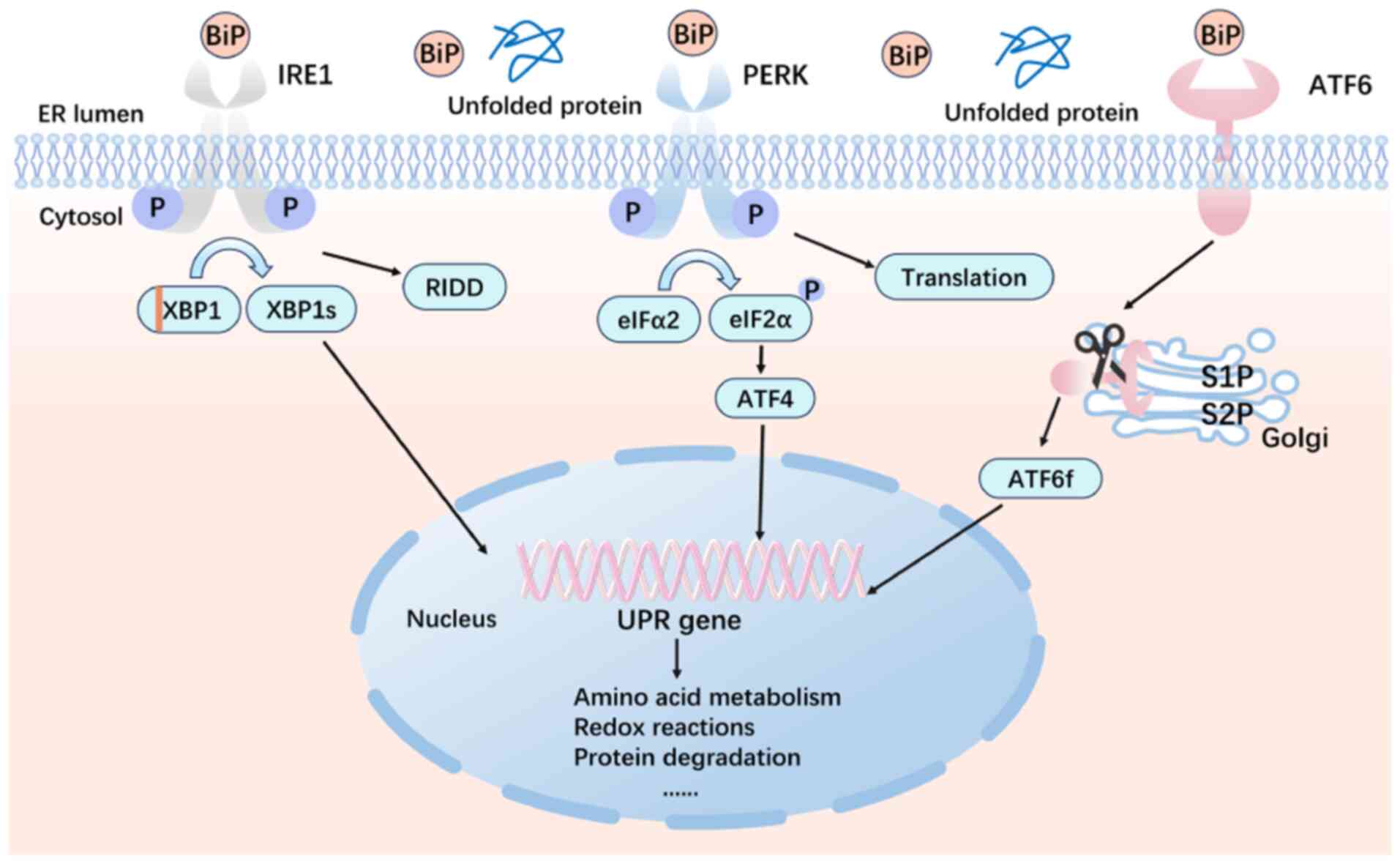
(6). As shown in Fig. 1, to maintain ER homeostasis and
restore ER function, the UPR mediates three transmembrane protein
pathways: Activating transcription factor 6 (ATF6), the protein
kinase RNA-like ER kinase (PERK) and inositol-requiring enzyme 1α
(IRE1α) (7). Furthermore, ERS
activation can result in either cell survival or death, depending
on the length and severity of the stress (4). This is because when cells are
exposed to ERS, the UPR shows two distinct results. However, cells
that are exposed to weak or short-term ERS will trigger adaptive
and pro-survival signalling, which will restore ER homeostasis to a
physiological level. Conversely, prolonged or severe ERS will
trigger autophagy and apoptosis-associated signalling across
several pathways, changing the UPR from a pro-survival network to a
pro-apoptotic system, which promotes apoptosis and eventually
causes illness (8,9).
EMT is a dynamic process in which epithelial cells
undergo a series of complex molecular modifications to
transdifferentiate into mesenchymal cells (2). Based on their biological context
and functions, EMTs are categorized into three subtypes: Type I EMT
is associated with embryonic development, Type II is linked to
tissue repair and organ fibrosis and Type III is related to cancer
invasion and metastasis. These subtypes contribute significantly to
various physiological and pathological conditions (2,10,11). EMT initiation depends on multiple
inducing signals and involves the activation of numerous signalling
pathways. Simultaneously, EMT is regulated by several key
transcription factors, such as Snail family transcriptional
repressor 1 (SNAIL1) and SNAIL2, zinc finger E-box-binding homeobox
1 (ZEB1) and ZEB2 and the TWIST transcription factor. These
transcription factors suppress the expression of epithelial marker
genes and activate genes associated with the mesenchymal phenotype,
playing crucial roles in development, fibrosis and cancer (11,12).
The induction of EMT involves complex interactions
between signalling pathways and transcription factors, and its core
regulatory mechanisms depend on the coordinated regulation of
signalling pathways such as transforming growth factor-β (TGF-β),
Wnt and Notch (13). Among
these, TGF-β1 critically induces EMT. TGF-β activates both
canonical (Smad-dependent) and non-canonical (Smad-independent)
signalling pathways by binding to its receptors TβRI and TβRII
(14). The canonical signalling
pathway is the primary route through which TGF-β regulates EMT:
TβRII recruits and phosphorylates TβRI, which in turn
phosphorylates the key signal transducers Smad2 and Smad3.
Subsequently, the phosphorylated Smad2/3 forms heterodimeric or
trimeric complexes with Smad4, which translocate into the nucleus
and recruit co-activators such as CREB-binding protein or P300 to
regulate the target genes' transcription (15). Additionally, TGF-β mediates
non-canonical signalling pathways through mechanisms such as
phosphorylation, acetylation, ubiquitination and protein-protein
interactions. These non-Smad signalling branches can either
independently execute TGF-β-mediated biological functions or
modulate the canonical Smad signalling pathway, thereby
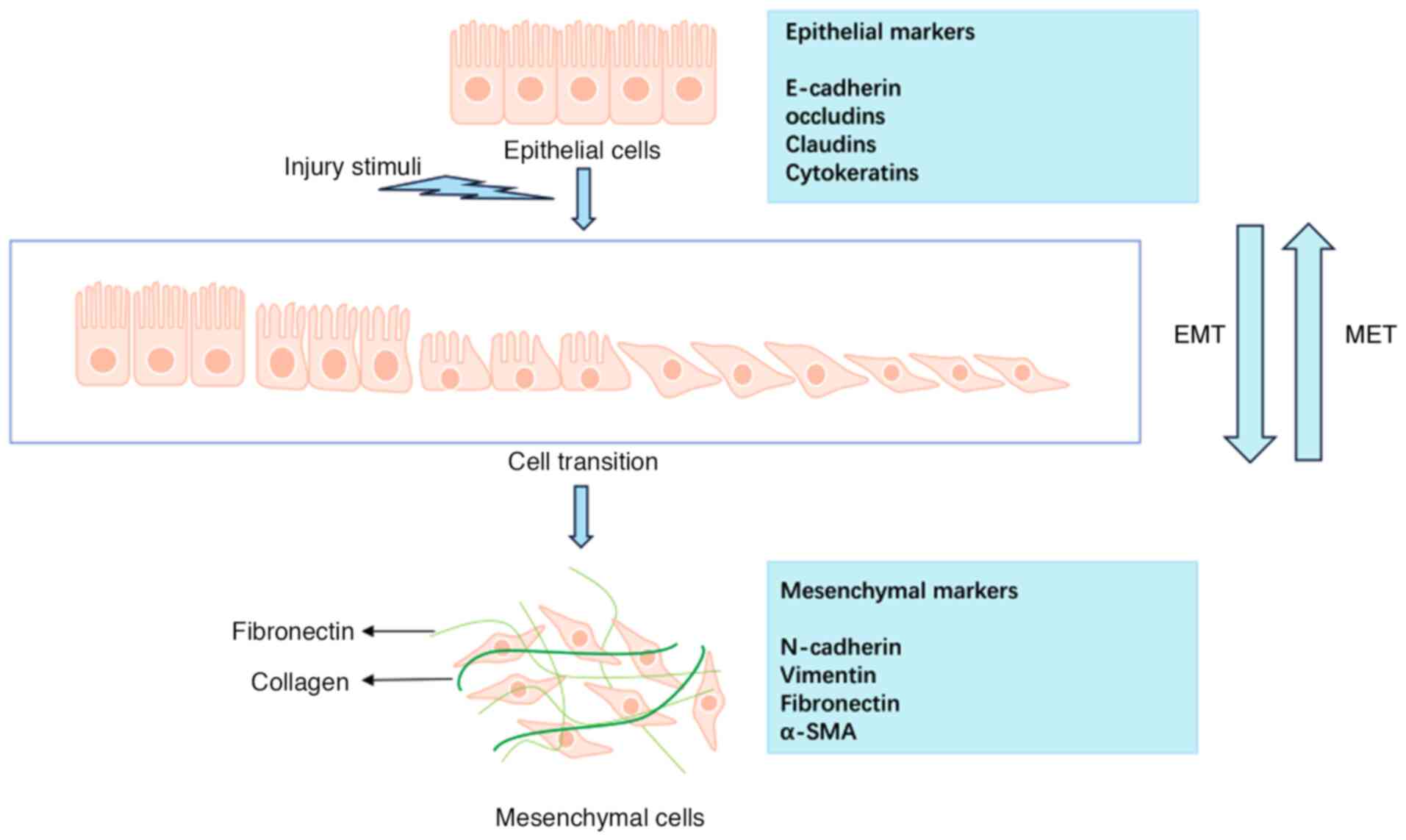
synergistically influencing the EMT process (16). As shown in Fig. 2, EMT is a reversible process in
which cells can be restored to the epithelial state by
mesenchymal-epithelial transformation (MET) (17), a feature that provides new
insights for developing related therapeutic strategies.
Necrosis of organ parenchymal cells and excessive
extracellular matrix deposition result in fibrosis, a pathological
process that causes connective tissue growth and, in certain cases,
organ sclerosis (18). Various
organs can be affected by fibrosis, and in developed nations,
mortality rates may reach 45% (19). Substantial evidence reveals that
ERS is associated with fibrosis in numerous organs. For instance,
ERS in fibrotic lung tissue is thought to contribute significantly
to aggravating the development of pulmonary fibrosis, a chronic
progressive disease in the lungs (20). A considerable increase in ERS
markers in the alveolar epithelial type II cells has been
progressively verified in patients in various studies with various
fibrotic lung disorders, such as interstitial lung disease,
asbestosis and silicosis (21).
Findings from a study showed that the UPR can accelerate the
development of liver fibrosis by inducing transporter and Golgi
organization 1-mediated hepatic stellate cell collagen I production
in an x-box binding protein 1 (XBP1)-dependent manner (22). Recently, in numerous
investigations, the pathogenic function of ERS in myocardial
fibrosis in the heart has been demonstrated. For instance, using
microarray and bioinformatics analysis, Li et al (23) investigated genetic changes in
cardiac fibrosis and discovered putative biomarkers linked to ERS.
In the hearts of pathologically cardiometabolically hypertrophied
mice, Zhang et al (24)
discovered that interferon gene stimulating drugs also induce
fibrosis and inflammation through ERS.
Currently, controlling ERS and chaperone function
may be a possible treatment strategy for fibrosis. Ghafoor et
al (25) revealed that lung
fibroblasts had higher expression levels of zinc finger CCCH-type
containing 4 protein (ZC3H4) and sigma 1 receptor (sigmar1), two
novel members of the zinc-finger protein family, and that
silica-enhanced fibroblast activation was reduced by ZC3H4
knockdown and ERS inhibition. Based on their findings, Sigmar1 and
ZC3H4 may be new therapeutic targets for silicosis (25). In addition, Chen et al
(26) showed that crystalline
silica-induced lung fibrosis is reduced when ERS is inhibited by
focusing on the IRE1α-thioredoxin domain containing 5 pathway. Sun
et al (20) reported that
lactic acid causes cellular fibrosis by activating caspase-12 via
the ATF4-C/EBP homologous protein (CHOP) axis mechanism. Since
caspase-12 induces apoptosis and promotes fibrosis, ERS inhibitors
can successfully prevent pulmonary fibrosis and alveolar epithelial
cell death, providing a new possible treatment target for pulmonary
fibrosis. Thalidomide has anti-inflammatory and immunomodulatory
properties. It reduces inflammatory responses and the ERS and
Toll-like receptor 4-nuclear factor κB (NF-κB) pathways in mice
with silicosis and silica-stimulated alveolar macrophage cell line
MH-S cells (27). Yang et
al (28) reported in their
study that ERS contributes to lung fibrosis by altering
lung-resident mesenchymal/stromal cells (LR-MSC) and converting
them into myofibroblasts. C/EBP homologous protein (CHOP) plays a
crucial role in this process, and the findings reveal that
medications that target CHOP or treatments involving the use of
CHOP to knock down LR-MSC may be effective ways to treat lung
fibrosis. Severe ERS is associated with liver fibrosis and
cirrhosis, which activate PERK and IRE1α, contributing to the
disease pathology. Therapeutic inhibition of the UPR pathway
reduces the severity of cirrhosis and liver fibrosis (29). Diacerein improves rat liver
damage caused by cholestasis by blocking the high-mobility group
box 1/receptor for advanced glycation end-products/NF-κB/JNK
pathway and ERS fibrosis (30).
Therefore, ERS modulation may be a promising therapeutic approach
to halt the advancement of fibrosis as the understanding of the
connection between ERS and fibrosis continues to develop.
EMT is a natural process of organ development and
wound repair through finely programmed processes (31). Reports show that numerous
fibrotic diseases are also associated with EMT, which has been
linked to inflammatory bowel disease, in which recurrent intestinal
inflammatory stimuli set off mucosal repair responses that result
in the deposition of extracellular matrix and the establishment of
fibrosis (32). The molecular
pathogenesis of subretinal fibrosis in neovascular age-related
macular degeneration is a complex process in which the EMT of
retinal pigment epithelium plays a critical role (33). Researchers have discovered that
fibrosis is considerably decreased when transcription factors or
signalling associated with EMT are inhibited (34). For instance, in a laser-induced
animal model, luteolin suppresses Smad2/3 and Yes-associated
protein signalling, thereby preventing EMT in the retinal pigment
epithelium (35). EMT in
diabetic nephropathy is driven by lactate through the
H3K14la/Kruppel-like factor (KLF)5 pathway. Furthermore,
renal-specific knockdown and pharmacological inhibition of KLF5
decreased EMT formation and mitigated the fibrosis associated with
diabetic nephropathy (36). The
catalytic subunit of the DNA-dependent protein kinase prevents EMT
in radiation-induced lung fibrosis through the ubiquitination and
degradation of Twist1 (37). By
controlling Smad-dependent and non-dependent signalling pathways in
proximal tubular cells that depend on TGF-β1 for EMT, chrysin has
been shown to prevent cyclosporine A-induced renal
tubulointerstitial fibrosis (38). Targeting TGF-β signalling appears
to be a promising therapeutic approach because of the crucial role
that TGF-β and its downstream molecules play in fibrosis and cancer
development (34).
ERS is essential for the development, spread and
treatment response of cancer. High levels of expression and
activation of ERS indicators, including IRE1, PERK, ATF6 and GRP78,
have been observed in various human malignancies, as reported in
several studies (39-43). ERS is produced when tumour cells
are exposed to both extrinsic and intrinsic factors that can change
protein homeostasis. Prolonged ERS of greater than moderate
intensity can, among other effects, promote cancer cell
transformation and proliferation, which in turn control tumour
growth and metastasis, among others (44). A current strategy to mitigate the
protective effects of ERS on cancer would be to target ERS
(44). The ERS-related IRE1/CHOP
pathway may be the mechanism by which Tongguanteng injection
combats osteosarcoma (45).
Through ERS and mitochondrial malfunction, laminarin, a
β-1,3-glucan obtained from brown algae, suppresses the development
of ovarian cancer cells (46).
Currently, numerous compounds are being created to target the three
UPR sensors. However, the elements that influence which sensors
behave as pro-or anti-apoptotic signals are unknown. Tumorigenesis,
progression, metastasis, immunological escape and radiation
resistance are all signs that cancer cells are using adaptive
responses to survive overstimulation. Nevertheless, tumour death
results from prolonged or severe stimulation (47). The complexity of the UPR as a
therapeutic target is highlighted by its dual roles in adaptation
and disease. Hence, effective modulation of these pathways may
delay disease progression and provide useful possibilities for
establishing novel therapeutic approaches by restoring ER function
or inducing controlled apoptosis in cancer cells.
EMT, in which tumour cells change from an epithelial
to a mesenchymal form, gaining motility, invasion and metastasis,
is a defining feature of tumour plasticity (48). Reportedly, the function of EMT is
driven by conserved zinc finger transcription factors, including
Snail, Zeb-1 and Slug, which are regulated by upstream TGF-β
activity. Cells that receive EMT grow and degrade from the
surrounding microenvironment before migrating from the original
site (49). Clinically, focusing
on EMT can have several advantages, including lowering chemotherapy
resistance and preventing metastases (50). Inhibiting important EMT
signalling pathways, preventing interactions with the tumour
microenvironment (TME) and reversing EMT by promoting MET are
possible therapeutic approaches (51). A well-known strategy to counter
EMT is to interfere with TGF β signalling. By blocking the EMT
process, galunisertib, a tiny TGF-βR1 inhibitor, can be potentially
used for treating several malignancies. Findings from clinical
trials have shown its effectiveness, e.g., when combined with
sorafenib for hepatocellular carcinoma and gemcitabine for advanced
pancreatic cancer (52). By
altering the matrix metalloproteinase and adenosine
monophosphate-activated protein kinase (AMPK)/NF-κB pathways,
specifically the upregulation of epithelial E-calmodulin and the
downregulation of mesenchymal waveform protein, the natural
compound methyl gallate prevents the proliferation, migration,
invasion and EMT of hepatocellular carcinoma cells (51). However, targeting
cancer-associated EMT specifically while preserving normal EMT
processes that are essential for tissue regeneration and repair is
a challenge; nonetheless, a thorough understanding of EMT
mechanisms may offer fresh insights to develop future medications
and therapeutic approaches.
ERS regulates the EMT process through three core
pathways of UPR (IRE1-XBP1, PERK-eIF2α and ATF6), and this
regulatory network demonstrates high conservation and specificity
across different disease models (60). Substantial evidence indicates a
complex regulatory network between ERS and EMT: Chemotherapeutic
agents such as cisplatin induce significant upregulation of ERS
markers (Bip, Chop, protein disulfide-isomerase) and EMT markers
(Vimentin, Snail) in multiple tissues (lung, liver, kidney), a
phenomenon also observed in primary lung adenocarcinoma patient
samples, confirming the universality of ERS-induced EMT (53). At the molecular level, lysyl
oxidase-like 2 overexpression activates the IRE1-XBP1 pathway,
leading to the spliced form of XBP1 (XBP1s) specifically
recognizing and binding to the unfolded protein response element
core motif (ACGTG) in the promoter regions of key EMT transcription
factors (SNAI1, SNAI2, ZEB2 and transcription factor 3 (TCF3).
Through chromatin immunoprecipitation and luciferase reporter
assays, researchers precisely mapped XBP1s binding sites at SNAI1
(-550 bp), SNAI2 (-526 bp), ZEB2 (-2,269/-658 bp) and TCF3 (-2,230
bp) promoters, demonstrating that this binding significantly
enhances transcriptional activity (61). Similar regulatory mechanisms have
been identified in various cancers: In breast cancer, XBP1 promotes
EMT and invasion by directly activating the Snail promoter and is
significantly associated with poor prognosis; in glioblastoma
multiforme, the IRE1-XBP1 axis not only upregulates EMT markers
(Vimentin, ZEB1) but also promotes the secretion of chemokines
(C-X-C motif chemokine ligand 2 and C-C motif chemokine ligand 2)
(62); in hepatocellular
carcinoma, a XBP1s/Twist/Snail cascade regulatory axis is formed
(63). Notably, viral infections
such as respiratory syncytial virus can also activate this pathway
by inducing SNAI1 expression (5.8-fold mRNA increase) through
IRE1α-XBP1s, a process specifically inhibited by the IRE1α
inhibitor KIRA8 (64). Beyond
the IRE1-XBP1 pathway, the PERK-eIF2α axis promotes EMT by
upregulating ZEB1 in fibrotic diseases (65,66), while the ATF6 pathway drives EMT
through distinct mechanisms in cervical and colorectal cancers
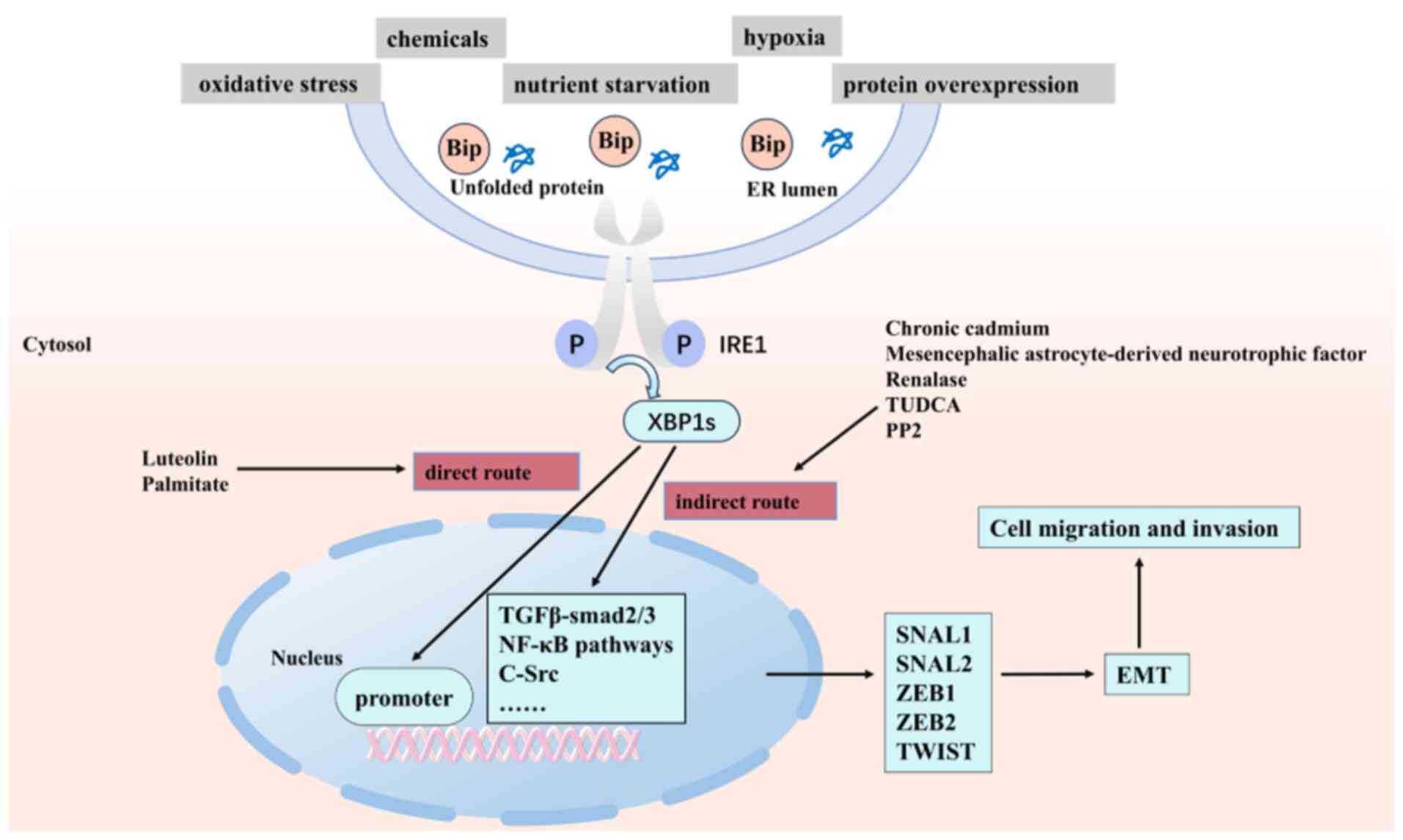
(67,68). As shown in Fig. 3, these findings systematically
revealed the core mechanism of the ERS-UPR-EMT regulatory network:
The XBP1s drive the EMT process by either directly binding to EMT
transcription factor promoters or regulating intermediate signaling
pathways (TGF-β/Smad, NF-κB, SRC, etc.). Based on these findings,
several targeted intervention strategies have shown therapeutic
potential, including ginsenoside's getinoblastoma 1-mediated
inhibition of the BiP/eIF2α/CHOP axis (69), mesenchymal stem cells modulating
the IRE1α branch (70) and
luteolin-induced EMT reversal through regulation of
E-cadherin/N-cadherin expression (71). These studies not only
systematically elucidate the core regulatory mechanisms of the
ERS-UPR-EMT network but also provide a crucial theoretical
foundation for developing precise therapeutic strategies for
fibrosis and cancer.
The TGF-β/Smad signaling pathway establishes a
complex bidirectional interaction network between ERS and EMT
through a multilevel molecular regulatory mechanism. When TGF-β
ligand binds to TβRII receptor, it induces the phosphorylation of
the GS structural domain of TβRI, which in turn specifically
catalyzes the phosphorylation of the C-terminal SSXS motif of
Smad2/3, promotes the formation of its complex with Smad4 and
translocates it to the nucleus, and regulates the expression of key
transcription factors of EMT (such as SNAIL1/2, ZEB1/2 and TWIST)
expression (72-74). Notably, ERS can activate this
pathway through multiple mechanisms: PM2.5 exposure induces
ERS-dependent TGF-β1/Smad3 signaling activation by decreasing
caveolin 1 protein levels (75);
Grp78 knockdown leads to its dissociation from TβRI, which
deregulates the inhibition of receptor activation (76); and cadmium exposure activates
Smad3 signaling directly by phosphorylating IRE1α. These findings
reveal an important role for ERS as an upstream regulator of the
TGF-β/Smad pathway (77).
However, the direction of regulation varies significantly in
different tissues: In endometrium and ovarian granulosa cells, ERS
explicitly acts as an upstream activator of the TGF-β/Smad pathway,
whereas in other models of fibrosis, a positive feedback loop may
develop, whereby activation of the TGF-β/Smad pathway in turn leads
to a further induction of ERS (78,79). This tissue specificity may arise
from cell-type-specific differences in the regulator's expression
or microenvironmental differences. Of particular interest is the
finding that SIRT7 activates the IRE1α-XBP1 axis to promote PD-L1
expression while inhibiting classical TGF-β signaling through
deacetylation of SMAD4, a finding that provides a new idea for the
development of combined therapeutic strategies targeting the
ERS-TGFβ-EMT network (80).
Future studies need to deeply resolve the molecular basis of the
differences in the direction of regulation in different tissues and
develop novel modulators that can specifically intervene in
pathological interactions without affecting physiological
functions.
Nrf2, a core regulator of cellular antioxidant
defence, maintains redox homeostasis by coordinating the dynamic
balance of the kelch-like ECH-associated protein 1 (Keap1)-E3
ubiquitin ligase scaffold cullin 3 (Cul3) ubiquitination system
(81). Under homeostatic
conditions, Keap1 promotes ubiquitination degradation of Nrf2 by
binding to the ETGE/DLG motifs of Nrf2 through its Kelch structural
domain (82); whereas, when the
PERK pathway is activated, it induces the expression of ATF4 by
phosphorylating eIF2α, which directly binds to the
antioxidant-responsive elements of the Nrf2 promoter and
upregulates Nrf2 transcription. Meanwhile, PERK kinase activity
also blocks Keap1 interactions with Nrf2 by phosphorylating the
Ser40 site of Nrf2, a dual mechanism that synergistically
stabilizes the Nrf2 protein (1,83). This protective mechanism is
hijacked in the TME: In lung cancer models, KRAS mutations enhance
Nrf2 nuclear translocation through activation of PI3K-AKT
signaling, leading to abnormally low ROS levels, which in turn
promotes EMT through the mechanisms of i) ROS-dependent
downregulation of the pro-apoptotic factor raf kinase inhibitor
protein, and ii) upregulation of the expression of SNAIL1 promoter
by direct binding to it via ATF4 to create a pro-metastatic
phenotype (84-86). ROS is not only a core mediator of
the ERS-EMT network, but its dynamic balance of production and
clearance determines cell fate. In a cadmium exposure-induced EMT
model of prostate cancer, chronic ROS accumulation activates the
ERS-Smad pathway through the following pathways: i) ROS induces
oxidative modification of the Cys931 site of the IRE1α kinase
structural domain, which triggers its autophosphorylation and tumor
necrosis factor receptor associated factor 2 (TRAF2) recruitment,
and then activates the JNK/NF-κB pathway to synergistically
upregulate TGF-β1; ii) mitochondrial ROS are activated through the
activation of the apoptosis signal-regulating kinase 1 (ASK1)-MAPK
kinase 4 pathway phosphorylates the linker region (Thr179/Ser204)
of Smad3, promoting its nuclear translocation and binding to the
Snail promoter. This TGF-β-independent Smad activation mechanism
can be specifically blocked by N-acetylcysteine (NAC), confirming
the critical role of the ROS-ERS axis (77). A similar mechanism is seen in
environmental toxicology models: the PM2.5 component 1-nitropyrene
activates the PERK-ATF4-CHOP axis via NADPH oxidase 4-dependent ROS
generation, inducing downregulation of GRP78 expression in alveolar
epithelial cells and dissociation of the E-cadherin/β-catenin
complex, leading to EMT and interstitial fibrosis, a process that
can be reversed by NAC or 4-PBA (ERS inhibitors) to reverse the
process (87). Therapeutic
strategies targeting this pathway need to take into account tissue
specificity: In KRAS-mutant tumors, lignans restore ROS levels and
induce pro-apoptotic signaling by competitively binding to the Neh1
structural domain of Nrf2 and disrupting its heterodimerization
with musculoaponeurotic fibrosarcoma proteins (86); whereas, in liver fibrosis, short
hairpin RNAs targeting mitochondrial calcium uniporter regulator 1
inhibit the ROS/Nrf2/Notch1 axis and the EMT processes (88). These findings systematically
reveal a sophisticated regulatory network between redox homeostasis
and EMT plasticity, providing a molecular basis for the development
of precision therapies based on the Nrf2-ROS-ERS node.
Programmed cell death is an actively regulated
cellular process initiated under specific physiological and
pathological conditions, primarily comprising three major forms:
Apoptosis, autophagy and ferroptosis (89). Apoptosis serves as a crucial
mechanism for eliminating abnormal or damaged cells (90), while autophagy represents a
highly conserved process that maintains intracellular homeostasis
by degrading dysfunctional cellular components such as protein/DNA
aggregates and abnormal organelles (91), and ferroptosis is a distinctive
form of iron-dependent regulated cell death driven by lipid
peroxidation (92). These cell
death modalities, together with ERS and EMT, constitute a complex
regulatory network that plays pivotal roles in fibrosis and tumor
progression.
Over time, the activated UPR engages multiple
cellular signaling pathways that largely determine cell fate
decisions, including autophagy, apoptosis, ferroptosis and
inflammation (93). Studies in
inflammatory stress models demonstrate synchronous activation of
all three UPR sensors (ATF6, IRE1α, PERK) in conjunctival
epithelial cells and fibroblasts, accompanied by upregulation of
inflammatory and apoptotic markers (IL-1β, BAX, caspase-3), along
with increased expression of autophagy-related protein light chain
3A/B in epithelial cells and lysosomal marker lysosome-associated
membrane protein 1 in fibroblasts. These findings confirm the
molecular mechanisms through which ERS remodels cell fate by
integrating multiple stress signaling pathways (94). Under physiological conditions,
the UPR precisely balances survival and death signals to determine
cellular outcomes. However, disruption of this equilibrium through
chronic UPR activation or functional impairment may contribute to
tumorigenesis (95).
In apoptosis regulation, persistent ERS activates
pro-apoptotic UPR pathways. Substantial evidence demonstrates that
two key UPR kinases, PERK and IRE1α, promote apoptosis through
distinct mechanisms (96,97).
Specifically, chronic ERS triggers pro-apoptotic programs via the
PERK-eIF2α-ATF4 signaling axis: ATF4 directly binds to C/EBP-ATF
response elements in the CHOP promoter to enhance its
transcription, while CHOP suppresses the anti-apoptotic protein
Bcl-2 and induces pro-apoptotic proteins building information
modelling/p53 upregulated modulator of apoptosis, ultimately
leading to mitochondrial membrane potential collapse and
caspase-9/3 cascade activation. Concurrently, IRE1α recruits TRAF2
through kinase domain autophosphorylation to form the
IRE1α-TRAF2-ASK1 complex, thereby activating JNK (Thr183/Tyr185
phosphorylation) and promoting c-Jun-mediated transcription of
pro-apoptotic genes, establishing a dual apoptotic driving
mechanism (98-100). Notably, certain natural
compounds such as the aqueous extract of Descuraniae Semen can
alleviate inflammation and apoptosis by modulating proteasomal
degradation and UPR pathways (101).
TGF-β serves as a central regulatory hub with dual
roles in EMT and apoptosis. Shenkang injection and rosiglitazone
ameliorate diabetic tubulopathy by inhibiting EMT and ERS-induced
apoptosis (102). The EMT
process is regulated by various molecular pathways that also
control autophagy. Research reveals that autophagy exhibits
bidirectional regulatory effects: It can facilitate EMT progression
(103), while also attenuating
EMT by suppressing SNAIL/SLUG overexpression and
ROS-NF-κB-hypoxia-inducible factor-1α pathway activation,
indicating the potential therapeutic value of autophagy modulators
(104).
Multiple pharmacological agents and nanomaterials
further demonstrate the complexity of this regulatory network.
Amlodipine exerts therapeutic effects by inducing ERS-mediated
apoptosis and inhibiting EMT (105). Piperine displays anticancer
activity in colorectal adenocarcinoma by modulating arf-like
protein 3 expression to influence cell cycle progression, EMT
pathways and ERS (106). Zinc
oxide nanoparticles suppress malignant progression and
chemoresistance in tumor cells by activating ERS and promoting
autophagy (107). Additionally,
C1q/TNF-related protein 4 alleviates high glucose-induced apoptosis
and ERS while normalizing EMT markers through
AMPK/autophagy-mediated signaling (108). ZC3H4, a member of the CCCH zinc
finger protein family, influences pulmonary fibrosis progression
via ERS and autophagy-induced endothelial-mesenchymal transition
(109).
Ferroptosis, as a newly discovered cell death
modality, shows close associations with both ERS and EMT. Altered
expression of key EMT transcription factors can modulate tumor cell
susceptibility to ferroptosis, while ERS participates in
ferroptosis regulation through oxidative stress and iron metabolism
(110,111). In diabetic nephropathy, the
ERS-XBP1-HMG-CoA reductase degradation 1-Nrf2 pathway has been
shown to promote EMT progression via ferroptosis (112), providing novel insights into
disease mechanisms. Ginsenoside Rg1 ameliorates cigarette
smoke-induced ferroptosis in chronic obstructive pulmonary disease
by suppressing ERS through modulation of the PERK/ATF4 axis
(113). Mechanistically,
ferroptosis can induce ERS and promote apoptosis through multiple
pathways, whereas persistent ERS leads to accumulation of ROS and
Fe2+ (essential for ferroptosis) via UPR and disrupts
redox homeostasis to enhance ferroptosis sensitivity (114,115).
In conclusion, the ERS-EMT-programmed cell death
network forms a sophisticated regulatory axis in various diseases.
Elucidating the dynamic mechanisms underlying this network and
developing novel therapeutic strategies that simultaneously target
multiple pathways will open new avenues for precision medicine.
Future research should focus on deciphering the intricate
interactions between different forms of programmed cell death and
exploring their clinical translation potential.
The TME is the environment that tumour cells live in
during tumorigenesis, development and metastasis. A hostile TME
changes the ER's ability to fold proteins, which promotes the
build-up of misfolded or unfolded proteins and, ultimately, ERS
(116). It can be caused by
either intrinsic tumour characteristics (such as high metabolic
demand, hypoxia, nutrient limitation and acidosis) or external
stressors (such as chemotherapy and radiation). By altering the
TME, ERS can reduce the effectiveness of anticancer therapies, such
as immunotherapy (117). In the
TME, ERS can impact immune cell function and tumour growth
(118). Through their influence
on cellular communication and immunological responses, tiny
extracellular vesicles can play a critical function in controlling
the TME, and ERS contributes to the evolution of hepatocellular
carcinoma (118). Different
stromal cell types in the TME, together with biochemical and
biophysical variables, have been shown to impact the EMT program,
influencing tumour growth (119,120). The release of catecholamines in
the TME can also regulate the EMT and metabolic changes in cancer
cells by activating EMT transcription factors such as ZEB1, Snail
or Slug/SNAI2 (121).
Maintaining a healthy TME and developing therapeutic or multidrug
resistance depends on the protein kinase C, Notch and TGF-β
signalling pathways (117). EMT
is reciprocally triggered by exosomes generated from the TME in
tumour cells, leading to metastasis and treatment resistance
(39). In vitro, enzymes
that support EMT, migration and invasion of cancer cells are
activated by extracellular ATP and adenosine in the TME, which bind
to specific receptors (122).
Findings from studies reveal that cancer therapy is made possible
by focusing on EMT-induced signals in the TME (119). There is a strong relationship
between TME and both ERS and tumour cell EMT.
Metabolic reprogramming within the TME is a critical
adaptive feature exhibited by tumour cells in response to
microenvironmental changes. This reprogramming serves as a key
driver of tumour progression (123). SEC63, a novel regulator of
metabolism in hepatocellular carcinoma, has been shown to
considerably contribute to maintaining ER homeostasis through its
mediation of metabolic reprogramming. In the nucleus, SEC63
activates UPR targets and increases the acetylation of SMAD3,
inducing Snail1 expression and promoting cancer cell metastasis
(124). In animal models of
chronic colitis transitioning to colon cancer, combining ERS and
metabolic reprogramming drives the malignant transformation of
chronic inflammation (125). In
colorectal cancer, the loss of stromal interaction molecule 2
alters ER Ca2+ levels and activates the cMyc and
PERK/ATF4 branches of the ERS pathway, leading to transcriptional
reprogramming and metabolic remodelling, which in turn drive tumour
growth and metastasis (126).
Activating STING, a stimulator of interferon genes, is closely
associated with ERS induction and related inflammatory responses.
The STING-dependent pathway is involved in the metabolic
reprogramming of macrophages and contributes to the establishment
and maintenance of a robust inflammatory phenotype (127). Metabolic reprogramming in
cancer cells alters the use of energy sources and regulates the
expression of genes associated with EMT (128). The mesenchymal stem cell-like
phenotype generated by EMT enables cancer cells to detach from the
primary tumour site, acquiring motility associated with metastatic
and invasive capabilities. Research findings indicate that
metabolic reprogramming is a core driver of EMT initiation and
propagation (129).
Additionally, changes in glycolysis, mitochondrial function, lipid
metabolism and choline metabolism functionally promote
TGF-β-induced EMT (130). EMT
reshapes metabolic pathways by switching metabolic reprogramming,
which supports transcription regulated by epigenetic mechanisms and
reduces the risk of ferroptosis (131). Cellular processes such as
metabolic reprogramming, EMT and ERS significantly increase the
complexity of cancer by driving uncontrolled cell proliferation,
metastasis and therapy resistance (132). Metabolic reprogramming, EMT and
ERS drive tumour progression through complex interactions within
the TME. However, their molecular mechanisms should be further
elucidated. In addition, their potential applications in cancer
therapy should be explored. Targeting these intricate cellular
processes may provide novel therapeutic strategies to halt cancer
progression.
From the perspective of molecular mechanisms, the
bidirectional regulation of EMT by ERS depends on the dynamic
balance of three key pathways: i) The IRE1α-XBP1 branch directly
recognizes the UPRE core motif (ACGTG) in the promoter regions of
EMT transcription factors through its spliced form XBP1s, with its
binding affinity regulated by the methylation status of CpG
islands; ii) the PERK-eIF2α-ATF4 axis requires sustained activation
to induce EMT via ZEB1; and iii) the ATF6 pathway exhibits a rapid
response in cervical cancer but requires prolonged activation in
colorectal cancer. This spatiotemporal specificity in regulation
provides an important basis for developing targeted drugs, as
exemplified by β-asarone, which selectively activates the ATF6
branch (without affecting the IRE1α and PERK pathways) to inhibit
bladder cancer EMT (133).
In terms of therapeutic strategies, a multi-tiered
intervention approach has been developed based on in-depth
understanding of the ERS-EMT network: First-generation ERS
modulators (e.g., 4-PBA, tauroursodeoxycholic acidnon-specifically
inhibit EMT by maintaining ER homeostasis); second-generation
targeted drugs (e.g., galunisertib) precisely block the kinase
domain of TGF-β receptor I; the most promising prospect lies in
achieving synergistic therapy by simultaneously modulating UPR
(e.g., inhibiting IRE1α RNase activity) and EMT signaling (e.g.,
blocking ZEB1 nuclear translocation), which would represent a
breakthrough in treating fibrosis, tumors and other diseases
(70,106,134-136). Notably, metabolic reprogramming
in the TME significantly impacts treatment efficacy, as
SEC63-mediated SMAD3 acetylation can increase tumor cell
sensitivity to ERS inhibitors by 3-5-fold (124).
Despite these advances, several key scientific
questions remain unanswered: The structural basis for the marked
selectivity of XBP1s in recognizing different EMT transcription
factor promoters (its binding affinity for SNAIL is 7-fold higher
than for TWIST) is still unclear; the crosstalk between the TGF-β
pathway and UPR exhibits tissue specificity, showing linear
activation in endometrial cells but forming a positive feedback
loop in pulmonary fibrosis and the molecular determinants of these
differences need to be elucidated; and the mechanisms by which
epigenetic regulation influences the reversibility of ERS and EMT
require further exploration.
This review provides an in-depth analysis of the
molecular regulatory network between ERS and EMT, revealing
multi-layered precise regulatory mechanisms. Based on these
molecular mechanisms, various targeted intervention strategies have
demonstrated therapeutic potential. For instance, ginsenoside Rb1
specifically inhibits the BiP/eIF2α/CHOP axis, luteolin modulates
E-cadherin/N-cadherin expression and lignan compounds competitively
bind to the Neh1 domain of Nrf2 (35,69,86). These interventions precisely
target key regulatory nodes at the molecular level. Future research
needs to further elucidate the structural basis of regulatory
direction differences in various tissue microenvironments and
develop novel modulators that can specifically intervene in
pathological interactions without affecting physiological
functions, thereby providing more effective strategies for
precision therapy.
Not applicable.
HC was the main contributor in writing the original
draft. SY performed investigation. YG and QH were responsible for
resources. WS acquired funding. All authors were involved in
reviewing and editing the manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The study was funded by the Basic Research Projects of Science
and Technology Department of Guizhou Province [grant no. Qian Ke
He-zk(2022)-659].
|
1
|
Santamaría PG, Mazón MJ, Eraso P and
Portillo F: UPR: An upstream signal to EMT induction in cancer. J
Clin Med. 8:6242019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marconi GD, Fonticoli L, Rajan TS,
Pierdomenico SD, Trubiani O, Pizzicannella J and Diomede F:
Epithelial-mesenchymal transition (EMT): The type-2 EMT in wound
healing, tissue regeneration and organ fibrosis. Cells.
10:15872021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brabletz S, Schuhwerk H, Brabletz T and
Stemmler MP: Dynamic EMT: A multi-tool for tumor progression. EMBO
J. 40:e1086472021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kropski JA and Blackwell TS: Endoplasmic
reticulum stress in the pathogenesis of fibrotic disease. J Clin
Invest. 128:64–73. 2018. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Uddin MS, Tewari D, Sharma G, Kabir MT,
Barreto GE, Bin-Jumah MN, Perveen A, Abdel-Daim MM and Ashraf GM:
Molecular mechanisms of ER stress and UPr in the pathogenesis of
Alzheimer's disease. Mol Neurobiol. 57:2902–2919. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yap KN, Yamada K, Zikeli S, Kiaris H and
Hood WR: Evaluating endoplasmic reticulum stress and unfolded
protein response through the lens of ecology and evolution. Biol
Rev Camb Philos Soc. 96:541–556. 2021. View Article : Google Scholar
|
|
7
|
Sims SG, Cisney RN, Lipscomb MM and Meares
GP: The role of endoplasmic reticulum stress in astrocytes. Glia.
70:5–19. 2022. View Article : Google Scholar :
|
|
8
|
Hu H, Tian M, Ding C and Yu S: The C/EBP
homologous protein (CHOP) transcription factor functions in
endoplasmic reticulum Stress-induced apoptosis and microbial
infection. Front Immunol. 9:30832018. View Article : Google Scholar
|
|
9
|
Wang P, Li J, Tao J and Sha B: The luminal
domain of the ER stress sensor protein PERK binds misfolded
proteins and thereby triggers PERK oligomerization. J Biol Chem.
293:4110–4121. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sato R, Semba T, Saya H and Arima Y:
Concise review: Stem cells and Epithelial-mesenchymal transition in
cancer: Biological implications and therapeutic targets. Stem
Cells. 34:1997–2007. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu R, Won JY, Kim CH, Kim DE and Yim H:
Roles of the phosphorylation of transcriptional factors in
Epithelial-mesenchymal transition. J Oncol. 2019:58104652019.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tomecka P, Kunachowicz D, Górczyńska J,
Gebuza M, Kuźnicki J, Skinderowicz K and Choromańska A: Factors
determining Epithelial-mesenchymal transition in cancer
progression. Int J Mol Sci. 25:89722024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin YT and Wu KJ: Epigenetic regulation of
epithelial-mesenchymal transition: Focusing on hypoxia and TGF-β
signaling. J Biomed Sci. 27:392020. View Article : Google Scholar
|
|
14
|
Sheikh KA, Amjad M, Irfan MT, Anjum S,
Majeed T, Riaz MU, Jassim AY, Sharif EAM and Ibrahim WN: Exploring
TGF-β signaling in cancer progression: Prospects and therapeutic
strategies. Onco Targets Ther. 18:233–262. 2025. View Article : Google Scholar :
|
|
15
|
Ding C, Liu B, Yu T, Wang Z, Peng J, Gu Y
and Li Z: SIRT7 protects against liver fibrosis by suppressing
stellate cell activation via TGF-β/SMAD2/3 pathway. Biomed
Pharmacother. 180:1174772024. View Article : Google Scholar
|
|
16
|
Zhang YE: Non-smad signaling pathways of
the TGF-β family. Cold Spring Harb Perspect Biol. 9:a0221292017.
View Article : Google Scholar
|
|
17
|
Kahlert UD, Joseph JV and Kruyt FAE:
EMT-and MET-related processes in nonepithelial tumors: Importance
for disease progression, prognosis, and therapeutic opportunities.
Mol Oncol. 11:860–877. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Long Y, Niu Y, Liang K and Du Y:
Mechanical communication in fibrosis progression. Trends Cell Biol.
32:70–90. 2022. View Article : Google Scholar
|
|
19
|
Huang C and Ogawa R: The vascular
involvement in soft tissue Fibrosis-lessons learned from
pathological scarring. Int J Mol Sci. 21:25422020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun Z, He W, Meng H, Ji Z, Qu J and Yu G:
Lactate activates ER stress to promote alveolar epithelial cells
apoptosis in pulmonary fibrosis. Respir Res. 25:4012024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bradley KL, Stokes CA, Marciniak SJ,
Parker LC and Condliffe AM: Role of unfolded proteins in lung
disease. Thorax. 76:92–99. 2021. View Article : Google Scholar
|
|
22
|
Maiers JL, Kostallari E, Mushref M, de
Assuncao TM, Li H, Jalan-Sakrikar N, Huebert RC, Cao S, Malhi H and
Shah VH: The unfolded protein response mediates fibrogenesis and
collagen I secretion through regulating TANGO1 in mice. Hepatology.
65:983–998. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li W, Liu P, Liu H, Zhang F and Fu Y:
Integrative analysis of genes reveals endoplasmic reticulum
stress-related immune responses involved in dilated cardiomyopathy
with fibrosis. Apoptosis. 28:14222023. View Article : Google Scholar
|
|
24
|
Zhang Y, Chen W and Wang Y: STING is an
essential regulator of heart inflammation and fibrosis in mice with
pathological cardiac hypertrophy via endoplasmic reticulum (ER)
stress. Biomed Pharmacother. 125:1100222020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ghafoor H, Chu H, Huang J, Chen M, Wang S,
Wang J and Chao J: ZC3H4 promotes pulmonary fibrosis via an ER
stress-related positive feedback loop. Toxicol Appl Pharmacol.
435:1158562022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen X, Li C, Liu J, He Y, Wei Y and Chen
J: Inhibition of ER stress by targeting the IRE1α-TXNDC5 pathway
alleviates crystalline silica-induced pulmonary fibrosis. Int
Immunopharmacol. 95:1075192021. View Article : Google Scholar
|
|
27
|
Li Y, Cai W, Jin F, Wang X, Liu W, Li T,
Yang X, Liu H, Xu H and Yang F: Thalidomide alleviates pulmonary
fibrosis induced by silica in mice by inhibiting ER stress and the
TLR4-NF-κB pathway. Int J Mol Sci. 23:56562022. View Article : Google Scholar
|
|
28
|
Yang X, Sun W, Jing X, Zhang Q, Huang H
and Xu Z: Endoplasmic reticulum stress modulates the fate of lung
resident mesenchymal stem cell to myofibroblast via C/EBP
homologous protein during pulmonary fibrosis. Stem Cell Res Ther.
13:2792022. View Article : Google Scholar
|
|
29
|
Ajoolabady A, Kaplowitz N, Lebeaupin C,
Kroemer G, Kaufman RJ, Malhi H and Ren J: Endoplasmic reticulum
stress in liver diseases. Hepatology. 77:619–639. 2023. View Article : Google Scholar
|
|
30
|
Abdelfattah AM, Mahmoud SS, El-Wafaey DI,
Abdelgeleel HM and Abdelhamid AM: Diacerein ameliorates
cholestasis-induced liver fibrosis in rat via modulating
HMGB1/RAGE/NF-κB/JNK pathway and endoplasmic reticulum stress. Sci
Rep. 13:114552023. View Article : Google Scholar
|
|
31
|
Wu F, Yang J, Liu J, Wang Y, Mu J, Zeng Q,
Deng S and Zhou H: Signaling pathways in cancer-associated
fibroblasts and targeted therapy for cancer. Signal Transduct
Target Ther. 6:2182021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wenxiu J, Mingyue Y, Fei H, Yuxin L,
Mengyao W, Chenyang L, Jia S, Hong Z, Shih DQ, Targan SR and
Xiaolan Z: Effect and mechanism of TL1A expression on
Epithelial-mesenchymal transition during chronic Colitis-related
intestinal fibrosis. Mediators Inflamm. 2021:59270642021.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu D, Zhang C and Zhang J, Xu GT and
Zhang J: Molecular pathogenesis of subretinal fibrosis in
neovascular AMD focusing on epithelial-mesenchymal transformation
of retinal pigment epithelium. Neurobiol Dis. 185:1062502023.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Peng D, Fu M, Wang M, Wei Y and Wei X:
Targeting TGF-β signal transduction for fibrosis and cancer
therapy. Mol Cancer. 21:1042022. View Article : Google Scholar
|
|
35
|
Zhang C, Zhang Y, Hu X, Zhao Z, Chen Z,
Wang X, Zhang Z, Jin H and Zhang J: Luteolin inhibits subretinal
fibrosis and epithelial-mesenchymal transition in laser-induced
mouse model via suppression of Smad2/3 and YAP signaling.
Phytomedicine. 116:1548652023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang X, Chen J, Lin R, Huang Y, Wang Z,
Xu S, Wang L, Chen F, Zhang J, Pan K and Yin Z: Lactate drives
epithelial-mesenchymal transition in diabetic kidney disease via
the H3K14la/KLF5 pathway. Redox Biol. 75:1032462024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yan Z, Zhu J, Liu Y, Li Z, Liang X, Zhou
S, Hou Y, Chen H, Zhou L, Wang P, et al: DNA-PKcs/AKT1 inhibits
epithelial-mesenchymal transition during radiation-induced
pulmonary fibrosis by inducing ubiquitination and degradation of
Twist1. Clin Transl Med. 14:e16902024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nagavally RR, Sunilkumar S, Akhtar M,
Trombetta LD and Ford SM: Chrysin ameliorates
Cyclosporine-A-induced renal fibrosis by inhibiting TGF-β1-Induced
Epithelial-mesenchymal transition. Int J Mol Sci. 22:102522021.
View Article : Google Scholar
|
|
39
|
Zhang W, Shi Y, Oyang L, Cui S, Li S, Li
J, Liu L, Li Y, Peng M, Tan S, et al: Endoplasmic reticulum
stress-a key guardian in cancer. Cell Death Discov. 10:3432024.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhao R, Lv Y, Feng T, Zhang R, Ge L, Pan
J, Han B, Song G and Wang L: ATF6α promotes prostate cancer
progression by enhancing PLA2G4A-mediated arachidonic acid
metabolism and protecting tumor cells against ferroptosis.
Prostate. 82:617–629. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ma X, Li Y and Zhao B: Ribosomal protein
L5 (RPL5)/E2F transcription factor 1 (E2F1) signaling suppresses
breast cancer progression via regulating endoplasmic reticulum
stress and autophagy. Bioengineered. 13:8076–8086. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pavlović N and Heindryckx F: Exploring the
role of endoplasmic reticulum stress in hepatocellular carcinoma
through mining of the human protein atlas. Biology (Basel).
10:6402021.
|
|
43
|
Chen J, Lei C, Zhang H, Huang X, Yang Y,
Liu J, Jia Y, Shi H, Zhang Y, Zhang J and Du J: RPL11 promotes
non-small cell lung cancer cell proliferation by regulating
endoplasmic reticulum stress and cell autophagy. BMC Mol Cell Biol.
24:72023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Oakes SA: Endoplasmic reticulum stress
signaling in cancer cells. Am J Pathol. 190:934–946. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xue XC, Zhou YY, Xu LY, Wei LY, Hu YJ,
Yang J, Zhang XQ, Wang MY, Han YL and Chen JJ: Tongguanteng
injection exerts anti-osteosarcoma effects through the ER
stress-associated IRE1/CHOP pathway. BMC Complement Med Ther.
24:4002024. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bae H, Song G, Lee JY, Hong T, Chang MJ
and Lim W: Laminarin-derived from brown algae suppresses the growth
of ovarian cancer cells via mitochondrial dysfunction and ER
stress. Mar Drugs. 18:1522020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Labrie M, Brugge JS, Mills GB and
Zervantonakis IK: Therapy resistance: Opportunities created by
adaptive responses to targeted therapies in cancer. Nat Rev Cancer.
22:323–339. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ang HL, Mohan CD, Shanmugam MK, Leong HC,
Makvandi P, Rangappa KS, Bishayee A, Kumar AP and Sethi G:
Mechanism of epithelial-mesenchymal transition in cancer and its
regulation by natural compounds. Med Res Rev. 43:1141–1200. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Huang Y, Hong W and Wei X: The molecular
mechanisms and therapeutic strategies of EMT in tumor progression
and metastasis. J Hematol Oncol. 15:1292022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Fontana R, Mestre-Farrera A and Yang J:
Update on Epithelial-mesenchymal plasticity in cancer progression.
Annu Rev Pathol. 19:133–156. 2024. View Article : Google Scholar :
|
|
51
|
Liang H, Chen Z, Yang R, Huang Q, Chen H,
Chen W, Zou L, Wei P, Wei S, Yang Y and Zhang Y: Methyl gallate
suppresses the migration, invasion, and Epithelial-mesenchymal
transition of hepatocellular carcinoma cells via the AMPK/NF-κB
signaling pathway in vitro and in vivo. Front Pharmacol.
13:8942852022. View Article : Google Scholar
|
|
52
|
Melisi D, Garcia-Carbonero R, Macarulla T,
Pezet D, Deplanque G, Fuchs M, Trojan J, Oettle H, Kozloff M,
Cleverly A, et al: Galunisertib plus gemcitabine vs. gemcitabine
for first-line treatment of patients with unresectable pancreatic
cancer. Br J Cancer. 119:1208–1214. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Shah PP, Dupre TV, Siskind LJ and Beverly
LJ: Common cytotoxic chemotherapeutics induce
epithelial-mesenchymal transition (EMT) downstream of ER stress.
Oncotarget. 8:22625–22639. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Delbrel E, Uzunhan Y, Soumare A, Gille T,
Marchant D, Planès C and Boncoeur E: ER stress is involved in
Epithelial-To-Mesenchymal transition of alveolar epithelial cells
exposed to a hypoxic micro-environment. Int J Mol Sci. 20:12992019.
View Article : Google Scholar
|
|
55
|
Gong L, Liu G, Zhu H, Li C, Li P, Liu C,
Tang H, Wu K, Wu J, Liu D, et al: IL-32 induces
epithelial-mesenchymal transition by triggering endoplasmic
reticulum stress in A549 cells. BMC Pulm Med. 20:2782020.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Liang X, Duan N, Wang Y, Shu S, Xiang X,
Guo T, Yang L, Zhang S, Tang X and Zhang J: Advanced oxidation
protein products induce endothelial-to-mesenchymal transition in
human renal glomerular endothelial cells through induction of
endoplasmic reticulum stress. J Diabetes Complications. 30:573–579.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Han J, Pang X, Shi X, Zhang Y, Peng Z and
Xing Y: Ginkgo biloba extract EGB761 ameliorates the extracellular
matrix accumulation and mesenchymal transformation of renal tubules
in diabetic kidney disease by inhibiting endoplasmic reticulum
stress. Biomed Res Int. 2021:66572062021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhou S, Yang J, Wang M, Zheng D and Liu Y:
Endoplasmic reticulum stress regulates epithelial-mesenchymal
transition in human lens epithelial cells. Mol Med Rep. 21:173–180.
2020.
|
|
59
|
Guo B, Cheng J, Jin X, He Y and Sun X:
Different calcium ion concentrations affect epithelial mesenchymal
transformation of human peritoneal mesothelial cells via
endoplasmic reticulum stress. Zhonghua Wei Zhong Bing Ji Jiu Yi
Xue. 36:50–55. 2024.In Chinese. PubMed/NCBI
|
|
60
|
Bartoszewska S, Cabaj A, Dąbrowski M,
Collawn JF and Bartoszewski R: miR-34c-5p modulates X-box-binding
protein 1 (XBP1) expression during the adaptive phase of the
unfolded protein response. FASEB J. 33:11541–11554. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Cuevas EP, Eraso P, Mazón MJ, Santos V,
Moreno-Bueno G, Cano A and Portillo F: LOXL2 drives
epithelial-mesenchymal transition via activation of IRE1-XBP1
signalling pathway. Sci Rep. 7:449882017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lhomond S, Avril T, Dejeans N, Voutetakis
K, Doultsinos D, McMahon M, Pineau R, Obacz J, Papadodima O, Jouan
F, et al: Dual IRE1 RNase functions dictate glioblastoma
development. EMBO Mol Med. 10:e79292018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wu S, Du R, Gao C, Kang J, Wen J and Sun
T: The role of XBP1s in the metastasis and prognosis of
hepatocellular carcinoma. Biochem Biophys Res Commun. 500:530–537.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Qiao D, Skibba M, Xu X, Garofalo RP, Zhao
Y and Brasier AR: Paramyxovirus replication induces the hexosamine
biosynthetic pathway and mesenchymal transition via the IRE1α-XBP1s
arm of the unfolded protein response. Am J Physiol Lung Cell Mol
Physiol. 321:L576–L594. 2021. View Article : Google Scholar
|
|
65
|
Zhu Y, Yang M, Li XH, Xu WJ, Gao W, Chen
YH, Li JD and Li Q: Nogo-B promotes epithelial-mesenchymal
transition in lung fibrosis via PERK branch of the endoplasmic
reticulum stress pathway. Ann Transl Med. 9:5632021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Meng X, Liu K, Xie H, Zhu Y, Jin W, Lu J
and Wang R: Endoplasmic reticulum stress promotes
epithelial-mesenchymal transition via the PERK signaling pathway in
paraquat-induced pulmonary fibrosis. Mol Med Rep. 24:5252021.
View Article : Google Scholar :
|
|
67
|
Liu F, Chang L and Hu J: Activating
transcription factor 6 regulated cell growth, migration and
inhibiteds cell apoptosis and autophagy via MAPK pathway in
cervical cancer. J Reprod Immunol. 139:1031202020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Li R, Zhou H, Li M, Mai Q, Fu Z, Jiang Y,
Li C, Gao Y, Fan Y, Wu K, et al: Gremlin-1 promotes colorectal
cancer cell metastasis by activating ATF6 and inhibiting ATF4
pathways. Cells. 11:21362022. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Ni YH, Deng HF, Zhou L, Huang CS, Wang NN,
Yue LX, Li GF, Yu HJ, Zhou W and Gao Y: Ginsenoside Rb1 ameliorated
Bavachin-induced renal fibrosis via Suppressing Bip/eIF2α/CHOP
Signaling-Mediated EMT. Front Pharmacol. 13:8724742022. View Article : Google Scholar
|
|
70
|
Luo R, Wei Y, Chen P, Zhang J, Wang L,
Wang W, Wang P and Tian W: Mesenchymal stem cells inhibit
Epithelial-to-Mesenchymal transition by modulating the IRE1α branch
of the endoplasmic reticulum stress response. Stem Cells Int.
2023:44837762023. View Article : Google Scholar
|
|
71
|
Imran M, Rauf A, Abu-Izneid T, Nadeem M,
Shariati MA, Khan IA, Imran A, Orhan IE, Rizwan M, Atif M, et al:
Luteolin, a flavonoid, as an anticancer agent: A review. Biomed
Pharmacothe. 112:1086122019. View Article : Google Scholar
|
|
72
|
Pan X, Phanish MK, Baines DL and Dockrell
MEC: High glucose-induced Smad3 linker phosphorylation and CCN2
expression are inhibited by dapagliflozin in a diabetic tubule
epithelial cell model. Biosci Rep. 41:BSR202039472021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Hao Y, Baker D and Ten Dijke P:
TGF-β-mediated epithelial-mesenchymal transition and cancer
metastasis. Int J Mol Sci. 202:7672019.
|
|
74
|
Noshita S, Kubo Y, Kajiwara K, Okuzaki D,
Nada S and Okada M: A TGF-β-responsive enhancer regulates SRC
expression and epithelial-mesenchymal transition-associated cell
migration. J Cell Sci. 136:jcs2610012023. View Article : Google Scholar
|
|
75
|
Liu H, Lai W, Nie H, Shi Y, Zhu L, Yang L,
Tian L, Li K, Bian L, Xi Z and Lin B: PM2.5 triggers autophagic
degradation of Caveolin-1 via endoplasmic reticulum stress (ERS) to
enhance the TGF-β1/Smad3 axis promoting pulmonary fibrosis. Environ
Int. 181:1082902023. View Article : Google Scholar
|
|
76
|
Borok Z, Horie M, Flodby P, Wang H, Liu Y,
Ganesh S, Firth AL, Minoo P, Li C, Beers MF, et al: Grp78 loss in
epithelial progenitors reveals an Age-linked role for endoplasmic
reticulum stress in pulmonary fibrosis. Am J Respir Crit Care Med.
201:198–211. 2020. View Article : Google Scholar :
|
|
77
|
Hu W, Xia M, Zhang C, Song B, Xia Z, Guo
C, Cui Y, Jiang W, Zhang S, Xu D and Fang J: Chronic cadmium
exposure induces epithelial mesenchymal transition in prostate
cancer cells through a TGF-β-independent, endoplasmic reticulum
stress induced pathway. Toxicol Lett. 353:107–117. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Bao M, Feng Q, Zou L, Huang J, Zhu C and
Xia W: Endoplasmic reticulum stress promotes endometrial fibrosis
through the TGF-β/SMAD pathway. Reproduction. 165:171–182. 2023.
View Article : Google Scholar
|
|
79
|
Takahashi N, Harada M, Hirota Y, Nose E,
Azhary JM, Koike H, Kunitomi C, Yoshino O, Izumi G, Hirata T, et
al: Activation of endoplasmic reticulum stress in granulosa cells
from patients with polycystic ovary syndrome contributes to ovarian
fibrosis. Sci Rep. 7:108242017. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yi X, Wang H, Yang Y, Wang H, Zhang H, Guo
S, Chen J, Du J, Tian Y, Ma J, et al: SIRT7 orchestrates melanoma
progression by simultaneously promoting cell survival and immune
evasion via UPR activation. Signal Transduct Target Ther.
8:1072023. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Adamson RJ, Payne NC, Bartual SG,
Mazitschek R and Bullock AN: Structural and biochemical
characterization establishes a detailed understanding of KEAP1-CUL3
complex assembly. Free Radic Biol Med. 215–225. 2004.
|
|
82
|
Raghunath A, Nagarajan R, Sundarraj K,
Palanisamy K and Perumal E: Identification of compounds that
inhibit the binding of Keap1a/Keap1b Kelch DGR domain with Nrf2
ETGE/DLG motifs in zebrafish. Basic Clin Pharmacol Toxicol.
125:259–270. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhang H, Feng Y, Si Y, Lu C, Wang J, Wang
S, Li L, Xie W, Yue Z, Yong J, et al: Shank3 ameliorates neuronal
injury after cerebral ischemia/reperfusion via inhibiting oxidative
stress and inflammation. Redox Biol. 69:1029832024. View Article : Google Scholar
|
|
84
|
Mukhopadhyay S, Goswami D, Adiseshaiah PP,
Burgan W, Yi M, Guerin TM, Kozlov SV, Nissley DV and McCormick F:
Undermining glutaminolysis bolsters chemotherapy while NRF2
promotes chemoresistance in KRAS-driven pancreatic cancers. Cancer
Res. 80:1630–1643. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Adachi Y, Kimura R, Hirade K and Ebi H:
Escaping KRAS: Gaining autonomy and resistance to KRAS inhibition
in KRAS mutant cancers. Cancers (Basel). 13:50812021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Ferino A, Rapozzi V and Xodo LE: The
ROS-KRAS-Nrf2 axis in the control of the redox homeostasis and the
intersection with survival-apoptosis pathways: Implications for
photodynamic therapy. J Photochem Photobiol B. 202:1116722020.
View Article : Google Scholar
|
|
87
|
Fu L, Zhao H, Xiang Y, Xiang HX, Hu B, Tan
ZX, Lu X, Gao L, Wang B, Wang H, et al: Reactive oxygen
species-evoked endoplasmic reticulum stress mediates
1-nitropyrene-induced epithelial-mesenchymal transition and
pulmonary fibrosis. Environ Pollut. 283:1171342021. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Jin M, Wang J, Ji X, Cao H, Zhu J, Chen Y,
Yang J, Zhao Z, Ren T and Xing J: MCUR1 facilitates
epithelial-mesenchymal transition and metastasis via the
mitochondrial calcium dependent ROS/Nrf2/Notch pathway in
hepatocellular carcinoma. J Exp Clin Cancer Res. 38:1362019.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Gundamaraju R, Lu W, Paul MK, Jha NK,
Gupta PK, Ojha S, Chattopadhyay I, Rao PV and Ghavami S: Autophagy
and EMT in cancer and metastasis: Who controls whom? Biochim
Biophys Acta Mol Basis Dis. 1868:1664312022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kapuy O: Mechanism of decision making
between autophagy and apoptosis induction upon endoplasmic
reticulum stress. Int J Mol Sci. 25:43682024. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Si L, Yang Z, Ding L and Zhang D:
Regulatory effects of lncRNAs and miRNAs on the crosstalk between
autophagy and EMT in cancer: A new era for cancer treatment. J
Cancer Res Clin Oncol. 148:547–564. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Chen X, Li J, Kang R, Klionsky DJ and Tang
D: Ferroptosis: Machinery and regulation. Autophagy. 17:2054–2081.
2021. View Article : Google Scholar :
|
|
93
|
Zhang Z, Zhang L, Zhou L, Lei Y, Zhang Y
and Huang C: Redox signaling and unfolded protein response
coordinate cell fate decisions under ER stress. Redox Biol.
25:1010472019. View Article : Google Scholar :
|
|
94
|
Leonardi A, Donato A, Rosani U, Di Stefano
A, Cavarzeran F and Brun P: Endoplasmic reticulum stress and
unfolded protein response in vernal keratoconjunctivitis. Invest
Ophthalmol Vis Sci. 65:232024. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Beilankouhi EAV, Sajadi MA, Alipourfard I,
Hassani P, Valilo M and Safaralizadeh R: Role of the ER-induced UPR
pathway, apoptosis, and autophagy in colorectal cancer. Pathol Res
Practice. 248:1547062023. View Article : Google Scholar
|
|
96
|
Chang TK, Lawrence DA, Lu M, Tan J,
Harnoss JM, Marsters SA, Liu P, Sandoval W, Martin SE and Ashkenazi
A: Coordination between two branches of the unfolded protein
response determines apoptotic cell fate. Mol Cell. 71:629–636.e5.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Hetz C and Papa FR: The unfolded protein
response and cell fate control. Mol Cell. 69:169–181. 2018.
View Article : Google Scholar
|
|
98
|
Zhao H, Liu T, Yang CE, Hu YH, Niu Y, Lei
SP, Chen L and Zhang MX: Poricoic acid A attenuates renal fibrosis
by inhibiting endoplasmic reticulum stress-mediated apoptosis. Braz
J Med Biol Res. 57:e142492024. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Eleftheriadis T, Pissas G, Golfinopoulos
S, Efthymiadi M, Poulianiti C, Polyzou Konsta MA, Liakopoulos V and
Stefanidis I: Routes of albumin overload toxicity in renal tubular
epithelial cells. Int J Mol Sci. 24:96402023. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Hetz C, Zhang K and Kaufman RJ:
Mechanisms, regulation and functions of the unfolded protein
response. Nat Rev Mol Cell Biol. 21:421–438. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Hsieh PC, Peng CK, Liu GT, Kuo CY, Tzeng
IS, Wang MC, Lan CC and Huang KL: Aqueous extract of descuraniae
semen attenuates lipopolysaccharide-induced inflammation and
apoptosis by regulating the proteasomal degradation and
IRE1α-dependent unfolded protein response in A549 cells. Front
Immunol. 13:9161022022. View Article : Google Scholar
|
|
102
|
Wang WW, Liu YL, Wang MZ, Li H, Liu BH, Tu
Y, Yuan CC, Fang QJ, Chen JX, Wang J, et al: Inhibition of renal
tubular epithelial mesenchymal transition and endoplasmic reticulum
stress-induced apoptosis with shenkang injection attenuates
diabetic tubulopathy. Front Pharmacol. 12:6627062021. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Strippoli R, Niayesh-Mehr R, Adelipour M,
Khosravi A, Cordani M, Zarrabi A and Allameh A: Contribution of
autophagy to epithelial mesenchymal transition induction during
cancer progression. Cancers. 16:8072024. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Chen HT, Liu H, Mao MJ, Tan Y, Mo XQ, Meng
XJ, Cao MT, Zhong CY, Liu Y, Shan H and Jiang GM: Crosstalk between
autophagy and epithelial-mesenchymal transition and its application
in cancer therapy. Mol Cancer. 18:1012019. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Chen YM, Yang WQ, Gu CW, Fan YY, Liu YZ
and Zhao BS: Amlodipine inhibits the proliferation and migration of
esophageal carcinoma cells through the induction of endoplasmic
reticulum stress. World J Gastroenterol. 30:367–380. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Wu C, Qian Y, Jiang J, Li D and Feng L:
Piperine inhibits the proliferation of colorectal adenocarcinoma by
regulating ARL3-mediated endoplasmic reticulum stress. Biomol
Biomed. 25:391–405. 2025. View Article : Google Scholar :
|
|
107
|
Gu W and Yang C: Zinc oxide nanoparticles
inhibit malignant progression and chemotherapy resistance of
ovarian cancer cells by activating endoplasmic reticulum stress and
promoting autophagy. Exp Ther Med. 26:5082023. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Cho W, Oh H, Choi SW, Abd El-Aty AM,
Birdal O, Jeong JH, Song JH and Jung TW: CTRP4 attenuates apoptosis
and epithelial-mesenchymal transition markers in podocytes through
an AMPK/autophagy-dependent pathway. Biochem Biophys Res Commun.
682:104–110. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Jiang R, Han L, Gao Q and Chao J: ZC3H4
mediates silica-induced EndoMT via ER stress and autophagy. Environ
Toxicol Pharmacol. 84:1036052021. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Han X, Duan X, Liu Z, Long Y, Liu C, Zhou
J, Li N, Qin J and Wang Y: ZEB1 directly inhibits GPX4
transcription contributing to ROS accumulation in breast cancer
cells. Breast Cancer Res Treat. 188:329–342. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Yuan L, Zhou M, Wasan HS, Zhang K, Li Z,
Guo K, Shen F, Shen M and Ruan S: Jiedu Sangen decoction inhibits
the invasion and metastasis of colorectal cancer cells by
regulating EMT through the hippo signaling pathway. Evid Based
Complement Alternat Med. 2019:14317262019. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Liu Z, Nan P, Gong Y, Tian L, Zheng Y and
Wu Z: Endoplasmic reticulum stress-triggered ferroptosis via the
XBP1-Hrd1-Nrf2 pathway induces EMT progression in diabetic
nephropathy. Biomed Pharmacotherapy. 164:1148972023. View Article : Google Scholar
|
|
113
|
Tan W, Liang Z, Tan X and Tan G:
Ginsenoside Rg1 improves cigarette smoke-induced ferroptosis in
COPD by regulating PERK/ATF4 axis to inhibit endoplasmic reticulum
stress. Biochem Biophys Res Commun. 739:1509462024. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Ao Q, Hu H and Huang Y: Ferroptosis and
endoplasmic reticulum stress in rheumatoid arthritis. Front
Immunol. 15:14388032024. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Zhou R, Wei K, Li X, Yan B and Li L:
Mechanisms of ferroptosis and the relationship between ferroptosis
and ER stress after JEV and HSV infection. Front Microbiol.
15:14154172024. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Nie Z, Chen M, Wen X, Gao Y, Huang D, Cao
H, Peng Y, Guo N, Ni J and Zhang S: Endoplasmic reticulum stress
and tumor microenvironment in bladder cancer: The missing link.
Front Cell Dev Biol. 9:6839402021. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Urra H, Aravena R, González-Johnson L and
Hetz C: The UPRising connection between endoplasmic reticulum
stress and the tumor microenvironment. Trends Cancer. 10:1161–1173.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Liu B, Yin X, Jiang G, Li Y, Jiang Z,
Qiang L, Chen N, Fan Y, Shen C, Dai L, et al: Identification of
endoplasmic reticulum stress-related subtypes, infiltration
analysis of tumor microenvironment, and construction of a
prognostic model in colorectal cancer. Cancers (Basel).
14:33262022. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Alvarez CL, Troncoso MF and Espelt MV:
Extracellular ATP and adenosine in tumor microenvironment: Roles in
epithelial-mesenchymal transition, cell migration, and invasion. J
Cell Physiol. 237:389–400. 2022. View Article : Google Scholar
|
|
120
|
Yang H, Li J, Niu Y, Zhou T, Zhang P, Liu
Y and Li Y: Interactions between the metabolic reprogramming of
liver cancer and tumor microenvironment. Front Immunol.
16:14947882025. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Daniel Y, Lelou E, Aninat C, Corlu A and
Cabillic F: Interplay between metabolism reprogramming and
Epithelial-to-Mesenchymal transition in cancer stem cells. Cancers
(Basel). 13:19732021. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Balaji S, Kim U, Muthukkaruppan V and
Vanniarajan A: Emerging role of tumor microenvironment derived
exosomes in therapeutic resistance and metastasis through
epithelial-to-mesenchymal transition. Life Sci. 280:1197502021.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Huang K, Han Y, Chen Y, Shen H, Zeng S and
Cai C: Tumor metabolic regulators: Key drivers of metabolic
reprogramming and the promising targets in cancer therapy. Mol
Cancer. 24:72025. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Hu C, Xin Z, Sun X, Hu Y, Zhang C, Yan R,
Wang Y, Lu M, Huang J, Du X, et al: Activation of ACLY by SEC63
deploys metabolic reprogramming to facilitate hepatocellular
carcinoma metastasis upon endoplasmic reticulum stress. J Exp Clin
Cancer Res. 42:1082023. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Tao J, Yin L, Wu A, Zhang J, Zhang J, Shi
H, Liu S, Niu L, Xu L, Feng Y, et al: PDIA2 bridges endoplasmic
reticulum stress and metabolic reprogramming during malignant
transformation of chronic colitis. Front Oncol. 12:8360872022.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Pathak T, Benson JC, Johnson MT, Xin P,
Abdelnaby AE, Walter V, Koltun WA, Yochum GS, Hempel N and Trebak
M: Loss of STIM2 in colorectal cancer drives growth and metastasis
through metabolic reprogramming and PERK-ATF4 endoplasmic reticulum
stress pathway. bioRxiv. Oct 3–2023.PubMed/NCBI
|
|
127
|
Guimarães ES, Marinho FV, de Queiroz N,
Antunes MM and Oliveira SC: Impact of STING inflammatory signaling
during intracellular bacterial infections. Cells. 11:742021.
View Article : Google Scholar
|
|
128
|
Kang H, Kim H, Lee S, Youn H and Youn B:
Role of metabolic reprogramming in Epithelial-Mesenchymal
transition (EMT). Int J Mol Sci. 20:20422019. View Article : Google Scholar
|
|
129
|
Bhattacharya D and Scimè A: Metabolic
regulation of epithelial to mesenchymal transition: Implications
for endocrine cancer. Front Endocrinol (Lausanne). 10:7732019.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Hua W, Ten Dijke P, Kostidis S, Giera M
and Hornsveld M: TGFβ-induced metabolic reprogramming during
epithelial-to-mesenchymal transition in cancer. Cell Mol Life Sci.
77:2103–2123. 2020. View Article : Google Scholar
|
|
131
|
Guo W, Duan Z, Wu J and Zhou BP:
Epithelial-mesenchymal transition promotes metabolic reprogramming
to suppress ferroptosis. Semin Cancer Biol. 112:20–35. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Kandasamy T, Sarkar S and Ghosh SS:
Harnessing drug repurposing to combat breast cancer by targeting
altered metabolism and Epithelial-to-Mesenchymal transition
pathways. ACS Pharmacol Transl Sci. 7:3780–3794. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Liu B, Dan W, Wei Y, Zhang Y, Wang C, Lei
Y, Hou T, Zhang Y and Gao Y: β-asarone inhibits the migration,
invasion, and EMT of bladder cancer through activating ER stress.
Cancer Med. 12:13610–13622. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Li CY, Chou TF and Lo YL: An innovative
nanoformulation utilizing tumor microenvironment-responsive
PEG-polyglutamic coating and dynamic charge adjustment for specific
targeting of ER stress inducer, microRNA, and immunoadjuvant in
pancreatic cancer: In vitro investigations. Int J Biol Macromol.
254:1279052024. View Article : Google Scholar
|
|
135
|
Granados-Principal S, Liu Y, Guevara ML,
Blanco E, Choi DS, Qian W, Patel T, Rodriguez AA, Cusimano J, Weiss
HL, et al: Inhibition of iNOS as a novel effective targeted therapy
against triple-negative breast cancer. Breast Cancer Res.
17:252025. View Article : Google Scholar
|
|
136
|
Wang W, Zhang Y, Wang Z, Liu X, Lu S and
Hu X: A native Drug-free macromolecular therapeutic to trigger
mutual reinforcing of endoplasmic reticulum stress and
mitochondrial dysfunction for cancer treatment. ACS Nano.
17:11023–11038. 2023. View Article : Google Scholar : PubMed/NCBI
|