Introduction
During malignant transformation from normal to
neoplastic tissues, tumors progressively develop distinct metabolic
adaptations to sustain their proliferative demands. In numerous
solid malignancies, dysregulated lipid homeostasis emerges as a
pathogenic driver or pathognomonic feature, exemplified by
hepatocellular carcinoma (HCC), colorectal adenocarcinoma, breast
cancer and clear cell renal cell carcinoma (ccRCC). This metabolic
rewiring positions lipid metabolism regulators as promising
therapeutic targets, with nuclear receptors constituting a major
crucial class of lipid-sensing molecules.
As ligand-activated transcription factors, nuclear
receptors interact with various cellular metabolites including
steroid hormones, vitamins and lipids. Through recruitment of
transcriptional co-regulators, these molecular sensors orchestrate
precise gene expression programs (1). Farnesoid X receptor (FXR),
initially described in 1995 as a retinoid X receptor
(RXR)-interacting partner, exemplifies this family (2). Though first identified in rat
hepatic, renal and adrenal tissues (2), FXR's designation as an orphan
receptor persisted until 1999 when physiological bile acid (BA)
concentrations were shown to activate it through ligand binding
(2,3). Subsequent investigations establish
FXR's central role in cholesterol homeostasis through the
regulation of BA synthesis enzymes, notably cholesterol 7α
hydroxylase, the rate-limiting enzyme of BA synthesis (3,4).
Expanding beyond hepatobiliary functions, clinical investigations
reveal FXR's modulatory effects on systemic lipid profiles through
molecular control of fatty acid (FA) synthesis, transport pathways
and β-oxidation processes. Thus, FXR-mediated signal transduction
is closely associated with multiple metabolic disorders, including
obesity, diabetes, atherosclerosis and non-alcoholic fatty liver
disease (NAFLD) (5-7).
The emerging paradigm of tumor metabolic
reprogramming, combined with FXR's pivotal functions in the
gut-liver axis has led to intense investigations into nuclear
receptor-targeted therapeutics. This review mainly focuses on FXR's
multifaceted roles in lipid metabolism regulation and its
involvement in lipid-rich tumor progression, providing conceptual
frameworks for advancing nuclear receptor-targeted therapeutic
strategies.
FXR
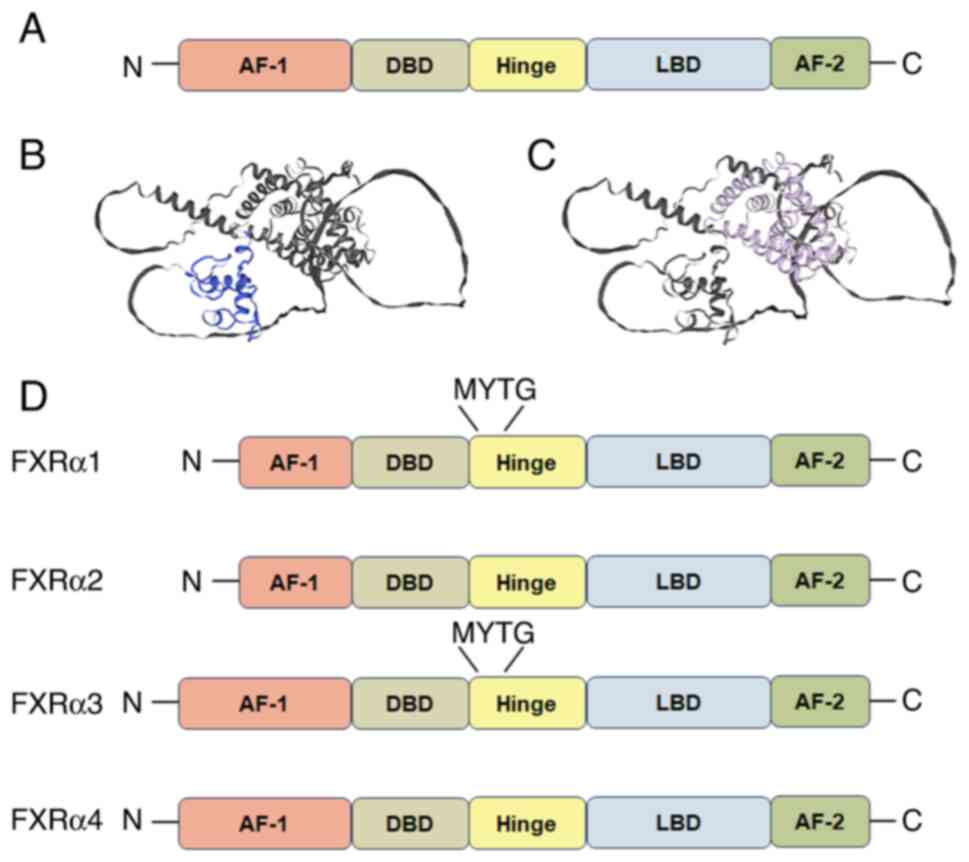
Structure of FXR
The nuclear receptor superfamily is categorized into
two classes based on DNA-binding properties: One class function as
homodimers, while another class forms heterodimers with RXR for DNA
interaction (4). FXR belongs to
the latter class and shares structural homology with other members,
including the retinoic acid receptor, the thyroid hormone receptor
and the vitamin D3 receptor (4). The structure of nuclear receptors
typically encompasses five domains: The amino terminal
transcriptional activation function-1, a DNA binding domain, a
hinge region, a ligand binding domain (LBD) and the carboxyl
terminal activation function-2 (AF-2). This architecture
facilitates RXR-mediated heterodimerization, as illustrated in
Fig. 1A-C (1,8).
FXR is originally cloned from rats using degenerate primers
corresponding to the semi-conserved DNA-binding domain of the
nuclear receptor (4). Early
functional characterization reveals weak farnesol-induced
activation; however, the absence of endogenous ligands under
physiological conditions initially classified FXR as an orphan
nuclear receptor. A paradigm shift occurred in 1999 when endogenous
BAs were demonstrated to robustly activate FXR at physiological
concentrations, triggering downstream transcriptional responses.
This discovery propels extensive investigations into FXR's
regulatory roles in BA homeostasis and hepato-enteric
pathophysiology. Structurally, FXR's LBD mirrors the characteristic
three-layered helical sandwich observed across nuclear receptors,
which was resolved in 2003 (Fig.
1C) (9,10). Of particular functional
significance is, H12, which harbors AF-2 and establishes
hydrophobic interactions with H3 and H4, stabilizing co-activator
binding through LXXLL motif engagement at the LBD interface
(9,10). In contrast to most ligands, BAs
occupy the FXR LBD binding site in an opposite direction, and there
are two sites binding to the LXXLL motif simultaneously. This
distinctive orientation enhances binding affinity compared with
single-motif co-activators. Comprehensive structural analyses of
FXR have been systematically documented in prior crystallographic
studies (11,12).
Subtypes and polymorphism of FXR
Two FXR genes have been identified: FXRα and FXRβ.
In rodents, rabbits and dogs, FXRβ has been shown to be activated
by lanosterol, while in humans and primates, it constitutes a
non-functional pseudogene (13).
Human physiology specifically expresses four functional FXRα
isoforms (FXRα1-4) differentiated by their amino-terminal domains,
as illustrated in Fig. 1D
(14). This is due to
differential promoter usage and alternative exon splicing patterns.
A four-amino acid residue insertion MYTG in the hinge region leads
to the difference between FXRα1/α3 and α2/α4, which potentially
impacts receptor functionality. In addition, tissue- and
species-specific expression profiles are observed in these isomers.
Murine models demonstrate predominant hepatic and intestinal
co-expression of both FXR subtypes, whereas renal tissue
preferentially expresses FXRβ and adrenal glands favor FXRα
(15). Human expression patterns
similarly mirror this compartmentalization, with hepatocytes
predominantly expressing FXRα1/α2 isoforms, while enterocytes show
preferential FXRα3/α4 expression (16). Furthermore, there is also
evidence that dynamic regulation of FXR isoform expression is
closely associated with the metabolic progress and physiological
states (17-19).
Genetic polymorphisms in nuclear receptor
superfamily members (vitamin D receptor, peroxisome
proliferator-activated receptors (PPARs), liver X receptors (LXRs),
small heterodimer partner (SHP), hepatocyte nuclear factor-1-α and
hepatocyte nuclear factor-4α (HNF4α) establish disease
predisposition through phenotypic modulation (20-27). Clinical investigations implicate
FXR genetic variation in hepatobiliary pathologies, exemplified by
four novel heterozygous FXR variants (-1g>t, M1V, W80R, M173T)
identified in intrahepatic cholestasis of pregnancy (28). The amino terminus contains two
ATG parts (M1 and M5), which could be identified as translational
initiation site. M1 plays the initiation role in humans, in
addition, -1g>t and M1V mutations impair protein expression and
reduce transcriptional activation of ileal BA binding protein
(IBABP)/bile salt export pump (BSEP) promoters, suggesting
compromised translational efficiency. M173T similarly affects IBABP
and BSEP promoter activation.
A study on FXR polymorphism in different races
conducted by Marzolini et al (20) found several meaningful single
nucleotide polymorphisms compared with an FXR reference sequence
from GenBank, including G-1T, C643T (H215Y) and G646T (A216S).
Furthermore, two synonymous polymorphisms (C783T, 261N; 1341T,
447H) are included in the aforementioned study as well, whose
locations predict amino acid differences within the hinge region of
FXR. The expression of SHP and organic anion transporting
polypeptide, two FXR target genes, was significantly decreased in
liver carrying FXR (G-1T) compared with FXR (-1G). However, BSEP,
another FXR target gene, is unassociated with an FXR polymorphism.
This differential regulation suggests target gene-specific
modulation mechanisms by FXR genetic variants, highlighting the
complexity of receptor-target gene interactions.
FXR response element (FXRE) and
ligands
As reviewed, FXR shares its structural homology with
classical nuclear receptors in its ability to form heterodimers
with RXR, subsequently binding DNA to transcriptionally activate
downstream target genes. The cognate DNA recognition, initially
characterized as FXRE, is subsequently designated the BA response
element, due to its activation by endogenous BA ligands (4). The canonical FXRE configuration
comprises a conserved (AGGTCA) hexamer arranged as an inverted
repeat with single nucleotide spacing (IR1) (29), representing both the most
prevalent and highest affinity DNA binding configuration for
nuclear receptor dimers (29,30). In addition, there are other FXRE,
such as IR0, IR8 (31), ER8
(32) and DR1 (33). Some of the FXRE sites are
tissue-specific, and a study of Chip-Seq in murine treated with
GW4064 showed only 11% of the sites were shared between the liver
and intestine (34).
Farnesoid derivatives are intermediates of
mevalonate pathway metabolism and can activate FXR at
supra-physiological dose concentrations (4). BAs, the natural ligand of FXR,
activate FXR at physiological concentrations. Comparative analyses
revealed chenodeoxycholic acid (CDCA) as the most potent endogenous
agonist, exhibiting activity superior to deoxycholic acid,
lithocholic acid (LCA) and cholic acid (2). However, a mechanistic exploration
of FXR's roles in sterol homeostasis has been constrained by
available pharmacological tools. While CDCA serves as the
archetypal endogenous ligand, its extensive interactions with BA
chaperone proteins, metabolism into LCA and non-specific cellular
effects render it suboptimal for experimental applications. These
limitations motivated the development of GW4064, a synthetically
optimized FXR agonist demonstrating superior specificity and
pharmacological stability (2).
Of note, multiple FXR-targeted therapeutics have advanced through
clinical development pipelines for cholestatic and metabolic
disorders including primary biliary cholangitis, primary sclerosing
cholangitis and non-alcoholic steatohepatitis (NASH) (Table I). Paralleling agonist
development, novel modulatory agents exhibiting distinct activation
profiles relative to CDCA and GW4064 are emerging, with comparative
pharmacodynamic characteristics outlined in Table II.
 | Table IFXR agonists in clinical trials. |
Table I
FXR agonists in clinical trials.
| FXR Agonists |
Condition/disease | Clinical trials
ID | Phase of study |
|---|
| OCA/INT-747 | PBC | NCT02308111 | IV |
| PBC | NCT01473524 | III |
| Biliary acid
diarrhea | NCT01585025 | II |
| PSC | NCT02177136 | II |
| NAFLD/NASH | NCT01265498 | II |
| NASH | NCT02633956 | II |
| NASH | NCT02548351 | III |
| NASH | NCT03439254 | |
| Barrett's
esophagus | NCT04939051 | II |
| Hepatic steatosis
and familial partial lipodystrophy | NCT02430077 | II |
| EDP-305 | PBC | NCT03394924 | II |
| NASH | NCT03421431 | II |
| NASH | NCT04378010 | II |
| MET409 | NASH/type 2
diabetes | NCT04702490 | II |
| TERN-101 | NASH | NCT04328077 | II |
|
Cilofexor/GS-9674 | PSC | NCT03890120 | III |
| PSC | NCT02943460 | II |
| PSC | NCT02943447 | II |
| NASH | NCT02781584 | II |
|
Tropifexor/LJN452 | PBC | NCT02516605 | II |
| NASH | NCT04065841 | II |
| NASH | NCT02855164 | II |
| Px-104 | NAFLD | NCT01999101 | II |
| EYP001a | NASH | NCT03812029 | II |
|
Nidufoxor/LMB763 | Diabetic
nephropathy | NCT03804879 | II |
| NASH | NCT02913105 | II |
| Table IIFXR modulates. |
Table II
FXR modulates.
| First author/s,
year | Compounds | Regulation of
target genes | Receptor
selectivity | (Refs.) |
|---|
| Urizar et
al, 2002; Burris et al, 2005 | Guggulsterone | HepG2: BSEP ↑, SHP
↓ Caco-2: IBABP ↓ | FXR, PXR, GR, MR,
AR, PR, ER | (131,132) |
| Bijsmans et
al, 2015 | Mometasone
furoate | HepG2: IL-8,
CXCL12, MCP-1 ↓ Reduced recruitment of p65 to IL-8, CXCL12 | FXR, GR | (133) |
| Dussault et
al, 2013 | AGN-34 | HepG2: CYP7A1 ↓
Caco-2: IBABP ↓ | FXR, RXR | (134) |
| Chang et al,
2016 | Xanthohumol | In KK-ay mice:
SREBP1, stearoyl-CoA desaturase-, PEPCK, G6Pase ↓ | FXR, CAR,
GABAA | (135) |
| Liu et al,
2010 | Oleanoic acid | HepG2: BSEP ↓ | FXR, PAPPA, CAR,
PXR | (136) |
| Fang et al,
2015 | Fexaramine | Intestinal
specificity; SHP, FGF19, IBABP, OSTA/B ↑ | FXR | (137) |
| Pellicciari et
al, 2016 | TC-100 | Intestinal
specificity; FGF15, SHP, ANG1 ↑ | FXR | (138) |
| Jin et al,
2015 | Ivermectin | Intestinal
specificity; FXR ↑ | FXR, CAR, LXRα,
PXR | (139) |
| Li et al,
2012 | EGCG | Intestinal
specificity; SHP, FGF15 ↑ | FXR | (140) |
Physiological functions of FXR
The regulation of FXR in vivo is complex, and
its physiological effects include several categories: i) It
maintains BA pool homeostasis, and reduces BA concentration in the
liver by increasing BA output and reducing BA synthesis; ii) it
reduces blood lipid levels and regulates plasma lipoproteins; iii)
it exerts liver protection and regeneration; iv) it regulates
glucose metabolism and gluconeogenesis (35); v) it regulates blood vessel
clotting and anticoagulation molecules (36,37); and vi) it is involved in urine
volume regulation and anti-renal fibrosis (38,39).
FXR and lipid metabolism
FXR and FA
FA homeostasis is governed by a balance between
uptake and elimination, with disruptions in this equilibrium
contributing to metabolic pathogenesis. In this section, the
regulatory roles of FXR in FA transport, de novo synthesis
and catabolism were systematically evaluated (Fig. 2).
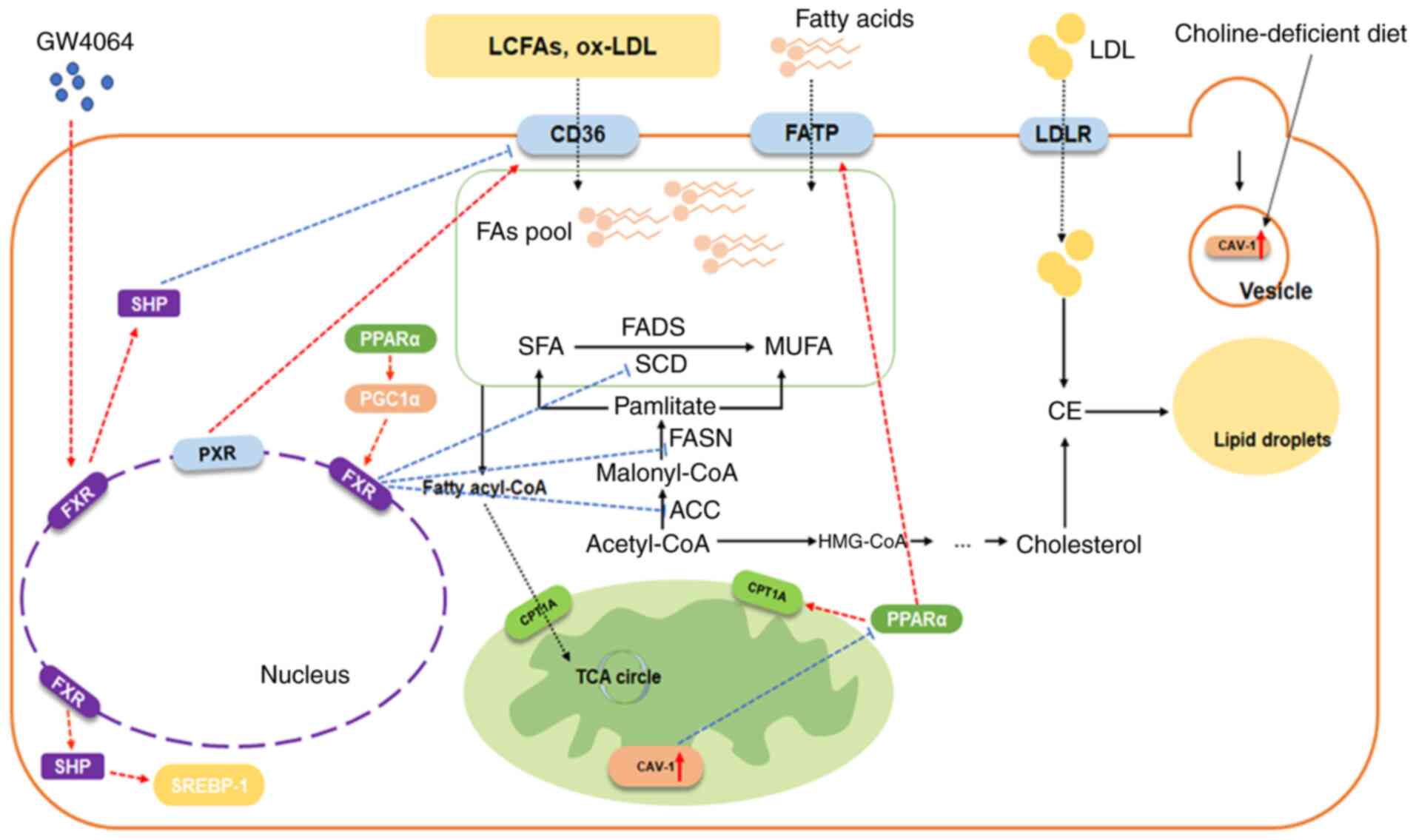 | Figure 2Regulation of FXR and FA metabolism.
The regulatory relationship indicates the regulation of adjacent
molecules, arrow-headed lines mean a promotion/activation, while
flat-headed lines mean an inhibition. FA, fatty acid; LCFA,
long-chain FAs; FXR, Farnesoid X receptor; SHP, small heterodimer
partner; PPARα, peroxisome proliferator-activated receptors α;
SREBP-1, sterol regulatory element binding protein 1; PGC1α,
peroxisome proliferator-activated receptor gamma coactivator-1
alpha; PXR, pregnane X receptor; FADS, FA desaturase; SCD,
stearoyl-CoA desaturase; SFA, saturated FA; MUFA, monounsaturated
FA; FASN, FA synthase; ACC, acetyl-CoA carboxylase; CAV-1,
caveolin-1; FATP, FA transport protein; LDL, low-density
lipoprotein; LDLR, LDL receptor; CPT1A, carnitine O-palmitoyl
transferase 1 A; TCA, tricarboxylic acid; ox-oxidized. |
FXR and FA uptake/transport
Cellular FA internalization is mediated by
specialized transporters, notably CD36, caveolins (CAVs) and the FA
transport protein (FATP) family (40).
CD36 (SR-B2/FAT) is a membrane-spanning glycoprotein
facilitating absorption of long-chain FAs and oxidized low-density
lipoprotein (LDL) uptake (40).
A mechanistic study demonstrated that hepatic CD36 overexpression
exacerbates FA influx in NAFLD murine models, whereas its genetic
silencing alleviates steatosis-associated lipid deposition
(41). As a transcriptional
target of PPARs and other nuclear receptors (40), CD36 expression has been shown to
be modulated by pregnane X receptor-mediated transcriptional
activation, driving hepatic triglyceride (TG) accumulation through
enhanced lipid uptake (42).
Small heterodimer partner is an atypical nuclear receptor that
lacks DNA-binding domain, which is regarded as the target gene of
FXR. GW4064 dose-dependently suppresses CD36 effectively reversing
hepatic steatosis in diet-induced obese mice (43). Complementary evidence from
diosgenin mechanistic studies confirms FXR/SHP-mediated CD36
downregulation as an anti-steatotic pathway (44). These findings collectively
established FXR/SHP/CD36 as a pivotal regulatory node in FA
homeostasis.
CAVs are essential components of caveolae (one site
of endocytosis) involved in several biological functional
processes, such as vesicle formation and lipid droplet formation.
CAV-1 has been extensively investigated in prior studies.
Preclinical investigations have revealed CAV-1 upregulation within
mitochondrial inner membranes and lipid droplets of choline
deficiency-induced fatty liver models (45), while CAV-1 knockout mice have
been shown to exhibit attenuated hepatic lipid storage capacity
(46), implicating its necessity
for physiological lipid storage. Further characterization of
CAV-1(−/−) mice reveals compromised PPARα signaling, manifested by
downregulated carnitine palmitoyl-transferase (CPT) and peroxisome
proliferator-activated receptor gamma coactivator 1-alpha. These
perturbations were found to be correlated with mitochondrial
dysfunction and to impair FA β-oxidation (47). In summary, CAV-1 deficiency
concurrently disrupts FXR signaling and BA metabolism, suggesting
its dual regulatory role-modulating both anabolic and catabolic
pathways-analogous to multifaceted functions of CD36.
The FATP family represents another critical
determinant of transmembrane FA flux. Motojima et al
(48) reported FATP is
upregulated in the liver of mice threated with the PPAR activator
Wy14643. FATP-mediated lipid metabolic reprogramming has been
extensively characterized in oncogenesis (49). FATP2 displays hepatorenal
expression, whereas FATP5 exhibits liver-specific localization
(50). A previous study on
knockout FATP2 or FATP5 mice found that FA uptake was reduced and
hepatic steatosis was reversed (51). Interestingly, when PPARα agonist
was administered to ApoE (−/−) FXR (−/−) mice, Lee et al
(52) observed that despite
lipid reduction which resulted in NAFLD and atherosclerosis
alleviated, the expression of FATP1 and CD36 was upregulated. This
suggests that FATP family members may not simply promote lipid
accumulation, but enhance lipid transport to drive intracellular
lipid metabolism pathways.
FXR and synthesis of Fas
FXR inhibits the de novo synthesis of FAs.
During the de novo synthesis of FAs in the body, acetyl-CoA
carboxylase (ACC) can carboxylate acetyl CoA to form malonyl CoA,
which is a rate-limiting enzymatic step in this pathway.
Experimental evidence demonstrates ACC's pivotal metabolic role.
ACC (−/−) mice have a lower weight than the wild-type (WT) group,
and using ACC inhibitors to treat mice induced by diet could
upregulate FA oxidation (FAO) and inhibit FA synthesis, thus
leading to NAFLD relief (53).
In addition, sterol regulatory element binding protein 1 (SREBP-1)
and carbohydrate reaction element binding protein (ChREBP) are also
essential transcriptional regulators in the lipid synthesis
pathway. In an ob/ob mouse model of obesity and insulin resistance,
the expression of SREBP-1 and ChREBP were significantly increased,
and the dual or selective inhibition of these factors effectively
alleviated the liver steatosis of the model mice (54,55).
In FXR deficient mice, SREBP-1, FA synthase (FASN)
and stearoyl-CoA desaturase-1 were significantly upregulated,
indicating that FXR might inhibit the synthesis of FA to improve
liver steatosis (56). On the
contrary, CA administration significantly downregulated SREBP-1 and
its transcriptional targets, while impairing lipogenic capacity in
murine hepatocytes (57). As one
of the key target genes of FXR, the role of SHP has been
investigated. Mechanistic investigations have revealed that SHP
mediated SREBP-1c promoter regulation. CDCA-induced SHP
upregulation has been shown to be inversely correlated with
SREBP-1c target gene suppression. CA or GW4064 could attenuate
serum TGs along with sterol regulatory element-binding protein 1
(SREBP1) decreases in SHP (+/+) mice; however, this was absent in
SHP (−/−) mice, establishing the FXR/SHP axis as a dual regulator
of FA transport and lipogenesis through SREBP-1c modulation.
ChREBP has been shown to regulate glucose-induced
fat production. Under hyperglycemic conditions, ChREBP combines
with Max-like protein and ChREBP to form two box-like motifs,
therefore promoting the expression of its target genes, such as
liver-type pyruvate kinase (LPK), FASN and ACC1, driving enhanced
FA and TG biosynthesis (58).
Given that FXR is negatively associated with the expression of LPK,
FASN and ACC, Caron et al (59) hypothesized that FXR interfered
with the transcriptional activity of ChREBP. Fasting-refeeding
studies revealed that INT-747, a synthetic FXR agonist,
significantly attenuated high-carbohydrate-induced increase in mRNA
expression of LPK in WT mice, with parallel observations in IHH and
HepaRG human hepatocytes. FXR did not affect ChREBP nuclear
translocation, but decreased transcription coactivator binding to
L4L3 region of LPK promoter to release ChREBP, meanwhile inducing
the recruitment of silencing mediator of retinoid and thyroid
hormone receptor (a transcriptional corepressor) leading to a
prevention of the FXR-dependent repression of LPK at a high glucose
concentration. This mechanism could be extended to other ChREBP
target genes under hyperglycemic conditions, collectively reducing
the generation of acetyl-CoA, suppressing de novo
lipogenesis, and potentially ameliorating NAFLD pathogenesis.
In addition, due to the enterohepatic cycle of BA
metabolism, the fibroblast growth factor (FGF) family members have
also been extensively investigated. The expression and excretion of
fibroblast growth factor-15/19 are induced by FXR in the intestine.
The intestinal activation of FXR stimulates the secretion of
FGF-19, which negatively regulates cholesterol 7α hydroxylase
(CYP7A1) transcription after binding to FGF4/klothoβ (60). FGF-19 inhibits insulin-induced FA
synthesis by i) decreasing the activity of SREBP-1c; ii) enhancing
STAT3 signaling and iii) downregulating PGC1β. Furthermore, the
FGF19/SHP/SREBP-1c axis suppresses lipogenic enzyme expression
while promoting FAO through ACC2 downregulation (61).
FXR and FAO
In FA metabolism, FA from acetyl-CoA and
malonyl-CoA, and FFA from TGs are catalyzed by fatty acyl-CoA
synthetase to form fatty acyl-CoA, which is the main step in FA
β-oxidation. Products are then transferred into the mitochondria by
carnitine CPT1a for the subsequent step. Accumulating evidence
demonstrates FXR's regulatory role in facilitating this metabolic
cascade.
Previous research on FXR-mediated FA transport has
revealed functional interplay with PPARα. The PPAR family is part
of the nuclear receptor sup-family, including PPAR-α, PPAR-β/δ and
PPAR-γ. PPARs serve as primary regulators of glucose and lipid
homeostasis, establishing them as therapeutic targets for metabolic
disorders and oncogenesis. Experimental evidence indicates that
PPARα-deficient mice develop pronounced hepatic steatosis (62), while FXR (−/−) ob/ob mice exhibit
significant PPARα downregulation (63). These observations suggest the
potential FXR-mediated regulation of FA β-oxidation through
PPARα-dependent mechanisms. Substantiating this hypothesis, Pineda
Torra et al (64) found
that the incubation of a human HepG2 cell line with CDCA or GW4064
led to a significant induction of PPARα in a transcriptional level.
Through mutation analysis, they found αFXRE in the human PPARα
promoter. In addition, induction of human PPARα by CDCA enhanced
the expression of PPARα target gene CPT1, which was not observed in
mice. This indicated that the underlying mechanism might be
species-specific, which is worth investigating further. In
addition, FXR could activate the transcription of liver
carboxylesterase 1, which enhanced FA β-oxidation through PPARα
(65). While reviewing the
previous article, it was also found that the FXR/SHP axis may
induce the AMPK-ACC-CPT1α mechanism to promote FA β-oxidation
(66).
Peroxisome proliferator-activated receptor γ
coactivator 1α (PGC1α) contributes to the regulation of certain
transcription factors, such as PPARα and -γ as the coactivator,
thus modulating several genes in metabolic pathways (67). In a study on the pharmacological
effect and mechanism of Nuciferine, it was found that Nuciferine
could attenuate hepatic steatosis in HFD/STZ-induced diabetic mice
through the PPARα/PGC1α pathway (68). In addition, FXR is activated by
PGC1α, which promotes PGC1α-mediated FA oxidation (69). In conclusion, FXR tended to
promote FA β-oxidation in the majority of studies.
FXR with cholesterol/Bas
Cholesterol undergoes hepatic biosynthesis with
subsequent conversion to BAs; this constitutes its principal
metabolic fate. BAs are divided into free BAs and conjugated acids
in structure, or primary BAs and secondary BAs in source. Given the
established role of FXR in regulating BA homeostasis, this section
focuses on its contribution to cholesterol metabolite dynamics and
enterohepatic BA cycling (Fig.
3).
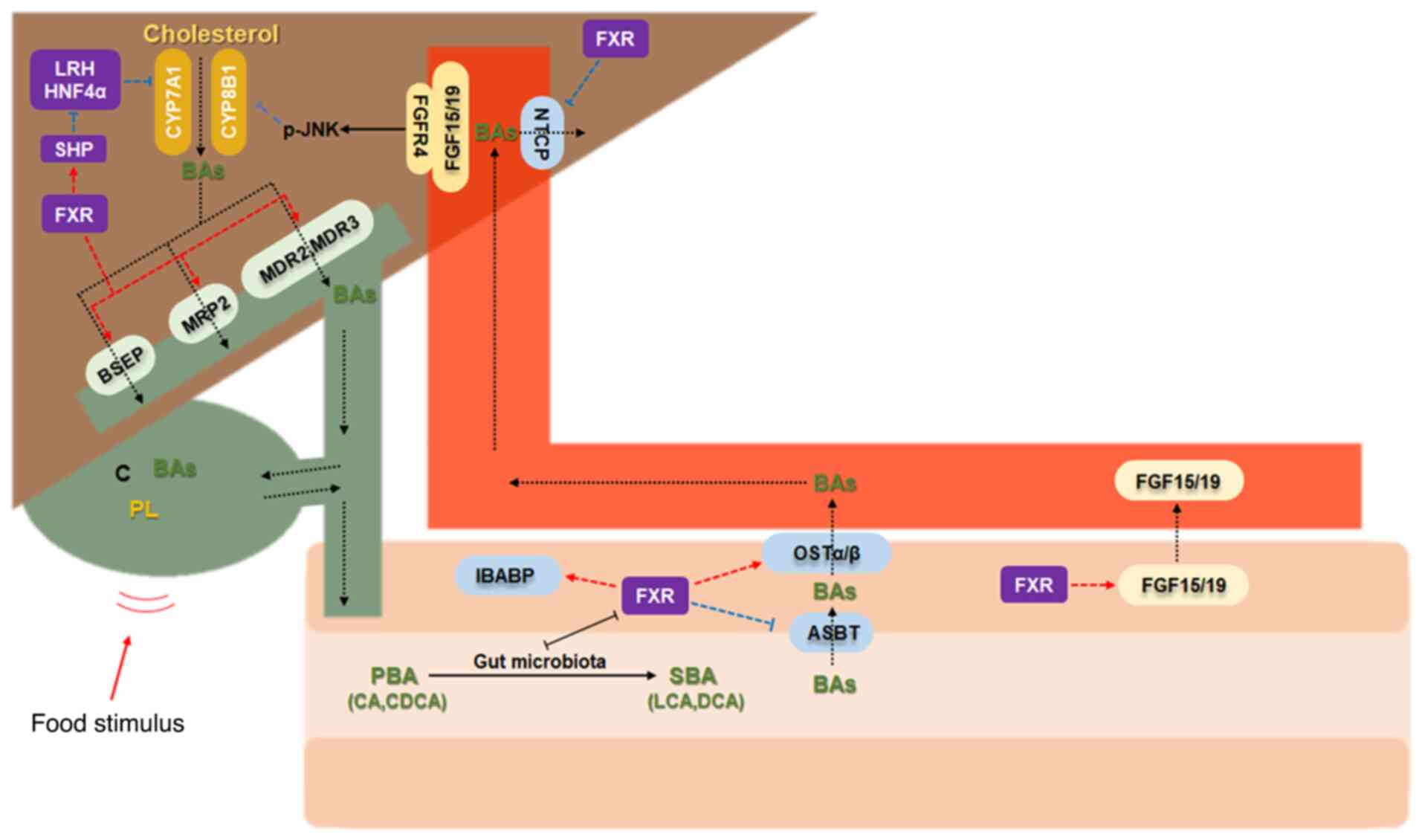 | Figure 3Regulation of FXR and bile acids. The
regulatory relationship indicates the regulation of adjacent
molecules, arrow-headed lines mean a promotion/activation, while
flat-headed lines mean an inhibition. FXR, farnesoid X receptor;
SHP, small heterodimer partner; BSEP/ABC11, bile salt export pump;
HNF4α, hepatocyte nuclear factor-4-alpha; IBABP, ileal bile acid
binding protein; CYP7A1, cholesterol 7α hydroxylase; CYP8B1,
cytochrome P450 8B1; MRP2, multidrug-resistant associated protein
2; MDR2/3, multidrug-resistant P-glycoprotein 2/3; FGF-15/19,
fibroblast growth factor-15/19; PBA, primary bile acids; SBA,
secondary bile acids; OSTα/β, organic solute transporter α/β; ASBT,
apical sodium-dependent bile acid transporter; NTCP, sodium
taurocholate co-transporting polypeptide; CA, cholic acid; CDCA,
chenodeoxycholic acid; LCA, lithocholic acid; DCA, deoxycholic
acid. |
Synthesis of Bas
Cholesterols could be catalyzed by CYP7A1 into 7α
hydroxycholesterol, then undergo a series of complicated enzymatic
reactions, finally being transformed into primary free BAs, such as
CA and CDCA, or combined with the corresponding glycine or taurine
to produce primary conjugated acids to discharge into the
intestine. CYP7A1 functions as the key enzyme of the classic BA
synthesis pathway (70).
12α-hydroxylase (CYP8B1) determines the ratio of CA and CDCA.
SHP, the target gene of FXR, can inhibit the
expression of CYP7A1 by blocking the trans-activation of the liver
activator liver receptor homolog-1 (LRH-1) and HNF4α (71,72). In addition, FXR can also reduce
BA synthesis by inhibiting CYP8B1, and the mechanism is similar
(70,73).
Transport of Bas
Following synthesis and conjugation with glycine or
taurine, BAs are secreted into bile canaliculi through BSEP, and
then enter the gallbladder for storage. Nutrient-responsive
gallbladder contraction initiates bile release into the duodenal
lumen. In the distal small intestine, BAs are reabsorbed into the
blood through apical sodium-dependent BA transporter. Next, in
intestinal epithelial cells, they are reabsorbed by IBABP and
organic solute transporter alpha/beta (OSTα/β) into portal
circulation. Hepatocyte reuptake completes this enterohepatic
cycling mechanism that is essential for maintaining BA pool
homeostasis. Almost all of these transporters involved in the
enterohepatic circulation of BAs are regulated by FXR (74,75). Others include multidrug-resistant
associated protein 2 (MRP2) (76), and multidrug-resistant
P-glycoprotein 3 (77), which
promote hepatic BA outflow.
FXR and plasma lipoprotein
Plasma lipoprotein is the blood component that
transports plasma lipids. It was reviewed independently to
elucidate the link between FXR and blood lipid (Fig. 4). This part actually has a
crosstalk with the FA metabolism and cholesterol metabolism. Very
low-density lipoprotein (VLDL), synthesized primarily in
hepatocytes, mediates the hepatic export of TG and cholesterol to
peripheral tissues. Upon entering circulation, nascent VLDL
acquires apolipoprotein C (apoC) from high-density lipoprotein
(HDL), where apoC-II activates endothelial lipoprotein lipase (LPL)
to hydrolyze VLDL-associated TG. Through progressive lipid exchange
with HDL, VLDL undergoes remodeling into cholesterol-enriched LDL,
with circulating VLDL and LDL levels serving as established
clinical biomarkers for the evaluation of dyslipidemia.
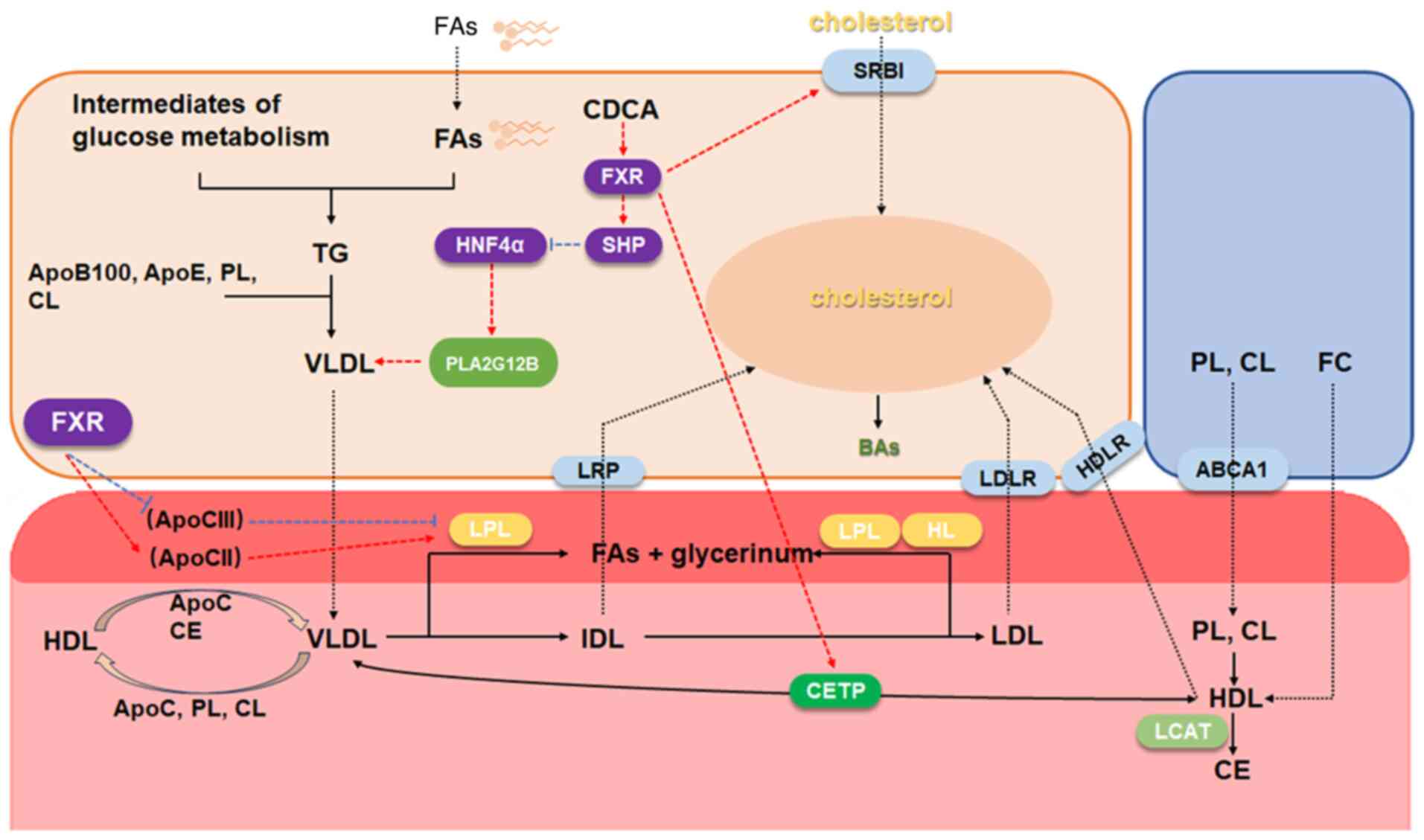 | Figure 4Regulation of FXR and plasma
lipoprotein. The regulatory relationship indicates the regulation
of adjacent molecules, arrow-headed lines mean a
promotion/activation, while flat-headed lines mean an inhibition.
FXR, farnesoid X receptor; SHP, small heterodimer partner; FA,
fatty acid; VLDL, very low-density lipoprotein; HDL, high-density
lipoprotein; IDL, intermediate density lipoprotein; LDL,
low-density lipoprotein; CETP, cholesterol ester transfer protein;
LCAT, phosphatidylcholine-cholesterol acyltransferase; LPL,
lipoprotein lipase; HL, hepatic lipase; PL, phospholipid; CL,
cardiolipin; FC, free cholesterol. |
Clinical investigations indicated that pharmacologic
FXR activation significantly reduces serum TGs, while aggravating
liver cholesterol accumulation and atherogenic serum lipoprotein
profiles (high LDL level) in FXR-deficient models (6,78). Consistently, CDCA was found to be
associated with decreased serum TG and VLDL production in patients
with hyperlipidemia, while colestyramine (BA chelating agent)
exhibited the opposite trends (79). The antilipidemic action of CDCA
involves multiple regulatory axes. One is the FXR/SHP/HNF4α axis
(80). Hirokane et al
(80) found that, following the
treatment of HepG2 cells with CDCA, the expression of microsomal TG
transfer protein (MTP) and apoB are attenuated as HNF-4 is
suppressed by SHP. Similarly, FXR/SHP/HNF4α can also reduce VLDL by
partially inhibiting the expression of phospholipase A2 group XIIB,
a mediator of VLDL (81). From
the aforementioned studies, the role of apoC II and LPL were
reported. The former activates LPL, and then the latter hydrolyzes
TG, leading to TGs clearing. ApoC III is also a component of
various lipoproteins, which is known as the inhibitor of LPL,
meanwhile affecting hepatic lipase, therefore impairing the
conversion of VLDL to IDL and LDL (82). FXR can promote TG clearing by
inducing hepatic apoC-II and inhibiting apoC-III and
angiopoietin-like protein 3 (83,84). CDCA could lead to FXR combined
promoter of apoC-II, but this does not exist in FXR-deficient
mice.
HDL is mainly synthesized in the liver and partly in
the small intestine, which is responsible for reverse cholesterol
transport-the process of shuttling peripheral cholesterol to
hepatic tissues for biliary excretion or direct intestinal
elimination. Lipid transfer proteins, particularly phospholipid
transfer protein (PLTP) and cholesteryl ester transfer protein
(CETP), govern lipoprotein remodeling through phospholipid and
cholesterol exchange between VLDL and HDL particles. FXR activation
by CDCA elevates PLTP activity and induces CETP expression through
ApoE-mediated pathways (85,86), thereby modulating lipoprotein
redistribution. Scavenger receptor class B type I (SRBI), a key HDL
receptor parallel to CD36 in lipid metabolism, demonstrates
FXR-dependent upregulation in murine with CA-containing diet,
whereas FXR deficiency abrogates this dietary response,
establishing SRBI as a regulatory node in FXR-mediated HDL
homeostasis.
FAs, cholesterol and plasma lipoproteins are
inextricably linked and convert each other, leading to the
crosstalk between metabolic mechanisms, which enables a more
comprehensive study of the role of FXR in lipid metabolism and BA
circulation.
FXR and lipid-rich tumors
Obesity constitutes one of the risk factors for
cancer. Patients with elevated lipid levels tend to have a higher
propensity to develop tumors. Gastrointestinal tumors, such as
liver and colon cancer, are typical examples. There are also
certain tumors, such as ccRCC, with intracellular accumulation of a
significant amount of lipid droplets as a typical pathological
feature, distinct from other subtypes. This section focuses on
these lipid-associated malignancies, elucidating FXR's pivotal
regulatory mechanisms in tumorigenesis and progression to inform
novel therapeutic strategies.
FXR and digestive system cancer
Liver cancer
The liver is the central hub for both lipid and BA
metabolism. Lipid homeostasis imbalance, alcohol, viral infection,
inflammation and cholestasis are all high-risk factors for
hepato-carcinogenesis. As aforementioned, the regulation of FXR
orchestrates numerous biological protective processes in the liver,
with its deficiency resulting in poor prognosis. Studies have shown
that FXR-null mice exhibit a significantly elevated incidence of
HCC (87,88). This observation is consistent
with clinical data showing the reduced FXR expression in human HCC
tissues being associated with multiple malignant pathological
characteristics, such as large tumor size, advanced Barcelona
Clinic Liver Cancer stage, poor differentiation and absence of
encapsulation (89). Of note,
organ-specific FXR modulation appears to exert different effects to
those of systemic FXR perturbation. An American study found that
~20% of mice with liver-specific FXR deficiency developed HCC,
which is well below the 90% of mice with global FXR knockouts
(90). Furthermore, the
tumor-modulating effects of FXR suggest tissue specificity. Kong
et al (91) reported that
hepatocyte-specific FXR deficiency was resistant to spontaneous HCC
development, but susceptible to CA-induced hepato-carcinogenesis.
The results of another study showed that intestine-specific FXR
reactivation could impair the spontaneous development of HCC
(92). These findings
underscored the critical role of systemic BA homeostasis regulated
by FXR in HCC pathogenesis, while highlighting pro- or
anti-tumorigenic functions of tissue-specific FXR.
The role of FXR/SHP axis in the segments of lipid
metabolism and BAs synthesis, including FXR/SHP/CD36,
FXR/SHP/SREBP1 and FXR/SHP/HNF4α has been summarized; however, the
relationship between FXR and SHP is not just linear. FXR (−/−) and
SHP (−/−) could result in tumorigenesis independently. As a strong
induced target, SHP could affect HCC in the liver of FXR (−/−)
mice. Li et al (93)
engineered FXR (−/−)/SHPTg mice to explore the
contribution of SHP. Their investigation revealed that SHP
overexpression reduced HCC proliferation while enhancing apoptosis,
without altering tumor incidence or dimensions. Although SHP
partially rescued FXR (−/−)-induced hepato-carcinogenesis, it
failed to inhibit JAK2/STAT3 pathway activation, suggesting a
complex crosstalk between inflammatory signaling and the FXR/SHP
axis during HCC development.
While cholestasis constitutes a recognized HCC risk
factor, recent evidence indicated that different BA species exhibit
differential carcinogenic potential, with hydrophobic BAs
demonstrating particular oncogenic properties (94). Chronic liver diseases, such as
cirrhosis and non-alcoholic steatohepatitis, frequently exhibit
compromised BA homeostasis due to diminished hepatic BA transporter
expression and impaired FXR signaling, creating permissive
microenvironments for HCC progression (71,72,74,75,94). As a BA-causing efflux and FXR
target gene, BSEP deficiency contributed to cholestasis and HCC
development. In addition to conventional regulatory mechanisms,
limited studies have shown that FXR subtypes affect BSEP
transcription. Chen et al (17) found that FXRα2 exhibited a
stronger transactivation activity of human BSEP than FXRα1.
Proinflammatory cytokines, such as IL-6 and TNF-α, were elevated in
HCC tissues. When Huh7 was treated by these inflammatory cytokines,
the ratio of FXR α1/FXR α2 was increased, while it was decreased
following BSEP treatment (17).
The aforementioned study showed that FXR subtypes affect bile
content, and thus the development of HCC.
On the other hand, FXR deficiency results in lipid
accumulation and attenuates FAO. Pharmacological FXR activation has
been shown to have a therapeutic potential in preclinical models of
HCC, notably ameliorating NAFLD and NASH. NAFLD pathophysiology
features elevated hepatic and circulating TG levels (95) and is converted to NASH if not
well controlled. Clinical evidence has revealed the consistent FXR
downregulation in both pediatric and adult NAFLD/NASH cohorts, as
compared with simple obesity or healthy hepatic tissue (96,97). The FXR agonist obeticholic acid
has been identified to be effective to NASH (98); however, there have been some
adverse reactions, prompting the intensive development of
next-generation FXR modulators. To address these concerns, multiple
FXR agonists in NAFLD and NASH have been explored (99-102). Lipocalin 13 (LCN13), recently
identified as an FXR-regulated hepatoprotective factor, mitigates
steatosis through the dual modulation of lipogenesis suppression
and FA β-oxidation enhancement. Its transcriptional downregulation
in patients with NASH, coupled with FXR-dependent activation,
positions LCN13 as both a novel FXR target and a promising
therapeutic candidate (103).
Beyond BA homeostasis and lipid modulation, emerging
mechanisms delineated in the study by Huang et al (104) implicated FXR in hepatic
regeneration, injury resolution, anti-inflammatory signaling and
direct neoplastic regulation. Crucially, these pathways have
exhibited extensive cross-talk, with collaborative interactions
ultimately driving hepato-carcinogenesis.
Colorectal cancer (CRC)
Due to its role in BA circulation and gut
microbiota, the development of intestinal tumors involves a complex
interaction with the FXR's expression and activity. Beyond the
direct regulation of BA synthesis/transport machinery, FXR-mediated
pathological mechanisms extend to bidirectional crosstalk with
microbial communities and immunomodulatory pathways.
The gut microbes have profound effects on BA
metabolism by promoting the decoupling, dehydrogenation and
dihydroxylation of primary BAs in the distal small intestine and
colon, thereby increasing the chemical diversity of Bas (105). Gut microbes and FXR, as two
factors in BA regulation, also interact with each other through the
change of BA pool. Sayin et al (105) analyzed the differences in BAs
between germ-free (GF) and conventionally raised (CONV-R) mice. The
BAs' composition spectrum showed organic differences between two
groups, and the BAs' pool of CONV-R was reduced by 71±2%. Further
intestinal segment analysis revealed the effect of the presence of
intestinal microbes on the BAs' profiles of different intestinal
segments; that is, the modification of BAs by microorganisms. Given
FXR's role in BA balance, they explored the activities of FXR and
found that FXR, SHP and FGF15 are upregulated in distal ileum of
CONV-R mice compared with the GF group. Furthermore, the
upregulation of SHP, FGF15 and IBABP was eliminated when
FXR−/− mice were rederived as GF. These findings
revealed that the effect of gut microbes on BAs is induced by FXR.
In patients with CRC, 16s rRNA shows multiple mucosal microbes
increased, including the genera Bacteroides, Curto
bacterium and Campylobacter (106). In addition, these microbes have
a stronger ability to produce DCA, which inhibits FXR and promotes
CRC. Consistent with this, the low expression of FXR was observed
in a different CRC study, meanwhile researchers found that FXR was
negatively correlated with the PI3K pathway and EMT (107). By contrast, FXR deficiency
results in the of BA profiles, which then affects gut PH and,
ultimately, enterotoxigenic Bacteroides Fragilis (ETBF)
mucosal colonization, contributing to the development of CRC
(108).
Immune microenvironment alterations emerge through
dual mechanisms. One reason for that is microbial imbalance.
Bacteroides fragilis toxins, produced by ETBF, have a
carcinogenic effect on colonic epithelial cells. It can induce
mucosal Th17 response, which in turn selectively leads to the
activation of the NF-κB pathway (109,110). In addition, Guo et al
(108) demonstrated that BA
levels affect IgA concentrations, which serve as the first line of
defense for intestinal immunity. Secretory IgA is increased in the
high-BA group, and decreased in the low-BA group. This change of
immune environment also plays a synergistic role in the
colonization of intestinal microbiota and CRC. Another reason for
that was that FXR directly influences immune cell function and
inflammation resolution. Colitis-associated cancer (CAC) is a
subtype of CRC associated with inflammatory bowel disease (IBD).
Macrophages are located in the intestinal epithelium sublaminar
propria, mediating the inflammatory response to foreign substances,
playing a role in defense, clearance and antigen presentation.
Previous studies have shown that the occurrence of inflammation
impaired BA homeostasis, and the activation of FXR exerted a
protective effect on the intestine (111,112). Compared with healthy tissues,
FXR and downstream gene expression was decreased in CAC tissues,
while proinflammatory factors were significantly increased. These
cytokines resulted in the upregulation of Paneth and goblet cell
markers, promoting organoid budding and branching, which suggests
that the stemness of intestinal stem cells was promoted, therefore
contributing to CAC. Since it has been previously suggested that
FXR reduces IL-17 and IFN, and Th17 cells secreting inflammatory
cytokines have been shown to be associated with CAC mouse models,
Dong et al (111)
explored the effect of FXR agonist Fexaramine D on tumor
progression. The results revealed that FXR activation enhanced the
polarization and maturation of macrophages, which further led to
the activation of Th17 cells (111). Furthermore, inflammation drove
an increase in innate lymphoid cells (ILC) in IBD, particularly
ILC3. The activation of FXR not only blocked the activation of
IL-17a and IL-17f in ILC but also regulated the maturation and
differentiation of ILC (112).
Collectively, intestinal carcinogenesis emerges from
dynamic FXR-mediated crosstalk between BA metabolism, microbial
ecology, barrier integrity and immune surveillance. As a primary
regulator of BA homeostasis, FXR represents both biomarker and
therapeutic target in CRC development, with pharmacological
activation holding promise for intercepting microbiome-driven
malignancy progression.
FXR and breast cancer
According to data from the World health Organization
(http://gco.iarc.who.int), breast cancer ranks
first in incidence and mortality among all cancer types in women.
Estrogen receptor, progesterone receptor and human epidermal growth
factor receptor 2 (HER2) status constitute critical determinants of
metastatic progression, with osseous metastasis being the most
frequent dissemination pattern. FXR has been found to be expressed
in primary breast cancers with bone metastases and plays an
important role in osteo-mimetism of tumor cells to result in the
osteo-tropism of breast cancer (113). Mechanistic investigations by
Silva et al (114)
revealed that bone-derived sodium deoxycholate enhanced the
metastatic potential of MDA-MB-231 cells through FXR upregulation
and nuclear translocation-an effect abrogated by
Z-guggulsterone-mediated FXR inhibition (114,115). However, another study showed
that FXR activators CDCA and GW4064 resulted in the apoptosis of
MCF-7 and MDA-MB-468 cell lines (116). Of note, FXR's oncogenic role
exhibits concentration-dependent duality. A low DCA concentration
(10 µmol/l) promotes metastasis, while a high DCA (100-150
µmol/l) causes apoptosis (114). Furthermore, it has been shown
that the mRNA expression of SHP, MRP2 and IBABP is induced in
breast cancer lines following treatment with a low concentration of
GW4060 (3 µmol/l) (116). The study by Alasmael et
al (117) also supported
that the activation of FXR induces apoptosis, without increasing
the migration potential. Discrepancies between these studies likely
reflect methodological variations in FXR activation thresholds and
cellular context dependencies (117).
For ER-α-positive breast cancer, tamoxifen (tam) is
the first-line endocrine therapy. However, drug resistance often
occurs on account of HER2+. Therefore, the combination
of endocrine therapy and targeted therapy is the optimal treatment
for patients with HR+HER2+ breast cancer.
Targeting HER2 is more critical. Giordano et al (118) found that the activation of FXR
by GW4064 and CDCA inhibited the growth of a tam-resistant MCF-7
TR1 cell line. Their results demonstrated that CDCA reduced HER2
expression and impaired EGF-mediated HER2 and p42/44 MAPK
phosphorylation. This suggested that the inhibition of HER2/MAPK
through FXR may offer a new therapy direction for
HR+HER2+ breast cancer.
Additional pathophysiological interactions emerge
through leptin signaling modulation (119). GW4064 inhibits the growth,
migration and invasion of breast cancer induced by leptin,
affecting tumor-promoting activities of cancer-associated
fibroblasts. The underlying mechanism showed that FXR increased the
expression of suppressor of the cytokine signaling 3, leading to
the inhibition of leptin signaling and downregulation of its target
genes.
These findings suggest that targeting FXR may
provide a new direction for breast cancer treatment but should take
into consider the biological context and dose effects.
FXR and urinary system cancer
Kidney cancer
RCC is the most common type of kidney cancer, with
ccRCC accounting for 60-70%. This histological subtype displays
characteristic metabolic aberrations manifested by cytoplasmic
lipid droplet and glycogen accumulation. In a multi-omics
investigation, Strauss et al (120) stratified patients with ccRCC
into non-progressing (NP) and low-risk tumors progressing within a
5-years average follow-up (P) cohorts based on tumor behavior
during a 5-year surveillance period. Comparative proteomic
profiling revealed an elevated expression of LXR/RXR- and
FXR/RXR-related proteins in the NP cohort compared with the P
cohort (121). One functional
validation study demonstrated that FXR knockdown suppressed ACHN
cell proliferation, although this antitumor effect was absent in
HK-2 proximal tubule cells (121). These findings were consistent
with those of Huang et al (122), who reported FXR upregulation in
specimens of patients with ccRCC and cell lines. FXR may influence
cell cycle through the cyclin E2/cyclin dependent kinase 2 pathway.
In conclusion, these results showed that FXR could affect the
development of ccRCC.
Most studies agree that the accumulation of FAs and
cholesterol leads to tumor progression, while its oxidative
metabolism inhibits tumor growth in ccRCC. Emerging evidence
positions FXR as a key metabolic regulator in ccRCC, characterized
by the upregulation of its transcriptional targets CD36 and ACC
alongside carnitine palmitoyl-transferase 1A downregulation
(123). Another FA transport
molecule, CAV-1, is upregulated and has been identified as a
promising therapeutic target (124). This apparent divergence from
the canonical FXR/SHP/CD36 and FXR/CAV-1 liver signaling pathways
raises thought-provoking questions about organ-specific metabolic
regulation, potentially reflecting the liver's unique lipid
homeostatic dominance.
SRBI promotes the influx of HDL into ccRCC,
resulting in the accumulation of cholesterol in ccRCC, which is
conducive to the growth of tumor cells (125). When inhibiting SRBI, the
proliferation of ccRCC is impaired (126). The expression levels of SRBI
and FXR are parallel in the liver (127). The evolving understanding of
renal vs. hepatic FXR signaling highlights fundamental differences
in organ-specific lipid regulatory networks.
Collectively, current evidence reveals a dualistic
role for FXR activation in ccRCC progression, simultaneously
modulating pro- and anti-tumorigenic pathways. The intricate
crosstalk between FXR-mediated lipid/cholesterol metabolism and
renal oncogenesis remains inadequately characterized, necessitating
systematic investigation.
Prostate cancer (PCa)
High cholesterol is a risk factor for PCa. Beyond BA
synthesis, cholesterol serves as the metabolic precursor for sex
hormone biosynthesis, particularly androgens that drive PCa
pathogenesis. While androgen receptor (AR/NR3C4) has been found
associated with lipid metabolism (128), emerging evidence reveals
complex regulatory cross-talk with nuclear receptors, including LXR
and FXR, acting as metabolic counterbalances. Two mechanistic
studies demonstrated that pharmacological FXR activation suppresses
PCa proliferation through phosphatase and tensin homolog deleted on
chromosome ten upregulation and SREBP1 downregulation (129,130). In addition, CDCA and GW4064,
FXR activators, both inhibit the expression of UGT2B15/17 and their
combination with androgens. This means FXR activators regulate
androgen metabolism in PCa as well. Despite growing evidence
implicating FXR in cholesterol and FA metabolic reprogramming
through SREBP1 modulation, the precise mechanistic interplay
between these pathways in PCa pathobiology remains incompletely
characterized and warrants systematic investigation.
Conclusion and perspectives
For most solid tumors, surgery is the first line of
treatment. The emergence of targeted drugs has brought the dawn of
precision treatment of tumors, but the invasive nature of surgery,
the resistance of targeted drugs and the recurrence of tumors have
brought new challenges, which require researchers to explore the
potential targets of tumor therapy from a new angle. An increasing
number of studies are focused on the key regulatory molecules of
metabolic reprogramming. For lipid-rich tumors, lipids play a
significant role in the process of pathological changes and become
one of the important driving factors for the transition from
pretumor to tumor stage. Thus, lipid metabolism is a good
breakthrough, and there are already a large number of small
molecule drugs targeting this aspect. In addition, lipid-lowering
agents, such as statins, have been used in clinical cohorts of
cancer patients to investigate the effect of serum lipid level on
tumor progression.
The present review showed that the interaction
between FXR and lipid metabolism involves a multifaceted regulatory
network of BA-cholesterol axes, FAs and lipoproteins, with profound
implications for lipid-rich tumors. The role of FXR in lipid
metabolism can be summarized as follows: First, FXR is activated by
physiological ligand CDCA or synthetic agonists (such as GW4064),
which inhibits LRH-1/HNF4α through SHP-dependent inhibition,
thereby inhibiting CYP7A1 and CYP8B1, and limiting BA synthesis. At
the same time, it upregulates BSEP and OSTα/β, coordinates BA
outflow and prevents cholestasis and lipid overload. Secondly, on
the one hand, FXR antagonizes fat production by inhibiting SREBP-1c
and ChREBP, thereby downregulating FASN and ACC1. On the other
hand, it enhances FAO through PPARα activation and CPT1A induction,
thereby controlling FA flux. Thirdly, FXR inhibits MTP/apoB through
SHP/HNF4α, thereby reducing VLDL secretion, and promotes
HDL-mediated cholesterol reverse transport for plasma lipoprotein
remodeling through PLTP and SRBI. In the tumors listed, FXR plays
both pro- and antitumor roles, depending on the heterogeneity of
the tumor.
Overall, the present review summarized the
relationship between FXR, lipids and tumors from the perspective of
lipid metabolic reprogramming, suggesting that FXR may be one of
the potential targets for tumor therapy, which provides new
implications for tumor therapy from the perspective of tumor
metabolic tendency.
Availability of data and materials
Not applicable.
Authors' contributions
YNS reviewed previous research on FXR, drafted the
manuscript and drew the figures. HJL and KS revised the manuscript
and helped with literature review. QHX revised the manuscript
critically for important intellectual content. The implementation
of this study was aided by the participation of all authors. All
authors read and approved the final version of the manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant nos. 82072816 and 82272713) and the
Natural Science Foundation of Shandong (grant no.
ZR2021LZY003).
References
|
1
|
Mangelsdorf DJ, Thummel C, Beato M,
Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M,
Chambon P and Evans RM: The nuclear receptor superfamily: The
second decade. Cell. 83:835–839. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Forman BM, Goode E, Chen J, Oro AE,
Bradley DJ, Perlmann T, Noonan DJ, Burka LT, McMorris T, Lamph WW,
et al: Identification of a nuclear receptor that is activated by
farnesol metabolites. Cell. 81:687–693. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Parks DJ, Blanchard SG, Bledsoe RK,
Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki
AM, Moore DD and Lehmann JM: Bile acids: Natural ligands for an
orphan nuclear receptor. Science. 284:1365–1368. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang H, Chen J, Hollister K, Sowers LC and
Forman BM: Endogenous bile acids are ligands for the nuclear
receptor FXR/BAR. Mol Cell. 3:543–553. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chávez-Talavera O, Tailleux A, Lefebvre P
and Staels B: Bile acid control of metabolism and inflammation in
obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty
liver disease. Gastroenterology. 152:1679–1694.e3. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mencarelli A and Fiorucci S: FXR an
emerging therapeutic target for the treatment of atherosclerosis. J
Cell Mol Med. 14:79–92. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu Q, Sun L, Hu X, Wang X, Xu F, Chen B,
Liang X, Xia J, Wang P, Aibara D, et al: Suppressing the intestinal
farnesoid X receptor/sphingomyelin phosphodiesterase 3 axis
decreases atherosclerosis. J Clin Invest. 131:e1428652021.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Glass CK: Differential recognition of
target genes by nuclear receptor monomers, dimers, and
heterodimers. Endocr Rev. 15:391–407. 1994.PubMed/NCBI
|
|
9
|
Downes M, Verdecia MA, Roecker AJ, Hughes
R, Hogenesch JB, Kast-Woelbern HR, Bowman ME, Ferrer JL, Anisfeld
AM, Edwards PA, et al: A chemical, genetic, and structural analysis
of the nuclear bile acid receptor FXR. Mol Cell. 11:1079–1092.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mi LZ, Devarakonda S, Harp JM, Han Q,
Pellicciari R, Willson TM, Khorasanizadeh S and Rastinejad F:
Structural basis for bile acid binding and activation of the
nuclear receptor FXR. Mol Cell. 11:1093–1100. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jiang L, Zhang H, Xiao D, Wei H and Chen
Y: Farnesoid X receptor (FXR): Structures and ligands. Comput
Struct Biotechnol J. 19:2148–2159. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tian SY, Chen SM, Pan CX and Li Y: FXR:
Structures, biology, and drug development for NASH and fibrosis
diseases. Acta Pharmacol Sin. 43:1120–1132. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Otte K, Kranz H, Kober I, Thompson P,
Hoefer M, Haubold B, Remmel B, Voss H, Kaiser C, Albers M, et al:
Identification of farnesoid X receptor beta as a novel mammalian
nuclear receptor sensing lanosterol. Mol Cell Biol. 23:864–872.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huber RM, Murphy K, Miao B, Link JR,
Cunningham MR, Rupar MJ, Gunyuzlu PL, Haws TF, Kassam A, Powell F,
et al: Generation of multiple farnesoid-X-receptor isoforms through
the use of alternative promoters. Gene. 290:35–43. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Y, Kast-Woelbern HR and Edwards PA:
Natural structural variants of the nuclear receptor farnesoid X
receptor affect transcriptional activation. J Biol Chem.
278:104–110. 2003. View Article : Google Scholar
|
|
16
|
Vaquero J, Monte MJ, Dominguez M, Muntané
J and Marin JJ: Differential activation of the human farnesoid X
receptor depends on the pattern of expressed isoforms and the bile
acid pool composition. Biochem Pharmacol. 86:926–939. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen Y, Song X, Valanejad L, Vasilenko A,
More V, Qiu X, Chen W, Lai Y, Slitt A, Stoner M, et al: Bile salt
export pump is dysregulated with altered farnesoid X receptor
isoform expression in patients with hepatocellular carcinoma.
Hepatology. 57:1530–1541. 2013. View Article : Google Scholar
|
|
18
|
Correia JC, Massart J, de Boer JF,
Porsmyr-Palmertz M, Martínez-Redondo V, Agudelo LZ, Sinha I,
Meierhofer D, Ribeiro V, Björnholm M, et al: Bioenergetic cues
shift FXR splicing towards FXRα2 to modulate hepatic lipolysis and
fatty acid metabolism. Mol Metab. 4:891–902. 2015. View Article : Google Scholar
|
|
19
|
Massafra V and van Mil SWC: Farnesoid X
receptor: A 'homeostat' for hepatic nutrient metabolism. Biochim
Biophys Acta Mol Basis Dis. 1864:45–59. 2018. View Article : Google Scholar
|
|
20
|
Marzolini C, Tirona RG, Gervasini G,
Poonkuzhali B, Assem M, Lee W, Leake BF, Schuetz JD, Schuetz EG and
Kim RB: A common polymorphism in the bile acid receptor farnesoid X
receptor is associated with decreased hepatic target gene
expression. Mol Endocrinol. 21:1769–1780. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ruscica M, Busnelli M, Runfola E, Corsini
A and Sirtori CR: Impact of PPAR-Alpha polymorphisms-the case of
metabolic disorders and atherosclerosis. Int J Mol Sci.
20:43782019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Meirhaeghe A and Amouyel P: Impact of
genetic variation of PPARgamma in humans. Mol Genet Metab.
83:93–102. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Valdivielso JM and Fernandez E: Vitamin D
receptor polymorphisms and diseases. Clin Chim Acta. 371:1–12.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dahlman I, Nilsson M, Jiao H, Hoffstedt J,
Lindgren CM, Humphreys K, Kere J, Gustafsson JA, Arner P and
Dahlman-Wright K: Liver X receptor gene polymorphisms and adipose
tissue expression levels in obesity. Pharmacogenet Genomics.
16:881–889. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nishigori H, Tomura H, Tonooka N, Kanamori
M, Yamada S, Sho K, Inoue I, Kikuchi N, Onigata K, Kojima I, et al:
Mutations in the small heterodimer partner gene are associated with
mild obesity in Japanese subjects. Proc Natl Acad Sci USA.
98:575–580. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vaxillaire M, Rouard M, Yamagata K, Oda N,
Kaisaki PJ, Boriraj VV, Chevre JC, Boccio V, Cox RD, Lathrop GM, et
al: Identification of nine novel mutations in the hepatocyte
nuclear factor 1 alpha gene associated with maturity-onset diabetes
of the young (MODY3). Hum Mol Genet. 6:583–586. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yamagata K, Furuta H, Oda N, Kaisaki PJ,
Menzel S, Cox NJ, Fajans SS, Signorini S, Stoffel M and Bell GI:
Mutations in the hepatocyte nuclear factor-4alpha gene in
maturity-onset diabetes of the young (MODY1). Nature. 384:458–460.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
van Mil SW, Milona A, Dixon PH, Mullenbach
R, Geenes VL, Chambers J, Shevchuk V, Moore GE, Lammert F, Glantz
AG, et al: Functional variants of the central bile acid sensor FXR
identified in intrahepatic cholestasis of pregnancy.
Gastroenterology. 133:507–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Edwards PA, Kast HR and Anisfeld AM:
BAREing it all: The adoption of LXR and FXR and their roles in
lipid homeostasis. J Lipid Res. 43:2–12. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Laffitte BA, Kast HR, Nguyen CM, Zavacki
AM, Moore DD and Edwards PA: Identification of the DNA binding
specificity and potential target genes for the farnesoid
X-activated receptor. J Biol Chem. 275:10638–10647. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen WD, Wang YD, Zhang L, Shiah S, Wang
M, Yang F, Yu D, Forman BM and Huang W: Farnesoid X receptor
alleviates age-related proliferation defects in regenerating mouse
livers by activating forkhead box m1b transcription. Hepatology.
51:953–962. 2010. View Article : Google Scholar
|
|
32
|
Gautier T, de Haan W, Grober J, Ye D, Bahr
MJ, Claudel T, Nijstad N, Van Berkel TJC, Havekes LM, Manns MP, et
al: Farnesoid X receptor activation increases cholesteryl ester
transfer protein expression in humans and transgenic mice. J Lipid
Res. 54:2195–2205. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Anisfeld AM, Kast-Woelbern HR, Meyer ME,
Jones SA, Zhang Y, Williams KJ, Willson T and Edwards PA:
Syndecan-1 expression is regulated in an isoform-specific manner by
the farnesoid-X receptor. J Biol Chem. 278:20420–20428. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Thomas AM, Hart SN, Kong B, Fang J, Zhong
XB and Guo GL: Genome-wide tissue-specific farnesoid X receptor
binding in mouse liver and intestine. Hepatology. 51:1410–1419.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Panzitt K and Wagner M: FXR in liver
physiology: Multiple faces to regulate liver metabolism. Biochim
Biophys Acta Mol Basis Dis. 1867:1661332021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Anisfeld AM, Kast-Woelbern HR, Lee H,
Zhang Y, Lee FY and Edwards PA: Activation of the nuclear receptor
FXR induces fibrinogen expression: A new role for bile acid
signaling. J Lipid Res. 46:458–468. 2005. View Article : Google Scholar
|
|
37
|
Zhao A, Lew JL, Huang L, Yu J, Zhang T,
Hrywna Y, Thompson JR, de Pedro N, Blevins RA, Peláez F, et al:
Human kininogen gene is transactivated by the farnesoid X receptor.
J Biol Chem. 278:28765–28770. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang X, Huang S, Gao M, Liu J, Jia X, Han
Q, Zheng S, Miao Y, Li S, Weng H, et al: Farnesoid X receptor (FXR)
gene deficiency impairs urine concentration in mice. Proc Natl Acad
Sci USA. 111:2277–2282. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jiang T, Wang XX, Scherzer P, Wilson P,
Tallman J, Takahashi H, Li J, Iwahashi M, Sutherland E, Arend L and
Levi M: Farnesoid X receptor modulates renal lipid metabolism,
fibrosis, and diabetic nephropathy. Diabetes. 56:2485–2493. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Glatz JFC and Luiken J: Dynamic role of
the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid
uptake and utilization. J Lipid Res. 59:1084–1093. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wilson CG, Tran JL, Erion DM, Vera NB,
Febbraio M and Weiss EJ: Hepatocyte-specific disruption of CD36
attenuates fatty liver and improves insulin sensitivity in HFD-fed
mice. Endocrinology. 157:570–585. 2016. View Article : Google Scholar :
|
|
42
|
Zhou J, Febbraio M, Wada T, Zhai Y, Kuruba
R, He J, Lee JH, Khadem S, Ren S, Li S, et al: Hepatic fatty acid
transporter Cd36 is a common target of LXR, PXR, and PPARgamma in
promoting steatosis. Gastroenterology. 134:556–567. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ma Y, Huang Y, Yan L, Gao M and Liu D:
Synthetic FXR agonist GW4064 prevents diet-induced hepatic
steatosis and insulin resistance. Pharm Res. 30:1447–1457. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen S, Sun S, Feng Y, Li X, Yin G, Liang
P, Yu W, Meng D, Zhang X, Liu H and Zhang F: Diosgenin attenuates
nonalcoholic hepatic steatosis through the hepatic
FXR-SHP-SREBP1C/PPARα/CD36 pathway. Eur J Pharmacol.
952:1758082023. View Article : Google Scholar
|
|
45
|
Mastrodonato M, Calamita G, Rossi R,
Mentino D, Bonfrate L, Portincasa P, Ferri D and Liquori GE:
Altered distribution of caveolin-1 in early liver steatosis. Eur J
Clin Invest. 41:642–651. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Fernández-Rojo MA, Restall C, Ferguson C,
Martel N, Martin S, Bosch M, Kassan A, Leong GM, Martin SD, McGee
SL, et al: Caveolin-1 orchestrates the balance between glucose and
lipid-dependent energy metabolism: Implications for liver
regeneration. Hepatology. 55:1574–1584. 2012. View Article : Google Scholar
|
|
47
|
Fernández-Rojo MA, Gongora M, Fitzsimmons
RL, Martel N, Martin SD, Nixon SJ, Brooks AJ, Ikonomopoulou MP,
Martin S, Lo HP, et al: Caveolin-1 is necessary for hepatic
oxidative lipid metabolism: Evidence for crosstalk between
caveolin-1 and bile acid signaling. Cell Rep. 4:238–247. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Motojima K, Passilly P, Peters JM,
Gonzalez FJ and Latruffe N: Expression of putative fatty acid
transporter genes are regulated by peroxisome
proliferator-activated receptor alpha and gamma activators in a
tissue- and inducer-specific manner. J Biol Chem. 273:16710–16744.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Acharya R, Shetty SS and Kumari NS: Fatty
acid transport proteins (FATPs) in cancer. Chem Phys Lipids.
250:1052692023. View Article : Google Scholar
|
|
50
|
Hirsch D, Stahl A and Lodish HF: A family
of fatty acid transporters conserved from mycobacterium to man.
Proc Natl Acad Sci USA. 95:8625–8629. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Falcon A, Doege H, Fluitt A, Tsang B,
Watson N, Kay MA and Stahl A: FATP2 is a hepatic fatty acid
transporter and peroxisomal very long-chain acyl-CoA synthetase. Am
J Physiol Endocrinol Metab. 299:E384–E393. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lee Y, Kim BR, Kang GH, Lee GJ, Park YJ,
Kim H, Jang HC and Choi SH: The effects of PPAR agonists on
atherosclerosis and nonalcoholic fatty liver disease in
ApoE-/-FXR-/- mice. Endocrinol Metab (Seoul). 36:1243–1253. 2021.
View Article : Google Scholar
|
|
53
|
Savage DB, Choi CS, Samuel VT, Liu ZX,
Zhang D, Wang A, Zhang XM, Cline GW, Yu XX, Geisler JG, et al:
Reversal of diet-induced hepatic steatosis and hepatic insulin
resistance by antisense oligonucleotide inhibitors of acetyl-CoA
carboxylases 1 and 2. J Clin Invest. 116:817–824. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Dentin R, Benhamed F, Hainault I, Fauveau
V, Foufelle F, Dyck JR, Girard J and Postic C: Liver-specific
inhibition of ChREBP improves hepatic steatosis and insulin
resistance in ob/ob mice. Diabetes. 55:2159–2170. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yahagi N, Shimano H, Hasty AH, Matsuzaka
T, Ide T, Yoshikawa T, Amemiya-Kudo M, Tomita S, Okazaki H, Tamura
Y, et al: Absence of sterol regulatory element-binding protein-1
(SREBP-1) ameliorates fatty livers but not obesity or insulin
resistance in Lep(ob)/Lep(ob) mice. J Biol Chem. 277:19353–19357.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ma K, Saha PK, Chan L and Moore DD:
Farnesoid X receptor is essential for normal glucose homeostasis. J
Clin Invest. 116:1102–1109. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Watanabe M, Houten SM, Wang L, Moschetta
A, Mangelsdorf DJ, Heyman RA, Moore DD and Auwerx J: Bile acids
lower triglyceride levels via a pathway involving FXR, SHP, and
SREBP-1c. J Clin Invest. 113:1408–1418. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Iizuka K, Takao K and Yabe D:
ChREBP-mediated regulation of lipid metabolism: Involvement of the
gut microbiota, liver, and adipose tissue. Front Endocrinol
(Lausanne). 11:5871892020. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Caron S, Huaman Samanez C, Dehondt H,
Ploton M, Briand O, Lien F, Dorchies E, Dumont J, Postic C, Cariou
B, et al: Farnesoid X receptor inhibits the transcriptional
activity of carbohydrate response element binding protein in human
hepatocytes. Mol Cell Biol. 33:2202–2211. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kliewer SA and Mangelsdorf DJ: Bile acids
as hormones: The FXR-FGF15/19 pathway. Dig Dis. 33:327–331. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Beenken A and Mohammadi M: The FGF family:
biology, pathophysiology and therapy. Nat Rev Drug Discov.
8:235–253. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Montagner A, Polizzi A, Fouché E, Ducheix
S, Lippi Y, Lasserre F, Barquissau V, Régnier M, Lukowicz C,
Benhamed F, et al: Liver PPARα is crucial for whole-body fatty acid
homeostasis and is protective against NAFLD. Gut. 65:1202–1214.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Prawitt J, Abdelkarim M, Stroeve JH,
Popescu I, Duez H, Velagapudi VR, Dumont J, Bouchaert E, van Dijk
TH, Lucas A, et al: Farnesoid X receptor deficiency improves
glucose homeostasis in mouse models of obesity. Diabetes.
60:1861–1871. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Pineda Torra I, Claudel T, Duval C, Kosykh
V, Fruchart JC and Staels B: Bile acids induce the expression of
the human peroxisome proliferator-activated receptor alpha gene via
activation of the farnesoid X receptor. Mol Endocrinol. 17:259–272.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Xu J, Li Y, Chen WD, Xu Y, Yin L, Ge X,
Jadhav K, Adorini L and Zhang Y: Hepatic carboxylesterase 1 is
essential for both normal and farnesoid X receptor-controlled lipid
homeostasis. Hepatology. 59:1761–1771. 2014. View Article : Google Scholar
|
|
66
|
Liu Y, Song A, Yang X, Zhen Y, Chen W,
Yang L, Wang C and Ma H: Farnesoid X receptor agonist decreases
lipid accumulation by promoting hepatic fatty acid oxidation in
db/db mice. Int J Mol Med. 42:1723–1731. 2018.PubMed/NCBI
|
|
67
|
Fernandez-Marcos PJ and Auwerx J:
Regulation of PGC-1alpha, a nodal regulator of mitochondrial
biogenesis. Am J Clin Nutr. 93:884s–890s. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhang C, Deng J, Liu D, Tuo X, Xiao L, Lai
B, Yao Q, Liu J, Yang H and Wang N: Nuciferine ameliorates hepatic
steatosis in high-fat diet/streptozocin-induced diabetic mice
through a PPARα/PPARγ coactivator-1α pathway. Br J Pharmacol.
175:4218–4228. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhang Y, Castellani LW, Sinal CJ, Gonzalez
FJ and Edwards PA: Peroxisome proliferator-activated receptor-gamma
coactivator 1alpha (PGC-1alpha) regulates triglyceride metabolism
by activation of the nuclear receptor FXR. Genes Dev. 18:157–169.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Rizzolo D, Kong B, Taylor RE, Brinker A,
Goedken M, Buckley B and Guo GL: Bile acid homeostasis in female
mice deficient in Cyp7a1 and Cyp27a1. Acta Pharm Sin B.
11:3847–3856. 2021. View Article : Google Scholar
|
|
71
|
Goodwin B, Jones SA, Price RR, Watson MA,
McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, et al:
A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1
represses bile acid biosynthesis. Mol Cell. 6:517–526. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Chiang JY: Bile acid regulation of gene
expression: Roles of nuclear hormone receptors. Endocr Rev.
23:443–463. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Kong B, Wang L, Chiang JY, Zhang Y,
Klaassen CD and Guo GL: Mechanism of tissue-specific farnesoid X
receptor in suppressing the expression of genes in bile-acid
synthesis in mice. Hepatology. 56:1034–1043. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Lee FY, Lee H, Hubbert ML, Edwards PA and
Zhang Y: FXR, a multipurpose nuclear receptor. Trends Biochem Sci.
31:572–580. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Cai SY and Boyer JL: FXR: A target for
cholestatic syndromes? Expert Opin Ther Targets. 10:409–421. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kast HR, Goodwin B, Tarr PT, Jones SA,
Anisfeld AM, Stoltz CM, Tontonoz P, Kliewer S, Willson TM and
Edwards PA: Regulation of multidrug resistance-associated protein 2
(ABCC2) by the nuclear receptors pregnane X receptor farnesoid
X-activated receptor and constitutive androstane receptor. J Biol
Chem. 277:2908–2915. 2002. View Article : Google Scholar
|
|
77
|
Huang L, Zhao A, Lew JL, Zhang T, Hrywna
Y, Thompson JR, de Pedro N, Royo I, Blevins RA, Peláez F, et al:
Farnesoid X receptor activates transcription of the phospholipid
pump MDR3. J Biol Chem. 278:51085–51090. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Sinal CJ, Tohkin M, Miyata M, Ward JM,
Lambert G and Gonzalez FJ: Targeted disruption of the nuclear
receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell.
102:731–744. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Denke MA and Grundy SM:
Hypertriglyceridemia: A relative contraindication to the use of
bile acid-binding resins? Hepatology. 8:974–975. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hirokane H, Nakahara M, Tachibana S,
Shimizu M and Sato R: Bile acid reduces the secretion of very low
density lipoprotein by repressing microsomal triglyceride transfer
protein gene expression mediated by hepatocyte nuclear factor-4. J
Biol Chem. 279:45685–45692. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Liu Q, Yang M, Fu X, Liu R, Sun C, Pan H,
Wong CW and Guan M: Activation of farnesoid X receptor promotes
triglycerides lowering by suppressing phospholipase A2 G12B
expression. Mol Cell Endocrinol. 436:93–101. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Giammanco A, Spina R, Cefalù AB and Averna
M: APOC-III: A gatekeeper in controlling triglyceride metabolism.
Curr Atheroscler Rep. 25:67–76. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Claudel T, Inoue Y, Barbier O,
Duran-Sandoval D, Kosykh V, Fruchart J, Fruchart JC, Gonzalez FJ
and Staels B: Farnesoid X receptor agonists suppress hepatic
apolipoprotein CIII expression. Gastroenterology. 125:544–555.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Kast HR, Nguyen CM, Sinal CJ, Jones SA,
Laffitte BA, Reue K, Gonzalez FJ, Willson TM and Edwards PA:
Farnesoid X-activated receptor induces apolipoprotein C-II
transcription: A molecular mechanism linking plasma triglyceride
levels to bile acids. Mol Endocrinol. 15:1720–1728. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Mak PA, Kast-Woelbern HR, Anisfeld AM and
Edwards PA: Identification of PLTP as an LXR target gene and apoE
as an FXR target gene reveals overlapping targets for the two
nuclear receptors. J Lipid Res. 43:2037–2041. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Urizar NL, Dowhan DH and Moore DD: The
farnesoid X-activated receptor mediates bile acid activation of
phospholipid transfer protein gene expression. J Biol Chem.
275:39313–39317. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Kim I, Morimura K, Shah Y, Yang Q, Ward JM
and Gonzalez FJ: Spontaneous hepatocarcinogenesis in farnesoid X
receptor-null mice. Carcinogenesis. 28:940–946. 2007. View Article : Google Scholar
|
|
88
|
Yang F, Huang X, Yi T, Yen Y, Moore DD and
Huang W: Spontaneous development of liver tumors in the absence of
the bile acid receptor farnesoid X receptor. Cancer Res.
67:863–867. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Su H, Ma C, Liu J, Li N, Gao M, Huang A,
Wang X, Huang W and Huang X: Downregulation of nuclear receptor FXR
is associated with multiple malignant clinicopathological
characteristics in human hepatocellular carcinoma. Am J Physiol
Gastrointest Liver Physiol. 303:G1245–G1253. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Takahashi S, Tanaka N, Fukami T, Xie C,
Yagai T, Kim D, Velenosi TJ, Yan T, Krausz KW, Levi M and Gonzalez
FJ: Role of Farnesoid X Receptor and Bile Acids in Hepatic Tumor
Development. Hepatol Commun. 2:1567–1582. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Kong B, Zhu Y, Li G, Williams JA, Buckley
K, Tawfik O, Luyendyk JP and Guo GL: Mice with hepatocyte-specific
FXR deficiency are resistant to spontaneous but susceptible to
cholic acid-induced hepatocarcinogenesis. Am J Physiol Gastrointest
Liver Physiol. 310:G295–G302. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Degirolamo C, Modica S, Vacca M, Di Tullio
G, Morgano A, D'Orazio A, Kannisto K, Parini P and Moschetta A:
Prevention of spontaneous hepatocarcinogenesis in farnesoid X
receptor-null mice by intestinal-specific farnesoid X receptor
reactivation. Hepatology. 61:161–170. 2015. View Article : Google Scholar
|
|
93
|
Li G, Kong B, Zhu Y, Zhan L, Williams JA,
Tawfik O, Kassel KM, Luyendyk JP, Wang L and Guo GL: Small
heterodimer partner overexpression partially protects against liver
tumor development in farnesoid X receptor knockout mice. Toxicol
Appl Pharmacol. 272:299–305. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Režen T, Rozman D, Kovács T, Kovács P,
Sipos A, Bai P and Mikó E: The role of bile acids in
carcinogenesis. Cell Mol Life Sci. 79:2432022. View Article : Google Scholar
|
|
95
|
Ooi GJ, Meikle PJ, Huynh K, Earnest A,
Roberts SK, Kemp W, Parker BL, Brown W, Burton P and Watt MJ:
Hepatic lipidomic remodeling in severe obesity manifests with
steatosis and does not evolve with non-alcoholic steatohepatitis. J
Hepatol. 75:524–535. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Nobili V, Alisi A, Mosca A, Della Corte C,
Veraldi S, De Vito R, De Stefanis C, D'Oria V, Jahnel J, Zohrer E,
et al: Hepatic farnesoid X receptor protein level and circulating
fibroblast growth factor 19 concentration in children with NAFLD.
Liver Int. 38:342–349. 2018. View Article : Google Scholar
|
|
97
|
Aguilar-Olivos NE, Carrillo-Córdova D,
Oria-Hernández J, Sánchez-Valle V, Ponciano-Rodríguez G,
Ramírez-Jaramillo M, Chablé-Montero F, Chávez-Tapia NC, Uribe M and
Méndez-Sánchez N: The nuclear receptor FXR, but not LXR,
up-regulates bile acid transporter expression in non-alcoholic
fatty liver disease. Ann Hepatol. 14:487–493. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Neuschwander-Tetri BA, Loomba R, Sanyal
AJ, Lavine JE, Van Natta ML, Abdelmalek MF, Chalasani N, Dasarathy
S, Diehl AM and Hameed B, et al: Farnesoid X nuclear receptor
ligand obeticholic acid for non-cirrhotic, non-alcoholic
steatohepatitis (FLINT): A multicentre, randomised,
placebo-controlled trial. Lancet. 385:956–965. 2015. View Article : Google Scholar :
|
|
99
|
Tully DC, Rucker PV, Chianelli D, Williams
J, Vidal A, Alper PB, Mutnick D, Bursulaya B, Schmeits J, Wu X, et
al: Discovery of tropifexor (LJN452), a highly potent non-bile acid
FXR agonist for the treatment of cholestatic liver diseases and
nonalcoholic steatohepatitis (NASH). J Med Chem. 60:9960–9973.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Zhang S, Wang J, Liu Q and Harnish DC:
Farnesoid X receptor agonist WAY-362450 attenuates liver
inflammation and fibrosis in murine model of non-alcoholic
steatohepatitis. J Hepatol. 51:380–388. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Wang J, Yang N and Xu Y: Natural products
in the modulation of farnesoid X receptor against nonalcoholic
fatty liver disease. Am J Chin Med. 52:291–314. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Huang W, Cao Z, Wang W, Yang Z, Jiao S,
Chen Y, Chen S, Zhang L and Li Z: Discovery of LH10, a novel
fexaramine-based FXR agonist for the treatment of liver disease.
Bioorg Chem. 143:1070712024. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Qin X, Tan Y, Ren W, Zhou W, Niu R, Liang
L, Li J, Cao K, Wei G, Zhu X and Huang M: Elevated expression of
LCN13 through FXR activation ameliorates hepatocellular lipid
accumulation and inflammation. Int Immunopharmacol. 131:1118122024.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Huang XF, Zhao WY and Huang WD: FXR and
liver carcinogenesis. Acta Pharmacol Sin. 36:37–43. 2015.
View Article : Google Scholar :
|
|
105
|
Sayin SI, Wahlström A, Felin J, Jäntti S,
Marschall HU, Bamberg K, Angelin B, Hyötyläinen T, Orešič M and
Bäckhed F: Gut microbiota regulates bile acid metabolism by
reducing the levels of tauro-beta-muricholic acid, a naturally
occurring FXR antagonist. Cell Metab. 17:225–235. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Ma Y, Zhang Y, Qu R, Zhou X, Sun L, Wang
K, Jiang C, Zhang Z and Fu W: Promotion of Deoxycholic acid effect
on colonic cancer cell lines in vitro by altering the mucosal
microbiota. Microorganisms. 10:24862022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Bailey AM, Zhan L, Maru D, Shureiqi I,
Pickering CR, Kiriakova G, Izzo J, He N, Wei C, Baladandayuthapani
V, et al: FXR silencing in human colon cancer by DNA methylation
and KRAS signaling. Am J Physiol Gastrointest Liver Physiol.
306:G48–G58. 2014. View Article : Google Scholar :
|
|
108
|
Guo S, Peng Y, Lou Y, Cao L, Liu J, Lin N,
Cai S, Kang Y, Zeng S and Yu L: Downregulation of the farnesoid X
receptor promotes colorectal tumorigenesis by facilitating
enterotoxigenic Bacteroides fragilis colonization. Pharmacol Res.
177:1061012022. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Chung L, Thiele Orberg E, Geis AL, Chan
JL, Fu K, DeStefano Shields CE, Dejea CM, Fathi P, Chen J, Finard
BB, et al: Bacteroides fragilis toxin coordinates a
pro-carcinogenic inflammatory cascade via targeting of colonic
epithelial cells. Cell Host Microbe. 23:203–214.e5. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Wu S, Rhee KJ, Albesiano E, Rabizadeh S,
Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, et al: A
human colonic commensal promotes colon tumorigenesis via activation
of T helper type 17 T cell responses. Nat Med. 15:1016–1022. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Dong X, Qi M, Cai C, Zhu Y, Li Y, Coulter
S, Sun F, Liddle C, Uboha NV, Halberg R, et al: Farnesoid X
receptor mediates macrophage-intrinsic responses to suppress
colitis-induced colon cancer progression. JCI Insight.
9:e1704282024. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Gadaleta RM, van Erpecum KJ, Oldenburg B,
Willemsen EC, Renooij W, Murzilli S, Klomp LW, Siersema PD,
Schipper ME, Danese S, et al: Farnesoid X receptor activation
inhibits inflammation and preserves the intestinal barrier in
inflammatory bowel disease. Gut. 60:463–472. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Absil L, Journé F, Larsimont D, Body JJ,
Tafforeau L and Nonclercq D: Farnesoid X receptor as marker of
osteotropism of breast cancers through its role in the
osteomimetism of tumor cells. BMC Cancer. 20:6402020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Silva J, Dasgupta S, Wang G, Krishnamurthy
K, Ritter E and Bieberich E: Lipids isolated from bone induce the
migration of human breast cancer cells. J Lipid Res. 47:724–733.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Krishnamurthy K, Wang G, Rokhfeld D and
Bieberich E: Deoxycholate promotes survival of breast cancer cells
by reducing the level of pro-apoptotic ceramide. Breast Cancer Res.
10:R1062008. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Swales KE, Korbonits M, Carpenter R, Walsh
DT, Warner TD and Bishop-Bailey D: The farnesoid X receptor is
expressed in breast cancer and regulates apoptosis and aromatase
expression. Cancer Res. 66:10120–10126. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Alasmael N, Mohan R, Meira LB, Swales KE
and Plant NJ: Activation of the Farnesoid X-receptor in breast
cancer cell lines results in cytotoxicity but not increased
migration potential. Cancer Lett. 370:250–259. 2016. View Article : Google Scholar
|
|
118
|
Giordano C, Catalano S, Panza S, Vizza D,
Barone I, Bonofiglio D, Gelsomino L, Rizza P, Fuqua SA and Andò S:
Farnesoid X receptor inhibits tamoxifen-resistant MCF-7 breast
cancer cell growth through downregulation of HER2 expression.
Oncogene. 30:4129–4140. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Giordano C, Barone I, Vircillo V, Panza S,
Malivindi R, Gelsomino L, Pellegrino M, Rago V, Mauro L, Lanzino M,
et al: Activated FXR inhibits leptin signaling and counteracts
tumor-promoting activities of cancer-associated fibroblasts in
breast malignancy. Sci Rep. 6:217822016. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Strauss P, Rivedal M, Scherer A, Eikrem Ø,
Nakken S, Beisland C, Bostad L, Flatberg A, Skandalou E, Beisvåg V,
et al: A multiomics disease progression signature of low-risk
ccRCC. Sci Rep. 12:135032022. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Fujino T, Sakamaki R, Ito H, Furusato Y,
Sakamoto N, Oshima T and Hayakawa M: Farnesoid X receptor regulates
the growth of renal adenocarcinoma cells without affecting that of
a normal renal cell-derived cell line. J Toxicol Sci. 42:259–265.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Huang S, Hou Y, Hu M, Hu J and Liu X:
Clinical significance and oncogenic function of NR1H4 in clear cell
renal cell carcinoma. BMC Cancer. 22:9952022. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Tan SK, Hougen HY, Merchan JR, Gonzalgo ML
and Welford SM: Fatty acid metabolism reprogramming in ccRCC:
Mechanisms and potential targets. Nat Rev Urol. 20:48–60. 2023.
View Article : Google Scholar
|
|
124
|
Zhang CJ, Zhu N, Wang YX, Liu LP, Zhao TJ,
Wu HT, Liao DF and Qin L: Celastrol attenuates lipid accumulation
and stemness of clear cell renal cell carcinoma via CAV-1/LOX-1
pathway. Front Pharmacol. 12:6580922021. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Xu GH, Lou N, Shi HC, Xu YC, Ruan HL, Xiao
W, Liu L, Li X, Xiao HB, Qiu B, et al: Up-regulation of SR-BI
promotes progression and serves as a prognostic biomarker in clear
cell renal cell carcinoma. BMC Cancer. 18:882018. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Riscal R, Bull CJ, Mesaros C, Finan JM,
Carens M, Ho ES, Xu JP, Godfrey J, Brennan P, Johansson M, et al:
Cholesterol auxotrophy as a targetable vulnerability in clear cell
renal cell carcinoma. Cancer Discov. 11:3106–3125. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Chao F, Gong W, Zheng Y, Li Y, Huang G,
Gao M, Li J, Kuruba R, Gao X, Li S and He F: Upregulation of
scavenger receptor class B type I expression by activation of FXR
in hepatocyte. Atherosclerosis. 213:443–448. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Cariello M, Ducheix S, Maqdasy S, Baron S,
Moschetta A and Lobaccaro JA: LXRs, SHP, and FXR in prostate
cancer: Enemies or ménage à quatre with AR? Nucl Recept Signal.
15:15507629188010702018. View Article : Google Scholar
|
|
129
|
Liu J, Tong SJ, Wang X and Qu LX:
Farnesoid X receptor inhibits LNcaP cell proliferation via the
upregulation of PTEN. Exp Ther Med. 8:1209–1212. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Liu N, Zhao J, Wang J, Teng H, Fu Y and
Yuan H: Farnesoid X receptor ligand CDCA suppresses human prostate
cancer cells growth by inhibiting lipid metabolism via targeting
sterol response element binding protein 1. Am J Transl Res.
8:5118–5124. 2016.PubMed/NCBI
|
|
131
|
Urizar NL, Liverman AB, Dodds DT, Silva
FV, Ordentlich P, Yan Y, Gonzalez FJ, Heyman RA, Mangelsdorf DJ and
Moore DD: A natural product that lowers cholesterol as an
antagonist ligand for FXR. Science. 296:1703–1706. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Burris TP, Montrose C, Houck KA, Osborne
HE, Bocchinfuso WP, Yaden BC, Cheng CC, Zink RW, Barr RJ, Hepler
CD, et al: The hypolipidemic natural product guggulsterone is a
promiscuous steroid receptor ligand. Mol Pharmacol. 67:948–954.
2005. View Article : Google Scholar
|
|
133
|
Bijsmans IT, Guercini C, Ramos Pittol JM,
Omta W, Milona A, Lelieveld D, Egan DA, Pellicciari R, Gioiello A
and van Mil SW: The glucocorticoid mometasone furoate is a novel
FXR ligand that decreases inflammatory but not metabolic gene
expression. Sci Rep. 5:140862015. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Dussault I, Beard R, Lin M, Hollister K,
Chen J, Xiao JH, Chandraratna R and Forman BM: Identification of
gene-selective modulators of the bile acid receptor FXR. J Biol
Chem. 278:7027–7033. 2003. View Article : Google Scholar
|
|
135
|
Chang Y, Lin TY, Lu CW, Huang SK, Wang YC
and Wang SJ: Xanthohumol-induced presynaptic reduction of glutamate
release in the rat hippocampus. Food Funct. 7:212–226. 2016.
View Article : Google Scholar
|
|
136
|
Liu W and Wong C: Oleanolic acid is a
selective farnesoid X receptor modulator. Phytother Res.
24:369–373. 2010. View Article : Google Scholar
|
|
137
|
Fang S, Suh JM, Reilly SM, Yu E, Osborn O,
Lackey D, Yoshihara E, Perino A, Jacinto S, Lukasheva Y, et al:
Intestinal FXR agonism promotes adipose tissue browning and reduces
obesity and insulin resistance. Nat Med. 21:159–165. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Pellicciari R, Passeri D, De Franco F,
Mostarda S, Filipponi P, Colliva C, Gadaleta RM, Franco P, Carotti
A, Macchiarulo A, et al: Discovery of
3α,7α,11β-Trihydroxy-6α-ethyl-5β-cholan-2 4-oic Acid (TC-100), a
novel bile acid as potent and highly selective FXR agonist for
enterohepatic disorders. J Med Chem. 59:9201–9214. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Jin L, Wang R, Zhu Y, Zheng W, Han Y, Guo
F, Ye FB and Li Y: Selective targeting of nuclear receptor FXR by
avermectin analogues with therapeutic effects on nonalcoholic fatty
liver disease. Sci Rep. 5:172882015. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Li G, Lin W, Araya JJ, Chen T, Timmermann
BN and Guo GL: A tea catechin, epigallocatechin-3-gallate, is a
unique modulator of the farnesoid X receptor. Toxicol Appl
Pharmacol. 258:268–274. 2012. View Article : Google Scholar :
|