Introduction
Enhancing mitochondrial function has been shown to
positively influence cellular and organismal longevity (1). However, the progressive
dysregulation of mitochondrial function over time constitutes a
hallmark of aging and contributes to the onset of age-related
diseases. Consequently, the increased risk of cancer development
with advancing age is closely associated with mitochondrial
dysfunction, which is driven by alterations in both mitochondrial
DNA (mtDNA) and nuclear-encoded mitochondrial genes (2). Aging and cancer share overlapping
mitochondrial pathways, including reactive oxygen species (ROS)
production, changes in mitochondrial biogenesis and mitophagy, and
the accumulation of mtDNA mutations (2). These alterations create a
biological environment conducive to both aging and oncogenesis.
While aging is typified by a gradual decline in mitochondrial
function, cancer cells frequently reprogram mitochondrial activity
to optimize survival and proliferative potential, highlighting the
paradoxical relationship between these two processes, which remains
a complex and unresolved scientific question.
Although mitochondrial dysfunction is intuitively
linked to the accumulation of mtDNA mutations, advances in
quantitative mtDNA mutation analysis have revealed that such
mutations are detectable across all age groups in healthy tissues
(3). This raises critical
questions, such as how young individuals maintain normal phenotypes
despite the presence of mtDNA mutations, and which mechanisms drive
the phenotypic shifts associated with aging and cancer. Moreover,
mitochondria engage in intricate interactions with nuclear genes to
regulate functionality. While the nuclear genome exerts regulatory
control over mitochondria, retrograde signaling from mitochondria
to the nucleus can either promote cellular adaptation to
environmental changes or, conversely, contribute to aging and
cancer progression by influencing nuclear gene expression (4). The mitochondrial genome accumulates
specific mutation patterns in various tissues during aging and
cancer (5). Ongoing research
aims to determine the molecular mechanisms underlying the dynamic
changes in mtDNA mutation patterns during aging and
oncogenesis.
The present review offers a detailed examination of
mtDNA mutations and their classifications in the contexts of aging
and cancer, and elucidates the role of mtDNA mutations in these
processes.
Mitochondrial function and structure
Cells house 100-20,000 mitochondria depending on
energy needs, each typically containing 2-10 circular mtDNA
molecules. Mitochondrial quantity and mtDNA content vary according
to cell type, energy demand and physiological condition (6). Essential for cellular metabolism
and homeostasis, mitochondria support ATP production, calcium
regulation, iron-sulfur cluster biosynthesis, apoptosis, metabolic
precursor synthesis and ROS generation (7). They proliferate and replicate
independently of the cell cycle to enable growth, division and
organelle repair (6,8,9).
Compared with nuclear DNA, mtDNA replicates faster, lacks histones,
has limited repair mechanisms and is 10-1,000-fold more prone to
mutations (10,11). Its coding density (~93%) far
exceeds that of nuclear DNA (1-2%), reflecting a streamlined
structure for energy production (12). This density heightens the impact
of mutations, leading to mitochondrial dysfunction, including
membrane potential loss (8,13). To preserve mitochondrial
integrity, cells use quality control mechanisms, such as mitophagy
and fusion/fission pathways (14), preventing mtDNA mutations from
exceeding thresholds that disrupt homeostasis (15). Unlike nuclear DNA, mtDNA lacks
robust repair systems.
Mitochondria contain double-stranded circular DNA of
bacterial origin, consisting of heavy and light chains (6,12). mtDNA spans 16.569 kb and encodes
37 genes, including 13 electron transport chain (ETC) polypeptides,
2 ribosomal RNA (rRNA) genes and 22 transfer RNAs (tRNAs) (12) (Fig. 1). Complex I of the ETC is
composed of 45 subunits in humans, including mtDNA-encoded
[mitochondrial-nicotinamide adenine dinucleotide (NADH)
dehydrogenase subunit (ND)1, ND2, ND3, ND4, ND4L, ND5 and ND6] and
nuclear DNA-encoded genes. Complex V is composed of two main parts:
F1 (catalytic) and FO (membrane-bound); it has 16 subunits in
humans, encoded by mtDNA (mitochondrial-ATP6 and ATP8) and nuclear
DNA. Unlike nuclear DNA, mtDNA is maternally inherited. It encodes
oxidative phosphorylation (OXPHOS)-related protein subunits (coding
regions) and respiratory control machinery (non-coding D-loops)
(6,16,17). The D-loop contains replication
origins and transcription promoters but does not code for proteins
or functional RNA. Heavy chains, which are rich in guanine, house
most protein-coding genes, whereas cytosine-rich light chains,
encode fewer genes. mtDNA, organized into nucleoid structures, is
anchored to the inner mitochondrial membrane (IMM) via proteins
such as mitochondrial transcription factor A (TFAM) and ATPase
family AAA domain-containing 3A (12). This tethering aids mtDNA
transcription and replication. Proximity to the IMM ensures
coordination of mtDNA-encoded protein integration and mitochondrial
function. Key replication proteins, such as polymerase γ (POLG) and
Twinkle helicase (TWINKLE), are IMM-anchored, enabling efficient
mtDNA replication, transcription and inheritance during
mitochondrial division (18,19).
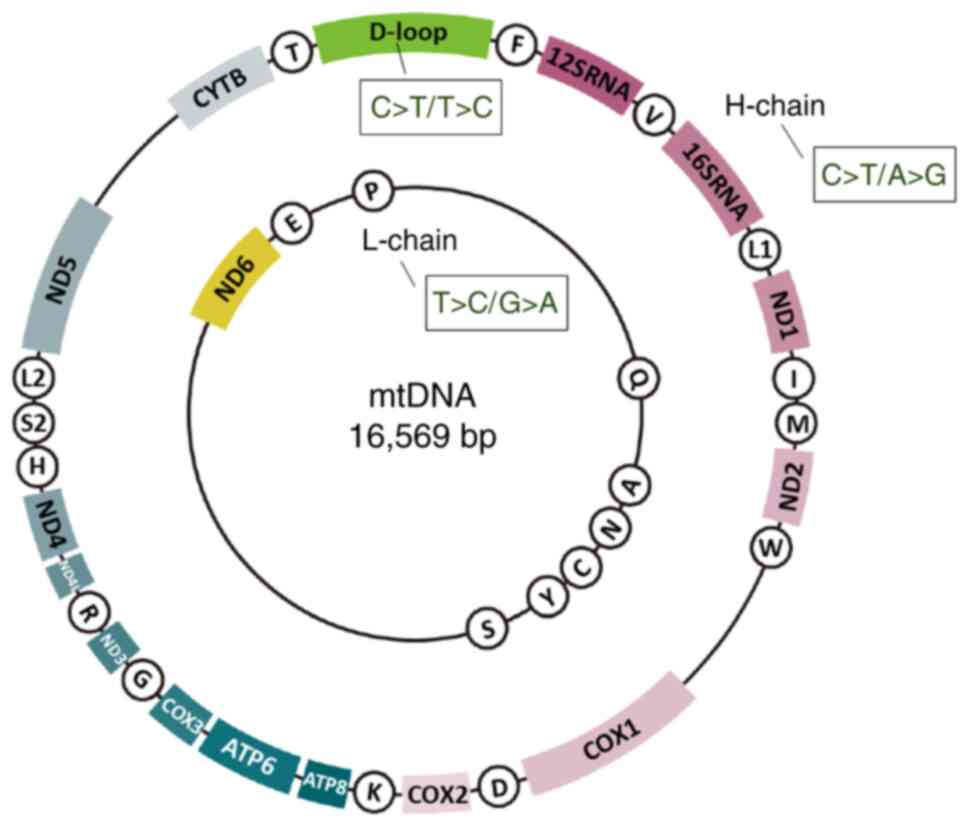 | Figure 1mtDNA structure and strand bias. This
figure is taken from Fig. 2 of
reference (19), with minor
modifications limited to the placement of the 'H-chain' label and
the depiction of strand bias; reprinted under license (https://creativecommons.org/licenses/by/4.0/). In
this version, only the strand-specific bias has been incorporated.
This dual-ring diagram depicts the organization of the
mitochondrial genome, with the outer ring representing the heavy
strand and the inner ring denoting the light strand. The 22
circular elements correspond to tRNA genes. The two labeled boxes,
12SRNA (small subunit) and 16SRNA (large subunit), represent rRNA
genes. The remaining boxes illustrate the 13 protein-coding genes
involved in the electron transport chain and the one D-loop region.
The 'C>T' notation within the boxes indicates a strand-specific
mutational bias. COX2, cytochrome c oxidase 2; mtDNA,
mitochondrial DNA; ND, nicotinamide adenine dinucleotide
dehydrogenase subunit; rRNA, ribosomal RNA; tRNA, transfer RNA. |
mtDNA variations
mtDNA variations are classified into haplotypes
(ancient lineages and adaptive polymorphisms), germline mutations
(heritable alterations within families) and somatic mutations
(arising in individual cells), each with unique traits and
implications (20-22). Haplotypes, maternally inherited
combinations of single nucleotide polymorphisms (SNPs) and genetic
markers, provide insights into ancestry, population genetics and
evolution. They represent normal genetic diversity and may
influence disease susceptibility without being inherently
pathological. Germline mutations occur in reproductive cells,
primarily oocytes, and are transmitted across generations (20-22). Even monozygotic twins may differ
in mtDNA variant proportions due to mutations during early
embryonic development, resulting in heteroplasmy. Heteroplasmy in
the context of mtDNA mutations describes the coexistence of
multiple mtDNA sequence variants; specifically, the presence of
both mutant and wild-type mtDNA molecules, within a single cell,
tissue or organism (10).
Somatic mutations arise from environmental factors, cellular
processes or mtDNA replication errors, accumulating with age and
contributing to disease. High mtDNA replication rates in
energy-demanding cells increase error risk. In addition, ROS
generated during OXPHOS damage mtDNA, causing mutations that
coexist with normal mtDNA, often as low-level heteroplasmy in
healthy individuals (21).
Heteroplasmy levels fluctuate due to genetic drift and selective
replication, which shape mtDNA mutation patterns over time
(23,24).
Crosstalk between nuclear and mitochondrial
genomes
Impact of nuclear DNA on mitochondrial
function
Nuclear genes regulate mtDNA because most
mitochondrial functions depend on nuclear-encoded proteins. Key
roles of nuclear genes include replication and repair, copy number
regulation, biogenesis, fusion/fission and antioxidant defense
(25). Firstly, nuclear-encoded
enzymes (such as POLG, TWINKLE and mitochondrial single stranded
DNA-binding protein) are essential for mtDNA replication (26) (Fig. 2). Repair enzymes address damage
caused by ROS and replication errors. Secondly, nuclear proteins
regulate mtDNA copy number through transcriptional and epigenetic
mechanisms, POLG, TFAM and TWINKLE genes, and pathways such as
peroxisome proliferator-activated receptor γ, coactivator 1α
(PGC-1α; a master regulator of mitochondrial biogenesis) and
AMP-activated protein kinase (AMPK) (27). PGC-1α activates TFAM via nuclear
respiratory factors 1 and 2 (28-32); this process supports rapid energy
generation, for example in cancer cells, which reprograms
metabolism from OXPHOS to glycolysis (Warburg effect) (33,34). Thirdly, oncogenes (MYC, RAS) and
loss of tumor suppressor genes [TP53, phosphatase and tensin
homolog (PTEN)] enhance glycolysis (34,35) and glutaminolysis (36). Hypoxia-inducible factor 1α
(HIF-1α) promotes glycolysis and inhibits mitochondrial activity in
hypoxic tumors (34). Mutations
in nuclear genes encoding components of the ETC, such as succinate
dehydrogenase (SDH) and fumarate hydratase (FH), can arise through
mutations in oncogenes or tumor suppressor genes (37). Loss-of-function mutations in SDH
or FH lead to the accumulation of succinate and fumarate,
respectively, which can stabilize HIF-1α and promote the Warburg
effect, forcing reliance on hexokinase 2-mediated glycolysis
(35,37-39). Fourthly, nuclear genes control
mitochondrial fusion/fission through mitofusin (MFN)1, MFN2 and
dynamin-related protein 1, ensuring mtDNA distribution and removing
defective mtDNA (40).
Mitophagy, mediated by PTEN-induced kinase 1 and Parkin, maintains
mitochondrial health (41).
Finally, nuclear genes encode antioxidants such as superoxide
dismutase 2 (SOD2) and glutathione peroxidase to minimize oxidative
mtDNA damage (42). This
nuclear-mitochondrial coordination supports cellular energy needs
and prevents mitochondrial dysfunction.
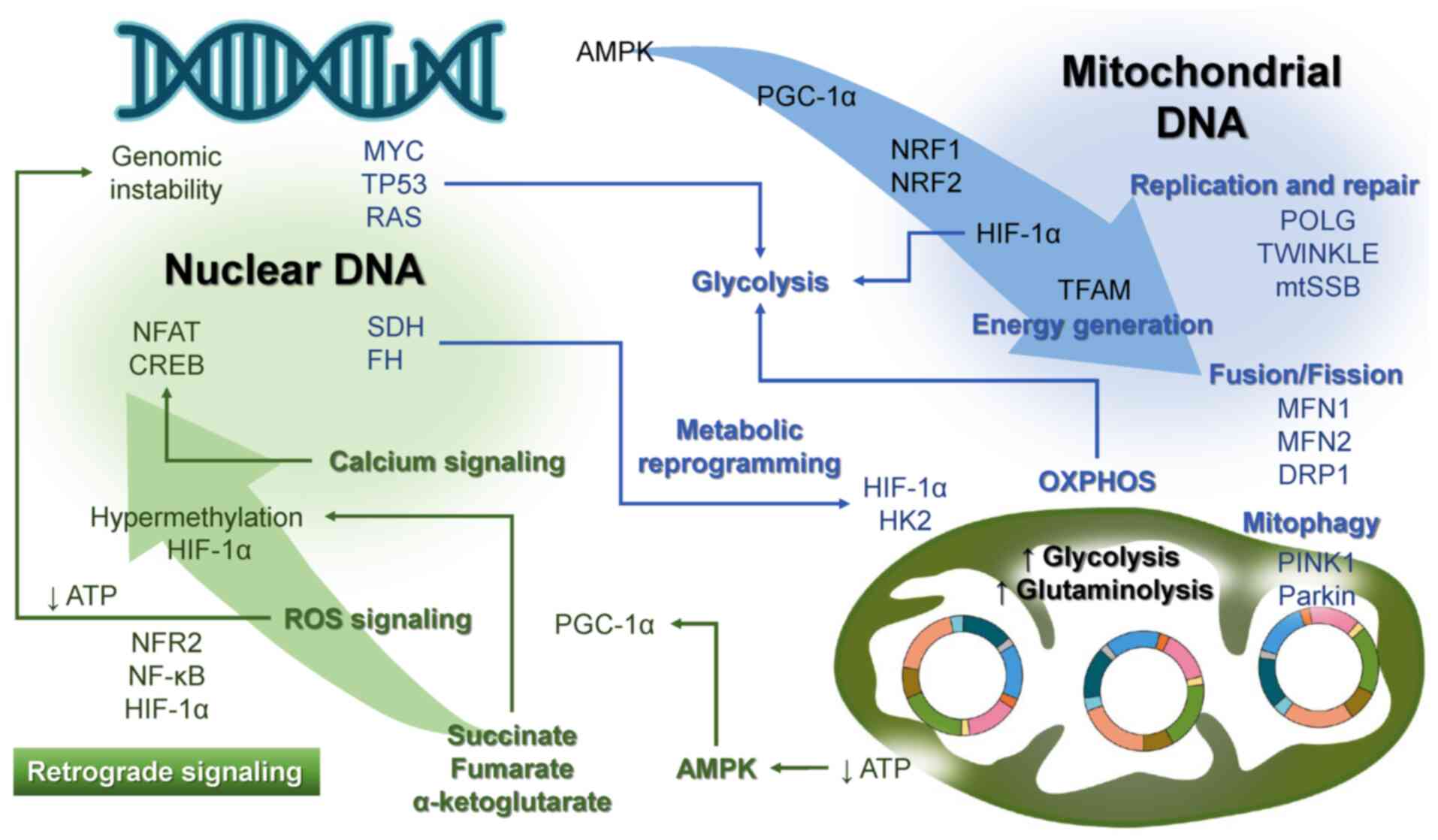 | Figure 2Crosstalk between nuclear and
mitochondrial genomes. The left portion of the figure represents
nuclear genes, whereas the right portion depicts mtDNA. Blue arrows
denote anterograde signaling from nuclear genes to mitochondria,
whereas green arrows illustrate retrograde signaling from
mitochondria to the nucleus. AMPK, AMP-activated protein kinase;
CREB, cyclic adenosine monophosphate response element binding
protein; DRP1, dynamin-related protein 1; FH, fumarate hydratase;
HIF-1α, hypoxia-inducible factor 1α; HK2, hexokinase 2; mtDNA,
mitochondrial DNA; MFN, mitofusin; mtSSB, mitochondrial single
stranded DNA-binding protein; NFAT, nuclear factor of activated T
cells; NRF, nuclear respiratory factor; OXPHOS, oxidative
phosphorylation; PGC-1α, peroxisome proliferator-activated receptor
γ, coactivator 1α; PINK1, PTEN-induced kinase 1; POLG, polymerase
γ; ROS, reactive oxygen species; SDH, succinate dehydrogenase;
TFAM, mitochondrial transcription factor A; TWINKLE, Twinkle
helicase. |
Retrograde signaling from mitochondria to
the nucleus
Retrograde signaling enables mitochondria to
influence nuclear gene expression in response to mitochondrial
function or stress (38,43). This pathway maintains cellular
homeostasis by aligning nuclear transcription with mitochondrial
states. Triggers include mitochondrial dysfunction, ROS, calcium
dysregulation, metabolic shifts and mtDNA mutations (38,43). Impaired OXPHOS and ATP production
activate AMPK, which restores energy balance by promoting catabolic
pathways and influencing nuclear transcription factors such as
PGC-1α to enhance mitochondrial biogenesis (44). Dysfunction mimics hypoxia,
stabilizing HIF-1α, which activates glycolysis and survival genes
(39). Mitochondrial stress,
particularly the increase in ROS and mutations in mtDNA, leads to
the activation of NF-κB as part of the cellular adaptive response
(45). ROS can activate the
NF-κB signaling pathway through the phosphorylation and subsequent
degradation of IκB, leading to the nuclear translocation of NF-κB.
The regulation of nuclear responses by NF-κB includes the
upregulation of antioxidant genes (such as SOD2 and catalase), the
induction of pro-inflammatory cytokines (including TNF-α and IL-6),
and the modulation of apoptosis and cell survival pathways
(43). Moderate concentrations
of ROS serve as pivotal signaling entities in a range of
physiological processes, encompassing cellular survival,
inflammatory regulation and immune system modulation (46). ROS also contribute to genomic
instability via inflammation, oxidative stress and DNA repair
suppression (43). Excess ROS
cause oxidative damage to nuclear DNA, impair repair mechanisms due
to ATP reduction, and disrupt deoxynucleotide triphosphate pools,
causing replication stress (38,47). Mitochondrial dysfunction alters
NAD+/NADH ratios and tricarboxylic acid cycle
intermediates, such as fumarate and succinate, which affect DNA and
histone methylation (48). These
metabolites inhibit α-ketoglutarate-dependent enzymes, causing
hypermethylation and stabilizing HIF-1α, thus promoting
tumorigenesis (34,38). Altered mitochondrial calcium
buffering affects nuclear calcium pathways, influencing factors
such as nuclear factor of activated T cells and cyclic adenosine
monophosphate response element binding protein, which are critical
for chromatin remodeling, DNA repair and cell survival (49). Taken together, retrograde
signaling enables mitochondria to adapt nuclear responses to
stress, metabolic shifts and damage, preserving cellular
function.
mtDNA replication and repair errors
Advances in the quantitative analysis of
mtDNA mutations
The frequency of mtDNA mutations varies widely
across individuals and studies due to differences in mutations
analyzed, detection methods and populations (50). Advances in DNA mutation detection
now provide notable sensitivity and accuracy (6,50-52). The field has transitioned from
low-throughput methods, such as Sanger sequencing, to
high-throughput, ultra-sensitive technologies capable of
identifying rare mutations (6,8,53-55). Techniques such as duplex
sequencing, digital PCR and single-molecule sequencing detect
mutations with unprecedented precision. Duplex sequencing reads
both DNA strands independently, confirming mutations only when
complementary changes occur in both strands, reducing the rate of
false positives (53). This type
of sequencing detects mutations with variant allele frequencies
(VAFs) as low as 1 in 10 million bases, aiding research on rare
mutations in cancer, aging and genetic disorders. Advances have
shed light on mtDNA mutation mechanisms in normal tissues (6,8,51,56). In 2023, double-stranded
sequencing revealed >89,000 somatic mtDNA mutations across eight
aged mouse tissues, uncovering tissue-specific mutational patterns
linked to aging (8). In humans,
similar mutational patterns accumulate over time, including in
cancer-affected tissues (5).
Characteristics of mtDNA mutation
patterns
Replication of mtDNA is initiated at the origin of
the heavy strand and proceeds through a strand-displacement
mechanism, wherein the parental heavy strand remains in a
single-stranded state until replication of the light strand is
subsequently initiated. The prolonged exposure of the heavy strand
in its single-stranded form renders it particularly susceptible to
spontaneous deamination events, notably the conversion of cytosine
to uracil, resulting in C-to-T transition mutations (57). This reflects the inherently
asymmetric nature of mtDNA replication, wherein the temporal delay
between heavy strand and light strand synthesis predisposes the
heavy strand to increased accumulation of base damage and
transition-type mutations. Strand bias in mtDNA mutations refers to
the asymmetric distribution of mutation frequency and types between
the heavy and light strands. The non-random distribution of
mutations observed during mtDNA replication, termed 'replication
bias', contributes to the emergence of distinct mutational
accumulation patterns across the mitochondrial genome. mtDNA
replication errors occur when polymerases incorporate incorrect
nucleotides or during slippage, causing small insertions or
deletions, especially in repetitive sequences (12,58). Repair errors happen when the
repair machinery incorrectly restores DNA damaged by internal
factors (including ROS) or external factors (such as ultraviolet
light). If uncorrected, replication errors lead to mutations in
daughter strands, causing genetic changes in the cell lineage.
Transition mutations (the replacement of one purine
with another purine, or one pyrimidine with another pyrimidine) are
the most common in mtDNA and result from replication errors or
spontaneous deamination, such as cytosine to uracil. Heavy chains
are linked to C>T and A>G transitions, whereas light chains
are associated with T>C and G>A transitions (54,59). These patterns are observed in
human tissues, tumors and model organisms such as Drosophila
(54,60,61). Transversions (the replacement of
purines with pyrimidines) are less common, and are often caused by
ROS or environmental mutagens (54,55,62). Transitions more frequently result
in synonymous changes, whereas transversions are more likely to
cause nonsynonymous mutations that impair protein function. The
majority of mtDNA mutations that accumulate during aging and
tumorigenesis are transition mutations, whereas transversion
mutations are relatively infrequent. This skewed mutation spectrum
can be attributed not only to the molecular mechanisms underlying
mutagenesis, but also to selective pressures at both the
mitochondrial quality control level and the cellular level, which
preferentially eliminate functionally deleterious mutations
(51,56,63). Notably, the subunits of the
OXPHOS system encoded by mtDNA are subject to stringent structural
and functional constraints, and transversion mutations are more
likely to induce non-conservative amino acid substitutions,
severely compromising protein conformation and enzymatic activity.
Consequently, such mutations can impair mitochondrial function by
reducing ATP production and increasing ROS generation, thereby
promoting the selective elimination of dysfunctional mitochondria
via quality control mechanisms such as mitophagy (51,56,63). Therefore, highly pathogenic
mutations, including transversions, are less likely to undergo
clonal expansion due to purifying selection, and their prevalence
remains low even in the context of aging and cancer progression.
Thus, mutation fate depends on genetic drift and natural selection,
influencing whether mutations are fixed, remain heteroplasmic or
are eliminated (56,64). Strand bias in mtDNA mutations
reflects asymmetric replication dynamics, oxidative stress
exposure, limited repair efficiency and selective pressures acting
on mutational events. Studying transition and transversion
mutations offers insights into mitochondrial dysfunction,
mutagenesis factors, and their potential as diagnostic or
therapeutic tools. Furthermore, the non-D-loop region exhibits high
mutation rates, primarily C>T and T>C transitions (65). Mutations in coding regions, such
as ND or cytochrome c oxidase genes, may be synonymous or
pathogenic, whereas mutations in tRNA genes can disrupt protein
synthesis (66). Common mtDNA
mutations in healthy individuals, such as C>T, T>C and
A>G, depend on factors including heteroplasmy and affected
genes. Additional influences on mtDNA mutations include bottleneck
effects, genetic drift, selection, mitophagy and mitochondrial
dynamics. The bottleneck theory explains rapid shifts in mtDNA
mutation prevalence during development due to reduced mtDNA copy
numbers (67). Genetic drift in
small mitochondrial populations can fix or eliminate mutations
regardless of selection. Selection acts on mutations based on
cellular fitness (68), with
mitophagy and mitochondrial dynamics further shaping mtDNA
integrity in health, aging and disease.
Characteristics of mtDNA mutations in aging
and cancer
Effect of aging on mtDNA mutations
mtDNA mutations are present in all human body fluids
and tissues, with frequencies influenced by age, environmental
exposure, oxidative stress, genetic factors and health status
(3,69). A 2008 study of umbilical cord
blood revealed that 0.5% of healthy newborns had pathogenic mtDNA
mutations, with the A3243G mutation in the tRNALeu(UUR)
gene being common. This mutation is associated with several
mitochondrial diseases, including mitochondrial encephalomyopathy,
lactic acidosis, and stroke-like episodes syndrome; maternally
inherited diabetes and deafness; and chronic progressive external
ophthalmoplegia (3). However,
the frequency of these mutations does not exceed the disease
threshold, likely due to their lack of health impact. Recently,
Hong et al (70) analyzed
mtDNA mutations from the UK Biobank, involving 500,000 participants
aged 40-69 years. The study revealed that 30.5% of 194,871
participants had heteroplasmic single nucleotide variants with a
VAF of 5%. Nonsynonymous mutations in complexes I and V of the ETC
occurred at frequencies of 46.5 and 65.8%, respectively. Despite
the high mutation rate in complex V, the low VAF suggested no
notable adverse effects on the ETC. Additionally, gene mutations
have been validated with single-cell sequencing by targeted
amplification of multiplex probes, revealing an average of 0.7
mutations per cell in blood cells obtained from a 76-year-old woman
(23). The same study found that
>60% of these mutations were in protein-coding genes, with
>70% classified as nonsynonymous, and over half of these
predicted to be highly pathogenic (23). In addition, despite being derived
from clinically healthy elderly individuals, 20% of detected
mutations exhibited a VAF exceeding 90% (23), suggesting that high-VAF clones
may proliferate due to less efficient mtDNA quality control with
age (71,72). Notably, these pathogenic mtDNAs
may be particularly well-adapted to the aging cellular environment
(73).
Clonal expansion of mtDNA mutations denotes the
process by which a particular mtDNA mutation, initially confined to
a limited number of mitochondrial genomes, progressively becomes
predominant within a cell or tissue over time. This clonal
expansion could increase genetic diversity under selective
pressure. Cote-L'Heureux et al (6) studied mtDNA mutations in tissues
from young (4.5 months) and old (26 months) mice, with human
equivalents being ~10 and 65 years old, respectively. Different
tissues showed distinct mtDNA mutation profiles, with the kidney
accumulating the most mutations. This study confirmed that mutated
mtDNA molecules may propagate through stochastic processes,
indicating clonal proliferation with age. Finally, a report
investigated mtDNA mutations in germ cells; in mice, mtDNA
mutations in oocytes increased with age (74). However, in macaques, mutations in
the liver and muscle have been reported to increase with age,
whereas oocyte mutations may plateau after 9 years of age (27 years
in humans), suggesting protection against the accumulation of
age-related mutations in oocytes (53).
mtDNA mutations in cancer
Studies on mtDNA mutations in cancer cells have been
ongoing since 1985, with notable discoveries made by the late 2000s
(38,75). Mutations at nucleotides 10398 and
16189 have been linked to breast and endometrial cancer (35). In early-stage breast cancer, two
tumor-specific heteroplasmic transitions (T2275C and A8601G) have
been identified (76). Earlier
studies identified less frequent detections, possibly due to the
use of less sensitive detection methods (35,76) or the relatively higher prevalence
of neutral SNPs (77,78). Systematic investigations of mtDNA
in various types of cancer have become increasingly prevalent, with
Lu et al reviewing 101 studies published between 1998 and
2008 (38). mtDNA mutations,
including point mutations, deletions and copy number changes, are
common in various types of cancer, such as gastric cancer, where
65% of patients have been reported to have at least one mutation
(30,79). Most gastric mutations occur in
the D-loop region, although complex I genes also exhibit mutations
(38). In endometrial and
pancreatic cancer, mutations have also been found in complex I and
other regions (22,80). Mutations in complex I can impair
the ETC and increase ROS production, leading to a higher risk of
B-cell lymphoma (81). Mutations
in complex V, especially ATP8, may hinder cancer cell proliferation
(82). Additionally, tRNA and
rRNA mutations have been observed in several types of cancer
(35,38,66,76,83), and the 4,977-bp deletion mutation
in mtDNA is common in gastric (84), lung (85) and liver cancer (29,38). Overall, the occurrence of mtDNA
mutations is not the primary cause of cancer development, but
similar mutations are commonly observed across different cancer
types (30,86).
Notably, advances in mtDNA mutation detection have
allowed for detailed analysis of mutation types, VAFs and
heteroplasmic variants. In 2021, whole-exome sequencing revealed
that pathogenic mtDNA mutations occur at frequencies similar to
cancer driver mutations in nuclear genes (17). Nonsynonymous mutations have been
shown to be common in complex I, whereas synonymous mutations are
frequent in complex V (17,22). A 2023 study using droplet digital
PCR showed that tissue-derived mtDNA has more heteroplasmic
mutations than whole blood mtDNA, suggesting that heteroplasmic
mutations contribute to carcinogenesis (87). Also in 2023, research on mtDNA in
extracellular vesicles in patients with colorectal cancer showed
higher mutation rates, and more missense and nonsense mutations
compared with whole blood mtDNA, highlighting the inadequacy of
whole blood mtDNA for cancer detection (88). Overall, unlike nuclear DNA
mutations that exhibit cancer type-specific signatures, mtDNA
mutations are largely consistent across various tumor types
(22,86), with no distinct mtDNA
corresponding to oncogenes or tumor suppressor genes.
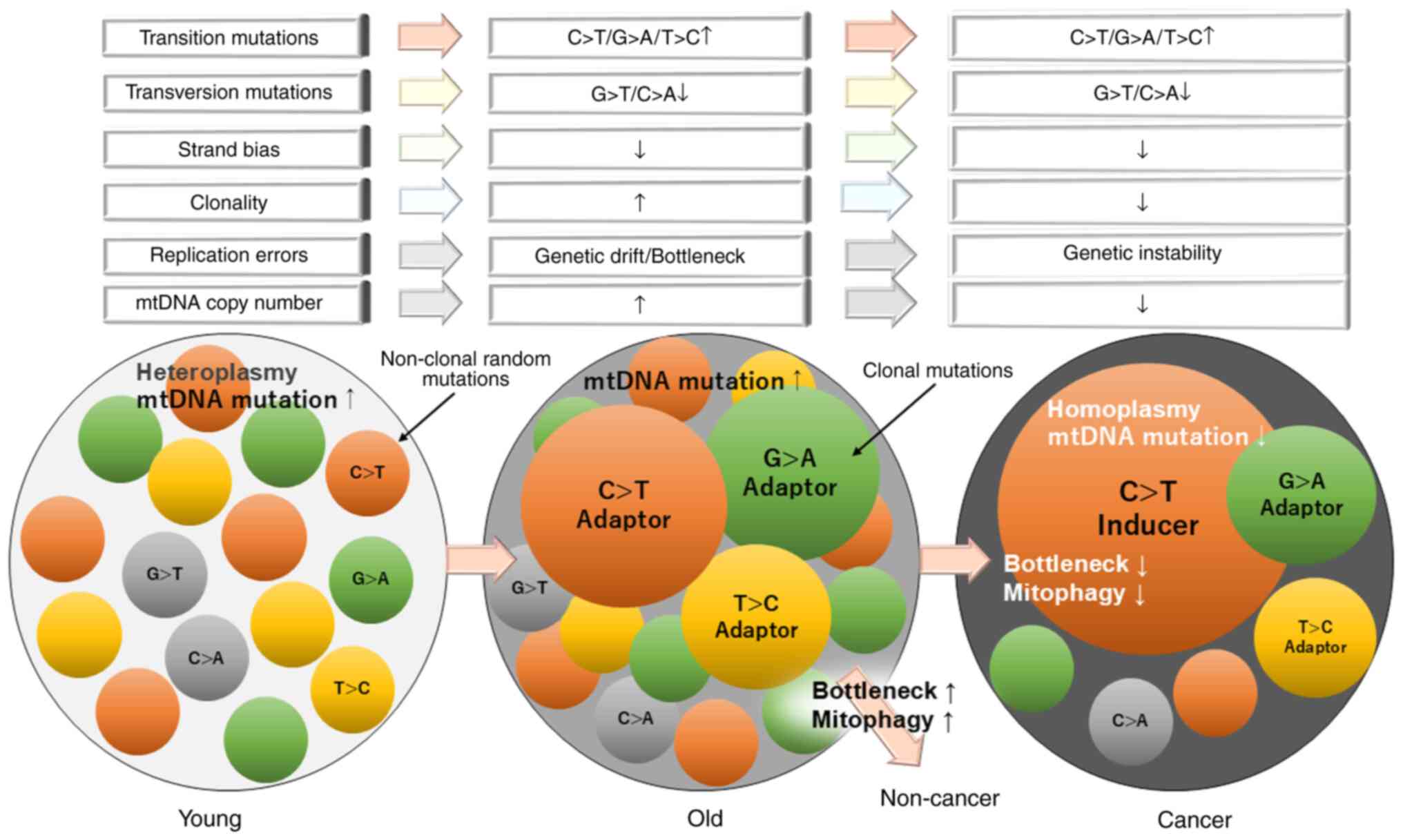
Carcinogenesis likely involves the transition from non-clonal
mutations to clonal mutations with high VAFs.
Changes in mtDNA mutation patterns and
dynamics of clonal expansion
Age-related changes
This subsection explores how mtDNA mutation types
and clonal expansion dynamics evolve with age. Recent findings have
highlighted that aging impacts mtDNA mutations, with data from the
UK Biobank (2023) showing a transition-to-transversion mutation
ratio of 28.7 among individuals aged 40-69 years, suggesting most
mtDNA mutations result from DNA POLG errors rather than oxidative
stress (70,89). Oxidative stress-induced
8-hydroxydeoxyguanosine causes G>T/C>A translocations;
however, studies in humans (89-91), mice (51,92) and Drosophila (93,94) have found limited evidence of such
mutations, indicating oxidative damage is not the primary cause of
age-related mtDNA mutations (8).
Even in patients with polycystic ovary syndrome, a condition linked
to oxidative stress, transition mutations dominate over
translocations (82.35 vs. 17.64%) (95). Additionally, a 2023 study in a
76-year-old woman revealed >95% of mtDNA mutations were
heteroplasmic transitions, predominantly G>A and T>C
(24). While these increase with
age, G>T and C>A translocations do not accumulate (56,57,65,90,96), suggesting they are either
eliminated or fail to persist (6,8,54). Although translocations are rare
in Drosophila (90),
high-resolution analyses in Caenorhabditis elegans (97) have linked them to oxidative
damage, and higher translocation frequencies in metabolically
active tissues indicate organ-specific mutation patterns (8). Furthermore, clonal expansion
mutations in mitotic tissues also increase with age (9). In healthy elderly individuals,
clonal mtDNA mutations are present in colonic epithelial crypts
(98), with expanding
OXPHOS-deficient mtDNA-mutant cells found in prostate epithelial
stem cells (99). In mice,
clonal expansion mutations are predominantly transitions, whereas
translocations fail to form clones (8). These findings suggest that
transition mutations proliferate with age, whereas oxidative
damage-related translocations, often nonsynonymous, are eliminated.
Transition mutations are thus more likely to undergo clonal
expansion as aging progresses.
Cancer-related changes
This subsection examines mtDNA mutation signatures
and clonal proliferation profiles in carcinogenesis. Since 2005,
progress has elucidated how mtDNA mutations influence tumor
initiation and progression (100). mtDNA mutation types in cancer
cells may vary by histological subtype. While translocation
mutations are present in some cancers, transition mutations
predominate (22,63). For example, 80% of medullary
thyroid carcinoma samples have been reported to display
nonsynonymous mutations, including both transition and
translocation mutations (101).
Rectal cancer shows relatively high translocation mutation
prevalence (102), whereas
gastric cancer exhibits T>C or G>A transitions and indels
associated with nucleotide repeat instability (30,79). Diffuse large B-cell lymphoma
displays random strand bias with increased C>T and A>G
transitions on the heavy strand (103). In endometrial cancer, most
G>A and T>C transition mutations occurred on the mtDNA light
strand, whereas C>T and A>G transitions have been
predominantly observed on the heavy strand, with transition
mutations occurring 24.4 times more frequently than translocation
mutations (22). A large-scale
analysis conducted by the International Cancer Genome Consortium
revealed that mtDNA mutation signatures are largely consistent
across tumor types, with transition mutations dominating (86). Further analysis of 1,675 human
cancers has demonstrated strand bias favoring the heavy chain,
primarily C>T and A>G transitions (63). Among 625 cancers, transition
mutations have been shown to be significantly more frequent than
translocation mutations, with most exhibiting homoplasmic
characteristics (66).
Furthermore, studies have linked mtDNA mutations to mitochondrial
dysfunction in cancer (65,103). Mutations in genes encoding
complex I components impair mitochondrial function, as seen in
thyroid tumors (100) and
triple-negative breast cancer (104). Approximately 12% of cancers
harbor truncating mtDNA mutations, primarily in ETC genes, reducing
OXPHOS (17). Conversely, large
mtDNA deletions, common in aging tissues (4), are less frequent in gastric cancer,
suggesting selective elimination during tumorigenesis (105). Heteroplasmic mtDNA mutations,
combining normal and mutated mtDNA, may mitigate deleterious
effects, preserving ATP synthesis and mitochondrial integrity
(22,106). This resilience likely enables
cancer cells to maintain ATP synthesis capacity and mitochondrial
integrity despite impairments in specific complexes (17,107). Colorectal cancer tissues show
fewer random mtDNA mutations than non-cancerous tissues, reflecting
a metabolic shift from OXPHOS to glycolysis (108). Similar shifts in mtDNA
mutational landscapes occur in head and neck squamous cell
carcinoma during cancer progression (109).
Profiling mtDNA mutations reveals distinct patterns
in cancer compared with adjacent tissues. Colon cancer exhibits
fewer non-clonal single base substitutions compared with adjacent
tissues, indicating reduced non-clonal mutations during cancer
progression (82,108). In liver cancer with hepatitis
B, mtDNA mutations in the D-loop region are reduced in tumor
tissues (82). Studies on breast
cancer progression have shown that specific transition mutations
are prevalent in normal cells but absent in transformed cells
(54,83,110). The predominant rare mutation
types identified in normal stem cells are C>T/G>A and
T>C/A>G transitions, whereas T>C/A>G transitions are
notably absent during transformation of human breast stem cells
into tumorigenic cells (54,110). Baker et al (65) provided a comprehensive analysis
of mtDNA mutations during the progression from normal colonic
epithelium to ulcerative colitis and colorectal cancer. This
previous study reported that clonal and subclonal mutations account
for 3.7% of all mutations but increase in frequency and
pathogenicity during dysplasia, subsequently decreasing in cancer
(65). These findings underscore
the role of clonal expansion and the loss of strand bias in
carcinogenesis. The loss of strand bias suggests a decline in the
fidelity of mtDNA replication and repair, or a breakdown in
regulatory mechanisms, potentially leading to genomic instability
and mitochondrial dysfunction (8,60). Consequently, cellular energy
metabolism and homeostasis may be compromised, thereby increasing
the risk of age-related diseases and tumorigenesis (8). Unlike nuclear gene-driven
carcinogenesis, mtDNA-driven processes emphasize loss of strand
bias and clonal changes over specific mutations (110). Non-clonal mtDNA mutations
decrease during carcinogenesis, whereas adaptive clonal mutations
are selectively retained (108,111). Aging, immortalization and stem
cell transformation favor the proliferation of environmentally
adapted clones, reducing genetic diversity and driving
carcinogenesis.
In summary, the mutational landscape of mtDNA within
cells undergoes dynamic changes with aging. Initially, a diverse
array of non-clonal mutations persists due to various factors,
including environmental exposure and replication errors. However,
as specific transition mutations confer a selective growth
advantage, cells harboring these advantageous (adaptive) mutations
undergo subclonal expansion, referred to as 'adapter clones',
ultimately stabilizing the mutational balance. Conversely, during
carcinogenesis, the prevalence of non-clonal mutations declines as
clones carrying driver mutations proliferate. This process fosters
tumor heterogeneity, characterized by dominant clones, termed
'inducer clones', with driver mutations against a backdrop of
subclonal variations (adapter clones). A comprehensive
understanding of these dynamics is crucial for elucidating the role
of mtDNA mutations in aging and carcinogenesis.
Alterations in mtDNA copy number
Beyond mutations and deletions, variations in mtDNA
copy number have been extensively studied across tumor types, and
the regulation of mtDNA copy number is closely tied to nuclear gene
activity (30). Nuclear-encoded
genes, such as TFAM, which are crucial for mitochondrial
biogenesis, maintain mtDNA replication and stability (29,30). Dysregulation or mutations in
oncogenes (such as MYC) (112)
and tumor suppressor genes (including TP53) (113) disrupt mitochondrial biogenesis
and replication, altering mtDNA copy numbers. Hypoxia, nutrient
deprivation and aberrant tumor microenvironment signaling also
influence mitochondrial dynamics and mtDNA copy number (112,113).
The relationship between aging and mtDNA copy number
is complex and varies across different tissues and individuals. In
normal tissues, mtDNA copy number variations reflect energy needs,
aging and stress, often serving adaptive functions.
High-metabolic-demand tissues, such as the myocardium, have
elevated mtDNA copy numbers, whereas low-demand tissues, including
blood and skin cells, exhibit fewer mitochondria and lower copy
numbers (114). Research has
indicated that mtDNA copy number tends to decrease with age in
certain tissues, such as blood and lymphocytes. For example, a
study conducted on a subset of the Italian population demonstrated
a modest but statistically significant age-associated decline in
mtDNA content within lymphocytes (114). The observed decrease in mtDNA
copy number in certain tissues has been associated with adverse
health outcomes. In older populations, lower mtDNA levels in
peripheral blood have been linked to higher mortality rates and
diminished health, including declines in cognitive and physical
performance (114). Conversely,
other tissues do not exhibit a clear age-related decline in mtDNA
copy number (114).
Investigations into skeletal muscle and heart tissues have reported
stable mtDNA levels across different age groups. While aging is
often accompanied by a reduction in mtDNA copy number in specific
tissues, this pattern is not universal. The decline in mtDNA levels
in certain tissues may contribute to age-related health issues. In
cancer, mtDNA copy number alterations show tissue-specific patterns
(30,77). Reviews by Chatterjee et al
(77) and Lee et al
(30) have revealed elevated
mtDNA copy numbers in head and neck squamous cell carcinoma,
papillary thyroid carcinoma and lung cancer, but reduced levels
(mtDNA depletion) in breast, kidney, liver, ovarian and gastric
cancer (30,77), possibly due to D-loop region
mutations (77). Reduced mtDNA
copy numbers may limit OXPHOS, shifting metabolism to glycolysis
and promoting tumor growth under hypoxia. Conversely, increased
mtDNA copy numbers enhance mitochondrial function, ROS production,
and signaling pathways supporting proliferation, survival and
metastasis. mtDNA is highly vulnerable to damage, potentially
triggering compensatory copy number changes (77). mtDNA copy number variations also
affect cancer prognosis in a cancer type-specific manner. Elevated
mtDNA copy numbers have been shown to be associated with lower
tumor grades and better outcomes in patients with glioma (115). However, higher mtDNA copy
numbers in peripheral blood predict poorer prognoses in
hepatocellular carcinoma, glioma, colorectal cancer, and head and
neck cancer (116). These
findings highlight the complex, cancer-specific prognostic
implications of mtDNA copy number variations.
Mechanisms of nuclear DNA underlying aging
and carcinogenesis
The role of the nuclear genome in aging
and cancer
Mutations in nuclear DNA contribute to both aging
and tumorigenesis through distinct yet interconnected molecular
pathways, including genomic instability, functional deterioration
of cellular processes and dysregulation of cell proliferation
control mechanisms (117).
These mutations accumulate over time as a result of DNA replication
errors, spontaneous base lesions, and exposure to endogenous and
exogenous stressors such as ultraviolet radiation and ROS, thereby
compromising genomic integrity, and driving hallmark features of
senescence and oncogenesis. Notably, certain mutations that confer
selective advantages can lead to the clonal expansion of mutant
cell populations (118).
Furthermore, nuclear genomic aberrations exert direct influence on
mtDNA mutational dynamics, an effect that becomes particularly
pronounced in aged tissues and tumor microenvironments. These
nuclear-mitochondrial interactions are mediated through
nuclear-encoded genes involved in mtDNA replication and repair,
mitochondrial quality control systems and OXPHOS functions
(26,27,38,43). Consequently, elucidating the
molecular mechanisms underlying nuclear DNA alterations in aging
and cancer may provide critical insights into the etiology and
propagation of mtDNA mutations. This subsection delineates the
principal characteristics of the nuclear genome that influence
aging and tumorigenesis.
While mutations in oncogenes and tumor suppressor
genes are closely linked to cancer initiation and progression,
alterations in the nuclear genome also accumulate with aging, even
in somatic cells devoid of disease (52). In the bone marrow of healthy
elderly individuals, mutations in genes such as DNA
methyltransferase 3 α (119),
tet methylcytosine dioxygenase 2 (116) and ASXL transcriptional
regulator 1 (120) can drive
clonal hematopoiesis of indeterminate potential, a phenomenon
associated with aging, and linked to elevated risks of
hematological malignancies and cardiovascular diseases (121). Beyond the hematopoietic system,
epithelial tissues also exemplify clonal expansion during aging. In
aged skin and esophageal epithelium, clones bearing mutations in
notch receptor 1 (122,123), TP53 (124) and cyclin-dependent kinase
inhibitor 2A (124) are
frequently observed. These mutations, common in non-cancerous aged
bone marrow and skin, are not inherently indicative of malignancy.
As aging diminishes cellular proliferative capacity in tissues such
as the bone marrow (121) and
skin (123), cells with driver
gene mutations gain a competitive edge over non-mutated clones.
Aging tissues often exhibit a mosaic of distinct clones, a
reflection of the cumulative accrual and selective expansion of
mutations, culminating in tissue mosaicism and clonal
heterogeneity. Certain clones expand during aging due to selective
advantages conferred by genetic or epigenetic modifications
(72,125). These expansions may arise from
mutations that enhance cellular survival or proliferation,
competitive disadvantages of neighboring cells, or the tissue
remodeling and turnover processes characteristic of aging.
Within dominant clones, subclones harboring
mutations that confer pronounced selective advantages, such as
increased proliferation, resistance to apoptosis or evasion of
immune surveillance, may emerge and undergo further selection.
Genomic instability, marked by the progressive accumulation of
mutations and chromosomal aberrations, is a defining feature of
both aging and cancer (72,125). As aging advances, hypoxic
conditions, immune pressures and competition for limited nutrients
create a highly selective microenvironment in which only clones
with advantageous mutations can thrive. This selective
proliferation often monopolizes resources such as nutrients and
spatial niches, inhibiting the growth of competing clones (125). Consequently, only the most
adaptive clones, often those with cancerous traits, dominate,
outcompeting less viable clones and reducing clonal diversity.
Although clonal proliferation may contribute to the maintenance of
tissue homeostasis in certain contexts, it simultaneously elevates
the risk of age-associated pathologies, including cancer and
functional decline.
Role of nuclear-mitochondrial crosstalk
in aging and cancer
Nuclear signaling pathways that directly modulate
mitochondrial function, particularly PGC-1α and AMPK, serve pivotal
roles in the pathophysiology of aging and cancer. PGC-1α is
indispensable for mitochondrial biogenesis and functional
maintenance, and its upregulation in mtDNA mutator mice, harboring
mutations in mtDNA POLG, has been shown to enhance mitochondrial
performance despite a modest elevation in mtDNA mutation load
(126). This indicates that
PGC-1α-induced mitochondrial biogenesis may mitigate the
deleterious functional consequences of mtDNA mutations, and
modulate clonal expansion and mutation selection. Furthermore, in
rat myoblasts treated with the ATP synthase inhibitor oligomycin,
AMPK is activated, subsequently promoting mtDNA replication
(127), suggesting a role for
AMPK in the regulation of mtDNA replication dynamics in response to
mitochondrial stress.
With advancing age, PGC-1α expression declines
across various tissues, including skeletal muscle, the brain and
bone marrow, contributing to reduced mitochondrial biogenesis,
diminished OXPHOS and impaired regenerative capacity (128). In aging bone tissue, decreased
PGC-1α levels are associated with compromised osteoblast
differentiation and enhanced adipogenic differentiation of
mesenchymal stem cells, factors implicated in the pathogenesis of
osteoporosis. In the central nervous system, diminished PGC-1α
expression is associated with neurodegenerative disorders such as
Parkinson's disease, exacerbating mitochondrial dysfunction and
increasing neuronal susceptibility to stress. In cancer, the role
of PGC-1α is highly context-dependent (128). In malignancies such as melanoma
and invasive breast carcinoma, PGC-1α expression is upregulated,
promoting mitochondrial biogenesis and OXPHOS, thereby facilitating
metastatic potential (129).
Conversely, in advanced thyroid cancer, particularly that harboring
BRAFV600E mutations, PGC-1α expression is suppressed,
contributing to mitochondrial dysfunction, heightened oxidative
stress and a metabolic shift toward aerobic glycolysis (the Warburg
effect), which collectively drive tumor progression (130). These dichotomous roles
underscore the relevance of PGC-1α in mitochondrial regulation and
highlight its potential as a therapeutic target.
AMPK, a master regulator of cellular energy
homeostasis and mitochondrial function, also exhibits
age-associated functional decline. In aged rat skeletal muscle, the
acute activation of AMPK-α2 by
5-aminoimidazole-4-carboxamide ribonucleotide (an AMPK activator)
or exercise is notably reduced compared with in younger
counterparts, leading to attenuated mitochondrial biogenesis
(131). In cancer, the role of
AMPK is likewise dualistic and context-specific. It can inhibit
tumorigenesis by enhancing OXPHOS and fatty acid oxidation while
suppressing glycolysis (132).
However, under glucose-restricted conditions, AMPK sustains
mitochondrial biogenesis via the p38/PGC-1α axis, thus supporting
cancer cell survival under metabolic duress (133). These findings highlight the
integral function of AMPK in mediating cell survival during
metabolic stress. Moreover, senescent cells in nutrient-poor
environments can maintain a state of stable cell cycle arrest and
reduced energy consumption via AMPK activation, contributing to
homeostatic aging. By contrast, nutrient-rich microenvironments
suppress AMPK activation, promoting cellular proliferation
(132,133). These observations suggest that
the fate of senescent cells, whether toward quiescence or malignant
transformation, may be governed by the metabolic characteristics of
their surrounding milieu.
Mechanisms of mtDNA mutations underlying
aging and carcinogenesis
In this section, the aforementioned key findings
are summarized, as illustrated in Fig. 3, and the mechanisms by which
mtDNA mutations contribute to aging and carcinogenesis are
investigated. mtDNA is continually influenced by endogenous and
exogenous factors. Most mtDNA mutations are random alterations
acquired early in life, typically without phenotypic effects. Even
when mutations expand clonally, their impact on cell generations is
minimized by genetic drift, bottleneck effects, mitochondrial
dynamics and mitophagy. This layered defense contrasts with the
reliance of the nuclear genome on robust repair mechanisms such as
double-strand break repair. Transition mutations, caused by DNA
POLG replication errors and base hydrolysis, are the primary source
of mtDNA mutations in aging and cancer (59,86). While transition mutations
increase with age, translocation mutations from oxidative stress
are rapidly repaired, preventing accumulation or clonal expansion.
The effects of carcinogens such as smoking (C:G>A:T), UV
radiation (C:G>T:A) and ROS (G:C>T:A) are rare in mtDNA from
lung and skin cancer (86),
although smoking-related C>A mutations are prominent in the
nuclear genome of patients with lung cancer (134). Unlike the nuclear genome,
mitochondria emphasize quality control through mitophagy and
dynamics over stringent repair. Advances in genetic analysis have
revealed a more complex landscape, encompassing transition and
translocation mutations. mtDNA displays conserved mutational
signatures regardless of cancer tissue origin (22,110), unlike oncogene and tumor
suppressor gene mutations in the nuclear genome.
Key distinctions between mtDNA in cancerous and
normal cells include the loss of strand bias and mutation type
variations, potentially driven by selective pressure, genetic drift
(23,24) or chance (135). Some clones act as 'adapters',
facilitating cellular adaptation to changing environments (21,22,35). Homoplasmic mtDNA mutations,
typically synonymous, are considered adaptive rather than
cancer-specific (77). Subclonal
proliferation of dysfunctional mitochondria is counteracted by
quality control mechanisms to preserve tissue function. Adaptive
mtDNA mutations confer a competitive edge over wild-type mtDNA.
When mutant clones meet equally fit other clones, a dynamic
equilibrium forms, maintaining homeostasis and reducing cancer
risk. In aging tissues, adaptive clones gradually accumulate
mutations with reduced strand bias. Among older individuals, mtDNA
processivity declines, exposing it to selective pressures favoring
mutations. This may disrupt equilibrium, selecting de novo
oncogenic 'inducers' from pre-existing 'adapters' (21). As selection shifts toward
'inducer' clones, cells adapt to new conditions, thrive and gain
advantages, potentially leading to tumorigenesis. Thus, the
transition in mtDNA diversity, from non-clonal mutations in young
tissues to clonal expansion of 'adapter' and 'inducer' mutations,
illustrates the selective pressures driving cancer cell emergence
with aging.
Conclusion
The present review highlights the relationship
between mtDNA mutations and their role in aging and cancer,
contrasting these with changes in the nuclear genome. Age-related
mtDNA mutation increases are more complex than previously
considered. Mitochondrial dysfunction results from reduced strand
bias, clonal expansion and contraction, tissue-specific dynamics
and regulatory mechanisms rather than a simple accumulation of
mutations. These findings have implications for understanding
mitochondrial biology and advancing treatments for age-related
diseases.
Availability of data and materials
Not applicable.
Authors' contributions
HK conceptualized the study, developed the
methodology, administered the project and prepared the original
draft of the manuscript. SI performed validation and provided the
necessary resources. SI and HK curated the data, created the
visualizations, and reviewed and edited the manuscript. Data
authentication is not applicable. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Figures were created by Mrs. Toyomi Kobayashi
(Ms.Clinic MayOne, Nara, Japan).
Funding
No funding was received.
References
|
1
|
Sun N, Youle RJ and Finkel T: The
mitochondrial basis of aging. Mol Cell. 61:654–666. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wallace DC: A mitochondrial paradigm of
metabolic and degenerative diseases, aging, and cancer: A dawn for
evolutionary medicine. Annu Rev Genet. 39:359–407. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Elliott HR, Samuels DC, Eden JA, Relton CL
and Chinnery PF: Pathogenic mitochondrial DNA mutations are common
in the general population. Am J Hum Genet. 83:254–260. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee HC, Chang CM and Chi CW: Somatic
mutations of mitochondrial DNA in aging and cancer progression.
Ageing Res Rev. 9(Suppl 1): S47–S58. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stewart JB and Chinnery PF: Extreme
heterogeneity of human mitochondrial DNA from organelles to
populations. Nat Rev Genet. 22:106–118. 2021. View Article : Google Scholar
|
|
6
|
Cote-L'Heureux A, Maithania YNK, Franco M
and Khrapko K: Are some mutations more equal than others? Elife.
12:e871942023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kowaltowski AJ: Alternative mitochondrial
functions in cell physiopathology: Beyond ATP production. Braz J
Med Biol Res. 33:241–250. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sanchez-Contreras M, Sweetwyne MT,
Tsantilas KA, Whitson JA, Campbell MD, Kohrn BF, Kim HJ, Hipp MJ,
Fredrickson J, Nguyen MM, et al: The multi-tissue landscape of
somatic mtDNA mutations indicates tissue-specific accumulation and
removal in aging. Elife. 12:e833952023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lawless C, Greaves L, Reeve AK, Turnbull
DM and Vincent AE: The rise and rise of mitochondrial DNA
mutations. Open Biol. 10:2000612020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pérez-Amado CJ, Bazan-Cordoba A,
Hidalgo-Miranda A and Jiménez-Morales S: Mitochondrial heteroplasmy
shifting as a potential biomarker of cancer progression. Int J Mol
Sci. 22:73692021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Khrapko K, Coller H, André P, Li XC, Foret
F, Belenky A, Karger BL and Thilly WG: Mutational spectrometry
without phenotypic selection: human mitochondrial DNA. Nucleic
Acids Res. 25:685–693. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shokolenko I and Alexeyev M: Mitochondrial
DNA: Consensuses and controversies. DNA (Basel). 2:131–148.
2022.PubMed/NCBI
|
|
13
|
Wallace DC: Mitochondrial diseases in man
and mouse. Science. 283:1482–1488. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Youle RJ and Narendra DP: Mechanisms of
mitophagy. Nat Rev Mol Cell Biol. 12:9–14. 2011. View Article : Google Scholar
|
|
15
|
Rossignol R, Faustin B, Rocher C, Malgat
M, Mazat JP and Letellier T: Mitochondrial threshold effects.
Biochem J. 370:751–762. 2003. View Article : Google Scholar
|
|
16
|
De Giorgi C and Saccone C: Mitochondrial
genome in animal cells. Structure, organization, and evolution.
Cell Biophys. 14:67–78. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gorelick AN, Kim M, Chatila WK, La K,
Hakimi AA, Berger MF, Taylor BS, Gammage PA and Reznik E:
Respiratory complex and tissue lineage drive recurrent mutations in
tumour mtDNA. Nat Metab. 3:558–570. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Young MJ and Copeland WC: Human
mitochondrial DNA replication machinery and disease. Curr Opin
Genet Dev. 38:52–62. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kobayashi H, Matsubara S, Yoshimoto C,
Shigetomi H and Imanaka S: A comprehensive review of the
contribution of mitochondrial DNA mutations and dysfunction in
polycystic ovary syndrome, supported by secondary database
analysis. Int J Mol Sci. 26:11722025. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zaidi AA, Wilton PR, Su MSW, Paul IM,
Arbeithuber B, Anthony K, Nekrutenko A, Nielsen R and Makova KD:
Bottleneck and selection in the germline and maternal age influence
transmission of mitochondrial DNA in human pedigrees. Proc Natl
Acad Sci USA. 116:25172–25178. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kopinski PK, Singh LN, Zhang S, Lott MT
and Wallace DC: Mitochondrial DNA variation and cancer. Nat Rev
Cancer. 21:431–445. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Khadka P, Young CKJ, Sachidanandam R,
Brard L and Young MJ: Our current understanding of the biological
impact of endometrial cancer mtDNA genome mutations and their
potential use as a biomarker. Front Oncol. 14:13946992024.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guo X, Xu W, Zhang W, Pan C,
Thalacker-Mercer AE, Zheng H and Gu Z: High-frequency and
functional mitochondrial DNA mutations at the single-cell level.
Proc Natl Acad Sci USA. 120:e22015181202023. View Article : Google Scholar :
|
|
24
|
Schaack S, Ho EKH and Macrae F:
Disentangling the intertwined roles of mutation, selection and
drift in the mitochondrial genome. Philos Trans R Soc Lond B Biol
Sci. 375:201901732020. View Article : Google Scholar :
|
|
25
|
Medeiros DM: Assessing mitochondria
biogenesis. Methods. 46:288–294. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
McKinney EA and Oliveira MT: Replicating
animal mitochondrial DNA. Genet Mol Biol. 36:308–315. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jornayvaz FR and Shulman GI: Regulation of
mitochondrial biogenesis. Essays Biochem. 47:69–84. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Scarpulla RC: Transcriptional paradigms in
mammalian mitochondrial biogenesis and function. Physiol Rev.
88:611–638. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yin PH, Lee HC, Chau GY, Wu YT, Li SH, Lui
WY, Wei YH, Liu TY and Chi CW: Alteration of the copy number and
deletion of mitochondrial DNA in human hepatocellular carcinoma. Br
J Cancer. 90:2390–2396. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee HC, Huang KH, Yeh TS and Chi CW:
Somatic alterations in mitochondrial DNA and mitochondrial
dysfunction in gastric cancer progression. World J Gastroenterol.
20:3950–3959. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qian L, Zhu Y, Deng C, Liang Z, Chen J,
Chen Y, Wang X, Liu Y, Tian Y and Yang Y: Peroxisome
proliferator-activated receptor gamma coactivator-1 (PGC-1) family
in physiological and pathophysiological process and diseases.
Signal Transduct Target Ther. 9:502024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
El-Hattab AW, Craigen WJ and Scaglia F:
Mitochondrial DNA maintenance defects. Biochim Biophys Acta Mol
Basis Dis. 1863:1539–1555. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Alberghina L: The Warburg effect
explained: integration of enhanced glycolysis with heterogeneous
mitochondria to promote cancer cell proliferation. Int J Mol Sci.
24:157872023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Soga T: Cancer metabolism: Key players in
metabolic reprogramming. Cancer Sci. 104:275–281. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Brandon M, Baldi P and Wallace DC:
Mitochondrial mutations in cancer. Oncogene. 25:4647–4662. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li T, Copeland C and Le A: Glutamine
metabolism in cancer. Adv Exp Med Biol. 1311:17–38. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Selak MA, Armour SM, MacKenzie ED,
Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB
and Gottlieb E: Succinate links TCA cycle dysfunction to
oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer
Cell. 7:77–85. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lu J, Sharma LK and Bai Y: Implications of
mitochondrial DNA mutations and mitochondrial dysfunction in
tumorigenesis. Cell Res. 19:802–815. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Reinsalu L, Puurand M, Chekulayev V,
Miller S, Shevchuk I, Tepp K, Rebane-Klemm E, Timohhina N, Terasmaa
A and Kaambre T: Energy metabolic plasticity of colorectal cancer
cells as a determinant of tumor growth and metastasis. Front Oncol.
11:6989512021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yapa NMB, Lisnyak V, Reljic B and Ryan MT:
Mitochondrial dynamics in health and disease. FEBS Lett.
595:1184–1204. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mao H, Chen W, Chen L and Li L: Potential
role of mitochondria-associated endoplasmic reticulum membrane
proteins in diseases. Biochem Pharmacol. 199:1150112022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
St-Pierre J, Drori S, Uldry M, Silvaggi
JM, Rhee J, Jäger S, Handschin C, Zheng K, Lin J, Yang W, et al:
Suppression of reactive oxygen species and neurodegeneration by the
PGC-1 transcriptional coactivators. Cell. 127:397–408. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Butow RA and Avadhani NG: Mitochondrial
signaling: The retrograde response. Mol Cell. 14:1–15. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ke R, Xu Q, Li C, Luo L and Huang D:
Mechanisms of AMPK in the maintenance of ATP balance during energy
metabolism. Cell Biol Int. 42:384–392. 2018. View Article : Google Scholar
|
|
45
|
Morgan MJ and Liu ZG: Crosstalk of
reactive oxygen species and NF-κB signaling. Cell Res. 21:103–115.
2011. View Article : Google Scholar
|
|
46
|
Murphy MP: How mitochondria produce
reactive oxygen species. Biochem J. 417:1–13. 2009. View Article : Google Scholar
|
|
47
|
Rampazzo C, Ferraro P, Pontarin G, Fabris
S, Reichard P and Bianchi V: Mitochondrial deoxyribonucleotides,
pool sizes, synthesis, and regulation. J Biol Chem.
279:17019–17026. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H,
Liu L, Liu Y, Yang C, Xu Y, et al: Inhibition of α-KG-dependent
histone and DNA demethylases by fumarate and succinate that are
accumulated in mutations of FH and SDH tumor suppressors. Genes
Dev. 26:1326–1338. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Macián F, López-Rodríguez C and Rao A:
Partners in transcription: NFAT and AP-1. Oncogene. 20:2476–2489.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Vijg J, Schumacher B, Abakir A, Antonov M,
Bradley C, Cagan A, Church G, Gladyshev VN, Gorbunova V, Maslov AY,
et al: Mitigating age-related somatic mutation burden. Trends Mol
Med. 29:530–540. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Arbeithuber B, Hester J, Cremona MA,
Stoler N, Zaidi A, Higgins B, Anthony K, Chiaromonte F, Diaz FJ and
Makova KD: Age-related accumulation of de novo mitochondrial
mutations in mammalian oocytes and somatic tissues. PLoS Biol.
18:e30007452020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Abascal F, Harvey LMR, Mitchell E, Lawson
ARJ, Lensing SV, Ellis P, Russell AJC, Alcantara RE, Baez-Ortega A,
Wang Y, et al: Somatic mutation landscapes at single-molecule
resolution. Nature. 593:405–410. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Arbeithuber B, Cremona MA, Hester J,
Barrett A, Higgins B, Anthony K, Chiaromonte F, Diaz FJ and Makova
KD: Advanced age increases frequencies of de novo mitochondrial
mutations in macaque oocytes and somatic tissues. Proc Natl Acad
Sci USA. 119:e21187401192022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ahn EH, Hirohata K, Kohrn BF, Fox EJ,
Chang CC and Loeb LA: Detection of ultra-rare mitochondrial
mutations in breast stem cells by duplex sequencing. PLoS One.
10:e01362162015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Salk JJ, Schmitt MW and Loeb LA: Enhancing
the accuracy of next-generation sequencing for detecting rare and
subclonal mutations. Nat Rev Genet. 19:269–285. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kennedy SR, Salk JJ, Schmitt MW and Loeb
LA: Ultra-sensitive sequencing reveals an age-related increase in
somatic mitochondrial mutations that are inconsistent with
oxidative damage. PLoS Genet. 9:e10037942013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sanchez-Contreras M, Sweetwyne MT, Kohrn
BF, Tsantilas KA, Hipp MJ, Schmidt EK, Fredrickson J, Whitson JA,
Campbell MD, Rabinovitch PS, et al: A replication-linked mutational
gradient drives somatic mutation accumulation and influences
germline polymorphisms and genome composition in mitochondrial DNA.
Nucleic Acids Res. 49:11103–11118. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Foury F, Hu J and Vanderstraeten S:
Mitochondrial DNA mutators. Cell Mol Life Sci. 61:2799–2811. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Matkarimov BT and Saparbaev MK: DNA repair
and mutagenesis in vertebrate mitochondria: Evidence for asymmetric
DNA strand inheritance. Adv Exp Med Biol. 1241:77–100. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tanaka M and Ozawa T: Strand asymmetry in
human mitochondrial DNA mutations. Genomics. 22:327–335. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Garesse R: Drosophila melanogaster
mitochondrial DNA: Gene organization and evolutionary
considerations. Genetics. 118:649–663. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Spelbrink JN, Toivonen JM, Hakkaart GA,
Kurkela JM, Cooper HM, Lehtinen SK, Lecrenier N, Back JW, Speijer
D, Foury F and Jacobs HT: In vivo functional analysis of the human
mitochondrial DNA polymerase POLG expressed in cultured human
cells. J Biol Chem. 275:24818–24828. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ju YS, Alexandrov LB, Gerstung M,
Martincorena I, Nik-Zainal S, Ramakrishna M, Davies HR,
Papaemmanuil E, Gundem G, Shlien A, et al: Origins and functional
consequences of somatic mitochondrial DNA mutations in human
cancer. Elife. 3:e029352014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
McMahon S and LaFramboise T: Mutational
patterns in the breast cancer mitochondrial genome, with clinical
correlates. Carcinogenesis. 35:1046–1054. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Baker KT, Nachmanson D, Kumar S, Emond MJ,
Ussakli C, Brentnall TA, Kennedy SR and Risques RA: Mitochondrial
DNA mutations are associated with ulcerative colitis preneoplasia
but tend to be negatively selected in cancer. Mol Cancer Res.
17:488–498. 2019. View Article : Google Scholar :
|
|
66
|
Chen XZ, Fang Y, Shi YH, Cui JH, Li LY, Xu
YC and Ling B: Deciphering the spectrum of somatic mutations in the
entire mitochondrial DNA genome. Genet Mol Res. 14:4331–4337. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Otten ABC and Smeets HJM: Evolutionary
defined role of the mitochondrial DNA in fertility, disease and
ageing. Hum Reprod Update. 21:671–689. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Suen DF, Narendra DP, Tanaka A, Manfredi G
and Youle RJ: Parkin overexpression selects against a deleterious
mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci
USA. 107:11835–11840. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Ferreira T and Rodriguez S: Mitochondrial
DNA: Inherent complexities relevant to genetic analyses. Genes
(Basel). 15:6172024. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Hong YS, Battle SL, Shi W, Puiu D,
Pillalamarri V, Xie J, Pankratz N, Lake NJ, Lek M, Rotter JI, et
al: Deleterious heteroplasmic mitochondrial mutations are
associated with an increased risk of overall and cancer-specific
mortality. Nat Commun. 14:61132023. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Ye K, Lu J, Ma F, Keinan A and Gu Z:
Extensive pathogenicity of mitochondrial heteroplasmy in healthy
human individuals. Proc Natl Acad Sci USA. 111:10654–10659. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
López-Otín C, Blasco MA, Partridge L,
Serrano M and Kroemer G: The hallmarks of aging. Cell.
153:1194–1217. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Walker MA, Lareau CA, Ludwig LS, Karaa A,
Sankaran VG, Regev A and Mootha VK: Purifying selection against
pathogenic mitochondrial DNA in human T cells. N Engl J Med.
383:1556–1563. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Arbeithuber B, Anthony K, Higgins B,
Oppelt P, Shebl O, Tiemann-Boege I, Chiaromonte F, Ebner T and
Makova KD: Mitochondrial DNA mutations in human oocytes undergo
frequency-dependent selection but do not increase with age. bioRxiv
[Preprint]: 2024.12.09.627454. 2024.
|
|
75
|
Monnat RJ Jr, Maxwell CL and Loeb LA:
Nucleotide sequence preservation of human leukemic mitochondrial
DNA. Cancer Res. 45:1809–1814. 1985.PubMed/NCBI
|
|
76
|
Wang CY, Wang HW, Yao YG, Kong QP and
Zhang YP: Somatic mutations of mitochondrial genome in early stage
breast cancer. Int J Cancer. 121:1253–1256. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Chatterjee A, Dasgupta S and Sidransky D:
Mitochondrial subversion in cancer. Cancer Prev Res (Phila).
4:638–654. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Bandelt HJ, Salas A and Bravi CM: What is
a 'novel' mtDNA mutation-and does 'novelty' really matter? J Hum
Genet. 51:1073–1082. 2006. View Article : Google Scholar
|
|
79
|
Hung WY, Wu CW, Yin PH, Chang CJ, Li AF,
Chi CW, Wei YH and Lee HC: Somatic mutations in mitochondrial
genome and their potential roles in the progression of human
gastric cancer. Biochim Biophys Acta. 1800:264–270. 2010.
View Article : Google Scholar
|
|
80
|
Kassauei K, Habbe N, Mullendore ME,
Karikari CA, Maitra A and Feldmann G: Mitochondrial DNA mutations
in pancreatic cancer. Int J Gastrointest Cancer. 37:57–64. 2006.
View Article : Google Scholar
|
|
81
|
Hashizume O, Shimizu A, Yokota M, Sugiyama
A, Nakada K, Miyoshi H, Itami M, Ohira M, Nagase H, Takenaga K and
Hayashi JI: Specific mitochondrial DNA mutation in mice regulates
diabetes and lymphoma development. Proc Natl Acad Sci USA.
109:10528–10533. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Yin C, Li DY, Guo X, Cao HY, Chen YB, Zhou
F, Ge NJ, Liu Y, Guo SS, Zhao Z, et al: NGS-based profiling reveals
a critical contributing role of somatic D-loop mtDNA mutations in
HBV-related hepatocarcinogenesis. Ann Oncol. 30:953–962. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Kwon S, Kim SS, Nebeck HE and Ahn EH:
Immortalization of different breast epithelial cell types results
in distinct mitochondrial mutagenesis. Int J Mol Sci. 20:28132019.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Kamalidehghan B, Houshmand M, Ismail P,
Panahi MSS and Akbari MHH: Delta mtDNA4977 is more common in
non-tumoral cells from gastric cancer sample. Arch Med Res.
37:730–735. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Dai JG, Xiao YB, Min JX, Zhang GQ, Yao K
and Zhou RJ: Mitochondrial DNA 4977 BP deletion mutations in lung
carcinoma. Indian J Cancer. 43:20–25. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Yuan Y, Ju YS, Kim Y, Li J, Wang Y, Yoon
CJ, Yang Y, Martincorena I, Creighton CJ, Weinstein JN, et al:
Comprehensive molecular characterization of mitochondrial genomes
in human cancers. Nat Genet. 52:342–352. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Li Y, Sundquist K, Vats S, Hong MG, Wang
X, Chen Y, Hedelius A, Saal LH, Sundquist J and Memon AA:
Mitochondrial heteroplasmic shifts reveal a positive selection of
breast cancer. J Transl Med. 21:6962023. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Bjørnetrø T, Bousquet PA, Redalen KR,
Trøseid AMS, Lüders T, Stang E, Sanabria AM, Johansen C, Fuglestad
AJ, Kersten C, et al: Next-generation sequencing reveals mitogenome
diversity in plasma extracellular vesicles from colorectal cancer
patients. BMC Cancer. 23:6502023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Zheng W, Khrapko K, Coller HA, Thilly WG
and Copeland WC: Origins of human mitochondrial point mutations as
DNA polymerase gamma-mediated errors. Mutat Res. 599:11–20. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Itsara LS, Kennedy SR, Fox EJ, Yu S,
Hewitt JJ, Sanchez-Contreras M, Cardozo-Pelaez F and Pallanck LJ:
Oxidative stress is not a major contributor to somatic
mitochondrial DNA mutations. PLoS Genet. 10:e10039742014.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Hoekstra JG, Hipp MJ, Montine TJ and
Kennedy SR: Mitochondrial DNA mutations increase in early stage
Alzheimer disease and are inconsistent with oxidative damage. Ann
Neurol. 80:301–306. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Ameur A, Stewart JB, Freyer C, Hagström E,
Ingman M, Larsson NG and Gyllensten U: Ultra-deep sequencing of
mouse mitochondrial DNA: Mutational patterns and their origins.
PLoS Genet. 7:e10020282011. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Samstag CL, Hoekstra JG, Huang CH,
Chaisson MJ, Youle RJ, Kennedy SR and Pallanck LJ: Deleterious
mitochondrial DNA point mutations are overrepresented in Drosophila
expressing a proofreading-defective DNA polymerase γ. PLoS Genet.
14:e10078052018. View Article : Google Scholar
|
|
94
|
Andreazza S, Samstag CL, Sanchez-Martinez
A, Fernandez-Vizarra E, Gomez-Duran A, Lee JJ, Tufi R, Hipp MJ,
Schmidt EK, Nicholls TJ, et al: Mitochondrially-targeted APOBEC1 is
a potent mtDNA mutator affecting mitochondrial function and
organismal fitness in Drosophila. Nat Commun. 10:32802019.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Shukla P, Mukherjee S and Patil A:
Identification of variants in mitochondrial D-loop and OriL region
and analysis of mitochondrial DNA copy number in women with
polycystic ovary syndrome. DNA Cell Biol. 39:1458–1466. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Toure S, Mbaye F, Gueye MD, Fall M, Dem A,
Lamy JB and Sembene M: Somatic mitochondrial mutations in oral
cavity cancers among senegalese patients. Asian Pac J Cancer Prev.
20:2203–2208. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Waneka G, Svendsen JM, Havird JC and Sloan
DB: Mitochondrial mutations in Caenorhabditis elegans show
signatures of oxidative damage and an AT-bias. Genetics.
219:iyab1162021. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Taylor RW, Barron MJ, Borthwick GM, Gospel
A, Chinnery PF, Samuels DC, Taylor GA, Plusa SM, Needham SJ,
Greaves LC, et al: Mitochondrial DNA mutations in human colonic
crypt stem cells. J Clin Invest. 112:1351–1360. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Blackwood JK, Williamson SC, Greaves LC,
Wilson L, Rigas AC, Sandher R, Pickard RS, Robson CN, Turnbull DM,
Taylor RW and Heer R: In situ lineage tracking of human prostatic
epithelial stem cell fate reveals a common clonal origin for basal
and luminal cells. J Pathol. 225:181–188. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Abu-Amero KK, Alzahrani AS, Zou M and Shi
Y: High frequency of somatic mitochondrial DNA mutations in human
thyroid carcinomas and complex I respiratory defect in thyroid
cancer cell lines. Oncogene. 24:1455–1460. 2005. View Article : Google Scholar
|
|
101
|
Abu-Amero KK, Alzahrani AS, Zou M and Shi
Y: Association of mitochondrial DNA transversion mutations with
familial medullary thyroid carcinoma/multiple endocrine neoplasia
type 2 syndrome. Oncogene. 25:677–684. 2006. View Article : Google Scholar
|
|
102
|
Wang B, Qiao L, Wang Y, Zeng J, Chen D,
Guo H and Zhang Y: Mitochondrial DNA D-loop lesions with the
enhancement of DNA repair contribute to gastrointestinal cancer
progression. Oncol Rep. 40:3694–3704. 2018.PubMed/NCBI
|
|
103
|
Zeng AGX, Leung ACY and Brooks-Wilson AR:
Somatic mitochondrial DNA mutations in diffuse large B-cell
lymphoma. Sci Rep. 8:36232018. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Vikramdeo KS, Anand S, Sudan SK, Pramanik
P, Singh S, Godwin AK, Singh AP and Dasgupta S: Profiling
mitochondrial DNA mutations in tumors and circulating extracellular
vesicles of triple-negative breast cancer patients for potential
biomarker development. FASEB Bioadv. 5:412–426. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Dani MAC, Dani SU, Lima SPG, Martinez A,
Rossi BM, Soares F, Zago MA and Simpson AJ: Less DeltamtDNA4977
than normal in various types of tumors suggests that cancer cells
are essentially free of this mutation. Genet Mol Res. 3:395–409.
2004.PubMed/NCBI
|
|
106
|
Young MJ, Sachidanandam R, Hales DB, Brard
L, Robinson K, Rahman MM, Khadka P, Groesch K and Young CKJ:
Identification of somatic mitochondrial DNA mutations,
heteroplasmy, and increased levels of catenanes in tumor specimens
obtained from three endometrial cancer patients. Life (Basel).
12:5622022.PubMed/NCBI
|
|
107
|
Martínez-Reyes I, Cardona LR, Kong H,
Vasan K, McElroy GS, Werner M, Kihshen H, Reczek CR, Weinberg SE,
Gao P, et al: Mitochondrial ubiquinol oxidation is necessary for
tumour growth. Nature. 585:288–292. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Ericson NG, Kulawiec M, Vermulst M,
Sheahan K, O'Sullivan J, Salk JJ and Bielas JH: Decreased
mitochondrial DNA mutagenesis in human colorectal cancer. PLoS
Genet. 8:e10026892012. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Mithani SK, Taube JM, Zhou S, Smith IM,
Koch WM, Westra WH and Califano JA: Mitochondrial mutations are a
late event in the progression of head and neck squamous cell
cancer. Clin Cancer Res. 13:4331–4335. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Ahn EH, Lee SH, Kim JY, Chang CC and Loeb
LA: Decreased mitochondrial mutagenesis during transformation of
human breast stem cells into tumorigenic cells. Cancer Res.
76:4569–4578. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Li D, Du X, Guo X, Zhan L, Li X, Yin C,
Chen C, Li M, Li B, Yang H and Xing J: Site-specific selection
reveals selective constraints and functionality of tumor somatic
mtDNA mutations. J Exp Clin Cancer Res. 36:1682017. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Chen J, Zheng Q, Hicks JL, Trabzonlu L,
Ozbek B, Jones T, Vaghasia AM, Larman TC, Wang R, Markowski MC, et
al: MYC-driven increases in mitochondrial DNA copy number occur
early and persist throughout prostatic cancer progression. JCI
Insight. 8:e1698682023. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Chang SC, Lin PC, Yang SH, Wang HS, Liang
WY and Lin JK: Mitochondrial D-loop mutation is a common event in
colorectal cancers with p53 mutations. Int J Colorectal Dis.
24:623–628. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Filograna R, Mennuni M, Alsina D and
Larsson NG: Mitochondrial DNA copy number in human disease: the
more the better? FEBS Lett. 595:976–1002. 2021. View Article : Google Scholar :
|
|
115
|
Zhang Y, Qu Y, Gao K, Yang Q, Shi B, Hou P
and Ji M: High copy number of mitochondrial DNA (mtDNA) predicts
good prognosis in glioma patients. Am J Cancer Res. 5:1207–1216.
2015.PubMed/NCBI
|
|
116
|
Hu L, Yao X and Shen Y: Altered
mitochondrial DNA copy number contributes to human cancer risk:
Evidence from an updated meta-analysis. Sci Rep. 6:358592016.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Maslov AY and Vijg J: Genome instability,
cancer and aging. Biochim Biophys Acta. 1790:963–969. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Hall MWJ, Jones PH and Hall BA: Relating
evolutionary selection and mutant clonal dynamics in normal
epithelia. J R Soc Interface. 16:201902302019. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Buscarlet M, Provost S, Zada YF, Barhdadi
A, Bourgoin V, Lépine G, Mollica L, Szuber N, Dubé MP and Busque L:
DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate
benign phenotypes and different genetic predispositions. Blood.
130:753–762. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Fujino T, Goyama S, Sugiura Y, Inoue D,
Asada S, Yamasaki S, Matsumoto A, Yamaguchi K, Isobe Y, Tsuchiya A,
et al: Mutant ASXL1 induces age-related expansion of phenotypic
hematopoietic stem cells through activation of Akt/mTOR pathway.
Nat Commun. 12:18262021. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Abelson S, Collord G, Ng SWK, Weissbrod O,
Mendelson Cohen N, Niemeyer E, Barda N, Zuzarte PC, Heisler L,
Sundaravadanam Y, et al: Prediction of acute myeloid leukaemia risk
in healthy individuals. Nature. 559:400–404. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Abby E, Dentro SC, Hall MWJ, Fowler JC,
Ong SH, Sood R, Herms A, Piedrafita G, Abnizova I, Siebel CW, et
al: Notch1 mutations drive clonal expansion in normal esophageal
epithelium but impair tumor growth. Nat Genet. 55:232–245. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Fowler JC, King C, Bryant C, Hall MWJ,
Sood R, Ong SH, Earp E, Fernandez-Antoran D, Koeppel J, Dentro SC,
et al: Selection of oncogenic mutant clones in normal human skin
varies with body site. Cancer Discov. 11:340–361. 2021. View Article : Google Scholar :
|
|
124
|
Testa U, Castelli G and Pelosi E: The
molecular characterization of genetic abnormalities in esophageal
squamous cell carcinoma may foster the development of targeted
therapies. Curr Oncol. 30:610–640. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Colom B, Alcolea MP, Piedrafita G, Hall
MWJ, Wabik A, Dentro SC, Fowler JC, Herms A, King C, Ong SH, et al:
Spatial competition shapes the dynamic mutational landscape of
normal esophageal epithelium. Nat Genet. 52:604–614. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Dillon LM, Williams SL, Hida A, Peacock
JD, Prolla TA, Lincoln J and Moraes CT: Increased mitochondrial
biogenesis in muscle improves aging phenotypes in the mtDNA mutator
mouse. Hum Mol Genet. 21:2288–2297. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Banik M and Adhya S: OXPHOS deficiency
induces mitochondrial DNA synthesis through non-canonical
AMPK-dependent mRNA compartmentalization. J Biosci. 47:672022.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Vernier M and Giguère V: Aging, senescence
and mitochondria: The PGC-1/ERR axis. J Mol Endocrinol. 66:R1–R14.
2021. View Article : Google Scholar
|
|
129
|
LeBleu VS, O'Connell JT, Gonzalez Herrera
KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A,
Domingos Chinen LT, Rocha RM, et al: PGC-1α mediates mitochondrial
biogenesis and oxidative phosphorylation in cancer cells to promote
metastasis. Nat Cell Biol. 16(992-1003): 1–15. 2014.
|
|
130
|
Wang Y, Peng J, Yang D, Xing Z, Jiang B,
Ding X, Jiang C, Ouyang B and Su L: From metabolism to malignancy:
The multifaceted role of PGC1α in cancer. Front Oncol.
14:13838092024. View Article : Google Scholar
|
|
131
|
Reznick RM, Zong H, Li J, Morino K, Moore
IK, Yu HJ, Liu ZX, Dong J, Mustard KJ, Hawley SA, et al:
Aging-associated reductions in AMP-activated protein kinase
activity and mitochondrial biogenesis. Cell Metab. 5:151–156. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Jiang S, Wang Y, Luo L, Shi F, Zou J, Lin
H, Ying Y, Luo Y, Zhan Z, Liu P, et al: AMP-activated protein
kinase regulates cancer cell growth and metabolism via nuclear and
mitochondria events. J Cell Mol Med. 23:3951–3961. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Chaube B, Malvi P, Singh SV, Mohammad N,
Viollet B and Bhat MK: AMPK maintains energy homeostasis and
survival in cancer cells via regulating p38/PGC-1α-mediated
mitochondrial biogenesis. Cell Death Discov. 1:150632015.
View Article : Google Scholar
|
|
134
|
Milholland B, Auton A, Suh Y and Vijg J:
Age-related somatic mutations in the cancer genome. Oncotarget.
6:24627–24635. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Coller HA, Khrapko K, Bodyak ND, Nekhaeva
E, Herrero-Jimenez P and Thilly WG: High frequency of homoplasmic
mitochondrial DNA mutations in human tumors can be explained
without selection. Nat Genet. 28:147–150. 2001. View Article : Google Scholar : PubMed/NCBI
|