Cyclin-dependent kinase (CDK)4/6 plays a key role in
cell cycle regulation, where aberrant activation of the cyclin
D-CDK4/6-retinoblastoma (Rb) gene axis often leads to unrestrained
cell proliferation, one of the central mechanisms of tumorigenesis
(1). Notably, CDK4/6 inhibitors,
such as palbociclib, ribociclib and abemaciclib, have shown marked
efficacy in the treatment of patients with hormone
receptor-positive/human epidermal growth factor receptor 2-negative
metastatic breast cancer (HR+/HER2− MBC)
(2). Traditionally, CDK4/6
inhibitors inhibit cell proliferation by inhibiting Rb
phosphorylation and inducing pre-DNA synthesis (G1) cell
cycle arrest in tumor cells (3).
However, it has also been shown that CDK4/6 inhibitors are
essential in modulating tumor immunity. For example, CDK4/6
inhibitors can promote tumor cytotoxic T-cell-mediated clearance by
affecting tumor cells and can reduce the activity of E2F-targeted
DNA methyltransferase 1 (DNMT1) in regulatory T cells (Tregs). In
addition, CDK4/6 inhibitors can enhance the therapeutic effects of
immune checkpoint inhibitors (ICIs) by upregulating antigen
presentation mechanisms and programmed death-ligand 1 (PD-L1)
expression, thus exhibiting good synergistic potential and
providing new perspectives for tumor immunotherapy (4).
Despite their potential, CDK4/6 inhibitors still
face challenges in the clinic. Clinical and preclinical studies
have shown that ~20% of patients with breast cancer (BC) have
innate resistance to CDK4/6 inhibitors (5), and >30% of patients gradually
develop acquired resistance during ongoing treatment (6), severely limiting the durability of
their efficacy and widespread use. The mechanisms of resistance,
whether innate or acquired, involve both compensatory activation of
cell cycle-related pathways and multiple non-cell cycle escape
strategies (7). In addition, the
hematological toxicity of CDK4/6 inhibitors should not be ignored.
In the PALOMA-3 study, 55.3% of patients developed grade 3
neutropenia and 9.7% developed grade 4 neutropenia, suggesting the
importance of regular patient monitoring and dose adjustment to
ensure safety (8). Therefore,
future studies should focus on overcoming resistance to CDK4/6
inhibitors, searching for predictive biomarkers of therapeutic
sensitivity, optimizing dosing regimens, and improving patient
monitoring and dose adjustment to improve therapeutic efficacy and
safety.
Based on the aforementioned information, the present
study aims to systematically review the mechanism of CDK4/6
inhibitors in cell cycle regulation and tumor immunomodulation, and
to thoroughly elaborate their drug resistance mechanisms, including
the compensatory activation of cell cycle-related pathways and
multiple non-cell cycle escape strategies. The review also
discusses the recent progress and future directions in overcoming
drug resistance and optimizing clinical application, with the aim
of providing a theoretical basis and practical guidance for the
future use of precision therapy with CDK4/6 inhibitors.
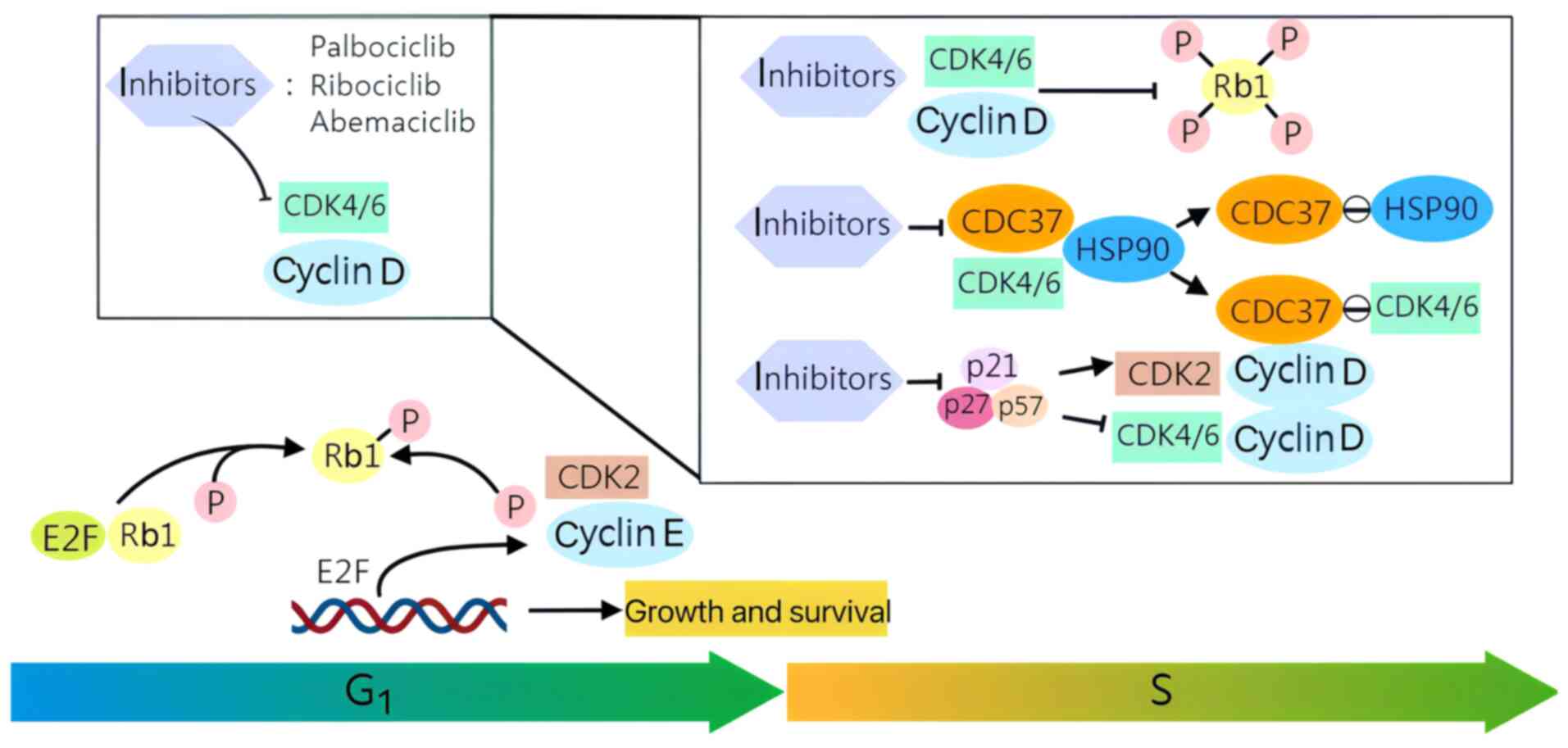
CDK4/6 are key regulators of the cell cycle via the
CDK4/6-Rb-E2F pathway, with CDK4/6 assuming a pivotal role in the
G1-S checkpoint of the cell cycle, which oversees genome
replication (9). The Rb
suppressor protein represents a principal cell cycle target of
CDK4/6. In normal cells, the phosphorylated state of Rb is
typically the consequence of a sequential interaction between
CDK4/6 and the D-type cyclin subunit, inhibiting Rb activity
(10). The D-type cyclin subunit
is generated by stimulating activation signals, including growth
factors and cell adhesion molecules. The phosphorylation of Rb
results in the deregulation of E2F inhibition, thereby enabling its
activation of downstream targets. This, in turn, facilitates the
recruitment of transcriptional activators and the irreversible
entry of quiescent cells into the DNA-synthesis (S) phase of the
cell cycle (1,11). CDK4/6 inhibitors induce complete
Rb dephosphorylation, resulting in the sequestration of the
transcription factor E2F and subsequent inhibition of cell cycle
progression, thereby causing cell cycle arrest in G1
(12). Common CDK4/6 inhibitors
include palbociclib, ribociclib and abemaciclib. The fundamental
mechanism of CDK4/6 inhibitors and their role in regulating the
cell cycle are elucidated in the present review.
CDK4/6 inhibitors prevent the formation of an active
CDK4/6 complex by binding to CDK4/6. The most significant function
of cyclin D-CDK4/6 in promoting cellular proliferation is the
phosphorylation of Rb, which results in a rapid reduction in the
phosphorylation of Rb1 at cyclin D-CDK4/6-dependent sites. This
indicates that CDK4/6 is subject to acute inhibition (13). Palbociclib, ribociclib and
abemaciclib have been demonstrated to impede the binding of CDK4
and CDK6 to cell division cycle 37, the kinase-targeting subunit of
heat shock protein 90 (HSP90). This prevents CDK4/6 from entering
the HSP90-chaperone system (14). CDK4/6 inhibitors have
demonstrated considerable efficacy in treating
HR+/HER2− MBC by binding to CDK4/6 (15). In addition to this mechanism, the
upregulation of cyclin D expression and formation of the cyclin
D-CDK4/6 complex results in the redistribution of kinase inhibitor
protein/CDK inhibitor protein inhibitors from the cyclin E-CDK2
complex to cyclin D-CDK4/6, which activates the kinase activity of
cyclin E-CDK2 and facilitates cell-cycle progression. This pathway
can also be blocked by CDK4/6 inhibitors (13).
The cell cycle is divided into interphase and
mitosis (M). Interphase encompasses G1, S and
pre-division (G2) phases. The cell cycle regulates
intricate, orchestrated interactions between cyclin-regulating
proteins and cell CDKs (16).
CDK4/6 are two major kinases promoting the G1-S phase
transition by phosphorylating and inactivating Rb, facilitating
S-phase entry and DNA replication. While the level of CDK remains
constant throughout the cell cycle, the expression of cyclin D is
subject to dynamic regulation at multiple levels (17). Targeting CDK4 and CDK6 with
CDK4/6 inhibitors blocks the binding of cyclin D to CDKs, thereby
halting the cell cycle at the G1 phase and preventing
cell proliferation (18). The
uncontrolled proliferation of cells, driven by aberrant cell cycle
progression, represents a pivotal characteristic of cancerous
growth. Consequently, the pursuit of pharmacological agents that
impede the cell cycle represents a rational strategy for the
treatment of cancer (19). The
cell cycle-blocking effect of CDK4/6 inhibitors has markedly
benefited the utilization of these pharmaceutical agents in
treating cancer (Fig. 1).
The hormones estrogen and progesterone have been
shown to trigger the process of breast morphogenesis through a
mechanism of action on a specific subset of mammary epithelial
cells that express their cognate receptors, known as ERα and
progesterone receptor (PR). Cyclin D1, a critical oncogene in BC,
is frequently amplified in ER- and PR-positive BC, which is
associated with a poor patient prognosis. Cyclin D1 is specifically
amplified in ERα+ BC and is a progesterone target gene
(20); thus, the process of
progesterone-induced cell proliferation may manifest through the
expression of cyclin D1. The binding between PR and progesterone
leads to cyclin D1 expression, which is connected to intracellular
signaling and activation of the mitotic cell cycle (21). Research findings have
demonstrated that in animals injected with ERα− cells, a
substantial decrease in tumor volume is observed following
treatment with adiponectin; conversely, in animals injected with
ERα+ cells, tumor growth is increased. In the absence of
ERα, the process of auto linker-mediated downregulation of cyclin
D1 has been shown to involve the recruitment of co-stressors; by
contrast, in the presence of ERα, the expression of cyclin D1 was
revealed to be induced by the activation complex (22). Cyclin D1 has been demonstrated to
regulate PR expression, thereby enhancing responses to estrogen and
progesterone. Meanwhile, progesterone can also regulate cyclin D1
through genetic effects or positive feedback loops. It has been
demonstrated that estrogen treatment of cyclin D1-transgenic mice
can lead to an increase in PR expression and the induction of
mammary hyperplasia. This process is stimulated by progesterone and
blocked by progesterone antagonists. Conversely, this previous
study observed a decrease in PR levels in cyclin D1-knockout mice.
The mechanism by which cyclin D1 regulates PR expression involves a
novel estrogen- and cyclin D1-response enhancer located in the
DNA-coding portion of the 3′untranslated region of the PR gene. The
expression of PR has been shown to be reduced in human BC cells by
small inhibitory RNA which targets at cyclin D1, which also
diminishes ER binding to the 3′-enhancing region. Given the
established role of estrogen and progesterone in regulating cyclin
D1, the involvement of cyclin D1 in the feed-forward loop may
potentially increase the risk of BC associated with the combination
of estrogen and progesterone (23). Furthermore, the ability of cyclin
D1 to regulate PRs suggests a novel function for cyclin D1 in
ovarian hormonal control of breast development and breast
carcinogenesis (23).
In addition to CDK4/6-Rb1 signaling, the
PI3K/AKT/mTOR signaling pathway is a vital growth stimulation
pathway in HR+/HER2− MBC (24). The PI3K family of proteins is
comprised of three classes of lipid kinases that catalyze the
phosphorylation of phosphatidylinositol 4,5-bisphosphate to
phosphatidylinositol 3,4,5-trisphosphate. The PI3K class 1 family,
a heterodimer with a regulatory and catalytic subunit, is the most
commonly involved with human cancer. Somatic mutations in genes
encoding components of the PI3K pathway occur in >70% of cases
of BC; these include mutations or amplifications of subunit p110α
(protein encoded by PIK3CA), subunit p110β and subunit p85α, which
are the catalytic subunits of PI3K, or mutations in PI3K
modulators, such as phosphatase and tensin homolog (PTEN), AKT and
mTOR (25). The most common
mechanism underlying PI3K pathway activation is via PIK3CA
mutations or amplifications, in which p110α (PIK3CA) serves a vital
role in BC tumorigenesis through extra-nuclear ER signaling and is
often responsible for endocrine therapy resistance (26). mTOR is a serine/threonine protein
kinase and a downstream effector of AKT, which consists of two
functionally distinct complexes: mTOR complex (mTORC)1 and mTORC2.
mTORC1 belongs to a network of regulatory feedback loops; once
activated, it limits the transmission of upstream effectors, such
as platelet-derived growth factor receptors α and β, to transmit
proliferative signals. This, in turn, results in the attenuation of
PI3K/AKT activity. Conversely, mTORC2 is implicated in the
regulation of AKT phosphorylation at S473 and the organization of
the cellular actin cytoskeleton. Furthermore, mTORC1 activation
results in a direct decrease in mTORC2 activity. The mTOR
downstream substrate S6 kinase can phosphorylate and activate
functional structural domains of ERs, consequently resulting in
ligand-independent receptor activation (27). Molecularly, estrogenic activity
has been shown to induce insulin-like growth factor, and activate
the PI3K/AKT and MAPK pathways, which have been observed to
downregulate ERs and PR gene expression at the cell surface. The
PI3K/AKT/mTOR pathway is the most frequently altered in MBC, and
the upregulation of this pathway has been demonstrated to promote
ER-dependent and -independent transcriptional activity, which
contributes to anti-estrogen resistance, leading to tumor cell
proliferation, survival, motility and metabolism. Drugs targeting
this pathway have demonstrated encouraging results in
HR+/HER2− MBC when combined with
anti-estrogens (28). In
addition, it has been shown that PI3K and mTOR inhibition can
enhance sensitivity to endocrine therapies in vivo,
providing a compelling rationale for the combination of these two
therapeutic modalities (29).
Similarly, mTORC1/2 inhibitors have demonstrated efficacy in
enhancing the sensitivity of drug-resistant HR+ BC cell
lines to CDK4/6 inhibitors (30).
The mechanisms facilitate the understanding of the
use of CDK4/6 inhibitors in clinically relevant tumors and in the
treatment of HR+/HER2− MBC, as mentioned in
the present review.
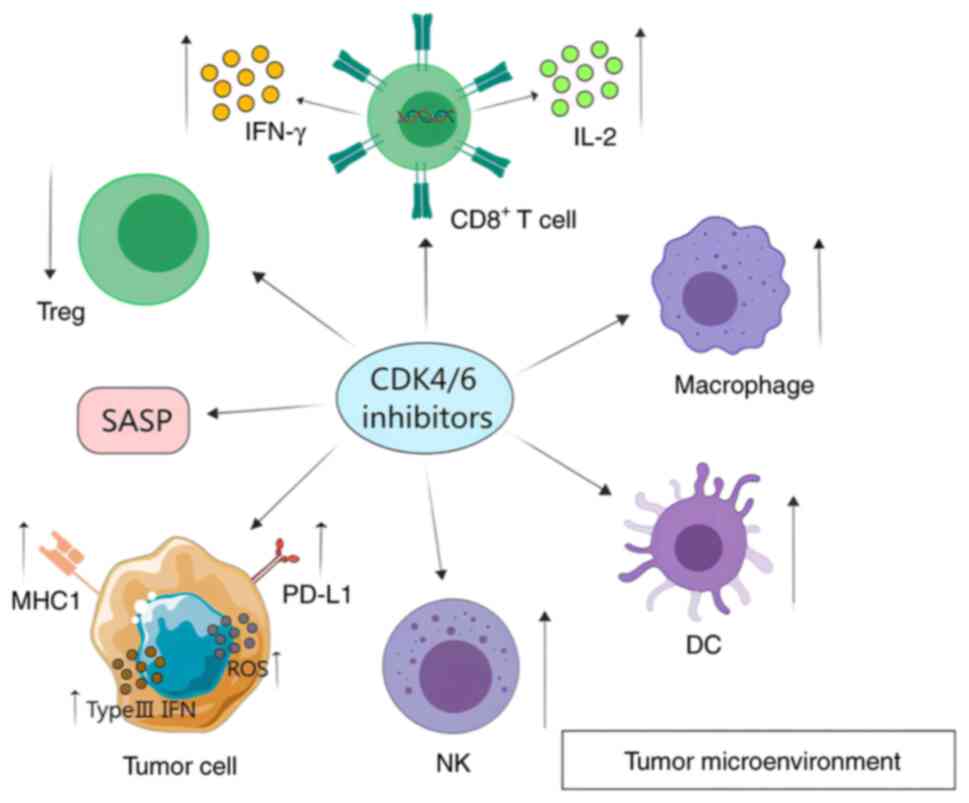
CDK4/6 inhibitors affect tumor immunity by altering
the immune microenvironment and modulating immune checkpoints
(Fig. 2).
CDK4/6 inhibitors exert a direct effect on tumors by
influencing the cell cycle, in addition to affecting cells within
the tumor immune microenvironment. CDK4/6 inhibitors enhance
cytokine secretion in both tumor cells and cytotoxic T lymphocytes
(CD8+ T cells), while the proliferation of
immunosuppressive Tregs is inhibited (31). In addition, CDK4/6 inhibitors
have been demonstrated to influence the functionality of immune
cells, including natural killer (NK) cells and macrophages, which
can consequently impact the tumor microenvironment.
Although CDK4/6 inhibitors have been demonstrated to
inhibit T-cell proliferation, they have also been shown to enhance
tumor infiltration and effector T-cell activation. Additionally,
tumor antigen-experienced T cells demonstrate a heightened
sensitivity to CDK4/6 inhibition relative to naive T cells
(32). Nuclear factor of
activated T cells 4 (NFAT4) is a well-documented regulator of IL-2
secretion, controlled by CDK6, and IL-2 is a well-established
marker of cellular activation. It has been demonstrated that brief
exposure to CDK4/6 inhibitors impedes CDK6-induced phosphorylation
of NFAT4, facilitates its nuclear translocation and augments its
transcriptional activity. This ultimately promotes IL-2 synthesis,
T-cell activation and cytokine production while reinforcing defense
against tumors (33). In a mouse
lung cancer model, CDK4/6 inhibitors have been observed to increase
the percentage of effector cells within the tumor microenvironment,
thereby enhancing the functionality of effector T cells. The
capacity of CDK4/6 inhibitors to induce long-term immune T-cell
memory in both mouse models and humans provides a robust foundation
for treating tumor recurrence (34).
Secondly, it has been observed that CDK4/6
inhibitors exert a considerable inhibitory effect on the
proliferation of Tregs. Tregs can impede the proliferation of
effector T cells and release cytokines, thereby facilitating tumor
cell immune evasion. It has been demonstrated that abemaciclib
enhances downstream p21 production via the Rb-E2F axis. p21 can
then compete with cyclin D for binding to CDK2, thereby regulating
the G1-S transition of the cell cycle and mediating cell
cycle arrest in Tregs. CDK4/6 inhibitors reduce immunosuppressive
Treg populations by inhibiting their proliferation. By inhibiting
the Rb-E2F axis and thereby reducing recombinant DNMT1 expression,
CDK4/6 inhibitors induce hypomethylation of genes that regulate
immune function. Selective inhibition of Treg proliferation may be
associated with the observation that Treg cells express higher
levels of Rb1 (4,34).
Administration of CDK4/6 inhibitors has been
observed to reduce the number of circulating Tregs, the proportion
of Tregs in tumors and their cellular activity. Notably, Tregs have
been observed to exhibit elevated CDK6 expression relative to other
T-cell subtypes (35). This
observation suggests that heightened levels of CDK6 and potential
increased reliance on CDK6 in Tregs could contribute to their
heightened sensitivity to CDK4/6 inhibitors. Furthermore, CDK2 has
been demonstrated to function as a negative regulator that CDK4/6
can modulate. CDK4/6 inhibitors influence cells by regulating CDK2
levels in FOXP3+ Tregs, thereby affecting Treg formation
(31). In addition, it has been
demonstrated that p21 can competitively bind to CDK2, thereby
regulating the G1-S transition of the cell cycle and
mediating the cycle block of Tregs. CDK4/6 inhibitors affect Treg
cycle block via the Rb-E2F-DNMT1 pathway, inhibiting Treg
proliferation, reducing cytotoxic T-lymphocyte depletion and
enhancing antitumor immunity (34).
Consequently, CDK4/6 inhibitors can modify the tumor
immune microenvironment, namely by enhancing the activation of
effector T cells, the capacity to generate immune T-cell memory
over time and the inhibition of Tregs proliferation.
CDK4/6 inhibitors have been demonstrated to enhance
antigen presentation. The genes encoding major histocompatibility
complex (MHC) class I molecules in cancer cells are upregulated by
CDK4/6 inhibitors, thereby strengthening the ability of immune
cells to process and present antigens. Inhibition of the CDK-Rb-E2F
axis by CDK4/6 inhibitors has been shown to attenuate methylation
of endogenous retroviral genes, increase levels of double-stranded
RNA and enhance type III interferon (IFN) synthesis. Notably,
elevated levels of type III IFN stimulate the expression of
IFN-sensitive genes and promote the transcription of downstream
genes encoding MHC class I molecules, thereby enhancing the
antitumor capacity of immune cells (36). It has been postulated that CDK4/6
inhibitors may exert direct immunomodulatory effects on tumor
cells. The number of MHC class I proteins on the surface of tumor
cells is increased by genes involved in peptide cleavage and
transport. This also enhances the activation of antigen-presenting
cells (APCs) and strengthens their ability to deliver antigens
(34). Drug-resistant tumors
adopt an immunosuppressive microenvironment characterized by
elevated MHC II-low macrophages and increased PD-L1 expression on
tumor cells. This phenomenon can be partially attributed to low
MHC-II levels in macrophages, which result in decreased antigen
presentation and T-cell activation (37). CDK4/6 inhibitors have the
potential to impede immune evasion by enhancing antigen
presentation to resistant tumors, thereby increasing MHC-II levels
in macrophages. Concurrently, CDK4/6 inhibitors elevate PD-L1
levels, which counteract the antitumor impact. Consequently, the
requirement for concomitant administration of ICIs arises.
The recruitment of NK cells in a
senescence-dependent manner has been demonstrated to mediate tumor
regression. Cellular senescence is a form of stable cell cycle
arrest, and a marked proportion of cancer cells retain the ability
to undergo this process. Consequently, the activation of senescence
represents a promising new approach to cancer intervention.
Palbociclib induces cell cycle arrest and leads to cellular
senescence (38). Following
dilution or washout from the extracellular medium, palbociclib is
released from intracellular acidic vesicles; this reversible drug
storage in acidic vesicles is often called lysosomal trapping. It
has been demonstrated that brief exposure of cells to palbociclib
is sufficient to produce stable cell cycle arrest and long-term
senescence (39). NK cells can
recognize and eliminate senescent cells that express activating
receptors, such as NK cell group 2D and DNAM-1 ligand, by releasing
granules containing perforin and granzyme. Combining CDK4/6
inhibitors with a targeted drug (trametinib) and hormone (hepcidin)
after endogenous tumor formation has been shown to result in a
notable reduction in Rb phosphorylation and tumor cell
proliferation, accumulation of SA-β-gal+ senescent tumor
cells, induction of senescence-associated secretory phenotype
factors (including TNF-α and intercellular cell adhesion
molecule-1) and subsequent NK cell recruitment in a KP GEMM mouse
model of lung adenocarcinoma (38).
Serum amyloid A1 (SAA1) serves a notable role in
inflating tumor cell senescence induced by palbociclib (4). This acute-phase protein is a serum
factor that exerts differential effects on the neutrophil
phenotype, enhancing the inflammatory response when neutrophils
arrive at the site of injury and promoting resolution during the
resolution phase of inflammation (40). Senescent cells coordinate
inflammatory processes and their resolution, partly by regulating
SAA1, through a coordinated program involving the transition of
neutrophils from an 'N1' to an 'N2' phenotype (41). Following tissue injury, induced
senescence prevents the propagation of damage by preventing the
proliferation of damaged cells (42). Furthermore, it causes tissue
remodeling by providing large amounts of bioactive factors in the
microenvironment. This perspective elucidates the capacity of
senescent cells to engage the immune system to facilitate their
clearance while promoting the stem cell signature of surrounding
cells to enhance tissue repair (43). Palbociclib confers reversible
phenotypic senescence on tumor cells, and it induces a senescent
phenotype with an inflammatory secretosome that recruits and
activates neutrophils by releasing different inflammatory factors,
such as IL-8 and SAA1. Meanwhile, activated neutrophils can cause
phagocytosis of senescent tumor cells (44).
Among the various immunotherapeutic strategies,
macrophage-based therapies have garnered marked interest in tumor
immunotherapy, given that macrophages represent a prominent
component of tumor-infiltrating immune cells. CDK4/6 inhibitors can
induce differentiation, maturation and antitumor activation of
macrophages, which can differentiate into the M1 type (antitumor
type). This subsequently opens up immune-suppressive signaling
pathways, as evidenced by an increase in the expression of PD-L1
and colony-stimulating factor 1 receptor, and a dependence on lipid
metabolism (45). Macrophages
are also implicated in the clearance of senescent cells. The
repolarization of macrophage subtypes from M2 to M1 demonstrates a
capacity to reverse tumor immunosuppression and promote antitumor
immunity within the tumor microenvironment (46). Additionally, macrophages have a
prolonged half-life in the circulation and can migrate specifically
to tumors. Consequently, macrophages can be utilized as drug
carriers for tumor therapy. It has been demonstrated that the
selective loading of the photosensitizer black phosphorus quantum
dots and abemaciclib into artificially assembled macrophages can
confer immunomodulatory and tumor-killing effects within the
microenvironment of malignant peripheral nerve sheath tumors
(47).
CDK4/6 inhibitors can enhance the antitumor effect
of DCs. However the loss of dendritic cells (DCs) within the tumor
microenvironment due to the damage by the tumor represents a
notable impediment to antitumor immunity following the
administration of CDK4/6 inhibitors and immune checkpoint blockade
(ICB). Consequently, restoring the DC compartment through the
adoptive transfer of ex vivo-differentiated DCs has been
shown to result in potent tumor suppression in CDK4/6 inhibitors-
and ICB-treated patients of BC. The addition of DCs can facilitate
tumor localization and induction of systemic CD4 T-cell responses
in patients treated with the CDK4/6 inhibitors-ICB-DC combination,
which is characterized by enrichment of programmed death-1 (PD-1),
and activating phenotype-1-negative T helper (Th)1 and Th2 cells.
Depletion of CD4 T cells can abrogate the antitumor benefit of the
CDK4/6 inhibitors-ICB-DC combination, and growing tumors exhibit an
increased proportion of terminally depleted CD8 T cells.
Furthermore, the restoration of DC function through the adoptive
transfer of exogenous mature bone marrow-derived DCs has been
reported to result in an enhanced tumor response to CDK4/6
inhibitors and ICB therapy. These findings provide substantial
evidence that the limited efficacy of CDK4/6 inhibitors and ICB
combination therapy observed in clinical trials may be attributable
to the inhibitory effect of CDK4/6 inhibitors on DCs. Furthermore,
DC therapy may be viable for augmenting CDK4/6 inhibitors and
immunotherapy responses in clinical settings (48).
Taken together, CDK4/6 inhibitors can modify the
tumor immune microenvironment by enhancing the capacity of immune
cells, including NK cells and macrophages, to present antigens,
induce tumor cell senescence and recruit neutrophils for tumor
destruction. Additionally, they can repolarize macrophage subtypes
from M2 to M1, thereby augmenting antitumor activity. Additionally,
DC therapy has been demonstrated to enhance the efficacy of CDK4/6
inhibitors.
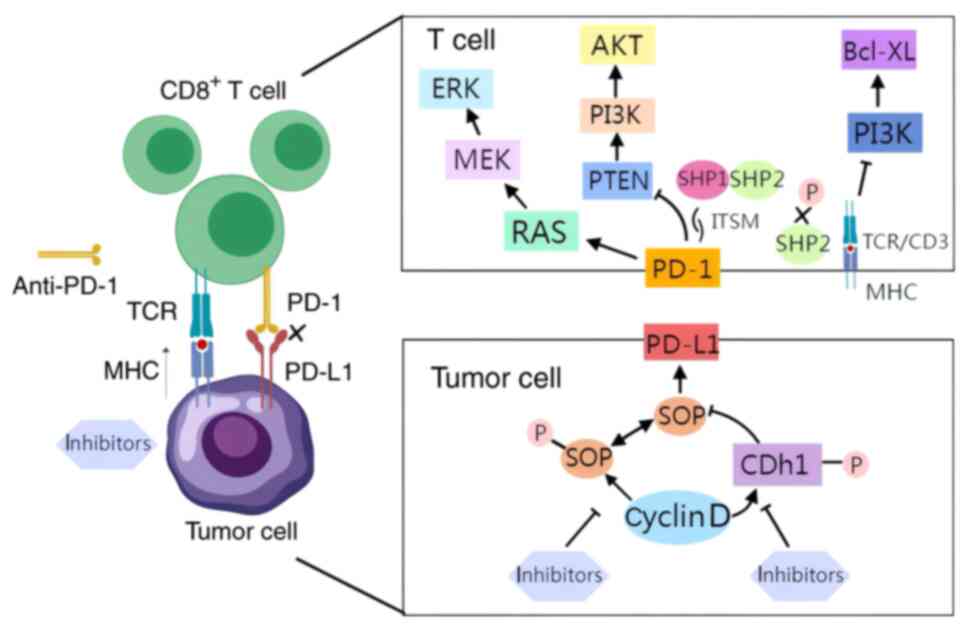
The administration of a single CDK4/6 inhibitor has
been demonstrated to elevate PD-L1 levels, thereby impeding the
response of the immune system to tumor cells. The molecular
mechanisms underlying this process have been the subject of
investigation. It has been reported that the abundance of the PD-L1
protein is regulated by the cyclin D-CDK4 and cullin 3 speckle-type
POZ protein (SPOP) E3 ubiquitin ligase via proteasome-mediated
degradation (49). In
vivo, the inhibition of CDK4/6 has been observed to increase
PD-L1 protein levels, mainly by inhibiting cyclin D-CDK4-mediated
phosphorylation of SPOP, which in turn promotes SPOP degradation by
anaphase-promoting complex/cyclosome bound to the regulatory
subunit Cdh1 (49). It has been
established that PD-1 is a pivotal immune checkpoint that modulates
the threshold of T- and B-cell response to antigens. As a crucial
regulatory checkpoint for T cells, PD-1 exerts a central role in
governing their cellular function. The interaction between PD-L1
and PD-1 induces T-cell depletion, thereby promoting immune evasion
(50). The immunoreceptor
tyrosine-based switch motif (ITSM) is an essential site of PD-1
biological function, and PD-1 is phosphorylated by binding to PD-L1
and further induces immunosuppression by activating a series of
intracellular pathways. The specific mechanisms by which PD-1
exerts its immunosuppressive effects differ between T and B
lymphocytes (51). Two signaling
pathways have been identified as being involved in the immune
response induced by T cells after pathogen invasion. The first
pathway is the binding of MHCs on the surface of APCs to the T-cell
receptor (TCR) (52). The second
pathway is the binding of immunostimulatory ligands expressed by
APCs to the TCR (52).
Consequently, this results in the transduction of activating or
inhibitory signals to T cells, modulating immune responses such as
T-cell activation and exhaustion. The engagement of PD-L1 and PD-1
in T cells leads to the recruitment of Src homology 2-containing
tyrosine phosphatase 1/2 (SHP-1/2) to the C-terminus of ITSM.
Subsequently, SHP-2 functions by dephosphorylating TCR-associated
CD-3ζ and ZAP70, inhibiting downstream signaling. Specifically, the
PI3K pathway has been shown to be inhibited, and the expression of
the cell survival gene BCL-XL is reduced.
PD-1 has also been shown to impede TCR-induced
PI3K/AKT pathway activation by stimulating PTEN. Moreover, PD-1 has
been shown to restrict the proliferation of T cells by hindering
the activation of the RAS/MEK/ERK pathway. Concurrently, PD-1
suppresses the activation of PKCδ, thereby diminishing the
secretion of cytokines, such as IFN-γ and IL-2, by T cells
(53). Furthermore, PD-1
signaling orchestrates T-cell metabolism by impeding glycolysis
while promoting lipolysis and fatty acid oxidation (54).
The PD-1/PD-L1 interaction has also been
demonstrated to inhibit B-cell activation. The binding of PD-L1 to
PD-1 results in the phosphorylation of two tyrosines located within
the ITSM of PD-L1 by the B-cell receptor. This, in turn, leads to
the recruitment of SHP-2 to the C-terminus of PD-1. Subsequently,
SHP-2 undergoes phosphorylation. Consequently, the
dephosphorylation of SHP-2 by the C-terminus of PD-1 leads to acute
Ca2+ disruption and long-term growth arrest.
Consequently, the PD-1-mediated suppression of the immune response
of B cells to antigens is impaired (55).
Notably, combining CDK4/6 inhibitors with PD-1/PD-L1
inhibitors has been demonstrated to augment the antitumor immune
response. The antitumor response has been reported to be further
enhanced when anti-PD-1 and anti-CTLA-4 are used in combination
with CDK4/6 and PI3K inhibitors, resulting in a more robust
antitumor effect in humans. This combination therapy can result in
enhanced T-cell activation and a reduction in the number of Tregs,
thereby enhancing cancer cell death (4). Schaer et al (56) demonstrated that the combination
of abemaciclib and anti-PD-L1 treatment results in the increased
expression of genes involved in antigen presentation and the innate
immune response, which in turn leads to an increase in the
expression of MHC class I and class II molecules in tumor cells. In
a BC mouse model, Goel et al (4) demonstrated that the combination of
CDK4/6 inhibitors and PD-1/PD-L1 inhibitors directly activates T
cells and modulates innate immunity by enhancing antigen
presentation. The combination therapy has been demonstrated to
present superior efficacy in inhibiting tumor growth compared with
abemaciclib monotherapy. Furthermore, combining CDK4/6 inhibitors
with anti-PD-1 antibodies can enhance tumor control in most mouse
models (34). Preclinical in
vivo BC studies have indicated that CDK4/6 inhibitors may
enhance tumor sensitivity to PD-1/PD-L1 blockers. Notably, Schaer
et al observed that treatment with a CDK4/6 inhibitor
followed by the staged use of anti-PD-L1 agents results in more
complete remission (31). In
conclusion, further investigation into the potential of combining
CDK4/6 inhibitors with ICIs to enhance antitumor immune responses
is warranted (Fig. 3).
PD-1 on immune cells and PD-L1 on tumor cells can
facilitate immune escape. The expression of PD-L1 on tumor cells
can be promoted by CDK4/6 inhibitors, which may encourage immune
evasion in a variety of tumor models (34). CDK4/6 inhibitors have been
demonstrated to elevate PD-L1 levels, thereby enhancing the
interaction of PD-L1 with the PD-1 receptor on the surface of T
cells and inhibiting T-cell activity (57); this allows tumor cells to evade
the immune system. Concurrently, CDK4/6 inhibitors also elevate the
number of negative immunodetection sites, PD-1 and CTLA-4, on T
lymphocytes, thereby exerting an adverse immunomodulatory
influence. However, CDK4/6 inhibitors can also induce cell cycle
blockade, trigger T cells, enhance antigen presentation, stimulate
the immune system, inhibit the growth of Tregs and reduce the
secretion of inhibitory cytokines. This results in a decrease in
the inhibitory effect on immune cells and an increase in the immune
response against tumor cells. Consequently, the impact of CDK4/6
inhibitors on immune evasion within the tumor microenvironment is
multifaceted. It is of the utmost importance to exercise caution
when developing antitumor drugs that utilize CDK4/6 inhibitors to
prevent immune evasion by tumor cells.
Given the prevalence of cell cycle gene
abnormalities in solid tumors, developing and approving CDK4/6
inhibitors for treating several solid tumors in recent years has
been a significant advancement in oncological therapy. This section
lists the most used CDK4/6 inhibitors approved for clinical
use.
In three large randomized phase III trials, the
CDK4/6 inhibitors palbociclib, ribociclib and abemaciclib were
investigated when each was combined with AIs in the first-line
treatment of postmenopausal patients with
HR+/HER2− MBC (59,65,69). The three CDK4/6 inhibitors
resulted in marked increased in PFS compared with AI treatment
alone, exhibiting nearly identical hazard ratios for PFS and
potentially analogous efficacy. In another three other large
randomized phase III trials, PALOMA-3 (palbociclib), MONALEESA-3
(ribociclib) and MONARCH-2 (abemaciclib), the combinations of the
three CDK4/6 inhibitors with fulvestrant were investigated in
patients with HR+/HER2− MBC as the second
line of therapy (63,74,75). In the CDK4/6 inhibitor group, all
three drugs showed prolonged PFS and OS benefits compared with
fulvestrant alone. The results from pertinent clinical studies are
provided in Table SI.
Palbociclib, ribociclib and abemaciclib have been
widely used in clinical practice, mostly in combination with
endocrine therapy for HR+/HER2− MBC. In
addition, extensive comparative studies on the efficacy and safety
of these three drugs have been conducted. Desnoyers et al
(76) performed a meta-analysis,
which showed that the OS and PFS of the three drugs in the
treatment of patients with HR+/HER2− MBC are
similar, and supplemented the relative safety and tolerability of
the three drugs, which may help guide clinicians in the
decision-making of drug selection. The results of this previous
study showed that ribociclib and abemaciclib are associated with
significantly lower rates of grade 3-4 neutropenia, but
significantly higher gastrointestinal toxicity when compared with
palbociclib. In addition, abemaciclib is less well tolerated, and
patients treated with this drug were reported to have a
significantly higher incidence of treatment interruptions due to
adverse events (AEs) compared with the other two drugs. In 2024, a
meta-analysis by Kappel et al (77) also showed similar results. Some
additional findings from this study were that palbociclib was shown
to be associated with a significantly higher incidence of
neutropenia, but a substantially lower risk of grade 3-4 infections
than ribociclib and abemaciclib. Cases of grade 3-4 transaminitis
and grade 3-4 neutropenia were also reported to be significantly
lower with abemaciclib than with ribociclib.
G1T38 is a novel CDK4/6 inhibitor developed by G1
Therapeutics, Inc. G1T38 has been demonstrated to block cells in
the G1 phase by reducing Rb phosphorylation and
inhibiting cell proliferation in CDK4/6-dependent oncogenic cell
lines, including those derived from BC, melanoma and leukemia.
Furthermore, G1T38 has been shown to possess selective and oral
bioavailability characteristics, with good selectivity. It is
currently undergoing phase II clinical trials and may become an
effective CDK4/6 inhibitor in the clinic in the future (81).
Narazaciclib (ON123300) is a CDK4/6 inhibitor
developed by Onconova Therapeutics, Inc. and is undergoing phase
I/II clinical studies. A previous study has shown that it exhibits
relatively high antitumor activity in mantle cell lymphoma cell
lines (82).
PF-06873600, a CDK2/4/6 inhibitor developed by
Pfizer, has been identified as a potential therapeutic agent for
cancer treatment and has been subjected to phase I clinical trials
for the treatment of patients with HR+/HER2−
BC, metastatic triple-negative BC (TNBC) or advanced
cisplatin-resistant epithelial ovarian/tubal carcinoma (83).
ETH-155008, developed by Shengke Pharmaceutical
(Jiangsu) Ltd., represents a novel dual inhibitor that may enable
effective acute myeloid leukemia (AML) therapy through dual
targeting of FLT3 and CDK4/6. By further blocking tumor cells at
the G1 stage, ETH-155008, through a synergistic effect,
to some extent avoids the development of CDK4/6 inhibitor
resistance. Notably, it shows a favorable safety and efficacy
profile in phase I clinical studies (84).
AMG925 (FLX925), developed by Amgen, has been shown
to act on FLT3 and CDK4/6 selectively. It is currently in phase I
clinical trials, primarily for the treatment of AML (80).
In addition to new CDK4/6 inhibitors entering
clinical studies, various drugs approved for clinical use,
including palbociclib, ribociclib and abemaciclib, are also under
investigation. In addition to HR+ BC, the clinical
studies of these drugs have primarily focused on other solid
tumors, including non-small cell lung cancer and prostate cancer.
In addition, further in-depth research may be conducted on the
therapeutic effect, safety, pharmacokinetics, dosage regimen and
other pertinent factors of these drugs. Table SIII provides a comprehensive
listing of clinical trials related to CDK4/6 inhibitors, as listed
on ClinicalTrials.gov (85-92).
Resistance to CDK4/6 inhibitors is an almost
inevitable biological event. From a therapeutic point of view, it
has become urgent to find biomarkers of sensitivity and resistance,
to understand the mechanisms of resistance to CDK4/6 inhibitors,
and to identify strategies to overcome them according to the
target. A trial of patients with HR+/HER2+
MBC treated with a combination of CDK4/6 inhibitors and endocrine
therapy, and analyzed by biopsy whole exome sequencing, identified
eight different mechanisms of CDK4/6 inhibitor resistance (93): i) Rb1 deletion, ii) AKT1
mutation/amplification, iii) RAS activation, iv) fibroblast growth
factor receptor (FGFR) activation, v) HER2 activation, vi) cell
cycle protein-E activation, vii) Aurora kinase A (AURKA)
amplification and viii) estrogen signaling deletion. These diverse
mechanisms (including alterations in cell cycle-specific or
non-specific mechanisms) have been observed in two-thirds of
resistant tumors examined. These mechanisms can be further
subdivided into the following categories: Rb function deficiency,
abnormalities in cell cycle regulatory factors, activation of
bypass signaling pathways, and dysfunction of other transcription
factors and hormones. The identification of these mechanisms has
the potential to yield novel therapeutic interventions for patients
with these tumors.
CDK4/6 inhibitors inhibit cell proliferation through
a mechanism of action dependent on Rb. Specifically, CDK4/6
inhibitors block CDK4/6 activity and prevent phosphorylation of the
Rb protein, which in turn maintains the inhibition of Rb on the E2F
transcription factor and prevents the cell cycle from moving from
the G1 phase into the S phase (94,95). However, it has been reported that
a variety of BC cell lines resistant to palbociclib possess reduced
levels of Rb expression compared with cells that had not received
the drug (96). This implies
that CDK4/6 inhibitors treatment may reduce Rb protein levels while
inhibiting proliferation. This phenomenon has been further
validated in clinical practice, where up to 10% of patients with
disease progression treated with CDK4/6 inhibitors have been shown
to possess alterations or deletions in both copies of the Rb1 gene,
a feature not seen in patients treated with endocrine therapy only
(97). Rb1 loss is a proven
resistance mechanism in vitro and in patient-derived tumor
xenograft models (24). The
level of Rb1 protein decline is further exacerbated by multiple
molecular causes (such as mutation, silencing or
hyperphosphorylation) (98).
Ultimately, the constitutive progression of the cell cycle
continues through the activation of other cell cycle mechanisms
(such as E2F amplification) and is not driven in a manner that
relies on CDK4/6 (99). However,
a simple decline in Rb levels is not sufficient to activate E2F
transcriptional activity necessary for cell cycle progression, nor
is it alone adequate to directly drive cell cycle deregulation
(100). In the process of
developing CDK4/6 inhibitor resistance, upregulation of mitogenic
or hormonal signaling, or activating mutations are also required to
stabilize c-Myc levels, which mediate the amplification of E2F
transcriptional activity, bypassing the inhibitory effects of
CDK4/6 inhibitors (96).
Therefore, for this group of patients, restoring their Rb levels or
inhibiting c-Myc to reduce E2F activity may be two critical methods
to overcome drug resistance.
Amplification of CDK4 or CDK6 is considered one of
the essential mechanisms of CDK4/6 inhibitor resistance, as this
amplification can drive cell cycle progression through activation
of the cyclin D-CDK4/6-Rb pathway, thereby impairing the effect of
CDK4/6 inhibitors on cell cycle arrest (101). CDK4 is widely expressed in
various cancer types and CDK4-upregulated cells have reduced
sensitivity to CDK4/6 inhibitors. In glioma cells, CDK4
overexpression can even lead to complete resistance to CDK4/6
inhibitors (102). In addition,
CDK4 overexpression has been identified as a potential biomarker
for predicting resistance to conventional chemotherapy in patients
with osteosarcoma (103).
Moreover, the overexpression of CDK4 may be attributed to the
enhancement of CDK4 activation by the cell-cycle regulator CDK7
through T-loop phosphorylation (104). Coombes et al (105) reported that the CDK7 inhibitor
samuraciclib exerts some clinical activity with an objective
remission rate of 36% (9/25 patients with
HR+/HER2− BC). In a population of patients
previously treated with a CDK4/6 inhibitor in combination with
fluvastatin, samuraciclib was associated with a median PFS time of
3.7 months (105). These data
suggest that samuraciclib may have potential clinical benefit in
managing disease progression associated with CDK4/6 inhibitor
resistance (105).
AURKA is a kinase that serves a vital role in cell
division, and has been associated with intrinsic and acquired
resistance to CDK4/6 inhibitors and endocrine therapies (93). AURKA amplification promotes cell
cycle progression directly or indirectly. For example, AURKA can
phosphorylate Rb proteins, weakening their inhibitory effect on E2F
transcription factors, thereby restoring E2F transcriptional
activity and driving the cell cycle from G1 to S phase
(116). In addition, AURKA is a
key regulatory component of the p53 pathway, particularly in the
checkpoint-response pathway necessary for oncogenic transformation
through phosphorylation and stabilization of p53, which confers
tumor cell resistance to CDK4/6 inhibitors (117). Together, these mechanisms
diminish the cell cycle-blocking effects of CDK4/6 inhibitors.
AURKA amplification is also highly associated with CDK4/6 inhibitor
treatment failure in clinical samples. Wander et al
(93) demonstrated that the
proportion of AURKA overexpression tends to be higher in patients
with disease progression after treatment with CDK4/6 inhibitors
than in patients with effective treatment. This finding suggests
that AURKA amplification may serve as a potential biomarker of
CDK4/6 inhibitor resistance to inform patient stratification and
treatment decisions.
The FGFR family consists of highly conserved
transmembrane receptors (FGFR1-4) involved in a number of key
mechanisms that lead to cancer, such as proliferation,
differentiation and cell survival (118). Post-progression analysis of
circulating tumor DNA from 34 patients treated with palbociclib in
combination with letrozole has been reported to show alterations in
FGFR, including FGFR1 amplification, FGFR2 amplification, and
activating FGFR1 and FGFR2 mutations (119). Amplification of FGFR1 is the
most common genomic alteration in BC, occurring in ~10% of BC cases
(120), and FGFR1 has been
identified as a potential mechanism of acquired resistance to
endocrine therapy in combination with CDK4/6 inhibitors (121,122). The development of this
resistance mechanism may be mediated by FGFR1 amplification, which
activates the PI3K/AKT/mTOR and RAS/MEK/ERK signaling pathways
(123). Genetic analysis of the
MONALEESA-2 study showed that patients with amplification of the
FGFR1/ZNF703 fusion gene had a PFS of 10.61 months, compared with
24.84 months for patients with wild-type FGFR1/ZNF703. In addition,
patients with high FGFR1 mRNA expression (50% of the median mRNA
expression) had a median PFS time of 22.21 months, whereas patients
with low FGFR1 mRNA expression did not reach a median PFS after 32
months of follow-up (65). These
results indicate that FGFR1 amplification and high expression may
be predictors for a poor prognosis in patients, and CDK4/6
inhibitors combined with blocking the FGFR pathway may be a
potential strategy used to overcome drug resistance and improve the
prognosis of patients.
PI3K/AKT/mTOR is hyperactivated or altered in
numerous types of cancer and regulates a wide range of cellular
processes, including proliferation, growth, metabolism,
angiogenesis and metastasis (124). In BC, inhibition of CDK4/6
promotes the adaptive rewiring of the PI3K/AKT/mTOR signaling
pathway in BC cells, which enhances their dependence on this axis,
promoting phosphorylation and activity of the PI3K/AKT/mTOR pathway
(101). Notably, overactivation
of the PI3K/AKT/mTOR pathway induces cyclin D1 and CDK4 protein
upregulation, whereas direct inhibition of the signaling pathway
silences cyclin D1 and CDK4, and leads to cell cycle arrest. These
findings confirm that activation of the PI3K/AKT/mTOR pathway and
resistance to CDK4/6 inhibitors may be interrelated (125). On the other hand, elevated
levels of phosphorylated AKT have been associated with prolonged
expression of the cyclin E2-CDK2 complex of cell cycle proteins,
which suggests that tumor cells can bypass the cyclin
D-CDK4/6-dependent mechanism of cell cycle regulation via an
alternative pathway to maintain cell cycle progression (24).
The MAPK signaling pathway, or RAS/MEK/ERK pathway,
is a signaling pathway downstream of FGFR1. Activation of the RAS
family of oncogenes (KRAS, HRAS, NRAS) has been observed in tumor
biopsies collected from patients with HR+ BC resistant
to CDK4/6 inhibitors (93). Most
patients with KRAS-mutant malignancies exhibit elevated levels of
expression of cyclin D1 (126).
Patients with high levels of cyclin D1 protein expression tend to
have a worse prognosis than those with low expression levels
(median OS 41.7 vs. 3.5 months) (127). This may be explained by the
fact that KRAS produces aberrant growth signals in the presence of
CDK4/6 inhibitors, leading to cellular resistance. In addition,
KRAS and RAF kinases regulate ERK1/2 via MEK. ERK1/2 is also highly
represented in the oncogenic profile of patient RNAseq data related
to the occurrence and development of cancer (128). Based on this, there have been
several clinically tested pharmacological agents that target the
MAPK pathway. For example, the FDA has approved MEK inhibitors for
certain types of cancer (such as melanoma) (128). In BC, the MEK1/2 inhibitor
selumetinib, in combination with fulvestrant and palbociclib, can
effectively inhibit the proliferation of BC cells resistant to
CDK4/6 inhibitors (119). These
trials have shown an association between acquired resistance to
CDK4/6 inhibitors and induced MAPK pathway activity, indicating
that selecting the correct target for targeted therapy is an
effective measure to address resistance.
In preclinical models, acquired CDK6 amplification
not only confers abemaciclib resistance by bypassing the activation
of alternative cell cycle checkpoints, but also leads to a notable
reduction in the mRNA and protein levels of ER and PR (106,111). Furthermore, it can suppress the
expression of ER-regulated genes, such as XBP1 and MYC (106). Reduced ER/PR levels impair
estrogen-dependent induction of cyclinD-CDK4/6 complex activity
(129). Notably, the absence of
ER/PR expression is closely associated with acquired insensitivity
to hormonal agents, such as the ER antagonists fulvestrant and
tamoxifen (130), suggesting
that the dual mechanisms of drug resistance involve cell cycle
dysregulation and endocrine therapy escape.
The AP-1 family is a key transcription factor for
cyclin D1, and includes Jun, Fos, and their related subfamilies of
activating transcription factors (ATF2, LRF1/ATF3 and B-ATF)
(131). In 20-40% of human
patients with BC, c-Jun exhibits high levels of activation. It has
been reported that c-Jun activation inhibits ER activity and
upregulates cyclin D1 transcription, thereby inducing resistance to
CDK4/6 inhibitors (132). To
the best of our knowledge, the efficacy of AP-1 inhibitors in
patients with CDK4/6 inhibitor-resistant BC has not yet been
thoroughly investigated. However, the results of this exploration
are of great clinical value.
MYC is a widespread oncogenic transcription factor
that promotes cancer cell progression (133). MYC-acquired mutations have been
identified in 8.8 and 20.1% of patients with BC in phase III
MONARCH-3 (NCT02246621) and phase II Next MONARCH-1 clinical trials
(NCT02747004) respectively (134,135). The Next MONARCH-1 clinical
trial has shown that acquired MYC genomic aberrations were enhanced
after abemaciclib monotherapy or combination therapy with a
nonsteroidal AI in patients with HR+ BC (136). In addition, Pandey et al
(137) has validated the
upregulation of MYC in palbociclib-resistant cells by reverse
transcription-quantitative PCR, suggesting that the MYC
upregulation may be another marker of acquired resistance to CDK4/6
inhibitors.
Despite the diversity of drug resistance
mechanisms, there are some commonalities and cross-regulatory
relationships among different drug resistance mechanisms. The
commonality of such a variety of drug resistance mechanisms lies in
the imbalance of the cell cycle regulation system. Whether through
loss of Rb function, amplification of CDK4/6 or activation of the
cyclin E-CDK2 complex, this ultimately leads to the bypass of the
G1-phase blocking signal. This co-imbalance makes the
cells no longer dependent on the CDK4/6-driven pathway, thus
reducing the effectiveness of CDK4/6 inhibitors (138). In addition, FGFR1 activation
and the abnormal transduction of upstream growth factor signaling
pathways (such as PI3K/AKT/mTOR and RAS/MAPK) affected by FGFR1 can
not only directly compensate for the loss of cell cycle regulation,
but also often interact with the regulation of intracellular cyclin
expression. This means that even when CDK4/6 is inhibited, these
alternative pathways can activate downstream cell proliferation
mechanisms (139). Part of the
resistance mechanism, while largely attributable to genetic
mutations (such as loss of Rb), is often also accompanied by
extensive epigenetic regulatory changes, such as the regulation of
AP-1 and MYC transcription factor activity. This interaction at the
genetic and epigenetic levels further underlies the commonality
between drug resistance mechanisms (140). These commonalities also suggest
that drug resistance can be predicted in a timely manner by
monitoring Rb status, cyclin E-CDK2 activity and related signaling
pathway changes in the early stage, so as to provide personalized
treatment. Combined treatment of CDK4/6 inhibitors with endocrine
therapy, chemotherapy and targeted therapy can simultaneously
target multiple pathways and may effectively overcome the problem
of drug resistance.
Targeting CDK4/6 is under active research in cancer
therapy as a promising cancer treatment. The present study
previously reviewed the resistance to CDK4/6 inhibitors in cancer
therapy, and addressing the resistance to CDK4/6 inhibitors has
profound implications for realizing their potential in cancer
therapy. Effective combination therapies involving CDK4/6
inhibitors have been shown to delay resistance while reducing the
side effects of other treatments, leading to favorable antitumor
effects (138). Despite the
current lack of effective remedies for low Rb1 expression, which is
the most fundamental cause of resistance, several drug combination
strategies have shown the potential to improve the drug sensitivity
of CDK4/6 inhibitors (141).
Drug combination strategies involving CDK4/6 inhibitors may include
endocrine therapy, chemotherapy, targeted therapy and
immunotherapy.
Currently, most international guidelines recommend
first-line CDK4/6 inhibitors for patients with newly diagnosed
HR+/HER2− MBC (148). However, the optimal timing of
CDK4/6 inhibitors has not been fully elucidated. The randomized
phase III SONIA trial (NCT03425838), conducted in the Netherlands,
randomized 1,050 patients who had not previously received treatment
for advanced BC to receive a CDK4/6 inhibitor in either the
first-line or second-line setting. This study aimed to address
whether delaying CDK4/6 inhibitors to second-line therapy after
initial endocrine therapy monotherapy affects efficacy, and also
assessed safety, quality of life and cost-effectiveness (149). The study aimed to assess
whether all patients should start treatment with CDK4/6 inhibitors.
The findings indicated that more research is required to identify
patients who truly benefit from first-line CDK4/6 inhibitor therapy
and those who do better when deferred to second-line treatment.
More recently, Sonke et al (150) found no statistically
significant benefit of using CDK4/6 inhibitors as first-line
therapy compared with second-line therapy (median 31.0 and 26.8
months, respectively; hazard ratio, 0.87; 95% confidence interval,
0.74-1.03; P=0.10). Health-related quality of life was also similar
in the two groups. In addition, first-line CDK4/6 inhibitor use was
associated with a longer duration of CDK4/6 inhibitor treatment and
more grade ≥3 AEs than second-line use. This study challenges past
perceptions of the need for first-line use of CDK4/6 inhibitors and
points to the need for robust sequencing trial data to determine
the best use of available therapies.
The commonly used chemotherapeutic agent paclitaxel
regulates microtubule proteins during cell cycle division, while
CDK4/6 inhibitors prevent phosphorylation of Rb1 during the
G0 phase of the cell cycle (153). The combination of paclitaxel
and CDK4/6 inhibitors can co-regulate the cell cycle and has been
shown to reduce the side effects of paclitaxel treatment, such as
hematopoietic stem cell death, and to improve its impact on TNBC
cells (154). Another study has
shown that CDK4/6 inhibitors and paclitaxel synergistically work
together by inducing apoptosis in KRAS-mutant lung adenocarcinoma
cells (155).
Platinum compounds, another widely used
chemotherapeutic agent for cancer treatment, often have side
effects such as acute kidney injury (AKI) when applied alone. Kim
et al (156)
demonstrated that CDK4/6 inhibitors can alleviate
platinum-associated AKI in an Rb1-dependent manner, but the exact
mechanism is unclear. Liu et al (157) reported that CDK4/6 inhibitors
can inhibit cell proliferation and induce apoptosis to reverse the
acquired resistance to platinum compounds in lung cancer cells. The
combination of the two in therapy provides a new therapeutic
strategy for patients with platinum compound-resistant lung
cancer.
Five studies have reported the efficacy of CDK4/6
inhibitors in neoadjuvant therapy, NeoPalAna (158), New MONARCH (74), N007 (159), CORALLEEN (160) and NeoPAL (161). Studies such as NeoPalAna, New
MONARCH and N007 mainly focused on the evaluation of the biological
effects of CDK4/6 inhibitors combined with endocrine therapy on
tumors, such as the reduction of Ki-67. These three studies
indicated that CDK4/6 inhibitors such as palbociclib and
abemaciclib combined with endocrine therapy can significantly
reduce Ki-67 levels and inhibit tumor proliferation, showing a good
biological response, and are potentially effective options for
neoadjuvant treatment of HR+/HER2− BC.
Although the treatments evaluated in these studies (74,158,159) were not directly compared with
chemotherapy, they laid the groundwork for subsequent comparative
trials, such as CORALLEEN (160) and NeoPAL (161), which directly assessed CDK4/6
inhibitors plus endocrine therapy vs. standard chemotherapy. The
latter two trials randomly assigned patients to receive neoadjuvant
endocrine therapy plus a CDK4/6 inhibitor or neoadjuvant
chemotherapy to investigate whether the addition of a CDK4/6
inhibitor to letrozole would provide at least as much clinical
benefit as standard chemotherapy. Overall, these two studies showed
that in selected patients with HR+/HER2− BC,
neoadjuvant therapy with a CDK4/6 inhibitor plus letrozole may
achieve some equivalent efficacy to conventional chemotherapy, but
gaps remain in endpoints such as the rate of pathological complete
response (160,161).
Increased PD-L1 expression in patients treated with
CDK4/6 inhibitors is a clinical problem currently being
encountered. It may be one of the potential mechanisms leading to
resistance to CDK4/6 inhibitors through evasion of
immune-surveillance checkpoints (162).
CDK4/6 inhibitors can be used in combination with
ICIs to transform immunologically 'cold' tumors into 'hot' tumors.
'Hot' tumors are more susceptible to immune cell infiltration and
are more easy to treat (163).
Results from preclinical studies have suggested that ICIs can be a
therapeutic strategy to overcome CDK4/6 resistance in BC (164,165). Meanwhile, ICI resistance is
associated with cyclin D1 amplification and CDKN2A deletion, which
can also be overcome by CDK4/6 inhibition (166-168). Schaer et al (56) and Jerby-Arnon et al
(169) observed synergistic
effects exhibited by sequential administration of CDK4/6 inhibitors
and ICIs in allogeneic tumor models. Schaer et al (56) showed that continuous
administration of abemaciclib followed by ICIs, divided into 28
days of treatment, resulted in improved antitumor activity than
14-day treatment or monotherapy. Jerby-Arnon et al (169) reported that, as compared with
other treatment approaches (vehicle, monotherapy, ICI + abemaciclib
Q3D, abemaciclib QD followed by ICI), staged combination therapy
(ICIs followed by ICIs plus abemaciclib) resulted in higher rates
of tumor suppression and survival in melanoma models.
Consistent with most preclinical data, combination
therapy with CDK4/6 inhibitors and ICIs have shown potential in
patients with tumors. Rugo et al (170), in a phase Ib study of
pembrolizumab combined with abemaciclib, found that this
combination showed good potential in preliminary clinical efficacy
and safety, although further validation is still needed. In an
early clinical study involving patients with metastatic disease,
Yuan et al (171)
revealed that palbociclib in combination with pembrolizumab showed
a complete remission rate of up to 31% in patients, with median PFS
and OS durations of 25.2 and 36.9 months, respectively.
Additionally, a previous case report demonstrated that a patient
with refractory SMARCA4-deficient small cell carcinoma of the ovary
hypercalcemic type experienced disease progression despite multiple
treatment regimens, including immunotherapy. Still, the combination
of a CDK4/6 inhibitor and ICIs demonstrated efficacy (172). The combination of a CDK4/6
inhibitor and ICIs has also been reported to be effective in
treating SMARCA4-deficient cancers and has demonstrated potential
(173). Bose et al
(174) similarly reported a
case in which a patient who resisted an ICI alone developed a
profound and long-lasting response to combination therapy with a
CDK4/6 inhibitor and an ICI.
Because combination therapies with CDK4/6
inhibitors and ICIs have been validated in preclinical studies in
overcoming drug resistance and have shown potential in cancer
treatment, a number of clinical trials evaluating the safety and
efficacy of CDK4/6 inhibitors in combination with ICIs for the
treatment of cancer are ongoing (Table SIV) (170,171,175-180). The PACE trial, a multicenter,
randomized, open-label phase II trial prospectively evaluated the
efficacy of palbociclib in combination with fluvastatin and
nivolumab in patients with ER+/HER2− MBC
after progression following treatment with CDK4/6 inhibitor and AI
combination therapy. The results demonstrated that combination
therapy with the PD-L1 antibody avelumab and CDK4/6 inhibitors
significantly improved PFS (176).
However, combination therapy with CDK4/6 inhibitors
and ICIs still has some limitations. Noticeable toxic reactions,
especially hematological toxicity such as neutropenia, have become
a hindrance limiting the potential of this therapy (171,181,182). Rugo et al (170) investigated the safety and
efficacy of abemaciclib plus pembrolizumab with/without anastrozole
in patients with HR+/HER− MBC and reported a
spectrum of toxicities with the combination therapy, including an
increased incidence and severity of hematological toxicities (such
as neutropenia) and immune-related AEs (such as hepatotoxicity and
pneumonia). Preliminary reports have indicated that the
toxicity-related symptom burden, including but not limited to
diarrhea and fatigue, results in a reduction of physical activity
scores in the short term. However, these symptoms can be managed
effectively, thereby restoring quality of life to a certain extent.
Another phase II clinical study reported that the combination of
nivolumab and abemaciclib, which has demonstrated greater clinical
activity compared with other CDK4/6 inhibitors, resulted in severe
and prolonged immune-associated AEs (183). A phase I clinical study by
Patnaik et al (180)
combined LY3300054, a PD-L1 inhibitor, with multiple targeted
agents, including abemaciclib. The study reported the impact of
hematological toxicity on the long-term prognosis of patients, with
some patients discontinuing treatment as a result. Combination
therapies with CDK4/6 inhibitors and ICIs currently present both
risks and opportunities. If toxicity can be effectively managed,
combination therapies have the potential to improve outcomes.
However, toxicity management strategies for this combination
therapy remain understudied and require further clinical
investigation.
Targeted therapies target specific cancer sites
(such as genes or protein molecules in tumor cells), thereby
interfering with tumor metastasis, inhibiting tumor cell
proliferation and differentiation, and inducing apoptosis of tumor
cells (184).
Extensive preclinical studies have demonstrated
that cyclin D1 acts as a common node to achieve crosstalk between
CDK4/6 and the PI3K/AKT/mTOR pathway (182). The binding of cyclin D1 to
CDK4/6 induces phosphorylation of Rb and its subsequent uncoupling
from E2F, which contributes to the progression of the cell cycle in
the G1-S phase (181). Aberrant PI3K/AKT/mTOR pathway
activation contributes to CDK4/6 inhibitor resistance (141). Clinical trials have examined
the effects of combining PI3K inhibitors and CDK4/6 inhibitors in
cancer therapy (Table SV)
(185-187). Notably, one trial showed that
the triple combination of the PIK3CA-isoform-specific inhibitor
alpelisib or the pan-PI3K inhibitor buparlisib with the CDK4/6
inhibitor ribociclib and fulvestrant had high toxicity (182). Another phase Ib trial tested a
triple combination therapy consisting of the CDK4/6 inhibitor
palbociclib, the PIK3CA-isoform-specific inhibitor taselisib, and
fulvestrant, which showed promising efficacy in
ER+/HER2− advanced BC with PIK3CA mutations
and demonstrated tolerability at pharmacologically active doses
(188).
AKT acts as a bridge between the PI3K and mTOR
signaling pathways. The TAKTIC study showed that AKT inhibitor
therapy in combination with CDK4/6 inhibitors and endocrine therapy
was efficacious in some patients and showed good tolerability
(189). Table SV includes information three
ongoing clinical trials on AKT inhibitors (NCT04862663, NCT04920708
and NCT03959891). Whether AKT inhibitors can be one of the
therapeutic options after CDK4/6 inhibitor resistance still awaits
validation in clinical trials.
The mTOR pathway is an important signaling pathway
downstream of PI3K, and thus, mTOR inhibitors have received as much
attention as PI3K inhibitors (190). Everolimus is a typical mTOR
inhibitor that has shown promising therapeutic efficacy in the
phase III, double-masked, randomized international BOLERO-2 trial
(median PFS extension of 4.6 months; P<0.0001) (191). For patients resistant to CDK4/6
inhibitors, mTOR inhibitors may provide satisfactory results.
PARP is a key enzyme involved in the repair of
single-stranded DNA gaps. Preclinical studies have shown that the
combination of PARP inhibitors, such as olaparib, and CDK4/6
inhibitors can synergistically inhibit the Rb1-E2F1 signaling
pathway at both the transcriptional and post-translational levels,
leading to cell cycle arrest and downregulation of the E2F1 gene
target, ultimately achieving inhibition of tumor cell
proliferation, induction of apoptosis and blocking of
neuroendocrine differentiation (192-194). Zhu et al (195) demonstrated that overexpression
of β-catenin, especially hyperphosphorylation of its Ser675 site,
activates the Wnt signaling pathway. This process mediates olaparib
resistance, which palbociclib significantly attenuates. Currently,
several PARP inhibitors, such as olaparib, have been approved by
the FDA for cancer treatment, and the potential of combination
therapies of PARP inhibitors and CDK4/6 inhibitors in treating
cancers such as TNBC has been reported (196).
Histone deacetylase (HDAC) inhibitors are also a
targeted agent that has received attention. A phase III clinical
trial (ACE; NCT02482753) evaluated the efficacy and safety of
chidamide combined with exemestane in the treatment of
postmenopausal patients with HR+ advanced BC. The
results showed that the modified PFS time was longer with chidamide
combined with exemestane (7.4 months) than with placebo combined
with exemestane (3.8 months) (197). The San Antonio Breast Cancer
Symposium meeting in 2021 reported that entinostat, another HDAC
inhibitor, was able to improve the PFS time in patients with
HR+/HER2− BC (198). HDAC inhibitors may therefore
offer new options for combination therapy with CDK4/6 inhibitors
and targeted agents.
BCL2 inhibitors are another option for combination
therapy with CDK4/6 inhibitors. An early phase I clinical trial
demonstrated that venetoclax, a BCL2 inhibitor, was highly
tolerated in combination with tamoxifen, and the high level of BCL2
expression in ~70% of patients with ER+ tumors led to
encouraging activity in both ER+ and BCL2+
metastatic BC (199). However,
a phase II clinical trial (Veronica; NCT03584009) showed that
venetoclax did not significantly improve PFS in patients who
progressed after treatment with CDK4/6 inhibitors (200). There is also an ongoing
clinical trial (NCT03900884) designed to evaluate the efficacy,
safety and tolerability of a BCL2 inhibitor (venetoclax) in
combination with fulvestrant in patients with
ER+/HER2− MBC. More clinical studies are
still needed regarding the therapeutic efficacy of BCL2 inhibitors
in combination with endocrine therapy and CDK4/6 inhibitors.
RT is one of the most widely used local therapeutic
options, with palliative RT and ablation commonly used for advanced
BC (202). Numerous preclinical
studies have shown that CDK4/6 inhibitors can enhance the
radiosensitivity of human cancer cell lines by inhibiting the
repair mechanism of double DNA scaffold breaks, enhancing apoptosis
and blocking cell cycle progression (203-207). This implies that the
combination of CDK4/6 inhibitors with RT may increase the antitumor
effect of RT but provides a theoretical basis for clinical
application.
The NATALEE study (NCT03701334) was a phase III
trial that demonstrated the benefit of this combination in patients
with stage II and stage III HR+/HER2− early
BC at risk of recurrence, including patients without lymph node
involvement (208). Currently
published data on the feasibility of concomitant RT and CDK4/6
inhibitors are based on small-scale retrospective series with low
evidence and a lack of prospective clinical data (209). There are currently several
ongoing clinical trials evaluating the efficacy of combination
therapy with CDK4/6 inhibitors and RT in BC (NCT03691493,
NCT03870919, NCT03750396, NCT04563507, NCT05664893 and
NCT04923542), almost all of which are in phase I or II. Of these,
the prospective phase II ASPIRE trial (NCT03691493) is evaluating
RT in combination with palbociclib and hormonal therapy in patients
with BC who have developed bone metastases. The PALATINE
prospective trial (NCT03870919) will evaluate localized breast RT
in combination with CDK4/6 inhibitors in advanced BC.
There is a need to characterize the safety issues
of combining CDK4/6 inhibitors and RT, and to help guide
therapeutic decisions. A systematic evaluation and meta-analysis
has previously been conducted, encompassing 15 studies with a total
of 1,133 patients diagnosed with HR+/HER2−
BC, 617 of whom received CDK4/6 inhibitors in conjunction with RT.
The results indicated a 29.4% incidence of severe hematological
toxicity, a 2.8% incidence of non-hematological toxicity, a 24.0%
CDK4/6 inhibitor dose reduction rate and a 2.3% discontinuation
rate. These findings are analogous to the toxicity rates observed
when CDK4/6 inhibitors are used as monotherapy, suggesting that
combination therapy does not appear to markedly augment toxicity.
However, it is important to note that the majority of the data were
derived from retrospective studies, which are subject to certain
limitations (202). Another
study analyzed 373 patients with MBC who received CDK4/6 inhibitors
and RT, and found similar results: The overall toxicity of this
combination therapy was shown to be limited, and it may be safe and
feasible (210). An
observational study revealed that the combination therapy led to an
elevated occurrence of grade ≥3 AEs up to 6 weeks following RT.
However, it exhibited a higher rate of local control, indicating
that the combination therapy achieves a balance between efficacy
and toxicity (211).
Conversely, a multicenter cohort study observed frequent moderate
toxicity reactions to RT with CDK4/6 inhibitors, thereby
underscoring the necessity for caution regarding this combination
therapy (212).
However, the preclinical rationale suggesting that
combined RT and CDK4/6 inhibitors may be unsafe is derived from the
cytostatic and immunomodulatory effects reported in studies testing
combination therapy in in vitro and in vivo models
(209). Selective inhibition of
CDK4/6 may exert its effect on the cell cycle by interfering with
the transition from G1 to S phase, decreasing the level
of phosphorylation of Rb proteins, and inducing G1-phase
cell cycle arrest. Irradiated normal cells may exhibit delayed
progression through the G1, S and G2 phases.
Cells demonstrate robust radiation tolerance in the G0,
early G1 and late S phases of the cell cycle, while
exhibiting heightened sensitivity in the late G1,
G2 and M phases. The combination of CDK4/6 inhibitors
and RT has been shown to induce a higher proportion of cells to
remain in the G2/M phase of the cell cycle (1,32,210,213,214).
Preclinical rationale and clinical data on the
potential synergistic toxicity of RT and CDK4/6 inhibitor are
limited and conflicting, and data on the feasibility are based on
low-level evidence from small retrospective series and are somewhat
heterogeneous; therefore, caution is recommended in the application
of this combination therapy (215).
CAR-T cell therapy is a novel and personalized
immunotherapeutic approach that has shown promising results in
treating hematological malignancies (216). However, the application of this
approach in solid tumors has been limited by a variety of factors
that may be related to antigenic escape and heterogeneity, limited
CAR-T cell transport and infiltration, T-cell depletion and lack of
persistence, associated with the immunosuppressive microenvironment
in solid tumors (217).
Combination therapies may be able to address these limitations. A
preclinical study by Lelliot et al (218) has shown that inhibition of
CDK4/6 activity improves the self-renewal and viability of
CD4+ and CD8+ CAR-T memory stem cells, which
can enhance the durability and therapeutic efficacy of CAR-T cell
therapies. This provides novel insight into the field of CDK4/6
inhibitor combination therapies. This idea is still in preclinical
development, and clinical trials combining CDK4/6 inhibitors and
CAR-T cell therapy for cancer treatment are lacking.
With the increasing use of CDK4/6 inhibitors in
combination with endocrine therapy, immunotherapy and targeted
therapies, clinicians must be aware of associated AEs and their
management. Drug-induced AEs often lead to dose reduction and
discontinuation, which may affect their efficacy. The safety
profile of some of the FDA-approved CDK4/6 inhibitors has been
described in the present review.
Nausea, vomiting, fatigue, and cytopenia are common
AEs of all CDK4/6 inhibitors (73,79,219,220). Neutropenia has been reported to
be a particularly significant AE in the cases of palbociclib and
ribociclib. In the PALOMA-2 trial, neutropenia was identified as
the most prevalent grade 3/4 AE associated with palbociclib in
combination with letrozole for the treatment of advanced BC
(59). The MONALEESA-2 study
revealed a substantial increase in neutropenia in the treatment of
HR+/HER− advanced BC with ribociclib in
combination with letrozole, with grade 3/4 AEs most frequently
characterized by neutropenia (65). The most prevalent AE associated
with the administration of abemaciclib was diarrhea, as documented
in the MONARCH 2 trial. In this trial, abemaciclib was administered
in combination with fulvestrant to patients diagnosed with
HR+/HER− MBC (68). Additionally, QTc prolongation and
hepatobiliary toxicity were identified as significant AEs
associated with ribociclib, as reported in the MONALEESA-2 trial
(221) of ribociclib in
combination with letrozole for the treatment of patients with
HR+/HER2− MBC, and in the MONALEESA-7 trial
(219), respectively.
Ribociclib in combination with endocrine therapy has been evaluated
in premenopausal women with HR+ advanced BC. Although
the rate of diarrhea was reported to be high among patients
receiving abemaciclib, it was usually managed with antidiarrheal
medications or dose reductions without discontinuation (222). Abemaciclib has also been
reported to be associated with a higher incidence of venous
thromboembolism events and a reversible increase in serum
creatinine levels (68,71). Hematological AEs are common with
CDK4/6 inhibitors, especially in response to combination therapy.
CDK6 is particularly important in promoting the proliferation of
hematologic precursors, and its inhibition results in decreased
production of hematopoietic cells (59,223). Unlike chemotherapy-induced
neutropenia, hematological AEs, such as neutropenia, caused by
CDK4/6 inhibitors can be rapidly reversed, reflecting cytostatic
effects on bone marrow neutrophil precursors (224). Most hematological abnormalities
caused by CDK4/6 inhibitors can be adequately managed with standard
supportive care and dose modifications (68). Currently, clinical management
strategies for the treatment of potential toxicities associated
with CDK4/6 inhibitors have been suggested. However, more research
is needed to better understand the toxicity profile, and to develop
management strategies to minimize drug interruptions and thus
optimize the highest treatment effect for patients with BC
(223).
CDK4/6 inhibitors are cell cycle-targeting drugs
designed to prevent tumor cell proliferation while restoring normal
cell cycle regulation (225).
Guidelines have recommended several CDK4/6 inhibitors as first- or
second-line drugs for various tumors. By exploring the clinical
application of the aforementioned CDK4/6 inhibitors, it may be
concluded that CDK4/6 inhibitors have notable potential in tumor
immunotherapy. This is particularly evident when combined with
endocrine therapy, chemotherapy, ICIs, targeted therapy, RT and CAR
T-cell therapy, as well as other drug combinations, where the
combination of related treatments can notably enhance therapeutic
efficacy.
However, CDK4/6 inhibitor resistance remains a
marked challenge in clinical practice, with several potential
resistance mechanisms (138).
To address this critical challenge, future research should assess
the full scope of the resistance mechanisms mediated by CDK4/6
inhibitors and the potential mechanisms of their synergistic
effects with various other therapies (such as endocrine therapy).
In addition, treatment options should be sought based on current
advances and the resistance mechanisms of CDK4/6 inhibitors in
combination with endocrine therapy should be determined through
appropriate clinical validation.
Protein degraders can increase selectivity, improve
efficacy and overcome drug resistance; they may also provide a
solution to the problem of CDK4/6 inhibitor resistance, which
limits the use of these inhibitors (226). The degradation products of CDK
can induce the ubiquitination and proteasomal degradation of CDK,
thereby reducing the expression levels and stability of CDKs and
achieving the purpose of CDK inhibition. Currently, the results of
preclinical studies have shown that in
ER+/HER2− BC models, compared with clinically
approved CDK4/6 inhibitors, CDK4/6 degraders exhibit excellent
single-agent antitumor activity both in vitro and in
vivo (227). However,
clinical trials using CDK4/6 degraders to overcome CDK4/6 inhibitor
resistance are at an early stage. ARV-471 is a selective, orally
available proteolysis-targeting chimera protein degrader targeting
wild-type and mutant ER. In the phase I/II VERITAC trial
(NCT04072952), ARV-471 demonstrated clinical activity in patients
with advanced HR+/HER2− BC who had received
prior treatment with a CDK4/6 inhibitor (228). In addition, the phase III
VERITAC-2 trial (NCT05654623) is ongoing to compare the efficacy of
ARV-471 with fulvestrant in patients who have failed treatment with
a CDK4/6 inhibitor (229).
CDK4/6 degraders show potential clinical value in overcoming drug
resistance, but their clinical application still requires more
research and trials to verify their safety and efficacy.
The development and testing of novel CDK4/6
inhibitors are also key research directions in the future. Given
that the activation of CDK2 represents a common mechanism of
resistance to CDK4/6 inhibitors, the development of compounds that
inhibit CDK4/6 and CDK2 may prevent or delay the occurrence of
resistance (13). Furthermore, a
novel CDK4/6 inhibitor compound with lower alkalinity may evade
lysosome isolation and be effective against drug-resistant cancer
types, such as TNBC (230).
Novel CDK inhibitor compounds from different synthetic frameworks
are currently under investigation, examples of such derivatives
include amidoyl, pyrazole, quinazoline and pyrimidine derivatives
(231). In the forthcoming
years, we may observe the advent of novel CDK4/6 inhibitors that
have exhibited substantial promise in preclinical studies and
clinical trials.
In addition, CDK4/6 inhibitors possess inherent
limitations in clinical practice. The most notable challenge
confronting these novel targeted therapies is the absence of
predictive molecular biomarkers. The clinical efficacy of CDK4/6
inhibitors in treatment is contingent upon developing predictive
biomarkers and biologically reasonable combination therapies
(232). In actual clinical
settings, the AEs caused by CDK4/6 inhibitors are also worth
consideration. Although there are currently clinical management
recommendations regarding the potential toxicity of CDK4/6
inhibitors, further research is still needed to understand their
toxicological characteristics and to continuously optimize
management strategies comprehensively. This will contribute to
reducing the occurrence of treatment interruptions and improving
the treatment efficacy and compliance of patients with BC (223).
The present study reviewed the regulatory role of
CDK4/6 inhibitors in tumor immunity and the potential value of
tumor immunotherapy. CDK4/6 inhibitors affect tumors by regulating
the cell cycle, changing the immune microenvironment and regulating
immune checkpoints, and they have garnered attention as clinical
drugs for the treatment of various types of cancer. Du et al
(233) thoroughly explored
their clinical application in malignant solid tumors, alongside
strategies for combining these inhibitors with endocrine or
targeted therapies. Sun et al (234) delved into the immunomodulatory
effects of targeting CDK4/6 in cancer, whereas Glaviano et
al (138) offered an
in-depth analysis of resistance mechanisms associated with CDK4/6
inhibitors. By contrast, the present review provides a more
systematic account of recent advances in CDK4/6 inhibitors in tumor
immunity and resistance mechanisms, with a comprehensive review of
recent advances in clinical trials. This integrated perspective may
reflect current research dynamics more comprehensively than the
existing literature, providing researchers with a unified frame of
reference.
In response to the problem of patient resistance to
CDK4/6 inhibitors, the current review further proposed a variety of
combination treatment strategies to enhance drug sensitivity and
improve efficacy. This direction has been less explored in the
existing literature and clinical trials. Although CDK4/6 inhibitors
show promising applications, the review concludes with an objective
analysis of current challenges and a clear direction for future
development, such as identifying predictive molecular biomarkers
and addressing safety issues, which need to be validated in more
clinical trials.
In summary, the present study systematically
reviewed the latest progress of CDK4/6 inhibitors in the field of
tumor immunity, proposed an innovative combination therapy strategy
and provided a clear direction for future research, showing its
unique contribution. These innovations not only provide novel ideas
for overcoming drug resistance and improving the effects of tumor
immunotherapy, but also provide suggestions for future follow-up
research and verification.
Not applicable.
JX and YW drafted and revised the manuscript. FH,
QZ, YC, SG, YX and RS collected the relevant papers and helped to
revise the manuscript. FH, QZ and YC designed the tables and
charts. JX and YW reviewed the article. Data authentication is not
applicable. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no known
competing interests.
Not applicable.
This research was supported by the National Natural Science
Foundation of China (grant no. 82103328), the Scientific Research
Project of Jiangsu Maternal and Child Health Association (grant no.
FYX202348 and the Jiangsu Graduate Research Innovation Program
(grant no. KYCX18_1453).
|
1
|
Xu H, Yu S, Liu Q, Yuan X, Mani S, Pestell
RG and Wu K: Recent advances of highly selective CDK4/6 inhibitors
in breast cancer. J Hematol Oncol. 10:972017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Braal CL, Jongbloed EM, Wilting SM,
Mathijssen RHJ, Koolen SLW and Jager A: Inhibiting CDK4/6 in breast
cancer with palbociclib, ribociclib, and abemaciclib: Similarities
and differences. Drugs. 81:317–331. 2021. View Article : Google Scholar :
|
|
3
|
CDK4/6 inhibitors induce antitumor
immunity. Cancer Discov. 7:10522017. View Article : Google Scholar
|
|
4
|
Goel S, DeCristo MJ, Watt AC, BrinJones H,
Sceneay J, Li BB, Khan N, Ubellacker JM, Xie S, Metzger-Filho O, et
al: CDK4/6 inhibition triggers anti-tumor immunity. Nature.
548:471–475. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kong T, Xue Y, Cencic R, Zhu X, Monast A,
Fu Z, Pilon V, Sangwan V, Guiot MC, Foulkes WD, et al: eIF4A
inhibitors suppress cell-cycle feedback response and acquired
resistance to CDK4/6 inhibition in cancer. Mol Cancer Ther.
18:2158–2170. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Finn RS, Crown JP, Lang I, Boer K,
Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, et
al: The cyclin-dependent kinase 4/6 inhibitor palbociclib in
combination with letrozole versus letrozole alone as first-line
treatment of oestrogen receptor-positive, HER2-negative, advanced
breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study.
Lancet Oncol. 16:25–35. 2015. View Article : Google Scholar
|
|
7
|
Lei ZN, Tian Q, Teng QX, Wurpel JND, Zeng
L, Pan Y and Chen ZS: Understanding and targeting resistance
mechanisms in cancer. MedComm (2020). 4:e2652023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Verma S, Bartlett CH, Schnell P, DeMichele
AM, Loi S, Ro J, Colleoni M, Iwata H, Harbeck N, Cristofanilli M,
et al: Palbociclib in combination with fulvestrant in women with
hormone receptor-Positive/HER2-Negative advanced metastatic breast
cancer: Detailed safety analysis from a multicenter, randomized,
Placebo-controlled, phase III study (PALOMA-3). Oncologist.
21:1165–1175. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang Y, Luo J, Chen X, Yang Z, Mei X, Ma
J, Zhang Z, Guo X and Yu X: CDK4/6 inhibitors: A novel strategy for
tumor radiosensitization. J Exp Clin Cancer Res. 39:1882020.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sobhani N, D'Angelo A, Pittacolo M,
Roviello G, Miccoli A, Corona SP, Bernocchi O, Generali D and Otto
T: Updates on the CDK4/6 inhibitory strategy and combinations in
breast cancer. Cells. 8:3212019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Abraham J, Coleman R, Elias A, Holmes FA,
Kalinsky K, Kittaneh M, Lower E, Mahtani R, Terry Mamounas E,
Pegram M, et al: Use of cyclin-dependent kinase (CDK) 4/6
inhibitors for hormone receptor-positive, human epidermal growth
factor receptor 2-negative, metastatic breast cancer: A roundtable
discussion by The Breast Cancer Therapy Expert Group (BCTEG).
Breast Cancer Res Treat. 171:11–20. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Johnson J, Thijssen B, McDermott U,
Garnett M, Wessels LFA and Bernards R: Targeting the RB-E2F pathway
in breast cancer. Oncogene. 35:4829–4835. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fassl A, Geng Y and Sicinski P: CDK4 and
CDK6 kinases: From basic science to cancer therapy. Science.
375:eabc14952022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hallett ST, Pastok MW, Morgan RML, Wittner
A, Blundell KLIM, Felletar I, Wedge SR, Prodromou C, Noble MEM,
Pearl LH and Endicott JA: Differential regulation of G1 CDK
complexes by the Hsp90-Cdc37 chaperone system. Cell Rep.
21:1386–1398. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kwapisz D: Cyclin-dependent kinase 4/6
inhibitors in breast cancer: Palbociclib, ribociclib, and
abemaciclib. Breast Cancer Res Treat. 166:41–54. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guo L, Qi J, Wang H, Jiang X and Liu Y:
Getting under the skin: The role of CDK4/6 in melanomas. Eur J Med
Chem. 204:1125312020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pan Q, Luo P, Hu K, Qiu Y, Liu G, Dai S,
Cui B, Yin D and Shi C: Periodic changes of cyclin D1 mRNA
stability are regulated by PC4 modifications in the cell cycle. J
Cell Biol. 223:e2023080662024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
O'Leary B, Finn RS and Turner NC: Treating
cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol.
13:417–430. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cai Z, Shi Q, Li Y, Jin L, Li S, Wong LL,
Wang J, Jiang X, Zhu M, Lin J, et al: LncRNA EILA promotes CDK4/6
inhibitor resistance in breast cancer by stabilizing cyclin E1
protein. Sci Adv. 9:eadi38212023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Beleut M, Rajaram RD, Caikovski M, Ayyanan
A, Germano D, Choi Y, Schneider P and Brisken C: Two distinct
mechanisms underlie progesterone-induced proliferation in the
mammary gland. Proc Natl Acad Sci USA. 107:2989–2994. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Błaszczak-Świątkiewicz K: New selective
progesterone receptor modulators and their impact on the RANK/RANKL
complex activity. Molecules. 25:13212020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mauro L, Pellegrino M, Giordano F, Ricchio
E, Rizza P, De Amicis F, Catalano S, Bonofiglio D, Panno ML and
Andò S: Estrogen receptor-α drives adiponectin effects on cyclin D1
expression in breast cancer cells. FASEB J. 29:2150–2160. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang C, Chen L, Li C, Lynch MC, Brisken C
and Schmidt EV: Cyclin D1 enhances the response to estrogen and
progesterone by regulating progesterone receptor expression. Mol
Cell Biol. 30:3111–3125. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Herrera-Abreu MT, Palafox M, Asghar U,
Rivas MA, Cutts RJ, Garcia-Murillas I, Pearson A, Guzman M,
Rodriguez O, Grueso J, et al: Early adaptation and acquired
resistance to CDK4/6 inhibition in estrogen receptor-positive
breast cancer. Cancer Res. 76:2301–2313. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang X, You L, Nepovimova E, Psotka M,
Malinak D, Valko M, Sivak L, Korabecny J, Heger Z, Adam V, et al:
Inhibitors of phosphoinositide 3-kinase (PI3K) and phosphoinositide
3-kinase-related protein kinase family (PIKK). J Enzyme Inhib Med
Chem. 38:22372092023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Presti D and Quaquarini E: The
PI3K/AKT/mTOR and CDK4/6 pathways in endocrine resistant
HR+/HER2-metastatic breast cancer: Biological mechanisms and new
treatments. Cancers (Basel). 11:12422019. View Article : Google Scholar
|
|
27
|
Hao C, Wei Y, Meng W, Zhang J and Yang X:
PI3K/AKT/mTOR inhibitors for hormone receptor-positive advanced
breast cancer. Cancer Treat Rev. 132:1028612025. View Article : Google Scholar
|
|
28
|
Liu Q, Liu Y, Li X, Wang D, Zhang A, Pang
J, He J, Chen X and Tang NJ: Perfluoroalkyl substances promote
breast cancer progression via ERα and GPER mediated PI3K/Akt and
MAPK/Erk signaling pathways. Ecotoxicol Environ Saf.
258:1149802023. View Article : Google Scholar
|
|
29
|
Luboff AJ and DeRemer DL: Capivasertib: A
novel AKT inhibitor approved for hormone-receptor-positive,
HER-2-negative metastatic breast cancer. Ann Pharmacother.
58:1229–1237. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Michaloglou C, Crafter C, Siersbaek R,
Delpuech O, Curwen JO, Carnevalli LS, Staniszewska AD, Polanska UM,
Cheraghchi-Bashi A, Lawson M, et al: Combined inhibition of mTOR
and CDK4/6 is required for optimal blockade of E2F function and
long-term growth inhibition in estrogen receptor-positive breast
cancer. Mol Cancer Ther. 17:908–920. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Teh JLF and Aplin AE: Arrested
developments: CDK4/6 inhibitor resistance and alterations in the
tumor immune microenvironment. Clin Cancer Res. 25:921–927. 2019.
View Article : Google Scholar :
|
|
32
|
Deng J, Wang ES, Jenkins RW, Li S, Dries
R, Yates K, Chhabra S, Huang W, Liu H, Aref AR, et al: CDK4/6
inhibition augments antitumor immunity by enhancing T-cell
activation. Cancer Discov. 8:216–233. 2018. View Article : Google Scholar
|
|
33
|
Mognol GP, Carneiro FRG, Robbs BK, Faget
DV and Viola JPB: Cell cycle and apoptosis regulation by NFAT
transcription factors: New roles for an old player. Cell Death Dis.
7:e21992016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu J, Cheng M, Xu J, Liang Y, Yin B and
Liang J: Effect of CDK4/6 inhibitors on tumor immune
microenvironment. Immunol Invest. 53:437–449. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Scirocchi F, Scagnoli S, Botticelli A, Di
Filippo A, Napoletano C, Zizzari IG, Strigari L, Tomao S, Cortesi
E, Rughetti A, et al: Immune effects of CDK4/6 inhibitors in
patients with HR+/HER2-metastatic breast cancer: Relief from
immunosuppression is associated with clinical response.
EBioMedicine. 79:1040102022. View Article : Google Scholar
|
|
36
|
Kent LN and Leone G: The broken cycle: E2F
dysfunction in cancer. Nat Rev Cancer. 19:326–338. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kohlmeyer JL, Lingo JJ, Kaemmer CA,
Scherer A, Warrier A, Voigt E, Raygoza Garay JA, McGivney GR,
Brockman QR, Tang A, et al: CDK4/6-MEK inhibition in MPNSTs causes
plasma cell infiltration, sensitization to PD-L1 blockade, and
tumor regression. Clin Cancer Res. 29:3484–3497. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ruscetti M, Leibold J, Bott MJ, Fennell M,
Kulick A, Salgado NR, Chen CC, Ho YJ, Sanchez-Rivera FJ, Feucht J,
et al: NK cell-mediated cytotoxicity contributes to tumor control
by a cytostatic drug combination. Science. 362:1416–1422. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Llanos S, Megias D, Blanco-Aparicio C,
Hernández-Encinas E, Rovira M, Pietrocola F and Serrano M:
Lysosomal trapping of palbociclib and its functional implications.
Oncogene. 38:3886–3902. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
De Buck M, Gouwy M, Wang JM, Van Snick J,
Opdenakker G, Struyf S and Van Damme J: Structure and expression of
different serum amyloid A (SAA) variants and their
concentration-dependent functions during host insults. Curr Med
Chem. 23:1725–1755. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sagiv A, Biran A, Yon M, Simon J, Lowe SW
and Krizhanovsky V: Granule exocytosis mediates immune surveillance
of senescent cells. Oncogene. 32:1971–1977. 2013. View Article : Google Scholar :
|
|
42
|
Ritschka B, Storer M, Mas A, Heinzmann F,
Ortells MC, Morton JP, Sansom OJ, Zender L and Keyes WM: The
senescence-associated secretory phenotype induces cellular
plasticity and tissue regeneration. Genes Dev. 31:172–183. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Muñoz-Espín D and Serrano M: Cellular
senescence: From physiology to pathology. Nat Rev Mol Cell Biol.
15:482–496. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Favaretto G, Rossi MN, Cuollo L,
Laffranchi M, Cervelli M, Soriani A, Sozzani S, Santoni A and
Antonangeli F: Neutrophil-activating secretome characterizes
palbociclib-induced senescence of breast cancer cells. Cancer
Immunol Immunother. 73:1132024. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mehta AK, Cheney EM, Hartl CA, Pantelidou
C, Oliwa M, Castrillon JA, Lin JR, Hurst KE, de Oliveira Taveira M,
Johnson NT, et al: Targeting immunosuppressive macrophages
overcomes PARP inhibitor resistance in BRCA1-associated
triple-negative breast cancer. Nat Cancer. 2:66–82. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gu M, Liu Y, Zheng W, Jing Z, Li X, Guo W,
Zhao Z, Yang X, Liu Z, Zhu X and Gao W: Combined targeting of
senescent cells and senescent macrophages: A new idea for
integrated treatment of lung cancer. Semin Cancer Biol.
106-107:43–57. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fang Y, Zhang Z, Liu Y, Gao T, Liang S,
Chu Q, Guan L, Mu W, Fu S, Yang H, et al: Artificial assembled
macrophage Co-deliver black phosphorus quantum dot and CDK4/6
inhibitor for colorectal cancer triple-therapy. ACS Appl Mater
Interfaces. 14:20628–20640. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kumar A, Ramani V, Bharti V, de Lima
Bellan D, Saleh N, Uzhachenko R, Shen C, Arteaga C, Richmond A,
Reddy SM and Vilgelm A: Dendritic cell therapy augments antitumor
immunity triggered by CDK4/6 inhibition and immune checkpoint
blockade by unleashing systemic CD4 T-cell responses. J Immunother
Cancer. 11:e0060192023. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang J, Bu X, Wang H, Zhu Y, Geng Y,
Nihira NT, Tan Y, Ci Y, Wu F, Dai X, et al: Cyclin D-CDK4 kinase
destabilizes PD-L1 via cullin 3-SPOP to control cancer immune
surveillance. Nature. 553:91–95. 2018. View Article : Google Scholar
|
|
50
|
Filippone A, Lanza M, Mannino D, Raciti G,
Colarossi C, Sciacca D, Cuzzocrea S and Paterniti I: PD1/PD-L1
immune checkpoint as a potential target for preventing brain tumor
progression. Cancer Immunol Immunother. 71:2067–2075. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hossain MA, Liu G, Dai B, Si Y, Yang Q,
Wazir J, Birnbaumer L and Yang Y: Reinvigorating exhausted
CD8+ cytotoxic T lymphocytes in the tumor
microenvironment and current strategies in cancer immunotherapy.
Med Res Rev. 41:156–201. 2021. View Article : Google Scholar
|
|
52
|
Szeto C, Lobos CA, Nguyen AT and Gras S:
TCR recognition of peptide-MHC-I: Rule makers and breakers. Int J
Mol Sci. 22:682020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wu X, Gu Z, Chen Y, Chen B, Chen W, Weng L
and Liu X: Application of PD-1 blockade in cancer immunotherapy.
Comput Struct Biotechnol J. 17:661–674. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wu M, Huang Q, Xie Y, Wu X, Ma H, Zhang Y
and Xia Y: Improvement of the anticancer efficacy of PD-1/PD-L1
blockade via combination therapy and PD-L1 regulation. J Hematol
Oncol. 15:242022. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Seto T, Sam D and Pan M: Mechanisms of
primary and secondary resistance to immune checkpoint inhibitors in
cancer. Med Sci (Basel). 7:142019.PubMed/NCBI
|
|
56
|
Schaer DA, Beckmann RP, Dempsey JA, Huber
L, Forest A, Amaladas N, Li Y, Wang YC, Rasmussen ER, Chin D, et
al: The CDK4/6 inhibitor abemaciclib induces a T cell inflamed
tumor microenvironment and enhances the efficacy of PD-L1
checkpoint blockade. Cell Rep. 22:2978–2994. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Shrestha M, Wang DY, Ben-David Y and
Zacksenhaus E: CDK4/6 inhibitors and the pRB-E2F1 axis suppress PVR
and PD-L1 expression in triple-negative breast cancer. Oncogenesis.
12:292023. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Beaver JA, Amiri-Kordestani L, Charlab R,
Chen W, Palmby T, Tilley A, Zirkelbach JF, Yu J, Liu Q, Zhao L, et
al: FDA Approval: Palbociclib for the treatment of postmenopausal
patients with estrogen Receptor-positive, HER2-Negative metastatic
breast cancer. Clin Cancer Res. 21:4760–4766. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Finn RS, Martin M, Rugo HS, Jones S, Im
SA, Gelmon K, Harbeck N, Lipatov ON, Walshe JM, Moulder S, et al:
Palbociclib and letrozole in advanced breast cancer. N Engl J Med.
375:1925–1936. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cristofanilli M, Turner NC, Bondarenko I,
Ro J, Im SA, Masuda N, Colleoni M, DeMichele A, Loi S, Verma S, et
al: Fulvestrant plus palbociclib versus fulvestrant plus placebo
for treatment of hormone-receptor-positive, HER2-negative
metastatic breast cancer that progressed on previous endocrine
therapy (PALOMA-3): Final analysis of the multicentre,
double-blind, phase 3 randomised controlled trial. Lancet Oncol.
17:425–439. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wedam S, Fashoyin-Aje L, Bloomquist E,
Tang S, Sridhara R, Goldberg KB, Theoret MR, Amiri-Kordestani L,
Pazdur R and Beaver JA: FDA approval summary: Palbociclib for male
patients with metastatic breast cancer. Clin Cancer Res.
26:1208–1212. 2020. View Article : Google Scholar
|
|
62
|
Finn RS, Dering J, Conklin D, Kalous O,
Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, et al: PD
0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially
inhibits proliferation of luminal estrogen receptor-positive human
breast cancer cell lines in vitro. Breast Cancer Res. 11:R772009.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Slamon DJ, Neven P, Chia S, Fasching PA,
De Laurentiis M, Im SA, Petrakova K, Bianchi GV, Esteva FJ, Martín
M, et al: Overall survival with ribociclib plus fulvestrant in
advanced breast cancer. N Engl J Med. 382:514–524. 2020. View Article : Google Scholar
|
|
64
|
Im SA, Lu YS, Bardia A, Harbeck N,
Colleoni M, Franke F, Chow L, Sohn J, Lee KS, Campos-Gomez S, et
al: Overall survival with ribociclib plus endocrine therapy in
breast cancer. N Engl J Med. 381:307–316. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hortobagyi GN, Stemmer SM, Burris HA, Yap
YS, Sonke GS, Paluch-Shimon S, Campone M, Blackwell KL, André F,
Winer EP, et al: Ribociclib as First-line therapy for HR-positive,
advanced breast cancer. N Engl J Med. 375:1738–1748. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Research C for DE and: FDA expands
ribociclib indication in HR-positive, HER2-negative advanced or
metastatic breast cancer. FDA. 2024.
|
|
67
|
Hortobagyi GN, Lacko A, Sohn J, Cruz F,
Ruiz Borrego M, Manikhas A, Hee Park Y, Stroyakovskiy D, Yardley
DA, Huang CS, et al: A phase III trial of adjuvant ribociclib plus
endocrine therapy versus endocrine therapy alone in patients with
HR-positive/HER2-negative early breast cancer: Final invasive
disease-free survival results from the NATALEE trial. Ann Oncol.
36:149–157. 2025. View Article : Google Scholar
|
|
68
|
Sledge GW, Toi M, Neven P, Sohn J, Inoue
K, Pivot X, Burdaeva O, Okera M, Masuda N, Kaufman PA, et al:
MONARCH 2: Abemaciclib in combination with fulvestrant in women
With HR+/HER2-Advanced breast cancer who had progressed while
receiving endocrine therapy. J Clin Oncol. 35:2875–2884. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Goetz MP, Toi M, Campone M, Sohn J,
Paluch-Shimon S, Huober J, Park IH, Trédan O, Chen SC, Manso L, et
al: MONARCH 3: Abemaciclib as initial therapy for advanced breast
cancer. J Clin Oncol. 35:3638–3646. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Dickler MN, Tolaney SM, Rugo HS, Cortés J,
Diéras V, Patt D, Wildiers H, Hudis CA, O'Shaughnessy J, Zamora E,
et al: MONARCH 1, A Phase II study of abemaciclib, a CDK4 and CDK6
inhibitor, as a single agent, in patients with refractory
HR+/HER2-Metastatic breast cancer. Clin Cancer Res. 23:5218–5224.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Johnston SRD, Harbeck N, Hegg R, Toi M,
Martin M, Shao ZM, Zhang QY, Martinez Rodriguez JL, Campone M,
Hamilton E, et al: Abemaciclib combined with endocrine therapy for
the adjuvant treatment of HR+, HER2-, Node-positive, High-risk,
early breast cancer (monarchE). J Clin Oncol. 38:3987–3998. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Harbeck N, Rastogi P, Martin M, Tolaney
SM, Shao ZM, Fasching PA, Huang CS, Jaliffe GG, Tryakin A, Goetz
MP, et al: Adjuvant abemaciclib combined with endocrine therapy for
high-risk early breast cancer: Updated efficacy and Ki-67 analysis
from the monarchE study. Ann Oncol. 32:1571–1581. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Royce M, Mulkey F, Osgood C, Bloomquist E
and Amiri-Kordestani L: US food and drug administration expanded
adjuvant indication of abemaciclib in High-risk early breast
cancer. J Clin Oncol. 41:3456–3457. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Sledge GW, Toi M, Neven P, Sohn J, Inoue
K, Pivot X, Burdaeva O, Okera M, Masuda N, Kaufman PA, et al: The
effect of abemaciclib plus fulvestrant on overall survival in
hormone Receptor-positive, ERBB2-Negative breast cancer that
progressed on endocrine Therapy-MONARCH 2: A randomized clinical
trial. JAMA Oncol. 6:116–124. 2020. View Article : Google Scholar
|
|
75
|
Turner NC, Slamon DJ, Ro J, Bondarenko I,
Im SA, Masuda N, Colleoni M, DeMichele A, Loi S, Verma S, et al:
Overall survival with palbociclib and fulvestrant in advanced
breast cancer. N Engl J Med. 379:1926–1936. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Desnoyers A, Nadler MB, Kumar V, Saleh R
and Amir E: Comparison of treatment-related adverse events of
different Cyclin-dependent kinase 4/6 inhibitors in metastatic
breast cancer: A network meta-analysis. Cancer Treat Rev.
90:1020862020. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Kappel C, Elliott MJ, Kumar V, Nadler MB,
Desnoyers A and Amir E: Comparative overall survival of CDK4/6
inhibitors in combination with endocrine therapy in advanced breast
cancer. Sci Rep. 14:31292024. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Slamon D, Lipatov O, Nowecki Z, McAndrew
N, Kukielka-Budny B, Stroyakovskiy D, Yardley DA, Huang CS,
Fasching PA, Crown J, et al: Ribociclib plus endocrine therapy in
early breast cancer. N Engl J Med. 390:1080–1091. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Johnston S, Martin M, Di Leo A, Im SA,
Awada A, Forrester T, Frenzel M, Hardebeck MC, Cox J, Barriga S, et
al: MONARCH 3 final PFS: A randomized study of abemaciclib as
initial therapy for advanced breast cancer. NPJ Breast Cancer.
5:52019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Carlino L, Christodoulou MS, Restelli V,
Caporuscio F, Foschi F, Semrau MS, Costanzi E, Tinivella A, Pinzi
L, Lo Presti L, et al: Structure-activity relationships of
hexahydrocyclopenta[c]quinoline derivatives as allosteric
inhibitors of CDK2 and EGFR. ChemMedChem. 13:2627–2634. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Bisi JE, Sorrentino JA, Jordan JL, Darr
DD, Roberts PJ, Tavares FX and Strum JC: Preclinical development of
G1T38: A novel, potent and selective inhibitor of cyclin dependent
kinases 4/6 for use as an oral antineoplastic in patients with
CDK4/6 sensitive tumors. Oncotarget. 8:42343–42358. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Profitós-Pelejà N, Ribeiro ML, Parra J,
Fernández-Serrano M, Marin-Escudero P, Makovski-Silverstein A,
Cosenza S, Esteller M and Roué G: Prolonged cell cycle arrest by
the CDK4/6 antagonist narazaciclib restores ibrutinib response in
preclinical models of BTKi-resistant mantle cell lymphoma.
Hematological Oncol. 41:553–554. 2023. View Article : Google Scholar
|
|
83
|
Freeman-Cook KD, Hoffman RL, Behenna DC,
Boras B, Carelli J, Diehl W, Ferre RA, He YA, Hui A, Huang B, et
al: Discovery of PF-06873600, a CDK2/4/6 inhibitor for the
treatment of cancer. J Med Chem. 64:9056–9077. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Liu C, Liu B, Xu C, Zhang P and Yu C:
ETH-155008, a novel selective dual inhibitor of FLT3 and CDK4/6 in
preclinical treatment of acute myeloid leukemia. Blood. 134:5141.
2019. View Article : Google Scholar
|
|
85
|
Gelbert LM, Cai S, Lin X, Sanchez-Martinez
C, Del Prado M, Lallena MJ, Torres R, Ajamie RT, Wishart GN, Flack
RS, et al: Preclinical characterization of the CDK4/6 inhibitor
LY2835219: In-vivo cell cycle-dependent/independent anti-tumor
activities alone/in combination with gemcitabine. Invest New Drugs.
32:825–837. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Goel S, Tan AR, Rugo HS, Aftimos P, Andrić
Z, Beelen A, Zhang J, Yi JS, Malik R and O'Shaughnessy J:
Trilaciclib prior to gemcitabine plus carboplatin for metastatic
triple-negative breast cancer: Phase III PRESERVE 2. Future Oncol.
18:3701–3711. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Lenz HJ, Liu T, Chen EY, Horváth Z,
Bondarenko I, Danielewicz I, Ghidini M, García-Alfonso P, Jones R,
Aapro M, et al: Trilaciclib prior to FOLFOXIRI/bevacizumab for
patients with untreated metastatic colorectal cancer: Phase 3
PRESERVE 1 trial. JNCI Cancer Spectr. 9:pkae1162025. View Article : Google Scholar :
|
|
88
|
Zhang J, Yang N, Ji D, Shen W, Li W, Han
R, Wang N, Tao H, Chapman SC, Sykes AK, et al: A Randomized phase i
study of abemaciclib in chinese patients with Advanced and/or
metastatic cancers. Target Oncol. 16:177–187. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
DeWire M, Fuller C, Hummel TR, Chow LML,
Salloum R, de Blank P, Pater L, Lawson S, Zhu X, Dexheimer P, et
al: A phase I/II study of ribociclib following radiation therapy in
children with newly diagnosed diffuse intrinsic pontine glioma
(DIPG). J Neurooncol. 149:511–522. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Hart LL, Ferrarotto R, Andric ZG, Beck JT,
Subramanian J, Radosavljevic DZ, Zaric B, Hanna WT, Aljumaily R,
Owonikoko TK, et al: Myelopreservation with trilaciclib in patients
receiving topotecan for small cell lung cancer: Results from a
randomized, double-blind, placebo-controlled phase II study. Adv
Ther. 38:350–365. 2021. View Article : Google Scholar :
|
|
91
|
Xu B, Li H, Zhang Q, Sun W, Yu Y, Li W,
Wang S, Liao N, Shen P, Liu Y, et al: Pharmacokinetics, safety,
activity, and biomarker analysis of palbociclib plus letrozole as
first-line treatment for ER+/HER2-advanced breast cancer in Chinese
women. Cancer Chemother Pharmacol. 88:131–141. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Miller TW, Traphagen NA, Li J, Lewis LD,
Lopes B, Asthagiri A, Loomba J, De Jong J, Schiff D, Patel SH, et
al: Tumor pharmacokinetics and pharmacodynamics of the CDK4/6
inhibitor ribociclib in patients with recurrent glioblastoma. J
Neurooncol. 144:563–572. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Wander SA, Cohen O, Gong X, Johnson GN,
Buendia-Buendia JE, Lloyd MR, Kim D, Luo F, Mao P, Helvie K, et al:
The genomic landscape of intrinsic and acquired resistance to
Cyclin-dependent kinase 4/6 inhibitors in patients with hormone
Receptor-positive metastatic breast cancer. Cancer Discov.
10:1174–1193. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Kim S, Leong A, Kim M and Yang HW: CDK4/6
initiates Rb inactivation and CDK2 activity coordinates cell-cycle
commitment and G1/S transition. Sci Rep. 12:168102022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Fisher RP: Getting to S: CDK functions and
targets on the path to cell-cycle commitment. F1000Res. 5:23742016.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kim S, Armand J, Safonov A, Zhang M, Soni
RK, Schwartz G, McGuinness JE, Hibshoosh H, Razavi P, Kim M, et al:
Sequential activation of E2F via Rb degradation and c-Myc drives
resistance to CDK4/6 inhibitors in breast cancer. Cell Rep.
42:1131982023. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
O'Leary B, Cutts RJ, Liu Y, Hrebien S,
Huang X, Fenwick K, André F, Loibl S, Loi S, Garcia-Murillas I, et
al: The genetic landscape and clonal evolution of breast cancer
resistance to palbociclib plus fulvestrant in the PALOMA-3 trial.
Cancer Discov. 8:1390–1403. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Sharma A, Comstock CES, Knudsen ES, Cao
KH, Hess-Wilson JK, Morey LM, Barrera J and Knudsen KE:
Retinoblastoma tumor suppressor status is a critical determinant of
therapeutic response in prostate cancer cells. Cancer Res.
67:6192–6203. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Malumbres M, Sotillo R, Santamaría D,
Galán J, Cerezo A, Ortega S, Dubus P and Barbacid M: Mammalian
cells cycle without the D-type cyclin-dependent kinases Cdk4 and
Cdk6. Cell. 118:493–504. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Pandey K, An HJ, Kim SK, Lee SA, Kim S,
Lim SM, Kim GM, Sohn J and Moon YW: Molecular mechanisms of
resistance to CDK4/6 inhibitors in breast cancer: A review. Int J
Cancer. 145:1179–1188. 2019. View Article : Google Scholar :
|
|
101
|
Goel S, Bergholz JS and Zhao JJ: Targeting
CDK4 and CDK6 in cancer. Nat Rev Cancer. 22:356–372. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Cen L, Carlson BL, Schroeder MA, Ostrem
JL, Kitange GJ, Mladek AC, Fink SR, Decker PA, Wu W, Kim JS, et al:
p16-Cdk4-Rb axis controls sensitivity to a cyclin-dependent kinase
inhibitor PD0332991 in glioblastoma xenograft cells. Neuro Oncol.
14:870–881. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Iwata S, Tatsumi Y, Yonemoto T, Araki A,
Itami M, Kamoda H, Tsukanishi T, Hagiwara Y, Kinoshita H, Ishii T,
et al: CDK4 overexpression is a predictive biomarker for resistance
to conventional chemotherapy in patients with osteosarcoma. Oncol
Rep. 46:1352021. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Schachter MM, Merrick KA, Larochelle S,
Hirschi A, Zhang C, Shokat KM, Rubin SM and Fisher RP: A Cdk7-Cdk4
T-loop phosphorylation cascade promotes G1 progression. Mol Cell.
50:250–260. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Coombes RC, Howell S, Lord SR, Kenny L,
Mansi J, Mitri Z, Palmieri C, Chap LI, Richards P, Gradishar W, et
al: Dose escalation and expansion cohorts in patients with advanced
breast cancer in a Phase I study of the CDK7-inhibitor
samuraciclib. Nat Commun. 14:44442023. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Yang C, Li Z, Bhatt T, Dickler M, Giri D,
Scaltriti M, Baselga J, Rosen N and Chandarlapaty S: Acquired CDK6
amplification promotes breast cancer resistance to CDK4/6
inhibitors and loss of ER signaling and dependence. Oncogene.
36:2255–2264. 2017. View Article : Google Scholar :
|
|
107
|
Schoninger SF and Blain SW: The ongoing
search for biomarkers of CDK4/6 inhibitor responsiveness in breast
cancer. Mol Cancer Ther. 19:3–12. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Li Z, Razavi P, Li Q, Toy W, Liu B, Ping
C, Hsieh W, Sanchez-Vega F, Brown DN, Da Cruz Paula AF, et al: Loss
of the FAT1 tumor suppressor promotes resistance to CDK4/6
inhibitors via the hippo pathway. Cancer Cell. 34:893–905.e8. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Cornell L, Wander SA, Visal T, Wagle N and
Shapiro GI: MicroRNA-mediated suppression of the TGF-β pathway
confers transmissible and reversible CDK4/6 inhibitor resistance.
Cell Rep. 26:2667–2680.e7. 2019. View Article : Google Scholar
|
|
110
|
Chu C, Geng Y, Zhou Y and Sicinski P:
Cyclin E in normal physiology and disease states. Trends Cell Biol.
31:732–746. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Watt AC and Goel S: Cellular mechanisms
underlying response and resistance to CDK4/6 inhibitors in the
treatment of hormone receptor-positive breast cancer. Breast Cancer
Res. 24:172022. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Chandarlapaty S and Razavi P: Cyclin E
mRNA: Assessing Cyclin-dependent kinase (CDK) activation state to
elucidate breast cancer resistance to CDK4/6 Inhibitors. J Clin
Oncol. 37:1148–1150. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Guarducci C, Bonechi M, Benelli M,
Biagioni C, Boccalini G, Romagnoli D, Verardo R, Schiff R, Osborne
CK, De Angelis C, et al: Cyclin E1 and Rb modulation as common
events at time of resistance to palbociclib in hormone
receptor-positive breast cancer. NPJ Breast Cancer. 4:382018.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Turner NC, Liu Y, Zhu Z, Loi S, Colleoni
M, Loibl S, DeMichele A, Harbeck N, André F, Bayar MA, et al:
Cyclin E1 expression and palbociclib efficacy in previously treated
hormone Receptor-positive metastatic breast cancer. J Clin Oncol.
37:1169–1178. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Al-Qasem AJ, Alves CL, Ehmsen S,
Tuttolomondo M, Terp MG, Johansen LE, Vever H, Hoeg LVA, Elias D,
Bak M and Ditzel HJ: Co-targeting CDK2 and CDK4/6 overcomes
resistance to aromatase and CDK4/6 inhibitors in ER+ breast cancer.
NPJ Precis Oncol. 6:682022. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Gong X, Du J, Parsons SH, Merzoug FF,
Webster Y, Iversen PW, Chio LC, Van Horn RD, Lin X, Blosser W, et
al: Aurora a kinase inhibition is synthetic lethal with loss of the
RB1 tumor suppressor gene. Cancer Discov. 9:248–263. 2019.
View Article : Google Scholar
|
|
117
|
Hsueh KW, Fu SL, Chang CB, Chang YL and
Lin CH: A novel Aurora-A-mediated phosphorylation of p53 inhibits
its interaction with MDM2. Biochim Biophys Acta. 1834:508–515.
2013. View Article : Google Scholar
|
|
118
|
Turner N and Grose R: Fibroblast growth
factor signalling: From development to cancer. Nat Rev Cancer.
10:116–129. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Formisano L, Lu Y, Servetto A, Hanker AB,
Jansen VM, Bauer JA, Sudhan DR, Guerrero-Zotano AL, Croessmann S,
Guo Y, et al: Aberrant FGFR signaling mediates resistance to CDK4/6
inhibitors in ER+ breast cancer. Nat Commun. 10:13732019.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Courjal F, Cuny M, Simony-Lafontaine J,
Louason G, Speiser P, Zeillinger R, Rodriguez C and Theillet C:
Mapping of DNA amplifications at 15 chromosomal localizations in
1875 breast tumors: Definition of phenotypic groups. Cancer Res.
57:4360–4367. 1997.PubMed/NCBI
|
|
121
|
Sharpe R, Pearson A, Herrera-Abreu MT,
Johnson D, Mackay A, Welti JC, Natrajan R, Reynolds AR, Reis-Filho
JS, Ashworth A and Turner NC: FGFR signaling promotes the growth of
triple-negative and basal-like breast cancer cell lines both in
vitro and in vivo. Clin Cancer Res. 17:5275–5286. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Mao P, Cohen O, Kowalski KJ, Kusiel JG,
Buendia-Buendia JE, Cuoco MS, Exman P, Wander SA, Waks AG, Nayar U,
et al: Acquired FGFR and FGF alterations confer resistance to
estrogen receptor (ER) targeted therapy in ER+ metastatic breast
cancer. Clin Cancer Res. 26:5974–5989. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Turner N, Pearson A, Sharpe R, Lambros M,
Geyer F, Lopez-Garcia MA, Natrajan R, Marchio C, Iorns E, Mackay A,
et al: FGFR1 amplification drives endocrine therapy resistance and
is a therapeutic target in breast cancer. Cancer Res. 70:2085–2094.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Ersahin T, Tuncbag N and Cetin-Atalay R:
The PI3K/AKT/mTOR interactive pathway. Mol Biosyst. 11:1946–1954.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Cai Z, Wang J, Li Y, Shi Q, Jin L, Li S,
Zhu M, Wang Q, Wong LL, Yang W, et al: Overexpressed Cyclin D1 and
CDK4 proteins are responsible for the resistance to CDK4/6
inhibitor in breast cancer that can be reversed by PI3K/mTOR
inhibitors. Sci China Life Sci. 66:94–109. 2023. View Article : Google Scholar
|
|
126
|
Hobbs GA, Wittinghofer A and Der CJ:
Selective targeting of the KRAS G12C mutant: Kicking KRAS when it's
down. Cancer Cell. 29:251–253. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Luangdilok S, Wanchaijiraboon P,
Chantranuwatana P, Teerapakpinyo C, Shuangshoti S and Sriuranpong
V: Cyclin D1 expression as a potential prognostic factor in
advanced KRAS-mutant non-small cell lung cancer. Transl Lung Cancer
Res. 8:959–966. 2019. View Article : Google Scholar
|
|
128
|
Tran KA, Cheng MY, Mitra A, Ogawa H, Shi
VY, Olney LP, Kloxin AM and Maverakis E: MEK inhibitors and their
potential in the treatment of advanced melanoma: The advantages of
combination therapy. Drug Des Devel Ther. 10:43–52. 2016.PubMed/NCBI
|
|
129
|
Finn RS, Aleshin A and Slamon DJ:
Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen
receptor-positive breast cancers. Breast Cancer Res. 18:172016.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Alves CL, Elias D, Lyng M, Bak M,
Kirkegaard T, Lykkesfeldt AE and Ditzel HJ: High CDK6 protects
cells from Fulvestrant-mediated apoptosis and is a predictor of
resistance to fulvestrant in estrogen Receptor-positive metastatic
breast cancer. Clin Cancer Res. 22:5514–5526. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Shaulian E and Karin M: AP-1 in cell
proliferation and survival. Oncogene. 20:2390–2400. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Shen Q, Uray IP, Li Y, Zhang Y, Hill J, Xu
XC, Young MR, Gunther EJ, Hilsenbeck SG, Colburn NH, et al:
Targeting the activator protein 1 transcription factor for the
prevention of estrogen receptor-negative mammary tumors. Cancer
Prev Res (Phila). 1:45–55. 2008. View Article : Google Scholar
|
|
133
|
Hydbring P, Castell A and Larsson LG: MYC
modulation around the CDK2/p27/SKP2 axis. Genes (Basel). 8:1742017.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Takahashi M, Tokunaga E, Mori J, Tanizawa
Y, van der Walt JS, Kawaguchi T, Goetz MP and Toi M: Japanese
subgroup analysis of the phase 3 MONARCH 3 study of abemaciclib as
initial therapy for patients with hormone receptor-positive, human
epidermal growth factor receptor 2-negative advanced breast cancer.
Breast Cancer. 29:174–184. 2022. View Article : Google Scholar
|
|
135
|
Hamilton E, Cortes J, Ozyilkan O, Chen SC,
Petrakova K, Manikhas A, Jerusalem G, Hegg R, Huober J, Zhang W, et
al: nextMONARCH Phase 2 randomized clinical trial: Overall survival
analysis of abemaciclib monotherapy or in combination with
tamoxifen in patients with endocrine-refractory HR +,
HER2-metastatic breast cancer. Breast Cancer Res Treat. 195:55–64.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Goetz MP, Hamilton EP, Campone M, Hurvitz
SA, Cortes J, Johnston S, Llombart-Cussac A, Kaufman PA, Toi M,
Jerusalem G, et al: Landscape of baseline and acquired genomic
alterations in circulating tumor DNA with abemaciclib alone or with
endocrine therapy in advanced breast cancer. Clin Cancer Res.
30:2233–2244. 2024. View Article : Google Scholar :
|
|
137
|
Pandey K, Park N, Park KS, Hur J, Cho YB,
Kang M, An HJ, Kim S, Hwang S and Moon YW: Combined CDK2 and CDK4/6
inhibition overcomes palbociclib resistance in breast cancer by
enhancing senescence. Cancers (Basel). 12:35662020. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Glaviano A, Wander SA, Baird RD, Yap KC,
Lam HY, Toi M, Carbone D, Geoerger B, Serra V, Jones RH, et al:
Mechanisms of sensitivity and resistance to CDK4/CDK6 inhibitors in
hormone receptor-positive breast cancer treatment. Drug Resist
Updat. 76:1011032024. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Mouron S, Manso L, Caleiras E,
Rodríguez-Peralto JL, Rueda OM, Caldas C, Colomer R,
Quintela-Fandino M and Bueno MJ: FGFR1 amplification or
overexpression and hormonal resistance in luminal breast cancer:
Rationale for a triple blockade of ER, CDK4/6, and FGFR1. Breast
Cancer Res. 23:212021. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Wang N, Ma T and Yu B: Targeting
epigenetic regulators to overcome drug resistance in cancers.
Signal Transduct Target Ther. 8:692023. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Xue Y and Zhai J: Strategy of combining
CDK4/6 inhibitors with other therapies and mechanisms of
resistance. Int J Clin Exp Pathol. 17:189–207. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Manohar PM and Davidson NE: Updates in
endocrine therapy for metastatic breast cancer. Cancer Biol Med.
19:202–212. 2022.
|
|
143
|
Finn RS, Dieras V, Rugo HS, Joy AA,
Moulder SL, Walshe JM, Mukai H, Shparyk YV, Park IH, Mori A, et al:
Palbociclib (PAL) + letrozole (L) as first-line (1L) therapy (tx)
in estrogen receptor-positive (ER+)/human epidermal growth factor
receptor 2-negative (HER2-) advanced breast cancer (ABC): Efficacy
and safety across patient (pt) subgroups. J Clin Oncol.
35:10392017. View Article : Google Scholar
|
|
144
|
Kalinsky K, Accordino MK, Chiuzan C, Mundi
PS, Trivedi MS, Novik Y, Tiersten A, Raptis G, Baer LN, Oh SY, et
al: A randomized, phase II trial of fulvestrant or exemestane with
or without ribociclib after progression on anti-estrogen therapy
plus cyclin-dependent kinase 4/6 inhibition (CDK 4/6i) in patients
(pts) with unresectable or hormone receptor-positive (HR+),
HER2-negative metastatic breast cancer (MBC): MAINTAIN trial. J
Clin Oncol. 40:LBA10042022. View Article : Google Scholar
|
|
145
|
Bidard FC, Kaklamani VG, Neven P, Streich
G, Montero AJ, Forget F, Mouret-Reynier MA, Sohn JH, Taylor D,
Harnden KK, et al: Elacestrant (oral selective estrogen receptor
degrader) versus standard endocrine therapy for Estrogen
Receptor-positive, human epidermal growth factor receptor
2-Negative advanced breast cancer: Results from the randomized
phase III EMERALD Trial. J Clin Oncol. 40:3246–3256. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Oliveira M, Pominchuck D, Nowecki Z,
Hamilton E, Kulyaba Y, Andabekov T, Hotko Y, Melkadze T, Nemsadze
G, Neven P, et al: Abstract GS3-02: GS3-02 Camizestrant, a next
generation oral SERD vs fulvestrant in post-menopausal women with
advanced ER-positive HER2-negative breast cancer: Results of the
randomized, multi-dose Phase 2 SERENA-2 trial. Cancer Res.
83:GS3–02. 2023. View Article : Google Scholar
|
|
147
|
Research C for DE and: FDA approves
elacestrant for ER-positive, HER2-negative, ESR1-mutated advanced
or metastatic breast cancer. FDA. 2023.
|
|
148
|
Magge T, Rajendran S, Brufsky AM and Foldi
J: CDK4/6 inhibitors: The Devil is in the detail. Curr Oncol Rep.
26:665–678. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Sonke GS, Van Ommen-Nijhof A, Wortelboer
N, Noort VVD, Swinkels AC, Blommestein HM, Beeker A, Beelen K,
Hamming LC, Heijns JB, et al: Primary outcome analysis of the phase
3 SONIA trial (BOOG 2017-03) on selecting the optimal position of
cyclin-dependent kinases 4 and 6 (CDK4/6) inhibitors for patients
with hormone receptor-positive (HR+), HER2-negative (HER2-)
advanced breast cancer (ABC). J Clin Oncol. 41:LBA10002023.
View Article : Google Scholar
|
|
150
|
Sonke GS, van Ommen-Nijhof A, Wortelboer
N, van der Noort V, Swinkels ACP, Blommestein HM, Guerrero Paez C,
Mol L, Beeker A, Beelene K, et al: Early versus deferred use of
CDK4/6 inhibitors in advanced breast cancer. Nature. 636:474–480.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Ogata R, Kishino E, Saitoh W, Koike Y and
Kurebayashi J: Resistance to cyclin-dependent kinase (CDK) 4/6
inhibitors confers cross-resistance to other CDK inhibitors but not
to chemotherapeutic agents in breast cancer cells. Breast Cancer.
28:206–215. 2021. View Article : Google Scholar
|
|
152
|
Rugo HS, Bardia A, Marmé F, Cortés J,
Schmid P, Loirat D, Trédan O, Ciruelos E, Dalenc F, Gómez Pardo P,
et al: Overall survival with sacituzumab govitecan in hormone
receptor-positive and human epidermal growth factor receptor
2-negative metastatic breast cancer (TROPiCS-02): A randomised,
open-label, multicentre, phase 3 trial. Lancet. 402:1423–1433.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Cao YN, Zheng LL, Wang D, Liang XX, Gao F
and Zhou XL: Recent advances in microtubule-stabilizing agents. Eur
J Med Chem. 143:806–828. 2018. View Article : Google Scholar
|
|
154
|
Cretella D, Fumarola C, Bonelli M, Alfieri
R, La Monica S, Digiacomo G, Cavazzoni A, Galetti M, Generali D and
Petronini PG: Pre-treatment with the CDK4/6 inhibitor palbociclib
improves the efficacy of paclitaxel in TNBC cells. Sci Rep.
9:130142019. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Zhang XH, Cheng Y, Shin JY, Kim JO, Oh JE
and Kang JH: A CDK4/6 inhibitor enhances cytotoxicity of paclitaxel
in lung adenocarcinoma cells harboring mutant KRAS as well as
wild-type KRAS. Cancer Biol Ther. 14:597–605. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Kim JY, Jayne LA, Bai Y, Feng MJHH, Clark
MA, Chung S, W Christman J, Cianciolo RE and Pabla NS: Ribociclib
mitigates cisplatin-associated kidney injury through
retinoblastoma-1 dependent mechanisms. Biochem Pharmaco.
177:1139392020. View Article : Google Scholar
|
|
157
|
Liu M, Cui L, Li X, Xia C, Li Y, Wang R,
Ren F, Liu H and Chen J: PD-0332991 combined with cisplatin
inhibits nonsmall cell lung cancer and reversal of cisplatin
resistance. Thorac Cancer. 12:924–931. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Ma CX, Gao F, Luo J, Northfelt DW, Goetz
M, Forero A, Hoog J, Naughton M, Ademuyiwa F and Suresh R:
NeoPalAna: Neoadjuvant palbociclib, a Cyclin-dependent kinase 4/6
inhibitor, and anastrozole for clinical stage 2 or 3 estrogen
receptor-positive breast cancer. Clin Cancer Res. 23:4055–4065.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Chow LWC, Morita S, Chow CYC, Ng WK and
Toi M: Neoadjuvant palbociclib on ER+ breast cancer (N007):
Clinical response and EndoPredict's value. Endocr Relat Cancer.
25:123–130. 2018. View Article : Google Scholar
|
|
160
|
Prat A, Saura C, Pascual T, Hernando C,
Muñoz M, Paré L, González Farré B, Fernández PL, Galván P, Chic N,
et al: Ribociclib plus letrozole versus chemotherapy for
postmenopausal women with hormone receptor-positive, HER2-negative,
luminal B breast cancer (CORALLEEN): An open-label, multicentre,
randomised, phase 2 trial. Lancet Oncol. 21:33–43. 2020. View Article : Google Scholar
|
|
161
|
Delaloge S, Dureau S, D'Hondt V,
Desmoulins I, Heudel PE, Duhoux FP, Levy C, Lerebours F,
Mouret-Reynier MA, Dalenc F, et al: Survival outcomes after
neoadjuvant letrozole and palbociclib versus third generation
chemotherapy for patients with high-risk oestrogen
receptor-positive HER2-negative breast cancer. Eur J Cancer.
166:300–308. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Bonelli M, La Monica S, Fumarola C and
Alfieri R: Multiple effects of CDK4/6 inhibition in cancer: From
cell cycle arrest to immunomodulation. Biochem Pharmacol.
170:1136762019. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Zhang S, Xu Q, Sun W and Zhou J and Zhou
J: Immunomodulatory effects of CDK4/6 inhibitors. Biochim Biophys
Acta Rev Cancer. 1878:1889122023. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
De Angelis C, Fu X, Cataldo ML, Nardone A,
Pereira R, Veeraraghavan J, Nanda S, Qin L, Sethunath V, Wang T, et
al: Activation of the IFN signaling pathway is associated with
resistance to CDK4/6 inhibitors and immune checkpoint activation in
ER-Positive breast cancer. Clin Cancer Res. 27:4870–4882. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Wang Q, Guldner IH, Golomb SM, Sun L,
Harris JA, Lu X and Zhang S: Single-cell profiling guided
combinatorial immunotherapy for fast-evolving CDK4/6
inhibitor-resistant HER2-positive breast cancer. Nat Commun.
10:38172019. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Wu CC, Wang YA, Livingston JA, Zhang J and
Futreal PA: Prediction of biomarkers and therapeutic combinations
for anti-PD-1 immunotherapy using the global gene network
association. Nat Commun. 13:422022. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Jang HJ, Truong CY, Lo EM, Holmes HM,
Ramos D, Ramineni M, Lee JS, Wang DY, Pietropaolo M, Ripley RT, et
al: Inhibition of cyclin dependent kinase 4/6 overcomes primary
resistance to programmed cell death 1 blockade in malignant
mesothelioma. Ann Thorac Surg. 114:1842–1852. 2022. View Article : Google Scholar
|
|
168
|
Litchfield K, Reading JL, Puttick C,
Thakkar K, Abbosh C, Bentham R, Watkins TBK, Rosenthal R, Biswas D,
Rowan A, et al: Meta-analysis of tumor- and T cell-intrinsic
mechanisms of sensitization to checkpoint inhibition. Cell.
184:596–614.e14. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Jerby-Arnon L, Shah P, Cuoco MS, Rodman C,
Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin JR, et al: A
cancer cell program promotes T cell exclusion and resistance to
checkpoint blockade. Cell. 175:984–997.e24. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Rugo HS, Kabos P, Beck JT, Jerusalem G,
Wildiers H, Sevillano E, Paz-Ares L, Chisamore MJ, Chapman SC,
Hossain AM, et al: Abemaciclib in combination with pembrolizumab
for HR+, HER2-metastatic breast cancer: Phase 1b study. NPJ Breast
Cancer. 8:1182022. View Article : Google Scholar
|
|
171
|
Yuan Y, Lee JS, Yost SE, Frankel PH, Ruel
C, Egelston CA, Guo W, Padam S, Tang A, Martinez N, et al: Phase
I/II trial of palbociclib, pembrolizumab and letrozole in patients
with hormone receptor-positive metastatic breast cancer. Eur J
Cancer. 154:11–20. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Lee EK, Esselen KM, Kolin DL, Lee LJ,
Matulonis UA and Konstantinopoulos PA: Combined CDK4/6 and PD-1
inhibition in refractory SMARCA4-deficient Small-cell carcinoma of
the ovary, hypercalcemic type. JCO Precis Oncol. 4:736–742. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Xue Y, Meehan B, Fu Z, Wang XQD, Fiset PO,
Rieker R, Levins C, Kong T, Zhu X, Morin G, et al: SMARCA4 loss is
synthetic lethal with CDK4/6 inhibition in non-small cell lung
cancer. Nat Commun. 10:5572019. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Bose S, Lee T, Choi S, Fazlollahi L,
Rasiej MJ, Schwartz GK and Ingham M: CDK4/6 inhibition with
Anti-PD-1 checkpoint blockade induces major response in aggressive
classic kaposi sarcoma after previous progression on Anti-PD-1
alone. JCO Precis Oncol. 6:e21005502022. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Daniel D, Kuchava V, Bondarenko I,
Ivashchuk O, Reddy S, Jaal J, Kudaba I, Hart L, Matitashvili A,
Pritchett Y, et al: Trilaciclib prior to chemotherapy and
atezolizumab in patients with newly diagnosed extensive-stage small
cell lung cancer: A multicentre, randomised, double-blind,
placebo-controlled Phase II trial. Int J Cancer. 148:2557–2570.
2021. View Article : Google Scholar :
|
|
176
|
Mayer EL, Ren Y, Wagle N, Mahtani R, Ma C,
DeMichele A, Cristofanilli M, Meisel J, Miller KD, Jolly T, et al:
Abstract GS3-06: GS3-06 Palbociclib After CDK4/6i and endocrine
therapy (PACE): A randomized phase II study of fulvestrant,
palbociclib, and avelumab for endocrine Pre-treated
ER+/HER2-metastatic breast cancer. Cancer Res. 83:GS3–06. 2023.
View Article : Google Scholar
|
|
177
|
Jerusalem G, Prat A, Salgado R, Reinisch
M, Saura C, Ruiz-Borrego M, Nikolinakos P, Ades F, Filian J, Huang
N, et al: Neoadjuvant nivolumab + palbociclib + anastrozole for
oestrogen receptor-positive/human epidermal growth factor receptor
2-negative primary breast cancer: Results from CheckMate 7A8.
Breast. 72:1035802023. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Dennis MJ, Sacco AG, Qi Y, Bykowski J,
Pittman E, Chen R, Messer K, Cohen EEW and Gold KA: A phase I study
of avelumab, palbociclib, and cetuximab in patients with recurrent
or metastatic head and neck squamous cell carcinoma. Oral Oncol.
135:1062192022. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Pujol JL, Vansteenkiste J, Paz-Ares
Rodríguez L, Gregorc V, Mazieres J, Awad M, Jänne PA, Chisamore M,
Hossain AM, Chen Y and Beck JT: Abemaciclib in combination with
pembrolizumab for stage IV KRAS-mutant or squamous NSCLC: A phase
1b study. JTO Clin Res Rep. 2:1002342021.PubMed/NCBI
|
|
180
|
Patnaik A, Yap TA, Chung HC, de Miguel MJ,
Bang YJ, Lin CC, Su WC, Italiano A, Chow KH, Szpurka AM, et al:
Safety and clinical activity of a new Anti-PD-L1 antibody as
monotherapy or combined with targeted therapy in advanced solid
tumors: The PACT Phase Ia/Ib trial. Clin Cancer Res. 27:1267–1277.
2021. View Article : Google Scholar
|
|
181
|
Garrido-Castro AC and Goel S: CDK4/6
inhibition in breast cancer: Mechanisms of response and treatment
failure. Curr Breast Cancer Rep. 9:26–33. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Clark AS, Makhlin I and DeMichele A:
Setting the pick: Can PI3K inhibitors circumvent CDK4/6 inhibitor
resistance? Clin Cancer Res. 27:371–373. 2021. View Article : Google Scholar
|
|
183
|
Masuda J, Sakai H, Tsurutani J, Tanabe Y,
Masuda N, Iwasa T, Takahashi M, Futamura M, Matsumoto K, Aogi K, et
al: Efficacy safety, and biomarker analysis of nivolumab in
combination with abemaciclib plus endocrine therapy in patients
with HR-positive HER2-negative metastatic breast cancer: A phase II
study (WJOG11418B NEWFLAME trial). J Immunother Cancer.
11:e0071262023. View Article : Google Scholar
|
|
184
|
Bedard PL, Hyman DM, Davids MS and Siu LL:
Small molecules, big impact: 20 years of targeted therapy in
oncology. Lancet. 395:1078–1088. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Gómez Tejeda Zañudo J, Barroso-Sousa R,
Jain E, Jin Q, Li T, Buendia-Buendia JE, Pereslete A, Abravanel DL,
Ferreira AR, Wrabel E, et al: Exemestane plus everolimus and
palbociclib in metastatic breast cancer: Clinical response and
genomic/transcriptomic determinants of resistance in a phase I/II
trial. Nat Commun. 15:24462024. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Jhaveri KL, Accordino MK, Bedard PL,
Cervantes A, Gambardella V, Hamilton E, Italiano A, Kalinsky K,
Krop IE, Oliveira M, et al: Phase I/Ib trial of inavolisib plus
palbociclib and endocrine therapy for PIK3CA-Mutated, hormone
Receptor-positive, human epidermal growth factor Receptor
2-Negative advanced or metastatic breast cancer. J Clin Oncol.
42:3947–3956. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Yardley DA, Noguchi S, Pritchard KI,
Burris HA III, Baselga J, Gnant M, Hortobagyi GN, Campone M,
Pistilli B, Piccart M, et al: Everolimus plus exemestane in
postmenopausal patients with HR(+) breast cancer: BOLERO-2 final
progression-free survival analysis. Adv Ther. 30:870–884. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Pascual J, Lim JSJ, Macpherson IR,
Armstrong AC, Ring A, Okines AFC, Cutts RJ, Herrera-Abreu MT,
Garcia-Murillas I, Pearson A, et al: Triplet therapy with
palbociclib, taselisib, and fulvestrant in PIK3CA-mutant breast
cancer and doublet palbociclib and taselisib in Pathway-mutant
solid cancers. Cancer Discov. 11:92–107. 2021. View Article : Google Scholar
|
|
189
|
Wander SA, Juric D, Supko JG, Micalizzi
DS, Spring S, Vidula N, Beeler M, Habin KR, Viscosi E, Fitzgerald
DM, et al: Phase Ib trial to evaluate safety and anti-tumor
activity of the AKT inhibitor, ipatasertib, in combination with
endocrine therapy and a CDK4/6 inhibitor for patients with hormone
receptor positive (HR+)/HER2 negative metastatic breast cancer
(MBC) (TAKTIC). J Clin Oncol. 38:10662020. View Article : Google Scholar
|
|
190
|
Huang J, Zheng L, Sun Z and Li J: CDK4/6
inhibitor resistance mechanisms and treatment strategies (review).
Int J Mol Med. 50:1282022. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Piccart M, Hortobagyi GN, Campone M,
Pritchard KI, Lebrun F, Ito Y, Noguchi S, Perez A, Rugo HS, Deleu
I, et al: Everolimus plus exemestane for hormone-receptor-positive,
human epidermal growth factor receptor-2-negative advanced breast
cancer: Overall survival results from BOLERO-2†. Ann Oncol.
25:2357–2362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Lord CJ and Ashworth A: PARP inhibitors:
Synthetic lethality in the clinic. Science. 355:1152–1158. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Guney Eskiler G, Ozman Z, Haciefendi A and
Cansaran-Duman D: Novel combination treatment of CDK 4/6 inhibitors
with PARP inhibitors in triple negative breast cancer cells. Naunyn
Schmiedebergs Arch Pharmacol. 396:1031–1041. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Wu C, Peng S, Pilié PG, Geng C, Park S,
Manyam GC, Lu Y, Yang G, Tang Z, Kondraganti S, et al: PARP and
CDK4/6 inhibitor combination therapy induces apoptosis and
suppresses neuroendocrine differentiation in prostate cancer. Mol
Cancer Ther. 20:1680–1691. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Zhu X, Chen L, Huang B, Li X, Yang L, Hu
X, Jiang Y, Shao Z and Wang Z: Efficacy and mechanism of the
combination of PARP and CDK4/6 inhibitors in the treatment of
triple-negative breast cancer. J Exp Clin Cancer Res. 40:1222021.
View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Curtin NJ and Szabo C: Poly(ADP-ribose)
polymerase inhibition: Past, present and future. Nat Rev Drug
Discov. 19:711–736. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Jiang Z, Li W, Hu X, Zhang Q, Sun T, Cui
S, Wang S, Ouyang Q, Yin Y, Geng C, et al: Tucidinostat plus
exemestane for postmenopausal patients with advanced, hormone
receptor-positive breast cancer (ACE): A randomised, double-blind,
placebo-controlled, phase 3 trial. Lancet Oncol. 20:806–815. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Reeves C: San Antonio breast cancer
symposium 2021. Lancet Oncol. 23:e182022. View Article : Google Scholar
|
|
199
|
Lok SW, Whittle JR, Vaillant F, The CE, Lo
LL, Policheni AN, Bergin ART, Desai J, Ftouni S, Gandolfo LC, et
al: A phase Ib dose-escalation and expansion study of the BCL2
inhibitor venetoclax combined with tamoxifen in ER and
BCL2-positive metastatic breast cancer. Cancer Discov. 9:354–369.
2019. View Article : Google Scholar
|
|
200
|
Lindeman GJ, Fernando TM, Bowen R, Jerzak
KJ, Song X, Decker T, Boyle F, McCune S, Armstrong A, Shannon C, et
al: VERONICA: Randomized phase II study of fulvestrant and
venetoclax in ER-positive metastatic breast cancer Post-CDK4/6
inhibitors-efficacy, safety, and biomarker results. Clin Cancer
Res. 28:3256–3267. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Benvenuti C, Grinda T, Rassy E,
Dixon-Douglas J, Ribeiro JM, Zambelli A, Santoro A and Pistilli B:
Unveiling the potential of Cyclin-dependent kinases 4 and 6
inhibitors beyond progression in hormone receptor positive/human
epidermal growth factor negative advanced breast cancer-a clinical
review. Curr Treat Options Oncol. 25:1517–1537. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Kubeczko M, Jarząb M, Gabryś D, Krzywon A,
Cortez AJ and Xu AJ: Safety and feasibility of CDK4/6 inhibitors
treatment combined with radiotherapy in patients with
HR-positive/HER2-negative breast cancer. A systematic review and
meta-analysis. Radiother Oncol. 187:1098392023. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Lee CL, Oh P, Xu ES, Ma Y, Kim Y, Daniel
AR and Kirsch DG: Blocking Cyclin-dependent kinase 4/6 during
single dose versus fractionated radiation therapy leads to opposite
effects on acute gastrointestinal toxicity in mice. Int J Radiat
Oncol Biol Phys. 102:1569–1576. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Naz S, Sowers A, Choudhuri R, Wissler M,
Gamson J, Mathias A, Cook JA and Mitchell JB: Abemaciclib, a
selective CDK4/6 inhibitor, enhances the radiosensitivity of
Non-small cell lung cancer in vitro and in vivo. Clin Cancer Res.
24:3994–4005. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Hagen KR, Zeng X, Lee MY, Tucker Kahn S,
Harrison Pitner MK, Zaky SS, Liu Y, O'Regan RM, Deng X and Saavedra
HI: Silencing CDK4 radiosensitizes breast cancer cells by promoting
apoptosis. Cell Div. 8:102013. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Whittaker S, Madani D, Joshi S, Chung SA,
Johns T, Day B, Khasraw M and McDonald KL: Combination of
palbociclib and radiotherapy for glioblastoma. Cell Death Discov.
3:170332017. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Huang CY, Hsieh FS, Wang CY, Chen LJ,
Chang SS, Tsai MH, Hung MH, Kuo CW, Shih CT, Chao TI and Chen KF:
Palbociclib enhances radiosensitivity of hepatocellular carcinoma
and cholangiocarcinoma via inhibiting ataxia telangiectasia-mutated
kinase-mediated DNA damage response. Eur J Cancer. 102:10–22. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Slamon DJ, Stroyakovskiy D, Yardley DA,
Huang C, Fasching PA, Crown J, Bardia A, Chia S, Im S, Martin M, et
al: Ribociclib and endocrine therapy as adjuvant treatment in
patients with HR+/HER2-early breast cancer: Primary results from
the phase III NATALEE trial. J Clin Oncol. 41:LBA5002023.
View Article : Google Scholar
|
|
209
|
Becherini C, Visani L, Caini S,
Bhattacharya IS, Kirby AM, Nader Marta G, Morgan G, Salvestrini V,
Coles CE, Cortes J, et al: Safety profile of cyclin-dependent
kinase (CDK) 4/6 inhibitors with concurrent radiation therapy: A
systematic review and meta-analysis. Cancer Treat Rev.
119:1025862023. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Franco R, Cao JQ, Yassa M and Hijal T:
Safety of CDK4/6 inhibitors combined with radiotherapy in patients
with metastatic breast cancer: A Review of the literature. Curr
Oncol. 30:5485–5496. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Ratosa I, Orazem M, Scoccimarro E,
Steinacher M, Dominici L, Aquilano M, Cerbai C, Desideri I,
Ribnikar D, Marinko T, et al: Cyclin-Dependent Kinase 4/6
inhibitors combined with radiotherapy for patients with metastatic
breast cancer. Clin Breast Cancer. 20:495–502. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Kawamoto T, Shikama N, Imano N, Kubota H,
Kosugi T, Sekii S, Harada H, Yamada K, Naoi Y, Miyazawa K, et al:
Incidence of and risk factors for non-hematologic toxicity with
combined radiotherapy and CDK4/6 inhibitors in metastatic breast
cancer using dose-volume parameters analysis: A multicenter cohort
study. Breast Cancer. 30:282–292. 2023. View Article : Google Scholar
|
|
213
|
Hashizume R, Zhang A, Mueller S, Prados
MD, Lulla RR, Goldman S, Saratsis AM, Mazar AP, Stegh AH, Cheng SY,
et al: Inhibition of DNA damage repair by the CDK4/6 inhibitor
palbociclib delays irradiated intracranial atypical teratoid
rhabdoid tumor and glioblastoma xenograft regrowth. Neuro Oncol.
18:1519–1528. 2016.PubMed/NCBI
|
|
214
|
Pesch AM, Hirsh NH, Chandler BC,
Michmerhuizen AR, Ritter CL, Androsiglio MP, Wilder-Romans K, Liu
M, Gersch CL, Larios JM, et al: Short-term CDK4/6 inhibition
radiosensitizes estrogen receptor-positive breast cancers. Clin
Cancer Res. 26:6568–6580. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Meattini I, Livi L, Lorito N, Becherini C,
Bacci M, Visani L, Fozza A, Belgioia L, Loi M, Mangoni M, et al:
Integrating radiation therapy with targeted treatments for breast
cancer: From bench to bedside. Cancer Treat Rev. 108:1024172022.
View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Zhang X, Zhu L, Zhang H, Chen S and Xiao
Y: CAR-T cell therapy in hematological malignancies: Current
opportunities and challenges. Front Immunol. 13:9271532022.
View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Zhang ZZ, Wang T, Wang XF, Zhang YQ, Song
SX and Ma CQ: Improving the ability of CAR-T cells to hit solid
tumors: Challenges and strategies. Pharmacol Res. 175:1060362022.
View Article : Google Scholar
|
|
218
|
Lelliott EJ, Kong IY, Zethoven M,
Ramsbottom KM, Martelotto LG, Meyran D, Zhu JJ, Costacurta M, Kirby
L, Sandow JJ, et al: CDK4/6 inhibition promotes antitumor immunity
through the induction of T-cell memory. Cancer Discov.
11:2582–2601. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Tripathy D, Im SA, Colleoni M, Franke F,
Bardia A, Harbeck N, Hurvitz SA, Chow L, Sohn J, Lee KS, et al:
Ribociclib plus endocrine therapy for premenopausal women with
hormone-receptor-positive, advanced breast cancer (MONALEESA-7): A
randomised phase 3 trial. Lancet Oncol. 19:904–915. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Turner NC, Ro J, André F, Loi S, Verma S,
Iwata H, Harbeck N, Loibl S, Huang Bartlett C, Zhang K, et al:
Palbociclib in Hormone-Receptor-positive advanced breast cancer. N
Engl J Med. 373:209–219. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Hortobagyi GN, Stemmer SM, Burris HA, Yap
YS, Sonke GS, Paluch-Shimon S, Campone M, Petrakova K, Blackwell
KL, Winer EP, et al: Updated results from MONALEESA-2, a phase III
trial of first-line ribociclib plus letrozole versus placebo plus
letrozole in hormone receptor-positive, HER2-negative advanced
breast cancer. Ann Oncol. 29:1541–1547. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Rugo HS, Huober J, García-Sáenz JA, Masuda
N, Sohn JH, Andre VAM, Barriga S, Cox J and Goetz M: Management of
Abemaciclib-associated adverse events in patients with hormone
Receptor-positive, human epidermal growth factor receptor
2-Negative advanced breast cancer: Safety analysis of MONARCH 2 and
MONARCH 3. Oncologist. 26:e53–e65. 2021. View Article : Google Scholar
|
|
223
|
Spring LM, Zangardi ML, Moy B and Bardia
A: Clinical management of potential toxicities and drug
interactions related to Cyclin-Dependent kinase 4/6 inhibitors in
breast cancer: Practical considerations and recommendations.
Oncologist. 22:1039–1048. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Asghar U, Witkiewicz AK, Turner NC and
Knudsen ES: The history and future of targeting cyclin-dependent
kinases in cancer therapy. Nat Rev Drug Discov. 14:130–146. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Hassanzadeh A, Shomali N, Kamrani A,
Soltani-Zangbar MS, Nasiri H and Akbari M: Cancer therapy by
cyclin-dependent kinase inhibitors (CDKIs): Bench to bedside. EXCLI
J. 23:862–882. 2024.PubMed/NCBI
|
|
226
|
Zheng M, Zhang XY, Chen W, Xia F, Yang H,
Yuan K and Yang P: Molecules inducing specific cyclin-dependent
kinase degradation and their possible use in cancer therapy. Future
Med Chem. 16:369–388. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Majeski H, Okano A, Pasis A, Carlson C,
Shakya A, Huang S, Hoskote Chourasia A, Fung LM and Liu Q:
Discovery of CDK4/6 bifunctional degraders for ER+/HER2-breast
cancer. J Clin Oncol. 14:10832023. View Article : Google Scholar
|
|
228
|
Schott AF, Hurvitz S, Ma C, Hamilton E,
Nanda R, Zahrah G, Hunter N, Tan AR, Telli M, Mesias JA, et al:
Abstract GS3-03: GS3-03 ARV-471, a PROTAC® estrogen
receptor (ER) degrader in advanced ER-positive/human epidermal
growth factor receptor 2 (HER2)-negative breast cancer: Phase 2
expansion (VERITAC) of a phase 1/2 study. Cancer Res. 83(Suppl 5):
GS3–03. 2023.
|
|
229
|
Bruls R: Treatment options beyond CDK4/6
inhibition. In: Medical Conferences; 2023
|
|
230
|
Fassl A, Brain C, Abu-Remaileh M, Stukan
I, Butter D, Stepien P, Feit AS, Bergholz J, Michowski W, Otto T,
et al: Increased lysosomal biomass is responsible for the
resistance of triple-negative breast cancers to CDK4/6 inhibition.
Sci Adv. 6:eabb22102020. View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Hassan MA-K and Ates-Alagoz Z:
Cyclin-dependent kinase 4/6 inhibitors against breast cancer. Mini
Rev Med Chem. 23:412–428. 2023. View Article : Google Scholar
|
|
232
|
Lv S, Yang J, Lin J, Huang X, Zhao H, Zhao
C and Yang L: CDK4/6 inhibitors in lung cancer: Current practice
and future directions. Eur Respir Rev. 33:2301452024. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Du Q, Guo X, Wang M, Li Y, Sun X and Li Q:
The application and prospect of CDK4/6 inhibitors in malignant
solid tumors. J Hematol Oncol. 13:412020. View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Sun M, Dong L, Wang Y, Liu C, Du J, Wang
B, Xing B, Yao X, Ren Y and Zhou X: The role of targeting CDK4/6 in
cancer immunotherapy. Holist Integ Oncol. 3:322024. View Article : Google Scholar
|