Introduction
Knee osteoarthritis (KOA), a progressive
degenerative joint disorder characterized by articular cartilage
degradation, synovial inflammation and osteophyte formation, is a
leading cause of age-associated mobility disability (1,2).
Epidemiological projections suggest OA prevalence will double by
2030 compared with the levels in the 2010s, reflecting global
demographic shifts toward aging populations (3). Studies suggest that chondrocyte
senescence, a terminal cell cycle arrest state, drives OA
progression (4,5). Senolytic elimination of senescent
chondrocytes preserves cartilage integrity in murine post-traumatic
OA models (6,7), while intra-articular injection of
senescent cells induces OA pathology (8). Mechanistically, senescent
chondrocytes exhibit high levels of senescence-associated secretory
phenotype (SASP) proteins, including MMPs, pro-inflammatory
cytokines and reactive oxygen species (ROS), which collectively
promote extracellular matrix (ECM) degradation and chronic
synovitis (9,10). However, the upstream molecular
mechanisms governing chondrocyte senescence remain incompletely
understood, hindering targeted OA therapy development.
Mitophagy, a selective autophagic process essential
for mitochondrial quality control, serves a key role in age-related
pathology research (11). OA
chondrocytes exhibit impaired mitophagic activity, leading to
pathological accumulation of dysfunctional mitochondria (12-14). This degradation mechanism is
notably suppressed in senescent cells and associated with abnormal
mitochondrial retention and elevated ROS generation (15). Notably, restoring mitophagic flux
attenuates cell senescence markers (16), while mitophagy activation reduces
oxidative stress in OA pathogenesis (17). These findings establish
mitochondrial homeostasis disruption as a key link between
chondrocyte senescence and OA progression.
PTEN-induced putative kinase 1 (PINK1), a master
regulator of mitochondrial quality control, serves critical roles
in age-related pathologies including Parkinson's disease and
cardiac hypertrophy (18,19).
PINK1 orchestrates mitochondrial homeostasis by phosphorylating
ubiquitin chains at Ser65 to recruit Parkin, initiating
ubiquitin-dependent clearance of damaged mitochondria (20). It also phosphorylates
mitochondrial Tu translation elongation factor (TUFm) at Ser222,
inhibiting autophagy-related 5-12 complex formation and suppressing
non-canonical mitophagy (21).
The role of PINK1 in chondrocyte senescence and K OA pathogenesis
remains controversial: While Shin et al (22) reported that PINK1-mediated
mitophagy contributes to cartilage degeneration, other studies
(23,24) have demonstrated that PINK1/Parkin
pathway activation exerts chondroprotective effects through
enhanced mitochondrial quality control. This paradox highlights
context-dependent regulatory functions of PINK1, potentially
influenced by disease stage and microenvironmental factors,
necessitating further mechanistic investigation.
The present study aimed to clarify PINK1-mediated
mitophagy in chondrocyte senescence during OA progression. Using
lipopolysaccharide (LPS)-induced inflammatory models, the PINK1
modulation effects on mitochondrial function, redox homeostasis and
senescence-related phenotypes were investigated. RNA-sequencing
(seq) with functional validation was performed to identify the
downstream effectors of PINK1, particularly its interaction with
the p38 MAPK/NF-κB signaling axis. The present findings may advance
molecular stratification of OA pathogenesis and establish a
framework for mitochondria-targeted therapy.
Materials and methods
Mouse model generation
A total of 20 male C57BL/6J mice (weight, 25±2 g;
age, 6 weeks) were purchased from Jiangsu Huachuang Xinnuo
Pharmaceutical Technology Co., Ltd. All experimental procedures
were approved by The Animal Ethics Committee of Nanjing University
of Chinese Medicine (Nanjing, China; approval no. 202409A051).
Animals were housed under specific pathogen-free conditions (25°C,
50% relative humidity) with a 12/12-h light/dark cycle, free access
to food and water and ≤5 mice/cage. Using SPSS software (IBM Corp.;
version 22.0), the mice were randomly assigned to the control or
the KOA model group (n=10/group). After a 7-day acclimation period,
destabilization of the medial meniscus (DMM) surgery was performed
on the right hind limb according to established methods (25). Anesthesia was induced with 3%
isoflurane and maintained with 1% isoflurane during surgery. The
surgical area was shaved and disinfected with 75% ethanol, and a
medial approach to the knee joint was used. After blunt layered
separation of the muscles, the medial meniscus was transected with
a scalpel to induce joint instability and establish the KOA model.
The control group underwent a sham procedure, involving all
surgical steps except for transection of the medial meniscus. All
animals were given a 14-day postoperative recovery period to ensure
model establishment. The animal experiment lasted a total of 3
weeks (7 days of acclimatization and 14 days for KOA model
induction). During this period, animals exhibiting severe decline
in mobility, difficulty moving or inability to access food and
water, indicative of extreme debilitation, were designated for
early euthanasia. No unexplained deaths occurred. Euthanasia was
performed by intraperitoneal injection of sodium pentobarbital (150
mg/kg). Death was confirmed based on the absence of spontaneous
respiration for >2 min, no palpable heartbeat, no response to
resuscitation and fixed dilated pupils with loss of the light
reflex. The knee joints were dissected and articular cartilage
specimens were snap-frozen in liquid nitrogen and stored at −80°C
for subsequent analysis.
Micro-computed tomography scanning
Micro-CT scanning (Skyscan; Bruker Corporation) was
performed on knee joint samples. The scanning parameters included a
voltage of 80 kV, current of 100 μA and resolution of 9
μm. The scan region was focused on the subchondral bone area
beneath the tibial plateau in the weight-bearing zone.
Subsequently, CTVox 3.3, Blue Scientific) to perform
three-dimensional reconstruction of the knee joint and conduct
structure model index of trabeculae) and bone volume fraction)
analyses to assess its structural characteristics.
Histological observation
Articular cartilage from the knee joint was
collected and fixed at room temperature in 4% paraformaldehyde for
48 h. The tissue was then decalcified in 10% EDTA) solution at
37°C. After paraffin embedding at 65°C, sections were prepared at a
thickness of 0.4 μm. The sections were stained at room
temperature with hematoxylin and eosin (H&E) and with Safranin
O-Fast Green (SO/FG). H&E staining was performed with
hematoxylin for 5-10 min and counterstain cytoplasm with eosin for
1-3 min. For SO/FG staining, with Fast Green for 5 min, then stain
with Safranin O for 30 min. The knee articular cartilage was
examined under a light microscope. OA Research Society
International (OARSI) score (26) was calculated to assess
pathological degeneration.
ELISA
The concentrations of pro-inflammatory cytokines,
including IL-1β, IL-6 and tumor necrosis factor-α (TNF-α), in serum
samples were quantified using commercially available mouse-specific
ELISA kits according to the manufacturers' instructions [Mouse
IL-1β (cat. no. MLB00C), IL-6 (cat. no. M6000B) and TNF-α ELISA kit
(cat. no. MTA00B; all R&D Systems, Inc.)]. Briefly, serum
samples were collected by centrifugation of whole blood at 3,000 ×
g for 10 min at 4°C and stored at −80°C until analysis. Standards
and samples were added to pre-coated 96-well plates in duplicate
and incubated for 2 h at room temperature. After washing five times
with wash buffer, biotinylated detection antibodies were added for
1 h at room temperature. The optical density was measured at 450
nm. Cytokine concentrations were calculated based on standard
curves generated for each assay. All samples were analyzed in
triplicate to ensure data reliability.
Cell isolation and culture
Immortalized human chondrocytes (SV40 cells; cat.
no. T0021) were purchased from Applied Biological Materials, Inc.
For primary cell culture, articular cartilage was aseptically
isolated from the tibial plateau and washed three times with PBS.
The cleaned cartilage fragments were minced into 1 mm3
pieces and digested in 0.2% type II collagenase at 37°C for 6 h
with gentle agitation. The cell suspension was filtered through a
70-μm cell strainer and centrifuged at 300 × g for 6 min at
room temperature to isolate chondrocytes. The harvested cells were
cultured in DMEM supplemented with 10% fetal bovine serum (both
Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin (Invitrogen; Thermo Fisher Scientific,
Inc.) under standard culture conditions (37°C, 5% CO2
and 95% humidity). Medium replacement was performed every 48 h
until 80-90% confluence was achieved. KOA conditions were modeled
by treating cells with 10 ng/ml IL-1β or 10 ng/ml TNF-α for 24 h;
or 1 μg/ml LPS for 24 h. For inhibition of the p38 MAPK
pathway, chondrocytes were treated with 1 μM talmapimod for
24 h. To activate the p38 MAPK signaling pathway, chondrocytes were
treated with 1 μg/ml diprovocim for 24 h. All treatments
were performed at 37°C.
PINK1 short hairpin (sh)RNA
transfection
shRNA designs were sourced from Sigma-Aldrich
website (27). Based on the
PINK1 gene (transcript ID: NM_032409.1), three shRNAs were
designed: TRCN0000007101 (PINK1-interference1) targeting
5'-CGGCTGGAGGAGTATCTGATA-3'), TRCN0000199193 (PINK1-i2) targeting
GAAGCCACCATGCCTACATTG and TRCN0000199446 (PINK1-i3) targeting
CGGACGCTGTTCCTCGTTATG-3'). Negative control) plasmid
(NC-interference) targeted TTCTCCGAACGTGTCACGTT (5'->3'). The
oligonucleotides (oligos) were designed using the stem-loop (loop)
sequence TCAAGAG The specific interference sequences are listed in
Table I. A third-generation
lentiviral system was used for transduction (lentiviral transfer
plasmid, packaging plasmid, and envelope plasmid are mixed at a
4:3:1 ratio (12:9:3 μg), employing a puromycin-resistant
lentiviral shRNA transfer vector and 293T packaging cells (both
Nanjing Kress Biotechnology Co., Ltd.). Transfection was performed
at 37°C using Lipo293™ (Beyotime Institute of Biotechnology; cat.
no. C0521) for 6 h, followed by a medium change 24 h
post-transfection. Viral supernatants were collected at 24 and 48 h
post-transfection, filtered through a 0.45 μm sterile
filter, concentrated and purified using a PEG Lentivirus
Purification kit (Chongqing Inovogen Biotechnology Co., Ltd.; cat.
no. P1201), then resuspended in DMEM. Aliquots of 200
μl/tube were prepared and stored at −80°C. Infection was
performed at an MOI of 70 at 37°C for 24 h, followed by selection
with 2 μg/ml puromycin for 48 h. Cells were then switched to
puromycin-free DMEM (Gibco, cat. no. 11965092.) for expansion, and
assessed knockdown efficiency using FITC labelling observed by
fluorescence microscopy and quantitative PCR (qPCR). qPCR was
performed on a LightCycler 480 system using the SYBR Green I Master
kit (Roche, cat. no. 04887352001). Primer sequences (5'→3') were as
follows: human PINK1 (forward: GCCTCATCGAGGAAAAACAGG; reverse:
GATCACTAGCCAGGGAACACG) and the housekeeping gene human GAPDH
(forward: GGAGCGAGATCCCTCCAAAAT; reverse: GGCTGTTGTCATACTTCTCATGG).
Thermal cycling conditions were as follows: initial denaturation at
95°C for 10 min, followed by 40 cycles of 95°C for 15 sec, 60°C for
30 sec (annealing), and 72°C for 30 sec. Relative gene expression
was calculated using the 2^-ΔΔCq method (28), normalized to GAPDH, and reported
as fold change relative to the control group. TRCN0000199446 was
selected as the shRNA interference sequence for subsequent
experiments.
 | Table IInterference sequences for PINK1
short hairpin RNA and empty vector plasmid. |
Table I
Interference sequences for PINK1
short hairpin RNA and empty vector plasmid.
| Name | Sequence
(5'->3') |
|---|
| NC-iF |
gatccgTTCTCCGAACGTGTCACGTTtcaagagAACGTGACACGTTCGGAGAAtttttt |
| NC-iR |
aattaaaaaaTTCTCCGAACGTGTCACGTTctcttgaAACGTGACACGTTCGGAGAAcg |
| PINK1-i1F |
gatccgCGGCTGGAGGAGTATCTGATAtcaagagTATCAGATACTCCTCCAGCCGtttttt |
| PINK1-i1R |
aattaaaaaaCGGCTGGAGGAGTATCTGATActcttgaTATCAGATACTCCTCCAGCCGcg |
| PINK1-i2F |
gatccGAAGCCACCATGCCTACATTGtcaagagCAATGTAGGCATGGTGGCTTCtttttt |
| PINK1-i2R |
aattaaaaaaGAAGCCACCATGCCTACATTGctcttgaCAATGTAGGCATGGTGGCTTCg |
| PINK1-i3F |
gatccgCGGACGCTGTTCCTCGTTATGtcaagagCATAACGAGGAACAGCGTCCGtttttt |
| PINK1-i3R |
aattaaaaaaCGGACGCTGTCCTCGTTATG ctcttga
CATAACGAGGAACAGCGTCCGcg |
Lentiviral transduction for PINK1
overexpression in primary mouse chondrocytes
For PINK1 overexpression, PINK1 gene oligos (IDs
B5469-1-38) were designed and amplified by Shanghai GenePharma Co.,
Ltd. and transduction was performed using a third-generation
lentiviral system. The control group received an empty-vector virus
with no insert, using the LV5 (EF-1aF/GFP&Puro) backbone. For
the overexpression group, the transcript ID was NM_026880.2 and the
same LV5 (EF-1aF/GFP&Puro) vector was used. The packaging
plasmids included pGag/Pol, pRev and the envelope plasmid pVSV-G.
All vectors/plasmids were supplied by Shanghai GenePharma Co., Ltd.
Packaging was performed in 293T producer cells (Shanghai GenePharma
Co., Ltd.). Cells were transfected at 37°C using the RNAi-mate
transfection reagent (Shanghai Genepharma Co., Ltd.) for 6 h, after
which the medium was replaced with DMEM containing 10% FBS. The
culture was maintained at 37°C for 72 h to collect viral
supernatant. Cell debris was removed by low-speed centrifugation at
4°C, 500 × g for 4 min, followed by filtration through a 0.45
μm sterile filter. Finally, the virus was concentrated by
ultracentrifugation at 4°C, 3,000 × g for 2 h, aliquoted, and
stored at −80°C. Primary mouse chondrocytes were used as target
cells. Infection was performed at an MOI of 90 at 37°C for 24 h,
followed by puromycin selection at 2 μg/ml for 48 h. The
cells were then expanded in puromycin-free medium for subsequent
assays. Lentiviral transfer plasmid, packaging and envelope plasmid
are mixed at a 4:3:1 ratio (12:9:3 μg).
RNA-seq
RNA quality and concentration were assessed using a
NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Inc.)
and an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.), with
RNA integrity number >8.0 considered acceptable for sequencing.
mRNA bearing poly (A) tails was enriched from 1-4 μg of
total RNA using 20 μl Dynabeads Oligo (dT)25 magnetic beads
(Invitrogen, cat. no. 61002). The mRNA was then fragmented into
200-300 bp short fragments by incubating at 94°C for 5 min in a
divalent cation-containing fragmentation buffer with the NEBNext
Magnesium RNA Fragmentation Module (New England Biolabs, cat. no.
E6150S). First-strand cDNA was synthesized using random hexamer
primers (50 μM; Invitrogen, cat. no. N8080127) and
SuperScript III reverse transcriptase (200 U/μl; both
Invitrogen, cat. no. 18080093) under the following conditions: 25°C
for 10 min, 50°C for 50 min, and inactivation at 85°C for 5 min.
Second-strand cDNA synthesis was performed with DNA Polymerase I
(10 U/μl; NEB, cat. no. M0209L) and RNase H (2 U/μl;
NEB, cat. no. M0297S) in a second-strand synthesis buffer
containing dNTPs (10 mM each), incubated at 16°C for 2.5 h. The
double-stranded cDNA was purified, end-repaired and ligated with
sequencing adapters. Libraries were constructed using the NEBNext
Ultra RNA Library Prep kit (cat. no. E7530L, New England BioLabs,
Inc.) and subjected to quality control using the Agilent 2100
Bioanalyzer. Final library concentrations were quantified by qPCR
and diluted to 150 pM. High-quality libraries were sequenced on an
Illumina NovaSeq 6000 platform (Illumina, Inc.) in paired-end mode
(150 bp reads). Raw sequencing data were preprocessed using FastQC
(v0.11.9) for quality assessment and Trimmomatic (v0.39) for
adapter trimming and low-quality base removal. Clean reads were
aligned to the reference genome (GRCh38) using HISAT2 (v2.2.1) and
gene expression levels were quantified using StringTie
(v2.2.1).
Transcriptome data analysis
Pearson correlation analysis were performed using
the cor()' function in R (version 4.3.0, The R Foundation for
Statistical Computing). Pearson's correlation coefficients (r) were
calculated for normally distributed continuous variables, while
Spearman rank correlation was used for nonparametric data. The
'corrplot' package (version 0.92, Comprehensive R Archive Network),
combined with hierarchical clustering, was used to visualize the
correlation matrix. Two-sided tests were applied, with P<0.05
indicating statistical significance, and Benjamini-Hochberg
correction was used for multiple comparisons. Principal component
analysis (PCA) was conducted with the 'prcomp()' function. Prior to
analysis, sampling adequacy was assessed with the
Kaiser-Meyer-Olkin test (threshold >0.6), and variable
correlations were evaluated with Bartlett's test of sphericity
(P<0.05). The number of principal components was determined
based on eigenvalues >1 (Kaiser criterion) and the scree plot.
The 'factoextra' package (version 1.0.7, Comprehensive R Archive
Network)) was used to generate biplots showing both sample scores
and variable loadings. The first two principal components (PC1 and
PC2) explaining the cumulative variance were selected for
visualization. Stability of the results was validated with 1,000
bootstrap iterations. Differential gene expression analysis was
performed using DESeq2 (1.40.2, Bioconductor Project), with a
significance threshold of |log2 fold-change|>1 and adjusted
P<0.05. Functional enrichment analysis of differentially
expressed genes was performed using Gene Ontology (GO, https://www.geneontology.org/) and Kyoto Encyclopedia
of Genes and Genomes (KEGG, genome.jp/kegg/) databases. Gene set
enrichment analysis was performed using the GSEA software (version
4.3.2, Broad Institute) in conjunction with the Molecular
Signatures Database (MSigDB v7.5.1, Broad Institute). A pre-ranked
gene list was generated based on log2 fold-change values from
differential expression analysis. The Hallmark gene sets (H) were
used for the analysis.
ROS
According to the manufacturer's instructions, the
intracellular levels of ROS were quantitatively measured using an
ROS Assay kit (cat. no. S0035M; Beyotime Institute of
Biotechnology). Briefly, chondrocytes were plated in 6-well culture
plates at a density of 1×106 cells/well and allowed to
adhere overnight at 37°C. Culture medium was replaced with fresh
DMEM (Gibco, cat. no. 11965092) containing the ROS-specific
fluorescent probe working solution, and the cells were maintained
at 37°C in a humidified 5% CO2 incubator for 30 min.
Cells were washed three times with PBS to remove excess probe
solution and stained with DAPI for 10 min at 37°C. Fluorescence
imaging for ROS detection was performed using an inverted
fluorescence microscope (Leica DMI8; Leica Microsystems GmbH)
equipped for excitation at 488 nm. All procedures were conducted
under standardized conditions to ensure consistency and
reproducibility of the results.
Measurement of mitochondrial membrane
potential
According to the manufacturer's instructions,
mitochondrial membrane potential changes were analyzed using the
Mitochondrial Membrane Potential Assay kit with JC-1 (Beyotime
Institute of Biotechnology). Chondrocytes were seeded at a density
of 1×106 cells/well in 6-well plates and incubated with
DMEM containing JC-1 working solution at 37°C for 20 min.
Fluorescence images were captured using a fluorescence microscope
(Leica DMI8; Leica Microsystems GmbH).
Senescence-associated β-galactosidase
(SA-β-gal) staining
Cell senescence was assessed using a SA-β-gal
Staining kit (cat. no. C0602; Beyotime Institute of Biotechnology)
following established protocols (25). Briefly, chondrocytes were rinsed
twice with PBS and fixed in 4% paraformaldehyde for 15 min at room
temperature. Cells were incubated with freshly prepared staining
solution containing X-gal in a humidified 37°C incubator for 16 h
under CO2-free conditions. Quantification was performed
by calculating the percentage of SA-β-gal-positive cells relative
to total cells in five randomly selected fields of view/sample
using ImageJ software (v1.54f, National Institutes of Health).
Protein extraction and western
blotting
Cells were lysed using RIPA buffer (Beyotime
Institute of Biotechnology) supplemented with a protease inhibitor
cocktail on ice for 30 min. The lysates were centrifuged at 12,000
× g for 15 min at 4°C, and the supernatants were collected. Protein
concentration was measured using the BCA protein assay. Equal
amounts of protein (20 μg/lane) were separated by 4-20%
SDS-PAGE and transferred onto polyvinylidene fluoride membranes
(MilliporeSigma). The membranes were blocked with 5% non-fat milk
for 1 h at room temperature and incubated overnight at 4°C with the
following primary antibodies: GAPDH (1:50,000; cat. no.
66004-1-Ig), β-tubulin (1:3,000; cat. no. 10094-1-AP), p65 (cat.
no. 10745-1-AP), phosphorylated (p-)p65 (cat. no. 82335-1-RR), p38
(cat. no. 14064-1-AP), p-p38 (all Proteintech Group, Inc.; cat. no.
28796-1-AP), MMP-3 (Affinity Biosciences; cat. no. AF0217), iNOS
(inducible nitric oxide synthase) (cat. no. 18985-1-AP), P16 (both
Proteintech Group, Inc.; cat. no. 10883-1-AP), P21 (Affinity
Biosciences; cat. no. AF6290), PINK1 (cat. no. 23274-1-AP), TUFm
(cat. no. 26730.1.AP), BNIP3L (Bcl-2) interacting protein 3-like)
(cat. no. 12986-1-AP), p62 (cat. no. 18420-1-AP) and LC3B
(Microtubule-associated protein 1 light chain 3B) (all 1:1,000; all
Proteintech Group, Inc.; cat. no. 18725.1-AP). Following washing
three times with 0.1% TBST, the membranes were incubated with
horseradish peroxidase-conjugated secondary antibodies for 1 h at
room temperature. The secondary antibodies were HRP-conjugated Goat
Anti-Rabbit IgG (H+L) (cat. no. SA00001-2, Proteintech Group, Inc.)
and HRP-conjugated Goat Anti-Mouse IgG (H+L) (both 1:5,000; cat.
no. SA00001-1, Proteintech Group, Inc.). Chemiluminescent detection
was performed with SuperSignal West Pico PLUS substrate (Thermo
Fisher Scientific, Inc.; cat. no. 34580). Protein bands were
visualized using an enhanced chemiluminescence detection system
(cat. no. 1708265, Bio-Rad Laboratories, Inc.) and analyzed using
ImageJ software (v1.53t, National Institutes of Health). All
experiments were performed in triplicate to ensure
reproducibility.
Statistical analysis
All data are presented as the mean ± standard
deviation of ≥3 independent experimental repeats for continuous
variables. Between-group comparisons were performed using one-way
ANOVA followed by Tukey's post hoc test or Bonferroni correction.
P<0.05 was considered to indicate a statistically significant
difference. All analyses and data visualization were conducted
using GraphPad Prism 5 (Dotmatics).
Results
Impaired mitochondrial autophagy and
enhanced cell senescence in KOA cartilage
Micro-CT analysis confirmed the successful
establishment of the KOA model. Compared with the sham-operated
group, the KOA group showed a significant increase in osteophyte
formation and more severe cartilage surface wear (Fig. 1A). Calculations of bone volume
fraction and the structure model index of trabeculae showed that
bone destruction was more pronounced in the KOA group than in the
sham group (Fig. 1A).
Histopathological evaluation revealed notable changes in the KOA
group, characterized by marked chondrocyte loss, a posterior shift
of the tidemark, and damage to the superficial cartilage structure.
Further assessment to evaluate cartilage degeneration showed that
the OARSI scores were significantly increased in the KOA group
(Fig. 1B). Quantitative ELISA
revealed significantly elevated levels of the pro-inflammatory
cytokines IL-1β, IL-6 and TNF-α in KOA compared with sham animals
(Fig. 1C). Western blot analysis
demonstrated a decline in mitophagy regulators (PINK1, TUFm, NIX,
p62 and LC3) concomitant with upregulation of senescence-associated
proteins (MMP3, iNOS, p21 and p16) in KOA cartilage compared with
sham tissue (Fig. 1D). These
protein alterations were conserved at the transcriptional level, as
evidenced by qPCR showing parallel modulation of corresponding mRNA
expression patterns (Fig. 1E).
Collectively, these findings demonstrated KOA progression is
mechanistically linked to impaired mitochondrial clearance and
exacerbated chondrocyte senescence in articular cartilage.
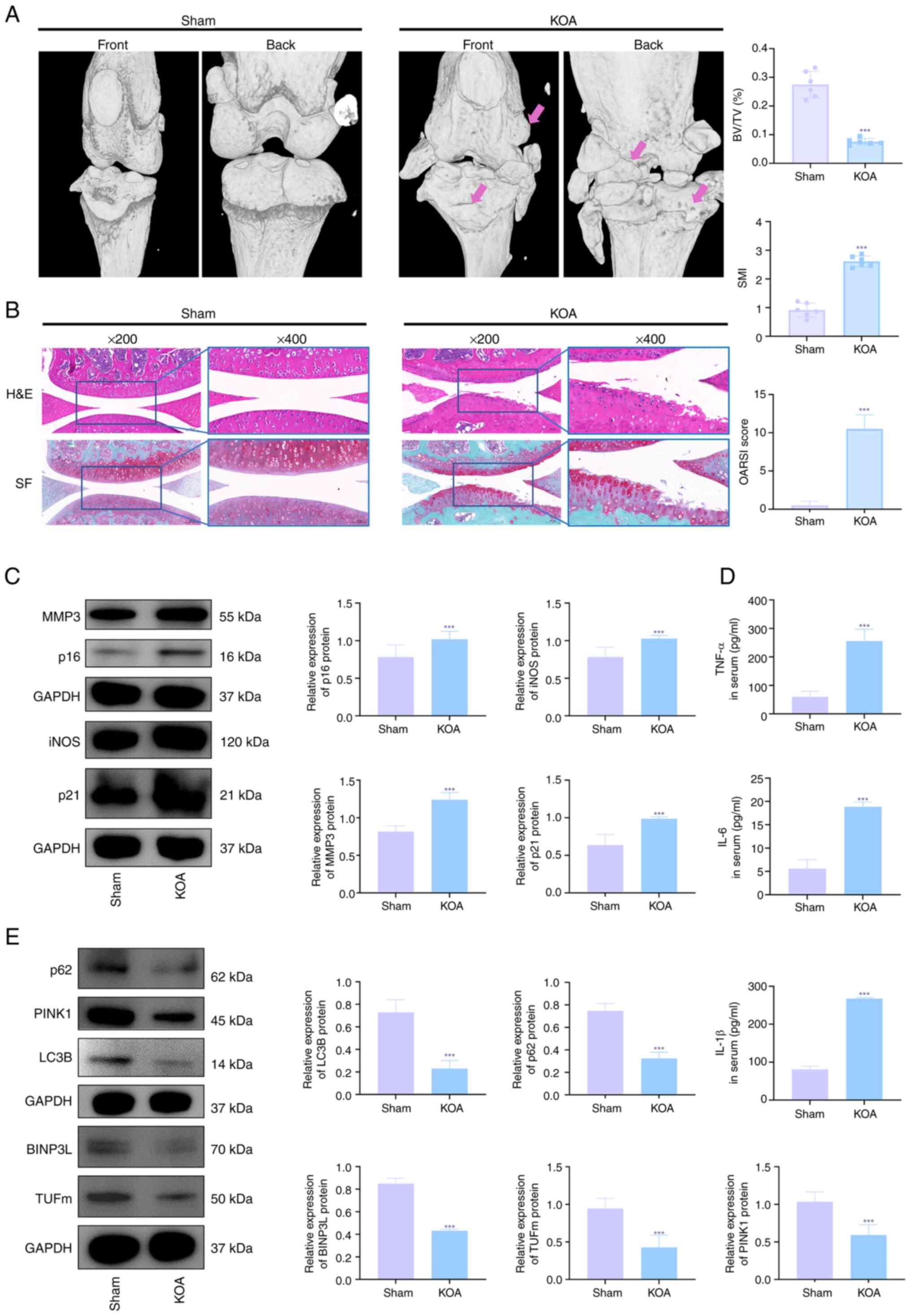 | Figure 1Impaired mitochondrial autophagy and
enhanced cell senescence in KOA cartilage. (A) Representative
microcomputed tomography 3D reconstructions of knee joints from
sham-operated and destabilization of the Medial Meniscus)induced
KOA mice 14 days post-surgery. (B) Histopathological evaluation and
OARSI scoring of knee joint sections (n=6), stained with HE and SF.
Scale bar, 50 μm. (C) Western blot analysis and densitometry
of senescence-associated markers (MMP3, iNOS, p21, p16) in
cartilage, normalized to GAPDH (n=3). (D) Serum levels of
proinflammatory cytokines (TNF-α, IL-6, IL-1β) measured by ELISA
using samples collected via cardiac puncture (n=6). (E) Western
blot analysis and densitometry of mitophagy-related proteins
(PINK1, LC3B ratio, p62, BNIP3L, TUFm) in cartilage, normalized to
GAPDH (n=3). ***P<0.001 vs. sham. KOA, knee
osteoarthritis; HE, hematoxylin and eosin; TNF-α, tumor necrosis
factor-α; PINK1, PTEN-induced putative kinase 1; TUFm, Tu
translation elongation factor; OARSI, Osteoarthritis Research
Society International; SO/FG, Safranine O-Fast Green; iNOS,
inducible nitric oxide synthase; BNIP3L, BCL2-interacting Protein 3
Like; SMI, structure model index of trabeculae; BV/TV, bone volume
fration. |
PINK1 overexpression decreases senescence
and promotes mitophagy in chondrocytes
In vitro, LPS, IL-1β and TNF-α significantly
upregulated the protein expression levels of the
senescence-associated markers p21, p16, iNOS and MMP3 (Fig. S1A), and increased the proportion
of SA-β-gal-positive cells (Fig.
S1B). To investigate the role of PINK1 in LPS-stimulated
chondrocytes, overexpression was performed using adenoviral
transfection. Western blot analysis revealed significant
upregulation of mitophagy-associated proteins (PINK1, TUFm, BINPL,
p62 and LC3B) in KOA chondrocytes compared with NC, which was
markedly reversed by Lv-PINK1 infection (Fig. 2A). JC-1 and ROS fluorescence
quantification demonstrated impaired mitochondrial membrane
potential and elevated ROS levels in KOA chondrocytes relative to
NC (Fig. 2B and C), both of
which were significantly ameliorated by Lv-PINK1 treatment.
SA-β-gal staining showed a significant increase in senescent
chondrocytes during KOA progression compared with blank controls,
with Lv-PINK1 infection decreasing senescence markers (Fig. 2D). Western blot analysis
confirmed decreased levels of senescence-associated proteins (MMP3,
iNOS, p21 and p16) following PINK1 overexpression (Fig. 2E). These coordinated changes
demonstrated that enhancing PINK1-mediated mitophagy mitigated
chondrocyte senescence through restored mitochondrial quality
control under inflammatory stress.
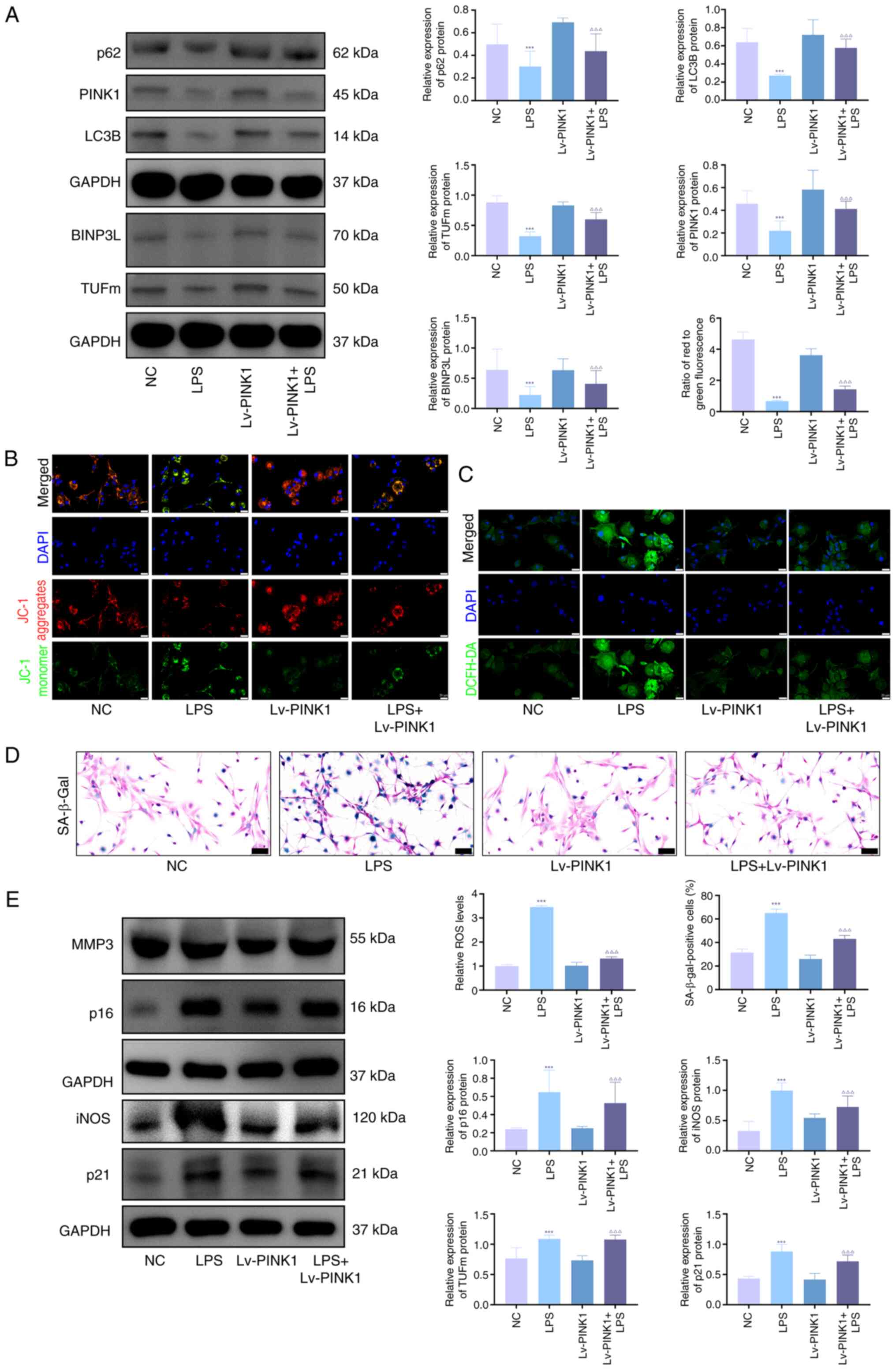 | Figure 2PINK1 overexpression decreases
senescence and promotes mitophagy in chondrocytes. (A) Western blot
analysis and densitometric quantification of mitophagy markers
(PINK1, LC3B, p62, BNIP3L, TUFm) in primary mouse chondrocytes
following transduction with Lv-PINK1, followed by LPS treatment (1
μg/ml, 24 h). Protein signals were normalized to GAPDH. (B)
Mitochondrial membrane potential was assessed using JC-1 staining.
Red, healthy mitochondria (J-aggregates); green, depolarized
mitochondria (J-monomers). (C) Levels of intracellular ROS were
detected using the DCFH-DA probe. Green fluorescence intensity was
quantified in ≥100 cells/group. Scale bar, 20 μm. (D)
Cellular senescence (blue) was evaluated by SA-β-gal staining. The
percentage of SA-β-gal-positive cells was quantified (scale bar, 50
μm). (E) Western blot quantification of
senescence-associated secretory phenotype markers (MMP3, iNOS) and
cell cycle inhibitors (p21, p16). Protein signals were normalized
to GAPDH. n=3. ***P<0.001 vs. NC;
∆∆∆P<0.001 vs. LPS. PINK1, PTEN-induced putative
kinase 1; TUFm, Tu translation elongation factor; ROS, reactive
oxygen species; SA-β-gal, senescence-associated β-galactosidase;
LPS, lipopolysaccharide; BNIP3L, BCL2-interacting Protein 3 Like;
Lv, Lentiviral Vector; iNOS, inducible nitric oxide synthase; NC,
negative Control. |
Transcriptomic analysis of PINK1
overexpression in OA chondrocytes
RNA-seq of chondrocytes revealed the anti-senescence
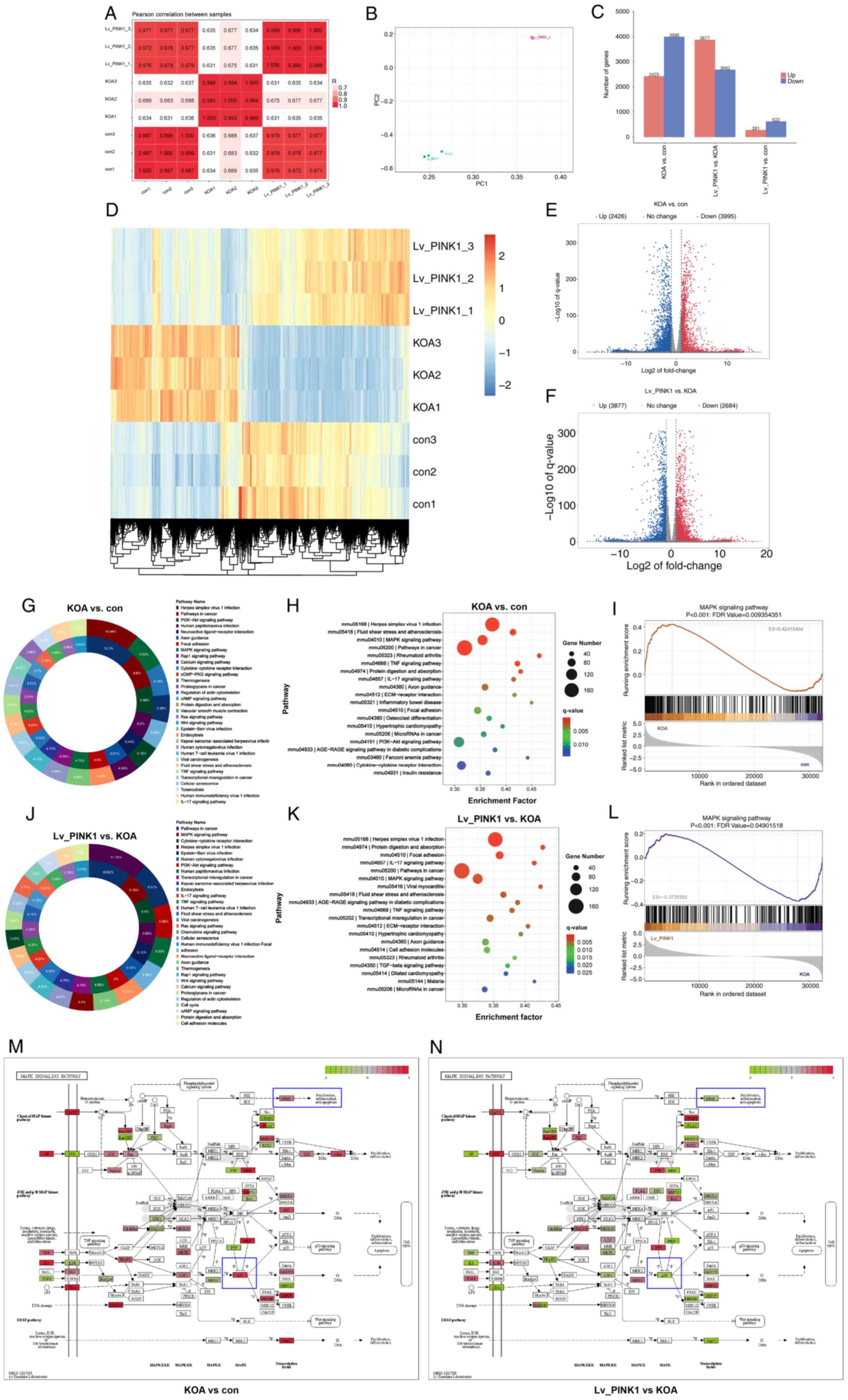
mechanism of PINK1. Correlation (Fig. 3A) and principal component
analysis (Fig. 3B) demonstrated
distinct clustering between KOA and control groups, while
Lv-PINK1-infected chondrocytes exhibited transcriptional profiles
comparable with controls. There were 6,420 dysregulated genes in
KOA vs. controls (2,425 up- and 3,995 downregulated), with Lv-PINK1
intervention altering 6,559 transcripts (3,877 up and 2,682
downregulated; Fig. 3C and D).
The volcano plots using criteria of |log2FC|>1 and P<0.05
confirmed these genome-wide alterations. KEGG pathway analysis
(Fig. 3G and J) and scatter
plots (Fig. 3H and K)
highlighted the top 10 pathways and revealed that 'MAPK signaling
pathway', 'TNF signaling pathway' and 'IL-17 signaling pathway'
were associated with KOA. Lv-PINK1 treatment predominantly
modulated 'focal adhesion' and 'IL-17 signaling pathway'. GSEA;
Fig. 3I and L) confirmed the
involvement of the MAPK signaling pathway in KOA development and
its modulation by Lv-PINK1. Changes in MAPK pathway genes (Fig. 3M and N) showed that p38
expression was increased during KOA but decreased by Lv-PINK1
infection. These multi-omics findings demonstrated that PINK1
overexpression may mitigate chondrocyte senescence and maintain
mitochondrial function via regulation of the MAPK signaling
pathway.
Activation of the p38 MAPK/NF-κB pathway
induces senescence and mitochondrial damage in LPS-treated
chondrocytes
To validate the transcriptomic findings, diprovocim,
a TLR (Toll like receptors)7/8 agonist that indirectly activates
p38 MAPK and NF-κB pathways, was used to investigate its effects on
KOA chondrocytes. ELISA demonstrated that diprovocim significantly
elevated IL-6 and TNF-α levels in the culture medium (Fig. 4A). Western blotting (Fig. 4B) confirmed that diprovocim
upregulated the expression of mitophagy-(PINK1, TUFm, BNIP3L, p62
and LC3B) and senescence-associated proteins (MMP3, iNOS, p21 and
p16), while promoting phosphorylation of p38 MAPK and NF-κB.
SA-β-gal staining revealed a marked increase in senescent cells
following diprovocim treatment (Fig.
4F). JC-1 staining (Fig. 4C)
and ROS assays (Fig. 4D) further
indicated decreased mitochondrial membrane potential and elevated
ROS accumulation in treated chondrocytes.
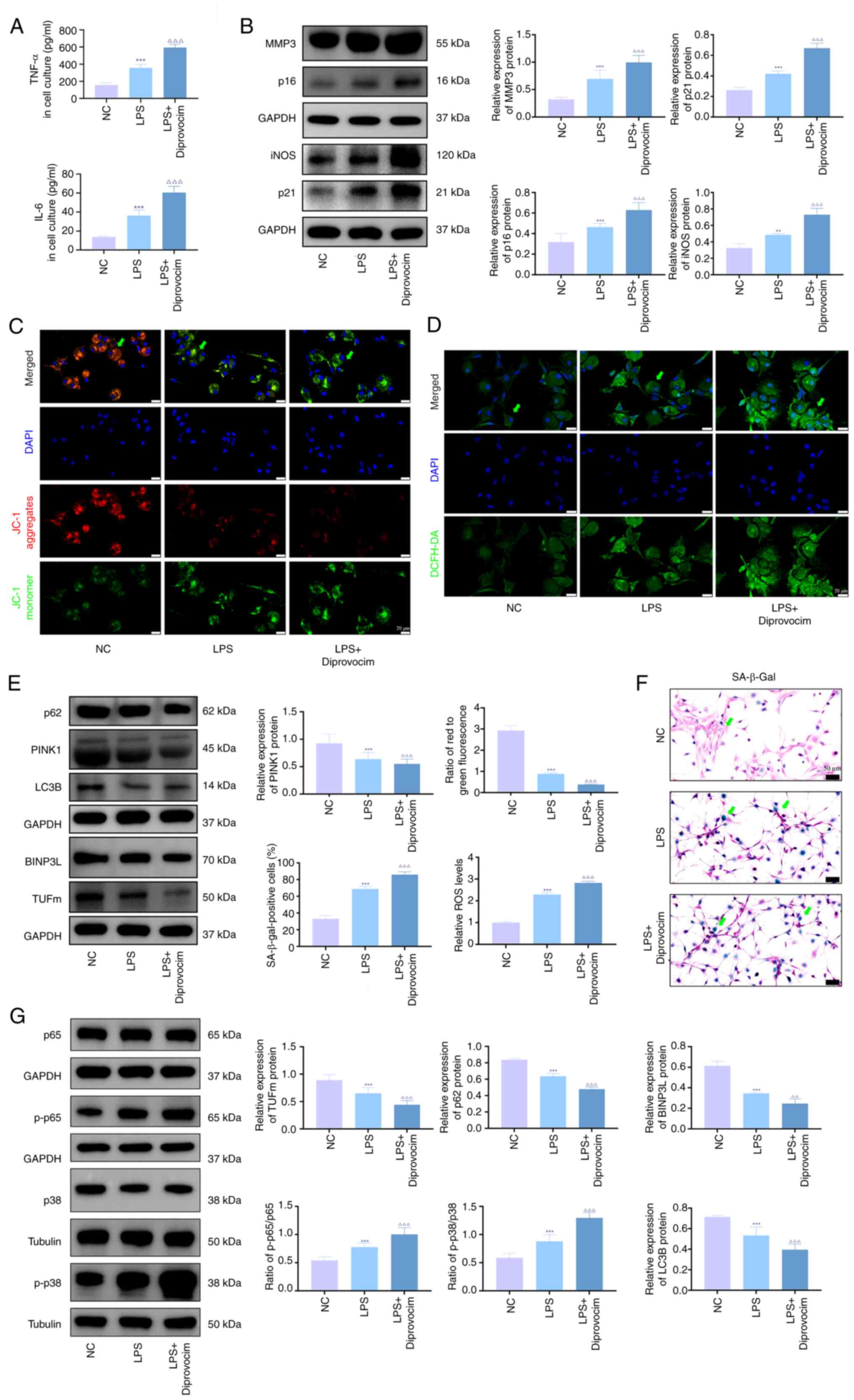 | Figure 4Activation of the p38 MAPK/NF-κB
pathway induces senescence and mitochondrial damage in LPS-treated
chondrocytes. (A) ELISA was performed to detect the levels of TNF-α
and IL-6 in the culture medium of immortalized human chondrocytes.
(B) Western blotting was performed to detect the protein expression
of MMP3, iNOS, p21 and p16 in immortalized human chondrocytes.
Representative (C) JC-1 staining showing mitochondrial membrane
potential in immortalized human chondrocytes. (D) DCFH-DA staining
showing ROS levels in immortalized human chondrocytes. (E) Western
blotting was performed to detect the protein expression of PINK1,
LC3B, p62, BINPL and TUFm in immortalized human chondrocytes. (F)
Representative SA-β-Gal staining showing senescence levels in each
group of immortalized human chondrocytes. (G) Western blotting was
performed to detect the protein expression levels of p-p65, p65,
p-p38 and p38. n=3. **P<0.01,
***P<0.001 vs. sham; ∆∆∆P<0.001 vs.
LPS. LPS, lipopolysaccharide; ROS, reactive oxygen species; PINK1,
PTEN-induced putative kinase 1; TUFm, Tu translation elongation
factor; SA-β-gal, senescence-associated β-galactosidase; iNOS,
inducible nitric oxide synthase; p-, phosphorylated; BINP3L, BCL2
Interacting Protein 3 Like. |
PINK1 gene knockdownenhances p38
MAPK/NF-κB phosphorylation and exacerbates chondrocyte
senescence
To validate the preliminary findings, PINK1
knockdown models in KOA chondrocytes were established to
investigate the activation of the p38 MAPK/NF-κB pathway and cell
senescence progression. Successful shRNA transfection was
demonstrated (Fig. 5A). Western
blot analysis demonstrated that PINK1 deficiency significantly
elevated phosphorylation levels of p38 MAPK and NF-κB compared with
the control (Fig. 5B). In
addition, senescence-associated proteins (MMP3, iNOS, p21 and p16)
were upregulated (Fig. 5C),
whereas mitochondrial autophagy markers (PINK1, TUFm, NIX, p62 and
LC3B) exhibited downregulation (Fig.
5D). LPS stimulation exacerbated these phenotypical alterations
and SA-β-gal staining revealed a pronounced increase in senescent
cells following PINK1 knockdown, particularly in LPS-treated cells
(Fig. 5E). JC-1 assays
demonstrated diminished mitochondrial membrane potential (Fig. 5F), while ROS quantification
confirmed elevated oxidative stress in PINK1-deficient cells
(Fig. 5G).
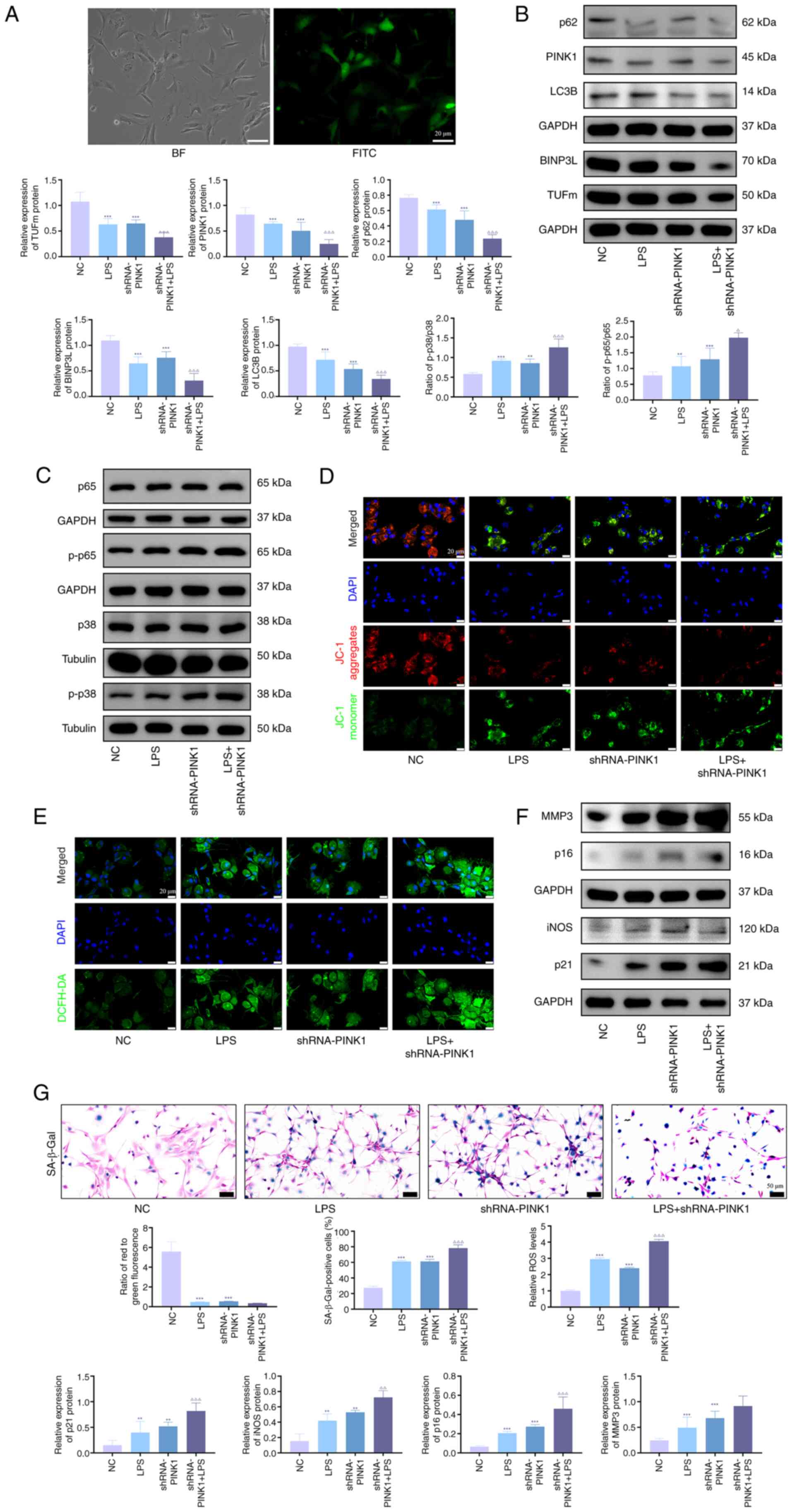 | Figure 5PINK1 gene knockdown enhances p38
MAPK/NF-κB phosphorylation and exacerbates chondrocyte senescence.
(A) Adenoviral infection efficiency of immortalized human
chondrocytes observed under a fluorescence microscope. Western blot
analysis of (B) PINK1, LC3B, p62, BINPL and TUFm and (C) p-p65,
p65, p-p38 and p38 protein expression. Representative (D) JC-1
staining showing mitochondrial membrane potential and (E) DCFH-DA
staining showing ROS levels in immortalized human chondrocytes. (F)
Western blot analysis of MMP3, iNOS, p21 and p16 protein expression
in immortalized human chondrocytes. (G) Representative SA-β-Gal
staining showing senescence levels in immortalized human
chondrocytes. n=3. **P<0.01, ***P<0.001
vs. sham; ∆P<0.05, ∆∆P<0.01,
∆∆∆P<0.001 vs. LPS. PINK1, PTEN-induced putative
kinase 1; TUFm, Tu translation elongation factor; ROS, reactive
oxygen species; SA-β-Gal, senescence-associated β-galactosidase;
BINP3L, BCL2-interacting Protein 3 Like; p-, Phosphorylation; iNOS,
inducible nitric oxide synthase; LPS, lipopolysaccharide; NC,
Negative Control; sh, short hairpin; BF, Bright field. |
Inhibition of the p38 MAPK/NF-κB pathway
ameliorates senescence and mitochondrial damage in PINK1-silenced
chondrocytes
To investigate whether PINK1 knockdown exacerbates
chondrocyte senescence via activation of the p38 MAPK/NF-κB
pathway, talmapimod, a specific p38 MAPK inhibitor, was used in
PINK1-silenced chondrocytes to evaluate its potential therapeutic
effects. Western blot analysis demonstrated that pharmacological
inhibition of p38 MAPK significantly decreased phosphorylation
levels of both p38 MAPK and NF-κB, while upregulating
senescence-associated proteins such as MMP3, iNOS, p21 and p16
(Fig. 6A and D). By contrast,
treatment with diprovocim produced opposing effects on these
biomarkers. Consistent with these findings, SA-β-gal staining
revealed a marked decrease in senescent cells following inhibitor
treatment, whereas agonist intervention significantly increased the
proportion of senescent cells (Fig.
6E). JC-1 staining indicated that p38 MAPK activation restored
mitochondrial membrane potential and markedly decreased ROS
accumulation (Fig. 6B and C).
Collectively, these results suggested modulation of the p38
MAPK/NF-κB pathway exerted regulatory effects on chondrocyte
senescence through the maintenance of mitochondrial homeostasis.
These results collectively indicated that compromised
PINK1-mediated mitophagy exacerbated mitochondrial dysfunction and
promoted cellular senescence under inflammatory conditions,
potentially via activation of the p38 MAPK/NF-κB pathway.
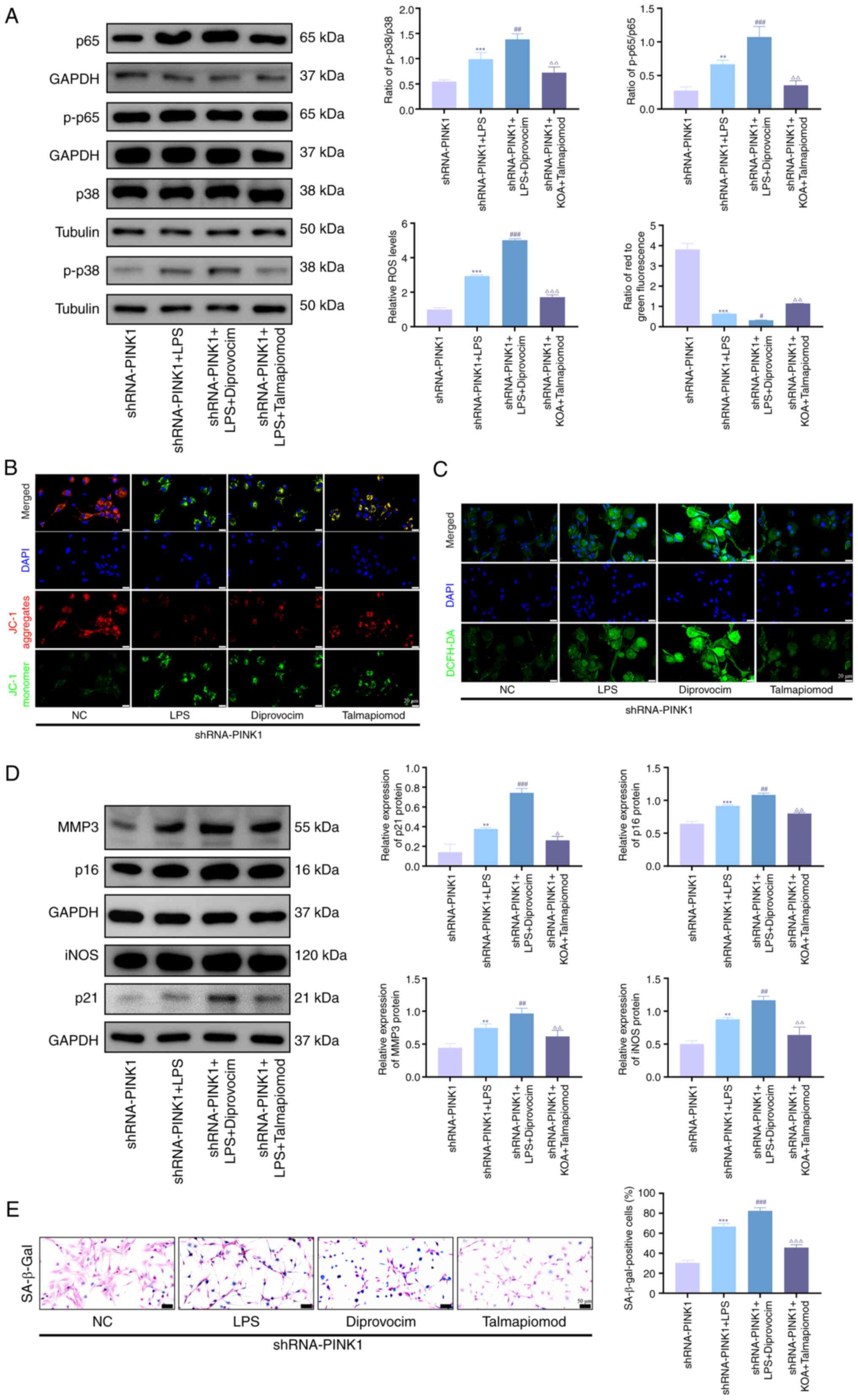 | Figure 6Inhibition of p38 MAPK/NF-κB pathway
ameliorates senescence and mitochondrial damage in PINK1-silenced
chondrocytes. (A) Western blot analysis of protein expression of
p-p65, p65, p-p38 and p38. (B) Representative JC-1 assay showing
mitochondrial membrane potential and (C) DCFH-DA staining showing
ROS levels, (D) Western blot analysis of protein expression of
MMP3, iNOS, p21 and p16 and (E) representative SA-β-Gal staining
showing senescence levels in immortalized human chondrocytes. n=3.
**P<0.01, ***P<0.001 vs. sham;
#P<0.05, ##P<0.01,
###P<0.001 vs. LPS; ∆P<0.05,
∆∆P<0.01, ∆∆∆P<0.001 vs. diprovocim.
ROS, reactive oxygen species; PINK1, PTEN-induced putative kinase
1; SA-β-gal, senescence-associated β-galactosidase; p-,
Phosphorylation; iNOS, inducible nitric oxide synthase; sh, short
hairpin; NC, Negative Control; LPS, Lipopolysaccharide. |
Discussion
As the sole resident cells of articular cartilage,
chondrocytes serve a key role in maintaining ECM homeostasis.
However, under chronic stress or inflammation, these cells
progressively transform into a senescent phenotype, culminating in
irreversible cell cycle arrest characterized by distinct
morphological changes including enlarged and flattened cell
morphology and functional decline (9). This senescence-associated metabolic
impairment compromises cartilage structural integrity, leading to
ECM degradation and tissue degeneration. Notably, senescent
chondrocytes exhibit paradoxical biological activity through
sustained secretion of proinflammatory cytokines (IL-1β, IL-6 and
TNF-α), MMPs and other catabolic mediators, as a phenomenon
collectively termed the SASP (29). The SASP propagates senescence via
paracrine signaling, disrupts ECM biosynthesis and activates
matrix-degrading enzymes, creating a self-amplifying loop that
accelerates cartilage destruction. Clinical evidence reveals
elevated senescence biomarkers, including telomere shortening and
increased senescent cell burden, in joint tissue of young patients
with post-traumatic OA (30).
Complementary studies demonstrate upregulation of SASP factors
across multiple articular compartments, particularly in subchondral
bone, synovium and infrapatellar fat pads (31-33). Here, male mice were used for DMM
modeling and mechanistic experiments to avoid hormone fluctuations
from the estrous cycle in females, which can confound inflammatory
phenotypes and metabolic endpoints. This approach enhances internal
consistency and decreases variability. The present study
corroborated the aforementioned clinical observations, showing
significant upregulation of cyclin-dependent kinase inhibitors p16
and p21 alongside marked elevation of serum IL-1β, IL-6 and TNF-α
levels compared with control groups. This multisystemic
manifestation of senescence markers across articular tissue
underscores the systemic nature of cell aging in K OA
pathogenesis.
The molecular mechanisms underlying cell senescence
have been extensively characterized, with mitochondrial dysfunction
emerging as a key determinant following Harman's free radical
theory of aging (34-36). Substantial evidence has
established that mitochondrial impairment orchestrates
intracellular ROS accumulation, disrupts adenosine triphosphate
biosynthesis and ultimately initiates cellular senescence through
defined molecular cascades (37-39). Evolutionary conservation has
equipped eukaryotic cells with sophisticated quality control
systems, including mitophagy, a selective autophagy mechanism
responsible for clearing depolarized mitochondria to preserve
organellar homeostasis (40).
Nevertheless, compromised mitophagic flux induces progressive
accumulation of dysfunctional mitochondria, culminating in
bioenergetic collapse and accelerated chondrocyte senescence. The
present study systematically investigated this mitophagy-senescence
axis through multimodal validation. Immunoblotting demonstrated
significant decreases in PINK1 and LC3-II expression in
osteoarthritic cartilage compared with healthy donor-matched
controls. Complementary in vitro analyses using LPS-induced
senescence models revealed mitochondrial membrane potential
dissipation accompanied by notable ROS elevation vs. untreated
chondrocytes. These mechanistically interlinked findings
substantiate impaired mitochondrial quality control as a
pathognomonic feature of K OA-associated chondrocyte senescence,
consistent with a prior demonstration of mitophagic failure in this
pathology (25).
PINK1 serves as a key regulator of mitophagy,
operating through both canonical and non-canonical molecular
pathways. In the canonical pathway, PINK1 orchestrates
mitochondrial quality control by phosphorylating ubiquitin chains
at Ser65 residues, thereby recruiting parkin to initiate
ubiquitination-mediated marking of damaged mitochondria (41,42). In vitro models of K OA
(25), demonstrated
PINK1-dependent PARKIN regulation, with specific inhibition
decreasing PARKIN expression, while PINK1 overexpression restored
physiological expression levels. These findings confirm the
essential role of the PINK1-PARKIN axis in OA pathogenesis
(23). Notably, the present
investigation revealed a novel non-canonical regulatory mechanism:
In LPS-induced senescent chondrocytes, PINK1 overexpression
upregulated TUFm expression, as determined by western blot
analysis. This finding aligns with the report by Lin et al
(21) in 293T cells, where
activated PINK1 promotes phosphorylation of TUFm at Ser222, which
hinders the formation of the Atg5-Atg12 complex, thereby
maintaining mitophagic flux homeostasis. Mechanistically, this
dual-axis regulatory mechanism explains the mechanism by which
PINK1 overexpression synchronously enhances mitochondrial clearance
efficiency while mitigating autophagic stress-induced cellular
damage.
Notably, the present study provided novel
mechanistic insights into the anti-senescence effects of PINK1
overexpression, demonstrating its dependence on suppression of the
p38 MAPK/NF-κB signaling pathway through integrated omics and
molecular analyses. Transcriptomic profiling revealed marked
upregulation of the MAPK pathway components in K OA progression,
which was attenuated by lentiviral-mediated PINK1 overexpression.
Molecular validation demonstrated elevated p38 phosphorylation
levels in KOA chondrocytes, a phenomenon exacerbated by PINK1
inhibition. This regulatory association corroborates recent
cardiovascular research where cardiac-specific PINK1 overexpression
suppressed p38 signaling activation, thereby ameliorating
pathological remodeling in heart failure models (43-45). Subsequent investigations have
revealed that PINK1 deficiency exacerbates LPS-induced pathological
cascades, characterized by ROS overproduction, aberrant activation
of the p38 MAPK/NF-κB signaling pathway and mitochondrial
dysfunction (46,47). This establishes a
self-perpetuating triad of oxidative stress, inflammatory
activation and autophagic suppression (48). Notably, pharmacological
inhibition of p38 signaling significantly ameliorated these
pathological manifestations and disrupted this degenerative cycle,
thereby underscoring the key role of PINK1 in mitigating senescence
through suppression of the p38 MAPK/NF-κB pathway. This mechanism
demonstrates cross-organ corroboration with the findings of Luo
et al (49) and Qigen
et al (50) in premature
testicular failure research, who demonstrated that p38 MAPK
mediates age- and obesity-induced Leydig cell senescence. The dual
regulatory capacity of PINK1, which simultaneously enhances
mitochondrial clearance and mitigates inflammatory stress,
positions it as a key coordinator of cell homeostasis.
Overexpression of PINK1 promotes cartilage repair
and regeneration through multiple mechanisms. As a key regulator of
mitochondrial quality control, PINK1 overexpression enhances
mitophagy in chondrocytes, clearing damaged mitochondria and
maintaining cellular energy homeostasis (51,52). Studies have shown that PINK1
activates the PARKIN-dependent mitophagy pathway, decreases the
accumulation of ROS, and inhibits chondrocyte apoptosis and matrix
degradation (25,53). PINK1 overexpression upregulates
the synthesis of type II collagen and proteoglycans, facilitating
reconstruction of the cartilage ECM (54). In addition, PINK1 modulates
inflammatory responses by lowering the expression of inflammatory
cytokines such as IL-1β and TNF-α, thereby creating a favorable
microenvironment for cartilage repair (55). These mechanisms make PINK1 a
potential therapeutic target for degenerative cartilage
disease.
PINK1-mediated mitophagy and its downstream p38
MAPK/NF-κB signaling pathway are highly conserved in mammals. This
evolutionary conservation provides a biological basis for
translating findings from animal models to human clinical
applications (56,57). The PINK1/PARKIN pathway, as a
core mechanism of mitochondrial quality control, has been shown to
play a serve role in multiple age-related diseases, including human
Parkinson's disease and cardiac disorder (58,59). These observations suggest that
the mechanism by which PINK1 alleviates chondrocyte senescence by
inhibiting the p38 MAPK/NF-κB signaling pathway, may be present in
human patients with KOA as well (60).
However, the present study had limitations and
potential constraints. First, it lacked direct validation using
human clinical samples. Although immortalized human chondrocytes
were used for in vitro experiments, these cell lines may
lose some physiological characteristics of primary chondrocytes
after multiple passages, making it difficult to reflect the true
pattern of PINK1 expression in cartilage from patients with primary
KOA and its association with disease severity. Second, the present
study primarily focused on changes in cartilage tissue, with
insufficient overall assessment of other joint tissue involved in
KOA and the whole-knee pathological changes. As KOA is a disease of
the entire joint, interactions between these tissues are key to
disease progression. In addition, the DMM model is a post-traumatic
OA model that primarily simulates secondary OA caused by mechanical
instability, which may not represent the pathophysiological
processes of age-related primary KOA, particularly with respect to
aging-associated metabolic and inflammatory mechanisms. Future
studies should integrate clinical sample analyses, extend the
observation period, increase sample size and adopt multi-species
validation strategies to evaluate the clinical value of PINK1 in
the prevention and treatment of human KOA.
In summary, the present study demonstrated that
PINK1, a critical regulator of mitophagy, significantly alleviated
the pathological changes in K OA by ameliorating chondrocyte
senescence. This effect was mediated by inhibition of the p38
MAPK/NF-κB signaling pathway. Further studies are warranted to
elucidate the detailed molecular mechanisms underlying the
regulation by PINK1 on this signaling pathway.
Supplementary Data
Availability of data and materials
The data generated in the present study may be found
in the National Center for Biotechnology Information) under
accession number PRJNA1331976 or at the following URL: dataview.ncbi.nlm.nih.gov/object/PRJNA1331976)
Authors' contributions
SY and PW confirm the authenticity of all the raw
data. LJ conceived and designed the study. YZ performed
experiments; JL performed experiments; ZH performed experiments; HF
performed experiments; ZH contributed to analysis and
interpretation of data; ZZ contributed to analysis and
interpretation of data; XS contributed to analysis and
interpretation of data; PW contributed to analysis and
interpretation of data. SY edited the manuscript. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
Ethical approval was obtained from Institutional
Animal Care and Use Committee at Nanjing University of Chinese
Medicine, Nanjing, China. (approval no. 202409A051).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 82104893), Jiangsu Provincial
Medical Key Discipline (Laboratory) Cultivation Unit (grant no.
JSDW202252), Clinical Medical Innovation Center for Knee
Osteoarthritis of Traditional Chinese Medicine, Jiangsu Provincial
Hospital of Chinese Medicine (grant no. Y2023zx05), Knee
Osteoarthritis Specialized Clinical Research Institute and Nanjing
University of Chinese Medicine (grant no. LCZBYJYZZ2024-003).
References
|
1
|
Arden NK, Perry TA, Bannuru RR, Bruyère O,
Cooper C, Haugen IK, Hochberg MC, McAlindon TE, Mobasheri A and
Reginster JY: Non-surgical management of knee osteoarthritis:
Comparison of ESCEO and OARSI 2019 guidelines. Nat Rev Rheumatol.
17:59–66. 2021. View Article : Google Scholar
|
|
2
|
Latourte A, Kloppenburg M and Richette P:
Emerging pharmaceutical therapies for osteoarthritis. Nat Rev
Rheumatol. 16:673–688. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wallace IJ, Worthington S, Felson DT,
Jurmain RD, Wren KT, Maijanen H, Woods RJ and Lieberman DE: Knee
osteoarthritis has doubled in prevalence since the mid-20th
century. Proc Natl Acad Sci USA. 114:9332–9336. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang X, Yang B, Xu X, Zhang Z, Tao Z,
Zhang W, Zhang Z and Zhou X: Cellular senescence in skeletal
diseases: A bibliometric analysis from 2007 to 2024. Exp Gerontol.
209:1128572025. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cao X, Luo P, Huang J, Liang C, He J, Wang
Z, Shan D, Peng C and Wu S: Intraarticular senescent chondrocytes
impair the cartilage regeneration capacity of mesenchymal stem
cells. Stem Cell Res Ther. 10:862019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jeon OH, Kim C, Laberge RM, Demaria M,
Rathod S, Vasserot AP, Chung JW, Kim DH, Poon Y, David N, et al:
Local clearance of senescent cells attenuates the development of
post-traumatic osteoarthritis and creates a pro-regenerative
environment. Nat Med. 23:775–781. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu Z, Wang C, Wei S, Wu R, Zhao W, Zhao
X, Li Y and Yang Y: Benzophenone-3 drives osteoarthritis
pathogenesis by regulating chondrocyte senescence. Chem Biol
Interact. 421:1117572025. View Article : Google Scholar
|
|
8
|
Xu M, Bradley EW, Weivoda MM, Hwang SM,
Pirtskhalava T, Decklever T, Curran GL, Ogrodnik M, Jurk D, Johnson
KO, et al: Transplanted senescent cells induce an
osteoarthritis-like condition in mice. J Gerontol A Biol Sci Med
Sci. 72:780–785. 2017.
|
|
9
|
Liu Y, Zhang Z, Li T, Xu H and Zhang H:
Senescence in osteoarthritis: From mechanism to potential
treatment. Arthritis Res Ther. 24:1742022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wakale S, Wu X, Sonar Y, Sun A, Fan X,
Crawford R and Prasadam I: How are aging and osteoarthritis
related? Aging Dis. 14:592–604. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Palikaras K, Lionaki E and Tavernarakis N:
Mechanisms of mitophagy in cellular homeostasis, physiology and
pathology. Nat Cell Biol. 20:1013–1022. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fang G, Wen X, Jiang Z, Du X, Liu R, Zhang
C, Huang G, Liao W and Zhang Z: FUNDC1/PFKP-mediated mitophagy
induced by KD025 ameliorates cartilage degeneration in
osteoarthritis. Mol Ther. 31:3594–3612. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang N, Xing B, Peng R, Shang J, Wu B,
Xiao P, Lin S, Xu X and Lu H: Inhibition of Cpt1a alleviates
oxidative stress-induced chondrocyte senescence via regulating
mitochondrial dysfunction and activating mitophagy. Mech Ageing
Dev. 205:1116882022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cao H, Zhou X, Xu B, Hu H, Guo J, Wang M,
Li N and Jun Z: Advances in the study of mitophagy in
osteoarthritis. J Zhejiang Univ Sci B. 25:197–211. 2024.In English,
Chinese. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang L, Li M, Li X, Liao T, Ma Z, Zhang
L, Xing R, Wang P and Mao J: Characteristics of sensory innervation
in synovium of rats within different knee osteoarthritis models and
the correlation between synovial fibrosis and hyperalgesia. J Adv
Res. 35:141–151. 2021. View Article : Google Scholar
|
|
16
|
Hu S, Zhang C, Ni L, Huang C, Chen D, Shi
K, Jin H, Zhang K, Li Y, Xie L, et al: Stabilization of HIF-1α
alleviates osteoarthritis via enhancing mitophagy. Cell Death Dis.
11:4812020. View Article : Google Scholar
|
|
17
|
Ansari MY, Khan NM, Ahmad I and Haqqi TM:
Parkin clearance of dysfunctional mitochondria regulates ROS levels
and increases survival of human chondrocytes. Osteoarthritis
Cartilage. 26:1087–1097. 2018. View Article : Google Scholar :
|
|
18
|
Barazzuol L, Giamogante F, Brini M and
Calì T: PINK1/Parkin mediated mitophagy, Ca2+
signalling, and ER-mitochondria contacts in Parkinson's disease.
Int J Mol Sci. 21:17722020. View Article : Google Scholar
|
|
19
|
Zhou H, Wang X, Xu T, Gan D, Ma Z, Zhang
H, Zhang J, Zeng Q and Xu D: PINK1-mediated mitophagy attenuates
pathological cardiac hypertrophy by suppressing the mtDNA
release-activated cGAS-STING pathway. Cardiovasc Res. 121:128–142.
2025. View Article : Google Scholar
|
|
20
|
Rakovic A, Grünewald A, Voges L, Hofmann
S, Orolicki S, Lohmann K and Klein C: PINK1-interacting proteins:
Proteomic analysis of overexpressed PINK1. Parkinsons Dis.
2011:1539792011.PubMed/NCBI
|
|
21
|
Lin J, Chen K, Chen W, Yao Y, Ni S, Ye M,
Zhuang G, Hu M, Gao J, Gao C, et al: Paradoxical mitophagy
regulation by PINK1 and TUFm. Mol Cell. 80:607–620.e12. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shin HJ, Park H, Shin N, Kwon HH, Yin Y,
Hwang JA, Song HJ, Kim J, Kim DW and Beom J: Pink1-mediated
chondrocytic mitophagy contributes to cartilage degeneration in
osteoarthritis. J Clin Med. 8:18492019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jin Z, Chang B, Wei Y, Yang Y, Zhang H,
Liu J, Piao L and Bai L: Curcumin exerts chondroprotective effects
against osteoarthritis by promoting AMPK/PINK1/Parkin-mediated
mitophagy. Biomed Pharmacother. 151:1130922022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhuang H, Ren X, Zhang Y, Li H and Zhou P:
β-Hydroxybutyrate enhances chondrocyte mitophagy and reduces
cartilage degeneration in osteoarthritis via the
HCAR2/AMPK/PINK1/Parkin pathway. Aging Cell. 23:e142942024.
View Article : Google Scholar
|
|
25
|
Jie L, Shi X, Kang J, Fu H, Yu L, Tian D,
Mei W and Yin S: Protocatechuic aldehyde attenuates chondrocyte
senescence via the regulation of PTEN-induced kinase
1/Parkin-mediated mitochondrial autophagy. Int J Immunopathol
Pharmacol. 38:39463202412717242024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Glasson SS, Chambers MG, Van Den Berg WB
and Little CB: The OARSI histopathology initiative-recommendations
for histological assessments of osteoarthritis in the mouse.
Osteoarthritis Cartilage. 18(Suppl 3): S17–S23. 2010. View Article : Google Scholar
|
|
27
|
Merck: Pre-designed shRNA. https://www.sigmaaldrich.cn/CN/zh/semi-configurators/shrna?activeLink=productSearch.
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2 (−Delta Delta C (T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
29
|
Rim YA, Nam Y and Ju JH: The role of
chondrocyte hypertrophy and senescence in osteoarthritis initiation
and progression. Int J Mol Sci. 21:23582020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kuszel L, Trzeciak T, Richter M and
Czarny-Ratajczak M: Osteoarthritis and telomere shortening. J Appl
Genet. 56:169–176. 2015. View Article : Google Scholar :
|
|
31
|
Diekman BO, Sessions GA, Collins JA,
Knecht AK, Strum SL, Mitin NK, Carlson CS, Loeser RF and Sharpless
NE: Expression of p16INK4a is a biomarker of chondrocyte
aging but does not cause osteoarthritis. Aging Cell. 17:e127712018.
View Article : Google Scholar
|
|
32
|
Liu Y, Duan J, Dang Y, Hao R, Wang H, Tan
E, Wang R, Li Y, Zhang S, Wang Y, et al: Remodeling of senescent
macrophages in synovium alleviates trauma- and aging-induced
osteoarthritis. Bioact Mater. 55:42–56. 2026.
|
|
33
|
Jiang T, Su S, Tian R, Jiao Y, Zheng S,
Liu T, Yu Y, Hua P, Cao X, Xing Y, et al: Immunoregulatory
orchestrations in osteoarthritis and mesenchymal stromal cells for
therapy. J Orthop Translat. 55:38–54. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Harman D: The biologic clock: The
mitochondria? J Am Geriatr Soc. 20:145–147. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xie Z, Zhang X, Li Y and Zhu R:
Mitochondrial dysfunction drives cellular senescence: Molecular
mechanisms of inter-organelle communication. Exp Gerontol.
211:1129132025. View Article : Google Scholar
|
|
36
|
Luo Y, Xu H, Xiong S and Ke J:
Understanding myalgic encephalomyelitis/chronic fatigue syndrome
physical fatigue through the perspective of immunosenescence. Compr
Physiol. 15:e700562025. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Abate M, Festa A, Falco M, Lombardi A,
Luce A, Grimaldi A, Zappavigna S, Sperlongano P, Irace C, Caraglia
M and Misso G: Mitochondria as playmakers of apoptosis, autophagy
and senescence. Semin Cell Dev Biol. 98:139–153. 2020. View Article : Google Scholar
|
|
38
|
Guo QQ, Wang SS, Jiang XY, Xie XC, Zou Y,
Liu JW, Guo Y, Li YH, Liu XY, Hao S, et al: Mitochondrial ROS
triggers mitophagy through activating the DNA damage response
signaling pathway. Proc Natl Acad Sci USA. 122:e25028411222025.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Abdeahad H, Moreno DG, Bloom S, Norman L,
Lesniewski LA and Donato AJ: MitoQ reduces senescence burden in
Doxorubicin-treated endothelial cells by reducing mitochondrial ROS
and DNA damage. Am J Physiol Heart Circ Physiol. September
30–2025.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hamacher-Brady A and Brady NR: Mitophagy
programs: Mechanisms and physiological implications of
mitochondrial targeting by autophagy. Cell Mol Life Sci.
73:775–795. 2016. View Article : Google Scholar :
|
|
41
|
Tufi R, Clark EH, Hoshikawa T, Tsagkaraki
C, Stanley J, Takeda K, Staddon JM and Briston T: High-content
phenotypic screen to identify small molecule enhancers of
Parkin-dependent ubiquitination and mitophagy. SLAS Discov.
28:73–87. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rusilowicz-Jones EV, Jardine J, Kallinos
A, Pinto-Fernandez A, Guenther F, Giurrandino M, Barone FG,
McCarron K, Burke CJ, Murad A, et al: USP30 sets a trigger
threshold for PINK1-PARKIN amplification of mitochondrial
ubiquitylation. Life Sci Alliance. 3:e2020007682020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang H, Xu T, Mei X, Zhao Q, Yang Q, Zeng
X, Ma Z, Zhou H, Zeng Q, Xu D and Ren H: PINK1 modulates Prdx2 to
reduce lipotoxicity-induced apoptosis and attenuate cardiac
dysfunction in heart failure mice with a preserved ejection
fraction. Clin Transl Med. 15:e701662025. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sedighi S, Liu T, O'Meally R, Cole RN,
O'Rourke B and Foster DB: Inhibition of cardiac p38 Highlights The
Role Of The Phosphoproteome In Heart Failure Progression. ACS
Omega. 10:36082–36097. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Meco M, Giustiniano E, Nisi F, Zulli P and
Agosteo E: MAPK, PI3K/Akt pathways, and GSK-3β activity in severe
acute heart failure in intensive care patients: An updated review.
J Cardiovasc Dev Dis. 12:2662025.
|
|
46
|
Su Z, Shu H, Huang X, Ding L, Liang F, Xu
Z, Zhu Z, Chen M, Wang X, Li G, et al: Rhapontigenin attenuates
neurodegeneration in a parkinson's disease model by downregulating
mtDNA-cGAS-STING-NF-κB-mediated neuroinflammation via
PINK1/DRP1-dependent microglial mitophagy. Cell Mol Life Sci.
82:3372025. View Article : Google Scholar
|
|
47
|
Cao B, Fang L, Zhang Y, Lin C, Liu P,
Zhang H, Fan O, Xu M, Qin Z and Wang C: MitoQ alleviates
m.3243A>G-induced mitochondrial dysfunction by stabilizing PINK1
and enhancing mitophagy. J Genet Genomics. S1673-8527(25)00229-2.
August 22–2025.Epub ahead of print. View Article : Google Scholar
|
|
48
|
Wang FS, Kuo CW, Ko JY, Chen YS, Wang SY,
Ke HJ, Kuo PC, Lee CH, Wu JC, Lu WB, et al: Irisin mitigates
oxidative stress, chondrocyte dysfunction and osteoarthritis
development through regulating mitochondrial integrity and
autophagy. Antioxidants (Basel). 9:8102020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Luo D, Qi X, Xu X, Yang L, Yu C and Guan
Q: Involvement of p38 MAPK in Leydig cell aging and age-related
decline in testosterone. Front Endocrinol (Lausanne).
14:10882492023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Qigen X, Haiming C, Kai X, Yong G and
Chunhua D: Prenatal DEHP exposure induces premature testicular
aging by promoting leydig cell senescence through the MAPK
signaling pathways. Adv Biol (Weinh). 7:e23001302023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang Z, Huang T, Chen X, Chen J, Yuan H,
Yi N, Miao C, Sun R and Ni S: Acetyl zingerone inhibits chondrocyte
pyroptosis and alleviates osteoarthritis progression by promoting
mitophagy through the PINK1/parkin signaling pathway. Int
Immunopharmacol. 161:1150552025. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bocheng L, Yongfu C, Junjie C, Ziqi L,
Lina G, Tingli Q, Li T and Qian Z: FoxO1/PINK1/Parkin-dependent
mitophagy mediates the chondroprotective effect of Guzhi Zengsheng
Zhitong decoction in osteoarthritis. Phytomedicine.
148:157322September 26–2025.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kang J, Jie L, Fu H, Zhang L, Lu G, Yu L,
Tian D, Liao T, Yin S, Xin R and Wang P: Adipose mesenchymal stem
cells derived exosomes ameliorates KOA Cartilage damage and
inflammation by activation of PINK1-mediated mitochondrial
autophagy. FASEB J. 39:e708112025. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wu T, Wang Y, Shen B, Guo K, Zhu Z, Liang
Y, Zeng J and Wu D: FBXO2 alleviates intervertebral disc
degeneration via dual mechanisms: Activating PINK1-Parkin mitophagy
and ubiquitinating LCN2 to suppress ferroptosis. Adv Sci (Weinh).
12:e061502025. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Li Q, Gu H, Song K, Kong X, Li Y, Liu Z,
Meng Q, Liu K, Li X, Xie Q, et al: Lentinan rewrites extracellular
matrix homeostasis by activating mitophagy via mTOR/PINK1/Parkin
pathway in cartilage to alleviating osteoarthritis. Int J Biol
Macromol. 322:1469002025. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Mohanan A, Washimkar KR and Mugale MN:
Unraveling the interplay between vital organelle stress and
oxidative stress in idiopathic pulmonary fibrosis. Biochim Biophys
Acta Mol Cell Res. 1871:1196762024. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Li Y, Chen H, Xie X, Yang B, Wang X, Zhang
J, Qiao T, Guan J, Qiu Y, Huang YX, et al: PINK1-mediated mitophagy
promotes oxidative phosphorylation and redox homeostasis to induce
drug-tolerant persister cancer cells. Cancer Res. 83:398–413. 2023.
View Article : Google Scholar
|
|
58
|
Zheng Y, Wei W, Wang Y, Li T, Wei Y and
Gao S: Gypenosides exert cardioprotective effects by promoting
mitophagy and activating PI3K/Akt/GSK-3β/Mcl-1 signaling. PeerJ.
12:e175382024. View Article : Google Scholar
|
|
59
|
Liu H, Ho PW, Leung CT, Pang SY, Chang
EES, Choi ZY, Kung MH, Ramsden DB and Ho SL: Aberrant mitochondrial
morphology and function associated with impaired mitophagy and
DNM1L-MAPK/ERK signaling are found in aged mutant Parkinsonian
LRRK2R1441G mice. Autophagy. 17:3196–3220. 2021.
View Article : Google Scholar
|
|
60
|
Wang W, Wang Q, Li W, Xu H, Liang X, Wang
W, Li N, Yang H, Xu Y, Bai J, et al: Targeting APJ drives
BNIP3-PINK1-PARKIN induced mitophagy and improves systemic
inflammatory bone loss. J Adv Res. 76:655–668. 2025. View Article : Google Scholar
|