Introduction
Proteins are fundamental components of cells and
form the essential material basis for sustaining and regulating
life processes. Disturbances in protein abundance, folding,
function or localization can disrupt physiological homeostasis,
underscoring the importance of maintaining protein homeostasis, or
proteostasis (1,2). Proteostasis is maintained by a
highly coordinated network of quality control mechanisms that
regulate protein synthesis, folding, modification, trafficking and
degradation, thereby ensuring the stability and functionality of
the proteome (3). When this
delicate balance is perturbed by genetic mutations, environmental
stress or aging, misfolded and aggregated proteins accumulate,
driving proteotoxic stress, organelle dysfunction and, ultimately,
disease (4). Proteostasis
collapse has been recognized as a unifying driver of diverse human
disorders, including neurodegenerative diseases, malignancies,
cardiovascular dysfunction and metabolic syndromes.
Against this backdrop, FK506-binding proteins
(FKBPs) have emerged as particularly intriguing regulators of
proteostasis. FKBPs are members of the immunophilin family, which
also comprises cyclophilins that share conserved peptidyl-prolyl
cis-trans isomerase activity but differ in structural domains and
functional specialization. Defined by a conserved peptidyl-prolyl
cis-trans isomerase (PPIase) domain and diversified by additional
modules conferring organelle targeting and chaperone interactions,
FKBPs extend far beyond their classical role as
immunosuppressant-binding proteins (5-7).
Increasing evidence shows that they actively shape proteostasis
networks by accelerating conformational maturation, scaffolding
protein complexes, modulating stress responses and directing
selective degradation, positioning them as key molecular links
between proteostasis regulation and disease pathogenesis (8-12).
Despite substantial progress in characterizing
individual FKBPs, no review has systematically integrated their
diverse roles in proteostasis regulation across multiple disease
contexts, yet, to the best of our knowledge. This article provides
the first unified framework, linking FKBP structure to proteostatic
function and disease relevance. In cancer, FKBPs adjust
translation, folding and degradation pathways to sustain the high
protein load of malignant cells, driving proteome remodeling and
tumor progression (13). In
neurodegenerative diseases, FKBPs influence the conformational fate
of pathogenic proteins by regulating folding and aggregation
processes, thereby determining neuronal survival and function
(14). In cardiovascular
disease, FKBPs stabilize the conformation of ion channel complexes
to preserve calcium signaling homeostasis, ensuring precise cardiac
contraction and electrical activity (15). In metabolic regulation, they act
as scaffolds to fine-tune kinase phosphorylation, integrating
energy sensing with metabolic signaling to maintain systemic
balance (16). These functions
not only underscore the multifaceted role of FKBPs as central
regulators of proteostasis but also establish them as critical
bridges linking proteostatic regulation to disease mechanisms. The
present study proposes targeting FKBPs as a novel target to correct
proteostasis imbalance and halt disease progression, thereby
opening new avenues for precision therapies across multiple organ
systems.
Overview of proteostasis
Proteostasis is the cellular process that maintains
a dynamic balance of protein synthesis, folding, modification,
trafficking, and degradation, ensuring proteins remain in the
proper quantity and functional conformation (8). Its core mechanisms are tightly
coordinated. Proteins are synthesized on ribosomes as nascent
polypeptides that are often unstable or partially folded, requiring
molecular chaperones and foldases for correct folding or assembly
into multimeric complexes (Fig.
1A). Before becoming functionally active, many proteins undergo
post-translational modifications such as phosphorylation,
acetylation, or ubiquitination, which regulate their stability,
activity, and interactions (Fig.
1B). Proteins must also be directed with precision to
organelles such as the endoplasmic reticulum (ER), mitochondria, or
nucleus to execute specific functions. When proteins misfold,
become damaged, or accumulate abnormally, two major degradation
systems maintain quality control: the ubiquitin-proteasome system
(UPS), which removes short-lived and defective proteins, and the
autophagy-lysosome pathway (ALP), which clears protein aggregates
and damaged organelles. Together, these mechanisms uphold the
dynamic equilibrium of the proteome (1,17).
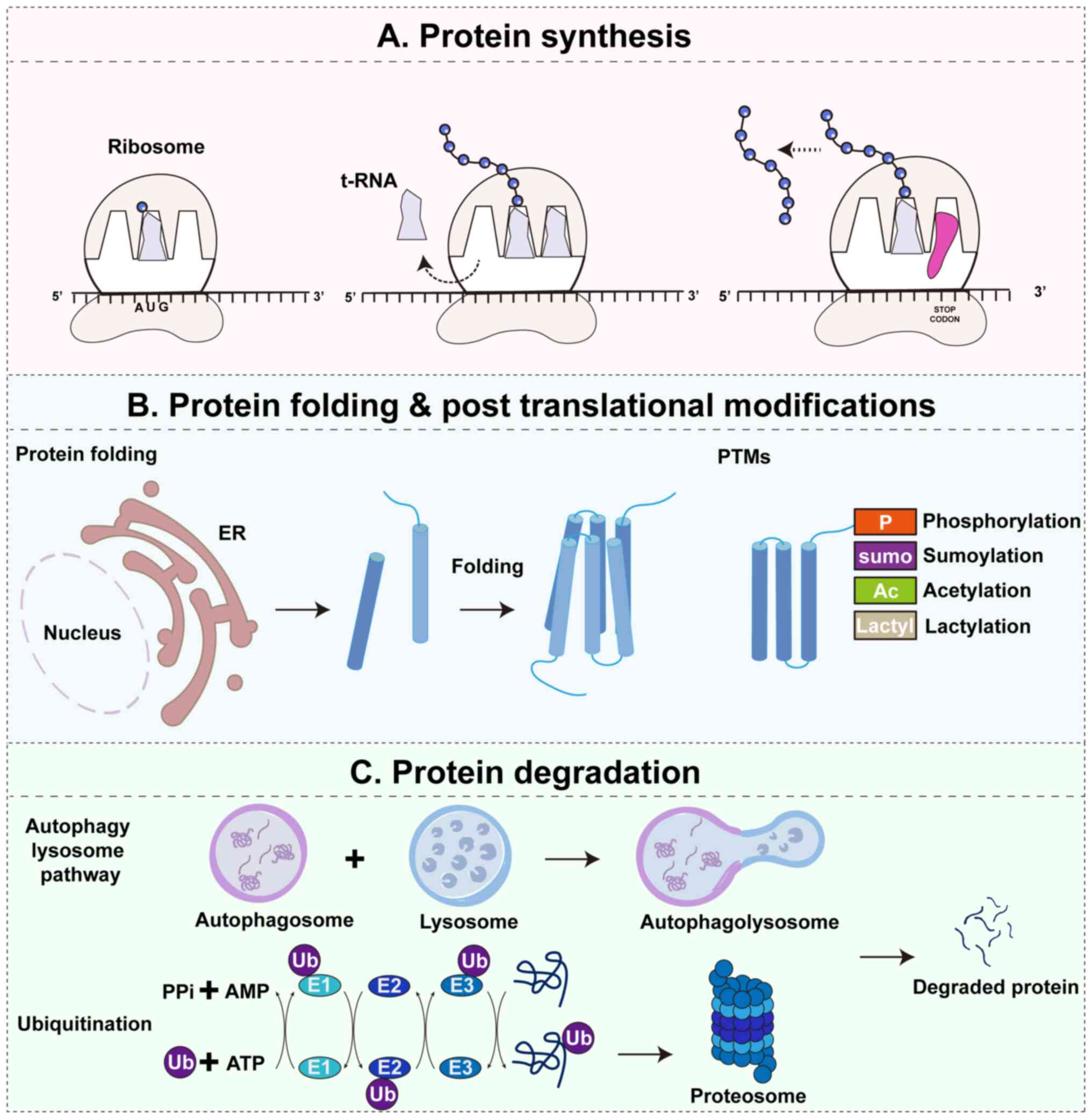 | Figure 1Overview of proteostasis and its core
pathways. Proteostasis is maintained through a dynamic balance of
protein synthesis, folding and post-translational modifications,
and degradation. (A) Protein synthesis. Ribosomes translate mRNA
into nascent polypeptide chains, which often require assistance
from molecular chaperones and foldases to achieve stable
structures. (B) Protein folding and post-translational
modifications. Newly synthesized proteins fold in the ER and
undergo PTMs, such as phosphorylation, sumoylation, acetylation and
lactylation, which regulate protein stability, activity and
interactions. (C) Protein degradation. Misfolded, damaged or
surplus proteins are eliminated through two major pathways: The
UPS, where substrates are sequentially ubiquitinated by E1, E2 and
E3 enzymes and degraded by the 26S proteasome into peptides; and
the ALP, where cytoplasmic substrates, including protein aggregates
and damaged organelles, are sequestered into autophagosomes, fused
with lysosomes and degraded into reusable biomolecules. Together,
these pathways establish a quality control cycle that preserves
proteome integrity and cellular homeostasis under both
physiological and stress conditions. P, phosphorylation; sumo,
sumoylation; Ac, acetylation; Lactyl, lactylation; UPS,
ubiquitin-proteasome system; ALP, autophagy-lysosome pathway; E1,
ubiquitin-activating enzyme; E2, ubiquitin-conjugating enzyme; E3,
ubiquitin ligase; ER, endoplasmic reticulum; AMP, adenosine
monophosphate; PPi, inorganic pyrophosphate; PTMs,
post-translational modifications; Ub, ubiquitin. |
The UPS primarily eliminates short-lived or
misfolded monomeric proteins. Substrates are first tagged with
ubiquitin chains through a cascade involving E1 activating enzymes,
E2 conjugating enzymes and E3 ligases. These ubiquitinated proteins
are then directed to the 26S proteasome, where they are unfolded
and degraded into short peptides, allowing rapid protein turnover
and removal of potentially toxic species (18). In parallel, the ALP is
responsible for the clearance of larger substrates, including
protein aggregates, damaged organelles and long-lived proteins.
During this process, isolation membranes form autophagosomes that
engulf the target substrates, which subsequently fuse with
lysosomes. Hydrolytic enzymes within the lysosome degrade the
contents into amino acids and lipids for cellular reuse (19). Together, UPS and ALP establish an
integrated quality control cycle that prevents harmful aggregate
accumulation while maintaining metabolic balance (Fig. 1C).
Under conditions of high translational load or
environmental stress, large amounts of misfolded or unfolded
proteins accumulate in the ER lumen, disturbing folding balance and
triggering the unfolded protein response (UPR) (20). The initiation of UPR depends on
the molecular chaperone glucose-regulated protein 78/binding
immunoglobulin protein (BiP), which serves as a central sensor.
Under normal conditions, BiP binds to the three principal ER stress
receptors, inositol-requiring enzyme 1α (IRE1α), PKR-like ER kinase
(PERK) and activating transcription factor 6 (ATF6), keeping them
inactive. When unfolded proteins accumulate, BiP preferentially
associates with these substrates and dissociates from the
receptors, thereby activating downstream signaling pathways.
Activated IRE1α mediates the unconventional splicing of spliced
X-box binding protein 1 (XBP1) mRNA, producing the transcription
factor XBP1s, which upregulates chaperones, foldases and
ER-associated degradation (ERAD) genes to enhance folding and
clearance capacity. PERK phosphorylates eIF2α to reduce global
translation, while selectively promoting ATF4 translation, which
activates antioxidant, autophagy and metabolic pathways. ATF6 is
transported to the Golgi apparatus, where proteolytic cleavage
releases its cytosolic fragment, which translocates to the nucleus
to induce transcription of chaperones and ERAD-related genes
(20). Together, these
mechanisms restore ER folding and degradation balance under
moderate stress, allowing cells to adapt. However, when stress is
excessive or persistent, UPR signaling shifts toward apoptosis,
e.g. through PERK-ATF4-C/EBP homologous protein (CHOP) induction,
thereby converting proteostatic imbalance into pathological injury
(20). Thus, the UPR functions
as a critical protective mechanism of proteostasis, but also as a
decisive switch between adaptation and cell death, with BiP acting
as the essential gatekeeper of this process.
Altogether, these processes form a highly integrated
proteostasis network that safeguards protein quality and cellular
function. Breakdown of this network leads to the accumulation of
toxic species, driving pathology in neurodegenerative, oncogenic,
cardiovascular and metabolic disorders. Against this backdrop,
FKBPs have emerged as critical regulators that interface with
multiple proteostatic pathways, highlighting their importance in
both physiological adaptation and disease progression.
Structural basis of FKBP functions in
protein homeostasis
The FKBP family shares a conserved core domain, the
PPIase domain (21). This domain
catalyzes the cis-trans isomerization of proline residues in
polypeptide chains, accelerating folding kinetics and ensuring that
nascent proteins rapidly reach their correct conformation (22). This activity is critical for
maintaining protein homeostasis, as proline isomerization often
represents a rate-limiting step in folding. Under stress or high
translational load, PPIase activity facilitates timely restoration
of folding equilibrium (23).
The smallest members, such as FKBP12 and FKBP12.6, consist almost
entirely of the PPIase domain and interact directly with substrates
or complexes, modulating their stability and function through
conformational control (24).
Larger members often contain multiple PPIase domains, for instance,
FKBP52 has two and FKBP65 has four, potentially broadening
substrate specificity or enabling cooperative folding of
multidomain proteins (25).
Beyond the PPIase core, numerous FKBPs possess
additional functional modules that expand their regulatory
capacity. The most prominent is the tetratricopeptide repeat (TPR)
domain, a 34-amino acid tandem repeat that mediates protein-protein
interactions and is found in proteins such as FKBP51 and FKBP52
(26). The TPR domain enables
specific binding to the molecular chaperone heat shock protein
(Hsp)90, integrating FKBPs into chaperone complexes. Hsp90 is a
highly conserved and ubiquitously expressed eukaryotic chaperone
that, unlike many other chaperones, primarily acts on partially or
fully folded proteins (27).
Through ATP-dependent conformational changes, Hsp90 maintains the
functional state of client proteins and stabilizes diverse
signaling proteins, receptors and transcription factors (28). This association allows FKBPs not
only to catalyze substrate folding independently but also to
contribute to protein maturation, conformational maintenance and
complex stability within the chaperone network (29). FKBP51 and FKBP52 act as molecular
scaffolds within this system. FKBP52 promotes hormone receptor
maturation and facilitates their active transport to the nucleus,
whereas FKBP51 modulates complex conformation or recruit
phosphatases to fine-tune signaling output (30,31). These activities directly
influence protein complex stability, subcellular localization and
signal transduction, thereby exerting precise control over protein
homeostasis.
Certain FKBPs contain other specialized domains that
confer distinct subcellular localization and regulatory
specificity. FKBP13, FKBP19, FKBP22, FKBP23, FKBP60 and FKBP65
possess N-terminal ER signal peptides and localize to the ER lumen,
where they regulate the folding and assembly of secretory and
membrane proteins (32-35). FKBP25 contains a DNA-binding
domain, enabling it to participate in transcriptional regulation
and chromatin remodeling (36).
FKBP38 features a transmembrane anchor that targets it to
mitochondria, where it recruits anti-apoptotic Bcl-2 family
proteins to regulate apoptosis and mitophagy (7,37). These additional domains extend
the influence of FKBPs to multiple organelles and signaling
networks, allowing them to maintain protein homeostasis across
diverse cellular contexts (Fig.
2).
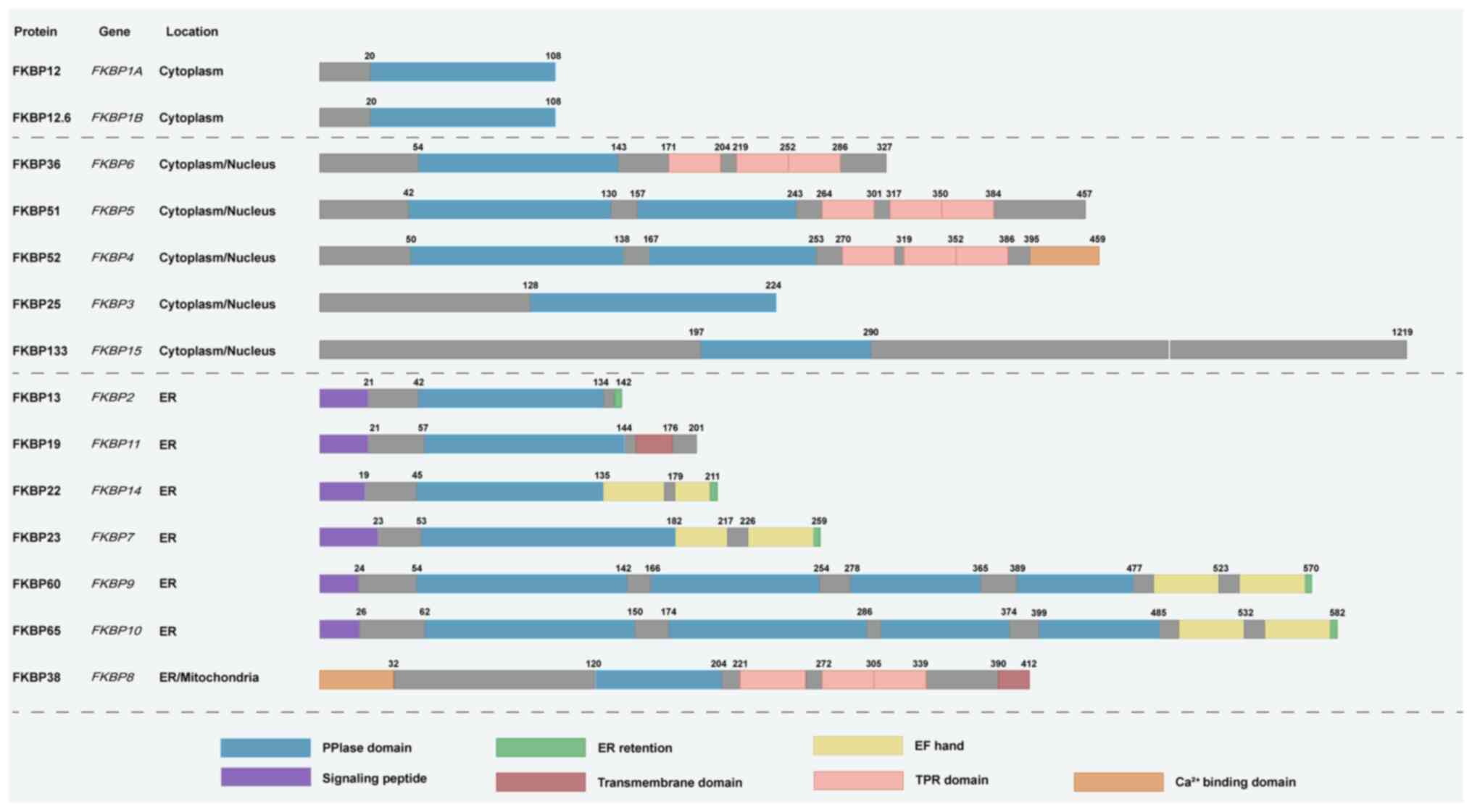 | Figure 2Structural diversity and subcellular
localization of FKBP family members. Schematic representation of
FKBPs, organized by their predominant localization in the
cytoplasm/nucleus or ER/mitochondria. The figure illustrates the
modular architecture of FKBPs and highlights how distinct domains
support their functional diversity in protein homeostasis. All
family members share the conserved PPIase domain, which catalyzes
proline isomerization to accelerate protein folding and
conformational stabilization. Several larger members, such as
FKBP51 and FKBP52, also contain TPR domains that mediate docking to
heat shock protein 90, enabling scaffold functions in protein
folding, complex stabilization and nuclear receptor trafficking.
Members including FKBP13, FKBP19, FKBP22, FKBP23, FKBP60 and FKBP65
harbor N-terminal ER signal peptides and ER retention motifs, which
target them to the ER lumen for the folding and assembly of
secretory and membrane proteins. FKBP38 contains a C-terminal
transmembrane anchor that localizes it to mitochondria and
facilitates anti-apoptotic Bcl-2 recruitment, linking FKBPs to
apoptosis and mitophagy regulation. Additional modules such as
EF-hand motifs and Ca2+-binding domains provide
responsiveness to calcium signaling, further expanding functional
versatility. Together, this structural heterogeneity enables FKBPs
to regulate protein folding, modification, trafficking and
degradation across diverse subcellular compartments. PPIase,
peptidyl-prolyl cis-trans isomerase; TPR, tetratricopeptide repeat;
ER, endoplasmic reticulum; EF-hand, helix-loop-helix
calcium-binding motif; FKBPs, FK506-binding proteins. |
In summary, the structural diversity of FKBPs
underpins their multilayered roles in protein homeostasis. The
PPIase domain directly modulates folding kinetics and
conformational stability. The TPR domain connects FKBPs to the
molecular chaperone network, mediating complex assembly and
maintenance. Specialized domains confer subcellular specificity and
functional diversification. Through coordinated action of these
modular elements, FKBPs regulate folding, complex stability,
localization and degradation, providing a structural and functional
basis for maintaining protein homeostasis in physiological and
stress conditions, as well as in disease states. In this review,
FKBP nomenclature refers to the protein products unless otherwise
specified. For clarity, gene symbols such as FKBP7,
FKBP9 and FKBP10 are used to denote their
corresponding protein products (FKBP7, FKBP9, FKBP10). This
convention is adopted to maintain consistency with other family
members (e.g., FKBP12, FKBP51, FKBP52), which are widely recognized
by their protein names.
FKBPs coordinate proteostasis networks to
drive tumor progression
Protein homeostasis is essential for tumor cells to
survive the proteotoxic stress generated by rapid proliferation and
harsh microenvironments. To cope with the increased burden of
protein synthesis and quality control, cancer cells rely on finely
tuned proteostasis networks that regulate folding, stability,
signaling and degradation (38).
Members of the FKBP family have emerged as central regulators in
this process, leveraging their structural modules to influence
distinct layers of proteostasis. Certain FKBPs within the ER
safeguard protein folding and translational balance, others
integrate post-translational modifications and selective
degradation to adapt the tumor microenvironment, while still others
stabilize nuclear receptors to amplify hormone-driven
proliferation. Together, these multifaceted functions highlight
FKBPs as key molecular nodes linking proteostasis to cancer
progression.
ER-resident FKBPs maintain ER
proteostasis to promote tumor invasion and metastasis
The ER is the principal site for protein synthesis,
folding and quality control, and its homeostasis is indispensable
for sustaining the rapid growth and survival of cancer cells.
Disruption of ER proteostasis caused by high translational demand
and oncogenic stress activates adaptive mechanisms such as the UPR,
which enables tumor cells to tolerate proteotoxic stress and resist
apoptosis. Several FKBPs localize to the ER lumen. Among them,
FKBP7, FKBP9 and FKBP10 (hereafter referring to the protein) have
been most extensively studied in the context of tumor biology.
FKBP9 and FKBP7 help tumor cells adapt to increased
protein synthesis and folding pressure by regulating the ER
proteostasis network. FKBP9 forms a complex with the molecular
chaperone BiP to support correct protein folding and assembly,
maintaining folding equilibrium within the ER. In glioblastoma,
FKBP9 inhibits the IRE1α-XBP1 signaling pathway and suppresses
CHOP-mediated apoptosis, preventing overactivation of the UPR and
enhancing resistance to ER stress (39). FKBP9 expression correlates
positively with BiP levels, and their co-expression is associated
with poor prognosis, underscoring its key role in ER homeostasis
(40).
FKBP7 contributes to proteostasis and extends its
influence to the tumor microenvironment. In pancreatic ductal
adenocarcinoma, FKBP7 is highly expressed in cancer-associated
fibroblasts (CAFs). By competing with BiP, FKBP7 alters the
secretion of collagen subtypes, reducing type I and increasing type
IV collagen. This promotes a dense extracellular matrix that
restricts immune infiltration and supports tumor invasion (41). These functions demonstrate that
FKBP7 modulates both ER stress adaptation and extracellular matrix
remodeling (Fig. 3).
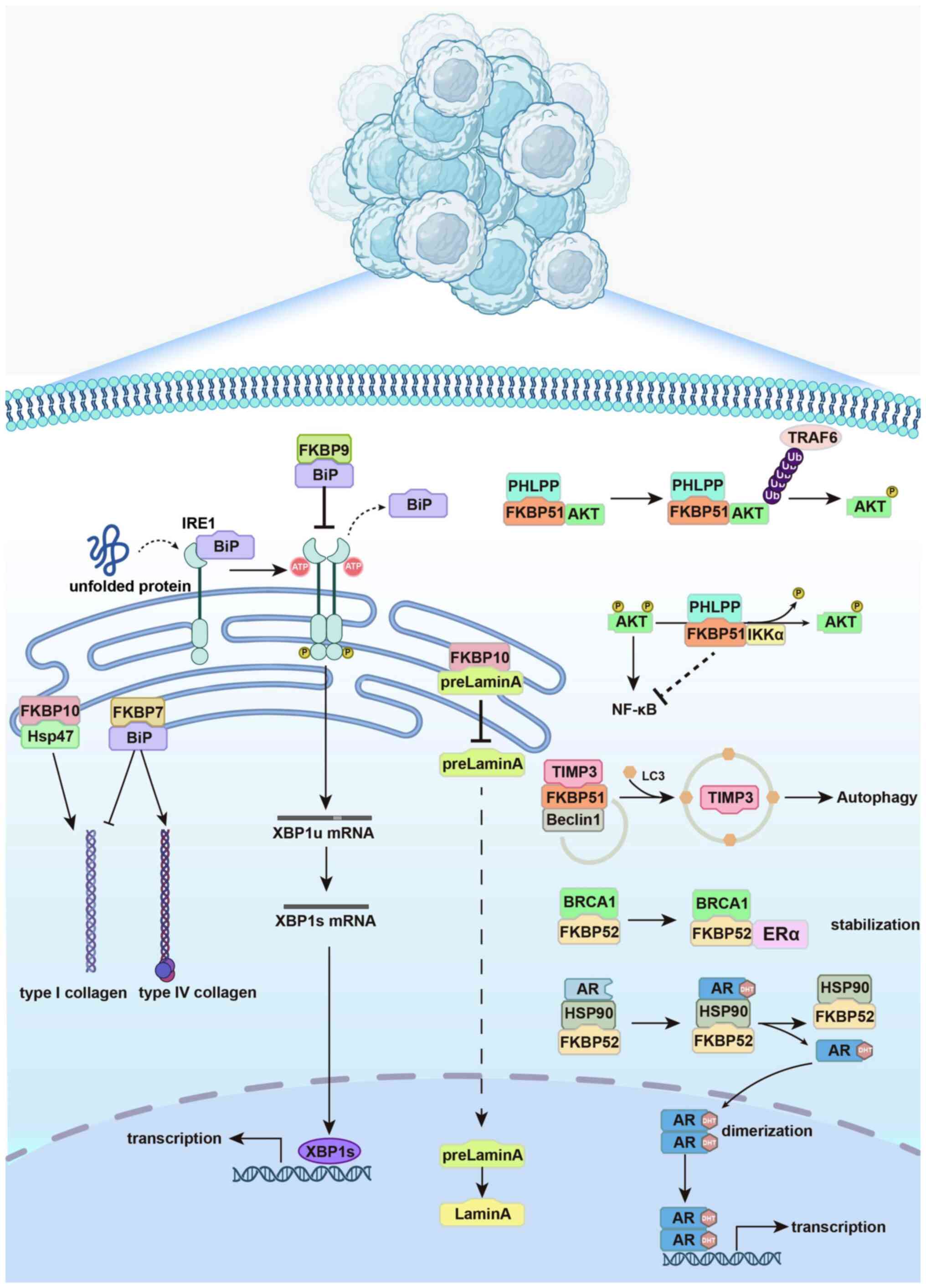 | Figure 3ER-resident and cytoplasmic FKBPs
regulate proteostasis to support tumor progression. Schematic
illustration of how representative FKBPs contribute to protein
homeostasis in cancer. In the ER, FKBP9 binds to BiP to maintain
folding equilibrium and suppress excessive IRE1α-XBP1 activation,
protecting cells from ER stress-induced apoptosis. FKBP7 interacts
with BiP in cancer-associated fibroblasts to modulate collagen
subtype secretion, favoring extracellular matrix remodeling and
tumor invasion. FKBP10 regulates substrate maturation and
localization, including retention of prelamin A in the ER and
stabilization of type I procollagen through cooperation with Hsp47,
and also supports translational efficiency at the ribosome. Outside
the ER, FKBP51 functions as a scaffold that shapes
post-translational modifications and autophagic turnover: It
recruits PHLPP to regulate AKT dephosphorylation, promotes Akt
ubiquitination via TRAF6 and directs TIMP3 degradation through the
Beclin1 complex, thereby modulating survival signaling and
microenvironment remodeling. FKBP52 acts as an Hsp90 co-chaperone
that stabilizes steroid hormone receptors such as AR and ERα,
facilitates their nuclear transport via dynein and enhances
transcriptional activation of oncogenic programs. Collectively,
these mechanisms highlight how distinct FKBPs integrate ER stress
responses, protein folding, post-translational regulation and
receptor signaling to maintain proteostasis and promote malignant
progression. FKBP, FK506-binding protein; ER, endoplasmic
reticulum; BiP, binding immunoglobulin protein (GRP78); IRE1α,
inositol-requiring enzyme 1α; XBP1s, X-box binding protein 1
spliced isoform; UPR, unfolded protein response; CAF,
cancer-associated fibroblast; PPIase, peptidyl-prolyl cis-trans
isomerase; AKT, protein kinase B; PHLPP, PH domain and leucine-rich
repeat protein phosphatase; TRAF6, TNF receptor-associated factor
6; TIMP3, tissue inhibitor of metalloproteinases 3; AR, androgen
receptor; ERα, estrogen receptor α; Hsp, heat shock protein; IKKα,
IκB kinase α; DHT, dihydrotestosterone; Ub, ubiquitin; BRCA1,
breast cancer 1, early onset. |
Unlike FKBP9 and FKBP7, FKBP10 plays a distinct role
in protein homeostasis that is less dependent on classical ER
stress signaling. FKBP10 primarily contributes to proteostasis by
regulating substrate folding and subcellular localization. Through
its PPIase domain, FKBP10 binds directly to specific client
proteins to influence their conformational maturation and
intracellular trafficking. In bladder cancer, FKBP10 binds prelamin
A, leading to its retention in the ER and preventing its
translocation into the nucleus, thereby disrupting Lamin A
formation and nuclear structure (42). This results in nuclear atypia and
enhances the migratory and invasive capacity of cancer cells. In
glioma, FKBP10 interacts with Hsp47 via its third PPIase domain to
promote the folding and stabilization of type I procollagen, which
directly facilitates extracellular matrix organization and the
establishment of a tumor-supportive microenvironment (10).
Beyond its role in folding and localization, FKBP10
also supports translational homeostasis in highly proliferative
cancer cells. Its conserved PPIase domain catalyzes the
isomerization of proline residues in nascent polypeptides,
accelerating translation elongation, particularly for proline-rich
ribosomal and structural proteins. Loss of FKBP10 impairs this
isomerization process, leading to ribosomal stalling at proline
motifs and reduced synthesis of proline-rich proteins, underscoring
the dependence of such proteins on FKBP10-mediated PPIase activity.
In non-small cell lung cancer (NSCLC), FKBP10 localizes to the
ribosomal catalytic center, reinforcing its function in supporting
translation (43). Loss of
FKBP10 or impairment of its enzymatic activity reduces translation
efficiency, impairs cell cycle progression and induces apoptosis.
FKBP10 is upregulated in multiple malignancies, including NSCLC,
colorectal cancer, renal cell carcinoma, bladder cancer and glioma,
and is associated with poor prognosis (44-46). Its knockdown not only inhibits
proliferation and migration but also sensitizes tumor cells to
chemotherapy and targeted therapies, highlighting its potential as
a therapeutic target (Fig.
3).
In conclusion, FKBP7, FKBP9 and FKBP10 are
ER-resident FKBPs that regulate proteostasis through distinct
mechanisms. FKBP9 and FKBP7 primarily participate in unfolded
protein responses and protein folding control, while FKBP10 governs
translational efficiency and substrate localization. These proteins
enable cancer cells to manage translational stress and maintain
proteome integrity under oncogenic pressure, linking ER
proteostasis to malignant progression. Targeting the functional
domains of these FKBPs may offer promising therapeutic strategies
for cancers characterized by dysregulated protein homeostasis and
elevated ER stress.
FKBP51 integrates post-translational
modifications (PTMs) and selective degradation to promote
microenvironment remodeling
PTMs are key regulatory mechanisms that control
protein function, stability and subcellular localization. They play
a critical role in maintaining cellular homeostasis and in
coordinating responses to external stimuli. In the tumor
microenvironment, PTMs are extensively involved in signal
transduction, cell cycle regulation, metabolic reprogramming and
immune evasion (47).
Dysregulation of PTMs is closely linked to tumor initiation,
progression and treatment resistance.
FKBP51 (also known as FKBP5) has emerged as a
critical regulator that integrates PTM-dependent signaling with
protein stability and cellular adaptation. FKBP51 contributes to
tumor progression by modulating phosphorylation, ubiquitination and
acetylation of key signaling proteins, thereby shaping the
functional output of oncogenic pathways.
One of the core functions of FKBP51 is to modulate
PTMs of key signaling proteins, maintaining the integrity of
signaling complexes and regulating downstream signaling outputs. A
well-characterized example is its bidirectional regulation of the
Akt pathway. In melanoma, FKBP51 interacts with Hsp90 to enhance
K63-linked ubiquitination of Akt, which increases Akt stability and
activity, thereby activating downstream effectors such as P70S6K
and Cyclin D1 to promote cell proliferation (48). By contrast, in prostate and
pancreatic cancers, FKBP51 enhances its interaction with PH domain
and leucine-rich repeat protein phosphatase (PHLPP), facilitating
dephosphorylation of Akt at Ser473 and attenuating Akt activity,
thereby suppressing survival signaling (49). The direction of FKBP51-mediated
Akt regulation depends on factors such as PHLPP expression, Hsp90
status and FKBP51's own PTM state, reflecting its functional
plasticity as a scaffold protein within signaling complexes
(Fig. 3).
Furthermore, in castration-resistant prostate
cancer, FKBP51 forms a complex with PHLPP and inhibitor of NF-κB
kinase subunit α to inhibit both Akt and NF-κB pathways (50). In melanoma, FKBP51 enhances
acetylation of the transcription factor YY1, which suppresses the
expression of the pro-apoptotic death receptor 5 and reduces
sensitivity to apoptosis triggered by TNF-related
apoptosis-inducing ligand (51).
These findings collectively indicate that FKBP51 modulates key
signaling pathways by coordinating phosphorylation, ubiquitination
and acetylation, thereby enabling tumor cells to adapt and survive
in hostile microenvironments.
In addition, FKBP51 regulates protein stability and
degradation by mediating selective autophagic turnover of specific
substrates. In clear cell renal cell carcinoma, FKBP51 binds to the
metalloproteinase inhibitor tissue inhibitor of metalloproteinases
3 (TIMP3) and recruits it to the Beclin1 autophagy complex,
promoting its lysosome-dependent degradation. Since TIMP3 inhibits
extracellular matrix degradation, its downregulation facilitates
tumor cell invasion (52). This
suggests that FKBP51 contributes to tumor microenvironment
remodeling by modulating the stability and degradation of specific
proteins (Fig. 3).
In summary, FKBP51 maintains protein homeostasis by
coordinating PTMs and selective degradation. Through its roles in
signal regulation, apoptosis resistance and microenvironment
adaptation, FKBP51 supports sustained proliferation, migration and
therapy resistance in tumor cells, highlighting its potential as a
key regulator of proteostasis and a promising therapeutic
target.
FKBP52 stabilizes and translocates
nuclear receptors to enhance hormone-driven tumor growth
FKBP52 (also known as FKBP4) is a key co-chaperone
within the Hsp90 complex. In various cancers, its oncogenic role is
closely linked to the regulation of nuclear receptor stability,
activity and subcellular localization. A central mechanism involves
its ability to assemble and stabilize hormone receptor-chaperone
complexes, enhance receptor conformational integrity and facilitate
their nuclear translocation. Through its TPR domain, FKBP52 binds
to Hsp90 and forms a stable chaperone complex (53). Its PPIase domain further
modulates the conformation of steroid receptors-such as androgen
receptor (AR), estrogen receptor α (ERα) and glucocorticoid
receptor (GR)-via prolyl isomerization, thereby enhancing ligand
binding and transcriptional activity (54,55). In breast and prostate cancers,
FKBP52 increases the abundance and activity of ERα and AR, and its
elevated expression is strongly associated with tumor progression
and poor prognosis (56,57).
Notably, FKBP52 plays a critical role in regulating
the subcellular trafficking of nuclear receptors. Upon ligand
binding, FKBP52 facilitates the recruitment of the dynein motor
complex to the receptor-Hsp90 complex. This interaction promotes
active transport of the receptor complex along microtubules toward
the nucleus, enabling efficient nuclear import through the nuclear
pore complex. This nuclear translocation is essential for
receptor-mediated transcriptional activation. For instance, FKBP52
enhances GR nuclear accumulation and transcriptional output by
supporting its interaction with the dynein complex (58). Similarly, FKBP52 promotes the
nuclear import of RelA (p65) in the NF-κB pathway by stabilizing
its association with Hsp70, thereby amplifying NF-κB
transcriptional activity and contributing to tumor proliferation
and inflammatory signaling (59)
(Fig. 3).
In summary, FKBP52 acts as a molecular scaffold that
regulates both the stability and nuclear localization of key
transcriptional regulators. By coordinating the chaperoning,
transport and activation of nuclear receptors and signaling
proteins, FKBP52 helps maintain protein homeostasis and promotes
cancer cell growth and adaptation. These findings highlight FKBP52
as a critical node in oncogenic signaling and a promising
therapeutic target.
FKBPs orchestrate the dual regulation of
pathogenic proteins in neurodegenerative diseases
Protein homeostasis is a central determinant of
neuronal survival, as the brain is particularly vulnerable to the
toxic effects of misfolded or aggregated proteins. In
neurodegenerative diseases, the collapse of proteostatic control
leads to the pathological accumulation of proteins such as
α-synuclein (α-Syn) and tau, which form aggregates that disrupt
synaptic integrity, impair intracellular trafficking and ultimately
drive neuronal death (60,61). Maintaining the balance between
protein folding, degradation and aggregation is therefore critical
for preventing neurotoxicity. FKBPs have emerged as important
regulators of pathogenic protein dynamics. By modulating
conformational states, post-translational processing and
degradation pathways, FKBPs directly shape the fate of
disease-related proteins, positioning them as key players in the
onset and progression of Parkinson's disease (PD) and Alzheimer's
disease (AD).
FKBP12 drives α-Syn misfolding and
aggregation to exacerbate PD pathogenesis
PD is a prevalent neurodegenerative disorder
primarily characterized by the selective degeneration of
dopaminergic neurons in the substantia nigra, leading to impaired
motor function (62,63). Although the precise mechanisms
underlying the disease remain incompletely elucidated, accumulating
evidence indicates that the aberrant aggregation of α-Syn
constitutes a critical pathological hallmark of PD (64). α-Syn is a widely expressed
cytoplasmic protein that normally participates in the regulation of
synaptic function. However, in PD, α-Syn undergoes pathological
aggregation into fibrillar structures within neurons, forming Lewy
bodies, which in turn induce neurotoxicity and contribute to
neurodegeneration (65).
Investigations have revealed that the PPIase
activity of FKBPs and their role in modulating protein folding are
intricately linked to the aggregation of α-Syn (66). FKBP12 contributes to the
pathogenesis of PD through multiple mechanisms that disrupt
proteostasis. It directly interferes with the folding and
aggregation of α-Syn by binding to its proline-rich C-terminal
region and catalyzing cis/trans isomerization of terminal prolines,
inducing pathogenic conformational changes in the monomer. This
markedly accelerates and alters aggregation kinetics, promoting the
formation of highly branched dendritic structures (66). In addition, FKBP12 forms a
complex with calcineurin under conditions of sustained cytosolic
Ca2+ elevation induced by α-Syn toxicity. This complex
drives pathological dephosphorylation of key presynaptic proteins
involved in vesicle trafficking, endocytosis and cytoskeletal
organization, including growth associated protein 43 and brain acid
soluble protein 1. The resulting synaptic dysfunction destabilizes
dopamine transporters at the plasma membrane, reduces dopamine
release and leads to neuronal death (67). Given FKBP12's pivotal role in the
disease process, targeting this protein represents a promising
strategy for disease-modifying therapeutic interventions. A recent
study demonstrated that rapamycin, through the inhibition of FKBP12
independent of the mTORC1 pathway, confers neuroprotective effects,
underscoring FKBP12 as a novel therapeutic target for PD (68). Furthermore, non-immunosuppressive
FKBP12 inhibitors, such as ElteN378, have shown efficacy in
preventing α-Syn aggregation, presenting a potential new class of
therapeutics for early-stage PD treatment (69) (Fig. 4A). However, challenges remain in
achieving adequate brain penetration and isoform selectivity for
these compounds, which may limit their translational applicability
and require further optimization.
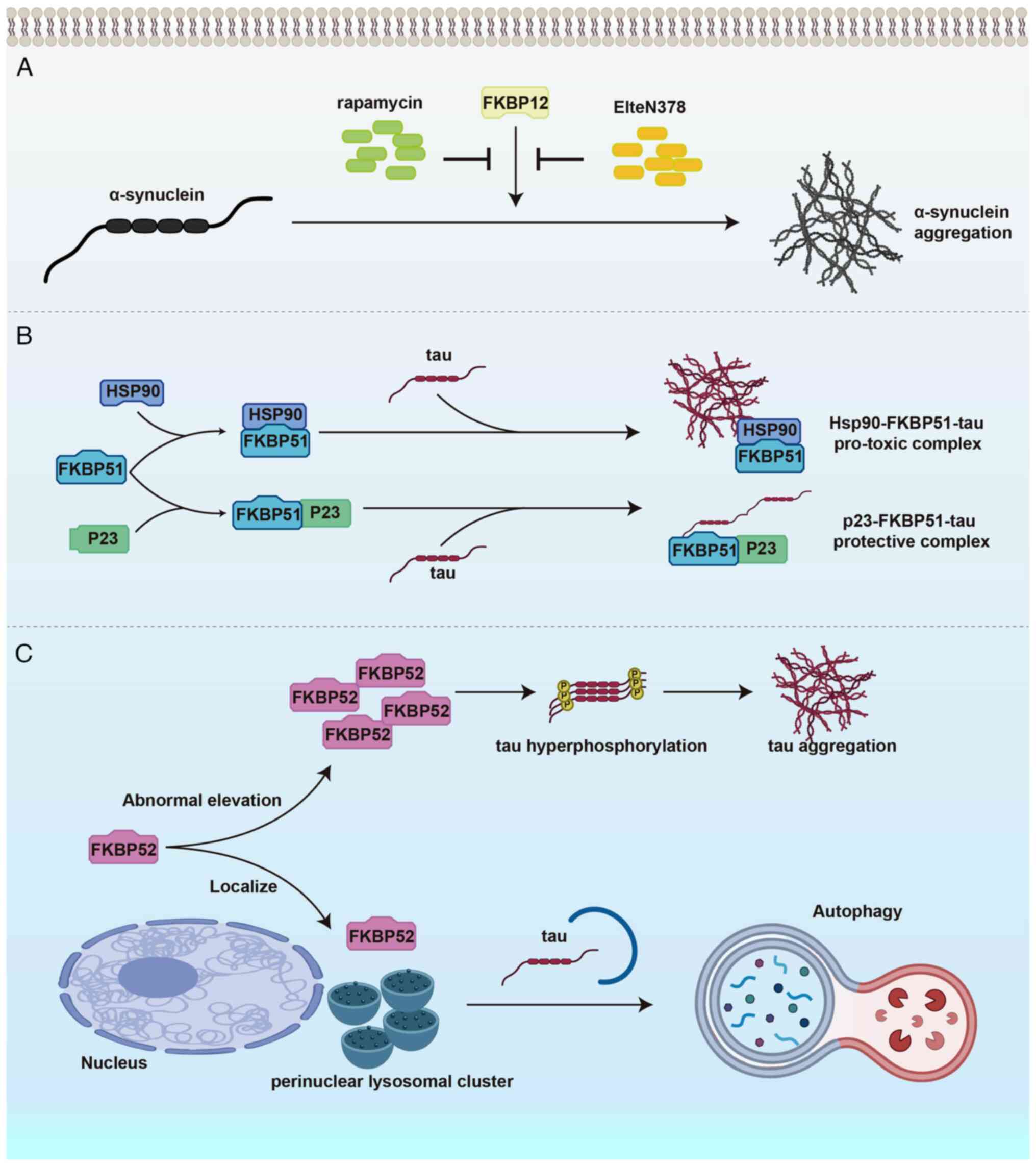 | Figure 4FKBPs regulate neuronal protein
homeostasis through distinct mechanisms in neurodegenerative
diseases. (A) FKBP12 accelerates α-synuclein misfolding and
aggregation via prolyl isomerization, promoting Parkinson's disease
pathology. Pharmacological inhibition of FKBP12 by rapamycin or the
non-immunosuppressive compound ElteN378 suppresses α-SYN
aggregation, conferring neuroprotective effects. (B) FKBP51
modulates tau proteostasis through the Hsp90 chaperone complex,
balancing tau aggregation and stabilization in Alzheimer's disease.
The Hsp90-FKBP51-tau complex promotes pathogenic tau
oligomerization, while the p23-FKBP51-tau complex stabilizes tau in
a non-aggregated state, exerting a protective effect. (C) Under
physiological conditions, FKBP52 facilitates tau degradation
through the autophagy-lysosome pathway. However, its abnormal
elevation enhances tau hyperphosphorylation and aggregation,
contributing to neurofibrillary tangle formation and
neurodegeneration. α-SYN, α-synuclein; PD, Parkinson's disease; AD,
Alzheimer's disease; FKBP, FK506-binding protein; Hsp, heat shock
protein. |
In summary, FKBP12, through its PPIase activity,
disrupts neuronal proteostasis via multiple pathways, including the
regulation of α-Syn conformation, alteration of its aggregation
dynamics and calcineurin-dependent dephosphorylation of synaptic
proteins. These processes collectively drive the onset and
progression of PD. The multifaceted role of FKBP12 in disease
pathogenesis not only reveals a new dimension of proteostasis
imbalance in PD but also provides a solid theoretical foundation
and potential avenues for the development of disease-modifying
therapies targeting FKBP12.
FKBP51 and FKBP52 modulate tau
aggregation-degradation balance to shape AD pathology
AD is a progressive neurodegenerative disorder
characterized by cognitive decline, with hallmark pathological
features including β-amyloid plaque deposition and neurofibrillary
tangles composed of hyperphosphorylated tau protein (70-72). Tau, a microtubule-associated
protein, undergoes conformational alterations and aggregation that
are considered central to the neuronal dysfunction and cell death
observed in AD (71,73-75). In recent years, FKBP51 and FKBP52
have garnered increasing attention for their regulatory roles in
tau pathology.
FKBP51 promotes tau oligomer formation through
cooperation with Hsp90. Hsp90 functions as a scaffold, precisely
positioning the proline-rich region of tau into the PPIase
catalytic pocket of FKBP51, thereby catalyzing proline cis/trans
isomerization. This process alters tau conformation and
phosphorylation status, accelerating oligomer accumulation. The
resulting changes enhance tau pathogenicity, disrupt neuronal
proteostasis and drive the progression of neurodegeneration
(76). By contrast, FKBP51 can
also form a complex with the Hsp90 co-chaperone p23, whose
negatively charged C-terminal tail binds to the positively charged,
aggregation-prone repeat domain of tau, inhibiting its
fibrillization kinetics. When p23 and FKBP51 are both present, a
p23-FKBP51-tau ternary complex may form, partially counteracting
the aggregation-promoting effect of FKBP51 and exerting a
protective regulatory influence on tau aggregation (77). These findings indicate that
FKBP51 can either exacerbate or suppress tau aggregation depending
on its interaction partners, with the functional outcome determined
by the composition and dynamic balance of the chaperone network
(Fig. 4B).
In parallel, FKBP52 plays a pivotal role in
regulating tau protein homeostasis, influencing both its
degradation and the formation of pathological aggregates (78). Under tau proteotoxic stress,
FKBP52 localizes to perinuclear lysosomal clusters and supports the
function of the ALP, promoting lysosomal degradation of tau and
preventing its abnormal secretion, thereby limiting extracellular
tau propagation (12). By
contrast, abnormally elevated FKBP52 levels markedly enhance tau
hyperphosphorylation and aggregation, with a more pronounced
pathological effect in the aged brain. This pro-aggregation
activity is not only associated with its regulation of tau
conformation and aggregation kinetics through the Hsp90 chaperone
network, but may also involve activation of glial cells and the
release of inflammatory mediators, creating a feedforward loop
between tau aggregation and neuroinflammation that accelerates
neuronal injury (79). These
findings indicate that FKBP52 exerts dual, context-dependent
effects on tau pathology, with its regulatory direction shifting
under different physiological and pathological conditions,
providing important insights into the progression of tauopathies
and potential therapeutic strategies (Fig. 4C).
Collectively, FKBP51 and FKBP52 play multifaceted
and context-dependent roles in tau proteostasis. They act through
distinct yet overlapping mechanisms, including PPIase activity,
Hsp90 co-chaperone interactions and regulation of the
autophagy-lysosome pathway. Depending on the cellular context, they
can either promote or restrain tau aggregation. This dual
regulation shifts the balance between neuronal resilience and
degeneration, highlighting their importance as modulators of AD
progression and as potential therapeutic targets. An important
unresolved question is what specific cellular or pathological cues
dictate whether FKBP51 and FKBP52 act to promote or suppress tau
aggregation, a topic that warrants further investigation.
FKBPs stabilize ion channel conformation to
protect against cardiac dysfunction
Protein homeostasis is central to cardiovascular
physiology, where the stability of ion channel complexes and
contractile proteins is indispensable for maintaining cardiac
excitability and pump function. Disruption of proteostasis under
pathological stress contributes to electrical instability, impaired
contractility and progressive remodeling, forming the basis of
numerous cardiovascular diseases (80,81). Within this framework, FKBPs have
emerged as critical modulators of protein conformation and complex
stability. Among them, FKBP12.6 plays a particularly important role
in governing ion channel regulation, positioning it as a key
determinant of cardiac function and a potential therapeutic target
in heart failure and arrhythmias.
FKBP12.6 stabilizes ryanodine receptor 2
(RyR2) conformation to prevent diastolic calcium leak in heart
failure (HF) and arrhythmia
In cardiomyocytes, RyR2 is the primary calcium
release channel on the sarcoplasmic reticulum (SR), and its protein
homeostasis is essential for regulating cardiac contractility and
rhythm (82,83). During excitation-contraction
coupling, RyR2 channels open in response to membrane
depolarization, releasing stored Ca2+ from the SR into
the cytosol to initiate contraction. In diastole, RyR2 channels are
expected to remain closed to prevent abnormal Ca2+
efflux and allow for Ca2+ reuptake and myocardial
relaxation (84,85). However, in HF and arrhythmias
such as atrial fibrillation (AF), dysregulation of the RyR2
macromolecular complex leads to pathological diastolic
Ca2+ leak, disrupting cytosolic Ca2+
homeostasis (86,87). This Ca2+ dysregulation
promotes delayed afterdepolarizations and triggered activity,
forming a shared pathological basis for HF and AF (88).
Within the RyR2 complex, FKBP12.6, also known as
calstabin2, acts as a key stabilizer of channel structure and
function. Cryo-electron microscopy studies reveal that RyR2
assembles as a homotetramer, with one FKBP12.6 molecule binding to
each protomer at specific interfacial regions. These sites are
located between the helical domain and SPRY domains, where FKBP12.6
stabilizes the closed conformation of the channel (89). Its binding extends the mean
closed time of RyR2, suppresses spontaneous openings and promotes
coupled gating between adjacent channels, thereby preventing
Ca2+ leakage in the resting state (90).
Under pathological conditions, particularly during
sustained sympathetic activation or oxidative stress, RyR2
homeostasis becomes disrupted. Post-translational modifications
such as protein kinase A-mediated hyperphosphorylation at Ser2808,
cysteine oxidation and S-nitrosylation result in the dissociation
of FKBP12.6 from the channel complex (15). This destabilizes the closed
conformation of RyR2, increases the open probability and promotes
diastolic Ca2+ leak, which in turn contributes to
Ca2+ overload, electrophysiological instability and
progressive cardiac dysfunction (91,92).
Multiple animal models support the central role of
FKBP12.6 in RyR2 regulation. In murine models of HF, RyR2 channels
exhibit increased Ser2808 phosphorylation and oxidative
modifications, accompanied by reduced FKBP12.6 binding. These
alterations correlate with enhanced Ca2+ leak and
reduced contractility (93).
Mice lacking FKBP12.6 develop spontaneous arrhythmias, whereas
FKBP12.6 overexpression or treatment with rycals such as S107
restores FKBP12.6 binding to RyR2, stabilizes channel closure,
reduces aberrant Ca2+ release and improves cardiac
function. Importantly, rycals do not directly block RyR2 openings
but enhance FKBP12.6 affinity for the channel, thereby stabilizing
the RyR2 macromolecular complex at a structural level (94). Similar mechanisms are observed in
AF, where atrial myocytes from patients with AF and animal models
show increased RyR2 phosphorylation and oxidation, decreased
FKBP12.6 binding and elevated diastolic Ca2+ spark
frequency. FKBP12.6-deficient mice, despite having structurally
normal hearts, exhibit enhanced susceptibility to pacing-induced
AF. Treatment with S107 suppresses this phenotype only in the
presence of FKBP12.6, indicating its essential role in the
therapeutic effect (94)
(Fig. 5).
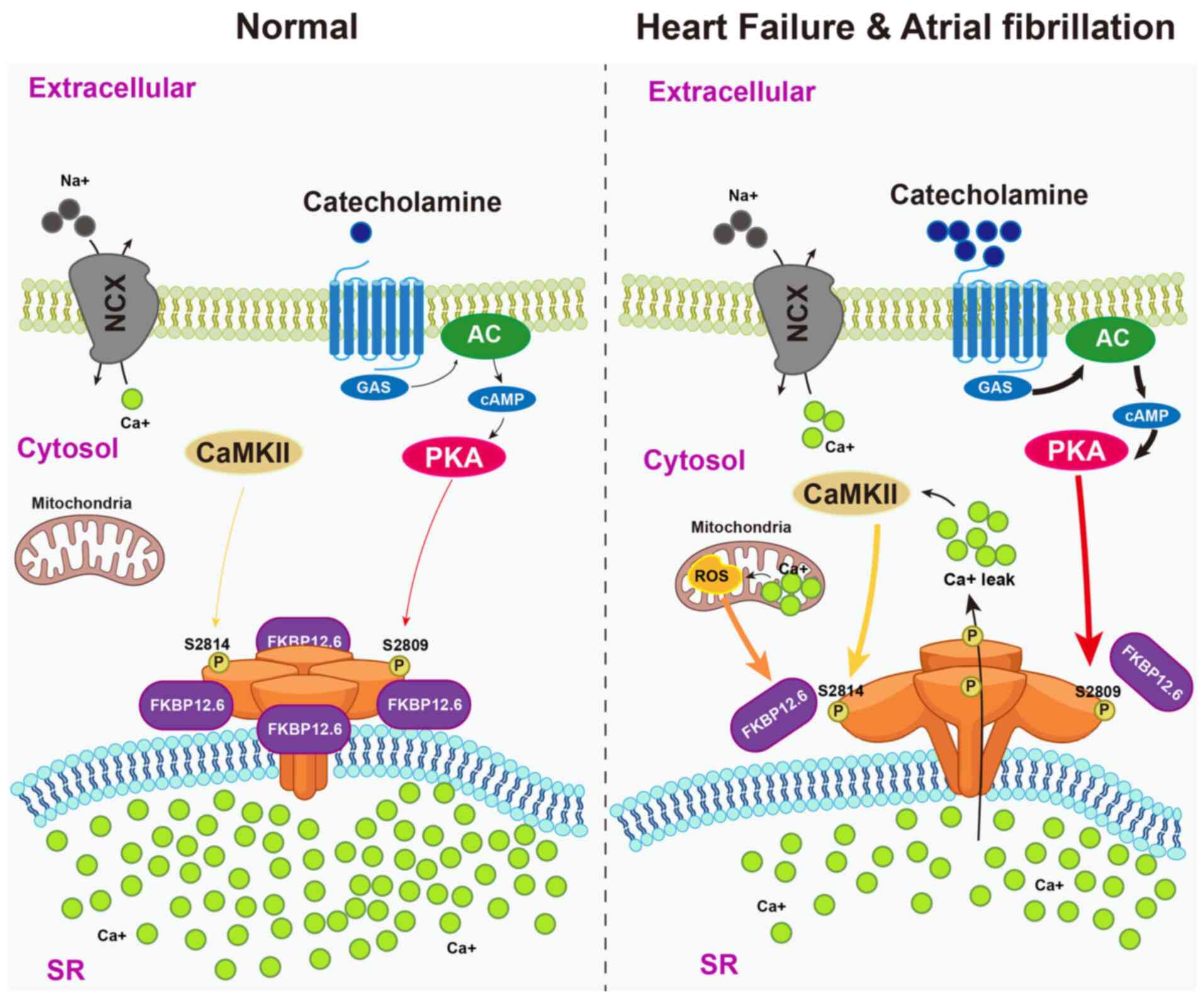 | Figure 5FKBP12.6 stabilizes RyR2 conformation
to prevent pathological Ca2+ leak in heart failure and
atrial fibrillation. Under physiological conditions (left),
catecholamine stimulation activates PKA and CaMKII, but RyR2
channels on the SR remain stabilized in the closed state by
FKBP12.6, preventing diastolic Ca2+ leak and preserving
Ca2+ cycling. In pathological settings such as heart
failure and atrial fibrillation (right), sustained sympathetic
drive and oxidative stress induce PKA hyperphosphorylation at
Ser2809, CaMKII phosphorylation at Ser2814 and oxidative
modifications of RyR2. These alterations disrupt FKBP12.6 binding,
destabilize the channel complex and promote aberrant
Ca2+ leak. The resulting cytosolic Ca2+
overload contributes to delayed afterdepolarizations,
arrhythmogenic activity and progressive cardiac dysfunction.
FKBP12.6, FK506-binding protein 12.6; RyR2, ryanodine receptor 2;
SR, sarcoplasmic reticulum; PKA, protein kinase A; CaMKII,
calcium/calmodulin-dependent protein kinase II; ROS, reactive
oxygen species; P, phosphorylation; GAS, GMP-AMP synthase; NCX,
sodium-calcium exchanger; AC, adenylyl cyclase. |
In summary, maintenance of RyR2 protein homeostasis
is critical for normal cardiac function. FKBP12.6 plays a pivotal
role in stabilizing the RyR2 complex and preventing pathological
Ca2+ leak. Therapeutic strategies targeting the
FKBP12.6-RyR2 interaction, particularly with rycal compounds, offer
a promising approach for precision treatment of HF and
arrhythmias.
FKBPs control metabolic kinase activity to
maintain metabolism balance
Metabolic homeostasis depends on the precise
regulation of key kinases that govern glucose utilization, lipid
turnover and energy sensing. The stability and activity of these
kinases are tightly controlled by proteostasis networks, which
ensure their correct folding, modification and timely degradation
(95,96). As pivotal regulators within this
system, FKBPs influence metabolic adaptation through multiple
proteostatic mechanisms. Among them, FKBP51 stands out for its role
as a molecular scaffold that shapes post-translational
modifications, particularly phosphorylation, thereby fine-tuning
the activity of central metabolic kinases and linking stress
responses to metabolic balance.
FKBP51 serves as a scaffold to modulate
kinase phosphorylation in metabolic regulation
In the central nervous system, FKBP51 influences
metabolic balance by modulating the activity of key kinases in the
autophagy pathway. Autophagy initiation requires activation of
AMP-activated protein kinase (AMPK) and is inhibited by mechanistic
target of rapamycin complex 1 (mTORC1) (97). AMPK activation depends on
phosphorylation of its upstream kinase liver kinase B1 (LKB1) at
Thr172, whereas mTORC1 activity is suppressed by the tuberous
sclerosis complex (TSC)1/2 complex (98). Members of the WD repeat domain
phosphoinositide-interacting (WIPI) protein family, WIPI4 and
WIPI3, act as scaffolds for LKB1-AMPK and TSC2, respectively,
coupling energy sensing to autophagy regulation (99,100). FKBP51 interacts with WIPI4 to
recruit LKB1 to the AMPK complex, enhancing Thr172 phosphorylation
and promoting UNC-51-like kinase 1 (ULK1) phosphorylation at Ser555
to initiate autophagy. In parallel, FKBP51 binds the WIPI3-TSC2
complex to cooperatively inhibit mTORC1 activity, further relieving
autophagy suppression (16).
The physiological relevance of these scaffold-based
mechanisms is supported by in vivo findings demonstrating
their dose-dependent effects on energy homeostasis and autophagy
regulation. In vivo, this regulation shows a clear dose
dependency. Mediobasal hypothalamus-specific deletion of FKBP51
reduces AMPK-ULK1 activation, enhances mTORC1 signaling, decreases
autophagy and leads to obesity, impaired glucose tolerance and
increased food intake. Moderate FKBP51 overexpression enhances AMPK
activity and autophagy in skeletal muscle and adipose tissue,
inhibits mTORC1 signaling, improves insulin sensitivity and limits
weight gain under high-fat diet conditions (101). By contrast, excessive FKBP51
expression activates AKT-mTORC1 signaling, suppresses autophagy and
disrupts proteostasis. This bidirectional effect of deficiency and
overexpression highlights FKBP51 as a dose-sensitive regulator of
autophagy and metabolic balance (102) (Fig. 6A). In glucose metabolism, FKBP51
modulates the AKT-forkhead box protein O1 (FOXO1) signaling axis in
pancreatic β cells to regulate cell function and survival. AKT is a
serine/threonine kinase activated by phosphorylation at Thr308 and
Ser473 downstream of insulin receptor-PI3K-pyruvate dehydrogenase
kinase 1/mTORC2 signaling (103). Activated AKT phosphorylates the
transcription factor FOXO1 at Ser256, promoting its nuclear export
and repressing the transcription of target genes (104,105). FOXO1 is essential for β-cell
differentiation, maturity and stress adaptation (106,107). As a scaffold protein, FKBP51
recruits the phosphatase PHLPP to AKT, facilitating
dephosphorylation at Ser473 and reducing AKT activity. This
decreases FOXO1 Ser256 phosphorylation, promotes its nuclear
retention and preserves transcriptional activity. Under
inflammatory stress, this mechanism helps maintain β-cell function,
enhance survival and sustain glucose-stimulated insulin secretion,
forming a protective FKBP51-PHLPP-AKT-FOXO1 regulatory pathway
(16,108) (Fig. 6B).
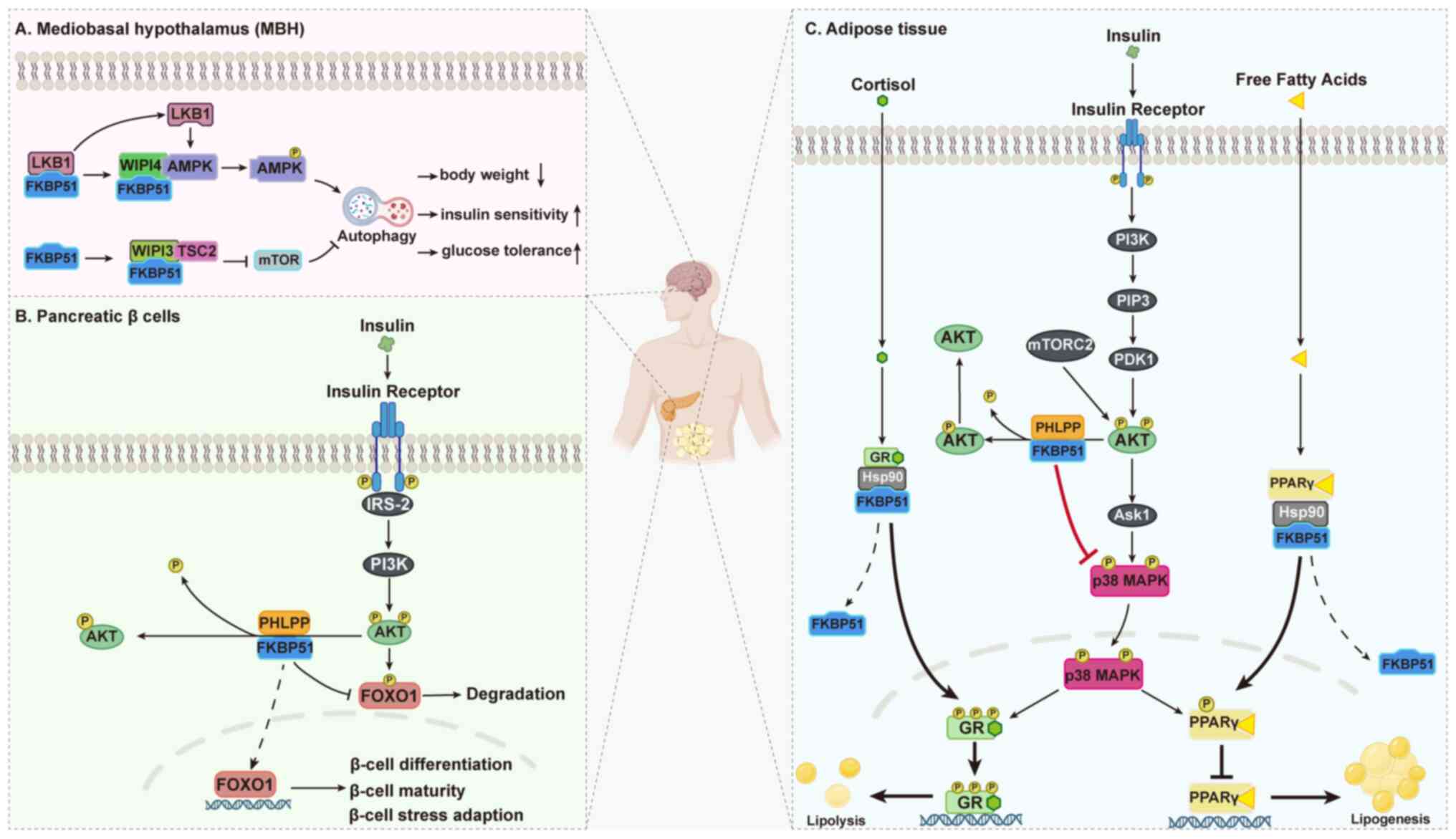 | Figure 6FKBP51 regulates metabolic
homeostasis by scaffolding kinase phosphorylation across multiple
tissues. Schematic representation of FKBP51-mediated regulation of
metabolic signaling. (A) In the MBH, FKBP51 interacts with WIPI4 to
recruit LKB1 to the AMPK complex, enhancing AMPK phosphorylation
and ULK1 activation to promote autophagy, while binding WIPI3-TSC2
to inhibit mTORC1 signaling. (B) In pancreatic β cells, FKBP51
scaffolds the phosphatase PHLPP to AKT, facilitating
dephosphorylation of AKT at Ser473 and decreasing FOXO1
phosphorylation, which preserves FOXO1 transcriptional activity and
supports β-cell differentiation, maturity and stress adaptation.
(C) In adipose tissue, FKBP51 suppresses AKT activity, indirectly
modulating p38 MAPK-mediated phosphorylation of PPARγ and GR,
thereby balancing lipogenesis and lipolysis. Together, these
mechanisms highlight FKBP51 as a dose-sensitive scaffold that
fine-tunes kinase phosphorylation to coordinate glucose
utilization, lipid storage and energy sensing, ultimately
maintaining systemic metabolic homeostasis. AMPK, AMP-activated
protein kinase; mTORC1, mechanistic target of rapamycin complex 1;
ULK1, UNC-51-like kinase 1; LKB1, liver kinase B1; WIPI, WD repeat
domain phosphoinositide-interacting protein; PHLPP, PH domain
leucine-rich repeat protein phosphatase; AKT, protein kinase B;
FOXO1, forkhead box protein O1; GR, glucocorticoid receptor; PPARγ,
peroxisome proliferator-activated receptor γ; MAPK,
mitogen-activated protein kinase; MBH, mediobasal hypothalamus;
HSP90, heat shock protein 90. |
In lipid metabolism, FKBP51 influences two key
nuclear receptors in adipocytes, peroxisome proliferator-activated
receptor γ (PPARγ) and glucocorticoid receptor (GR), to balance
lipogenesis and lipolysis. The p38 mitogen-activated protein kinase
phosphorylates PPARγ at Ser112, decreasing its transcriptional
activity and suppressing lipogenesis, while phosphorylation of GR
enhances its transcriptional activity, promoting lipolysis
(109). FKBP51 suppresses AKT
activity, which indirectly reduces p38 activation, thereby lowering
GR phosphorylation and lipolytic gene transcription while relieving
inhibitory phosphorylation of PPARγ to enhance lipogenic activity
(16). FKBP51, as part of the
Hsp90 chaperone complex, also retains GR and PPARγ in the
cytoplasm, preventing their nuclear translocation and
phosphorylation (31). Upon
ligand binding, FKBP52 replaces FKBP51 in the GR complex, enabling
GR nuclear import, whereas PPARγ is released from FKBP51 by protein
phosphatase 5 to dephosphorylate Ser112 and restore activity
(16). Notably, during early
adipocyte differentiation, FKBP51 translocates from mitochondria to
the nucleus, where it binds GRα and inhibits its transcriptional
activity, linking stress signaling to lipid metabolic gene
expression in a time-dependent manner (110) (Fig. 6C).
Collectively, these findings establish FKBP51 as a
central scaffold that orchestrates kinase phosphorylation to
fine-tune metabolic signaling. By bridging phosphatases and kinases
such as AKT, p38, AMPK and mTORC1, FKBP51 exerts precise control
over glucose utilization, lipid storage and autophagy. This
scaffold-dependent regulation allows cells to dynamically adapt to
nutritional and stress cues, thereby safeguarding systemic
metabolic balance. Importantly, the dose-sensitive nature of FKBP51
underscores its dual capacity to either maintain homeostasis or
drive metabolic dysfunction, highlighting its significance as a
pivotal regulator of proteostasis within energy metabolism.
Therapeutic targeting of FKBPs to restore
proteostasis
Targeting FKBPs offers a novel and unifying
therapeutic strategy to correct proteostasis imbalance across
diverse diseases. Given their structural modularity and central
positioning in protein quality control, FKBPs provide actionable
nodes for pharmacological intervention. As summarized in Table I, representative FKBP members
participate in distinct pathological contexts-from
neurodegeneration and cancer to cardiovascular and metabolic
disorders-through diverse proteostasis-related mechanisms and
corresponding therapeutic strategies. In neurodegenerative
diseases, FKBP12 accelerates α-Syn misfolding and aggregation in
PD, making it a candidate for non-immunosuppressive inhibitors such
as ElteN378, which block its interaction with α-Syn and thereby
mitigate proteotoxic stress (69). FKBP51 and FKBP52, on the other
hand, regulate tau conformational fate in AD. Ligands or interface
inhibitors that selectively modulate FKBP51-Hsp90 or FKBP52-tau
complexes may restore tau homeostasis and attenuate neurotoxicity
(12,77).
 | Table IFKBPs as therapeutic targets to
restore proteostasis. |
Table I
FKBPs as therapeutic targets to
restore proteostasis.
| FKBP (protein;
gene) | Disease
context | Proteostasis axis
leveraged | Therapeutic
strategy | (Refs.) |
|---|
| FKBP12
(FKBP1A) | Parkinson's disease
(α-synuclein aggregation) | Protein folding and
aggregation control |
Non-immunosuppressive PPIase inhibitors
(e.g., ElteN378), peptidomimetics disrupting FKBP12-α-syn
interaction | (66,67,69) |
| FKBP51
(FKBP5) | Alzheimer's disease
(tau aggregation) | Chaperone-dependent
folding and oligomer stabilization | Selective FKBP51
ligands; inhibitors of FKBP51-Hsp90-tau interface | (77) |
| Cancer (melanoma,
prostate, pancreatic) | Post-translational
modification scaffolding (Akt/PTM balance) | Scaffold
modulators; PROTACs/ASOs to downregulate overexpression | (50-52) |
| Metabolic disorders
(obesity, insulin resistance) | Kinase
phosphorylation scaffolding (AKT, AMPK, mTORC1) | Small-molecule
FKBP51 modulators; PROTACs; siRNA/ASO therapies | (16,101,102) |
| FKBP52
(FKBP4) | Alzheimer's disease
(tau degradation/aggregation) | Autophagy-lysosome
pathway and Hsp90 co-chaperone network | Allosteric
inhibitors of PPIase pocket; inhibitors disrupting FKBP52-tau/Hsp90
interaction | (12,78,79) |
| Prostate/breast
cancers (hormone-driven growth) | Nuclear receptor
folding and trafficking | Inhibitors of
FKBP52-dynein interaction; blockade of FKBP52-Hsp90-AR/ER
complexes | (56-58) |
| FKBP12.6
(FKBP1B) | Heart failure,
atrial fibrillation (RyR2 destabilization) | Ion channel complex
stabilization | Rycals (e.g., S107)
to strengthen FKBP12.6-RyR2 binding | (94) |
| FKBP9/FKBP60
(FKBP9) | Glioblastoma (ER
stress adaptation) | ER proteostasis
(BiP-mediated folding and UPR tuning) | RNAi/ASO knockdown;
inhibitors of FKBP9-BiP interface | (40) |
| FKBP7/FKBP23
(FKBP7) | Pancreatic ductal
adenocarcinoma (CAF-mediated ECM remodeling) | ER proteostasis and
extracellular matrix modulation | Inhibitors
preventing FKBP7-BiP competition; stromal-targeted strategies | (41) |
| FKBP10/FKBP65
(FKBP10) | NSCLC, CRC, RCC,
bladder cancer, glioma | Translational
homeostasis and collagen/protein maturation | PPIase inhibitors;
blockade of FKBP10-Hsp47 or FKBP10-prelamin A interactions;
RNAi/ASO knockdown | (10,42,45,46) |
In oncology, ER-resident proteins FKBP9, FKBP7 and
FKBP10 enable tumor cells to adapt to translational overload and ER
stress. FKBP9 sustains glioblastoma growth by restraining
IRE1α-XBP1 signaling, while FKBP7 in pancreatic cancer-associated
fibroblasts remodels collagen deposition to enhance invasion
(40,41). FKBP10, overexpressed in multiple
solid tumors, supports ribosomal translation and stabilizes
structural proteins (10,42).
These functions highlight FKBPs as tractable targets for either
enzymatic inhibition, interference with client binding (e.g., BiP,
Hsp47), or RNA interference/antisense oligonucleotide-based
knockdown to impair tumor proteostasis and sensitize cancers to
therapy.
In the cardiovascular system, FKBP12.6 stabilizes
the RyR2 calcium channel and prevents diastolic calcium leak. Small
molecules such as rycals (e.g., S107) that strengthen FKBP12.6-RyR2
binding have shown promise in preclinical HF and arrhythmia models
by restoring calcium homeostasis (94). In metabolism, FKBP51 acts as a
scaffold to fine-tune kinase phosphorylation, balancing AKT, AMPK
and mTORC1 signaling. Selective FKBP51 modulators, degraders or
oligonucleotide-based strategies represent emerging tools to
correct its dose-dependent bidirectional effects on glucose
tolerance, lipid balance and autophagy (16,100,101) (Table I).
Collectively, these examples establish FKBPs as a
novel class of proteostasis regulators that bridge molecular
folding networks with system-level disease mechanisms. Therapeutic
strategies tailored to individual family members and disease
contexts not only expand our understanding of the
proteostasis-disease axis but also provide a foundation for
precision interventions targeting multi-organ disorders.
Proteostasis-regulating roles of
less-studied FKBPs across disease contexts
While much attention in the literature has focused
on canonical FKBPs such as FKBP51, FKBP52 and FKBP12, emerging
evidence highlights the functional significance of other,
less-studied FKBP family members in regulating proteostasis across
diverse disease contexts. FKBP38 (FKBP8) regulates epithelial
barrier integrity in inflammatory bowel disease by recruiting MLCK1
to the perijunctional actomyosin ring, and also modulates Bcl-2
stability and autophagy in glioblastoma to preserve tumor cell
proteostasis (111,112). FKBPL has been shown to interact
with DLK to inhibit its kinase activity and promote
lysosome-dependent degradation, thereby maintaining axonal
proteostasis in neuronal injury (113). In connective tissue disorders
such as vascular Ehlers-Danlos syndrome, FKBP22 (FKBP14) functions
as an ER-resident PPIase that ensures proper collagen folding and
secretion (33). FKBP25 (FKBP3)
influences proteostasis by suppressing mTORC1 activity and
promoting autophagy, while its stability is tightly regulated by
ubiquitination (36). Table II summarizes the current
understanding of these underexplored FKBPs, their associated
pathologies and the key mechanisms by which they influence protein
homeostasis.
| Table IIProteostasis-regulating roles of
less-studied FKBPs across disease contexts. |
Table II
Proteostasis-regulating roles of
less-studied FKBPs across disease contexts.
| FKBP (protein;
gene) | Disease
context | Mechanism of
proteostasis regulation | (Refs.) |
|---|
| FKBP38
(FKBP8) | Inflammatory bowel
disease | Binds MLCK1 to
facilitate its recruitment to the perijunctional actomyosin ring,
regulating epithelial barrier protein stability | (111) |
| Glioblastoma | Modulates Bcl-2
stability and participates in protein folding and autophagy,
maintaining tumor cell proteostasis | (112) |
| FKBPL | Neuronal injury
(axon degeneration, RGC death) | Interacts with DLK
to inhibit its kinase activity and promotes lysosome-dependent
degradation, regulating axonal proteostasis | (113) |
| FKBP22
(FKBP14) | Connective tissue
disorders (vascular Ehlers-Danlos syndrome) | ER-resident PPIase
catalyzing proline isomerization in collagen, ensuring proper
collagen folding and secretion | (33) |
| FKBP25
(FKBP3) | Cancer | Suppresses mTORC1
activity and promotes autophagy; its stability is regulated by
ubiquitination, influencing protein degradation and tumor
growth | (36) |
| FKBP51
(FKBP5) | Huntington's
disease | PPIase activity
modulates folding and aggregation of mHTT; inhibition enhances
autophagic clearance of mHTT | (9) |
Conclusion
Protein homeostasis is fundamental to the
preservation of cellular integrity and its disruption represents a
common pathological axis across cancer, neurodegeneration,
cardiovascular dysfunction and metabolic disorders. This review
highlights FKBPs as versatile modulators of proteostasis, acting
through diverse mechanisms including conformational control,
chaperone-assisted folding, complex stabilization, stress
adaptation and selective degradation. By integrating these
processes, FKBPs emerge not merely as accessory factors but as
central regulators that shape the fate of disease-related proteins
and determine cellular resilience or vulnerability.
The synthesis of the present review demonstrates
that distinct FKBP isoforms exert disease-specific functions:
ER-resident members such as FKBP7/9/10 orchestrate tumor proteome
remodeling, FKBP12 drives pathogenic protein aggregation in PD,
FKBP51 and FKBP52 modulate tau balance in AD, FKBP12.6 safeguards
cardiac rhythm by stabilizing RyR2 and FKBP51 fine-tunes metabolic
kinase signaling to maintain systemic balance. These findings
collectively establish FKBPs as critical bridges linking
proteostasis to multi-system pathogenesis.
Importantly, their structural modularity and
context-dependent functions position FKBPs as novel and druggable
nodes within proteostasis networks. Therapeutic strategies
targeting FKBP-client interactions, their scaffold functions or
their enzymatic activity provide unprecedented opportunities to
restore proteostasis in disease states. Moving forward, integrating
chemical biology, structural proteomics and translational studies
will be essential to exploit FKBPs as precision targets. By
reframing FKBPs within the proteostasis paradigm, this review not
only expands our conceptual understanding of disease biology but
also charts a new course for therapeutic innovation across multiple
organ systems.
Availability of data and materials
Not applicable.
Authors' contributions
ZL was involved in the conceptualization of the
study and in writing-original draft, writing-review and editing. XL
contributed to the writing-review and editing, performed
visualization and provided supervision. HZ obtained resources,
acquired funding and was involved in writing-review and editing.
Data authentication is not applicable. All authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Declaration of generative AI in scientific
writing
During the preparation of this work, artificial
intelligence tools ChatGPT (GPT-5; 2025 version; OpenAI) were used
to improve the readability and language of the manuscript or to
generate images, and subsequently, the authors revised and edited
the content produced by the artificial intelligence tools as
necessary, taking full responsibility for the ultimate content of
the present manuscript'
Abbreviations:
|
AD
|
Alzheimer's disease
|
|
AF
|
atrial fibrillation
|
|
AKT
|
protein kinase B
|
|
AMPK
|
AMP-activated protein kinase
|
|
AR
|
androgen receptor
|
|
ASO
|
antisense oligonucleotide
|
|
BiP
|
binding immunoglobulin protein
|
|
CAF
|
cancer-associated fibroblast
|
|
CaMKII
|
calcium/calmodulin-dependent protein
kinase II
|
|
CRC
|
colorectal cancer
|
|
ER
|
endoplasmic reticulum
|
|
FKBP
|
FK506-binding protein
|
|
HF
|
heart failure
|
|
Hsp90
|
heat shock protein 90
|
|
mTORC1
|
mechanistic target of rapamycin
complex 1
|
|
NF-κB
|
nuclear factor κ-light-chain-enhancer
of activated B cells
|
|
NSCLC
|
non-small cell lung cancer
|
|
PD
|
Parkinson's disease
|
|
PPIase
|
peptidyl-prolyl cis-trans
isomerase
|
|
PROTAC
|
proteolysis targeting chimera
|
|
PTM
|
post-translational modification
|
|
ROS
|
reactive oxygen species
|
|
RyR2
|
ryanodine receptor 2
|
|
Ub
|
ubiquitin
|
|
UPR
|
unfolded protein response
|
Acknowledgements
The figures of this paper were prepared with
Figdraw 2.0 and BioRender (https://www.biorender.com).
Funding
This research was supported by grants from the National Natural
Science Foundation of China (grant no. 81873523).
References
|
1
|
Díaz-Hung ML and Hetz C: Proteostasis and
resilience: On the interphase between individual's and
intracellular stress. Trends Endocrinol Metab. 33:305–317. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hetz C: Adapting the proteostasis capacity
to sustain brain healthspan. Cell. 184:1545–1560. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weinberg J, Gaur M, Swaroop A and Taylor
A: Proteostasis in aging-associated ocular disease. Mol Aspects
Med. 88:1011572022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gressler AE, Leng H, Zinecker H and Simon
AK: Proteostasis in T cell aging. Semin Immunol. 70:1018382023.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lu Q, Qin X, Chen C, Yu W, Lin J, Liu X,
Guo R, Reiter RJ, Ashrafizadeh M, Yuan M and Ren J: Elevated levels
of alcohol dehydrogenase aggravate ethanol-evoked cardiac
remodeling and contractile anomalies through FKBP5-yap-mediated
regulation of ferroptosis and ER stress. Life Sci. 343:1225082024.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jeanne X, Török Z, Vigh L and Prodromou C:
The role of the FKBP51-Hsp90 complex in Alzheimer's disease: An
emerging new drug target. Cell Stress Chaperones. 29:792–804. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nakamura K, Aoyama-Ishiwatari S, Nagao T,
Paaran M, Obara CJ, Sakurai-Saito Y, Johnston J, Du Y, Suga S,
Tsuboi M, et al: Mitochondrial complexity is regulated at
ER-mitochondria contact sites via PDZD8-FKBP8 tethering. Nat
Commun. 16:34012025. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Akbar M, Toppo P and Nazir A: Ageing,
proteostasis, and the gut: Insights into neurological health and
disease. Ageing Res Rev. 101:1025042024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bailus BJ, Scheeler SM, Simons J, Sanchez
MA, Tshilenge KT, Creus-Muncunill J, Naphade S, Lopez-Ramirez A,
Zhang N, Lakshika Madushani K, et al: Modulating FKBP5/FKBP51 and
autophagy lowers HTT (huntingtin) levels. Autophagy. 17:4119–4140.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cai HQ, Zhang MJ, Cheng ZJ, Yu J, Yuan Q,
Zhang J, Cai Y, Yang LY, Zhang Y, Hao JJ, et al: FKBP10 promotes
proliferation of glioma cells via activating AKT-CREB-PCNA axis. J
Biomed Sci. 28:132021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mei L, Zheng YM, Song T, Yadav VR, Joseph
LC, Truong L, Kandhi S, Barroso MM, Takeshima H, Judson MA and Wang
YX: Rieske iron-sulfur protein induces FKBP12.6/RyR2 complex
remodeling and subsequent pulmonary hypertension through
NF-κB/cyclin D1 pathway. Nat Commun. 11:35272020. View Article : Google Scholar
|
|
12
|
Chambraud B, Daguinot C, Guillemeau K,
Genet M, Dounane O, Meduri G, Poüs C, Baulieu EE and Giustiniani J:
Decrease of neuronal FKBP4/FKBP52 modulates perinuclear lysosomal
positioning and MAPT/Tau behavior during MAPT/Tau-induced
proteotoxic stress. Autophagy. 17:3491–3510. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ke H, Chen Z, Chen L, Zhang H, Wang Y,
Song T, Bi A, Li Q, Sheng H, Jia Y, et al: FK506-binding proteins:
Emerging target and therapeutic opportunity in multiple tumors. Int
J Biol Macromol. 307:1419142025. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang L, Chakraborty P, Zhang L, Wong M,
Hill SE, Webber CJ, Libera J, Blair LJ, Wolozin B and Zweckstetter
M: Chaperoning of specific tau structure by immunophilin FKBP12
regulates the neuronal resilience to extracellular stress. Sci Adv.
9:eadd97892023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Richardson SJ, Thekkedam CG, Casarotto MG,
Beard NA and Dulhunty AF: FKBP12 binds to the cardiac ryanodine
receptor with negative cooperativity: Implications for heart muscle
physiology in health and disease. Philos Trans R Soc Lond B Biol
Sci. 378:202201692023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Smedlund KB, Sanchez ER and Hinds TD Jr:
FKBP51 and the molecular chaperoning of metabolism. Trends
Endocrinol Metab. 32:862–874. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cozachenco D, Ribeiro FC and Ferreira ST:
Defective proteostasis in Alzheimer's disease. Ageing Res Rev.
85:1018622023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dikic I and Schulman BA: An expanded
lexicon for the ubiquitin code. Nat Rev Mol Cell Biol. 24:273–287.
2023. View Article : Google Scholar
|
|
19
|
Nixon RA and Rubinsztein DC: Mechanisms of
autophagy-lysosome dysfunction in neurodegenerative diseases. Nat
Rev Mol Cell Biol. 25:926–946. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen X, Shi C, He M, Xiong S and Xia X:
Endoplasmic reticulum stress: Molecular mechanism and therapeutic
targets. Signal Transduct Target Ther. 8:3522023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Agam G, Atawna B, Damri O and Azab AN: The
Role of FKBPs in complex disorders: Neuropsychiatric diseases,
cancer, and type 2 diabetes mellitus. Cells. 13:8012024. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Stauffer WT, Goodman AZ and Gallay PA:
Cyclophilin inhibition as a strategy for the treatment of human
disease. Front Pharmacol. 15:14179452024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhuang S, Chakraborty P and Zweckstetter
M: Regulation of tau by peptidyl-prolyl isomerases. Curr Opin
Struct Biol. 84:1027392024. View Article : Google Scholar
|
|
24
|
Deutscher RCE, Meyners C, Repity ML,
Sugiarto WO, Kolos JM, Maciel EVS, Heymann T, Geiger TM, Knapp S,
Lermyte F and Hausch F: Discovery of fully synthetic FKBP12-mTOR
molecular glues. Chem Sci. 16:4256–4263. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hanaki S and Shimada M: Impact of FKBP52
on cell proliferation and hormone-dependent cancers. Cancer Sci.
114:2729–2738. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Soto OB, Ramirez CS, Koyani R,
Rodriguez-Palomares IA, Dirmeyer JR, Grajeda B, Roy S and Cox MB:
Structure and function of the TPR-domain immunophilins FKBP51 and
FKBP52 in normal physiology and disease. J Cell Biochem.
125:e304062024. View Article : Google Scholar
|
|
27
|
Singh MK, Shin Y, Ju S, Han S, Choe W,
Yoon KS, Kim SS and Kang I: Heat shock response and heat shock
proteins: Current understanding and future opportunities in human
diseases. Int J Mol Sci. 25:42092024. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gu J, He Y, He C, Zhang Q, Huang Q, Bai S,
Wang R, You Q and Wang L: Advances in the structures, mechanisms
and targeting of molecular chaperones. Signal Transduct Target
Ther. 10:842025. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pokhrel S, Devi S and Gestwicki JE:
Chaperone-dependent and chaperone-independent functions of
carboxylate clamp tetratricopeptide repeat (CC-TPR) proteins.
Trends Biochem Sci. 50:121–133. 2025. View Article : Google Scholar
|
|
30
|
Baischew A, Engel S, Taubert MC, Geiger TM
and Hausch F: Large-scale, in-cell photocrosslinking at
single-residue resolution reveals the molecular basis for
glucocorticoid receptor regulation by immunophilins. Nat Struct Mol
Biol. 30:1857–1866. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Noddings CM, Johnson JL and Agard DA:
Cryo-EM reveals how Hsp90 and FKBP immunophilins co-regulate the
glucocorticoid receptor. Nat Struct Mol Biol. 30:1867–1877. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jarayseh T, Debaenst S, De Saffel H,
Rosseel T, Milazzo M, Bek JW, Hudson DM, Van Nieuwerburgh F,
Gansemans Y, Josipovic I, et al: Bmpr1aa modulates the severity of
the skeletal phenotype in an fkbp10-deficient Bruck syndrome
zebrafish model. J Bone Miner Res. 40:154–166. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ishikawa Y, Bonna A, Gould DB and Farndale
RW: Local net charge state of collagen triple helix is a
determinant of FKBP22 binding to collagen III. Int J Mol Sci.
24:151562023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Herrema H, Guan D, Choi JW, Feng X,
Salazar Hernandez MA, Faruk F, Auen T, Boudett E, Tao R, Chun H and
Ozcan U: FKBP11 rewires UPR signaling to promote glucose
homeostasis in type 2 diabetes and obesity. Cell Metab.
34:1004–1022.e8. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Akintade DD and Chaudhuri B: FK506-binding
protein 2 (FKBP13) inhibit Bax-induced apoptosis in Saccharomyces
cerevisiae (yeast). Cell Biol Toxicol. 39:719–728. 2023. View Article : Google Scholar :
|
|
36
|
Yang Y, Chen X, Yao W, Cui X, Li N, Lin Z,
Zhao B and Miao J: Esterase D stabilizes FKBP25 to suppress mTORC1.
Cell Mol Biol Lett. 26:502021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yao RQ, Ren C, Xia ZF and Yao YM:
Organelle-specific autophagy in inflammatory diseases: A potential
therapeutic target underlying the quality control of multiple
organelles. Autophagy. 17:385–401. 2021. View Article : Google Scholar :
|
|
38
|
López-Otín C, Blasco MA, Partridge L,
Serrano M and Kroemer G: Hallmarks of aging: An expanding universe.
Cell. 186:243–278. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li S, Xia W, Sun B, Peng W, Yang D, Gao J,
He S, Yang H, Zhu Y, Zhou H, et al: The stability of FKBP9
maintained by BiP is crucial for glioma progression. Genes Dis.
11:1011232023. View Article : Google Scholar
|
|
40
|
Xu H, Liu P, Yan Y, Fang K, Liang D, Hou
X, Zhang X, Wu S, Ma J, Wang R, et al: FKBP9 promotes the malignant
behavior of glioblastoma cells and confers resistance to
endoplasmic reticulum stress inducers. J Exp Clin Cancer Res.
39:442020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Quemerais C, Jean C, Brunel A, Decaup E,
Labrousse G, Audureau H, Raffenne J, Belhabib I, Cros J, Perraud A,
et al: Unveiling FKBP7 as an early endoplasmic reticulum sentinel
in pancreatic stellate cell activation, collagen remodeling and
tumor progression. Cancer Lett. 614:2175382025. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhao X, Wang J, Tian S, Tang L, Cao S, Ye
J, Cai T, Xuan Y, Zhang X, Li X and Li H: FKBP10 promotes the
muscle invasion of bladder cancer via lamin A dysregulation. Int J
Biol Sci. 21:758–771. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ramadori G, Ioris RM, Villanyi Z, Firnkes
R, Panasenko OO, Allen G, Konstantinidou G, Aras E, Brenachot X,
Biscotti T, et al: FKBP10 regulates protein translation to sustain
lung cancer growth. Cell Rep. 30:3851–3863.e6. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ma W, Li X, Yang L, Pan J, Chen Y, Lu Y,
Dong X, Li D and Gan W: High VSX1 expression promotes the
aggressiveness of clear cell renal cell carcinoma by
transcriptionally regulating FKBP10. J Transl Med. 20:5542022.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu R, Zou Z, Chen L, Feng Y, Ye J, Deng
Y, Zhu X, Zhang Y, Lin J, Cai S, et al: FKBP10 promotes clear cell
renal cell carcinoma progression and regulates sensitivity to the
HIF2α blockade by facilitating LDHA phosphorylation. Cell Death
Dis. 15:642024. View Article : Google Scholar
|
|
46
|
Fu Y, Chen J, Ma X, Chang W, Zhang X, Liu
Y, Liu Y, Shen H, Hu X and Ren AJ: Subcellular expression patterns
of FKBP prolyl isomerase 10 (FKBP10) in colorectal cancer and its
clinical significance. Int J Mol Sci. 24:114152023. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li W, Li F, Zhang X, Lin HK and Xu C:
Insights into the post-translational modification and its emerging
role in shaping the tumor microenvironment. Signal Transduct Target
Ther. 6:4222021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tufano M, Marrone L, D'Ambrosio C, Di
Giacomo V, Urzini S, Xiao Y, Matuozzo M, Scaloni A, Romano MF and
Romano S: FKBP51 plays an essential role in Akt ubiquitination that
requires Hsp90 and PHLPP. Cell Death Dis. 14:1162023. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Luo K, Li Y, Yin Y, Li L, Wu C, Chen Y,
Nowsheen S, Hu Q, Zhang L, Lou Z and Yuan J: USP49 negatively
regulates tumorigenesis and chemoresistance through FKBP51-AKT
signaling. EMBO J. 36:1434–1446. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Shang Z, Yu J, Sun L, Tian J, Zhu S, Zhang
B, Dong Q, Jiang N, Flores-Morales A, Chang C and Niu Y: LncRNA
PCAT1 activates AKT and NF-κB signaling in castration-resistant
prostate cancer by regulating the PHLPP/FKBP51/IKKα complex.
Nucleic Acids Res. 47:4211–4225. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tufano M, Cesaro E, Martinelli R, Pacelli
R, Romano S and Romano MF: FKBP51 affects TNF-related apoptosis
inducing ligand response in melanoma. Front Cell Dev Biol.
9:7189472021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mao S, Zhang D, Chen L, Tan J, Chu Y,
Huang S, Zhou W, Qin H, Xia Q, Zhao Y, et al: FKBP51 promotes
invasion and migration by increasing the autophagic degradation of
TIMP3 in clear cell renal cell carcinoma. Cell Death Dis.
12:8992021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Batko J, Antosz K, Miśków W, Pszczołowska
M, Walczak K and Leszek J: Chaperones-A new class of potential
therapeutic targets in Alzheimer's disease. Int J Mol Sci.
25:34012024. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Maeda K, Habara M, Kawaguchi M, Matsumoto
H, Hanaki S, Masaki T, Sato Y, Matsuyama H, Kunieda K, Nakagawa H
and Shimada M: FKBP51 and FKBP52 regulate androgen receptor
dimerization and proliferation in prostate cancer cells. Mol Oncol.
16:940–956. 2022. View Article : Google Scholar :
|
|
55
|
Habara M, Sato Y, Goshima T, Sakurai M,
Imai H, Shimizu H, Katayama Y, Hanaki S, Masaki T, Morimoto M, et
al: FKBP52 and FKBP51 differentially regulate the stability of
estrogen receptor in breast cancer. Proc Natl Acad Sci USA.
119:e21102561192022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Xiong H, Chen Z, Lin B, Xie B, Liu X, Chen
C, Li Z, Jia Y, Wu Z, Yang M, et al: Naringenin regulates
FKBP4/NR3C1/NRF2 axis in autophagy and proliferation of breast
cancer and differentiation and maturation of dendritic cell. Front
Immunol. 12:7451112022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Mangé A, Coyaud E, Desmetz C, Laurent E,
Béganton B, Coopman P, Raught B and Solassol J: FKBP4 connects
mTORC2 and PI3K to activate the PDK1/Akt-dependent cell
proliferation signaling in breast cancer. Theranostics.
9:7003–7015. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kim MJ, Choi GE, Chae CW, Lim JR, Jung YH,
Yoon JH, Park JY and Han HJ: Melatonin-mediated FKBP4
downregulation protects against stress-induced neuronal
mitochondria dysfunctions by blocking nuclear translocation of GR.
Cell Death Dis. 14:1462023. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zong S, Jiao Y, Liu X, Mu W, Yuan X, Qu Y,
Xia Y, Liu S, Sun H, Wang L, et al: FKBP4 integrates
FKBP4/Hsp90/IKK with FKBP4/Hsp70/RelA complex to promote lung
adenocarcinoma progression via IKK/NF-κB signaling. Cell Death Dis.
12:6022021. View Article : Google Scholar
|
|
60
|
Wilson DM III, Cookson MR, Van Den Bosch
L, Zetterberg H, Holtzman DM and Dewachter I: Hallmarks of
neurodegenerative diseases. Cell. 186:693–714. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Creekmore BC, Watanabe R and Lee EB:
Neurodegenerative disease tauopathies. Annu Rev Pathol. 19:345–370.
2024. View Article : Google Scholar :
|
|
62
|
Weintraub D, Aarsland D, Chaudhuri KR,
Dobkin RD, Leentjens AF, Rodriguez-Violante M and Schrag A: The
neuropsychiatry of Parkinson's disease: Advances and challenges.
Lancet Neurol. 21:89–102. 2022. View Article : Google Scholar :
|
|
63
|
Morris HR, Spillantini MG, Sue CM and
Williams-Gray CH: The pathogenesis of Parkinson's disease. Lancet.
403:293–304. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Ye H, Robak LA, Yu M, Cykowski M and
Shulman JM: Genetics and pathogenesis of Parkinson's syndrome. Annu
Rev Pathol. 18:95–121. 2023. View Article : Google Scholar
|
|
65
|
Ding XB, Wang XX, Xia DH, Liu H, Tian HY,
Fu Y, Chen YK, Qin C, Wang JQ, Xiang Z, et al: Impaired meningeal
lymphatic drainage in patients with idiopathic Parkinson's disease.
Nat Med. 27:411–418. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Caminati G and Procacci P: Mounting
evidence of FKBP12 implication in neurodegeneration. Neural Regen
Res. 15:2195–2202. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Caraveo G, Soste M, Cappelleti V, Fanning
S, van Rossum DB, Whitesell L, Huang Y, Chung CY, Baru V, Zaichick
S, et al: FKBP12 contributes to α-synuclein toxicity by regulating
the calcineurin-dependent phosphoproteome. Proc Natl Acad Sci USA.
114:E11313–E11322. 2017. View Article : Google Scholar
|
|
68
|
Zhang Z, Shen Z, Xie S, Li J, Zhang Z,
Zhang S, Peng B and Huang Q, Li M, Ma S and Huang Q: Rapamycin
exerts neuroprotective effects by inhibiting FKBP12 instead of
mTORC1 in the mouse model of Parkinson's disease.
Neuropharmacology. 275:1105042025. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Caminati G, Martina MR, Menichetti S and
Procacci P: Blocking the FKBP12 induced dendrimeric burst in
aberrant aggregation of α-synuclein by using the ElteN378 synthetic
inhibitor. J Enzyme Inhib Med Chem. 34:1711–1715. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Scheltens P, De Strooper B, Kivipelto M,
Holstege H, Chételat G, Teunissen CE, Cummings J and van der Flier
WM: Alzheimer's disease. Lancet. 397:1577–1590. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Liu E, Zhang Y and Wang JZ: Updates in
Alzheimer's disease: From basic research to diagnosis and
therapies. Transl Neurodegener. 13:452024. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zheng Q and Wang X: Alzheimer's disease:
Insights into pathology, molecular mechanisms, and therapy. Protein
Cell. 16:83–120. 2025. View Article : Google Scholar
|
|
73
|
Ossenkoppele R, van der Kant R and Hansson
O: Tau biomarkers in Alzheimer's disease: Towards implementation in
clinical practice and trials. Lancet Neurol. 21:726–734. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Carter SF, Herholz K, Rosa-Neto P,
Pellerin L, Nordberg A and Zimmer ER: Astrocyte biomarkers in
Alzheimer's disease. Trends Mol Med. 25:77–95. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Graff-Radford J, Yong KXX, Apostolova LG,
Bouwman FH, Carrillo M, Dickerson BC, Rabinovici GD, Schott JM,
Jones DT and Murray ME: New insights into atypical Alzheimer's
disease in the era of biomarkers. Lancet Neurol. 20:222–234. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Oroz J, Chang BJ, Wysoczanski P, Lee CT,
Pérez-Lara Á, Chakraborty P, Hofele RV, Baker JD, Blair LJ, Biernat
J, et al: Structure and pro-toxic mechanism of the human
Hsp90/PPIase/Tau complex. Nat Commun. 9:45322018. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Chakraborty P and Zweckstetter M:
Interplay of p23 with FKBP51 and their chaperone complex in
regulating tau aggregation. Nat Commun. 16:6692025. View Article : Google Scholar
|
|
78
|
Chambraud B, Byrne C, Meduri G, Baulieu EE
and Giustiniani J: FKBP52 in neuronal signaling and
neurodegenerative diseases: A microtubule story. Int J Mol Sci.
23:17382022. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Criado-Marrero M, Gebru NT, Blazier DM,
Gould LA, Baker JD, Beaulieu-Abdelahad D and Blair LJ: Hsp90
co-chaperones, FKBP52 and Aha1, promote tau pathogenesis in aged
wild-type mice. Acta Neuropathol Commun. 9:652021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ren J, Bi Y, Sowers JR, Hetz C and Zhang
Y: Endoplasmic reticulum stress and unfolded protein response in
cardiovascular diseases. Nat Rev Cardiol. 18:499–521. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Abdellatif M, Rainer PP, Sedej S and
Kroemer G: Hallmarks of cardiovascular ageing. Nat Rev Cardiol.
20:754–777. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Marks AR: Targeting ryanodine receptors to
treat human diseases. J Clin Invest. 133:e1628912023. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Keefe JA, Garber R, McCauley MD and
Wehrens XHT: Tachycardia and atrial fibrillation-related
cardiomyopathies: Potential mechanisms and current therapies. JACC
Heart Fail. 12:605–615. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Do TQ and Knollmann BC: Inhibitors of
intracellular RyR2 calcium release channels as therapeutic agents
in arrhythmogenic heart diseases. Annu Rev Pharmacol Toxicol.
65:443–463. 2025. View Article : Google Scholar
|
|
85
|
Grisorio L, Bongianino R, Gianeselli M and
Priori SG: Gene therapy for cardiac diseases: Methods, challenges,
and future directions. Cardiovasc Res. 120:1664–1682. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Papa A, Kushner J and Marx SO: Adrenergic
regulation of calcium channels in the heart. Annu Rev Physiol.
84:285–306. 2022. View Article : Google Scholar :
|
|
87
|
Shemarova I: The dysfunction of
Ca2+ channels in hereditary and chronic human heart
diseases and experimental animal models. Int J Mol Sci.
24:156822023. View Article : Google Scholar
|
|
88
|
Keefe JA, Moore OM, Ho KS and Wehrens XHT:
Role of Ca2+ in healthy and pathologic cardiac function:
From normal excitation-contraction coupling to mutations that cause
inherited arrhythmia. Arch Toxicol. 97:73–92. 2023. View Article : Google Scholar
|
|
89
|
Fowler ED and Zissimopoulos S: Molecular,
subcellular, and arrhythmogenic mechanisms in genetic RyR2 disease.
Biomolecules. 12:10302022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Do TQ and Knollmann BC: RYR2 as new target
for antiarrhythmic therapy: Harnessing the power of existing
chemical entities for drug discovery. Heart Rhythm. 22:1372–1373.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Szentandrássy N, Magyar Z, Hevesi J,
Bányász T, Nánási PP and Almássy J: Therapeutic approaches of
ryanodine receptor-associated heart diseases. Int J Mol Sci.
23:44352022. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Benitah JP, Perrier R, Mercadier JJ,
Pereira L and Gómez AM: RyR2 and calcium release in heart failure.
Front Physiol. 12:7342102021. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Chi X, Gong D, Ren K, Zhou G, Huang G, Lei
J, Zhou Q and Yan N: Molecular basis for allosteric regulation of
the type 2 ryanodine receptor channel gating by key modulators.
Proc Natl Acad Sci USA. 116:25575–25582. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Dridi H, Kushnir A, Zalk R, Yuan Q,
Melville Z and Marks AR: Intracellular calcium leak in heart
failure and atrial fibrillation: A unifying mechanism and
therapeutic target. Nat Rev Cardiol. 17:732–747. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Steinberg GR and Hardie DG: New insights
into activation and function of the AMPK. Nat Rev Mol Cell Biol.
24:255–272. 2023. View Article : Google Scholar
|
|
96
|
Szwed A, Kim E and Jacinto E: Regulation
and metabolic functions of mTORC1 and mTORC2. Physiol Rev.
101:1371–1426. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Al-Kuraishy HM, Sulaiman GM, Mohsin MH,
Mohammed HA, Dawood RA, Albuhadily AK, Al-Gareeb AI, Albukhaty S
and Abomughaid MM: Targeting of AMPK/MTOR signaling in the
management of atherosclerosis: Outmost leveraging. Int J Biol
Macromol. 309:1429332025. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Zhang D, Lu C and Sang K: Exercise as a
metabolic regulator: Targeting AMPK/mTOR-autophagy crosstalk to
counteract sarcopenic obesity. Aging Dis. Jun 7–2025.Epub ahead of
print.
|
|
99
|
Cong Y, So V, Tijssen MAJ, Verbeek DS,
Reggiori F and Mauthe M: WDR45, one gene associated with multiple
neurodevelopmental disorders. Autophagy. 17:3908–3923. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Almannai M, Marafi D and El-Hattab AW:
WIPI proteins: Biological functions and related syndromes. Front
Mol Neurosci. 15:10119182022. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Bajaj T, Häusl AS, Schmidt MV and Gassen
NC: FKBP5/FKBP51 on weight watch: Central FKBP5 links regulatory
WIPI protein networks to autophagy and metabolic control.
Autophagy. 18:2756–2858. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Häusl AS, Bajaj T, Brix LM, Pöhlmann ML,
Hafner K, De Angelis M, Nagler J, Dethloff F, Balsevich G, Schramm
KW, et al: Mediobasal hypothalamic FKBP51 acts as a molecular
switch linking autophagy to whole-body metabolism. Sci Adv.
8:eabi47972022. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Campbell IH, Frye MA and Campbell H:
Metabolic plasticity: An evolutionary perspective on metabolic and
circadian dysregulation in bipolar disorder. Mol Psychiatry.
30:5600–5612. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Tian S, Wu T, Zhang Z, Lv S, Ji X, Zhao Z,
Ma X, Wang J and Bi Y: Activation of central Angiotensin-(1-7)/Mas
receptor alleviates synaptic damage in diabetes-associated
cognitive impairment via modulating AKT/FOXO1/PACAP axis. Int J
Biol Sci. 21:2824–2842. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Yin B, Qian B, Yu H, Ke S, Li Z, Hua Y, Lu
S, Wang C, Li M, Guo S, et al: NNMT/1-MNA protects against hepatic
ischemia-reperfusion injury through the AKT/FOXO1/ANGPT2/JNK axis.
Nat Commun. 16:47792025. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Wang H, Bai R and Wang Y, Qu M, Zhou Y,
Gao Z and Wang Y: The multifaceted function of FoxO1 in pancreatic
β-cell dysfunction and insulin resistance: Therapeutic potential
for type 2 diabetes. Life Sci. 364:1233842025. View Article : Google Scholar
|
|
107
|
Lees J, Hay J, Moles MW and Michie AM: The
discrete roles of individual FOXO transcription factor family
members in B-cell malignancies. Front Immunol. 14:11791012023.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Liu N, Li R, Cao J, Song X, Ma W, Liu T,
Liang R, Zheng R and Wang S: The inhibition of FKBP5 protects
β-cell survival under inflammation stress via AKT/FOXO1 signaling.
Cell Death Discov. 9:2472023. View Article : Google Scholar
|
|
109
|
Salama SA and Elshafey MM: Cross-talk
between PPARγ, NF-κB, and p38 MAPK signaling mediates the
ameliorating effects of bergenin against the iron overload-induced
hepatotoxicity. Chem Biol Interact. 368:1102072022. View Article : Google Scholar
|
|
110
|
Kokkinopoulou I and Moutsatsou P:
Mitochondrial glucocorticoid receptors and their actions. Int J Mol
Sci. 22:60542021. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Zuo L, Kuo WT, Cao F, Chanez-Paredes SD,
Zeve D, Mannam P, Jean-François L, Day A, Vallen Graham W, Sweat
YY, et al: Tacrolimus-binding protein FKBP8 directs myosin light
chain kinase-dependent barrier regulation and is a potential
therapeutic target in Crohn's disease. Gut. 72:870–881. 2023.
View Article : Google Scholar
|
|
112
|
Dowling AL, Walbridge S, Ertekin C,
Namagiri S, Camacho K, Chowdhury A, Bryant JP, Kohut E, Heiss JD,
Brown DA, et al: FKBP38 regulates self-renewal and survival of GBM
neurospheres. Cells. 12:25622023. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Lee B, Oh Y, Cho E, DiAntonio A, Cavalli
V, Shin JE, Choi HW and Cho Y: FK506-binding protein-like and
FK506-binding protein 8 regulate dual leucine zipper kinase
degradation and neuronal responses to axon injury. J Biol Chem.
298:1016472022. View Article : Google Scholar : PubMed/NCBI
|














