Inosine monophosphate (IMP), as a crucial
intermediate in the metabolism of purine nucleotides, is essential
for both de novo synthesis and salvage synthesis pathways
(1). The body effectively
recovers ~90% of free bases in the salvage pathway through
hypoxanthine-guanine phosphoribosyltransferase, using resources to
synthesize the necessary IMP and guanine monophosphate. The regular
demands of the body are mainly satisfied by this mechanism
(2). However, salvage synthesis
is unable to keep up with the growing need for purines due to the
rapid proliferation of tumor cells. The primary mechanism at this
stage is de novo synthesis (3,4),
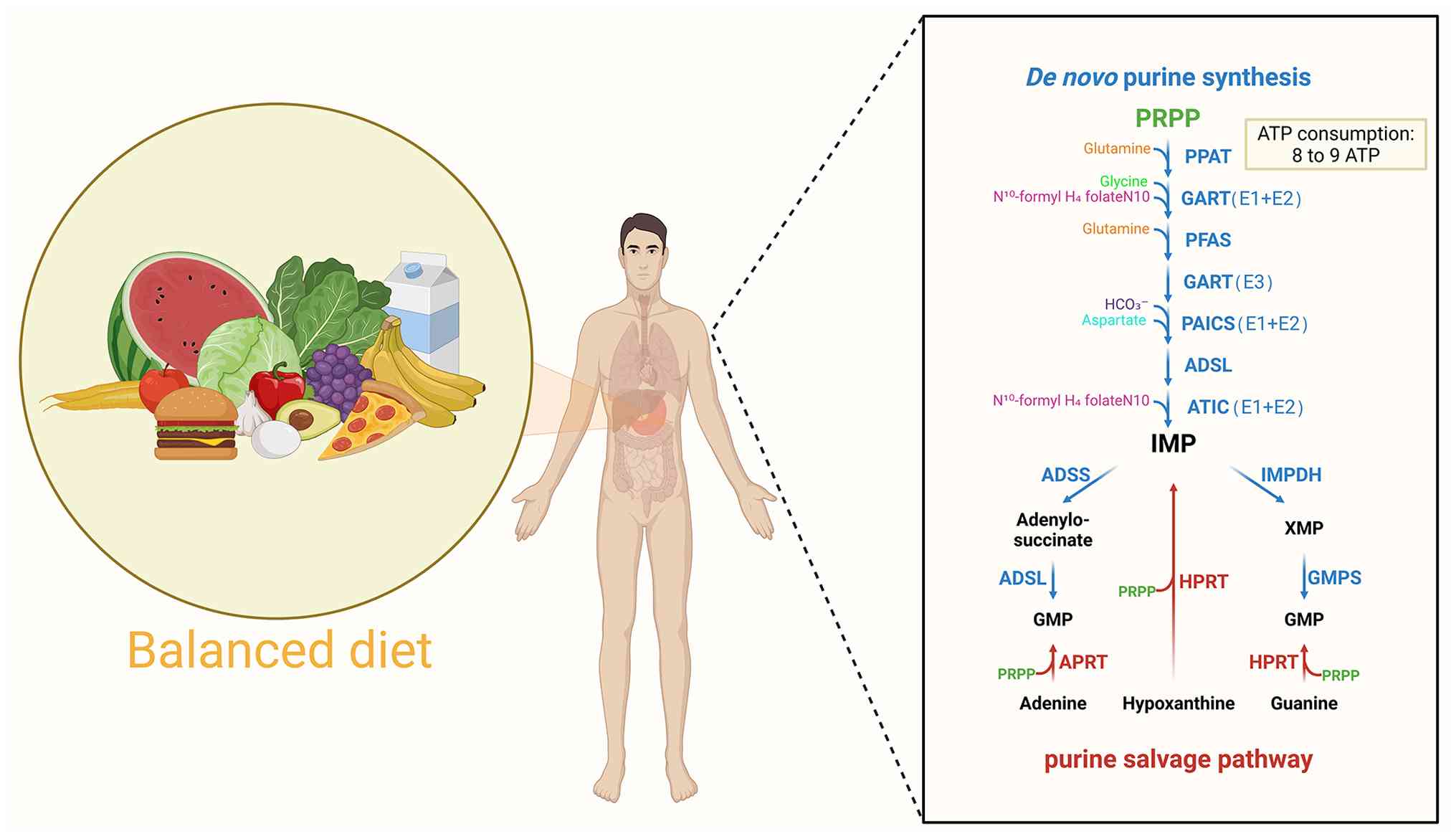
which uses 5'-ribose-5'-phosphate as its absolute starting
substrate (a common precursor for all purine nucleotide synthesis,
usually not listed separately) and catalyzes reactions through
trifunctional enzymes [trifunctional glycinamide nucleoside
transcarboxylase (GART) protein containing glycinamide nucleoside
synthase, GART and aminoimidazole nucleoside synthase domains],
bifunctional enzymes [phosphoribosylaminoimidazole carboxylase and
succinocarboxamide synthetase (PAICS) containing
carboxyaminoimidazole ribonucleotide and
succinylaminoimidazolecarboxamide ribonucleotide domains],
5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP
cyclohydrolase (ATIC) containing 5-aminoimidazole-4-carboxamide
ribonucleotide (AICAR) and IMP cyclohydrolase domains] and
monofunctional enzymes [phosphoribosyl pyrophosphate
amidotransferase (PPAT), formylglycinamide ribonucleotide synthase
(FGAMS) and adenylosuccinate lyase (ADSL)] (5-7).
This process consumes large amounts of ATP, glutamine, formate,
glycine, aspartate and carbon dioxide to synthesize the necessary
purine nucleotides. Among these, IMP is crucial for adenine and
guanine monophosphate, which are indicators of cellular
proliferation and development (5,8).
Furthermore, the need for purine synthesis precursors is
significantly increased because of the rapid proliferation
characteristics of tumor cells. This drives the regulation of
related metabolic pathways, such as reprogramming of glucose
metabolism, regulation of serine and glutamine metabolism, the
one-carbon cycle and mitochondrial function. Collectively, these
processes constitute an extensive metabolic network that surrounds
the primary axis of IMP metabolism (9-11). Major metabolic pathways are
intricately linked and mutually reinforced within the dysregulated
tumor cell, continuously delivering IMP to support its rampant
proliferation (11). These
pathways also include RAS-ERK, PI3K/AKT-mTORC1 and
Hippo-Yes-associated protein (YAP), which modulate various enzymes
and metabolic changes through demand-driven and feedback-regulated
mechanisms, thereby maintaining the uncontrolled growth of tumor
cells. Numerous enzymes, including hypoxanthine dehydrogenase,
purinosomes, which are multi-enzyme complexes responsible for de
novo purine biosynthesis, and modifications in the immune
microenvironment linked to hypoxanthine metabolism, have been
identified as important targets for preventing tumors or
suppressing pathogens. Although it has been thoroughly examined in
disciplines such as protozoology and immunology, research on its
function in tumor cells has accelerated recently. The objective is
to develop novel IMP inhibitors for treating tumors (12-14) (Fig. 1).
Numerous previous studies have suggested targeting
IMP metabolism as a highly promising cancer therapeutic strategy,
precisely because of the critical role of hypoxanthine. We have
developed an integrated conceptual framework that includes
precursor supply, dynamic enzyme regulation and signaling pathway
modulation based on a series of metabolic reprogramming events in
tumor cells. This framework has excellent potential to understand
and exploit IMP metabolism in tumor cells. Additionally, targeted
inhibition strategies for hypoxanthine dehydrogenase and the
effects of IMP metabolism on the immune microenvironment were
systematically reviewed. Certain techniques have demonstrated
successful reduction of tumor cell proliferation in preclinical
models. These studies shed light on the physiological significance
of IMP synthesis regulation and offer more precise targets and
theoretical foundations for developing IMP metabolism-targeted
cancer therapies.
Aerobic glycolysis in tumor cells serves as the
foundation for producing IMP synthesis precursors. Most cancer
cells adhere to the Warburg effect, which prioritizes glycolysis to
convert glucose into lactate even in the presence of abundant
oxygen, whereas normal cells usually generate energy by
mitochondrial oxidative phosphorylation. According to the theory by
early Warburg, tumor cells prefer aerobic glycolysis, an
inefficient ATP production method that yields only 2 ATP molecules,
whereas oxidative phosphorylation produces 36 ATP. Warburg
explained that this was a passive adaptation due to mitochondrial
dysfunction caused by hypoxia, lactate accumulation and other
factors, which results in irreversible respiratory impairment
(15). However, subsequent
studies revealed that most cancer cells have normal mitochondrial
function and are still capable of oxidative phosphorylation; their
metabolic activity has changed to glycolysis (16-18). It has been found that cancer
cells preferentially use aerobic glycolysis to meet the
biosynthetic demands of proliferation by allocating glucose-derived
carbon toward biosynthesis rather than complete oxidative
phosphorylation. The pentose phosphate pathway (PPP) converts ~10%
of glucose into 5-phosphoribose to supply nucleotide synthesis;
another portion is metabolized into acetyl-CoA for lipid synthesis;
and only a small fraction is completely oxidized to carbon dioxide
(CO2) to meet basic ATP requirements. Cancer cells
coordinate the PPP with glutamine metabolism in an integrated
process, in addition to allocating carbon through aerobic
glycolysis (19). A total of
three vital enzymes are critical to the aerobic glycolysis of tumor
cells: Hexokinase 2 (HK2), pyruvate kinase M2 (PKM2) and lactate
dehydrogenase A (LDH-A) (20).
HK2 is highly expressed in many tumors and localizes to the outer
mitochondrial membrane to bind with the voltage-dependent anion
channel (VDAC) along with hexokinase 1 and hexokinase domain
component 1. This arrangement allows direct use of VDAC-derived ATP
for glucose phosphorylation, thereby improving hexokinase activity,
accelerating glucose-6-phosphate (G-6-P) synthesis, modulating
glycolytic flux and inhibiting G-6-P accumulation (21). By contrast, LDH-A inhibition
significantly suppresses cell proliferation. LDH-A-specific
inhibitors (such as oxaloacetate) block lactate production in tumor
cells, causing intracellular carbon accumulation, impaired
diversion of glycolytic intermediates toward nucleotide and lipid
synthesis, decreased NAD+ regeneration, reduced
glycolytic flux and diminished NADPH production, because glutamine
metabolism depends on glycolysis-coupled NAD+
regeneration, ultimately leading to growth arrest or apoptosis.
Therefore, LDH-A is crucial for preserving the precursors needed
for IMP synthesis (22).
Furthermore, proliferative signals inhibit the activity of PKM2, a
tumor-specific isoform. Glycolytic intermediates, such as
3-phosphoglycerate (3-PG) and phosphoenolpyruvate (PEP), in their
low-activity dimeric forms, are inefficiently converted to pyruvate
and are instead redirected into anabolic pathways. Particularly,
3-PG and fructose-6-phosphate enter the non-oxidative branch of the
PPP to produce ribose-5-phosphate, whereas PEP facilitates the
biosynthesis of alanine and serine (23,24).
The PPP, which is an essential part of carbohydrate
metabolism, is primarily responsible for producing
5'-phosphoribulose and NADPH. The former serves as a critical
precursor for IMP synthesis within the body, whereas the latter is
essential for maintaining redox homeostasis (25). The PPP consists of two
functionally complementary branches: Oxidative and non-oxidative.
The oxidative branch in tumor cells, driven mainly by
glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate
dehydrogenase, produces NADPH to support antioxidant defense and
lipid biosynthesis, while the non-oxidative branch is adapted to
hypoxic and highly proliferative conditions. This branch
circumvents the limitations of oxidative phosphorylation under
hypoxia by converting glycolytic intermediates into 5-phosphoribose
independently of mitochondria. It is catalyzed by
transketolase-like 1 (TKTL1) and transaldolase. This reversible
pathway supplies >60% of the 5-phosphoribose required by tumor
cells and dynamically connects glycolysis to biosynthetic pathways
to distribute carbon in accordance with energetic and anabolic
requirements (26,27). These two branches are
interconnected rather than independent: The oxidative branch
produces NADPH, which is generated by the oxidative branch,
protects non-oxidative enzymes such as TKTL1 from reactive oxygen
species (ROS)-mediated damage, whereas the non-oxidative branch
replenishes G-6-P for the oxidative pathway by recycling
intermediates into glycolysis, forming a metabolic cycle that
collectively supports tumor proliferation, apoptosis resistance and
metastatic potential (26). It
is now recognized that tumor cells exhibit upregulation of the PPP
pathway, particularly in rapidly growing tumors such as
glioblastoma. Increased expression of enzymes such as G6PD
indicates enhanced PPP activity, providing an abundance of
ribose-5-phosphate and promoting de novo IMP synthesis.
Elevated IMP, adenosine monophosphate (AMP) and guanosine
monophosphate (GMP) levels were verified in five patient-derived
glioblastoma models, maintaining cellular proliferation and
stemness. Furthermore, concurrent NADPH synthesis also improves
invasive phenotypes by maintaining tumor cell redox homeostasis. In
conclusion, tumor cells modulate the PPP pathway to boost
5-phosphoribose needed for IMP synthesis and NADPH to maintain a
stable tumor microenvironment (28). It has been demonstrated that
combining therapy with G6PD and glycolysis inhibitors significantly
improves radiosensitivity in human gliomas, suggesting that this
combination may be a novel targeted treatment approach (29).
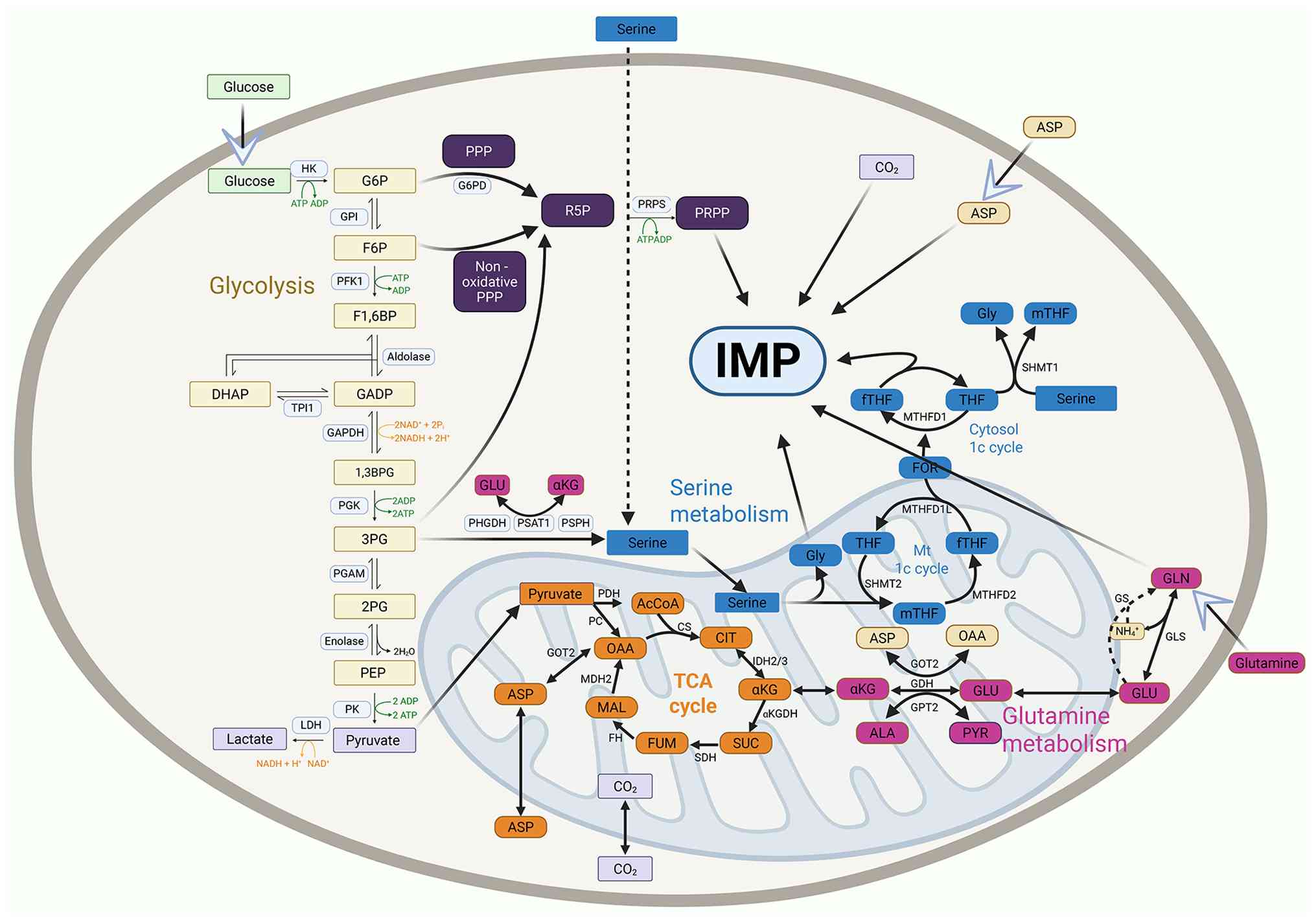
Glucose is taken up by cancer cells and metabolized
into precursors such as serine and glycine, which are essential for
one-carbon metabolism and provide the carbon units needed to
synthesize IMP and glycine (30). The primary source of cytoplasmic
serine is extracellular uptake through the
alanine/serine/cysteine/threonine transporter 1 and the diversion
of ~10% of the glycolytic intermediate 3-PG into de novo
serine synthesis, which is catalyzed by phosphoglycerate
dehydrogenase (PHGDH). Serine is subsequently produced through
transamination by phosphoserine aminotransferase 1 and
dephosphorylated by phosphoserine phosphatase. Cytoplasmic serine
hydroxymethyltransferase 1 (SHMT1) or mitochondrial serine
hydroxymethyltransferase 2 (SHMT2) then catalyze the combination
with tetrahydrofolate (THF) to produce glycine and
5,10-methylenetetrahydrofolate (31-33). Most cancer cells preferentially
use mitochondrial SHMT2 to boost mitochondrial folate synthesis
rather than cytoplasmic SHMT1 to convert serine to glycine; SHMT2
knockout leads to increased accumulation of AICAR, an intermediate
in IMP synthesis (34). It is
interesting to note that SHMT2 generates S-adenosylmethionine (SAM)
through one-carbon metabolism, increasing N6-methyladenosine
(m6A) modification of PPAT mRNA. This modification
enables recognition and stabilization by insulin-like growth factor
(IGF) 2 mRNA-binding protein 2, which eventually increases PPAT
expression to promote renal cell cancer development (35). Tumor cells synthesize required
IMP through extracellular serine uptake when serine availability is
limited. This finding suggests that lowering IMP synthesis may
enhance the efficacy of therapies in specific tumor cells (36). The primary carbon source for
purine ring formation in the cancerous tissues of non-small cell
lung cancer (NSCLC) is glucose. This process involves the
activation and compartmentalization of the glucose-to-serine
pathway in the cytoplasm, as well as an enhanced reverse one-carbon
flux that reduces the incorporation of external serine into purine
synthesis (30,37). Numerous types of tumor cell have
been shown to exhibit a significant increase in gene expression
linked to serine metabolism. For instance, PHGDH is markedly
overexpressed in melanoma, breast cancer and NSCLC, particularly in
70% of triple-negative breast cancers, which is associated with
poor prognosis (37,38).
Serine metabolism and glycolysis are closely related
due to complex feedback regulation. Elevated serine levels activate
the low-activity M2 isoform of PKM2, which catalyzes the conversion
of phosphoenolpyruvate to pyruvate in the final stage of glycolysis
(39). This process maintains
glycolytic flux while restricting diversion of 3-PG into de
novo serine synthesis. Serine depletion lowers PKM2 activity,
impairs the terminal glycolytic step and results in 3-PG
accumulation that is redirected into the serine biosynthetic
pathway. Simultaneously, elevated 2-phosphoglycerate (2-PG)
activates PHGDH, a crucial enzyme in serine and glycine synthesis,
thereby increasing de novo serine production; 2-PG is
converted to 3-PG by phosphoglycerate mutase, which is immediately
upstream of PHGDH. As a result, rapid tumor proliferation and high
IMP consumption cause serine deficiency, inhibit PKM2 activity,
increase 3-PG accumulation and promote compensatory serine
synthesis. This feedback circuit partially explains the limited
effectiveness of the purine synthesis inhibitor methotrexate.
Furthermore, ribonucleotides-purine biosynthetic intermediates-can
allosterically activate PKM2 under glucose-restricted conditions,
offering an additional adaptive mechanism for cancer cell survival
(40). Thus, focusing on the
association between serine metabolism and glycolysis may provide a
viable strategy for managing cell growth and developing cancer
therapies.
Glutamine is necessary for cancer cell proliferation
and, together with its metabolite aspartate, serves as a crucial
precursor for IMP synthesis. Rapid tumor proliferation diverts
vital tricarboxylic acid cycle (TCA cycle) intermediates toward
biosynthesis, disrupting the balance of the TCA cycle. Glutamine is
converted to glutamate through the glutaminase pathway and then
metabolized by glutamate dehydrogenase 1 into α-ketoglutarate,
which enters the TCA cycle to replenish carbon sources (41,42). Furthermore, α-ketoglutarate can
be further oxidized to produce succinate and fumarate. These
metabolites assist the TCA cycle in cancer cells by providing ATP,
NADH and FADH2. They also act as essential tumor
metabolites that promote rapid tumor cell proliferation (41,43,44). Additionally, glutamate is
converted by glutamic-oxaloacetic transaminase (GOT) and
glutamic-pyruvic transaminase (GPT) into alanine and aspartate,
which provide tumor cells with nitrogen sources. In particular, GPT
catalyzes the conversion of glutamate and pyruvate into
α-ketoglutarate and alanine, with α-ketoglutarate replenishing the
TCA cycle and alanine supporting protein synthesis. It has been
demonstrated that GPT2 has a crucial role in breast cancer,
glioblastoma and KRAS-driven colorectal cancer (45-47). GOT catalyzes the transformation
of glutamate and oxaloacetate into α-ketoglutarate and aspartate.
Endogenous synthesis is essential since cancer cells have a limited
ability to absorb exogenous aspartate. Aspartate production exerts
a central oncogenic function by promoting nucleotide and protein
synthesis and contributing to the malate-aspartate shuttle, which
produces NADPH to maintain tumor cell redox homeostasis (43,48). Additionally, asparagine
synthetase (ASNS) utilizes glutamate as a substrate to produce
asparagine. This process consumes significant amounts of aspartate
and stimulates its synthesis in tumor cells (49). Research has revealed the
involvement of ASNS in tumor development and metastasis, in
addition to its link to poor survival rates in several cancers,
including NSCLC (50). Notably,
glutamate catalyzed from glutamine serves as an important precursor
for glutathione (GSH). It produces GSH, an essential reducing agent
that scavenges ROS and protects cancer cells from apoptosis,
together with cysteine and glycine (51). Furthermore, the ability of this
metabolic pathway to convert glutamine into biosynthetic precursors
is particularly critical in hypoxic conditions (52).
Mitochondria, as the primary metabolic organelles in
cells, coordinate cellular energy production and provide the raw
materials needed for cell proliferation, given the marked demand of
tumor cells for TCA cycle precursors and the energy produced by
de novo IMP synthesis. Their critical role in the IMP
metabolic pathway is undeniable. Mitochondria replenish TCA cycle
intermediate metabolites by oxidizing glutamine-derived
α-ketoglutarate or acetyl-CoA and maintain TCA cycle activity
(53). Furthermore,
mitochondrial respiration produces aspartate, which increases IMP
synthesis in low-oxygen environments, promoting tumor growth and
the progression of cancer (54,55). The blocking of mitochondrial
respiration reduces intracellular aspartate levels, which in turn
restricts the production of asparagine, which is derived from
aspartate (56). Interestingly,
fibroblasts associated with cancer serve as an additional source of
aspartate, which promotes the proliferation of tumor cells
(54). Furthermore, SHMT2
converts serine into glycine and one-carbon units in mitochondria,
whereas mitochondria produce ATP and CO2 by oxidative
phosphorylation (57).
Therefore, mitochondria serve as factories for the synthesis of
de novo IMP precursors and are crucial for IMP metabolism.
Additionally, purinosomes involved in IMP synthesis are
intrinsically linked to mitochondria. Targeting mitochondrial
function may be a viable strategy for inhibiting purine
biosynthesis due to this mitochondrial dependence (8) (Fig.
2).
The rate-limiting enzyme is the one that catalyzes
the slowest step in a metabolic pathway; its activity controls the
overall flux of the pathway and is a crucial component of metabolic
regulation. Tumor cells have been demonstrated to stimulate
proliferation by enzymatically modulating the metabolism of
hypoxanthine nucleotides. Rate-limiting enzymes such as
phosphoribosyl pyrophosphate (PRPP) synthetase (PRPS), PPAT and
IMPDH are important players. Additionally, purinosomes, functional
assemblies of enzymes involved in de novo purine synthesis,
are critical for the metabolic reprogramming of nucleotides in
tumors. This review systematically summarizes how these enzymes
dynamically modulate IMP production to satisfy tumor proliferative
demands and explores variations in IMPDH dependency among tumor
cell types (58-72) (Table I).
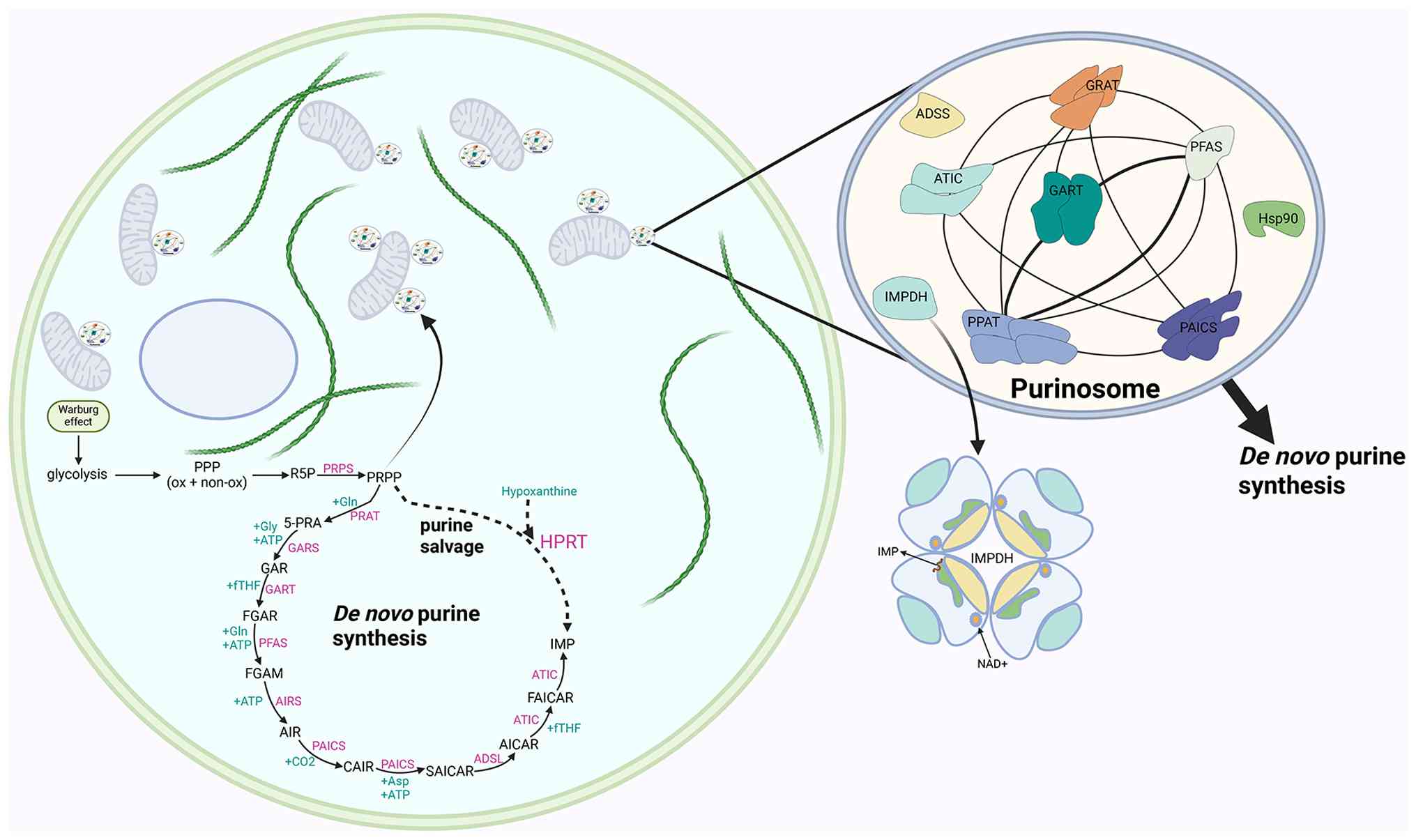
Purinosomes, initially identified in 2008, are
dynamic intracellular structures made up of six enzymes involved in
de novo purine synthesis. The core scaffold comprises PPAT,
GART and FGAMS, while PAICS, ADSL and ATIC are associated with
peripheral structures. This spatial organization facilitates
metabolic channeling in order to produce IMP and downstream purines
to meet cellular demands effectively. Notably, purinosomes are
functionally coupled to mitochondria, suggesting a link between
de novo purine synthesis and cellular energy metabolism. The
primary function of purinosomes in growth-related pathways makes
them attractive therapeutic targets, despite the fact that their
significance in cancer is still unclear (8,73-75). Its formation is driven by reduced
cellular purine levels and increased metabolic demand. Enzymes of
the de novo synthesis pathway assemble into purinosomes when
cells require high purine levels, such as during
G1/S-phase proliferation or when salvage pathways are
compromised. This organization improves metabolic efficiency by
channeling substrates, limiting diffusion or degradation of
intermediates, and increasing de novo synthesis flux by
~50%, thereby increasing IMP production threefold as compared to
normal growth conditions. Pyrimidine body production rises by 25%
in cells with impaired salvage pathways, such as HGPRT knockout
cells, thereby compensating to maintain purine supply. Pathway flux
may be markedly more noticeable during the G1 phase of
cell proliferation because these investigations are asynchronous
(8,76,77). Functional loss of any purinosomes
component disrupts its stability and structure (78,79), and purine supplementation causes
complete or significant depletion of purinosomes (8). According to time-lapse fluorescence
microscopy analysis, the proportion of purinosome-positive cells
peaked during the G1 phase, providing essential purine
nucleotides for cell growth, and progressively decreases throughout
the course of the remaining cell cycle. Interestingly, expression
levels of de novo purine synthesis enzymes did not change
across the cell cycle, suggesting that mechanisms other than
purinosome assembly and disassembly may also regulate this process
(77). Available evidence
suggests that a significant portion of purinosomes in
purinosome-positive cells localize to mitochondria rather than
being randomly distributed throughout the cytoplasm, indicating a
close mitochondrial association due to the high energetic needs of
purine synthesis. Microtubules provide a structural foundation for
purinosome function, as evidenced by the spatial regulation of
purinosome assembly being dependent on microtubules. Purinosomes
travel along the microtubule network, and microtubule
depolymerization disrupts their mitochondrial colocalization,
leading to a roughly 50% reduction in the flux of de novo
purine synthesis. Super-resolution fluorescence microscopy further
revealed colocalization of purinosomes with mitochondria, with at
least nine purinosome components located at the
microtubule-mitochondria interface. This organization allows
purinosomes to directly access ATP, one-carbon units (formate),
aspartate and glutamine, thereby maximizing the efficiency of
purine synthesis. Notably, purinosome formation is increased by
defects in mitochondrial functions, including electron transport or
oxidative phosphorylation, whereas excessive purinosome
accumulation can impair mitochondrial function (5,80,81). Although the role of purine
synthase in cancer has not been extensively studied, it is a
promising target for possible treatments due to its crucial
involvement in notable cellular growth pathways. Furthermore, its
dependence on mitochondria suggests that targeting mitochondria can
be a strategy to inhibit purine biosynthesis (8). Additionally, molecular chaperones
heat shock protein (HSP70)/HSP90 actively contribute to purinosomes
assembly, stability maintenance and functional execution by direct
binding, folding assistance and degradation regulation. They serve
as essential regulators of the biophysical properties and
structural integrity of purinosomes, and they also provide a
potential therapeutic strategy targeting purinosomes by interfering
with chaperone function (82-84). Tumor cells use mechanisms to
regulate the assembly and disassembly of purinosomes or their
interactions with other organelles to meet their high IMP demand.
Tumor cells use microtubules to assemble purinosomes when IMP is
deficient, a crucial adaptive mechanism that enables cancer cells
to respond to purine scarcity. The process can be inhibited by
docetaxel, a microtubule-stabilizing chemotherapeutic agent
(85) (Fig. 3).
Numerous vital concerns about the structure and
function of purinosomes remain unresolved despite initial
observations. The present review summarized the main knowledge gaps
and emerging hypotheses in purine vesicle biology to highlight
their potential for cancer research.
Purinosomes play a central role in cellular
metabolism, but little is known about their regulatory mechanisms
and functional properties in the tumor context. First, whether
various tumor types exhibit distinct patterns of purinosome
assembly remains unknown. For instance, although myelocytomatosis
oncogene (MYC)-driven tumors frequently exhibit increased de
novo purine synthesis flux (86), systematic studies of assembly
principles unique to tumor subtypes are limited. Second,
purinosomes are markedly enriched in the vicinity of mitochondria;
nevertheless, the functional mechanism responsible for this spatial
coupling remains undefined. Additionally, purinosomes may receive
essential substrates from mitochondria, such as ATP, formate and
aspartate (8). Can purinosomes
specifically inhibit tumor cells by disrupting mitochondrial
colocalization, potentially developing a new therapeutic
avenue?
Furthermore, the assembly of purinosomes depends on
molecular chaperones such as HSP90/HSP70, indicating that they
constitute functional targets for chaperone inhibitors (83). However, the extent of their
dependence on the chaperone system and their specificity in tumor
cells remains to be confirmed. Finally, the morphological, dynamic
and functional traits of the purinosome closely resemble those of
liquid-liquid phase separation (LLPS)-formed aggregates (82,87,88). Although LLPS is the most favored
proposed mechanism to explain its development and characteristics,
there is still limited evidence. There are likely other/further
mechanisms.
Overall, these unresolved issues constitute
important areas of research in purine metabolism. An in-depth
investigation of their mechanisms may help clarify patterns of
nucleotide metabolism in tumors and provide crucial information for
developing novel metabolic-targeted treatment approaches.
The human genome encodes two isoforms of IMPDH:
IMPDH1, located on chromosome 7, and IMPDH2, located on chromosome
3. These isoforms are essential enzymes that catalyze the
conversion of IMP to xanthosine monophosphate, regulating the
influx of guanine nucleotides. Interestingly, they are found in
nearly all organisms (100).
The assembly of intracellular IMPDH filaments forms rod-like and
ring-like structures. This process enables proliferating cells to
maintain high levels of guanine nucleotides, which are necessary
for rapid cell division. Furthermore, IMPDH is frequently
overexpressed in numerous types of tumor cells (101).
According to available data, IMPDH is highly
expressed in a variety of tumor types, with IMPDH2 expression being
particularly high. Increased IMPDH activity enhances the GMP
biosynthetic pathway, leading to markedly elevated GMP and GTP
production. This elevated nucleotide output meets tumor cell
demands for purines, energy and signaling necessary for DNA and RNA
synthesis, thereby promoting proliferation (102). For instance, increased
expression of IMPDH in colorectal cancer encourages cell
proliferation, invasion and migration. Similar results were
observed in acute myeloid leukemia, glioblastoma and small-cell
lung cancer (67,70,103,104). There is evidence that the
conversion of IMP to GMP consumes NAD+ and produces
NADH. This process is accelerated by high IMPDH expression in tumor
cells, which increases NADH synthesis and significantly lowers the
cytoplasmic NAD+/NADH ratio. This change impairs GAPDH
activity and disrupts glycolytic flux because the glycolytic enzyme
GAPDH needs NAD+ as a cofactor. As a result,
glucose-derived intermediates are redirected toward the PPP,
thereby increasing 5-phosphoribose synthesis and accelerating de
novo nucleotide synthesis in tumor cells (105,106). Furthermore, IMPDH is essential
in the tumor microenvironment. De novo purine synthesis is
inhibited when important substrates such as 5'-ribose-5-phosphate
and glucose are limited; under these conditions, increased IMPDH
expression compensates by using salvage pathway-derived IMP to
maintain GMP and GTP production (1). According to recent research,
cellular hypoxia also increases IMPDH expression, which encourages
GMP production to sustain basal proliferation in hypoxic conditions
(107). However, further
research on this mechanism is still needed.
Malignant tumors of the hematopoietic system exhibit
a high degree of dependence on IMPDH2. This dependency arises from
their high rates of proliferation, reduced ability to recycle
purines and a MYC-driven increase in purine synthesis metabolism.
For instance, acute myeloid leukemia (AML) with mixed lineage
leukemia (MLL) rearrangements exhibits high sensitivity to IMPDH
inhibition, highlighting the crucial role of IMPDH2 in the
initiation and maintenance of leukemia (72). Multiple myeloma and certain acute
lymphoblastic leukemia subtypes also depend on IMPDH2 to maintain
GTP supply, thereby sustaining their proliferative capacity
(109,110). These tumors frequently struggle
to maintain nucleotide homeostasis through metabolic compensation
pathways when IMPDH is inhibited, making IMPDH inhibition a unique
metabolic vulnerability in such tumors.
By contrast, solid tumors exhibit subtype-specific
dependence on IMPDH. IMPDH2 is consistently increased in
glioblastoma and is involved in ribosome biosynthesis and DNA
damage repair. Inhibiting IMPDH2 improves the effectiveness of
radiotherapy and Temozolomide chemotherapy, indicating a distinct
metabolic therapeutic window (70). IMPDH2 is also markedly elevated
in highly proliferative solid tumors such as ovarian cancer, and is
associated with disease progression and poor prognosis. Its
suppression influences purine synthesis and may also regulate
microenvironmental processes, including inflammation and
angiogenesis (111,112). Despite having higher IMPDH2
levels, colorectal cancer is more sensitive to local
pharmacokinetic features (104). Meanwhile, IMPDH1 expression can
be increased in certain solid tumors under conditions such as
hypoxia or nutritional deprivation to maintain oxidative metabolism
and NADPH homeostasis, thereby reducing sensitivity to pure D-type
IMPDH inhibitors. This implies that combination strategies must be
considered (70).
Furthermore, IMPDH2 upregulation and elevated
nucleolar activity are observed in certain virus-associated or
immune-coupled tumors (such as Epstein-Barr virus-associated
lymphoproliferative disorders), a dependency resulting from GTP
metabolic requirements and aberrant immune regulation (108,113). IMPDH inhibition may have both
antiproliferative and immunomodulatory effects in these diseases,
providing unique therapeutic potential.
Overall, the dependencies of various tumors on IMPDH
vary widely, ranging from high-proliferation-dependent types
centered on IMPDH2 to adaptive dependency types regulated by the
microenvironment and pharmacokinetics, and finally to
metabolism-immune coupling types. These differences imply that
IMPDH-targeted therapies should be customized through specialized
investigation of tumor type, subtype characteristics, IMPDH1/2
expression patterns and metabolic compensatory capacity to improve
treatment selectivity and clinical translation potential.
Significant progress is being made in studying the
regulation of purine synthesis through multiple signaling pathways.
Currently, the most commonly acknowledged signaling pathways
implicated in IMP synthesis are RAS-ERK, PI3K/AKT-mTORC1 and
Hippo-YAP.
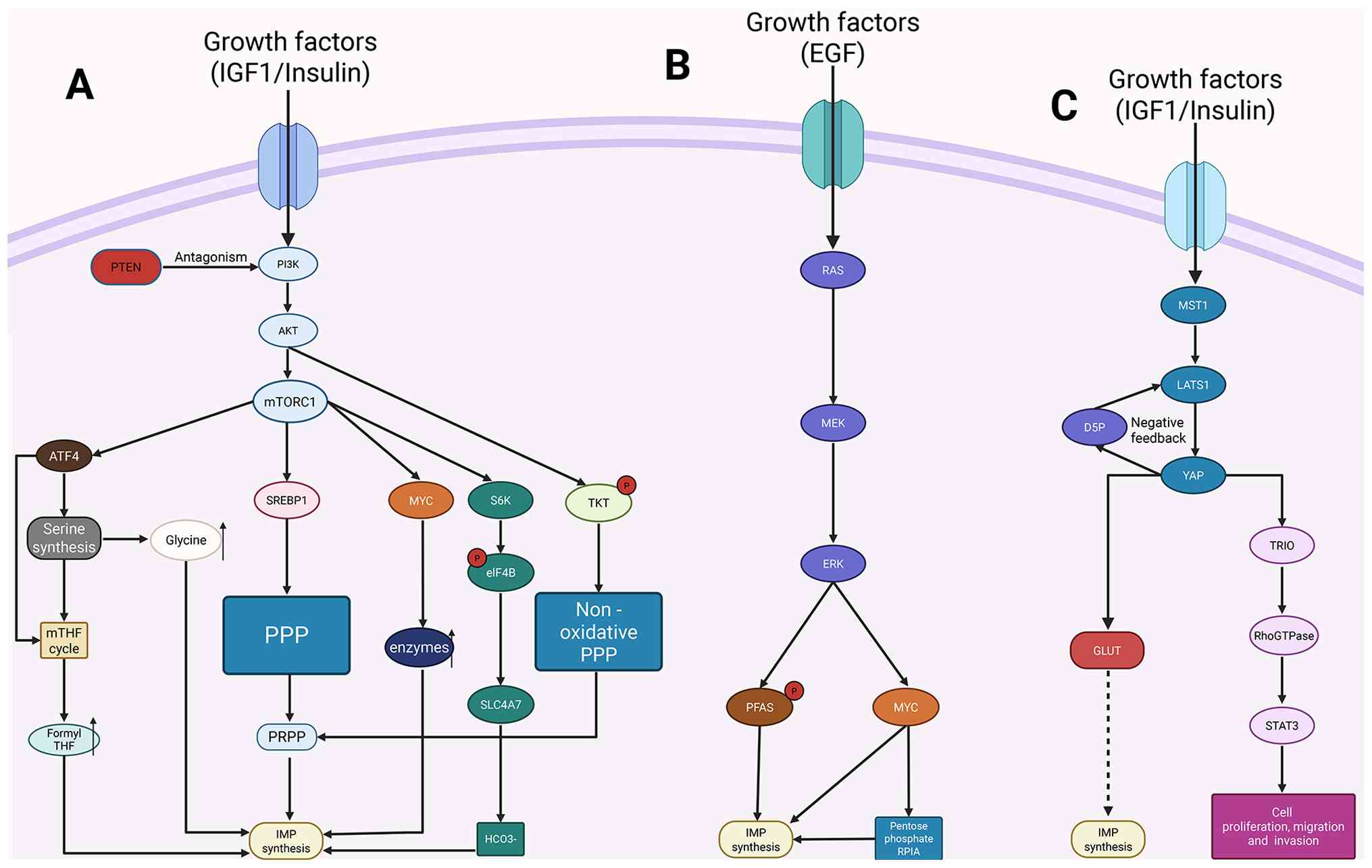
Additionally, mTORC1 serves as a major hub for
signaling and metabolism and regulates the balance between anabolic
and catabolic processes in cells (114). Growth factors enhance mTORC1
signaling by activating the PI3K/Akt pathway, and establishing the
PI3K/AKT-mTORC1 signaling axis (Fig.
4A) (115). Furthermore,
mTORC1 signaling triggers the synthesis of de novo IMP
through its regulatory mechanisms by activating the transcription
factors activating transcription factor 4 (ATF4) and MYC (116,117). ATF4, a metabolic effector of
mTORC1, increases the synthesis of enzymes in the serine/glycine
synthesis pathway and the mitochondrial THF cycle upon induction.
These pathways respectively generate glycine and one-carbon acyl
units for de novo IMP synthesis (116). The production of activated
ribose needed for nucleotide synthesis is also increased by the
regulation of the mTORC1's oxidative branch of the PPP through the
transcription factor sterol regulatory element-binding protein 1
(118). Growth signals activate
TKT through Akt signaling, which in turn activates the
non-oxidative PPP pathway to promote PRPP production (119). Notably, ATF4 increases the
expression of the cystine transporter solute carrier family 7
member 11 in its mTORC1 function, which improves cellular cystine
uptake. After being converted to cysteine, cystine acts as a direct
precursor for the synthesis of GSH, which reduces oxidative stress
in tumor cells driven by rapid nucleotide and protein synthesis
(120).
Furthermore, mTORC1 activation stimulates MYC-driven
purine biosynthesis by upregulating de novo purine synthase.
MYC plays a crucial function in regulating genes involved in
nucleotide synthesis by directly binding to the promoters of most
nucleotide metabolism genes and increasing their expression. For
instance, MYC upregulates the expression of genes such as PPAT,
carbamoyl-phosphate synthetase 2/aspartate
transcarbamylase/dihydroorotase and IMPDH by directly binding to
the promoters (121). MYC
increases the expression levels of IMPDH1 and IMPDH2 in both an
in vitro human B-lymphocyte model and an in vivo
mouse inducible MYC transgenic hepatocarcinoma model. MYC directly
binds to the promoter regions of these two genes, recruiting
histone acetyltransferases and chromatin remodeling complexes, such
as transketolase, to induce histone acetylation, alter chromatin
structure and initiate transcription (122,123). Furthermore, MYC regulates the
expression of GPN-loop GTPase 1 and 3, leveraging their connections
with GTP and RNA polymerase I (Pol I), while improving Pol II
assembly efficiency to fulfill global transcription demands,
thereby modulating related enzymes and ribosomal biogenesis
pathways (124). Furthermore,
growth factors (such as insulin, IGF1 and EGF) activate mTORC1 and
selectively promote mRNA translation of the bicarbonate
cotransporter solute carrier family 4 member 7 (SLC4A7) via
S6K-dependent eukaryotic translation initiation factor 4B
phosphorylation, thereby increasing intracellular
HCO3− levels. Additionally, SLC4A7 deficiency
reduces HCO3− uptake and de novo IMP
synthesis in tumor cells without affecting intracellular pH.
Simultaneously, SLC4A7 deficiency increases tumor sensitivity to
the mTORC1 inhibitor rapamycin, demonstrating that mTORC1 regulates
HCO3− metabolism through SLC4A7 to promote
IMP synthesis and cellular growth (125).
Purinosomes assembly in tumor cells has been linked
to mTORC1, which has been demonstrated to be essential for its
localization to mitochondria. Nevertheless, the precise mechanism
by which mTORC1 regulates this process remains unclear. It will be
fascinating to investigate the possible mechanisms behind
mTOR-mediated purinosomes-mitochondrial localization and
purinosomes assembly in cancer. Additionally, the increase in
lysosomes within tumor cells is a particular response to purine
imbalance, with mTORC1 functioning as a crucial kinase that
connects purinosomes and lysosomal activity (126). Furthermore, the tumor
suppressor phosphatase and tensin homolog (PTEN) inhibits the
activation of PI3K/AKT, and the PI3K/AKT pathway is activated in
the absence of PTEN. This then improves purine and pyrimidine de
novo synthesis pathways through mTORC1, including upregulating
IMPDH translation (127,128).
It remains elusive how mTORC1 activation integrates diverse
metabolic pathways to consistently stimulate metabolic output and
maintain cell proliferation during tumor metabolic reprogramming.
Furthermore, translational opportunities for cancer treatment are
provided by the collaborative mechanisms between mTORC1 and
signaling pathways such as RAS/ERK.
The ERK pathway regulates the extended expression of
genes involved in purine and pyrimidine synthesis by activating the
MYC transcription factor. This acts as an important mechanism for
the continuous increase in nucleotide production in cancer cells, a
process that is now commonly recognized (Fig. 4B) (129). The RAF-MEK-ERK pathway
phosphorylates downstream 90 kDa ribosomal S6 kinase on RAS
activation, which further stimulates the transcriptional activity
of MYC (130). MYC, the primary
regulator of nucleotide synthesis, directly binds to the promoters
of essential purine synthesis enzymes such as PPAT, PAICS and ATIC,
upregulating their expression to activate the nucleotide synthesis
pathway fully (28,131,132). Additionally,
phosphofructokinase-platelet has been demonstrated to enhance
ERK-mediated c-Myc stability (133). Furthermore, mutant K-RAS
activates MYC through ERK, which upregulates ribose-5-phosphate
isomerase-A, a crucial enzyme in the non-oxidative PPP. This
reroutes glucose intermediates into the PPP and increases PRPP
synthesis (134-137), while concurrently regulating
glutamine metabolism to supply nitrogen sources for nucleotide
synthesis, maintaining elevated intracellular nucleotide levels
(138,139). MYC contributes to tumor growth
in two ways: Promoting nucleotide synthesis and introducing a
distinct oncogenic stress through activation of RNA degradation and
nucleotide catabolism. Novel therapeutic approaches for MYC-driven
cancers can be achieved by targeting the compensatory mechanisms of
this process (140). The acute
regulation of IMP synthesis by the ERK pathway is still being
investigated. It is hypothesized that RAS-ERK may directly modulate
the activity of purine synthase enzymes such as PPAT and PAICS
because IMP synthesis is significantly higher in ERK-hyperactivated
cancers than in normal cells. This mechanism guides the development
of direct ERK-targeting drugs that support purine synthesis, even
though the specific phosphorylation sites remain unknown. It has
been revealed that ERK2 increases purine synthesis rates by
directly adding a phosphate group to threonine T619 at position 619
of phosphoribosylformylglycinamidine synthase (PFAS), an important
enzyme in the purine synthesis pathway (141). This implies that the ERK2-PFAS
axis may be a metabolic vulnerability in RAS pathway-driven cancer
cells. Concurrently targeting the purine synthesis pathway and ERK
signaling may emerge as a novel treatment strategy for
RAS/RAF-mutated cancers.
YAP acts as a key effector in the Hippo pathway,
playing an essential role in organ size regulation and
tumorigenesis (Fig. 4C). YAP
improves glutamate ammonia ligase (GLUL) by directly binding and
transcriptionally upregulating GLUL, thereby promoting de
novo nucleotide synthesis. This provides the raw materials for
rapid cell proliferation, which promotes liver growth and
tumorigenesis (142). Yap1
stimulates de novo nucleotide synthesis by inducing glucose
transporter glucose transporter 1, thereby boosting glucose uptake
and anabolic utilization (143,144). Furthermore, YAP regulates
deoxyribonucleoside triphosphate synthesis by upregulating key
enzymes required for their production, such as ribonucleotide
reductase regulatory subunit M2 and deoxythymidine kinase (145). YAP regulates the Ras-related C3
botulinum toxin substrate 1/Ras homolog family member A switch and
STAT3 by directly activating triple functional domain protein
(TRIO), forming the YAP-TRIO-RhoGTPase-STAT3 signaling network that
controls cell migration and invasion (146). Additionally, it has been
demonstrated that YAP is regulated by cell-cell interactions and
mechanical signals in addition to serving as a crucial sensor of
the cellular metabolic state, being directly controlled by the
metabolite D-ribose-5-phosphate (D5P). Myosin heavy chain 9-large
tumor suppressor kinase 1 (LATS1) complex assembly is induced by a
decrease in intracellular D5P, which results in LATS1 degradation
and subsequent YAP activation. As a result, purine nucleoside
phosphorylase levels rise, causing purine nucleoside degradation
and establishing a negative feedback loop that elevates the D5P
concentration. This research places D5P at the center stage,
establishing it as a key metabolic node that connects glucose and
nucleotide metabolism with the Hippo-YAP signaling pathway
(147). D5P or its precursors
may serve as novel anticancer metabolites or, in conjunction with
current metabolic treatments, such as GLUT inhibitors, offer new
therapeutic approaches for cancers with high YAP activity.
In conclusion, while originating from distinct
oncogene activations, the mTORC1, ERK and YAP pathways exhibit
notable functional convergence in de novo IMP synthesis.
First, all three pathways improve the supply of carbon, nitrogen
and one-carbon units required for IMP synthesis by regulating
glucose metabolism, serine/one-carbon metabolism and glutamine use.
Second, the ERK-MYC axis and mTORC1 upregulate the expression or
activity of necessary rate-limiting enzymes (PPAT, PFAS and IMPDH),
increasing the flux of purine de novo synthesis. Third,
mTORC1 and YAP work together to increase purinosomes assembly and
mitochondrial localization, achieving spatial coupling of
nucleotide synthesis with energy/substrate supply at the
subcellular level. Additionally, the D5P-YAP feedback signal
establishes a dynamic equilibrium between purine degradation and
resynthesis, providing an adaptive mechanism for cancer cells to
maintain nucleotide homeostasis under nutritional changes.
Therefore, these three mechanisms ultimately promote rapid
proliferation, maintain RNA/DNA synthesis and guarantee GTP-driven
signaling by enhancing IMP synthesis while having differing
drivers. Accordingly, IMP/GTP metabolism appears to be a downstream
metabolic vulnerability shared by several signaling pathways.
IMP metabolism is a critical node in purine
metabolism. The downstream conversion of its metabolites, GMP/GTP,
directly or indirectly impacts T-cell activation, myeloid cell
polarization and the transmission of adenosine signaling pathways,
thereby exerting significant effects on the human immune
system.
First, T cells exhibit a marked rise in GTP demand
during activation and proliferation. GTP possesses dual roles as
both an essential metabolic substrate and signaling molecule. It
serves as a crucial substrate for RNA synthesis, supporting T-cell
clonal expansion, while also functioning as a key molecule in
receptor signaling mediated by small GTPases. GTP is involved in
vital activities, including immunological synapse formation, T-cell
receptor signal amplification, cytoskeletal remodeling and integrin
activation (148-151). Furthermore, it is the only
precursor needed to synthesize tetrahydrobiopterin (BH4). BH4 is
essential for T-cell mitochondrial electron transport, ATP supply
maintenance and cell cycle progression. It affects nucleotide
synthesis, ultimately resulting in decreased T-cell proliferation
(152). Thus, a substantial GTP
pool is a metabolic requirement for T cells to progress from
initial activation to effector differentiation. T cells demonstrate
functional suppression when IMPDH is inhibited because
intracellular GTP levels significantly decrease, and T cells are
unable to maintain normal clonal expansion and effector
differentiation. These mechanisms explain why IMPDH inhibitors
cause immunosuppression in clinical settings (138) and suggest that the metabolic
imbalance of IMPs in the tumor microenvironment may weaken
antitumor immunity by limiting GTP availability, providing an
important theoretical foundation for targeting IMP metabolism to
boost immune responses (153).
Second, IMP metabolism is closely associated with
macrophage polarization. An adequate supply of purine nucleotides
significantly increases the pro-inflammatory gene expression,
migration, phagocytosis and anti-pathogen capabilities of
classically activated macrophages under inflammatory conditions,
suggesting that IMP/GMP/GTP levels are essential for M1 macrophage
function (154). Increased
purine degradation and uric acid production in the tumor
microenvironment cause macrophages to adopt an immunosuppressive
tumor-associated macrophage phenotype with high programmed cell
death ligand 1 (PD-L1) expression, which can be re-polarized by
inhibiting IMP metabolism (155). Furthermore, impaired purine
degradation in monocyte-derived macrophages raises isocitrate
dehydrogenase 3 activity and causes increased production of α33sdz5
d-ketoglutarate, which promotes M2-like polarization (156).
Furthermore, the body maintains equilibrium by an
intriguing cross-regulation mechanism in which high levels of AMP
and GMP block their own synthesis, while GTP stimulates AMP
synthesis. By contrast, ATP promotes the synthesis of GMP.
Accordingly, increased IMP/GMP/GTP metabolism in tumor cells leads
to enhanced IMP/AMP/ATP metabolism. Subsequently, AMP/ATP can
catalyze the production of additional adenosine using
ectonucleoside triphosphate diphosphohydrolase
1/ecto-5'-nucleotidase (157,158). The role of adenosine in
immunology is unmatched. It is an essential class of
immunosuppressive signaling molecules in immune regulation that
maintains immune homeostasis, limits inflammatory responses and
modulates the tumor immune microenvironment (156,159,160). Consequently, the balance of
IMP/GMP/GTP metabolism affects the operation of the adenosine
signaling pathway, eventually affecting the immune
microenvironment, including tumor cells.
In conclusion, immune system function depends on
the equilibrium of IMP/GMP/GTP metabolism. It exhibits significant
associations with T cells, macrophages, adenosine and other
components and exerts an unparalleled impact on the immune
microenvironment. Scientific investigation into the immunological
implications of this extraordinarily complex process has never
stopped.
Current IMPDH-targeting strategies in tumors
primarily take advantage of the metabolic dependence of cancer
cells on the synthesis of guanine nucleotides from scratch. These
methods disrupt DNA/RNA or GTP synthesis, causing apoptosis, by
inhibiting the rate-limiting enzyme IMPDH to prevent GMP
production.
Targeted therapies developed specifically against
IMPDH can cause the production of highly potent IMPDH inhibitors.
First, it is possible to exploit tumor cell heterogeneity, which
reflects the unique specificity of IMPDH across various tumor cell
types. For instance, IMPDH2 is markedly expressed in cells in
breast cancer and melanoma. BMS-566419 and AVN944 were designed to
target this characteristic, significantly inhibiting the growth of
tumors with high IMPDH2 expression. Recent studies have also
revealed that berberine, a natural product, inhibits the
progression of colorectal cancer by targeting IMPDH2. However, the
structural basis for IMPDH2 selective inhibition remains unclear,
necessitating further structural optimization to improve activity
(161). By contrast, IMPDH1 is
significantly elevated in high-risk groups of tumors like head and
neck squamous cell carcinoma (HNSCC), with the purine biosynthesis
pathway being the most notably increased metabolic pathway. MPA/MMF
markedly reduced the viability of HNSCC cells, inhibited
proliferation, increased apoptosis rates, and suppressed migration
and invasion (162).
Additionally, targeted drugs that only inhibit IMPDH activity in
urothelial carcinoma associated 1 (UCA1)-overexpressing cells can
be developed to reduce toxicity to normal cells by leveraging the
IMPDH1/2-dependent regulation specificity of UCA1/twist family bHLH
transcription factor 1 in bladder cancer (163). Furthermore, a thorough
examination of the structural characteristics of IMPDH can
completely clarify its distinct role in purine synthesis, which
will help develop more specific inhibitors (100,164,165). For instance, highly specific
inhibitors targeting the cystathionine β-synthase (CBS) domain
could be developed due to the substantial homology between IMPDH1
and IMPDH2, their tetrameric structure and the presence of CBS
domains capable of binding GTP/GDP for negative feedback regulation
(166). Furthermore,
investigating associations between IMP synthesis precursors and
other metabolic pathways can lead to the identification of dual- or
multi-target inhibitors. For instance, the dual-covalent inhibitor
HA344 blocks both metabolic pathways to target and eradicate tumor
cells by covalently binding to both PKM2, the essential enzyme in
glycolysis, and IMPDH, the crucial enzyme in de novo purine
production (167). When used in
combination with immune checkpoint inhibitors (ICIs), it increases
immune activation because the antitumor effect of IMPDH inhibitors
balances the immunosuppressive effect of PD-L1 upregulation.
Co-administration with ICIs does not affect their antitumor
effectiveness (68).
Importantly, IMPDH inhibitors can reduce tumor cell
progression when combined with other targeted therapies. Studies
have revealed that toll-like receptor 1/2 agonists induce
differentiation in MLL-fusion AML cells, complementing the
mechanism of IMPDH inhibitors with significant synergistic effects
(72). In vitro
experiments verified synergistic inhibition of AML cell growth when
coupled with the B-cell lymphoma 2 inhibitor venetoclax (168). When co-administered with Ataxia
telangiectasia and Rad3-related protein inhibitors, they cause
p53-independent replication catastrophe, significantly raising
in vitro replication stress levels and markedly inhibiting
tumor growth in MKL-1 xenograft models, suggesting a potential
treatment approach for Merkel cell carcinoma (169). Even though the Food and Drug
Administration has not yet approved IMPDH inhibitors for oncology
indications, ongoing preclinical studies and exploratory clinical
trials are still being conducted to improve their therapeutic
potential (109,110,170-195) (Table II).
IMP metabolism is an essential part of tumor cell
metabolic reprogramming that plays an important role in cell
proliferation, energy balance and nucleotide supply. The metabolic
network that supplies precursors for IMP synthesis provides a
continuous material basis for tumor cell proliferation within the
framework presented in this study. Dynamic regulation of crucial
enzymes, such as PRPS, PPAT, IMPDH and purinosomes, ensures
effective de novo IMP synthesis across a variety of tumor
types. Simultaneously, signaling pathways such as RAS-ERK,
PI3K/AKT-mTORC1 and Hippo-YAP improve the adaptability of IMP
metabolism, allowing sustained tumor cell proliferation in complex
microenvironments. IMPDH inhibition reveals notable differences in
dependence across hematologic malignancies and some solid tumors,
offering potential for precision therapies. Additionally, the
interplay between IMP metabolism and the immune microenvironment
provides a theoretical framework for combined metabolic-immune
therapies.
IMP metabolism in tumor cells remains under
investigation. According to previous studies, targeting this
pathway for cancer treatment holds great potential: i) Integrating
metabolic networks based on precursor supply, such as combining IMP
metabolism with aerobic glycolysis to uncover deeper mechanisms;
ii) developing combined intervention strategies by focusing on
intersection points between IMP metabolism and signaling pathways;
iii) current IMPDH inhibitors face challenges in cancer treatment
because of high therapeutic doses, notable interindividual
differences and limited effectiveness against certain cancers. New,
highly effective IMPDH inhibitors can be developed based on
structural specificity, such as targeting the CBS domain.
Furthermore, exploring combination therapies with other drugs or
optimizing IMPDH inhibitor delivery routes is warranted; iv)
identifying IMPDH-dependent tumors using multi-omics and metabolic
imaging technologies to enable personalized treatment; v) examining
synergistic effects between IMPDH inhibition and immune modulation
to advance metabolic-immune combination treatment strategies.
Future comprehensive investigation of IMP metabolism in tumor
cellular metabolic reprogramming will contribute to building
fundamental knowledge of tumor metabolism, thereby advancing the
field of tumor treatment engineering.
Not applicable.
HZ, HW and XL wrote the original draft. XL was
involved in the conceptualization of the study. YW, DY, WZ and QT
contributed to manuscript editing. ZS and JS provided supervision
and reviewed and edited the manuscript. All authors reviewed the
manuscript and have read and approved the final manuscript. Data
authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
No funding was received.
|
1
|
Tran DH, Kim D, Kesavan R, Brown H, Dey T,
Soflaee MH, Vu HS, Tasdogan A, Guo J, Bezwada D, et al: De novo and
salvage purine synthesis pathways across tissues and tumors. Cell.
187:3602–3618.e20. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zarrella S, Miranda MR, Covelli V, Restivo
I, Novi S, Pepe G, Tesoriere L, Rodriquez M, Bertamino A, Campiglia
P, et al: Endoplasmic reticulum stress and its role in metabolic
reprogramming of cancer. Metabolites. 15:2212025. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Villa E, Ali ES, Sahu U and Ben-Sahra I:
Cancer cells tune the signaling pathways to empower de novo
synthesis of nucleotides. Cancers (Basel). 11:6882019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Murray AW: The biological significance of
purine salvage. Annu Rev Biochem. 40:811–826. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pareek V, Pedley AM and Benkovic SJ: Human
de novo purine biosynthesis. Crit Rev Biochem Mol Biol. 56:1–16.
2021. View Article : Google Scholar
|
|
6
|
Camici M, Garcia-Gil M, Pesi R, Allegrini
S and Tozzi MG: Purine-metabolising enzymes and apoptosis in
cancer. Cancers (Basel). 11:13542019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
He J, Zou LN, Pareek V and Benkovic SJ:
Multienzyme interactions of the de novo purine biosynthetic protein
PAICS facilitate purinosome formation and metabolic channeling. J
Biol Chem. 298:1018532022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pedley AM and Benkovic SJ: A new view into
the regulation of purine metabolism: The purinosome. Trends Biochem
Sci. 42:141–154. 2017. View Article : Google Scholar
|
|
9
|
Lu M, Wu Y, Xia M and Zhang Y: The role of
metabolic reprogramming in liver cancer and its clinical
perspectives. Front Oncol. 14:14541612024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hon KW, Zainal Abidin SA, Othman I and
Naidu R: The crosstalk between signaling pathways and cancer
metabolism in colorectal cancer. Front Pharmacol. 12:7688612021.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pandey S, Singh R, Habib N, Tripathi RM,
Kushwaha R and Mahdi AA: Regulation of hypoxia dependent
reprogramming of cancer metabolism: Role of HIF-1 and its potential
therapeutic implications in leukemia. Asian Pac J Cancer Prev.
25:1121–1134. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shi JZ, Zhu YJ, Zhang MJ, Yan Y, Zhao LL,
Zhang HD, Liu Y, Wu WH, Cheng Z, Qiu CG, et al: Hypoxanthine
promotes pulmonary vascular remodeling and adenosine deaminase is a
therapeutic target for pulmonary hypertension. JACC Basic Transl
Sci. 10:1012732025. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cuny GD, Suebsuwong C and Ray SS:
Inosine-5'-monophosphate dehydrogenase (IMPDH) inhibitors: A patent
and scientific literature review (2002-2016). Expert Opin Ther Pat.
27:677–690. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cholewiński G, Iwaszkiewicz-Grześ D, Prejs
M, Głowacka A and Dzierzbicka K: Synthesis of the inosine
5'-monophosphate dehydrogenase (IMPDH) inhibitors. J Enzyme Inhib
Med Chem. 30:550–563. 2015. View Article : Google Scholar
|
|
15
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Weinhouse S: The Warburg hypothesis fifty
years later. Z Krebsforsch Klin Onkol Cancer Res Clin Oncol.
87:115–126. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moreno-Sánchez R, Rodríguez-Enríquez S,
Marín-Hernández A and Saavedra E: Energy metabolism in tumor cells.
FEBS J. 274:1393–418. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Moreno-Sánchez R, Rodríguez-Enríquez S,
Saavedra E, Marín-Hernández A and Gallardo-Pérez JC: The
bioenergetics of cancer: Is glycolysis the main ATP supplier in all
tumor cells? Biofactors. 35:209–225. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jing Z, Liu Q, He X, Jia Z, Xu Z, Yang B
and Liu P: NCAPD3 enhances Warburg effect through c-myc and E2F1
and promotes the occurrence and progression of colorectal cancer. J
Exp Clin Cancer Res. 41:1982022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fang J, Luo S and Lu Z: HK2: Gatekeeping
microglial activity by tuning glucose metabolism and mitochondrial
functions. Mol Cell. 83:829–831. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fantin VR, St-Pierre J and Leder P:
Attenuation of LDH-A expression uncovers a link between glycolysis,
mitochondrial physiology, and tumor maintenance. Cancer Cell.
9:425–434. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Christofk HR, Vander Heiden MG, Harris MH,
Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL and
Cantley LC: The M2 splice isoform of pyruvate kinase is important
for cancer metabolism and tumour growth. Nature. 452:230–233. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Christofk HR, Vander Heiden MG, Wu N,
Asara JM and Cantley LC: Pyruvate kinase M2 is a
phosphotyrosine-binding protein. Nature. 452:181–186. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Patra KC and Hay N: The pentose phosphate
pathway and cancer. Trends Biochem Sci. 39:347–354. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Riganti C, Gazzano E, Polimeni M, Aldieri
E and Ghigo D: The pentose phosphate pathway: An antioxidant
defense and a crossroad in tumor cell fate. Free Radical Biol Med.
53:421–436. 2012. View Article : Google Scholar
|
|
27
|
TeSlaa T, Ralser M, Fan J and Rabinowitz
JD: The pentose phosphate pathway in health and disease. Nat Metab.
5:1275–1289. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang X, Yang K, Xie Q, Wu Q, Mack SC, Shi
Y, Kim LJY, Prager BC, Flavahan WA, Liu X, et al: Purine synthesis
promotes maintenance of brain tumor initiating cells in glioma. Nat
Neurosci. 20:661–673. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Manganelli G, Masullo U, Passarelli S and
Filosa S: Glucose-6-phosphate dehydrogenase deficiency:
Disadvantages and possible benefits. Cardiovasc Hematol Disord Drug
Targets. 13:73–82. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fan TWM, Bruntz RC, Yang Y, Song H,
Chernyavskaya Y, Deng P, Zhang Y, Shah PP, Beverly LJ, Qi Z, et al:
De novo synthesis of serine and glycine fuels purine nucleotide
biosynthesis in human lung cancer tissues. J Biol Chem.
294:13464–13477. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu D, Zhang K, Khan FA, Pandupuspitasari
NS, Guan K, Sun F and Huang C: A comprehensive review on signaling
attributes of serine and serine metabolism in health and disease.
Int J Biol Macromol. 260:1296072024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shunxi W, Xiaoxue Y, Guanbin S, Li Y,
Junyu J and Wanqian L: Serine metabolic reprogramming in
tumorigenesis, tumor immunity, and clinical treatment. Adv Nutr.
14:1050–1066. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Geeraerts SL, Heylen E, De Keersmaecker K
and Kampen KR: The ins and outs of serine and glycine metabolism in
cancer. Nat Metab. 3:131–141. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tibbetts AS and Appling DR:
Compartmentalization of Mammalian folate-mediated one-carbon
metabolism. Annu Rev Nutr. 30:57–81. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huo FC, Xie M, Zhu ZM, Zheng JN and Pei
DS: SHMT2 promotes the tumorigenesis of renal cell carcinoma by
regulating the m6A modification of PPAT. Genomics. 114:1104242022.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hennequart M, Labuschagne CF, Tajan M,
Pilley SE, Cheung EC, Legrave NM, Driscoll PC and Vousden KH: The
impact of physiological metabolite levels on serine uptake,
synthesis and utilization in cancer cells. Nat Commun. 12:61762021.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
DeNicola GM, Chen PH, Mullarky E, Sudderth
JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, et al: NRF2
regulates serine biosynthesis in non-small cell lung cancer. Nat
Genet. 47:1475–1481. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Locasale JW, Grassian AR, Melman T,
Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen
T, Sharfi H, et al: Phosphoglycerate dehydrogenase diverts
glycolytic flux and contributes to oncogenesis. Nat Genet.
43:869–874. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
39
|
Locasale JW: Serine, glycine and
one-carbon units: Cancer metabolism in full circle. Nat Rev Cancer.
13:572–583. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kamynina E, Lachenauer ER, DiRisio AC,
Liebenthal RP, Field MS and Stover PJ: Arsenic trioxide targets
MTHFD1 and SUMO-dependent nuclear de novo thymidylate biosynthesis.
Proc Natl Acad Sci USA. 114:E2319–E2326. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jin L, Li D, Alesi GN, Fan J, Kang HB, Lu
Z, Boggon TJ, Jin P, Yi H, Wright ER, et al: Glutamate
dehydrogenase 1 signals through antioxidant glutathione peroxidase
1 to regulate redox homeostasis and tumor growth. Cancer Cell.
27:257–270. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yelamanchi SD, Jayaram S, Thomas JK,
Gundimeda S, Khan AA, Singhal A, Keshava Prasad TS, Pandey A,
Somani BL and Gowda H: A pathway map of glutamate metabolism. J
Cell Commun Signal. 10:69–75. 2016. View Article : Google Scholar :
|
|
43
|
Choi YK and Park KG: Targeting glutamine
metabolism for cancer treatment. Biomol Ther (Seoul). 26:19–28.
2018. View Article : Google Scholar :
|
|
44
|
Vanhove K, Derveaux E, Graulus GJ,
Mesotten L, Thomeer M, Noben JP, Guedens W and Adriaensens P:
Glutamine addiction and therapeutic strategies in lung cancer. Int
J Mol Sci. 20:2522019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang B, Chen Y, Bao L and Luo W: GPT2 is
induced by hypoxia-inducible factor (HIF)-2 and promotes
glioblastoma growth. Cells. 11:25972022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kim M, Gwak J, Hwang S, Yang S and Jeong
SM: Mitochondrial GPT2 plays a pivotal role in metabolic adaptation
to the perturbation of mitochondrial glutamine metabolism.
Oncogene. 38:4729–4738. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shen Y, Huang Q, Zhang Y, Hsueh CY and
Zhou L: A novel signature derived from metabolism-related genes GPT
and SMS to predict prognosis of laryngeal squamous cell carcinoma.
Cancer Cell Int. 22:2262022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ahn CS and Metallo CM: Mitochondria as
biosynthetic factories for cancer proliferation. Cancer Metab.
3:12015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Krall AS, Xu S, Graeber TG, Braas D and
Christofk HR: Asparagine promotes cancer cell proliferation through
use as an amino acid exchange factor. Nat Commun. 7:114572016.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cai DJ, Zhang ZY, Bu Y, Li L, Deng YZ, Sun
LQ, Hu CP and Li M: Asparagine synthetase regulates lung-cancer
metastasis by stabilizing the β-catenin complex and modulating
mitochondrial response. Cell Death Dis. 13:5662022. View Article : Google Scholar
|
|
51
|
Ma G, Zhang Z, Li P, Zhang Z, Zeng M,
Liang Z, Li D, Wang L, Chen Y, Liang Y and Niu H: Reprogramming of
glutamine metabolism and its impact on immune response in the tumor
microenvironment. Cell Commun Signal. 20:1142022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Metallo CM, Gameiro PA, Bell EL, Mattaini
KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L,
et al: Reductive glutamine metabolism by IDH1 mediates lipogenesis
under hypoxia. Nature. 481:380–384. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Spinelli JB and Haigis MC: The
multifaceted contributions of mitochondria to cellular metabolism.
Nat Cell Biol. 20:745–754. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bertero T, Oldham WM, Grasset EM, Bourget
I, Boulter E, Pisano S, Hofman P, Bellvert F, Meneguzzi G, Bulavin
DV, et al: Tumor-stroma mechanics coordinate amino acid
availability to sustain tumor growth and malignancy. Cell Metab.
29:124–140.e10. 2019. View Article : Google Scholar
|
|
55
|
Sullivan LB, Gui DY, Hosios AM, Bush LN,
Freinkman E and Vander Heiden MG: Supporting aspartate biosynthesis
is an essential function of respiration in proliferating cells.
Cell. 162:552–563. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Krall AS, Mullen PJ, Surjono F, Momcilovic
M, Schmid EW, Halbrook CJ, Thambundit A, Mittelman SD, Lyssiotis
CA, Shackelford DB, et al: Asparagine couples mitochondrial
respiration to ATF4 activity and tumor growth. Cell Metab.
33:1013–1026.e6. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Newman AC and Maddocks ODK: One-carbon
metabolism in cancer. Br J Cancer. 116:1499–1504. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Li T, Song LL, Zhang YW, Han YL, Zhan ZY,
Xv Z, Li Y, Tang Y, Yang Y, Wang S, et al: Molecular mechanism of
c-Myc and PRPS1/2 against thiopurine resistance in Burkitt's
lymphoma. J Cell Mol Med. 24:6704–6715. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Macmillan AC, Karki B, Yang JC, Gertz KR,
Zumwalde S, Patel JG, Czyzyk-Krzeska MF, Meller J and Cunningham
JT: PRPS activity tunes redox homeostasis in Myc-driven lymphoma.
Redox Biol. 84:1036492025. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Liu T, Wang Z, Ye L, Duan Y, Jiang H, He
H, Xiao L, Wu Q, Xia Y, Yang M, et al: Nucleus-exported CLOCK
acetylates PRPS to promote de novo nucleotide synthesis and liver
tumour growth. Nat Cell Biol. 25:273–284. 2023.PubMed/NCBI
|
|
61
|
Song L, Li P, Sun H, Ding L, Wang J, Li B,
Zhou BS, Feng H and Li Y: PRPS2 mutations drive acute lymphoblastic
leukemia relapse through influencing PRPS1/2 hexamer stability.
Blood Sci. 5:39–50. 2022. View Article : Google Scholar
|
|
62
|
Lv Y, Wang X, Li X, Xu G, Bai Y, Wu J,
Piao Y, Shi Y, Xiang R and Wang L: Nucleotide de novo synthesis
increases breast cancer stemness and metastasis via cGMP-PKG-MAPK
signaling pathway. PLoS Biol. 18:e30008722020. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Goswami MT, Chen G, Chakravarthi BV, Pathi
SS, Anand SK, Carskadon SL, Giordano TJ, Chinnaiyan AM, Thomas DG,
Palanisamy N, et al: Role and regulation of coordinately expressed
de novo purine biosynthetic enzymes PPAT and PAICS in lung cancer.
Oncotarget. 6:23445–23461. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Liu B, Song M, Qin H, Zhang B, Liu Y, Sun
Y, Ma Y and Shi T: Phosphoribosyl Pyrophosphate amidotransferase
promotes the progression of thyroid cancer via regulating pyruvate
kinase M2. Onco Targets Ther. 13:7629–7639. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kitagawa Y, Kondo S, Fukuyo M, Wakae K,
Dochi H, Mizokami H, Komura S, Kobayashi E, Hirai N, Ueno T, et al:
Phosphoribosyl pyrophosphate amidotransferase: Novel biomarker and
therapeutic target for nasopharyngeal carcinoma. Cancer Sci.
115:3587–3595. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ding M, Ma C, Lin Y, Fang H, Xu Y, Wang S,
Chen Y, Zhou J, Gao H, Shan Y, et al: Therapeutic targeting de novo
purine biosynthesis driven by β-catenin-dependent PPAT upregulation
in hepatoblastoma. Cell Death Dis. 16:1792025. View Article : Google Scholar
|
|
67
|
Huang F, Ni M, Chalishazar MD, Huffman KE,
Kim J, Cai L, Shi X, Cai F, Zacharias LG, Ireland AS, et al:
Inosine monophosphate dehydrogenase dependence in a subset of small
cell lung cancers. Cell Metab. 28:369–382.e5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zheng MM, Li JY, Guo HJ, Zhang J, Wang LS,
Jiang KF, Wu HH, He QJ, Ding L and Yang B: IMPDH inhibitors
upregulate PD-L1 in cancer cells without impairing immune
checkpoint inhibitor efficacy. Acta Pharmacol Sin. 46:1058–1067.
2025. View Article : Google Scholar
|
|
69
|
Zhao JZ, Wang W, Liu T, Zhang L, Lin DZ,
Yao JY, Peng X, Jin G, Ma TT, Gao JB, et al: MYBL2 regulates de
novo purine synthesis by transcriptionally activating IMPDH1 in
hepatocellular carcinoma cells. BMC Cancer. 22:12902022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kofuji S, Hirayama A, Eberhardt AO,
Kawaguchi R, Sugiura Y, Sampetrean O, Ikeda Y, Warren M, Sakamoto
N, Kitahara S, et al: IMP dehydrogenase-2 drives aberrant nucleolar
activity and promotes tumorigenesis in glioblastoma. Nat Cell Biol.
21:1003–1014. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Liu X, Wang J, Wu LJ, Trinh B and Tsai
RYL: IMPDH inhibition decreases TERT expression and synergizes the
cytotoxic effect of chemotherapeutic agents in glioblastoma cells.
Int J Mol Sci. 25:59922024. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Liu X, Sato N, Yabushita T, Li J, Jia Y,
Tamura M, Asada S, Fujino T, Fukushima T, Yonezawa T, et al: IMPDH
inhibition activates TLR-VCAM1 pathway and suppresses the
development of MLL-fusion leukemia. EMBO Mol Med. 15:e156312023.
View Article : Google Scholar :
|
|
73
|
An S, Kumar R, Sheets ED and Benkovic SJ:
Reversible compartmentalization of de novo purine biosynthetic
complexes in living cells. Science. 320:103–106. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Deng Y, Gam J, French JB, Zhao H, An S and
Benkovic SJ: Mapping protein-protein proximity in the purinosome. J
Biol Chem. 287:36201–36207. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wu D, Yang S, Yuan C, Zhang K, Tan J, Guan
K, Zeng H and Huang C: Targeting purine metabolism-related enzymes
for therapeutic intervention: A review from molecular mechanism to
therapeutic breakthrough. Int J Biol Macromol. 282:1368282024.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Pedley AM, Pareek V and Benkovic SJ: The
purinosome: A case study for a mammalian metabolon. Annu Rev
Biochem. 91:89–106. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Chan CY, Zhao H, Pugh RJ, Pedley AM,
French J, Jones SA, Zhuang X, Jinnah H, Huang TJ and Benkovic SJ:
Purinosome formation as a function of the cell cycle. Proc Natl
Acad Sci USA. 112:1368–1373. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Baresova V, Krijt M, Skopova V, Souckova
O, Kmoch S and Zikanova M: CRISPR-Cas9 induced mutations along de
novo purine synthesis in HeLa cells result in accumulation of
individual enzyme substrates and affect purinosome formation. Mol
Genet Metab. 119:270–277. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Pelet A, Skopova V, Steuerwald U, Baresova
V, Zarhrate M, Plaza JM, Hnizda A, Krijt M, Souckova O, Wibrand F,
et al: PAICS deficiency, a new defect of de novo purine synthesis
resulting in multiple congenital anomalies and fatal outcome. Hum
Mol Genet. 28:3805–3814. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Chan CY, Pedley AM, Kim D, Xia C, Zhuang X
and Benkovic SJ: Microtubule-directed transport of purine
metabolons drives their cytosolic transit to mitochondria. Proc
Natl Acad Sci USA. 115:13009–13014. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
French JB, Jones SA, Deng H, Pedley AM,
Kim D, Chan CY, Hu H, Pugh RJ, Zhao H, Zhang Y, et al: Spatial
colocalization and functional link of purinosomes with
mitochondria. Science. 351:733–737. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Pedley AM, Boylan JP, Chan CY, Kennedy EL,
Kyoung M and Benkovic SJ: Purine biosynthetic enzymes assemble into
liquid-like condensates dependent on the activity of chaperone
protein HSP90. J Biol Chem. 298:1018452022. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
French JB, Zhao H, An SO, Niessen S, Deng
YJ, Cravatt BF and Benkovic SJ: Hsp70/Hsp90 chaperone machinery is
involved in the assembly of the purinosome. Proc Natl Acad Sci USA.
110:2528–2533. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Pedley AM, Karras GI, Zhang X, Lindquist S
and Benkovic SJ: Role of HSP90 in the regulation of de novo purine
biosynthesis. Biochemistry. 57:3217–3221. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Yu J, Jin C, Su C, Moon D, Sun M, Zhang H,
Jiang X, Zhang F, Tserentsoodol N, Bowie ML, et al: Resilience and
vulnerabilities of tumor cells under purine shortage stress. Clin
Cancer Res. 31:4345–4360. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Stine ZE, Walton ZE, Altman BJ, Hsieh AL
and Dang CV: MYC, metabolism, and cancer. Cancer Discov.
5:1024–1039. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Wunder T and Mueller-Cajar O: Biomolecular
condensates in photosynthesis and metabolism. Curr Opin Plant Biol.
58:1–7. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Chou MC, Wang YH, Chen FY, Kung CY, Wu KP,
Kuo JC, Chan SJ, Cheng ML, Lin CY, Chou YC, et al: PAICS
ubiquitination recruits UBAP2 to trigger phase separation for
purinosome assembly. Mol Cell. 83:4123–4140.e12. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Cunningham JT, Moreno MV, Lodi A, Ronen SM
and Ruggero D: Protein and nucleotide biosynthesis are coupled by a
single rate-limiting enzyme, PRPS2, to drive cancer. Cell.
157:1088–1103. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Camicia R, Winkler HC and Hassa PO: Novel
drug targets for personalized precision medicine in
relapsed/refractory diffuse large B-cell lymphoma: A comprehensive
review. Mol Cancer. 14:2072015. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Delos Santos K, Kwon E and Moon NS:
PRPS-associated disorders and the drosophila model of arts
syndrome. Int J Mol Sci. 21:48242020. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zhang L, Zhao X, Hu J, Li T, Chen HZ,
Zhang A, Wang H, Yu J and Zhang L: PRPS2 enhances RNA
m6A methylation by stimulating SAM synthesis through
enzyme-dependent and independent mechanisms. Nat Commun.
16:39662025. View Article : Google Scholar
|
|
93
|
Jing XQ, Wang XJ, Zhang T, Zhu WC, Fang Y,
Wu HX, Liu X, Ma D, Ji X, Jiang Y, et al: Cell-cycle-dependent
phosphorylation of PRPS1 fuels nucleotide synthesis and promotes
tumorigenesis. Cancer Res. 79:4650–4664. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Zhuo W, Xia H, Lan B, Chen Y, Wang X and
Liu J: Signature of immune-related metabolic genes predicts the
prognosis of hepatocellular carcinoma. Front Immunol.
15:14813312024. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Liu C, Knudsen GM, Pedley AM, He J,
Johnson JL, Yaron TM, Cantley LC and Benkovic SJ: Mapping
post-translational modifications of de novo purine biosynthetic
enzymes: Implications for pathway regulation. J Proteome Res.
18:2078–2087. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kodama M, Oshikawa K, Shimizu H, Yoshioka
S, Takahashi M, Izumi Y, Bamba T, Tateishi C, Tomonaga T, Matsumoto
M and Nakayama KI: A shift in glutamine nitrogen metabolism
contributes to the malignant progression of cancer. Nat Commun.
11:13202020. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Xiang H, Kasajima R, Azuma K, Tagami T,
Hagiwara A, Nakahara Y, Saito H, Igarashi Y, Wei F, Ban T, et al:
Multi-omics analysis-based clinical and functional significance of
a novel prognostic and immunotherapeutic gene signature derived
from amino acid metabolism pathways in lung adenocarcinoma. Front
Immunol. 15:13619922024. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Loftus SK, Baxter LL, Cronin JC, Fufa TD;
NISC Comparative Sequencing Program; Pavan WJ: Hypoxia-induced
HIF1α targets in melanocytes reveal a molecular profile associated
with poor melanoma prognosis. Pigment Cell Melanoma Res.
30:339–532. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Wu Z, Nguyen PT, Sondhi V, Yao RW, Lu Z,
Dai T, Chiang JC, Cai F, Williams IM, Blatt EB, et al: NUDT5
regulates purine metabolism and thiopurine sensitivity by
interacting with PPAT. Science. 390:1134–1142. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Hedstrom L: IMP dehydrogenase: Structure,
mechanism, and inhibition. Chem Rev. 109:2903–2928. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Calise SJ and Chan EKL: Anti-rods/rings
autoantibody and IMPDH filaments: An update after fifteen years of
discovery. Autoimmun Rev. 19:1026432020. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Kim HR, Roe JS, Lee JE, Hwang IY, Cho EJ
and Youn HD: A p53-inducible microRNA-34a downregulates Ras
signaling by targeting IMPDH. Biochem Biophys Res Commun.
418:682–688. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Duan S, Huang W, Liu X, Liu X, Chen N, Xu
Q, Hu Y, Song W and Zhou J: IMPDH2 promotes colorectal cancer
progression through activation of the PI3K/AKT/mTOR and
PI3K/AKT/FOXO1 signaling pathways. J Exp Clin Cancer Res.
37:3042018. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Casado P, Rio-Machin A, Miettinen JJ,
Bewicke-Copley F, Rouault-Pierre K, Krizsan S, Parsons A, Rajeeve
V, Miraki-Moud F, Taussig DC, et al: Integrative phosphoproteomics
defines two biologically distinct groups of KMT2A rearranged acute
myeloid leukaemia with different drug response phenotypes. Signal
Transduct Target Ther. 8:802023. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Liu W, Li J, Xu S, Wang Y, Li J, Wang S,
Fu L, Jiang M and Bai G: Phillyrin and its metabolites exert
antipyretic effects by targeting the NAD+ binding domain
of GAPDH, MDH2 and IDH2. Phytomedicine. 134:1559552024. View Article : Google Scholar
|
|
106
|
Kunjithapatham R and Ganapathy-Kanniappan
S: GAPDH with NAD+-binding site mutation competitively
inhibits the wild-type and affects glucose metabolism in cancer.
Biochim Biophys Acta Gen Subj. 1862:2555–2563. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Bie M, Tang Y, Xia Y, Zhang Q, Tian Y,
Cheng C, Li X, Qi X and Kang F: HIF-1α mediates osteoclast-induced
disuse osteoporosis via cytoophidia in the femur of mice. Bone.
168:1166482023. View Article : Google Scholar
|
|
108
|
Ogawa-Iio A, Takeuchi K, Shigemi K,
Genoveso MJ, Niitsu H, Koh I, Ota Y, Yamane K, Hinoi T, Osaka N, et
al: IMPDH and GTP metabolism in cancer: Mechanisms, regulation, and
translational scope. Cancer Sci. 116:3250–3265. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Ishitsuka K, Hideshima T, Hamasaki M, Raje
N, Kumar S, Podar K, Le Gouill S, Shiraishi N, Yasui H, Roccaro AM,
et al: Novel inosine monophosphate dehydrogenase inhibitor VX-944
induces apoptosis in multiple myeloma cells primarily via
caspase-independent AIF/Endo G pathway. Oncogene. 24:5888–5896.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Tzoneva G, Dieck CL, Oshima K,
Ambesi-Impiombato A, Sánchez-Martín M, Madubata CJ, Khiabanian H,
Yu J, Waanders E, Iacobucci I, et al: Clonal evolution mechanisms
in NT5C2 mutant-relapsed acute lymphoblastic leukaemia. Nature.
553:511–514. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Grusch M, Rosenberger G, Fuhrmann G, Braun
K, Titscher B, Szekeres T, Fritzer-Skekeres M, Oberhuber G, Krohn
K, Hengstschlaeger M, et al: Benzamide riboside induces apoptosis
independent of Cdc25A expression in human ovarian carcinoma N.1
cells. Cell Death Differ. 6:736–744. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Tian Y, Zhang J, Chen L and Zhang X: The
expression and prognostic role of IMPDH2 in ovarian cancer. Ann
Diagn Pathol. 46:1515112020. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Sugimoto A, Watanabe T, Matsuoka K, Okuno
Y, Yanagi Y, Narita Y, Mabuchi S, Nobusue H, Sugihara E, Hirayama
M, et al: Growth transformation of B cells by epstein-barr virus
requires IMPDH2 induction and nucleolar hypertrophy. Microbiol
Spectr. 11:e00440232023. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Szwed A, Kim E and Jacinto E: Regulation
and metabolic functions of mTORC1 and mTORC2. Physiol Rev.
101:1371–1426. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Demetriades C, Plescher M and Teleman AA:
Lysosomal recruitment of TSC2 is a universal response to cellular
stress. Nat Commun. 7:106622016. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara
JM and Manning BD: mTORC1 induces purine synthesis through control
of the mitochondrial tetrahydrofolate cycle. Science. 351:728–733.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Fontana F, Giannitti G, Marchesi S and
Limonta P: The PI3K/Akt pathway and glucose metabolism: A dangerous
liaison in cancer. Int J Biol Sci. 20:3113–3125. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Düvel K, Yecies JL, Menon S, Raman P,
Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S,
et al: Activation of a metabolic gene regulatory network downstream
of mTOR complex 1. Mol Cell. 39:171–183. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Saha A, Connelly S, Jiang J, Zhuang S,
Amador DT, Phan T, Pilz RB and Boss GR: Akt phosphorylation and
regulation of transketolase is a nodal point for amino acid control
of purine synthesis. Mol Cell. 55:264–276. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Torrence ME, MacArthur MR, Hosios AM,
Valvezan AJ, Asara JM, Mitchell JR and Manning BD: The
mTORC1-mediated activation of ATF4 promotes protein and glutathione
synthesis downstream of growth signals. Elife. 10:e633262021.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Mollaoglu G, Guthrie MR, Böhm S,
Brägelmann J, Can I, Ballieu PM, Marx A, George J, Heinen C,
Chalishazar MD, et al: MYC drives progression of small cell lung
cancer to a variant neuroendocrine subtype with vulnerability to
aurora kinase inhibition. Cancer Cell. 31:270–285. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Liu YC, Li F, Handler J, Huang CR, Xiang
Y, Neretti N, Sedivy JM, Zeller KI and Dang CV: Global regulation
of nucleotide biosynthetic genes by c-Myc. PLoS One. 3:e27222008.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Dang CV: MYC on the path to cancer. Cell.
149:22–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Huang F, Huffman KE, Wang Z, Wang X, Li K,
Cai F, Yang C, Cai L, Shih TS, Zacharias LG, et al: Guanosine
triphosphate links MYC-dependent metabolic and ribosome programs in
small-cell lung cancer. J Clin Invest. 131:e1399292021. View Article : Google Scholar :
|
|
125
|
Ali ES, Lipońska A, O'Hara BP, Amici DR,
Torno MD, Gao P, Asara JM, Yap MF, Mendillo ML and Ben-Sahra I: The
mTORC1-SLC4A7 axis stimulates bicarbonate import to enhance de novo
nucleotide synthesis. Mol Cell. 82:3284–3298.e7. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Liu Y, Pareek V, Bhowmik D, Zhang X and
Benkovic SJ: Purinosomes and lysosomes interact to maintain the
purine pools. Int J Biochem Cell Biol. 186:1068302025. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Valvezan AJ, Turner M, Belaid A, Lam HC,
Miller SK, McNamara MC, Baglini C, Housden BE, Perrimon N,
Kwiatkowski DJ, et al: mTORC1 couples nucleotide synthesis to
nucleotide demand resulting in a targetable metabolic
vulnerability. Cancer Cell. 32:6242017. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Worby CA and Dixon JE: PTEN. Annu Rev
Biochem. 83:641–669. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Zuo Z, Liu J, Sun Z, Cheng YW, Ewing M,
Bugge TH, Finkel T, Leppla SH and Liu S: ERK and c-Myc signaling in
host-derived tumor endothelial cells is essential for solid tumor
growth. Proc Natl Acad Sci USA. 120:e22119271202023. View Article : Google Scholar :
|
|
130
|
Liefwalker DF, Ryan M, Wang Z, Pathak KV,
Plaisier S, Shah V, Babra B, Dewson GS, Lai IK, Mosley AR, et al:
Metabolic convergence on lipogenesis in RAS, BCR-ABL, and
MYC-driven lymphoid malignancies. Cancer Metab. 9:312021.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Mannava S, Grachtchouk V, Wheeler LJ, Im
M, Zhuang D, Slavina EG, Mathews CK, Shewach DS and Nikiforov MA:
Direct role of nucleotide metabolism in C-MYC-dependent
proliferation of melanoma cells. Cell Cycle. 7:2392–2400. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Trejo-Solís C, Castillo-Rodríguez RA,
Serrano-García N, Silva-Adaya D, Vargas-Cruz S, Chávez-Cortéz EG,
Gallardo-Pérez JC, Zavala-Vega S, Cruz-Salgado A and
Magaña-Maldonado R: Metabolic roles of HIF1, c-Myc, and p53 in
glioma cells. Metabolites. 14:2492024. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Liu W, Ding Z, Tao Y, Liu S, Jiang M, Yi
F, Wang Z, Han Y, Zong H, Li D, et al: A positive feedback loop
between PFKP and c-Myc drives head and neck squamous cell carcinoma
progression. Mol Cancer. 23:1412024. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Ying H, Kimmelman AC, Lyssiotis CA, Hua S,
Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff
JL, et al: Oncogenic Kras maintains pancreatic tumors through
regulation of anabolic glucose metabolism. Cell. 149:656–670. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Santana-Codina N, Roeth AA, Zhang Y, Yang
A, Mashadova O, Asara JM, Wang X, Bronson RT, Lyssiotis CA, Ying H
and Kimmelman AC: Oncogenic KRAS supports pancreatic cancer through
regulation of nucleotide synthesis. Nat Commun. 9:49452018.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Jiang X, Wang T, Zhao B, Sun H, Dong Y, Ma
Y, Li Z, Wu Y, Wang K, Guan X, et al: KRASG12D-driven
pentose phosphate pathway remodeling imparts a targetable
vulnerability synergizing with MRTX1133 for durable remissions in
PDAC. Cell Rep Med. 6:1019662025. View Article : Google Scholar
|
|
137
|
Gao W, Xu Y, Chen T, Du Z, Liu X, Hu Z,
Wei D, Gao C, Zhang W and Li Q: Targeting oxidative pentose
phosphate pathway prevents recurrence in mutant Kras colorectal
carcinomas. PLoS Biol. 17:e30004252019. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Romero R, Sayin VI, Davidson SM, Bauer MR,
Singh SX, LeBoeuf SE, Karakousi TR, Ellis DC, Bhutkar A,
Sánchez-Rivera FJ, et al: Keap1 loss promotes Kras-driven lung
cancer and results in dependence on glutaminolysis. Nat Med.
23:1362–1368. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Krauß D, Moreno-Viedma V, Adachi-Fernandez
E, de Sá Fernandes C, Genger JW, Fari O, Blauensteiner B,
Kirchhofer D, Bradaric N, Gushchina V, et al: EGFR controls
transcriptional and metabolic rewiring in KRAS(G12D) colorectal
cancer. EMBO Mol Med. 17:1355–1392. 2025. View Article : Google Scholar
|
|
140
|
Meena JK, Wang JH, Neill NJ, Keough D,
Putluri N, Katsonis P, Koire AM, Lee H, Bowling EA, Tyagi S, et al:
MYC induces oncogenic stress through RNA decay and ribonucleotide
catabolism in breast cancer. Cancer Discov. 14:1699–1716. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Ali ES, Sahu U, Villa E, O'Hara BP, Gao P,
Beaudet C, Wood AW, Asara JM and Ben-Sahra I: ERK2 phosphorylates
PFAS to mediate posttranslational control of de novo purine
synthesis. Mol Cell. 78:1178–1191.e6. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Cox AG, Hwang KL, Brown KK, Evason K,
Beltz S, Tsomides A, O'Connor K, Galli GG, Yimlamai D, Chhangawala
S, et al: Yap reprograms glutamine metabolism to increase
nucleotide biosynthesis and enable liver growth. Nat Cell Biol.
18:886–896. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Fujita M, Orisaka M, Mizutani T, Fujita Y,
Onuma T, Tsuyoshi H and Yoshida Y: YAP/TAZ Promote GLUT1 expression
and are associated with prognosis in endometrial cancer. Cancers
(Basel). 17:25542025. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Gao C and Wang Y: YAP: The nexus between
metabolism and cardiac remodeling. J Clin Invest. 132:e1576642022.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Yu H, Tang H, Deng C, Lin Q, Yu P, Chen S
and Ruan J: RRM2 improves cardiomyocyte proliferation after
myocardial ischemia reperfusion Injury through the Hippo-YAP
pathway. Dis Markers. 2021:50898722021. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Shah SR, Ren C, Tippens ND, Park J,
Mohyeldin A, Wang S, Vela G, Martinez-Gutierrez JC, Margolis SS,
Schmidt S, et al: YAP controls cell migration and invasion through
a Rho GTPase switch. Sci Signal. 18:eadu37942025.PubMed/NCBI
|
|
147
|
Tu CE, Liu YF, Liu HW, Jiao CM, Liu Q,
Hung MC, Li P, Wan XB, Fan XJ and Wang YL: D-ribose-5-phosphate
inactivates YAP and functions as a metabolic checkpoint. J Hematol
Oncol. 18:22025. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Zhang H, Guo Z, Yi J, Wu J, Wang Y, Ren T,
Zhang Y, Zhao H, Wu N, Wei G and Zhang D: Thymic egress and
peripheral T cell homeostasis regulated by Rho GTPase-activating
protein 30. Cell Death Differ. 32:2294–2308. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Ahmad Mokhtar AM, Salikin NH, Haron AS,
Amin-Nordin S, Hashim IF, Mohd Zaini Makhtar M, Zulfigar SB and
Ismail NI: RhoG's role in T cell activation and function. Front
Immunol. 13:8450642022. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Cantrell DA: GTPases and T cell
activation. Immunol Rev. 192:122–130. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Chen L and Flies DB: Molecular mechanisms
of T cell co-stimulation and co-inhibition. Nat Rev Immunol.
13:227–242. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Cronin SJF, Seehus C, Weidinger A, Talbot
S, Reissig S, Seifert M, Pierson Y, McNeill E, Longhi MS, Turnes
BL, et al: The metabolite BH4 controls T cell proliferation in
autoimmunity and cancer. Nature. 563:564–568. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Madsen HB, Peeters MJ, Straten PT and
Desler C: Nucleotide metabolism in the regulation of tumor
microenvironment and immune cell function. Curr Opin Biotechnol.
84:1030082023. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
John SV, Seim GL, Erazo-Flores BJ, Votava
JA, Urquiza US, Arp NL, Steill J, Freeman J, Carnevale LN, Roberts
I, et al: Classically activated macrophages undergo functionally
significant nucleotide metabolism remodelling driven by nitric
oxide. Nat Metab. 7:1681–1702. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Yang L, Li A, Yu W, Wang H, Zhang L, Wang
D, Wang Y, Zhang R, Lei Q, Liu Z, et al: Blockade of purine
metabolism reverses macrophage immunosuppression and enhances
anti-tumor immunity in non-small cell lung cancer. Drug Resist
Updat. 78:1011752025. View Article : Google Scholar
|
|
156
|
Lu Y, Sun Q, Guan Q, Zhang Z, He Q, He J,
Ji Z, Tian W, Xu X, Liu Y, et al: The XOR-IDH3α axis controls
macrophage polarization in hepatocellular carcinoma. J Hepatol.
79:1172–1184. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Shen J, Liao B, Gong L, Li S, Zhao J, Yang
H, Gong Y and Li Y: CD39 and CD73: Biological functions, diseases
and therapy. Mol Biomed. 6:972025. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Zhang Y, Jin W, Deng Z, Gao B, Zhu Y, Fu
J, Xu C, Wang W, Bai T, Jiao L, et al: Metabolic reprogramming
nanomedicine potentiates colon cancer sonodynamic immunotherapy by
inhibiting the CD39/CD73/ADO pathway. Acta Pharm Sin B.
15:2655–2672. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Allard D, Cormery J, Bricha S, Fuselier C,
Abbas Aghababazadeh F, Giraud L, Skora E, Haibe-Kains B and Stagg
J: Adenosine uptake through the nucleoside transporter ENT1
suppresses antitumor immunity and T-cell pyrimidine synthesis.
Cancer Res. 85:692–703. 2025. View Article : Google Scholar
|
|
160
|
Yang H, Lei MML, Xie L, Shou Y and Lee
TKW: Deciphering adenosine signaling in hepatocellular carcinoma:
Pathways, prognostic models, and therapeutic implications. Clin Mol
Hepatol. 31:706–729. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
He X, Cui J, Ma H, Abuduaini N, Huang Y,
Tang L, Wang W, Zhang Y, Wang Y, Lu W, et al: Berberrubine is a
novel and selective IMPDH2 inhibitor that impairs the growth of
colorectal cancer. Biochem Pharmacol. 218:1158682023. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Wang L, Yang R, Kong Y, Zhou J, Chen Y, Li
R, Chen C, Tang X, Chen X, Xia J, et al: Integrative single-cell
and bulk transcriptomes analyses reveals heterogeneity of
serine-glycine-one-carbon metabolism with distinct prognoses and
therapeutic vulnerabilities in HNSCC. Int J Oral Sci. 16:442024.
View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Liu SS, Li JS, Xue M, Wu WJ, Li X and Chen
W: LncRNA UCA1 participates in de novo synthesis of guanine
nucleotides in bladder cancer by recruiting TWIST1 to increase
IMPDH1/2. Int J Biol Sci. 19:2599–2612. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Burrell AL and Kollman JM: IMPDH
dysregulation in disease: A mini review. Biochem Soc Trans.
50:71–82. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Jia HW, Yang HL, Xiong ZL, Deng MH, Wang
T, Liu Y and Cheng M: Design, synthesis and antitumor activity
evaluation of novel indole acrylamide derivatives as IMPDH
inhibitors. Bioorg Chem. 129:1062132022. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Naffouje R, Grover P, Yu H, Sendilnathan
A, Wolfe K, Majd N, Smith EP, Takeuchi K, Senda T, Kofuji S and
Sasaki AT: Anti-tumor potential of IMP dehydrogenase inhibitors: A
century-long story. Cancers (Basel). 11:13462019. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Zerhouni M, Martin AR, Furstoss N,
Gutierrez VS, Jaune E, Tekaya N, Beranger GE, Abbe P, Regazzetti C,
Amdouni H, et al: Dual covalent inhibition of PKM and IMPDH targets
metabolism in cutaneous metastatic melanoma. Cancer Res.
81:3806–3821. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Sezaki M and Huang G: Repurposing
immunosuppressants for antileukemia therapy. EMBO Mol Med.
15:e170422023. View Article : Google Scholar :
|
|
169
|
Schnabel JL, Frost TC, Wang AC,
Ananthapadmanabhan V, Gurram S, Soroko KM, Gokhale PC and DeCaprio
JA: IMPDH inhibition induces DNA replication stress and ATR
sensitivity in Merkel cell carcinoma. iScience. 28:1125672025.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Tuncyurek P, Mayer JM, Klug F, Dillmann S,
Henne-Bruns D, Keller F and Stracke S: Everolimus and mycophenolate
mofetil sensitize human pancreatic cancer cells to gemcitabine in
vitro: A novel adjunct to standard chemotherapy? Eur Surg Res.
39:380–387. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Floryk D and Huberman E: Mycophenolic
acid-induced replication arrest, differentiation markers and cell
death of androgen-independent prostate cancer cells DU145. Cancer
Lett. 231:20–29. 2006. View Article : Google Scholar
|
|
172
|
Messina E, Micheli V and Giacomello A:
Guanine nucleotide depletion induces differentiation and aberrant
neurite outgrowth in human dopaminergic neuroblastoma lines: A
model for basal ganglia dysfunction in Lesch-Nyhan disease.
Neurosci Lett. 375:97–100. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Dun B, Sharma A, Teng Y, Liu H, Purohit S,
Xu H, Zeng L and She JX: Mycophenolic acid inhibits migration and
invasion of gastric cancer cells via multiple molecular pathways.
PLoS One. 8:e817022013. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Takebe N, Cheng X, Wu S, Bauer K,
Goloubeva OG, Fenton RG, Heyman M, Rapoport AP, Badros A,
Shaughnessy J, et al: Phase I clinical trial of the inosine
monophosphate dehydrogenase inhibitor mycophenolate mofetil
(cellcept) in advanced multiple myeloma patients. Clin Cancer Res.
10:8301–8308. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Rodríguez-Pascual J, Sha P, García-García
E, Rajeshkumar NV, De Vicente E, Quijano Y, Cubillo A, Angulo B,
Hernando O and Hidalgo M: A preclinical and clinical study of
mycophenolate mofetil in pancreatic cancer. Invest New Drugs.
31:14–19. 2013. View Article : Google Scholar
|
|
176
|
Koonrungsesomboon N, Teeyakasem P, Dukaew
N, Sirikaew N, Thongkumkoon P, Yongpitakwattana P, Thepbundit V,
Chaiyawat P, Klangjorhor J, Sakuludomkan C, et al: Efficacy and
safety of mycophenolate mofetil in patients with high-grade locally
advanced or metastatic osteosarcoma (ESMMO): A multicenter, phase
II clinical trial. Int J Clin Oncol. 30:2362–2374. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Torres-Diz M, Reglero C, Falkenstein CD,
Castro A, Hayer KE, Radens CM, Quesnel-Vallières M, Ang Z, Sehgal
P, Li MM, et al: An alternatively spliced gain-of-function NT5C2
isoform contributes to chemoresistance in acute lymphoblastic
leukemia. Cancer Res. 84:3327–3336. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
De Clercq E: C-nucleosides to be
revisited. J Med Chem. 59:2301–2311. 2016. View Article : Google Scholar
|
|
179
|
Malek K, Boosalis MS, Waraska K, Mitchell
BS and Wright DG: Effects of the IMP-dehydrogenase inhibitor,
Tiazofurin, in bcr-abl positive acute myelogenous leukemia. Part I.
In vivo studies. Leukemia Res. 28:1125–1136. 2004. View Article : Google Scholar
|
|
180
|
Tricot GJ, Jayaram HN, Lapis E, Natsumeda
Y, Nichols CR, Kneebone P, Heerema N, Weber G and Hoffman R:
Biochemically directed therapy of leukemia with tiazofurin, a
selective blocker of inosine 5'-phosphate dehydrogenase activity.
Cancer Res. 49:3696–3701. 1989.PubMed/NCBI
|
|
181
|
Melink TJ, Von Hoff DD, Kuhn JG, Hersh MR,
Sternson LA, Patton TF, Siegler R, Boldt DH and Clark GM: Phase I
evaluation and pharmacokinetics of tiazofurin
(2-beta-D-ribofuranosylthiazole-4-carboxamide, NSC 286193). Cancer
Res. 45:2859–2865. 1985.PubMed/NCBI
|
|
182
|
Maroun JA and Stewart DJ: Phase I study of
tiazofurin (2-beta-D-ribofuranosylthiazole-4-carboxamide, NSC
286193). Invest New Drugs. 8(Suppl 1): S33–S39. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Stewart DJ, Eisenhauer E, Macdonald DR,
Cairncross JG and Langleben A: Phase II study of tiazofurin in
gliomas in adults. A national cancer institute of Canada study. J
Neurooncol. 15:175–179. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Jayaram HN, Yalowitz JA, Arguello F and
Greene JF Jr: Toxicity and efficacy of benzamide riboside in cancer
chemotherapy models. Curr Med Chem. 9:787–792. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Szekeres T, Sedlak J and Novotny L:
Benzamide riboside, a recent inhibitor of inosine 5'-monophosphate
dehydrogenase induces transferrin receptors in cancer cells. Curr
Med Chem. 9:759–764. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Gharehbaghi K, Sreenath A, Hao Z, Paull
KD, Szekeres T, Cooney DA, Krohn K and Jayaram HN: Comparison of
biochemical parameters of benzamide riboside, a new inhibitor of
IMP dehydrogenase, with tiazofurin and selenazofurin. Biochem
Pharmacol. 48:1413–1419. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Jain J, Ma JG, Furey B, Recher C, Demur C,
Poondru S, Zhang J, Li S, Firestone B, Olson K, et al: The IMPDH
inhibitor VX-944 demonstrates in vivo efficacy in an aggressive
leukemia model. Blood. 104:25302004. View Article : Google Scholar
|
|
188
|
Hamilton JM, Harding MW, Genna T and Bol
DK: A phase I dose-ranging study of the pharmacokinetics,
pharmacodynamics, safety, and tolerability of AVN944, an IMPDH
inhibitor, in healthy male volunteers. J Clin Pharmacol. 49:30–38.
2009. View Article : Google Scholar
|
|
189
|
Garcia-Manero G, Pemmaraju N, Alvarado Y,
Naqvi K, Ravandi F, Jabbour E, De Lumpa R, Kantarjian H, Advani A,
Mukherjee S, et al: Results of a phase 1/2a dose-escalation study
of FF-10501-01, an IMPDH inhibitor, in patients with acute myeloid
leukemia or myelodysplastic syndromes. Leuk Lymphoma. 61:1943–1953.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Yang H, Fang Z, Wei Y, Bohannan ZS,
Gañán-Gómez I, Pierola AA, Paradiso LJ, Iwamura H and Garcia-Manero
G: Preclinical activity of FF-10501-01, a novel
inosine-5'-monophosphate dehydrogenase inhibitor, in acute myeloid
leukemia. Leuk Res. 59:85–92. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Ichii M, Oritani K, Murase M, Komatsu K,
Yamazaki M, Kyoden R, Kito N, Nozaki Y, Saito M, Iwamura H and
Kanakura Y: Molecular targeting of inosine-5'-monophosphate
dehydrogenase by FF-10501 promotes erythropoiesis via ROS/MAPK
pathway. Leuk Lymphoma. 59:448–459. 2018. View Article : Google Scholar
|
|
192
|
Levine AM, Shimodaira S and Lai MMC:
Treatment of HCV-related mantle-cell lymphoma with ribavirin and
pegylated interferon alfa. New Engl J Med. 349:2078–2079. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Borden KLB and Culjkovic-Kraljacic B:
Ribavirin as an anti-cancer therapy: Acute myeloid leukemia and
beyond? Leuk Lymphoma. 51:1805–1815. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Dunn LA, Fury MG, Sherman EJ, Ho AA,
Katabi N, Haque SS and Pfister DG: Phase I study of induction
chemotherapy with afatinib, ribavirin, and weekly carboplatin and
paclitaxel for stage IVA/IVB human papillomavirus-associated
oropharyngeal squamous cell cancer. Head Neck. 40:233–241. 2018.
View Article : Google Scholar
|
|
195
|
Casaos J, Gorelick NL, Huq S, Choi J, Xia
Y, Serra R, Felder R, Lott T, Kast RE, Suk I, et al: The use of
ribavirin as an anticancer therapeutic: Will it go viral? Mol
Cancer Ther. 18:1185–1194. 2019. View Article : Google Scholar : PubMed/NCBI
|