Diabetic cardiomyopathy (DCM) is a metabolic
disorder characterized by pathological features including
triglyceride accumulation in cardiomyocytes, reduced glucose
utilization, and structural alterations such as cardiac steatosis,
myocardial fibrosis, hypertrophy and cardiomyocyte death. According
to the 2024 ESC guidelines, DCM can be diagnosed in diabetic
patients based on evidence of systolic or diastolic dysfunction,
such as ventricular hypertrophy or diffuse myocardial fibrosis,
when other cardiovascular risk factors or comorbidities are not
present. DCM encompasses a spectrum of cardiac abnormalities,
including coronary artery disease and heart failure (HF) (1). Its prevalence among diabetic
patients reaches up to 67%, and it is a recognized precursor to HF
(2).
In the diabetic heart, energy metabolism shifts
toward fatty acid oxidation (FAO) as the predominant adenosine
triphosphate (ATP) source. Despite elevated circulating free fatty
acids (FFAs), mitochondrial efficiency in ATP production is
impaired. Current therapeutic strategies remain limited, although
glycemic control is traditionally emphasized, intensive
glucose-lowering has not significantly reduced cardiovascular
mortality in clinical trials. Intriguingly, recent studies
challenge the conventional view of glucotoxicity as the initiating
factor, positioning lipotoxicity as a primordial event in DCM
pathogenesis (3). Given that
both lipids and glucose are the primary energy sources for
maintaining cardiac energy homeostasis, targeting cellular
metabolic dysregulation represents a promising therapeutic
direction.
When cellular metabolic homeostasis is disrupted,
excess fatty acids (FAs) cannot be efficiently oxidized, leading to
further lipid accumulation and the subsequent biogenesis of lipid
droplets (LDs). Typically, surplus lipids in cardiomyocytes,
macrophages, fibroblasts and neurons are stored within LDs as
neutral lipids that consist of triglycerides (TGs), cholesterol
esters (CEs), or retinol esters. Structurally, LDs are
evolutionarily conserved organelles composed of a neutral lipid
core surrounded by a phospholipid (PL) monolayer, that binds
specific proteins and cholesterol (4). The types and functions of LDs lead
to differences in their protein and lipid composition.
Nevertheless, the precise regulatory role of LD dynamics in overall
lipid metabolism remains incompletely understood.
Insulin resistance exacerbates lipolysis of adipose
tissue, leading to excessive systemic release of FAs. This elevated
FA flux promotes the expression of LD associated protein Plin5,
increases triacylglycerol (TAG) levels and enlarges LD size in
insulin-sensitive tissues such as the heart (5). Plin5 enrichment at LD-mitochondria
contact interface, implicates these inter-organellar contacts in
governing FA trafficking and cellular metabolic balance. At this
contact interface, Plin5 restricts mitochondrial respiration and
lipolysis through inhibiting adipose triglyceride lipase (ATGL), as
well as the decrease in lipolysis levels promotes dynamin-related
protein 1 (Drp1) to mediate mitochondrial fission (6,7).
Although insulin resistance impairs myocardial glucose uptake,
emerging evidence indicates that LDs also regulate glucose
metabolism (8). Restricting LD
accumulation has been shown to ameliorate insulin resistance
(9). Besides, LDs in
cardiomyocytes and fibroblasts confer cytoprotective effects
against lipotoxicity damage through lipase-mediated lipolysis and
autophagy-dependent lipophagy, thereby attenuating energy
metabolism dysregulation and structural cardiac damage.
Furthermore, LD biogenesis is facilitated through LD-endoplasmic
reticulum (ER) contact sites, where the conversion of
non-esterified FAs (NEFAs) into neutral lipids sequestered within
LDs serves to alleviate ER stress (10).
Beyond their metabolic functions, LDs serve as
multifunctional organelles that orchestrate diverse cellular
processes. LDs regulate substrate availability, mitigate oxidative
stress, modulate inflammatory responses, and function as critical
mediators in the immune signaling and cellular quality control
mechanisms, including autophagy and ferroptosis. Through these
pleiotropic roles, LDs facilitate energy production and alleviate
ER stress under physiological conditions. However, abnormal
accumulation of LDs is causally linked to mitochondrial
dysfunction, ER stress, and broader metabolic perturbations
(11,12). Collectively, these LD-associated
alterations disrupt cardiac homeostasis and contribute to
characteristic pathological features of DCM, including lipotoxic
injury, plaque formation and cardiac steatosis. Therefore, the
authors propose that LDs represent a pivotal therapeutic target for
DCM.
In the present review, LD biology was
comprehensively examined in the context of DCM, encompassing the
biogenesis of LDs, lipolytic pathways and lipophagy. It was further
analyzed how LD associated proteins modulate the processes of DCM.
Particular emphasis is placed on cell type-specific and
stage-dependent LD characteristics throughout disease progression.
Finally, existing pharmacological interventions targeting LD
dynamics and lipid metabolism were evaluated, proposing that
targeting LDs may offer a novel avenue for DCM treatment.
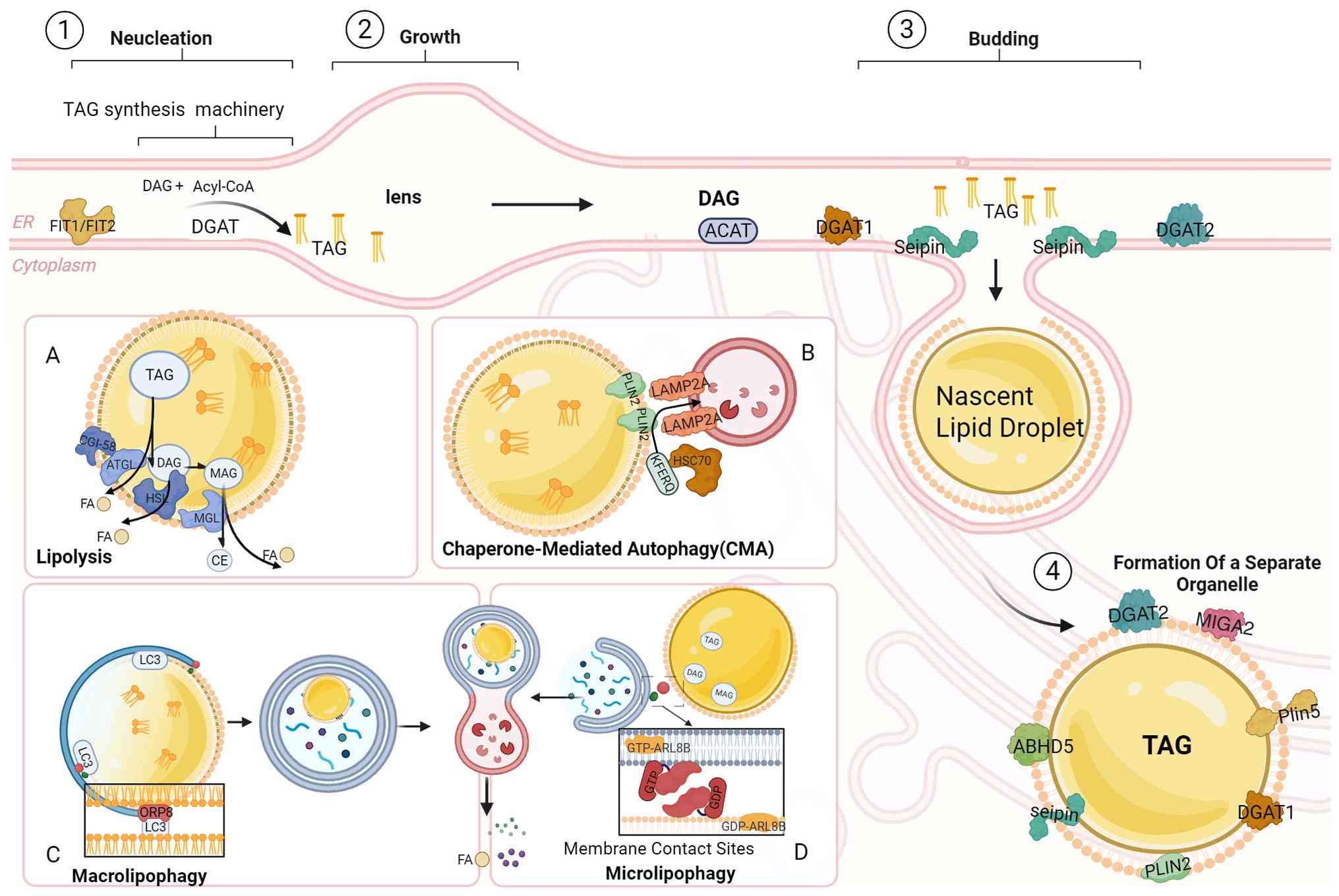
In cardiac tissue, the ER serves as the site of LD
biogenesis, where neutral lipid synthesis precedes their assembly
into nascent LDs (13). The ER
harbors essential enzymes responsible for synthesizing the two
major neutral lipid species: Acyl-CoA: cholesterol
O-acyltransferases 1 (ACAT1, as ACAT2 is not expressed in the
heart) primarily catalyzes CEs (14), and TAGs, synthesized by
diacylglycerol acyltransferase (DGAT) 1 and DGAT2 (15). Whereby deficiency in specific
lipid synthesis enzymes can be compensated by alternative pathways
to maintain neutral lipid production and LD formation (16). DGAT1 localizes exclusively to the
ER membrane, whereas DGAT2 displays dynamic subcellular
distribution, localizing to ER membrane and LD interface. When
intracellular FA concentrations increase, DGAT2 translocates to the
LD surface, thereby facilitating TAG storage and LD expansion
(15).
Beyond the canonical DGAT-mediated pathway,
alternative mechanisms contribute to TAG synthesis, including
transmembrane thioredoxin 1, which catalyzes storage lipid
production independently of DGAT1/2, though the mechanism requires
further investigation (17). LD
nucleation occurs when TAG and CEs accumulate to concentrations of
5-10 mmol/l within the ER bilayer, forming lipid lenses that
coalesce under the action of PL acid and diacylglycerol (DAG)
(4,18-20). The subsequent LD budding is
orchestrated by ER-intra TAG concentration and the scaffolding
protein Seipin which localizes to nascent lipid lenses to drive LD
growth and facilitate the detachment of the PL monolayer-enveloped
LD through ER-to-cytoplasm (21). LD budding directionality and size
are determined by the asymmetric distribution of the PL monolayer
cap, which is governed by ER membrane asymmetry, lipid composition
and the spatial positioning of budding site (22,23). Perturbations in neutral lipids
trafficking to the ER membrane or aberrant retention on the
cytoplasmic leaflet impair the clearance of misfolded proteins,
thereby triggering ER stress and inflammatory responses that
necessitate continuous PL supplementation to sustain budding
(24). Notably, a fundamental
distinction exists between mammalian and yeast LD biology; whereas
mammalian LDs exist as independent cytoplasmic organelles, yeast
LDs maintain persistent ER membrane continuity. This evolutionary
divergence reflects species-specific adaptations in lipid storage
compartmentalization and ER-LD membrane dynamics (25,26).
To meet cardiac energy demands, stored LDs undergo
catabolism through lipolysis and lipohagy, processes that hydrolyze
TAG into FA for subsequent oxidation. Lipolysis proceeds through a
sequential three-step enzymatic cascade: (i) ATGL mediates
rate-limiting TAG hydrolysis at the sn-2 position, generating DAG
and FAs. (ii) Hormone-sensitive lipase (HSL) subsequently cleaves
DAG into FAs and monoacylglycerol (MAG). (iii) MAG lipase (MGL)
completes the process by liberating the final FAs and MAG (27,28). The specificity and efficiency of
ATGL-mediated lipolysis are enhanced by its cofactor comparative
gene identification 58/Alpha beta hydrolase domain-containing
protein 5, which redirects ATGL activity toward the sn-1 position
and cooperates with DGAT2 to generate SN1,3DAG. SN1,3DAG is a
stereoisomer that bypasses protein kinase C activation but serves
as the optimal HSL substrate for continued lipolytic flux (27,28). CGI 58 inhibited the lipolysis
rate after interacting with Plin1, and the inhibitory effect
disappeared after PKA phosphorylation of Plin1 (29,30). Similarly, Plin5 phosphorylation
orchestrates a functional switch from a lipolysis barrier to a
promoter of TAG breakdown, as phosphorylated PLIN5 recruits both
CGI 58 and ATGL, facilitating the coordinated hydrolysis of TAG
into glycerol and Fas (31,32). This phosphorylation-dependent
regulation of perilipin-lipase interactions represents a critical
control node integrating hormonal signals with cellular energy
status to modulate cardiac lipid catabolism.
LDs are partially or completely decomposed into
glycerol and FAs in a process known as lipophagy. Lipophagy mainly
degrades neutral lipids such as TAGs and CEs through three
autophagy modes, including molecular chaperon-mediated autophagy
(CMA), macro-autophagy and micro-autophagy, each characterized by
specific molecular mechanisms. In CMA, the 71 kDa heat shock
cognate protein (HSC70) proteins directly recognizes Plin2, Plin3
and Plin5 via the five-peptide motif protein KFERQ like
pentapeptide motifs, facilitating their selective degradation, and
subsequently eliminating their protective shielding of ATGL at the
LD surface (33). Following
HSC70-mediated recognition, the KFERQ-bearing perilipins binds to
lysosome-associated membrane protein 2A (LAMP2A), assembling into a
translocation complex that delivers these cargo proteins into the
lysosomal lumen for degradation. The CMA-mediated removal of Plin2
and Plin3 consequently enables ATGL accessibility to LDs, thereby
promoting both ATGL-mediated lipolysis and subsequent lipophagy
(34). During macro-autophagy,
LDs recruit autophagy related proteins to initiate the biogenesis
of autophagosomes, facilitating the selective sequestration and
lysosomal degradation of lipid cargo (35). Micro-autophagy mediates direct LD
engulfment by lysosomes through membrane contact site tethering
mechanisms, wherein LD contents are transferred to the lysosomal
lumen via direct injection or membrane transfer remains unclear,
potentially regulated by membrane fusion associated small GTPases,
and hydrolyzed by lysosomal acid lipase independent of cytoplasmic
lipases and autophagy (36).
It should be noted that the degradation of LD
through lipolysis or lipohagy is determined by the size of the LD.
Lipohagy can only degrade smaller LDs in liver, while LDs exceeding
the degradation capacity of lipohagy will trigger ATGL-mediated
cytoplasmic neutral lipolysis under normal conditions, with
starvation or cellular stress inducing acid lipolysis to release
FAs for FAO. Thus, the liberation of lipids and TGs from LDs for
energy production is facilitated by neutral and acidic lipolysis
(37). Whether similar
size-dependent and pathway-selective mechanisms operate across DCM
warrants further investigation (Fig.
1).
The dynamic stability and regulatory function of LDs
are significantly reliant on surface-binding proteins. The
categorization of these LD-associated proteins remains
insufficient: One perspective separates them according to the
mechanism of metastasis. Class I proteins such as DGAT2, ACSL3 and
GPAT4 are translocated from the ER to LDs through the ERTOLD and
include hydrophobic hairpin motifs that directly engage with LDs
(38).
The CYTOLD pathway mediates the direct transport of
Class II proteins from the cytosol to LDs. These proteins include
Plins, members of the Perilipin family that possess an amphipathic
helix, as well as cell death-inducing DNA Fragmentation Factor
Alpha (DFFA)-like effector (CIDE) family of proteins. Conversely,
CGI58, which is associated with LDs via protein-protein
interactions, engages with proteins, including histones and the
small GTPase Rab18 (RAB18), which is anchored to LDs through lipid
modification (39,40).
Based on their roles, these proteins can be
categorized into LD structural proteins/resident proteins, lipid
metabolism enzymes, and transport/signal transduction proteins.
Such proteins are produced through fusion or localized lipid
synthesis.
DGAT2: LD biogenesis involves functional coupling of
FA transporter protein 1 on the ER membrane and DGAT2 at nascent LD
surfaces, which coordinately promote TAG synthesis and lipid
transfer (41). However, DGAT2
expression must be tightly controlled, as overexpression
compromises ER membrane stability (42). Post-translational modification
provides additional regulation, H2S intervention
enhances ubiquitin-mediated degradation of DGAT1 and DGAT2 and
enhanced the expression of the E3 ligase Hrd1 to reduce LDs
accumulation (43). The small
GTPase RAB1B dynamically regulates DGAT2 subcellular distribution
by facilitating its trafficking from ER to LDs via secretory
pathways, promoting the expansion and growth of LDs (44). Collectively, these mechanisms
encompassing protein interactions, expression control, proteolytic
regulation and vesicular trafficking converge to fine-tune LD
dynamics in response to metabolic demands.
The perilipin family represents the major
LD-resident proteins in mammalian cells. In cardiac tissue, PLIN2
and PLIN5 predominate; whereas Plin1 is mainly located in
adipocytes, Plin5 facilitates TAG storage. During the metabolic
transition from glycolysis to β-oxidation, Plin2 upregulation
activates the peroxisome proliferator-activated receptors (PPARs)
signaling pathway (45,46). Plin2 regulates the shape, size
and morphology of LDs, while both Plin2 and Plin5 inhibit ATGL
mediated lipolysis, thereby modulating lipid accumulation (47,48).
ABHD5/CGI 58: CGI 58, a highly conserved regulator,
is characterized by its interaction with Plin and ATGL to function
as a coactivator in ATGL-mediated lipolysis (52). PKA-mediated phosphorylation of
Plin5 in the heart enhances the expression of CGI 58. By contrast,
non-phosphorylated Plin5 binds CGI 58 and inhibits lipase activity.
Beyond Plin regulation, cellular homeostasis also requires
coordinated action of other key metabolic enzymes. Lipolysis is
primarily mediated by three enzymes: ATGL, HSL and MGL.
Upregulation of these key lipolysis enzymes ameliorate cardiac
steatosis and myocardial fibrosis (53). Decreased expression of lipolysis
enzymes aggravates mitochondrial dysfunction (54).
ATGL acts as the rate-limiting enzyme for TAG
hydrolysis in LDs, ATGL deficiency reduces PPARα expression and FAO
levels, resulting in human neutral lipid storage disorder and
cardiomyopathy (55).
Restricting lipid absorption alone fails to reverse ATGL deficiency
induced cardiomyotoxicity (56).
Yet ATGL overexpression ameliorates myocardial injury by enhancing
lipolysis (55). This indicates
that ATGL deficiency extends beyond mere lipid accumulation, as its
mediated lipolysis liberates PPAR agonists that activate downstream
PPARα/PGC-1α-regulated FAO and mitochondrial biogenesis (57). Although pharmacological
interventions partially restore FAO, the signaling function of ATGL
remains indispensable for cardiac protection.
HSL: Overexpression of Plin2 markedly reduces HSL
expression, indicating that Plin2 as a functional inhibitor of HSL
(53). HSL exerts a
cardioprotective effect that precedes the overt development of
cardiac lipotoxicity. In the context of DCM, overexpression of HSL
does not influence VLDL absorption but effectively downregulates
key profibrotic factors, including collagen, TGF-β and matrix
metalloproteinase (MMP) 2. This suppression alleviates the
lipotoxicity and LDs accumulation driven by TAG and DAG (58).
LDs sequester excess histones, preventing histone
toxicity, and supplies histones for chromatin assembly during DNA
replication (59,60). Multiple proteins govern stability
of LDs while concurrently engaging in membrane transport and signal
transduction, thereby influencing cellular metabolic remodeling
(61). Proteins of the LD
proteome related to LD biogenesis in DCM are included in Table I.
LDs establish contact sites with multiple
organelles, most notably mitochondria; the adult heart derives ~95%
of its ATP from mitochondrial oxidative phosphorylation, with FAO
contributing 40-60% of reduction equivalents. Mitochondrial
dysfunction generates lipotoxicity intermediates that promote
intracellular lipid accumulation. LDs counteract lipotoxicity by
sequestering excessive FAs and lipid intermediates. During energy
stress such as fasting or β-adrenergic stimulation, increased
cardiac energy demand and elevated adipocyte lipolysis lead to
heightened expression of LDs, Plin5, and mitochondrial LDs contact
sites in cardiac tissue. This metabolic demand promotes formation
of 'peridroplet mitochondria (PDM)', a specialized mitochondrial
subpopulation with unique functional, ridge-structured, and
proteomically distinct mitochondrial compartment attached to LDs
(5,69). Plin5 mediates these
LD-mitochondria contacts (LDMCs) and preferentially targets PDMs
(5). In cardiomyocytes, enhanced
Plin5-dependent LDMC improve mitochondrial respiratory capacity and
metabolic flexibility. Paradoxically, FAO remains independent of
LDMC (70). This may relate to
PDM primarily promoting FA activation and TAG synthesis with
stronger oxidative phosphorylation and ATP supply capacity, which
are not mainly responsible for FAO (71).
Previous studies have revealed a link between LDs
and mitophagy. Mitophagy leads lysosomes to phagocytose PDM and
release FAs, which aggravates intracellular lipid peroxidation and
iron accumulation and eventually cause ferroptosis. Inhibiting
mitophagy significantly reduces LD accumulation and FFA levels, yet
DGAT1 expression remains elevated and continues to drive nascent
LDs formation (72).
PLIN2 regulates LD homeostasis and mitochondrial
remodeling through epigenetic mechanisms. By suppressing lipolysis,
Plin2 elevates acetyl-CoA and histone acetylation levels,
maintaining embryonic stem cell pluripotency (73). Loss of Plin2 shifts cellular
metabolism from glycolysis to oxidative phosphorylation and
accelerates pluripotency exit. Under these conditions, Plin2 is
recognized by Hsc70 and degraded through CMA, promoting
mobilization of LDs (74). This
is similar to the metabolic transition of cardiomyocytes from
embryonic to adult stages. Although short-term glucose deprivation
alters Plin2 conformation, weakening its interaction with ATGL and
enhancing lipolysis, though the impact on cardiac metabolism
remains uncertain (75).
In fact, the link between LDs and mitochondria is
largely established by specific molecules or pathways that serve
multiple functions in both LD dynamics and mitochondrial operation.
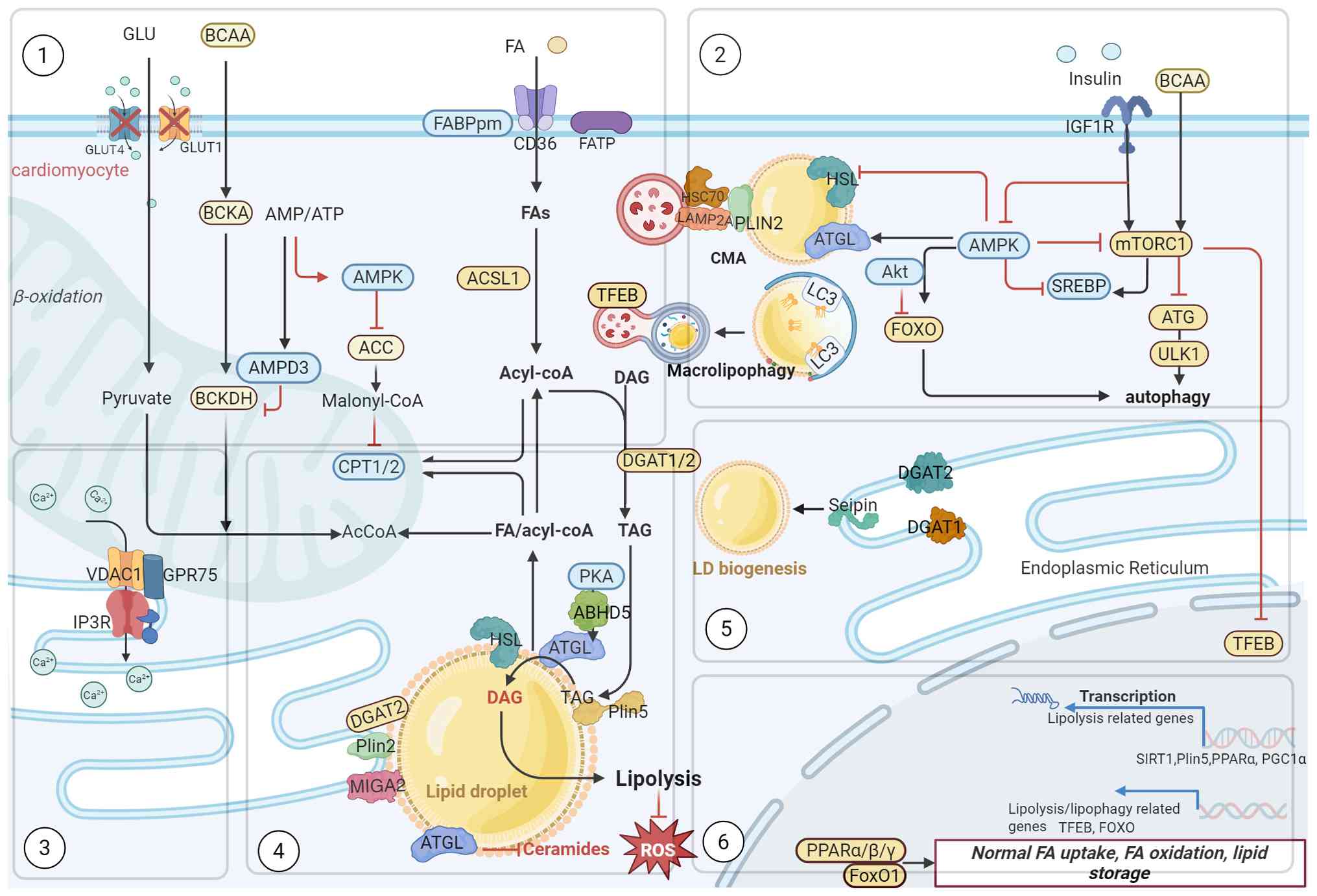
Under nutrient excess conditions, mitophagy becomes dysregulated
and AMPK activity declines (76). Only part of AMPK-mediated
phosphorylation of ACC1 and ACC2 fails to effectively inhibit FA
synthesis, paradoxically promoting LD formation. AMPK activates
PPARs γ coactivator 1 alpha (PGC1α) is blocked, disrupting the
formation of complexes between PGC-1α, Plin5 and silent information
regulator 1 (SIRT1), and impairing mitochondrial biosynthesis
(77).
Concurrently, excessive mammalian target of
rapamycin complex 1 (mTORC1) signaling stimulates lipid
accumulation by activating its key downstream effector sterol
regulatory element binding protein 1c (SREBP1c). Autophagy is
inhibited by Akt-mediated suppression of FOXO transcription and
further impaired by elevated branched-chain amino acids (BCAAs),
which suppress the mTORC1/autophagy-related protein (ATG)/ULK
pathway (78,79). SIRT1 mediated deacetylation
activates FOXO1, whose deacetylation status regulates the
expression of ATGL and PPARα (80).
Under energy-deficient conditions, FOXO1 also
recruits Rab7 to sustain autophagy, a process involved in the
micro-lipophagy degradation of LDs. In DCM, the inhibitory effect
of FOXO on transcription factor EB (TFEB) is weakened (81). It is worth noting that PKA
promotes lipolysis mediated by Plin5, CGI58 and ATGL (8). Therefore, LD metabolism is
coordinately regulated by upstream kinases, for instance, PKA,
mTOR, as well as AMPK and transcription factors including silent
information regulator 1 (SIRT1), PGC-1α, FOXO, SREBP1, PPARs and
TFEB (31).
LDs represent the principal cellular compartment
specialized for neutral lipid storage, distinguished from other
organelles by both structural architecture and functional capacity.
The ER despite serving as the site of lipid biosynthesis,
incorporates lipids within its PL bilayer rather than sequestering
them in concentrated deposits, limiting its storage capacity and
potentially compromising membrane function under lipid excess
(4). Mitochondria maintain
membrane lipids essential for bioenergetics but actively oxidize
rather than store FAs, making them vulnerable to lipotoxic damage
when substrate availability exceeds oxidative capacity. Peroxisomes
metabolize very-long-chain and BCAAs through β-oxidation yet lack
mechanisms for TAG or CEs accumulation (72). Lysosomes degrade lipids delivered
via endocytosis or autophagy but function as catabolic rather than
storage compartments.
By contrast, LDs possess a unique PL monolayer
encompassing a hydrophobic neutral lipid core, an architecture that
enables massive expansion without membrane stress or dysfunction of
organelles (4). This specialized
structure allows LDs to accumulate energy-dense TAGs and CEs while
protecting other organelles from lipotoxic intermediates. The
dynamic nature of LD expansion, lipophagy and lipolysis regulate
lipid mobilization coordinated with metabolic demands, whereas
lipid accumulation in other organelles typically signals
pathological dysfunction rather than adaptive storage.
Consequently, LDs serve as the primary defense against abnormal
lipid deposition, protecting mitochondria, ER, and plasma membrane
integrity from lipotoxicity-induced cellular damage.
Beyond their canonical storage function, LDs protect
cells from lipotoxicity by limiting lipid peroxidation and
buffering excess FAs during metabolic dysregulation. LDs regulate
diverse metabolic substrates, including BCAAs and glucose, both
central to DCM pathogenesis. LDs participate in intermembrane lipid
trafficking, store precursor molecules for lipids, mitigate
oxidative damage, and modulate inflammatory responses. By
coordinating these diverse functions, LDs protect mitochondria and
other organelles while regulating cellular processes such as
autophagy, ferroptosis, and cell division (11,12).
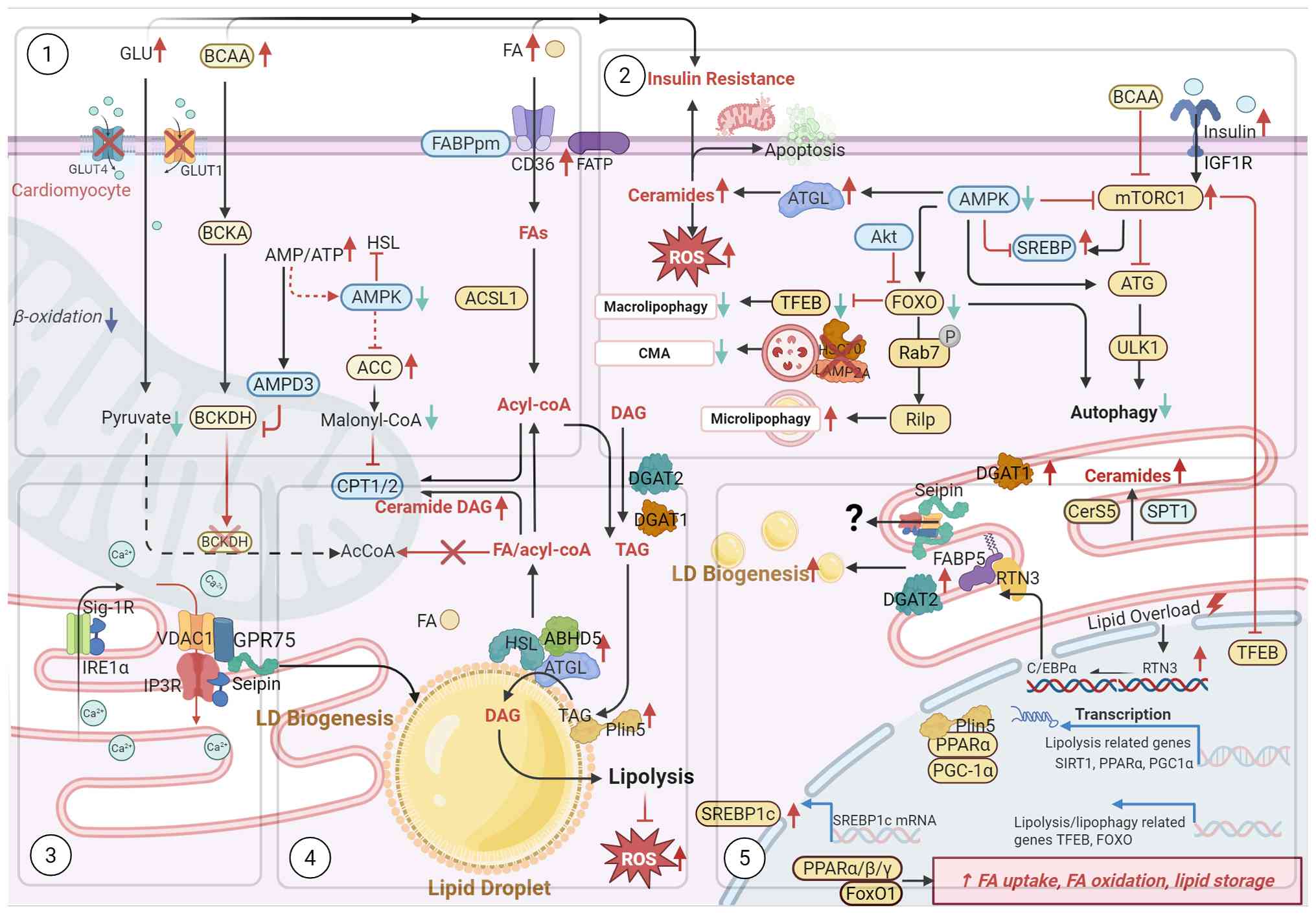
Hyperglycemia induces cardiac lipid overload and
metabolic dysregulation, with insulin resistance exacerbating
lipotoxic injury to cardiomyocytes, disrupts metabolic homeostasis,
manifesting as mitochondrial dysfunction, ER stress and calcium
overload. LDs likely play multiple roles in modulating these
pathological responses through their capacity to store and mobilize
lipids. Under conditions of sustained hyperglycemia, excessive FA
influx overwhelms oxidative capacity, triggering abnormal lipid
accumulation that compromises cellular homeostasis. Insulin
resistance further impairs metabolic flexibility, preventing
adaptive substrate switching and intensifying reliance on FAO
despite limited mitochondrial capacity (82). The resultant accumulation of
lipotoxic intermediates, including DAG, ceramides and
acyl-carnitines, directly impairs mitochondrial respiration, induce
calcium dysregulation and ER stress.
LDs limit FAs' incorporation into membrane PLs and
reducing susceptibility to peroxidative damage. This antioxidant
function preserves organellar integrity and prevents ferroptosis
driven by lipid peroxidation (72). Conversely, LDs mobilize stored
lipids through two pathways: Neutral lipase-mediated lipolysis and
autophagy-dependent acid lipolysis. Lipolysis liberates FAs for FAO
during energy demand. Insulin resistance impairs autophagy, a
mechanism essential for maintaining cellular homeostasis.
Lipophagy, representing a cargo-selective autophagy pathway,
specifically targets LDs for lysosomal degradation. Current
evidence indicates that both CMA and micro-autophagy contribute to
metabolic regulation in DCM, although the role of macro-autophagy
remains controversial (81). The
proteins and PLs on the surface of LDs interact with specific
enzymes to exert important functions. The balance between LD
storage and degradation thus determines lipid flux, organelle
function, and influence cellular metabolic homeostasis in DCM.
Cardiomyocytes mainly rely on oxidative
phosphorylation for energy supply and can also generate energy
through glycolysis and other ways. Nowadays, the change of FAO in
DCM remains controversial (83).
It may be related to the stage of DCM. A multimeric complex
consisting of extended synaptotagmin 1 (ESYT1), ESYT2, VAMP
Associated Protein B and C localizes to ER-LD-mitochondria contact
sites. Loss of this complex reduces LD-derived FAO by 67% and
promotes LDs accumulation, indicating that the complex facilitates
lipid mobilization and prevents lipotoxicity (84,85). This reduced oxidation capacity
appears paradoxical given previous studies of elevated FAO in DCM,
suggesting compensatory mechanisms may operate under different
metabolic conditions (57).
These findings demonstrate that LD accumulation and the resulting
imbalance in FAO are important pathogenic mechanisms in DCM.
Normally, acyl-CoA synthetase long chain family
member 1 activates FAs to fatty acyl-CoA for subsequent
β-oxidation, which directly generates acetyl-CoA for the TCA cycle.
Long-chain FAs require CPT1/2-mediated mitochondrial transport
before oxidation (88). In DCM,
long term overload of FAO promotes electron leakage, generating
reactive oxygen species (ROS) and aggravates oxidative stress.
Plin5 suppresses the ROS-activated Phosphoinositide 3-kinase
(PI3K)/protein kinase B (AKT) pathway and the mitogen-activated
protein kinase pathway (p38/ERK/JNK), hence preventing oxidative
stress and apoptosis (89,90).
LDs protect against ferroptosis through antioxidant
mechanisms. FSP1, located in LDs, and maintains antioxidant
capacity by reducing Coenzyme Q10, preventing peroxidation of
neutral lipids. Impairment of FSP1 function induces the
peroxidation of polyunsaturated FA-LDs (PUFA-LDs), triggering
ferroptosis of cardiomyocytes (91). In type 1 diabetic patients,
glucolipotoxicity reduces LAMP2A, Hsc70 and Hsc90 expression,
inhibits the clearance of autophagosomes and inactivates TFEB,
causing autophagy disorders and rendering the myocardium vulnerable
to ER stress and metabolic injury (92).
Cardiomyocytes maintain lipid homeostasis by
balancing the FAO rate with LD biosynthesis. Under nutrient excess,
insulin activates acetyl-CoA carboxylase (ACC) to generate
malonyl-CoA, which inhibits the acetylation level of CPT1 and MPC2,
thereby restricting FAO (93,94). Studies in CPT1b-deficient
skeletal muscle confirm that reducing FAO alleviates mitochondrial
lipid burden during energy surplus (95). Conversely, ACC2 knockdown
maintains FAO and reduced cardiac hypertrophy, demonstrating that
preserving oxidative capacity protects cardiac function (96).
DGAT1 and GPAT4 are upregulated to enhance TAG
synthesis, and the elevation of TAG levels is positively correlated
with systolic dysfunction, suggesting that excessive accumulation
of LDs adversely affects cardiac structure (97).
Downregulated TGR5 fails to inhibit DHHC4, which
mediates the palmitoylation of CD36, consequently promoting CD36
overexpression, the abnormal FAO, and the elevated LDs'
accumulation (98,99). Exceeding the LDs' buffering
capacity results in surplus FAs, leading to cellular lipotoxicity
and disruption of energy supply. Concurrently, the inhibitory
effect of ATGL on ceramide will be weakened, resulting in increased
ROS production and triggers mitochondrial dysfunction, ultimately
inducing apoptosis and aggravating insulin resistance (100,101) (Figs. 2 and 3).
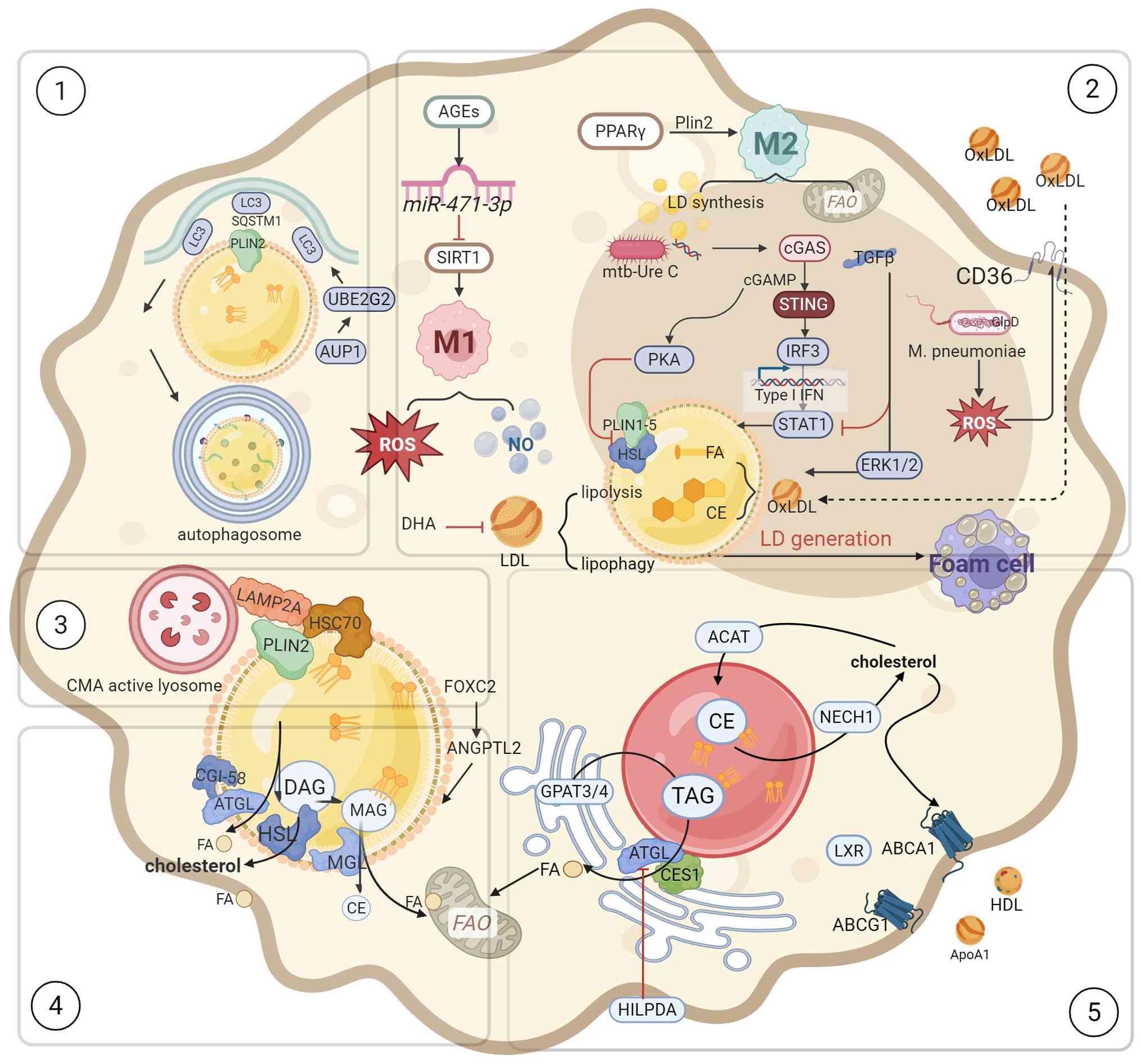
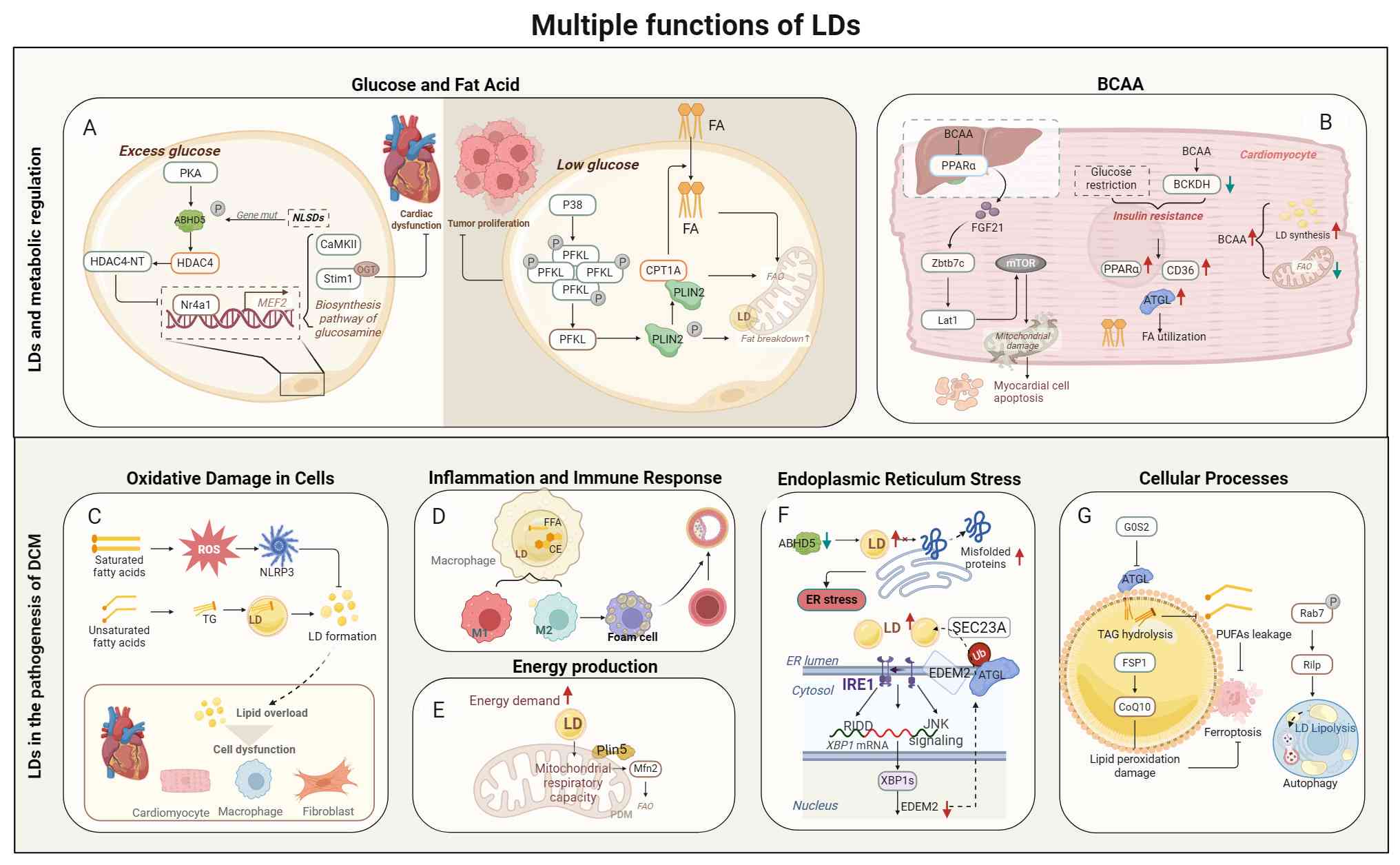
Macrophages primarily orchestrate inflammatory and
immunological responses to sustain cellular metabolic homeostasis.
While numerous studies have researched lipotoxicity in
cardiomyocytes, studies on lipotoxicity in macrophages remain
insufficient. Macrophages modulate their energy acquisition through
dynamic remodeling of membrane receptors coupled with metabolic
reprogramming of glucose and lipid pathways. These cells meet their
lipid requirements by engulfing apoptotic cells, lipoprotein
particles and LDs.
Low-density lipoprotein (LDL) is oxidized to OxLDL,
which macrophages internalize via CD36-mediated endocytosis.
Lysosomal hydrolysis releases FFAs and TC which are stored in LDs
as CE. Reduced lipid autophagy and cholesterol efflux further lead
to LDs accumulation, ultimately impairing phagocytic capacity and
transforming into foam cells (102).
Macrophages form LDs under conditions of
inflammation and oxidative stress. With different phenotypic
switches, the expression levels of LD-related proteins are also
different. Plin1 and Plin2 regulate both polarization of
macrophages and LD size. Plin1 was found to promote larger LDs when
overexpressed in M1 macrophages. With Plin1 overexpression
promoting larger LDs in M1 macrophages, Plin1 associates with the
inflammatory phenotype whereas Plin2 correlates with the
anti-inflammatory phenotype and collectively governing lipid
storage (103). Current
investigations of LD biology in macrophage subtypes remain confined
to classic M1 macrophages and M2 macrophages despite the
identification of diverse macrophage phenotypes. Nevertheless, LDs
serve critical functions in maintaining cellular homeostasis and
macrophage function, processes fundamentally linked to
atherosclerosis (104).
Advanced glycation end products in the diabetic
cardiac microenvironment promote M1 macrophage polarization through
the miR-471-3p/SIRT1 pathway. M1 macrophages enhance glycolysis
while suppressing FAO, thereby activating the pentose phosphate
pathway to generate pro-inflammatory and bactericidal mediators
(105). M2 macrophages, by
contrast, exhibit greater LD accumulation and elevated FAO levels,
maintaining their anti-inflammatory phenotype through PPARγ
(101). TGF-β promotes LDs
synthesis via the ERK1/2 pathway and attenuates STAT1 to inhibit M1
polarization (106). Activation
of the PPAR pathway upregulates CD36 and Plin2, thereby promoting
LD formation, and influences immunophenotypic switching (107,108). CX3CR1 signaling appears to
inhibit M1 polarization while promoting foam cell formation
(109). The expanding lipid
core subsequently elevates the levels of MMP, nitric oxide and
endothelin, and eventually causes plaque rupture (110).
Macrophage-derived LD-associated hydrolase
upregulates Abca1 and Abcg1 expression through an LXR dependent
mechanism, thereby attenuating the inflammatory response induced by
SE accumulation (111).
Although conventional perspective considers that accumulation of
LDs fosters insulin resistance or inflammation, protective
regulatory mechanisms exist. Forkhead box protein C2 (foxc2)
suppresses the angiopoietin-like 2 (angptl2)/NOD-like receptor
thermal protein domain associated protein 3 (NLRP3) inflammasome
pathway, reducing the expression of NLRP3, ASC and caspase-1. Foxc2
also modulates Bcl-2/Bax to diminish apoptosis, oxidative stress
and LDs' accumulation in macrophages (112,113).
Foam cell formation is regulated by lipophagy
factors including HSPA5, UBE2G2 and AUP1. Upon autophagy
activation, LDs initiate lipophagy by recruiting LC3 and adaptor
protein Sequestosome 1 (SQSTM1). Sirt6-dependent chromatin
remodeling factor SNF2H inhibits Wnt family member 1/β-catenin
pathway to accelerate lipophagy and autophagosome lysosome mediated
lipolysis and accelerating autophagosome-lysosome-mediated
lipolysis and FA transfer to mitochondria for FAO. A key unresolved
question concerns the specific mechanism through which sirtuin6
regulates this transfer (114).
In vascular smooth muscle cells, Plin2 similarly facilitates
lipophagy through elevated LC3II levels and enhanced autophagosomes
biogenesis, which impedes foam cell formation (115). Targeting lipophagy may
represent a promising strategy for stabilizing vulnerable plaques
(Fig. 4).
Cardiac fibroblasts exhibit region-specific
distribution patterns and undergo distinct phenotypic transitions
during pathological remodeling. Following acute myocardial
infarction (MI) and HF, F9 (FAP/POSTN+) fibroblasts
differentiate into the POSTN lineage through PI3K-Akt and AGE-RAGE
signaling rather than adopting an ACTA2+ myofibroblasts
phenotype (116). The
transition of CD34+ cells to FABP4+
fibroblasts represents a critical fibrotic process, enhancing lipid
storage via the PPARγ/Akt/Gsk3β pathway activation (117). Therapeutically targeting the
NF-κB and Wnt/β-catenin/GSK3β pathways may mitigate pathological
cardiac remodeling, including cardiac hypertrophy and fibrosis
(118), underscoring how
inflammatory signals govern fibroblast activation and
differentiation. Lipotoxic intermediates, for instance, long-chain
acyl-CoA, ceramide, acylcarnitine and DAG, can directly induce
myocardial fibrosis (119).
Hypoxia and cellular stress stimulate intracellular LD accumulation
in fibroblasts. During early stage of myocardial fibrosis, cardiac
fibroblasts proliferate excessively and excessive secretion of
α-smooth muscle actin and alongside extracellular matrix (ECM)
proteins generated in large quantities (116).
Fibroblasts-derived kinesin-like protein 23 (Kif23)
activates the Ras homolog family member a pathway to enhance
proliferation while suppressing LD hydrolase carboxylesterase 1d
through ROCK1, thereby impairing FAO and promoting lipid
accumulation in fibroblasts, and facilitating myofibroblast
transformation (120). Thus,
targeting Kif23 may represent a new direction for attenuating
myocardial fibrosis. Nevertheless, additional investigation is
required to define how LDs in fibroblasts contribute to myocardial
fibrosis. Fully exploring the effects of cellular stress,
inflammation, and LD accumulation on cardiac lipotoxicity
represents a novel target for fibroblasts in DCM.
Elevated glucose levels promote the production of
LDs, while excessive glucose and FFAs can damage peripheral
neurons, disrupting the sympathetic nervous system's control over
the heart (121). The
conventional viewpoint points out that lipotoxicity is considered a
key driver of metabolic heart disease. Nevertheless, certain
investigations indicate that cardiac dysfunction in neutral lipid
storage disorders resulting from genetic mutations in CGI58 and
ATGL is predominantly influenced by CGI58 rather than ATGL. This
indicates that LDs may have a possible correlation with glucose
metabolism. CGI58, serving as a serine protease, responds to
phosphorylation by PKA and subsequently cleaves the epigenetic
regulator histone deacetylase 4 (HDAC4), producing the N-terminal
polypeptide of HDAC4 (HDAC4-NT), HDAC4-NT transcriptionally repress
Nr4a1 expression, which interacts with myocyte enhancer factor 2
(MEF2). Nr4a1 prevents CaMKII from driving HDAC4 to activate MEF2.
This blocks MEF2 from subsequently activating the hexosamine
biosynthesis pathway, a metabolism linked to protein
O-GlcNAcylation. Consequently, the calcium-handling protein Stim1
avoids undergoing O-GlcNAcylation, thereby protecting cardiac
function (8,31,57,122). ATGL is blocked by Plin5;
however, the ablation of Plin5 does not affect gene expression
involved in glucose metabolism, FAO, along with glycogen metabolism
(123).
Glucose deprivation activates P38 to phosphorylate
glycolytic enzyme phosphofructokinase, liver type (PFKL) at Thr331,
converting it from a tetramer to a monomer. PFKL then acts as a
protein kinase, phosphorylating Plin2 to enhance its interaction
with CPT1A, therefore facilitating FA transfer, FAO and lipolysis.
PFKL, which inhibits glycolysis during low glucose availability, is
posited to promote the anchoring of PLIN2 to LDs' phosphorylation
and mitochondria to modulate lipolysis. This direct transfer of FAs
from glucose to LDs may prevent lipid accumulation and mitigate
cellular lipotoxic damage (75).
Clinical studies report elevated plasma
concentrations of fibroblast growth factor 21 (FGF21) in patients
with DCM. Animal experiments show that inhibiting FGF21 reduces
PGC1α expression and upregulates CD36, leading to LD accumulation
and cardiac dysfunction. PGC1α is a downstream factor of FGF21,
effectively reduces blood glucose as well as lipid levels while
enhancing insulin sensitivity. Under dual intervention of knocking
down FGF21 and fasting, glucose production and ketone body
synthesis are damaged, suggesting that FGF21 holds promise as a
potent therapeutic target for DCM management (124,125). Alterations in BCAA and FGF21
signaling establish metabolic links between the heart and the
liver.
BCAAs inhibit the PPARα-FGF21 axis in the liver and
reduce FGF21 secretion in the liver, thereby upregulating the
content of BCAA in the heart and the expression levels of
zinc-finger and BTB domain-containing 7B and Lat1, activating mTOR
signaling and inducing mitochondrial damage and cardiomyocyte
apoptosis, ultimately accelerating myocardial fibrosis in mice with
type 1 diabetes (126).
Knockout of PPARα in mice induced a metabolic shift towards
glycolysis, HIF1α, as well as augmented glucose metabolism
alongside ECM remodeling, finally decreasing FAO (127). PGC1α is also involved in
regulating the rate of glycolysis BRM-associated factor 60c
facilitates the transition of oxidative muscle fibers to glycolytic
muscle fibers via the PGC1α-PPARα-mTOR pathway, thereby preserving
oxidative metabolism/glycolysis balance and enhancing mitochondrial
function in rats with HF (128). The metabolic disorder of BCAA
in cardiomyocytes results into an abnormal rise in the biosynthesis
of LD as well as a reduction in FAO rate, which coincides with
suppression of branched-chain alpha-ketoacid dehydrogenase (BCKDH)
located in the ER and the excessive expression of AMPD3.
Interestingly, there is a mutual repulsion between BCAA oxidation
and glucose metabolism (83). In
the DCM state, BCKDH cannot restrict the upregulation of AMPD3.
Subsequently, the uncoupling of AMPD3 activation induces excessive
adenine nucleotide degradation and ROS production. After knocking
out BCKDH-E1α in cardiomyocytes, the expression levels of PPARα,
CD36 and ATGL significantly increase, making the utilization of
cardiac substrates more inclined towards FA, which is similar to
the rat model of obese type 2 diabetes state (129).
FFAs derived from food are transported to
cardiomyocytes, with excess lipids stored in LDs and VLDL. VLDL and
LDL transfer lipids to the heart and adipose tissue. As TAG
reserve, LDs maintain the supply of circulating FAs and maintain
cardiac lipid homeostasis and energy demand (130). Most of the TAGs constituting
LDs come from lipoproteins and NEFAs, and a small amount of FA is
provided by the heart via de novo lipogenesis (DNL) pathway.
This process activates transcription factors such as
insulin-responsive SREBP1 and Farnesoid X Receptor (FXR), enhancing
adipogenic gene expression to drive FA trafficking into the ER for
neutral lipid synthesis (TAG and CE). Increased expression of
DGAT2, which regulates TAG synthesis in prediabetes and type II
diabetes, leads to abnormal nerve lipid signaling (131). In the presence of
electronegative LDL (-), the intracellular levels of NEFA, TAG and
ROS in cardiomyocytes lacking plin5 are substantially increased
(132). Acetylcholine may
reduce cardiomyocyte apoptosis through PLIN5, activating the
interaction between LDL and mitochondria (133).
The biogenesis and degradation of LDs finely
regulate lipid homeostasis. Prolonged lipid excess in diabetic
heart triggers the dysfunction in cardiomyocytes, macrophages,
along with vascular endothelial cells, with lipotoxic injury being
the primary cause. The response of cardiomyocytes to different
kinds of FAs remains incompletely understood. Evidence suggests
that lipotoxicity is closely tied to FA saturation levels,
saturated FAs (SFAs), such as palmitate, hinder LD formation and
inflict significant cellular damage. The activation of the ULK1
autophagy signaling cascade may be linked to the ROS-dependent
NLRP3, driving IL-1β/IL-18 production, leading to subsequent
insulin signaling disruption. Conversely, unsaturated FAs such as
oleic acid encourage the accumulation of TAG in LDs and store
excess palmitic acid in the TAG pool, thereby preventing palmitic
acid from triggering apoptosis and consequently reducing lipotoxic
cellular damage (134). The
previously mentioned investigations have overlooked the potential
excess of FAs, while an appropriate amount of palmitic acid can
actually promote the formation of LDs.
The accumulation of TAG and stearoyl-CoA desaturase
1 in LDs enables endothelial cells to sequester palmitic acid and
prevent the palmitoylation of ciliary proteins, which would
otherwise lead to lipotoxic injury. Upon reinstating the supply of
PA, ciliary homeostasis is restored, subsequently delaying AS
(135). PA also activates the
Toll-like receptor (TLR) 4/MyD88 pathway in macrophages, triggering
expression of TNF-α and intercellular adhesion molecule-1 (136,137).
Impaired TAG synthesis within cells can trigger
lipotoxicity, which is associated with the synthesis of lipotoxic
intermediates. Elevated DGAT2 expression in type 2 diabetic mice
promotes TAG synthesis (131).
Myocardial-specific knockout of DGAT1 significantly reduces LD
formation, leading to DAG and ceramide accumulation, which activate
PKCα and induce cardiac dysfunction (138); conversely, overexpression of
DGAT1 promotes TAG storage in LDs without harming the heart
(139). Lipid overload
upregulates CCAAT/enhancer-binding protein α (C/EBPα), increasing
the transcription of reticulon 3 (RTN3), subsequently driving LD
accumulation and cardiac dysfunction through the DGAT2-dependent
RTN3-FABP5 pathway (140).
Therefore, isolating TAG and promoting LD formation represent
potential strategies to alleviate lipotoxicity.
Lipotoxicity is closely associated with ROS; ROS
induces post-translational modifications of a-kinase anchor protein
121 (AKAP121), DRP1 as well as optic atrophy 1, enhancing
mitochondrial fission and leading to mitochondrial dysfunction
(141). Increased ceramide DAG
levels activate NADPH oxidase to damage mtDNA and reduce ATP
synthesis (142), ultimately
impairing left ventricular compensatory function (143). Targeting Sirt5 to mediate CPT2
dessuccinylation and C1q/tumor necrosis factor (TNF)-related
protein 3 to inhibit mitochondrial apoptosis can reduce
lipotoxicity (144,145). CPT2 is a regulator of FA
uptake, and further studies have confirmed the relationship between
CD36 and lipotoxicity and ER stress. Endogenous H2S
stimulates Hrd1 to mediate S-sulfation to regulate VAMP3
ubiquitination and inhibit CD36 membrane translocation to reduce FA
uptake. Exogenous H2S can reduce ER stress and
lipotoxicity (146,147).
SREBP1c represents another potential target for
reducing lipotoxicity, p21-activated kinase 3 in the myocardium
promotes SREBP1c expression to induce lipid peroxidation deposition
through the mTOR/S6 kinase 1 pathway (148,149). SREBP1c activation linked to
myocardial injury in patients with aortic stenosis and metabolic
syndrome, and statins can trigger lipotoxic injury through the DNL
pathway (149,150).
LDs first convert stored constituent lipids into
active lipid mediator precursors, which then generate active lipid
mediators that interact with cells to interact with cells and
participate in inflammatory responses. FAs stored in LDs include
SFAs, monounsaturated FAs and PUFAs, of which PUFAs are precursors
of two key lipid mediators-pro-inflammatory eicosanoid compounds
are mainly derived from ω6-PUFAs, while specific pro-resolved
mediators (SPMs) are derived from ω3-PUFAs (151).
Active lipid precursors on the monolayer PL shells
of LDs rely on cytosolic phospholipase A2α to produce lysoPLs and
PUFAs such as arachidonic acid, eicosapentaenoic acid (EPA), along
with docosahexaenoic acid. TAG releases PUFAs through the lipolytic
pathway mediated by ATGL and HSL. During ATGL-regulated lipolysis,
released FFAs act as agonists for FFA receptor 4 (FFA4), which in
turn inhibits adenylate cyclase via Gi/o protein and
reduces high levels of cyclic adenosine monophosphate (cAMP) in LDs
and ER regions, thus balancing energy mobilization and storage.
FFA4 thus regulates the lipolysis process at LDs through cellular
endocrine signaling (152).
EPA inhibits TGFβ-mediated myocardial fibrosis via
FFA4, possibly through its ability to activate PKC and induce
phosphorylation of endothelial nitric oxide synthase (eNOS)
(153). Cyclooxygenase (COX)
co-localizes with 5-lipoxygenase on the LDs' surface, cooperatively
regulating the generation of lipid mediators (154). Although eicosanoids and SPMs
produced by COX and LOX have partial anti-inflammatory properties,
immune and inflammatory responses can also be stimulated by these
mediators via their interactions with receptors such as PPARs and
TLRs (155). However,
hypoxia-induced overexpression of the LD-associated protein
(HILPDA) inhibits ATGL-mediated lipolysis and obstructs the
generation of prostaglandin E2 as well as interleukin-6 (156). Similarly, the removal of HILPDA
in adipose tissue reduced LDs' accumulation and lipid excess,
although did not ameliorate inflammation, possibly due to tissue
specificity (157).
Research shows that viruses and bacteria can target
and interact with LDs. The integration of ORF6 into the LD
monolayer is mediated by its two amphipathic helices. ORF6 inhibits
UBXD8-Plin2 binding in LD-mitochondrial interactions to enhance
lipolysis, and it also promotes lipid synthesis by binding to
DGAT1/2 at LD-ER contact sites. Finally, accelerated FAO provides
energy for virus replication (84). It has been recently indicated
that LDs are not just energy storage units but also help regulate
immune responses in cells like macrophages. Pneumococcus
accelerates LDs' accumulation in macrophages by producing ROS
through glycerol-3-phosphate oxidase (158), Mycobacterium
tuberculosis protein (Mtb) Ure C inhibits DNA repair in
macrophages, genomic instability and intracellular micronucleus
stimulation activate the cGAS-STING pathway, phosphorylating TBK1
and IRF3 to induce IFN-β to upregulate the scavenger receptor SR-A1
as well as promote the formation of LDs that provides energy for
pathogen survival (114).
Subsequently, Mtb activates the anti-glycolysis G protein-coupled
receptor 109a, shifting macrophages from glycolysis to the ketone
body synthesis pathway and accelerating foamy transformation
(159). Thus, targeting the
composition and formation of LDs may be a new direction for
immunotherapy.
ROS are generated by the cytosolic nicotinamide
adenine dinucleotide phosphate (NADPH) oxidase (NOX) as well as
mitochondrial ETC complexes Ⅰ/Ⅱ/Ⅲ. Large amounts of ROS damage
mitochondria and affect cellular homeostasis. Therefore, the
presence of LDs makes it possible for cells to store excess FFAs
and also relieves oxidative stress and reduces the accumulation of
lipotoxic intermediates; storage of FAs occurs through their
incorporation into neutral lipids' pools. In times of energy demand
such as starvation and cellular nutrient deprivation, it is
supplied to mitochondria to maintain energy production.
During energy stress, PDM facilitates targeted
transfer of FA from LDs to mitochondria, rapidly replenishing
energy deficits while mitigating lipotoxicity damage caused by FA
accumulation (75). Besides PDM,
LDs can transport FAs to lysosomes via an autophagy-dependent
pathway for cytoplasmic release and mitochondrial energy supply.
This mechanism appears better suited for energy-deficient states
such as starvation (160,161).
Mitofusin 2 (Mfn2) enrichment after aerobic
exercise reduces LD accumulation and increases FAO levels in PDM.
Mfn2 acts as a mitochondrial outer membrane protein that enables
mitochondria to link both LD and ER (162,163). It is hypothesized that PDM may
promote FAO through a special pattern related to Mfn2 (164). After the interaction between
Mfn2 and ATGL, the lipolysis of liver LDs, FA transport and the
formation of more PDM are enhanced, and the ATP synthesis capacity
and high FAO level of PDM are maintained (165). Mfn2 as well as LD-localized
Hsc70 assemble into a complex to promote FA transfer in myocardial
LDMC. Upregulation of Mfn2 after lipid overload may restore the
coupling between LDs and mitochondria and reduce
lipotoxicity-induced myocardial damage via the ubiquitin-proteasome
degradation pathway (166).
However, the mechanisms of lipids' transfer from
LDs to mitochondria remain uncertain. Investigations show that
mitochondria and ER contact sites enlist seipin to accelerate LDs'
biosynthesis and lipid transfer, mitigating neutral lipid
accumulation. Mutations in the seipin protein can result in
irregularities at the LD-ER contact interface, suggesting its
potential role in regulating the construction of membrane bridges
by stabilizing this contact interface (167). Seipin keeps the level of
phosphatidic acid in the ER under control and achieves protein
transport through LD-ER contact, transferring TAG to nascent LDs
and counteracting the shrinkage of small LDs induced by LD
maturation (168,169).
Seipin forms a complex with Oxysterol binding
protein-related protein 5 (ORP5) and ORP8 within the PA-enriched
mitochondrial-associated membrane subdomain. ORP8 enhances
interaction with ORP5 via a coiled-coil domain, maintaining lipid
transport across the mitochondrial ER-LD tri-interface and
promoting LD biogenesis (67).
This mechanism is widely conserved in mammals. Additionally, there
are two necessary factors for Plin5 transferring FAs from LDs to
mitochondria: Plin5 requires PKA-mediated phosphorylation at serine
155 and an intact C-terminal domain (31). Its interaction with the chaperone
ACSL4 at the MCS enables the conversion of FA into acyl-CoA for
FAO, completing the lipid movement from LDs to mitochondria
(170,171).
ER stress, featured by insufficient protein folding
capacity, calcium ion imbalance as well as lipid metabolism
disorder, is a key metabolic feature of DCM (172). Increased intracellular lipid
accumulation induces ER stress and LD formation. It has been
suggested that ER stress can degrade Plin2 and inhibit the
generation of LDs (48),
therefore it can be concluded that LDs and ER stress influence each
other. Researchers point out that ROS-triggered ER stress in DCM
induces cardiomyocyte apoptosis mainly through protein kinase
RNA-like ER kinase (PERK) pathway, but not IRE1 or activating
transcription factor (ATF) 6 signaling pathways (173). At this time, G protein-coupled
receptor 78 (GPR78) dissociates from PERK, and PERK activates the
eukaryotic translation initiation factor 2 subunit α/ATF4 pathway
to enhance cardiac autophagy and alter normal myocardial morphology
(174).
LDs likely represent a common therapeutic for
mitigating ER stress and preserving lipid homeostasis. Patients
with HF with preserved ejection fraction exhibit decreased LDs and
PDMs, abnormal LD dynamics, lipid overload, and downregulation of
X-box binding protein 1 (XBP1), which affects the ER degradation
enhancing alpha mannosidase-like protein 2 (EDEM2), which regulates
protein glycosylation, endoplasmic reticulum-associated
degradation, and maintains ER stability (175). The absence of XBP1 or EDEM2 in
cardiomyocytes leads to decreased cardiac systolic function and
exacerbates LD formation and lipid disorders. Under excessive FFA
stimulation, EDEM2 interacts with SEC23A in an ATGL-dependent
manner to efficiently alleviate lipotoxic damage, including
myocardial LDs' accumulation and elevated TAG and DAG levels
(176). These data suggest that
enhancing cellular lipid oxidation and accumulating comparatively
'non-toxic' lipids to partially regulate lipid flow may be a viable
approach to reduce lipotoxicity (177).
LDs participate in the regulation of normal cell
division, ferroptosis, autophagy as well as other cellular
processes and indirectly affect the progression of DCM. LD
regulates normal cell division and induces DGAT-dependent LD
biosynthesis during cell cycle arrest. The G0/G1 switch 2 gene
(G0S2) inhibits ATGL-mediated TAG hydrolysis and prevents excessive
leakage of PUFAs within TAG, which is essential for LDs to protect
cells against ferroptosis (178). During iron depletion, cells
enhance LD biosynthesis through the DGAT1-dependent pathway to
regulate mitophagy and maintain cellular homeostasis (179).
During the process of DCM micro-autophagy, Rab7 is
phosphorylated at the 183 site of tyrosine and interacts with
Rab-interacting lysosome protein (Rilp) to accelerate the lipolysis
of LDs via the lysosomal pathway (81). Additionally, RAB18 may regulate
the process of autophagy by downregulating adipogenic genes'
expression and reducing TC and TAG levels. RAB18 knockout did not
affect lipolysis gene expression, but the liver lipophagy was
weakened and LD accumulation was observed in mice (180). While RNA is mainly involved in
the transmission of genetic information, it was recently revealed
that RNA regulates lipid metabolism (181). Previous experiments have shown
that overexpression of LIPTER can alleviate cardiac dysfunction and
ameliorate cardiomyopathy in mice, and that LIPTER in
cardiomyocytes connects long non-coding RNAs to strengthen the
connection between LDs and actin by binding
PA/Phosphatidylinositol-4-phosphate and interacting with myosin
heavy chain 10 protein, facilitating LD transfer (182) (Fig. 5).
Abnormal cardiac LD accumulation constitutes a key
feature of cardiac steatosis in diabetic patients. It is associated
with a large accumulation of TAG and an enhanced lipolytic barrier
in the heart after overexpression of Plin5 (64,183). This imbalance in LD dynamics
accelerates ventricular remodeling and cardiac dysfunction. Studies
in PPARα knockout mice have shown reduced TAG accumulation after
fasting, while normal mice fasting for 16-48 h exhibit an
upregulation of Plin2 expression and a concurrent increase in
accumulation of LDs (184),
suggesting that LDs can provide energy supply. Cardiac lipid
accumulation is independent of systemic metabolism, and PPARα
downregulation itself does not induce cardiac pathology (185).
During starvation, vacuolar protein
sorting-associated protein 13D (VPS13D), endosomal sorting complex
required for transport protein tumor susceptibility 101, along with
adaptor-binding domain of VPS13, facilitate FAs transport at the
LDMC site to achieve high-efficiency oxidation (186). This pathway may become
dysregulated in diabetic hearts due to chronic energy excess.
LD-associated proteins form a complex regulatory
network in the heart. Reduced expression levels of ATGL and CGI58
jointly impair lipolysis, which instead increases TG synthesis,
promotes cardiac steatosis, and raises oxidative stress levels
(65). Plin2 is involved in
regulating LDL hydrolysis, and the increased TAG accumulation
following Plin2 knockout may be associated with lipophagy.
Overexpression of Plin2 reduces HSL levels, suggesting that energy
stress during metabolic remodeling stimulates Plin2 to enhance
lipase affinity for LDs, consequently exacerbating cardiac
steatosis and dysfunction (53,187).
Plin5 inhibits lipolysis to preserve TAG storage
within LDs, thereby limiting the rate of FAO and reducing excessive
oxidative stress in heart. Upon Plin5 knockout, the upregulation of
PPARα as well as PGC-1α induces mitochondrial proliferation,
resulting in higher FAO and lipolysis, which can worsen cardiac
hypertrophy potentially leading to HF (188). The deletion of Plin5 exerted no
notable impact on heart function. During fasting, Plin5 associates
with Rab8a in skeletal muscle and collaborates with ATGL, this
process couples LCFAs transfer from LDs to mitochondria, indicating
that Plin5 may have a cardioprotective effect through preserving
mitochondrial integrity (64,189).
Myocardial steatosis and fibrosis are the basis of
ventricular remodeling in DCM. Computed tomography scans of
patients with MI have revealed substantial lipid deposition
(190). Reduced expression of
eNOS at the infarct site exacerbates abnormal myocardial lipid
accumulation. This in turn promotes LD formation, increases
oxidative stress, and activates the TGFβ/TβRII pathway,
phosphorylating Smad2/3, inducing Smad4 to modulate transcription
of ECM genes and hindering myocardial repair (190,191).
Macrophages also help regulate fibroblast
activation through IL-1β and its downstream target mesenchyme
homeobox 1 (116). In fact, the
process of macrophage-to-myofibroblast transition (MMT) may involve
LD regulation. In chronic pancreatitis, high expression of
mitochondrial uncoupling protein 2 (UCP2) regulates ACSL3 to
promote acinar cell release of LDs. Stimulation of SIRT1 and
activation of downstream TGF-β/Smad3 signal transduction affect MMT
and fibrosis process, while whether UCP2 has a similar regulatory
effect still needs to be explored (192).
In this process, ATGL-dependent lipolysis promotes
FA accumulation and inhibits ECM remodeling in fibroblasts
(193). Low levels of CGI58 in
the heart decrease the lipolysis rate and aggravate oxidative
stress and myocardial steatosis (65). This indicates that the regulation
of lipolysis is particular to cell type.
In addition to lipid metabolic disorders,
alterations in cardiomyocyte energy metabolism are also considered
to indirectly affect fibrosis progression (194). Wilms' tumor 1-associating
protein promotes AR methylation in a YTHDF2-dependent way,
subsequently promoting FAO proliferation, along with cardiac
fibroblasts migration (195).
Direct investigations of cardiac fibroblasts and
LDs are limited; however, alternative cellular models offer
insights. In mouse embryonic fibroblasts, FAs generated by
non-specific autophagy can be directed to LDs. This process is
considered a protective regulatory technique, as LDs sequester
excess FFAs to mitigate lipotoxicity (196). DGAT1 can channel FFAs generated
during mTORC1-regulated autophagy to TAG-rich LDs, which then hold
onto the FAs and reduce mitochondrial damage caused by
acylcarnitine accumulation (160). TANGO2 is associated with LDs
and the ER to modulate acyl-CoA metabolism. The cardiomyopathy and
arrhythmia caused by the TANGO2 mutation may result in disturbance
of LDs' morphology, function, and neutral lipid metabolism
homeostasis. Consequently, lipid peroxide levels in
TanGO2-deficient fibroblasts were raised metabolized, and LDs
increased (197). Patients with
neutral lipid storage disease with myopathy exhibit muscular,
cardiac and hepatic impairment due to defective LD functions,
similar to cells deficient in ATGL or CGI58, therefore providing a
suitable model for investigating LD function (198). Collectively, these findings
suggest that LDs may influence cardiac steatosis and myocardial
fibrosis.
Cardiac lipid-lowering therapeutics have gained
substantial research attention over recent decades. Emerging
strategies now target multiple approaches of cellular lipid
homeostasis, including biosynthesis, uptake, trafficking and
catabolism, with the dynamic regulation of LDs representing a
critical target. Currently, pharmacological approaches targeting
LDs encompass three principal mechanisms: Modulating lipid
metabolism to prevent excessive accumulation, indirectly
suppressing LD biogenesis, and promoting LD degradation through
enhanced lipophagy or lipolysis pathways (197,199).
Statins lower blood lipids by blocking the
mevalonate pathway, inhibiting Rho/Ras/Rac, and activating PPARα/β,
resulting in anti-inflammatory pleiotropic effects (200,201). They also downregulate the
expression of Plin5 in hepatocytes (202). However, long-term usage of
activated cardiac SREBP1 to speed up the process of DCM may make
myocardial lipid deposition and lipid peroxidation worse (149). Niacin reduces the progression
of atherosclerosis by diminishing the synthesis of proinflammatory
cytokines by macrophages (203). It also facilitates reverse
cholesterol transport through adipocyte lipolysis via the cAMP/PKA
and 15d-PGJ2/PPARγ pathways, inhibits the expression of Plin4 in
neuroblastoma to prevent the formation of LDs and promotes
macrophage polarization to the anti-inflammatory M2 type (204,205). However, the regulating
mechanism of cardiac LDs remains ambiguous. CL2-57, an ABCA1
inducer that targets cholesterol transport, promotes cholesterol
efflux from macrophages, reduces inflammation, and prevents lipid
abnormalities caused by LXR agonists (206,207). The MEK1/2 inhibitor (U0126) in
conjunction with the LXR ligand (T0901317) modulates lipid
metabolism through lipolysis and FAO to reduce LXR-associated
adverse effects (208). IL4 via
the STAT6/PPARγ axis, boosts ABCA1/ABCG1 expression, decreases
lipid overload and foam cell formation, and stimulates M2
polarization to control late adipogenic phase suppression, which
prevents inflammation and formation of LDs (199). Notably, IL4 did not influence
C/EBPβ and PPARγ in lipogenesis and lipid metabolism during the
first phase of lipid synthesis, nor did it affect HSL. Moreover,
ezetimibe impedes cholesterol absorption (209,210). In addition, ezetimibe can
accelerate LDs synthesis through LD-mitochondrial membrane contact
sites, store excess FAs in LDs, reduce lipotoxic damage, and at the
same time, LDs degradation may also be accelerated, thus alleviates
podocyte apoptosis (211). It
indicates that LD-organelle interaction may serve as an effective
target for intervention. Proprotein convertase subtilisin kexin 9
(PCSK9) inhibitors enhance LDL-LDLR binding by preventing LDLR
degradation and promoting lipophagy to eliminate lipids (212), with PCSK9 mediating lipid
degradation independently of LDLR (213). Inclisiran, a small interfering
RNA targeting PCSK9, significantly reduces LDL-C levels in patients
with cardiovascular risk (214). This effect may be associated
with the suppression of AKT activation, affecting downstream mTOR
and the facilitation of ULK1 kinase complex formation, which
regulates Beclin1 and enhances the degradation of intracellular
lipids via lipophagy (215).
Short-term continuous and excessive administration
of prednisolone substantially elevates the mRNA levels of CGI-58,
CEIDE-C and CEIDE-A in serum FA and adipose tissue and reduce the
expression of G0S2 mRNA. CIDE proteins block ATGL-mediated
lipolysis and promote lipid storage (216,217), indicating that prednisolone is
involved in both lipolysis and lipid storage through complex
pathways and alleviates insulin resistance by inhibiting insulin
receptor phosphorylation and Akt activation (218,219).
Fibrates, such as fenofibrate, activate PPARα to
reduce blood glucose and lipid levels, modulate insulin resistance,
markedly enhance cardiac function, and maintain glucose metabolism
via the downstream Nr4a1/hexokinase 2 pathway. They also enhance
the sirtuin 3/superoxide dismutase 2 pathway to achieve
anti-oxidative stress (122,220). Furthermore, the elevation of
LC3 and SQSTM1 in a diabetic condition hinders the heart's
autophagic capacity, potentially linked to the formation of LD and
macrophage foam cells. Fenofibrate restores autophagy ability
through FGF21/SIRT1 to alleviate macrophage foam cell formation and
myocardial fibrosis caused by LD accumulation (221).
Pemafibrate, a novel PPARα agonist, also improves
cardiac diastolic function in diabetic patients (222). Thiazolidinediones, such as
troglitazone, reduce lipotoxicity through AMPK activation, but
full-acting PPARγ agonists are associated with obesity and cardiac
risk (223,224). However, the incomplete agonist
dihydrosanguinarine is safer, reducing LD accumulation and
enhancing glucose uptake (225). SGLT2i inhibitors exhibit
multidimensional cardioprotection: Empagliflozin regulates
autophagy balance through AMPK/GS3Kβ and mTOR/ULK1 pathways
(79). In vitro models of
SGLT2i inhibitors all increase LD biosynthesis in a DGAT1-dependent
manner, but the specific pathways differ. Canagliflozin does not
reduce LD formation in vascular endothelium via an AMPK-dependent
pathway, while empagliflozin may reduce lipogenesis through the
SREBP1/PPARγ pathway (226,227). In the meantime, empagliflozin
also reprograms energy metabolism in podocytes, inhibiting glucose
utilization and switching to FAs for energy, enhancing lipolysis
and ketone body formation, providing additional fuel for the heart,
and alleviating ER stress and lipotoxicity, which is extremely
similar to metabolism shift observed in the diabetic heart
(228-230). In summary, as the intersection
of metabolism and inflammation, the formation and decomposition of
LDs and the targeted intervention of cellular homeostasis
constitute an emerging therapeutic frontier in DCM.
Despite gaining substantial progress,
investigations of LD biology in DCM exhibit critical knowledge
gaps. Studies on LDs in cardiac fibroblasts and vascular
endothelial cells remain markedly underdeveloped, while the
existing studies on macrophages have predominantly focused on M1
and M2 polarization states, neglecting other types related to DCM
pathogenesis. The present review predominantly explores the role of
LDs in DCM and does not comprehensively address their involvement
in other heart diseases, such as atherosclerosis and HF. Exploring
dynamic functions of LD across diverse cardiovascular diseases
would provide comprehensive insights into their regulatory
mechanisms and therapeutic potential in the heart.
Contemporary understanding of LD evolved beyond
their classical role as inert lipid reservoirs to recognize their
multiple regulatory functions in metabolic substrates, oxidative
stress responses, inflammation, immunity, and cellular processes
such as autophagy and ferroptosis, ER stress.
LDs sequester excess lipids through diverse
mechanisms, effectively mitigating the accumulation of lipotoxic
intermediates that trigger cellular oxidative damage. In addition,
LDs catabolize stored lipids to supply FAs for oxidation and delay
cardiac energy depletion. Under pathological conditions, lipophagy
attenuates myocardial injury induced by glucotoxicity and
lipotoxicity while delaying myocardial fibrosis remodeling and
macrophage foam cell formation. LDs participate extensively in cell
proliferation, development and signal transduction, with
LD-associated proteins maintaining cellular metabolic homeostasis
through protein-protein interactions that modulate signaling
cascades. However, research on macrophages and fibroblasts remains
in its infancy. As central hubs of lipid metabolism, LDs critically
influence metabolic homeostasis, and their dysregulation drives
metabolic imbalance in DCM. Therapeutic strategies targeting LD
accumulation include modulating lipid metabolism, interfering with
Plin2 or DGAT1 to inhibit biogenesis, and promoting lipophagy and
lipolysis to accelerate degradation. PDM localize at LD surfaces
and establish functional coupling that facilitates metabolic
exchange, suggesting that mitochondrial-directed interventions,
including regulation of FAO rate or antioxidant treatment, may
alleviate LDs accumulation. However, the safety profiles and
off-target effects of these approaches remain incompletely
characterized, and existing pharmacological agents exhibit
significant adverse effects that necessitate mechanistic
clarification for rational drug development. Understanding of the
regulation of heart function by LDs remains limited. The mechanism
of action of LDs in each stage of DCM still needs to be studied,
and the impact of the cellular heterogeneity of LDs on the
occurrence of DCM remains unclear. The development of related drugs
targeting LD to reduce lipid deposition holds great promise for the
treatment of DCM.
The data generated in the present study may be
requested from the corresponding author.
YL was responsible for conceptualization of the
review, literature search, drafting of the manuscript, and
visualization of table and graphs. XF conceived the study, wrote
the original manuscript and prepared the figures. XG, YJ, WS, XSu,
JG and GY curated data and collected references. BZ, ZZ, YK and XSh
organized the table, JZ acquired funding and contributed to writing
review and editing. All authors read and approved the final version
of the manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by the National Natural Science
Foundation of China (grant nos. 8257152346 and 82574965), the QI
HUANG Scholars (Junping Zhang) Special Funding [National TCM
People's Education Letter (2022) No. 6] and the Inheritance and
Innovation Team for Traditional Chinese Medicine Prevention and
Treatment of Atherosclerosis (grant no. 4042502037).
|
1
|
Seferović PM, Paulus WJ, Rosano G,
Polovina M, Petrie MC, Jhund PS, Tschöpe C, Sattar N, Piepoli M,
Papp Z, et al: Diabetic myocardial disorder. A clinical consensus
statement of the heart failure association of the ESC and the ESC
working group on myocardial & pericardial diseases. Eur J Heart
Fail. 26:1893–1903. 2024. View Article : Google Scholar
|
|
2
|
Segar MW, Khan MS, Patel KV, Butler J,
Tang WHW, Vaduganathan M, Lam CSP, Verma S, McGuire DK and Pandey
A: Prevalence and prognostic implications of diabetes with
cardiomyopathy in community-dwelling adults. J Am Coll Cardiol.
78:1587–1598. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lewis GF, Carpentier A, Adeli K and Giacca
A: Disordered fat storage and mobilization in the pathogenesis of
insulin resistance and type 2 diabetes. Endocr Rev. 23:201–229.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Olzmann JA and Carvalho P: Dynamics and
functions of lipid droplets. Nat Rev Mol Cell Biol. 20:137–155.
2019. View Article : Google Scholar
|
|
5
|
Varghese M, Kimler VA, Ghazi FR, Rathore
GK, Perkins GA, Ellisman MH and Granneman JG: Adipocyte lipolysis
affects Perilipin 5 and cristae organization at the cardiac lipid
droplet-mitochondrial interface. Sci Rep. 9:47342019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kuramoto K, Sakai F, Yoshinori N, Nakamura
TY, Wakabayashi S, Kojidani T, Haraguchi T, Hirose F and Osumi T:
Deficiency of a lipid droplet protein, perilipin 5, suppresses
myocardial lipid accumulation, thereby preventing type 1
diabetes-induced heart malfunction. Mol Cell Biol. 34:2721–2731.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kolleritsch S, Kien B, Schoiswohl G,
Diwoky C, Schreiber R, Heier C, Maresch LK, Schweiger M, Eichmann
TO, Stryeck S, et al: Low cardiac lipolysis reduces mitochondrial
fission and prevents lipotoxic heart dysfunction in perilipin 5
mutant mice. Cardiovasc Res. 116:339–352. 2020.
|
|
8
|
Jebessa ZH, Shanmukha KD, Dewenter M,
Lehmann LH, Xu C, Schreiter F, Siede D, Gong XM, Worst BC, Federico
G, et al: The lipid-droplet-associated protein ABHD5 protects the
heart through proteolysis of HDAC4. Nat Metab. 1:1157–1167. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kuo CY, Tsou SH, Kornelius E, Chan KC,
Chang KW, Li JC, Huang CN and Lin CL: The protective effects of
liraglutide in reducing lipid droplets accumulation and myocardial
fibrosis in diabetic cardiomyopathy. Cell Mol Life Sci. 82:392025.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bosma M, Dapito DH, Drosatos-Tampakaki Z,
Huiping-Son N, Huang LS, Kersten S, Drosatos K and Goldberg IJ:
Sequestration of fatty acids in triglycerides prevents endoplasmic
reticulum stress in an in vitro model of cardiomyocyte
lipotoxicity. Biochim Biophys Acta. 1841:1648–1655. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Renne MF and Hariri H: Lipid
droplet-organelle contact sites as hubs for fatty acid metabolism,
trafficking, and metabolic channeling. Front Cell Dev Biol.
9:7262612021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zheng X, Ho QWC, Chua M, Stelmashenko O,
Yeo XY, Muralidharan S, Torta F, Chew EGY, Lian MM, Foo JN, et al:
Destabilization of β Cell FIT2 by saturated fatty acids alter lipid
droplet numbers and contribute to ER stress and diabetes. Proc Natl
Acad Sci USA. 119:e21130741192022. View Article : Google Scholar
|
|
13
|
Gao M, Huang X, Song BL and Yang H: The
biogenesis of lipid droplets: Lipids take center stage. Prog Lipid
Res. 75:1009892019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Parini P, Davis M, Lada AT, Erickson SK,
Wright TL, Gustafsson U, Sahlin S, Einarsson C, Eriksson M, Angelin
B, et al: ACAT2 is localized to hepatocytes and is the major
cholesterol-esterifying enzyme in human liver. Circulation.
110:2017–2023. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wilfling F, Wang H, Haas JT, Krahmer N,
Gould TJ, Uchida A, Cheng JX, Graham M, Christiano R, Fröhlich F,
et al: Triacylglycerol synthesis enzymes mediate lipid droplet
growth by relocalizing from the ER to lipid droplets. Dev Cell.
24:384–399. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Petschnigg J, Wolinski H, Kolb D, Zellnig
G, Kurat CF, Natter K and Kohlwein SD: Good fat, essential cellular
requirements for triacylglycerol synthesis to maintain membrane
homeostasis in yeast. J Biol Chem. 284:30981–30993. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
McLelland GL, Lopez-Osias M, Verzijl CRC,
Ellenbroek BD, Oliveira RA, Boon NJ, Dekker M, Van Den Hengel LG,
Ali R, Janssen H, et al: Identification of an alternative
triglyceride biosynthesis pathway. Nature. 621:171–178. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Choudhary V, Ojha N, Golden A and Prinz
WA: A conserved family of proteins facilitates nascent lipid
droplet budding from the ER. J Cell Biol. 211:261–271. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Duelund L, Jensen GV, Hannibal-Bach HK,
Ejsing CS, Pedersen JS, Pakkanen KI and Ipsen JH: Composition,
structure and properties of POPC-triolein mixtures. Evidence of
triglyceride domains in phospholipid bilayers. Biochim Biophys
Acta. 1828:1909–1917. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Khandelia H, Duelund L, Pakkanen KI and
Ipsen JH: Triglyceride blisters in lipid bilayers: Implications for
lipid droplet biogenesis and the mobile lipid signal in cancer cell
membranes. PLoS One. 5:e128112010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Holzhütter HG: Dynamical modelling of
lipid droplet formation suggests a key function of membrane
phospholipids. FFEBS J. 292:206–225. 2025. View Article : Google Scholar
|
|
22
|
Jarc E and Petan T: Lipid droplets and the
management of cellular stress. Yale J Biol Med. 92:435–452.
2019.PubMed/NCBI
|
|
23
|
Ploegh HL: A lipid-based model for the
creation of an escape hatch from the endoplasmic reticulum. Nature.
448:435–438. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chorlay A, Monticelli L, Veríssimo
Ferreira J, Ben M'barek K, Ajjaji D, Wang S, Johnson E, Beck R,
Omrane M, Beller M, et al: Membrane asymmetry imposes
directionality on lipid droplet emergence from the ER. Dev Cell.
50:25–42.e7. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ohsaki Y, Kawai T, Yoshikawa Y, Cheng J,
Jokitalo E and Fujimoto T: PML isoform II plays a critical role in
nuclear lipid droplet formation. J Cell Biol. 212:29–38. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Romanauska A and Köhler A: The inner
nuclear membrane is a metabolically active territory that generates
nuclear lipid droplets. Cell. 174:700–715.e18. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Eichmann TO, Kumari M, Haas JT, Farese RV
Jr, Zimmermann R, Lass A and Zechner R: Studies on the substrate
and stereo/regioselectivity of adipose triglyceride lipase,
hormone-sensitive lipase, and diacylglycerol-O-acyltransferases. J
Biol Chem. 287:41446–41457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Goeritzer M, Schlager S, Radovic B,
Madreiter CT, Rainer S, Thomas G, Lord CC, Sacks J, Brown AL, Vujic
N, et al: Deletion of CGI-58 or adipose triglyceride lipase
differently affects macrophage function and atherosclerosis. J
Lipid Res. 55:2562–2575. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kohlmayr JM, Grabner GF, Nusser A, Höll A,
Manojlović V, Halwachs B, Masser S, Jany-Luig E, Engelke H,
Zimmermann R and Stelzl U: Mutational scanning pinpoints distinct
binding sites of key ATGL regulators in lipolysis. Nat Commun.
15:25162024. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zierler KA, Jaeger D, Pollak NM, Eder S,
Rechberger GN, Radner FPW, Woelkart G, Kolb D, Schmidt A, Kumari M,
et al: Functional cardiac lipolysis in mice critically depends on
comparative gene identification-58. J Biol Chem. 288:9892–9904.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pollak NM, Jaeger D, Kolleritsch S,
Zimmermann R, Zechner R, Lass A and Haemmerle G: The interplay of
protein kinase A and perilipin 5 regulates cardiac lipolysis. J
Biol Chem. 290:1295–1306. 2015. View Article : Google Scholar
|
|
32
|
Zhang X, Xu W, Xu R, Wang Z, Zhang X, Wang
P, Peng K, Li M, Li J, Tan Y, et al: Plin5 bidirectionally
regulates lipid metabolism in oxidative tissues. Oxid Med Cell
Longev. 2022:45949562022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ma SY, Sun KS, Zhang M, Zhou X, Zheng XH,
Tian SY, Liu YS, Chen L, Gao X, Ye J, et al: Disruption of Plin5
degradation by CMA causes lipid homeostasis imbalance in NAFLD.
Liver Int. 40:2427–2438. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kaushik S and Cuervo AM: Degradation of
lipid droplet-associated proteins by chaperone-mediated autophagy
facilitates lipolysis. Nat Cell Biol. 17:759–770. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hu R, Chen B, Wang Z, Qin A, Zhao Z, Lou X
and Tang BZ: Intriguing 'chameleon' fluorescent bioprobes for the
visualization of lipid droplet-lysosome interplay. Biomaterials.
203:43–51. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schulze RJ, Krueger EW, Weller SG, Johnson
KM, Casey CA, Schott MB and McNiven MA: Direct lysosome-based
autophagy of lipid droplets in hepatocytes. Proc Natl Acad Sci USA.
117:32443–32452. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schott MB, Weller SG, Schulze RJ, Krueger
EW, Drizyte-Miller K, Casey CA and McNiven MA: Lipid droplet size
directs lipolysis and lipophagy catabolism in hepatocytes. J Cell
Biol. 218:3320–3335. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bersuker K and Olzmann JA: Establishing
the lipid droplet proteome: Mechanisms of lipid droplet protein
targeting and degradation. Biochim Biophys Acta Mol Cell Biol
Lipids. 1862(10 Pt B): 1166–1177. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Quiroga IY, Pellon-Maison M, Suchanek AL,
Coleman RA and Gonzalez-Baro MR: Glycerol-3-phosphate
acyltransferases 3 and 4 direct glycerolipid synthesis and affect
functionality in activated macrophages. Biochem J. 476:85–99. 2019.
View Article : Google Scholar
|
|
40
|
Harris CA, Haas JT, Streeper RS, Stone SJ,
Kumari M, Yang K, Han X, Brownell N, Gross RW, Zechner R and Farese
RV Jr: DGAT enzymes are required for triacylglycerol synthesis and
lipid droplets in adipocytes. J Lipid Res. 52:657–667. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xu N, Zhang SO, Cole RA, McKinney SA, Guo
F, Haas JT, Bobba S, Farese RV Jr and Mak HY: The FATP1-DGAT2
complex facilitates lipid droplet expansion at the ER-lipid droplet
interface. J Cell Biol. 198:895–911. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Reichert I, Lee JY, Weber L, Fuh MM,
Schlaeger L, Rößler S, Kinast V, Schlienkamp S, Conradi J, Vondran
FWR, et al: The triglyceride-synthesizing enzyme diacylglycerol
acyltransferase 2 modulates the formation of the hepatitis C virus
replication organelle. PLoS Pathog. 20:e10125092024. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sun Y, Zhang L, Lu B, Wen J, Wang M, Zhang
S, Li Q, Shu F, Lu F, Liu N, et al: Hydrogen sulphide reduced the
accumulation of lipid droplets in cardiac tissues of db/db mice via
Hrd1 S-sulfhydration. J Cell Mol Med. 25:9154–9167. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Malis Y, Armoza-Eilat S, Nevo-Yassaf I,
Dukhovny A, Sklan EH and Hirschberg K: Rab1b facilitates lipid
droplet growth by ER-to-lipid droplet targeting of DGAT2. Sci Adv.
10:eade77532024. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li X, Wang N, Gui M, Wang C, Ding Y, Bai
B, Li C, Zhang J and Fang L: Quantitative proteomics reveals PPAR
signaling pathway regulates the cardiomyocyte activity of neonatal
mouse heart. Proteomics. 23:e22003302023. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Griseti E, Bello AA, Bieth E, Sabbagh B,
Iacovoni JS, Bigay J, Laurell H and Čopič A: Molecular mechanisms
of perilipin protein function in lipid droplet metabolism. FEBS
Lett. 598:1170–1198. 2024. View Article : Google Scholar
|
|
47
|
Najt CP, Adhikari S, Heden TD, Cui W,
Gansemer ER, Rauckhorst AJ, Markowski TW, Higgins L, Kerr EW, Boyum
MD, et al: Organelle interactions compartmentalize hepatic fatty
acid trafficking and metabolism. Cell Rep. 42:1124352023.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Akoumi A, Haffar T, Mousterji M, Kiss RS
and Bousette N: Palmitate mediated diacylglycerol accumulation
causes endoplasmic reticulum stress, Plin2 degradation, and cell
death in H9C2 cardiomyoblasts. Exp Cell Res. 354:85–94. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Najt CP, Lwande JS, McIntosh AL,
Senthivinayagam S, Gupta S, Kuhn LA and Atshaves BP: Structural and
functional assessment of perilipin 2 lipid binding domain(s).
Biochemistry. 53:7051–7066. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Deng Y, Zhou C, Mirza AH, Bamigbade AT,
Zhang S, Xu S and Liu P: Rab18 binds PLIN2 and ACSL3 to mediate
lipid droplet dynamics. Biochim Biophys Acta Mol Cell Biol Lipids.
1866:1589232021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Xu D, Li Y, Wu L, Li Y, Zhao D, Yu J,
Huang T, Ferguson C, Parton RG, Yang H and Li P: Rab18 promotes
lipid droplet (LD) growth by tethering the ER to LDs through SNARE
and NRZ interactions. J Cell Biol. 217:975–995. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tseng YY, Sanders MA, Zhang H, Zhou L,
Chou CY and Granneman JG: Structural and functional insights into
ABHD5, a ligand-regulated lipase co-activator. Sci Rep.
12:25652022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ueno M, Suzuki J, Hirose M, Sato S,
Imagawa M, Zenimaru Y, Takahashi S, Ikuyama S, Koizumi T, Konoshita
T, et al: Cardiac overexpression of perilipin 2 induces dynamic
steatosis: Prevention by hormone-sensitive lipase. Am J Physiol
Endocrinol Metab. 313:E699–E709. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang B, Li X, Liu G, Zhang C, Zhang X,
Shen Q, Sun G and Sun X: Peroxiredomin-4 ameliorates
lipotoxicity-induced oxidative stress and apoptosis in diabetic
cardiomyopathy. Biomed Pharmacother. 141:1117802021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Pulinilkunnil T, Kienesberger PC,
Nagendran J, Waller TJ, Young ME, Kershaw EE, Korbutt G, Haemmerle
G, Zechner R and Dyck JR: Myocardial adipose triglyceride lipase
overexpression protects diabetic mice from the development of
lipotoxic cardiomyopathy. Diabetes. 62:1464–1477. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Oluwadare J, Cabodevilla AG, Son NH, Hu Y,
Mullick AE, Verano M, Alemán JO, Ramasamy R and Goldberg IJ:
Blocking lipid uptake pathways does not prevent toxicity in adipose
triglyceride lipase (ATGL) deficiency. J Lipid Res. 63:1002742022.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Haemmerle G, Moustafa T, Woelkart G,
Büttner S, Schmidt A, Van De Weijer T, Hesselink M, Jaeger D,
Kienesberger PC, Zierler K, et al: ATGL-mediated fat catabolism
regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat
Med. 17:1076–1085. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ueno M, Suzuki J, Zenimaru Y, Takahashi S,
Koizumi T, Noriki S, Yamaguchi O, Otsu K, Shen WJ, Kraemer FB and
Miyamori I: Cardiac overexpression of hormone-sensitive lipase
inhibits myocardial steatosis and fibrosis in streptozotocin
diabetic mice. Am J Physiol Endocrinol Metab. 294:E1109–E1118.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Li Z, Thiel K, Thul PJ, Beller M, Kühnlein
RP and Welte MA: Lipid droplets control the maternal histone supply
of drosophila embryos. Curr Biol. 22:2104–2113. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Stephenson RA, Thomalla JM, Chen L,
Kolkhof P, White RP, Beller M and Welte MA: Sequestration to lipid
droplets promotes histone availability by preventing turnover of
excess histones. Development. 148:dev1993812021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zehmer JK, Huang Y, Peng G, Pu J, Anderson
RG and Liu P: A role for lipid droplets in inter-membrane lipid
traffic. Proteomics. 9:914–921. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Pham TK, Nguyen THT, Yi JM, Kim GS, Yun
HR, Kim HK and Won JC: Evogliptin, a DPP-4 inhibitor, prevents
diabetic cardiomyopathy by alleviating cardiac lipotoxicity in
db/db mice. Exp Mol Med. 55:767–778. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Roe ND, Handzlik MK, Li T and Tian R: The
role of diacylglycerol acyltransferase (DGAT) 1 and 2 in cardiac
metabolism and function. Sci Rep. 8:49832018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Pollak NM, Schweiger M, Jaeger D, Kolb D,
Kumari M, Schreiber R, Kolleritsch S, Markolin P, Grabner GF, Heier
C, et al: Cardiac-specific overexpression of perilipin 5 provokes
severe cardiac steatosis via the formation of a lipolytic barrier.
J Lipid Res. 54:1092–1102. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Inoue T, Kobayashi K, Inoguchi T, Sonoda
N, Maeda Y, Hirata E, Fujimura Y, Miura D, Hirano K and Takayanagi
R: Downregulation of adipose triglyceride lipase in the heart
aggravates diabetic cardiomyopathy in db/db mice. Biochem Biophys
Res Commun. 438:224–229. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Magré J and Prieur X: Seipin deficiency as
a model of severe adipocyte dysfunction: Lessons from rodent models
and teaching for human disease. Int J Mol Sci. 23:7402022.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Guyard V, Monteiro-Cardoso VF, Omrane M,
Sauvanet C, Houcine A, Boulogne C, Ben Mbarek K, Vitale N, Faklaris
O, El Khallouki N, et al: ORP5 and ORP8 orchestrate lipid droplet
biogenesis and maintenance at ER-mitochondria contact sites. J Cell
Biol. 221:e2021121072022. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Bai B, Yang W, Fu Y, Foon HL, Tay WT, Yang
K, Luo C, Gunaratne J, Lee P, Zile MR, et al: Seipin knockout mice
develop heart failure with preserved ejection fraction. JACC Basic
Transl Sci. 4:924–937. 2019. View Article : Google Scholar
|
|
69
|
Benador IY, Veliova M, Mahdaviani K,
Petcherski A, Wikstrom JD, Assali EA, Acín-Pérez R, Shum M,
Oliveira MF, Cinti S, et al: Mitochondria bound to lipid droplets
have unique bioenergetics, composition, and dynamics that support
lipid droplet expansion. Cell Metab. 27:869–885.e6. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kien B, Kolleritsch S, Kunowska N, Heier
C, Chalhoub G, Tilp A, Wolinski H, Stelzl U and Haemmerle G: Lipid
droplet-mitochondria coupling via perilipin 5 augments respiratory
capacity but is dispensable for FA oxidation. J Lipid Res.
63:1001722022. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Talari NK, Mattam U, Meher NK, Paripati
AK, Mahadev K, Krishnamoorthy T and Sepuri NBV: Lipid-droplet
associated mitochondria promote fatty-acid oxidation through a
distinct bioenergetic pattern in male wistar rats. Nat Commun.
14:7662023. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Yang P, Li J, Zhang T, Ren Y, Zhang Q, Liu
R, Li H, Hua J, Wang WA, Wang J and Zhou H: Ionizing
radiation-induced mitophagy promotes ferroptosis by increasing
intracellular free fatty acids. Cell Death Differ. 30:2432–2445.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wu Y, Chen K, Li L, Hao Z, Wang T, Liu Y,
Xing G, Liu Z, Li H, Yuan H, et al: Plin2-mediated lipid droplet
mobilization accelerates exit from pluripotency by lipidomic
remodeling and histone acetylation. Cell Death Differ.
29:2316–2331. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Moussaieff A, Rouleau M, Kitsberg D, Cohen
M, Levy G, Barasch D, Nemirovski A, Shen-Orr S, Laevsky I, Amit M,
et al: Glycolysis-mediated changes in acetyl-CoA and histone
acetylation control the early differentiation of embryonic stem
cells. Cell Metab. 21:392–402. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Meng Y, Guo D, Lin L, Zhao H, Xu W, Luo S,
Jiang X, Li S, He X, Zhu R, et al: Glycolytic enzyme PFKL governs
lipolysis by promoting lipid droplet-mitochondria tethering to
enhance β-oxidation and tumor cell proliferation. Nat Metab.
6:1092–1107. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Wang LY and Chen C: Energy metabolism
homeostasis in cardiovascular diseases. J Geriatr Cardiol.
18:1044–1057. 2021.
|
|
77
|
Gallardo-Montejano VI, Saxena G, Kusminski
CM, Yang C, McAfee JL, Hahner L, Hoch K, Dubinsky W, Narkar VA and
Bickel PE: Nuclear perilipin 5 integrates lipid droplet lipolysis
with PGC-1α/SIRT1-dependent transcriptional regulation of
mitochondrial function. Nat Commun. 7:127232016. View Article : Google Scholar
|
|
78
|
Wang Q and Ren J: mTOR-independent
autophagy inducer trehalose rescues against insulin
resistance-induced myocardial contractile anomalies: Role of p38
MAPK and Foxo1. Pharmacol Res. 111:357–373. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhang L, Zhang H, Xie X, Tie R, Shang X,
Zhao Q, Xu J, Jin L, Zhang J and Ye P: Empagliflozin ameliorates
diabetic cardiomyopathy via regulated branched-chain amino acid
metabolism and mTOR/p-ULK1 signaling pathway-mediated autophagy.
Diabetol Metab Syndr. 15:932023. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hariharan N, Maejima Y, Nakae J, Paik J,
Depinho RA and Sadoshima J: Deacetylation of FoxO by Sirt1 plays an
essential role in mediating starvation-induced autophagy in cardiac
myocytes. Circ Res. 107:1470–1482. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Ke J, Pan J, Lin H, Huang S, Zhang J, Wang
C, Chang ACY and Gu J: Targeting Rab7-Rilp mediated microlipophagy
alleviates lipid toxicity in diabetic cardiomyopathy. Adv Sci
(Weinh). 11:e24016762024. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Fan Y, Yan Z, Li T, Li A, Fan X, Qi Z and
Zhang J: Primordial drivers of diabetes heart disease:
Comprehensive insights into insulin resistance. Diabetes Metab J.
48:19–36. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Walejko JM, Christopher BA, Crown SB,
Zhang GF, Pickar-Oliver A, Yoneshiro T, Foster MW, Page S, Van
Vliet S, Ilkayeva O, et al: Branched-chain α-ketoacids are
preferentially reaminated and activate protein synthesis in the
heart. Nat Commun. 12:16802021. View Article : Google Scholar
|
|
84
|
Yue M, Hu B, Li J, Chen R, Yuan Z, Xiao H,
Chang H, Jiu Y, Cai K and Ding B: Coronaviral ORF6 protein mediates
inter-organelle contacts and modulates host cell lipid flux for
virus production. EMBO J. 42:e1125422023. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Eke C, Babcock S, Gaston G, Elizondo G,
Chung H, Asal A, Chatfield KC, Sparagna GC, DeBarber AE, Packwood
W, et al: Cardiomyopathy in a c.1528G>C hadha mouse is
associated with cardiac tissue lipotoxicity and altered cardiolipin
species. J Lipid Res. 66:1007922025. View Article : Google Scholar
|
|
86
|
Chi YJ, Bai ZY, Feng GL, Lai XH and Song
YF: ER-mitochondria contact sites regulate hepatic lipogenesis via
Ip3r-Grp75-vdac complex recruiting Seipin. Cell Commun Signal.
22:4642024. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Lv Y, Cheng L and Peng F: Compositions and
functions of mitochondria-associated endoplasmic reticulum
membranes and their contribution to cardioprotection by exercise
preconditioning. Front Physiol. 13:9104522022. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Schlaepfer IR and Joshi M: CPT1A-mediated
fat oxidation, mechanisms, and therapeutic potential.
Endocrinology. 161:bqz0462020. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Zhou PU, Li M, Han XW, Bi YH, Zhang WG, Wu
ZY and Wu G: Perilipin 5 deficiency promotes atherosclerosis
progression through accelerating inflammation, apoptosis, and
oxidative stress. J Cell Biochem. 120:19107–19123. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Bragado P, Armesilla A, Silva A and Porras
A: Apoptosis by cisplatin requires p53 mediated p38alpha MAPK
activation through ROS generation. Apoptosis. 12:1733–1742. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Lange M, Wölk M, Li VW, Doubravsky CE,
Hendricks JM, Kato S, Otoki Y, Styler B, Johnson SL, Harris CA, et
al: FSP1-mediated lipid droplet quality control prevents neutral
lipid peroxidation and ferroptosis. Nat Cell Biol. 27:1902–1913.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Trivedi PC, Bartlett JJ, Perez LJ, Brunt
KR, Legare JF, Hassan A, Kienesberger PC and Pulinilkunnil T:
Glucolipotoxicity diminishes cardiomyocyte TFEB and inhibits
lysosomal autophagy during obesity and diabetes. Biochim Biophys
Acta. 1861(12 Pt A): 1893–1910. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Song X, Fan C, Wei C, Yu W, Tang J, Ma F,
Chen Y and Wu B: Mitochondria fission accentuates oxidative stress
in hyperglycemia-induced H9c2 cardiomyoblasts in vitro by
regulating fatty acid oxidation. Cell Biol Int. 48:1378–1391. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Saddik M, Gamble J, Witters LA and
Lopaschuk GD: Acetyl-CoA carboxylase regulation of fatty acid
oxidation in the heart. J Biol Chem. 268:25836–25845. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Ghosh S, Wicks SE, Vandanmagsar B, Mendoza
TM, Bayless DS, Salbaum JM, Dearth SP, Campagna SR, Mynatt RL and
Noland RC: Extensive metabolic remodeling after limiting
mitochondrial lipid burden is consistent with an improved metabolic
health profile. J Biol Chem. 294:12313–12327. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kolwicz SC Jr, Olson DP, Marney LC,
Garcia-Menendez L, Synovec RE and Tian R: Cardiac-specific deletion
of acetyl CoA carboxylase 2 prevents metabolic remodeling during
pressure-overload hypertrophy. Circ Res. 111:728–738. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Lopaschuk GD and Ussher JR: Evolving
concepts of myocardial energy metabolism: More than just fats and
carbohydrates. Circ Res. 119:1173–1176. 2016. View Article : Google Scholar
|
|
98
|
Wang H, Wang J, Cui H, Fan C, Xue Y, Liu
H, Li H, Li J, Li H, Sun Y, et al: Inhibition of fatty acid uptake
by TGR5 prevents diabetic cardiomyopathy. Nat Metab. 6:1161–1177.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Glatz JFC, Heather LC and Luiken JJFP:
CD36 as a gatekeeper of myocardial lipid metabolism and therapeutic
target for metabolic disease. Physiol Rev. 104:727–764. 2024.
View Article : Google Scholar
|
|
100
|
Chathoth S, Ismail MH, Alghamdi HM,
Zakaria HM, Hassan KA, Alshomimi S, Vatte C, Cyrus C, Alsaif HS,
Mostafa A, et al: Insulin resistance induced by de novo
pathway-generated C16-ceramide is associated with type 2 diabetes
in an obese population. Lipids Health Dis. 21:242022. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Law BA, Liao X, Moore KS, Southard A,
Roddy P, Ji R, Szulc Z, Bielawska A, Schulze PC and Cowart LA:
Lipotoxic very-long-chain ceramides cause mitochondrial
dysfunction, oxidative stress, and cell death in cardiomyocytes.
FASEB J. 32:1403–1416. 2018. View Article : Google Scholar
|
|
102
|
Jeong SJ, Lee MN and Oh GT: The role of
macrophage lipophagy in reverse cholesterol transport. Endocrinol
Metab (Seoul). 32:41–46. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Cho KY, Miyoshi H, Nakamura A, Greenberg
AS and Atsumi T: Lipid droplet protein PLIN1 regulates inflammatory
polarity in human macrophages and is involved in atherosclerotic
plaque development by promoting stable lipid storage. J Atheroscler
Thromb. 30:170–181. 2023. View Article : Google Scholar :
|
|
104
|
Robichaud S, Fairman G, Vijithakumar V,
Mak E, Cook DP, Pelletier AR, Huard S, Vanderhyden BC, Figeys D,
Lavallée-Adam M, et al: Identification of novel lipid droplet
factors that regulate lipophagy and cholesterol efflux in
macrophage foam cells. Autophagy. 17:3671–3689. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Liu G, Yan D, Yang L, Sun Y, Zhan L, Lu L,
Jin Z, Zhang C, Long P, Chen J and Yuan Q: The effect of miR-471-3p
on macrophage polarization in the development of diabetic
cardiomyopathy. Life Sci. 268:1189892021. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Bose D, Banerjee S, Chatterjee N, Das S,
Saha M and Saha KD: Inhibition of TGF-β induced lipid droplets
switches M2 macrophages to M1 phenotype. Toxicol In Vitro.
58:207–214. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Kimmel AR and Sztalryd C: The perilipins:
Major cytosolic lipid droplet-associated proteins and their roles
in cellular lipid storage, mobilization, and systemic homeostasis.
Annu Rev Nutr. 36:471–509. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Yang T, Chen J, Gao L, Huang Y, Liao G and
Cao Y: Induction of lipid droplets in THP-1 macrophages by
multi-walled carbon nanotubes in a diameter-dependent manner: A
transcriptomic study. Toxicol Lett. 332:65–73. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Ni Y, Zhuge F, Ni L, Nagata N, Yamashita
T, Mukaida N, Kaneko S, Ota T and Nagashimada M: CX3CL1/CX3CR1
interaction protects against lipotoxicity-induced nonalcoholic
steatohepatitis by regulating macrophage migration and M1/M2
status. Metabolism. 136:1552722022. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Keeter WC, Ma S, Stahr N, Moriarty AK and
Galkina EV: Atherosclerosis and multi-organ-associated pathologies.
Semin Immunopathol. 44:363–374. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Goo YH, Plakkal Ayyappan J, Cheeran FD,
Bangru S, Saha PK, Baar P, Schulz S, Lydic TA, Spengler B, Wagner
AH, et al: Lipid droplet-associated hydrolase mobilizes stores of
liver X receptor sterol ligands and protects against
atherosclerosis. Nat Commun. 15:65402024. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Yang L, Li T and Zha L: Foxc2 alleviates
ox-LDL-induced lipid accumulation, inflammation, and apoptosis of
macrophage via regulating the expression of Angptl2. Inflammation.
43:1397–1410. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Tabata M, Kadomatsu T, Fukuhara S, Miyata
K, Ito Y, Endo M, Urano T, Zhu HJ, Tsukano H, Tazume H, et al:
Angiopoietin-like protein 2 promotes chronic adipose tissue
inflammation and obesity-related systemic insulin resistance. Cell
Metab. 10:178–188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Wang T, Cheng Z, Zhao R, Cheng J, Ren H,
Zhang P, Liu P, Hao Q, Zhang Q, Yu X, et al: Sirt6 enhances
macrophage lipophagy and improves lipid metabolism disorder by
regulating the Wnt1/β-catenin pathway in atherosclerosis. Lipids
Health Dis. 22:1562023. View Article : Google Scholar
|
|
115
|
Bao Y, Zhu L, Wang Y, Liu J, Liu Z, Li Z,
Zhou A and Wu H: Gualou-Xiebai herb pair and its active ingredients
act against atherosclerosis by suppressing VSMC-derived foam cell
formation via regulating P2RY12-mediated lipophagy. Phytomedicine.
128:1553412024. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Amrute JM, Luo X, Penna V, Yang S,
Yamawaki T, Hayat S, Bredemeyer A, Jung IH, Kadyrov FF, Heo GS, et
al: Targeting immune-fibroblast cell communication in heart
failure. Nature. 635:423–433. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Du L, Wang X, Guo Y, Tao T, Wu H, Xu X,
Zhang B, Chen T, Xu Q and Guo X: Altered lipid metabolism promoting
cardiac fibrosis is mediated by CD34+ cell-derived FABP4+
fibroblasts. Exp Mol Med. 56:1869–1886. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Liu JJ, Shentu LM, Ma N, Wang LY, Zhang
GM, Sun Y, Wang Y, Li J and Mu YL: Inhibition of NF-κB and
Wnt/β-catenin/GSK3β signaling pathways ameliorates cardiomyocyte
hypertrophy and fibrosis in streptozotocin (STZ)-induced type 1
diabetic rats. Curr Med Sci. 40:35–47. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Soppert J, Lehrke M, Marx N, Jankowski J
and Noels H: Lipoproteins and lipids in cardiovascular disease:
From mechanistic insights to therapeutic targeting. Adv Drug Deliv
Rev. 159:4–33. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Chen H, Xu J, Huang Q, Zhao J, Hu Y, Wang
C, Zhong W, Zhang Y, Fan C, Chang J and Liu X: Kif23 promotes
myocardial fibrosis by suppressing Ces1d-dependent lipid
metabolism. Hypertension. 83:116–129. 2026. View Article : Google Scholar
|
|
121
|
Berezin AE, Berezin AA and Lichtenauer M:
Myokines and heart failure: Challenging role in adverse cardiac
remodeling, myopathy, and clinical outcomes. Dis Markers.
2021:66446312021. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Men L, Hui W, Guan X, Song T, Wang X,
Zhang S and Chen X: Cardiac transcriptome analysis reveals Nr4a1
mediated glucose metabolism dysregulation in response to high-fat
diet. Genes (Basel). 11:7202020. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Li Y, Torp MK, Norheim F, Khanal P, Kimmel
AR, Stensløkken KO, Vaage J and Dalen KT: Isolated Plin5-deficient
cardiomyocytes store less lipid droplets than normal, but without
increased sensitivity to hypoxia. Biochim Biophys Acta Mol Cell
Biol Lipids. 1866:1588732021. View Article : Google Scholar
|
|
124
|
Potthoff MJ, Inagaki T, Satapati S, Ding
X, He T, Goetz R, Mohammadi M, Finck BN, Mangelsdorf DJ, Kliewer SA
and Burgess SC: FGF21 induces PGC-1alpha and regulates carbohydrate
and fatty acid metabolism during the adaptive starvation response.
Proc Natl Acad Sci USA. 106:10853–10858. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Chen C, Meng Z, Zheng Y, Hu B and Shen E:
Fibroblast growth factor 21 inhibition aggravates cardiac
dysfunction in diabetic cardiomyopathy by improving lipid
accumulation. Exp Ther Med. 15:75–84. 2018.PubMed/NCBI
|
|
126
|
Zheng H, Zhang X, Li C, Wang D, Shen Y, Lu
J, Zhao L, Li X and Gao H: BCAA mediated microbiota-liver-heart
crosstalk regulates diabetic cardiomyopathy via FGF21. Microbiome.
12:1572024. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Wang X, Zhu XX, Jiao SY, Qi D, Yu BQ, Xie
GM, Liu Y, Song YT, Xu Q, Xu QB, et al: Cardiomyocyte peroxisome
proliferator-activated receptor α is essential for energy
metabolism and extracellular matrix homeostasis during pressure
overload-induced cardiac remodeling. Acta Pharmacol Sin.
43:1231–1242. 2022. View Article : Google Scholar
|
|
128
|
Chen Q, Chen L, Jian J, Li J and Zhang X:
The mechanism behind BAF60c in myocardial metabolism in rats with
heart failure is through the PGC1alpha-PPARalpha-mTOR signaling
pathway. Biochem Cell Biol. 100:93–103. 2022. View Article : Google Scholar
|
|
129
|
Ogawa T, Kouzu H, Osanami A, Tatekoshi Y,
Sato T, Kuno A, Fujita Y, Ino S, Shimizu M, Toda Y, et al:
Downregulation of extramitochondrial BCKDH and its uncoupling from
AMP deaminase in type 2 diabetic OLETF rat hearts. Physiol Rep.
11:e156082023. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Kienesberger PC, Pulinilkunnil T,
Nagendran J and Dyck JRB: Myocardial triacylglycerol metabolism. J
Mol Cell Cardiol. 55:101–110. 2013. View Article : Google Scholar
|
|
131
|
O'Brien PD, Guo K, Eid SA, Rumora AE,
Hinder LM, Hayes JM, Mendelson FE, Hur J and Feldman EL: Integrated
lipidomic and transcriptomic analyses identify altered nerve
triglycerides in mouse models of prediabetes and type 2 diabetes.
Dis Models Mech. 13:dmm0421012020. View Article : Google Scholar
|
|
132
|
Revuelta-López E, Cal R, Julve J, Rull A,
Martínez-Bujidos M, Perez-Cuellar M, Ordoñez-Llanos J, Badimon L,
Sanchez-Quesada JL and Llorente-Cortés V: Hypoxia worsens the
impact of intracellular triglyceride accumulation promoted by
electronegative low-density lipoprotein in cardiomyocytes by
impairing perilipin 5 upregulation. Int J Biochem Cell Biol.
65:257–267. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Wu Q, Zhao M, He X, Xue R, Li D, Yu X,
Wang S and Zang W: Acetylcholine reduces palmitate-induced
cardiomyocyte apoptosis by promoting lipid droplet lipolysis and
perilipin 5-mediated lipid droplet-mitochondria interaction. Cell
Cycle. 20:1890–1906. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Listenberger LL, Han X, Lewis SE, Cases S,
Farese RV Jr, Ory DS and Schaffer JE: Triglyceride accumulation
protects against fatty acid-induced lipotoxicity. Proc Natl Acad
Sci USA. 100:3077–3082. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Tan Y, Huang Z, Jin Y, Wang J, Fan H, Liu
Y, Zhang L, Wu Y, Liu P, Li T, et al: Lipid droplets sequester
palmitic acid to disrupt endothelial ciliation and exacerbate
atherosclerosis in male mice. Nat Commun. 15:82732024. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Zheng XY, Sun CC, Liu Q, Lu XY, Fu LL,
Liang G, Zhang XH and Chen GZ: Compound LM9, a novel MyD88
inhibitor, efficiently mitigates inflammatory responses and
fibrosis in obesity-induced cardiomyopathy. Acta Pharmacol Sin.
41:1093–1101. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Moore KJ, Sheedy FJ and Fisher EA:
Macrophages in atherosclerosis: A dynamic balance. Nat Rev Immunol.
13:709–721. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Liu L, Trent CM, Fang X, Son NH, Jiang H,
Blaner WS, Hu Y, Yin YX, Farese RV Jr, Homma S, et al:
Cardiomyocyte-specific loss of diacylglycerol acyltransferase 1
(DGAT1) reproduces the abnormalities in lipids found in severe
heart failure. J Biol Chem. 289:29881–29891. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Liu Q, Siloto RM, Lehner R, Stone SJ and
Weselake RJ: Acyl-CoA:diacylglycerol acyltransferase: Molecular
biology, biochemistry and biotechnology. Prog Lipid Res.
51:350–377. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Guo D, Zhang M, Qi B, Peng T, Liu M, Li Z,
Fu F, Guo Y, Li C, Wang Y, et al: Lipid overload-induced RTN3
activation leads to cardiac dysfunction by promoting lipid droplet
biogenesis. Cell Death Differ. 31:292–308. 2024. View Article : Google Scholar :
|
|
141
|
Tsushima K, Bugger H, Wende AR, Soto J,
Jenson GA, Tor AR, McGlauflin R, Kenny HC, Zhang Y, Souvenir R, et
al: Mitochondrial reactive oxygen species in lipotoxic hearts
induce post-translational modifications of AKAP121, DRP1, and OPA1
that promote mitochondrial fission. Circ Res. 122:58–73. 2018.
View Article : Google Scholar
|
|
142
|
Demine S, Renard P and Arnould T:
Mitochondrial uncoupling: A key controller of biological processes
in physiology and diseases. Cells. 8:7952019. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Jelenik T, Flögel U, Álvarez-Hernández E,
Scheiber D, Zweck E, Ding Z, Rothe M, Mastrototaro L, Kohlhaas V,
Kotzka J, et al: Insulin resistance and vulnerability to cardiac
ischemia. Diabetes. 67:2695–2702. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Wang Q, Chi J, Wang C, Yuan Y, Tian R,
Yang Y and Chen X: CTRP3 attenuates myocardial lipotoxicity via
suppression of lipid accumulation, inflammation, apoptosis, and
mitochondrial oxidative stress. Front Cardiovasc Med.
12:15759292025. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Wu M, Tan J, Cao Z, Cai Y, Huang Z, Chen
Z, He W, Liu X, Jiang Y, Gao Q, et al: Sirt5 improves
cardiomyocytes fatty acid metabolism and ameliorates cardiac
lipotoxicity in diabetic cardiomyopathy via CPT2 de-succinylation.
Redox Biol. 73:1031842024. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Guo R, Wu Z, Jiang J, Liu C, Wu B, Li X,
Li T, Mo H, He S, Li S, et al: New mechanism of lipotoxicity in
diabetic cardiomyopathy: Deficiency of Endogenous H 2 S Production
and ER stress. Mech Ageing Dev. 162:46–52. 2017. View Article : Google Scholar
|
|
147
|
Yu M, Du H, Wang B, Chen J, Lu F, Peng S,
Sun Y, Liu N, Sun X, Shiyun D, et al: Exogenous H2S Induces Hrd1
S-sulfhydration and prevents CD36 translocation via VAMP3
ubiquitylation in diabetic hearts. Aging Dis. 11:286–300. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Chen X, Ruiz-Velasco A, Zou Z, Hille SS,
Ross C, Fonseka O, Gare SR, Alatawi NHO, Raja R, Zhang J, et al:
PAK3 exacerbates cardiac lipotoxicity via SREBP1c in obesity
cardiomyopathy. Diabetes. 73:1805–1820. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Huang TS, Wu T, Fu XL, Ren HL, He XD,
Zheng DH, Tan J, Shen CH, Xiong SJ, Qian J, et al: SREBP1 induction
mediates long-term statins therapy related myocardial lipid
peroxidation and lipid deposition in TIIDM mice. Redox Biol.
78:1034122024. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Marfella R, Di Filippo C, Portoghese M,
Barbieri M, Ferraraccio F, Siniscalchi M, Cacciapuoti F, Rossi F,
D'Amico M and Paolisso G: Myocardial lipid accumulation in patients
with pressure-overloaded heart and metabolic syndrome. J Lipid Res.
50:2314–2323. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Wahli W and Michalik L: PPARs at the
crossroads of lipid signaling and inflammation. Trends Endocrinol
Metab. 23:351–363. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
O'Brien SL, Tripp E, Barki N,
Blondel-Tepaz E, Smith G, Boufersaoui A, Roberts J, Pike JA,
Correia J, Miljus T, et al: Intracrine FFA4 signaling controls
lipolysis at lipid droplets. Nat Chem Biol. 22:109–119. 2026.
View Article : Google Scholar
|
|
153
|
O'Connell TD, Block RC, Huang SP and
Shearer GC: ω3-polyunsaturated fatty acids for heart failure:
Effects of dose on efficacy and novel signaling through free fatty
acid receptor 4. J Mol Cell Cardiol. 103:74–92. 2017. View Article : Google Scholar
|
|
154
|
Moreira LS, Piva B, Gentile LB,
Mesquita-Santos FP, D'Avila H, Maya-Monteiro CM, Bozza PT,
Bandeira-Melo C and Diaz BL: Cytosolic phospholipase A2-driven PGE2
synthesis within unsaturated fatty acids-induced lipid bodies of
epithelial cells. Biochim Biophys Acta. 1791:156–165. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Jarc E and Petan T: A twist of FATe: Lipid
droplets and inflammatory lipid mediators. Biochimie. 169:69–87.
2020. View Article : Google Scholar
|
|
156
|
Van Dierendonck XAMH, Vrieling F,
Smeehuijzen L, Deng L, Boogaard JP, Croes CA, Temmerman L, Wetzels
S, Biessen E, Kersten S and Stienstra R: Triglyceride breakdown
from lipid droplets regulates the inflammatory response in
macrophages. Proc Natl Acad Sci USA. 119:e21147391192022.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
van Dierendonck XAMH, De La Rosa Rodriguez
MA, Georgiadi A, Mattijssen F, Dijk W, Van Weeghel M, Singh R,
Borst JW, Stienstra R and Kersten S: HILPDA uncouples lipid droplet
accumulation in adipose tissue macrophages from inflammation and
metabolic dysregulation. Cell Rep. 30:1811–1822.e6. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Yamamoto T, Okuno M, Kuwano K and Ogura Y:
Mycoplasma pneumoniae drives macrophage lipid uptake via
GlpD-mediated oxidation, facilitating foam cell formation. Int J
Med Microbiol. 318:1516462025. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Singh V, Jamwal S, Jain R, Verma P,
Gokhale R and Rao KV: Mycobacterium tuberculosis-driven targeted
recalibration of macrophage lipid homeostasis promotes the foamy
phenotype. Cell Host Microbe. 12:669–681. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Nguyen TB, Louie SM, Daniele JR, Tran Q,
Dillin A, Zoncu R, Nomura DK and Olzmann JA: DGAT1-Dependent lipid
droplet biogenesis protects mitochondrial function during
starvation-induced autophagy. Dev Cell. 42:9–21.e5. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Wu X, Geng F, Cheng X, Guo Q, Zhong Y,
Cloughesy TF, Yong WH, Chakravarti A and Guo D: Lipid droplets
maintain energy homeostasis and glioblastoma growth via autophagic
release of stored fatty acids. Iscience. 23:1015692020. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
de Brito OM and Scorrano L: Mitofusin 2
tethers endoplasmic reticulum to mitochondria. Nature. 456:605–610.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Boutant M, Kulkarni SS, Joffraud M,
Ratajczak J, Valera-Alberni M, Combe R, Zorzano A and Cantó C: Mfn2
is critical for brown adipose tissue thermogenic function. EMBO J.
36:1543–1558. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Bórquez JC, Díaz-Castro F, La Fuente FP,
Espinoza K, Figueroa AM, Martínez-Ruíz I, Hernández V,
López-Soldado I, Ventura R, Domingo JC, et al: Mitofusin-2 induced
by exercise modifies lipid droplet-mitochondria communication,
promoting fatty acid oxidation in male mice with NAFLD. Metabolism.
152:1557652024. View Article : Google Scholar
|
|
165
|
Wang LJ, Lai XH, Luo Z, Feng GL and Song
YF: Diallyl disulfide alleviates hepatic steatosis by the
conservative mechanism from fish to tetrapod: Augment
Mfn2/Atgl-Mediated lipid droplet-mitochondria coupling. Redox Biol.
77:1033952024. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Hu L, Tang D, Qi B, Guo D, Wang Y, Geng J,
Zhang X, Song L, Chang P, Chen W, et al: Mfn2/Hsc70 complex
mediates the formation of mitochondria-lipid droplets membrane
contact and regulates myocardial lipid metabolism. Adv Sci (Weinh).
11:e23077492024. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Salo VT, Li S, Vihinen H, Hölttä-Vuori M,
Szkalisity A, Horvath P, Belevich I, Peränen J, Thiele C,
Somerharju P, et al: Seipin facilitates triglyceride flow to lipid
droplet and counteracts droplet ripening via endoplasmic reticulum
contact. Dev Cell. 50:478–493.e9. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Wang H, Becuwe M, Housden BE, Chitraju C,
Porras AJ, Graham MM, Liu XN, Thiam AR, Savage DB, Agarwal AK, et
al: Seipin is required for converting nascent to mature lipid
droplets. Elife. 5:e165822016. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Cottier S and Schneiter R: Lipid droplets
form a network interconnected by the endoplasmic reticulum through
which their proteins equilibrate. J Cell Sci. 135:jcs2588192022.
View Article : Google Scholar
|
|
170
|
Miner GE, So CM, Edwards W, Ragusa JV,
Wine JT, Wong Gutierrez D, Airola MV, Herring LE, Coleman RA, Klett
EL and Cohen S: PLIN5 interacts with FATP4 at membrane contact
sites to promote lipid droplet-to-mitochondria fatty acid
transport. Dev Cell. 58:1250–1265.e6. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Herrmann T, Buchkremer F, Gosch I, Hall
AM, Bernlohr DA and Stremmel W: Mouse fatty acid transport protein
4 (FATP4): Characterization of the gene and functional assessment
as a very long chain acyl-CoA synthetase. Gene. 270:31–40. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Lemmer IL, Willemsen N, Hilal N and
Bartelt A: A guide to understanding endoplasmic reticulum stress in
metabolic disorders. Mol Metab. 47:1011692021. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Liu ZW, Zhu HT, Chen KL, Dong X, Wei J,
Qiu C and Xue JH: Protein kinase RNA-like endoplasmic reticulum
kinase (PERK) signaling pathway plays a major role in reactive
oxygen species (ROS)-mediated endoplasmic reticulum stress-induced
apoptosis in diabetic cardiomyopathy. Cardiovasc Diabetol.
12:1582013. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Vanhoutte D, Schips TG, Vo A, Grimes KM,
Baldwin TA, Brody MJ, Accornero F, Sargent MA and Molkentin JD:
Thbs1 induces lethal cardiac atrophy through PERK-ATF4 regulated
autophagy. Nat Commun. 12:39282021. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Munteanu CVA, Chirițoiu GN, Chirițoiu M,
Ghenea S, Petrescu AJ and Petrescu ŞM: Affinity proteomics and
deglycoproteomics uncover novel EDEM2 endogenous substrates and an
integrative ERAD network. Mol Cell Proteomics. 20:1001252021.
View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Fonseka O, Raja R, Ross C, Gare SR, Zhang
J, Hille SS, King K, Ruiz-Velasco A, Kaur N, Chen X, et al:
XBP1s-EDEM2 prevents the onset and development of HFpEF by
ameliorating cardiac lipotoxicity. Circulation. 151:1583–1605.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Son NH, Yu S, Tuinei J, Arai K, Hamai H,
Homma S, Shulman GI, Abel ED and Goldberg IJ: PPARγ-induced
cardiolipotoxicity in mice is ameliorated by PPARα deficiency
despite increases in fatty acid oxidation. J Clin Invest.
120:3443–3454. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Lee H, Horbath A, Kondiparthi L, Meena JK,
Lei G, Dasgupta S, Liu X, Zhuang L, Koppula P, Li M, et al: Cell
cycle arrest induces lipid droplet formation and confers
ferroptosis resistance. Nat Commun. 15:792024. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Long M and McWilliams TG: Lipid droplets
promote efficient mitophagy. Autophagy. 19:724–725. 2023.
View Article : Google Scholar :
|
|
180
|
Zhang LW, Han YQ, Yang Y, Li YJ, Ma YX and
Ming SL: RAB18 deficiency disrupts lipid metabolism and autophagy
in mice. Biochem Biophys Res Commun. 760:1516732025. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Tran KV, Brown EL, DeSouza T, Jespersen
NZ, Nandrup-Bus C, Yang Q, Yang Z, Desai A, Min SY, Rojas-Rodriguez
R, et al: Human thermogenic adipocyte regulation by the long
noncoding RNA LINC00473. Nat Metab. 2:397–412. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Han L, Huang D, Wu S, Liu S, Wang C, Sheng
Y, Lu X, Broxmeyer HE, Wan J and Yang L: Lipid droplet-associated
lncRNA LIPTER preserves cardiac lipid metabolism. Nat Cell Biol.
25:1033–1046. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Nakamura K, Miyoshi T, Yoshida M, Akagi S,
Saito Y, Ejiri K, Matsuo N, Ichikawa K, Iwasaki K, Naito T, et al:
Pathophysiology and treatment of diabetic cardiomyopathy and heart
failure in patients with diabetes mellitus. Int J Mol Sci.
23:35872022. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Suzuki J, Shen WJ, Nelson BD, Selwood SP,
Murphy GM Jr, Kanefara H, Takahashi S, Oida K, Miyamori I and
Kraemer FB: Cardiac gene expression profile and lipid accumulation
in response to starvation. Am J Physiol Endocrinol Metab.
283:E94–E102. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Fillmore N, Hou V, Sun J, Springer D and
Murphy E: Cardiac specific knock-down of peroxisome proliferator
activated receptor α prevents fasting-induced cardiac lipid
accumulation and reduces perilipin 2. PLoS One. 17:e02650072022.
View Article : Google Scholar
|
|
186
|
Wang J, Fang N, Xiong J, Du Y, Cao Y and
Ji WK: An ESCRT-dependent step in fatty acid transfer from lipid
droplets to mitochondria through VPS13D-TSG101 interactions. Nat
Commun. 12:12522021. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Mardani I, Tomas Dalen K, Drevinge C,
Miljanovic A, Ståhlman M, Klevstig M, Scharin Täng M, Fogelstrand
P, Levin M, Ekstrand M, et al: Plin2-deficiency reduces lipophagy
and results in increased lipid accumulation in the heart. Sci Rep.
9:69092019. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Wang C, Yuan Y, Wu J, Zhao Y, Gao X, Chen
Y, Sun C, Xiao L, Zheng P, Hu P, et al: Plin5 deficiency
exacerbates pressure overload-induced cardiac hypertrophy and heart
failure by enhancing myocardial fatty acid oxidation and oxidative
stress. Free Radic Biol Med. 141:372–382. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Ouyang Q, Chen Q, Ke S, Ding L, Yang X,
Rong P, Feng W, Cao Y, Wang Q, Li M, et al: Rab8a as a
mitochondrial receptor for lipid droplets in skeletal muscle. Dev
Cell. 58:289–305.e6. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Ichikawa Y, Kitagawa K, Chino S, Ishida M,
Matsuoka K, Tanigawa T, Nakamura T, Hirano T, Takeda K and Sakuma
H: Adipose tissue detected by multislice computed tomography in
patients after myocardial infarction. JACC: Cardiovasc Imaging.
2:548–555. 2009.PubMed/NCBI
|
|
191
|
Vilahur G, Casani L, Juan-Babot O, Guerra
JM and Badimon L: Infiltrated cardiac lipids impair
myofibroblast-induced healing of the myocardial scar
post-myocardial infarction. Atherosclerosis. 224:368–376. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Wang K, Zhang L, Deng B, Jiang W, Kuang T,
Chen C, Zhao K, Shi Q, He J and Wang W: UCP2 Upregulates ACSL3 to
enhance lipid droplet release from acinar cells and modulates the
Sirt1/Smad3 pathway to promote macrophage-to-myofibroblast
transition in chronic pancreatitis. Adv Sci (Weinh). 12:e125712025.
View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Caves E, Jussila A, Forni MF, Benvie A,
Lei V, King D, Edelman H, Hamdan M, Odell ID, Hinchcliff M, et al:
Atgl-Dependent adipocyte lipolysis promotes lipodystrophy and
restrains fibrogenic responses during skin fibrosis. J Invest
Dermatol. 145:1896–1909.e5. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Mühlfeld C, Pfeiffer C, Schneider V,
Bornemann M and Schipke J: Voluntary activity reverses
spermidine-induced myocardial fibrosis and lipid accumulation in
the obese male mouse. Histochem Cell Biol. 155:75–88. 2021.
View Article : Google Scholar :
|
|
195
|
Song K, Sun H, Tu B, Zhou Y, Lin LC, Liu
ZY, Li R, Yang JJ, Zhang Y, Zhao JY and Tao H: WTAP boosts lipid
oxidation and induces diabetic cardiac fibrosis by enhancing AR
methylation. iScience. 26:1079312023. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Rambold AS, Cohen S and
Lippincott-Schwartz J: Fatty acid trafficking in starved cells:
Regulation by lipid droplet lipolysis, autophagy, and mitochondrial
fusion dynamics. Dev Cell. 32:678–692. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Lujan AL, Foresti O, Sugden C, Brouwers N,
Farre AM, Vignoli A, Azamian M, Turner A, Wojnacki J and Malhotra
V: Defects in lipid homeostasis reflect the function of TANGO2 in
phospholipid and neutral lipid metabolism. Elife. 12:e853452023.
View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Missaglia S, Coleman RA, Mordente A and
Tavian D: Neutral lipid storage diseases as cellular model to study
lipid droplet function. Cells. 8:1872019. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Ho KT, Chu FY, Lin YK, Chin HH, Yang SC,
Yang CP and Chang YH: Interleukin-4 ameliorates macrophage lipid
stress through promoting cholesterol efflux and lipid homeostasis.
Cytokine. 188:1568692025. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Oesterle A, Laufs U and Liao JK:
Pleiotropic effects of statins on the cardiovascular system. Circ
Res. 120:229–243. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Climent E, Benaiges D and Pedro-Botet J:
Hydrophilic or lipophilic statins? Front Cardiovasc Med.
8:6875852021. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Langhi C, Marquart TJ, Allen RM and Baldán
Á: Perilipin-5 is regulated by statins and controls triglyceride
contents in the hepatocyte. J Hepatol. 61:358–365. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Lipszyc PS, Cremaschi GA,
Zorrilla-Zubilete M, Bertolino ML, Capani F, Genaro AM and Wald MR:
Niacin modulates pro-inflammatory cytokine secretion. A potential
mechanism involved in its anti-atherosclerotic effect. Open
Cardiovasc Med J. 7:90–98. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Bernier F, Kuhara T and Xiao J: Probiotic
Bifidobacterium breve MCC1274 protects against oxidative stress and
neuronal lipid droplet formation via PLIN4 gene regulation.
Microorganisms. 11:7912023. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Rubic T, Trottmann M and Lorenz RL:
Stimulation of CD36 and the key effector of reverse cholesterol
transport ATP-binding cassette A1 in monocytoid cells by niacin.
Biochem Pharmacol. 67:411–419. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Lewandowski CT, Khan MW, BenAissa M,
Dubrovskyi O, Ackerman-Berrier M, LaDu MJ, Layden BT and Thatcher
GRJ: Metabolomic analysis of a selective ABCA1 inducer in
obesogenic challenge provides a rationale for therapeutic
development. Ebiomedicine. 66:1032872021. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Quinet EM, Savio DA, Halpern AR, Chen L,
Miller CP and Nambi P: Gene-selective modulation by a synthetic
oxysterol ligand of the liver X receptor. J Lipid Res.
45:1929–1942. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Chen Y, Duan Y, Yang X, Sun L, Liu M, Wang
Q, Ma X, Zhang W, Li X, Hu W, et al: Inhibition of ERK1/2 and
activation of LXR synergistically reduce atherosclerotic lesions in
ApoE-deficient mice. Arterioscler, Thromb, Vasc Biol. 35:948–959.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Wang J, Ai XB, Wang F, Zou YW, Li L and Yi
XL: Efficacy of ezetimibe combined with atorvastatin in the
treatment of carotid artery plaque in patients with type 2 diabetes
mellitus complicated with coronary heart disease. Int Angiol.
36:467–473. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Giugliano RP, Wiviott SD, Blazing MA, De
Ferrari GM, Park JG, Murphy SA, White JA, Tershakovec AM, Cannon CP
and Braunwald E: Long-term safety and efficacy of achieving very
low levels of low-density lipoprotein cholesterol: A prespecified
analysis of the IMPROVE-IT trial. JAMA Cardiol. 2:547–555. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Kim JJ, Yang EJ, Molina David J, Cho S,
Ficarella M, Pape N, Schiffer JE, Njeim R, Kim SS, Lo Re C, et al:
Ezetimibe enhances lipid droplet and mitochondria contact
formation, improving fatty acid transfer and reducing lipotoxicity
in alport syndrome podocytes. Int J Mol Sci. 25:131342024.
View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Sun H, Samarghandi A, Zhang N, Yao Z,
Xiong M and Teng BB: Proprotein convertase subtilisin/kexin type 9
interacts with apolipoprotein B and prevents its intracellular
degradation, irrespective of the low-density lipoprotein receptor.
Arterioscler Thromb Vasc Biol. 32:1585–1595. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Sun H, Krauss RM, Chang JT and Teng BB:
PCSK9 deficiency reduces atherosclerosis, apolipoprotein B
secretion, and endothelial dysfunction. J Lipid Res. 59:207–223.
2018. View Article : Google Scholar :
|
|
214
|
Wiegman A, Peterson AL, Hegele RA,
Bruckert E, Schweizer A, Lesogor A, Wang Y and Defesche J: Efficacy
and safety of inclisiran in adolescents with genetically confirmed
homozygous familial hypercholesterolemia: Results from the
double-blind, placebo-controlled part of the ORION-13 randomized
trial. Circulation. 151:1758–1766. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Xiao J, Deng YM, Liu XR, Cao JP, Zhou M,
Tang YL, Xiong WH, Jiang ZS, Tang ZH and Liu LS: PCSK9: A new
participant in lipophagy in regulating atherosclerosis? Clin Chim
Acta. 495:358–364. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Singh M, Kaur R, Lee MJ, Pickering RT,
Sharma VM, Puri V and Kandror KV: Fat-specific protein 27 inhibits
lipolysis by facilitating the inhibitory effect of transcription
factor Egr1 on transcription of adipose triglyceride lipase. J Biol
Chem. 289:14481–14487. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Grahn THM, Kaur R, Yin J, Schweiger M,
Sharma VM, Lee MJ, Ido Y, Smas CM, Zechner R, Lass A, et al:
Fat-specific protein 27 (FSP27) interacts with adipose triglyceride
lipase (ATGL) to regulate lipolysis and insulin sensitivity in
human adipocytes. J Biol Chem. 289:12029–12039. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Ramshanker N, Jessen N, Voss TS, Pedersen
SB, Jørgensen JOL, Nielsen TS, Frystyk J and Møller N: Effects of
short-term prednisolone treatment on indices of lipolysis and
lipase signaling in abdominal adipose tissue in healthy humans.
Metabolism. 99:1–10. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Yu CY, Mayba O, Lee JV, Tran J, Harris C,
Speed TP and Wang JC: Genome-wide analysis of glucocorticoid
receptor binding regions in adipocytes reveal gene network involved
in triglyceride homeostasis. PLoS One. 5:e151882010. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Park J, Song H, Moon S, Kim Y, Cho S, Han
K, Park CY, Cho SW and Oh CM: Cardiometabolic benefits of
fenofibrate in heart failure related to obesity and diabetes.
Cardiovasc Diabetol. 23:3432024. View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Zhang J, Cheng Y, Gu J, Wang S, Zhou S,
Wang Y, Tan Y, Feng W, Fu Y, Mellen N, et al: Fenofibrate increases
cardiac autophagy via FGF21/SIRT1 and prevents fibrosis and
inflammation in the hearts of type 1 diabetic mice. Clin Sci
(Lond). 130:625–641. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Yamamoto K, Ohta Y, Taguchi A, Akiyama M,
Nakabayashi H, Nagao Y, Ryoko H, Wada Y, Yamamoto T, Yano M and
Tanizawa Y: Effects of pemafibrate on left ventricular diastolic
function in patients with type 2 diabetes mellitus: A pilot study.
Diabetol Int. 14:434–439. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Montaigne D, Butruille L and Staels B:
PPAR control of metabolism and cardiovascular functions. Nat Rev
Cardiol. 18:809–823. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Wang MY and Unger RH: Role of PP2C in
cardiac lipid accumulation in obese rodents and its prevention by
troglitazone. Am J Physiol Endocrinol Metab. 288:E216–E221. 2005.
View Article : Google Scholar
|
|
225
|
Chow YL, Iwata Y and Sato F:
Dihydrosanguinarine enhances glucose uptake in mouse 3T3-L1 cells.
ACS Omega. 2:6916–6925. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Miklankova D, Markova I, Hüttl M and
Malinska H: Empagliflozin alters lipid metabolism in the myocardium
and liver in a prediabetes model with severe dyslipidemia. Front
Pharmacol. 15:13939462024. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Dabour MS, Abdelgawad IY, Grant MKO,
El-Sawaf ES and Zordoky BN: Canagliflozin mitigates
carfilzomib-induced endothelial apoptosis via an AMPK-dependent
pathway. Biomed Pharmacother. 164:1149072023. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Cai L, Zhao Y, Li Z, Xiao L, Wu Y, Wang S,
Liu Q, Ye Y, Guo Y and Zhang D: A human engineered heart
tissue-derived lipotoxic diabetic cardiomyopathy model revealed
early benefits of empagliflozin. Adv Sci (Weinh). 12:e031732025.
View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Tsai HH, Hsiao FC, Yu AL, Juang JH, Yu J
and Chu PH: Empagliflozin reduces high glucose-induced
cardiomyopathy in hiPSC-derived cardiomyocytes: Glucose-induced
lipotoxicity in hiPSC-derived cardiomyocytes. Stem Cell Rev Rep.
21:849–858. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Vallon V: How can inhibition of glucose
and sodium transport in the early proximal tubule protect the
cardiorenal system? Nephrol Dial Transplant. 39:1565–1573. 2024.
View Article : Google Scholar : PubMed/NCBI
|