Introduction
GC is a global health issue. According to 2022
GLOBOCAN data, GC ranks as the 5th most common malignancy worldwide
and is a leading cause of cancer-related mortality (1). In China, the incidence of GC is the
5th highest compared with all types of cancer (2,3).
Helicobacter pylori (H. pylori) infection is
recognized as one of the most pronounced pathogenic factors for GC.
In 1994, the International Agency for Research on Cancer of the
World Health Organization classified H. pylori as a Group 1
(definite) carcinogen associated with GC (4). Among individuals infected with
H. pylori, ~60% of the general population is infected or has
been previously infected with H. pylori, with the infection
rate reaching as ≤84% among patients with GC (5). Despite improvements in living
standards and sanitation leading to a decline in the global GC
incidence and mortality, it remains one of the major causes of
mortality (6).
Glycolysis is a fundamental metabolic pathway widely
studied in cancer, especially within tumor cell metabolism, where
it serves as a key component. The present review emphasizes that
glycolysis should not be rigidly classified as 'aerobic' or
'anaerobic' since lactate may be the end product of glycolysis
regardless of oxygen availability (7). Unless otherwise specified, the term
'glycolysis' in the present review predominantly refers to aerobic
glycolysis, the Warburg effect, in which cancer cells
preferentially metabolize glucose to lactate even in the presence
of sufficient oxygen. This process is distinct from anaerobic
glycolysis, which occurs under hypoxic conditions as a compensatory
energy pathway when oxidative phosphorylation is limited. While
both processes share the common endpoint of converting glucose into
lactate, aerobic glycolysis is a hallmark of several types of
cancer, enabling rapid ATP generation and supporting the
biosynthetic demands necessary for tumor cell proliferation. GC
cells typically exhibit enhanced glycolytic activity. During cancer
cell proliferation, the metabolic phenotype shifts to increased
glycolysis and lactate production to meet the elevated energy and
biosynthesis requirements (8,9).
Currently, research primarily focuses on the role of glycolysis in
gastrointestinal stromal tumors and colorectal cancer (10,11). However, the specific mechanisms
of glycolysis in GC and their impact on GC progression remain
insufficiently understood.
The present review aims to investigate the specific
mechanisms which drive glycolysis in the development and
progression of GC. By analyzing the relationship between the
expression of glycolysis-related enzymes and the prognosis of GC,
the present review aims to elucidate the effects of glycolytic
pathways on the growth, invasion and drug resistance of GC cells.
The present study also focuses on the impact of H. pylori
infection on the glycolytic pathways in GC. Given the key role of
metabolic reprogramming in GC, therapeutic strategies targeting
these pathways, collectively termed metabolism-targeted therapy,
hold potential. Such approaches may include inhibiting key
glycolytic enzymes, disrupting H. pylori-induced metabolic
adaptations or modulating the tumor microenvironment (TME), with
the potential to improve outcomes and overcome drug resistance.
Combined effect of H. pylori and
hyperglycemia in the pathogenesis of GC
H. pylori and the pathogenic mechanisms
of GC
Current research indicates that H. pylori
exerts carcinogenic effects on the gastric mucosa through a complex
interaction of bacterial, host and environmental factors. H.
pylori-induced carcinogenesis can be broadly divided into two
mechanisms: i) Those based on the induction of chronic inflammation
and ii) those associated with H. pylori-specific virulence
factors.
H. pylori-induced carcinogenesis via
chronic inflammation
Infection by H. pylori and the ensuing
chronic inflammation of the gastric mucosa represent key steps in
the initiation and progression of GC. H. pylori infection
can upregulate various pro-inflammatory cytokines, which trigger an
inflammatory response. Among these cytokines, the activation of
NF-κB and the upregulation of IL-8 are considered key mechanisms
underlying H. pylori-induced chronic inflammation and GC
development (12). Activation of
Jak1/Stat3 mediates NF-κB activation and increases IL-8
upregulation in AGS cells infected by H. pylori (13).
Carcinogenesis via specific virulence
factors
Among all virulence factors, CagA pathogenicity
island and VacA are considered the most important. Both
phosphorylated and non-phosphorylated CagA can extensively interact
with numerous host proteins to activate downstream signaling
pathways, leading to disorganization of gastric epithelial cell
polarity and initiation of pro-inflammatory responses (12,14).
Additionally, VacA exhibits various biological
activities such as binding to and being internalized by host cells
to induce notable cellular vacuolization. It also disrupts
epithelial tight junctions, and inhibits T lymphocyte activation
and proliferation within the lamina propria, and releases
pro-inflammatory factors such as IL-6, IL-8 and TNF-α further
intensifying the local inflammatory environment (15,16) (Fig. 1). Through the pathogenic effects
of CagA or the chronic inflammation it induces, H. pylori
can promote GC progression by exacerbating aberrant DNA methylation
patterns, especially hypermethylation of tumor suppressor genes
(17).
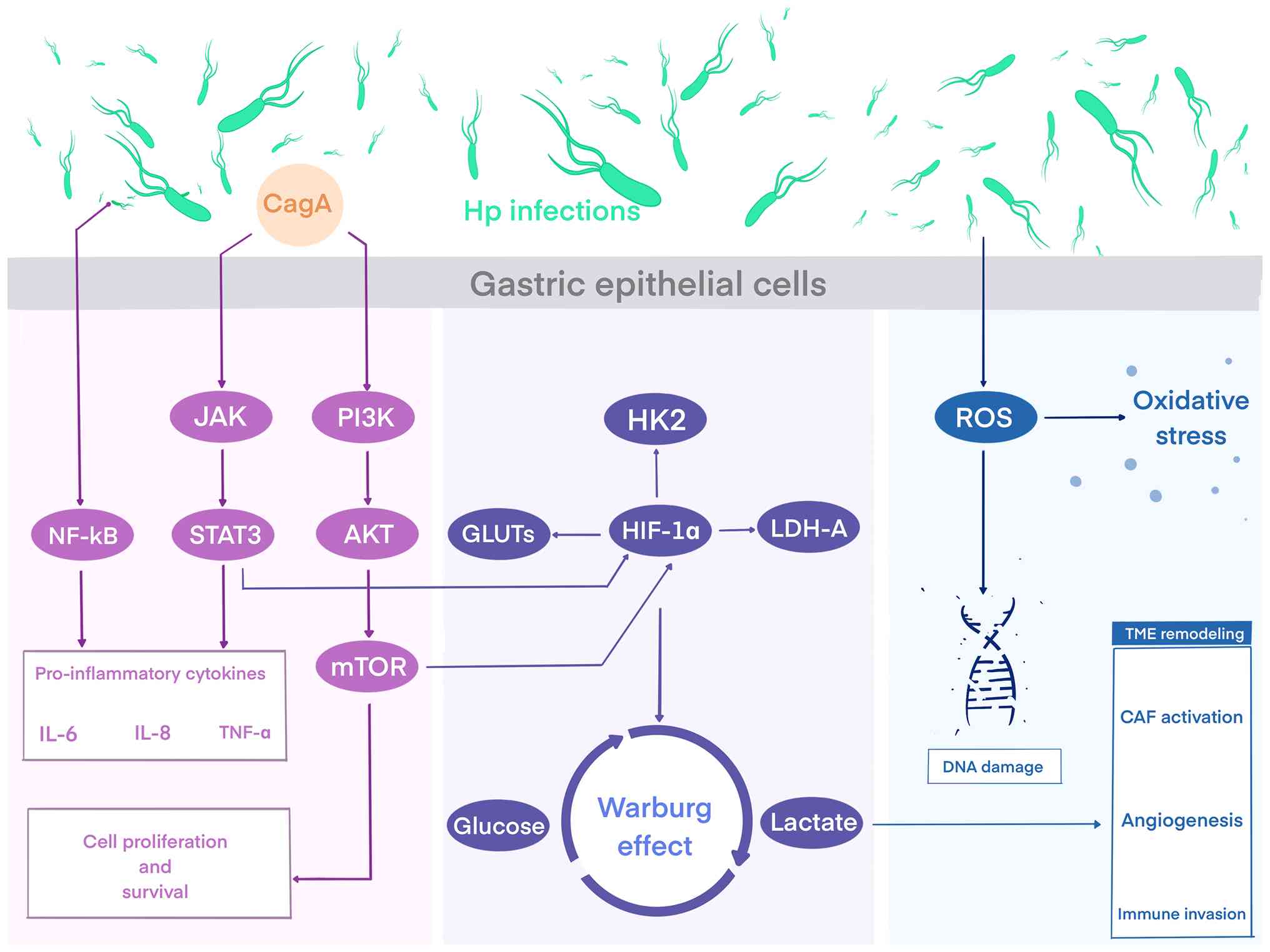 | Figure 1H. pylori-induced inflammatory
and oxidative pathways converge on glycolytic reprogramming to
drive GC progression. Pathogenic mechanisms of H. pylori
infection in GC. The virulence factor CagA activates JAK/STAT3,
NF-κB and PI3K/AKT/mTOR pathways, inducing pro-inflammatory
cytokines (IL-6, IL-8 and TNF-α) and promoting cell proliferation.
These signals upregulate glycolytic regulators (HIF-1α, GLUTs, HK2
and LDH-A), driving the Warburg effect with increased glucose
uptake and lactate production. Concurrently, H. pylori
stimulates ROS generation, leading to oxidative stress, DNA damage
and tumor microenvironment remodeling, including CAF activation,
angiogenesis and immune cell infiltration. Figure created using
Procreate: Savage Interactive Pty Ltd (Version 5.4.7) and Adobe
Illustrator (Adobe Inc 2025; Version 29.x). H. pylori,
Helicobacter pylori; GC, gastric cancer; CagA,
cytotoxin-associated gene A; HK2, Hexokinase 2; ROS, reactive
oxygen species; GLUTs, glucose transporters; HIF-1α,
hypoxia-inducible factor 1-α; LDH-A, lactate dehydrogenase A;
TNF-α, tumor necrosis factor α; CAF, cancer-associated
fibroblasts. |
Glycolytic activity has been shown to upregulate
immune checkpoint expression, suggesting that glycolysis may
enhance responses to immunotherapy and predict its efficacy
(18). Hypoxia inducible factor
(HIF)-1α, a transcriptional regulator of immune and oncogenic
responses, associates with the severity of gastric lesions. H.
pylori infection markedly upregulates HIF-1α expression and
increases reactive oxygen species (ROS) in human gastric epithelial
cells, promoting cell cycle arrest in G0/G1
and slowing the progression of precancerous gastric lesions
(19-21).
Hyperglycemia contributes to H. pylori
colonization and susceptibility
Hyperglycemia is associated with the release of
pro-inflammatory factors, oxidative stress, impaired immune
function and increased insulin secretion (20,21). H. pylori infection induces
metabolic disturbances mediated by inflammatory cytokines produced
in the gastric mucosa. Evidence suggests that hyperglycemia and GC
share common risk factors, which may synergize with H.
pylori infection to promote GC (22).
Under high-glucose conditions, H. pylori can
maintain growth and viability for up to 48 h. The growth and
adhesion abilities of H. pylori are enhanced, further
promoting the expression of virulence factors associated with its
type IV secretion system (23).
Glycated hemoglobin (HbA1c) is the most valuable indicator of
long-term blood glucose control. An epidemiological study by Ikeda
et al assessed the interaction between HbA1c levels and the
incidence of GC, indicating that hyperglycemia increases the risk
of GC associated with H. pylori infection (24). A study reported that persistently
impaired fasting glucose is associated with an increased risk of
several types of cancer, with gastrointestinal types of cancer
showing a dose-dependent association (25). Among patients without a diabetes
diagnosis, research has found a positive association between blood
glucose levels and cancer mortality rates, with hyperglycemia
promoting GC cell proliferation and reducing sensitivity to
chemotherapeutic agents (26).
The enhanced colonization of H. pylori in
hyperglycemic conditions is associated with several factors:
Diabetes-induced impairments in cellular and humoral immunity may
increase susceptibility to H. pylori infection; prolonged
H. pylori infection combined with high glucose levels
disrupts the gastrointestinal microbiota, reduces gastric acid
secretion and slows gastrointestinal motility, creating an
environment conducive to H. pylori colonization (27); changes in glucose metabolism may
alter the gastric mucosa, facilitating H. pylori
colonization (23) and diabetic
patients have higher healthcare utilization rates, implying more
frequent exposure to pathogens (28).
H. pylori increases insulin
resistance
H. pylori infection may lead to chronic
inflammatory responses in the gastrointestinal tract, participating
in immune responses by producing inflammatory factors. Studies
indicate that inflammation may trigger and ultimately increase
insulin resistance. Compared with patients that are H.
pylori-negative, those infected with H. pylori show
elevated levels of IL-6 and TNF-α (29-31). The pro-inflammatory cytokine,
IL-6, has been recognized as one of the earliest predictors or
pathogenic mediators of insulin resistance, while TNF-α has been
shown to exacerbate insulin resistance (32). H. pylori employs its
virulence factors, CagA and VacA, to upregulate pro-inflammatory
cytokines and activate the NF-κB signaling cascade (33). Under normal conditions, NF-κB
remains inactive; however, factors such as insulin resistance,
inflammatory mediators and disruptions in glucose and lipid
metabolism can trigger the NF-κB signaling cascade within cells
(34). NF-κB directly induces
the production of pro-inflammatory cytokines, including TNF-α and
IL-6, thereby initiating inflammatory responses (35). Through its signaling pathways,
NF-κB impacts various interconnected organs, influencing glucose
metabolism disorders (36).
H. pylori increases gastric hormone
secretion
The presence of H. pylori influences
metabolism by regulating hormones and markedly altering the gut
microbiota. Ghrelin and leptin are key hormones in human energy
homeostasis: Leptin suppresses food intake in response to satiety,
while ghrelin stimulates appetite (37). H. pylori not only
indirectly regulates energy homeostasis by altering gut microbiota
composition but also directly modulates leptin and ghrelin
secretion (38,39). H. pylori-induced gastric
mucosal damage leads to increased leptin and gastrin levels, while
plasma ghrelin levels decrease (40).
Research by Açbay et al showed that H.
pylori acts as a physiological amplifier of insulin release,
potentially enhancing insulin secretion in response to glucose and
dietary stimulation by increasing gastrin secretion (41), while also inhibiting glucose
absorption in the small intestine (42). However, Verhulst and Depoortere
(43) found a negative feedback
mechanism between ghrelin and insulin, which raises circulating
glucose levels (44).
Similarly, in the gastric mucosa of H.
pylori-infected Mongolian gerbils, ghrelin levels decreased due
to infection (45). These
findings suggest that H. pylori infection negatively impacts
ghrelin dynamics in the stomach and plasma, increasing insulin
secretion by lowering serum ghrelin concentrations, which
subsequently inhibits insulin release.
Additionally, a clinical study indicated higher
insulin resistance in H. pylori-infected individuals
(46). This suggests an
association between H. pylori infection,-decreased ghrelin,
and increased leptin levels, associated with disrupted energy
homeostasis, elevated fasting insulin levels and reduced insulin
sensitivity.
H. pylori activates signaling
pathways
In exploring the mechanisms underlying the
progression of GC, the present review identified multiple key
molecular targets and signaling pathways. The innate immune
response triggered by H. pylori in gastric mucosa involves
various cellular signaling pathways associated with metabolic
regulation, providing directions for novel therapeutic
strategies.
GC induced by H. pylori is associated with
chronic inflammation, characterized by neutrophil and macrophage
infiltration in gastric epithelial cells, which facilitates the
accumulation of pro-inflammatory cytokines and reactive ROS
(47). The release of the
virulence factor CagA upon infection activates downstream pathways,
including and cytokine-stimulated transduction JAK-STAT signaling
(48). This activation triggers
expression of insulin growth factor (IGF)-1 and inflammatory
pathways, further enhanced by cytokines such as IL-8, IL-1β, IL-6
and TNF-α (49).
H. pylori induces NADPH oxidase activation,
producing ROS that activate NF-κB and Jak1-Stat3 pathways in
gastric epithelial cells (13).
Jak1-Stat3 activation serves as an upstream signal for NF-κB
activation, inducing IL-8 expression in H. pylori-infected
gastric cells (50). IL-8
transcription requires NF-κB activation, which is essential for the
observed increase in IL-8 mRNA in infected cells (51).
Additionally, elevated serum IGF-1 levels, combined
with H. pylori infection, may increase GC risk, H.
pylori-induced PI3K/Akt/mTOR signaling regulates glycolysis and
protein synthesis, supporting cellular growth and metabolism
(52). Both H. pylori and
IGF-1 activate the PI3K/Akt-mTOR pathway (49). The insulin pathway interacts with
two tyrosine kinase receptors activated by insulin, IGF-1 and IGF-2
(18).
Insulin receptors (IRs) for IGF-1 and IGF-2 are
highly expressed in GC cells (53,54). Exogenous IGF-1 and IGF-2 markedly
stimulate GC cell proliferation and trigger downstream responses
within the insulin pathway. Upregulation of IGF-1R may activate the
PI3K/AKT/mTOR signaling cascade, promoting GC cell migration and
invasion (49,55). Conversely, downregulating IGF-1
expression may disrupt IGF-1/IGF-2-IGF-1R signaling, inhibiting GC
cell proliferation and invasion (56).
Hyperglycemia often coincides with insulin
resistance and excessive activation of insulin signaling pathways
may contribute to GC development. Insulin resistance can stimulate
inflammatory responses and activate NF-κB, carrying out a key role
in GC occurrence and progression (57). Hyperinsulinemia is associated
with an increased incidence of GC and associates with elevated
expression levels of IR and IGF1R (57). Increased expression of IGF-1,
induced by hyperinsulinemia, acts as a mitogen, reducing apoptosis
in tumor cells (22).
IGF1-mediated signaling regulates multiple processes in GC
(58), with interferon-induced
transmembrane protein (IFITM)2, considered a tumor suppressor that
is highly expressed in GC (58).
The study indicated that IGF1R activation upregulates IFITM2
expression in GC and is associated with IL-6 secretion (58). STAT3, a transcription factor
activated by IGF1/IGF1R signaling, carries out a key role in tumor
progression (59) (Fig. 2); phosphorylated (p)-STAT3 is
important in GC initiation and progression, promoting cell
survival, proliferation, angiogenesis and metastasis (60,61). The IGF-2/IR-A signaling pathway
has also been shown to notably promote malignancy in diabetic and
prediabetic populations (62).
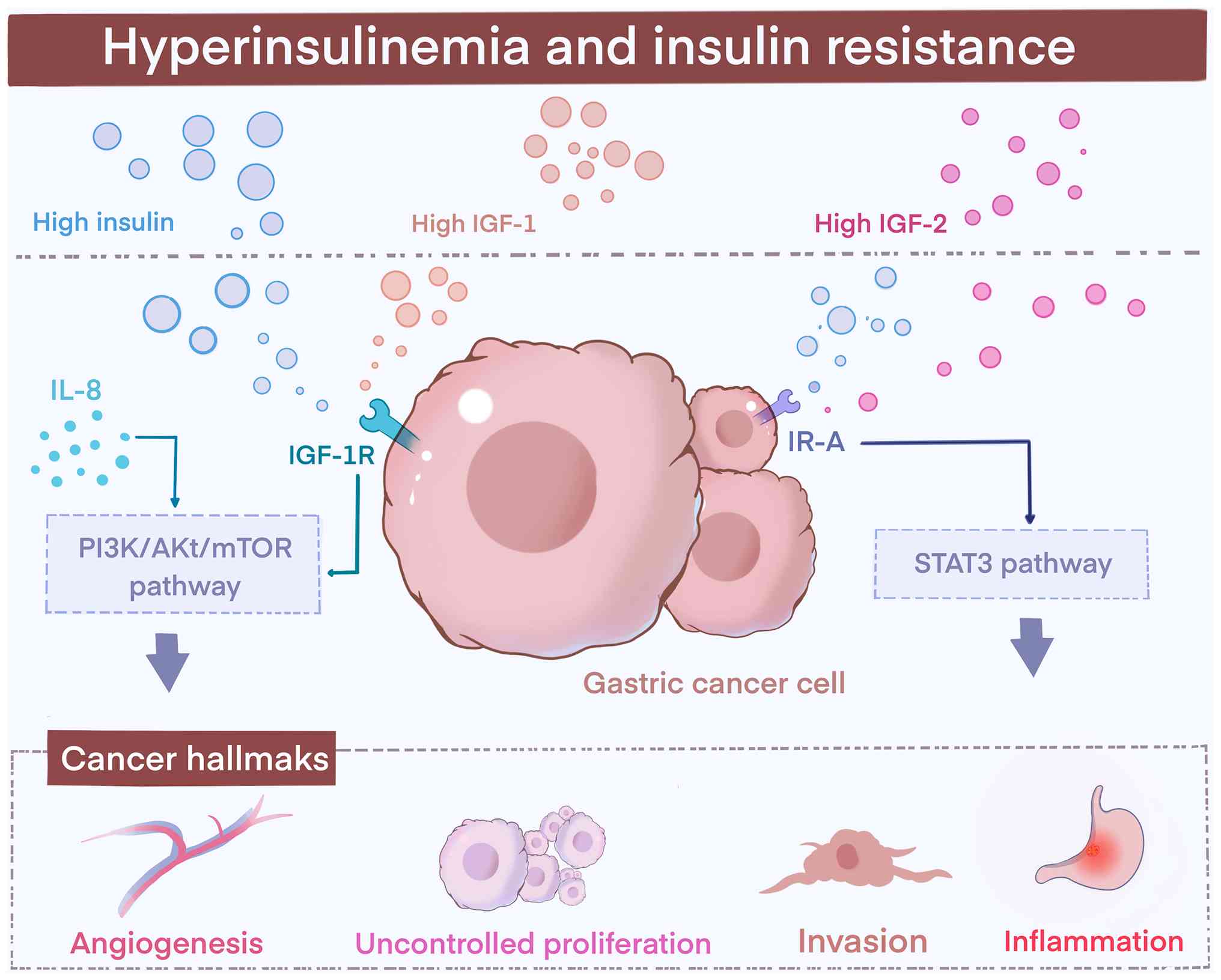 | Figure 2H. pylori infection under
hyperglycemic conditions. H. pylori infection releases CagA
and inflammatory cytokines (IL-8, IL-1β, IL-6 and TNF-α),
activating IGF-1R-mediated PI3K/AKT/mTOR and IR-A/STAT3 pathways.
Hyperinsulinemia elevates IGF-1/IGF-2 levels, further stimulating
these cascades. Together, these signals promote glycolysis,
proliferation, angiogenesis, invasion and chronic inflammation,
reinforcing the Warburg effect in GC cells. Figure created using
Procreate, Savage Interactive Pty Ltd (Version 5.4.7) and Adobe
Illustrator, Adobe Inc 2025 (Version 29.x). H. pylori,
Helicobacter pylori; CagA, Cytotoxin-associated gene A; GC,
gastric cancer; IGF-1/2, insulin-like growth factor-1/2; IR-A,
insulin receptor isoform A. |
These results indicate an association between
hyperglycemia and H. pylori-induced GC. The synergistic
effect of hyperglycemia and H. pylori infection in GC
development can be explained by hyperinsulinemia or insulin
resistance. Hyperglycemia itself may act as a carcinogenic factor,
excessive ROS production under hyperglycemic conditions leads to
DNA strand breaks in nuclear or mitochondrial DNA, resulting in
mutations of proto-oncogenes and tumor suppressor genes.
Additionally, hyperglycemia-induced oxidative damage in the gastric
mucosa may enhance the proliferative effects of H. pylori on
epithelial cells, leading to genetic or epigenetic changes that
promote GC development (24,63). A major feature of GC is altered
glucose metabolism, with upregulated glycolysis requiring
substantial glucose intake for cell proliferation and
differentiation. Hyperglycemia promotes the growth and migration of
cancer cells, facilitating GC progression (64).
TME
The TME is the cellular milieu in which tumor or
cancer stem cells reside. The TME is a highly complex network
composed of structurally and functionally essential elements in the
typical matrix, including fibroblasts, myofibroblasts,
neuroendocrine cells, immune and inflammatory cells, the blood and
lymphatic vascular networks, and the extracellular matrix (ECM),
which serves as a supportive framework (65). Due to the inherent proliferative
characteristics of tumors, the TME provides a unique
physicochemical environment and fosters complex metabolic patterns.
The TME contains various cell types and secretory elements that can
serve as potential targets for cancer therapy. Increasing evidence
indicates that understanding the relationship between glucose
metabolism and the TME represents a promising new direction for
targeted therapies (66-69).
Role of the TME in GC
The acidic conditions and unique endocrine system in
the stomach make the TME of GC distinct. Metabolites and cytokines
secreted by these cell types, including GC cells, also constitute
essential elements of the TME. Each component of the GC TME carries
out a role in inducing immune tolerance, thereby promoting GC
progression (70).
GC is associated with the TME. Typically, cancer
cells exhibit dysregulated metabolism characterized by increased
glucose consumption to fulfill anabolic demands. Emerging evidence
indicates that cancer cell metabolism influences the immune status
of the TME. When both the TCA cycle and glycolysis pathways are
activated, the GC TME displays increased infiltration of anti-tumor
effector cells, suggesting an enhancement in anti-tumor immune
responses that may contribute to immune evasion and resistance to
immunotherapy (71).
The TME is composed of diverse immune cells,
including T lymphocytes, macrophages, NK cells and dendritic cells
(72). Tumor-associated
macrophages (TAMs), a notable TME component, substantially
influence the interactions between cancer cells and their
surroundings. TAMs exhibit notable functional plasticity,
transforming into TAMs through reprogramming of monocytes that
migrate to the tumor in response to recruitment signals. This
transformation accelerates neovascularization and suppresses
anti-tumor immune responses, promoting tumor growth, malignant
transformation and invasiveness while diminishing anti-tumor T cell
activity.
TAMs can be categorized into different subtypes
based on functional states, mainly reflected in two polarization
modes: M1 and M2. M1-polarized macrophages exhibit anti-cancer
potential, producing pro-inflammatory cytokines such as IL-6, IL-8,
IL-12 and TNF-α, which collectively inhibit tumor growth (65,66). Conversely, M2-polarized
macrophages secrete anti-inflammatory cytokines such as IL-4, IL-10
and IL-13, which are key for tumor progression, metastasis and
angiogenesis, forming an essential component of the pro-tumorigenic
microenvironment (73,74).
H. pylori may decrease M1 macrophage
differentiation while promoting M2 macrophage differentiation or
M1-to-M2 macrophage transdifferentiation, thereby facilitating
tumor progression and invasion through the induction of
angiogenesis in solid tumors and the mediation of immunosuppressive
signals (75). In the early
stages of GC, TAMs are recruited to the tumor site via chemokine
signaling, contributing to tumor initiation. Notably, hypoxic
environments enhance glycolytic pathways, which reduce the number
of M1 macrophages, highlighting the role of metabolic reprogramming
in the TME (76). The majority
of TAMs adopt the M2 phenotype, which supports tumor expansion and
metastasis by limiting antigen presentation and anti-tumor immune
responses.
Studies indicate that TAMs compete with tumor cells
within the TME for nutrients such as glucose (77,78). Carcinogenic and anti-cancer
signals interact in this metabolic process, leading to glycolytic
pathway activation and aerobic respiration inhibition. Glucose
metabolism is regulated by various carcinogenic and
tumor-suppressive signals, with activated glycolysis and impaired
aerobic respiration shaping the altered glucose metabolism observed
in GC (79). The acidic TME also
profoundly affects TAM polarization. Lactic acid produced during
high-glucose glycolysis intensifies TME acidification, inducing TAM
polarization towards the M2 phenotype and strengthening Treg
function (80). Consequently,
TAMs undergo glucose metabolism similar to that of tumor cells,
contributing to metabolic reprogramming through activated
glycolysis. Tumor progression may be hindered by reversing M2 TAM
polarization back to the M1 state, thereby stimulating the immune
system with pro-inflammatory characteristics (81).
TAMs secrete growth factors such as vascular
endothelial growth factor and transform TGF-β, actively promoting
vascular network formation and tissue structure remodeling. In
advanced and metastatic GC, TAMs construct an immunosuppressive
microenvironment through mechanisms that include T cell inhibition,
regulatory Treg recruitment and the release of anti-inflammatory
factors such as IL-10, IL-6 and TGF-β (82). These cumulative effects weaken
the anti-cancer immune response of the host, leading to treatment
resistance in advanced GC (83).
Adjacent to immune components, the metabolism of
non-tumor cells in the TME also carries out a key role in cancer
progression and treatment response. For instance, cancer-associated
fibroblasts (CAFs) are primary components of the TME, secreting
cytokines and ECM components that promote tumor cell proliferation,
invasion and metastasis.
To survive under hypoxic tumor conditions, CAFs
adopt a glycolytic metabolic pattern, supporting cancer cells by
secreting growth factors and modifying the ECM, thereby undergoing
metabolic reprogramming to meet TME demands. A study on secretomics
revealed that H. pylori infection induces mesenchymal stem
cell differentiation into CAF-like cells, reprogramming normal
epithelial cells toward a pro-tumorigenic, invasive phenotype
through an EMT associated with cancer stem cells (84). This transition disrupts cell
junctions, enhances migration, reduces apoptosis and increases
oncogenic potential.
TGF-β signaling in CAFs drives ECM remodeling and
alters the physical properties of ECM fibers (82,85). Activation of TGF-β1/Smad2/3
signaling in CAFs upregulates glycolysis (86) (Fig. 3). Furthermore, ROS from cancer
cells can induce oxidative stress in CAFs (87), leading to a metabolic shift from
anaerobic respiration to glycolysis. This shift results in
energy-rich metabolite production, including pyruvate, lactate and
fatty acids, which nutritionally support other cancer cells
(88).
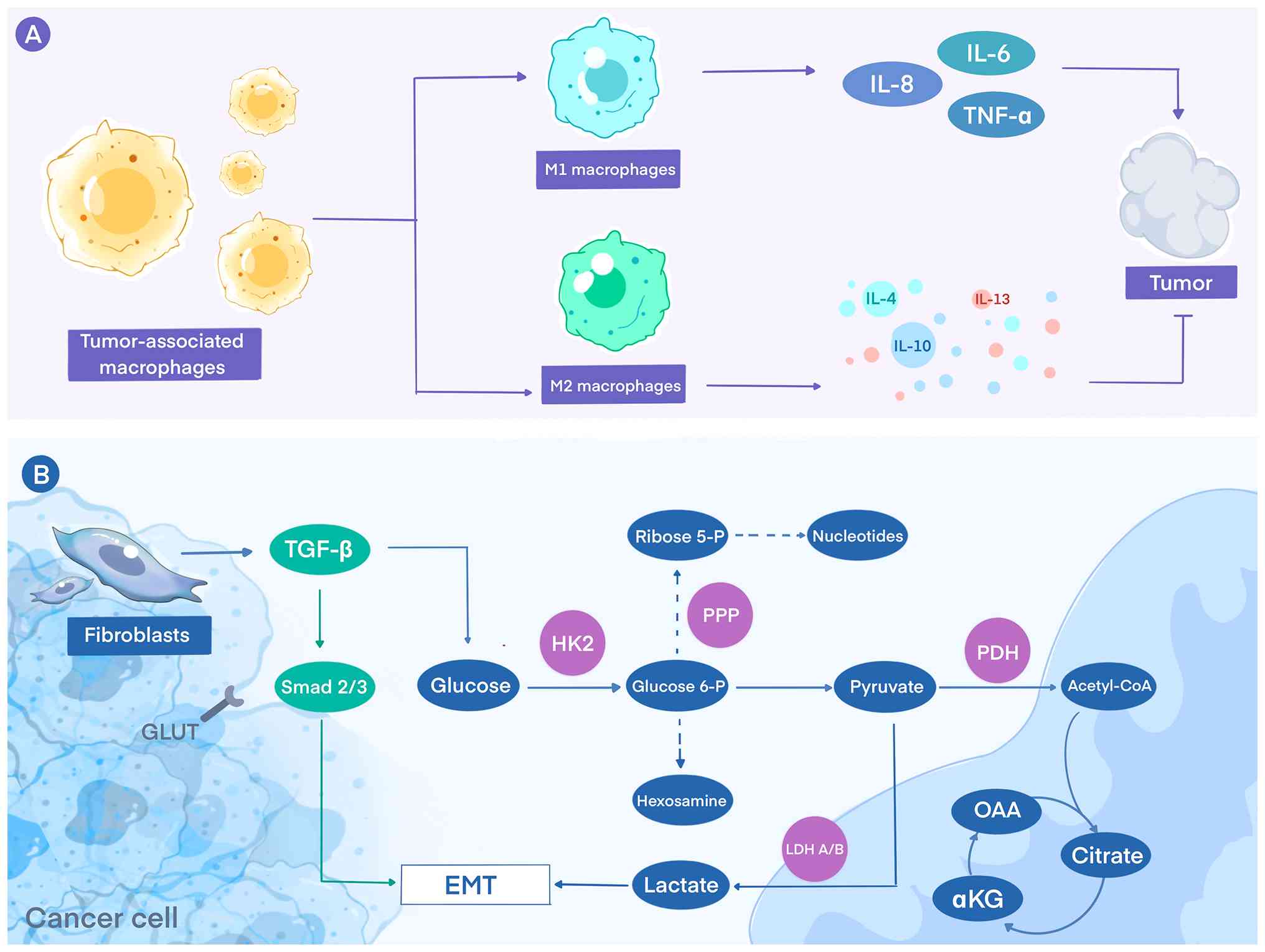 | Figure 3TME regulated by H. pylori
infection. (A) The polarization of macrophages into M1 and M2
phenotypes, which is influenced by hypoxia inducible factor-1 and
lactic acid. M1 macrophages produce pro-inflammatory cytokines,
including IL-6, IL-8 and TNF-α, which contribute to anti-tumor
immunity. By contrast, M2 macrophages secrete anti-inflammatory
cytokines such as IL-4, IL-10 and IL-13, promoting tumor growth and
immune evasion. (B) Glycolytic Pathway Alterations in H.
pylori-Associated GC Cells. CAFs positioned in the tumor
microenvironment secrete TGF-β, which activates Smad 2/3 signaling
in GC cells. This signaling enhances HK2-mediated glycolysis,
leading to increased production of glucose-6-phosphate, diversion
into the PPP for ribose-5-phosphate and nucleotide synthesis, and
conversion to lactate via LDH-A/B. Lactate is exported from CAFs or
cancer cells, acidifying the microenvironment and promoting EMT.
Meanwhile, pyruvate generated during glycolysis enters the TCA
cycle through PDH, producing acetyl-CoA, citrate, OAA and αKG. This
schematic illustrates the metabolic cross-talk between H.
pylori-associated GC cells and CAFs, highlighting how
stromal-derived signaling and metabolites reinforce tumor
glycolysis and biosynthetic activity within the 'reverse Warburg
effect' framework. Figure created using Procreate (Version 5.4.7);
Savage Interactive Pty Ltd and Adobe Illustrator, Adobe Inc 2025
(Version 29.x). H. pylori, Helicobacter pylori. GC,
gastric cancer; TNF-α, tumor necrosis factor α; TGF-β, transforming
growth factor-β; HK2, Hexokinase 2; Glucose 6-P,
Glucose-6-Phosphate; PPP, pentose phosphate pathway; PDH, pyruvate
dehydrogenase; EMT, epithelial-mesenchymal transition; LDH A/B,
lactate dehydrogenase A/B; OAA, oxaloacetate; αKG,
α-ketoglutarate. |
CAFs secrete lactate, which tumors utilize for
energy and intermediate products, a phenomenon known as the reverse
Warburg effect (89). Lactate
acts as both an energy source and a signaling molecule,
underscoring the complexity of metabolic interactions within the
TME. Understanding and targeting TME components holds potential for
GC treatment, offering strategies to inhibit tumor growth and
improve treatment efficacy.
Glucose metabolism in the TME
During growth, tumor cells adjust a range of
metabolic processes to adapt to the nutrient-deprived TME, meeting
the energy demands required for rapid proliferation. To support
tumorigenesis and development, cells within the TME maintain a
dynamic balance in interaction with cancer cells. As cancer cells
proliferate abnormally, their oxygen demand sharply increases,
triggering hypoxia and the formation of an acidic microenvironment
within the tumor. In this context, the uncontrolled proliferation
of tumor cells is closely associated with hypoxia and acidosis
(8,90).
The study suggests that lactic acidosis can shift
the predominant Warburg effect toward a non-glycolytic phenotype,
suppressing glycolysis and enhancing mitochondrial respiration in
cancer cells, potentially enabling cells to utilize glucose more
efficiently in a metabolically restricted environment (91). Hypoxia not only directly
influences tumor cell metabolic patterns but also disrupts the
normal physiological structure of the gastric mucosa. Hypoxia is
primarily mediated by HIFs, which enhance the 'Warburg effect' by
upregulating glycolytic genes such as hexokinase, LDH-A and GLUT
(92). In glycolysis, glucose or
glycogen is broken down into pyruvate, ATP, NADH and lactate.
Lactate, a byproduct of glycolysis, is present in the GC
microenvironment and promotes macrophage polarization through
HIF-1α-mediated angiogenic signals within the GC microenvironment
(93). HIF-1 enhances glycolysis
by promoting mitochondrial fission under hypoxic conditions,
thereby facilitating malignant transformation of the gastric mucosa
(94). These findings highlight
the intricate and interdependent relationship between cancer cell
metabolism and the TME.
H. pylori and glycolysis
H. pylori infection can lead to alterations
in host metabolic reprogramming. In this section, unless otherwise
specified, 'glycolysis' refers to aerobic glycolysis (Warburg
effect) as defined in the Introduction. Glycolysis (commonly
referred to as the Warburg effect in tumor metabolism) is a
cellular phenomenon, which is also a key indicator of tumor
progression, and there is an association between glycolysis and GC
(95). The Warburg effect
confers various survival advantages to cancer cells and markedly
impacts the metabolic state and functional regulation of
neighboring cells within the TME. The majority of the energy needed
for tumor cell proliferation is derived from glycolysis, which is
the main driver of the Warburg effect (96).
Tumor cells utilize glycolysis, oxidative
phosphorylation (OXPHOS) and fatty acid oxidation as energy
sources, adapting to microenvironmental changes and ensuring
survival through metabolic reprogramming (97). H. pylori-induced
glycolytic abnormalities not only increase energy consumption in
host cells but also promote cytokine release and inflammatory
responses. The elevated glycolytic rate provides a rich nutrient
source for the bacteria, exacerbating gastric mucosal damage and
establishing a vicious cycle. Consequently, targeting the
glycolytic pathway has emerged as a novel strategy for treating
H. pylori infection.
Characterization of glycolysis in GC
In normal, differentiated cells, OXPHOS serves as
the primary energy pathway. Mitochondria are the main site of
cellular OXPHOS, where NADH and FADH2 transfer electrons
to oxygen through the electron transport chain (ETC), generating a
proton gradient that drives ATP synthase to produce ATP, the most
efficient cellular energy production method (8). By contrast, tumor cells exhibit a
marked preference for glycolysis, even in oxygen-rich conditions,
displaying a high capacity for glucose uptake and lactate
accumulation. This metabolic shift is considered an adaptive
response to the high energy demands of rapidly proliferating tumor
cells, enabling cancer cells to meet their metabolic requirements
during rapid growth (98,99).
Under aerobic conditions, GC cells exhibit abnormal
metabolic characteristics, primarily converting pyruvate to
lactate. Although this process is less efficient in terms of ATP
production, it effectively generates the intermediate metabolites
necessary for rapid cell proliferation, resulting in a
proliferation rate distinct from that of normal cells.
Additionally, lactate is the final product of the Warburg effect,
and its accumulation during glycolysis creates an acidic
environment that destabilizes the extracellular matrix, promoting
tumor cell metastasis. Lactate can also facilitate immune evasion
by directly inhibiting the cytotoxicity and proliferation of immune
cells (100).
H. pylori is a primary factor in GC
development The study indicates that it promotes GC by enhancing
glycolysis and inducing mitochondrial dysfunction (101). Mitochondrial functional defects
often result from elevated ROS levels during mitochondrial electron
transport, which halts OXPHOS and promotes a switch to glycolysis
in tumor cells (102). This
abnormal mitochondrial metabolic pattern supplies the tumor with a
steady flow of nutrients and adaptability, enabling cancer cells to
survive and proliferate rapidly. The underlying mechanisms involve
the reversibility of several reactions within the TCA cycle and the
presence of multiple anaerobic pathways centered around
mitochondria, which ensure metabolic flexibility (103,104). The Warburg effect facilitates
efficient energy acquisition in tumor cells through glycolysis and
lactic acid fermentation, providing a metabolic advantage conducive
to growth and proliferation. Meanwhile, mitochondrial metabolism
remains essential for sustained tumor growth, coordinating the TCA
cycle and ETC to offer a stable biosynthetic source for tumor
cells.
Challenges and clinical considerations in
targeting glycolysis for cancer therapy
Although inhibition of glycolysis has emerged as a
promising anti-cancer strategy, multiple challenges hinder its
clinical translation. Metabolic plasticity in tumors allows cancer
cells to adapt to glycolytic blockade by switching to alternative
energy pathways, such as oxidative phosphorylation or
glutaminolysis, thereby reducing the efficacy of therapy (105,106). Glycolysis is also indispensable
for certain normal rapidly proliferating cells, including activated
immune cells, raising concerns regarding systemic toxicity when it
is broadly inhibited (107).
Moreover, marked metabolic heterogeneity exists both among tumor
types and within distinct intratumoral regions; hypoxic areas rely
predominantly on glycolysis, whereas normoxic regions favor
oxidative metabolism, making uniform targeting less effective
(96,108). Prolonged inhibition of
glycolysis may activate compensatory signaling networks and induce
metabolic reprogramming, resulting in acquired drug resistance
(109).
Despite these challenges, the dependence of H.
pylori-associated GC on specific glycolytic enzymes such as
HK2, PKM2, LDHA and PDK1 presents actionable vulnerabilities. The
following section provides a comprehensive overview of these
glycolysis-related therapeutic targets, summarizing their
regulatory mechanisms, prognostic relevance and current
pharmacological strategies, including synthetic inhibitors,
repurposed drugs and phytochemicals, to guide future translational
research.
Association between the expression of
glycolysis-related enzymes and the prognosis of GC
The Warburg effect can enhance the expression of
key glycolytic enzymes such as hexokinase (HK), glucose transporter
(GLUT), lactate dehydrogenase (LDH), pyruvate kinase (PK) and
3-phosphoinositide-dependent protein kinase 1 (PDK1), increasing
tumor cell tolerance to hypoxic conditions and promoting the
invasion and metastasis of malignancies (95). Therefore, the state of glycolysis
may also serve as a potential biomarker for cancer prognosis.
HK2 carries out a central role in the Warburg
effect and the development of malignant tumors. As a downstream
effector of Akt in cancerous environments, HK2 exhibits a high
affinity for glucose (110).
HK2 is anchored to the outer mitochondrial membrane through its
interaction with the mitochondrial protein voltage-dependent anion
channel (VDAC), shielding it from inhibition by
glucose-6-phosphate, the end product of its catalysis. This
anchoring markedly increases the glycolysis rate, enhancing ATP
production efficiency (111).
The ample ATP supplied by mitochondria accelerates
HK2-catalyzed, rate-limiting steps in glycolysis. During H.
pylori infection, dysregulation of glycolysis in the gastric
mucosa is observed, Immunohistochemistry analyses confirmed
elevated HK2 expression in patients infected with H. pylori,
with markedly increased levels observed in the gastric mucosa
(112,113).
Hexokinase domain-containing 1 (HKDC1) is involved
in mediating aerobic glycolysis and contributes to tumorigenesis in
various types of cancer. In H. pylori-induced GC, H.
pylori modulates the EMT pathway by upregulating TGF-β1
expression via HKDC1. In vitro and in vivo
experiments demonstrated that H. pylori infection increases
the expression of TGF-β1 and p-Smad2, thereby activating the EMT
pathway, whereas HK1 and HK2, which are mitochondrial-associated
proteins, exhibit an upward trend in expression (113). Current eradication therapies
still rely mainly on proton pump inhibitors (PPIs) and antibiotics.
Rabeprazole, a second-generation PPI commonly used in peptic ulcer
treatment, has been shown to exert effects beyond acid suppression.
As demonstrated by Zhou et al (112), rabeprazole suppresses STAT3
phosphorylation and nuclear translocation, thereby reducing STAT3
binding to the HK2 promoter and downregulating HK2 transcription.
Furthermore, direct metabolic intervention represents another
strategic approach. Notably, thyroid hormone T3 therapy can drive a
metabolic shift from HK2-driven glycolysis to mitochondrial OXPHOS,
which has been demonstrated to inhibit tumor development (101). The HK2 inhibitor
3-bromopyruvate and the phosphofructokinase inhibitor sodium
citrate suppress glycolysis and promote mitochondrial-associated
apoptosis in GC through the downregulation of the anti-apoptotic
protein Bcl-2 and the upregulation of the pro-apoptotic protein Bax
(114). Consequently, these
agents markedly inhibit tumor cell proliferation and glycolytic
activity in orthotopic xenograft models of GC. Together, these
findings underscore the broad potential of targeting cellular
metabolism, positioning the STAT3/HK2 axis and glycolytic pathway
as promising targets for improving the prognosis of patients
infected with H. pylori (115).
The PI3K/Akt signaling pathway, one of the commonly
dysregulated pathways in tumors, extensively regulates cellular
metabolic reprogramming by upregulating glucose transporters and
glycolytic enzymes. Cellular glucose uptake largely depends on
membrane transporter concentration, mainly by the glucose
transporter family, with GLUT1 being a key rate-limiting step in
glucose uptake (116). GLUT1 is
highly expressed in primary GC and is associated with disease stage
and prognosis, suggesting its role in glucose homeostasis in GC
cells (116,117). During H. pylori
infection, HIF-1α and bromodomain-containing protein 4 (BRD4)
cooperate to increase glycolysis by transcriptionally activating
GLUT1 and HK2. BRD4 deficiency reduces HIF-1α-dependent GLUT1
expression and HK2 levels, thereby impairing glucose uptake and
glycolytic capacity (118). By
targeting Na+/K+-ATPase, cardiac glycosides
(CGs), including ouabain, oleandrin and digoxin, inhibit glycolytic
metabolism in GC cells. In MKN45 cells, CGs downregulate plasma
membrane GLUT1 expression, with ouabain being effective at
sub-inhibitory concentrations for the α1 subunit. This leads to
suppressed 2-deoxy-D-glucose uptake, decreased lactate production
and impaired cell proliferation (119). Therefore, GLUT1 may serve as a
potential therapeutic target for GC.
LDHA, a key glycolytic enzyme, converts pyruvate to
lactate and is overexpressed in gastrointestinal tumors,
particularly GC. GC cells rely on high GLUT expression for
increased glucose uptake and energy metabolism. Lactic acid
accumulation in GC cells promotes glucose metabolism and glycolysis
rates and enhances cancer progression and invasiveness by inducing
histone lactylation (120). In
GC, LDHA is markedly upregulated by transcription factors such as
HIF-1α and c-Myc. This enhanced expression promotes the conversion
of pyruvate to lactate, which not only sustains high glycolytic
flux by regenerating NAD+ but also results in lactate
accumulation, thereby fostering an acidic TME. High LDH expression
is a key factor in the development of GC. A proteomics study
revealed that H. pylori infection promotes the expression of
this gene in GC tissues (121).
These changes collectively enhance tumor cell proliferation,
invasion and immune evasion. Targeting this metabolic pathway with
LDH-A inhibitors has emerged as a promising therapeutic strategy.
Among them, oxamate, a well-established LDH inhibitor and pyruvate
analog, competitively binds to the catalytic site of LDH-A, thereby
blocking pyruvate-to-lactate conversion, impairing NAD+
regeneration, and ultimately leading to suppressed glycolysis,
reduced ATP production and apoptosis induction (122). In a distinct manner, silibinin,
a polyphenolic compound derived from Silybum marianum, impedes
glycolytic metabolism by inhibiting the HIF-1α/LDH-A axis, thereby
enhancing antitumor immune responses (123-126). Both compounds represent
promising candidates for anticancer therapy through targeting
glycolysis. 5-FU-resistant GC cells show enhanced glycolysis, along
with upregulation of key glycolytic enzymes such as HK2 and LDHA.
CagA activates the Akt pathway, enhancing glycolysis in GC cells
and leading to 5-FU resistance. Inhibiting glycolysis or the Akt
pathway can overcome this resistance (127).
The expression of PKM2, a glycolysis-related enzyme
associated with tumor progression, is upregulated in GC cells due
to CagA, which induces increased PKM2 expression and increases with
GC progression. PKM2 has been shown to regulate cancer-specific
metabolic pathways that drive gastric cancer cell proliferation and
tumor growth by enhancing glycolytic flux, highlighting its key
role in metabolic reprogramming in GC (128). CagA promotes GC development by
inducing PKM2 expression via the Erk pathway. Furthermore, PKM2
carries out an essential role in regulating the final stage of
glycolysis, facilitating glycolysis in cancer cells by catalyzing
the transfer of a phosphate group from phosphoenolpyruvate to ADP,
forming ATP and pyruvate (129). At present, there is no direct
evidence supporting an association between PKM2 and H.
pylori infection. Shikonin, a natural product derived from the
roots of medicinal herbs such as Lithospermum erythrorhizon,
Arnebia euchroma and Onosma paniculata, exerts anticancer
effects partly through suppression of glycolysis in tumor cells.
Its mechanism involves the inhibition of PKM2 phosphorylation,
thereby reducing PKM2 activity. This finding is supported by
evidence that both PKM2 inhibitors and activators can alter the
effect of shikonin on glycolysis (130). The PI3K/Akt/mTOR signaling
pathway, which is known to regulate cell proliferation, apoptosis
and glucose metabolism, appears to be relevant in this context. For
instance, the specific PI3K inhibitor LY294002 has been shown to
suppress GC cell proliferation, induce early apoptosis and markedly
reduce lactate dehydrogenase activity and lactate production,
effects partly attributed to the inhibition of PKM2 expression
(131,132). Downregulation of PKM2
subsequently decreased the expression of Glut-1 and LDHA,
attenuating the Warburg effect. In parallel, pantoprazole (PPZ), a
third-generation proton pump inhibitor identified as a PKM2
inhibitor, suppresses the Akt/GSK-3β/β-catenin pathway and restores
chemosensitivity in GC cells (101,133). PPZ also reverses
chemoresistance in SGC7901 cells by downregulating
V-ATPase/mTOR/HIF-1α-mediated P-gp and MRP1 signaling.
Collectively, these findings highlight PKM2 as a promising target
for metabolic intervention in GC.
PDK-1 is a key enzyme that reduces mitochondrial
ROS. It acts as a direct target of HIF-1α. PDK-1 blocks the
conversion of pyruvate to acetyl-CoA, thereby preventing pyruvate
from entering the TCA cycle and inhibiting its metabolism within
the cycle. This shift, regulated by HIF-1α-targeted PDK-1, from
aerobic oxidation to glycolytic glucose metabolism, reduces
mitochondrial oxygen consumption and limits ROS accumulation,
ultimately promoting tumor growth (93). H. pylori infection induces
PDK1 dephosphorylation, resulting in aberrant Akt phosphorylation
and degradation, which disrupts cell survival signaling and the
balance between apoptosis and proliferation. In CagA-positive-GC
cells, inhibition of Akt phosphorylation or key glycolytic enzymes
(HK2/LDHA) synergistically enhances the cytotoxicity of 5-FU and
markedly reverses drug resistance (105). In vitro data indicate
that high PDK1 expression in GC cells is associated with reduced
sensitivity to 5-FU, suggesting that PDK1 overexpression is a
potential marker of poor prognosis (134,135). In gastric AGS cells, H.
pylori infection leads to PDK-1 dephosphorylation, which
disrupts cyclic PI3K signaling pathways related to cell survival
post-infection. This dephosphorylation of PDK-1, along with changes
in Akt phosphorylation, is one of the mechanisms by which H.
pylori infection alters the balance between apoptosis and cell
proliferation, potentially contributing to H. pylori-induced
GC development (136).
Dichloroacetate (DCA), a non-specific mitochondrial PDK-1
inhibitor, has been demonstrated to reactivate pyruvate
dehydrogenase, thereby shifting pyruvate metabolism from glycolysis
toward mitochondrial oxidative phosphorylation and subsequently
suppressing lactate production. This metabolic reprogramming not
only disrupts glycolytic flux but also may enhance cancer cell
sensitivity to conventional therapies (137,138). On the basis of this evidence,
the present review reviewed published studies and evaluated the
effects of DCA monotherapy alone and in combination with 5-FU in GC
cell lines exhibiting high PDK-1 expression and elevated glycolytic
activity. Hur et al (135) were the first to demonstrate
that low-dose DCA induces marked metabolic alterations in these
PDK-1-overexpressing cells with minimal effects on cell viability.
Therefore, the observed synergistic effect between DCA and 5-FU
suggests that DCA could serve as a promising adjunctive agent to
conventional chemotherapy, particularly for patients with
treatment-resistant disease and poor prognosis.
Comprehensive overview of glycolysis-related
therapeutic targets
To the best of our knowledge, research on the use
of various molecules associated with abnormal glucose metabolism as
prognostic biomarkers in GC is limited. Furthermore, the
therapeutic potential of drugs targeting glycolysis for treating GC
remains largely unexplored. The present review systematically
synthesized published studies on agents targeting glucose
metabolism in human GC cell lines and summarized their reported
efficacy, offering a novel perspective for GC treatment. H.
pylori infection reprograms gastric cell metabolism toward
enhanced glycolysis, markedly upregulating HK2, PKM2, LDHA and PDK1
expression. These alterations, driven by inflammation and
hypoxia-related signaling, promote tumor progression and represent
promising therapeutic targets.
In addition to synthetic inhibitors and repurposed
drugs such as 2-deoxy-D-glucose and rabeprazole, several
plant-derived phytochemicals have demonstrated notable
glycolysis-inhibiting activity in GC models, offering multi-target
modulation with generally favorable safety profiles. These natural
compounds interfere with diverse oncogenic and metabolic signaling
cascades, ultimately reducing lactate production and impairing
tumor cell bioenergetics (139). Rosmarinic acid, a phenolic
compound abundantly found in aromatic plants, suppresses glycolysis
in GC cells by inhibiting the IL-6/STAT3 pathway (140). Curcumin, the main polyphenol in
turmeric, downregulates HK2, PKM2 and LDHA expression via
inhibition of the PI3K/Akt/mTOR and HIF-1α signaling pathways,
leading to reduced lactate generation (141). Resveratrol, a stilbene found in
grapes, diminishes HK2 and LDHA expression through inhibition of
HIF-1α and c-Myc, and blocks GLUT1-mediated glucose uptake.
Epigallocatechin gallate, the major catechin in green tea, targets
both GLUT1 and LDHA, thereby suppressing lactate production and
increasing oxidative metabolism (142). Quercetin, a flavonoid widely
distributed in edible plants, interferes with the Akt/mTOR pathway,
resulting in decreased glycolytic enzyme activity and reduced
lactate levels (127). These
alterations, driven by inflammation- and hypoxia-related signaling,
promote tumor progression and represent promising therapeutic
targets. Table I (101,110,111,122,123,126,127,130,131,133,135,137-140,142-156) summarizes their regulation,
prognostic significance, representative inhibitors, mechanisms of
action and current development status, providing a concise
reference for future translational research.
 | Table IKey glycolytic enzymes as potential
therapeutic targets in H. pylori-associated GC. |
Table I
Key glycolytic enzymes as potential
therapeutic targets in H. pylori-associated GC.
| Glycolytic enzyme
and target | Expression and
regulation in H. pylori-related GC | Therapeutic
strategy/specific drugs | Mechanism of
action | Current research
stage/clinical evidence | (Refs.) |
|---|
| HK2 | Infection with
H. pylori activates the PI3K/Akt/mTOR signaling pathway,
which increases HIF-1α expression and consequently upregulates HK2.
NF-κB-mediated inflammatory signaling also enhances HK2 expression,
promoting glycolysis and inhibiting apoptosis. | Chemical
inhibitors: 2-DG, 3-BrPA, SCT13; repurposed drugs: Rabeprazole,
Noradrenaline, T3; natural compounds: Licochalcone A, Baicalein,
Curcumin, Resveratrol, Quercetin. | Inhibition of HK2
decreases the formation of glucose-6-phosphate, which reduces ATP
and lactate production and induces apoptosis. Rabeprazole blocks
STAT3 phosphorylation, thereby decreasing HK2 transcription.
Natural compounds inhibit the Akt/mTOR/HIF-1α axis, which leads to
decreased HK2 expression. | 2-DG: Phase I/II
trial in advanced solid tumors (NCT00096707);
Rabeprazole-preclinical GC models. | (101,110,111,127,139,140,143-148,152-154) |
| PKM2 | PKM2 is upregulated
in GC, regulating metabolic pathways that support tumor growth.
HIF-1α/c-Myc enhance PKM2 transcription, and PI3K/Akt/mTOR
signaling promotes its nuclear localization. | Chemical
inhibitors: Shikonin, TEPP-46; Repurposed drugs: Pantoprazole,
LY294002; natural compounds: Baicalein, Licochalcone A,
Curcumin. | Inhibition of PKM2
blocks the conversion of PEP to pyruvate, which suppresses
glycolysis. Pantoprazole reduces Akt/GSK-3β/β-catenin signaling,
leading to decreased PKM2 expression. | Preclinical;
controversy on PKM2's non-metabolic nuclear functions in tumor
regulation. | (127,130,131,133,139,140,144,145) |
| LDHA | HIF-1α together
with c-Myc increases LDHA expression, leading to elevated lactate
production and acidification of the tumor microenvironment. | Chemical
inhibitors: Oxamate, FX11; repurposed drugs: Natural compounds:
Silibinin, Licochalcone A, Baicalein, Curcumin, Resveratrol, EGCG,
Quercetin, Noradrenaline. | Inhibition of LDHA
reduces lactate accumulation and NAD+ regeneration,
resulting in decreased glycolytic flux. Silibinin inhibits the
HIF-1α/LDHA axis, which lowers LDHA expression. | Oxamate,
preclinical (murine models). | (122,123,126,127,142,139,144,145,149,150) |
| PDK-1 | H.
pylori-induced HIF-1α expression upregulates PDK1, which
inhibits PDH. This shifts metabolism toward increased lactate
production and contributes to chemoresistance. | Chemical
inhibitors: DCA; repurposed drugs: Natural compounds: Baicalein,
Curcumin, Resveratrol, EGCG, Quercetin, Licochalcone A. | Inhibition of PDK1
reactivates PDH, facilitating entry of pyruvate into the TCA cycle.
This reduces lactate production and increases
chemosensitivity. | DCA: Phase I/II
trials in glioblastoma (NCT00566410); limited trial data are
available for GC. | (127,135,137-139,144,145,151,155,156) |
Summary and future research directions
H. pylori infection drives gastric
carcinogenesis by inducing a profound metabolic shift toward
glycolysis (the Warburg effect). Mechanistically, virulence factors
activate oncogenic signaling, including the PI3K/Akt/mTOR and STAT3
pathways. These pathways converge on the level of the transcription
factor HIF-1α, which upregulates key glycolytic enzymes such as
HK2, PKM2 and LDHA. This metabolic reprogramming provides the
bioenergetic and biosynthetic substrates essential for cell
proliferation, survival and invasion.
This reliance on glycolysis, however, represents a
key therapeutic vulnerability. 2-Deoxy-D-glucose (2-DG), a
glycolysis inhibitor studied since the late 1950s, showed limited
effects in early monotherapy trials, indicating that cancer cells
can metabolize alternative substrates via oxidative
phosphorylation. In the first trial combining 2-DG with weekly
docetaxel (NCT00096707), the regimen was well tolerated without
pharmacokinetic interaction, achieving disease control in 32% of 34
patients (1 partial response; 11 stable disease ≥8 weeks).
Preclinical evidence of enhanced chemotherapy cytotoxicity supports
further Phase II evaluation (152). Preclinical evidence has
indicated DCA can reverse the Warburg effect and inhibit tumor
growth in cancer models. In the clinical setting, a Phase I/II
trial in recurrent glioblastoma (NCT00566410) explored positron
emission tomography (PET) scanning using 18F-FDG uptake as a
potential biomarker of response; in some patients, prolonged DCA
administration was associated with stable tumor size and reduced
18F-FDG uptake. Given its non-cytotoxic mechanism and slow onset of
action, the preclinical study suggests that DCA may be more
effective as an apoptosis-sensitizer in combination with cytotoxic
or targeted therapies compared with as monotherapy (156). Furthermore, the identification
of predictive biomarkers, such as the expression levels of key
glycolytic enzymes, is key for patient stratification and the
development of a precision medicine approach for H.
pylori-associated GC.
Availability of data and materials
Not applicable.
Authors' contributions
YL, FW, YD, YH, FS, JY, GG, MW, CL, XL, QD, JX and
RX contributed to conceiving the review; acquiring and analyzing
data; drafting, reviewing, revising and approving the manuscript;
and the decision to submit it for publication. No datasets were
generated or analyzed for the present study. Data authentication
not applicable. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
H. pylori
|
Helicobacter pylori
|
|
GC
|
gastric cancer
|
|
CagA
|
Cytotoxin-associated gene A
|
|
Cag PAI
|
Cag pathogenicity island
|
|
VacA
|
vacuolating cytotoxin A
|
|
IGF
|
insulin-like growth factor
|
|
IR-A
|
insulin receptor isoform A
|
|
ROS
|
reactive oxygen species
|
|
TME
|
tumor microenvironment
|
|
ECM
|
extracellular matrix
|
|
TAMs
|
tumor-associated macrophages
|
|
PDK1
|
3-phosphoinositide-dependent protein
kinase 1
|
|
CAF
|
cancer-associated fibroblasts
|
|
EMT
|
epithelial-mesenchymal transition
|
|
GLUT
|
glucose transporter
|
|
HK2
|
hexokinase 2
|
|
OXPHOS
|
oxidative phosphorylation
|
|
ETC
|
electron transport chain
|
|
TCA cycle
|
tricarboxylic acid cycle
|
|
LDH
|
lactate dehydrogenase
|
|
PKM2
|
pyruvate kinase M2
|
|
5-FU
|
5-fluorouracil
|
Acknowledgements
Not applicable.
Funding
Research grants were provided by the National Natural Science
Foundation of China (grant nos. 82170628, 32160208, 82560124, and
82570604), the Science and Technology Plan Project of Guizhou
Province [grant no. QIAN KE HE JI CHU-ZK (2023) YI BAN 556] and the
Guizhou Provincial Department of Science and Technology [grant no.
QIAN KE HE PING TAI REN CAI-GCC (2023) 040].
References
|
1
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263.
2024.PubMed/NCBI
|
|
2
|
Arnold M, Abnet CC, Neale RE, Vignat J,
Giovannucci EL, McGlynn KA and Bray F: Global burden of 5 major
types of gastrointestinal cancer. Gastroenterology.
159:335–349.e15. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhou Y, Song K, Chen Y, Zhang Y, Dai M, Wu
D and Chen H: Burden of six major types of digestive system cancers
globally and in China. Chin Med J (Engl). 137:1957–1964. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sexton RE, Al Hallak MN, Diab M and Azmi
AS: Gastric cancer: A comprehensive review of current and future
treatment strategies. Cancer Metastasis Rev. 39:1179–1203. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sitarz R, Skierucha M, Mielko J, Offerhaus
GJA, Maciejewski R and Polkowski WP: Gastric cancer: Epidemiology,
prevention, classification, and treatment. Cancer Manag Res.
10:239–248. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Niu P, Zhang F, Ma D, Zhou X, Zhu Y, Luan
X, Zhao L, Wang W, Zhang X, Han X, et al: Trends of older gastric
cancer incidence, mortality, and survival in the highest gastric
cancer risk area in China: 2010-2019 and prediction to 2024. BMC
Public Health. 24:24492024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu Y, Ma W, Liu W and Zhang S: Lactate: A
pearl dropped in the ocean: An overlooked signal molecule in
physiology and pathology. Cell Biol Int. 47:295–307. 2023.
View Article : Google Scholar
|
|
8
|
Barba I, Carrillo-Bosch L and Seoane J:
Targeting the warburg effect in cancer: Where do we stand? Int J
Mol Sci. 25:31422024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Paul S, Ghosh S and Kumar S: Tumor
glycolysis, an essential sweet tooth of tumor cells. Semin Cancer
Biol. 86:1216–1230. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shima T, Taniguchi K, Inomata Y, Arima J
and Lee SW: Glycolysis in gastrointestinal stromal tumor: A brief
overview. Neoplasia. 55:1010222024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xia Y, Zhang L, Ocansey DKW, Tu Q, Mao F
and Sheng X: Role of glycolysis in inflammatory bowel disease and
its associated colorectal cancer. Front Endocrinol (Lausanne).
14:12429912023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang F, Meng W, Wang B and Qiao L:
Helicobacter pylori-induced gastric inflammation and gastric
cancer. Cancer Lett. 345:196–202. 2014. View Article : Google Scholar
|
|
13
|
Cha B, Lim JW and Kim H: Jak1/Stat3 is an
upstream signaling of NF-κB activation in Helicobacter
pylori-induced IL-8 production in gastric epithelial AGS cells.
Yonsei Med J. 56:862–866. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Salvatori S, Marafini I, Laudisi F,
Monteleone G and Stolfi C: Helicobacter pylori and gastric cancer:
Pathogenetic mechanisms. Int J Mol Sci. 24:28952023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang SW, Kwon HS, Sohn IS, Kim YJ and
Hwang HS: Association of Vac A- and Cag A-specific Helicobacter
pylori strain infection with spontaneous preterm birth. J Matern
Fetal Neonatal Med. 30:995–1000. 2017. View Article : Google Scholar
|
|
16
|
D'Elios MM, Manghetti M, De Carli M, Costa
F, Baldari CT, Burroni D, Telford JL, Romagnani S and Del Prete G:
T helper 1 effector cells specific for Helicobacter pylori in the
gastric antrum of patients with peptic ulcer disease. J Immunol.
158:962–967. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kang GH, Lee HJ, Hwang KS, Lee S, Kim JH
and Kim JS: Aberrant CpG island hypermethylation of chronic
gastritis, in relation to aging, gender, intestinal metaplasia, and
chronic inflammation. Am J Pathol. 163:1551–1556. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kwon HJ, Park MI, Park SJ, Moon W, Kim SE,
Kim JH, Choi YJ and Lee SK: Insulin resistance is associated with
early gastric cancer: A prospective multicenter case control study.
Gut Liver. 13:154–160. 2019. View Article : Google Scholar :
|
|
19
|
Noto JM, Piazuelo MB, Romero-Gallo J,
Delgado AG, Suarez G, Akritidou K, Girod Hoffman M, Roa JC, Taylor
CT and Peek RM Jr: Targeting hypoxia-inducible factor-1 alpha
suppresses Helicobacter pylori-induced gastric injury via
attenuation of both cag-mediated microbial virulence and
proinflammatory host responses. Gut Microbes. 15:22639362023.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang M, Zhong J, Song Z, Xu Q, Chen Y and
Zhang Z: Regulatory mechanisms and potential therapeutic targets in
precancerous lesions of gastric cancer: A comprehensive review.
Biomed Pharmacother. 177:1170682024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Canales J, Valenzuela M, Bravo J,
Cerda-Opazo P, Jorquera C, Toledo H, Bravo D and Quest AF:
Helicobacter pylori induced phosphatidylinositol-3-OH kinase/mTOR
activation increases hypoxia inducible factor-1α to promote loss of
cyclin D1 and G0/G1 cell cycle arrest in human gastric cells. Front
Cell Infect Microbiol. 7:922017. View Article : Google Scholar
|
|
22
|
Wang L and Zhang Z: Diabetes mellitus and
gastric cancer: Correlation and potential mechanisms. J Diabetes
Res. 2023:43884372023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sheu SM, Cheng H, Kao CY, Yang YJ, Wu JJ
and Sheu BS: Higher glucose level can enhance the H pylori adhesion
and virulence related with type IV secretion system in AGS cells. J
Biomed Sci. 21:962014. View Article : Google Scholar
|
|
24
|
Ikeda F, Doi Y, Yonemoto K, Ninomiya T,
Kubo M, Shikata K, Hata J, Tanizaki Y, Matsumoto T, Iida M and
Kiyohara Y: Hyperglycemia increases risk of gastric cancer posed by
Helicobacter pylori infection: A population-based cohort study.
Gastroenterology. 136:1234–1241. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ahn BY, Kim B, Park S, Kim SG, Han K and
Cho SJ: Cumulative exposure to impaired fasting glucose and
gastrointestinal cancer risk: A nationwide cohort study. Cancer.
130:1807–1015. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tseng CH: The relationship between
diabetes mellitus and gastric cancer and the potential benefits of
metformin: An extensive review of the literature. Biomolecules.
11:10222021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mansori K, Dehghanbanadaki H, Naderpour S,
Rashti R, Moghaddam AB and Moradi Y: A systematic review and
meta-analysis of the prevalence of Helicobacter pylori in patients
with diabetes. Diabetes Metab Syndr. 14:601–607. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Grant RW, Buse JB and Meigs JB; University
HealthSystem Consortium (UHC) Diabetes Benchmarking Project Team:
Quality of diabetes care in U.S. academic medical centers: Low
rates of medical regimen change. Diabetes Care. 28:337–442. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang X, Zhao G, Shao S and Yao Y:
Helicobacter pylori triggers inflammation and oncogenic
transformation by perturbing the immune microenvironment. Biochim
Biophys Acta Rev Cancer. 1879:1891392024. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Walduck A, Andersen LP and Raghavan S:
Inflammation, immunity, and vaccines for Helicobacter pylori
infection. Helicobacter. 20(Suppl 1): S17–S25. 2015. View Article : Google Scholar
|
|
31
|
Yang XT, Niu PQ, Li XF, Sun MM, Wei W,
Chen YQ and Zheng JY: Differential cytokine expression in gastric
tissues highlights Helicobacter pylori's role in gastritis. Sci
Rep. 14:76832024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shoelson SE, Herrero L and Naaz A:
Obesity, inflammation, and insulin resistance. Gastroenterology.
132:2169–2180. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen B, Liu X, Yu P, Xie F, Kwan JSH, Chan
WN, Fang C, Zhang J, Cheung AHK, Chow C, et al: H: pylori-induced
NF-κB-PIEZO1-YAP1-CTGF axis drives gastric cancer progression and
cancer-associated fibroblast-mediated tumour microenvironment
remodelling. Clin Transl Med. 13:e14812023. View Article : Google Scholar
|
|
34
|
Gong P, Wang J, Wang S, Yang W, Yao W, Li
N, Wang J, Zhao Y, Chen F, Xie J, et al: Metabolomic analysis of
the Puerarin hypoglycemic activity via AMPK-mTOR and PPARγ-NF-κB
signaling pathways. Phytomedicine. 130:1555462024. View Article : Google Scholar
|
|
35
|
Yu H, Lin L, Zhang Z, Zhang H and Hu H:
Targeting NF-κB pathway for the therapy of diseases: Mechanism and
clinical study. Signal Transduct Target Ther. 5:2092020. View Article : Google Scholar
|
|
36
|
de Lima EP, Moretti RC Jr, Torres Pomini
K, Laurindo LF, Sloan KP, Sloan LA, Castro MVM, Baldi E Jr, Ferraz
BFR, de Souza Bast os Mazuqueli Pereira E, et al: Glycolipid
metabolic disorders, metainflammation, oxidative stress, and
cardiovascular diseases: Unraveling pathways. Biology (Basel).
13:5192024.PubMed/NCBI
|
|
37
|
Espinoza García AS, Martínez Moreno AG and
Reyes Castillo Z: The role of ghrelin and leptin in feeding
behavior: Genetic and molecular evidence. Endocrinol Diabetes Nutr
(Engl Ed). 68:654–663. 2021.PubMed/NCBI
|
|
38
|
Khosravi Y, Bunte RM, Chiow KH, Tan TL,
Wong WY, Poh QH, Doli Sentosa IM, Seow SW, Amoyo AA, Pettersson S,
et al: Helicobacter pylori and gut microbiota modulate energy
homeostasis prior to inducing histopathological changes in mice.
Gut Microbes. 7:48–53. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Roper J, Francois F, Shue PL, Mourad MS,
Pei Z, Olivares de Perez AZ, Perez-Perez GI, Tseng CH and Blaser
MJ: Leptin and ghrelin in relation to Helicobacter pylori status in
adult males. J Clin Endocrinol Metab. 93:2350–2357. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Franceschi F, Annalisa T, Teresa DR,
Giovanna D, Ianiro G, Franco S, Viviana G, Valentina T, Riccardo LL
and Antonio G: Role of Helicobacter pylori infection on nutrition
and metabolism. World J Gastroenterol. 20:12809–12817. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Açbay O, Celik AF and Gündoğdu S: Does
Helicobacter pylori-induced gastritis enhance food-stimulated
insulin release? Dig Dis Sci. 41:1327–1331. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Calam J, Gibbons A, Healey ZV, Bliss P and
Arebi N: How does Helicobacter pylori cause mucosal damage? Its
effect on acid and gastrin physiology. Gastroenterology. 113(6
Suppl): S43–S49; discussion S50. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Verhulst PJ and Depoortere I: Ghrelin's
second life: From appetite stimulator to glucose regulator. World J
Gastroenterol. 18:3183–3195. 2012.PubMed/NCBI
|
|
44
|
Isomoto H, Ueno H, Nishi Y, Wen CY,
Nakazato M and Kohno S: Impact of Helicobacter pylori infection on
ghrelin and various neuroendocrine hormones in plasma. World J
Gastroenterol. 11:1644–1648. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Suzuki H, Masaoka T, Hosoda H, Ota T,
Minegishi Y, Nomura S, Kangawa K and Ishii H: Helicobacter pylori
infection modifies gastric and plasma ghrelin dynamics in Mongolian
gerbils. Gut. 53:187–194. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhou X, Liu W, Gu M, Zhou H and Zhang G:
Helicobacter pylori infection causes hepatic insulin resistance by
the c-Jun/miR-203/SOCS3 signaling pathway. J Gastroenterol.
50:1027–1040. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Valenzuela MA, Canales J, Corvalán AH and
Quest AF: Helicobacter pylori-induced inflammation and epigenetic
changes during gastric carcinogenesis. World J Gastroenterol.
21:12742–12756. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xi Y, Zhang XL, Luo QX, Gan HN, Liu YS,
Shao SH and Mao XH: Helicobacter pylori regulates stomach diseases
by activating cell pathways and DNA methylation of host cells.
Front Cell Dev Biol. 11:11876382023. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ghafari F, Alizadeh AM, Agah S, Irani S
and Mokhtare M: Insulin-like growth factor 1 serum levels in
different stages of gastric cancer and their association with
Helicobacter pylori status. Peptides. 158:1708922022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chichirau BE, Diechler S, Posselt G and
Wessler S: Tyrosine kinases in Helicobacter pylori infections and
gastric cancer. Toxins (Basel). 11:5912019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Choi JH, Cho SO and Kim H: α-Lipoic acid
inhibits expression of IL-8 by suppressing activation of MAPK,
Jak/Stat, and NF-κB in H. pylori-Infected gastric epithelial AGS
cells. Yonsei Med J. 57:260–264. 2016. View Article : Google Scholar :
|
|
52
|
Sokolova O, Vieth M, Gnad T, Bozko PM and
Naumann M: Helicobacter pylori promotes eukaryotic protein
translation by activating phosphatidylinositol 3 kinase/mTOR. Int J
Biochem Cell Biol. 55:157–163. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Heckl SM, Wiesener V, Behrens HM, Ulase D,
Krüger S and Röcken C: The expression of the insulin receptor in
gastric cancer correlates with the HER2 status and may have
putative therapeutic implications. Gastric Cancer. 22:1130–1142.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yi HK, Hwang PH, Yang DH, Kang CW and Lee
DY: Expression of the insulin-like growth factors (IGFs) and the
IGF-binding proteins (IGFBPs) in human gastric cancer cells. Eur J
Cancer. 37:2257–2263. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Guo C, Chu H, Gong Z, Zhang B, Li C, Chen
J and Huang L: HOXB13 promotes gastric cancer cell migration and
invasion via IGF-1R upregulation and subsequent activation of
PI3K/AKT/mTOR signaling pathway. Life Sci. 278:1195222021.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ge J and Chen Z, Huang J, Yuan W, Den Z
and Chen Z: Silencing insulin-like growth factor-1 receptor
expression inhibits gastric cancer cell proliferation and invasion.
Mol Med Rep. 11:633–638. 2015. View Article : Google Scholar
|
|
57
|
Hidaka A, Sasazuki S, Goto A, Sawada N,
Shimazu T, Yamaji T, Iwasaki M, Inoue M, Noda M, Tajiri H, et al:
Plasma insulin, C-peptide and blood glucose and the risk of gastric
cancer: The Japan public health center-based prospective study. Int
J Cancer. 136:1402–1410. 2015. View Article : Google Scholar
|
|
58
|
Xu L, Zhou R, Yuan L, Wang S, Li X, Ma H,
Zhou M, Pan C, Zhang J, Huang N, et al: IGF1/IGF1R/STAT3
signaling-inducible IFITM2 promotes gastric cancer growth and
metastasis. Cancer Lett. 393:76–85. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zong CS, Chan J, Levy DE, Horvath C,
Sadowski HB and Wang LH: Mechanism of STAT3 activation by
insulin-like growth factor I receptor. J Biol Chem.
275:15099–15105. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kanda N, Seno H, Konda Y, Marusawa H,
Kanai M, Nakajima T, Kawashima T, Nanakin A, Sawabu T, Uenoyama Y,
et al: STAT3 is constitutively activated and supports cell survival
in association with survivin expression in gastric cancer cells.
Oncogene. 23:4921–4929. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Judd LM, Bredin K, Kalantzis A, Jenkins
BJ, Ernst M and Giraud AS: STAT3 activation regulates growth,
inflammation, and vascularization in a mouse model of gastric
tumorigenesis. Gastroenterology. 131:1073–1085. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Scalia P, Giordano A, Martini C and
Williams SJ: Isoform- and paralog-switching in IR-Signaling: When
diabetes opens the gates to cancer. Biomolecules. 10:16172020.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Papachristoforou E, Lambadiari V, Maratou
E and Makrilakis K: Association of glycemic indices (Hyperglycemia,
glucose variability, and hypoglycemia) with oxidative stress and
diabetic complications. J Diabetes Res. 2020:74897952020.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhou Y, Wang Y, Wang S and Shen L:
Hyperglycemia promotes human gastric carcinoma progression via
aquaporin 3. Dig Dis Sci. 60:2338–2345. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Chen F, Zhuang X, Lin L, Yu P, Wang Y, Shi
Y, Hu G and Sun Y: New horizons in tumor microenvironment biology:
Challenges and opportunities. BMC Med. 13:452015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Liu J and Cao X: Glucose metabolism of
TAMs in tumor chemoresistance and metastasis. Trends Cell Biol.
33:967–978. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Arner EN and Rathmell JC: Metabolic
programming and immune suppression in the tumor microenvironment.
Cancer Cell. 41:421–433. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Reinfeld BI, Madden MZ, Wolf MM, Chytil A,
Bader JE, Patterson AR, Sugiura A, Cohen AS, Ali A, Do BT, et al:
Cell-programmed nutrient partitioning in the tumour
microenvironment. Nature. 593:282–288. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wang JX, Choi SYC, Niu X, Kang N, Xue H,
Killam J and Wang Y: Lactic acid and an acidic tumor
microenvironment suppress anticancer immunity. Int J Mol Sci.
21:83632020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Liu Y, Li C, Lu Y, Liu C and Yang W: Tumor
microenvironment-mediated immune tolerance in development and
treatment of gastric cancer. Front Immunol. 13:10168172022.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Shi J, Wu Z, Wu X, Huangfu L, Guo T, Cheng
X, Han J, Li Z, Xing X and Ji J: Characterization of
glycometabolism and tumor immune microenvironment for predicting
clinical outcomes in gastric cancer. iScience. 26:1062142023.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Laplane L, Duluc D, Larmonier N, Pradeu T
and Bikfalvi A: The multiple layers of the tumor environment.
Trends Cancer. 4:802–809. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Boutilier AJ and Elsawa SF: Macrophage
polarization states in the tumor microenvironment. Int J Mol Sci.
22:69952021. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Ren J, Xu B, Ren J, Liu Z, Cai L, Zhang X,
Wang W, Li S, Jin L and Ding L: The Importance of M1-and
M2-polarized macrophages in glioma and as potential treatment
targets. Brain Sci. 13:12692023. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Gambardella V, Castillo J, Tarazona N,
Gimeno-Valiente F, Martínez-Ciarpaglini C, Cabeza-Segura M, Roselló
S, Roda D, Huerta M, Cervantes A and Fleitas T: The role of
tumor-associated macrophages in gastric cancer development and
their potential as a therapeutic target. Cancer Treat Rev.
86:1020152020. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhihua Y, Yulin T, Yibo W, Wei D, Yin C,
Jiahao X, Runqiu J and Xuezhong X: Hypoxia decreases macrophage
glycolysis and M1 percentage by targeting microRNA-30c and mTOR in
human gastric cancer. Cancer Sci. 110:2368–2377. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Vitale I, Manic G, Coussens LM, Kroemer G
and Galluzzi L: Macrophages and metabolism in the tumor
microenvironment. Cell Metab. 30:36–50. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Cheng K, Cai N, Zhu J, Yang X, Liang H and
Zhang W: Tumor-associated macrophages in liver cancer: From
mechanisms to therapy. Cancer Commun (Lond). 42:1112–1140. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Wang Y, Zhang J, Shi H, Wang M, Yu D, Fu
M, Qian Y, Zhang X, Ji R, Wang S, et al: M2 Tumor-associated
macrophages-derived exosomal MALAT1 promotes glycolysis and gastric
cancer progression. Adv Sci (Weinh). 11:e23092982024. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Wang ZH, Peng WB, Zhang P, Yang XP and
Zhou Q: Lactate in the tumour microenvironment: From immune
modulation to therapy. EBioMedicine. 73:1036272021. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Gao J, Liang Y and Wang L: Shaping
polarization of tumor-associated macrophages in cancer
immunotherapy. Front Immunol. 13:8887132022. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Tao L, Huang G, Song H, Chen Y and Chen L:
Cancer associated fibroblasts: An essential role in the tumor
microenvironment. Oncol Lett. 14:2611–2620. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Qiu Y, Lu G, Li N, Hu Y, Tan H and Jiang
C: Exosome-mediated communication between gastric cancer cells and
macrophages: Implications for tumor microenvironment. Front
Immunol. 15:13272812024. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Krzysiek-Maczka G, Targosz A, Wrobel T,
Paw M, Szczyrk U, Opila J, Strzalka M, Wierdak M, Major P,
Brzozowski T, et al: Time-extended exposure of gastric epithelial
cells to secretome of Helicobacter pylori-activated fibroblasts
induces reprogramming of gastric epithelium towards
pre-cancerogenic and pro-invasive phenotype. Am J Cancer Res.
12:1337–1371. 2022.PubMed/NCBI
|
|
85
|
Liu Y, Zhang X, Gu W, Su H, Wang X, Wang
X, Zhang J, Xu M and Sheng W: Unlocking the crucial role of
cancer-associated fibroblasts in tumor metastasis: Mechanisms and
therapeutic prospects. J Adv Res. 71:399–413. 2025. View Article : Google Scholar :
|
|
86
|
Jena BC, Das CK, Banerjee I, Bharadwaj D,
Majumder R, Das S, Biswas A, Kundu M, Roy PK, Kundu CN and Mandal
M: TGF-β1 induced autophagy in cancer associated fibroblasts during
hypoxia contributes EMT and glycolysis via MCT4 upregulation. Exp
Cell Res. 417:1131952022. View Article : Google Scholar
|
|
87
|
Sahai E, Astsaturov I, Cukierman E,
DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR,
Hunter T, et al: A framework for advancing our understanding of
cancer-associated fibroblasts. Nat Rev Cancer. 20:174–186. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Orimo A, Gupta PB, Sgroi DC,
Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL
and Weinberg RA: Stromal fibroblasts present in invasive human
breast carcinomas promote tumor growth and angiogenesis through
elevated SDF-1/CXCL12 secretion. Cell. 121:335–348. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Pavlides S, Whitaker-Menezes D,
Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro
MC, Wang C, Fortina P, Addya S, et al: The reverse Warburg effect:
Aerobic glycolysis in cancer associated fibroblasts and the tumor
stroma. Cell Cycle. 8:3984–4001. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Chen L, Huang L, Gu Y, Cang W, Sun P and
Xiang Y: Lactate-Lactylation hands between metabolic reprogramming
and immunosuppression. Int J Mol Sci. 23:119432022. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Xie J, Wu H, Dai C, Pan Q, Ding Z, Hu D,
Ji B, Luo Y and Hu X: Beyond Warburg effect-dual metabolic nature
of cancer cells. Sci Rep. 4:49272014. View Article : Google Scholar
|
|
92
|
Dang CV: The interplay between MYC and HIF
in the Warburg effect. Ernst Schering Found Symp Proc. 4:35–53.
2007.
|
|
93
|
Kim JW, Tchernyshyov I, Semenza GL and
Dang CV: HIF-1-mediated expression of pyruvate dehydrogenase
kinase: A metabolic switch required for cellular adaptation to
hypoxia. Cell Metab. 3:177–185. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Xiao Y, Liu X, Xie K, Luo J, Zhang Y,
Huang X, Luo J and Tan S: Mitochondrial dysfunction induced by
HIF-1α under hypoxia contributes to the development of gastric
mucosal lesions. Clin Transl Med. 14:e16532024. View Article : Google Scholar
|
|
95
|
Liao M, Yao D, Wu L, Luo C, Wang Z, Zhang
J and Liu B: Targeting the Warburg effect: A revisited perspective
from molecular mechanisms to traditional and innovative therapeutic
strategies in cancer. Acta Pharm Sin B. 14:953–1008. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Liberti MV and Locasale JW: The warburg
effect: How does it benefit cancer cells? Trends Biochem Sci.
41:211–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Kooshan Z, Cárdenas-Piedra L, Clements J
and Batra J: Glycolysis, the sweet appetite of the tumor
microenvironment. Cancer Lett. 600:2171562024. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Yoshida GJ: Metabolic reprogramming: The
emerging concept and associated therapeutic strategies. J Exp Clin
Cancer Res. 34:1112015. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Koppenol WH, Bounds PL and Dang CV: Otto
Warburg's contributions to current concepts of cancer metabolism.
Nat Rev Cancer. 11:325–337. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Hayes C, Donohoe CL, Davern M and Donlon
NE: The oncogenic and clinical implications of lactate induced
immunosuppression in the tumour microenvironment. Cancer Lett.
500:75–86. 2021. View Article : Google Scholar
|
|
101
|
Liu Y, Zhang Z, Wang J, Chen C, Tang X,
Zhu J and Liu J: Metabolic reprogramming results in abnormal
glycolysis in gastric cancer: A review. Onco Targets Ther.
12:1195–1204. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Papandreou I, Cairns RA, Fontana L, Lim AL
and Denko NC: HIF-1 mediates adaptation to hypoxia by actively
downregulating mitochondrial oxygen consumption. Cell Metab.
3:187–197. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Lu J, Tan M and Cai Q: The Warburg effect
in tumor progression: Mitochondrial oxidative metabolism as an
anti-metastasis mechanism. Cancer Lett. 356(2 Pt A): 156–164. 2015.
View Article : Google Scholar
|
|
104
|
Yu Y, Jiang Y, Glandorff C and Sun M:
Exploring the mystery of tumor metabolism: Warburg effect and
mitochondrial metabolism fighting side by side. Cell Signal.
120:1112392024. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Liu T, Zhao X, Cai T, Li W and Zhang M:
Metabolic reprogramming in Helicobacter pylori infection: From
mechanisms to therapeutics. Front Cell Infect Microbiol.
15:16780442025. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Ganapathy-Kanniappan S and Geschwind JF:
Tumor glycolysis as a target for cancer therapy: Progress and
prospects. Mol Cancer. 12:1522013. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Zhao Y, Liu H, Riker AI, Fodstad O, Ledoux
SP, Wilson GL and Tan M: Emerging metabolic targets in cancer
therapy. Front Biosci (Landmark Ed). 16:1844–1860. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Ward PS and Thompson CB: Metabolic
reprogramming: A cancer hallmark even warburg did not anticipate.
Cancer Cell. 21:297–308. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Zhang D, Li J, Wang F, Hu J, Wang S and
Sun Y: 2-Deoxy-D-glucose targeting of glucose metabolism in cancer
cells as a potential therapy. Cancer Lett. 355:176–183. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Fontana F, Giannitti G, Marchesi S and
Limonta P: The PI3K/Akt pathway and glucose metabolism: A dangerous
liaison in cancer. Int J Biol Sci. 20:3113–3125. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Liu Q, Liu Z, Zhang X, Zeng A and Song L:
Revisiting of cancer immunotherapy: Insight from the dialogue
between glycolysis and PD-1/PD-L1 axis in the tumor
microenvironment. Int J Biol Sci. 21:1202–1221. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Zhou Y, Chen S, Yang F, Zhang Y, Xiong L,
Zhao J, Huang L, Chen P, Ren L, Li H, et al: Rabeprazole suppresses
cell proliferation in gastric epithelial cells by targeting
STAT3-mediated glycolysis. Biochem Pharmacol. 188:1145252021.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Fang Z, Zhang W, Wang H, Zhang C, Li J,
Chen W, Xu X, Wang L, Ma M, Zhang S and Li Y: Helicobacter pylori
promotes gastric cancer progression by activating the
TGF-β/Smad2/EMT pathway through HKDC1. Cell Mol Life Sci.
81:4532024. View Article : Google Scholar
|
|
114
|
Wang TA, Xian SL, Guo XY, Zhang XD and Lu
YF: Combined 18F-FDG PET/CT imaging and a gastric orthotopic
xenograft model in nude mice are used to evaluate the efficacy of
glycolysis-targeted therapy. Oncol Rep. 39:271–279. 2018.
|
|
115
|
Pu Z, Xu M, Yuan X, Xie H and Zhao J:
Circular RNA circCUL3 accelerates the warburg effect progression of
gastric cancer through regulating the STAT3/HK2 Axis. Mol Ther
Nucleic Acids. 22:310–318. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Mueckler M and Thorens B: The SLC2 (GLUT)
family of membrane transporters. Mol Aspects Med. 34:121–138. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Fenske W, Völker HU, Adam P, Hahner S,
Johanssen S, Wortmann S, Schmidt M, Morcos M, Müller-Hermelink HK,
Allolio B and Fassnacht M: Glucose transporter GLUT1 expression is
an stage-independent predictor of clinical outcome in
adrenocortical carcinoma. Endocr Relat Cancer. 16:919–928. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Modi N, Chen YH, Dong XC, Hu XM, Lau GW,
Wilson KT, Peek RM Jr and Chen LF: BRD4 regulates
glycolysis-dependent Nos2 expression in macrophages upon H pylori
infection. Cell Mol Gastroenterol Hepatol. 17:292–308.e1. 2024.
View Article : Google Scholar :
|
|
119
|
Fujii T, Katoh M, Ootsubo M, Nguyen OTT,
Iguchi M, Shimizu T, Tabuchi Y, Shimizu Y, Takeshima H and Sakai H:
Cardiac glycosides stimulate endocytosis of GLUT1 via intracellular
Na+,K+-ATPase α3-isoform in human cancer cells. J Cell Physiol.
237:2980–2991. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Kumagai S, Koyama S, Itahashi K,
Tanegashima T, Lin YT, Togashi Y, Kamada T, Irie T, Okumura G, Kono
H, et al: Lactic acid promotes PD-1 expression in regulatory T
cells in highly glycolytic tumor microenvironments. Cancer Cell.
40:201–218.e9. 2022. View Article : Google Scholar
|
|
121
|
Zhou J, Wang W, Xie Y, Zhao Y, Chen X, Xu
W, Wang Y and Guan Z: Proteomics-based identification and analysis
of proteins associated with Helicobacter pylori in gastric cancer.
PLoS One. 11:e01465212016. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Liu X, Yang Z, Chen Z, Chen R, Zhao D,
Zhou Y and Qiao L: Effects of the suppression of lactate
dehydrogenase A on the growth and invasion of human gastric cancer
cells. Oncol Rep. 33:157–162. 2015. View Article : Google Scholar
|
|
123
|
Zhao Z, Han F, Yang S, Wu J and Zhan W:
Oxamate-mediated inhibition of lactate dehydrogenase induces
protective autophagy in gastric cancer cells: Involvement of the
Akt-mTOR signaling pathway. Cancer Lett. 358:17–26. 2015.
View Article : Google Scholar
|
|
124
|
Fiume L, Vettraino M, Manerba M and Di
Stefano G: Inhibition of lactic dehydrogenase as a way to increase
the anti-proliferative effect of multi-targeted kinase inhibitors.
Pharmacol Res. 63:328–334. 2011. View Article : Google Scholar
|
|
125
|
Fujiwara S, Kawano Y, Yuki H, Okuno Y,
Nosaka K, Mitsuya H and Hata H: PDK1 inhibition is a novel
therapeutic target in multiple myeloma. Br J Cancer. 108:170–178.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Deng X, Zhao J, Qu L, Duan Z, Fu R, Zhu C
and Fan D: Ginsenoside Rh4 suppresses aerobic glycolysis and the
expression of PD-L1 via targeting AKT in esophageal cancer. Biochem
Pharmacol. 178:1140382020. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Jia L, Huang S, Yin X, Zan Y, Guo Y and
Han L: Quercetin suppresses the mobility of breast cancer by
suppressing glycolysis through Akt-mTOR pathway mediated autophagy
induction. Life Sci. 208:123–130. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Shiroki T, Yokoyama M, Tanuma N, Maejima
R, Tamai K, Yamaguchi K, Oikawa T, Noguchi T, Miura K, Fujiya T, et
al: Enhanced expression of the M2 isoform of pyruvate kinase is
involved in gastric cancer development by regulating
cancer-specific metabolism. Cancer Sci. 108:931–940. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Wang P, Sun C, Zhu T and Xu Y: Structural
insight into mechanisms for dynamic regulation of PKM2. Protein
Cell. 6:275–287. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Zhao X, Zhu Y, Hu J, Jiang L, Li L, Jia S
and Zen K: Shikonin inhibits tumor growth in mice by suppressing
pyruvate Kinase M2-mediated aerobic glycolysis. Sci Rep.
8:145172018. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Lu J, Chen M, Gao S, Yuan J, Zhu Z and Zou
X: LY294002 inhibits the Warburg effect in gastric cancer cells by
downregulating pyruvate kinase M2. Oncol Lett. 15:4358–4364.
2018.PubMed/NCBI
|
|
132
|
Chen S, Fisher RC, Signs S, Molina LA,
Shenoy AK, Lopez MC, Baker HV, Koomen JM, Chen Y, Gittleman H, et
al: Inhibition of PI3K/Akt/mTOR signaling in PI3KR2-overexpressing
colon cancer stem cells reduces tumor growth due to apoptosis.
Oncotarget. 8:50476–50488. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Shen Y, Chen M, Huang S and Zou X:
Pantoprazole inhibits human gastric adenocarcinoma SGC-7901 cells
by downregulating the expression of pyruvate kinase M2. Oncol Lett.
11:717–722. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Gao S, Song D, Liu Y, Yan H and Chen X:
Helicobacter pylori CagA protein attenuates 5-Fu sensitivity of
gastric cancer cells through upregulating cellular glucose
metabolism. Onco Targets Ther. 13:6339–6349. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Hur H, Xuan Y, Kim YB, Lee G, Shim W, Yun
J, Ham IH and Han SU: Expression of pyruvate dehydrogenase kinase-1
in gastric cancer as a potential therapeutic target. Int J Oncol.
42:44–54. 2013. View Article : Google Scholar :
|
|
136
|
King CC and Obonyo M: Helicobacter pylori
modulates host cell survival regulation through the
serine-threonine kinase, 3-phosphoinositide dependent kinase 1
(PDK-1). BMC Microbiol. 15:2222015. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Tong J, Xie G, He J, Li J, Pan F and Liang
H: Synergistic antitumor effect of dichloroacetate in combination
with 5- fluorouracil in colorectal cancer. J Biomed Biotechnol.
2011:7405642011.
|
|
138
|
Xuan Y, Hur H, Ham IH, Yun J, Lee JY, Shim
W, Kim YB, Lee G, Han SU and Cho YK: Dichloroacetate attenuates
hypoxia-induced resistance to 5-fluorouracil in gastric cancer
through the regulation of glucose metabolism. Exp Cell Res.
321:219–230. 2014. View Article : Google Scholar
|
|
139
|
Zhao M, Wei F, Sun G, Wen Y, Xiang J, Su
F, Zhan L, Nian Q, Chen Y and Zeng J: Natural compounds targeting
glycolysis as promising therapeutics for gastric cancer: A review.
Front Pharmacol. 13:10043832022. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Han S, Yang S, Cai Z, Pan D, Li Z, Huang
Z, Zhang P, Zhu H, Lei L and Wang W: Anti-Warburg effect of
rosmarinic acid via miR-155 in gastric cancer cells. Drug Des Devel
Ther. 9:2695–2703. 2015.PubMed/NCBI
|
|
141
|
Ai Y, Zhao Z, Wang H, Zhang X, Qin W, Guo
Y, Zhao M, Tang J, Ma X and Zeng J: Pull the plug:
Anti-angiogenesis potential of natural products in gastrointestinal
cancer therapy. Phytother Res. 36:3371–3393. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Han JH, Kim M, Kim HJ, Jang SB, Bae SJ,
Lee IK, Ryu D and Ha KT: Targeting lactate dehydrogenase a with
catechin resensitizes SNU620/5FU gastric cancer cells to
5-fluorouracil. Int J Mol Sci. 22:54062021. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Wang TA, Zhang XD, Guo XY, Xian SL and Lu
YF: 3-Bromopyruvate and sodium citrate target glycolysis, suppress
survivi n, and induce mitochondrial-mediated apoptosis in gastric
cancer cells and inhibit gastric orthotopic transplantation tumor
growth. Oncol Rep. 35:1287–1296. 2016. View Article : Google Scholar :
|
|
144
|
Chen F, Zhuang M, Zhong C, Peng J, Wang X,
Li J, Chen Z and Huang Y: Baicalein reverses hypoxia-induced 5-FU
resistance in gastric cancer AGS cells through suppression of
glycolysis and the PTEN/Akt/HIF-1α signaling pathway. Oncol Rep.
33:457–463. 2015. View Article : Google Scholar
|
|
145
|
Wu J, Zhang X, Wang Y, Sun Q, Chen M, Liu
S and Zou X: Licochalcone A suppresses hexokinase 2-mediated tumor
glycolysis in gastric cancer via downregulation of the Akt
signaling pathway. Oncol Rep. 39:1181–1190. 2018.
|
|
146
|
Xu Y, Wang Q, Zhang L and Zheng M:
2-Deoxy-D-glucose enhances TRAIL-induced apoptosis in human gastric
cancer cells through downregulating JNK-mediated cytoprotective
autophagy. Cancer Chemother Pharmacol. 81:555–564. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Wang Y, Wang S, Yang Q, Li J, Yu F and
Zhao E: Norepinephrine enhances aerobic glycolysis and may act as a
predictive factor for immunotherapy in gastric cancer. J Immunol
Res. 2021:55806722021.PubMed/NCBI
|
|
148
|
García-Carracedo D, Villaronga MÁ,
Álvarez-Teijeiro S, Hermida-Prado F, Santamaría I, Allonca E,
Suárez-Fernández L, Gonzalez MV, Balbín M, Astudillo A, et al:
Impact of PI3K/AKT/mTOR pathway activation on the prognosis of
patients with head and neck squamous cell carcinomas. Oncotarget.
7:29780–29793. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Sellam LS, Zappasodi R, Chettibi F,
Djennaoui D, Yahi-Ait Mesbah N, Amir-Tidadini ZC, Touil-Boukoffa C,
Ouahioune W, Merghoub T and Bourouba M: Silibinin down-regulates
PD-L1 expression in nasopharyngeal carcinoma by interfering with
tumor cell glycolytic metabolism. Arch Biochem Biophys.
690:1084792020. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Zhang B, Yang Y, Shi X, Liao W, Chen M,
Cheng AS, Yan H, Fang C, Zhang S, Xu G, et al: Proton pump
inhibitor pantoprazole abrogates adriamycin-resistant gastric
cancer cell invasiveness via suppression of Akt/GSK-β/β-catenin
signaling and epithelial-mesenchymal transition. Cancer Lett.
356(2, Part B): 704–712. 2015. View Article : Google Scholar
|
|
151
|
Stacpoole PW: Review of the pharmacologic
and therapeutic effects of diisopropylammonium dichloroacetate
(DIPA). J Clin Pharmacol J New Drugs. 9:282–291. 1969. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Raez LE, Papadopoulos K, Ricart AD,
Chiorean EG, DiPaola RS, Stein MN, Rocha Lima CM, Schlesselman JJ,
Tolba K, Langmuir VK, et al: A phase I dose-escalation trial of
2-deoxy-D-glucose alone or combined with docetaxel in patients with
advanced solid tumors. Cancer Chemother Pharmacol. 71:523–530.
2013. View Article : Google Scholar
|
|
153
|
Maher JC, Krishan A and Lampidis TJ:
Greater cell cycle inhibition and cytotoxicity induced by
2-deoxy-d-glucose in tumor cells treated under hypoxic vs aerobic
conditions. Cancer Chemother Pharmacol. 53:116–122. 2004.
View Article : Google Scholar
|
|
154
|
Novack DV: The FOX(O1) blasts off. Cell
Metab. 11:175–176. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Michelakis ED, Sutendra G, Dromparis P,
Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR,
Fulton D, et al: Metabolic modulation of glioblastoma with
dichloroacetate. Sci Transl Med. 2:31ra342010. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Chu QS, Sangha R, Spratlin J, Vos LJ,
Mackey JR, McEwan AJ, Venner P and Michelakis ED: A phase I
open-labeled, single-arm, dose-escalation, study of dichloroacetate
(DCA) in patients with advanced solid tumors. Invest New Drugs.
33:603–610. 2015. View Article : Google Scholar : PubMed/NCBI
|














