Intracerebral hemorrhage (ICH) accounts for 10-15%
of all stroke cases, with >2 million patients worldwide
annually. Characterized by complex pathophysiology and a one-month
mortality rate of ~70%, ICH poses a severe clinical challenge
(1-7). Globally, the incidence of ICH has
demonstrated an upward trend with advancing medical understanding
(8,9), driven by factors such as the
increasing elderly population, widespread use of anticoagulants,
antiplatelets and thrombolytics (10,11). While preclinical research using
animal ICH models has yielded promising developments in potential
treatments (12-15), the absence of evidence-based
therapeutic strategies in clinical settings remains a critical
barrier to improving ICH prognosis. This gap underscores the urgent
need for translating preclinical advancements into clinically
viable interventions to address the unmet therapeutic needs in ICH
management.
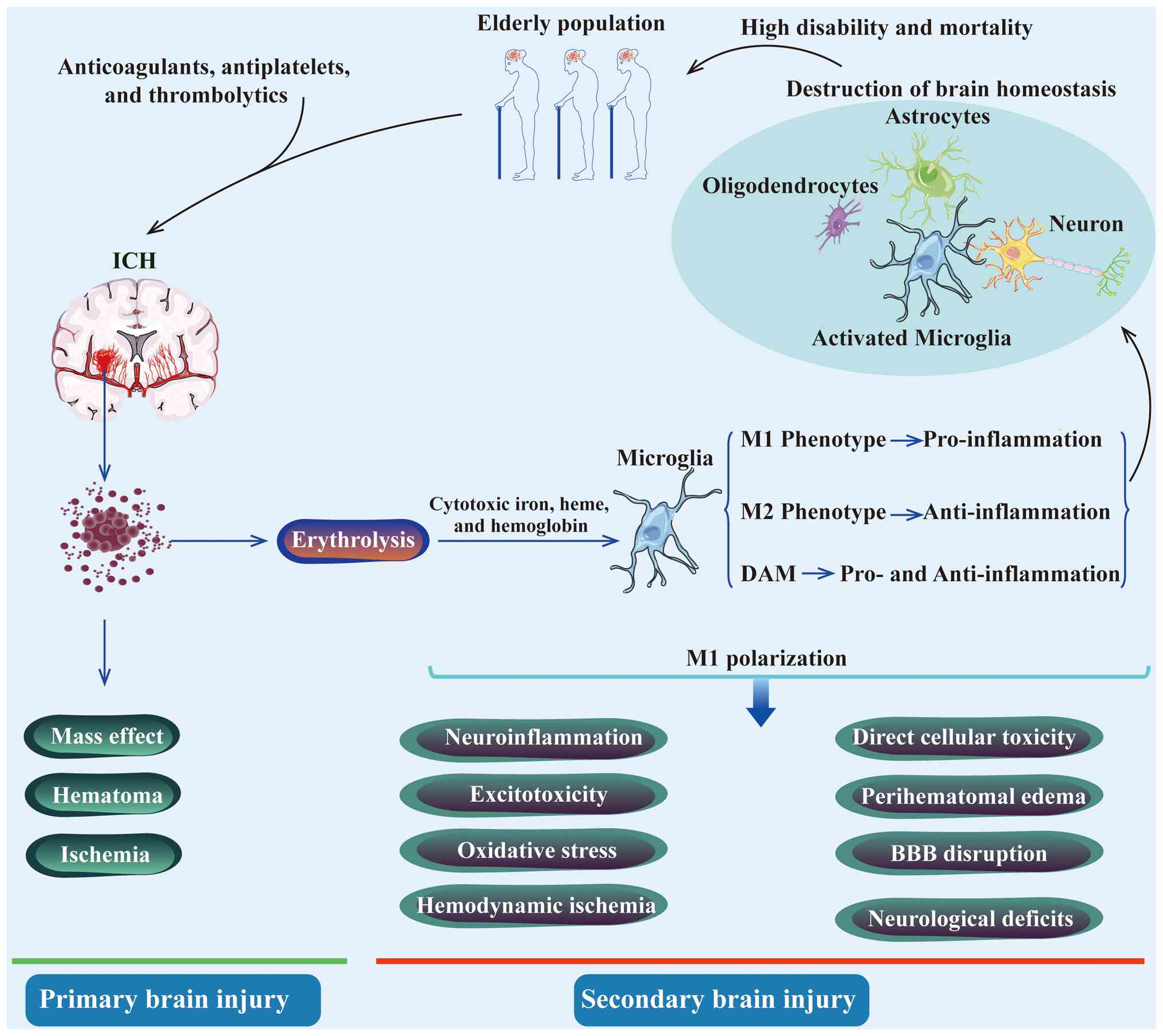
Following ICH, rupture of brain parenchymal vessels
leads to erythrocytes accumulation and hematoma formation, which
directly compresses brain tissue and causes primary brain injury
(PBI). In the hematoma, erythrocyte hemolysis releases toxic
hemolytic products that induce secondary brain injury (SBI) and
irreversible neurological deficits (16,17). Mounting evidence indicates that
inflammatory responses are central to the pathophysiology of SBI
(18). During this process,
circulating monocyte-derived macrophages and resident microglia in
the central nervous system (CNS) infiltrate the hemorrhagic site.
These microglia/macrophages serve as key regulators of
neuroinflammation and hematoma resolution mitigation in SBI
(19,20), highlighting their critical role
in shaping post-ICH neuroinflammatory trajectories and potential as
therapeutic targets.
Microglia, the primary immune cells of the CNS,
constitute 5-10% of brain cells and are often referred to as the
'resident macrophages' of the brain (21,22). They interact dynamically with
neurons, astrocytes and oligodendrocytes under physiological
conditions, playing a fundamental role in sustaining brain
homeostasis (23). The highly
plastic nature of microglia, including their phenotypic diversity,
hinges on the type of stressor or neuropathology that they
encounter (24). Specifically,
during distinct pathological phases of ICH, microglia polarization
generates pro-inflammatory (M1-like) or anti-inflammatory (M2-like)
mediators, which directly influence the progression of ICH and
neurological outcomes (20,25). This dual functionality
underscores the central role of microglia in modulating
neuroinflammation and tissue repair after ICH.
The hematoma and hemolysis resulting from ICH
trigger robust microglial activation, exacerbating
neuroinflammation (26,27). Chang et al (28) demonstrated that microglia rapidly
respond to hemorrhagic insult within 1-1.5 h after ICH onset by
initially adopting a protective phenotype, in which IL-10 mediates
microglial phagocytosis of hematoma components and promotes
hematoma resolution, thereby highlighting the early protective role
of microglia in mitigating SBI. By contrast, microglial depletion
exacerbates brain damage after ICH, manifesting as brain swelling,
neuronal degeneration and worsened neurological impairments
(29-31). Critically, emerging evidence
suggests that microglia do not act in isolation. Therefore, the
present review will discuss whether the pathological and reparative
processes following ICH are determined not by any single cell type,
but by a microglia-centered, spatiotemporally dynamic neuroimmune
network. The present review systematically evaluated the microglial
inflammatory response and its therapeutic targets after ICH, while
integrating current understanding of their anti-inflammatory
therapeutic potential. Notably, key hurdles to clinical translation
were emphasized, including ICH-induced iron overload, mass effect,
persistent inflammatory responses and oxidative damage.
ICH-induced brain injury, characterized by a high
risk of recurrent bleeding and ischemia, typically unfolds as a
cascade of pathological processes comprising two successive stages:
PBI and SBI (32,33). During the PBI stage, blood from
ruptured vessels forms a hematoma within the first few hours,
exerting a mass effect through mechanical compression of brain
parenchymal structures. This process leads to elevated intracranial
pressure and potential brain herniation, often managed clinically
through advanced surgical interventions (33,34). The SBI stage follows, driven by
hemolytic products from erythrolysis, including hemoglobin, heme,
free iron, along with neuroinflammation, excitotoxicity and
oxidative stress (35-38). Concomitant pathological changes
include hemodynamic ischemia, cerebral edema exacerbation, direct
cellular toxicity and blood-brain barrier (BBB) disruption
(1,15,18,35,39,40). Hemolytic byproducts infiltrate
the perihematomal region, activating microglia/macrophages for
hematoma clearance (7,39,41,42), while thrombin activation
contributes to vasculogenic edema via endothelial cell and BBB
damage (43). Perihematomal
edema (PHE), a hallmark of SBI linked to poorer prognosis, has
emerged as a key therapeutic target in post-ICH pathophysiology
(44-46). Additionally, intracranial
hematomas can expand into adjacent brain tissue via perivascular
spaces, white matter tracts and perineurium, distorting and
disrupting nerve fibers beyond repair (7,47,48). Collectively, these SBI-related
pathological changes culminate in permanent brain tissue damage and
severe neurological deficits, underscoring the critical need for
continuous focus on mitigating SBI mechanisms in ICH management
(43,49).
The neuroinflammatory response following ICH is a
highly dynamic and temporally regulated process, in which
microglial polarization plays a central role. Post-ICH
neuroinflammation evolves through distinct yet overlapping phases,
each characterized by specific pathological drivers, shifting
microglial phenotypes and unique molecular signatures. This
chronological progression dictates the functional outcome of
microglia, transforming them from sentinels of acute damage to
modifiers of chronic recovery. Understanding this temporal
regulation is therefore central for deciphering the dual roles of
microglia in injury exacerbation and resolution, and for designing
stage-specific therapeutic interventions.
ICH-induced hematoma represents a primary driver of
brain injury. A rapid neuroinflammatory reaction emerges within
minutes to hours post-ICH ictus, characterized by glial population
activation and the production of cytotoxic factors, reactive oxygen
species, matrix metalloproteinases and related pro-inflammatory
mediators (50). Single-cell RNA
sequencing of peri-injury cortical tissue from patients with
traumatic brain injury (TBI) and ICH identified five microglial
subpopulations: (i) Homeostatic; (ii) pro-inflammatory; (iii)
stressed; (iv) ribosome biogenesis; and (v) phagocytic (51). Specific knockout of microglial
C5aR1 ameliorated neurological outcomes in mice with TBI and ICH,
attenuating neuroinflammatory responses and reducing cerebral edema
(51). Concurrently, within
hours to days, extravasated erythrocytes in the hematoma release
neurotoxic blood products, including cytotoxic iron, heme and
hemoglobin, triggering SBI and inducing persistent cerebral edema
and progressive tissue damage (52,53). Emerging evidence highlights
regulatory mechanisms that may mitigate this process by modulating
the CCR4/ERK/Nrf2 signaling pathway and activating peroxisome
proliferator-activated receptor γ (PPAR-γ). These mechanisms
facilitate microglial phagocytosis and phenotypic polarization
toward immune homeostasis, offering potential therapeutic
strategies to counteract SBI (34,54,55).
The breakdown of the BBB and subsequent cerebral
edema represent life-threatening events in ICH (56,57). Microglia, through nuclear
receptor subfamily 4 group A member 1, can alleviate
neuroinflammation in the acute phase after stroke and ameliorate
ischemic brain injury (58).
Accumulating evidence highlights the key role of microglial
activation in driving SBI after ICH (59,60). Microglial M1 polarization
promotes the secretion of pro-inflammatory cytokines, exacerbating
neuroinflammation (61,62). Chen et al (63) demonstrated that TNF-α derived
from M1-type microglia induces endothelial necroptosis, resulting
in BBB disruption. In ischemic stroke models, anti-TNF-α therapy
significantly mitigated endothelial necroptosis, BBB damage and
neurological deficits, underscoring the causal link between
microglial inflammation and vascular pathology. Neuroprotective
agents have been shown to attenuate lipopolysaccharide-induced NO
release, TNF-α secretion and NF-κB activation in microglia, while
promoting anti-inflammatory cytokine production to protect the BBB
and facilitate neural repair (64-66). Similarly, cytokines such as IL-4
and IL-10 promote microglial polarization toward an
anti-inflammatory phenotype termed M2, emerging as potential
therapeutic targets for modulating BBB integrity after ICH
(67).
Previously, microglial subtype termed
disease-associated microglia (DAM) was identified through
transcriptional single-cell analysis in the brains of Alzheimer's
disease (AD)-transgenic (Tg-AD) mice and triggering receptor
expressed on myeloid cells 2 (TREM2)−/− Tg-AD mice
(68-70). The differentiation of microglia
into DAM occurs in two distinct phases, including an initial
TREM2-independent activation program, followed by a TREM2-dependent
maturation stage. This specialized microglial subset exhibits the
potential to limit neurodegeneration, offering critical insights
for future therapy in AD and other brain disorders (70,71). Subsequent genome-wide
transcriptomic analyses revealed the presence of DAM-like states
across diverse pathological contexts, including aging and ALS
(70). While DAM shares partial
molecular overlap with the classical M1 pro-inflammatory phenotype,
their transcriptional signatures exhibit distinct differences
(72). Notably, transcriptomic
profiling of microglial activation in AD and aging models has
revealed that DAM co-expresses anti- and pro-inflammatory
sub-programs, highlighting their dual functional complexity
(73,74). The progressive transition from
homeostatic microglia to reactive DAM is critically dependent on
TREM2, a receptor predominantly expressed on microglial surfaces
(75-77). After ICH, activated TREM2 in
perihematomal regions mitigates neuronal loss, neuroinflammation
and neurological deficits through the activation of the PI3K/Akt
signaling pathway (78). The
activation of TREM2 can significantly alter the phagocytic and
inflammatory functions of microglial cells, making it a critical
regulator of DAM polarization (79). Additionally, exosomes derived
from human-induced pluripotent stem cell-derived neural stem cells
(hiPSC-NSC-Exos) have been shown to suppress pro-inflammatory
cascades in DAM within AD mouse models (80). Wang et al (81) further demonstrated that
hiPSC-NSC-Exos stabilize the BBB by downregulating monocyte
chemoattractant protein-1 secretion in astrocytes via the PI3K/Akt
pathway activation, underscoring the therapeutic versatility of
DAM-targeted interventions across neurological diseases (81). Moreover, Gao et al
(74) reported that
transcription factors such as IRF1, CEBPα and LXRβ modulated anti-
and pro-inflammatory DAM gene expression through regulating the Erk
signaling pathway. Collectively, these findings underscore the
notable sensitivity of microglia to dynamic pathological cues in
brain tissue, emphasizing their role as highly plastic sensors and
effectors of CNS injury and disease.
Notably, several studies have explored the
spatiotemporal specialization of microglial subsets during
evolution and disease, leveraging single-cell analyses to define
precise molecular signatures and cellular dynamics (82,83). Olah et al (84) identified that 9/10 myeloid
clusters express high levels of microglia-enriched genes (such as
C1QA, C1QB, C1QC and GPR34), and they identified these nine
clusters as distinct microglial clusters. Ochocka et al
(85) demonstrated microglial
cellular and functional heterogeneity through scRNA-seq and flow
cytometry by isolating distinct microglial clusters with unique
gene expression profiles and functional roles, revealing that in
naïve and GL261 glioma-bearing mice, reactive microglia exhibited
spatially segregated distributions when analyzing CD11b+
myeloid cells. Furthermore, Hammond et al (86) and Li et al (87) revealed that microglial
heterogeneity reaches its peak during early development, indicating
that microglia exhibit diverse transcriptional states across
development, homeostasis, aging and disease (88). These findings not only identify
microglial subclasses linked to neurodegenerative diseases and
behavioral disorders but also provide a framework to dissect the
multifaceted roles of microglia in human brain pathologies
(89).
Following ICH, modulators of microglial activation
and polarization, including key signaling pathways, transcription
factors and specific markers of M1/M2 subclasses, hold clinical and
translational importance, providing a basis for modulating
microglial function to mitigate brain injury (38). While the classical M1/M2
dichotomy has provided a practical conceptual framework for
describing pro-inflammatory vs. anti-inflammatory microglial
responses following ICH, it has become increasingly clear that this
binary model oversimplifies the diversity of microglial states
in vivo. Following ICH onset, pro-inflammatory
microglia-derived mediators often overwhelm the reparative capacity
of anti-inflammatory microglia (20). Thus, identifying these modulators
and influencing factors is essential for promoting microglial
polarization toward neuroprotective subclasses, offering novel
insights into alleviating post-ICH microglial pathology. Based on
the aforementioned analyses of microglial polarization in ICH and
investigations into surface markers of microglial subtypes,
shifting microglia from a pro-inflammatory to an anti-inflammatory
state represents a critical strategy to improve outcomes after SBI
(Fig. 1).
By integrating the microglial response in ICH within
a dynamic continuum framework, the present review offers distinct
added value over traditional classification perspectives. First,
this framework can reconcile and unify seemingly contradictory
findings in the literature. For example, the differential
expression of the same marker in the injury core vs. remote
regions, or at different time points, can be interpreted as
positional shifts of the same cell population along the state
continuum. Second, it emphasizes the critical importance of
spatiotemporal dimensions by explicitly linking microglial states
to proximity to the hematoma and specific disease stages. This
directly suggests the existence of key time windows and spatial
targets for therapeutic intervention. Finally, this paradigm points
to new directions for future research: Encouraging the use of
spatial transcriptomics to map the whole-brain landscape of
microglial states post-ICH; employing single-cell multi-omics to
identify key regulatory nodes driving state transitions; and
ultimately designing next-generation smart therapies aimed at
dynamically steering microglia toward beneficial functional states,
rather than simply suppressing or activating them. Therefore,
adopting a dynamic continuum perspective not only more accurately
reflects the biological reality but also serves as a conceptual
prerequisite for advancing ICH immunotherapy from broad
interventions toward precise modulation.
Microglia do not act in isolation but orchestrate a
complex multicellular network with astrocytes, infiltrating immune
cells and endothelial cells. This crosstalk constitutes a critical
amplification loop of SBI and a potential therapeutic target after
ICH.
Activated microglia and astrocytes form a
bidirectional regulatory loop to modulate neuroinflammation and
glial scar formation. Astrocytes promote an anti-inflammatory
milieu by secreting costimulatory molecules such as CD200 and
interacting with microglia to activate anti-inflammatory cytokines
(90). In preclinical models,
C5aR1 on microglia integrates local and peripheral C5a signals,
triggering a cascade of neuroinflammation amplification, neurotoxic
astrocyte polarization and neutrophil recruitment that culminates
in cerebral edema (51). In ICH,
the C3-C3aR signaling axis, formed by astrocytic C3 and microglial
C3aR, mediates the inhibitory effect of A1 astrocytes on the
phagocytosis of myelin debris (91). The release of IL-15 from
astrocytes triggers a shift in microglia toward a pro-inflammatory
phenotype, creating a feedback loop that exacerbates
neuroinflammation following ICH (92). TRPA1 in astrocytes regulates
neuroinflammation and neurological deficits post-ICH, by
suppressing the MAPK/NF-κB signaling pathway, driving a shift
toward the neuroprotective A2 astrocytic phenotype and enhancing
the proliferation of phagocytic microglia (93). Bidirectional regulation between
microglia and astrocytes serves as a key mechanism modulating the
inflammatory response after ICH, primarily through influencing
cytokine profiles and cellular phenotypic transitions. The
crosstalk between microglia and astrocytes represents a focal point
and cutting-edge area in immunology research, with their
bidirectional modulation of phenotypic and functional states
warranting particular attention in the field of ICH.
Microglia serve as a bridge linking central and
peripheral immunity after ICH. IL-10 modulates both microglial
phagocytic function and the infiltration of monocyte-derived
macrophages following ICH, with CD36 serving as a key phagocytic
effector downstream of IL-10 signaling (94). Activin-A, derived from M2
microglia/macrophages, plays a crucial role in promoting
oligodendrocyte differentiation and thereby fostering myelin
regeneration, highlighting its potential as a therapeutic target
for white matter injury following cerebral hemorrhage (95). In PHE, crosstalk between
microglia-derived osteopontin and CD44-positive cells plays a
critical role in regulating the local immune milieu, thereby
influencing the pathological progression (96). Extracellular vesicles secreted by
bone marrow-derived macrophages have the potential to boost
microglial phagocytosis in the aftermath of ICH (97). Microglia act as an immune bridge,
regulating phagocytic function and repair processes through
specific molecules and responding to external signals such as
extracellular vesicles. However, current findings largely remain as
isolated mechanisms, and the mechanisms by which they integrate
into a coordinated spatiotemporal network within the dynamically
evolving ICH milieu remains unclear.
The communication between microglia and endothelial
cells constitutes a critical axis within the neurovascular unit,
fundamentally influencing the pathophysiology of ICH. This
bidirectional crosstalk regulates central processes including BBB
integrity, neuroinflammation and vascular repair, making it a
pivotal focus for therapeutic intervention. Their functional
interplay is facilitated by spatial proximity and involves both
contact-dependent and soluble mediators. Activated microglia can
directly influence BBB permeability through the release of
inflammatory factors and proteases interacting with endothelial
cells (67). In the context of
ICH, endothelial cells lining the choroid plexus are prone to
pyroptosis at the 24-h time point (98). ICH-induced pyroptosis in
endothelial cells is accompanied by the release of pro-inflammatory
cytokines IL-6 and IL-1β, which engage with microglia to activate
the NF-κB signaling pathway and ultimately lead to increased
transcriptional expression of Lcn2 and Msr1 (98). This initiates a cycle of
endothelial damage activating microglia, whose response further
exacerbates vascular dysfunction and inflammation. Hence,
therapeutic strategies aimed at the microglia-endothelial axis,
such as inhibiting key inflammatory pathways or protecting
endothelial health, offer a promising avenue to alleviate SBI
post-ICH.
Microglia play a dual role as both drivers of
secondary injury and promoters of repair, which presents both
challenges and opportunities for the treatment of ICH. This dual
role not only poses complex challenges for the treatment of ICH but
also identifies novel opportunities to achieve neuroprotection by
modulating microglial functions. At present, although numerous
molecular targets have been proven to regulate microglial
functions, translating such findings from preclinical models to
successful clinical applications faces a core issue: The need to
shift from merely enumerating discrete mechanisms to prioritizing
the development of therapeutic strategies based on translational
feasibility, target specificity and their integration into the
dynamic pathophysiology of ICH.
The pro-damaging properties of microglia have been
widely validated. Soluble epoxide hydrolase expression is
upregulated in microglia following ICH and potently drives
neuroinflammatory responses, whereas its inhibition suppresses
microglia-mediated inflammation (99). Additionally, deficiency of
TWIK-related K+ channel 1 exacerbates microglial and
neutrophil recruitment and pro-inflammatory factor production in
ICH-induced SBI (100).
Furthermore, microglia-expressed integrin Mac-1, acting in
conjunction with the endocytic receptor LRP1 in the neurovascular
unit, promotes thrombolytic tissue plasminogen activator
(tPA)-induced activation of platelet-derived growth factor-cc,
thereby increasing BBB permeability in ischemic stroke (56). These studies collectively
indicate that targeting the pro-inflammatory pathways of microglia
represents a viable therapeutic direction for alleviating
early-stage injury.
The anti-inflammatory and reparative potential of
microglia is equally prominent. The crosstalk between regulatory T
lymphocytes and microglial neuroinflammation has been well
documented. Studies have shown that regulatory T lymphocytes
relieve ICH-induced neuroinflammation by enhancing a phenotypic
shift from M1 to M2 microglia via modulation of the
IL-10/GSK3β/PTEN signaling pathway (101-103). This Treg-mediated
immunomodulation highlights the therapeutic potential of harnessing
microglial plasticity to drive neurorepair following ICH. However,
both Treg-based and M2-polarization strategies face notable
translational hurdles in clinical translation, including the lack
of robust, non-invasive biomarkers of microglial phenotype in
humans, the potential risk of systemic immunosuppression and
substantial uncertainty regarding the optimal intervention timing.
Simple functional inhibition or promotion may be insufficient to
address the spatiotemporal complexity of microglial dynamics
following ICH. Therefore, future development of therapeutic
strategies must undergo a fundamental shift in logic. It is
hypothesized that prioritizing and integrating intrinsic regulatory
targets that possess relative microglia selectivity, high
druggability and well-defined therapeutic time windows. This
approach should aim to develop precision interventions capable of
adapting to the pathological progression of ICH, rather than merely
describing isolated mechanisms.
Following ICH, the anti-inflammatory functions of
microglia are mediated by diverse signaling axes that form complex
networks intertwined with multiple biological processes.
Elucidating these signaling pathways and their molecular
foundations is critical for identifying promising therapeutic
targets to mitigate neuropathological deficits in ICH. Among them,
targeting intrinsic regulatory axes is particularly promising. This
approach directly modulates the cell-autonomous mechanisms of
microglia, including phagocytosis, polarization and inflammatory
signaling, by acting on their intrinsic signaling pathways. Due to
this cell-specific mode of action, it offers high target
specificity with a potentially favorable systemic side-effect
profile, representing the most promising direction for current
clinical translation efforts in post-ICH neuroinflammation.
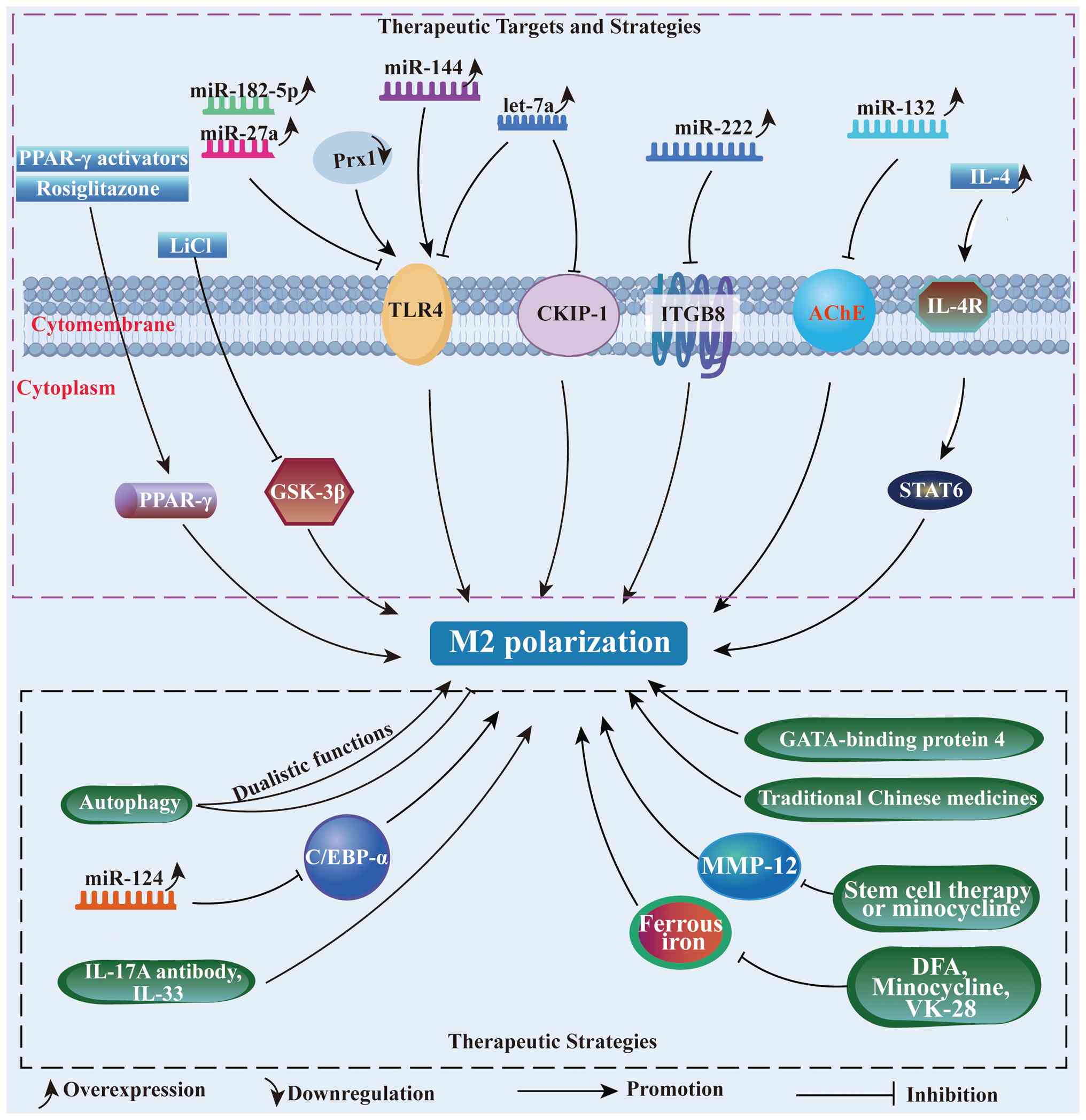
Studies have demonstrated that
rosiglitazone-mediated activation of PPAR-γ protects against BBB
damage and ameliorates hemorrhage transformation, potentially by
promoting anti-inflammatory polarization of microglia (104,105). Phagocytosis is critical for
enhancing hematoma resolution, promoting healing after ICH injury.
Zhao et al (106) found
that PPAR-γ activators significantly upregulated PPAR-γ-regulated
genes such as CD36 catalase, while downregulating pro-inflammatory
genes and alleviating neuronal injury by enhancing microglial
phagocytosis, whereas PPAR-γ gene knockdown or anti-CD36 antibody
treatment significantly impaired this phagocytic function following
ICH. PPAR-γ activation also enhances the phagocytic capacity of
anti-inflammatory microglia via CD36 (52). Building on preclinical research,
PPAR-γ activators have entered clinical applications.
Simvastatin-activated PPAR-γ improves microglia-induced erythrocyte
phagocytosis and exhibits neuroprotective effects in patients with
ICH (107,108). These findings highlight PPAR-γ
as a pivotal regulator of microglial phagocytosis and inflammation,
with therapeutic activators offering promise for clinical
translation in ICH.
Adenosine monophosphate-activated protein kinase
(AMPK) regulates the balance between pro- and anti-inflammatory
microglial subclasses by acting as a central sensor of brain injury
(109,110). Adiponectin receptor 1
(AdipoR1), constitutively expressed in microglia, is activated by
endogenous C1q/TNF-related protein 9 (CTRP9), which exhibits
increased expression within the first 24 h after ICH. CTRP9
treatment upregulates AdipoR1 and phosphorylated (p-) AMPK protein
levels while reducing inflammatory cytokines, thereby decreasing
neuroinflammation by modulating the AdipoR1/AMPK/NF-κB signaling
pathway (111). Following ICH,
CTRP9-activated AdipoR1 further mitigates neuropathological damage
and BBB dysfunction by engaging APPL1/AMPK/Nrf2 signaling,
highlighting CTRP9 as a promising therapeutic agent for improving
BBB integrity (112,113). Similarly, activation of
melanocortin receptor 4 alleviates neuropathological deficits by
regulating the AMPK signaling pathway (114). Future research should focus on
developing brain-specific AMPK activators to enhance translational
feasibility.
Inhibition of glycogen synthase kinase-3β (GSK-3β)
confers neuroprotective effects in in vivo experiments
(115-117). By enhancing microglial
phagocytosis and promoting M2-phenotype microglial differentiation,
GSK-3β inhibition significantly improves hematoma resolution and
cognitive deficits in rats with ICH (118,119). Additionally, lithium chloride
treatment downregulated GSK-3β activity, reducing mature
oligodendrocytes death and upregulating brain-derived neurotrophic
factor expression (120). Thus,
GSK-3β is classified as a medium-priority target, requiring
time-window stratified studies and dose-optimization research to
confirm its clinical applicability.
As aforementioned, regulatory T lymphocytes suppress
the microglia-induced inflammatory response and improve
neurological deficits by inhibiting NF-κB activation via the
JNK/ERK pathway (38,101,121). Hyperbaric oxygen therapy has
been shown to reduce the expression levels of pro-inflammatory
cytokines and p-JNK, proposing a link between JNK signaling and
downregulation of M1 microglial immune activity (122,123). Current clinical management of
ICH faces notable limitations, including concerns about treatment
efficacy, surgical trauma, complications and drug side effects,
with few standardized interventions available. Hyperbaric oxygen
therapy (HOT) emerges as a promising alternative approach, although
the specific mechanisms by which HOT modulates ICH pathophysiology
require further investigation and validation. These findings
highlight the need to explore non-pharmacological, pathway-targeted
strategies such as HOT to address unmet clinical needs in ICH
treatment.
Intrinsic regulatory targets with high druggability
and existing clinical repurposing potential should be prioritized
for subsequent translational studies. AMPK and GSK-3β, while
mechanistically promising, require specificity enhancement and
clinical feasibility validation to advance toward clinical
application.
Intercepting detrimental extracellular signals
targeting microglial pattern recognition receptors (PRRs) is a key
upstream intervention strategy. This approach aims to block the
initial 'trigger' of microglial activation, thereby inhibiting
downstream inflammatory cascades. However, PRRs are often widely
expressed in myeloid cells, leading to potential systemic immune
suppression, which poses a major translational issue for this class
of targets.
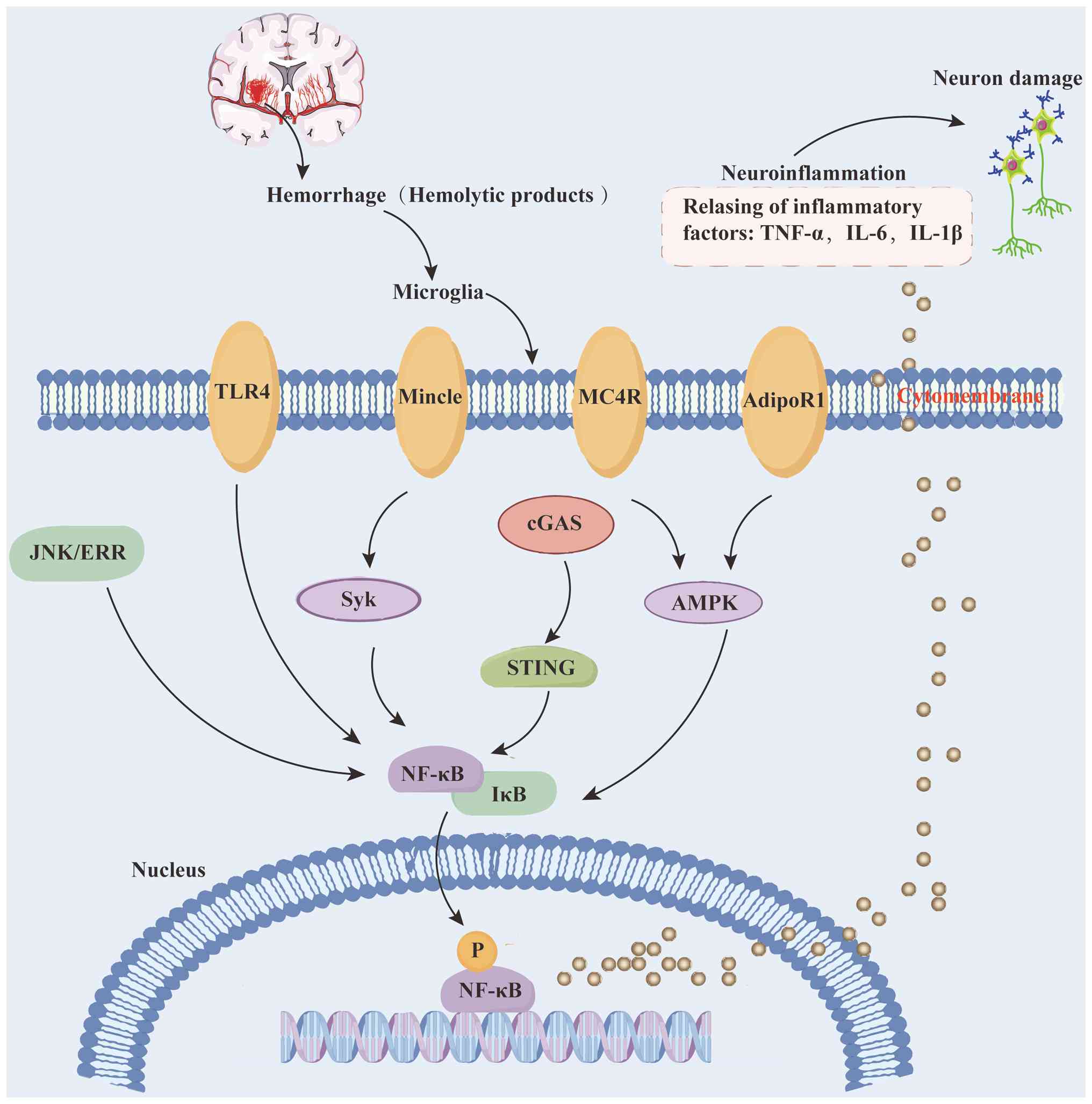
Toll-like receptor 4 (TLR4), a central player in
innate immune responses, has been recognized as a key pattern
recognition receptor (124).
TLR4 deficiency attenuated perihematomal inflammation by reducing
the infiltration of pro-inflammatory microglia in in vivo
ICH experiments (125,126). By contrast, TLR4 activation
impairs microglial phagocytosis of erythrocytes, delaying
CD36-mediated hematoma resolution and exacerbating neurological
deficits in ICH (127,128). Additionally, TLR4-driven
microglial autophagy has been linked to exacerbated
neuroinflammation in ICH mouse models (129). Collectively, these studies
highlight the dual role of TLR4 in modulating both inflammatory
recruitment and hematoma clearance after ICH. Targeting TLR4
signaling represents a promising therapeutic strategy to balance
anti-inflammatory effects with enhanced phagocytic function,
positioning it as a potential candidate for future ICH
interventions.
DNA damage initiates the innate immune response,
with cGAS, a key DNA sensor, detecting disease-associated damaged
DNA to activate its downstream effector, STING. This cascade
results in the upregulation of type I interferon production and
phosphorylation of interferon regulatory factor 3 (130). cGAS acts as a critical
regulator of inflammation and autophagy, with upregulated cGAS in
striatal brain damage mediating inflammation via increased
expression of pro-inflammatory genes (Ccl5 and Cxcl10) and
activating autophagy through the major initiators LC3A and LC3B
(131). In ischemic stroke
models, tPA therapy enhances neutrophil extracellular trap
expression in plasma and brain tissue. DNase I treatment or
peptidyl-arginine deiminase 4 deficiency, both of which inhibit
neutrophil extracellular trap formation, reverse tPA-induced cGAS
upregulation. By contrast, in tPA-treated mice, cGAMP (a
cGAS-derived second messenger) suppresses DNase I-induced
anti-hemorrhagic effects by dampening STING and type I interferon
signaling (132). Jiang et
al (133) further
demonstrated that cGAS knockdown furthers M2 microglial
polarization and attenuates the inflammatory response by disrupting
the cGAS-STING axis in stroke mice. Shi et al (134) extended these findings,
demonstrating that a cGAS inhibitor delivered via immunosuppressive
nanoparticles reduces microglial inflammation and enhances
anti-inflammatory polarization in post-stroke rats. IronQ
pretreatment confers mesenchymal stem cells (MSCs) with a
synergistic effect to alleviate neuroinflammation and improve
neurological function, achieved by inhibiting the cGAS-STING
signaling pathway (135).
C-type lectin-like receptors, a family of
transmembrane PRRs predominantly expressed in myeloid cells, play a
critical role in innate immunity (136). Dysregulation of C-type
lectin-like receptors following tissue injury can drive excessive
production of inflammatory mediators and exacerbate inflammatory
progression. Among C-type lectin-like receptors, microglial
macrophage-inducible C-type lectin (Mincle), widely expressed on
macrophages, binds nuclear spliceosome-associated protein 130
released from necrotic cells, potently enhancing the inflammatory
response (137,138). In alcohol-induced liver injury
models, Mincle activation via its downstream effector spleen
tyrosine kinase (Syk) upregulates the expression levels of
pro-inflammatory genes (139),
while in Crohn's disease, the Mincle/Syk axis exacerbates
intestinal mucosal inflammation through macrophage pyroptosis
(140). Inhibiting this pathway
attenuates inflammatory responses, highlighting its therapeutic
potential. Preclinical studies across multiple neurological
disorders have demonstrated that blocking the Mincle/Syk signaling
axis confers neuroprotection (141). For example, interventions
including the Syk inhibitor BAY61-3606, acupuncture and MSC
transplantation suppress Mincle/Syk-mediated microglial activation,
reducing neuroinflammation in in vivo stroke models
(138,141-145). Collectively, these findings
establish the Mincle/Syk pathway as a promising therapeutic target
for ICH, offering a rationale for developing novel immunomodulatory
strategies to mitigate post-ICH neuroinflammation and promote
neurological recovery.
To date, autophagy exhibits dualistic functions,
making it challenging to assess whether its effects after ICH are
predominantly harmful or beneficial. Excessive autophagy
exacerbates endoplasmic reticulum stress (ERS)-induced brain injury
as early as 6 h following ICH. By contrast, at day 7 post-ICH in
rats, autophagy strengthens ERS protective functions by clearing
cellular debris (146). Tan
et al (147)
demonstrated that enhanced autophagy attenuates oxidative stress
injury following ICH by upregulating antioxidant protein
expression, while autophagy inhibitors reverse this neuroprotective
effect. Previous studies further indicate that autophagy positively
regulates the inflammatory response in the context of ICH (148,149).
The levels of ILs during ICH progression are
modulated by microglial functions. Xu et al (150) demonstrated that IL-4-loaded
nanoparticles activating the IL-4/STAT6 axis promoted hematoma
resolution and functional recovery in mice with ICH. By contrast,
IL-15, a pro-inflammatory cytokine, regulates homeostasis and
microglial immunoreactivity following CNS inflammatory events
(92). Yu et al (151) showed that an
IL-17A-neutralizing antibody attenuates microglial activation and
blocks ICH-mediated pro-inflammatory cytokines. Shi et al
(152) further revealed that
IL-17A enhances microglial autophagy and neuroinflammation.
Administration of an IL-17A-neutralizing antibody significantly
reduced brain edema and improved neurological outcomes in mice with
ICH, while genetic inhibition of autophagy-related genes ATG5 and
ATG7 suppressed microglial autophagy and inflammation (153). Intraventricular injection of
IL-33 alleviated white matter and neuronal injury by promoting
microglial M2 polarization following ICH (154). Additionally, inhibiting the
NF-κB signaling pathway through diverse interventions, including
microRNAs (miRNAs or miRs), GATA-binding protein 4 and traditional
Chinese medicines, can reduce neuroinflammation in ICH therapy
(155-159). These findings highlight the
bidirectional role of ILs and NF-κB in regulating microglial
phenotypes and underscore their therapeutic potential in modulating
neuroinflammation after ICH.
Intercepting extracellular signals via PRRs
represents a rational upstream therapeutic strategy, yet its
translational potential is constrained by insufficient target
specificity and limited validation in ICH-specific models.
Therefore, future research should focus on developing
microglia-specific PRR modulators and conduct in-depth validation
in clinically relevant ICH models. In summary, although extensive
preclinical studies have made notable progress at the mechanistic
level, successfully translating these findings into clinical
applications requires further systematic investigation of promising
interventions (Fig. 2).
Exosomes and miRNAs represent emerging precision
therapeutic tools for modulating microglia after ICH, offering
advantages such as low immunogenicity, high target specificity and
the ability to regulate multiple pathways simultaneously, thereby
effectively addressing the spatiotemporal complexity of microglial
dynamics post-ICH. To translate these promising tools into precise
therapies, a rational framework is required. This involves
identifying key molecular targets within microglial dynamic
continuum, categorizing them based on their pathological function,
and employing engineered exosomes for spatiotemporally controlled
delivery. Current evidence points to three strategic categories of
targets: (i) Inflammatory initiators (for example, TLR4), the
suppression of which quenches upstream danger signaling; (ii)
polarization nodes (for example, CKIP-1 and C/EBP-α), the
modulation of which actively reprogrammes microglia toward a
reparative phenotype; and (iii) Cellular stress regulators (for
example, mTOR), the intervention of which maintains microglial
homeostasis. The convergence of exosome biology and miRNA
targetology offers a unique path to address these targets in a
combined and specific manner.
Given their minimal risks of immunogenicity and
tumorigenicity, exosomes secreted by human umbilical cord
mesenchymal stem cells (hUCMSC-exo) represent a promising
alternative to cell-based therapies. Evidence indicates that
hUCMSC-exo administration can alleviate astrocyte inflammation and
mitochondrial damage associated with ICH by inhibiting the
TLR4/NF-κB signaling pathway (160). Exosomal PTEN-induced kinase 1
from hUCMSCs attenuates neurological deficits, dysregulated
microglial M1/M2 polarization and inflammatory responses after ICH
in mice (161). Exosomes
derived from human adipose-derived mesenchymal stem cells
(hADSCs-Exo) were successfully internalized by microglia cells and
exerted robust anti-inflammatory actions by suppressing the exerted
robust anti-inflammatory actions of inflammatory factors and
promoting M1 to M2 transition (162). Exosomes, which are nanoscale
vesicles measuring 30-150 nm in diameter, encapsulate a diverse
cargo encompassing mRNA, miRNA, proteins and growth factors
(163,164). Beyond serving as delivery
vehicles, the specific contents of exosomes, especially miRNAs, are
pivotal for their immunomodulatory effects. Consequently,
identifying key miRNAs and elucidating their mechanisms in
microglial polarization has become a major research frontier in
developing RNA-based therapeutics for ICH.
Increasing genetic and epigenetic evidence
highlights the critical role of miRNAs in modulating gene
expression and microglial polarization following ICH (165). For example, let-7a modulates
microglial M2 polarization by targeting CKIP-1 at day 3 post-ICH in
mice. In the aforementioned study, let-7a overexpression reduced
CKIP-1 protein levels, promoted M2 polarization characterized by
increased expression of IL-10 and Arg-1, and alleviated
neuroinflammation. By contrast, let-7a inhibition upregulated
CKIP-1, driving M1 polarization (165). Similarly, miR-7 overexpression
suppresses TLR4 protein expression, attenuating microglial
inflammation in both ICH rats and lipoprotein-stimulated microglial
inflammation models (166).
Further research has demonstrated that targeting TLR4 through the
inhibition of the Prx1/TLR4/NF-κB signaling axis at day 3 post-ICH
provides neuroprotection against ICH-induced brain injury,
presenting a promising anti-neuroinflammatory strategy (155). In stroke in vivo and
in vitro experiments, miR-182-5p and miR-27a modulate
inflammation by targeting TLR4, and their overexpression
downregulates TLR4 protein levels while reducing the release of
pro-inflammatory factors (167,168).
Previous evidence identified that integrin subunit
β8, a direct target of miR-222, can be negatively regulated by
miR-222 to alleviate microglial inflammation and apoptosis
(169). Previously, miR-132 was
shown to inhibit the cholinergic inflammatory response by targeting
acetylcholinesterase. Zhang et al (170) reported that lentivirus-mediated
miR-132 overexpression in the right caudate nucleus 14 days before
autologous blood-induced ICH suppressed pro-inflammatory microglial
activation, alleviated BBB dysfunction and reduced neuronal
reduction at day 3 post-ICH. miRNAs also act as key mediators of
autophagy-induced microglial inflammation, post-transcriptionally
suppressing gene expression and function (171). It has been demonstrated that
miR-144 enhances hemoglobin-mediated microglial autophagic
inflammation by directly targeting the 3' untranslated regions
(UTRs) of mTOR (171,172). Similarly, miR-124 promotes
microglial M2 polarization and alleviates inflammation by targeting
the 3'-UTR of C/EBP-α. In ICH mice, miR-124 mimic administration
significantly reduced neurological deficits, brain water content
(BWC) and C/EBP-α expression at day 3, compared with miR-124
inhibitor treatment. Consistent negative regulation of C/EBP-α by
miR-124 was also observed in erythrocyte lysate-stimulated
microglia transduced with miR-124 mimics or inhibitors (173).
Exosomes and miRNAs represent more than just a list
of promising entities; they form the core of an emerging precision
immunomodulation paradigm for ICH. Moving beyond the generic
suppression of neuroinflammation, this paradigm is defined by the
rational selection of targets across the microglial functional
spectrum (for example, TLR4, CKIP-1 and mTOR) and their precise
manipulation via engineered exosomes loaded with specific miRNA
combinations. The primary translational challenge no longer lies
solely in target identification, but in optimizing these
intelligent delivery systems to ensure their stability, targeting
efficiency, and controlled cargo release, and in validating their
synergistic efficacy in advanced disease models. This approach
heralds a shift from broad anti-inflammatory strategies toward
context-sensitive immune remodeling tailored to the spatiotemporal
dynamics of ICH.
Modulating downstream effector molecules targets
the terminal pathological processes of microglial activation,
aiming to block the final 'damage cascade' rather than regulating
microglial function directly. This strategy is characterized by
clear mechanisms of action but may lack specificity due to the
widespread involvement of effector molecules in normal
physiological processes.
Matrix metalloproteinases (MMPs), a universal
superfamily of structurally related zinc-dependent endopeptidases,
are upregulated following ICH and contribute to extracellular
matrix degradation. The role of MMPs in ECM disruption driven by
inflammation serves as a pathological basis for stroke, as
evidenced in numerous studies (174). Wells et al (175) investigated MMP function in ICH
mice and found that MMP-12 was the most significantly elevated
isoform, with experiments revealing that MMP-12 knockout mice
exhibited substantial forelimb motor recovery compared with WT
mice, while WT mice showed increased recruitment of
Iba1+ cells with macrophage morphology to the injury
site, indicating that MMP-12 exacerbates SBI after ICH (175). Subsequent studies demonstrated
that stem cell therapy or minocycline administration following ICH
reduced perihematomal MMP-12 expression and attenuated
microglia-infiltrated inflammatory responses (176,177), positioning MMP-induced
microglial activation as a potential therapeutic target. For
example, minocycline promotes the microglial phenotypic shift from
pro-inflammatory M1 to anti-inflammatory M2 (178). However, while minocycline
therapy effectively mitigates the early elevation of MMP-12 and
TNF-α, its therapeutic efficacy diminishes by 1-week post-ICH.
Additional studies have revealed that stem cell therapy notably
reduces microglial infiltration and MMP-12 expression in
perihematomal regions after ICH (177,179).
Increasing evidence indicates that ferrous iron
released from hemolysis represents a key pathogenic factor in
hematoma following ICH (180).
Iron toxicity-mediated microglial activation and pro-inflammatory
response are major contributors to brain injury after ICH.
Deferoxamine, an iron chelator capable of crossing the BBB to bind
iron, has been identified to moderately modulate microglial
activation following ICH when administered intraperitoneally to
decrease iron accumulation (181-183). As a microglial activation
inhibitor, minocycline can also reduce iron levels to prevent
neuronal death after ICH (184,185). Similarly, VK-28, a
brain-permeable iron chelator, promotes microglial M2 phenotype
polarization, reduces BWC, alleviates white matter injury and
improves neurobehavioral deficits after ICH (186,187). Observational studies highlight
the complexity of regulatory systems governing microglial function,
underscoring the critical need to decipher phenotypic and genotypic
variations to develop promising therapeutic strategies for ICH.
Modulating downstream effector molecules offers
mechanism-specific intervention for post-ICH injury. Iron chelation
is a high-priority strategy due to its ICH-specific mechanisms and
existing clinical drug availability, while MMP-12 is limited by
narrow intervention windows and poor specificity. For downstream
targets, defining precise intervention timelines (aligned with
pathological progression) is critical for balancing therapeutic
efficacy and safety. Therefore, advancing these downstream
strategies into the clinic critically depends on translational
research that rigorously defines their optimal therapeutic windows
in complex, time-course models (Fig.
3).
The core academic contribution of this review lies
in the establishment of an innovative conceptual framework for the
hierarchical targeted regulation of microglial neuroinflammation,
which systematically categorizes regulatory strategies into four
dimensions: Targeting the intrinsic regulatory axes of microglia,
intercepting detrimental extracellular signals, implementing
precise interventions with exosomes and miRNAs, and regulating
downstream effector molecules, thus achieving a logical upgrade
from mechanistic description to translation-oriented research. Its
unique research perspective is reflected in taking translational
feasibility, targeting specificity and pathological dynamic
adaptability as the core criteria to clarify the regulatory
priority and clinical translational potential of each signaling
axis; it combines with the time dependence of pathological
progression in ICH to distinguish the intervention windows and
applicable scenarios of different regulatory strategies, and
meanwhile focuses on the key challenges in translational medicine
to analyze the clinical translational bottlenecks and optimization
directions of various strategies. In summary, the present review
systematically integrates the latest regulatory mechanisms of
microglial neuroinflammation following ICH.
ICH is distinguished by the concurrent presence of
hyperdense and hypodense areas within a single hematoma, resulting
in a 'blended' appearance, with hyperdense areas typically
indicative of more coagulated or organized clots and hypodense
areas often signifying relatively newly extravasated, incompletely
coagulated blood (188,189). The upregulation of MAPT, CYCS,
GAP43 and MAP1B, coupled with the enrichment of ferroptotic,
mitochondrial dysfunction-related, oxidative stress-associated,
inflammatory and cytoskeletal pathways, suggests that the intricate
biological processes occurring within hematomas (190). While this association
establishes a theoretical bridge between radiology and molecular
biology, direct causal evidence verifying the mechanisms by which
these genetic and pathway alterations drive the formation of
hyperdense-hypodense regions remains scarce.
In terms of diagnostic and discriminative value,
multiple molecular biomarkers exhibit promising performance in ICH
management. Serum levels of matrix MMP-9, VEGF, GFAP and S100
calcium-binding protein B are significantly higher in patients with
ICH, and the serum levels of these molecular biomarkers are
correlated with a larger volume of PHE. The biomarkers GFAP, MMP-9
and APO-C1 independently discriminated between ischemic stroke and
ICH within 24 h and markedly boosted the discriminative capacity of
predictive models (191). The
cerebral blood flow-based neurological score serves as an
independent predictor of post-treatment ICH risk in adult patients
with moyamoya disease (192).
Despite these encouraging results, the specificity of these
biomarkers remains incompletely validated. Notably, key
inflammatory mediators such as MMP-9 and VEGF are also elevated in
other acute cerebrovascular events and systemic inflammatory
conditions. This overlap may limit their discriminative accuracy,
especially in clinically heterogeneous patient populations.
Furthermore, most supporting evidence originates from single-center
studies with limited sample sizes. Therefore, multicenter,
prospective validation is essential to confirm their true clinical
applicability.
The association between molecular signatures and
ICH prognosis further highlights the translational potential of
these biomarkers. Elevation of inflammatory biomarkers is
associated with adverse outcomes in ICH (193), and serum YKL-40 levels may
serve as a candidate prognostic biomarker for the recurrence of
cerebral amyloid angiopathy-related ICH (194). Moreover, the onset of acute
anemia following ICH is prevalent, rapidly progressive and closely
linked to inflammatory processes (195). Given that anemia development is
associated with unfavorable clinical outcomes, it is proposed as a
potential modifiable target to improve patient prognosis. The
current understanding of anemia as a therapeutic target remains
superficial as most studies only confirm the correlation between
anemia and poor outcomes but lack interventional trials to verify
whether correcting anemia can ameliorate ICH prognosis.
For early diagnosis and prevention, a panel of
novel biomarkers has emerged as complementary tools when computed
tomography and/or magnetic resonance imaging (MRI) are either
unavailable or fail to reveal distinct signs of potential vascular
rupture. To facilitate early diagnosis, ICH prevention and predict
patient prognosis following hemorrhagic events, several parameters
can be assessed including: Red cell distribution width, Red cell
distribution width to lymphocyte ratio, resolvin D2 levels,
C-reactive protein to albumin ratio levels, nucleotide-binding
oligomerization domain-like receptor family pyrin domain-containing
1 levels, and the precise and reliable assessment of circulating
miRNAs, long non-coding RNA, circular RNA and mRNA expression in
biological fluids (196-202).
Furthermore, an in-depth investigation into differentially
expressed inflammatory proteins, serum secretoneurin, and levels of
soluble TLR4 and TLR2 in the early stage of ICH will facilitate an
improved understanding of the pathogenic mechanisms underlying ICH,
enable more accurate prediction of patient prognosis and facilitate
the exploration of novel therapeutic strategies (198,203-205). A major difficulty in
translating these biomarkers to clinical practice is the lack of
standardized detection protocols, as different laboratories adopt
varying assay methods, leading to inconsistent results that hinder
cross-study comparison. Additionally, the optimal combination of
these biomarkers for multi-marker diagnosis has not been
systematically determined, and cost-effectiveness analysis is
needed to evaluate their feasibility in resource-limited clinical
settings.
Emerging evidence indicates that pro-inflammatory
and anti-inflammatory microglial phenotypes exhibit divergent
functions and implications, offering mechanistic insights into
microglial regulation through intracellular and extracellular
signaling pathways. Post-ICH brain injury and repair are not driven
by isolated cellular responses, but are orchestrated by a highly
dynamic, microglia-centered neuroimmune network. The functional
regulation of microglia after ICH is a central therapeutic target
for neuroinflammation. In the present review, a comprehensive
overview of the cellular and molecular mechanisms governing
microglial activation after ICH was provided. Substantial progress
has been made in identifying signaling molecular pathways linked to
SBI following ICH in preclinical studies. PPAR-γ agonists,
microglia-targeted exosomes and TLR4-specific inhibitors may
represent the most promising directions in terms of current
translational potential. However, extended clinical trials are
essential for further validation.
The present review had several critical challenges
which must be addressed. First, although animal models are widely
used to investigate ICH etiology and pathophysiology, current
models, predominantly rodent-based, do not fully recapitulate the
complex etiology of human spontaneous ICH. Rodents exhibit robust
spontaneous sensorimotor recovery and limited cognitive
dysfunction, failing to reflect multifaceted clinical risk factors
(such as hypertension and anticoagulant use). Second, experimental
models do not fully replicate the pathological complexity of human
ICH. To address this, preclinical research increasingly employs
large animal models and advanced 3.0T MRI techniques to
characterize key parameters such as hematoma expansion, white
matter injury and hematoma evacuation, enabling cross-species
comparisons with human disease. Third, most studies on ICH-induced
brain injury focus on single-factor interventions. Future research
should prioritize developing multi-targeted agents or combinatorial
therapies to address the interconnected pathways driving
neuroinflammation and tissue damage. While the current landscape of
robust clinical trials remains limited and molecular genetic
investigations into microglial phenotypic transitions are lacking,
the present review offers an optimistic perspective on practical
intervention strategies targeting microglial function in ICH.
Further research into therapeutic strategies addressing microglial
activation-driven neuroinflammation is critical for evaluating the
translational potential of these promising approaches.
Not applicable.
GY, GC and XB conceptualized the study. XF, CP, LZ
and OW wrote and prepared the original draft of the manuscript. DL
and BZ wrote, reviewed and edited the manuscript. GY and XF
prepared figures. XF, GY and XB acquired funding. All authors read
and approved the final version of the manuscript. Data
authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by The National Natural Science
Foundation of China (grant no. 82200361), the Sichuan Science and
Technology Program (grant no. 2025NSFSC2140), the Health Commission
of Sichuan Province Medical Science and Technology Program (grant
no. 24QNMP012), the Brain Disease Innovation Team of the Affiliated
Traditional Chinese Medicine Hospital of Southwest Medical
University (grant no. 2022-CXTD-05), the Luzhou Science and
Technology Program (grant no. 2025LZXNYDJC53), the Xuyong People's
Hospital-Southwest Medical University Strategic Cooperation in
Science and Technology (grant no. 2025XYXNYD13) and the Southwest
Medical University Technology Program (grant no. 2024ZXYZX14).
|
1
|
Keep RF, Hua Y and Xi G: Intracerebral
haemorrhage: Mechanisms of injury and therapeutic targets. Lancet
Neurol. 11:720–731. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schrag M and Kirshner H: Management of
intracerebral hemorrhage: JACC focus seminar. J Am Coll Cardiol.
75:1819–1831. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rajashekar D and Liang JW: Intracerebral
Hemorrhage. StatPearls; Treasure Island, FL: 2021
|
|
4
|
Jain A, Malhotra A and Payabvash S:
Imaging of spontaneous intracerebral hemorrhage. Neuroimaging Clin
N Am. 31:193–203. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Witsch J, Siegerink B, Nolte CH, Sprügel
M, Steiner T, Endres M and Huttner HB: Prognostication after
intracerebral hemorrhage: A review. Neurol Res Pract. 3:222021.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Biffi A, Anderson CD, Battey TW, Ayres AM,
Greenberg SM, Viswanathan A and Rosand J: Association between blood
pressure control and risk of recurrent intracerebral hemorrhage.
JAMA. 314:904–912. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bosche B, Mergenthaler P, Doeppner TR,
Hescheler J and Molcanyi M: Complex clearance mechanisms after
intraventricular hemorrhage and rt-PA treatment-a review on
clinical trials. Transl Stroke Res. 11:337–344. 2020. View Article : Google Scholar
|
|
8
|
Wu S and Anderson CS: A need to re-focus
efforts to improve long-term prognosis after stroke in China.
Lancet Glob Health. 8:e468–e469. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
van Asch CJ, Luitse MJ, Rinkel GJ, van der
Tweel I, Algra A and Klijn CJ: Incidence, case fatality, and
functional outcome of intracerebral haemorrhage over time,
according to age, sex, and ethnic origin: a systematic review and
meta-analysis. Lancet Neurol. 9:167–176. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Carpenter AM, Singh IP, Gandhi CD and
Prestigiacomo CJ: Genetic risk factors for spontaneous
intracerebral haemorrhage. Nat Rev Neurol. 12:40–49. 2016.
View Article : Google Scholar
|
|
11
|
Wu S, Wu B, Liu M, Chen Z, Wang W,
Anderson CS, Sandercock P, Wang Y, Huang Y, Cui L, et al: Stroke in
China: Advances and challenges in epidemiology, prevention, and
management. Lancet Neurol. 18:394–405. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Alharbi BM, Tso MK and Macdonald RL:
Animal models of spontaneous intracerebral hemorrhage. Neurol Res.
38:448–455. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen-Roetling J, Li Y, Cao Y, Yan Z, Lu X
and Regan RF: Effect of hemopexin treatment on outcome after
intracerebral hemorrhage in mice. Brain Res. 1765:1475072021.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bentz K, Molcanyi M, Schneider A, Riess P,
Maegele M, Bosche B, Hampl JA, Hescheler J, Patz S and Schäfer U:
Extract derived from rat brains in the acute phase following
traumatic brain injury impairs survival of undifferentiated stem
cells and induces rapid differentiation of surviving cells. Cell
Physiol Biochem. 26:821–830. 2010. View Article : Google Scholar
|
|
15
|
Xiong XY, Wang J, Qian ZM and Yang QW:
Iron and intracerebral hemorrhage: From mechanism to translation.
Transl Stroke Res. 5:429–441. 2014. View Article : Google Scholar
|
|
16
|
Ziai WC: Hematology and inflammatory
signaling of intracerebral hemorrhage. Stroke. 44(Suppl 1):
S74–S78. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chiang PT, Tsai LK and Tsai HH: New
targets in spontaneous intracerebral hemorrhage. Curr Opin Neurol.
38:10–17. 2025. View Article : Google Scholar :
|
|
18
|
Chen S, Yang Q, Chen G and Zhang JH: An
update on inflammation in the acute phase of intracerebral
hemorrhage. Transl Stroke Res. 6:4–8. 2015. View Article : Google Scholar
|
|
19
|
Liu J, Zhu Z and Leung GK:
Erythrophagocytosis by microglia/macrophage in intracerebral
hemorrhage: From mechanisms to translation. Front Cell Neurosci.
16:8186022022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bai Q, Xue M and Yong VW: Microglia and
macrophage phenotypes in intracerebral haemorrhage injury:
Therapeutic opportunities. Brain. 143:1297–1314. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Eldahshan W, Fagan SC and Ergul A:
Inflammation within the neurovascular unit: Focus on microglia for
stroke injury and recovery. Pharmacol Res. 147:1043492019.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bian Z, Gong Y, Huang T, Lee CZW, Bian L,
Bai Z, Shi H, Zeng Y, Liu C, He J, et al: Deciphering human
macrophage development at single-cell resolution. Nature.
582:571–576. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Prinz M, Jung S and Priller J: Microglia
biology: One century of evolving concepts. Cell. 179:292–311. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wolf SA, Boddeke HW and Kettenmann H:
Microglia in physiology and disease. Annu Rev Physiol. 79:619–643.
2017. View Article : Google Scholar
|
|
25
|
Friedman BA, Srinivasan K, Ayalon G,
Meilandt WJ, Lin H, Huntley MA, Cao Y, Lee SH, Haddick PCG, Ngu H,
et al: Diverse brain myeloid expression profiles reveal distinct
microglial activation states and aspects of Alzheimer's disease not
evident in mouse models. Cell Rep. 22:832–847. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang M, Hua Y, Keep RF, Wan S, Novakovic N
and Xi G: Complement inhibition attenuates early erythrolysis in
the hematoma and brain injury in aged rats. Stroke. 50:1859–1868.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vinukonda G, Liao Y, Hu F, Ivanova L,
Purohit D, Finkel DA, Giri P, Bapatla L, Shah S, Zia MT, et al:
Human cord blood-derived unrestricted somatic stem cell infusion
improves neurobehavioral outcome in a rabbit model of
intraventricular hemorrhage. Stem Cells Transl Med. 8:1157–1169.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chang CF, Wan J, Li Q, Renfroe SC, Heller
NM and Wang J: Alternative activation-skewed microglia/macrophages
promote hematoma resolution in experimental intracerebral
hemorrhage. Neurobiol Dis. 103:54–69. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sekerdag E, Solaroglu I and Gursoy-Ozdemir
Y: Cell death mechanisms in stroke and novel molecular and cellular
treatment options. Curr Neuropharmacol. 16:1396–1415. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jin WN, Shi SX, Li Z, Li M, Wood K,
Gonzales RJ and Liu Q: Depletion of microglia exacerbates
postischemic inflammation and brain injury. J Cereb Blood Flow
Metab. 37:2224–2236. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang X, Ren H, Wood K, Li M, Qiu S, Shi
FD, Ma C and Liu Q: Depletion of microglia augments the
dopaminergic neurotoxicity of MPTP. FASEB J. 32:3336–3345. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Baang HY and Sheth KN: Stroke prevention
after intracerebral hemorrhage: Where are we now? Curr Cardiol Rep.
23:1622021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Huang AP, Hsu YH, Wu MS, Tsai HH, Su CY,
Ling TY, Hsu SH and Lai DM: Potential of stem cell therapy in
intracerebral hemorrhage. Mol Biol Rep. 47:4671–4680. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tschoe C, Bushnell CD, Duncan PW,
Alexander-Miller MA and Wolfe SQ: Neuroinflammation after
intracerebral hemorrhage and potential therapeutic targets. J
Stroke. 22:29–46. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Aronowski J and Zhao X: Molecular
pathophysiology of cerebral hemorrhage: Secondary brain injury.
Stroke. 42:1781–1786. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Babu R, Bagley JH, Di C, Friedman AH and
Adamson C: Thrombin and hemin as central factors in the mechanisms
of intracerebral hemorrhage-induced secondary brain injury and as
potential targets for intervention. Neurosurg Focus. 32:E82012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Duan X, Wen Z, Shen H, Shen M and Chen G:
Intracerebral hemorrhage, oxidative stress, and antioxidant
therapy. Oxid Med Cell Longev. 2016:12032852016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lan X, Han X, Li Q, Yang QW and Wang J:
Modulators of microglial activation and polarization after
intracerebral haemorrhage. Nat Rev Neurol. 13:420–433. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zheng Y, Tan X and Cao S: The critical
role of erythrolysis and microglia/macrophages in clot resolution
after intracerebral hemorrhage: A review of the mechanisms and
potential therapeutic targets. Cell Mol Neurobiol. 43:59–67. 2023.
View Article : Google Scholar
|
|
40
|
Mohammed Thangameeran SI, Tsai ST, Hung
HY, Hu WF, Pang CY, Chen SY and Liew HK: A role for endoplasmic
reticulum stress in intracerebral hemorrhage. Cells. 9:7502020.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lua J, Ekanayake K, Fangman M and Dore S:
Potential role of soluble toll-like receptors 2 and 4 as
therapeutic agents in stroke and brain hemorrhage. Int J Mol Sci.
22:99772021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wei Y, Song X, Gao Y, Gao Y, Li Y and Gu
L: Iron toxicity in intracerebral hemorrhage: Physiopathological
and therapeutic implications. Brain Res Bull. 178:144–154. 2022.
View Article : Google Scholar
|
|
43
|
Wilkinson DA, Pandey AS, Thompson BG, Keep
RF, Hua Y and Xi G: Injury mechanisms in acute intracerebral
hemorrhage. Neuropharmacology. 134:240–248. 2018. View Article : Google Scholar
|
|
44
|
Chen Y, Chen S, Chang J, Wei J, Feng M and
Wang R: perihematomal edema after intracerebral hemorrhage: An
update on pathogenesis, risk factors, and therapeutic advances.
Front Immunol. 12:7406322021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bautista W, Adelson PD, Bicher N,
Themistocleous M, Tsivgoulis G and Chang JJ: Secondary mechanisms
of injury and viable pathophysiological targets in intracerebral
hemorrhage. Ther Adv Neurol Disord. 14:175628642110492082021.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Brouwers HB and Greenberg SM: Hematoma
expansion following acute intracerebral hemorrhage. Cerebrovasc
Dis. 35:195–201. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yin J, Lu TM, Qiu G, Huang RY, Fang M,
Wang YY, Xiao D and Liu XJ: Intracerebral hematoma extends via
perivascular spaces and perineurium. Tohoku J Exp Med. 230:133–139.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fu X, Zhou G, Zhuang J, Xu C, Zhou H, Peng
Y, Cao Y, Zeng H, Li J, Yan F, et al: White matter injury after
intracerebral hemorrhage. Front Neurol. 12:5620902021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Qureshi AI, Mendelow AD and Hanley DF:
Intracerebral haemorrhage. Lancet. 373:1632–1644. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chen W, Guo C, Huang S, Jia Z, Wang J,
Zhong J, Ge H, Yuan J, Chen T, Liu X, et al: MitoQ attenuates brain
damage by polarizing microglia towards the M2 phenotype through
inhibition of the NLRP3 inflammasome after ICH. Pharmacol Res.
161:1051222020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhou J, Ma S, Feng D, Tian Y, Li L, Guo H,
Shi Y, Cui W, Dong J, Hao S, et al: C5aR1(+) microglia exacerbate
neuroinflammation and cerebral edema in acute brain injury. Neuron.
114:444–462.e9. 2026. View Article : Google Scholar
|
|
52
|
Zhao X, Grotta J, Gonzales N and Aronowski
J: Hematoma resolution as a therapeutic target: The role of
microglia/macrophages. Stroke. 40(Suppl 3): S92–S94. 2009.
View Article : Google Scholar
|
|
53
|
Wang G, Wang L, Sun XG and Tang J:
Haematoma scavenging in intracerebral haemorrhage: from mechanisms
to the clinic. J Cell Mol Med. 22:768–777. 2018. View Article : Google Scholar :
|
|
54
|
Deng S, Sherchan P, Jin P, Huang L, Travis
Z, Zhang JH, Gong Y and Tang J: Recombinant CCL17 enhances hematoma
resolution and activation of CCR4/ERK/Nrf2/CD163 signaling pathway
after intracerebral hemorrhage in mice. Neurotherapeutics.
17:1940–1953. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhuang J, Peng Y, Gu C, Chen H, Lin Z,
Zhou H, Wu X, Li J, Yu X, Cao Y, et al: Wogonin accelerates
hematoma clearance and improves neurological outcome via the PPAR-ү
pathway after intracerebral hemorrhage. Transl Stroke Res.
12:660–675. 2021. View Article : Google Scholar
|
|
56
|
Su EJ, Cao C, Fredriksson L, Nilsson I,
Stefanitsch C, Stevenson TK, Zhao J, Ragsdale M, Sun YY, Yepes M,
et al: Microglial-mediated PDGF-CC activation increases
cerebrovascular permeability during ischemic stroke. Acta
Neuropathol. 134:585–604. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Fang Y, Gao S, Wang X, Cao Y, Lu J, Chen
S, Lenahan C, Zhang JH, Shao A and Zhang J: Programmed cell deaths
and potential crosstalk with blood-brain barrier dysfunction after
hemorrhagic stroke. Front Cell Neurosci. 14:682020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Liu P, Chen Y, Zhang Z, Yuan Z, Sun JG,
Xia S, Cao X, Chen J, Zhang CJ, Chen Y, et al: Noncanonical
contribution of microglial transcription factor NR4A1 to
post-stroke recovery through TNF mRNA destabilization. PLoS Biol.
21:e30021992023. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Carson MJ, Doose JM, Melchior B, Schmid CD
and Ploix CC: CNS immune privilege: Hiding in plain sight. Immunol
Rev. 213:48–65. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Gong Y, Li H, Cui H and Gong Y: Microglial
mechanisms and therapeutic potential in brain injury
post-intracerebral hemorrhage. J Inflamm Res. 18:2955–2973. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wang J: Preclinical and clinical research
on inflammation after intracerebral hemorrhage. Prog Neurobiol.
92:463–477. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhou Y, Wang Y, Wang J, Anne Stetler R and
Yang QW: Inflammation in intracerebral hemorrhage: From mechanisms
to clinical translation. Prog Neurobiol. 115:25–44. 2014.
View Article : Google Scholar
|
|
63
|
Chen AQ, Fang Z, Chen XL, Yang S, Zhou YF,
Mao L, Xia YP, Jin HJ, Li YN, You MF, et al: Microglia-derived
TNF-alpha mediates endothelial necroptosis aggravating blood
brain-barrier disruption after ischemic stroke. Cell Death Dis.
10:4872019. View Article : Google Scholar
|
|
64
|
Zlokovic BV: The blood-brain barrier in
health and chronic neurodegenerative disorders. Neuron. 57:178–201.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Liu X, Chen X, Zhu Y, Wang K and Wang Y:
Effect of magnolol on cerebral injury and blood brain barrier
dysfunction induced by ischemia-reperfusion in vivo and in vitro.
Metab Brain Dis. 32:1109–1118. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chen J, Jin H, Xu H, Peng Y, Jie L, Xu D,
Chen L, Li T, Fan L, He P, et al: The neuroprotective effects of
necrostatin-1 on subarachnoid hemorrhage in rats are possibly
mediated by preventing blood-brain barrier disruption and
RIP3-Mediated necroptosis. Cell Transplant. 28:1358–1372. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Ronaldson PT and Davis TP: Regulation of
blood-brain barrier integrity by microglia in health and disease: A
therapeutic opportunity. J Cereb Blood Flow Metab. 40(Suppl 1):
S6–S24. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ozaki E, Delaney C, Campbell M and Doyle
SL: Minocycline suppresses disease-associated microglia (DAM) in a
model of photoreceptor cell degeneration. Exp Eye Res.
217:1089532022. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Da Mesquita S and Kipnis J: DAMed in
(Trem) 2 Steps. Cell. 169:1172–1174. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Keren-Shaul H, Spinrad A, Weiner A,
Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch
K, Lara-Astaiso D, Toth B, et al: A unique microglia type
associated with restricting development of Alzheimer's disease.
Cell. 169:1276–1290 e17. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Deczkowska A, Keren-Shaul H, Weiner A,
Colonna M, Schwartz M and Amit I: Disease-associated microglia: A
universal immune sensor of neurodegeneration. Cell. 173:1073–1081.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Garcia-Revilla J, Alonso-Bellido IM,
Burguillos MA, Herrera AJ, Espinosa-Oliva AM, Ruiz R,
Cruz-Hernández L, García-Domínguez I, Roca-Ceballos MA, Santiago M,
et al: Reformulating pro-oxidant microglia in neurodegeneration. J
Clin Med. 8:17192019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Rangaraju S, Dammer EB, Raza SA,
Rathakrishnan P, Xiao H, Gao T, Duong DM, Pennington MW, Lah JJ,
Seyfried NT and Levey AI: Identification and therapeutic modulation
of a pro-inflammatory subset of disease-associated-microglia in
Alzheimer's disease. Mol Neurodegener. 13:242018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Gao T, Jernigan J, Raza SA, Dammer EB,
Xiao H, Seyfried NT, Levey AI and Rangaraju S: Transcriptional
regulation of homeostatic and disease-associated-microglial genes
by IRF1, LXRβ, and CEBPα. Glia. 67:1958–1975. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Mecca C, Giambanco I, Donato R and Arcuri
C: Microglia and aging: The role of the TREM2-DAP12 and
CX3CL1-CX3CR1 Axes. Int J Mol Sci. 19:3182018. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Arcuri C, Mecca C, Bianchi R, Giambanco I
and Donato R: The pathophysiological role of microglia in dynamic
surveillance, phagocytosis and structural remodeling of the
developing CNS. Front Mol Neurosci. 10:1912017. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Xu YJ, Au NPB and Ma CHE: Functional and
phenotypic diversity of microglia: implication for microglia-based
therapies for Alzheimer's disease. Front Aging Neurosci.
14:8968522022. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Chen S, Peng J, Sherchan P, Ma Y, Xiang S,
Yan F, Zhao H, Jiang Y, Wang N, Zhang JH and Zhang H: TREM2
activation attenuates neuroinflammation and neuronal apoptosis via
PI3K/Akt pathway after intracerebral hemorrhage in mice. J
Neuroinflammation. 17:1682020. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Brown GC and St George-Hyslop PH:
Deciphering microglial diversity in Alzheimer's disease. Science.
356:1123–1124. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Madhu LN, Kodali M, Upadhya R, Rao S,
Somayaji Y, Attaluri S, Shuai B, Kirmani M, Gupta S, Maness N, et
al: Extracellular vesicles from human-induced pluripotent stem
cell-derived neural stem cells alleviate proinflammatory cascades
within disease-associated microglia in Alzheimer's disease. J
Extracell Vesicles. 13:e125192024. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wang C, Cheng F, Han Z, Yan B, Liao P, Yin
Z, Ge X, Li D, Zhong R, Liu Q, et al: Human-induced pluripotent
stem cell-derived neural stem cell exosomes improve blood-brain
barrier function after intracerebral hemorrhage by activating
astrocytes via PI3K/AKT/MCP-1 axis. Neural Regen Res. 20:518–532.
2025. View Article : Google Scholar
|
|
82
|
Prinz M, Erny D and Hagemeyer N: Ontogeny
and homeostasis of CNS myeloid cells. Nat Immunol. 18:385–392.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Masuda T, Sankowski R, Staszewski O,
Böttcher C, Amann L, Sagar, Scheiwe C, Nessler S, Kunz P, van Loo
G, et al: Spatial and temporal heterogeneity of mouse and human
microglia at single-cell resolution. Nature. 566:388–392. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Olah M, Menon V, Habib N, Taga MF, Ma Y,
Yung CJ, Cimpean M, Khairallah A, Coronas-Samano G, Sankowski R, et
al: Single cell RNA sequencing of human microglia uncovers a subset
associated with Alzheimer's disease. Nat Commun. 11:61292020.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Ochocka N, Segit P, Walentynowicz KA,
Wojnicki K, Cyranowski S, Swatler J, Mieczkowski J and Kaminska B:
Single-cell RNA sequencing reveals functional heterogeneity of
glioma-associated brain macrophages. Nat Commun. 12:11512021.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Hammond TR, Dufort C, Dissing-Olesen L,
Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh
J, et al: Single-Cell RNA sequencing of microglia throughout the
mouse lifespan and in the injured brain reveals complex cell-state
changes. Immunity. 50:253–271 e6. 2019. View Article : Google Scholar
|
|
87
|
Li Q, Cheng Z, Zhou L, Darmanis S, Neff
NF, Okamoto J, Gulati G, Bennett ML, Sun LO, Clarke LE, et al:
Developmental heterogeneity of microglia and brain myeloid cells
revealed by deep single-cell RNA sequencing. Neuron. 101:207–223
e10. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Dorman LC and Molofsky AV: Demystifying
microglia: And now the work begins. Immunity. 50:11–13. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Gosselin D, Skola D, Coufal NG, Holtman
IR, Schlachetzki JCM, Sajti E, Jaeger BN, O'Connor C, Fitzpatrick
C, Pasillas MP, et al: An environment-dependent transcriptional
network specifies human microglia identity. Science.
356:eaal32222017. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Varnum MM, Kiyota T, Ingraham KL, Ikezu S
and Ikezu T: The anti-inflammatory glycoprotein, CD200, restores
neurogenesis and enhances amyloid phagocytosis in a mouse model of
Alzheimer's disease. Neurobiol Aging. 36:2995–3007. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Zheng J, Lu J, Mei S, Wu H, Sun Z, Fang Y,
Xu S, Wang X, Shi L, Xu W, et al: Ceria nanoparticles ameliorate
white matter injury after intracerebral hemorrhage:
microglia-astrocyte involvement in remyelination. J
Neuroinflammation. 18:432021. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Shi SX, Li YJ, Shi K, Wood K, Ducruet AF
and Liu Q: IL (Interleukin)-15 bridges astrocyte-microglia
crosstalk and exacerbates brain injury following intracerebral
hemorrhage. Stroke. 51:967–974. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Dong H, Wen X, Zhang BW, Wu Z and Zou W:
Astrocytes in intracerebral hemorrhage: Impact and therapeutic
objectives. Front Mol Neurosci. 17:13274722024. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Li Q, Lan X, Han X, Durham F, Wan J,
Weiland A, Koehler RC and Wang J: Microglia-derived interleukin-10
accelerates post-intracerebral hemorrhage hematoma clearance by
regulating CD36. Brain Behav Immun. 94:437–457. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh
JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin
RJM and Ffrench-Constant C: M2 microglia and macrophages drive
oligodendrocyte differentiation during CNS remyelination. Nat
Neurosci. 16:1211–1218. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Zhang P, Gao C, Guo Q, Yang D, Zhang G, Lu
H, Zhang L, Zhang G and Li D: Single-cell RNA sequencing reveals
the evolution of the immune landscape during perihematomal edema
progression after intracerebral hemorrhage. J Neuroinflammation.
21:1402024. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Hu L, Chen Z, Lu J, Jiang S, Lin H, Zhou
J, Wang N, Ding C, Ni W, Peng H, et al: Extracellular vesicles from
bone marrow-derived macrophages enriched in ARG1 enhance microglial
phagocytosis and haematoma clearance following intracerebral
haemorrhage. J Extracell Vesicles. 14:e700412025. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Gu L, Chen H, Geng R, Liang T, Chen Y,
Wang Z, Ye L, Sun M, Shi Q, Wan G, et al: Endothelial
pyroptosis-driven microglial activation in choroid plexus mediates
neuronal apoptosis in hemorrhagic stroke rats. Neurobiol Dis.
201:1066952024. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Wu CH, Shyue SK, Hung TH, Wen S, Lin CC,
Chang CF and Chen SF: Genetic deletion or pharmacological
inhibition of soluble epoxide hydrolase reduces brain damage and
attenuates neuroinflammation after intracerebral hemorrhage. J
Neuroinflammation. 14:2302017. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Fang Y, Tian Y, Huang Q, Wan Y, Xu L, Wang
W, Pan D, Zhu S and Xie M: Deficiency of TREK-1 potassium channel
exacerbates blood-brain barrier damage and neuroinflammation after
intracerebral hemorrhage in mice. J Neuroinflammation. 16:962019.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Zhou K, Zhong Q, Wang YC, Xiong XY, Meng
ZY, Zhao T, Zhu WY, Liao MF, Wu LR, Yang YR, et al: Regulatory T
cells ameliorate intracerebral hemorrhage-induced inflammatory
injury by modulating microglia/macrophage polarization through the
IL-10/GSK3beta/PTEN axis. J Cereb Blood Flow Metab. 37:967–979.
2017. View Article : Google Scholar
|
|
102
|
Taylor RA, Chang CF, Goods BA, Hammond MD,
Mac Grory B, Ai Y, Steinschneider AF, Renfroe SC, Askenase MH,
McCullough LD, et al: TGF-β1 modulates microglial phenotype and
promotes recovery after intracerebral hemorrhage. J Clin Invest.
127:280–292. 2017. View Article : Google Scholar :
|
|
103
|
Wang G, Shi Y, Jiang X, Leak RK, Hu X, Wu
Y, Pu H, Li WW, Tang B, Wang Y, et al: HDAC inhibition prevents
white matter injury by modulating microglia/macrophage polarization
through the GSK3β/PTEN/Akt axis. Proc Natl Acad Sci USA.
112:2853–2858. 2015. View Article : Google Scholar
|
|
104
|
Li Y, Zhu ZY, Lu BW, Huang TT, Zhang YM,
Zhou NY, Xuan W, Chen ZA, Wen DX, Yu WF and Li PY: Rosiglitazone
ameliorates tissue plasminogen activator-induced brain hemorrhage
after stroke. CNS Neurosci Ther. 25:1343–1352. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Luo Y, Yin W, Signore AP, Zhang F, Hong Z,
Wang S, Graham SH and Chen J: Neuroprotection against focal
ischemic brain injury by the peroxisome proliferator-activated
receptor-gamma agonist rosiglitazone. J Neurochem. 97:435–448.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zhao X, Sun G, Zhang J, Strong R, Song W,
Gonzales N, Grotta JC and Aronowski J: Hematoma resolution as a
target for intracerebral hemorrhage treatment: role for peroxisome
proliferator-activated receptor gamma in microglia/macrophages. Ann
Neurol. 61:352–362. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Wang Y, Chen Q, Tan Q, Feng Z, He Z, Tang
J, Feng H, Zhu G and Chen Z: Simvastatin accelerates hematoma
resolution after intracerebral hemorrhage in a PPARү-dependent
manner. Neuropharmacology. 128:244–254. 2018. View Article : Google Scholar
|
|
108
|
Chen Q, Shi X, Tan Q, Feng Z, Wang Y, Yuan
Q, Tao Y, Zhang J, Tan L, Zhu G, et al: Simvastatin promotes
hematoma absorption and reduces hydrocephalus following
intraventricular hemorrhage in part by upregulating CD36. Transl
Stroke Res. 8:362–373. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Ohnishi M, Katsuki H, Fujimoto S, Takagi
M, Kume T and Akaike A: Involvement of thrombin and
mitogen-activated protein kinase pathways in hemorrhagic brain
injury. Exp Neurol. 206:43–52. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Saito M, Saito M and Das BC: Involvement
of AMP-activated protein kinase in neuroinflammation and
neurodegeneration in the adult and developing brain. Int J Dev
Neurosci. 77:48–59. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Zhao L, Chen S, Sherchan P, Ding Y, Zhao
W, Guo Z, Yu J, Tang J and Zhang JH: Recombinant CTRP9
administration attenuates neuroinflammation via activating
adiponectin receptor 1 after intracerebral hemorrhage in mice. J
Neuroinflammation. 15:2152018. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Zhao W, Kong F, Gong X, Guo Z, Zhao L and
Wang S: Activation of AdipoR1 with rCTRP9 preserves BBB integrity
through the APPL1/AMPK/Nrf2 signaling pathway in ICH Mice. Oxid Med
Cell Longev. 2021:28012632021. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Xu N, Zhang Y, Doycheva DM, Ding Y, Zhang
Y, Tang J, Guo H and Zhang JH: Adiponectin attenuates neuronal
apoptosis induced by hypoxia-ischemia via the activation of
AdipoR1/APPL1/LKB1/AMPK pathway in neonatal rats.
Neuropharmacology. 133:415–428. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Chen S, Zhao L, Sherchan P, Ding Y, Yu J,
Nowrangi D, Tang J, Xia Y and Zhang JH: Activation of melanocortin
receptor 4 with RO27-3225 attenuates neuroinflammation through
AMPK/JNK/p38 MAPK pathway after intracerebral hemorrhage in mice. J
Neuroinflammation. 15:1062018. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Zheng J, Liu Z, Li W, Tang J, Zhang D and
Tang X: Lithium posttreatment confers neuroprotection through
glycogen synthase kinase-3β inhibition in intracerebral hemorrhage
rats. J Neurosurg. 127:716–724. 2017. View Article : Google Scholar
|
|
116
|
Zhao S, Liu Z, Yu Z, Wu X, Li R and Tang
X: BIO alleviates inflammation through inhibition of GSK-3β in a
rat model of intracerebral hemorrhage. J Neurosurg. 133:383–391.
2019. View Article : Google Scholar
|
|
117
|
Zhao Y, Wei ZZ, Zhang JY, Zhang Y, Won S,
Sun J, Yu SP, Li J and Wei L: GSK-3β inhibition induced
neuroprotection, regeneration, and functional recovery after
intracerebral hemorrhagic stroke. Cell Transplant. 26:395–407.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Li R, Liu Z, Wu X, Yu Z, Zhao S and Tang
X: Lithium chloride promoted hematoma resolution after
intracerebral hemorrhage through GSK-3β-mediated pathways-dependent
microglia phagocytosis and M2-phenotype differentiation,
angiogenesis and neurogenesis in a rat model. Brain Res Bull.
152:117–127. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Liu Z, Li R, Jiang C, Zhao S, Li W and
Tang X: The neuroprotective effect of lithium chloride on cognitive
impairment through glycogen synthase kinase-3β inhibition in
intracerebral hemorrhage rats. Eur J Pharmacol. 840:50–59. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Li M, Xia M, Chen W, Wang J, Yin Y, Guo C,
Li C, Tang X, Zhao H, Tan Q, et al: Lithium treatment mitigates
white matter injury after intracerebral hemorrhage through
brain-derived neurotrophic factor signaling in mice. Transl Res.
217:61–74. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Yang Z, Yu A, Liu Y, Shen H, Lin C, Lin L,
Wang S and Yuan B: Regulatory T cells inhibit microglia activation
and protect against inflammatory injury in intracerebral
hemorrhage. Int Immunopharmacol. 22:522–525. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Wang M, Cheng L, Chen ZL, Mungur R, Xu SH,
Wu J, Liu XL and Wan S: Hyperbaric oxygen preconditioning
attenuates brain injury after intracerebral hemorrhage by
regulating microglia polarization in rats. CNS Neurosci Ther.
25:1126–1133. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Yang L, Tang J, Chen Q, Jiang B, Zhang B,
Tao Y, Li L, Chen Z and Zhu G: Hyperbaric oxygen preconditioning
attenuates neuroinflammation after intracerebral hemorrhage in rats
by regulating microglia characteristics. Brain Res. 1627:21–30.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Fang H, Wang PF, Zhou Y, Wang YC and Yang
QW: Toll-like receptor 4 signaling in intracerebral
hemorrhage-induced inflammation and injury. J Neuroinflammation.
10:272013. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Sansing LH, Harris TH, Welsh FA, Kasner
SE, Hunter CA and Kariko K: Toll-like receptor 4 contributes to
poor outcome after intracerebral hemorrhage. Ann Neurol.
70:646–656. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Wang YC, Wang PF, Fang H, Chen J, Xiong XY
and Yang QW: Toll-like receptor 4 antagonist attenuates
intracerebral hemorrhage-induced brain injury. Stroke.
44:2545–2552. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Fang H, Chen J, Lin S, Wang P, Wang Y,
Xiong X and Yang Q: CD36-mediated hematoma absorption following
intracerebral hemorrhage: negative regulation by TLR4 signaling. J
Immunol. 192:5984–5992. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Lyon AR, Citro R, Schneider B, Morel O,
Ghadri JR, Templin C and Omerovic E: Pathophysiology of takotsubo
syndrome: JACC state-of-the-art review. J Am Coll Cardiol.
77:902–921. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Yang Z, Liu B, Zhong L, Shen H, Lin C, Lin
L, Zhang N and Yuan B: Toll-like receptor-4-mediated autophagy
contributes to microglial activation and inflammatory injury in
mouse models of intracerebral haemorrhage. Neuropathol Appl
Neurobiol. 41:e95–e106. 2015. View Article : Google Scholar
|
|
130
|
Lei C, Tan Y, Ni D, Peng J and Yi G:
cGAS-STING signaling in ischemic diseases. Clin Chim Acta.
531:177–182. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Sharma M, Rajendrarao S, Shahani N,
Ramirez-Jarquin UN and Subramaniam S: Cyclic GMP-AMP synthase
promotes the inflammatory and autophagy responses in Huntington
disease. Proc Natl Acad Sci USA. 117:15989–15999. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Wang R, Zhu Y, Liu Z, Chang L, Bai X, Kang
L, Cao Y, Yang X, Yu H, Shi MJ, et al: Neutrophil extracellular
traps promote tPA-induced brain hemorrhage via cGAS in mice with
stroke. Blood. 138:91–103. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Jiang GL, Yang XL, Zhou HJ, Long J, Liu B,
Zhang LM and Lu D: cGAS knockdown promotes microglial M2
polarization to alleviate neuroinflammation by inhibiting
cGAS-STING signaling pathway in cerebral ischemic stroke. Brain Res
Bull. 171:183–195. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Shi J, Yang Y, Yin N, Liu C, Zhao Y, Cheng
H, Zhou T, Zhang Z and Zhang K: Engineering CXCL12 biomimetic
decoy-integrated versatile immunosuppressive nanoparticle for
ischemic stroke therapy with management of overactivated brain
immune microenvironment. Small Methods. 6:e21011582022. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Yang G, Kantapan J, Mazhar M, Hu Q, Bai X,
Zou Y, Wang H, Yang S, Wang L and Dechsupa N: Pretreated MSCs with
IronQ transplantation attenuate microglia neuroinflammation via the
cGAS-STING signaling pathway. J Inflamm Res. 17:1643–1658. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Drouin M, Saenz J and Chiffoleau E: C-type
lectin-like receptors: Head or tail in cell death immunity. Front
Immunol. 11:2512020. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Del Fresno C, Cueto FJ and Sancho D:
Sensing tissue damage by myeloid C-Type lectin receptors. Curr Top
Microbiol Immunol. 429:117–145. 2020.PubMed/NCBI
|
|
138
|
He X, Huang Y, Liu Y, Zhang X, Yue P, Ma
X, Miao Z, Long X, Yang Y, Wan X, et al: BAY61-3606 attenuates
neuroinflammation and neurofunctional damage by inhibiting
microglial Mincle/Syk signaling response after traumatic brain
injury. Int J Mol Med. 49:52022. View Article : Google Scholar
|
|
139
|
Kim JW, Roh YS, Jeong H, Yi HK, Lee MH,
Lim CW and Kim B: Spliceosome-associated protein 130 exacerbates
alcohol-induced liver injury by inducing NLRP3
inflammasome-mediated IL-1β in mice. Am J Pathol. 188:967–980.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Gong W, Zheng T, Guo K, Fang M, Xie H, Li
W, Tang Q, Hong Z, Ren H, Gu G, et al: Mincle/Syk signalling
promotes intestinal mucosal inflammation through induction of
macrophage pyroptosis in Crohn's disease. J Crohns Colitis.
14:1734–1747. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Yang G, Kantapan J, Mazhar M, Bai X, Zou
Y, Wang H, Huang B, Yang S, Dechsupa N and Wang L: Mesenchymal stem
cells transplantation combined with IronQ attenuates ICH-induced
inflammation response via Mincle/syk signaling pathway. Stem Cell
Res Ther. 14:1312023. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Liu XY, Dai XH, Zou W, Yu XP, Teng W, Wang
Y, Yu WW, Ma HH, Chen QX, Liu P, et al: Acupuncture through Baihui
(DU20) to Qubin (GB7) mitigates neurological impairment after
intracerebral hemorrhage. Neural Regen Res. 13:1425–1432. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Li Y, Dong Y, Ran Y, Zhang Y, Wu B, Xie J,
Cao Y, Mo M, Li S, Deng H, et al: Three-dimensional cultured
mesenchymal stem cells enhance repair of ischemic stroke through
inhibition of microglia. Stem Cell Res Ther. 12:3582021. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
He Y, Xu L, Li B, Guo ZN, Hu Q, Guo Z,
Tang J, Chen Y, Zhang Y, Tang J and Zhang JH: Macrophage-inducible
C-type lectin/spleen tyrosine kinase signaling pathway contributes
to neuroinflammation after subarachnoid hemorrhage in rats. Stroke.
46:2277–2286. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
de Rivero Vaccari JC, Brand FJ III, Berti
AF, Alonso OF, Bullock MR and de Rivero Vaccari JP: Mincle
signaling in the innate immune response after traumatic brain
injury. J Neurotrauma. 32:228–236. 2015. View Article : Google Scholar
|
|
146
|
Duan XC, Wang W, Feng DX, Yin J, Zuo G,
Chen DD, Chen ZQ, Li HY, Wang Z and Chen G: Roles of autophagy and
endoplasmic reticulum stress in intracerebral hemorrhage-induced
secondary brain injury in rats. CNS Neurosci Ther. 23:554–566.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Tan X, Yang Y, Xu J, Zhang P, Deng R, Mao
Y, He J, Chen Y, Zhang Y, Ding J, et al: Luteolin exerts
neuroprotection via modulation of the p62/Keap1/Nrf2 pathway in
intracerebral hemorrhage. Front Pharmacol. 10:15512020. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Xiao H, Chen H, Jiang R, Zhang L, Wang L,
Gan H, Jiang N, Zhao J, Zhai X and Liang P: NLRP6 contributes to
inflammation and brain injury following intracerebral haemorrhage
by activating autophagy. J Mol Med (Berl). 98:1319–1331. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Ghadri JR, Wittstein IS, Prasad A, Sharkey
S, Dote K, Akashi YJ, Cammann VL, Crea F, Galiuto L, Desmet W, et
al: International expert consensus document on takotsubo Syndrome
(Part I): Clinical characteristics, diagnostic criteria, and
pathophysiology. Eur Heart J. 39:2032–2046. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Xu J, Chen Z, Yu F, Liu H, Ma C, Xie D, Hu
X, Leak RK, Chou SHY, Stetler RA, et al: IL-4/STAT6 signaling
facilitates innate hematoma resolution and neurological recovery
after hemorrhagic stroke in mice. Proc Natl Acad Sci USA.
117:32679–32690. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Yu A, Duan H, Zhang T, Pan Y, Kou Z, Zhang
X, Lu Y, Wang S and Yang Z: IL-17A promotes microglial activation
and neuroinflammation in mouse models of intracerebral haemorrhage.
Mol Immunol. 73:151–157. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Shi H, Wang J, Wang J, Huang Z and Yang Z:
IL-17A induces autophagy and promotes microglial neuroinflammation
through ATG5 and ATG7 in intracerebral hemorrhage. J Neuroimmunol.
323:143–151. 2018. View Article : Google Scholar
|
|
153
|
Yuan B, Shen H, Lin L, Su T, Zhong L and
Yang Z: Autophagy promotes microglia activation through
Beclin-1-Atg5 pathway in intracerebral hemorrhage. Mol Neurobiol.
54:115–124. 2017. View Article : Google Scholar
|
|
154
|
Chen Z, Xu N, Dai X, Zhao C, Wu X, Shankar
S, Huang H and Wang Z: Interleukin-33 reduces neuronal damage and
white matter injury via selective microglia M2 polarization after
intracerebral hemorrhage in rats. Brain Res Bull. 150:127–135.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Liu DL, Zhao LX, Zhang S and Du JR:
Peroxiredoxin 1-mediated activation of TLR4/NF-ĸB pathway
contributes to neuroinflammatory injury in intracerebral
hemorrhage. Int Immunopharmacol. 41:82–89. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Shang Y, Dai S, Chen X, Wen W and Liu X:
MicroRNA-93 regulates the neurological function, cerebral edema and
neuronal apoptosis of rats with intracerebral hemorrhage through
TLR4/NF-kappaB signaling pathway. Cell Cycle. 18:3160–3176. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Xu H, Cao J, Xu J, Li H, Shen H, Li X,
Wang Z, Wu J and Chen G: GATA-4 regulates neuronal apoptosis after
intracerebral hemorrhage via the NF-ĸB/Bax/Caspase-3 pathway both
in vivo and in vitro. Exp Neurol. 315:21–31. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Hu YY, Huang M, Dong XQ, Xu QP, Yu WH and
Zhang ZY: Ginkgolide B reduces neuronal cell apoptosis in the
hemorrhagic rat brain: possible involvement of Toll-like receptor
4/nuclear factor-kappa B pathway. J Ethnopharmacol. 137:1462–1468.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Dong XQ, Yu WH, Hu YY, Zhang ZY and Huang
M: Oxymatrine reduces neuronal cell apoptosis by inhibiting
Toll-like receptor 4/nuclear factor kappa-B-dependent inflammatory
responses in traumatic rat brain injury. Inflamm Res. 60:533–539.
2011. View Article : Google Scholar
|
|
160
|
Nan C, Zhang Y, Zhang A, Shi Y, Yan D, Sun
Z, Jin Q, Huo H, Zhuo Y and Zhao Z: Exosomes derived from human
umbilical cord mesenchymal stem cells decrease neuroinflammation
and facilitate the restoration of nerve function in rats suffering
from intracerebral hemorrhage. Mol Cell Biochem. 480:309–323. 2025.
View Article : Google Scholar :
|
|
161
|
Li J, Yang L, Zhao L and Li J: Exosomal
PINK1 from human umbilical cord mesenchymal stem cells attenuates
neurological deficits and inflammatory responses after
intracerebral hemorrhage in mice. ACS Chem Neurosci. 16:619–627.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Zhao L and Li J: Microglial uptake of
hADSCs-Exo mitigates neuroinflammation in ICH. Cell Signal.
119:1111462024. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
El Andaloussi S, Mager I, Breakefield XO
and Wood MJ: Extracellular vesicles: Biology and emerging
therapeutic opportunities. Nat Rev Drug Discov. 12:347–357. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Phinney DG and Pittenger MF: Concise
review: MSC-derived exosomes for cell-free therapy. Stem Cells.
35:851–858. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Yang Z, Jiang X, Zhang J, Huang X, Zhang
X, Wang J, Shi H and Yu A: Let-7a promotes microglia M2
polarization by targeting CKIP-1 following ICH. Immunol Lett.
202:1–7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Zhang XD, Fan QY, Qiu Z and Chen S: MiR-7
alleviates secondary inflammatory response of microglia caused by
cerebral hemorrhage through inhibiting TLR4 expression. Eur Rev Med
Pharmacol Sci. 22:5597–5604. 2018.PubMed/NCBI
|
|
167
|
Wang J, Xu Z, Chen X, Li Y, Chen C, Wang
C, Zhu J, Wang Z, Chen W, Xiao Z and Xu R: MicroRNA-182-5p
attenuates cerebral ischemia-reperfusion injury by targeting
Toll-like receptor 4. Biochem Biophys Res Commun. 505:677–684.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Lv YN, Ou-Yang AJ and Fu LS: MicroRNA-27a
negatively modulates the inflammatory response in
lipopolysaccharide-stimulated microglia by targeting TLR4 and
IRAK4. Cell Mol Neurobiol. 37:195–210. 2017. View Article : Google Scholar
|
|
169
|
Bai YY and Niu JZ: miR222 regulates brain
injury and inflammation following intracerebral hemorrhage by
targeting ITGB8. Mol Med Rep. 21:1145–1153. 2020.PubMed/NCBI
|
|
170
|
Zhang Y, Han B, He Y, Li D, Ma X, Liu Q
and Hao J: MicroRNA-132 attenuates neurobehavioral and
neuropathological changes associated with intracerebral hemorrhage
in mice. Neurochem Int. 107:182–190. 2017. View Article : Google Scholar
|
|
171
|
Wang Z, Yuan B, Fu F, Huang S and Yang Z:
Hemoglobin enhances miRNA-144 expression and autophagic activation
mediated inflammation of microglia via mTOR pathway. Sci Rep.
7:118612017. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Yu A, Zhang T, Zhong W, Duan H, Wang S, Ye
P, Wang J, Zhong S and Yang Z: miRNA-144 induces microglial
autophagy and inflammation following intracerebral hemorrhage.
Immunol Lett. 182:18–23. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Yu A, Zhang T, Duan H, Pan Y, Zhang X,
Yang G, Wang J, Deng Y and Yang Z: MiR-124 contributes to M2
polarization of microglia and confers brain inflammatory protection
via the C/EBP-α pathway in intracerebral hemorrhage. Immunol Lett.
182:1–11. 2017. View Article : Google Scholar
|
|
174
|
Florczak-Rzepka M, Grond-Ginsbach C,
Montaner J and Steiner T: Matrix metalloproteinases in human
spontaneous intracerebral hemorrhage: An update. Cerebrovasc Dis.
34:249–262. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Wells JE, Biernaskie J, Szymanska A,
Larsen PH, Yong VW and Corbett D: Matrix metalloproteinase (MMP)-12
expression has a negative impact on sensorimotor function following
intracerebral haemorrhage in mice. Eur J Neurosci. 21:187–196.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Wasserman JK and Schlichter LC:
Minocycline protects the blood-brain barrier and reduces edema
following intracerebral hemorrhage in the rat. Exp Neurol.
207:227–237. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Liang H, Guan D, Gao A, Yin Y, Jing M,
Yang L, Ma W, Hu E and Zhang X: Human amniotic epithelial stem
cells inhibit microglia activation through downregulation of tumor
necrosis factor-alpha, interleukin-1β and matrix
metalloproteinase-12 in vitro and in a rat model of intracerebral
hemorrhage. Cytotherapy. 16:523–534. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Miao H, Li R, Han C, Lu X and Zhang H:
Minocycline promotes posthemorrhagic neurogenesis via M2 microglia
polarization via upregulation of the TrkB/BDNF pathway in rats. J
Neurophysiol. 120:1307–1317. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Chen M, Li X, Zhang X, He X, Lai L, Liu Y,
Zhu G, Li W, Li H, Fang Q, et al: The inhibitory effect of
mesenchymal stem cell on blood-brain barrier disruption following
intracerebral hemorrhage in rats: Contribution of TSG-6. J
Neuroinflammation. 12:612015. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Li Z, Liu Y, Wei R, Khan S, Zhang R, Zhang
Y, Yong VW and Xue M: Iron neurotoxicity and protection by
deferoxamine in intracerebral hemorrhage. Front Mol Neurosci.
15:9273342022. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Wu H, Wu T, Xu X and Wang J and Wang J:
Iron toxicity in mice with collagenase-induced intracerebral
hemorrhage. J Cereb Blood Flow Metab. 31:1243–1250. 2011.
View Article : Google Scholar
|
|
182
|
Hatakeyama T, Okauchi M, Hua Y, Keep RF
and Xi G: Deferoxamine reduces neuronal death and hematoma lysis
after intracerebral hemorrhage in aged rats. Transl Stroke Res.
4:546–553. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Hu S, Hua Y, Keep RF, Feng H and Xi G:
Deferoxamine therapy reduces brain hemin accumulation after
intracerebral hemorrhage in piglets. Exp Neurol. 318:244–250. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Zhao F, Hua Y, He Y, Keep RF and Xi G:
Minocycline-induced attenuation of iron overload and brain injury
after experimental intracerebral hemorrhage. Stroke. 42:3587–3593.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Cao S, Hua Y, Keep RF, Chaudhary N and Xi
G: Minocycline effects on intracerebral hemorrhage-induced iron
overload in aged rats: Brain iron quantification with magnetic
resonance imaging. Stroke. 49:995–1002. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Li Q, Wan J, Lan X, Han X, Wang Z and Wang
J: Neuroprotection of brain-permeable iron chelator VK-28 against
intracerebral hemorrhage in mice. J Cereb Blood Flow Metab.
37:3110–3123. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Dai S, Hua Y, Keep RF, Novakovic N, Fei Z
and Xi G: Minocycline attenuates brain injury and iron overload
after intracerebral hemorrhage in aged female rats. Neurobiol Dis.
126:76–84. 2019. View Article : Google Scholar
|
|
188
|
Li Q, Zhang G, Huang YJ, Dong MX, Lv FJ,
Wei X, Chen JJ, Zhang LJ, Qin XY and Xie P: Blend sign on computed
tomography: novel and reliable predictor for early hematoma growth
in patients with intracerebral hemorrhage. Stroke. 46:2119–2123.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Li Y, Ren S, Wang L, Mao Y, Wu G, Li Q and
Tang Z: Is the CT blend sign composed of two parts of blood with
different age? Neurocrit Care. 35:367–378. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Hang H, Liu S, Wang L, Zhang L, Liu C, Xu
B, Zhao H and Wu G: Proteomic insights into the pathogenesis of
intracerebral hemorrhage: The role of blend sign in hematoma
expansion. Proteomics Clin Appl. 20:e700282026. View Article : Google Scholar
|
|
191
|
Misra S, Singh P, Sengupta S, Kushwaha M,
Rahman Z, Bhalla D, Talwar P, Nath M, Chakraborty R, Kumar P, et
al: Subtyping strokes using blood-based protein biomarkers: A
high-throughput proteomics and machine learning approach. Eur J
Clin Invest. 55:e143722025. View Article : Google Scholar
|
|
192
|
Li B, Liu Y, Du X, Liu S, Wang X, Wang S,
Han C, Zhao X, Sheng F, Zhang H, et al: Champagne bottle neck sign
predicts intracranial hemorrhage in Moyamoya disease: A long-term
follow-up study. Clin Neurol Neurosurg. 260:1092332026. View Article : Google Scholar
|
|
193
|
Bader ER, Pana TA, Barlas RS, Metcalf AK,
Potter JF and Myint PK: Elevated inflammatory biomarkers and poor
outcomes in intracerebral hemorrhage. J Neurol. 269:6330–6341.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Xu F, Xu J, Wang Q, Gao F, Fu J, Yan T,
Dong Q, Su Y and Cheng X: Serum YKL-40 as a predictive biomarker of
cerebral amyloid angiopathy-related intracerebral hemorrhage
recurrence. J Alzheimers Dis. 99:503–511. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Roh DJ, Poyraz FC, Mao E, Shen Q, Kansara
V, Cottarelli A, Song S, Nemkov T, Kumar A, Hudson KE, et al:
Anemia from inflammation after intracerebral hemorrhage and
relationships with outcome. J Am Heart Assoc. 13:e0355242024.
View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Gareev I, Beylerli O and Zhao B: MiRNAs as
potential therapeutic targets and biomarkers for non-traumatic
intracerebral hemorrhage. Biomark Res. 12:172024. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Bai C, Liu X, Wang F, Sun Y, Wang J and
Liu J, Hao X, Zhou L, Yuan Y and Liu J: Identification of
immune-related biomarkers for intracerebral hemorrhage diagnosis
based on RNA sequencing and machine learning. Front Immunol.
15:14219422024. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Chen K, Cheng X, Yuan S, Sun Y, Hao J, Tan
Q, Lin Y, Li S and Yang J: Signature and function of plasma
exosome-derived circular RNAs in patients with hypertensive
intracerebral hemorrhage. Mol Genet Genomics. 299:502024.
View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Guilan MB, Bagheri SR, Roshani R and
Alimohammadi E: Red cell distribution width to lymphocyte ratio
could serve as a new inflammatory biomarker for predicting hematoma
expansion in patients with intracerebral hemorrhage. BMC Neurol.
24:1622024. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Li W, Wang J, Tang C, Lv X and Zhu S: A
Prospective cohort study of elevated serum NLRP1 levels to
prognosticate neurological outcome after acute intracerebral
hemorrhage at a single academic institution. Neuropsychiatr Dis
Treat. 20:737–753. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
He J, Zhang Y, Hao P, Li T, Xiao Y, Peng
L, Feng Y, Cheng X, Deng H, Wang P, et al: Association between red
blood cell distribution width and long-term mortality in patients
with intracerebral hemorrhage. Neurocrit Care. 40:1059–1069. 2024.
View Article : Google Scholar
|
|
202
|
Yang W, Lu T, Shan H, Zou S, Ye Z, Zhang
K, Lin Q, Dai J, Cai J, Yu W, et al: RVD2 emerges as a serological
marker in relation to severity and six-month clinical outcome
following acute intracerebral hemorrhage: A prospective cohort
study from a single academic institution. Clin Chim Acta.
565:1199882025. View Article : Google Scholar
|
|
203
|
Cheng X, Hu D, Wang C, Lu T, Ning Z, Li K,
Ren Z, Huang Y, Zhou L, Chung SK, et al: Plasma inflammation
markers linked to complications and outcomes after spontaneous
intracerebral hemorrhage. J Proteome Res. 23:4369–4383. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Mansoor M, Nadeem M and Ahmed M:
'Enhancing prognostic accuracy in intracerebral hemorrhage: The
role of serum secretoneurin and emerging biomarkers'. Neurosurg
Rev. 47:4782024. View Article : Google Scholar
|
|
205
|
Lei C, Chen K, Gu Y, Li Y, Zhu X, Li H,
Xue R, Chang X and Yang X: The association between TLR2/4 and
clinical outcome in intracerebral hemorrhage. Clin Neurol
Neurosurg. 244:1084402024. View Article : Google Scholar : PubMed/NCBI
|