Introduction
Pulmonary fibrosis (PF) is a fatal interstitial lung
disease of unknown etiology, ultimately leading to the loss of
pulmonary function and respiratory failure (1). Epidemiological data show that
idiopathic PF (IPF), the main form of PF, affects ~3 million
individuals worldwide (2), with
a higher incidence observed in North America compared to Asia and
Europe, and its burden is increasing despite regional variability
(3). Although current therapies
(e.g., Nintedanib and Pirfenidone) can slow the progression of PF,
these treatments are not curative and their efficacy remains
limited. The pathophysiology for PF is complex, involving damage to
alveolar epithelial cells, abnormal activation of fibroblasts and
myofibroblasts and the promotion of fibrosis through multiple
signaling pathways (including TGF-β, Wnt/β-catenin and PI3K/Akt)
(4). Consequently, there is a
pressing rationale to explore beyond conventional antifibrotic
strategies and critically evaluate the most promising emerging
therapeutic paradigms.
Recent advances in regenerative and molecular
medicine have opened new avenues. Stem cell therapies, particularly
airway basal stem cells and induced pluripotent stem cells (iPSCs),
have demonstrated that these cells can differentiate into various
lung cell types in preclinical models (5-7),
promoting the repair of damaged lung tissue. Furthermore, iPSC
technology has made it possible to generate patient-specific stem
cells, overcoming issues related to cell sourcing and
transplantation compatibility (8,9).
Gene editing technologies, such as CRISPR-Cas9, have shown immense
potential in repairing gene mutations associated with PF and
regulating the expression of fibrosis-related genes (8). Targeted drugs are considered the
mainstay of PF treatment, and numerous novel targeted agents have
been developed to inhibit key signaling pathways of PF. Research
into epigenetics has provided new insights into PF therapy.
Epigenetic modifications, including DNA methylation, histone
modification and regulation by non-coding RNAs, have been verified
to play crucial roles in the onset and progression of PF.
Furthermore, RNA delivery technologies, such as small interfering
RNA (siRNA) and mRNA, are rapidly emerging as novel therapeutic
strategies that can target fibrosis-inducing genes and inhibit the
fibrotic response. Finally, the potential application of AI
technologies in predicting therapeutic targets for PF was
discussed, particularly in optimizing personalized treatment
regimens.
This review aims to analyze these interconnected
advances, thereby providing a comprehensive overview of the
pathobiological underpinnings of PF and a critical appraisal of the
next generation of therapeutic strategies - from stem cells and
targeted agents to epigenetic modulators, nucleic acid delivery and
AI-driven solutions. The convergence of these disciplines holds the
promise of shifting the treatment paradigm from palliative
management to true disease modification, ultimately aiming to
improve patient prognosis and quality of life.
Methodology
This narrative review was conducted based on a
comprehensive literature search performed in PubMed (https://pubmed.ncbi.nlm.nih.gov/), one of the
most widely used biomedical databases. This work aimed to identify
relevant studies and reviews related to PF and its emerging
therapeutic strategies. The following keywords and medical subject
headings terms were used in various combinations: 'pulmonary
fibrosis', 'lung fibrosis', 'interstitial lung disease', 'pulmonary
fibrosis treatment', 'anti-fibrotic therapy', 'stem cell therapy',
'induced pluripotent stem cells', 'gene therapy', 'epigenetic
modifications', 'epigenetic interventions', 'targeted therapy',
'precision medicine' and 'artificial intelligence'. Boolean
operators (AND, OR) were applied to combine terms and maximize
retrieval sensitivity. The search was limited to English-language
publications, with no restrictions on publication date to ensure
inclusion of foundational and recent advances. Articles were
screened by title and abstract for relevance, followed by full-text
evaluation for final inclusion. Additional references were
identified through manual searching of cited references in key
articles.
Pathogenesis of PF
PF is a chronic, irreversible disease characterized
by progressive scarring of lung tissue. The core pathological
change involves repeated injury to alveolar epithelial cells,
leading to abnormal repair mechanisms. This in turn causes
excessive activation of fibroblasts and extensive deposition of
extracellular matrix (ECM), and ultimately disrupts the normal
structure of the lung parenchyma and severely impairs gas exchange
function. According to epidemiological data, the median survival
time of patients diagnosed with PF is typically only 3-5 years
(10), with a poor prognosis and
a five-year survival rate of <30%, a figure lower than that of
most malignant cancers. Although significant progress has been made
in recent years in understanding the etiology of PF, its exact
pathogenesis remains incompletely understood. Currently, it is
widely accepted that PF results from a complex disease process in
genetically predisposed individuals under the long-term influence
of various environmental factors. This process involves multiple
layers of interaction, including genetic background, epigenetic
regulation, age-related changes, environmental exposures and immune
system abnormalities (Fig.
1).
![Overview of pathogenesis and
pathophysiological progression in pulmonary fibrosis. The schematic
illustrates the multi-layered interactions-including genetic
predisposition, environmental exposures, age-related decline and
immune dysregulation - that collectively drive disease initiation.
The interplay of these factors triggers a sequential pathogenic
cascade beginning with alveolar epithelial cell injury, followed by
dysregulated inflammation and immune activation, leading to
fibroblast recruitment and myofibroblast differentiation and
culminating in excessive extracellular matrix deposition and tissue
remodeling. ROS, reactive oxygen species; 1-NP, 1-nitropyrene; BaP,
benzo[a]pyrene; MUC5B, mucin 5B, oligomeric mucus/gel-forming;
TERT, telomerase reverse transcriptase; TERC, telomerase RNA
component; rs35705950, a single nucleotide polymorphism in the
MUC5B promoter region; TOLLIP, toll interacting protein.](/article_images/ijmm/57/5/ijmm-57-05-05783-g00.jpg) | Figure 1Overview of pathogenesis and
pathophysiological progression in pulmonary fibrosis. The schematic
illustrates the multi-layered interactions-including genetic
predisposition, environmental exposures, age-related decline and
immune dysregulation - that collectively drive disease initiation.
The interplay of these factors triggers a sequential pathogenic
cascade beginning with alveolar epithelial cell injury, followed by
dysregulated inflammation and immune activation, leading to
fibroblast recruitment and myofibroblast differentiation and
culminating in excessive extracellular matrix deposition and tissue
remodeling. ROS, reactive oxygen species; 1-NP, 1-nitropyrene; BaP,
benzo[a]pyrene; MUC5B, mucin 5B, oligomeric mucus/gel-forming;
TERT, telomerase reverse transcriptase; TERC, telomerase RNA
component; rs35705950, a single nucleotide polymorphism in the
MUC5B promoter region; TOLLIP, toll interacting protein. |
Genetic susceptibility plays a
foundational role in PF pathogenesis
In terms of genetic factors, genome-wide association
studies (GWAS) have been performed to identify several
susceptibility loci for PF. Notably, a single nucleotide
polymorphism, rs35705950, located in the promoter region of the
mucin 5B gene (MUC5B), was confirmed as the strongest genetic risk
factor, with this variant increasing the risk of developing PF by
4-7 times, as validated in multiple independent cohorts (11-14). MUC5B, a key component of airway
mucus, may lead to abnormal mucus secretion due to mutations in its
promoter region, thereby affecting the homeostasis of the alveolar
microenvironment (15). In
addition to MUC5B, mutations in telomerase-associated genes, such
as telomerase reverse transcriptase (TERT) and telomerase RNA
component (TERC), are closely associated with familial PF. These
mutations cause telomere shortening, accelerating the aging process
of alveolar epithelial cells (16-18). Polymorphisms in the
Toll-interacting protein gene may influence the intensity of the
inflammatory response of lung tissue to injury by regulating the
Toll-like receptor signaling pathway (19,20). Notably, these genetic risk
factors often exhibit a pronounced age-dependent penetrance, among
which aging can lead to a decline in the repair capacity of
alveolar epithelial cells, telomere shortening and mitochondrial
dysfunction. This helps explain why PF primarily affects
middle-aged and elderly populations (21).
Aging is a key biological driver of
PF
Aging is one of the core biological processes in the
occurrence and development of PF. It has been reported that
pulmonary fibroblasts isolated from patients with PF often exhibit
accelerated aging phenotypes, including increased β-galactosidase
activity, elevated expression of p21, p16, p53 and cytokines
related to the senescence-associated secretory phenotype (SASP), as
well as reduced proliferative capacity (22). Abnormal transcription factors
related to endothelial cell senescence have been identified as the
contributors for the sustained activation of fibroblasts
responsible for collagen production during PF progression (23,24). Raslan et al (25) performed single-cell RNA
sequencing on the lungs of young and aged mice with
bleomycin-induced lung injury and then demonstrated that
endothelial activation resolved in the lungs of young mice but
persisted in the lungs of aged mice. The abnormal state of
activated pulmonary endothelial cells in vivo may lead to
impaired lung injury repair and sustained fibrosis. Additionally,
the decline in tissue stem cell function due to aging is a key
factor in the development of PF. Specifically, the decreased
self-renewal and differentiation capacity of alveolar epithelial
type II (AEC2) cells, which serve as progenitor cells for alveolar
epithelium, directly impacts the repair capacity of lung tissue. Lu
et al (26) designed a
gene-editing nanoparticle platform, incorporating a CRISPR-Cas9
system, to effectively clear reactive oxygen species (ROS) while
reducing SASP factors by knocking down the senescence-promoting
gene lysine acetyltransferase 7, thereby providing robust
antioxidant and anti-aging effects for alveolar epithelial type II
(AT2) cells.
Environmental exposure: A trigger in PF
pathogenesis
Air pollutants, particularly fine particulate matter
(PM2.5) and inhalable particles (PM10), have been confirmed to be
highly correlated with the incidence and mortality of PF (27,28). These particles promote fibrosis
development through various mechanisms, such as inducing oxidative
stress, mitochondrial dysfunction, telomere attrition, initiating
chronic inflammation and directly damaging alveolar epithelium
(29). There is a close link
between metabolic products and IPF. Air pollution may induce
changes in metabolites. For instance, Wang et al (30) found that each
pollutant-associated metabolic feature was positively correlated
with the risk of IPF. At the molecular level, PM10 exposure
upregulates the expression of matrix metalloproteinases (MMPs) and
disrupts the integrity of the basement membrane (25,27-29,31). PM2.5 exposure increases the
expression of TGF-β1, α-smooth muscle actin and type I collagen in
mouse lungs, while activating the TGF-β/Smad signaling pathway and
promoting the transformation of fibroblasts into myofibroblasts
(32). Gaseous pollutants, such
as nitrogen dioxide, primarily contribute to PF pathogenesis by
inducing respiratory inflammation and increasing vascular
permeability (33). Tobacco
smoke is another major environmental risk factor, and active and
passive smoking increase the risk of PF by 2-3 times (34). Thousands of chemicals in
cigarettes (e.g., benzopyrene, ROS) can cause DNA damage and
abnormal apoptosis in alveolar epithelial cells, while activating
pulmonary fibroblasts (35,36). It is worth noting that the
widespread use of e-cigarettes has caused new public health
concerns. It was found that heavy metal particles (e.g., lead,
cadmium) and volatile organic compounds (e.g., formaldehyde) in
e-cigarette vapor may promote PF development through inducing
oxidative stress and inflammatory responses (37). These occupational exposures (such
as silica dust, asbestos and coal dust) have been proven to be the
direct causes of certain types of PF, such as pneumoconiosis. These
occupational dusts often accumulate in the lungs over many years.
After inhalation, silica particles are phagocytosed by macrophages
but resist degradation, triggering excessive ROS production and
inflammatory signaling pathways, continuously stimulating local
inflammation and fibrosis (38,39). Histone deacetylase 10 has become
a key regulator of oxidative stress and inflammation in silicosis.
Furthermore, certain chemicals, such as herbicides and chemotherapy
drugs, are related to the onset of PF, potentially through direct
cytotoxic or immune-modulating mechanisms. Importantly,
environmental exposure can induce changes in epigenetic patterns,
thereby altering gene expression profiles and contributing to the
development and progression of PF.
Role of microbial infections
In recent years, the relationship between microbial
infections and PF has garnered increasing attention. Compared to
control groups, several bacterial species, such as Haemophilus
influenzae, Streptococcus and Moraxella
catarrhalis, were found to be more prevalent in the lung tissue
of patients with PF (40,41).
These bacteria may directly cause damage to airway epithelial cells
by inducing host immune responses or through the activation of
chronic low-level antigen stimulation, which triggers a
wound-healing cascade. Additionally, they may indirectly promote
fibrosis by inducing persistent inflammation (40). Viral infections, particularly the
PF caused by severe acute respiratory syndrome-coronavirus
(SARS-CoV)-2 infection, have become a clinical focus. SARS-CoV-2
infection can trigger a cytokine storm, leading to pro-fibrotic
responses and resulting in significant fibrotic changes in the
lungs, which are associated with respiratory failure and high
mortality rates (42,43). Furthermore, various viral
infections, including those caused by human T-cell leukemia virus,
cytomegalovirus, human immunodeficiency virus and Epstein-Barr
virus, are associated with the development of PF. These viral
infections induce immune-mediated damage resulting in the
accumulation of macrophages, neutrophils, eosinophils and type 2
T-helper (Th2) cells at the site of injury, thereby releasing large
amounts of pro-inflammatory and profibrotic cytokines (44).
In summary, the pathogenesis of PF is not caused by
a single factor but results from the combined effects of genetic
predisposition, aging and various environmental exposures
(including air pollutants, smoking and microbial infections). These
factors lay the foundation or provide the initial 'hit' for
sustained alveolar epithelial injury and abnormal repair,
disrupting pulmonary homeostasis and thereby initiating a complex
pathophysiological process. The core features of this process
include abnormal cellular behavior and molecular signaling
activation under the interaction of genetic and environmental
factors, ultimately leading to irreversible fibrosis.
Pathophysiological process of PF
Alveolar epithelial cell injury and
abnormal activation
The pathophysiology of PF is initiated by alveolar
epithelial cell injury, particularly involving alveolar epithelial
type I (AT1) and AT2 cells (45). Repeated lung injury damages AT2
cells, which subsequently become hyperactivated, leading to the
overactivation of Wnt/β-catenin and SHH signaling pathways and the
secretion of TGF-β. Furthermore, injured AT2 cells secrete SASP
factors, leukotrienes and prostaglandins, adopting a senescent
phenotype that promotes the proliferation of myofibroblasts.
Role of inflammation and immune
cells
After epithelial injury, abnormal inflammatory and
immune responses are rapidly activated. Injured epithelial cells
release pro-inflammatory cytokines (e.g., TNF-α, IL-1β) and
chemokines (e.g., C-C motif chemokine ligand 2, C-X-C motif
chemokine ligand 12), recruiting neutrophils, monocytes and
lymphocytes to the site of damage. In PF, this inflammatory
response is often chronic and dysregulated. Macrophages play a dual
role in PF progression: Initially, they are predominantly
pro-inflammatory M1-type, secreting cytokines such as IL-1β and
IL-6; later, they gradually polarize toward an anti-inflammatory
and pro-fibrotic M2-type, secreting cytokines like TGF-β and IL-10
(46,47). Other immune cells, such as Th2
cells, promote fibroblast activation and ECM production through the
secretion of IL-4 and IL-13, while the regulatory function of
regulatory T cells may become imbalanced, collectively contributing
to the formation of a fibrotic microenvironment (48).
Activation of fibroblasts/myofibroblasts
and ECM deposition
The combined effects of the aforementioned injury
and inflammatory microenvironment drive the core execution phase of
fibrosis. Under continuous stimulation by injury signals and
inflammatory factors (especially TGF-β), fibroblasts in the lung
interstitium are activated and differentiate into myofibroblasts.
These cells highly express α-smooth muscle actin (α-SMA), possess
strong contractile ability, and exhibit a high capacity for ECM
secretion. Myofibroblasts originate from diverse sources, including
the activation of local fibroblasts, recruitment of bone
marrow-derived fibrocytes, epithelial-mesenchymal transition (EMT),
endothelial-mesenchymal transition and pericyte
transdifferentiation.
TGF-β has been verified to be the most central
pro-fibrotic factor (49). It
promotes fibroblast proliferation and differentiation into
myofibroblasts via both Smad-dependent (e.g., Smad2/3
phosphorylation) and non-Smad-dependent (e.g., MAPK, PI3K/Akt)
signaling pathways. Besides, it stimulates the synthesis of large
amounts of ECM components (primarily type I and III collagen),
while suppressing the activity of MMPs and increasing the
expression of their tissue inhibitors, resulting in reduced ECM
degradation and net increased deposition.
ECM remodeling and mechanosignaling
feedback
As the disease progresses, excessive ECM abnormally
accumulates in the lung interstitium, leading to alveolar wall
thickening, structural destruction and the formation of a
'honeycomb lung.' This structural remodeling not only impairs gas
exchange but also alters the mechanical properties of the lung
tissue (e.g., increased stiffness). The altered mechanical
environment itself can further activate mechanosensitive signaling
pathways in fibroblasts, such as such as Yes-associated protein
(YAP)/transcriptional co-activator with PDZ-binding motif (TAZ),
thereby continuously driving their activation and pro-fibrotic gene
expression, forming a self-reinforcing vicious cycle (50,51). Myofibroblasts also secrete
various ECM-modifying enzymes (e.g., MMP1, MMP3, MMP7, MMP9) and
components [e.g., collagen type III α1 chain (COL3A1), COL6A1],
exacerbating the pathological remodeling of the ECM (52).
In summary, the pathophysiology of PF is a dynamic,
multi-stage network process. It begins with epithelial injury,
progresses through complex intercellular communication and abnormal
activation of signaling pathways (centered on TGF-β and integrating
multiple pathways such as Wnt, Hedgehog and YAP/TAZ (53), and ultimately leads to
irreversible destruction of lung structure characterized by
myofibroblast aggregation and excessive ECM deposition.
Importantly, these complex molecular and cellular events are
largely subject to precise regulation at the epigenetic level.
Epigenetic changes in PF
Epigenetics is the study of gene expression and
functional changes that do not involve changes to the DNA sequence
but are regulated by other molecular mechanisms.
Epigenetic changes include abnormalities in DNA
methylation, dysregulation of histone modifications and
disturbances in non-coding RNA expression. DNA methylation changes
typically occur in CpG dinucleotide clusters within the gene
promoter regions (also known as 'CpG islands'), and these changes
are closely related to the transcriptional silencing of the
affected genes. By contrast, DNA demethylation leads to the
reactivation and expression of genes, a process that is of great
significance in epigenetics. Studies have shown that half of the
loci in GWAS gene regions exhibit methylation changes in patients
with IPF, and DNA methylation plays a crucial role in the
expression of specific genes in PF lungs (54-56). McErlean et al (57) compared the DNA methylation
characteristics of primary airway macrophages (Ams) obtained from
patients with IPF and healthy donors and found that the changes in
the DNA methylome were related to the differentiation and phenotype
of Ams in the process of PF. They suggested that the metabolic
functions of AMs are involved in the pathogenesis of PF. The
epigenetic changes in lipid and glucose metabolism-related genes
were associated with the clinical severity of PF. Wang et al
(50) showed that in patients
with IPF and bleomycin (BLM)-induced PF mouse models,
overexpression of methyl-CpG-binding domain 2 protein in
myofibroblasts inhibited the expression of the erythroid
differentiation regulator 1 promoter. These epigenetic changes
promoted the differentiation of fibroblasts into myofibroblasts,
thereby exacerbating the progression of PF.
Histone modification is another important mechanism
in epigenetics, where histones undergo modifications including
acetylation, methylation and phosphorylation, altering chromatin
structure and thereby affecting gene expression. Histone
deacetylases (HDACs) are key enzymes in regulating chromatin
remodeling and gene transcription, and increasing evidence suggests
that the HDAC family is closely associated with the progression of
chronic fibrotic diseases (58).
Hua et al (59) found
that the HDAC2/SIN3 transcription regulator family member A
(Sin3A)/methyl-CpG-binding protein (MeCP) 2 complex acts as an
endogenous inhibitor of connective tissue growth factor in lung
fibroblasts. Jeong et al (60) discovered that HDAC3 promotes
alveolar EMT and fibroblast migration under hypoxic conditions. RNA
epigenetic modifications play a crucial role in PF, particularly
the roles of N6-methyladenosine (m6A) and 5-methylcytosine (m5C) -
two prevalent RNA modifications that regulate gene expression - in
the fibrosis process. YTH domain-containing protein 1 (YTHDC1),
which is primarily expressed in AECII cells, exhibits significantly
reduced expression in these cells during PF. This downregulation of
YTHDC1 contributes to disease progression, as YTHDC1 has been shown
to counteract stress-induced pulmonary senescence and fibrosis
through a non-canonical mechanism independent of its m6A-binding
ability. Mechanistically, YTHDC1 promotes the interaction between
TopBP1 and MRE11, thereby activating ATR and facilitating DNA
damage repair (61-63). Additionally, m5C modification has
been shown to play a critical role in fibroblast activation and
inflammatory pathways, and acts as a key factor in the development
of PF (64,65).
Urgent need for new potential treatment
options for PF
In recent years, an in-depth understanding of the
pathophysiological mechanisms and epigenetic regulation of PF has
pointed the way toward developing novel therapies that go beyond
traditional anti-fibrotic drugs. However, current clinical
treatment options remain limited. Currently, the standard treatment
involves medication, with pirfenidone and nintedanib being the two
approved drugs for PF. These medications can slow the progression
of the disease and are selected for palliative treatment in the
later stages of PF. The two antifibrotic drugs have similar
efficacy: Pirfenidone primarily works by inhibiting fibroblast
proliferation and collagen synthesis, mainly through the regulation
of TGFβ. Its side effects include gastrointestinal reactions and
skin sensitivity or allergies. Nintedanib is a tyrosine kinase
inhibitor targeting platelet-derived growth factor (PDGF),
fibroblast growth factor and VEGF receptors, which interferes with
active processes in fibrosis, such as fibroblast proliferation,
migration, differentiation and ECM secretion (66). However, its side effects are more
prominent, particularly diarrhea, nausea and vomiting. The
treatment effectiveness of both drugs generally depends on their
tolerance by patients, with discontinuation rates ranging from 10
to 20% (67). Although these
drugs have demonstrated some clinical efficacy, their primary role
is to slow disease progression rather than reverse the fibrotic
process or existing fibrotic damage, and their impact on improving
lung function is limited.
Other treatment options may include oxygen therapy,
pulmonary rehabilitation and lung transplantation in advanced
stages, but these treatments are not suitable for all patients and
have specific limitations. New drugs targeting novel pathways are
currently under development, such as NADPH oxidase 1/4 inhibitors
(68), pirfenidone analogs
(69), translation initiation
factor 3A modulators (70),
antifibrotic agents (71) and
senolytics (72). However, their
efficacy and clinical applications remain to be validated.
At present, although the use of antifibrotic drugs
has slowed the decline in lung function in patients with IPF,
neither drug has shown effectiveness in relieving symptoms, and few
patients experience safety and tolerability issues, mainly
gastrointestinal. Importantly, comorbidities and complications,
such as acute exacerbations, pulmonary hypertension (PH),
cardiovascular diseases, gastroesophageal reflux disease and lung
cancer, further exacerbate the disease burden and contribute to the
high mortality rate of IPF (73). When all drug treatments fail,
lung transplantation becomes the only treatment option. However,
given the limited availability of lung donors and the clinical
issues associated with post-transplant immune rejection, current
treatment options for PF are highly limited. Therefore, there is an
urgent need to develop new treatment strategies for PF.
Emergence of novel treatment approaches
As our understanding of the pathophysiological
mechanisms of PF has deepened, treatment strategies have also been
continuously innovated. Traditional therapeutic methods have
gradually shown their limitations. Therefore, an increasing number
of emerging treatment approaches are being explored and applied.
These new therapies emphasize precision medicine and focus on
individual patient differences, driving the treatment of PF toward
a multidisciplinary and personalized approach. In addition to
seeking drugs that effectively inhibit disease progression, there
is a growing demand to alleviate treatment side effects and improve
the cost-effectiveness of therapies. The rise of cutting-edge
technologies, such as stem cell therapy, nucleic acid delivery
techniques, targeted drugs and epigenetic interventions, has
brought new hope for patients with PF and provided diversified
options and prospects for clinical treatment. To systematically
elaborate on the core characteristics and clinical prospects of
these emerging strategies, this article summarizes their mechanisms
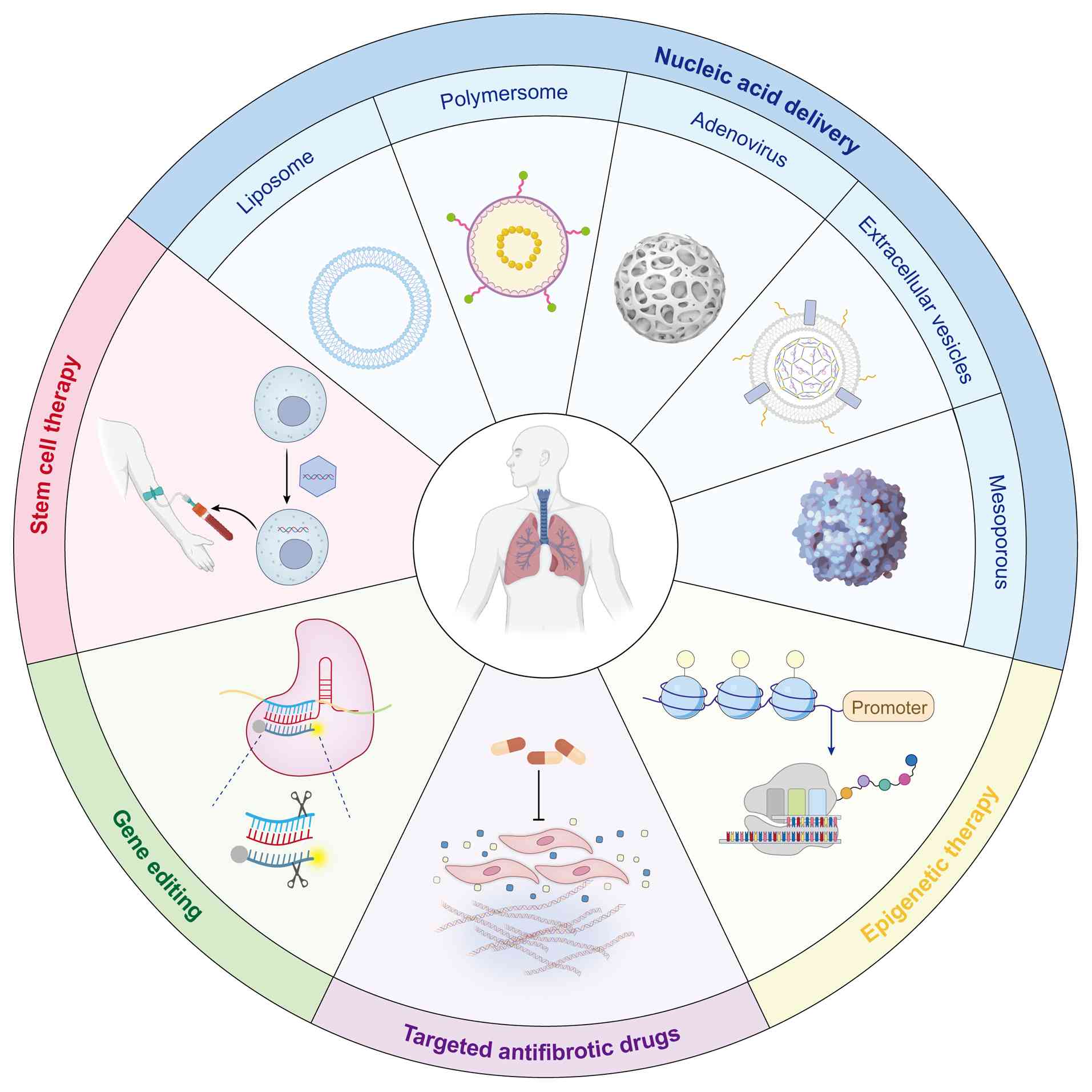
of action, advantages and challenges (Fig. 2 and Table I).
 | Table IComparison of emerging therapeutic
strategies for pulmonary fibrosis. |
Table I
Comparison of emerging therapeutic
strategies for pulmonary fibrosis.
| Therapeutic
category | Representative
agent/approach | Development
stage | Key advantages | Major
challenges | (Refs.) |
|---|
| Stem cell
therapy | 1. Lung-derived
stem cells (e.g., AT2 cells, basal cells)
2. MSCs (from bone marrow, adipose tissue, etc.)
3. iPSCs | Preclinical (some
phase I trials ongoing) | Multimodal repair,
low immunogenicity (EVs), disease modeling | Tumorigenicity risk
(iPSCs), scalable production, delivery efficiency high (especially
for MSC-EVs) | (74-99) |
| Targeted drug
therapy | 1. Pathway
Inhibitors: TGF-β (e.g., ALK5 inhibitors), Wnt, PI3K/Akt (e.g.,
Omipalisib), Hippo-YAP pathway inhibitors
2. Inflammatory factor antagonists: IL-13, CCL2, LIGHT/TL1A
monoclonal antibodies, etc. | Phase I/II clinical
trials | High specificity,
combinable with existing drugs | Patient
stratification needed, resistance possible | (100-123) |
| Epigenetic
therapy | 1. DNA methylation
modulation: DNMT inhibitors (e.g., 5-AZA)
2. Histone modification modulation: HDAC inhibitors
3. Non-coding RNA regulation: miRNA agonists/antagonists
4. Epigenome editing: CRISPR/dCas9 systems | Preclinical to
early clinical | Reverses pathogenic
memory, synergistic potential | Off-target effects,
toxicity (5-AZA) | (128-146) |
| Nucleic acid
delivery therapy | 1. Delivery
Systems: LNPs, polymeric nanoparticles, AAV, EVs
2. Therapeutic molecules: siRNA, mRNA, miRNA, CRISPR
components | Rapidly developing
(siRNA drugs already approved) | Precision at the
genetic level, modular and flexible platform, suitability for local
lung delivery | Efficient,
cell-specific delivery to the lungs; stability and immunogenicity
of nucleic acid drugs; long-term safety of delivery vehicles | (92,93,149-158) |
| AI | AI-driven platforms
& tools: e.g., AlphaFold (structure prediction), deep learning
models (drug design, image analysis) | Early development
(target discovery phase) | Accelerates
R&D, predicts novel targets | Validation in
vivo required, data dependency | (159-164) |
Stem cell therapy
Stem cell therapy for IPF is an important component
of translational medicine, offering emerging regenerative medical
treatments for this disease (74,75). Stem cells used in the treatment
of IPF include pulmonary-derived stem cells, mesenchymal stem cells
(MSCs) derived from bone marrow (BMSCs), adipose tissue and
placenta, iPSCs and embryonic stem cells (75). The therapeutic mechanisms of
these stem cells mainly focus on their ability to exert immune
modulation, alleviating pulmonary inflammation (76,77). These processes are essential for
inhibiting the progression and worsening of PF. Additionally, stem
cells can repair lung tissue by secreting anti-fibrotic factors and
angiogenesis factors that promote tissue healing (78), while differentiating into various
cell types to replace dysfunctional cells, thus recovering lung
function (79).
Another promising approach is using natural or
synthetic scaffolds to generate bioengineered functional lung
tissue for medical purposes. To date, various decellularization
techniques have been applied to lung tissue. For instance,
decellularized 3D hydrogel-based organoids can be used to assess
the impact of the ECM microenvironment on fibroblast phenotypes,
tissue homeostasis and disease function, and can potentially be
transplanted into damaged lung tissue (80). However, the effectiveness of
these methods still requires further validation.
Pulmonary-derived stem cells mainly include basal
cells from the nasal epithelium (5), proximal trachea and bronchial
cells, and AT2 cells from the alveolar region (81). AT2 cells are considered the
progenitor cells of AT1 cells. In a BLM-induced PF rat model,
transplantation of AT2 cells reduced PF, decreased the lung scar
area, accelerated body weight recovery and lowered the
hydroxyproline content (82).
Furthermore, AT2 cell transplantation effectively alleviated
pulmonary edema, collagen deposition and immune cell infiltration,
all of which contribute to the progression of PF. Numerous studies
have also shown that basal stem cell transplantation has a certain
therapeutic effect on PF (83,84).
Although autologous lung stem cells have therapeutic
potential, there are issues such as limited donor availability,
reduced self-renewal capacity due to increased donor age, and
interference with genetic and epigenetic memory. These issues have,
to a certain extent, limited their clinical application (85,86). Autologous lung stem cells are
difficult to expand and their biological characteristics gradually
change with successive passages. To date, no method has been
established that allows for widespread ex vivo proliferation
of lung stem cells while maintaining their self-renewal and
differentiation potential. Considering the inherent challenges in
obtaining and expanding primary lung stem cells, current studies
focus on establishing functional induced lung stem cells in
vitro.
MSCs can be expanded in vitro and recruited
to injured areas after allogeneic transplantation, promoting
epithelial tissue repair and exhibiting immune-regulatory functions
(87). MSC-based therapies
include both systemic and local administration (88). However, challenges such as low
cell survival rates, dependency on dosage and frequency for
therapeutic efficacy, and issues like graft rejection and
tumorigenicity, have limited their widespread use (89-91). In recent years, MSC-derived
exosomes (MSC-EVs) have attracted attention as a promising
alternative treatment. MSC-EVs carry bioactive molecules such as
microRNAs (miRNAs), proteins and lipids, which can specifically
regulate the pathological processes of IPF while avoiding the risks
associated with traditional cell transplantation. Zhou et al
(92) demonstrated that BMSC-EVs
could delay the progression of IPF in a mouse model by delivering
miR-186. Wan et al (93)
demonstrated that EVs derived from BMSCs overexpressing miR-29b-5p
can alleviate IPF via the frizzled 6 pathway. Nevertheless, the
clinical translation of MSC EVs still faces challenges, including
the effective separation, modification and delivery of exosomes,
which require further optimization.
As a breakthrough technology, iPSCs transform
somatic cells into PSCs by genetic reprogramming (94), which can avoid the ethical
problems and immune rejection caused by embryonic stem cells, and
thus hold immense application potential. This gives iPSCs great
potential for therapeutic applications. iPSCs can serve as
effective cellular substitutes in IPF models and improve the
pathological manifestations of PF. However, iPSCs still face
several technical challenges in clinical treatment, including low
differentiation efficiency and cellular heterogeneity during
differentiation (95,96). In response, researchers have
proposed various optimization strategies. For example, Soh et
al (6) improved the
efficiency of iPSC-derived lung progenitor cells through a two-step
differentiation protocol. In 2014, Huang et al (7) developed an efficient method to
direct human human pluripotent stem cells (hPSCs) to differentiate
into lung and airway epithelial cells, including basal cells,
goblet cells, Clara cells, ciliated cells and both AT. Later,
Yamamoto et al (97)
reported a long-term expansion method for alveolar organoids
containing iPS-derived alveolar stem cells (SFTPC+), showing that
the differentiation process and cellular heterogeneity of SFTPC+
cells and their progenitors closely resemble those of AT2 cells. In
2025, Pezet et al (98)
successfully converted hPSCs into expandable spheres, named induced
respiratory progenitor cells, achieving a 95% purity of induced AT1
cells.
Furthermore, the use of CRISPR/Cas9 technology, zinc
finger nucleases for homology-directed repair, small/short DNA
fragments and sequence-specific transcription activator-like
effector nucleases for gene editing enables the precise correction
of mutations associated with genetic diseases such as cystic
fibrosis (8,99). In summary, researchers use
induced or gene-edited iPSCs to create disease models that simulate
human disease pathology. By investigating these models, researchers
can gain a deeper understanding of the underlying mechanisms of the
disease and identify potential drug targets, providing new
directions for personalized treatment of PF.
Targeted drug therapy
Scholars have identified several important signaling
pathways in the progression of PF, including the TGF-β, Wnt,
PI3K-Akt and Hippo-YAP pathways. TGF-β is considered one of the
most important molecules in the fibrosis process. TGF-β regulates
cell proliferation, fibroblast activation, ECM deposition and
myofibroblast differentiation through both its classic Smad and
non-Smad signaling pathways. In particular, TGF-β1 plays a central
role in PF by promoting the transformation of fibroblasts into
myofibroblasts, thereby advancing fibrosis progression (49). To inhibit TGF-β1 activation,
several strategies have been developed in clinical and experimental
studies, such as using antisense oligonucleotides to block TGF-β
synthesis, targeting TGF-β ligands with proteoglycans or soluble
TGF-β receptors, or inhibiting TGF-β receptor activity via activin
receptor-like kinase 5 inhibitors (100). Studies have also found that the
Sloan-Kettering Institute proto-oncoprotein, a negative regulator
of the TGF-β signaling pathway, can effectively modulate
TGF-β1/Smad signaling, slowing fibroblast proliferation and EMT,
providing a new strategy for treating PF (101-103). It is well known that integrins
promote TGF-β activation, and avβ6 integrin is one of the most
extensively studied potential therapeutic targets in IPF. However,
the phase II clinical trial of the anti-integrin drug BG00001
showed no significant change in forced vital capacity between the
treatment and placebo groups, and certain patients even experienced
acute exacerbations (104).
Pentoxifylline-2 (PTX-2) inhibits the production of TGF-β, possibly
by suppressing the differentiation of monocytes into macrophages,
which in turn reduces TGF-β levels. Zinc pentoxifylline α is a
recombinant form of human PTX-2 (rhPTX-2). In phase II trials
(NCT04552899, NCT02550873), rhPTX-2 showed significant efficacy in
restoring lung function (104,105). However, in the 52-week phase
III randomized controlled trial STARSCAPE, no significant
differences were observed between rhPTX-2 and the placebo in
patients with IPF (104).
Galectin-3 (Gal-3), a lectin that binds to β-galactosides, is
significantly elevated in various fibrotic diseases. This
β-galactoside-binding lectin promotes fibrosis progression by
modulating the expression of TGF-β receptors. A phase I/IIa
clinical trial showed that individuals receiving the Gal-3
inhibitor TD139 exhibited reduced expression of Gal-3 compared to
control subjects. The results suggested that inhibition of Gal-3
expression in the lungs correlates with a reduction in plasma
biomarkers. This is closely related to the pathological biology of
IPF (PDGF-BB, plasminogen activator inhibitor-1, Gal-3, CCL18 and
chitinase-3-like protein 1) (106).
The Wnt signaling pathway plays a crucial role in
various fibrotic diseases, particularly in regulating the
proliferation and differentiation of fibroblasts. Wnt signaling
promotes fibroblast activation and accelerates fibrosis progression
by activating the downstream β-catenin-dependent pathway (107). A study showed that betulinic
acid (BA), a pentacyclic triterpenoid, can alleviate the effects of
BLM-induced PF in mice, particularly in reducing collagen
deposition and improving lung function. BA effectively reduces the
expression of fibrotic markers, including fibronectin, collagen I
and α-SMA. Further research indicates that BA can inhibit
Wnt3a-induced fibroblast activation and subsequent Wnt/β-catenin
pathway activation, resulting in reduced nuclear accumulation of
β-catenin and phosphorylation of key signaling proteins such as
low-density lipoprotein receptor-related protein 6 and Dishevelled
2 (108). Another study
explored the use of SBC-115076 as a potential treatment for pPH
associated with PF (109). The
results demonstrated that proprotein convertase subtilisin/kexin
type 9 (PCSK9) inhibition reduced pulmonary artery thickening and
right ventricular remodeling in a BLM-induced PF animal model. More
importantly, SBC-115076 treatment also diminished the activation of
the Wnt/β-catenin pathway, a core driver of fibrosis and vascular
remodeling. These findings suggest that PCSK9 inhibitors serve as
promising therapeutic agents for PF and PH by modulating key
fibrotic and vascular remodeling pathways. These studies provide a
theoretical basis for targeting the Wnt signaling pathway as a
potential therapeutic target.
The PI3K-Akt pathway plays a pivotal role in the
pathogenesis of PF (110). PI3K
is a group of membrane-associated lipid kinases classified into
three classes based on their molecular structure. Class I PI3Ks are
the most widely studied in various diseases. PI3Kα is commonly
upregulated or mutated in lung-related diseases (111), PI3Kγ is often overexpressed in
IPF lungs and fibroblasts (112), and class III PI3K is involved
in the formation of autophagosome membranes, potentially affecting
PF (113). AKT is a
serine/threonine protein kinase with three subtypes, and research
on PF has primarily focused on AKT1 and AKT2. AKT1-mediated mitotic
cells help alveolar macrophages resist apoptosis (114). This is essential for the
development of PF, while AKT2 regulates PF by inducing macrophages
to produce TGF-β1 and IL-13. AKT2-deficient mice are protected from
BLM-induced PF and inflammation (115). Evidence suggests that the
PI3K/Akt signaling pathway can promote PF by activating multiple
key factors involved in cell proliferation, survival, and ECM
deposition (116,117). Inhibitors targeting the
PI3K/Akt pathway, such as omipalisib and rapamycin, have shown
potential efficacy for PF in clinical trials. These studies
indicate that the PI3K/Akt pathway is an important therapeutic
target for PF.
Hippo-YAP signaling pathway also plays an essential
role in the pathogenesis of fibrosis. Liu et al (53) demonstrated that YAP, a homolog of
Drosophila Yki, and TAZ, transcriptional co-activators, regulate
fibroblast activation and matrix synthesis. Inhibition of the Hippo
signaling pathway can reduce BLM-induced PF (118). Qing et al (119) used a G protein-coupled receptor
(GPCR) ligand screening system to identify a dopamine receptor D2
antagonist that selectively blocks YAP in macrophages. By targeting
GPCRs, drugs can modulate the Hippo-YAP signaling pathway,
achieving anti-fibrotic effects. Zeyada et al (120) found that trigonelline (Trig), a
natural plant alkaloid with multiple pharmacological effects, can
attenuate the sphingosine kinase 1/sphingosine-1-phosphate axis in
the lungs and its downstream Hippo targets YAP-1 and TAZ. After
Trig administration, EMT in BLM-induced mouse lung tissue was
reversed. These strategies provide new insights for developing
novel targeted therapeutic drugs.
In addition to the signaling pathways, inflammation
plays a critical role in the onset and progression of PF.
Particularly during acute exacerbations and in certain subtypes of
PF, inflammatory responses exacerbate the fibrotic process. Studies
have shown that cytokines (such as IL-13, IL-6), chemokines (such
as CCL2), and growth factors (such as TGF-β) are key factors
driving fibroblast activation and ECM deposition (121,122), making them important targets
for anti-inflammatory therapies. Recent research has indicated that
inhibition of TNF superfamily (TNFSF) members, such as
lymphotoxin-like, exhibits inducible expression, and competes with
HSV glycoprotein D for HVEM (LIGHT/TNFSF14) and TNF-like ligand 1A
(TNFSF15), can significantly reverse fibrosis in a preclinical
model providing a new direction for inflammation-targeted therapies
(123).
However, the widespread use of corticosteroids and
immunosuppressants for anti-inflammatory treatment often yields
limited effectiveness and may even worsen the condition,
highlighting the need for more precise, mechanism-based therapeutic
approaches. Although clinical applications of combined
anti-inflammatory and anti-fibrotic treatments face several
challenges, including patient selection, drug side effects and
ongoing immune modulation requirements, these recent findings lay a
solid foundation for the future development of personalized,
inflammation-targeted therapies for PF.
Epigenetic therapy
Epigenetic therapy has gained significant attention
as a novel treatment strategy, particularly for complex diseases
such as cancer, genetic disorders, cardiovascular diseases and
chronic diseases like fibrosis (124-127). DNA methylation regulates gene
expression by directly affecting the transcriptional activity of
genes. Numerous studies have indicated that DNA methylation plays a
pivotal role in the progression of PF (54,55). Currently, treatment strategies
for DNA methylation focus primarily on DNA demethylating agents.
DNA methyltransferases (DNMTs) are key catalytic enzymes in the
process of DNA methylation and studies have shown that
pharmacological inhibitors targeting DNMTs can effectively suppress
the expression of fibrotic genes. For example, TGF-β1 significantly
induced the methylation of the Thy-1 promoter in lung fibroblasts,
while DNMT inhibitors such as 5-azacytidine (5-AZA) could
downregulate the expression of fibrotic-related genes, including
α-SMA and collagen type I, significantly inhibiting collagen
deposition (128). Wei et
al (129) showed that 5-AZA
and glycyrrhizic acid alleviated peroxisome proliferator-activated
receptor γ (PPARγ)-mediated fibrosis suppression through mechanisms
that increased sensitivity to DNMTs, suggesting that targeting the
DNMT/PPARγ axis could benefit patients with PF. The DNA methylation
inhibitors 5-AZA (Vidaza®) and its deoxy derivative
5-aza-2'-deoxycytidine (Dacogen®) are widely used in
cancer treatment, particularly for hematological malignancies,
where they have been proven effective in reactivating and
upregulating tumor suppressor genes (130,131). However, the use of 5-AZA and
similar drugs requires careful attention to their potential toxic
effects. Some reports indicate that DNMT inhibitors may cause
hepatotoxicity, with certain patients experiencing adverse effects
such as interstitial lung fibrosis, pneumonia and acute lung injury
(132,133). Therefore, it is of great
significance to develop DNA methylation inhibitors with lower
toxicity to ensure the safety and efficacy of these treatments in
clinical applications.
In recent years, the widespread use of CRISPR/dCas9
technology has introduced a new approach for treating PF through
epigenome editing. Wang et al (134) employed CRISPR/Cas9-mediated
homology-directed repair to target the important epigenetic
biomarker O6-methylguanine-DNA methyltransferase for de novo
methylation, resulting in stable upregulation of DNMTs in edited
HeLa cells. Qu et al (135) used CRISPR/dCas9-Dnmt3A-mediated
epigenome editing to effectively reverse matrix stiffness-induced
overexpression of desmoplakin. Wu et al (136) investigated its impact on
ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) expression and DNA
methylation patterns in human bronchial epithelial cells. After
using CRISPR/dCas-zeste homolog 2 to epigenetically downregulate
UCHL1, they observed a reduction in mRNA expression of COL1A1 and
fibronectin. These studies demonstrate the significant potential of
CRISPR/Cas9-directed epigenetic silencing in functional and
therapeutic research. However, off-target effects remain a major
hurdle in the clinical application of CRISPR/Cas9 technology.
Compared to other gene delivery platforms, such as recombinant
adeno-associated viruses (AAVs) (137), CRISPR/Cas9 has conceptual
advantages, but targeting specific cell types remains a
challenge.
In the study of PF, the increased activity of HDACs
is considered a significant factor in promoting fibrosis (138). HDACs inhibit gene transcription
by removing acetyl groups from histones, leading to a more compact
chromatin structure. To date, the US Food and Drug Administration
(FDA) has approved four HDAC inhibitors for cancer treatment
(139), but none have been
approved for fibrotic diseases. Emerging in vitro and in
vivo preclinical evidence increasingly suggests that HDACs play
a beneficial role in preventing or reversing fibrosis (138,140). Rubio et al (141) found that EP300 reduced nuclear
HDAC activity and interfered with the ribonucleoprotein complex
function in IPF, and inhibiting EP300 significantly reduced
fibrosis markers, providing a basis for more effective treatments
targeting the etiology of IPF. For patients with IPF, HDAC3
expression has been found to significantly increase and to be
closely correlated with the progression of fibrosis (142). Inhibiting HDAC3 activity was
shown to restore the expression of fibrosis-related genes and
reduce the degree of PF. Yu et al (143) designed and synthesized 24 novel
HDAC6, HDAC8 or dual HDAC6/8 inhibitors, identifying five HDAC
inhibitors that can alleviate TGF-β-induced PF. Future HDAC
inhibitors could be combined with other drugs or therapies as
potential strategies for treating PF.
MiRNAs regulate mRNA to alter the expression of
target genes and dynamically regulate DNA methylation and other
non-coding RNAs, including themselves. These regulatory mechanisms
lead to changes in miRNAs, triggering chain reactions in the
TGF-β1/Smad, MAPK and PI3K/AKT pathways, ultimately resulting in
the expression of a fibrotic phenotype. Animal studies have shown
that miRNA-29 can target various fibrosis-related genes, including
COL1A1, COL3A1 and TGF-β1, and inhibiting these genes can slow down
the occurrence of fibrosis (144). Designing miRNAs at the genetic
level represents a promising therapeutic strategy and miRNA-based
drugs integrated into in vivo and in vitro
applications have the potential to yield the most direct clinical
outcomes. Furthermore, the use of extracellular vesicles (EVs),
particularly those derived from MSCs, as delivery systems for
miRNAs has shown promising therapeutic effects in the treatment of
PF (92,93). However, a significant challenge
for targeting miRNAs in IPF is the potential difference in gene
regulation by individual miRNAs between mice and humans. Therefore,
future research should focus on the continuous improvement of
existing models and emphasize the testing and validation of
hypotheses across multiple model systems.
Long non-coding, RNAs (lncRNAs) represent a key
aspect of epigenetic therapy. LncRNAs play crucial roles in
regulating gene expression, chromatin modifications and cell fate
determination. Savary et al (145) found that lncRNA dynamin 3
opposite strand was one of the most strongly induced lncRNAs in a
human lung fibroblast cell line (MRC-5) stimulated with TGF-β1,
through RNA sequencing and small RNA sequencing. Xia et al
(146) showed that lncRNA SYISL
promotes fibroblast-to-myofibroblast transformation via
miR-23a-mediated regulation of TRIO and F-actin binding protein.
In vivo delivery of SYISL-targeted short hairpin RNA
significantly reduced collagen deposition, hydroxyproline content
and fibrosis marker expression in BLM-induced mice. These studies
further demonstrate the potential of targeting lncRNAs as a
therapeutic strategy for IPF.
One important feature of epigenetic therapy is its
reversibility, implying that by modulating epigenetic markers, it
is possible to partially restore the normal expression of genes.
This offers new hope for treating numerous incurable diseases.
Although epigenetic therapy has achieved some positive results in
basic research, it still faces numerous challenges in clinical
applications, such as selecting appropriate treatment targets,
developing delivery systems and ensuring the long-term
effectiveness of treatments. In the future, as research in
epigenetics advances, these therapeutic approaches are expected to
gradually move toward clinical use, potentially bringing new
breakthroughs in the treatment of diseases like fibrosis.
Nucleic acid delivery therapy
Nucleic acid delivery therapies have fundamentally
transformed the treatment of a wide range of diseases, including
genetic disorders, infectious diseases and malignant tumors. By
delivering nucleic acid molecules such as plasmid DNA, small
interfering siRNA, miRNA and circular RNA, it is possible to
regulate key signaling pathways associated with specific diseases
at the molecular level. This can directly or indirectly influence
gene expression, gene silencing or gene deletion, thereby
inhibiting or repairing the production of abnormal proteins.
Compared to traditional drug treatments, nucleic acid delivery
offers more precise gene modification capabilities, enabling
targeted therapy that effectively avoids systemic side effects.
The unique physiological structure of the lungs
makes them an ideal route for drug delivery, facilitating both
systemic drug therapy and localized treatment of lung diseases. The
lung's distinct structure allows it to serve as an ideal target for
drug delivery, capable of delivering both systemic therapies and
local treatments for pulmonary diseases. Recent advancements have
been made in the development of delivery systems targeting
pulmonary capillary endothelial cells and specific lung cells,
particularly the inhalation method, which has proven to be an
effective approach for treating lung diseases. The outbreak of
COVID-19 significantly accelerated the development and
commercialization of mRNA-based vaccines. This in turn boosted the
application of nucleic acid drugs in pulmonary delivery, opening
new prospects for vaccine development and gene therapy (147,148).
However, the unique characteristics of the lungs
present challenges. Due to continuous exposure to external air, the
lungs are susceptible to infections and inflammation, which
necessitates a heightened focus on the safety of nucleic acid
delivery carriers. There are two primary routes for nucleic acid
delivery to the lungs: Systemic intravenous injection and localized
inhalation. Both routes face distinct challenges, especially when
nucleic acids must cross the cell membrane. Due to their large
molecular structure and negative charge, nucleic acids typically
struggle to penetrate the cell membrane directly, and RNA is prone
to degradation, which limits the delivery effectiveness. Therefore,
enhancing nucleic acid stability, improving intracellular
expression and ensuring the stability of aerosolization have become
key research areas in lung nucleic acid delivery.
To overcome these challenges, researchers have
developed various innovative carrier systems, such as liposomes,
polymer NPs and LNPs. These carriers effectively enhance the
stability of RNA, preventing degradation in external environments
and by nucleases, while improving delivery efficiency. As delivery
technologies continue to advance, the prospects for pulmonary
nucleic acid drug delivery are increasingly promising, offering new
hope for the treatment of pulmonary diseases.
Applications of liposomes and polymer
NPs
As an effective delivery vehicle, liposomes have
been extensively explored and applied for nucleic acid drug
delivery. In 2018, the US FDA approved the first siRNA drug
formulated with liposomes. Liposomes are used for siRNA delivery
and can decorate collagen-binding peptides and collagenases, target
fibrotic lung tissue and aid in the restoration of normal lung
structure (149,150). Zhao et al (151) developed a non-inflammatory LNP
exhibited a 40-fold enhancement in pulmonary protein expression
without inducing significant inflammatory responses compared to
traditional LNP formulations. Furthermore, they developed ursolic
acid-incorporated phosphoramide-based LNPs encapsulating mRNA
encoding nuclear receptor subfamily 1 group D member 1. These
non-inflammatory LNPs effectively combat pulmonary fibrosis by
reducing inflammation and oxidative stress, promoting fibroblast
conversion, and enhancing angiogenesis. Polymer NPs also serve as
an effective drug delivery system, improving drug loading
efficiency and solubility. Polysaccharide-based natural products,
such as chitosan, are of particular interest due to their excellent
biocompatibility and biodegradability, and have been widely studied
for gene drug delivery. Vlasova et al (152) proposed a method for efficiently
synthesizing cationic poly(ethyleneimine) derivatives, using a
fission-Ugi reaction to synthesize ionizable polymers for lung
delivery of various sizes of RNA and gene editing tools. Compared
to in vivo polyethylenimine, lipid-polymer-lipid hybrid NPs
showed a 300-fold increase in efficiency for systemic mRNA delivery
to the lungs. These NPs were also able to deliver complex
CRISPR-Cas9 gene RNA to the lungs, achieving ~6% gene editing in
lung tissue. Bai et al (153) developed an inhalable and
mucosal-penetrating NP system that was formulated from PLGA-PEG and
G0-C14 (termed PPGC), which enabled efficient mucosal delivery of
siIL11. In a mouse model, siIL11@PPGC NPs significantly reduced
fibrosis development and improved lung function without inducing
systemic toxicity.
Application of AAVs
AAV, a small single-stranded DNA virus, has been
studied for pulmonary nucleic acid drug delivery. AAV exhibits low
immunogenicity and has been used in clinical trials to treat Leber
congenital amaurosis and spinal muscular atrophy (154). Studies show that AAV can bind
to glycosaminoglycan receptors in bronchial mucus and effectively
penetrate the mucus layer for pulmonary gene delivery.
Additionally, AAV6 has been used to deliver miRNA-21-5p in a
hyperoxic acute lung injury rat model to prevent apoptosis of AT2
cells (155). Wang et al
(156) found that AAV6-mediated
knockdown of nestin inhibited TGF-β signaling and significantly
alleviated PF in mouse models and patients with IPF. Although AAV
application faces challenges in safety and immune response, it
still shows great potential as a gene therapy tool for lung
diseases, particularly for fibrosis treatment.
Exosome delivery
EVs, composed of phospholipid bilayers, possess
excellent biocompatibility and targeting capabilities, making them
potential carriers for nucleic acid delivery (92,93). EVs can maintain lung health by
modulating immune responses, inducing tissue repair and maintaining
pulmonary homeostasis. They can be detected in lung tissue and
biological fluids such as bronchoalveolar lavage fluid and blood,
providing information about disease progression and serving as
biomarkers for diseases (157).
The efficacy of exosomes derived from umbilical MSCs (hUCMSC-EVs)
has been validated in clinical trials (158) (MR-46-22-004531,
ChiCTR2300075466). Patients who received nebulized hUCMSC-EVs as an
adjunct therapy showed degenerative changes in lung fibrosis on
consecutive CT scans, compared to those who only received standard
treatment. Due to their natural membrane protection and targeting
abilities, EVs have broad application potential in gene therapy and
drug delivery.
In summary, research on nucleic acid delivery
therapies and their carrier systems is continuously advancing
towards improving delivery efficiency, overcoming physiological
barriers and enhancing therapeutic efficacy. By using various
carrier systems such as liposomes, polymer NPs, adeno-associated
viruses and exosomes, the effective delivery of nucleic acid drugs
can be achieved, showing great potential in the treatment of both
local and systemic diseases.
AI rise
The development of new drugs is a multi-step,
lengthy and costly process. Although traditional drug discovery
methods are valuable, they face numerous challenges, including low
success rates and inefficient predictions of drug properties,
toxicity and drug-target interactions. In recent years,
advancements in AI have provided rapid and effective strategies to
gradually overcome these obstacles, significantly reshaping the
landscape of drug discovery and development. AI has already
achieved remarkable results in various therapeutic fields,
including oncology, infectious diseases, neurology and rare
diseases. By utilizing machine learning (ML), deep learning (DL)
and natural language processing, various stages of the drug
development process, including target identification, drug
screening, drug design, drug-target interactions, drug repurposing,
prediction of physicochemical properties, toxicity evaluation and
pharmacokinetic forecasting, have been significantly enhanced.
Through the analysis of large datasets, AI can identify potential
drug molecules and accurately predict their biological activity,
thereby accelerating the drug development process.
With the continuous progress of AI technologies,
scientists can identify potential therapeutic targets for diseases
such as PF more quickly and design effective drugs targeting these
specific targets. AI can mine key biomarkers of diseases from large
biomedical datasets and discover numerous potential molecular
mechanisms, providing strong support for the development of new
drugs. Various AI models and AI-driven tools have now been
developed to screen, design, discover and optimize drugs, such as
AlphaFold, Chemistry42, Clinico and ProTox-II. Utilizing
AI-supported platforms, Pun et al (159) identified therapeutic targets
for amyotrophic lateral sclerosis. Xu et al (160) successfully identified Traf2 and
Nck-interacting kinase (TNIK) as key regulators in the pathology of
IPF. Furthermore, AI can optimize clinical trials by analyzing
patient data and recruitment patterns. Xu et al (160) reported the first clinical trial
of TNIK-targeting inhibitors (NCT05938920), where they used AI to
streamline preclinical candidate nomination to just 18 months and
reduced the completion time of Phase 0/1 clinical trials by at
least 30 months from target discovery. This is the first reported
case where an AI platform successfully identified disease-related
targets and compounds, marking a revolutionary shift toward
streamlining drug discovery.
DL plays a multifaceted role in drug discovery,
with applications ranging from predicting compound activity,
generating new chemical structures and predicting chemical
reactions, to calculating ligand-protein interactions and analyzing
biomedical imaging. Witten et al (161) created a dataset containing
>9,000 LNP activity measurements and used it to train a directed
information transfer neural network for predicting nucleic acid
delivery with various lipid structures. They evaluated 1.6 million
lipids in silico and identified two structures, FO-32 and
FO-35, that exhibited localized mRNA delivery to mouse muscle and
nasal mucosa. Gerckens et al (162) developed an end-to-end deep
learning model by analyzing thousands of immunofluorescence-stained
ECM images obtained through automated high-throughput microscopy.
This model screened a small molecule drug repurposing library to
inhibit ECM deposition, with AI-driven fiber pattern detection
identifying Tranilast as an effective anti-fibrotic inhibitor.
ML, a branch of AI, involves deriving models or
rule sets from an initial training set. These models are then used
to assess new datasets. ML has made significant contributions to
the analysis of high-resolution X-ray computed tomography scans for
predicting the progression of IPF (163). Muratov et al (164) explored the application of ML in
identifying transcriptomic changes related to lung diseases caused
by titanium dioxide (TiO2)-NPs. This approach has
markedly contributed to understanding the potential effects of
TiO2-NP inhalation, advancing the field of
nanotoxicology, and supporting the development of safer
nanotechnology applications. As omics platforms increasingly
integrate with machine learning, one major obstacle remains the
cost-effectiveness of using these expensive assays and complex
machine learning techniques for diagnostics. However, as these
technologies become more mainstream and affordable, this may no
longer pose a significant issue. Proteomics and machine learning
may uncover new relationships that could revolutionize the
diagnosis and management of future patients.
Despite the enormous potential AI holds for drug
development, there are still several challenges to address. Issues
such as data accessibility, the integration of diverse datasets and
the interpretability of AI models remain pressing concerns. The
'black box' nature of AI makes it difficult for researchers to
understand the decision-making process of AI models, which could
impact transparency and trust in drug development. As technology
continues to evolve, improvements in algorithms, interdisciplinary
data integration and stronger collaboration between laboratories
and AI technologies will further drive the application of AI in
treating diseases like PF.
Furthermore, AI has made it possible to explore
vast chemical spaces, optimize clinical trials and identify new
therapeutic targets, thus paving the way for the development of
precision medicine. However, challenges such as limited data
availability, integration of diverse datasets, AI model
interpretability and ethical concerns remain key obstacles.
Overcoming these limitations through improved algorithms,
standardized databases and interdisciplinary collaboration is
crucial. Overall, AI is continuously reshaping drug discovery by
shortening timelines, increasing success rates and driving the
development of innovative and accessible therapies to address unmet
medical needs.
Conclusion and outlook
Significant progress has been made in the treatment
of PF over the past few decades, yet its complex pathological
mechanisms and treatment challenges continue to pose a substantial
clinical hurdle. Although existing treatments such as anti-fibrotic
drugs and lung transplantation have helped slow disease progression
to a certain extent, their effectiveness remains limited and they
cannot cure the disease. With the development of cutting-edge
technologies, including stem cell therapy, targeted drugs,
epigenetic therapy, nucleic acid delivery and AI, the treatment of
PF is poised to enter a new era.
Stem cell therapy, as a potential regenerative
medicine approach, has demonstrated promise in experimental studies
for repairing lung tissue and reversing the fibrotic process.
However, improving stem cell survival, targeting and long-term
effects remains a significant challenge for clinical applications.
Targeted drug therapies, which precisely inhibit key
fibrosis-related molecules such as TGF-β and PDGF, have shown some
clinical success. However, the effectiveness of single-target
treatments is often hindered by resistance and pathological
remodeling, necessitating the combination with other therapeutic
strategies for more effective combination therapies.
Epigenetic therapy offers a novel treatment
approach by regulating gene expression and repairing epigenetic
changes caused by genetic damage or environmental factors. While
the development of epigenetic drugs is still in its early stages,
their potential to intervene in the fibrotic process and restore
normal gene function should not be underestimated. Nucleic acid
delivery technologies, utilizing small RNA molecules and gene
editing techniques, enable direct regulation of gene expression and
the repair of disease-causing genes, providing a more precise and
personalized treatment option for PF. Despite challenges such as
efficient delivery, targeting and side effects, advancements in
delivery technology hold the promise of breakthroughs in this
field. The application of AI in PF treatment has also become an
emerging research focus. AI can leverage big data analysis, machine
learning and other technologies to precisely screen drugs, predict
disease progression and optimize individualized treatment plans,
markedly enhancing treatment efficiency. Nevertheless, the future
of PF treatment remains fraught with challenges, particularly in
effectively integrating these advanced technologies into clinical
practice.
Given the significant heterogeneity of PF and the
possible variation in the disease mechanisms across different
patients, future treatment strategies should focus more on
individualized and precision therapies. Additionally, how to
effectively combine various treatment approaches to overcome the
limitations of single therapies remains a critical issue. Overall,
the future treatment of PF will rely on interdisciplinary
collaboration, combining stem cells, gene therapy, targeted drugs,
epigenetic repair and AI, offering new hope for the treatment of
this disease. However, further clinical validation and
technological breakthroughs are needed before these innovations can
be widely applied.
Limitations
Although this review aims to systematically
elaborate on the progress and prospects of emerging therapeutic
strategies for PF, it is subject to several limitations. First,
most of the advanced therapies discussed (e.g., iPSC-based cell
therapy, epigenetic editing and novel nucleic acid delivery
systems) are currently primarily in the preclinical or early
clinical trial stages. Their long-term efficacy and safety in human
patients have not yet been fully validated. Second, existing
studies exhibit heterogeneity across different model systems (e.g.,
various animal models, cell lines) and patient populations, which
limits the extrapolation and generalizability of some conclusions.
Additionally, the discussion was performed largely based on
published literature, which may not cover all the latest or
unpublished clinical trial data, and the literature selection may
have been influenced by the authors' search strategy and database
scope. Furthermore, although this review emphasizes the importance
of multi-target and combination therapies, understanding of the
optimal combination regimens, timing and underlying interaction
mechanisms remains incomplete. PF itself is highly heterogeneous
(e.g., different etiologies, disease stages), yet most current
research on novel therapies still tends to treat IPF as a
homogeneous entity; the efficacy for specific subtypes requires
further exploration. Finally, achieving truly individualized and
precise treatment depends on a robust biomarker system, and
research in this area is still in its early stages. Acknowledging
these limitations helps in viewing the current research findings
more objectively and points the way toward key breakthroughs needed
in the future. Before these innovations can be widely applied,
further clinical validation and technological breakthroughs are
required.
Availability of data and materials
Not applicable.
Authors' contributions
SL and YL performed data acquisition and data
analysis and wrote the manuscript. XL and QY conceived and
supervised the work. Data authentication is not applicable. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
PF
|
pulmonary fibrosis
|
|
IPF
|
idiopathic PF
|
|
AI
|
artificial intelligence
|
|
siRNA
|
small interfering RNA
|
|
BSCs
|
airway basal stem cells
|
|
iPSCs
|
induced pluripotent stem cells
|
|
ECM
|
extracellular matrix
|
|
TGF
|
transforming growth factor
|
|
EMT
|
epithelial-mesenchymal transition
|
|
GWAS
|
genome-wide association studies
|
|
MUC5B
|
mucin 5B gene
|
|
AEC2
|
alveolar epithelial type II
|
|
ROS
|
reactive oxygen species
|
|
MMPs
|
matrix metalloproteinases
|
|
AT1
|
type I alveolar epithelial cells
|
|
SASP
|
senescence-associated secretory
phenotype
|
|
BLM
|
bleomycin
|
|
HDACs
|
histone deacetylases
|
|
5mC
|
5-methylcytosine
|
|
m6A
|
N6-methylcytosine
|
|
MSCs
|
mesenchymal stem cells
|
|
5-AZA
|
5-azacytidine
|
|
PH
|
pulmonary hypertension
|
|
AAVs
|
adeno-associated viruses
|
|
EVs
|
extracellular vesicles
|
|
LNPs
|
lipid nanoparticles
|
|
ML
|
machine learning
|
|
DL
|
deep learning
|
Acknowledgements
Not applicable.
Funding
This work was supported by the China Foundation for
International Medical Exchange (grant no. z-2016-23-2101-05).
References
|
1
|
Selman M, King TE and Pardo A; American
Thoracic Society; European Respiratory Society; American College of
Chest Physicians: Idiopathic pulmonary fibrosis: Prevailing and
evolving hypotheses about its pathogenesis and implications for
therapy. Ann Intern Med. 134:136–151. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Senhaji L, Senhaji N, Abbassi M, Karhate
M, Serraj M, El Biaze M, Benjelloun MC, Ouldim K, Bouguenouch L and
Amara B: Idiopathic pulmonary fibrosis: A comprehensive review of
risk factors, genetics, diagnosis, and therapeutic approaches.
Biomedicines. 14:902026. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Golchin N, Patel A, Scheuring J, Wan V,
Hofer K, Collet JP, Elpers B and Lesperance T: Incidence and
prevalence of idiopathic pulmonary fibrosis: A systematic iterature
review and meta-analysis. BMC Pulm Med. 25:3782025. View Article : Google Scholar
|
|
4
|
Moss BJ, Ryter SW and Rosas IO: Pathogenic
mechanisms underlying idiopathic pulmonary fibrosis. Annu Rev
Pathol. 17:515–546. 2022. View Article : Google Scholar
|
|
5
|
Zhao X, Yu F, Li C, Li Y, Chao SS, Loh WS,
Pan X, Shi L and Wang DY: The use of nasal epithelial
stem/progenitor cells to produce functioning ciliated cells in
vitro. Am J Rhinol Allergy. 26:345–350. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Soh BS, Zheng D, Li Yeo JS, Yang HH, Ng
SY, Wong LH, Zhang W, Li P, Nichane M, Asmat A, et al: CD166(pos)
subpopulation from differentiated human ES and iPS cells support
repair of acute lung injury. Mol Ther. 20:2335–2346. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang SX, Islam MN, O'Neill J, Hu Z, Yang
YG, Chen YW, Mumau M, Green MD, Vunjak-Novakovic G, Bhattacharya J
and Snoeck HW: Efficient generation of lung and airway epithelial
cells from human pluripotent stem cells. Nat Biotechnol. 32:84–91.
2014. View Article : Google Scholar :
|
|
8
|
Firth AL, Menon T, Parker GS, Qualls SJ,
Lewis BM, Ke E, Dargitz CT, Wright R, Khanna A, Gage FH and Verma
IM: Functional gene correction for cystic fibrosis in lung
epithelial cells generated from patient iPSCs. Cell Rep.
12:1385–1390. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fleischer A, Lorenzo IM, Palomino E, Aasen
T, Gómez F, Servera M, Asensio VJ, Gálvez V, Izpisúa-Belmonte JC
and Bachiller D: Generation of two induced pluripotent stem cell
(iPSC) lines from p.F508del cystic fibrosis patients. Stem Cell
Res. 29:1–5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lederer DJ and Martinez FJ: Idiopathic
pulmonary fibrosis. N Engl J Med. 378:1811–1823. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nakano Y, Yang IV, Walts AD, Watson AM,
Helling BA, Fletcher AA, Lara AR, Schwarz MI, Evans CM and Schwartz
DA: MUC5B promoter variant rs35705950 affects MUC5B expression in
the distal airways in idiopathic pulmonary fibrosis. Am J Respir
Crit Care Med. 193:464–466. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Borie R, Crestani B, Dieude P, Nunes H,
Allanore Y, Kannengiesser C, Airo P, Matucci-Cerinic M, Wallaert B,
Israel-Biet D, et al: The MUC5B variant is associated with
idiopathic pulmonary fibrosis but not with systemic sclerosis
interstitial lung disease in the European Caucasian population.
PLoS One. 8:e706212013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mouawad A, Chouery E, Chebly A, Salem N,
Corbani S, Safieddine M and Dabar G: Association of the MUC5B
promoter polymorphism with idiopathic pulmonary fibrosis in a
lebanese cohort. Front Genet. 16:15448642025. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Plender EG, Prodanov T, Hsieh P, Nizamis
E, Harvey WT, Sulovari A, Munson KM, Kaufman EJ, O'Neal WK,
Valdmanis PN, et al: Structural and genetic diversity in the
secreted mucins, MUC5AC and MUC5B. Am J Hum Genet. 111:1700–1716.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Herrera JA, Maslanka M, Blumhagen RZ,
Blomberg R, Lwin NY, Brancato J, Cool CD, Huber JP, Kurche JS,
Magin CM, et al: The MUC5B promoter variant results in proteomic
changes in the nonfibrotic lung. JCI Insight. 10:e1896362025.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Duckworth A, Gibbons MA, Allen RJ, Almond
H, Beaumont RN, Wood AR, Lunnon K, Lindsay MA, Wain LV, Tyrrell J
and Scotton CJ: Telomere length and risk of idiopathic pulmonary
fibrosis and chronic obstructive pulmonary disease: A mendelian
randomisation study. Lancet Respir Med. 9:285–294. 2021. View Article : Google Scholar
|
|
17
|
Allen RJ, Stockwell A, Oldham JM,
Guillen-Guio B, Schwartz DA, Maher TM, Flores C, Noth I, Yaspan BL,
Jenkins RG, et al: Genome-wide association study across five
cohorts identifies five novel loci associated with idiopathic
pulmonary fibrosis. Thorax. 77:829–833. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Newton CA, Oldham JM, Ley B, Anand V,
Adegunsoye A, Liu G, Batra K, Torrealba J, Kozlitina J, Glazer C,
et al: Telomere length and genetic variant associations with
interstitial lung disease progression and survival. Eur Respir J.
53:18016412019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Michalski JE and Schwartz DA: Genetic risk
factors for idiopathic pulmonary fibrosis: Insights into
immunopathogenesis. J Inflamm Res. 13:1305–1318. 2020. View Article : Google Scholar
|
|
20
|
Oldham JM, Ma SF, Martinez FJ, Anstrom KJ,
Raghu G, Schwartz DA, Valenzi E, Witt L, Lee C, Vij R, et al:
TOLLIP, MUC5B, and the Response to N-Acetylcysteine among
individuals with idiopathic pulmonary fibrosis. Am J Respir Crit
Care Med. 192:1475–1482. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
López-Otín C, Blasco MA, Partridge L,
Serrano M and Kroemer G: The hallmarks of aging. Cell.
153:1194–1217. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Álvarez D, Cárdenes N, Sellarés J, Bueno
M, Corey C, Hanumanthu VS, Peng Y, D'Cunha H, Sembrat J, Nouraie M,
et al: IPF lung fibroblasts have a senescent phenotype. Am J
Physiol Lung Cell Mol Physiol. 313:L1164–L1173. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cho SJ and Stout-Delgado HW: Aging and
lung disease. Annu Rev Physiol. 82:433–459. 2020. View Article : Google Scholar
|
|
24
|
Caporarello N, Meridew JA, Aravamudhan A,
Jones DL, Austin SA, Pham TX, Haak AJ, Moo Choi K, Tan Q, Haresi A,
et al: Vascular dysfunction in aged mice contributes to persistent
lung fibrosis. Aging Cell. 19:e131962020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Raslan AA, Pham TX, Lee J, Kontodimas K,
Tilston-Lunel A, Schmottlach J, Hong J, Dinc T, Bujor AM,
Caporarello N, et al: Lung injury-induced activated endothelial
cell states persist in aging-associated progressive fibrosis. Nat
Commun. 15:54492024. View Article : Google Scholar
|
|
26
|
Lu Q, Ye C, Mao W, Zeng F, Liu Z, Chen R,
Cheng X, Gao Y, Wang M, Liu M, et al: Targeting senescent alveolar
type 2 cells with a gene-editable FePt Dual-atom catalyst for
mitigating idiopathic pulmonary fibrosis. ACS Nano. 19:23162–23176.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sesé L, Annesi-Maesano I, Cavalin C and
Nunes H: Air pollution and poverty: A deadly combination in
idiopathic pulmonary fibrosis? Eur Respir J. 58:21017142021.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cui F, Sun Y, Xie J, Li D, Wu M, Song L,
Hu Y and Tian Y: Air pollutants, genetic susceptibility and risk of
incident idiopathic pulmonary fibrosis. Eur Respir J.
61:22007772023. View Article : Google Scholar
|
|
29
|
Majewski S and Piotrowski WJ: Air
pollution-an overlooked risk factor for idiopathic pulmonary
fibrosis. J Clin Med. 10:772020. View Article : Google Scholar
|
|
30
|
Wang Y, Tian F, Qian ZM, Feng J, Wang X,
McMillin SE, Howard SW and Lin H: Air pollution, metabolic
signatures, and the risk of idiopathic pulmonary fibrosis. Sci
Total Environ. 964:1784092025. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Morales-Bárcenas R, Chirino YI,
Sánchez-Pérez Y, Osornio-Vargas ÁR, Melendez-Zajgla J, Rosas I and
García-Cuellar CM: Particulate matter (PM10) induces
metalloprotease activity and invasion in airway epithelial cells.
Toxicol Lett. 237:167–173. 2015. View Article : Google Scholar
|
|
32
|
Cahill EF, Kennelly H, Carty F, Mahon BP
and English K: Hepatocyte growth factor is required for mesenchymal
stromal cell protection against bleomycin-induced pulmonary
fibrosis. Stem Cells Transl Med. 5:1307–1318. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yoon HY, Kim SY, Kim OJ and Song JW:
Nitrogen dioxide increases the risk of mortality in idiopathic
pulmonary fibrosis. Eur Respir J. 57:20018772021. View Article : Google Scholar
|
|
34
|
Pardo A and Selman M: The interplay of the
genetic architecture, aging, and environmental factors in the
pathogenesis of idiopathic pulmonary fibrosis. Am J Respir Cell Mol
Biol. 64:163–172. 2021. View Article : Google Scholar
|
|
35
|
Zhang Y, Huang W, Zheng Z, Wang W, Yuan Y,
Hong Q, Lin J, Li X and Meng Y: Cigarette smoke-inactivated SIRT1
promotes autophagy-dependent senescence of alveolar epithelial type
2 cells to induce pulmonary fibrosis. Free Radic Biol Med.
166:116–127. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Stancil IT, Michalski JE, Davis-Hall D,
Chu HW, Park JA, Magin CM, Yang IV, Smith BJ, Dobrinskikh E and
Schwartz DA: Pulmonary fibrosis distal airway epithelia are
dynamically and structurally dysfunctional. Nat Commun.
12:45662021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Poschenrieder F, Rotter M, Gschwendtner A
and Hamer OW: E-cigarette-induced lung disease: From acute to
chronic. Lancet. 396:5642020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Spagnolo P, Ryerson CJ, Guler S, Feary J,
Churg A, Fontenot AP, Piciucchi S, Udwadia Z, Corte TJ, Wuyts WA,
et al: Occupational interstitial lung diseases. J Intern Med.
294:798–815. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Murgia N, Akgun M, Blanc PD, Costa JT,
Moitra S, Muñoz X, Toren K and Ferreira AJ: Issue 3-The
occupational burden of respiratory diseases, an update.
Pulmonology. 31:24168082025. View Article : Google Scholar
|
|
40
|
Spagnolo P, Molyneaux PL, Bernardinello N,
Cocconcelli E, Biondini D, Fracasso F, Tiné M, Saetta M, Maher TM
and Balestro E: The role of the lung's microbiome in the
pathogenesis and progression of idiopathic pulmonary fibrosis. Int
J Mol Sci. 20:56182019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Molyneaux PL, Cox MJ, Willis-Owen SA,
Mallia P, Russell KE, Russell AM, Murphy E, Johnston SL, Schwartz
DA, Wells AU, et al: The role of bacteria in the pathogenesis and
progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care
Med. 190:906–913. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Thompson BT, Chambers RC and Liu KD: Acute
respiratory distress syndrome. N Engl J Med. 377:562–572. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wendisch D, Dietrich O, Mari T, von
Stillfried S, Ibarra IL, Mittermaier M, Mache C, Chua RL, Knoll R,
Timm S, et al: SARS-CoV-2 infection triggers profibrotic macrophage
responses and lung fibrosis. Cell. 184:6243–6261. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Huang WJ and Tang XX: Virus infection
induced pulmonary fibrosis. J Transl Med. 19:4962021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sakai N and Tager AM: Fibrosis of two:
Epithelial cell-fibroblast interactions in pulmonary fibrosis.
Biochim Biophys Acta. 1832:911–921. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cheng P, Li S and Chen H: Macrophages in
lung injury, repair, and fibrosis. Cells. 10:4362021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yao Y, Wang Y, Zhang Z, He L, Zhu J, Zhang
M, He X, Cheng Z, Ao Q, Cao Y, et al: Chop deficiency protects mice
against bleomycin-induced pulmonary fibrosis by attenuating M2
macrophage production. Mol Ther. 24:915–925. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gieseck RL III, Wilson MS and Wynn TA:
Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol.
18:62–76. 2018. View Article : Google Scholar
|
|
49
|
Meng XM, Nikolic-Paterson DJ and Lan HY:
TGF-β: The master regulator of fibrosis. Nat Rev Nephrol.
12:325–338. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang Y, Zhang L, Huang T, Wu GR, Zhou Q,
Wang FX, Chen LM, Sun F, Lv Y, Xiong F, et al: The
methyl-CpG-binding domain 2 facilitates pulmonary fibrosis by
orchestrating fibroblast to myofibroblast differentiation. Eur
Respir J. 60:20036972022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Haak AJ, Kostallari E, Sicard D, Ligresti
G, Choi KM, Caporarello N, Jones DL, Tan Q, Meridew J, Diaz
Espinosa AM, et al: Selective YAP/TAZ inhibition in fibroblasts via
dopamine receptor D1 agonism reverses fibrosis. Sci Transl Med.
11:eaau62962019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Confalonieri P, Volpe MC, Jacob J,
Maiocchi S, Salton F, Ruaro B, Confalonieri M and Braga L:
Regeneration or repair? The role of alveolar epithelial cells in
the pathogenesis of idiopathic pulmonary fibrosis (IPF). Cells.
11:20952022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liu F, Lagares D, Choi KM, Stopfer L,
Marinković A, Vrbanac V, Probst CK, Hiemer SE, Sisson TH, Horowitz
JC, et al: Mechanosignaling through YAP and TAZ drives fibroblast
activation and fibrosis. Am J Physiol Lung Cell Mol Physiol.
308:L344–L357. 2015. View Article : Google Scholar
|
|
54
|
Yang IV, Pedersen BS, Rabinovich E,
Hennessy CE, Davidson EJ, Murphy E, Guardela BJ, Tedrow JR, Zhang
Y, Singh MK, et al: Relationship of DNA methylation and gene
expression in idiopathic pulmonary fibrosis. Am J Respir Crit Care
Med. 190:1263–1272. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Rabinovich EI, Kapetanaki MG, Steinfeld I,
Gibson KF, Pandit KV, Yu G, Yakhini Z and Kaminski N: Global
methylation patterns in idiopathic pulmonary fibrosis. PLoS One.
7:e337702012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sanders YY, Ambalavanan N, Halloran B,
Zhang X, Liu H, Crossman DK, Bray M, Zhang K, Thannickal VJ and
Hagood JS: Altered DNA methylation profile in idiopathic pulmonary
fibrosis. Am J Respir Crit Care Med. 186:525–535. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
McErlean P, Bell CG, Hewitt RJ, Busharat
Z, Ogger PP, Ghai P, Albers GJ, Calamita E, Kingston S, Molyneaux
PL, et al: DNA methylome alterations are associated with airway
macrophage differentiation and phenotype during lung fibrosis. Am J
Respir Crit Care Med. 204:954–966. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Korfei M, Mahavadi P and Guenther A:
Targeting histone deacetylases in idiopathic pulmonary fibrosis: A
future therapeutic option. Cells. 11:16262022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hua HS, Wen HC, Lee HS, Weng CM, Yuliani
FS, Kuo HP, Chen BC and Lin CH: Endothelin-1 induces connective
tissue growth factor expression in human lung fibroblasts by
disrupting HDAC2/Sin3A/MeCP2 corepressor complex. J Biomed Sci.
30:402023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jeong SH, Son ES, Lee YE, Kyung SY, Park
JW and Kim SH: Histone deacetylase 3 promotes alveolar
epithelial-mesenchymal transition and fibroblast migration under
hypoxic conditions. Exp Mol Med. 54:922–931. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhang C, Chen L, Xie C, Wang F, Wang J,
Zhou H, Liu Q, Zeng Z, Li N, Huang J, et al: YTHDC1 delays cellular
senescence and pulmonary fibrosis by activating ATR in an
m6A-independent manner. EMBO J. 43:61–86. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Liu M, Sheng Y, Li M, Pan T, Jiang W,
Zhang Y, Pan X, Huang C, Li J and Wang Y: METTL3-Dependent YTHDF2
Mediates TSC1 expression to regulate alveolar epithelial
mesenchymal transition and promote idiopathic pulmonary fibrosis. J
Cell Physiol. 240:e314732025. View Article : Google Scholar
|
|
63
|
Chen S, Wang Y, Zhang J, Liu B, Liu W, Cao
G, Li R, Li H, Zhai N, Song X, et al: YTHDC1 phase separation
drives the nuclear export of m(6)A-modified lncNONMMUT062668.2
through the transport complex SRSF3-ALYREF-XPO5 to aggravate
pulmonary fibrosis. Cell Death Dis. 16:2792025. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhou Y, Hu Z, Sun Q and Dong Y:
5-methyladenosine regulators play a crucial role in development of
chronic hypersensitivity pneumonitis and idiopathic pulmonary
fibrosis. Sci Rep. 13:59412023. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Tian L, Song W, Wu J, Lan Y and Chen L:
Diagnostic and predictive values of m5C-associated genes in
idiopathic pulmonary fibrosis. Mol Med Rep. 31:532025. View Article : Google Scholar
|
|
66
|
Wollin L, Wex E, Pautsch A, Schnapp G,
Hostettler KE, Stowasser S and Kolb M: Mode of action of nintedanib
in the treatment of idiopathic pulmonary fibrosis. Eur Respir J.
45:1434–1445. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hayton C and Chaudhuri N: Managing
idiopathic pulmonary fibrosis: Which drug for which patient? Drugs
Aging. 34:647–653. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Thannickal VJ, Jandeleit-Dahm K,
Szyndralewiez C and Török NJ: Pre-clinical evidence of a dual NADPH
oxidase 1/4 inhibitor (setanaxib) in liver, kidney and lung
fibrosis. J Cell Mol Med. 27:471–481. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Nandawadekar LD, Kambhampati V, Kunuru SK,
Kuncha M, Andugulapati SB and Reddy DS: Design and synthesis of
novel pirfenidone analogues for targeting fibroblast
differentiation via transforming growth factor-β/Smad pathway using
in vitro and in vivo models. ACS Pharmacol Transl Sci. 8:3101–3125.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wu YH, Li XW, Li WQ, Li XH, Li YJ, Hu GY,
Liu ZQ and Li D: Fluorofenidone attenuates bleomycin-induced
pulmonary fibrosis by inhibiting eukaryotic translation initiation
factor 3a (eIF3a) in rats. Eur J Pharmacol. 773:42–50. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Corte TJ, Behr J, Cottin V, Glassberg MK,
Kreuter M, Martinez FJ, Ogura T, Suda T, Wijsenbeek M, Berkowitz E,
et al: Efficacy and safety of admilparant, an LPA(1) antagonist, in
pulmonary fibrosis: A phase 2 randomized clinical trial. Am J
Respir Crit Care Med. 211:230–238. 2025. View Article : Google Scholar
|
|
72
|
Nambiar A, Kellogg D III, Justice J, Goros
M, Gelfond J, Pascual R, Hashmi S, Masternak M, Prata L, LeBrasseur
N, et al: Senolytics dasatinib and quercetin in idiopathic
pulmonary fibrosis: results of a phase I, single-blind,
single-center, randomized, placebo-controlled pilot trial on
feasibility and tolerability. Ebiomedicine. 90:1044812023.
View Article : Google Scholar
|
|
73
|
Giulianelli G, Cocconcelli E, Fiorentù G,
Bernardinello N, Balestro E and Spagnolo P: Idiopathic pulmonary
fibrosis, today and tomorrow: Certainties and new therapeutic
horizons. Pulm Ther. 11:195–234. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Nakamura Y, Niho S and Shimizu Y:
Cell-Based therapy for fibrosing interstitial lung diseases,
current status, and potential applications of iPSC-Derived cells.
Cells. 13:8932024. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Xu L, Hou X, Zhou X and Jiang M: Advances
in the research and application of stem cell therapies for
idiopathic pulmonary fibrosis. Am J Clin Exp Immunol. 14:300–312.
2025. View Article : Google Scholar
|
|
76
|
Barczyk M, Schmidt M and Mattoli S: Stem
cell-based therapy in idiopathic pulmonary fibrosis. Stem Cell Rev
Rep. 11:598–620. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Doherty DF, Roets L and Krasnodembskaya
AD: The role of lung resident mesenchymal stromal cells in the
pathogenesis and repair of chronic lung disease. Stem cells.
41:431–443. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wang X, Deng Z, Wang Z, Gan S, Xu L and
Zhang X: Engineered MSC-exosomes delivering mirnas for respiratory
disease diagnostics and therapy: Opportunities and challenges. Int
J Nanomedicine. 21:5607372026. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Yu F, Liu F, Liang X, Duan L, Li Q, Pan G,
Ma C, Liu M, Li M, Wang P and Zhao X: iPSC-Derived airway
epithelial cells: progress, promise, and challenges. Stem Cells.
41:1–10. 2023. View Article : Google Scholar
|
|
80
|
Phogat S, Thiam F, Al Yazeedi S, Abokor FA
and Osei ET: 3D in vitro hydrogel models to study the human lung
extracellular matrix and fibroblast function. Respir Res.
24:2422023. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Barkauskas CE, Cronce MJ, Rackley CR,
Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW and Hogan BL:
Type 2 alveolar cells are stem cells in adult lung. J Clin Invest.
123:3025–3036. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Serrano-Mollar A, Nacher M, Gay-Jordi G,
Closa D, Xaubet A and Bulbena O: Intratracheal transplantation of
alveolar type II cells reverses bleomycin-induced lung fibrosis. Am
J Respir Crit Care Med. 176:1261–1268. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Ma Q, Ma Y, Dai X, Ren T, Fu Y, Liu W, Han
Y, Wu Y, Cheng Y, Zhang T and Zuo W: Regeneration of functional
alveoli by adult human SOX9(+) airway basal cell transplantation.
Protein Cell. 9:267–282. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Liu H, Liu S, Jiang J, Zhang Y, Luo Y,
Zhao J, Xu J, Xie Y, Liao W, Wang W, et al: CoQ10 enhances the
efficacy of airway basal stem cell transplantation on
bleomycin-induced idiopathic pulmonary fibrosis in mice. Respir
Res. 23:392022. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Poetsch MS, Strano A and Guan K: Human
induced pluripotent stem cells: From cell origin, genomic
stability, and epigenetic memory to translational medicine. Stem
Cells. 40:546–555. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Huang W, Zhou C, Yu Y, Qin S, Chen L, Lin
H, Zhang S, Xia L and Liang W: Functionalized mesenchymal stem
cells for enhanced bone regeneration: Advances and challenges. Stem
Cell Res Ther. 16:6002025. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Lin T, Yang Y and Chen X: A review of the
application of mesenchymal stem cells in the field of hematopoietic
stem cell transplantation. Eur J Med Res. 28:2682023. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Khalili MR, Ahmadloo S, Mousavi SA,
Joghataei MT, Brouki Milan P, Naderi Gharahgheshlagh S, Mohebi SL,
Haramshahi SMA and Hosseinpour Sarmadi V: Navigating mesenchymal
stem cells doses and delivery routes in heart disease trials: A
comprehensive overview. Regen Ther. 29:117–127. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Pak J, Lee JH and Lee SH: Regenerative
repair of damaged meniscus with autologous adipose tissue-derived
stem cells. Biomed Res Int. 2014:4360292014. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Hocking AM: Mesenchymal stem cell therapy
for cutaneous wounds. Adv Wound Care (New Rochelle). 1:166–171.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Velikova T, Dekova T and Miteva DG:
Controversies regarding transplantation of mesenchymal stem cells.
World J Transplant. 14:905542024. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zhou J, Lin Y, Kang X, Liu Z, Zhang W and
Xu F: microRNA-186 in extracellular vesicles from bone marrow
mesenchymal stem cells alleviates idiopathic pulmonary fibrosis via
interaction with SOX4 and DKK1. Stem Cell Res Ther. 12:962021.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Wan X, Chen S, Fang Y, Zuo W, Cui J and
Xie S: Mesenchymal stem cell-derived extracellular vesicles
suppress the fibroblast proliferation by downregulating FZD6
expression in fibroblasts via micrRNA-29b-3p in idiopathic
pulmonary fibrosis. J Cell Physiol. 235:8613–8625. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Takahashi K and Yamanaka S: Induction of
pluripotent stem cells from mouse embryonic and adult fibroblast
cultures by defined factors. Cell. 126:663–676. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Roszell B, Mondrinos MJ, Seaton A, Simons
DM, Koutzaki SH, Fong GH, Lelkes PI and Finck CM: Efficient
derivation of alveolar type II cells from embryonic stem cells for
in vivo application. Tissue Eng Part A. 15:3351–3365. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Wang D, Morales JE, Calame DG, Alcorn JL
and Wetsel RA: Transplantation of human embryonic stem cell-derived
alveolar epithelial type II cells abrogates acute lung injury in
mice. Mol Ther. 18:625–634. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Yamamoto Y, Gotoh S, Korogi Y, Seki M,
Konishi S, Ikeo S, Sone N, Nagasaki T, Matsumoto H, Muro S, et al:
Long-term expansion of alveolar stem cells derived from human iPS
cells in organoids. Nat Methods. 14:1097–1106. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Pezet MG, Torres JA, Thimraj TA, Matkovic
I, Schrode N, Murray JW, Saqi A, Beaumont KG and Snoeck HW: Human
respiratory airway progenitors derived from pluripotent cells
generate alveolar epithelial cells and model pulmonary fibrosis.
Nat Biotechnol. Feb 24–2025.Epub ahead of print. View Article : Google Scholar
|
|
99
|
Merkert S, Bednarski C, Göhring G,
Cathomen T and Martin U: Generation of a gene-corrected isogenic
control iPSC line from cystic fibrosis patient-specific iPSCs
homozygous for p.Phe508del mutation mediated by TALENs and ssODN.
Stem Cell Res. 23:95–97. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Ong CH, Tham CL, Harith HH, Firdaus N and
Israf DA: TGF-β-induced fibrosis: A review on the underlying
mechanism and potential therapeutic strategies. Eur J Pharmacol.
911:1745102021. View Article : Google Scholar
|
|
101
|
Schepers D, Doyle AJ, Oswald G, Sparks E,
Myers L, Willems PJ, Mansour S, Simpson MA, Frysira H, Maat-Kievit
A, et al: The SMAD-binding domain of SKI: a hotspot for de novo
mutations causing Shprintzen-Goldberg syndrome. Eur J Hum Genet.
23:224–228. 2015. View Article : Google Scholar
|
|
102
|
Li P, Wang QS, Zhai Y, Xiong RP, Chen X,
Liu P, Peng Y, Zhao Y, Ning YL, Yang N and Zhou YG: Ski mediates
TGF-β1-induced fibrosarcoma cell proliferation and promotes tumor
growth. J Cancer. 11:5929–5940. 2020. View Article : Google Scholar
|
|
103
|
Yang H, Zhan L, Yang T, Wang L, Li C, Zhao
J, Lei Z, Li X and Zhang HT: Ski prevents TGF-β-induced EMT and
cell invasion by repressing SMAD-dependent signaling in non-small
cell lung cancer. Oncol Rep. 34:87–94. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Richeldi L, Schiffman C, Behr J, Inoue Y,
Corte TJ, Cottin V, Jenkins RG, Nathan SD, Raghu G, Walsh SLF, et
al: Zinpentraxin alfa for idiopathic pulmonary fibrosis: The
randomized phase III STARSCAPE trial. Am J Respir Crit Care Med.
209:1132–1140. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Raghu G, van den Blink B, Hamblin MJ,
Brown AW, Golden JA, Ho LA, Wijsenbeek MS, Vasakova M, Pesci A,
Antin-Ozerkis DE, et al: Effect of recombinant human pentraxin 2 vs
placebo on change in forced vital capacity in patients with
idiopathic pulmonary fibrosis: A randomized clinical trial. JAMA.
319:2299–2307. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Hirani N, MacKinnon AC, Nicol L, Ford P,
Schambye H, Pedersen A, Nilsson UJ, Leffler H, Sethi T, Tantawi S,
et al: Target inhibition of galectin-3 by inhaled TD139 in patients
with idiopathic pulmonary fibrosis. Eur Respir J. 57:20025592021.
View Article : Google Scholar :
|
|
107
|
Chilosi M, Poletti V, Zamò A, Montagna L,
Piccoli P, Pedron S, Bertaso M, Scarpa A, Murer B, Cancellieri A,
et al: Aberrant Wnt/beta-catenin pathway activation in idiopathic
pulmonary fibrosis. Am J Pathol. 162:1495–1502. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Li X, Liu X, Deng R, Gao S, Jiang Q, Liu
R, Li H, Miao Y, Zhai Y, Zhang S, et al: Betulinic acid attenuated
bleomycin-induced pulmonary fibrosis by effectively intervening
Wnt/β-catenin signaling. Phytomedicine. 81:1534282021. View Article : Google Scholar
|
|
109
|
Lin J, Pan Z, Sun J, Wang X, Yin D, Huo C
and Guo Q: PCSK9 inhibitor alleviates experimental pulmonary
fibrosis-induced pulmonary hypertension via attenuating
epithelial-mesenchymal transition by suppressing Wnt/β-catenin
signaling in vivo and in vitro. Front Med (Lausanne).
11:15091682024. View Article : Google Scholar
|
|
110
|
Wang J, Hu K, Cai X, Yang B, He Q, Wang J
and Weng Q: Targeting PI3K/AKT signaling for treatment of
idiopathic pulmonary fibrosis. Acta Pharm Sin B. 12:18–32. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Green S, Trejo CL and McMahon M:
PIK3CA(H1047R) accelerates and enhances KRAS(G12D)-Driven lung
tumorigenesis. Cancer Res. 75:5378–5391. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Conte E, Gili E, Fruciano M, Korfei M,
Fagone E, Iemmolo M, Lo Furno D, Giuffrida R, Crimi N, Guenther A
and Vancheri C: PI3K p110γ overexpression in idiopathic pulmonary
fibrosis lung tissue and fibroblast cells: In vitro effects of its
inhibition. Lab Invest. 93:566–576. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Zhai C, Cheng J, Mujahid H, Wang H, Kong
J, Yin Y, Li J, Zhang Y, Ji X and Chen W: Selective inhibition of
PI3K/Akt/mTOR signaling pathway regulates autophagy of macrophage
and vulnerability of atherosclerotic plaque. PLoS One.
9:e905632014. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Larson-Casey JL, Deshane JS, Ryan AJ,
Thannickal VJ and Carter AB: Macrophage Akt1 kinase-mediated
mitophagy modulates apoptosis resistance and pulmonary fibrosis.
Immunity. 44:582–596. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Nie Y, Sun L, Wu Y, Yang Y, Wang J, He H,
Hu Y, Chang Y, Liang Q, Zhu J, et al: AKT2 regulates pulmonary
inflammation and fibrosis via modulating macrophage activation. J
Immunol. 198:4470–4480. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Hu X, Xu Q, Wan H, Hu Y, Xing S, Yang H,
Gao Y and He Z: PI3K-Akt-mTOR/PFKFB3 pathway mediated lung
fibroblast aerobic glycolysis and collagen synthesis in
lipopolysaccharide-induced pulmonary fibrosis. Lab Invest.
100:801–811. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Lu Y, Azad N, Wang L, Iyer AK, Castranova
V, Jiang BH and Rojanasakul Y: Phosphatidylinositol-3-kinase/akt
regulates bleomycin-induced fibroblast proliferation and collagen
production. Am J Respir Cell Mol Biol. 42:432–441. 2010. View Article : Google Scholar
|
|
118
|
Du W, Tang Z, Yang F, Liu X and Dong J:
Icariin attenuates bleomycin-induced pulmonary fibrosis by
targeting Hippo/YAP pathway. Biomed Pharmacother. 143:1121522021.
View Article : Google Scholar
|
|
119
|
Qing J, Ren Y, Zhang Y, Yan M, Zhang H, Wu
D, Ma Y, Chen Y, Huang X, Wu Q, et al: Dopamine receptor D2
antagonism normalizes profibrotic macrophage-endothelial crosstalk
in non-alcoholic steatohepatitis. J Hepatol. 76:394–406. 2022.
View Article : Google Scholar
|
|
120
|
Zeyada MS, Eraky SM and El-Shishtawy MM:
Trigonelline mitigates bleomycin-induced pulmonary inflammation and
fibrosis: Insight into NLRP3 inflammasome and SPHK1/S1P/Hippo
signaling modulation. Life Sci. 336:1222722024. View Article : Google Scholar
|
|
121
|
Bringardner BD, Baran CP, Eubank TD and
Marsh CB: The role of inflammation in the pathogenesis of
idiopathic pulmonary fibrosis. Antioxid Redox Signal. 10:287–301.
2008. View Article : Google Scholar
|
|
122
|
Ma H, Liu S, Li S and Xia Y: Targeting
growth factor and cytokine pathways to treat idiopathic pulmonary
fibrosis. Front Pharmacol. 13:9187712022. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Steele H, Willicut A, Dell G, Ghastine A,
Nguyen XX, Lembicz P, Doerflein H, Suchoski T, Kato E,
Feghali-Bostwick C, et al: Combination therapy blocking TNF
superfamily members 14 and 15 reverses pulmonary fibrosis. J
Immunol. 214:808–817. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Song J, Yang P, Chen C, Ding W, Tillement
O, Bai H and Zhang S: Targeting epigenetic regulators as a
promising avenue to overcome cancer therapy resistance. Signal
Transduct Target Ther. 10:2192025. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Shi Y, Zhang H, Huang S, Yin L, Wang F,
Luo P and Huang H: Epigenetic regulation in cardiovascular disease:
Mechanisms and advances in clinical trials. Signal Transduct Target
Ther. 7:2002022. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Kumar S, Shanker OR, Banerjee J, Tripathi
M, Chandra PS and Dixit AB: Epigenetics in epilepsy. Prog Mol Biol
Transl Sci. 198:249–269. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Huang R, Fu P and Ma L: Kidney fibrosis:
From mechanisms to therapeutic medicines. Signal Transduct Target
Ther. 8:1292023. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Neveu WA, Mills ST, Staitieh BS and
Sueblinvong V: TGF-β1 epigenetically modifies Thy-1 expression in
primary lung fibroblasts. Am J Physiol Cell Physiol. 309:C616–C626.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Wei A, Gao Q, Chen F, Zhu X, Chen X, Zhang
L, Su X, Dai J, Shi Y and Cao W: Inhibition of DNA methylation
de-represses peroxisome proliferator-activated receptor-γ and
attenuates pulmonary fibrosis. Br J Pharmacol. 179:1304–1318. 2022.
View Article : Google Scholar
|
|
130
|
Christman JK: 5-Azacytidine and
5-aza-2'-deoxycytidine as inhibitors of DNA methylation:
Mechanistic studies and their implications for cancer therapy.
Oncogene. 21:5483–5495. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Klutstein M, Nejman D, Greenfield R and
Cedar H: DNA methylation in cancer and aging. Cancer Res.
76:3446–3450. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Hu C, Liu X, Zeng Y, Liu J and Wu F: DNA
methyltransferase inhibitors combination therapy for the treatment
of solid tumor: Mechanism and clinical application. Clin
Epigenetics. 13:1662021. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Alyamany R, Alnughmush A, Almutlaq M,
Alyamany M and Alfayez M: Azacitidine induced lung injury: Report
and contemporary discussion on diagnosis and management. Front
Oncol. 14:13454922024. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Wang J, Li D, Yang J, Chang L, Zhang R and
Li J: CRISPR/Cas9-mediated epigenetic editing tool: An optimized
strategy for targeting de novo DNA methylation with stable status
via homology directed repair pathway. Biochimie. 202:190–205. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Qu J, Zhu L, Zhou Z, Chen P, Liu S, Locy
ML, Thannickal VJ and Zhou Y: Reversing Mechanoinductive DSP
Expression by CRISPR/dCas9-mediated Epigenome Editing. Am J Respir
Crit Care Med. 198:599–609. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Wu DD, Lau ATY, Xu YM, Reinders-Luinge M,
Koncz M, Kiss A, Timens W, Rots MG and Hylkema MN: Targeted
epigenetic silencing of UCHL1 expression suppresses collagen-1
production in human lung epithelial cells. Epigenetics.
18:21755222023. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Wu X, Yu Y, Wang M, Dai D, Yin J, Liu W,
Kong D, Tang S, Meng M, Gao T, et al: AAV-delivered muscone-induced
transgene system for treating chronic diseases in mice via
inhalation. Nat Commun. 15:11222024. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Lyu X, Hu M, Peng J, Zhang X and Sanders
YY: HDAC inhibitors as antifibrotic drugs in cardiac and pulmonary
fibrosis. Ther Adv Chronic Dis. 10:20406223198626972019. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Li Y and Seto E: HDACs and HDAC inhibitors
in cancer development and therapy. Cold Spring Harb Perspect Med.
6:a0268312016. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Royce SG, Moodley Y and Samuel CS: Novel
therapeutic strategies for lung disorders associated with airway
remodelling and fibrosis. Pharmacol Ther. 141:250–260. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Rubio K, Singh I, Dobersch S, Sarvari P,
Günther S, Cordero J, Mehta A, Wujak L, Cabrera-Fuentes H, Chao CM,
et al: Inactivation of nuclear histone deacetylases by EP300
disrupts the MiCEE complex in idiopathic pulmonary fibrosis. Nat
Commun. 10:22292019. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Yu H, Liu S, Wang S and Gu X: The
involvement of HDAC3 in the pathogenesis of lung injury and
pulmonary fibrosis. Front Immunol. 15:13921452024. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Yu WC, Yeh TY, Ye CH, Chong PCT, Ho YH, So
DK, Yap KY, Peng GR, Shao CH, Jagtap AD, et al: Discovery of HDAC6,
HDAC8, and 6/8 inhibitors and development of cell-based drug
screening models for the treatment of TGF-β-Induced idiopathic
pulmonary fibrosis. J Med Chem. 66:10528–10557. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Wang M, Huo Z, He X, Liu F, Liang J, Wu L
and Yang D: The role of MiR-29 in the mechanism of fibrosis. Mini
Rev Med Chem. 23:1846–1858. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Savary G, Dewaeles E, Diazzi S, Buscot M,
Nottet N, Fassy J, Courcot E, Henaoui IS, Lemaire J, Martis N, et
al: The long noncoding RNA DNM3OS is a reservoir of fibromirs with
major functions in lung fibroblast response to TGF-β and pulmonary
fibrosis. Am J Respir Crit Care Med. 200:184–198. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Xia C, Cheng L, Zhao W, Chang A, Wang Z,
Liu H, Pan X, Li W, Koji S, Li Z, et al: LncRNA SYISL promotes
fibroblast myofibroblast transition via miR-23a-mediated TRIOBP
regulation. Cell Mol Life Sci. 82:2142025. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Chow MYT, Qiu Y and Lam JKW: Inhaled RNA
therapy: From promise to reality. Trends Pharmacol Sci. 41:715–729.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Kim J, Jozic A, Lin Y, Eygeris Y, Bloom E,
Tan X, Acosta C, MacDonald KD, Welsher KD and Sahay G: Engineering
lipid nanoparticles for enhanced intracellular delivery of mRNA
through inhalation. ACS Nano. 16:14792–14806. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Pan T, Zhou Q, Miao K, Zhang L, Wu G, Yu
J, Xu Y, Xiong W, Li Y and Wang Y: Suppressing Sart1 to modulate
macrophage polarization by siRNA-loaded liposomes: A promising
therapeutic strategy for pulmonary fibrosis. Theranostics.
11:1192–1206. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Cheng D, Wang Y, Li Z, Xiong H, Sun W, Xi
S, Zhou S, Liu Y and Ni C: Liposomal UHRF1 siRNA shows lung
fibrosis treatment potential through regulation of fibroblast
activation. JCI Insight. 7:e1628312022. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Zhao Z, Shan X, Ding J, Ma B, Li B, Huang
W, Yang Q, Fang Y, Chen J, Song C, et al: Boosting RNA
nanotherapeutics with V-ATPase activating non-inflammatory lipid
nanoparticles to treat chronic lung injury. Nat Commun.
16:64772025. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Vlasova KY, Kerr A, Pennock ND, Jozic A,
Sahel DK, Gautam M, Murthy NTV, Roberts A, Ali MW, MacDonald KD, et
al: Synthesis of ionizable lipopolymers using split-Ugi reaction
for pulmonary delivery of various size RNAs and gene editing.
bioRxiv [Preprint] 2024.06.11.598497. 2024.
|
|
153
|
Bai X, Zhao G, Chen Q, Li Z, Gao M, Ho W,
Xu X and Zhang XQ: Inhaled siRNA nanoparticles targeting IL11
inhibit lung fibrosis and improve pulmonary function post-bleomycin
challenge. Sci Adv. 8:eabn71622022. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Duncan GA, Kim N, Colon-Cortes Y,
Rodriguez J, Mazur M, Birket SE, Rowe SM, West NE, Livraghi-Butrico
A, Boucher RC, et al: An adeno-associated viral vector capable of
penetrating the mucus barrier to inhaled gene therapy. Mol Ther
Methods Clin Dev. 9:296–304. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Qin S, Wang H, Liu G, Mei H and Chen M:
miR-21-5p ameliorates hyperoxic acute lung injury and decreases
apoptosis of AEC II cells via PTEN/AKT signaling in rats. Mol Med
Rep. 20:4953–4962. 2019.PubMed/NCBI
|
|
156
|
Wang J, Lai X, Yao S, Chen H, Cai J, Luo
Y, Wang Y, Qiu Y, Huang Y, Wei X, et al: Nestin promotes pulmonary
fibrosis via facilitating recycling of TGF-β receptor I. Eur Respir
J. 59:20037212022. View Article : Google Scholar
|
|
157
|
Park KS, Lässer C and Lötvall J:
Extracellular vesicles and the lung: from disease pathogenesis to
biomarkers and treatments. Physiol Rev. 105:1733–1821. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Li M, Huang H, Wei X, Li H, Li J, Xie B,
Yang Y, Fang X, Wang L, Zhang X, et al: Clinical investigation on
nebulized human umbilical cord MSC-derived extracellular vesicles
for pulmonary fibrosis treatment. Signal Transduct Target Ther.
10:1792025. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Pun FW, Liu BHM, Long X, Leung HW, Leung
GHD, Mewborne QT, Gao J, Shneyderman A, Ozerov IV, Wang J, et al:
Identification of therapeutic targets for amyotrophic lateral
sclerosis using PandaOmics - An AI-Enabled biological target
discovery platform. Front Aging Neurosci. 14:9140172022. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Xu Z, Ren F, Wang P, Cao J, Tan C, Ma D,
Zhao L, Dai J, Ding Y, Fang H, et al: A generative AI-discovered
TNIK inhibitor for idiopathic pulmonary fibrosis: A randomized
phase 2a trial. Nat Med. 31:2602–2610. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Witten J, Raji I, Manan RS, Beyer E,
Bartlett S, Tang Y, Ebadi M, Lei J, Nguyen D, Oladimeji F, et al:
Artificial intelligence-guided design of lipid nanoparticles for
pulmonary gene therapy. Nat Biotechnol. 43:1790–1799. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Gerckens M, Schorpp K, Pelizza F, Wögrath
M, Reichau K, Ma H, Dworsky AM, Sengupta A, Stoleriu MG,
Heinzelmann K, et al: Phenotypic drug screening in a human fibrosis
model identified a novel class of antifibrotic therapeutics. Sci
Adv. 7:eabb36732021. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Moodley Y: The analysis of proteomics by
machine learning in separating idiopathic pulmonary fibrosis from
connective tissue disease-interstitial lung disease. Am J Respir
Crit Care Med. 210:378–380. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Muratov V, Jagiello K, Mikolajczyk A,
Danielsen PH, Halappanavar S, Vogel U and Puzyn T: The role of
machine learning in predicting titanium dioxide nanoparticles
induced pulmonary pathology using transcriptomic biomarkers. J
Hazard Mater. 493:1382402025. View Article : Google Scholar : PubMed/NCBI
|