Introduction
Iron is an essential element of life and plays a
central role in biological processes (1). Iron is required for the proper
functioning of numerous enzymes. However, free ferrous iron
(Fe2+) exerts toxicity despite its biological
importance. Dysregulation of intracellular iron homeostasis, a
critical etiological factor in neurodegenerative diseases, triggers
destructive effects via Fenton reaction-derived reactive oxygen
species (ROS), ultimately leading to ferroptosis (2,3).
Ferroptosis causes the oxidative destruction of the cell membrane
through three key mechanisms: Dysregulation of the antioxidant
system, disruption of iron metabolism and accelerated lipid
peroxidation. To prevent detrimental effects, cellular systems
tightly regulate iron levels through coordinated mechanisms,
including the hepcidin-ferroportin (FPN) axis, the divalent metal
transporter 1 (DMT1)-transferrin (Tf) system, and the
ferritin-nuclear receptor coactivator 4 (NCOA4) pathway (1,2).
Ferritin is a highly conserved iron-containing
storage protein ubiquitously distributed in living organisms. Its
unique three-dimensional structure composed of a protein shell and
an inorganic mineral core enables it to oxidize and store iron. The
protein shell forms a hollow cavity assembled from two functionally
distinct subunits: The ferritin heavy chain (FTH) with ferroxidase
activity and the ferritin light chain (FTL), which facilitates iron
mineralization. The mineral core sequesters up to 4,000 ferric iron
(Fe3+) ions. Ferritin, the primary iron storage protein
in biological systems, stores iron predominantly in the iron
oxyhydroxide form, potentially incorporating phosphorus-containing
components (4). However, the
precise mineral structure of this core remains unclear. The iron
sequestration mechanism relies on the ferroxidase activity of FTH
and the specialized chemical microenvironment within the cavity,
which collectively promotes iron ion recruitment and
mineralization. Importantly, excess free iron that is not properly
incorporated into the ferritin core may induce oxidative stress
(OS) by catalyzing ROS generation, leading to cellular damage
(5). Ferritinophagy, a selective
autophagy process mediated by NCOA4, plays a pivotal role in the
regulation of systemic iron homeostasis. Under conditions of iron
overload, ferritinophagy is activated, leading to the release of
stored Fe2+ from ferritin. This increase in the labile
iron pool can exacerbate ROS production via the Fenton reaction
and, when coupled with insufficient antioxidant capacity, may
trigger ferroptosis. The interconnected pathway between
ferritinophagy and ferroptosis has been implicated in the
pathogenesis of diverse human diseases, including metabolic
disorders, neurodegenerative diseases, malignancies and infectious
conditions. In this context, as iron overload and OS represent
common pathological hallmarks across multiple disease entities, Jin
et al (6) postulated that
ferritinophagy may exhibit a broader disease relevance through its
dual regulatory effects on iron homeostasis and ROS dynamics, which
directly intersect with ferroptotic pathways.
Neurodegenerative diseases are characterized by
progressive neuronal dysfunction and death due to hallmark
pathologies, such as abnormal protein aggregation, synaptic loss
and region-specific neurodegeneration. Major disorders include
Alzheimer's disease (AD), which affects ~54.6 million patients in
China alone (representing ~3.9% of its population), and is marked
by deposits of β-amyloid (Aβ) plaques and tau tangles (7). The prevalence of AD increases with
age, with an estimated incidence of 35% in adults aged 85 years and
above (8). Furthermore, typical
representative disorders comprise Parkinson's disease (PD),
characterized by the aggregation of α-synuclein (α-Syn) leading to
the loss of dopaminergic neurons - with projections indicating the
global number of individuals living with PD will reach 25.2 million
by 2050, more than double the 2021 figure - and amyotrophic lateral
sclerosis (ALS), characterized by the degeneration of motor neurons
(9). The etiology of
neurodegenerative diseases involves multifactorial interactions
such as genetic mutations, OS, mitochondrial dysfunction and
neuroinflammation. Despite advances in the understanding of the
molecular mechanisms, therapies that effectively halt disease
progression remain unavailable. Current research focuses on early
biomarker discovery, pathogenic protein clearance strategies and
neuroprotective interventions, and breakthroughs are urgently
needed to address the growing global burden on aging
populations.
This review provides a comprehensive overview of the
relationship between brain iron homeostasis and neurodegenerative
diseases. Specifically, the review aimed to analyze the key
molecular pathways of ferroptosis, elucidate the pathological
consequences of ferritin dysregulation, discuss the diagnostic and
translational potential of ferritin as a biomarker and evaluate the
application of magnetic resonance imaging (MRI) for dynamic brain
iron monitoring.
The role of ferritin in modulating cerebral
iron homeostasis
Ferritin, a critical intracellular iron storage
protein, plays a pivotal role in maintaining cellular iron
homeostasis through its multi-subunit cavity structure, which
mediates Fe2+ oxidation and iron core deposition.
However, it plays two opposing roles: It protects cells from
oxidative damage by sequestering iron, yet it can promote
ferroptosis when ferritinophagy releases Fe2+ for the
Fenton reaction. Once iron enters the brain parenchyma, it is
distributed among neurons, astrocytes, microglia and
oligodendrocytes, each of which has distinct iron requirements and
regulatory mechanisms. In neuropathy, neuroinflammation frequently
coexists with ferroptosis. This process involves the activation of
primary immunocompetent cells such as microglia and astrocytes.
Combined treatment with iron agents and lipopolysaccharides
significantly increases the branch/process length of microglia and
enhances astrocyte immunoreactivity in the hippocampal and cortical
regions (10). The release of
common inflammatory mediators during neuroinflammation is closely
linked to the regulation of cellular iron metabolism. Microglia,
which are the resident immune cells of the central nervous system
(CNS), play a central role in neuroinflammation. Conventionally,
iron accumulation in microglia has been recognized to trigger
pro-inflammatory activation. Iron overload markedly upregulates the
expression of cofilin, a key protein regulating actin dynamics in
microglia, suggesting that cofilin is involved in the iron
overload-induced pro-inflammatory phenotypic transformation of
these cells (11). Astrocytes, a
key glial cell type, are the central regulators of iron homeostasis
in the brain. In the hippocampal CA1 region, FTH1 and FTL1 mRNAs
exhibit preferential localization to distal astrocytic
compartments, such as the fine perisynaptic processes that
ensheathe synapses, with FTH1 mRNA abundance significantly
exceeding that of FTL1 (12).
Aged mice demonstrate an ~1.8-fold increase in the FTH1/FTL1 ratio
and redistribution of FTH1 mRNA towards these peripheral astrocytic
processes (12). This shift
toward a higher proportion of FTH1, which possesses ferroxidase
activity, is likely a compensatory response to elevated OS in the
aging brain, enhancing the capacity to sequester iron in a less
reactive form. The functions of oligodendrocytes extend beyond
synthesizing myelin sheaths to enable saltatory nerve conduction.
One study proposed that oligodendrocytes participate in iron
detoxification by secreting FTH1, a critical component of the
neuronal antioxidant defense system (13).
Precise regulation of brain iron homeostasis is
crucial for neuronal survival, with ferritin occupying a central
position in this network as the primary intracellular iron storage
protein. Cellular iron metabolism begins with the binding of
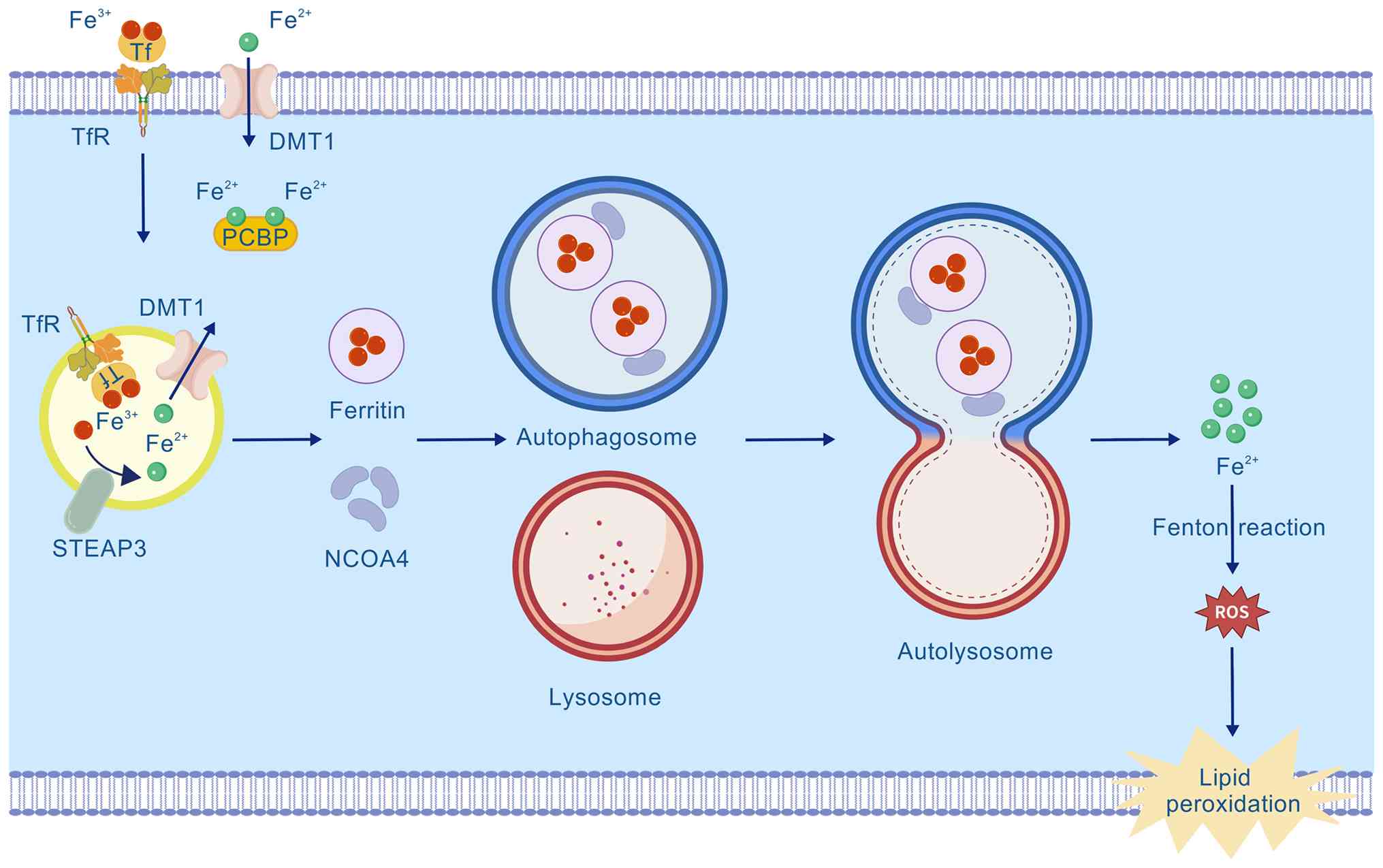
Tf-bound Fe3+ to the Tf receptor 1 (TfR1), which is
followed by endocytosis (Fig. 1)
(14,15). Within the endosome,
Fe3+ is reduced to Fe2+ by the
six-transmembrane epithelial antigen prostate 3 and then
transported into the cytosol via DMT1. Cytosolic Fe2+ is
chaperoned by proteins such as poly(rC)-RNA-binding protein (PCBP)
and delivered to ferritin for storage in a biocompatible form,
thereby effectively preventing the neurotoxicity of labile iron
(Fig. 1) (16). Dynamic ferritin expression is key
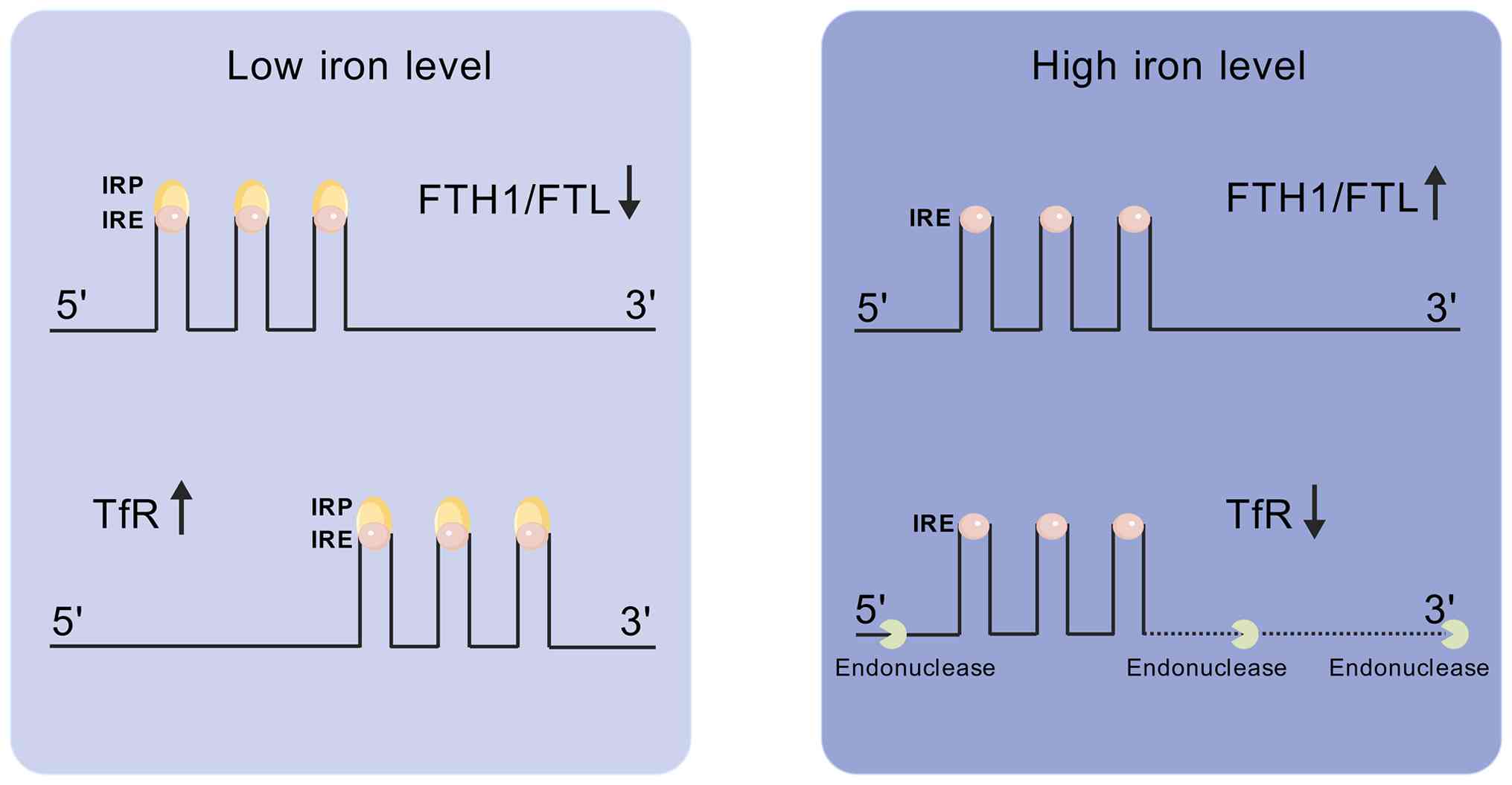
to cellular iron-buffering systems. This process is precisely
controlled by the iron regulatory protein (IRP)/iron-responsive
element (IRE) system (Fig. 2).
IRPs bind to IREs located in the 5' or 3' untranslated regions of
iron-related mRNAs, thereby regulating the expression of iron
uptake (TfR1) and storage (FTH1/FTL) proteins. Under iron-deficient
conditions, IRPs bind to IREs with high affinity, repressing
ferritin mRNA translation and stabilizing TfR1 mRNA. Conversely,
when iron is sufficient, IRP binding is reduced, promoting ferritin
synthesis and facilitating endonucleolytic degradation of TfR1 mRNA
(17). This IRP/IRE-mediated
regulation operates on a relatively rapid timescale, enabling cells
to swiftly adapt to fluctuations in iron availability. This rapid
response is particularly crucial in acute brain iron challenges,
such as hemorrhage, where it serves as an immediate defense against
iron toxicity. Conversely, in chronic conditions like
neurodegenerative diseases, persistent iron overload may overwhelm
or dysregulate this adaptive system, contributing to long-term iron
accumulation and neuronal vulnerability. This core regulatory
network is further integrated with upstream stress-signaling
pathways. For example, the transcription factor EB, a master
regulator of the autophagy-lysosome system, confers dual protection
against ferroptosis. It upregulates TfR1 and promotes its lysosomal
membrane localization to facilitate clearance of the labile iron
pool, and cooperatively enhances the synthesis of FTL and FTH via a
TfR1-dependent pathway, thereby reinforcing safe iron storage
(18). Similarly, activation of
peroxisome proliferator-activated receptor δ modulates the
expression of DMT1, FPN1 and ferritin by regulating IRP1,
ultimately restoring iron homeostasis and preventing neurotoxicity
(19). Collectively, these
findings reveal that ferritin is a key effector molecule involved
in multiple neuroprotective pathways. Supporting this notion, a
study using AD models demonstrated that TfR1 knockdown restored
iron homeostasis and ameliorated mitochondrial dysfunction, a
process possibly involving ferritin, suggesting that TfR1 is a
potential target for mitigating iron overload in AD by modulating
this network (20). Finally,
cellular iron balance relies on efflux. FPN1, the sole known
cellular iron exporter, mediates iron efflux during iron overload.
This process is negatively regulated by hepcidin through its
interaction with FPN1 (Fig. 3),
working in concert with the storage function of ferritin to
maintain precise intracellular iron homeostasis.
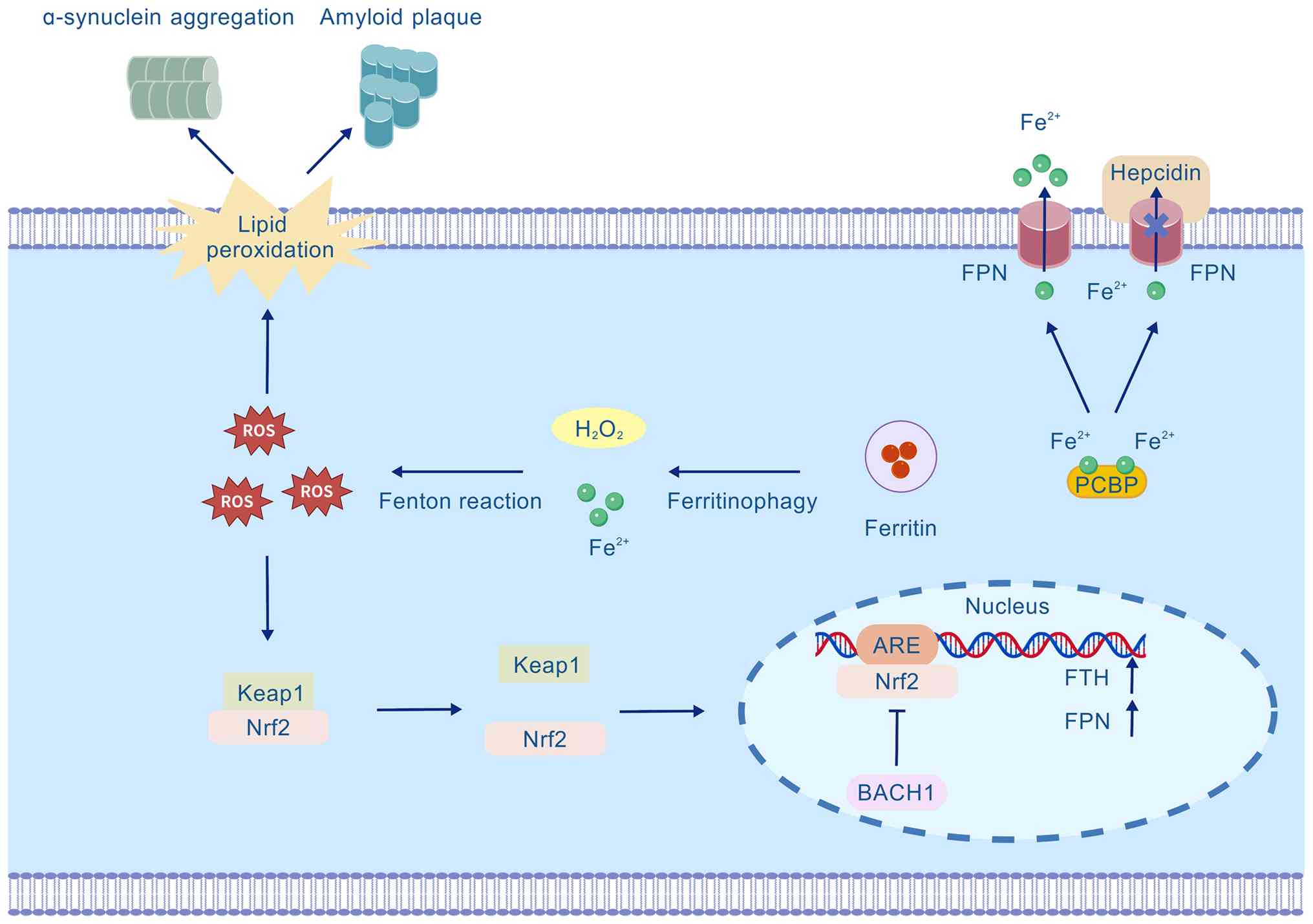 | Figure 3The Keap1/Nrf2/ARE axis in cellular
iron homeostasis. FPN mediates cellular iron efflux, a process
negatively regulated by hepcidin. Cytosolic oxidative stress such
as ROS generated from the Fenton reaction between Fe2+
and H2O2 triggers the dissociation of Keap1
from Nrf2, thus enabling Nrf2 nuclear translocation. Nuclear Nrf2
binds to ARE and initiates the transcription of cytoprotective
genes FTH and FPN. BACH1 antagonizes Nrf2 by competing for ARE
binding. Fenton reaction-derived ROS also stimulates lipid
peroxidation, contributing to neurodegenerative pathologies such as
amyloid plaques and α-synuclein aggregates. ARE, antioxidant
response element; BACH1, BTB and CNC homology 1; FPN, ferroportin;
Keap1, Kelch-like ECH-associated protein 1; Nrf2, nuclear factor
erythroid 2-related factor 2; PCBP, poly(rC)-binding protein; ROS,
reactive oxygen species. |
Ferritinophagy is a form of selective autophagy in
which the receptor NCOA4 binds to FTH1, delivering ferritin to the
autophagosome for degradation and the consequent release of
redox-active Fe2+. In essence, this NCOA4-mediated
process degrades the cytosolic iron storage complex via
autophagosomes to release bound iron into the labile iron pool.
Excessive ferritinophagy activation disrupts iron metabolism,
induces cerebral iron accumulation and triggers neuronal
ferroptosis (Fig. 1) (21). This molecular mechanism has
recently been implicated in the pathogenesis of multiple
neurodegenerative diseases, and has emerged as a critical area of
therapeutic target research (6).
It is crucial to note that while ferritinophagy can be involved in
basal iron recycling, its excessive activation - which is
pathogenic and leads to ferroptosis - is primarily triggered by
cellular iron overload and various stress signals, not merely by
iron starvation. In NCOA4-deficient HT22 cells, alterations in the
molecular markers associated with functional iron deficiency have
been identified (22).
Concurrently, differential gene expression linked to key neuronal
processes, including development, mitochondrial function, apoptosis
and neurodegenerative diseases, have been observed (22). These in vitro data
suggested that NCOA4-mediated ferritinophagy may serve as a
potential molecular target for the prevention of neurodegenerative
disorders. Currently, the development of small-molecule inhibitors
that block the NCOA4-FTH1 interaction has emerged as a novel
therapeutic strategy for multiple diseases. However, a lack of
structural information has forced drug discovery to rely on
phenotypic screening rather than rational drug design, with the
reported lead compounds primarily targeting NCOA4 (23). Through structural analyses,
Hoelzgen et al (24)
delineated critical features on the FTH1 surface essential for the
formation of the NCOA4·FTH1 complex, providing precise targets for
designing inhibitors of this complex. ADP-ribosylation-like factor
6 interacting protein 5 (JWA) is an all-trans retinoic
acid-responsive gene that plays a multifaceted role in cellular
homeostasis. JWA protein is critical for the survival of
dopaminergic neurons in PD (25). However, whether JWA regulates
ferroptosis in dopaminergic neurons remains unclear. Furthermore,
JWA blocks ferritinophagy by occupying the FTH1-binding site of
NCOA4, establishing JWA as a novel ferroptosis regulator that
specifically inhibits NCOA4-mediated ferritinophagy (26).
Mechanisms of ferroptosis
Ferroptosis is triggered by the disruption of the
intricate intracellular redox balance. Its core mechanisms revolve
around three interconnected axes: i) The failure of the
cystine/glutamate antiporter system Xc−/glutathione
(GSH)/GSH peroxidase (GPX)4 antioxidant axis, which eliminates the
primary defense against lipid peroxides; ii) the presence of
iron-driven lipid peroxidation, which supplies the lethal
substrates; and iii) the Kelch-like ECH-associated protein 1
(Keap1)/nuclear factor erythroid 2-related factor 2
(Nrf2)/antioxidant response element (ARE) signaling pathway as a
central regulatory hub. Critically, ferroptosis execution requires
the simultaneous convergence of the first two axes: A compromised
antioxidant capacity must coincide with active, iron-facilitated
lipid peroxidation.
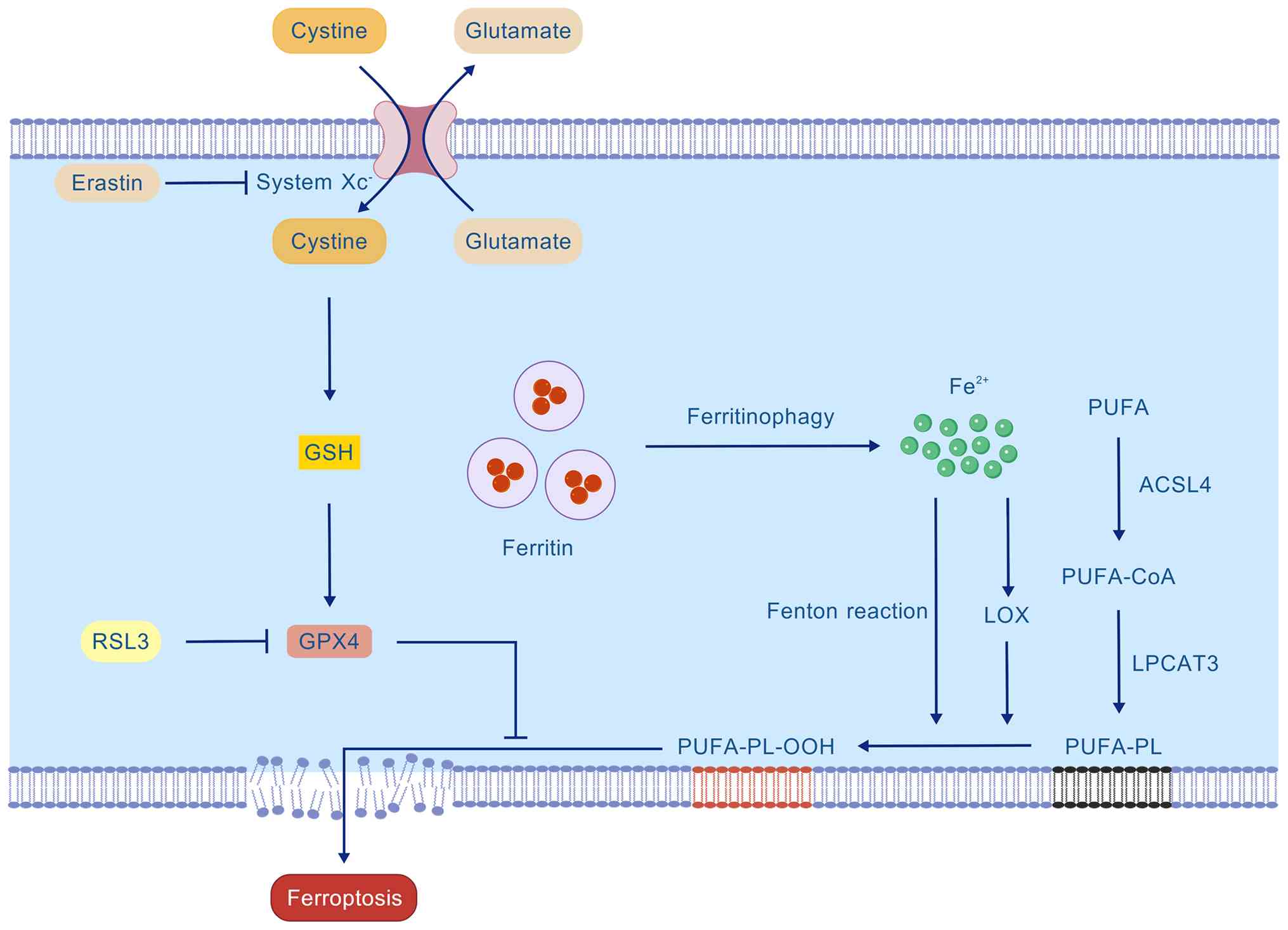
Xc−-GSH-GPX4 axis
The Xc−/GSH/GPX4 axis constitutes the
primary intracellular antioxidant defense mechanism against
ferroptosis. This axis comprises the cystine/glutamate antiporter
system Xc−, GSH and GPX4, which function sequentially to
maintain phospholipid redox homeostasis (Fig. 4). System Xc− serves as
the initial step of this defense axis. It is a heterodimeric
transport complex composed of heavy chain solute carrier family 3
member 2 (SLC3A2; 4F2hc) and light chain SLC7A11 (xCT) (27). It exchanges intracellular
glutamate with extracellular cystine in a 1:1 ratio. Upon cellular
uptake, cystine is reduced to cysteine, which is a precursor for
GSH synthesis. The selenoenzyme GPX4 utilizes GSH as a substrate to
reduce toxic lipid hydroperoxides into nontoxic lipid alcohols,
thereby playing a critical role in defending against ferroptosis
(28). Impairment of any
component of this protective axis markedly increases cellular
susceptibility to ferroptosis. For instance, erastin inhibits
system Xc− activity, thereby blocking cystine uptake,
depleting GSH and inducing ferroptosis (29). Conversely, the RAS-selective
lethal compound 3 directly inhibits GPX4 activity, leading to
uncontrolled lipid peroxide accumulation, membrane damage and
ferroptosis (30).
Lipid peroxidation
Oxidation of polyunsaturated fatty acids (PUFAs)
containing bis-allylic hydrogen atoms, such as arachidonic acid and
adrenic acid, is a hallmark of ferroptosis (Fig. 4). First, PUFAs must be activated
and esterified into membrane phospholipids. This process is
initiated by acyl-CoA synthetase long-chain family member 4
(ACSL4), which converts free PUFAs into their acyl-CoA esters
(PUFA-CoAs), a prerequisite for their incorporation into membrane
lipids (31). As the primary
enzyme that loads PUFAs into phospholipids, ACSL4 determines the
cellular abundance of PUFA-containing membrane lipids, thereby
establishing the membrane's intrinsic sensitivity to lipid
peroxidation and ferroptosis. Lysophosphatidylcholine
acyltransferase 3 catalyzes the remodeling of PUFA-CoAs into
membrane phospholipids, generating PUFA-containing phospholipids
(PUFA-PLs) (32).
Membrane-integrated PUFA-PLs serve as direct substrates for lipid
peroxidation. Lipoxygenases (LOXs), particularly 15-LOX, are highly
selective for PUFA-PLs, including arachidonoyl phospholipids, and
oxidize them to phospholipid hydroperoxides (PL-OOH). By contrast,
large quantities of ferrous iron released from ferritin via
ferritinophagy can efficiently promote the oxidation of PUFA-PLs to
PL-OOH by generating high levels of ROS through the Fenton
reaction. GPX4 is the key enzyme that prevents ferroptosis by
catalyzing the reduction of PL-OOH to their corresponding non-toxic
PL-OH, using GSH as a cofactor. Once formed, if not promptly
reduced by GPX4, PL-OOH reacts with adjacent PUFA-PLs, initiating a
self-amplifying positive feedback cycle of lipid peroxidation. This
uncontrolled chain reaction ultimately leads to irreversible
disruption of membrane integrity and cell death (30).
The Keap1/Nrf2/ARE axis
Nrf2 is a key transcription factor regulating the
cellular OS response, and many of its downstream target genes are
directly involved in the regulation of ferroptosis (Fig. 3). These genes encompass proteins
related to iron metabolism (such as FTH1 and FPN) and core
antioxidant components (including GPX4, SLC7A11 and the GSH
synthesis pathway). Under homeostatic conditions, the Neh2 domain
of Nrf2 interacts with Keap1 via its ETGE and DLG motifs, leading
to constitutive ubiquitination and degradation of Nrf2 and negative
regulation of this pathway (33). When the cell encounters OS, such
as that triggered by the Fenton reaction between cytosolic free
Fe2+ and H2O2, which generates
highly reactive hydroxyl radicals (·OH), Keap1 dissociates from
Nrf2, allowing stable Nrf2 to accumulate and translocate into the
nucleus. Inside the nucleus, Nrf2 binds to the ARE and recruits
transcriptional coactivators such as cAMP response element-binding
protein-binding protein/p300. Specifically, the Neh4 and Neh5
domains of Nrf2 interact with the TAZ1 and TAZ2 domains of
CBP/p300, acting as a scaffold to facilitate chromatin remodeling
and assembly of the transcription machinery (34). This interaction robustly
initiates the transcription of its target genes. By upregulating
FTH (enhancing iron storage) and FPN (promoting iron efflux), Nrf2
effectively reduces the intracellular labile iron pool. These two
responses are not contradictory but form a balanced, coordinated
strategy to mitigate iron toxicity. Simultaneously, Nrf2 directly
enhances the cellular antioxidant capacity by positively regulating
GPX4, thereby synergistically suppressing neuronal ferroptosis. The
activity of this pathway is finely regulated by BTB and CNC
homology 1 (BACH1). BACH1 competes with Nrf2 to bind ARE sequences,
thereby repressing the transcription of Nrf2 target genes (35). In OS, BACH1 is inactivated, which
relieves its transcriptional repression and consequently promotes
an Nrf2-driven antioxidant response.
Pathophysiological contributions of ferritin
dysregulation in neurodegenerative disorders
Neurodegenerative diseases are closely linked to
dysregulation of ferritin-mediated iron homeostasis. Studies have
indicated that these disorders are characterized by cerebral iron
dyshomeostasis, with excessive iron deposition in specific regions.
Iron overload exacerbates OS and lipid peroxidation via Fenton
reactions, thereby driving neuronal damage. Ferritin, a critical
iron storage protein, may exhibit compensatory upregulation early
in the disease to sequester labile iron. However, persistent
overload or impaired degradation ultimately promotes pathological
iron release. Such dysregulation facilitates toxic protein
aggregation (e.g., Aβ, α-Syn) and activates cell death pathways
like ferroptosis.
AD
AD neuropathology is characterized by extracellular
Aβ plaques and intracellular neurofibrillary tangles (NFTs)
composed of hyperphosphorylated tau. Aβ plaques arise from
aggregation of Aβ1-40/42 peptides derived from Aβ protein precursor
cleavage, whereas NFTs result from the accumulation of
phosphorylated tau (p-tau). Dysregulated iron metabolism in AD
brains leads to pathological iron deposition in regions such as the
hippocampus and cortex. Excess iron induces OS via Fenton
reactions, promoting Aβ aggregation and tau hyperphosphorylation.
Lactylation of the tau protein at the K677 site was found to confer
neuroprotection in an AD mouse model. This modification acts by
suppressing p38 mitogen-activated protein kinase-mediated
ferritinophagy, thereby reducing iron release and mitigating
ferroptosis and neuroinflammation (36). It should be noted that this
protective effect has so far been demonstrated only in a mouse
model. Thus, validation in human post-mortem brain tissues and
other experimental systems is essential to confirm its
pathophysiological relevance in patients with AD. These findings
indicate that tau lactylation is a promising therapeutic target. Aβ
is generated via β-secretase and γ-secretase cleavage of amyloid
precursor protein. Mutations in presenilin (PSEN), which are
linked to familial AD, impair γ-secretase activity. PSEN
mutations exacerbate oxidative damage by blocking
γ-secretase-mediated upregulation of FTH/FTL under iron challenge,
implicating γ-secretase in IRP-IRE-regulated iron metabolism
(37).
Iron is essential for myelination, synaptic
plasticity, oxidative metabolism and neurotransmitter synthesis in
the CNS (38). Iron overload
contributes to ferroptosis, OS and neuroinflammation, disrupting
the CNS microenvironment. During this process, hepcidin, which is
primarily secreted by hepatocytes, maintains systemic iron
homeostasis by suppressing the iron export function of FPN, the
only known iron exporter. This regulatory process is corroborated
in the context of AD. Analysis of the cingulate cortex from Braak
stage III-VI patients with AD by Chaudhary et al (39) demonstrated that upregulated
hepcidin transcription leads to decreased FPN expression, resulting
in increased ferritin levels and elevated total brain iron content.
It is generally thought that the upregulation of hepcidin is
triggered by IL-6 released from activated glial cells or
peripherally derived IL-6 transport - that is, inflammation induces
hepcidin upregulation (40).
However, it has not been ruled out that hepcidin upregulation may
represent a physiological feedback response to early, localized
iron overload.
Ferritin co-localizes with Aβ plaques in AD brains
(41). In healthy brains, FTH is
predominantly found in the oligodendrocytes and astrocytes
(41). In AD mouse hippocampi,
FTH1 mRNA accumulates in astrocytic somata, whereas FTL1 mRNA
redistributes to thickened astrocytic processes near Aβ deposits,
suggesting spatial regulation of iron homeostasis under
physiological and pathophysiological conditions (12).
Activated microglia located near neuritic plaques
exhibit robust ferritin expression (42). Neuritic plaques comprise neurons,
microglia and Aβ, though their pathogenesis is incompletely
understood. Microglial ferritin (iron sequestration) and Aβ (iron
chelation) may synergistically promote plaque formation (43). FTL+ ionized
calcium-binding adapter molecule 1+ microglia, a major
Aβ-plaque-infiltrating subset, are elevated in patients with AD
(44). Microglial activation
coincides with ferritin upregulation, suggesting that iron storage
modulates neuroinflammatory microenvironments in AD (45). Although AD involves concurrent
iron accumulation and neuroinflammation, it remains unclear whether
microglial iron retention and ferritin expression result from
elevated iron levels, inflammatory activation or both. A study
using human induced pluripotent stem cell-derived microglia
revealed that microglial ferritin is regulated by iron rather than
by inflammation; iron-laden microglia suppress pro-inflammatory
responses but induce OS (46).
Apolipoprotein E (ApoE) is a major genetic risk
factor for AD, and its role in the disease process may be closely
linked to intracellular iron metabolism and tau pathology. Mice
with the ApoE4 genotype exhibit significantly diminished ferritin
expression in the CA1 and hippocampal regions and reduced FPN
levels in the hippocampus (47).
The decrease in ferritin levels may compromise the intracellular
iron-binding capacity, whereas reduction in FPN restricts cellular
iron export. Collectively, these alterations contribute to neuronal
iron overload, which, in turn, drives the toxic accumulation of
hyperphosphorylated tau. ApoE can activate the PI3K/AKT signaling
pathway and inhibit the autophagic degradation of ferritin, a
process known as ferritinophagy (48). This process reduces
iron-dependent lipid peroxidation and maintains cellular iron
homeostasis. By contrast, p-tau, a major component of NFTs, is
significantly elevated in the cerebrospinal fluid (CSF) of patients
with AD. Increased ferritin levels in the CSF of these patients are
correlated with p-tau181, and this association is significantly
mediated by CSF ApoE levels (49). These findings suggest that ApoE
may indirectly influence tau phosphorylation and aggregation by
regulating iron metabolism pathways. However, the precise molecular
mechanisms underlying the interactions between ApoE, ferritin and
tau hyperphosphorylation require further investigation.
PD
PD is characterized by the progressive loss of
dopaminergic neurons in the substantia nigra pars compacta, and
iron dysregulation plays a central role in this process.
Pathological iron deposition and α-Syn aggregation form a vicious
cycle in the brain of patients with PD and drives neuronal damage.
Furthermore, toxic interactions between iron and α-Syn can induce
cellular senescence, a process that may precede overt neuronal loss
(50). At the molecular level,
this interaction triggers ferroptosis, an iron-dependent form of
regulated cell death. For instance, microRNA (miR)-335 exacerbates
ferroptosis by suppressing FTH1 expression, thereby increasing
intracellular Fe2+ levels (51). However, the precise extent of
FTH1 reduction by miR-335 remains to be quantified. By contrast,
the neuroprotective hormone melatonin (MT) counteracts
α-Syn-induced ferroptosis through MT1 receptor-mediated activation
of the Sirtuin 1/Nrf2/heme oxygenase-1/GPX4 signaling axis
(52).
Iron dysregulation exhibits cell-type-specific
manifestations. In microglia, iron accumulation is closely
correlated with neuroinflammation, as evidenced by the positive
association between microgliosis and iron load in the postmortem PD
substantia nigra (53).
Pathogenic mutations in leucine-rich repeat kinase 2, such as
G2019S, disrupt iron handling by impairing the function of its
substrate Ras-associated binding protein 8a, leading to
significantly reduced FTH1 transcription in oligodendrocytes,
astrocytes and microglia (54,55). These abnormalities are associated
with, and may potentially contribute to, the formation of Lewy
bodies, where both iron and ferritin are co-deposited (41). However, a direct causal link
remains to be established.
The spatiotemporal dynamics of iron distribution are
critical in the pathology of PD. The newly developed
immunomicroprobe particle-induced X-ray emission technique, which
enables the quantification of iron in specific structures such as
Lewy bodies at a micron-level resolution, is a powerful tool for
elucidating the fine spatiotemporal characteristics of iron
metabolism (56). Clinically,
CSF profiles from patients with PD experiencing excessive daytime
sleepiness reveal a distinct pattern of elevated iron, reduced
ferritin and increased IL-1β, suggesting that iron overload may
promote excessive daytime sleepiness via microglial activation and
neuroinflammation (57).
It is important to note that iron-related mechanisms
in PD are context dependent. Although restless leg syndrome (RLS)
in the general population is often associated with iron deficiency
in the brain, a meta-analysis found no association between PD-RLS
and systemic iron parameters, indicating that PD-RLS may involve a
pathophysiology distinct from that of the classical iron deficiency
hypothesis (58).
ALS
ALS, the most common motor neuron disease, is
characterized by the progressive degeneration of the upper and
lower motor neurons, leading to muscle atrophy, functional
impairment and death. Most patients with ALS share the pathological
hallmark of the abnormal cytoplasmic aggregation of TAR DNA-binding
protein 43 (TDP-43) in motor neurons. As resident immune cells in
the brain, microglia exhibit unclear responses to TDP-43 in ALS.
Phosphorylated TDP-43 pathology was found to drive the microglial
transition from a phagocytically active state, marked by
early-stage neuroprotective phagocytic activity via elevated CD68
expression, to a dysfunctional state characterized by late-phase
L-ferritin accumulation, thus offering novel insights into ALS
pathogenesis (59). Significant
spatial and functional associations between microglial activation
and abnormal ferritin accumulation underscore their critical roles
in ALS progression (60). Given
that iron dysmetabolism also occurs during ALS progression, iron is
likely to play a pivotal role in ALS pathology. Decreased activity
of the AKT signaling pathway may be involved in the dysregulation
of iron metabolism in ALS. Specifically, ALS downregulates AKT
protein expression and upregulates iron import and storage-related
proteins (TfR1, PCBP, FTL and FTH), whereas suppressing the
expression of the iron exporter FPN1 may ultimately lead to
increased iron accumulation in the skeletal muscle (61). Human spinal cord gene
co-expression network analysis identified two key modules enriched
for ALS genetic risk: SC.M4 (regulating RNA processing and
epigenetics) and SC.M2 (enriched for oligodendrocyte-specific
intracellular transport and autophagy-related genes) (62). Within the SC.M2 module, NCOA4
emerged as a computationally inferred hub gene based on
co-expression and genome-wide association study data. This position
identifies NCOA4 as a statistical risk association for ALS
(58), implicating iron
dysmetabolism and autophagic imbalance as potential contributors to
neurodegeneration.
Aceruloplasminemia (ACP)
ACP is a rare autosomal recessive disorder caused by
mutations in the ceruloplasmin gene that lead to reduced
ferroxidase activity. Patients with ACP typically exhibit a triad
of clinical manifestations, including neurological symptoms
(cognitive decline, neuropsychiatric abnormalities and movement
disorders), retinal degeneration and diabetes mellitus (63). In ACP, Fe3+ can be
transported to plasma Tf and delivered to other cells. The impaired
ferroxidase function of ceruloplasmin in ACP disrupts the
conversion of Fe2+ to Fe3+, thereby
inhibiting iron release from the storage sites and reducing
systemic iron bioavailability. To meet the iron demands for
neurotransmitter synthesis, neurons excessively uptake non-Tf-bound
iron, a highly toxic, free iron species that exacerbates neuronal
death. FTH, which possesses intrinsic ferroxidase activity, is
upregulated in ceruloplasmin-knockout mouse models. This
mouse-specific finding suggests a potential compensatory role in
maintaining iron homeostasis following ceruloplasmin deficiency
(64). Whether FTH is similarly
upregulated in patients with ACP remains to be confirmed.
Iron-related neurodegeneration in ACP is primarily linked to the
abnormal accumulation of magnetic Fe3+ within
ferritin/hemosiderin cores, predominantly in the form of
ferrihydrite-iron, which serves as the principal driver of
iron-sensitive MRI contrast (65). Given the rarity of this
condition, case reports and individual case analyses remain
important resources for investigating its clinical features,
genetic mechanisms and therapeutic strategies (63,66).
Other neurodegenerative diseases
Friedreich ataxia (FA) is a rare inherited
neurodegenerative disorder caused by mutations in the frataxin
gene, leading to reduced frataxin protein levels. The most common
mutation is a homozygous GAA trinucleotide repeat expansion in the
first intron of the frataxin gene. Frataxin is a mitochondrial
protein that is essential for mitochondrial function, and its
deficiency results in mitochondrial dysfunction, iron accumulation
and OS. The degeneration of large sensory neurons in the dorsal
root ganglia is an early event in FA; however, the mechanism
underlying their heightened vulnerability to frataxin deficiency
remains unclear. Reduced ferritin levels (particularly FTH1) are
critical for frataxin deficiency-induced ferroptosis, which
exacerbates free iron toxicity and oxidative damage to drive
neuronal death (67). Targeting
ferritin expression or function, such as by enhancing FTH1
activity, may represent a potential therapeutic strategy for FA. In
patients with FA, pathological damage to the dentate nucleus is
characterized by the progressive atrophy of large neurons. Abnormal
iron and ferritin accumulation in white matter oligodendrocytes may
represent secondary pathological changes following neuronal atrophy
(68). Patients with FA exhibit
systemic iron storage depletion and intracellular alterations
resembling iron starvation responses, indicating that iron
dysregulation in FA reflects an intercompartmental iron
redistribution imbalance rather than an absolute overload (69).
Hereditary ferritinopathy, a subtype of
neurodegeneration with brain iron accumulation (NBIA), is primarily
caused by frameshift mutations in the FTL gene. Its pathology
involves the age-dependent amplification of cerebral iron/ferritin
deposition. The core mechanism may not involve iron-mediated OS but
rather aggregation of mutant FTL, ubiquitination and proteostasis
imbalance, triggering cell death, a pathway shared with multiple
neurodegenerative diseases (70).
TDP-43 pathological inclusions are hallmark features
of limbic-predominant age-related TDP-43 encephalopathy
neuropathological changes. Phosphorylated TDP-43 inclusions
localized on the surface or interior of small blood vessels are
termed Lin bodies. Shahidehpour et al (71) observed frequent co-localization
of Lin bodies with ferritin, suggesting that post-hemorrhagic
erythrophagocytosis elevates intracellular iron and iron storage
proteins, including ferritin.
Progressive supranuclear palsy (PSP), a rare
neurodegenerative disease, is characterized by the abnormal
aggregation of 4-repeat tau and pathological tau structures (e.g.,
NFT, globular tangles). In patients with PSP, mitochondrial
ferritin accumulates abnormally within substantia nigra neurons.
Light chain 3 (LC3), an autophagosome marker, reflects the
mitophagy status. The colocalization of LC3 with mitochondrial
ferritin in the substantia nigra neurons of patients with PSP
supports a model wherein the accumulation of mitochondrial ferritin
initially exerts protective effects by chelating iron and reducing
ROS levels (72). However, under
sustained stress, this protective mechanism fails, leading to
accumulated mitochondrial damage, enhanced mitophagy (marked by LC3
accumulation) and cell death (72).
Multiple sclerosis (MS) is a CNS autoimmune disease
characterized by inflammatory demyelination and secondary
neurodegeneration. Oligodendrocytes in the CNS produce myelin
sheaths to wrap axons, enabling rapid saltatory conduction of nerve
impulses and maintaining axonal structural integrity. A study using
experimental autoimmune encephalomyelitis mice revealed that
ferritinophagy controls the generation of lipid ROS via the Fenton
reaction, inducing oligodendrocyte death and demyelination at the
peak stage of the disease (73).
Multiple system atrophy (MSA), a rare
neurodegenerative synucleinopathy, features α-Syn-positive
cytoplasmic inclusions in oligodendrocytes, central to
neurodegeneration. In an aged proteolipid protein-α-Syn mouse
model, which overexpresses α-Syn in oligodendrocytes, iron
accumulation and disrupted iron-ferritin interactions were observed
in the substantia nigra, putamen and cerebellum (74). Iron elevation in MSA mice may
involve ceruloplasmin dysfunction; therefore, targeting iron
metabolism represents a potential therapeutic strategy (74).
Wilson's disease, also known as hepatolenticular
degeneration, is a rare autosomal recessive disorder caused by
ATPase copper transporting β mutations (13q14) that impair copper
transport and cause copper accumulation in the liver, brain and
kidneys. Of note, patients with Wilson's disease exhibit both
copper and iron dysregulation. Elevated ferritin levels in Wilson's
disease, which are partially reversible with copper chelation
therapy, suggest that iron metabolism disturbances are linked to
inflammation and copper toxicity, which require multidimensional
interventions (75).
Other diseases
Ferroptosis, an iron-dependent, lipid
peroxidation-driven form of cell death, has pathological
significance extending beyond single diseases and is emerging as a
core mechanism in diverse tissue injuries and chronic disorders.
Understanding ferroptosis mechanisms in other diseases will
facilitate the precise elucidation of its specificity in
neurodegenerative pathologies.
With the global surge in the prevalence of type 2
diabetes (T2D), the associated cognitive dysfunction has become a
research focus in neurometabolism, owing to its substantial impact
on the quality of life of patients and as a public health burden.
Elevated TfR levels alongside decreased ferritin, GPX4 and SLC7A11
levels in the hippocampal neurons of T2D model mice confirm that
hippocampal neuronal ferroptosis activation is a crucial mechanism
for cognitive impairment (76).
Iron accumulation and ROS synergistically induce
neuronal dysfunction, which may constitute a critical mechanism of
epileptogenesis. A previous study revealed that, compared with
postmortem controls, ferritin expression in epileptic brains
exhibited a significant relocation from microglia and
oligodendrocytes to astrocytes (77). This finding suggests that
strategies to reduce astrocytic iron uptake (thereby attenuating
pro-inflammatory responses) may represent a novel therapeutic
strategy for treating epilepsy (77).
Comparative analysis of ferritin
dysregulation across neurodegenerative diseases
Neurodegenerative diseases share common pathways in
iron dyshomeostasis, yet exhibit distinct, disease-specific
manifestations. The common core lies in pathological iron
accumulation within specific brain regions, which, through
mechanisms such as OS and ferritinophagy, ultimately converges on
ferroptosis, a shared cell death pathway. However, key features
differ markedly among diseases: Regarding interacting pathological
proteins, disrupted iron metabolism forms a vicious cycle with
disease-specific protein aggregates: Aβ/tau in AD, α-Syn in PD and
TDP-43 in ALS. In terms of predominantly affected cell types,
ferritin responses in microglia are particularly prominent in AD
and ALS, whereas oligodendrocyte dysfunction plays a more critical
role in demyelinating disorders such as MS. In summary,
ferritin-mediated iron dysregulation represents a shared
pathological nexus in neurodegeneration, yet it is modulated by
disease-specific molecular and cellular contexts. This
understanding holds significant implications for developing
broad-spectrum neuroprotective strategies and precision therapies
targeted to specific diseases.
Ferritin as a biomarker for the diagnosis of
neurodegenerative diseases
Early diagnosis and monitoring of neurodegenerative
disorders, such as AD, PD and ALS, pose major challenges in
clinical neuroscience. Dysregulated brain iron metabolism plays a
pivotal role in neurodegenerative pathologies by mediating
pathological processes, such as OS, protein misfolding and
aggregation, and neuronal degeneration. As the core protein for
intracellular iron storage, fluctuations in ferritin levels not
only reflect the dynamic equilibrium of brain iron homeostasis but
also highlight its double-edged sword nature; while buffering iron
toxicity, ferritin may simultaneously signal iron overload. This
dual characteristic suggests that ferritin may be a promising
multidimensional biomarker. Its disease-specific expression
patterns in the CSF, peripheral blood and neuroimaging in
heterogeneous conditions, such as AD, PD and ALS, offer new
perspectives to overcome the spatiotemporal limitations of
traditional biomarkers.
AD
AD is an irreversible neurodegenerative disorder
characterized by chronic progressive dementia. Most patients with
AD are diagnosed at advanced stages, and no effective therapies
currently exist to halt or reverse its progression. Notably, AD has
a prolonged preclinical phase before symptom onset; however,
current diagnosis primarily relies on clinical observation, which
underscores the urgent need for the early identification of
specific biomarkers to enable precise diagnosis and intervention.
AD is associated with chronic inflammation, OS, mitochondrial
dysfunction and neurotoxicity. Studies have suggested that
disrupted iron homeostasis may be another potential pathogenic
factor in AD. Cerebral iron deposition exacerbates oxidative damage
through the Fenton reaction, directly contributing to AD
pathological processes. Iron homeostasis-associated proteins (e.g.,
ferritin, Tf), which are responsible for iron storage and
transport, may reflect early-stage metabolic disturbances in AD
through changes in their expression levels. Consequently, iron and
iron-related proteins have emerged as potential biomarker
candidates. However, its role in AD remains elusive.
CSF ferritin, a biomarker implicated in iron
metabolism regulation and inflammatory responses, shows significant
potential for the early detection of AD. CSF ferritin levels are
significantly elevated during the preclinical stages of AD and
correlate with complement activation and other inflammatory
markers, suggesting its role as a key bridging molecule that links
neuroinflammation and neurodegeneration (78,79). A recent systematic review and
meta-analysis, encompassing 25 studies and 3,469 participants,
confirmed that CSF ferritin levels are significantly elevated in
patients with AD compared to controls (pooled standardized mean
difference=0.44, 95% CI: 0.02-0.86) (80). This population-level evidence
strengthens the case for ferritin as a quantifiable biomarker
linked to AD pathology.
Regarding classic AD pathology, ferritin shows a
particularly strong association with tau pathology. A positive
correlation between CSF ferritin and p-tau levels was revealed and
it was mediated by ApoE (49).
Furthermore, regardless of the cerebral Aβ deposition status, CSF
ferritin levels are consistently elevated in subjects with high
total tau and correlate with other neuronal injury markers such as
fatty acid-binding protein 3, collectively pointing to a tau-driven
neurodegenerative pathway (38,81). However, CSF ferritin levels are
not significantly associated with cerebral amyloid pathology
(38). Taken together, these
findings position CSF ferritin primarily as a marker related to tau
pathology and consequent neurodegeneration, rather than a general
marker of AD. This characteristic supports its utility in
differential diagnosis. For instance, in cerebral amyloid
angiopathy, which also features Aβ deposition, CSF ferritin
correlates negatively with both Aβ-40 and Aβ-42 levels, a pattern
distinct from that observed in AD, potentially aiding in
distinguishing these frequently co-occurring disorders (82). It should be noted, however, that
these findings are based on limited data and small sample sizes,
necessitating further validation.
Serum ferritin levels in peripheral blood are also
clinically relevant. Serum ferritin levels positively correlate
with AD severity, and its combination with homocysteine and
C-reactive protein significantly improves the diagnostic efficacy
for AD with mild cognitive impairment (83). However, a longitudinal analysis
of multiorgan blood parameters reported a negative correlation
between plasma ferritin levels and AD severity, suggesting that its
dynamic changes may be more complex and potentially useful as a
marker of disease progression (84). Beyond AD-specific pathology,
systemic factors such as chronic heart failure have been found to
influence phosphorylated tau, possibly through the modulation of
serum ferritin levels, revealing a potential interaction between
systemic circulation and CNS pathology (85).
Finally, individual differences, including genotype
and long-term dietary habits, significantly modulated the
expression of AD biomarkers in the hippocampus (47). This adds a layer of complexity to
the clinical application of ferritin and other biomarkers,
emphasizing the necessity for a comprehensive perspective in future
research and clinical interpretation that situates biomarkers
within an individual's overall physiological and pathological
contexts.
PD
Iron is essential for ROS generation, which induces
OS and subsequent damage to neurons in the substantia nigra of
patients with PD. Various techniques have been developed to assess
iron and related biomarker concentrations. Biochemical analyses,
histopathological studies and neuroimaging have demonstrated that
iron deposition in patients with PD is primarily localized to the
substantia nigra. However, investigations on iron levels across
diverse biological fluids from patients with PD have yielded
conflicting results regarding both the direction of change
(increase, decrease or no difference) and their correlation with
clinical features (86-88).
Emerging evidence suggests that ferritin levels are
associated with disease progression and specific clinical
manifestations of PD. In the CSF, a longitudinal study revealed a
significant increase in total iron and a decrease in ferritin
levels over time, indicating its potential as a dynamic marker of
disease progression (87).
Furthermore, elevated CSF ferritin levels negatively correlate with
Mini-Mental State Examination scores in patients with PD dementia,
supporting the detrimental role of cerebral iron accumulation in
cognitive function (89).
Furthermore, altered serum ferritin levels have been observed in
patients with PD compared with healthy controls (90). Serum ferritin levels have also
been positively correlated with the volumes of key brain
structures, including the caudate nucleus and putamen, suggesting
that serum ferritin levels may serve as a biomarker reflecting
disease burden or the extent of neurodegeneration in PD (88). However, it is important to note
that peripheral ferritin levels may not directly correlate with
brain iron content as measured by specialized MRI techniques, and
thus, their interpretation requires caution. Combining it with
neuroimaging features could enhance this assessment.
The establishment of ferritin as a reliable
biomarker faces significant challenges. First, study findings have
been inconsistent. Some reports have found no significant
differences in serum ferritin or Tf levels between patients and
controls (91). A systematic
meta-analysis further highlighted that although neuroimaging
techniques consistently show increased iron concentrations in the
substantia nigra, serum and CSF iron-related parameters lack
consistent changes (92).
Second, ferritin lacks specificity, as plasma levels have been
proven ineffective in predicting PD risk in individuals carrying
pathogenic mutations in the gene encoding the enzyme
β-glucocerebrosidase (93). More
importantly, the correlation between peripheral iron indices and
iron accumulation in the brain remains elusive. No significant
association between subcortical iron and blood iron markers has
been reported, suggesting that cerebral iron accumulation in
neurodegeneration may be independent of systemic iron status and
potentially regulated by local inflammatory processes (94).
Altogether, the value of ferritin as a biomarker is
primarily related to disease progression and certain clinical
symptoms. However, its specificity, sensitivity and complex
relationship with the peripheral iron status are major obstacles. A
Mendelian randomization study offered a new perspective, revealing
a genetic causal link between lower serum iron levels (but not
ferritin, total iron-binding capacity or Tf saturation) and
increased PD risk (95). This
Mendelian randomization study specifically links genetically
lowered serum iron to PD risk. The absence of a link for ferritin
in this analysis likely means that the genetic variants used are
not strong drivers of ferritin levels; however, ferritin is not
biologically unimportant in PD. This underscores the need to
precisely define the roles of different iron metabolism parameters
in future studies. Overall, ferritin is more likely to serve as a
component of a multimodal biomarker panel than as a standalone
diagnostic tool.
ALS
Emerging evidence highlights the critical role of
ferritin as a biomarker of neurodegenerative disorders. Although
dysregulated iron metabolism in PD is closely linked to nigral
neurodegeneration and cognitive decline, similar iron homeostasis
imbalances, marked by elevated serum ferritin levels, have been
implicated in the pathophysiology of ALS, reflecting the shared yet
distinct mechanisms of OS and neuroinflammation. A large-scale,
multicenter, longitudinal, multimodal body fluid biomarker cohort
study systematically evaluated the utility of multiple candidate
biomarkers for ALS stratification and potential therapeutic
evaluation, and demonstrated significantly elevated serum ferritin
levels in patients with ALS (96). A systematic review and
meta-analysis confirmed elevated serum ferritin levels in patients
with ALS, and serum ferritin levels negatively correlated with
survival (hazard ratio=1.38; 95% CI, 1.02-1.88; P=0.039) (97). These findings provide new
evidence for the involvement of energy metabolism dysregulation,
OS-mediated iron homeostasis imbalance and immune dysregulation in
ALS pathophysiology.
Positioning and future directions of
ferritin as a biomarker
Although ferritin levels in CSF and blood are
altered (increased or decreased) in AD, PD and ALS, its use as an
independent diagnostic biomarker with high sensitivity and
specificity faces significant challenges. Most existing studies
report associations but lack large-scale validation with unified,
well-defined diagnostic cutoff values. Furthermore, the majority of
research uses clinical diagnosis rather than more precise gold
standards, such as amyloid-β positron emission tomography (Aβ-PET),
tau-PET or neuropathology, as the reference, which introduces bias
when evaluating diagnostic performance. Thus, the core clinical
value of ferritin may not lie in replacing established core
diagnostic markers, but rather in serving a complementary role. The
most promising direction lies in combining ferritin with
neurofilament light chain, neuroimaging measures and other
inflammatory indicators to construct multimodal diagnostic
models.
Therapeutic potential of ferritin in
neurodegenerative diseases
Pathological progression of neurodegenerative
diseases is closely linked to disrupted iron homeostasis in the
brain. Ferritin, a key intracellular iron storage protein,
mitigates OS by chelating free Fe3+, thereby effectively
reducing iron-mediated Fenton reactions and suppressing ROS
generation. Studies have identified numerous compounds that target
iron homeostasis for the treatment of neurodegenerative diseases
(Table I) (98-109). For instance, ebselen, a
synthetic organoselenium compound, reduces iron deposition by
downregulating ferritin light chain expression, thereby suppressing
oxidative stress and reversing cellular senescence (102). Donepezil, an
acetylcholinesterase inhibitor used in Alzheimer's disease, has
been shown to stabilize ferritin structure through high-affinity
binding, suggesting a potential role in modulating cerebral iron
metabolism beyond its cholinergic effects (103). However, none of these compounds
have yet progressed to clinical trials. Therefore, it is important
to note that their journey toward clinical application faces
several recognized hurdles, which are critical areas for future
research. Future efforts should focus on addressing these
pharmacokinetic and safety challenges to advance the most viable
candidates into the clinical development pipeline.
 | Table ICompounds targeting iron homeostasis
for the treatment of neurodegenerative diseases and their
mechanisms of action. |
Table I
Compounds targeting iron homeostasis
for the treatment of neurodegenerative diseases and their
mechanisms of action.
| Name | Disease | Mechanism | Model | (Refs.) |
|---|
| Moschus | AD | Activates the
Keap1/Nrf2 pathway to upregulate FTH1 expression, maintaining
neuronal iron balance and inhibiting ferroptosis. | HT22 cells | (98) |
| SSF | AD | Significantly
upregulates ferritin expression and reduces cerebral free iron
levels, thereby inhibiting oxidative stress and ameliorating
synaptic dysfunction and neuroplasticity damage. | Rats | (99) |
| Quercetin | AD | Specifically binds
to key functional sites of human ferritin, stabilizing its
conformation to regulate iron ion homeostasis. | In silico | (100) |
| Naringenin | AD | Specifically binds
to key functional sites of human ferritin, stabilizing its
conformation to regulate iron ion homeostasis. | In silico | (100) |
| Bryostatin 1 | AD | Inhibits
pathological iron accumulation through stable binding to
ferritin. | In silico | (101) |
| Ebselen | AD | Reduces iron
deposition by downregulating the FTL and reverses senescent
cellular phenotypes. | SH-SY5Y cells | (102) |
| Donepezil | AD | Stabilizes the
ferritin structure through high-affinity binding, potentially
modulating cerebral iron metabolism. | In silico | (103) |
| EGCG | PD | Reduces iron influx
by inhibiting the expression of the iron transporter Malvolio and
promotes upregulation of the iron storage protein ferritin, thereby
decreasing free iron levels and restoring cerebral iron
homeostasis. | Drosophila | (104) |
| Morroniside | PD | Significantly
reduced free iron levels by activating the nuclear factor Nrf2/ARE
pathway and upregulating the expression of FTH1 and FPN. | Mice | (105) |
| Ginsenoside
Rg1 | PD | Regulates the
expression of FTH and FTL in oligodendrocytes, restores cerebral
iron metabolism homeostasis and suppresses lipid peroxidation,
thereby enhancing dopaminergic neuron survival. | Mice | (106) |
| α-Lipoic acid | PD | Activates the
SIRT1/Nrf2 signaling pathway, subsequently promoting the expression
of FTH1 and GPX4, leading to significant improvement of motor
dysfunction. | PC12 cells and
mice | (107) |
| Ganoderic acid
A | PD | Significantly
alleviates dopaminergic neuron ferroptosis and motor impairments by
specifically inhibiting the NCOA4-mediated ferritinophagy
pathway. | Mice | (108) |
| Clozapine | PD | Its metabolite,
clozapine-N-oxide, effectively blocks dopaminergic neuron
ferroptosis by inhibiting ferritinophagy, thereby reducing
intracellular iron release and lipid peroxidation. | SH-SY5Y cells and
mice | (109) |
Promising preclinical data notwithstanding, several
key challenges have hindered the clinical translation of
ferroptosis inhibitors (e.g., ferrostatin-1) for neurodegenerative
diseases (110). Key challenges
include poor blood-brain barrier penetration, uncertain long-term
safety given the physiological roles of iron and lipid metabolism
and the coexistence of other cell death mechanisms (e.g.,
apoptosis), which may reduce the benefit of inhibiting ferroptosis
alone. Advances in brain delivery, safety profiling and combination
strategies are needed to move these agents toward clinical
trials.
Furthermore, the unique hollow nanocage structure of
ferritin makes it a promising nanocarrier for the targeted delivery
of neuroprotective agents (111). Ferritin nanocages overcome the
limitations of traditional delivery systems, owing to their
superior biocompatibility, highly efficient hydrophobic
molecule-loading capacity and targeted delivery properties.
However, efficient crossing of the blood-brain barrier remains a
significant challenge for these ~12-nm structures, with most work
to date conducted in vitro or in peripheral cells (e.g.,
retina, peripheral blood mononuclear cells) rather than in the
human brain in vivo (112,113). Ferritin nanocages enable the
precise detection of pathological biomarkers, such as tau protein,
and targeted drug delivery, providing innovative tools for
non-invasive diagnosis and disease modulation. In one study,
humanized ferritin nanocages were used to efficiently deliver the
hydrophobic BODIPY fluorescent probe BT1, thus resolving the
challenges of probe solubility and targeted delivery. This achieved
high-sensitivity and low-toxicity detection of pathological tau
proteins in live human retinal cells, establishing a novel
nanotechnology platform for early non-invasive AD diagnosis
(112). Peripheral blood
mononuclear cells (PBMCs) from patients with AD exhibit neuron-like
pathological features (e.g., mitochondrial dysfunction, OS) and
serve as critical models for exploring neuroinflammation and
apoptosis mechanisms. In addition, as a convenient in vitro
platform, PBMCs are widely used to screen the therapeutic effects
of drug candidates, offering vital insights for the development of
AD treatment strategies. Researchers have developed H-subunit
ferritin nanocages loaded with bisdemethoxycurcumin, which
significantly improves water solubility, stability and blood-brain
barrier penetration capability. This nanocarrier demonstrated
targeted modulation of inflammation-related gene expression in
PBMCs from patients with AD, reversing the imbalance between
pro-inflammatory and anti-inflammatory genes, thereby providing a
novel delivery strategy for neuroinflammatory intervention
(113). Another study developed
a sequence-targeted nanodelivery system based on recombinant human
H-ferritin, which significantly enhanced lycopene enrichment in
neurons through receptor-mediated blood-brain barrier penetration
and mitochondrial targeting, activating pro-survival mitophagy and
clearing pathological α-Syn aggregates. This study demonstrated the
unique advantages of ferritin nanocarriers for neurodegenerative
disease treatment, including intrinsic biocompatibility,
non-invasive delivery capability and multi-mechanism coordination,
providing an innovative paradigm for next-generation brain-targeted
nano-therapeutics (114).
MRI-based evaluation of brain iron
homeostasis in neurodegenerative disorders
Dysregulated cerebral iron homeostasis is a
pathological hallmark of neurodegenerative diseases. Exploiting the
paramagnetic properties of iron, MRI is the only effective modality
for in vivo assessment of brain iron deposition (65). Although conventional MRI
techniques are sensitive to changes in total iron concentration,
they lack the chemical specificity needed to distinguish between
different iron pools, such as toxic redox-active iron and
protective iron safely stored within proteins like ferritin
(115). Recent advancements in
novel MRI methodologies have shifted the paradigm from measuring
iron deposition towards assessing the more biologically relevant
iron homeostasis.
Emerging evidence indicates that the analysis of
r1-r2* relaxometry enables
non-invasive evaluation of brain iron homeostasis in vivo.
This technique successfully delineated the characteristic
paramagnetic profiles of ferritin, Tf and free Fe2+,
revealing the spatial heterogeneity in the iron mobilization
capacity across brain regions and its dynamic changes during aging
(116). These findings suggest
that novel biomarkers can be used for early diagnosis. The physical
basis for this advancement lies in pathological alterations in the
ferritin mineral core. Under physiological conditions, the ferritin
core is predominantly composed of superparamagnetic ferrihydrite,
whereas under pathological conditions, it is enriched with strongly
magnetic magnetite. A comparative study evaluating relaxivity
differences between native ferritin and magnetoferritin under a 7T
magnetic field validated the ability of MRI to distinguish
mineralogical features. This provides a potential tool for the
non-invasive diagnosis of ferritin-related iron accumulation and
pathogenic magnetization processes, as observed in
neurodegenerative diseases (115). Complementarily, off-resonance
saturation (ORS) MRI has achieved the specific quantification of
ferritin-bound iron in postmortem human brain tissue. The
distribution of this iron was highly co-localized with pathological
iron deposits, further confirming that the magnetic properties of
ferritin as superparamagnetic nanoparticles can be precisely
captured (117).
In patients with PD, quantitative susceptibility
mapping (QSM) and susceptibility weighted imaging (SWI) offer
complementary functions; QSM provides an accurate three-dimensional
quantification of the iron concentration in the substantia nigra,
whereas SWI delivers a high-contrast visualization of lesions.
Together, these studies reveal the core features of abnormally
elevated iron concentrations in the substantia nigra pars compacta
of patients with PD (92).
Notably, iron overload in PD is initiated in the nigrosome-1
region, with most the iron bound to ferritin. Although neuromelanin
has a lower total iron load, it is a major contributor to the
R2* relaxation signal, indicating
differential contributions of various iron pools to the MRI signal
(118). However, it remains
elusive whether this regional iron accumulation directly
contributes to neuronal death or represents a compensatory
mechanism. Furthermore, the strong correlation between QSM magnetic
susceptibility and transcranial sonography (TCS) measures of
substantia nigra echogenicity suggests that iron accumulation is
the underlying mechanism of nigral hyperechogenicity. The combined
application of QSM and TCS may provide a multimodal clinical
assessment framework for PD (119,120).
In summary, novel MRI techniques such as
r1-r2* relaxometry, QSM, SWI and
ORS are progressively deciphering the mechanisms of disrupted
cerebral iron metabolism by revealing chemically specific changes
in ferritin levels. These non-invasive imaging biomarkers hold
significant promise as key players in the early diagnosis, subtype
classification and therapeutic monitoring of neurodegenerative
diseases.
Conclusions and future perspectives
Emerging evidence indicates that various
environmental stressors, particularly airborne pollutants, such as
formaldehyde and fine particulate matter (PM), can exert neurotoxic
effects by dysregulating ferritinophagy, a selective form of
autophagy for ferritin degradation. This dysregulation subsequently
disrupts cellular iron homeostasis, implicating ferritinophagy as a
critical mechanistic link in pollutant-induced neurotoxicity.
Formaldehyde is neurotoxic and can trigger neurodegenerative
diseases (121). Formaldehyde
induced hippocampal neuronal cell damage by promoting
ferritinophagy, while hydrogen sulfide (H2S) exerted
protective effects against formaldehyde-induced neurotoxicity in
HT22 cells (a murine hippocampal neuronal cell line) (121,122). Furthermore, H2S
antagonizes formaldehyde neurotoxicity by upregulating growth
differentiation factor 11, which suppresses ferritinophagy and
ferroptosis (122). PM2.5
refers to particulate matter with an aerodynamic diameter ≤2.5
micrometers. Accumulating evidence indicates that chronic PM2.5
exposure induces neuroinflammation and neurodegeneration. In mouse
neuroblastoma N2a cells, PM2.5 exposure disrupts autophagic flux by
impairing lysosomal function. Autophagic dysfunction hinders the
intracellular degradation of ferroptosis-related proteins, such as
GPX4 and ferritin, thereby inhibiting ferroptosis (123). Conversely, a study in human
neuroblastoma SH-SY5Y cells demonstrated that PM2.5 induces
autophagy-dependent ferroptosis via endoplasmic reticulum stress,
which is potentially linked to NCOA4-mediated ferritinophagy-driven
iron accumulation (124). These
findings reveal opposite effects of PM2.5 on autophagy and
ferroptosis across different neuronal cell lines (inhibition vs.
induction), underscoring the cell-type-specific nature of the
response to this environmental stressor. Based on the shared
mechanism by which formaldehyde and PM2.5 disrupt ferritinophagy to
exert neurotoxic effects, future research on neurodegenerative
diseases in human populations must systematically integrate
long-term individual environmental exposure data. Employing
molecular epidemiological approaches to validate the environmental
exposure-disrupted iron metabolism-disease onset pathway in human
populations will provide crucial scientific evidence for the
precise prevention and early identification of these diseases.
Emerging evidence suggests that both intrinsic
physiological states and extrinsic environmental factors are
critical modulators of iron homeostasis in the brain and influence
the pathophysiology of neurodegenerative diseases. For instance, in
a Drosophila phosphatase and tensin homolog induced kinase 1
knockdown model, mating behavior was found to alleviate OS via a
dual mechanism: Suppression of iron import and upregulation of
ferritin (125). Dietary iron
intake is another potent regulator, with complex outcomes. Although
an iron-restricted diet in adult rats triggers systemic metabolic
adaptations that maintained brain iron levels (126), a high-iron diet in aged mice
directly promotes the accumulation of AD-related pathological
proteins (e.g., phosphorylated tau and Aβ1-42) in the hippocampus
and cortex (127). These
compelling findings highlight a significant and often-overlooked
source of experimental variability. Therefore, future studies
utilizing animal models of neurodegenerative diseases must
systematically incorporate and report key parameters, such as
mating status and dietary composition, as standard practice.
Acknowledging and controlling these variables are essential for
enhancing the reproducibility, interpretability and translational
value of preclinical research.
β-propeller protein-associated neurodegeneration
(BPAN), a recently identified subtype of NBIA, is the only X-linked
dominant subtype caused by mutations in the WD repeat domain 45
(WDR45)/WIPI4 gene (128). WDR45 knockout in SH-SY5Y
neuroblastoma cells does not alter NCOA4 levels, suggesting that
WDR45 deficiency impairs amphisome-lysosome fusion or
lysosomal function, rather than autophagosome formation (129). However, Tsukida et al
(130) observed significantly
reduced NCOA4 expression in patient-derived cells harboring
WDR45 variants, leading to ferritinophagy dysfunction and
subsequent dysregulation of iron metabolism. The temporal
discrepancy between these studies warrants attention: NCOA4
downregulation observed by Tsukida et al (130) may represent a secondary effect
of chronic lysosomal dysfunction, whereas acute WDR45
knockout in SH-SY5Y models may not yet reach the threshold for
triggering NCOA4 degradation. Further studies are required to
elucidate how WDR45 variants affect ferritinophagy
inpatients exhibiting BPAN.
Insulin-like growth factor binding protein-2
(IGFBP-2) is a regulatory factor in diabetes that primarily
modulates insulin metabolism via the IGF signaling pathway. miRNAs
promote neuroinflammation and neuronal damage in AD by regulating
IGFBP-2 overexpression and neuronal ferritin deposition, thereby
disrupting the balance of the Nrf2/SLC7A11/GPX4 pathway and
exacerbating OS and ferroptosis (131). These findings reveal a
potential mechanistic link between diabetes and AD; IGFBP-2, a key
regulatory factor in diabetes, may also drive AD neurodegeneration
by mediating ferroptosis. This suggests that diabetes-related
insulin resistance and metabolic disturbances may exacerbate
cerebral OS and iron dyshomeostasis through shared molecules, such
as IGFBP-2, thereby increasing the risk of AD. Elucidating this
connection is important for understanding the comorbid mechanisms
of these two diseases.
Although this review synthesizes substantial
evidence linking dysregulated ferritin dynamics to
neurodegenerative pathology, the findings primarily reflect
correlational observations. A key knowledge gap remains regarding
the direction of causality: It remains to be determined whether
alterations in ferritin dynamics, such as aberrant expression or
ferritinophagy, act as primary drivers of neuronal loss or
represent secondary consequences of upstream pathological
processes, including mitochondrial dysfunction and
neuroinflammation. Future studies should prioritize research
designs and analytical methods that strengthen causal inference.
For example, Mendelian randomization analysis, which uses genetic
variants as instrumental variables for iron homeostasis traits like
serum ferritin and iron levels, can help estimate the potential
causal effects of these traits on neurodegenerative disease risk
while minimizing confounding from environmental factors (132). In addition to Mendelian
randomization, future research should adopt longitudinal study
designs to establish the temporality of ferritin dysregulation,
determining whether these changes precede the onset of clinical
symptoms. Investigating the dose-response relationship between
brain iron accumulation and the rate of cognitive or motor decline
will further support a causal link. In addition, evaluating these
associations against established causal frameworks, such as the
Bradford Hill criteria, can provide a structured approach to weigh
the totality of evidence - from the strength and consistency of the
observed associations across different studies to the biological
plausibility and experimental evidence derived from in vitro
and in vivo models of ferroptosis inhibition (133).
Iron dyshomeostasis participates profoundly in the
pathological progression of neurodegenerative diseases, including
PD, AD and ALS. As a core iron-storage protein, ferritin plays a
critical role in the regulation of dynamic iron homeostasis in the
brain. However, significant knowledge gaps persist regarding the
transmembrane transport mechanisms between neurons and glial cells,
as well as their functional transformation within disease
microenvironments. Ferritinophagy, a central pathway for
ferritin-mediated iron release, requires the systematic elucidation
of its molecular regulatory networks and spatiotemporally specific
activation patterns in neurodegenerative pathologies. Ferroptosis,
an iron-dependent, lipid peroxidation-driven form of cell death, is
increasingly implicated in neurodegenerative diseases. Of
particular interest, advances in understanding ferroptosis
regulatory pathways in other diseases, such as T2D and epilepsy,
have provided novel perspectives on the molecular interplay between
cerebral iron dysregulation and neuronal degeneration. From a
clinical translation standpoint, ferritin has potential as a
biomarker for early diagnosis of neurodegenerative diseases,
although its sensitivity and specificity require further validation
through multicenter large-scale cohort studies. In therapeutic
development, ferritin-targeted interventions, such as modulating
its synthesis/degradation balance or blocking pathological iron
release, have shown preliminary efficacy in delaying disease
progression by restoring cerebral iron homeostasis. Nonetheless,
overcoming technical challenges, including blood-brain-barrier
delivery efficiency and cell-specific targeting, remains crucial.
Future studies should integrate multiomics technologies, organoid
models and live imaging approaches to systematically unravel the
spatiotemporal regulatory dynamics of ferritin networks in
neurodegeneration, thereby advancing the application of precision
diagnostics and therapeutic strategies.
Availability of data and materials
Not applicable.
Authors' contributions
WC, HT and RW wrote the manuscript. XC created the
figures and table. YJ supervised the review, revised the
manuscript, obtained financial support, conceptualized the review
and performed the literature search. All the authors have read and
approved the final version of this manuscript. Data authentication
is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
ApoE
|
apolipoprotein E
|
|
Aβ
|
β-amyloid
|
|
ACP
|
aceruloplasminemia
|
|
ACSL4
|
acyl-CoA synthetase long-chain family
member 4
|
|
AD
|
Alzheimer's disease
|
|
ALS
|
amyotrophic lateral sclerosis
|
|
ARE
|
antioxidant response element
|
|
BACH1
|
BTB and CNC homolog 1
|
|
BPAN
|
β-propeller protein-associated
neurodegeneration
|
|
CSF
|
cerebrospinal fluid
|
|
CNS
|
central nervous system
|
|
DMT1
|
divalent metal transporter 1
|
|
FA
|
Friedreich ataxia
|
|
FPN
|
ferroportin
|
|
FTH
|
ferritin heavy chain
|
|
FTL
|
ferritin light chain
|
|
GSH
|
glutathione
|
|
GPX4
|
glutathione peroxidase 4
|
|
HO-1
|
heme oxygenase-1
|
|
Iba1
|
ionized calcium-binding adapter
molecule 1
|
|
IGFBP-2
|
insulin-like growth factor binding
protein-2
|
|
IRP
|
iron regulatory protein
|
|
IRE
|
iron-responsive element
|
|
Keap1
|
Kelch-like ECH-associated protein
1
|
|
LOX
|
lipoxygenase
|
|
LC3
|
light chain 3
|
|
MS
|
multiple sclerosis
|
|
MSA
|
multiple system atrophy
|
|
MT
|
melatonin
|
|
MRI
|
magnetic resonance imaging
|
|
NBIA
|
neurodegeneration with brain iron
accumulation
|
|
NFT
|
neurofibrillary tangle
|
|
Nrf2
|
nuclear factor erythroid 2-related
factor 2
|
|
NCOA4
|
nuclear receptor coactivator 4
|
|
OS
|
oxidative stress
|
|
PM
|
particulate matter
|
|
PBMC
|
peripheral blood mononuclear cell
|
|
PI3K
|
phosphatidylinositol 3-kinase
|
|
p-tau
|
phosphorylated tau
|
|
PSP
|
progressive supranuclear palsy
|
|
PSEN
|
presenilin
|
|
PUFA
|
polyunsaturated fatty acid
|
|
PL
|
phospholipid
|
|
PPARδ
|
peroxisome proliferator-activated
receptor δ
|
|
PCBP
|
poly(rC)-RNA-binding protein
|
|
PD
|
Parkinson's disease
|
|
QSM
|
quantitative susceptibility
mapping
|
|
RLS
|
restless legs syndrome
|
|
ROS
|
reactive oxygen species
|
|
SWI
|
susceptibility weighted imaging
|
|
STEAP3
|
six-transmembrane epithelial antigen
of prostate 3
|
|
T2D
|
type 2 diabetes
|
|
TfR
|
transferrin receptor
|
|
TFEB
|
transcription factor EB
|
|
TDP-43
|
TAR DNA-binding protein 43
|
Acknowledgements
The figures were created using BioGDP.com (134).
Funding
This study was supported by the Natural Science Research Project
of Anhui Educational Committee (grant no. 2024AH050702) and the
Anhui Traditional Chinese Medicine Inheritance and Innovation
Research Project (grant no. 2024cccx288).
References
|
1
|
Billesbolle CB, Azumaya CM, Kretsch RC,
Powers AS, Gonen S, Schneider S, Arvedson T, Dror RO, Cheng Y and
Manglik A: Structure of hepcidin-bound ferroportin reveals iron
homeostatic mechanisms. Nature. 586:807–811. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee J and Hyun DH: The interplay between
intracellular iron homeostasis and neuroinflammation in
neurodegenerative diseases. Antioxidants (Basel). 12:9182023.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wei S, Qiu T, Yao X, Wang N, Jiang L, Jia
X, Tao Y, Wang Z, Pei P, Zhang J, et al: Arsenic induces pancreatic
dysfunction and ferroptosis via mitochondrial
ROS-autophagy-lysosomal pathway. J Hazard Mater. 384:1213902020.
View Article : Google Scholar
|
|
4
|
Bossoni L, Labra-Munoz JA, van der Zant
HSJ, Čaluković V, Lefering A, Egli R and Huber M: In-depth
magnetometry and EPR analysis of the spin structure of human-liver
ferritin: from DC to 9 GHz. Phys Chem Chem Phys. 25:27694–27717.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Arosio P, Elia L and Poli M: Ferritin,
cellular iron storage and regulation. IUBMB Life. 69:414–422. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jin X, Jiang C, Zou Z, Huang H, Li X, Xu S
and Tan R: Ferritinophagy in the etiopathogenic mechanism of
related diseases. J Nutr Biochem. 117:1093392023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jia L, Du Y, Chu L, Zhang Z, Li F, Lyu D,
Li Y, Li Y, Zhu M, Jiao H, et al: Prevalence, risk factors, and
management of dementia and mild cognitive impairment in adults aged
60 years or older in China: a cross-sectional study. Lancet Public
Health. 5:e661–e671. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Oh ES: Dementia. Ann Intern Med.
177:ITC161–ITC176. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Su D, Cui Y, He C, Yin P, Bai R, Zhu J,
Lam JST, Zhang J, Yan R, Zheng X, et al: Projections for prevalence
of Parkinson's disease and its driving factors in 195 countries and
territories to 2050: Modelling study of Global Burden of disease
study 2021. BMJ. 388:e0809522025. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ashraf AA, Aljuhani M, Hubens CJ,
Jeandriens J, Parkes HG, Geraki K, Mahmood A, Herlihy AH and So PW:
Inflammation subsequent to mild iron excess differentially alters
regional brain iron metabolism, oxidation and neuroinflammation
status in mice. Front Aging Neurosci. 16:13933512024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shehjar F, James AW, Mahajan R and Shah
ZA: Inhibition of iron-induced cofilin activation and inflammation
in microglia by a novel cofilin inhibitor. J Neurochem.
169:e162602025. View Article : Google Scholar :
|
|
12
|
Tortuyaux R, Avila-Gutierrez K, Oudart M,
Mazaré N, Mailly P, Deschemin JC, Vaulont S, Escartin C and
Cohen-Salmon M: Physiopathological changes of ferritin mRNA density
and distribution in hippocampal astrocytes in the mouse brain. J
Neurochem. 164:847–857. 2023. View Article : Google Scholar
|
|
13
|
Mukherjee C, Kling T, Russo B, Miebach K,
Kess E, Schifferer M, Pedro LD, Weikert U, Fard MK, Kannaiyan N, et
al: Oligodendrocytes provide antioxidant defense function for
neurons by secreting ferritin heavy chain. Cell Metab. 32:259–272
e10. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baringer SL, Simpson IA and Connor JR:
Brain iron acquisition: An overview of homeostatic regulation and
disease dysregulation. J Neurochem. 165:625–642. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Aisen P: Transferrin receptor 1. Int J
Biochem Cell Biol. 36:2137–2143. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yanatori I, Richardson DR, Toyokuni S and
Kishi F: The new role of poly (rC)-binding proteins as iron
transport chaperones: Proteins that could couple with
inter-organelle interactions to safely traffic iron. Biochim
Biophys Acta Gen Subj. 1864:1296852020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Santos MCFD, Anderson CP, Neschen S,
Zumbrennen-Bullough KB, Romney SJ, Kahle-Stephan M, Rathkolb B,
Gailus-Durner V, Fuchs H, Wolf E, et al: Irp2 regulates insulin
production through iron-mediated Cdkal1-catalyzed tRNA
modification. Nat Commun. 11:2962020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen L, Ma Y, Ma X, Liu L, Jv X, Li A,
Shen Q, Jia W, Qu L, Shi L and Xie J: TFEB regulates cellular
labile iron and prevents ferroptosis in a TfR1-dependent manner.
Free Radic Biol Med. 208:445–457. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee WJ, Lee HG, Hur J, Lee GH, Won JP, Kim
E, Hwang JS and Seo HG: PPARδ activation mitigates 6-OHDA-Induced
neuronal damage by regulating intracellular iron levels.
Antioxidants (Basel). 11:8102022. View Article : Google Scholar
|
|
20
|
Kang T, Han Z, Zhu L and Cao B: TFR1
knockdown alleviates iron overload and mitochondrial dysfunction
during neural differentiation of Alzheimer's disease-derived
induced pluripotent stem cells by interacting with GSK3B. Eur J Med
Res. 29:1012024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhai X, Yan W, Liu S, Tian L, Zhang Y,
Zhao Y, Ni Y, Shen H, Wang J, Wan Z, et al: Silver nanoparticles
induce iron accumulation-associated cognitive impairment via
modulating neuronal ferroptosis. Environ Pollut. 346:1235552024.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bengson EF, Guggisberg CA, Bastian TW,
Georgieff MK and Ryu MS: Quantitative omics analyses of NCOA4
deficiency reveal an integral role of ferritinophagy in iron
homeostasis of hippocampal neuronal HT22 cells. Front Nutr.
10:10548522023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fang Y, Chen X, Tan Q, Zhou H, Xu J and Gu
Q: Inhibiting ferroptosis through disrupting the NCOA4-FTH1
Interaction: A new mechanism of action. ACS Cent Sci. 7:980–989.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hoelzgen F, Nguyen TTP, Klukin E, Boumaiza
M, Srivastava AK, Kim EY, Zalk R, Shahar A, Cohen-Schwartz S,
Meyron-Holtz EG, et al: Structural basis for the intracellular
regulation of ferritin degradation. Nat Commun. 15:38022024.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Miao SH, Sun HB, Ye Y, Yang JJ, Shi YW, Lu
M, Hu G and Zhou JW: Astrocytic JWA expression is essential to
dopaminergic neuron survival in the pathogenesis of Parkinson's
disease. CNS Neurosci Ther. 20:754–762. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao X, Kang Z, Han R, Wang M, Wang Y, Sun
X, Wang C, Zhou J, Cao L and Lu M: JWA binding to NCOA4 alleviates
degeneration in dopaminergic neurons through suppression of
ferritinophagy in Parkinson's disease. Redox Biol. 73:1031902024.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu MR, Zhu WT and Pei DS: System Xc(-): A
key regulatory target of ferroptosis in cancer. Invest New Drugs.
39:1123–1131. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Conrad M and Friedmann Angeli JP:
Glutathione peroxidase 4 (Gpx4) and ferroptosis: What's so special
about it? Mol Cell Oncol. 2:e9950472015. View Article : Google Scholar
|
|
29
|
Chen L, Li X, Liu L, Yu B, Xue Y and Liu
Y: Erastin sensitizes glioblastoma cells to temozolomide by
restraining xCT and cystathionine-ү-lyase function. Oncol Rep.
33:1465–1474. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kuwata H and Hara S: Role of acyl-CoA
synthetase ACSL4 in arachidonic acid metabolism. Prostaglandins
Other Lipid Mediat. 144:1063632019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kagan VE, Mao G, Qu F, Angeli JP, Doll S,
Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al: Oxidized
arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem
Biol. 13:81–90. 2017. View Article : Google Scholar :
|
|
33
|
Suzuki T, Motohashi H and Yamamoto M:
Toward clinical application of the Keap1-Nrf2 pathway. Trends
Pharmacol Sci. 34:340–346. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Katoh Y, Itoh K, Yoshida E, Miyagishi M,
Fukamizu A and Yamamoto M: Two domains of Nrf2 cooperatively bind
CBP, a CREB binding protein, and synergistically activate
transcription. Genes Cells. 6:857–868. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Warnatz HJ, Schmidt D, Manke T, Piccini I,
Sultan M, Borodina T, Balzereit D, Wruck W, Soldatov A, Vingron M,
et al: The BTB and CNC homology 1 (BACH1) target genes are involved
in the oxidative stress response and in control of the cell cycle.
J Biol Chem. 286:23521–23532. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
An X, He J, Xie P, Li C, Xia M, Guo D, Bi
B, Wu G, Xu J, Yu W and Ren Z: The effect of tau K677 lactylation
on ferritinophagy and ferroptosis in Alzheimer's disease. Free
Radic Biol Med. 224:685–706. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zou K, Islam S, Sun Y, Gao Y, Nakamura T,
Komano H, Tomita T and Michikawa M: Presenilin deficiency increases
susceptibility to oxidative damage in fibroblasts. Front Aging
Neurosci. 14:9025252022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pan R, Luo S, Huang Q, Li W, Cai T, Lai K
and Shi X; Alzheimer's Disease Neuroimaging Initiative: The
associations of cerebrospinal fluid ferritin with neurodegeneration
and neuroinflammation along the Alzheimer's disease continuum. J
Alzheimers Dis. 88:1115–1125. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chaudhary S, Ashok A, McDonald D, Wise AS,
Kritikos AE, Rana NA, Harding CV and Singh N: Upregulation of local
hepcidin contributes to iron accumulation in Alzheimer's disease
brains. J Alzheimers Dis. 82:1487–1497. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nemeth E, Rivera S, Gabayan V, Keller C,
Taudorf S, Pedersen BK and Ganz T: IL-6 mediates hypoferremia of
inflammation by inducing the synthesis of the iron regulatory
hormone hepcidin. J Clin Invest. 113:1271–1276. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Raha AA, Biswas A, Henderson J,
Chakraborty S, Holland A, Friedland RP, Mukaetova-Ladinska E, Zaman
S and Raha-Chowdhury R: Interplay of ferritin accumulation and
ferroportin loss in ageing brain: Implication for protein
aggregation in down syndrome dementia, Alzheimer's, and Parkinson's
diseases. Int J Mol Sci. 23:10602022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mayr E, Rotter J, Kuhrt H, Winter K,
Stassart RM, Streit WJ and Bechmann I: Detection of molecular
markers of ferroptosis in human Alzheimer's disease brains. J
Alzheimers Dis. 102:1133–1154. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Streit WJ, Rotter J, Winter K, Muller W,
Khoshbouei H and Bechmann I: Droplet degeneration of hippocampal
and cortical neurons signifies the beginning of neuritic plaque
formation. J Alzheimers Dis. 85:1701–1720. 2022. View Article : Google Scholar
|
|
44
|
Kenkhuis B, Somarakis A, de Haan L,
Dzyubachyk O, Ijsselsteijn ME, de Miranda NFCC, Lelieveldt BPF,
Dijkstra J, van Roon-Mom WMC, Höllt T and van der Weerd L: Iron
loading is a prominent feature of activated microglia in
Alzheimer's disease patients. Acta Neuropathol Commun. 9:272021.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Munoz-Castro C, Noori A, Magdamo CG, Li Z,
Marks JD, Frosch MP, Das S, Hyman BT and Serrano-Pozo A: Cyclic
multiplex fluorescent immunohistochemistry and machine learning
reveal distinct states of astrocytes and microglia in normal aging
and Alzheimer's disease. J Neuroinflammation. 19:302022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kenkhuis B, van Eekeren M, Parfitt DA,
Ariyurek Y, Banerjee P, Priller J, van der Weerd L and van Roon-Mom
WMC: Iron accumulation induces oxidative stress, while depressing
inflammatory polarization in human iPSC-derived microglia. Stem
Cell Reports. 17:1351–1365. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Black E, Rasch A, Wimmer T, Li A, Araujo
A, Cieslak S, Steed KS, Adhikari RD, Wisco JJ and Hutchinson BA:
The effects of age, genotype and diet on hippocampal subfield iron
dysregulation and Alzheimer's disease biomarkers in an ApoE mouse
model. Eur J Neurosci. 57:1033–1047. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Belaidi AA, Masaldan S, Southon A,
Kalinowski P, Acevedo K, Appukuttan AT, Portbury S, Lei P, Agarwal
P, Leurgans SE, et al: Apolipoprotein E potently inhibits
ferroptosis by blocking ferritinophagy. Mol Psychiatry. 29:211–220.
2024. View Article : Google Scholar
|
|
49
|
Ayton S, Janelidze S, Kalinowski P,
Palmqvist S, Belaidi AA, Stomrud E, Roberts A, Roberts B, Hansson O
and Bush AI: CSF ferritin in the clinicopathological progression of
Alzheimer's disease and associations with APOE and inflammation
biomarkers. J Neurol Neurosurg Psychiatry. 94:211–219. 2023.
View Article : Google Scholar
|
|
50
|
Shen QQ, Jv XH, Ma XZ, Li C, Liu L, Jia
WT, Qu L, Chen LL and Xie JX: Cell senescence induced by toxic
interaction between alpha-synuclein and iron precedes nigral
dopaminergic neuron loss in a mouse model of Parkinson's disease.
Acta Pharmacol Sin. 45:268–281. 2024. View Article : Google Scholar
|
|
51
|
Li X, Si W, Li Z, Tian Y, Liu X, Ye S,
Huang Z, Ji Y, Zhao C, Hao X, et al: miR-335 promotes ferroptosis
by targeting ferritin heavy chain 1 in in vivo and in vitro models
of Parkinson's disease. Int J Mol Med. 47:612021. View Article : Google Scholar
|
|
52
|
Lv QK, Tao KX, Yao XY, Pang MZ, Cao BE,
Liu CF and Wang F: Melatonin MT1 receptors regulate the
Sirt1/Nrf2/Ho-1/Gpx4 pathway to prevent α-synuclein-induced
ferroptosis in Parkinson's disease. J Pineal Res. 76:e129482024.
View Article : Google Scholar
|
|
53
|
Martin-Bastida A, Tilley BS, Bansal S,
Gentleman SM, Dexter DT and Ward RJ: Iron and inflammation: In vivo
and post-mortem studies in Parkinson's disease. J Neural Transm
(Vienna). 128:15–25. 2021. View Article : Google Scholar
|
|
54
|
Khan SS, Jaimon E, Lin YE, Nikoloff J,
Tonelli F, Alessi DR and Pfeffer SR: Loss of primary cilia and
dopaminergic neuroprotection in pathogenic LRRK2-driven and
idiopathic Parkinson's disease. Proc Natl Acad Sci USA.
121:e24022061212024. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Mamais A, Kluss JH, Bonet-Ponce L, Landeck
N, Langston RG, Smith N, Beilina A, Kaganovich A, Ghosh MC,
Pellegrini L, et al: Mutations in LRRK2 linked to Parkinson disease
sequester Rab8a to damaged lysosomes and regulate
transferrin-mediated iron uptake in microglia. PLoS Biol.
19:e30014802021. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Friedrich I, Reimann K, Jankuhn S,
Kirilina E, Stieler J, Sonntag M, Meijer J, Weiskopf N, Reinert T,
Arendt T and Morawski M: Cell specific quantitative iron mapping on
brain slices by immuno-μPIXE in healthy elderly and Parkinson's
disease. Acta Neuropathol Commun. 9:472021. View Article : Google Scholar
|
|
57
|
Hu Y, Guo P, Lian TH, Zuo LJ, Yu SY, Liu
L, Jin Z, Yu QJ, Wang RD, Li LX, et al: Clinical characteristics,
iron metabolism and neuroinflammation: New insight into excessive
daytime sleepiness in Parkinson's disease. Neuropsychiatr Dis
Treat. 17:2041–2051. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Maggi G, Barone A, Mastromarino C,
Santangelo G and Vitale C: Prevalence and clinical profile of
patients with restless legs syndrome in Parkinson's disease: A
meta-analysis. Sleep Med. 121:275–286. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Swanson MEV, Mrkela M, Murray HC, Cao MC,
Turner C, Curtis MA, Faull RLM, Walker AK and Scotter EL:
Microglial CD68 and L-ferritin upregulation in response to
phosphorylated-TDP-43 pathology in the amyotrophic lateral
sclerosis brain. Acta Neuropathol Commun. 11:692023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Gao J, Okolo O, Siedlak SL, Friedland RP
and Wang X: Ferritin is closely associated with microglia in
amyotrophic lateral sclerosis. J Neuropathol Exp Neurol.
83:917–926. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Halon-Golabek M, Flis DJ, Zischka H,
Akdogan B, Wieckowski MR, Antosiewicz J and Ziolkowski W:
Amyotrophic lateral sclerosis associated disturbance of iron
metabolism is blunted by swim training-role of AKT signaling
pathway. Biochim Biophys Acta Mol Basis Dis. 1870:1670142024.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wang JC, Ramaswami G and Geschwind DH:
Gene co-expression network analysis in human spinal cord highlights
mechanisms underlying amyotrophic lateral sclerosis susceptibility.
Sci Rep. 11:57482021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Caplan DN, Rapalino O, Karaa A, Rosovsky
RP and Uljon S: Case 35-2020: A 59-year-old woman with type 1
diabetes mellitus and obtundation. N Engl J Med. 383:1974–1983.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Niu L, Zhou Y, Lu L, Su A and Guo X:
Ceruloplasmin deficiency impaired brain iron metabolism and
behavior in mice. Cell Biochem Biophys. 80:385–393. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Vroegindeweij LHP, Bossoni L, Boon AJW,
Wilson JHP, Bulk M, Labra-Muñoz J, Huber M, Webb A, van der Weerd L
and Langendonk JG: Quantification of different iron forms in the
aceruloplasminemia brain to explore iron-related neurodegeneration.
Neuroimage Clin. 30:1026572021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lobbes H, Reynaud Q, Mainbourg S,
Savy-Stortz C, Ropert M, Bardou-Jacquet E and Durupt S: A new
pathogenic missense variant in a consanguineous north-african
family responsible for a highly variable aceruloplasminemia
phenotype: A case-report. Front Neurosci. 16:9063602022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Sanz-Alcazar A, Portillo-Carrasquer M,
Delaspre F, Pazos-Gil M, Tamarit J, Ros J and Cabiscol E:
Deciphering the ferroptosis pathways in dorsal root ganglia of
Friedreich ataxia models. The role of LKB1/AMPK, KEAP1, and GSK3β
in the impairment of the NRF2 response. Redox Biol. 76:1033392024.
View Article : Google Scholar
|
|
68
|
Koeppen AH: Tissue iron in friedreich
ataxia. J Integr Neurosci. 23:42024. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Grander M, Haschka D, Indelicato E,
Kremser C, Amprosi M, Nachbauer W, Henninger B, Stefani A, Högl B,
Fischer C, et al: Genetic determined iron starvation signature in
friedreich's ataxia. Mov Disord. 39:1088–1098. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kurzawa-Akanbi M, Keogh M, Tsefou E,
Ramsay L, Johnson M, Keers S, Wsa Ochieng L, McNair A, Singh P,
Khan A, et al: Neuropathological and biochemical investigation of
Hereditary Ferritinopathy cases with ferritin light chain mutation:
Prominent protein aggregation in the absence of major mitochondrial
or oxidative stress. Neuropathol Appl Neurobiol. 47:26–42. 2021.
View Article : Google Scholar
|
|
71
|
Shahidehpour RK, Nelson PT and Bachstetter
AD: A pathologic study of Perivascular pTDP-43 Lin bodies in
LATE-NC. Acta Neuropathol Commun. 12:1142024. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Abu Bakar ZH, Bellier JP, Yanagisawa D,
Kato T, Mukaisho KI and Tooyama I: LC3/FtMt colocalization patterns
reveal the progression of FtMt accumulation in nigral neurons of
patients with progressive supranuclear palsy. Int J Mol Sci.
23:5372022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Li X, Chu Y, Ma R, Dou M, Li S, Song Y, Lv
Y and Zhu L: Ferroptosis as a mechanism of oligodendrocyte loss and
demyelination in experimental autoimmune encephalomyelitis. J
Neuroimmunol. 373:5779952022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Shukla JJ, Stefanova N, Bush AI, McColl G,
Finkelstein DI and McAllum EJ: Therapeutic potential of iron
modulating drugs in a mouse model of multiple system atrophy.
Neurobiol Dis. 159:1055092021. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Gromadzka G, Wierzbicka D, Litwin T and
Przybylkowski A: Iron metabolism is disturbed and anti-copper
treatment improves but does not normalize iron metabolism in
Wilson's disease. Biometals. 34:407–414. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Xie Z, Wang X, Luo X, Yan J, Zhang J, Sun
R, Luo A and Li S: Activated AMPK mitigates diabetes-related
cognitive dysfunction by inhibiting hippocampal ferroptosis.
Biochem Pharmacol. 207:1153742023. View Article : Google Scholar
|
|
77
|
Zimmer TS, David B, Broekaart DWM,
Schidlowski M, Ruffolo G, Korotkov A, van der Wel NN, van Rijen PC,
Mühlebner A, van Hecke W, et al: Seizure-mediated iron accumulation
and dysregulated iron metabolism after status epilepticus and in
temporal lobe epilepsy. Acta Neuropathol. 142:729–759. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Brosseron F, Maass A, Kleineidam L,
Ravichandran KA, González PG, McManus RM, Ising C, Santarelli F,
Kolbe CC, Häsler LM, et al: Soluble TAM receptors sAXL and sTyro3
predict structural and functional protection in Alzheimer's
disease. Neuron. 110:1009–1022 e4. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ayton S, Janelidze S, Roberts B, Palmqvist
S, Kalinowski P, Diouf I, Belaidi AA, Stomrud E, Bush AI and
Hansson O: Acute phase markers in CSF reveal inflammatory changes
in Alzheimer's disease that intersect with pathology, APOE
epsilon4, sex and age. Prog Neurobiol. 198:1019042021. View Article : Google Scholar
|
|
80
|
Gong L, Sun J and Cong S: Levels of iron
and iron-related proteins in Alzheimer's disease: A systematic
review and meta-analysis. J Trace Elem Med Biol. 80:1273042023.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Brosseron F, Kleemann K, Kolbe CC,
Santarelli F, Castro-Gomez S, Tacik P, Latz E, Jessen F and Heneka
MT: Interrelations of Alzheimer s disease candidate biomarkers
neurogranin, fatty acid-binding protein 3 and ferritin to
neurodegeneration and neuroinflammation. J Neurochem.
157:2210–2224. 2021. View Article : Google Scholar
|
|
82
|
Banerjee G, Forsgard N, Ambler G, Keshavan
A, Paterson RW, Foiani MS, Toombs J, Heslegrave A, Thompson EJ,
Lunn MP, et al: Cerebrospinal fluid metallomics in cerebral amyloid
angiopathy: An exploratory analysis. J Neurol. 269:1470–1475. 2022.
View Article : Google Scholar :
|
|
83
|
Li Y, Duan L, Shi Y, Qi J and Li T:
Diagnostic accuracy of combined measurement of serum homocysteine,
C-Reactive protein, and serum ferritin in mild cognitive
impairments and Alzheimer's disease. J Coll Physicians Surg Pak.
34:1303–1308. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Baldini A, Greco A, Lomi M, Giannelli R,
Canale P, Diana A, Dolciotti C, Del Carratore R and Bongioanni P:
Blood analytes as biomarkers of mechanisms involved in Alzheimer's
disease progression. Int J Mol Sci. 23:132892022. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Traub J, Otto M, Sell R, Göpfert D, Homola
G, Steinacker P, Oeckl P, Morbach C, Frantz S, Pham M, et al: Serum
phosphorylated tau protein 181 and neurofilament light chain in
cognitively impaired heart failure patients. Alzheimers Res Ther.
14:1492022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Jimenez-Jimenez FJ, Alonso-Navarro H,
Garcia-Martin E and Agundez JAG: Biological fluid levels of iron
and iron-related proteins in Parkinson's disease: Review and
meta-analysis. Eur J Neurol. 28:1041–1055. 2021. View Article : Google Scholar
|
|
87
|
Maass F, Michalke B, Willkommen D,
Canaslan S, Schmitz M, Bähr M, Zerr I and Lingor P: Cerebrospinal
fluid iron-ferritin ratio as a potential progression marker for
Parkinson's disease. Mov Disord. 36:2967–2969. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Melek IM, Kus B, Kaptan Z and Petekkaya E:
Correlation of metal ions with specific brain region volumes in
neurodegenerative diseases. Turk J Med Sci. 53:1465–1475. 2023.
View Article : Google Scholar
|
|
89
|
Ayton S, Hall S, Janelidze S, Kalinowski
P, Palmqvist S, Belaidi AA, Roberts B, Roberts A, Stomrud E, Bush
AI and Hansson O: The neuroinflammatory acute phase response in
parkinsonian-related disorders. Mov Disord. 37:993–1003. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Tripathi CB, Gangania M, Kushwaha S and
Agarwal R: Evidence-Based discriminant analysis: A new insight into
iron profile for the diagnosis of Parkinson's disease. Ann Indian
Acad Neurol. 24:234–238. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Barmaki H, Morovati A, Eydivandi Z, Jafari
Naleshkenani F, Saedi S, Musavi H, Abbasi M and Hemmati-Dinarvand
M: The Association between Serum Oxidative Stress Indexes and
Pathogenesis of Parkinson's disease in the northwest of iran. Iran
J Public Health. 50:606–615. 2021.PubMed/NCBI
|
|
92
|
Xu Y, Huang X, Geng X and Wang F:
Meta-analysis of iron metabolism markers levels of Parkinson's
disease patients determined by fluid and MRI measurements. J Trace
Elem Med Biol. 78:1271902023. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Galper J, Balwani M, Fahn S, Waters C,
Krohn L, Gan-Or Z, Dzamko N and Alcalay RN: Cytokines and gaucher
biomarkers in glucocerebrosidase carriers with and without
parkinson disease. Mov Disord. 36:1451–1455. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Spence H, Mengoa-Fleming S, Sneddon AA,
McNeil CJ and Waiter GD: Associations between sex, systemic iron
and inflammatory status and subcortical brain iron. Eur J Neurosci.
60:5069–5085. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Chen H, Wang X, Chang Z, Zhang J and Xie
D: Evidence for genetic causality between iron homeostasis and
Parkinson's disease: A two-sample Mendelian randomization study. J
Trace Elem Med Biol. 84:1274302024. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Thompson AG, Gray E, Verber N, Bobeva Y,
Lombardi V, Shepheard SR, Yildiz O, Feneberg E, Farrimond L,
Dharmadasa T, et al: Multicentre appraisal of amyotrophic lateral
sclerosis biofluid biomarkers shows primacy of blood neurofilament
light chain. Brain Commun. 4:fcac0292022. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Cheng Y, Chen Y and Shang H: Aberrations
of biochemical indicators in amyotrophic lateral sclerosis: A
systematic review and meta-analysis. Transl Neurodegener. 10:32021.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Song C, Chu Z, Dai J, Xie D, Qin T, Xie L,
Zhai Z, Huang S, Xu Y and Sun T: Water extract of moschus
alleviates erastin-induced ferroptosis by regulating the Keap1/Nrf2
pathway in HT22 cells. J Ethnopharmacol. 326:1179372024. View Article : Google Scholar
|
|
99
|
Zhang H, Liu QQ, Ding SK, Li H and Shang
YZ: Flavonoids from stems and leaves of scutellaria baicalensis
georgi improve composited Aβ-Induced Alzheimer's disease model
rats' memory and neuroplasticity disorders. Comb Chem High
Throughput Screen. 26:1519–1532. 2023. View Article : Google Scholar
|
|
100
|
Shamsi A, Shahwan M, Khan MS, Husain FM,
Alhumaydhi FA, Aljohani ASM, Rehman MT, Hassan MI and Islam A:
Elucidating the interaction of human ferritin with quercetin and
naringenin: Implication of natural products in neurodegenerative
diseases: molecular docking and dynamics simulation insight. ACS
Omega. 6:7922–7930. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Shahwan M, Alhumaydhi FA, Sharaf SE,
Alghamdi BS, Baeesa S, Tayeb HO, Ashraf GM and Shamsi A:
Computational insight into the binding of bryostatin 1 with
ferritin: Implication of natural compounds in Alzheimer's disease
therapeutics. J Biomol Struct Dyn. 41:5635–5645. 2023. View Article : Google Scholar
|
|
102
|
Mukem S, Sayoh I, Maungchanburi S and
Thongbuakaew T: Ebselen, iron uptake inhibitor, alleviates iron
overload-induced senescence-like neuronal cells SH-SY5Y via
suppressing the mTORC1 signaling pathway. Adv Pharmacol Pharm Sci.
2023:66413472023.PubMed/NCBI
|
|
103
|
Shahwan M, Khan MS, Husain FM and Shamsi
A: Understanding binding between donepezil and human ferritin:
molecular docking and molecular dynamics simulation approach. J
Biomol Struct Dyn. 40:3871–3879. 2022. View Article : Google Scholar
|
|
104
|
Xia Y, Wang H, Xie Z, Liu ZH and Wang HL:
Inhibition of ferroptosis underlies EGCG mediated protection
against Parkinson's disease in a Drosophila model. Free Radic Biol
Med. 211:63–76. 2024. View Article : Google Scholar
|
|
105
|
Li M, Zhang J, Jiang L, Wang W, Feng X,
Liu M and Yang D: Neuroprotective effects of morroniside from
Cornus officinalis sieb. Et zucc against Parkinson's disease via
inhibiting oxidative stress and ferroptosis. BMC Complement Med
Ther. 23:2182023. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Chen Y, Li YY, Wang S, Zhou T, Chen NH and
Yuan YH: Ginsenoside Rg1 plays a neuroprotective role in regulating
the iron-regulated proteins and against lipid peroxidation in
oligodendrocytes. Neurochem Res. 47:1721–1735. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Zheng Q, Ma P, Yang P, Zhai S, He M, Zhang
X, Tu Q, Jiao L, Ye L, Feng Z and Zhang C: Alpha lipoic acid
ameliorates motor deficits by inhibiting ferroptosis in Parkinson's
disease. Neurosci Lett. 810:1373462023. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Li QM, Wu SZ, Zha XQ, Zang DD, Zhang FY
and Luo JP: Ganoderic acid A mitigates dopaminergic neuron
ferroptosis via inhibiting NCOA4-mediated ferritinophagy in
Parkinson's disease mice. J Ethnopharmacol. 332:1183632024.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Sun Q, Wang Y, Hou L, Li S, Hong JS, Wang
Q and Zhao J: Clozapine-N-oxide protects dopaminergic neurons
against rotenone-induced neurotoxicity by preventing
ferritinophagy-mediated ferroptosis. Free Radic Biol Med.
212:384–402. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Scarpellini C, Klejborowska G, Lanthier C,
Hassannia B, Vanden Berghe T and Augustyns K: Beyond ferrostatin-1:
A comprehensive review of ferroptosis inhibitors. Trends Pharmacol
Sci. 44:902–916. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Gu C, Zhang T, Lv C, Liu Y, Wang Y and
Zhao G: His-mediated reversible self-assembly of ferritin nanocages
through two different switches for encapsulation of cargo
molecules. ACS Nano. 14:17080–17090. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Barolo L, Gigante Y, Mautone L, Ghirga S,
Soloperto A, Giorgi A, Ghirga F, Pitea M, Incocciati A, Mura F, et
al: Ferritin nanocage-enabled detection of pathological tau in
living human retinal cells. Sci Rep. 14:115332024. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Morasso C, Truffi M, Tinelli V,
Stivaktakis P, Di Gerlando R, Francesca D, Perini G, Faisal M, Aid
J, Noridov B, et al: Exploring the anti-inflammatory effects of
curcumin encapsulated within ferritin nanocages: A comprehensive in
vivo and in vitro study in Alzheimer's disease. J
Nanobiotechnology. 22:7182024. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Xia X, Li H, Xu X, Zhao G and Du M:
Facilitating pro-survival mitophagy for alleviating Parkinson's
disease via sequence-targeted lycopene nanodots. ACS Nano.
17:17979–17995. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Strbak O, Balejcikova L, Kmetova M, Gombos
J, Kovac J, Dobrota D and Kopcansky P: Longitudinal and transverse
relaxivity analysis of native ferritin and magnetoferritin at 7 T
MRI. Int J Mol Sci. 22:84872021. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Filo S, Shaharabani R, Bar Hanin D, Adam
M, Ben-David E, Schoffman H, Margalit N, Habib N, Shahar T and
Mezer AA: Non-invasive assessment of normal and impaired iron
homeostasis in the brain. Nat Commun. 14:54672023. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Bossoni L, Hegeman-Kleinn I, van Duinen
SG, Bulk M, Vroegindeweij LHP, Langendonk JG, Hirschler L, Webb A
and van der Weerd L: Off-resonance saturation as an MRI method to
quantify mineral-iron in the post-mortem brain. Magn Reson Med.
87:1276–1288. 2022. View Article : Google Scholar :
|
|
118
|
Brammerloh M, Morawski M, Friedrich I,
Reinert T, Lange C, Pelicon P, Vavpetič P, Jankuhn S, Jäger C,
Alkemade A, et al: Measuring the iron content of dopaminergic
neurons in substantia nigra with MRI relaxometry. Neuroimage.
239:1182552021. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Luyken AK, Lappe C, Viard R, Löhle M,
Kleinlein HR, Kuchcinski G, Langner S, Wenzel AM, Walter M, Weber
MA, et al: High correlation of quantitative susceptibility mapping
and echo intensity measurements of nigral iron overload in
Parkinson's disease. J Neural Transm (Vienna). 132:407–417. 2025.
View Article : Google Scholar :
|
|
120
|
Ying CC, Wang CS, Ren YK, Ding CW, Zhang
YC, Wu J, Yang M, Zhang Y, Mao P, Sheng YJ, et al: Correlation
between substantia nigra hyperechogenicity and iron metabolism in
the postural instability gait difficulty subtype of Parkinson's
disease. Ultrasound Med Biol. 49:2422–2427. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Hu Y, Wu L, Yang SQ, Wei HJ, Wang CY, Kang
X, Jiang JM, Zhang P and Tang XQ: Formaldehyde induces
ferritinophagy to damage hippocampal neuronal cells. Toxicol Ind
Health. 37:685–694. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Tang YH, Wu L, Huang HL, Zhang PP, Zou W,
Tang XQ and Tang YY: Hydrogen sulfide antagonizes
formaldehyde-induced ferroptosis via preventing ferritinophagy by
upregulation of GDF11 in HT22 cells. Toxicology. 491:1535172023.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Xiong Q, Tian X, Xu C, Ma B, Liu W, Sun B,
Ru Q and Shu X: PM(2.5) exposure-induced ferroptosis in neuronal
cells via inhibiting ERK/CREB pathway. Environ Toxicol.
37:2201–2213. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Guo CC, Xu ZM, Lyu Y, Li XH, Li ZF, He H,
Tian FJ and Zheng JP: PM(2.5) induces autophagy-dependent
ferroptosis by endoplasmic reticulum stress in SH-SY5Y cells. J
Appl Toxicol. 43:1013–1025. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Liu ZH, Zhai Y, Xia Y and Liao Q: Mating
modifies oxidative stress in the brain and confers protection
against Parkinson's disease in a Drosophila model. Biochem Biophys
Res Commun. 737:1509112024. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Pino JMV, Nishiduka ES, da Luz MHM, Silva
VF, Antunes HKM, Tashima AK, Guedes PLR, de Souza AAL and Lee KS:
Iron-deficient diet induces distinct protein profile related to
energy metabolism in the striatum and hippocampus of adult rats.
Nutr Neurosci. 25:207–218. 2022. View Article : Google Scholar
|
|
127
|
Chen M, Xu E, Zeng C, Zhu W, Zheng J and
Chen H: High dietary iron has a greater impact on brain iron
homeostasis and cognitive function in old compared with young
C57BL/6J male mice. J Nutr. 151:2835–2842. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Susgun S, Demirel M, Yalcin Cakmakli G,
Salman B, Oguz KK, Elibol B, Ugur Iseri SA and Yapici Z: Targeted
resequencing reveals high-level mosaicism for a novel frameshift
variant in WDR45 associated with beta-propeller protein-associated
neurodegeneration. Int J Neurosci. 134:1040–1045. 2024. View Article : Google Scholar
|
|
129
|
Aring L, Choi EK, Kopera H, Lanigan T,
Iwase S, Klionsky DJ and Seo YA: A neurodegeneration gene, WDR45,
links impaired ferritinophagy to iron accumulation. J Neurochem.
160:356–375. 2022. View Article : Google Scholar :
|
|
130
|
Tsukida K, Muramatsu SI, Osaka H, Yamagata
T and Muramatsu K: WDR45 variants cause ferrous iron loss due to
impaired ferritinophagy associated with nuclear receptor
coactivator 4 and WD repeat domain phosphoinositide interacting
protein 4 reduction. Brain Commun. 4:fcac3042022. View Article : Google Scholar
|
|
131
|
Luo C, Wu G, Xiao Z, Hu R, Qiao M, Li W,
Liu C, Li Z, Lan C and Huang Z: Role of miRNA regulation in IGFBP-2
overexpression and neuronal ferroptosis: Insights into the
Nrf2/SLC7A11/GPX4 pathway in Alzheimer's disease. Int J Biol
Macromol. 287:1385372025. View Article : Google Scholar
|
|
132
|
Sanderson E, Glymour MM, Holmes MV, Kang
H, Morrison J, Munafò MR, Palmer T, Schooling CM, Wallace C, Zhao Q
and Smith GD: Mendelian randomization. Nat Rev Methods Primers.
2:62022. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Fedak KM, Bernal A, Capshaw ZA and Gross
S: Applying the bradford hill criteria in the 21st century: How
data integration has changed causal inference in molecular
epidemiology. Emerg Themes Epidemiol. 12:142015. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Jiang S, Li H, Zhang L, Mu W, Zhang Y,
Chen T, Wu J, Tang H, Zheng S, Liu Y, et al: Generic diagramming
platform (GDP): A comprehensive database of high-quality biomedical
graphics. Nucleic Acids Res. 53:D1670–D1676. 2025. View Article : Google Scholar :
|