Introduction
As public awareness of health issues grows,
traditional Chinese medicine (TCM) has gained increasing
recognition for its unique role in preventive healthcare and
disease treatment. In particular, Chinese herbal formulas have
garnered broad market acceptance due to their proven efficacy and
relatively low side effects (1,2).
Concurrently, active herbal monomers have emerged as a key research
focus within TCM, attracting considerable attention from the
scientific community (1).
Methodological advances have enabled researchers to isolate
high-purity herbal monomers from medicinal herbs with greater
precision, utilizing advanced separation and purification
techniques, thus providing a strong foundation for further
exploration (3,4). In the realm of disease treatment,
herbal monomers have demonstrated notable therapeutic efficacy
(5,6).
Cardiovascular and cerebrovascular diseases (CCVDs)
refer to disorders affecting the heart and brain vasculature,
including ischemic and hemorrhagic conditions in these areas, as
well as in systemic tissues. These conditions are primarily caused
by factors such as hyperlipidemia, blood hyperviscosity,
atherosclerosis (AS) and hypertension (7,8),
posing a major threat to human health (9,10). Although medications and surgical
interventions can alleviate symptoms, they do not support tissue
regeneration or functional recovery (9,10). Consequently, the development of
novel drugs and therapeutic targets for CCVDs is of key importance
(11). The TCM Salvia
miltiorrhiza (Danshen), known for its ability to promote blood
circulation, resolve stasis, dredge collaterals and relieve pain,
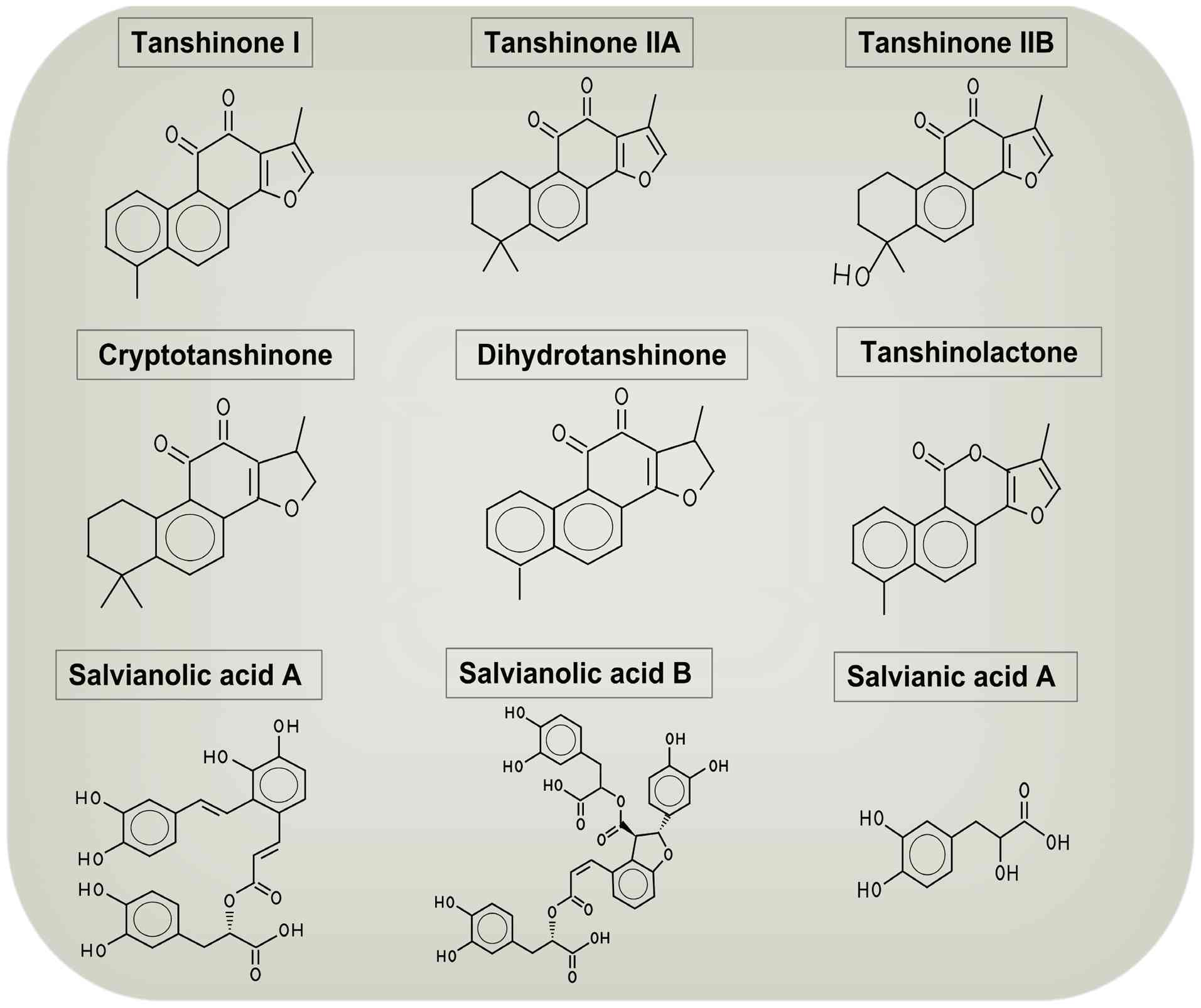
is widely used in the treatment of CCVDs (12-15). Modern research has identified
>40 lipophilic compounds and 50 hydrophilic active components in
Danshen (15-18). The lipophilic components,
primarily tanshinones, include tanshinone I, tanshinone IIA (TSA),
tanshinone IIB, cryptotanshinone and dihydrotanshinone. The
hydrophilic components, such as phenolic acids, include alvianolic
acids A, B and C (16-18) (Fig. 1). Among these, TSA is the most
abundant and pharmacologically active lipophilic compound in
Danshen and has been approved for treating cardiovascular diseases
(19). Extensive studies confirm
that TSA exhibits anti-inflammatory, antioxidant, antitumor,
vasodilatory and neuroprotective effects (20-22). It has demonstrated therapeutic
potential in a range of conditions, including myocardial injury,
pulmonary injury, non-alcoholic fatty liver disease, hepatic
fibrosis, gastritis, glomerulonephritis, diabetes, depression,
Alzheimer's disease and cancer (for example, breast, liver and lung
cancer) (23-29).
Understanding the molecular targets and mechanisms
of action of TCM not only offers scientific explanations for its
role in disease prevention and treatment but also aids in the
identification of novel therapeutic targets and lead compounds. The
present discusses the structure and pharmacological effects of TSA,
with a particular emphasis on recent research advancements
regarding its molecular mechanisms in the treatment of CCVDs.
Additionally, the present review highlights challenges in TSA
research and proposes strategies to overcome them. TSA shows
considerable therapeutic potential, and the present review aims to
provide a comprehensive overview of the current research landscape,
offering valuable insights for future studies. Furthermore, it is
hoped that the present review will contribute to a deeper
understanding of the pathological mechanisms underlying CCVDs and
promote the development of precision medicine.
Chemical structure of TSA
Tanshinone compounds, a class of diterpenoids
(30), include tanshinone I,
TSA, tanshinone IIB, cryptotanshinone, dihydrotanshinone and
tanshinlactone. TSA features a polycyclic aromatic hydrocarbon
skeleton with a phenanthrenequinone group and multiple ring
structures, forming the basis of its biological activity (17,30). Its molecular formula is C19H18O3,
with a molecular weight of 294.33. TSA appears as orange-red
needle-like crystals, soluble in ethanol but poorly soluble in
water, exhibiting strong lipophilicity (30). It melts at 209-210°C and is
sensitive to light, darkening upon exposure to heat or light
(17,31), requiring storage in airtight,
light-protected conditions. Its low water solubility limits its
bioavailability (30,31).
Pharmacological activities and functions of
TSA
TSA demonstrates a wide range of pharmacological
effects, including anti-inflammatory, antioxidant,
anti-mitochondrial apoptosis, anti-allergic, anti-thrombotic,
anti-fibrotic, anti-tumor, immunomodulatory, vasodilatory
(modulating the renin-angiotensin system) and neuroprotective
activities (28,32-39) (Fig. 2). These mechanisms have been
substantiated through various studies.
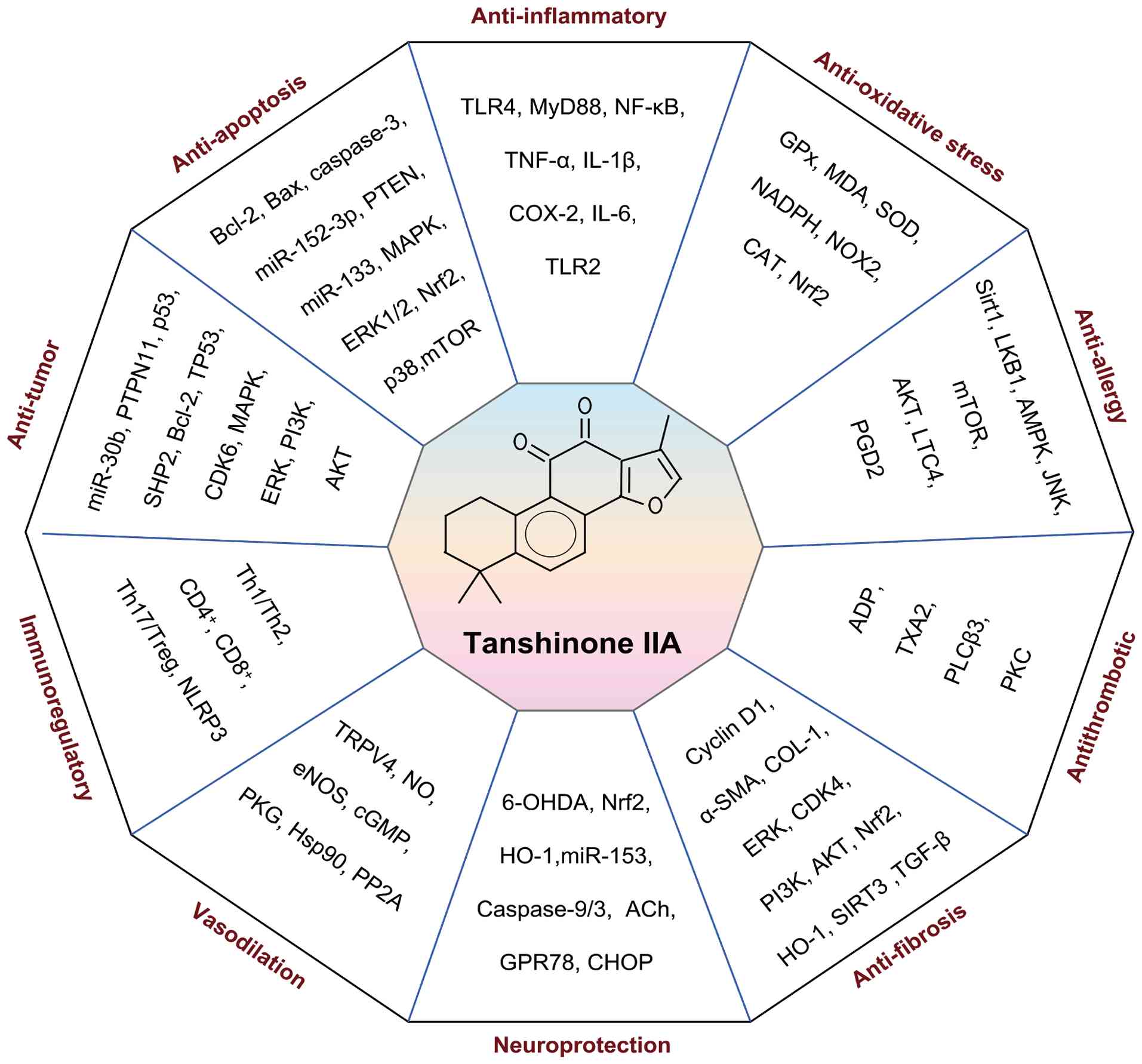 | Figure 2Pharmacological activities and
functions of Tanshinone IIA. Tanshinone IIA exerts multiple
pharmacological effects through modulation of various molecular
pathways, including anti-inflammatory, antioxidant,
anti-mitochondrial apoptosis, antiallergic, antithrombotic,
antifibrotic, antitumor, immunomodulatory, vasodilatory (via
regulation of the RAS system) and neuroprotective activities. ACh,
acetylcholine; ADP, adenosine diphosphate; AKT, protein kinase B;
α-SMA, alpha-smooth muscle actin; AMPK, AMP activated protein
kinase; CAT, catalase; CHOP, CCAAT-enhancer-binding protein
homologous protein (C/EBP homologous protein); CDK4, cyclin,
dependent kinase 4; cGMP, cyclic guanosine monophosphate; COX2,
cyclooxygenase2; COL-1, collagen type 1; eNOS, endothelial nitric
oxide synthase; ERK, extracellular signal-regulated kinase; Gpx,
glutathione peroxidase; JNK, c-Jun N-terminal kinase; GPR78,
glucose-regulated protein 78; HO-1, heme oxygenase 1; Hsp90, heat
shock protein 90; IL-6, interleukin-6; LKB1, liver kinase B1; LTC4,
leukotriene C4; MDA, malondialdehyde; MyD88, myeloid
differentiation primary response gene 88; mTOR, mechanistic target
of rapamycin; NF-κB, nuclear factor kappa-light-chain-enhancer of
activated B cells; NLRP3, NLR family pyrin domain-containing 3;
NOX2, NADPH oxidase 2; Nrf2, nuclear factor erythroid 2-related
factor 2; PI3K, phosphatidylinositol 3 kinase; PGD2, prostaglandin
D2; PKC, protein kinase C; PKG, protein kinase G; PLCβ3,
phosphorylation of phospholipase cβ3; PP2A, protein phosphatase 2A;
PTEN, phosphatase and tensin homolog; PTPN, protein tyrosine
phosphatase non-receptor type; SHP, Src homology 2
domain-containing protein tyrosine phosphatase; SIRT1, sirtuin 1;
SOD, superoxide dismutase; TP53, tumor protein 53; TGF-β,
transforming growth factor-β; TLR4, toll like receptor 4; TNF-α,
tumor necrosis factor-alpha; TRPV4, transient receptor potential
vanilloid 4; TXA2, thromboxane A2; 6-OHDA,6-hydroxydopamine. |
Anti-inflammatory
Inflammatory responses induced by cytokines and
chemokines are implicated in several inflammatory diseases,
highlighting the importance of anti-inflammatory therapies
(40). In lipopolysaccharide
(LPS)-induced RAW264.7 macrophages, TSA markedly inhibited
LPS-induced mRNA expression of pro-inflammatory factors such as
TNF-α, IL-1β and cyclo-oxygenase (COX)-2 (32). Mechanistically, TSA suppressed
inflammatory signaling by downregulating the activation of key
proteins [toll-like receptor (TLR) 4, myeloid differentiation
primary response 88 (MyD88) and NF-κB] in the TLR4-MyD88-NF-κB
pathway (32). A separate study
showed that TSA reduced Propionibacterium acnes-induced expression
of pro-inflammatory factors (IL-1β, IL-8, and TNF-α) and activation
of key proteins [TLR2, NF-κB and intercellular adhesion molecule 1
(ICAM-1)] in the TLR2/NF-κB signaling pathway in THP-1 cells,
suggesting that TSA alleviates inflammation by blocking this
pathway (41). Additionally, TSA
was found to downregulate microRNA-33 expression in THP-1
macrophages, thereby reducing oxidized low-density lipoprotein
(ox-LDL)-induced expression of pro-inflammatory cytokines IL-1β,
IL-6 and TNF-α (42).
Anti-oxidative stress
Oxidative stress is a key factor in the development
of various pathological conditions, disrupting the redox balance
and causing excessive free radical accumulation, which damages
cellular structures and macromolecules (43). TSA upregulates both mRNA
expression and enzymatic activity of glutathione (GSH) peroxidase
(GPx), reducing hydrogen peroxide
(H2O2)-induced apoptosis in J774 macrophages
(33). Furthermore, TSA
decreases intracellular reactive oxygen species (ROS) production,
inhibiting macrophage uptake of ox-LDL and foam cell formation
(44). In transverse aortic
constriction (TAC) model rats, TSA markedly reduced malondialdehyde
(MDA) levels in myocardial tissue and decreased pro-inflammatory
cytokine levels (TNF-α and IL-6), while increasing superoxide
dismutase (SOD) activity, ultimately attenuating pressure
overload-induced cardiac remodeling (45). In LPS-induced brain injury mouse
models, TSA alleviated oxidative stress and inflammatory responses
by reducing serum levels of TNF-α and IL-1β, while enhancing SOD
activity and decreasing MDA content, thus improving pathological
brain damage (46).
Anti-apoptotic effects
Mitochondrial apoptosis, a form of programmed cell
death, is a normal process during development and aging, serving to
maintain homeostasis within tissue cell populations (47). However, dysregulated
mitochondrial apoptosis can accelerate disease progression
(47). TSA exerts anti-cardiac
remodeling effects by activating the sirtuin (SIRT) 1 signaling
pathway, which upregulates the anti-apoptotic protein Bcl-2 while
downregulating pro-apoptotic proteins Bax and Caspase-3 (45). TSA markedly reduces angiotensin
II (AngII)-induced apoptosis in H9C2 rat cardiomyocytes by inducing
microRNA-152-3p expression, which in turn downregulates PTEN and
suppresses cardiomyocyte apoptosis (48). Another study demonstrated that
TSA mitigates H2O2 and doxorubicin
(DOX)-induced apoptosis in H9C2 cardiomyocytes by upregulating
microRNA (miR)-133, leading to the inhibition of Caspase-9
(49). TSA has also been shown
to activate the MAPK ERK1/2 pathway, which upregulates miR-133
expression, thereby protecting neonatal rat cardiomyocytes from
apoptosis under hypoxic conditions (50). Moreover, TSA inhibits oxidative
stress-induced cardiomyocyte apoptosis by binding to Kelch-like
ECH-associated protein 1 (Keap1) and promoting its degradation,
which enhances nuclear factor erythroid 2-related factor 2 (Nrf2)
gene transcription and stimulates Nrf2-driven antioxidant gene
expression. Additionally, TSA ameliorates
H2O2-induced Caspase-3/9 activation and
mitochondrial dysfunction by inhibiting the p38 and mTOR signaling
pathways, ultimately reducing oxidative stress-induced
cardiomyocyte apoptosis (51).
Anti-allergy effects
In allergic and inflammatory diseases, mast cells
play a pivotal role by releasing histamine, leukotrienes,
prostaglandins and various cytokines, which interact
synergistically to exacerbate allergic and inflammatory symptoms
(52). TSA can suppress
FcεRI-mediated mast cell signaling and allergic responses through
activation of Sirt1/LKB1/AMPK pathway (34). The anti-allergic properties of
TSA hold considerable clinical potential, particularly in allergic
rhinitis and allergic asthma, where mast cell activation in the
nasal mucosa and airway inflammation leading to bronchospasm are
key pathological factors (53-57). TSA may alleviate symptoms such as
sneezing, rhinorrhea, nasal congestion and itching by inhibiting
mast cell degranulation and reducing the release of mediators such
as histamine and leukotrienes, thus serving as a potential novel
intranasal spray or oral therapeutic agent (53,57). Alternatively, through its
anti-allergic and anti-inflammatory properties, it may help control
chronic airway inflammation in asthma and reduce acute
exacerbations, acting as a complementary or alternative therapy to
existing inhaled corticosteroids or leukotriene receptor
antagonists (55). However,
further research is still required to explore this potential.
Anti-thrombotic effects
Thrombi present considerable risks to cardiovascular
and cerebrovascular health by causing vascular occlusion, thereby
disrupting blood flow to important organs such as the heart and
brain, which can lead to conditions such as myocardial infarction
and cerebral infarction (58).
TSA has been shown to inhibit ADP (3 μM)-induced reversible
platelet aggregation in rats. Mechanistic studies indicate that TSA
suppresses platelet activation by modulating tubulin acetylation
and inhibiting ERK-2 phosphorylation (35). However, in vivo
experiments reveal that TSA (10 mg/kg) notably prolongs murine
bleeding time by 58% compared with controls (35). In a permanent middle cerebral
artery occlusion rat model, TSA exhibited antiplatelet effects by
inhibiting platelet aggregation, reducing thromboxane A2 release
and downregulating phosphorylation of phospholipase Cβ3 and protein
kinase C (PKC), thereby blocking the PLC/PKC signaling pathway
(59). These results suggest
that TSA holds potential as a therapeutic agent for improving blood
viscosity, microcirculation and preventing CCVDs (59).
Anti-fibrosis effects
TSA also demonstrates antifibrotic properties by
reducing collagen deposition (60). Studies have shown that TSA
downregulates extracellular matrix (ECM) gene transcription and
collagen expression in dermal fibroblasts by inhibiting the
TGF-β/Smad and MAPK/ERK signaling pathways, contributing to its
antifibrotic effects (60,61). One study indicated that TSA
markedly reduced serum levels of alanine aminotransferase (ALT),
aspartate aminotransferase (AST), lactate dehydrogenase (LDH) and
γ-GT in carbon tetrachloride (CCl4)-induced hepatic
fibrosis mice, diminished collagen deposition and downregulated the
expression of fibrosis markers such as α-smooth muscle acting
(α-SMA) and type I collagen, alleviating hepatic fibrosis (62). Mechanistic analysis revealed that
TSA inhibited ERK phosphorylation, reduced cyclin D1 and CDK4
expression, and blocked the formation of cyclin D1-CDK4 complexes,
thereby decreasing Smad3 linker region phosphorylation and
inhibiting hepatic stellate cell (HSC) proliferation, resulting in
cell cycle arrest at the G1 phase (62). Another study demonstrated that
TSA hindered HSC activation by inhibiting KRAS protein and
modulating the PI3K/AKT and Nrf2/heme oxygenase-1 (HO-1) pathways,
reducing collagen deposition and reversing the progression of
CCl4-induced hepatic fibrosis in mice (63). Additionally, TSA upregulated
SIRT3 expression to inhibit TGF-β1 activation and its downstream
target TSP-1, thereby decreasing collagen deposition,
downregulating the fibrosis marker fibronectin and improving
DOX-induced renal fibrosis in mice (36).
Anti-tumor effects
TSA exerts antitumor effects by inhibiting cancer
cell proliferation, inducing apoptosis, arresting cell cycle
progression and suppressing invasion and migration (64-66). Specifically, TSA suppresses HepG2
cell proliferation by modulating the miR-30b-p53-PTPN11/SHP2
pathway (64). TSA also induces
apoptosis in triple-negative breast cancer by downregulating Bcl-2
and upregulating TP53 expression (65). Furthermore, TSA inhibits tumor
cell proliferation, migration and invasion through regulation of
the MAPK/ERK/TRIB3 and PI3K/AKT signaling pathways (28,66). These findings highlight the
antitumor potential of TSA.
Immunomodulation
A study using cecal ligation and puncture
(CLP)-induced septic mice models explored the immunomodulatory
effects of TSA on sepsis-induced immunosuppression (67). TSA at different doses (5, 15 and
45 mg/kg, i.p.) were used at different time-points (0, 3, 6 and 12
h) after treatment with CLP to evaluate its effect on the survival
of septic mice. Results demonstrated that TSA markedly improved
survival rates in a dose- and time-dependent manner (67). Immunologically, TSA reversed
CLP-induced reductions in splenic CD4+ and
CD8+ T lymphocyte counts, mitigated their apoptosis and
suppressed regulatory T cell (Treg) expansion. It also restored
T-helper (Th) 1/Th2 cytokine secretion by increasing IFN-γ and IL-2
levels, while decreasing IL-4 and IL-10 levels (67). Additionally, TSA improved T cell
function by reducing serum levels of high-mobility group box 1
(HMGB1), enhancing macrophage phagocytic activity and promoting
bacterial clearance (67).
Furthermore, TSA alleviated coxsackievirus B3-induced myocardial
inflammation by inhibiting pro-inflammatory Th1 cytokines (IFN-γ
and IL-2) and promoting anti-inflammatory Th2 cytokines (IL-4 and
IL-10), thereby modulating the Th1/Th2 immune balance (37). Another in vitro study
showed that TSA enhanced IL-15-driven NK cell differentiation
through activation of the p38 MAPK phosphorylation pathway
(68). Another study
demonstrated that TSA exerted anti-inflammatory and
cardioprotective effects by inhibiting NLRP3 inflammasome
activation and its downstream effectors (Caspase-1, IL-1β and
IL-18), while modulating Th17/Treg cell balance (69). Collectively, these findings
indicate that TSA improves immune status by regulating immune cell
activity and function, thereby enhancing both anti-infective and
antitumor responses.
Promotion of vasodilation
Endothelial function is vital for maintaining
cardiovascular and cerebrovascular health, with nitric oxide (NO)
released by vascular endothelium playing a key role in promoting
vasodilation (70). TSA markedly
enhances NO production in vascular endothelial cells (ECs)
(71). Research by Wang et
al (38) revealed that TSA
enhances transient receptor potential vanilloid 4 (TRPV4) channel
currents by reducing TRPV4 protein degradation and increasing its
expression, thereby promoting endothelial NO synthase (eNOS)
expression and NO production. NO activates soluble guanylate
cyclase, elevating cGMP and protein kinase G (PKG) levels, which
ultimately leads to vascular smooth muscle relaxation (38). These findings highlight the key
role of the TRPV4-NO-PKG signaling pathway in TSA-induced
vasodilation. A study examining the protective effects of TSA on
diabetes-induced endothelial dysfunction revealed that high glucose
environments reduce eNOS expression and NO production, impairing
vasodilation (72). TSA
ameliorates these abnormalities through multiple
post-transcriptional mechanisms: i) Prolonging eNOS mRNA half-life;
ii) inhibiting eNOS protein degradation and enhancing its
stability; iii) reducing eNOS uncoupling by increasing
tetrahydrobiopterin concentration and Hsp90/eNOS interaction; and
iv) inhibiting the translocation of protein phosphatase 2A (PP2A)
subunit PP2A-A, blocking PP2A-A/eNOS interaction and preventing
eNOS dephosphorylation at serine 1177 (72). This research demonstrates that
TSA enhances the eNOS/NO pathway through multiple mechanisms,
providing novel strategies for treating diabetic cardiovascular
complications.
Neuroprotection
A previous study investigated the protective effects
of TSA on dopaminergic neurons in a 6-hydroxydopamine
(6-OHDA)-induced Parkinson's disease model (73). In vitro experiments
demonstrated that TSA considerably reduced 6-OHDA-induced LDH
release and ROS generation in SH-SY5Y cells, promoted nuclear
translocation of Nrf2 and enhanced the expression of antioxidant
response element (ARE)-regulated genes (for example, HO-1 and
glutamate-cysteine ligase catalytic subunit/glutamate-cysteine
ligase modifier subunit), while inhibiting mitochondrial membrane
potential damage, cytochrome c (cyt c) release and
activation of Caspase-9/3 (73).
In vivo experiments showed that TSA improved rotational
behavior in 6-OHDA-treated rats, reduced dopaminergic neuron loss
in the substantia nigrostriatal pathway and increased striatal
dopamine and its metabolites. The specific molecular mechanism
involved TSA maintaining Nrf2/ARE pathway activity by inhibiting
6-OHDA-induced upregulation of miR-153, which targets the
3'-untranslated region of Nrf2, thereby exerting neuroprotective
effects (73). Another study
demonstrated that TSA protected against hippocampal-dependent
cognitive impairment in diabetic rats by attenuating endoplasmic
reticulum stress-induced apoptosis (74). TSA alleviated oxidative stress by
enhancing SOD activity and reducing ROS and MDA levels, while
inhibiting endoplasmic reticulum stress markers GRP78 and CHOP and
reducing hippocampal neuronal apoptosis (Caspase-3) (74). Additionally, TSA-pretreated
mesenchymal stem cells (TSA-MSCs) notably improved spatial learning
and memory deficits in Alzheimer's disease rats modeled by
unilateral intrahippocampal injection of β-amyloid (25-35) peptide, showing superior efficacy
when compared with untreated MSCs (39). This effect was achieved by
downregulating pro-inflammatory cytokines (IL-1, IL-6 and TNF-α)
and upregulating neurotransmitter the acetylcholine (ACh) levels,
thereby reducing hippocampal neuronal damage (39). These findings collectively
indicate the potent neuroprotective effects of TSA, highlighting
its ability to protect neuronal cells from injury.
Comparative pharmacological
characteristics of major tanshinones
Tanshinone I, salvianolic acid A, TSA and tanshinone
IIB are the core components of Salvia miltiorrhiza, collectively
contributing to its cardiocerebrovascular protective effects
through synergistic actions (Table
I) (13,21,24,75-96). All four components possess
anti-inflammatory and antioxidant properties, improve
microcirculation and inhibit thrombosis, thereby synergistically
protecting the cardiovascular and cerebrovascular systems (13,75-78). However, they differ in their
specific pharmacological profiles and clinical applications.
 | Table IComparison of pharmacological
properties of tanshinone I, salvianolic acid A, tanshinone IIB and
tanshinone IIA. |
Table I
Comparison of pharmacological
properties of tanshinone I, salvianolic acid A, tanshinone IIB and
tanshinone IIA.
| Component | Chemical type | Relative
content | Isolation and
purification | Common
properties | Core
advantages | Applicable
scenarios | (Refs.) |
|---|
| Tanshinone I | Liposoluble
diterpenoid quinone | Medium | Moderate | Anti-inflammatory
and anti-oxidant, improves microcirculation, inhibits thrombosis,
synergistically protects cardiovascular and cerebrovascular
systems | Anti-bacterial-and
anti inflammatory, immunomodulatory, anti-tumor (gastric cancer,
liver cancer) | Cardiovascular
diseases with infection risk, adjuvant tumor therapy | (76,77,79-83) |
| Salvianolic acid
A | Water-soluble
phenolic acid | Very low | Difficult | | Anti-oxidant,
inhibits thrombosis, protects organs (heart, brain, liver,
kidney) | Hyperlipidemia,
fibrosis, tumor prevention | (78-90) |
| Tanshinone IIB | Liposoluble
diterpenoid quinone | Very low | Difficult | | Regulates blood
lipids, neuroprotective, cardioprotective | Hyperlipidemia,
stroke | (13,91-93) |
| Tanshinone IIA | Liposoluble
diterpenoid quinone | Very high | Easy | | Dilates coronary
arteries, anti-myocardial ischemia, inhibits thrombosis | Coronary heart
disease angina pectoris, recovery phase of cerebral infarction,
heart failure | (21,24,75,78,94-96) |
Tanshinone I has a moderate content and is
relatively easy to isolate and purify. It has been shown to inhibit
pathogens such as Staphylococcus aureus and Streptococcus
hemolyticus (79,80) and enhance immune function, aiding
in anti-infection (81). It also
demonstrates inhibitory effects on tumor cells, including those of
gastric and liver types of cancer (82,83). Thus, it is more suitable for
cardiovascular diseases with infection risks and as an adjunct in
tumor therapy. Salvianolic acid A, although hydrophilic, has low
content and is prone to oxidative degradation, limiting its
application (78,84). It scavenges free radicals,
inhibits inflammatory mediators such as TNF-α and IL-6, and
suppresses platelet aggregation, reducing blood viscosity (85-87). It also protects myocardial cells
and liver and kidney functions (88-90). Therefore, it is suitable for
hyperlipidemia, fibrosis and tumor prevention. Tanshinone IIB is
present in very low quantities in Salvia miltiorrhiza, and its
isolation and purification are difficult and expensive, limiting
large-scale research (13).
Existing studies indicate that tanshinone IIB has antioxidant,
lipid-regulating, neuroprotective and cardioprotective effects
(91-93). Therefore, it is suitable for the
treatment of hyperlipidemia and stroke. TSA, with a high content,
is the most abundant lipophilic component in Salvia miltiorrhiza,
making it easy to extract (78).
It dilates coronary arteries, increases myocardial blood flow and
reduces blood viscosity (21,24,94). It also scavenges oxygen free
radicals, alleviates ischemia-reperfusion injury and reduces
myocardial infarction area (95,96). Therefore, it is more suitable for
the treatment of coronary heart disease, angina, the recovery
period of cerebral infarction and HF. In summary, TSA offers unique
advantages in the treatment of CCVDs and is the preferred component
for further development.
The molecular mechanisms of TSA in the
progression of CCVDs
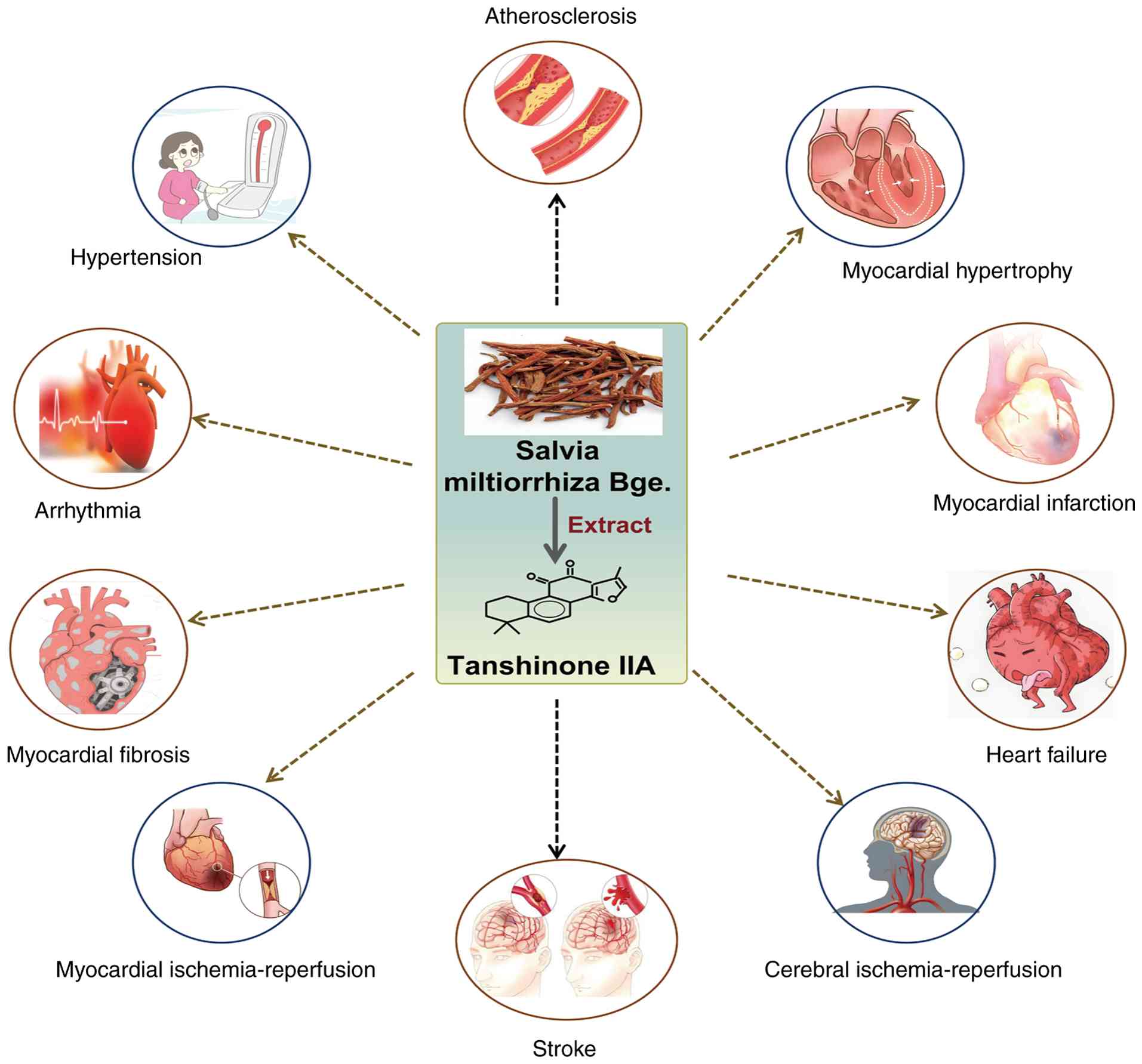
TSA exhibits various pharmacological effects,
including anti-inflammatory, antioxidant, antitumor, vasodilatory
and neuroprotective properties. It carries out a therapeutic role
in multiple CCVDs, such as AS, cardiac hypertrophy, myocardial
infarction, myocardial ischemia-reperfusion, cerebral
ischemia-reperfusion, HF, hypertension, myocardial fibrosis,
arrhythmia and stroke (Fig. 3
and Table II) (42,46,48,49,69, 97-144).
| Table IITSA is involved in alleviating the
development of CCVDs. |
Table II
TSA is involved in alleviating the
development of CCVDs.
| Disease | Mechanism | (Refs.) |
|---|
|
Atherosclerosis | TSA decreases
miR-33 expression, resulting in reduced ox-LDL levels. This leads
to decreased expression of IL-1β, IL-6 and TNF-α, thereby reducing
inflammatory responses. | (42) |
| TSA downregulates
ApoB, which leads to upregulation of SREBP-1. This, in turn,
results in downregulation of MTP, ultimately causing a decrease in
TG levels. | (97) |
| TSA inhibits
phosphorylation of ERK1/2, JNK, p38 and NF-κB signaling pathways.
This suppression reduces expression of pro-inflammatory cytokines
IL-1β, IL-6 and TNF-α, ultimately attenuating inflammatory
responses. | (98) |
| TSA activates PI3K
signaling, leading to increased eNOS expression and enhanced NO
production. Elevated NO levels induce ATF3 expression, which
subsequently downregulates ET-1 expression. | (99) |
| TSA downregulates
METTL3 expression, leading to decreased SIRT5 levels. This
subsequently reduces PERK, CHOP and ATF5 expression, ultimately
attenuating ERS responses. | (100) |
| TSA suppresses
NF-κB activation, resulting in upregulation of LOX-1 receptor
expression. Increased LOX-1 facilitates reduced ox-LDL uptake,
thereby decreasing foam cell formation. TSA reduces miR-375
expression, leading to upregulation of KLF4 transcription
factor. | (101) |
| Increased KLF4
promotes STAT6 activation and M2 polarization of macrophages, while
simultaneously suppressing NF-κB signaling. This results in
decreased expression of IL-6, TNF-α and reduced inflammatory
responses. | (102) |
| TSA increases
miR-13b expression, which downregulates WNT5A signaling. Reduced
WNT5A activity leads to decreased ox-LDL levels and subsequent
downregulation of IL-1β, IL-6 and TNF-α expression, ultimately
attenuating inflammatory responses. | (103) |
| TSA reduces NADPH,
NOX2, MDA, IL-6, TNF-α and MCP-1 expression while increasing SOD
activity, resulting in decreased ROS production. | (104) |
| TSA decreases TG,
TC and LDL levels while increasing HDL expression, leading to
overall lipid homeostasis improvement. | (105) |
| TSA reduces ox-LDL
and MDA levels while increasing Cu/Zn SOD expression, resulting in
decreased reactive oxygen species production. | (106) |
| Hypertension | TSA decreases
Caspase-3 expression while increasing Bcl-2/Bax ratio, resulting in
reduced apoptosis. | (108) |
| TSA reduces MMP2
and TIMP2 expression, leading to decreased fibrosis
progression. | (108,109) |
| TSA reduces NADPH
levels, which subsequently downregulates NOX2, NOX4 and p47phox
expression. This leads to decreased MDA levels and increased SOD
activity, ultimately reducing ROS production. | (110) |
| TSA downregulates
PDK1 expression, resulting in decreased AKT signaling. This
subsequently inhibits smooth muscle cell proliferation. | (111) |
| Myocardial
hypertrophy | TSA reduces Cys-C
levels, leading to decreased Wnt signaling activation. This
suppression reduces β-catenin and WISP-1 expression levels. | (107) |
| TSA activates PI3K
signaling, leading to increased AKT phosphorylation. Enhanced AKT
activity inhibits calcineurin activity, which subsequently reduces
NFATc3 nuclear translocation. This results in decreased expression
of ANP, BNP and β-MHC. | (112) |
| TSA downregulates
ALKBH5 expression, resulting in decreased galectin expression. This
subsequently reduces ANP, BNP and β-MHC levels. | (113) |
| Myocardial
infarction | TSA inhibits
p38MAPK signaling, leading to decreased SRF and MEF2 expression.
This results in reduced miR-1 levels and increased Cx43 expression,
ultimately protecting against myocardial injury. | (114) |
| TSA reduces TNF-α
levels, which suppresses NF-κB activation and subsequently
decreases MCP-1 expression, leading to reduced inflammatory
responses. | (115) |
| TSA increases PGK1
expression, resulting in decreased PDHK1 levels. This promotes M1
to M2 macrophage conversion and subsequently reduces IL-1β, IL-6
and TNF-α expression, attenuating inflammation. | (116) |
| TSA downregulates
TGF-β signaling, leading to decreased Smad3 activation. This
results in increased miR-29b expression and subsequent
downregulation of TGF-β1, Col1α1 and α-SMA, ultimately reducing
fibrosis. | (117) |
| TSA inhibits TLR4
expression, which suppresses NF-κB activation and subsequently
reduces NLRP3 inflammasome formation. This leads to decreased IL-1β
and IL-18 expression, attenuating inflammatory responses. | (118) |
| TSA reduces NOX4,
NADPH and MDA levels while increasing SOD activity, resulting in
decreased ROS production. | (119) |
| Myocardial
ischemia-reperfusion | TSA reduces NLRP3
inflammasome formation, leading to decreased Caspase-1, IL-1β and
IL-18 expression. This results in increased Th17/Treg ratio and
reduced inflammatory responses. | (69) |
| TSA activates PI3K
signaling, leading to increased AKT phosphorylation. Enhanced AKT
activity suppresses NF-κB activation and subsequently reduces IL-6
and TNF-α expression, attenuating inflammatory responses. | (120) |
| TSA activates PI3K
signaling, resulting in increased AKT phosphorylation and mTOR
activation. This leads to decreased MDA, SDH and COX expression
while increasing Bax and Bcl-2 levels, ultimately reducing
oxidation and apoptosis. | (121) |
| TSA decreases LDH
levels while increasing 14-3-3η expression. This promotes Bcl-2
upregulation and mPTP inhibition, subsequently reducing ROS and
cytochrome c levels and decreasing Caspase-3 expression, thereby
suppressing apoptosis. | (122) |
| TSA downregulates
HSA2 expression, resulting in decreased FGF9 levels. This leads to
reduced Bax and Caspase-3 expression, ultimately suppressing
apoptosis. | (123) |
| TSA increases HDAC1
expression, leading to Nrf2 activation and HO-1 upregulation. This
results in decreased Bax/Bcl-2 ratio and reduced TNF-α and IL-1β
expression, while decreasing MDA, Fe2+ and increasing
GSH and Gpx4 levels, ultimately attenuating apoptosis, inflammation
and ferroptosis. | (124) |
| TSA increases ATM
expression, leading to GADD45 upregulation and ORC activation. This
enhances DNA damage repair and DNA biosynthesis processes. | (125) |
| TSA-MSCexo
increases miR-223-5p expression, which downregulates CCR2 and
subsequently reduces inflammatory responses. | (126) |
| TSA combined with
Astragaloside IV suppresses STING pathway activation, leading to
decreased Bax, Caspase-3 and increased Bcl-2 expression, ultimately
reducing apoptosis. | (127) |
| TSA combined with
Astragaloside IV inhibits STING pathway signaling, resulting in
decreased IL-1β, IL-6, TNF-α and iNOS expression, thereby
attenuating inflammatory responses. | (127) |
| TSA combined with
Astragaloside IV suppresses STING pathway activation, leading to
decreased MDA levels and increased GSH and SOD expression,
ultimately reducing reactive oxygen species production. | (127) |
| Heart failure | TSA increases
miR-152-3p expression, resulting in decreased PTEN levels and
reduced apoptosis. | (48) |
| TSA increases
miR-133 expression, leading to decreased Caspase-3 and Caspase-9
expression, ultimately reducing apoptosis. | (49) |
| TSA activates PI3K
signaling, leading to increased AKT phosphorylation. Enhanced AKT
activity results in decreased Caspase-3 expression and increased
Bcl-2 levels, ultimately reducing apoptosis. | (128) |
| TSA increases DAXX,
MEK and ERK1/2 expression while upregulating p38, Caspase-3 and
Caspase-8 expression. This leads to decreased apoptosis through
multiple pathways. | (129) |
| TSA activates AMPK
signaling, resulting in decreased mTOR activity. This leads to
increased LC3, Beclin1 and Bcl-2/Bax ratio while decreasing p62,
Caspase-3 and Caspase-9 expression, ultimately reducing both
autophagy and apoptosis. | (130) |
| Myocardial
fibrosis | TSA upregulates
GPER expression, leading to decreased COL-1 and increased MMP-1
expression. This results in reduced fibrosis progression. | (131) |
| TSA increases GPER
expression, which activates PKA signaling and subsequently
upregulates CREB expression. Enhanced CREB activity promotes
elastin expression while reducing MMP2/9 levels, ultimately
attenuating fibrosis. | (131) |
| TSA increases
miR-618 expression, resulting in decreased α-SMA, Col-1 and TIMP1/4
levels. This leads to reduced fibrosis progression. | (132) |
| TSA reduces NADPH
levels, which subsequently downregulates Colα1, MMP2/9, TIMP1/2,
NOX2 and p67phox expression. This results in decreased fibrosis and
ROS production. | (133) |
| Arrhythmia | TSA reduces SRF
expression, leading to decreased miR-1 levels. This subsequently
downregulates Kir2.1 expression and reduces IK1 current, ultimately
affecting cardiac repolarization. | (134) |
| Cerebral
ischemia-reperfusion | TSA upregulates
Nrf2 expression, leading to increased HO-1 levels. This results in
upregulation of Claudin-5 and ZO-1 expression, ultimately reducing
inflammation and ROS production. | (46) |
| TSA reduces HMGB1
expression, which suppresses NF-κB activation and subsequently
decreases inflammatory and apoptotic responses. | (135) |
| TSA increases
miR-124-5p expression, resulting in decreased FoxO1 levels. This
leads to reduced IL-1β, IL-6 and TNF-α expression, along with
decreased Bax and Caspase-3 while increasing Bcl-2 expression,
ultimately attenuating inflammation and apoptosis. | (136) |
| TSA downregulates
TLR4 expression, resulting in decreased MyD88 levels and subsequent
NF-κB suppression, ultimately reducing inflammatory responses. | (137) |
| TSA reduces TGM2
expression, leading to decreased PANX1 levels and subsequently
attenuating inflammatory responses. | (138) |
| TSA increases
miR-449a expression, leading to decreased ACSL4 levels and reduced
ferroptosis. | (139) |
| Stroke | TSA increases
Bcl-2/Bax ratio, leading to reduced apoptosis. | (140) |
| TSA upregulates
Nrf2 expression, resulting in increased SOD and CAT levels while
decreasing MDA and 8-OHdG expression, ultimately reducing ROS
production. | (141) |
| TSA activates TORC1
signaling, leading to increased pCREB expression and subsequently
upregulating BDNF levels, which enhances neuroprotection. | (142) |
| TSA combined with
Puerarin increases Nrf2 expression, activating ARE signaling and
upregulating HO-1 and NQO1 while decreasing Keap1 levels. This
results in increased T-AOC, CAT, SOD and GSH levels while reducing
GSSG and MDA, ultimately decreasing ROS. | (143) |
| TSA combined with
Puerarin upregulates Nrf2 expression, activating ARE signaling and
increasing HO-1 and NQO1 while decreasing Keap1 levels. This leads
to reduced IL-6, TNF-α, ICAM-1 and COX-2 expression, attenuating
inflammatory responses. | (143) |
| TSA inhibits NF-κB
activation, resulting in decreased NLRP3 inflammasome formation and
subsequently reducing IL-1β and IL-18 expression, ultimately
attenuating inflammatory responses. | (144) |
AS
AS is a disease characterized by lipid accumulation
and inflammation, serving as a notable risk factor for CCVDs
(145). AS is a chronic
immune-inflammatory condition driven by pro-inflammatory molecules
that act on various cell types, including ECs, vascular smooth
muscle cells (VSMCs) and monocytes/macrophages (145). TSA, a bioactive compound with
multifaceted properties, effectively inhibits the progression of AS
through mechanisms such as suppressing adipogenesis, exerting
anti-inflammatory and antioxidant effects (97,98,146), and inhibiting autophagy,
demonstrating notable therapeutic potential in cardiovascular
disease management (Fig. 4).
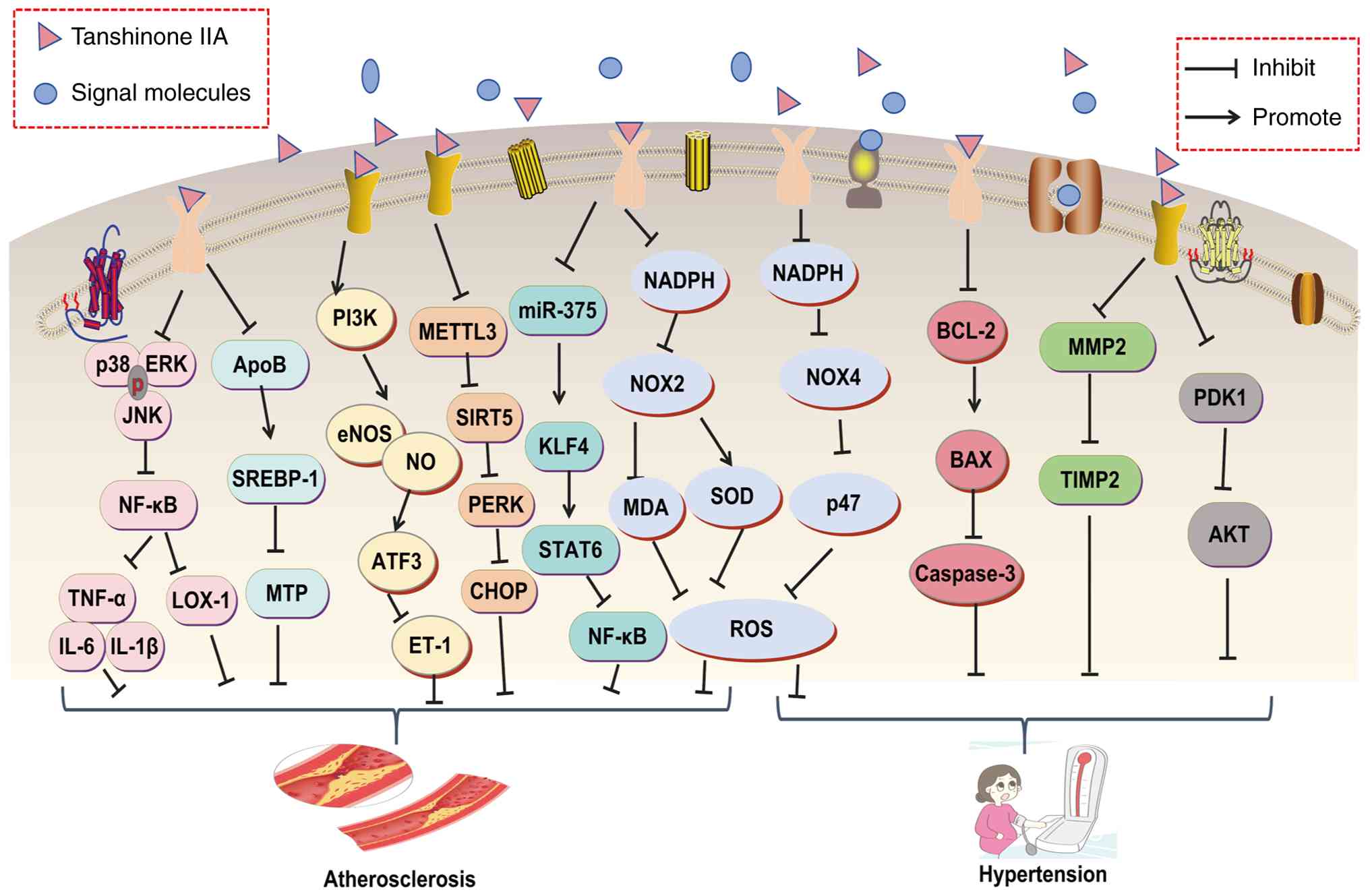 | Figure 4Molecular mechanisms of Tanshinone
IIA in the treatment of atherosclerosis and hypertension. AKT,
protein kinase B; APOB, apolipoprotein B; ATF3, activating
transcription factor 3; eNOS, endothelial nitric oxide synthase;
ET-1, endothelin 1; KLF4, Krüppel-like Factor 4; LOX-1, lectin-like
oxidized LDL receptor 1; MDA, malondialdehyde; MMP2, matrix
metalloproteinase 2; MTP, microsomal triglyceride transfer protein;
NO, nitric oxide; PDK1, pyruvate dehydrogenase Kinase 1; PI3K,
phosphatidylinositol 3 kinase; ROS, reactive oxygen species; SOD,
superoxide dismutase; STAT6, signal transducer and activator of
transcription 6; TIMP2, tissue inhibitor of metalloproteinases 2;
TNF-α, tumor necrosis factor-α; TSA, tanshinone IIA; ERK,
extracellular signal-regulated kinase; JNK, c-Jun N-terminal
kinase; NF-κB, nuclear factor κ-light-chain-enhancer of activated B
cells; IL-6, interleukin-6. |
TSA dose-dependently inhibits the secretion of ApoB
and triglycerides (TGs) in HepG2 cells by downregulating the
transcriptional levels of microsomal triglyceride transfer protein
(MTP), thereby suppressing lipoprotein assembly and reducing ApoB
secretion (97). Additionally,
TSA enhances ApoB degradation via the ubiquitin-proteasome pathway
and promotes nuclear translocation of the transcription factor
sterol regulatory element-binding protein 1, further inhibiting MTP
expression (97). In a study
utilizing a low density lipoprotein receptor knockout mouse model
induced by a high-fat diet and LPS-stimulated RAW264.7 macrophages,
TSA alleviated AS by markedly reducing atherosclerotic plaque area,
lowering serum and hepatic lipid levels (TCs and TGs), improving
liver function (AST and ALT) and decreasing serum and tissue levels
of inflammatory cytokines (IL-1β, IL-6 and TNF-α) (98). Furthermore, TSA inhibited the
phosphorylation of ERK1/2, JNK, p38, NF-κB and p65, thereby
modulating the MAPKs/NF-κB signaling pathway and attenuating
inflammatory responses during AS progression (98). These findings suggest that TSA
holds therapeutic potential in alleviating AS and treating
cardiovascular diseases by inhibiting lipid secretion and
inflammation.
ECs form the interface between vascular walls and
blood, allowing permeability for the exchange of oxygen and
nutrients between blood and tissues (70). ECs carry out a key role in
inflammatory responses, anti-(pro-)thrombotic regulation and
vascular tone modulation, all of which are associated with the
onset and progression of cardiovascular diseases (147). NO, produced by eNOS, is
essential for maintaining cardiovascular homeostasis, and its
dysregulation often leads to endothelial dysfunction and
cardiovascular disorders. TSA markedly enhances NO production in
human vascular ECs, potentially through the activation of eNOS
(71). Research by Hong et
al (99) revealed that TSA
markedly inhibits endothelin-1 (ET-1) mRNA expression and protein
secretion in HUVECs, while increasing NO production and eNOS
phosphorylation. ET-1, a potent vasoconstrictive peptide, carries
out a key role in cardiovascular diseases such as AS, ischemic
heart disease and stroke (148,149). Mechanistic studies suggest that
TSA promotes NO generation by activating the PI3K/eNOS pathway,
upregulating the expression of activating transcription factor
(ATF) 3 and ultimately suppressing ET-1 expression (99). A recent study demonstrated that
TSA protects myocardial ECs by stabilizing mitochondrial membrane
potential, regulating calcium homeostasis and inhibiting
endoplasmic reticulum stress markers (CHOP, PERK and ATF5) through
enhancing the interaction between SIRT5 and methyl transferase-like
3 (100).
Macrophages play a pivotal role in the initiation
and progression of AS (150).
Upon engulfing ox-LDL, macrophages transform into foam cells, a key
event in atherosclerotic plaque formation. The ox-LDL not only
exerts pro-inflammatory effects but also induces macrophages to
release a variety of inflammatory cytokines, thereby exacerbating
vascular inflammation (151).
Lectin-like, oxidized low-density lipoprotein receptor (LOX)-1, a
scavenger receptor expressed in vascular ECs, smooth muscle cells
and macrophages, is responsible for recognizing and internalizing
ox-LDL. It carries out a key role in endothelial dysfunction,
inflammatory responses and foam cell formation during AS (101). Studies by Xu et al
(44) demonstrated that TSA
downregulates LOX-1 expression in macrophages (both the RAW 264.7
cell line and primary mouse peritoneal macrophages) by inhibiting
the NF-κB signaling pathway, thus reducing ox-LDL uptake and foam
cell formation, and ultimately exerting anti-atherosclerotic
effects. In vivo experiments further confirmed that TSA
decreases ROS generation, blocks the nuclear translocation and DNA
binding activity of NF-κB p65 subunit and lowers LOX-1 expression
and lesion area in atherosclerotic plaques of apolipoprotein E
knockout (ApoE−/−) mice (44). This study identified a novel
mechanism through which TSA exerts anti-atherosclerotic effects via
the LOX-1/NF-κB axis.
Macrophage autophagy and polarization also carry out
key roles in AS development (151). Research by Chen et al
(102) showed that TSA markedly
reduces plasma levels of TG, TC and LDL in high-fat diet-fed
ApoE−/− mice, while decreasing aortic plaque area and
lipid deposition. Moreover, TSA promotes M2 macrophage polarization
by increasing CD206 and anti-inflammatory cytokine IL-10/Arg-1
expression and inhibits M1 polarization by reducing CD197 and
pro-inflammatory cytokines (TNF-α/IL-6). TSA also enhances the
expression of autophagy markers, including LC3-II and Beclin1
(102). Mechanistically, TSA
upregulates transcription factor, Krüppel-like factor (KLF4) by
inhibiting miR-375, activating the STAT6/monocyte chemotactic
protein-induced protein pathway to promote M2 polarization, and
simultaneously suppressing the NF-κB pathway to reduce
inflammation. In vitro experiments with ox-LDL-induced
RAW264.7 cells revealed that miR-375 directly targets KLF4. These
findings suggest that TSA regulates macrophage autophagy and
polarization balance through the miR-375/KLF4 axis, offering a
novel therapeutic target for AS (102).
miRNAs have been implicated in the development of
AS. One study demonstrated that TSA inhibits adipogenesis and
inflammatory responses in ox-LDL-induced human monocyte-derived
macrophages (THP-1 cells) (103). The primary mechanism involves
TSA upregulating miR-130b, which suppresses WNT5A protein
expression, thereby reducing ox-LDL-induced lipid accumulation and
decreasing the mRNA levels of the pro-inflammatory cytokines IL-1β,
IL-6 and TNF-α (103). Another
study revealed that TSA attenuates ox-LDL-induced inflammatory
responses in THP-1 macrophages by downregulating miR-33, inhibiting
the secretion of pro-inflammatory cytokines (IL-1β, IL-6 and TNF-α)
and alleviating atherosclerotic inflammation (42). These findings provide novel
mechanistic insights into the potential of TSA as a therapeutic
agent for AS.
Oxidative stress can lead to vascular EC damage,
promoting monocyte adhesion and migration into the subendothelial
space, where they differentiate into macrophages (152). A study by Xu et al
(104) showed that TSA
mitigates high-cholesterol diet-induced atherosclerotic plaque
formation and enhances plaque stability in ApoE−/− mice
by suppressing the NF-κB signaling pathway, thereby reducing
oxidative stress and inflammatory responses. In vitro
experiments confirmed that TSA inhibits ox-LDL-induced ROS
generation in macrophages by downregulating the gp91phox subunit of
NADPH oxidase, as well as reducing pro-inflammatory cytokine
expression (IL-6, TNF-α and MCP-1) and matrix metalloproteinase
(MMP) -9 activity, independent of lipid regulation (104). These findings suggest that TSA
exerts anti-atherosclerotic effects through dual antioxidant and
anti-inflammatory mechanisms. Another study investigated the
anti-inflammatory and antioxidant effects of TSA in an
ovariectomized ApoE−/− mouse model of AS (105). The results showed that TSA
markedly reduced aortic lipid deposition, lowered serum levels of
total cholesterol, TG and LDL, while increasing high-density
lipoprotein levels (105).
Additionally, TSA enhanced SOD activity, reduced MDA content and
suppressed the expression of inflammatory factors such as NF-κB,
AP-1, sICAM-1 and E-selectin in serum. These effects resembled
those of estrogen (17β-estradiol) and were partially inhibited by
an estrogen receptor (ER) antagonist (ICI182780) (105). The study further revealed that
TSA exerts its protective effects by inhibiting p-ERK1/2 protein
expression in the ERK signaling pathway without directly affecting
serum estrogen levels (105).
These results suggest that TSA, as a phytoestrogen, may have
therapeutic potential for cardiovascular diseases in postmenopausal
women by activating ERs and the ERK signaling pathway.
Atherosclerotic calcification (AC), a severe
pathological manifestation of AS, is characterized by abnormal
deposition of calcium salts in the arterial wall, leading to
reduced vascular elasticity, lumen stenosis and increased risk of
cardiovascular and cerebrovascular events (153). A study investigated the
inhibitory effect of TSA on vitamin D2- and high-cholesterol
diet-induced AC in rats, exploring its underlying mechanisms
(106). The research showed
that TSA mitigates AC by reducing serum levels of ox-LDL,
decreasing superoxide anion production and MDA content in blood
vessels, while simultaneously enhancing Cu/Zn SOD activity and its
mRNA and protein expression, thereby suppressing ox-LDL generation.
The study concluded that TSA alleviates AC through antioxidant
stress and upregulation of Cu/Zn SOD expression, providing
experimental support for the application of natural antioxidants in
cardiovascular disease treatment (106).
Hypertension
Hypertension is a prevalent cardiovascular condition
characterized by persistently elevated arterial blood pressure,
which can lead to long-term damage to vital organs such as the
heart, blood vessels and kidneys (154). Vasodilation effectively reduces
blood pressure and protects target organs through several
mechanisms, including ATP-sensitive potassium (KATP) channel
activation, improved endothelial function and modulation of calcium
signaling (154,155). TSA selectively activates KATP
channels, causing membrane hyperpolarization in VSMCs. This process
reduces intracellular calcium ion concentration (Ca2+),
inducing vasodilation and lowering systolic blood pressure (SBP) in
spontaneously hypertensive rats (SHRs). This research identifies
KATP channel activation and calcium signaling regulation as the
primary mechanisms underlying the antihypertensive and vasodilatory
effects of TSA (156). Another
study revealed that TSA inhibits the Cys-C/Wnt signaling pathway,
reducing SBP in SHR while also protecting against myocardial
hypertrophy (107). Myocardial
hypertrophy, a frequent complication of hypertension, increases
cardiac oxygen demand, disrupts diastolic-systolic coordination and
worsens cardiac dysfunction (157). Pang et al (108) demonstrated that TSA mitigates
hypertension-induced left ventricular hypertrophy through multiple
mechanisms, including antioxidant, anti-apoptotic and anti-fibrotic
effects. Specifically, TSA reduces cardiomyocyte apoptosis by
decreasing TUNEL-positive cell proportion, downregulating Caspase-3
activity and lowering the Bax/Bcl-2 ratio (108). It alleviates oxidative stress
by reducing MDA levels and enhancing SOD activity in cardiomyocytes
(108). Additionally, TSA
suppresses cardiac fibrosis by modulating paracrine factors (for
example, decreasing TGF-β1 and increasing bFGF), regulating the
TGF-β/Smads signaling pathway and altering matrix MMP-2 and tissue
inhibitor of metalloproteinase-2 (TIMP2) expression, which reduces
the MMP2/TIMP2 ratio (108).
TSA also regulates the apelin-apelin receptor (APJ) system by
increasing plasma apelin levels while downregulating APJ expression
(108).
TSA improves myocardial contractility and reduces
ECM deposition, without notably affecting blood pressure (108). Myocardial fibrosis, a common
complication of hypertension, can also be alleviated by TSA
(158). Fang et al
(109) demonstrated that TSA,
while not notably lowering blood pressure, effectively alleviated
cardiac interstitial fibrosis and improved cardiac function in
renovascular hypertensive rats (2K2C model). Its mechanism likely
involves modulating the transcriptional balance of MMPs/TIMPs by
inhibiting MMP-2/9 and TIMP-2 (109). Supporting this, Wang et
al (110) found that TSA
notably improved cardiac function in two-kidney, two-clip (2K2C)
hypertensive rats [reducing left ventricular end-diastolic pressure
and increasing ejection fraction and left ventricular fractional
shortening (LVFS)], while attenuating myocardial hypertrophy and
fibrosis, independent of its antihypertensive effect (no notable
impact on blood pressure) (110). TSA achieved these effects by
inhibiting NADPH oxidase (Nox) activity and downregulating the
expression of its subunits (Nox2, Nox4 and p47phox), reducing
superoxide anion (O2−) generation in the
heart and aorta. This ultimately ameliorated cardiac remodeling
through its antioxidant properties (110). These findings suggest a novel
therapeutic strategy for managing hypertension-related myocardial
pathology.
Excessive proliferation of VSMCs leads to arterial
wall thickening and lumen narrowing, increasing peripheral vascular
resistance and cardiac afterload. Long-term consequences may
include cardiac hypertrophy and HF (159,160). This process also disrupts
vascular tone regulation, destabilizes hemodynamics, exacerbates
blood pressure fluctuations and creates a vicious cycle that
elevates the risk of cardiovascular events (159,160). A study demonstrated that TSA at
doses of 0.5/5 mg/kg administered for 2 weeks markedly reduced
blood pressure in hypertensive rats (2K1C model) and inhibited
ET-1-induced proliferation of basilar artery smooth muscle cells,
though without affecting PI3K phosphorylation (111). The primary mechanism involves
TSA targeting phosphoinositide-dependent kinase-1 (PDK1) to
suppress the PI3K/PDK1/AKT pathway, inhibiting arterial smooth
muscle cell proliferation and alleviating hypertension-associated
cerebrovascular remodeling (111). As a multi-targeted agent, TSA
demonstrates promising potential in mitigating cardiac fibrosis,
suppressing arterial smooth muscle cell proliferation and improving
cardiac function. These findings offer valuable insights into the
treatment of hypertension-related myocardial pathology (Fig. 4).
Myocardial hypertrophy
Myocardial hypertrophy is a complex adaptive
response characterized by increased cardiomyocyte volume and
ventricular wall thickening, with both physiological and
pathological manifestations (161). Pathological myocardial
hypertrophy not only impairs diastolic and systolic cardiac
function but also predisposes individuals to severe complications
such as arrhythmias and HF, presenting notable health risks
(161). In an earlier study, a
rat model of myocardial hypertrophy was established by constricting
the thoracic aorta. Compared with the model group, treatment groups
showed marked reductions in heart mass index, left ventricular mass
index, left ventricular posterior wall thickness, interventricular
septal thickness and myocardial fiber diameter as observed in
H&E staining. The primary molecular mechanism underlying these
effects involved TSA inhibiting the AKT signaling pathway, thus
preventing myocardial hypertrophy (162). Another study explored the
protective effects of TSA against isoproterenol (ISO)-induced
myocardial hypertrophy and its underlying mechanisms (163). TSA markedly inhibited
ISO-induced increases in cardiomyocyte surface area and
downregulated mRNA and protein expression of hypertrophy markers
such as atrial natriuretic peptide (ANP), brain natriuretic peptide
(BNP) and β-myosin heavy chain (β-MHC). Additionally, TSA exerted
anti-hypertrophic effects by suppressing intracellular calcium
transients and the calcineurin/nuclear factor of activated T cells
(Calcineurin/NFATc3) signaling pathway (163). This research provided new
pharmacological evidence supporting TSA as a treatment for
pathological myocardial hypertrophy. Further supporting evidence
came from Weng et al (112), who found that TSA enhanced the
activation of the PI3K/AKT signaling pathway by binding to ER,
inhibiting Leu27IGF-II-induced calcineurin activation and
preventing NFATc3 nuclear translocation, ultimately alleviating
cardiomyocyte hypertrophy (112). Moreover, TSA reduced cardiac
hypertrophy in SHRs by inhibiting the Cys-C/Wnt signaling pathway,
decreasing protein expression of Cys-C, Wnt2, β-catenin and WISP-1
(107). Zhang et al
(113) discovered that TSA
alleviated myocardial hypertrophy through m6A modification of
galectin-3. Using both an Ang II-induced in vitro
cardiomyocyte hypertrophy model and a TAC-induced in vivo
cardiac hypertrophy model, they observed that TSA markedly
inhibited cardiomyocyte hypertrophy and downregulated hypertrophy
markers such as ANP, BNP and β-MHC. TSA reduced galectin-3 mRNA
stability and protein expression by inhibiting the RNA demethylase
ALKBH5, thereby enhancing m6A modification of galectin-3 mRNA
(113). This study revealed a
novel mechanism through which TSA regulates galectin-3 expression
via ALKBH5-mediated m6A modification, providing a potential
therapeutic target for cardiac hypertrophy (Fig. 5).
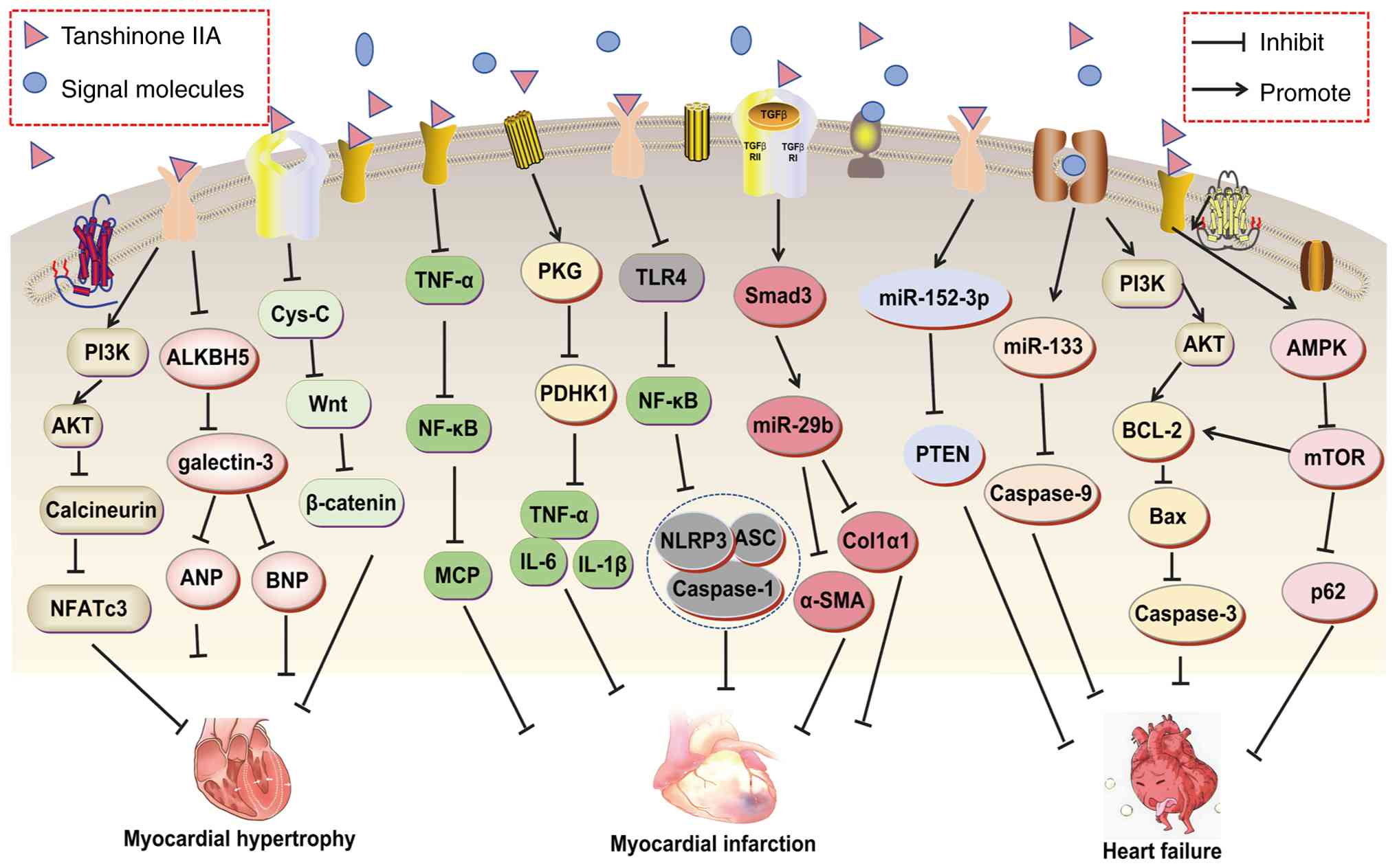 | Figure 5Molecular mechanisms of Tanshinone
IIA in the treatment of myocardial hypertrophy, myocardial
infarction, and heart failure. α-SMA, α-smooth muscle actin; AKT,
protein kinase B; ALKBH5, AlkB Homolog 5; ASC, Apoptosis-associated
speck-like protein containing a CARD; AMPK, AMP activated protein
kinase; ANP, atrial natriuretic peptide; BNP, brain natriuretic
peptide; COL-1, collagen type 1; Cys-C, cystatin c; IL-6,
interleukin-6; mTOR, mechanistic target of rapamycin; NF-κB,
nuclear factor kappa-light-chain-enhancer of activated B cells;
NLRP3, NLR family pyrin domain-containing 3; PDHK1, pyruvate
dehydrogenase kinase 1; PI3K, phosphatidylinositol 3 kinase; PKG,
protein kinase G; PTEN, phosphatase and tensin homolog; TLR4, toll
like receptor 4; TNF-α, tumor necrosis factor-α; NF-κB, nuclear
factor κ-light-chain-enhancer of activated B cells. |
Myocardial infarction
Myocardial infarction is a severe cardiovascular
condition that causes extensive cardiomyocyte necrosis, impairs
cardiac function and can lead to HF (164). Ischemia and hypoxia are central
to the pathophysiology of myocardial infarction. Acute coronary
artery occlusion induces regional myocardial ischemia and hypoxia,
triggering complex intracellular biochemical reactions in
cardiomyocytes, leading to energy metabolic dysfunction, calcium
ion overload, and ultimately cardiomyocyte injury and death
(164). In a study by Zhang
et al (114), a rat
myocardial infarction model (ligation of the left anterior
descending coronary artery; LAD) and an in vitro hypoxic
cardiomyocyte culture system were used to demonstrate that TSA
protects cardiomyocytes from ischemic and hypoxic injury. TSA
achieves this by inhibiting p38 MAPK phosphorylation,
downregulating the expression of cardiac-specific transcription
factors serum response factor (SRF) and myocyte enhancer factor 2,
reducing excessive miR-1 expression and restoring its target
protein, connexin-43 levels (114).
Ischemia and hypoxia activate inflammatory
signaling pathways in the body, promoting the infiltration of
inflammatory cells into the infarcted area and releasing large
amounts of inflammatory cytokines, which exacerbate myocardial
injury and contribute to the development of HF (165). A study by Ren et al
(115) investigated the
inhibitory effect of TSA on post-myocardial infarction inflammatory
responses and its underlying mechanisms. Using an in vivo
rat myocardial infarction model (LAD ligation), they found that TSA
notably improved cardiac function, reduced infarct size and
collagen deposition, and downregulated the expression of MCP-1 and
TGF-β1, while also reducing macrophage infiltration (115). In vitro experiments
demonstrated that TSA inhibited MCP-1 and TGF-β1 secretion in
TNF-α-stimulated rat primary cardiac fibroblasts (CFs) but had no
notable effect on rat primary cardiomyocytes. The study suggested
that the cardioprotective effects of TSA might be achieved by
suppressing TNF-α/NF-κB signaling-mediated inflammatory responses,
with CFs as the primary target (115). Another study using a mouse
myocardial infarction model revealed that TSA improved cardiac
function, reduced inflammatory cell infiltration and fibrosis, and
decreased pro-inflammatory cytokine expression (for example IL-1β,
TNF-α or IL-6) and increased anti-inflammatory IL-10 secretion.
Additionally, TSA promoted M2 macrophage polarization and inhibited
M1 polarization (116).
Mechanistically, TSA targeted PGK1 (a key glycolytic enzyme) to
inhibit its binding to mitochondria, reducing PDHK1 (pyruvate
dehydrogenase kinase 1) activity, restoring mitochondrial function
and reprogramming macrophage metabolism. This reprogramming shifted
macrophage metabolism from glycolysis to oxidative phosphorylation,
driving a shift from M1 pro-inflammatory to M2 anti-inflammatory
phenotypes (116). This study
identified the PGK1-PDHK1 axis as a novel target of TSA in
regulating the immune microenvironment through metabolic
reprogramming, offering a new innovative energy metabolism-based
strategy for anti-inflammatory therapy in myocardial
infarction.
After myocardial infarction, cardiomyocyte necrosis
triggers a repair process in which fibroblasts proliferate and
produce ECM, leading to scar formation (166). This excessive fibrosis alters
cardiac structure, including ventricular wall thickening and
chamber dilation, impairing both systolic and diastolic function
(166). Therefore, myocardial
infarction is associated with fibrosis, a key factor in cardiac
dysfunction. Studying this relationship is essential for developing
therapeutic strategies. A study by Yang et al (117), using a rat acute myocardial
infarction model and in vitro CF experiments, demonstrated
that medium-to-high doses of TSA notably improved left ventricular
function, reduced collagen deposition and downregulated TGF-β1,
Col1a1, Col3a1 and α-SMA expression while upregulating miR-29b
(117). Further experiments
showed that miR-29b inhibition prevented the antifibrotic effects
of TSA, whereas Smad3 small interfering (si)RNA treatment
suppressed miR-29b expression, confirming that TSA exerts its
antifibrotic effects by upregulating miR-29b via the TGF-β-Smad3
signaling pathway (117). To
the best of our knowledge, this study is the first to identify
miR-29b as a direct effector molecule mediating the antifibrotic
effects of TSA, unveiling a novel pathway through which TCM active
components target miRNAs to modulate cardiac remodeling. It also
offers a potential therapeutic strategy for post-myocardial
infarction fibrosis based on the TGF-β/miR-29b axis (117).
Ischemia-hypoxia injury triggers a complex
pathophysiological cascade, with pyroptosis emerging as a form of
programmed cell death (167).
Pyroptosis involves cellular swelling followed by rupture,
resulting in the release of cellular contents that initiate
inflammatory responses (168).
In myocardial infarction, pyroptosis contributes to cardiomyocyte
loss, activates immune cells and exacerbates inflammation and
fibrosis (168). In a study
using a rat HF model and an H9C2 cardiomyocyte
hypoxia-reoxygenation model, TSA notably improved cardiac function
[increasing left ventricular ejection fraction (LVEF) and LVFS],
reduced serum NT-pro-BNP, IL-1β and IL-18 levels, and suppressed
NLRP3 inflammasome activation by inhibiting the TLR4/NF-κB p65
pathway (118). This led to
decreased Caspase-1-dependent GSDMD-N cleavage and IL-1β/IL-18
maturation, alleviating myocardial inflammation and pyroptosis
(118). This study is the first
to demonstrate that TSA inhibits cardiomyocyte pyroptosis by
targeting the TLR4/NF-κB p65 axis, offering a novel therapeutic
target for TSA-mediated regulation of programmed cell death in
post-myocardial infarction HF.
Myocardial infarction is a leading cause of HF,
with post-myocardial infarction inflammatory responses, oxidative
stress, fibrotic processes and neuroendocrine activation playing
key roles in the progression of HF (169). Chen et al (119) established an myocardial
infarction rat model by ligating the LAD and found that TSA
treatment prevented myocardial infarction-induced declines in
cardiac function, including LVEF and LVFS, while alleviating
structural abnormalities such as left ventricular end-diastolic
volume (LVEDV) and left ventricular end-systolic volume (LVESV).
TSA also suppressed the upregulation of collagen I, collagen III,
TGF-β, α-SMA, MMP2 and MMP9 in myocardial infarction rat hearts and
mitigated Ang II-induced CF fibrosis. Additionally, TSA reduced
oxidative stress by enhancing SOD activity and decreasing MDA,
superoxide anion levels and Nox4 activity (119). Further experiments revealed
that Nox4 overexpression attenuated TSA-mediated improvements in
cardiac function and fibrosis in HF rats, suggesting that Nox4
critically regulates the inhibitory effects of TSA on HF and
cardiac fibrosis (119). This
study is the first to demonstrate that TSA ameliorates
post-myocardial infarction HF-related cardiac remodeling by
targeting Nox4-dependent oxidative stress pathways (119).
A study by Zhu et al (170) found that TSA exerts
cardioprotective effects by modulating the gut-brain axis following
myocardial infarction. Their research demonstrated that TSA
markedly improved cardiac function in mice after MI (increasing
LVEF and LVFS, decreasing LVEDV and LVESV), alleviated myocardial
hypertrophy, fibrosis and cardiomyocyte apoptosis, and suppressed
inflammatory responses in cardiac tissue (reducing TNF-α, IL-1β and
p-NF-κB p65 expression) (170).
Additionally, TSA improved myocardial infarction-induced intestinal
pathological damage, including increasing villus length, reducing
intestinal edema and fibrosis, and enhancing intestinal barrier
integrity (upregulating ZO-1, Occludin and Cingulin expression).
16S rDNA sequencing revealed that TSA reshapes the gut microbiota,
increasing α-diversity (Chao1 and Shannon indices) and altering
β-diversity. Furthermore, TSA reduced serum levels of LPS,
inhibited microglial activation and neuroinflammation in the
paraventricular nucleus (PVN; reducing IL-6 and TNF-α expression)
and decreased sympathetic nervous system activity (reducing
tyrosine hydroxylase [TH] and norepinephrine [NE] levels) (170). Consequently, it was concluded
that TSA alleviates myocardial injury by improving gut microbiota
and barrier function, reducing LPS release into the bloodstream,
thereby inhibiting PVN neuroinflammation and sympathetic nervous
system overactivation (170).
However, the study did not definitively conclude whether the
cardioprotective effects of TSA are independent of the gut
microbiota, as it did not use antibiotics to deplete the microbiota
or fecal microbiota transplantation to validate causality, which
remains to be explored in future research (Fig. 5).
Myocardial ischemia-reperfusion
Myocardial ischemia-reperfusion is a common
clinical condition characterized by the restoration of blood flow
to the myocardium after a period of ischemia (171). During this process, myocardial
cells undergo pathophysiological changes that cause notable damage.
Upon reperfusion, excessive oxygen-free radicals trigger oxidative
stress, damaging myocardial cell membranes, proteins and nucleic
acids, which disrupts cellular structure and function (171). Simultaneously,
ischemia-reperfusion activates inflammatory responses, leading to
infiltration of inflammatory cells into myocardial tissue and the
release of inflammatory cytokines, further exacerbating myocardial
injury and increasing cell necrosis and apoptosis (171). TSA mitigates myocardial
ischemia-reperfusion injury (MIRI) through multiple mechanisms,
including antioxidant effects (elevating SOD and reducing MDA),
anti-inflammatory actions (inhibiting TNF-α and IL-6),
anti-apoptotic effects (regulating Bcl-2/Bax and Caspase-3) and
improving energy metabolism (enhancing ATP production) (172,173).
One study found that TSA notably reduced the
myocardial infarction area, improved left ventricular ejection
fraction and decreased cardiomyocyte apoptosis in
streptozotocin-induced diabetic rats. Additionally, TSA enhanced
AKT phosphorylation, inhibited NF-κB phosphorylation and
downregulated inflammatory cytokine expression (TNF-α and IL-6),
thereby alleviating MIRI in rats (120). These results suggested that TSA
exerts anti-apoptotic, anti-inflammatory and cardioprotective
effects by activating the PI3K/AKT-dependent pathway (120).
Another study demonstrated that TSA notably reduced
the myocardial infarction area, serum CK-MB and LDH levels in a rat
model of myocardial ischemia-reperfusion induced by left anterior
descending coronary artery ligation. TSA also decreased
mitochondrial oxidative stress markers (MDA and
H2O2) while increasing SOD activity and
SDH/COX levels, and inhibiting cardiomyocyte apoptosis (increased
Bcl-2 expression and decreased Bax expression) (121). Mechanistic studies revealed
that TSA exerts its protective effects by upregulating PI3K,
p-AKT/AKT, p-mTOR/mTOR and p-eNOS/eNOS expression, suggesting that
TSA activates the PI3K/AKT/mTOR pathway to mitigate oxidative
stress and apoptosis, thus protecting against MIRI (121). Furthermore, Li et al
(69) demonstrated through in
vivo experiments that TSA reduced myocardial infarction area,
improved cardiac contractile function and decreased serum levels of
myocardial injury markers such as LDH and creatine kinase in a rat
model of MIRI. Additionally, TSA exerted anti-inflammatory and
cardioprotective effects by inhibiting the NLRP3 inflammasome and
its downstream targets (Caspase-1, IL-1β and IL-18), while
modulating Th17/Treg cell balance (69).
Myocardial ischemia leads to hypoxia in cardiac
tissue, and after reperfusion, the myocardium regains oxygen,
entering the reoxygenation phase. However, reperfusion and
reoxygenation induce notable changes in cellular redox status and
trigger inflammatory responses (174). A study demonstrated that TSA
pretreatment (8 μM for 48 h) notably enhanced the survival
rate of H9c2 cells subjected to hypoxia/reoxygenation (A/R) injury,
reduced LDH activity and upregulated 14-3-3η protein expression
(122). This promoted the
translocation of Bcl-2 to the outer mitochondrial membrane, where
it interacted with voltage-dependent anion channel 1, inhibiting
mitochondrial permeability transition pore (mPTP) opening. As a
result, TSA reduced ROS generation and cyt c release,
ultimately suppressing Caspase-3 activation and apoptosis (122). The protective effect of TSA was
comparable to ischemic preconditioning, suggesting that TSA may
serve as a nutritional preconditioning strategy to mitigate MIRI
(122). TSA can also inhibit
the expression of the HAS2/FGF9 axis, alleviating hypoxia-induced
apoptosis in AC16 cardiomyocytes (reducing Cleaved Caspase-3 and
Bax expression), inflammation (reducing IL-6, IL-1β and TNF-α) and
oxidative damage (reducing MDA levels) (123). Yan et al (124) established an in vitro
oxygen-glucose deprivation/reoxygenation (OGD/R) cardiomyocyte
model and an in vivo rat myocardial ischemia-reperfusion
model. Their results showed that TSA pretreatment downregulated
histone deacetylase 1 (HDAC1) expression, thereby promoting nuclear
translocation of Nrf2 and upregulating antioxidant proteins,
including HO-1, cystine transporter xCT and GSH peroxidase 4
(Gpx4). This led to reduced cardiomyocyte apoptosis (decreased
Bax/Bcl-2 ratio), lower inflammatory cytokine release (TNF-α and
IL-1β) and inhibition of ferroptosis, as evidenced by reduced MDA
and Fe2+ levels, along with increased GSH levels
(124). These findings suggest
that TSA exerts anti-inflammatory, anti-apoptotic and
anti-ferroptotic effects by activating the Nrf2-xCT/Gpx4/HO-1 axis,
which is otherwise suppressed by HDAC1, thereby ameliorating MIRI
(124). Additionally, TSA
pretreatment markedly improved the survival rate of H9c2
cardiomyocytes in an A/R model, reduced LDH activity, MDA levels,
total iron content and ROS levels, while upregulating GSH and Gpx4
expression. TSA also inhibited VDAC1-mediated mPTP opening and
apoptosis (96). This study
revealed that TSA exerts dual cardioprotective mechanisms by
targeting VDAC1 to simultaneously inhibit ferroptosis and
apoptosis, providing novel molecular targets and a theoretical
basis for using TSA, an active component of Salvia miltiorrhiza, in
treating ischemic heart disease (96). The study also found that TSA
activates the ATM/GADD45/ORC pathway to promote DNA damage repair
and DNA biosynthesis, thereby alleviating injury in H9C2
cardiomyocytes induced by OGD/R (125). However, the majority of these
studies are based solely on in vitro cell models, lacking
validation from animal models or clinical data, and further
exploration is still required.
A study by Li et al (126) demonstrated that TSA enhances
the therapeutic efficacy of MSC-derived exosomes (MSCexo) in MIRI
by upregulating miR-223-5p within these exosomes.
TSA-preconditioned MSCexo (TSA-MSCexo) notably improved cardiac
function and reduced infarct size in rats compared with untreated
MSCexo. TSA-MSCexo also suppressed CCR2 activation, thereby
reducing monocyte infiltration and promoting angiogenesis (126). Mechanistically, TSA-MSCexo
delivered miR-223-5p to target CCR2 expression, modulating
inflammatory responses and enhancing vascular repair (126). These findings provide
experimental evidence for the development of a cell-free
exosome-based therapy combining TSA and MSCs, offering a novel
approach for the clinical treatment of ischemic cardiomyopathy.
In the exploration of therapeutic strategies for
MIRI, combination drug therapy has shown distinct advantages
(175,176). In addition to the
aforementioned combination of TSA and MSCexo, the synergistic use
of multiple drugs or bioactive substances has gained increasing
attention (175,176). For example, a study
investigated the protective effects and molecular mechanisms of
combining TSA and astragaloside IV (As-IV) in the treatment of MIRI
(127). The results revealed
that this combination therapy (Co) exhibited notably enhanced
efficacy when compared with either TSA or As-IV alone, both in
vivo and in vitro. In a MIRI mouse model, Co more
effectively reduced myocardial infarct size, decreased myocardial
enzyme levels (CK, CK-MB and LDH), improved cardiac function (LVEF
and LVFS) and alleviated myocardial pathological damage (127). In vitro experiments
showed that Co provided stronger protection against HR and
H2O2-induced HL-1 cell injury. It
considerably inhibited apoptosis (reducing Bax and cleaved
Caspase-3 while increasing Bcl-2), mitigated oxidative stress
(reducing ROS and MDA levels while enhancing GSH and SOD activity)
and suppressed inflammatory responses (downregulating IL-6, IL-1β,
TNF-α and iNOS mRNA expression) (127). A mechanistic study indicated
that the combined administration of TSA and As-IV exerted
anti-apoptotic, antioxidant and anti-inflammatory effects in MIRI
by synergistically inhibiting the STING pathway, with superior
efficacy compared with monotherapy (127). These findings provide
theoretical understanding for the use of TSA in treating MIRI,
confirming its multi-target mechanisms in mitigating reperfusion
injury and laying the foundation for clinical translation. However,
further high-quality research is needed to validate its
standardized application.
HF
HF often results from extensive cardiomyocyte
apoptosis, as these cells are essential functional units of the
heart and their loss directly impairs both contractile and
diastolic function (177).
Ischemic or hypoxic conditions can induce cardiomyocyte apoptosis,
leading to notable negative impacts on cardiac structure and
function (178,179). Therefore, identifying effective
strategies to inhibit cardiomyocyte apoptosis is important for
preventing and treating HF. Previous research has shown that TSA
can prevent DOX-induced cardiomyocyte apoptosis in rats by
suppressing ROS generation, reducing Caspase-3 activation and cyt
c release, increasing Bcl-2 expression and enhancing AKT
phosphorylation via the PI3K/AKT signaling pathway, thereby
protecting cardiomyocytes from DOX-induced toxicity (128). A previous study further
revealed that TSA inhibits DOX-induced cardiomyocyte apoptosis in
HF mice by activating the DAXX/MEK/ERK1/2 pathway (increasing
phosphorylation of p-ERK1/2 and p-MEK) and downregulating
Caspase-8, p-P38 and Caspase-3 expression (129).
Additionally, two studies have shown that miRNAs
play a role in TSA-mediated inhibition of cardiomyocyte apoptosis
(48,49). TSA upregulates miR-152-3p,
reducing PTEN expression and notably suppressing Ang II-induced
apoptosis in rat cardiomyocyte H9C2 cells (48). TSA also upregulates miR-133,
inhibiting the activation of Caspase-9 and its downstream apoptotic
effectors (including Caspase-3 and PARP), thereby ameliorating
H2O2- and DOX-induced apoptosis in H9C2 cells
(49). Oxidative stress is
another key factor in cardiomyocyte apoptosis. A study using
H2O2 to induce oxidative stress demonstrated
that the miR-133-Caspase-9 pathway serves as a core mechanism by
which TSA counters oxidative stress-induced myocardial injury
(49).
Autophagy, an intracellular degradation and
recycling process, plays a key role in HF development (180). During HF, cardiomyocytes
encounter various stressors, triggering autophagy to remove damaged
organelles and proteins, thus maintaining cellular homeostasis.
However, dysregulated autophagy, whether excessive or insufficient,
can impair cardiomyocytes and exacerbate HF (181). Modulating autophagy levels may
therefore provide a novel therapeutic approach for HF. A study by
Zhang et al (130)
demonstrated that TSA notably improved cardiac function in HF rats
(induced by LAD ligation), reduced cardiomyocyte apoptosis and
promoted autophagy by activating AMPK (increasing p-AMPK
phosphorylation) while inhibiting mTOR signaling (reducing mTOR
phosphorylation). This regulation led to altered expression of key
autophagy-related proteins (upregulated LC3/Beclin-1, downregulated
p62) and apoptosis-related proteins (increased Bcl-2/Bax ratio,
reduced Caspase-3/7 levels) (130). This study first elucidated that
TSA improves cardiac function through dual mechanisms, autophagy
activation and apoptosis inhibition, providing a novel therapeutic
target for HF (Fig. 5).
Myocardial fibrosis
Myocardial fibrosis plays a pivotal role in cardiac
remodeling, marked by the abnormal accumulation of collagen and ECM
components (182). This
fibrotic transition, often associated to aging (183), obesity (184) and diabetes (185), markedly impairs cardiac
function through multiple mechanisms (182). TSA reduces collagen deposition
and promotes elastic fiber formation in human CFs (131). Specifically, TSA selectively
activates the G protein-coupled ER (GPER) in human CFs, notably
suppressing COL-1 gene expression, protein synthesis and
deposition, while upregulating matrix MMP-1 expression (131). Additionally, TSA triggers the
PKA/CREB phosphorylation pathway via GPER, enhancing elastin
transcription, secretion and cross-linking (by upregulating
elastin-binding protein EBP and lysyl oxidase LOX), while
inhibiting the elastase activity of MMP-2/9 (131). This study is the first to
demonstrate that TSA coordinates collagen reduction with elastic
fiber generation through the GPER-PKA-CREB axis, offering a novel
approach for treating cardiac fibrosis. Another study found that
TSA considerably inhibits Ang II-induced activation of CFs and
α-SMA production in vitro, while reducing collagen I/II and
TIMP1/4 expression (132). In a
TAC rat model, TSA (10 mg/kg) improved cardiac function, alleviated
cardiac hypertrophy and decreased collagen deposition, effects
dependent on miR-618 induction (inhibition of miR-618 abolished the
protective effects of TSA) (132). This study is the first to
reveal that TSA suppresses myocardial fibrosis via the
miR-618/TIMPs axis, providing new molecular evidence for TSA-based
cardiovascular therapy (132).
Furthermore, Huang et al (133) investigated the protective
effects of TSA on subclinical LPS-induced cardiac fibrosis in mice
and its underlying mechanisms. The results showed that TSA
effectively alleviated LPS-induced myocardial fibrosis by
inhibiting NADPH oxidase activity, suppressing the expression of
fibrosis-related genes (Col1α1, Col3α1, MMP2, MMP9, TIMP1 and
TIMP2), reducing protein levels of NOX2 and p67phox, and decreasing
ROS generation (133). These
findings suggest potential drug targets and strategies for
preventing and treating subclinical LPS-associated cardiac
fibrosis.
Arrhythmia
Arrhythmia is a prevalent cardiovascular disorder
associated with cardiocerebrovascular function (186). It can lead to abnormal cardiac
pumping, disrupt blood circulation and potentially trigger or
worsen various cardiocerebrovascular diseases, such as coronary
artery disease, cardiomyopathy and hypertensive heart disease,
thereby increasing the risk of severe events such as stroke and HF
(186). A study explored the
activating effects of TSA on human KCNQ1/KCNE1 (IKs) potassium
channels expressed in HEK 293 cells (134). The results demonstrated that
TSA concentration-dependently enhanced IKs current amplitude
(EC50=64.5 μM), accelerated channel activation kinetics,
slowed inactivation and shifted the voltage dependence of IKs
activation toward hyperpolarization (134). Notably, the effects of TSA were
independent of β-adrenergic stimulation (for example,
isoproterenol), NO donor (for example, sodium nitroprusside),
protein kinase A inhibitor (for example, H-89 dihydrochloride) or
soluble guanylate cyclase inhibitor (1H-[1,2,4]Oxadiazolo[4,3-a]
quinoxalin-1-one), indicating its direct and specific action on
channel dynamics (134).
Furthermore, TSA showed no notable effects on hERG, Kv1.5 or IK1
channels expressed in HEK 293 cells (134). These findings suggest that TSA
may specifically enhance IKs to treat arrhythmias associated with
IK dysfunction, particularly during cardiac electrical remodeling.
Another study revealed that chronic TSA administration (10 mg/kg/d
for 3 months) markedly reduced the incidence and mortality of acute
myocardial ischemia-induced arrhythmias in rats (left anterior
descending coronary artery ligation model) (187). Electrophysiological analysis
showed that TSA restored the reduced inward rectifier potassium
current (IK1) density and Kir2.1 channel protein expression
post-myocardial infarction, thereby reversing resting membrane
potential depolarization (187). Mechanistically, myocardial
infarction upregulated miR-1 expression by 2.9-fold, which targeted
the 3'-untranslated region of KCNJ2 (encoding Kir2.1) to reduce
Kir2.1 protein levels without affecting mRNA (187). TSA normalized miR-1 levels by
suppressing SRF, a transcriptional activator of miR-1, thereby
alleviating translational inhibition of Kir2.1 (187). To the best of our knowledge,
this study is the first to identify the SRF/miR-1/Kir2.1/IK1
signaling axis as a novel therapeutic target for ischemic
arrhythmias.
He et al (188) investigated the preventive
effects of TSA on atrial fibrillation (AF) and its
electrophysiological mechanisms using a rabbit model of chronic HF
(CHF) induced by rapid ventricular pacing. The results showed that
combined perfusion with ACh (1 μM) and ISO (1 μM)
induced AF in all failing hearts and 11 out of 12 control hearts.
However, co-perfusion with TSA (10 μM) notably reduced AF
inducibility by 50% in both control and CHF groups (188). Electrophysiological analysis
revealed that TSA did not alter atrial action potential duration
but notably prolonged the effective refractory period, increased
atrial post-repolarization refractoriness and moderately prolonged
interatrial conduction time (188). Mechanistically, TSA exerted
antiarrhythmic effects by inhibiting Na+ channel recovery and
modulating calcium handling (for example suppressing RyR2-mediated
calcium leak) (188). This
study first demonstrated that TSA suppresses AF through dual
modulation of prolonged post-repolarization refractoriness and
conduction slowing, offering a novel therapeutic strategy for AF in
patients with CHF.
Cerebral ischemia-reperfusion
Cerebral ischemia-reperfusion refers to the
restoration of blood flow to brain tissue after ischemia (189,190). This process induces changes in
blood-brain barrier (BBB) permeability, leading to barrier
dysfunction and enabling substances that typically cannot cross the
BBB to infiltrate, triggering complex pathological responses. These
alterations disrupt normal brain metabolism and hinder functional
recovery, potentially worsening cerebral injury (189,190). TSA can inhibit the increase in
BBB permeability, reduce oxidative stress and inflammation, enhance
the expression of tight junction proteins such as Claudin-5 and
ZO-1, and activate the Nrf2/HO-1 signaling pathway, thereby
protecting brain microvascular EC function (46). Chen et al (191) showed that both in vitro
using a rat brain microvascular EC model and in vivo with a
rat brain perfusion model, PgP or Mrp1/2 inhibitors (such as
verapamil or quinidine) markedly enhanced TSA brain penetration,
indicating that PgP and Mrp1/2 carry out a key role in restricting
the passage of TSA across the BBB (191).
Cerebral ischemia-reperfusion therapy aims to
mitigate or reverse brain injury but may paradoxically induce
oxidative stress, inflammation and apoptosis, thereby exacerbating
cerebral damage (189). Wang
et al (135)
demonstrated that TSA markedly reduced cerebral infarct volume,
decreased HMGB1 protein levels, inhibited NF-κB nuclear
translocation, downregulated glial fibrillary acidic protein (GFAP)
expression and suppressed apoptosis, thereby alleviating
ischemia-reperfusion injury in a rat MCAO model. Their findings
indicated that TSA exerts anti-inflammatory and anti-apoptotic
effects by inhibiting the activation of the HMGB1/NF-κB signaling
pathway (135). Further studies
confirmed that TSA markedly reduced infarct volume, decreased brain
water content, improved neurological function scores and suppressed
inflammatory responses and neuronal apoptosis (136-138). Using both an MCAO/R rat models
and an in vitro OGD/R cell models (136,137), researchers found that TSA
inhibited the expression of pro-inflammatory cytokines (IL-1β, IL-6
and TNF-α), reduced levels of pro-apoptotic proteins (Bax and
Cleaved-Caspase-3) and enhanced the expression of the
anti-apoptotic protein Bcl-2. Notably, TSA downregulated the mRNA
and protein expression of TLR4, MyD88 and NF-κB (p105) in brain
tissue, inhibited the phosphorylation of IκB-α and NF-κB p65,
thereby stabilizing IκB-α and p65 proteins (137). These findings demonstrate that
TSA exerts neuroprotective effects by targeting the inhibition of
the TLR4/NF-κB signaling cascade, alleviating neuroinflammation
following cerebral ischemia-reperfusion injury (137). Additionally, research by Yu
et al (138) suggests
that TSA alleviates cerebral ischemia-reperfusion injury by
downregulating TGM2, inhibiting PANX1 expression and suppressing
microglial activation and neuroinflammation (TNF-α, IL-1β and
IL-6), highlighting the multi-target regulation of TSA in the same
disease.
Moreover, studies indicate that TSA can improve
cerebral ischemia-reperfusion injury by regulating miRNA expression
(136,139). TSA exerts neuroprotective
effects by upregulating miR-124-5p and inhibiting the expression of
FoxO1 (136). Research by Yu
et al (139) found
similar therapeutic effects (137,138), where TSA alleviates cerebral
ischemia-reperfusion injury in a rat MCAO/R model and in SH-SY5Y
cells subjected to OGD/R by promoting the expression of miR-449a.
This upregulation of miR-449a inhibits the expression of its target
gene ACSL4, thereby suppressing neuronal ferroptosis (139). These findings provide
theoretical support for the potential application of TSA in
treating cerebral ischemia-reperfusion injury (Fig. 6).
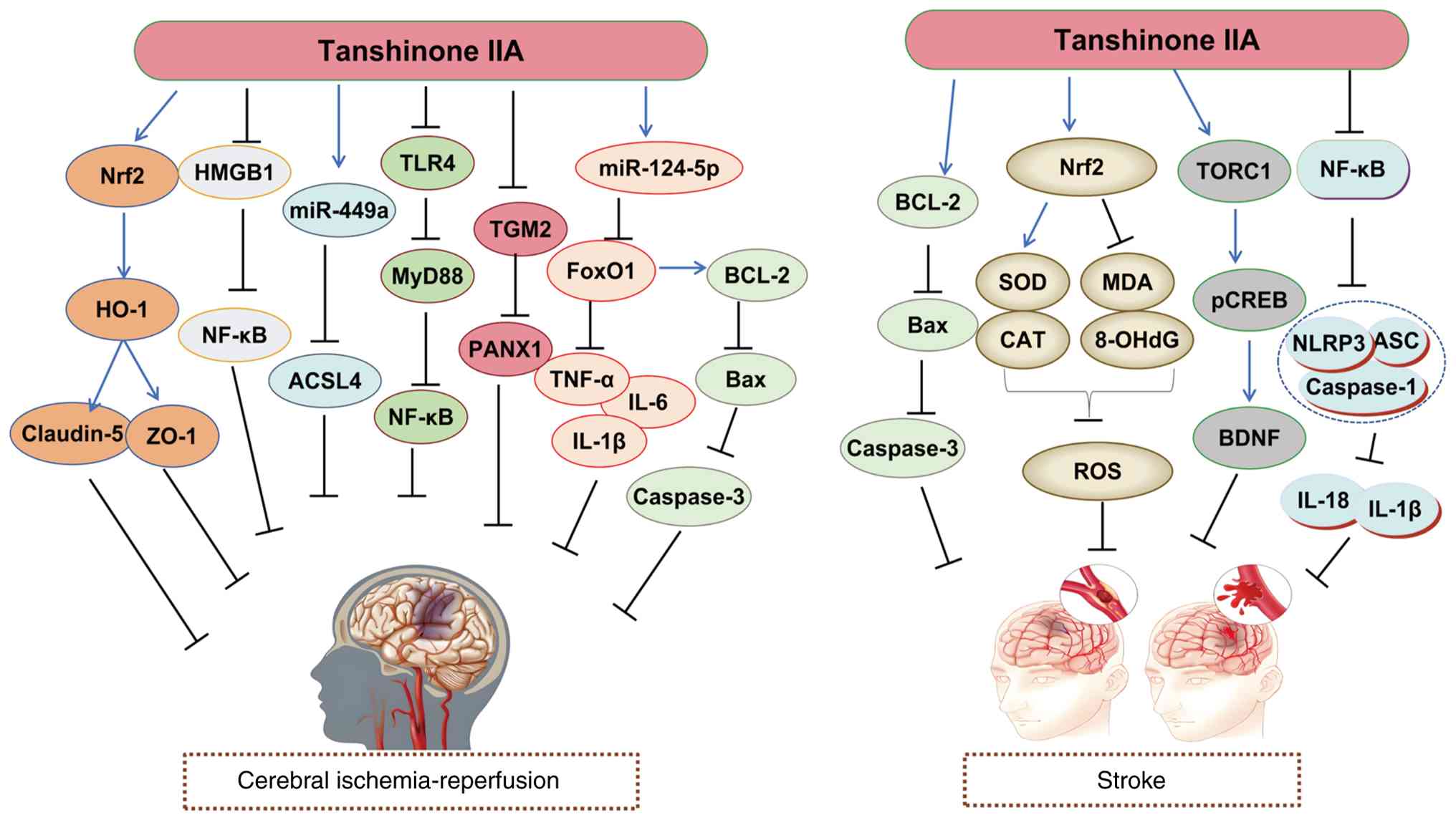 | Figure 6Molecular mechanisms of Tanshinone
IIA in the treatment of cerebral ischemia-reperfusion injury and
stroke. BDNF, brain-derived neurotrophic factor; CAT, catalase;
CREB, CAMP response element binding protein; FoxO3, forkhead box
O3; HO-1, heme oxygenase 1; IL-6, interleukin-6; HMGB1, high
mobility group box 1; MDA, malondialdehyde; NF-κB, nuclear factor
kappa-light-chain-enhancer of activated B cells; Nrf2, nuclear
factor erythroid 2-related factor 2; PI3K, phosphatidylinositol 3
kinase; ROS, reactive oxygen species; SOD, superoxide dismutase;
TNF-α, tumor necrosis factor-α; TORC1, target of rapamycin complex
1; 8-OHdG, 8-Hydroxy-2'-deoxyguanosine. |
Stroke
Stroke is an acute cerebrovascular disease caused
by vascular rupture or occlusion, which disrupts blood flow to the
brain and leads to subsequent tissue damage. It encompasses both
ischemic and hemorrhagic strokes, with ischemic stroke being the
more prevalent form (192). The
pathological mechanisms of stroke are multifaceted: Ischemic stroke
results from vascular occlusion, leading to cerebral hypoperfusion
(ischemia-hypoxia), which triggers energy metabolic dysfunction,
calcium overload, oxidative stress and inflammatory responses,
further exacerbating neuronal injury and mortality (193). By contrast, hemorrhagic stroke
involves vascular rupture, resulting in hematoma formation that
causes direct toxic effects and mass effects on brain tissue,
leading to severe damage (194). Additionally, stroke induces
secondary complications such as cerebral edema and BBB disruption,
impairing neurological function and resulting in cognitive and
motor deficits (193).
Emerging evidence suggests that TSA may alleviate
oxidative stress-related neuronal injury. One study shows that TSA
notably reduces H2O2-induced cytotoxicity in
primary rat cortical neurons by inhibiting intracellular calcium
elevation and modulating the Bcl-2/Bax protein ratio to suppress
apoptosis (140). Furthermore,
TSA reverses H2O2-induced long-term
potentiation (LTP) impairment in hippocampal neurons without
affecting basal synaptic transmission or LTP induction (140). These findings suggest that TSA
may exert antioxidant and anti-apoptotic effects, making it a
potential therapeutic agent for neurodegenerative diseases such as
stroke and AD (140). Another
study suggested neuroprotective effects of TSA via antioxidant
pathways (141). Using a MCAO
mouse model, TSA treatment notably improved neurological function
scores, reduced infarct volume and inhibited apoptosis (141). Mechanistically, TSA upregulates
Nrf2 mRNA expression, promotes Nrf2 nuclear translocation and
enhances antioxidant enzyme activity (for example, SOD and
catalase), while reducing oxidative byproducts such as MDA and
8-hydroxy-2'-deoxyguanosine (8-OHdG) (141). Notably, Nrf2 knockout and small
interfering RNA-mediated knockdown experiments revealed that Nrf2
deficiency abrogates the neuroprotective and antioxidant effects of
TSA, confirming that Nrf2 activation is the key mechanism
underlying the efficacy of TSA (141).
Additionally, an earlier study demonstrated that
TSA exerts neuroprotective effects in ischemic stroke rats (MCAO
model) by considerably reducing neurological deficit scores, brain
water content and infarct volume, while markedly enhancing the
nuclear accumulation of TORC1 and upregulating the expression of
TORC1, pCREB and BDNF (142).
Another study explored the neuroprotective effects and synergistic
mechanisms of puerarin (Pue) combined with TSA in rats with
ischemic stroke (143). The
results showed that the Pue-TSA combination therapy considerably
reduced neurological scores, infarct volume and levels of S-100b
and NSE in MCAO rats, indicating effective mitigation of ischemic
brain injury. Furthermore, the combined treatment markedly enhanced
total antioxidant capacity and the activities of CAT, SOD and
reduced GSH, while decreasing oxidized GSH activity and MDA levels.
It also reduced the levels of inflammatory mediators such as TNF-α,
IL-6, ICAM-1 and COX-2 (143).
A mechanistic study revealed that Pue-TSA combination therapy
markedly activated the Nrf2/ARE signaling pathway, increased the
nuclear expression of Nrf2, decreased its cytoplasmic expression
and markedly upregulated HO-1 and NAD(P)H oxidoreductase 1, while
downregulating Keap1 (143).
These findings suggest that the combined use of Pue and TSA exerts
synergistic neuroprotective effects by activating the Nrf2/ARE
pathway, inhibiting oxidative stress and inflammatory responses,
providing experimental evidence for clinical application (143).
Subarachnoid hemorrhage (SAH), a type of
hemorrhagic stroke, has also been shown to benefit from TSA
treatment. Yang et al (144) demonstrated that TSA markedly
improved neurological deficits, reduced cerebral edema and
inhibited neuronal apoptosis in SAH rats (established by the
endovascular perforation method). It also downregulated the
expression of inflammatory factors such as IL-6, MCP-1, TNF-α,
IL-1β and IL-18. Mechanistically, TSA inhibited the phosphorylation
of NF-κB and the activation of NLRP3 inflammasome-related proteins
(NLRP3, Caspase-1, IL-1β and IL-18) (144). In vitro experiments
using oxygenated hemoglobin-induced primary neuronal models further
confirmed that TSA protected neurons from SAH injury by blocking
the NF-κB/NLRP3 pathway (144).
This study provides a potential therapeutic strategy for the
treatment of SAH (Fig. 6).
Multi-targeting of TSA and pathway
crosstalk
TSA exhibits multifaceted pharmacological
functions, suggesting its multi-target characteristics. For
example, in MIRI, TSA suppresses disease progression by activating
the PI3K/AKT and Nrf2/HO-1 pathways. However, due to limitations in
existing research, the interplay between these pathways within the
same study remains insufficiently understood. Based on previous
reports, it is hypothesized that AKT may phosphorylate Keap1,
reducing its binding to Nrf2 and thereby enhancing Nrf2 nuclear
translocation and the expression of downstream antioxidant genes.
Such cross-talk is particularly important in cancer contexts,
where, for example, PI3K/AKT-driven tumor cells activate Nrf2/HO-1
to evade oxidative stress-induced apoptosis, fostering drug
resistance (28,195,196). Conversely, Nrf2 activation can
inhibit excessive PI3K/AKT pathway activity by modulating ROS
levels, indirectly affecting AKT phosphorylation status and
preventing excessive cellular proliferation. This negative feedback
mechanism may protect neurons from metabolic disturbances in
neurodegenerative diseases (197,198). However, the specific
interaction mechanisms between PI3K/AKT and Nrf2/HO-1 in
cardiovascular diseases warrant further investigation in future
studies.
Notably, in hypertension, cancer and hepatic
fibrosis, TSA alleviates disease progression by inhibiting the
PI3K/AKT pathway (28,63,111). Conversely, in diseases such as
AS, cardiac hypertrophy, HF and MIRI, TSA mitigates disease
progression by activating the PI3K/AKT pathway (99,112,121,128). This differential regulation of
the PI3K/AKT pathway by TSA across various diseases is primarily
the result of the combined effects of disease-specific pathological
mechanisms and the multi-target nature of the drug. First,
disease-specific pathological mechanisms determine the functional
role of the PI3K/AKT pathway. The biological functions of the
PI3K/AKT pathway are highly context-dependent, with its effects
varying dynamically according to disease type, cell type and
pathological stage. In hypertension, sustained activation of
PI3K/AKT promotes vascular smooth muscle cell proliferation and
migration, leading to vascular wall thickening and, ultimately,
vascular remodeling (111). In
cancer, PI3K/AKT drives tumor angiogenesis by upregulating factors
such as vascular endothelial growth factor and hypoxia inducible
factor-1α, supporting tumor growth and metastasis (28). In fibrosis (for example, hepatic
and pulmonary fibrosis), PI3K/AKT activation promotes myofibroblast
transdifferentiation and collagen deposition (63). In these contexts, inhibiting this
pathway can block the pathological process. However, in AS, cardiac
hypertrophy, HF and ischemia-reperfusion injury (99,112,121,128), the protective function of the
PI3K/AKT pathway is impaired or insufficiently upregulated, making
it an ally that requires activation. Under physiological
conditions, PI3K/AKT inhibits cardiomyocyte apoptosis, reduces
oxidative stress and improves energy metabolism, thereby exerting
cardioprotective effects (199,200). Furthermore, activating PI3K/AKT
can promote the differentiation of endothelial progenitor cells and
repair vascular endothelial damage (for example in AS) (99). In these scenarios, activating
this pathway enhances cellular protective mechanisms.
Second, the multi-target nature of TSA enables its
bidirectional regulatory effects of inhibition or activation. TSA
does not simply activate or inhibit a single pathway; instead, it
dynamically modulates PI3K/AKT activity by influencing upstream and
downstream signaling molecules and their interactive networks. In
hypertension, TSA inhibits PDK1 or the PI3K p110 subunit, blocking
AKT phosphorylation (111). In
AS, TSA inhibits NF-κB or the NLRP3 inflammasome, reducing the
release of inflammatory cytokines (TNF-α and IL-1β), thereby
alleviating their inhibitory effect on the PI3K/AKT pathway
(99). In HF, TSA inhibits
pro-apoptotic factors downstream of AKT or upregulates Bcl-2 (an
anti-apoptotic protein), synergistically enhancing the
anti-apoptotic effect of AKT (128). In MIRI, TSA scavenges ROS,
preventing ROS-mediated PTEN activation and subsequent inhibition
of the PI3K/AKT pathway (121).
Depending on the dysregulation of the PI3K/AKT pathway
(overactivation or inhibition) in a specific disease state, TSA can
recalibrate pathway activity to a physiological equilibrium point
by modulating its upstream and downstream molecular network,
thereby alleviating the pathological process in a targeted manner
(99,111,121).
In summary, the pathological mechanisms of CCVDs
primarily involve endothelial dysfunction, inflammatory responses,
oxidative stress and cell necrosis or apoptosis (201-203). TSA can inhibit the progression
of these diseases by modulating various signaling pathways. The
underlying mechanisms of these core targets stem from its
pharmacological functions. For instance, the NF-κB pathway, which
contributes to the development of AS (98), myocardial infarction (115), MIRI (120), cerebral ischemia-reperfusion
injury (135) and stroke
(144), serve as a key
regulator of inflammation in cardiovascular diseases. Its
activation triggers the release of pro-inflammatory cytokines, such
as TNF-α and IL-6, promoting inflammation. Given the
anti-inflammatory properties of TSA, it can suppress inflammation
in these diseases by inhibiting the NF-κB pathway. Notably,
intervention of TSA is not simply inhibition; rather, it involves
context-dependent modulation, either directly blocking activation
or indirectly regulating activity through upstream pathways,
demonstrating the multi-target therapeutic characteristics of
TCM.
Research on nanodelivery systems for
TSA
TSA suffers from poor water solubility and a short
half-life (30,31). The development of nanodelivery
systems with specific structural features can considerably enhance
the water solubility of TSA and prolong its in vivo
circulation half-life (204,205). Additionally, surface
modification of these nanocarriers enables targeted drug delivery,
improving both bioavailability and therapeutic specificity
(204,205).
A TSA-loaded reconstituted high-density lipoprotein
(TA-rHDL) system was developed, with spherical and discoidal
TA-rHDL structures prepared and characterized to investigate their
targeting mechanisms in foam cell models and pharmacokinetics in
rabbits (206,207). Results indicated that spherical
TA-rHDL targeted foam cells via scavenger receptor class B type I
and cholesteryl ester-triglyceride exchange pathways, while
discoidal TA-rHDL could reconvert into spherical structures,
enhancing targeting efficiency through similar mechanisms (206). TA-rHDL markedly prolonged
circulation time, with a 4- to 13-fold increase in area under the
curve (AUC) and apolipoprotein modification further optimized
pharmacokinetic parameters. TA-rHDL serves both as a drug delivery
vehicle and an anti-atherogenic carrier, offering a long-acting,
targeted delivery strategy for hydrophobic cardiovascular drugs
(206). Moreover, by leveraging
the inherent anti-atherosclerotic properties of rHDL, this system
facilitates drug-carrier synergy therapy, presenting a novel
strategy for treating cardiovascular disease.
Zhang et al (208) developed a lipid-polymer hybrid
nanoparticle (LPN) system for mitochondrial-targeted TSA delivery.
The study involved synthesizing a targeting ligand,
triphenylphosphonium (TPP)-Lys-D-α-tocopheryl polyethylene glycol
1000 succinate (TPGS), by conjugating TPGS with TPP cations.
TPP-TPGS/TSA/LPNs were then prepared using nanoprecipitation. In
vitro experiments showed sustained release, with >80% of TSA
released within 60 h, considerably enhancing uptake efficiency in
cardiomyocytes (208). In
vivo pharmacokinetic studies revealed that TPP-TPGS/TSA/LPNs
achieved an AUC of 129.27 mg/l h, notably higher when compared with
unmodified TSA/LPNs (42.27 mg/l h) and TSA/NPs (20.46 mg/l h)
(208). In a rat myocardial
infarction model, the TPP-TPGS/TSA/LPNs treatment group reduced
infarct size to 28%, outperforming unmodified groups (37%) and free
TSA groups (51%) (208). These
findings suggest that TPP-TPGS/TSA/LPNs are an advanced nanocarrier
system capable of improving the aqueous solubility, bioavailability
and mitochondrial targeting in myocardial tissue of TSA, offering a
promising drug delivery strategy for cardiovascular diseases
(208). Furthermore, Fan et
al (209) developed a
composite material, sodium alginate (SA)/liquid metal (LM)/TSA, by
ultrasonically dispersing LM nanoparticles and the drug TSA into a
SA solution for intrapericardial injection in the treatment of
myocardial infarction. This material exhibits low viscosity,
allowing smooth injection via syringe, and is X-ray opaque
due to the incorporation of LM. In vitro experiments showed
that SA/LM/TSA possesses excellent biocompatibility and
hemocompatibility, enabling the slow and stable release of TA. In a
rat myocardial infarction model, intrapericardial injection of
SA/LM/TSA proved to be a safe and effective administration method
(209). After 7 days of
treatment, compared with the control and TSA direct injection
groups, the SA/LM/TSA group demonstrated considerable improvements
in cardiac function (LVEF and LVFS), recovery of ST-segment
elevation on electrocardiogram and substantial reductions in
infarct size and fibrotic area (209). Concurrently, myocardial injury
markers (cTnT, cTnI, CK, CK-MB and LDH) decreased, pro-inflammatory
cytokine levels (TNF-α and TGF-β) declined and anti-inflammatory
cytokine levels (IL-4 and IL-10) increased, indicating that
SA/LM/TSA exerts synergistic anti-inflammatory and cardioprotective
effects (209). However, the
study did not explore the specific molecular mechanisms by which
SA/LM/TSA improves cardiac function, such as the involvement of
specific signaling pathways. Additionally, only a rat model with a
short observation period (7 days) was used, lacking long-term
efficacy and safety assessments. Further large animal experiments
and pharmacokinetic studies are needed for validation (209).
In ischemic stroke research, Ma et al
(210) developed a TSA
microemulsion (TSA ME), a reformulated delivery system utilizing
microemulsion technology to enhance bioavailability and targeting.
The study demonstrated that TSA ME notably improved neurological
deficits, reduced infarct volume in a rat MCAO model and inhibited
histone deacetylase activity in an OGD/R hippocampal neuron model
(210). Mechanistically, TSA ME
upregulated H3K18ac and H4K8ac expression, downregulated
excitotoxicity-related NMDAR1 protein and apoptosis-related
Caspase-3 and concurrently promoted neuronal cytoskeletal protein
MAP-2 expression (210).
Compared with the conventional TSA solution, the microemulsion
markedly improved drug delivery efficiency, with efficacy
comparable to valproic acid (positive control) (210). These findings provide
experimental evidence for TSA ME as a therapeutic candidate for
ischemic stroke, highlighting its neuroprotective role via
epigenetic modulation in cerebral ischemia-reperfusion injury.
In another study, the effects of cationized bovine
serum albumin-conjugated PEGylated TSA nanoparticles
(CBSA-PEG-TSA-NPs) on neuronal signaling pathways and
neuroprotection in cerebral ischemia were investigated (211). CBSA-PEG-TSA-NPs considerably
prolonged the circulation time, increased plasma concentration and
enhanced brain accumulation of TSA (211). The nanoparticles effectively
reduced infarct volume, neurological dysfunction, neutrophil
infiltration and neuronal apoptosis, while suppressing
pro-inflammatory cytokines (TNF-α and IL-8) and upregulating
anti-inflammatory cytokines (IL-10 and TGF-β1) (211). Additionally, CBSA-PEG-TSA-NPs
markedly downregulated GFAP, MMP-9 and COX-2 protein levels and
reduced phosphorylation of ERK1/2, p38MAPK and JNK (211). The study concluded that
CBSA-PEG-TSA-NPs exert neuroprotective effects in ischemic stroke
by modulating MAPK signaling pathways (211). Further research explored the
neuroprotective effects of TSA-loaded PLGA nanoparticles (TSA-NPs)
in a porcine ischemic stroke model (204). The study evaluated the
acute-phase therapeutic efficacy of TSA-NPs on cerebral
pathological damage and motor dysfunction (204). TSA-NPs markedly reduced
hemispheric swelling, midline shift and ischemic lesion volume,
while alleviating cytotoxic edema, white matter injury and cerebral
hemorrhage. Moreover, TSA-NPs improved post-stroke gait parameters
(for example, step frequency and stride length) by inhibiting
oxidative stress and inflammation (for example, reducing
circulating neutrophil proportion) (204). These studies address clinical
translation challenges of TSA, including its short half-life, poor
solubility and limited BBB penetration, offering efficient targeted
delivery solutions for ischemic stroke (204). Waters et al (204) work validated the
neuroprotective mechanisms of nanocarrier systems in large animal
models, providing preclinical evidence closer to human
pathology.
A recent study explored the therapeutic efficacy of
ROS-responsive TPP-modified TSA micelles (TK-TPP-TSA@ Ms) in
mitigating DOX-induced HF (205). The research developed a
microenvironment-responsive micellar system targeting myocardial
mitochondria by encapsulating TSA within micelles and conjugating
TPP-modified DSPE-PEG2000 as a targeting moiety, with ROS-cleavable
TK bonds serving as linkers. In vitro experiments
demonstrated that TK-TPP-TSA@ Ms exhibited robust uptake in H9c2
cells, considerably reducing ROS levels induced by DOX, suppressing
inflammatory cytokine secretion, inhibiting apoptosis and restoring
mitochondrial membrane potential (205). In vivo studies further
confirmed that TK-TPP-TSA@Ms effectively alleviated DOX-induced
myocardial tissue damage, reduced apoptosis and inflammatory
cytokine expression, mitigated oxidative stress and inhibited HF
progression in mice. Fluorescence microscopy and flow cytometry
analyses validated the targeting capability and ROS responsiveness
of micelles. Under HF conditions, ROS cleaved the TK bonds,
exposing TPP targeting ligands and enhancing micelle accumulation
in damaged cardiomyocytes (205). Additionally, TK-TPP-TSA@ Ms
demonstrated favorable biocompatibility and stability, with
negligible hemolytic effects, making them suitable for clinical
applications. The study concluded that TK-TPP-TSA@ Ms represent a
safe and effective targeted therapeutic strategy for cardiac
diseases, with potential as a promising treatment for HF (205).
In summary, nanomedicine delivery systems offer
notable advantages in treating CCVDs. They enhance drug solubility
and stability, addressing the challenges of poor water solubility
and short half-life of TSA, thus improving bioavailability and
ensuring greater drug accumulation at target sites. Additionally,
these systems enable precise lesion targeting, reducing drug
distribution to non-target tissues and minimizing adverse effects
while enhancing therapeutic efficacy. Moreover, nanocarriers
facilitate sustained or controlled drug release, prolonging the
duration of action and maintaining effective concentrations, which
further improves treatment outcomes.
Conclusion and future prospects
In recent years, active components of TCM have
gained recognition for their unique advantages and potential in
treating CCVDs. Various TCM monomers, such as Panax notoginseng
saponins (212), astragaloside
(213) and ginsenosides
(214), effectively alleviate
symptoms and reduce the risk of complications in CCVDs through
multiple mechanisms, including lipid regulation, antioxidant
effects, anti-inflammatory actions and the improvement of vascular
endothelial function (212-214). TSA, a monomer derived from
Salvia miltiorrhiza, is a prime example. It not only exhibits these
therapeutic mechanisms but also shows notable protective effects
against MIRI and cerebral infarction in numerous preclinical
studies and preliminary clinical trials. Its mechanisms include
regulating multiple signaling pathways, such as inhibiting
inflammation-related pathways and activating antioxidant
stress-related pathways, thereby improving cardiovascular and
cerebrovascular function through diverse channels (26,143,215).
However, due to insufficient foundational research,
the molecular mechanisms underlying the multi-target effects of TSA
remain incompletely understood, particularly its interactions with
ERs and regulatory networks involving specific miRNAs. Future
studies could integrate single-cell sequencing technology to
analyze the target sites of TSA in specific cell types, such as
cardiomyocytes and neurons. Combining proteomics to explore the
interaction networks of TSA with ERs and miRNAs (for example,
miR-29b and miR-133) and utilizing gene editing techniques, such as
CRISPR-Cas9, to validate key pathways (for example, Nrf2/HO-1 or
PI3K/AKT/mTOR) by knocking out the ALKBH5 gene would help clarify
the role of m6A modification in the anti-myocardial hypertrophy
effects of TSA.
Moreover, the interactions between TSA and other
drugs remain largely unexplored. While TSA is often used in
combination with other medications, the mechanisms behind these
interactions and their impact on efficacy and safety remain
unclear, creating challenges for rational clinical drug use. Future
research could investigate the synergistic effects of TSA with
other Chinese herbal components (for example, salvianolic acid B)
to develop compound formulations. Interdisciplinary collaboration
will be essential to overcome current limitations and improve
formulation development.
Finally, in terms of clinical translation, the
lipophilic nature of TSA limits its oral absorption, making it
difficult to achieve effective plasma concentrations and hindering
its utilization (21). Thus, the
majority of clinical studies have been small-scale trials lacking
the support of large-scale, multicenter data (216-218). Additionally, the study periods
are short, and the limited duration of observations is insufficient
to evaluate long-term efficacy and safety. Currently, the most
studied clinical formulations are Danshen injection and TSA
sulfonate sodium injection (218-220). TSA is converted into TSA
sulfonate sodium through a sulfonation reaction (221). This process involves specific
chemical conditions that promote the binding of functional groups
in the TSA molecule to sulfonate ions, resulting in the formation
of TSA sulfonate sodium, which exhibits increased water solubility
and enhanced bioactivity (221). This transformation not only
improves the pharmacokinetic properties of TSA but also boosts its
stability and efficacy in vivo (221). TSA sulfonate sodium can improve
myocardial ischemia, inhibit platelet aggregation and provide
antioxidative, anti-inflammatory and cardioprotective effects
(222-225). It has been used clinically to
treat CCVDs, such as coronary heart disease (226), angina pectoris (220) and stroke (227). Although clinical research still
has limitations, including variable trial quality, limited sample
sources and a lack of long-term follow-up data, the positive
efficacy and favorable safety profile demonstrated by TSA sulfonate
sodium injection have laid a solid foundation for its clinical
application.
Through sulfonation structural modification and
nanodrug delivery systems (such as rHDL (206), TPP-TPGS/LPNs (208), and CBSA-PEG-TSA-NPs (211), TSA's water solubility has been
enhanced, its half-life extended and its targeting ability
improved. As a result, TSA has transitioned from traditional
applications to precision therapy, making notable strides in areas
such as cardiovascular diseases (94,118), diabetic nephropathy (228,229) and tumor-targeted therapy
(29,230). However, several challenges
remain in achieving broader clinical application and precision
therapy for TSA. In the treatment of cardiovascular diseases, while
TSA sulfonate sodium injection has shown efficacy, individual
patient variability may lead to inconsistent therapeutic outcomes
(231-233). Future research should focus on
investigating genetic characteristics, metabolic status and other
factors to enable personalized, precision medicine. In diabetic
nephropathy, research is still in its early stages, and the precise
mechanisms through which TSA acts on renal lesions require further
exploration (228,229). A deeper understanding of its
mechanism will facilitate the optimization of treatment regimens
and improve therapeutic efficacy. In tumor-targeted therapy,
although nanodrug delivery systems have improved the targeting
ability of TSA (208), tumor
cell heterogeneity and the complex tumor microenvironment
complicate treatment (234).
Continuous optimization of nanodrug delivery systems is necessary
to enhance their specificity for tumor cells and efficiency in
targeted delivery. Additionally, combining TSA with other tumor
therapies should be explored to leverage its synergistic effects in
comprehensive cancer treatment. To further promote clinical
translation of TSA, interdisciplinary collaboration among experts
in medicine, pharmacology, biology and other fields is essential
for conducting large-scale, high-quality clinical trials. A robust
long-term follow-up system must be established to accumulate more
clinical data and evaluate the long-term efficacy and safety of
TSA. Moreover, the standardization of drug development should be
strengthened to ensure the effectiveness and safety of TSA-related
drugs in clinical practice.
With ongoing technological advancements and deeper
research, TSA is poised to carry out a pivotal role in treating
more diseases. Overcoming existing challenges will expand treatment
options and improve therapeutic outcomes for patients. The
potential of TSA in precision therapy is expected to be fully
realized, paving the way for new breakthroughs and transformative
changes in clinical treatments.
Availability of data and materials
Not applicable.
Authors' contributions
WP was responsible for conceptualization,
validation and writing the original draft of the manuscript. PL
revised and edited the manuscript. CD contributed to literature
collection and the design of the manuscript framework. YoLi
contributed to investigation, methodology and revision. WP and YiLi
were responsible for the acquisition of funding and YiLi was
responsible for the project administration and formal analysis. All
authors read and approved the final version of the manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests
Acknowledgements
Not applicable.
Funding
This work was supported by Jing'an District Traditional Chinese
Medicine Key Clinical Specialty Construction Project (grant no.
JA2024-Z003) and Shanghai Municipal Health Technology Commission
Traditional Chinese Medicine Project (grant no. 2024BJ017).
References
|
1
|
Miao K, Liu W, Xu J, Qian Z and Zhang Q:
Harnessing the power of traditional Chinese medicine monomers and
compound prescriptions to boost cancer immunotherapy. Front
Immunol. 14:12772432023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang K, Chen Q, Shao Y, Yin S, Liu C, Liu
Y, Wang R, Wang T, Qiu Y and Yu H: Anticancer activities of TCM and
their active components against tumor metastasis. Biomed
Pharmacother. 133:1110442021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang H and Gu J: Progress of experimental
study on treatment of psoriasis by Chinese medicinal monomer and
single or compound recipe in Chinese materia medica. Chin J Integr
Med. 13:312–316. 2007. View Article : Google Scholar
|
|
4
|
Liang J, Zhu Y, Liu S, Kuang B, Tian Z,
Zhang L, Yang S, Lin M, Chen N, Liu X, et al: Progress of exosomal
MicroRNAs and traditional Chinese medicine monomers in
neurodegenerative diseases. Phytother Res. 38:5323–5349. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lv D, Liu Y, Tang R, Fu S, Kong S, Liao Q,
Li H and Lin L: Analysis of clinical trials using anti-tumor
traditional Chinese medicine monomers. Drug Des Devel Ther.
18:1997–2020. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu X, Zhang Y, Yi F, Geng Z, Guo M, Ling
X, Li J and Li L: Anti-inflammatory and barrier repair mechanisms
of active components in Daemonorops draco Bl. for UVB-induced skin
damage. Sci Rep. 15:171242025. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pan Y, Wu W, Jiang X and Liu Y:
Mesenchymal stem cell-derived exosomes in cardiovascular and
cerebrovascular diseases: From mechanisms to therapy. Biomed
Pharmacother. 163:1148172023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen C, Ai Q, Tian H and Wei Y: CKLF1 in
cardiovascular and cerebrovascular diseases. Int Immunopharmacol.
139:1127182024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu C, Du L, Wang S, Kong L, Zhang S, Li
S, Zhang W and Du G: Differences in the prevention and control of
cardiovascular and cerebrovascular diseases. Pharmacol Res.
170:1057372021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang S, Yin X, Jiang T, Xu J and Wang D:
Impact of cardiovascular and cerebrovascular diseases mortality on
life expectancy in tianjin, 2004 and 2020. Asia Pac J Public
Health. 36:455–462. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Luan J and Zhai G: Targeted drug delivery
for cardiovascular and cerebrovascular diseases. Curr Drug Targets.
17:467–474. 2016. View Article : Google Scholar
|
|
12
|
Lan Q, Chen L, Chen MT, Wan ZX, Peng T,
Mazhar M, Liu P, Luo G, Jiang Y and Liu MN: Salvia miltiorrhiza:
Insights on the protective effect and mechanism of myocardial
ischemia-reperfusion injury. Braz J Med Biol Res. 58:e147232025.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li ZM, Xu SW and Liu PQ: Salvia
miltiorrhizaBurge (Danshen): A golden herbal medicine in
cardiovascular therapeutics. Acta Pharmacol Sin. 39:802–824. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wei B, Sun C, Wan H, Shou Q, Han B, Sheng
M, Li L and Kai G: Bioactive components and molecular mechanisms of
Salvia miltiorrhiza Bunge in promoting blood circulation to remove
blood stasis. J Ethnopharmacol. 317:1166972023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim MS, Bang JH, Lee J, Kim HW, Sung SH,
Han JS and Jeon WK: Salvia miltiorrhiza extract protects white
matter and the hippocampus from damage induced by chronic cerebral
hypoperfusion in rats. BMC Complement Altern Med. 15:4152015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiang Z, Gao W and Huang L: Tanshinones,
critical pharmacological components in salvia miltiorrhiza. Front
Pharmacol. 10:2022019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pang H, Wu L, Tang Y, Zhou G, Qu C and
Duan JA: Chemical analysis of the herbal medicine salviae
miltiorrhizae radix et rhizoma (Danshen). Molecules. 21:512016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ma XH, Ma Y, Tang JF, He YL, Liu YC, Ma
XJ, Shen Y, Cui GH, Lin HX, Rong QX, et al: The biosynthetic
pathways of tanshinones and phenolic acids in Salvia miltiorrhiza.
Molecules. 20:16235–16254. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang X, Yang Y, Liu X and Gao X:
Pharmacological properties of tanshinones, the natural products
from Salvia miltiorrhiza. Adv Pharmacol. 87:43–70. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shi MJ, Dong BS, Yang WN, Su SB and Zhang
H: Preventive and therapeutic role of Tanshinone ⅡA in hepatology.
Biomed Pharmacother. 112:1086762019. View Article : Google Scholar
|
|
21
|
Guo R, Li L, Su J, Li S, Duncan SE, Liu Z
and Fan G: Pharmacological activity and mechanism of tanshinone IIA
in related diseases. Drug Des Devel Ther. 14:4735–4748. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhong C, Lin Z, Ke L, Shi P, Li S, Huang
L, Lin X and Yao H: Recent research progress (2015-2021) and
perspectives on the pharmacological effects and mechanisms of
tanshinone IIA. Front Pharmacol. 12:7788472021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li Z, Zou J, Cao D and Ma X:
Pharmacological basis of tanshinone and new insights into
tanshinone as a multitarget natural product for multifaceted
diseases. Biomed Pharmacother. 130:1105992020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Du L, Guan C, Zhang H, Jia H and Wan Q:
Harnessing the therapeutic value of Tanshinone IIA: A breakthrough
therapy in cardiovascular diseases. Front Pharmacol.
16:16201522025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fu L, Han B, Zhou Y, Ren J, Cao W, Patel
G, Kai G and Zhang J: The anticancer properties of tanshinones and
the pharmacological effects of their active ingredients. Front
Pharmacol. 11:1932020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lu TC, Wu YH, Chen WY and Hung YC:
Targeting oxidative stress and endothelial dysfunction using
tanshinone IIA for the treatment of tissue inflammation and
fibrosis. Oxid Med Cell Longev. 2022:28117892022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cao Y, Tang H, Wang G, Li P, Song Z, Li W,
Sun X, Zhong X, Yu Q, Zhu S and Zhu L: Targeting survivin with
Tanshinone IIA inhibits tumor growth and overcomes chemoresistance
in colorectal cancer. Cell Death Discov. 9:3512023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jiang Y, Bi Y, Zhou L, Zheng S, Jian T and
Chen J: Tanshinone IIA inhibits proliferation and migration by
downregulation of the PI3K/Akt pathway in small cell lung cancer
cells. BMC Complement Med Ther. 24:682024. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Luo N, Zhang K, Li X, Hu Y and Guo L:
Tanshinone IIA destabilizes SLC7A11 by regulating PIAS4-mediated
SUMOylation of SLC7A11 through KDM1A, and promotes ferroptosis in
breast cancer. J Adv Res. 69:313–327. 2025. View Article : Google Scholar :
|
|
30
|
Lee AR, Wu WL, Chang WL, Lin HC and King
ML: Isolation and bioactivity of new tanshinones. J Nat Prod.
50:157–160. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu WY and Wang YP: Pharmacological actions
and therapeutic applications of Salvia miltiorrhiza depside salt
and its active components. Acta Pharmacol Sin. 33:1119–1130. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fan G, Jiang X, Wu X, Fordjour PA, Miao L,
Zhang H, Zhu Y and Gao X: Anti-inflammatory activity of tanshinone
IIA in LPS-Stimulated RAW264.7 Macrophages via miRNAs and
TLR4-NF-κB pathway. Inflammation. 39:375–384. 2016. View Article : Google Scholar
|
|
33
|
Li YI, Elmer G and Leboeuf RC: Tanshinone
IIA reduces macrophage death induced by hydrogen peroxide by
upregulating glutathione peroxidase. Life Sci. 83:557–562. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li X, Park SJ, Jin F, Deng Y, Yang JH,
Chang JH, Kim DY, Kim JA, Lee YJ, Murakami M, et al: Tanshinone IIA
suppresses FcεRI-mediated mast cell signaling and anaphylaxis by
activation of the Sirt1/LKB1/AMPK pathway. Biochem Pharmacol.
152:362–372. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Maione F, De Feo V, Caiazzo E, De Martino
L, Cicala C and Mascolo N: Tanshinone IIA, a major component of
Salvia milthorriza Bunge, inhibits platelet activation via Erk-2
signaling pathway. J Ethnopharmacol. 155:1236–1242. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fan Y, Kang S, Shao T, Xu L and Chen J:
Activation of SIRT3 by Tanshinone IIA ameliorates renal fibrosis by
suppressing the TGF-β/TSP-1 pathway and attenuating oxidative
stress. Cell Signal. 122:1113482024. View Article : Google Scholar
|
|
37
|
Guo G, Zhao Q, Wang Q and Li E: Tanshinone
IIA ameliorate coxsackie virus B3-induced viral myocarditis through
the inhibition of inflammation and modulation T Helper 1/T Helper 2
balance in mice. Pharmacology. 103:136–142. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang P, Gu Y, Lu J, Song M, Hou W, Li P,
Sun Y, Wang J and Chen X: Endothelial TRPV4 channel mediates the
vasodilation induced by Tanshinone IIA. Chem Biol Interact.
402:1111812024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huang N, Li Y, Zhou Y, Zhou Y, Feng F, Shi
S, Ba Z and Luo Y: Neuroprotective effect of tanshinone
IIA-incubated mesenchymal stem cells on Aβ(25-35)-induced
neuroinflammation. Behav Brain Res. 365:48–55. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dinarello CA: Anti-inflammatory agents:
Present and future. Cell. 140:935–950. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li Y and Zhou Y: The therapeutic effect of
tanshinone IIA on propionibacterium acnes-induced inflammation in
vitro. Dermatol Ther. 31:e127162018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang C, Lei X and Li J: Tanshinone IIA
reduces oxidized low-density lipoprotein-induced inflammatory
responses by downregulating microRNA-33 in THP-1 macrophages. Int J
Clin Exp Pathol. 12:3791–3798. 2019.
|
|
43
|
Filomeni G, De Zio D and Cecconi F:
Oxidative stress and autophagy: the clash between damage and
metabolic needs. Cell Death Differ. 22:377–388. 2015. View Article : Google Scholar :
|
|
44
|
Xu S, Liu Z, Huang Y, Le K, Tang F, Huang
H, Ogura S, Little PJ, Shen X and Liu P: Tanshinone II-A inhibits
oxidized LDL-induced LOX-1 expression in macrophages by reducing
intracellular superoxide radical generation and NF-κB activation.
Transl Res. 160:114–124. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Feng J, Li S and Chen H: Tanshinone IIA
inhibits myocardial remodeling induced by pressure overload via
suppressing oxidative stress and inflammation: Possible role of
silent information regulator 1. Eur J Pharmacol. 791:632–639. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang X, Wang WM, Han H, Zhang Y, Liu JL,
Yu JY, Liu HM, Liu XT, Shan H and Wu SC: Tanshinone IIA protected
against lipopolysaccharide-induced brain injury through the
protective effect of the blood-brain barrier and the suppression of
oxidant stress and inflammatory response. Food Funct. 13:8304–8312.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang Z, Li Y, Sheng C, Yang C, Chen L and
Sun J: Tanshinone IIA inhibits apoptosis in the myocardium by
inducing microRNA-152-3p expression and thereby downregulating
PTEN. Am J Transl Res. 8:3124–3132. 2016.PubMed/NCBI
|
|
49
|
Song T, Yao Y, Wang T, Huang H and Xia H:
Tanshinone IIA ameliorates apoptosis of myocardiocytes by
up-regulation of miR-133 and suppression of Caspase-9. Eur J
Pharmacol. 815:343–350. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang L, Wu Y, Li Y, Xu C, Li X, Zhu D,
Zhang Y, Xing S, Wang H, Zhang Z and Shan H: Tanshinone IIA
improves miR-133 expression through MAPK ERK1/2 pathway in hypoxic
cardiac myocytes. Cell Physiol Biochem. 30:843–852. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yan SH, Zhao NW, Geng ZR, Shen JY, Liu FM,
Yan D, Zhou J, Nie C, Huang CC and Fang ZY: Modulations of
Keap1-Nrf2 signaling axis by TIIA ameliorated the oxidative
stress-induced myocardial apoptosis. Free Radic Biol Med.
115:191–201. 2018. View Article : Google Scholar
|
|
52
|
Bellanti JA and Settipane RA: Allergy and
immunology: At the crossroad of inflammation and disease. Allergy
Asthma Proc. 44:1–2. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Costanzo G, Marchetti M, Ledda AG,
Sambugaro G, Bullita M, Paoletti G, Heffler E, Firinu D and
Costanzo GAML: Mast cells in allergic and non-allergic upper
airways diseases: Sentinel in the watchtower. Int J Mol Sci.
25:126152024. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kaminuma O, Nishimura T, Saeki M, Mori A
and Hiroi T: T cell-mediated nasal hyperresponsiveness in allergic
rhinitis. Biol Pharm Bull. 43:36–40. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Méndez-Enríquez E and Hallgren J: Mast
cells and their progenitors in allergic asthma. Front Immunol.
10:8212019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Thangam EB, Jemima EA, Singh H, Baig MS,
Khan M, Mathias CB, Church MK and Saluja R: The role of histamine
and histamine receptors in mast cell-mediated allergy and
inflammation: The hunt for new therapeutic targets. Front Immunol.
9:18732018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhang J, Xie X, Ma R and Liu P: The role
of mast cells in allergic rhinitis. PeerJ. 13:e197342025.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Asada Y, Yamashita A, Sato Y and
Hatakeyama K: Thrombus formation and propagation in the onset of
cardiovascular events. J Atheroscler Thromb. 25:653–664. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Fei YX, Wang SQ, Yang LJ, Qiu YY, Li YZ,
Liu WY, Xi T, Fang WR and Li YM: Salvia miltiorrhiza Bunge
(Danshen) extract attenuates permanent cerebral ischemia through
inhibiting platelet activation in rats. J Ethnopharmacol.
207:57–66. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jiang Y, Hu F, Li M and Li Q: Tanshinone
IIA ameliorates the development of dermal fibrosis in systemic
sclerosis. Clin Exp Pharmacol Physiol. 51:e138342024. View Article : Google Scholar
|
|
61
|
Wang DT, Huang RH, Cheng X, Zhang ZH, Yang
YJ and Lin X: Tanshinone IIA attenuates renal fibrosis and
inflammation via altering expression of TGF-β/Smad and NF-κB
signaling pathway in 5/6 nephrectomized rats. Int Immunopharmacol.
26:4–12. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Liao W, Wu F, Hao Z, Ye R, Liu C, Wu J, Wu
M, Zhou X, Sun M, Liu Y and Fang M: Tanshinone IIA alleviates liver
fibrosis by suppressing hepatic stellate cell proliferation via
ERK/cyclin D1/p-Smad3L signaling axis. Iran J Basic Med Sci.
28:638–646. 2025.PubMed/NCBI
|
|
63
|
Li Q, Huang D, Liao W, Su X, Li J, Zhang
J, Fang M and Liu Y: Tanshinone IIA regulates CCl(4) induced liver
fibrosis in C57BL/6J mice via the PI3K/Akt and Nrf2/HO-1 signaling
pathways. J Biochem Mol Toxicol. 38:e236482024. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Ren X, Wang C, Xie B, Hu L, Chai H, Ding
L, Tang L, Xia Y and Dou X: Tanshinone IIA induced cell death via
miR30b-p53-PTPN11/SHP2 signaling pathway in human hepatocellular
carcinoma cells. Eur J Pharmacol. 796:233–241. 2017. View Article : Google Scholar
|
|
65
|
Liu J, Zhang C, Liu S, Wang X, Wu X and
Hao J: Tanshinone IIA promotes apoptosis by downregulating BCL2 and
upregulating TP53 in triple-negative breast cancer. Naunyn
Schmiedebergs Arch Pharmacol. 396:365–374. 2023. View Article : Google Scholar
|
|
66
|
Zhang W, Liu M, Ji Y, Yu D, Ma C, Zhao J
and Qu P: Tanshinone IIA inhibits endometrial carcinoma growth
through the MAPK/ERK/TRIB3 pathway. Arch Biochem Biophys.
743:1096552023. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Gao M, Ou H, Jiang Y, Wang K, Peng Y,
Zhang H, Yang M and Xiao X: Tanshinone IIA attenuates
sepsis-induced immunosuppression and improves survival rate in a
mice peritonitis model. Biomed Pharmacother. 112:1086092019.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kim WS, Kim DO, Yoon SJ, Kim MJ, Yoon SR,
Park YJ, Jung H, Kim TD, Kwon BM and Choi I: Cryptotanshinone and
tanshinone IIA enhance IL-15-induced natural killer cell
differentiation. Biochem Biophys Res Commun. 425:340–347. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Li D, Yang Z, Gao S, Zhang H and Fan G:
Tanshinone IIA ameliorates myocardial ischemia/reperfusion injury
in rats by regulation of NLRP3 inflammasome activation and Th17
cells differentiation. Acta Cir Bras. 37:e3707012022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Godo S and Shimokawa H: Endothelial
functions. Arterioscler Thromb Vasc Biol. 37:e108–e114. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Huang KJ, Wang H, Xie WZ and Zhang HS:
Investigation of the effect of tanshinone IIA on nitric oxide
production in human vascular endothelial cells by fluorescence
imaging. Spectrochim Acta A Mol Biomol Spectrosc. 68:1180–1186.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Li YH, Xu Q, Xu WH, Guo XH, Zhang S and
Chen YD: Mechanisms of protection against diabetes-induced
impairment of endothelium-dependent vasorelaxation by Tanshinone
IIA. Biochim Biophys Acta. 1850:813–823. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zhang XS, Ha S, Wang XL, Shi YL, Duan SS
and Li ZA: Tanshinone IIA protects dopaminergic neurons against
6-hydroxydopamine-induced neurotoxicity through
miR-153/NF-E2-related factor 2/antioxidant response element
signaling pathway. Neuroscience. 303:489–502. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Chen J, Bi Y, Chen L, Zhang Q and Xu L:
Tanshinone IIA exerts neuroprotective effects on
hippocampus-dependent cognitive impairments in diabetic rats by
attenuating ER stress-induced apoptosis. Biomed Pharmacother.
104:530–536. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Huang X, Deng H, Shen QK and Quan ZS:
Tanshinone IIA: Pharmacology, total synthesis, and progress in
structure-modifications. Curr Med Chem. 29:1959–1989. 2022.
View Article : Google Scholar
|
|
76
|
Huang X, Jin L, Deng H, Wu D, Shen QK,
Quan ZS, Zhang CH and Guo HY: Research and development of natural
product tanshinone I: Pharmacology, total synthesis, and structure
modifications. Front Pharmacol. 13:9204112022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Ke L, Zhong C, Chen Z, Zheng Z, Li S, Chen
B, Wu Q and Yao H: Tanshinone I: Pharmacological activities,
molecular mechanisms against diseases and future perspectives.
Phytomedicine. 110:1546322023. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Luo L, Xue J, Shao Z, Zhou Z, Tang W, Liu
J, Hu H and Yang F: Recent developments in Salvia miltiorrhiza
polysaccharides: Isolation, purification, structural
characteristics and biological activities. Front Pharmacol.
14:11392012023. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Wang D, Lu C, Sun F, Cui M, Mu H, Duan J
and Geng H: A tanshinone I derivative enhances the activities of
antibiotics against Staphylococcus aureus in vitro and in vivo. Res
Microbiol. 168:46–54. 2017. View Article : Google Scholar
|
|
80
|
Zhao J, Liu H, Hong Z, Luo W, Mu W, Hou X,
Xu G, Fang Z, Ren L, Liu T, et al: Tanshinone I specifically
suppresses NLRP3 inflammasome activation by disrupting the
association of NLRP3 and ASC. Mol Med. 29:842023. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Hu C, He X, Zhang H, Hu X, Liao L, Cai M,
Lin Z, Xiang J, Jia X, Lu G, et al: Tanshinone I limits
inflammasome activation of macrophage via docking into Syk to
alleviate DSS-induced colitis in mice. Mol Immunol. 173:88–98.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Jing X, Xu Y, Cheng W, Guo S, Zou Y and He
L: Tanshinone I induces apoptosis and pro-survival autophagy in
gastric cancers. Cancer Chemother Pharmacol. 77:1171–1181. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Qian Z, Feng N and Geng A: Tanshinone I
suppresses hepatocellular carcinoma cells growth through targeting
DNA double-strand break repair. Cancer Biol Ther. 24:22299582023.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Ho JH and Hong CY: Salvianolic acids:
Small compounds with multiple mechanisms for cardiovascular
protection. J Biomed Sci. 18:302011. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Feng S, Cong H and Ji L: Salvianolic acid
a exhibits anti-inflammatory and antiarthritic effects via
inhibiting NF-κB and p38/MAPK pathways. Drug Des Devel Ther.
14:1771–1778. 2020. View Article : Google Scholar
|
|
86
|
Wu Y, Wang Z, Lin Z, Fu X, Zhan J and Yu
K: Salvianolic acid a has anti-osteoarthritis effect in vitro and
in vivo. Front Pharmacol. 11:6822020. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zhou AM, Xiang YJ, Liu EQ, Cai CH, Wu YH,
Yang LB and Zeng CL: Salvianolic acid a inhibits platelet
activation and aggregation in patients with type 2 diabetes
mellitus. BMC Cardiovasc Disord. 20:152020. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Wang R, Song F, Li S, Wu B, Gu Y and Yuan
Y: Salvianolic acid A attenuates CCl(4)-induced liver fibrosis by
regulating the PI3K/AKT/mTOR, Bcl-2/Bax and caspase-3/cleaved
caspase-3 signaling pathways. Drug Des Devel Ther. 13:1889–1900.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Yang D, Xia X and Xi S: Salvianolic acid A
attenuates arsenic-induced ferroptosis and kidney injury via
HIF-2α/DUOX1/GPX4 and iron homeostasis. Sci Total Environ.
907:1680732024. View Article : Google Scholar
|
|
90
|
Zhu J, Guo J, Liu Z, Liu J, Yuan A, Chen
H, Qiu J, Dou X, Lu D and Le Y: Salvianolic acid A attenuates
non-alcoholic fatty liver disease by regulating the AMPK-IGFBP1
pathway. Chem Biol Interact. 400:1111622024. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Han JY, Fan JY, Horie Y, Miura S, Cui DH,
Ishii H, Hibi T, Tsuneki H and Kimura I: Ameliorating effects of
compounds derived from Salvia miltiorrhiza root extract on
microcirculatory disturbance and target organ injury by ischemia
and reperfusion. Pharmacol Ther. 117:280–295. 2008. View Article : Google Scholar
|
|
92
|
Yu XQ, Xue CC, Zhou ZW, Li CG and Zhou SF:
Tanshinone IIB, a primary active constituent from Salvia
miltiorrhiza, exerts neuroprotective effect via inhibition of
neuronal apoptosis in vitro. Phytother Res. 22:846–850. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Yu XY, Lin SG, Zhou ZW, Chen X, Liang J,
Duan W, Yu XQ, Wen JY, Chowbay B, Li CG, et al: Tanshinone IIB, a
primary active constituent from Salvia miltiorrhza, exhibits
neuro-protective activity in experimentally stroked rats. Neurosci
Lett. 417:261–265. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Ma X, Zhang L, Gao F, Jia W and Li C:
Salvia miltiorrhiza and Tanshinone IIA reduce endothelial
inflammation and atherosclerotic plaque formation through
inhibiting COX-2. Biomed Pharmacother. 167:1155012023. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Fang Y, Duan C, Chen S, Liu Z, Jiang B, Ai
W, Wang L, Xie P and Fang H: Tanshinone-IIA inhibits myocardial
infarct via decreasing of the mitochondrial apoptotic signaling
pathway in myocardiocytes. Int J Mol Med. 48:1582021. View Article : Google Scholar
|
|
96
|
Hu T, Zou HX, Le SY, Wang YR, Qiao YM,
Yuan Y, Liu JC, Lai SQ and Huang H: Tanshinone IIA confers
protection against myocardial ischemia/reperfusion injury by
inhibiting ferroptosis and apoptosis via VDAC1. Int J Mol Med.
52:1092023. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Kang YJ, Jin UH, Chang HW, Son JK, Lee SH,
Son KH, Chang YC, Lee YC and Kim CH: Inhibition of microsomal
triglyceride transfer protein expression and atherogenic risk
factor apolipoprotein B100 secretion by tanshinone IIA in HepG2
cells. Phytother Res. 22:1640–1645. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Zhang Y, Wang J, Yang S, Kou H and Liu P:
Tanshinone IIA alleviate atherosclerosis and hepatic steatosis via
down-regulation of MAPKs/NF-κB signaling pathway. Int
Immunopharmacol. 152:1144652025. View Article : Google Scholar
|
|
99
|
Hong HJ, Hsu FL, Tsai SC, Lin CH, Liu JC,
Chen JJ, Cheng TH and Chan P: Tanshinone IIA attenuates cyclic
strain-induced endothelin-1 expression in human umbilical vein
endothelial cells. Clin Exp Pharmacol Physiol. 39:63–68. 2012.
View Article : Google Scholar
|
|
100
|
Pu X, Wu Q, Yan Z, Zhou S, Zhang Q, Zhang
X, Cai Y, Liu Z, Liu R and Chang X: Tanshinone IIA modulates Sirt5
and Metll3 interaction to govern mitochondria-endoplasmic reticulum
unfolded protein response in coronary microvascular injury.
Phytomedicine. 145:1569822025. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Jin P and Cong S: LOX-1 and
atherosclerotic-related diseases. Clin Chim Acta. 491:24–29. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Chen W, Li X, Guo S, Song N, Wang J, Jia L
and Zhu A: Tanshinone IIA harmonizes the crosstalk of autophagy and
polarization in macrophages via miR-375/KLF4 pathway to attenuate
atherosclerosis. Int Immunopharmacol. 70:486–497. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Yuan L, Li Q, Zhang Z, Liu Q, Wang X and
Fan L: Tanshinone IIA inhibits the adipogenesis and inflammatory
response in ox-LDL-challenged human monocyte-derived macrophages
via regulating miR-130b/WNT5A. J Cell Biochem. 121:1400–1408. 2020.
View Article : Google Scholar
|
|
104
|
Xu S, Little PJ, Lan T, Huang Y, Le K, Wu
X, Shen X, Huang H, Cai Y, Tang F, et al: Tanshinone II-A
attenuates and stabilizes atherosclerotic plaques in
apolipoprotein-E knockout mice fed a high cholesterol diet. Arch
Biochem Biophys. 515:72–79. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Liu X, Guo CY, Ma XJ, Wu CF, Zhang Y, Sun
MY, Pan YT and Yin HJ: Anti-inflammatory effects of tanshinone IIA
on atherosclerostic vessels of ovariectomized ApoE mice are
mediated by estrogen receptor activation and through the ERK
signaling pathway. Cell Physiol Biochem. 35:1744–1755. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Tang F, Wu X, Wang T, Wang P, Li R, Zhang
H, Gao J, Chen S, Bao L, Huang H and Liu P: Tanshinone II A
attenuates atherosclerotic calcification in rat model by inhibition
of oxidative stress. Vascul Pharmacol. 46:427–438. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Feng J, Chen HW, Pi LJ, Wang J and Zhan
DQ: Protective effect of tanshinone IIA against cardiac hypertrophy
in spontaneously hypertensive rats through inhibiting the Cys-C/Wnt
signaling pathway. Oncotarget. 8:10161–10170. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Pang H, Han B, Yu T and Peng Z: The
complex regulation of tanshinone IIA in rats with
hypertension-induced left ventricular hypertrophy. PLoS One.
9:e922162014. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Fang J, Xu SW, Wang P, Tang FT, Zhou SG,
Gao J, Chen JW, Huang HQ and Liu PQ: Tanshinone II-A attenuates
cardiac fibrosis and modulates collagen metabolism in rats with
renovascular hypertension. Phytomedicine. 18:58–64. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Wang P, Wu X, Bao Y, Fang J, Zhou S, Gao
J, Pi R, Mou YG and Liu P: Tanshinone IIA prevents cardiac
remodeling through attenuating NAD (P)H oxidase-derived reactive
oxygen species production in hypertensive rats. Pharmazie.
66:517–524. 2011.PubMed/NCBI
|
|
111
|
Yu ZL, Wang JN, Wu XH, Xie HJ, Han Y, Guan
YT, Qin Y and Jiang JM: Tanshinone IIA Prevents rat basilar artery
smooth muscle cells proliferation by inactivation of PDK1 during
the development of hypertension. J Cardiovasc Pharmacol Ther.
20:563–571. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Weng YS, Wang HF, Pai PY, Jong GP, Lai CH,
Chung LC, Hsieh DJ, HsuanDay C, Kuo WW and Huang CY: Tanshinone IIA
Prevents Leu27IGF-II-induced cardiomyocyte hypertrophy mediated by
estrogen receptor and subsequent Akt activation. Am J Chin Med.
43:1567–1591. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Zhang M, Chen Y, Chen H, Shen Y, Pang L,
Wu W and Yu Z: Tanshinone IIA alleviates cardiac hypertrophy
through m6A modification of galectin-3. Bioengineered.
13:4260–4270. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Zhang Y, Zhang L, Chu W, Wang B, Zhang J,
Zhao M, Li X, Li B, Lu Y, Yang B and Shan H: Tanshinone IIA
inhibits miR-1 expression through p38 MAPK signal pathway in
post-infarction rat cardiomyocytes. Cell Physiol Biochem.
26:991–998. 2010. View Article : Google Scholar
|
|
115
|
Ren ZH, Tong YH, Xu W, Ma J and Chen Y:
Tanshinone II A attenuates inflammatory responses of rats with
myocardial infarction by reducing MCP-1 expression. Phytomedicine.
17:212–218. 2010. View Article : Google Scholar
|
|
116
|
Gao S, Yang Z, Li D, Wang B, Zheng X, Li C
and Fan G: Intervention of Tanshinone IIA on the PGK1-PDHK1 pathway
to reprogram macrophage phenotype after myocardial infarction.
Cardiovasc Drugs Ther. 38:1359–1373. 2024. View Article : Google Scholar
|
|
117
|
Yang F, Li P, Li H, Shi Q, Li S and Zhao
L: microRNA-29b mediates the antifibrotic effect of tanshinone IIA
in postinfarct cardiac remodeling. J Cardiovasc Pharmacol.
65:456–464. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Chai R, Ye Z, Xue W, Shi S, Wei Y, Hu Y
and Wu H: Tanshinone IIA inhibits cardiomyocyte pyroptosis through
TLR4/NF-κB p65 pathway after acute myocardial infarction. Front
Cell Dev Biol. 11:12529422023. View Article : Google Scholar
|
|
119
|
Chen R, Chen W, Huang X and Rui Q:
Tanshinone IIA attenuates heart failure via inhibiting oxidative
stress in myocardial infarction rats. Mol Med Rep. 23:4042021.
View Article : Google Scholar
|
|
120
|
Zhang Y, Wei L, Sun D, Cao F, Gao H, Zhao
L, Du J, Li Y and Wang H: Tanshinone IIA pretreatment protects
myocardium against ischaemia/reperfusion injury through the
phosphatidylinositol 3-kinase/Akt-dependent pathway in diabetic
rats. Diabetes Obes Metab. 12:316–322. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Li Q, Shen L, Wang Z, Jiang HP and Liu LX:
Tanshinone IIA protects against myocardial ischemia reperfusion
injury by activating the PI3K/Akt/mTOR signaling pathway. Biomed
Pharmacother. 84:106–114. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Zhang Z, He H, Qiao Y, Huang J, Wu Z, Xu
P, Yin D and He M: Tanshinone IIA Pretreatment Protects H9c2 cells
against anoxia/reoxygenation injury: Involvement of the
translocation of Bcl-2 to mitochondria mediated by 14-3-3η. Oxid
Med Cell Longev. 2018:35839212018. View Article : Google Scholar
|
|
123
|
Wang Y, Sun W, Shen L, Yu P, Shen Q, Zhou
Y, Yao L and Chen X: Tanshinone IIA protects
ischemia/reperfusion-induced cardiomyocyte injury by inhibiting the
HAS2/FGF9 Axis. Cardiol Res Pract. 2024:25816382024. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Yan K, Yu S, Gao X, Li L and Ding L:
Tanshinone IIA ameliorates myocardial ischemia-reperfusion injury
via activating HDAC1-Repressed Nrf2-xCT/Gpx4/HO-1 Axis. Chem Biol
Drug Des. 105:e700952025. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Sang Y, Du J, Zulikala D and Sang Z:
Mechanistic analysis of Tanshinone IIA's regulation of the
ATM/GADD45/ORC signaling pathway to reduce myocardial
ischemia-reperfusion injury. Front Pharmacol. 15:15103802024.
View Article : Google Scholar
|
|
126
|
Li S, Yang K, Cao W, Guo R, Liu Z, Zhang
J, Fan A, Huang Y, Ma C, Li L and Fan G: Tanshinone IIA enhances
the therapeutic efficacy of mesenchymal stem cells derived exosomes
in myocardial ischemia/reperfusion injury via up-regulating
miR-223-5p. J Control Release. 358:13–26. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Zhai P, Chen Q, Wang X, Ouyang X, Yang M,
Dong Y, Li J, Li Y, Luo S, Liu Y, et al: The combination of
Tanshinone IIA and Astragaloside IV attenuates myocardial
ischemia-reperfusion injury by inhibiting the STING pathway. Chin
Med. 19:342024. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Hong HJ, Liu JC, Chen PY, Chen JJ, Chan P
and Cheng TH: Tanshinone IIA prevents doxorubicin-induced
cardiomyocyte apoptosis through Akt-dependent pathway. Int J
Cardiol. 157:174–179. 2012. View Article : Google Scholar
|
|
129
|
Xu L, He D, Wu Y, Shen L, Wang Y and Xu Y:
Tanshinone IIA inhibits cardiomyocyte apoptosis and rescues cardiac
function during doxorubicin-induced cardiotoxicity by activating
the DAXX/MEK/ERK1/2 pathway. Phytomedicine. 107:1544712022.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Zhang X, Wang Q, Wang X, Chen X, Shao M,
Zhang Q, Guo D, Wu Y, Li C, Wang W and Wang Y: Tanshinone IIA
protects against heart failure post-myocardial infarction via
AMPKs/mTOR-dependent autophagy pathway. Biomed Pharmacother.
112:1085992019. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Mao S, Wang Y, Zhang M and Hinek A:
Phytoestrogen, tanshinone IIA diminishes collagen deposition and
stimulates new elastogenesis in cultures of human cardiac
fibroblasts. Exp Cell Res. 323:189–197. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Yan N, Xiao C, Wang X, Xu Z and Yang J:
Tanshinone IIA from Salvia miltiorrhiza exerts anti-fibrotic
effects on cardiac fibroblasts and rat heart tissues by suppressing
the levels of pro-fibrotic factors: The key role of miR-618. J Food
Biochem. 46:e140782022. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Huang L, Zhu J, Zheng M, Zou R, Zhou Y and
Zhu M: Tanshinone IIA protects against subclinical
lipopolysaccharide induced cardiac fibrosis in mice through
inhibition of NADPH oxidase. Int Immunopharmacol. 60:59–63. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Sun DD, Wang HC, Wang XB, Luo Y, Jin ZX,
Li ZC, Li GR and Dong MQ: Tanshinone IIA: A new activator of human
cardiac KCNQ1/KCNE1 (I(Ks)) potassium channels. Eur J Pharmacol.
590:317–321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Wang JG, Bondy SC, Zhou L, Yang FZ, Ding
ZG, Hu Y, Tian Y, Wen PY, Luo H, Wang F, et al: Protective effect
of Tanshinone IIA against infarct size and increased HMGB1, NFκB,
GFAP and apoptosis consequent to transient middle cerebral artery
occlusion. Neurochem Res. 39:295–304. 2014. View Article : Google Scholar
|
|
136
|
Su W, Lv M, Wang D, He Y, Han H, Zhang Y,
Zhang X, Lv S and Yao L: Tanshinone IIA alleviates traumatic brain
injury by reducing ischemia-reperfusion via the miR-124-5p/FoxO1
Axis. Mediators Inflamm. 2024:74590542024. View Article : Google Scholar
|
|
137
|
Zhou C, Yu Z, Chen T, Chen Q, Zhang Y, Cai
J, Xu C and Yu L: Tanshinone IIA attenuates
cerebral-ischemia-reperfusion-induced neuroinflammation by
inhibiting the TLR4/NF-κB signaling cascade: A study integrating
network pharmacology, bioinformatics, and experimental validation.
Phytomedicine. 149:1575482025. View Article : Google Scholar
|
|
138
|
Yu H, Zhang R, Wang Q, Zhang H, Lei Z, Li
R, Shen W, Zhang Y, Lv S and Qian Y: Tanshinone IIA inhibits
microglial activation and inflammation and relieves cerebral
ischemia-reperfusion injury through TGM2/PANX1. Biochem Genet. Dec
6–2025.Epub ahead of print. View Article : Google Scholar
|
|
139
|
Yu H, Li Y, Yang Y and Qian Y, Gao X, Wang
X, Zhan P, Tang D, Qin M and Qian Y: Tanshinone IIA inhibits
neuronal ferroptosis and relieves cerebral ischemia-reperfusion
injury by regulating miR-449a/ACSL4. Metab Brain Dis. 40:2312025.
View Article : Google Scholar
|
|
140
|
Wang W, Zheng LL, Wang F, Hu ZL, Wu WN, Gu
J and Chen JG: Tanshinone IIA attenuates neuronal damage and the
impairment of long-term potentiation induced by hydrogen peroxide.
J Ethnopharmacol. 134:147–155. 2011. View Article : Google Scholar
|
|
141
|
Cai M, Guo Y, Wang S, Wei H, Sun S, Zhao G
and Dong H: Tanshinone IIA elicits neuroprotective effect through
activating the nuclear factor erythroid 2-related factor-dependent
antioxidant response. Rejuvenation Res. 20:286–297. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Liu L, Zhang X, Wang L, Yang R, Cui L, Li
M, Du W and Wang S: The neuroprotective effects of Tanshinone IIA
are associated with induced nuclear translocation of TORC1 and
upregulated expression of TORC1, pCREB and BDNF in the acute stage
of ischemic stroke. Brain Res Bull. 82:228–233. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Miao Q, Wang R, Sun X, Du S and Liu L:
Combination of puerarin and tanshinone IIA alleviates ischaemic
stroke injury in rats via activating the Nrf2/ARE signalling
pathway. Pharm Biol. 60:1022–1031. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Yang F, Ma N, Li S, Chen F, Huang X, Zhao
L and Cao L: Tanshinone IIA alleviates early brain injury after
subarachnoid hemorrhage in rats by inhibiting the activation of
NF-κB/NLRP3 inflammasome. Biol Pharm Bull. 47:279–291. 2024.
View Article : Google Scholar
|
|
145
|
Falk E: Pathogenesis of atherosclerosis. J
Am Coll Cardiol. 47(Suppl 8): C7–C12. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Xuan Y, Gao Y, Huang H, Wang X, Cai Y and
Luan QX: Tanshinone IIA attenuates atherosclerosis in
apolipoprotein E knockout mice infected with porphyromonas
gingivalis. Inflammation. 40:1631–1642. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Trepels T, Zeiher AM and Fichtlscherer S:
The endothelium and inflammation. Endothelium. 13:423–429. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Maguire JJ, Kuc RE and Davenport AP:
Vasoconstrictor activity of novel endothelin peptide, ET-1(1-31),
in human mammary and coronary arteries in vitro. Br J Pharmacol.
134:1360–1366. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Wang Y, Ruan Y and Wu S: ET-1 regulates
the human umbilical vein endothelial cell cycle by adjusting the
ERβ/FOXN1 signaling pathway. Ann Transl Med. 8:14992020. View Article : Google Scholar
|
|
150
|
Shapouri-Moghaddam A, Mohammadian S,
Vazini H, Taghadosi M, Esmaeili SA, Mardani F, Seifi B, Mohammadi
A, Afshari JT and Sahebkar A: Macrophage plasticity, polarization,
and function in health and disease. J Cell Physiol. 233:6425–6440.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Cao H, Jia Q, Yan L, Chen C, Xing S and
Shen D: Quercetin suppresses the progression of atherosclerosis by
regulating MST1-Mediated Autophagy in ox-LDL-Induced RAW264.7
macrophage foam cells. Int J Mol Sci. 20:60932019. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Rendra E, Riabov V, Mossel DM,
Sevastyanova T, Harmsen MC and Kzhyshkowska J: Reactive oxygen
species (ROS) in macrophage activation and function in diabetes.
Immunobiology. 224:242–253. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Durham AL, Speer MY, Scatena M, Giachelli
CM and Shanahan CM: Role of smooth muscle cells in vascular
calcification: Implications in atherosclerosis and arterial
stiffness. Cardiovasc Res. 114:590–600. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Elliott WJ: Systemic hypertension. Curr
Probl Cardiol. 32:201–259. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Zicha J and Vaněčková I: Altered balance
between vasoconstrictor and vasodilator systems in experimental
hypertension. Physiol Res. 73:901–928. 2024. View Article : Google Scholar
|
|
156
|
Chan P, Liu IM, Li YX, Yu WJ and Cheng JT:
Antihypertension induced by tanshinone IIA isolated from the roots
of salvia miltiorrhiza. Evid Based Complement Alternat Med.
2011:3926272011. View Article : Google Scholar :
|
|
157
|
Musumeci MB, Mastromarino V, Casenghi M,
Tini G, Francia P, Maruotti A, Romaniello A, Magrì D, Lillo R,
Adduci C, et al: Pulmonary hypertension and clinical correlates in
hypertrophic cardiomyopathy. Int J Cardiol. 248:326–332. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Zhang Z, Tang J, Song J, Xie M, Liu Y,
Dong Z, Liu X, Li X, Zhang M, Chen Y, et al: Elabela alleviates
ferroptosis, myocardial remodeling, fibrosis and heart dysfunction
in hypertensive mice by modulating the IL-6/STAT3/GPX4 signaling.
Free Radic Biol Med. 181:130–142. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Deng HW, Ye ZM, Hu RT and Qin C: The role
of long noncoding RNAs in vascular smooth muscle cell phenotype and
the pathogenesis of cardiovascular and cerebrovascular aneurysms. J
Cardiovasc Pharmacol. 84:125–135. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Gao M, Ma MM, Lu FT, Huang CC, Sun L, Lv
XF, Zhang B, Wang GL and Guan YY: Low chloride-regulated ClC-5
contributes to arterial smooth muscle cell proliferation and
cerebrovascular remodeling. Hypertension. 79:e73–e85. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Nakamura M and Sadoshima J: Mechanisms of
physiological and pathological cardiac hypertrophy. Nat Rev
Cardiol. 15:387–407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Tu EY, Zhou YG, Wang ZH, Liang QS and Yang
GT: Effects of tanshinone II A on the myocardial hypertrophy signal
transduction system protein kinase B in rats. Chin J Integr Med.
15:365–370. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Tan X, Li J, Wang X, Chen N, Cai B, Wang
G, Shan H, Dong D, Liu Y, Li X, et al: Tanshinone IIA protects
against cardiac hypertrophy via inhibiting calcineurin/NFATc3
pathway. Int J Biol Sci. 7:383–389. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Lu L, Liu M, Sun R, Zheng Y and Zhang P:
Myocardial infarction: Symptoms and treatments. Cell Biochem
Biophys. 72:865–867. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Liu X, Zhang W, Luo J, Shi W, Zhang X, Li
Z, Qin X, Liu B and Wei Y: TRIM21 deficiency protects against
atrial inflammation and remodeling post myocardial infarction by
attenuating oxidative stress. Redox Biol. 62:1026792023. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Prabhu SD and Frangogiannis NG: The
biological basis for cardiac repair after myocardial infarction:
From inflammation to fibrosis. Circ Res. 119:91–112. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Vasudevan SO, Behl B and Rathinam VA:
Pyroptosis-induced inflammation and tissue damage. Semin Immunol.
69:1017812023. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Zhang J, Huang L, Shi X, Yang L, Hua F, Ma
J, Zhu W, Liu X, Xuan R, Shen Y, et al: Metformin protects against
myocardial ischemia-reperfusion injury and cell pyroptosis via
AMPK/NLRP3 inflammasome pathway. Aging (Albany NY). 12:24270–24287.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Bahit MC, Kochar A and Granger CB:
Post-Myocardial infarction heart failure. JACC Heart Fail.
6:179–186. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Zhu T, Chen J, Zhang M, Tang Z, Tong J,
Hao X, Li H, Xu J and Yang J: Tanshinone IIA exerts
cardioprotective effects through improving gut-brain axis
post-myocardial infarction. Cardiovasc Toxicol. 24:1317–1334. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Algoet M, Janssens S, Himmelreich U, Gsell
W, Pusovnik M, Van den Eynde J and Oosterlinck W: Myocardial
ischemia-reperfusion injury and the influence of inflammation.
Trends Cardiovasc Med. 33:357–366. 2023. View Article : Google Scholar
|
|
172
|
Zhu PC, Shen J, Qian RY, Xu J, Liu C, Hu
WM, Zhang Y and Lv LC: Effect of tanshinone IIA for myocardial
ischemia/reperfusion injury in animal model: preclinical evidence
and possible mechanisms. Front Pharmacol. 14:11652122023.
View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Zhang X, Jiang H, Zhang L, Chen C, Xing M,
Du D, Li Y, Ma Y, Ma Y and Li C: Efficacy of tanshinone IIA in rat
models with myocardial ischemia-reperfusion injury: A systematic
mini-review and meta-analysis. PeerJ. 12:e178852024. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Wang L, Qiu S, Li X, Zhang Y, Huo M and
Shi J: Myocardial-Targeting tannic cerium nanocatalyst attenuates
ischemia/reperfusion injury. Angew Chem Int Ed Engl.
62:e2023055762023. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Ma T, Wang J, Sun G, Li K, Qu H, Wang Y,
Li S and Wu B: Irisin and liraglutide combination therapy mitigates
myocardial reperfusion injury in obese rats: Role of endoplasmic
reticulum stress and apoptosis. Arch Biochem Biophys.
768:1103702025. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Dong Z, Yang L, Jiao J, Jiang Y, Li H, Yin
G, Yang P and Sun L: Aspirin in combination with gastrodin protects
cardiac function and mitigates gastric mucosal injury in response
to myocardial ischemia/reperfusion. Front Pharmacol. 13:9951022022.
View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Rogers C and Bush N: Heart failure:
Pathophysiology, diagnosis, medical treatment guidelines, and
nursing management. Nurs Clin North Am. 50:787–799. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Tanai E and Frantz S: Pathophysiology of
heart failure. Compr Physiol. 6:187–214. 2015. View Article : Google Scholar
|
|
179
|
Boorsma EM, Ter Maaten JM, Damman K, Dinh
W, Gustafsson F, Goldsmith S, Burkhoff D, Zannad F, Udelson JE and
Voors AA: Congestion in heart failure: A contemporary look at
physiology, diagnosis and treatment. Nat Rev Cardiol. 17:641–655.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Du J, Liu Y and Fu J: Autophagy and heart
failure. Adv Exp Med Biol. 1207:223–227. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Shirakabe A, Zhai P, Ikeda Y, Saito T,
Maejima Y, Hsu CP, Nomura M, Egashira K, Levine B and Sadoshima J:
Drp1-Dependent mitochondrial autophagy plays a protective role
against pressure overload-induced mitochondrial dysfunction and
heart failure. Circulation. 133:1249–1263. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Espeland T, Lunde IG, Amundsen BA,
Gullestad L and Aakhus S: Myocardial fibrosis. Tidsskr Nor
Laegeforen. Oct 12–2018.Epub ahead of print. View Article : Google Scholar
|
|
183
|
Tamiato A, Tombor LS, Fischer A,
Muhly-Reinholz M, Vanicek LR, Toğru BN, Neitz J, Glaser SF, Merten
M, Rodriguez Morales D, et al: Age-dependent RGS5 loss in pericytes
induces cardiac dysfunction and fibrosis. Circ Res. 134:1240–1255.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Deng Y, Liu X, Xie M, Zhao R, Ji L, Tang
K, Yang W, Ou W, Xie M and Li T: Obesity enables NLRP3 activation
and induces myocardial fibrosis via hyperacetylation of HADHa.
Diabetes. 72:1597–1608. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Li XM, Jiang L, Liu X, Guo YK, Li Y, Gao
Y, Han PL, Xia CC, Peng WL, Hu BY, et al: Myocardial interstitial
fibrosis and subclinical left ventricular systolic dysfunction in
type 2 diabetes mellitus patients with obesity: A 3.0 T MRI study.
Cardiovasc Diabetol. 25:222025. View Article : Google Scholar
|
|
186
|
Fu DG: Cardiac arrhythmias: Diagnosis,
symptoms, and treatments. Cell Biochem Biophys. 73:291–296. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Shan H, Li X, Pan Z, Zhang L, Cai B, Zhang
Y, Xu C, Chu W, Qiao G, Li B, et al: Tanshinone IIA protects
against sudden cardiac death induced by lethal arrhythmias via
repression of microRNA-1. Br J Pharmacol. 158:1227–1235. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
188
|
He Z, Sun C, Xu Y and Cheng D: Reduction
of atrial fibrillation by Tanshinone IIA in chronic heart failure.
Biomed Pharmacother. 84:1760–1767. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Jurcau A and Simion A: Neuroinflammation
in cerebral ischemia and ischemia/reperfusion injuries: From
pathophysiology to therapeutic strategies. Int J Mol Sci.
23:142021. View Article : Google Scholar
|
|
190
|
Jiang F, Xu C, Fan X, Yang S, Fan W, Li M,
Song J, Wei W, Chen H, Zhong D and Li G: MyD88 inhibition
attenuates cerebral ischemia-reperfusion injury by regulating the
inflammatory response and reducing blood-brain barrier damage.
Neuroscience. 549:121–137. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Chen X, Zhou ZW, Xue CC, Li XX and Zhou
SF: Role of P-glycoprotein in restricting the brain penetration of
tanshinone IIA, a major active constituent from the root of Salvia
miltiorrhiza Bunge, across the blood-brain barrier. Xenobiotica.
37:635–678. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Guzik A and Bushnell C: Stroke
epidemiology and risk factor management. Continuum (Minneap Minn).
23(1, Cerebrovascular Disease): 15–39. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Zhao Y, Zhang X, Chen X and Wei Y:
Neuronal injuries in cerebral infarction and ischemic stroke: From
mechanisms to treatment (Review). Int J Mol Med. 49:152022.
View Article : Google Scholar :
|
|
194
|
Campbell BCV, De Silva DA, Macleod MR,
Coutts SB, Schwamm LH, Davis SM and Donnan GA: Ischaemic stroke.
Nat Rev Dis Primers. 5:702019. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Fang M, Huang DR, Zhang JW, Liao WJ, Wu F
and Liu YW: Tanshinone Ⅱ_A exerts anti-hepatocellular carcinoma
effects by inhibiting oxidative stress via PI3K/Akt and Nrf2/HO-1
signaling pathway. Zhongguo Zhong Yao Za Zhi. 49:6724–6734. 2024.In
Chinese.
|
|
196
|
Liu H, Wang L, Hu M, Hua J, Lian X, Lian C
and Zhang J: Brusatol regulates ferroptosis of ovarian cancer
through the Nrf2/HO-1/NQO1 and AKT/mTOR double signaling pathways.
Curr Pharm Des. Aug 19–2025.Epub ahead of print. View Article : Google Scholar
|
|
197
|
Fu C, Wu Y, Liu S, Luo C, Lu Y, Liu M,
Wang L, Zhang Y and Liu X: Rehmannioside A improves cognitive
impairment and alleviates ferroptosis via activating PI3K/AKT/Nrf2
and SLC7A11/GPX4 signaling pathway after ischemia. J
Ethnopharmacol. 289:1150212022. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Wu X, Wang J, Song L, Guan Y, Cao C, Cui
Y, Zhang Y and Liu C: Catalpol weakens depressive-like behavior in
mice with streptozotocin-induced hyperglycemia via
PI3K/AKT/Nrf2/HO-1 signaling pathway. Neuroscience. 473:102–118.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Chen Q, Wang Q, Zhao Y, Xie X, Feng H, Wei
Y, Yu M, Pan X and Hu T: Flavonoids from Polygonum hydropiper L.
regulate PCV2-induced oxidative stress of RAW264.7 cells via
Pi3k/AKT and Nrf2/HO-1 signaling pathways. Sci Rep. 15:291122025.
View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Huang Q, Zhang C, Dong S, Han J, Qu S, Xie
T, Zhao H and Shi Y: Asafoetida exerts neuroprotective effect on
oxidative stress induced apoptosis through PI3K/Akt/GSK3β/Nrf2/HO-1
pathway. Chin Med. 17:832022. View Article : Google Scholar
|
|
201
|
Hannawi Y: Cerebral small vessel disease:
A review of the pathophysiological mechanisms. Transl Stroke Res.
15:1050–1069. 2024. View Article : Google Scholar
|
|
202
|
Katusic ZS, d'Uscio LV and He T:
Cerebrovascular endothelial dysfunction: Role of BACE1.
Arterioscler Thromb Vasc Biol. 44:1737–1747. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
You N, Liu G, Yu M, Chen W, Fei X, Sun T,
Han M, Qin Z, Wei Z and Wang D: Reconceptualizing
Endothelial-to-mesenchymal transition in atherosclerosis: Signaling
pathways and prospective targeting strategies. J Adv Res.
77:419–441. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Waters ES, Kaiser EE, Yang X, Fagan MM,
Scheulin KM, Jeon JH, Shin SK, Kinder HA, Kumar A, Platt SR, et al:
Intracisternal administration of tanshinone IIA-loaded
nanoparticles leads to reduced tissue injury and functional
deficits in a porcine model of ischemic stroke. IBRO Neurosci Rep.
10:18–30. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Zhao Y, Wang J, Zhang Z, Kong L, Liu M,
Chen M and Gao L: A ROS-responsive TPP-modified tanshinone IIA
micelle improves DOX-induced heart failure. Int J Pharm.
672:1253182025. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Zhang W, Li J, Liu J, Wu Z, Xu Y and Wang
J: Tanshinone IIA-loaded reconstituted high density lipoproteins:
Atherosclerotic plaque targeting mechanism in a foam cell model and
pharmacokinetics in rabbits. Pharmazie. 67:324–330. 2012.PubMed/NCBI
|
|
207
|
Zhang W, He H, Liu J, Wang J, Zhang S,
Zhang S and Wu Z: Pharmacokinetics and atherosclerotic lesions
targeting effects of tanshinone IIA discoidal and spherical
biomimetic high density lipoproteins. Biomaterials. 34:306–319.
2013. View Article : Google Scholar
|
|
208
|
Zhang S, Li J, Hu S, Wu F and Zhang X:
Triphenylphosphonium and D-α-tocopheryl polyethylene glycol 1000
succinate-modified, tanshinone IIA-loaded lipid-polymeric
nanocarriers for the targeted therapy of myocardial infarction. Int
J Nanomedicine. 13:4045–4057. 2018. View Article : Google Scholar
|
|
209
|
Fan L, Qu H, Wang B, Li HZ, Yang WW, Guo
H, Zhang SS, Long LZ, Liu Y, Zhou G, et al: Delivery of liquid
metal particles and tanshinone IIA into the pericardial cavity for
myocardial infarction treatment. J Mater Chem B. 12:11916–11925.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Ma H, Hu ZC, Long Y, Cheng LC, Zhao CY and
Shao MK: Tanshinone IIA microemulsion protects against cerebral
ischemia reperfusion injury via regulating H3K18ac and H4K8ac in
vivo and in vitro. Am J Chin Med. 50:1845–1868. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Liu X, Ye M, An C, Pan L and Ji L: The
effect of cationic albumin-conjugated PEGylated tanshinone IIA
nanoparticles on neuronal signal pathways and neuroprotection in
cerebral ischemia. Biomaterials. 34:6893–6905. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Wang D, Lv L, Xu Y, Jiang K, Chen F, Qian
J, Chen M, Liu G and Xiang Y: Cardioprotection of Panax notoginseng
saponins against acute myocardial infarction and heart failure
through inducing autophagy. Biomed Pharmacother. 136:1112872021.
View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Liang Y, Chen B, Liang D, Quan X, Gu R,
Meng Z, Gan H, Wu Z, Sun Y, Liu S and Dou G: Pharmacological
effects of astragaloside IV: A review. Molecules. 28:61182023.
View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Zheng Q, Bao XY, Zhu PC, Tong Q, Zheng GQ
and Wang Y: Ginsenoside Rb1 for myocardial ischemia/reperfusion
injury: Preclinical evidence and possible mechanisms. Oxid Med Cell
Longev. 2017:63136252017. View Article : Google Scholar
|
|
215
|
Ma J, Hou D, Wei Z, Zhu J, Lu H, Li Z,
Wang X, Li Y, Qiao G and Liu N: Tanshinone IIA attenuates cerebral
aneurysm formation by inhibiting the NF-κB-mediated inflammatory
response. Mol Med Rep. 20:1621–1628. 2019.PubMed/NCBI
|
|
216
|
Lu Y, Yan Y and Liu X: Effects of
alprostadil combined with tanshinone IIa injection on
microcirculation disorder, outcomes, and cardiac function in AMI
patients after PCI. Ann Palliat Med. 10:97–103. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Xu Q, Li H, Zhang X, Ding H and Cao J:
Tanshinone IIA elevates serum soluble klotho levels and decreases
cardiovascular events in patients on maintenance hemodialysis: a
prospective before-after study. Ann Palliat Med. 9:2131–2140. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Zhang M, Liu Y, Sun X, Zhang X and Hua L:
Salvia miltiorrhiza adjuvant therapy facilitates cardiac function
recovery in patients with myocardial infarction. Pak J Pharm Sci.
38:45–53. 2025.PubMed/NCBI
|
|
219
|
Mao S, Taylor S, Chen Q, Zhang M and Hinek
A: Sodium tanshinone IIA sulfonate prevents the adverse left
ventricular remodelling: Focus on polymorphonuclear
neutrophil-derived granule components. J Cell Mol Med.
23:4592–4600. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Mao S, Wang L, Zhao X, Guo L, Lin Q, Wang
X, Dai X, Shang H, Zhang M and Hinek A: Efficacy of sodium
tanshinone IIA sulfonate in patients with Non-ST elevation acute
coronary syndrome undergoing percutaneous coronary intervention:
results from a multicentre, controlled, randomized trial.
Cardiovasc Drugs Ther. 35:321–329. 2021. View Article : Google Scholar
|
|
221
|
Zhou ZY, Zhao WR, Zhang J, Chen XL and
Tang JY: Sodium tanshinone IIA sulfonate: A review of
pharmacological activity and pharmacokinetics. Biomed Pharmacother.
118:1093622019. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Guan R, Yao H, Li Z, Qian J, Yuan L, Cai
Z, Ding M, Liu W, Xu J, Li Y, et al: Sodium tanshinone IIA
sulfonate attenuates cigarette smoke extract-induced mitochondrial
dysfunction, oxidative stress, and apoptosis in alveolar epithelial
cells by enhancing SIRT1 pathway. Toxicol Sci. 183:352–362. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Lan J, Li K, Gresham A and Miao J:
Tanshinone IIA sodium sulfonate attenuates inflammation by
upregulating circ-Sirt1 and inhibiting the entry of NF-κB into the
nucleus. Eur J Pharmacol. 914:1746932022. View Article : Google Scholar
|
|
224
|
Wu P, Du Y, Xu Z, Zhang S, Liu J, Aa N and
Yang Z: Protective effects of sodium tanshinone IIA sulfonate on
cardiac function after myocardial infarction in mice. Am J Transl
Res. 11:351–360. 2019.PubMed/NCBI
|
|
225
|
Zhong L, Ding W, Zeng Q, He B, Zhang H,
Wang L, Fan J, He S, Zhang Y and Wei A: Sodium tanshinone IIA
sulfonate attenuates erectile dysfunction in rats with
hyperlipidemia. Oxid Med Cell Longev. 2020:72869582020.PubMed/NCBI
|
|
226
|
Li S, Jiao Y, Wang H, Shang Q, Lu F, Huang
L, Liu J, Xu H and Chen K: Sodium tanshinone IIA sulfate adjunct
therapy reduces high-sensitivity C-reactive protein level in
coronary artery disease patients: A randomized controlled trial.
Sci Rep. 7:174512017. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Ji B, Zhou F, Han L, Yang J, Fan H, Li S,
Li J, Zhang X, Wang X, Chen X and Xu Y: Sodium tanshinone IIA
sulfonate enhances effectiveness Rt-PA treatment in acute ischemic
stroke patients associated with ameliorating blood-brain barrier
damage. Transl Stroke Res. 8:334–340. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Wu Q, Guan YB, Zhang KJ, Li L and Zhou Y:
Tanshinone IIA mediates protection from diabetes kidney disease by
inhibiting oxidative stress induced pyroptosis. J Ethnopharmacol.
316:1166672023. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Xu Z, Cai K, Su SL, Zhu Y, Liu F and Duan
JA: Salvianolic acid B and tanshinone IIA synergistically improve
early diabetic nephropathy through regulating PI3K/Akt/NF-κB
signaling pathway. J Ethnopharmacol. 319:1173562024. View Article : Google Scholar
|
|
230
|
Chen S, Li P, Shi K, Tang S, Zhang W, Peng
C, Li T, Xie H, Liu C and Zhou J: Tanshinone IIA promotes
ferroptosis in cutaneous melanoma via STAT1-mediated upregulation
of PTGS2 expression. Phytomedicine. 141:1567022025. View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Zhang H, Long M, Wu Z, Han X and Yu Y:
Sodium tanshinone IIA silate as an add-on therapy in patients with
unstable angina pectoris. J Thorac Dis. 6:1794–1799. 2014.
|
|
232
|
Wu X, Fan M, Wei S and Guo D: The efficacy
and safety of sodium tanshinone ⅡA sulfonate injection in the
treatment of unstable angina pectoris: A systematic review and
meta-analysis. PLoS One. 18:e02908412023. View Article : Google Scholar
|
|
233
|
Liu F, Zhao Q, Sun L, Yan M, Gao M, Wang
H, Li Z, Zhou D, Li C, Li T, et al: A systematic review and
meta-analysis of randomized controlled trials on the efficacy and
safety of tanshinone IIA sodium sulfonate injection as adjunctive
therapy for stroke. J Ethnopharmacol. 353:1204132025. View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Zhao R, Wang H, Li S, Wang L, Lei X, Peng
P, Du L and Dong J: Tanshinone IIA@β-cyclodextrin encapsulated with
Eucommia ulmoides rubber/acetylated starch film as novel oral
delivery system for therapy of orthotopic colon cancer. Carbohydr
Polym. 353:1232952025. View Article : Google Scholar
|