Introduction
Fetal growth restriction (FGR) refers to a condition
in which the birth weight of a fetus is below two standard
deviations of the mean weight for the same gestational age, or
below the 10th percentile of the normal weight for that gestational
age (1). FGR is one of the major
complications in perinatal medicine, with a mortality rate four to
six times higher than that of normal neonates (2). The etiology of FGR is
multifactorial and complex, and it remains incompletely elucidated.
Current evidence suggests that key contributing factors include
maternal malnutrition or selective eating habits (3), gestational hypertension (4), intrauterine infections (5), fetal chromosomal abnormalities
(6), reduced levels of certain
hormones (7), umbilical cord
torsion (8) and diverse
placental pathologies, including preeclampsia, FGR, maternal and
fetal vascular malperfusion, and chronic villitis (9). Collectively, these factors result
in reduced placental blood flow and diminished perfusion, which
ultimately compromise the nutrient supply to the fetus (1). FGR not only hampers fetal
development, leading to outcomes such as stillbirth and neonatal
asphyxia, but also exerts long-term effects on physical and
cognitive development during childhood and adolescence (7). Furthermore, individuals who
experienced FGR in utero are at a higher risk of cardiovascular,
neurological and metabolic disorders in adulthood compared with
their peers (2). Consequently,
the prevention and management of FGR are of considerable clinical
importance, with implications for maternal and child health as well
as the promotion of favorable perinatal outcomes.
Oxidative stress arises from an imbalance between
the generation of reactive oxygen species (ROS) and the biological
system's capacity to neutralize or repair their effects (10). ROS, also referred to as free
radicals, are highly reactive oxygen-containing molecules with
unpaired electrons. They readily trigger chain oxidation reactions
that damage DNA, RNA, lipids and proteins, which in turn leads to
cellular dysfunction and, in severe cases, organ failure.
Antioxidants exert a protective role by scavenging ROS or reducing
free radicals (10). During
pregnancy, the placenta, due to its high metabolic rate and
mitochondrial activity, constitutes a major source of ROS (11). While moderate oxidative stress is
essential for placental function, particularly during early
gestation when it facilitates trophoblast invasion, placental
remodeling and angiogenesis, as well as in late pregnancy when it
participates in the initiation of parturition (12), excessive oxidative stress can
disrupt placental formation. This imbalance may trigger immune
dysregulation and functional impairment, which are closely
associated with pregnancy complications, including FGR (13).
The present review outlines the role of redox
imbalance in FGR, highlighting the principal sources of oxidative
stress and discussing its regulatory and promoting effects on the
pathogenesis of FGR from three perspectives: Endoplasmic reticulum
(ER) stress, metabolic reprogramming and epigenetic regulation
(Fig. 1). Finally, based on
current evidence, the present review summarizes advances in
preclinical and clinical research on antioxidant interventions for
FGR, with the aim of providing insights for translational
applications and future therapeutic strategies.
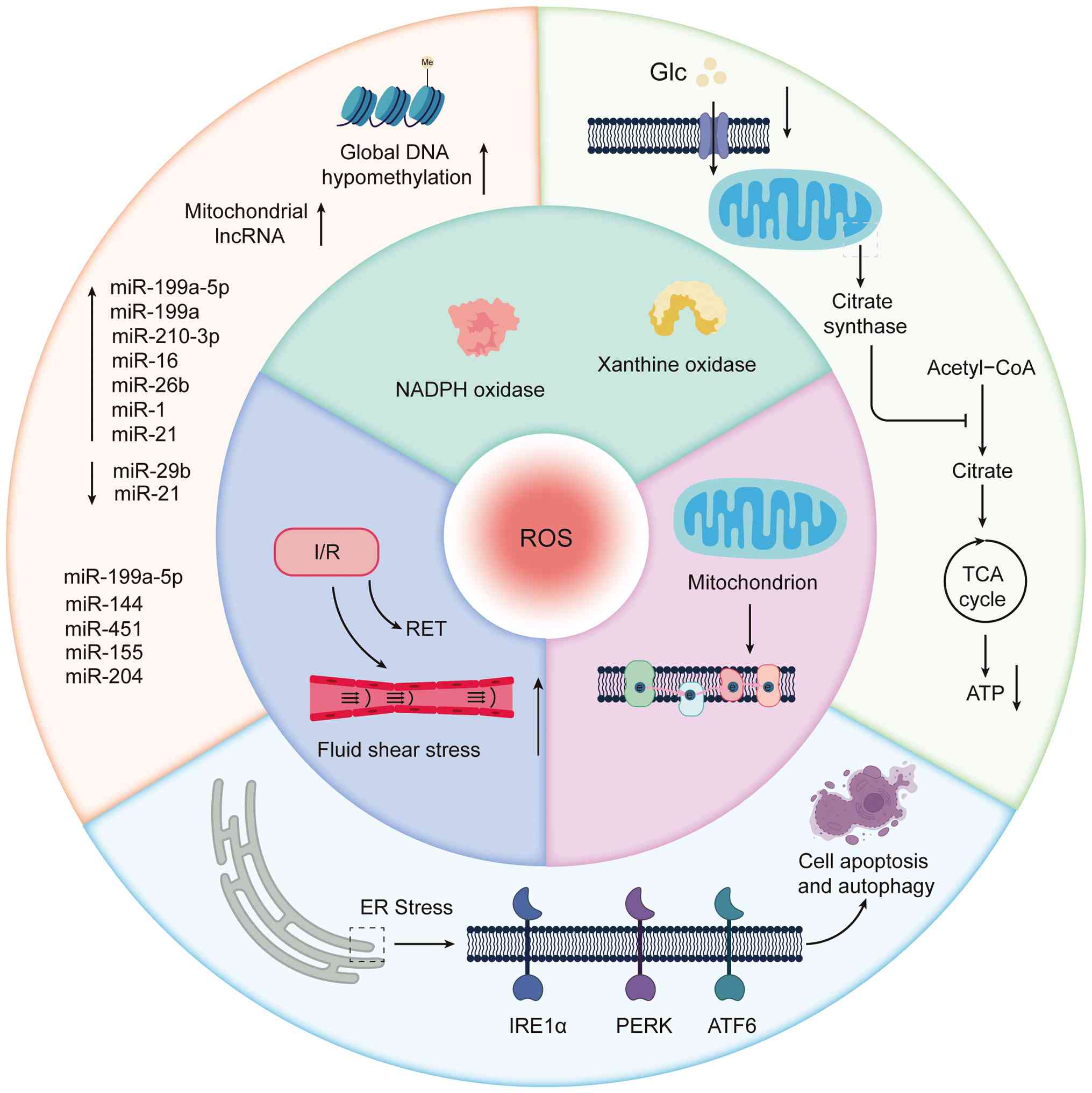 | Figure 1Major sources of ROS and their
molecular roles in the pathogenesis of FGR. ROS are mainly
generated from three sources: Enzyme-derived ROS, mitochondrial ROS
and I/R-induced ROS. Enzyme-derived ROS are mainly produced by NOX
and xanthine oxidase, while mitochondrial ROS mainly result from
electron leakage at complexes I and III of the electron transport
chain. During I/R, RET occurs at complex I. ER stress regulates
cell apoptosis and autophagy via the IRE1α, PERK and ATF6 signaling
pathways, thereby contributing to the progression of FGR. In FGR,
glucose uptake is reduced, the mitochondrial structure is
disrupted, and the TCA cycle is impaired, ultimately resulting in
decreased ATP production. Furthermore, epigenetic regulation also
serves a key role in FGR. The placenta of growth-restricted fetuses
exhibits global hypomethylation in hypoxia-related pathways,
accompanied by marked upregulation of mitochondrial lncRNAs.
Several miRNAs, including miR-199a-5p, miR-155, miR-16, miR-29b,
miR-204, miR-1 and miR-21, are also implicated in placental
development and are dynamically regulated by oxidative stress. ROS,
reactive oxygen species; I/R, ischemia-reperfusion; NOX, NADPH
oxidase; RET, reverse electron transport; ER, endoplasmic
reticulum; FGR, fetal growth restriction; TCA, tricarboxylic acid;
lncRNA/lnc, long noncoding RNA; miRNA/miR, microRNA; PERK, protein
kinase RNA-like endoplasmic reticulum kinase; ATF6, activating
transcription factor 6; IRE1α, inositol-requiring enzyme 1α; Glc,
glucose. |
Origins and mechanisms of oxidative
stress
Placental redox imbalance and oxidative
stress markers
Placental oxidative stress is a central pathogenic
mechanism underlying placental dysfunction in pregnancies
complicated by FGR (14). During
early gestation, a mild degree of oxidative stress occurs naturally
in the placenta. This physiological phenomenon is critical for
vascular formation and angiogenesis within the placenta. Evidence
indicates that excessive use of potent antioxidants during this
stage, which reduces ROS levels, can suppress angiogenesis and
increase the risk of FGR (14).
However, when oxidative stress exceeds the physiological range, the
remodeling of spiral arteries becomes impaired, thereby limiting
trophoblast invasion. Failure of spiral artery remodeling leads to
local ischemia, followed by reperfusion and reoxygenation, which
together create a cyclic hypoxia-reoxygenation process. This
process is considered a major trigger of enhanced local oxidative
stress and can activate the mitochondrial respiratory chain as well
as ROS-generating enzymes such as NADPH oxidase (NOX) and xanthine
oxidase (XO) (15). The major
enzymatic, mitochondrial and ischemia-reperfusion-related sources
of ROS and their downstream effects on ER stress, cellular
metabolism and epigenetic regulation in FGR are summarized in
Fig. 1. Increased activity of
these enzymes promotes excessive ROS production, disrupting the
redox balance of the placenta and ultimately inducing damage to
DNA, proteins and lipids (16).
Furthermore, oxidative metabolites released by the placenta,
particularly lipid peroxidation products, may enter the maternal
circulation. This process enhances oxidation of maternal
low-density lipoprotein, sustaining lipid peroxidation cascades and
resulting in maternal vascular dysfunction (17,18). To provide a more direct
representation of the pathological trends, previous studies
(19-23) have summarized key oxidative
stress biomarkers in FGR, as shown in Table I.
 | Table IAlterations in oxidative stress
biomarkers in FGR. |
Table I
Alterations in oxidative stress
biomarkers in FGR.
| Authors, year | Biomarker | Sample | Model | Observed
change | (Refs.) |
|---|
| Biri et al,
2007 | MDA | Maternal plasma,
cord blood, placenta | Human FGR | Higher in maternal
plasma, cord plasma and placenta compared with controls | (19) |
| Zhao et al,
2020 | MDA | Placenta | FGR mouse
model | Placental MDA
increased; fetal and placental weights reduced; placental function
impaired | (23) |
| Biri et al,
2007 | Xanthine
oxidase | Maternal plasma,
cord blood, placenta | Human FGR all three
samples | XO activity was
increased in | (19) |
| Biri et al,
2007 | Total antioxidant
capacity | Maternal plasma,
cord blood, placenta | Human FGR | Total antioxidant
capacity was decreased in all three samples | (19) |
| Biri et al,
2007 | Superoxide
dismutase | Maternal plasma,
cord blood | Human FGR | SOD activity was
increased in maternal and cord blood | (19) |
| Biri et al,
2007 | Glutathione
peroxidase | Maternal blood,
placenta | Human FGR | Enzyme activity
increased | (19) |
| Biri et al,
2007 | Catalase | Cord blood,
placenta | Human FGR | Enzyme activity
reduced | (19) |
| Kimura et
al, 2013 | d-ROMs | Maternal blood | Preeclampsia with
or without FGR, human | Elevated in PE with
or without FGR | (22) |
| Fujimaki et
al, 2011 | d-ROMs | Umbilical cord
blood | Preeclampsia with
FGR, human | Increased only when
FGR coexisted with PE | (21) |
| Fujimaki et
al, 2011; Kimura et al, 2013 | 8-OHdG | Placenta | Preeclampsia with
or without FGR, human | Oxidative DNA
damage marker elevated | (21,22) |
| Fujimaki et
al, 2011 | Ref-1 | Placenta | Preeclampsia with
or without FGR, human | Increased in
preeclampsia without FGR; not increased in preeclampsia with FGR,
suggesting limited repair response | (21) |
| Tasta et al,
2021 | γH2AX | Placenta | FGR mouse
model | Elevated in FGR
mouse | (20) |
| Tasta et al,
2021 | 4HNE protein
adducts | Placenta | FGR mouse model;
human trophoblasts in vitro | SIRT1-4HNE adducts
detected in FGR mouse placentas; 4HNE induced SA-β-gal activity in
HTR-8/SVneo cells | (20) |
| Kimura et
al, 2013 | GSH | Placenta | FGR mouse
model | Decreased | (22) |
Compared with normal pregnancies, higher levels of
malondialdehyde (MDA) have been detected in maternal blood, cord
blood and placental tissue from FGR-related pregnancies (19). A recent meta-analysis including
48 studies and 4,684 newborns reported a moderate pooled effect
size for cord blood MDA levels in FGR neonates compared with
controls, supporting a robust association between elevated MDA and
FGR at the population level; however, because nearly all included
studies used cross-sectional or case-control designs, these data
cannot demonstrate from an epidemiological standpoint that
increased MDA levels are a direct causal driver of FGR (24). The same meta-analysis further
demonstrated that the direction and magnitude of MDA differences
varied substantially across studies according to the diagnostic
criteria for FGR, the presence or absence of concomitant
preeclampsia, and the gestational age at sampling, resulting in
considerable heterogeneity in the sensitivity and specificity of
MDA when used as a stand-alone screening marker. Therefore, MDA
appears more suitable as a biomarker for risk stratification of
fetal exposure to an oxidative lipid milieu rather than as an
independent diagnostic or predictive tool (24). In a human FGR cohort, XO activity
was increased, whereas total antioxidant capacity was decreased in
maternal plasma, cord blood and placental tissue, further
indicating a systemic shift toward a pro-oxidant state (19). In preeclampsia, the
concentrations of MDA and 4-hydroxynonenal (4HNE) in both placenta
and plasma are also markedly elevated (25). 4HNE can form adducts with
proteins, including sirtuin 1 (SIRT1), thereby mediating protein
modification under oxidative stress (20). Interactions between SIRT1 and
4HNE have also been observed in placental tissue from mouse models
of FGR (20). An in vitro
study has further demonstrated that 4HNE induces increased
senescence-associated-β-galactosi dase activity and acetylated
protein accumulation in human trophoblast cells (HTR-8/SVneo),
consistent with the formation of 4HNE-SIRT1 adducts (20). Collectively, these findings
indicate that the accumulation of 4HNE protein adducts, including
4HNE-SIRT1 conjugates, is a characteristic feature of placental
oxidative injury in FGR and related disorders (20).
Derivatives of reactive oxygen metabolites (d-ROMs)
represent another oxidative stress indicator and are elevated in
preeclamptic pregnancies regardless of the presence of FGR
(21,22). In cord blood, increased d-ROM
levels have been detected only in preeclamptic cases complicated by
FGR, but not in isolated preeclampsia (21). In the same placental specimens,
expression of redox factor-1, a redox-sensitive DNA repair protein,
was increased in preeclampsia without FGR but not in preeclampsia
complicated by FGR, suggesting a relatively limited activation of
redox-regulated repair pathways when FGR coexists (21). DNA oxidative damage markers such
as 8-hydroxy-2'-deoxyguanosine (8-OHdG) are also upregulated in
placentas from patients with preeclampsia, with or without FGR
(21,22). In addition, elevated levels of
histone H2A.X phosphorylated at serine 139, a marker of DNA
double-strand breaks, have been reported in placental tissue from
mouse models of FGR (20). A
recent scoping review of multiple cord blood oxidative stress
markers found that reports linking d-ROMs to FGR are few in number
and based on relatively small sample sizes, suggesting that its
stand-alone diagnostic and predictive utility in clinical practice
may be limited (26).
The occurrence of FGR is also closely linked to
dysregulation of the placental antioxidant defense system (19). In FGR pregnancies, increased
activity of superoxide dismutase (SOD) and glutathione peroxidase
(GPx) has been observed in maternal blood, cord blood and placental
tissue, whereas catalase (CAT) levels are reduced (19). A recent meta-analysis
demonstrated that SOD and CAT activities in FGR neonates are
overall lower than in controls, with effect sizes even exceeding
those observed for MDA, suggesting that under conditions of more
severe disease or prolonged exposure, antioxidant enzyme activity
may shift from an early compensatory upregulation to a later-stage
depletion, thereby amplifying lipid peroxidation and DNA damage
(24). The pathogenic role of
oxidative stress in FGR and preeclampsia has also been validated in
animal models. For example, microcystin-LR (MC-LR), a
cyanobacterial toxin, reduced fetal and placental weights in mice
(23). Histopathological
findings revealed decreased vascular density in the placental
labyrinth layer of MC-LR-treated mice, accompanied by reduced
expression of key placental factors such as VEGFA and placental
growth factor (PlGF), as well as nutrient transporters, including
glucose transporter 1 and proton-coupled folate transporter. These
changes were associated with increased MDA levels and diminished
antioxidant capacity, including reduced glutathione (GSH) levels,
total antioxidant capacity and enzymatic activity, which are
indicative of oxidative stress-mediated injury (23). Furthermore, ER stress signaling
was activated in placentas, further impairing placental development
and reducing fetal weight (23).
Similarly, Toblli et al (27) demonstrated that iron-deficiency
anemia in rat models increased placental MDA and SOD1 activity,
while reducing GSH, CAT and GPx activity, leading to FGR and
smaller litter sizes. In another study, Huang et al
(28) showed that mangiferin, a
natural antioxidant, effectively alleviated hypertension and
proteinuria, improved fetal weight, and reduced placental and
maternal oxidative stress in a preeclampsia mouse model complicated
by FGR. These effects, reflected by decreased MDA levels and
increased SOD, GPx and GSH levels, may involve activation of the
mTOR signaling pathway, which helps regulate oxidative stress
responses (28). A prospective
clinical study demonstrated that, in women with severe FGR
complicated by preeclampsia, systemic free thiol levels were
markedly reduced, whereas plasma ischemia modified albumin (IMA)
concentrations were increased and associated with blood pressure
and the extent of placental histopathological damage, suggesting
that systemic oxidative markers measured in maternal blood may
provide incremental information on disease severity and complement
placenta- or fetus-derived indices, although their predictive
performance still requires validation in larger cohorts (29). A recent scoping review of cord
blood oxidative stress biomarkers further indicated that indices of
overall oxidative status, CAT, GSH and IMA showed relatively
consistent associations with FGR across multiple studies, whereas
results for individual ROS or single antioxidant enzymes were often
highly variable, supporting the notion that the integrated status
of the antioxidant network is more clinically interpretable than
any single marker alone (25).
Overall, extensive evidence from human studies and
animal models consistently indicates that enhanced oxidative stress
combined with impaired antioxidant defense serves a pivotal role in
the pathogenesis and progression of FGR.
Major sources of oxidative stress in the
placenta
Enzymatic drivers of ROS generation:
NOXs and XO
i) NOXs. The NOX family comprises transmembrane
proteins that transfer electrons to O2, thereby
generating superoxide anions (O2•−) (Fig. 2) (30). This family includes seven
members, namely NOX1-5 and dual oxidase 1-2 (30). Matsubara and Sato (31) have detected NOX enzymatic
activity in the microvillous membrane of human
syncytiotrophoblasts, with activity most evident at 25 weeks of
gestation but undetectable before 12 weeks. The absence of activity
in early pregnancy may reflect the low-oxygen environment of the
placenta at that stage (31).
Their study also compared NOX activity between FGR placentas and
normal placentas, and found no significant differences.
Nevertheless, subtle or qualitative changes cannot be excluded
because enzyme histochemistry may lack sensitivity to detect minor
alterations (31). Experimental
evidence has demonstrated that NOX1, NOX2 and NOX5 are present in
the chorionic villi, mainly localized in trophoblast cells
(32). In early villi, NOX
enzymes are the predominant source of O2•− before 10
weeks of gestation, with p47-phagocyte oxidase (phox) localized at
the apical membrane of syncytiotrophoblasts (33). Increased superoxide levels are
associated with activation of the p38MAPK pathway, suggesting that
NOX may contribute to early placental development through MAPK
signaling (33). Clinical and
experimental data have indicated that NOX1 expression is elevated
in preeclampsia during early pregnancy, resulting in excessive
superoxide production, which promotes disease progression (34). In addition, NOX subunits,
including p22phox, p47phox and p67phox, are upregulated in
preeclamptic placentas (35).
Poinsignon et al (36)
reported that NOX4 expression was also increased in preeclampsia.
Unlike NOX1 and NOX2, NOX4 predominantly produces hydrogen peroxide
(H2O2), which may function as a redox
signaling mediator (36). In
control placentas, NOX4 in syncytiotrophoblasts is predominantly
localized to the nucleus, whereas in early-onset preeclampsia it
shows stronger cytoplasmic and membrane staining (36). This pattern may reflect an
adaptive mechanism by which trophoblasts respond to maternal
hypertension by releasing H2O2 into the
intervillous space to activate antioxidant and vasodilatory
pathways (36). However, excess
H2O2 can be converted to hydroxyl radicals
(•OH) through the Fenton reaction, which induces lipid peroxidation
and cellular damage (36).
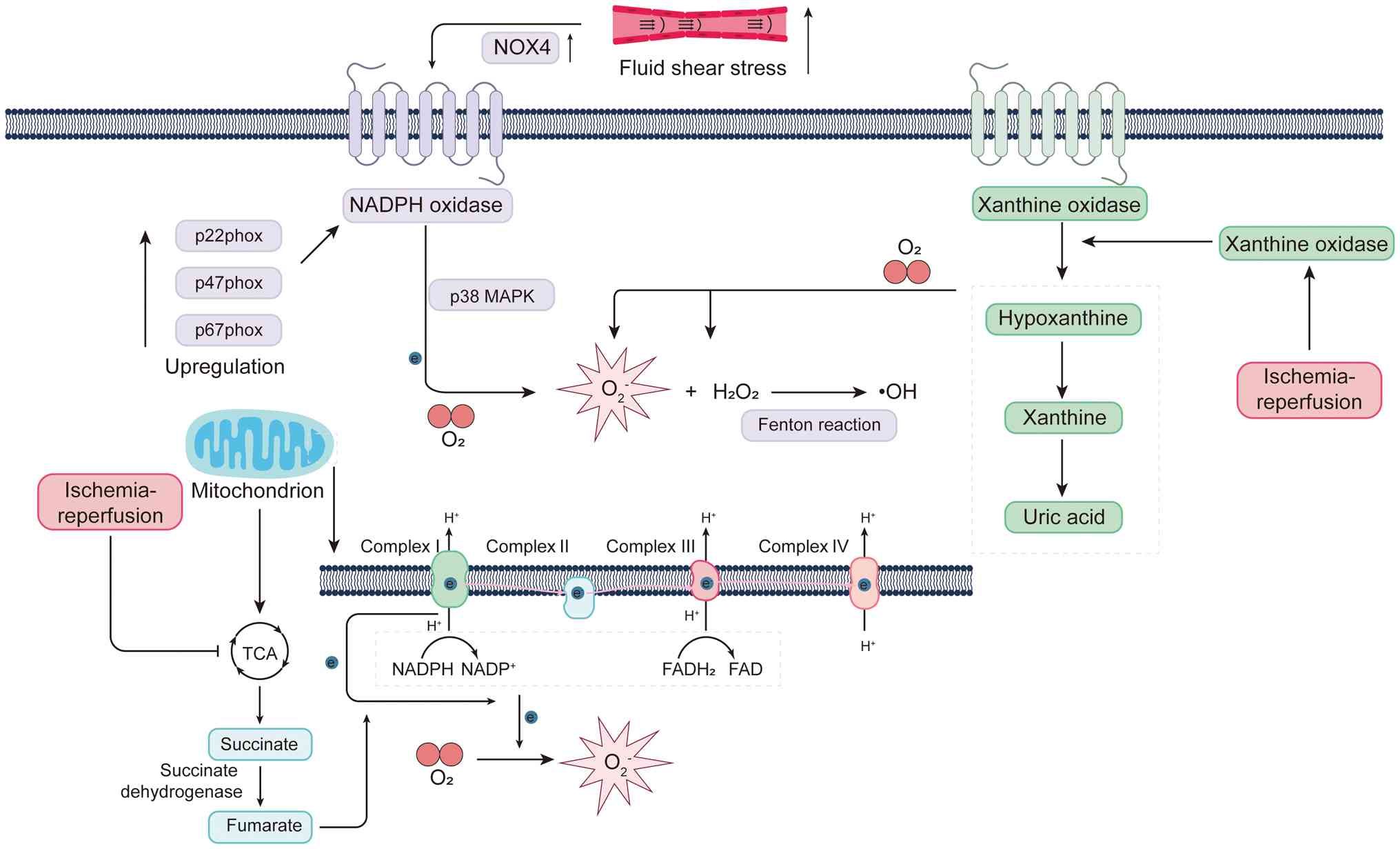 | Figure 2Major sources and molecular
mechanisms of ROS generation. NOX is a major source of
O2•−, and elevated O2•−
levels are associated with activation of the p38 MAPK pathway. The
NOX subunits p22phox, p47phox and
p67phox are highly expressed in the placenta. Excess
H2O2 can be further converted into •OH via
the Fenton reaction. XO catalyzes the stepwise oxidation of
hypoxanthine to xanthine and uric acid, accompanied by the
generation of H2O2 and O2. The
electron transport chain, consisting of five multi-subunit protein
complexes (I-V), resides in the mitochondrial inner membrane.
Complexes I, III and IV pump protons (H+) from the
mitochondrial matrix into the intermembrane space. During oxidative
phosphorylation, O2 undergoes partial reduction by
electrons leaking from complexes I and III, giving rise to
O2•−. In the matrix,
O2•− is dismutated to
H2O2. H2O2 may
subsequently undergo the Fenton reaction with
Fe2+/Cu+ to produce •OH. During reperfusion,
the TCA cycle intermediate succinate selectively accumulates. The
accumulated succinate is rapidly re-oxidized by succinate
dehydrogenase, driving reverse electron transport at complex I and
triggering a burst of ROS. Ischemia-reperfusion also facilitates
the conversion of xanthine dehydrogenase to XO, which in its
oxidase form uses O2 as an electron acceptor to
continuously generate oxygen radicals. Increased FSS further
induces NOX4 expression, thereby enhancing
H2O2 production. NOX, NADPH oxidase; XO,
xanthine oxidase; TCA, tricarboxylic acid cycle; ROS, reactive
oxygen species; FSS, fluid shear stress; phox, phagocyte oxidase;
FAD, flavin adenine dinucleotide. |
ii) XO. XO, an enzymatic form of xanthine
oxidoreductase (XOR), serves a central role in purine metabolism by
catalyzing the sequential oxidation of hypoxanthine to xanthine and
xanthine to uric acid. These reactions are accompanied by the
generation of ROS such as H2O2 and
O2•−, positioning XO as a key regulator of redox
homeostasis and oxidative stress (37). Evidence has highlighted the
critical role of XO in pregnancy-related disorders. This enzyme is
not only the major source of uric acid but also an important
mediator of oxidative stress responses during gestation. Under
ischemic conditions, the dehydrogenase form of xanthine
oxidoreductase is converted into XO through sulfhydryl oxidation or
limited proteolysis. Upon reperfusion, when oxygen supply is
restored and hypoxanthine/xanthine have accumulated, XO uses
O2 as an electron acceptor and generates large amounts
of superoxide and H2O2, thereby producing a
burst of ROS characteristic of ischemia-reperfusion injury
(Fig. 2) (37).
An animal study has indicated that exposure to
electronic cigarettes during pregnancy reduces fetal and placental
weights while markedly increasing placental XO activity, suggesting
that environmental exposures may impair placental function through
XO upregulation (38).
Similarly, maternal inhalation of titanium dioxide nanoparticles
has been shown to elevate XOR activity in the placental labyrinth
of fetal mice. This was accompanied by decreased antioxidant
defenses, such as reduced CAT activity, along with increased
prostaglandin and thromboxane synthesis, favoring
H2O2 accumulation and a more pro-oxidant,
inflammatory milieu in the labyrinth zone, and thus, aggravating
placental oxidative stress (39,40). In addition, high fructose intake,
serving as a model of metabolic stress, has been found to enhance
XO activity and uric acid production in the placenta, leading to
functional impairment and FGR (41). These findings collectively
suggest that excessive XO activation represents a convergent
pathway through which diverse exogenous stressors compromise
placental function.
In human pregnancy, XO activity is also markedly
elevated under pathological conditions. In preeclampsia, maternal
serum XO activity and uric acid levels are increased even when
renal function remains normal, indicating that hyperuricemia is not
solely attributable to impaired renal clearance but is closely
linked to XO-mediated uric acid overproduction (42). XO levels are simultaneously
elevated in the fetal circulation, further underscoring the active
role of the placenta-fetal unit in pathological hyperuricemia
(42). An experimental study has
demonstrated that the XO inhibitor allopurinol effectively lowered
placental uric acid levels and improved placental function,
supporting the causal involvement of XO in gestational oxidative
injury (43).
Clinical evidence provides further support. In FGR
pregnancies, maternal plasma, cord plasma and placental tissues
exhibit higher XO activity, accompanied by increased lipid
peroxidation products, such as MDA, and decreased antioxidant
capacity, which together implicate XO activation as a major
contributor to placental oxidative injury (19). A large cohort study involving 665
women with suspected or confirmed placental insufficiency
demonstrated that maternal uric acid levels were inversely
associated with neonatal birth weight, with the association being
particularly pronounced in infants whose weight fell below the
third percentile (44). These
findings not only highlight the pathogenic role of XO-derived
products in FGR but also suggest their potential as early
predictive biomarkers and therapeutic targets.
Mitochondrial electron transport chain
(ETC) as a central source of ROS
Mitochondria are multifunctional organelles that
serve essential roles in cellular metabolism, calcium
(Ca2+) homeostasis, redox balance and cell fate
determination. They serve as the cellular powerhouses by generating
ATP through oxidative phosphorylation. In addition to ATP,
mitochondria also provide metabolic intermediates required for the
synthesis of macromolecules such as DNA, RNA, proteins and lipids
(45). They are further involved
in maintaining intracellular Ca2+ homeostasis (46,47) and regulate cell death by
releasing cytochrome c and activating apoptotic factors such as
caspases (48). ROS are
by-products of oxidative phosphorylation and participate in the
regulation of redox homeostasis (49).
The mitochondrial ETC is composed of five
multi-subunit protein complexes (complexes I-V) located in the
inner mitochondrial membrane. Enzymatic reactions of the
tricarboxylic acid (TCA) cycle produce reducing equivalents in the
form of NADH and reduced flavin adenine dinucleotide
(FADH2), which transfer electrons to the ETC through
complexes I and III, respectively. During electron transfer from
NADH or FADH2 to O2, complexes I, III and IV
pump protons (H+) from the mitochondrial matrix into the
intermembrane space (50).
Molecular oxygen is reduced to water at complex IV, serving as the
terminal electron acceptor (51). The proton gradient created across
the inner membrane drives protons back into the matrix through
complex V (ATP synthase), coupling this energy to the synthesis of
ATP from ADP and inorganic phosphate (52). ROS generation is an intrinsic
consequence of mitochondrial oxidative metabolism. Approximately
1-2% of O2 consumed during oxidative phosphorylation
undergoes partial reduction by electrons leaking from complexes I
and III, producing O2•− (50-52). Given the high level of
O2 consumption by mitochondria, a substantial proportion
of O2•− arises during this process.
Superoxide generated at complex I is released exclusively into the
mitochondrial matrix, whereas complex III produces superoxide
released into both the matrix and intermembrane space (50). In the matrix,
O2•− is dismutated by SOD2 (Mn-SOD) to
H2O2, while in the intermembrane space SOD1
(Cu,Zn-SOD) performs the same reaction (51,53). H2O2 may
further react with Fe2+ or Cu+ via the Fenton
reaction to generate OH• (54),
and O2•− can react with nitric oxide (NO) to
form peroxynitrite (ONOO−) (55). Both OH• and ONOO− are
highly reactive oxidants. To counterbalance these species,
mitochondria possess intrinsic antioxidant systems, in which
H2O2 is detoxified primarily by GPX1/4 and
the mitochondrial peroxiredoxins peroxiredoxin 3 and peroxiredoxin
5 (53,56).
In the context of gestational hypoxia, placental
mitochondria undergo metabolic reprogramming to maintain energy
homeostasis. This involves structural and functional adjustments of
ETC complexes, a predominance of mitochondrial fission and
activation of the non-canonical unfolded protein response (UPR),
highlighting the close relationship between oxidative stress
adaptation and placental function (57). A clinical study in monochorionic
twin pregnancies complicated by selective FGR (sFGR) has revealed
placental ultrastructural abnormalities, elevated ROS levels,
reduced energy reserves, and mitochondrial genomic and epigenetic
alterations, supporting the coexistence of mitochondrial
dysfunction and oxidative stress (57). A functional study has further
demonstrated that impaired zinc finger protein 554 activity in the
placenta and trophoblasts reduced the antioxidant capacity and
triggered mitochondria-derived ROS-mediated apoptosis and
autophagy. Administration of N-acetylcysteine (NAC) partially
reversed these phenotypes, underscoring a causal link between
mitochondrial ROS, cell death and placental dysfunction (58). Advanced maternal age is also
associated with higher placental lipid peroxidation and apoptosis,
with more pronounced effects in male fetuses, suggesting that
maternal age may increase the risk of FGR through mitochondrial and
oxidative stress pathways (59).
A study at high altitude has demonstrated that placental
mitochondrial oxidative capacity was closely related to fetal
oxygen delivery, while in preeclampsia this capacity was
suppressed, indicating the pathophysiological significance of
placental mitochondrial metabolism under hypoxic conditions
(60). Animal experiments
provide mechanistic and therapeutic insights. In rodent models of
hypoxic pregnancy, the mitochondria-targeted antioxidant
mitoquinone mesylate (MitoQ) improved uterine artery reactivity and
vascular remodeling, suggesting that attenuation of oxidative
damage may enhance placental perfusion, although direct benefits to
fetal growth remain to be established (61). Environmental pollutants, such as
1-nitropyrene, can inhibit progesterone synthesis and induce
intrauterine growth restriction through mitochondrial ROS-driven
signaling, whereas treatment with MitoQ or general control
nonderepressible 2 inhibitors partially alleviates these effects
(62). A human trophoblast study
has shown that hydrogen sulfide regulated mitochondrial dynamics
via sulfhydration of Miro2, thereby promoting cell migration and
invasion, which links mitochondrial plasticity to deep placental
invasion and adverse pregnancy outcomes (63). Furthermore, 25-hydroxycholesterol
induces mitochondrial ROS accumulation, loss of membrane potential
and lipid peroxidation, and triggers apoptosis, ferroptosis and
autophagy, further establishing a connection between
oxysterol-mediated mitochondrial oxidative stress and placental
dysfunction (64). The impact of
mitochondrial dysfunction on fetal organs also deserves attention.
In an ovine FGR model, the mitochondrial oxidative phosphorylation
capacity was reduced compared with that in appropriately grown
control fetuses, accompanied by decreased expression of genes
related to lipid metabolism, with differences observed between
cardiac ventricles. These findings suggest a link between
intrauterine growth restriction, cardiac maturation and abnormal
energy metabolism (65). Another
study demonstrated that limited substrate supply in utero reduced
the number of fetal cardiac mitochondria and the content of ETC
complexes, while reshaping metabolic pathways. This provides a
mechanistic basis for the association between intrauterine growth
restriction and increased long-term cardiovascular risk (66).
Impaired perfusion and
ischemia-reperfusion-induced ROS bursts
During normal pregnancy, maternal spiral arteries
undergo extensive physiological remodeling. This process involves
the progressive loss of vascular smooth muscle cells and elastic
membranes, which allows the vessels to dilate and relax, thereby
ensuring a steady, low-resistance and continuous blood supply to
the placenta. When this remodeling process is impaired and smooth
muscle cells are retained, the affected tissue becomes highly
susceptible to ischemia-reperfusion injury, a condition that is
closely associated with excessive generation of ROS (67). Metabolomic studies have shown
that succinate, an intermediate of the TCA cycle, selectively
accumulates during the reperfusion phase. The rapid re-oxidation of
accumulated succinate by succinate dehydrogenase drives a burst of
ROS production through reverse electron transfer at mitochondrial
complex I (68,69). Ischaemic succinate accumulation
can, in part, be fueled by fumarate generated via the purine
nucleotide cycle and the malate-aspartate shuttle, thereby linking
fumarate to this ROS-generating pathway (68). At the same time,
ischemia-reperfusion promotes the conversion of xanthine
dehydrogenase into XO. In this oxidase state, XO utilizes oxygen as
an electron acceptor, leading to sustained generation of oxygen
radicals (70). From a
hemodynamic perspective, failure of spiral artery remodeling causes
maternal blood to enter the intervillous space in turbulent jets at
abnormally high velocities of 1-2 m/sec (67). This abnormal increase in fluid
shear stress (FSS) has been demonstrated to markedly upregulate
PlGF in both coculture systems and intact vascular models (67). Elevated FSS also induces an
increase in Nox4 mRNA expression in endothelial and smooth muscle
cells, which in turn elevates H2O2 levels
(71). Notably, knockdown of
Nox4 in endothelial cells abrogates the effects of FSS on both
H2O2 production and PlGF expression (71). Soluble fms-like tyrosine kinase 1
(sFlt-1) secretion is also regulated by NOX. The use of the Nox
inhibitor diphenyleneiodonium reduces sFlt-1 release (72). A study has revealed that the p38
MAPK signaling pathway is not only involved in sFlt-1 secretion but
also mediates Nox activation. Inhibition of p38 MAPK
phosphorylation decreases both sFlt-1 secretion and superoxide
production (72). p38 MAPK has
been reported to be activated under hypoxia-reoxygenation
conditions (73), suggesting
that it may serve as a key regulator of ROS production and
angiogenic factor expression in the context of ischemia-reperfusion
injury.
Oxidative stress, ER stress and FGR
ER stress and the dual role of the
UPR
The ER is a critical intracellular organelle
responsible for the synthesis of transmembrane and secretory
proteins, as well as for Ca2+ storage and lipid
production (74). Under normal
physiological conditions, the ER ensures correct protein folding
and the maturation of nascent proteins into their native
conformations, which are subsequently transported to the
extracellular space or other cellular compartments (75). However, the ER is highly
sensitive to oxidative stress. When oxidative stress occurs,
misfolded proteins accumulate in the ER lumen, triggering ER stress
and activating the UPR (75).
The UPR leads to a general reduction in protein synthesis, the
upregulation of molecular chaperones and enhanced expression of
components associated with ER-associated protein degradation (ERAD)
(76).
In homeostasis, the major UPR sensors, protein
kinase RNA-like ER kinase (PERK), activating transcription factor 6
(ATF6) and inositol-requiring enzyme 1α (IRE1α), remain inactive
through binding of their luminal domains to glucose-regulated
protein 78 (GRP78) (Fig. 3)
(75). GRP78 itself functions as
an important chaperone protein that assists in protein folding.
Under stress conditions, such as hypoxia, GRP78 dissociates from
these complexes to support protein folding, while indirectly or
directly enabling activation of PERK, ATF6 and IRE1α, thereby
initiating the UPR (75). PERK
is a transmembrane kinase that associates with GRP78 under resting
conditions. Under stress conditions, dissociation allows PERK to
oligomerize and undergo autophosphorylation, thereby activating its
kinase activity (76). Activated
PERK phosphorylates the transcription factor nuclear factor
erythroid 2-related factor 2 (NRF2), releasing it from the Keap1
complex and promoting the transcription of antioxidant genes. PERK
also phosphorylates the translation initiation factor eukaryotic
initiation factor 2α (eIF2α), which reduces global protein
synthesis (77). Paradoxically,
phosphorylation of eIF2α enhances the translation of ATF4. ATF4 in
turn regulates the transcription of genes involved in cell fate,
including C/EBP homologous protein (CHOP), growth arrest and DNA
damage-inducible protein 34 (GADD34), ATF3 and autophagy-related
genes (78). ATF6 also remains
bound to GRP78 in homeostasis. Following stress-induced
dissociation, ATF6 is transported to the Golgi apparatus, where it
undergoes sequential cleavage by site-1 protease and site-2
protease, generating an N-terminal fragment with transcriptional
activity. This fragment translocates into the nucleus and binds to
cis-regulatory elements such as cAMP response element and ER stress
response element-1, thereby activating the transcription of
chaperone proteins, including GRP78 and glucose-regulated protein
94, as well as cell fate regulators such as CHOP (79). IRE1α is a transmembrane protein
with both kinase and endoribonuclease activities. At rest, it is
bound to GRP78 in an inactive state. Upon accumulation of ROS in
the ER lumen, GRP78 dissociates and facilitates folding, while
IRE1α directly interacts with misfolded proteins, triggering its
activation (75). Activation
requires oligomerization and autophosphorylation (80). Active IRE1α exerts multiple RNA
processing functions, including regulation of its own mRNA
stability (81), cleavage of
specific microRNAs (82), and
most prominently, splicing of X-box binding protein 1 (XBP1) mRNA.
The spliced XBP1 protein enhances the expression of ERAD-related
genes and chaperones, thereby restoring protein homeostasis
(83). In addition, IRE1α can
promote autophagy via the JNK pathway (84). Notably, under prolonged stress,
IRE1α shifts toward pro-apoptotic functions. Persistent activation
recruits TNF receptor-associated factor 2, which subsequently
activates apoptosis signal-regulating kinase 1 and initiates
downstream p38 MAPK and JNK cascades, two major pro-apoptotic
signaling pathways (85). In
parallel, increased CHOP expression exerts multiple pro-apoptotic
effects. These include activation of GADD34, which mediates eIF2α
dephosphorylation, restoring translation initiation and promoting
synthesis of pro-apoptotic proteins, thereby exacerbating ER stress
(86). CHOP also induces ER
oxidase 1α, which enhances Ca2+ transfer and triggers
mitochondrial cytochrome c release, leading to caspase
activation and ultimately apoptosis (87). Furthermore, CHOP modulates
nuclear transcription by downregulating the anti-apoptotic protein
Bcl-2 and upregulating the pro-apoptotic protein Bim, further
promoting apoptosis (88).
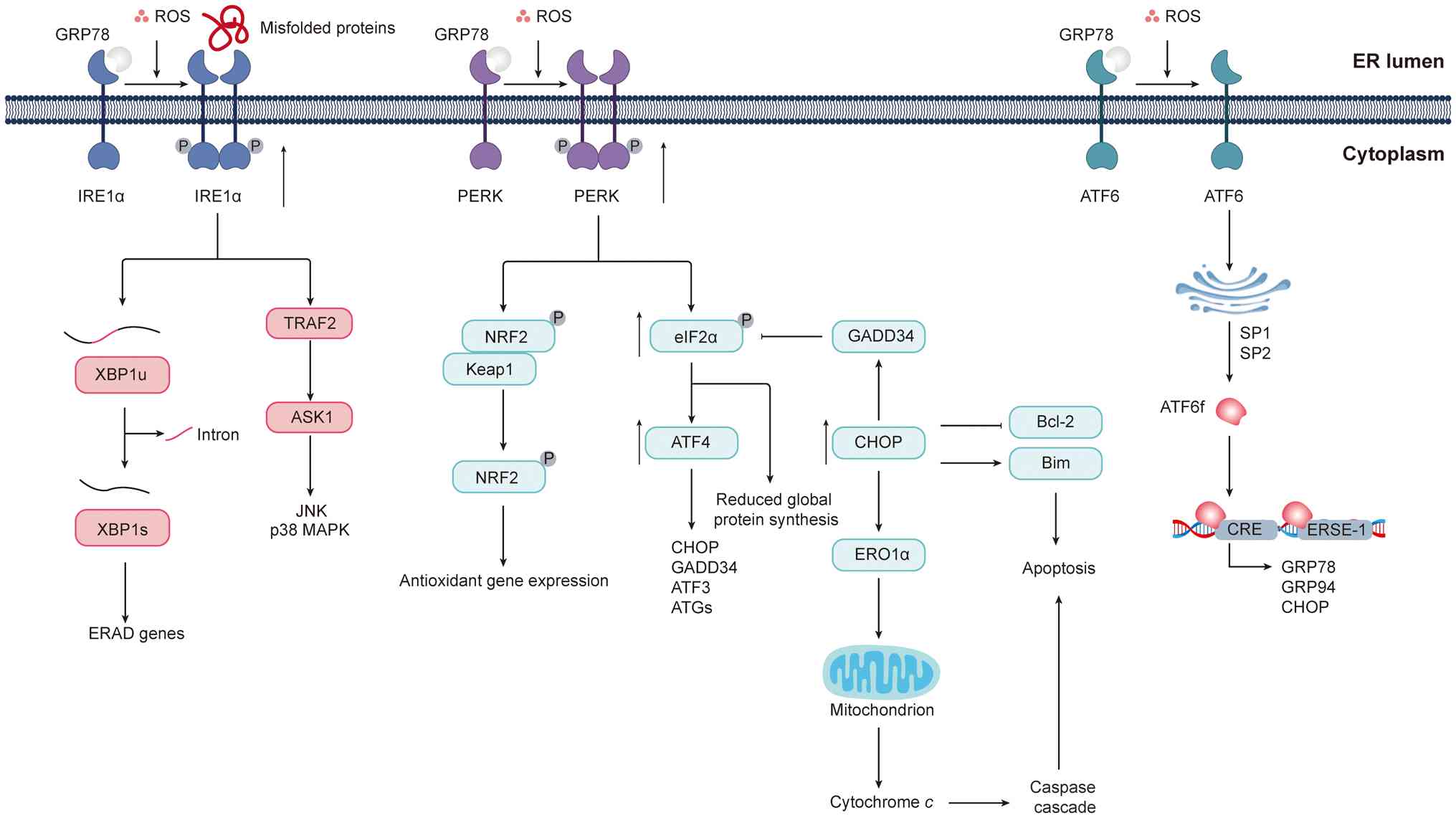 | Figure 3Oxidative stress and ROS signaling in
ER stress and the UPR. The ER is highly sensitive to oxidative
stress. When redox imbalance leads to the accumulation of misfolded
proteins within the ER lumen, it triggers ER stress and activates
the UPR. Under homeostatic conditions, the three major signaling
branches of the UPR (PERK, ATF6 and IRE1α) remain inactive through
their interaction with the molecular chaperone GRP78 at their
luminal domains. Upon stress induction, GRP78 dissociates from
these sensors, facilitating the oligomerization and
autophosphorylation of PERK, the translocation and cleavage of
ATF6, and the activation of IRE1α. Activated PERK phosphorylates
the transcription factor NRF2, leading to its release from the
Keap1 complex and the subsequent induction of antioxidant gene
expression. PERK also phosphorylates the translation initiation
factor eIF2α, thereby globally inhibiting protein synthesis while
selectively enhancing the translation of ATF4. ATF4 regulates a
series of cell fate-related genes, including CHOP, GADD34, ATF3 and
ATGs. Upon CHOP upregulation, GADD34-mediated dephosphorylation of
eIF2α restores translation initiation. CHOP also induces the
expression of the ER oxidoreductase ERO1α, which triggers the
release of cytochrome c and activates the caspase-dependent
apoptotic signaling pathway. In addition, CHOP can
transcriptionally downregulate Bcl-2 and upregulate Bim, further
promoting apoptosis through multiple mechanisms. ATF6 is
transported to the Golgi apparatus, where it is cleaved by SP1 and
SP2 proteases to release its N-terminal transcriptionally active
fragment. This fragment enters the nucleus and binds to
cis-regulatory elements such as CRE and ERSE, inducing the
transcription of chaperone proteins, including GRP78 and GRP94, as
well as cell fate regulators such as CHOP. The activation of IRE1α
also depends on oligomerization and autophosphorylation. Its
ribonuclease activity then mediates the splicing of XBP1 mRNA,
generating the active XBP1s protein, which enhances the expression
of ERAD components and molecular chaperones. However, under
prolonged stress conditions, IRE1α recruits TRAF2, leading to the
activation of ASK1 and subsequent stimulation of the p38 MAPK and
JNK signaling cascades. ROS, reactive oxygen species; ER,
endoplasmic reticulum; UPR, unfolded protein response; PERK,
PKR-like endoplasmic reticulum kinase; ATF, activating
transcription factor; IRE1α, inositol-requiring enzyme 1α; GRP78,
glucose-regulated protein 78; NRF2, nuclear factor erythroid
2–related factor 2; Keap1, Kelch-like ECH-associated protein 1;
eIF2α, eukaryotic initiation factor 2α; CHOP, C/EBP homologous
protein; GADD34, growth arrest and DNA damage-inducible protein 34;
ATGs, autophagy-related genes; ERO1α, endoplasmic reticulum
oxidoreductin 1α; SP1, site-1 protease; SP2, site-2 protease; CRE,
cAMP response element; ERSE, ER stress response element; GRP94,
glucose-regulated protein 94; XBP1, X-box binding protein 1; XBP1s,
spliced X-box binding protein 1; ERAD, endoplasmic
reticulum-associated degradation; TRAF2, TNF receptor-associated
factor 2; ASK1, apoptosis signal-regulating kinase 1; P, phosphate
group; ATF6f, activating transcription factor 6 fragment; XBP1u,
X-box binding protein 1 (unspliced); Bim, Bcl-2 interacting
mediator of cell death. |
ER stress signaling pathways in placental
pathophysiology IRE1-XBP1 axis in placental stress responses
Aberrant ER stress and excessive activation of the
IRE1 signaling pathway are closely linked to abnormal placental
development and adverse pregnancy outcomes. These alterations are
most evident in preeclampsia and in pregnancies complicated by FGR
(89). Compared with placentas
from normotensive pregnancies, placental tissues from patients with
preeclampsia, particularly in early-onset cases, exhibit increased
IRE1 phosphorylation, enhanced XBP1 splicing and dilation of the ER
lumen (89). Similarly, decidual
tissues from patients with preeclampsia, with or without FGR, show
higher XBP1 expression than those from normotensive patients with
uncomplicated pregnancies (90).
Endometrial biopsies from patients with recurrent miscarriage or
implantation failure also exhibit elevated IRE1 expression compared
with controls, whereas spliced X-box binding protein 1 (XBP1s)
expression is lower, suggesting that dysregulated IRE1 activation
may contribute to these complications (91). Phosphorylated IRE1 was not
examined in decidual tissue samples from women with preeclampsia
(with or without FGR) and normotensive controls, and additional
studies are needed to determine whether IRE1 activity itself is
altered. In vitro work has demonstrated that exposing
trophoblast cells to hypoxia followed by reoxygenation, which
models conditions in early-onset preeclampsia, markedly increased
IRE1 phosphorylation together with other ER stress and apoptotic
markers (92,93). These results support a role for
IRE1 signaling in stress responses under pathological conditions.
Other pregnancy complications associated with ER stress and IRE1
activation include intrahepatic cholestasis of pregnancy,
characterized by accumulation of bile acids in the liver and
systemic circulation. In a mouse model of intrahepatic cholestasis,
heightened IRE1 activation in placental tissue coincided with
apoptosis and FGR. Treatment with the IRE1 inhibitor 4 μ8c
prevented trophoblast apoptosis and improved fetal growth (94). Similarly, in HTR 8/SVneo cells,
inhibition of IRE1 signaling prevented deoxycholic acid-induced
cell death (94). Physiological
and environmental stressors can also trigger ER stress and activate
IRE1 signaling, thereby increasing the risk of pregnancy
complications. Maternal obesity is a well-known risk factor, and
placental tissues from obese pregnancies exhibit higher levels of
phosphorylated IRE1 and XBP1s than those from women with normal
weight (95,96). Palmitic acid, which is the most
abundant saturated fatty acid in circulation and commonly elevated
in obesity, disrupts ER morphology, reduces trophoblast
invasiveness, and induces ER stress and apoptotic signaling
(97,98). Additional stressors that
contribute to trophoblast ER stress and IRE1 overactivation include
viral infections, such as Zika virus, and exposure to toxins, such
as nicotine and ethanol (99,100). In these clinical and
experimental settings, IRE1 signaling has been evaluated mainly in
endpoint samples (for example, term placental tissues from
complicated pregnancies or trophoblast cultures harvested hours
after exposure), and usually using static measurements of protein
abundance rather than dynamic read-outs of IRE1 phosphorylation or
XBP1 mRNA splicing. As a result, it remains uncertain whether
sustained activation of the IRE1-XBP1s signaling axis is a primary
driver of placental dysfunction or a downstream consequence of
these stressors.
PERK-eIF2α/ATF4 pathway and regulation of
cell fate
The PERK pathway likely serves a key role in shaping
trophoblast function and cell fate under pathological placental
conditions (101). In
preeclamptic placentas, higher levels of phosphorylated PERK,
phosphorylated eIF2α, ATF4 and CHOP have been reported, with
signals mainly confined to the syncytiotrophoblast layer (101,102). Elevated phosphorylated PERK
levels have also been detected in decidual tissues (103). In decidua collected at delivery
from pregnancies complicated by FGR, the levels of phosphorylated
eIF2α and ATF4 were increased compared with those in decidua from
gestational age-matched uncomplicated pregnancies (104). These observations indicate that
altered PERK signaling in the decidua may hinder decidualization
and promote pregnancy complications. Maternal serum from women with
preeclampsia activates the PERK pathway in placental explants and
HTR-8/SVneo cells, causing increased phosphorylation of eIF2α and
induction of CHOP. Cell death follows serum exposure, although the
extent to which PERK drives this cytotoxicity has not been fully
determined (105).
PERK-dependent trophoblast death in preeclampsia may be tied to
deficiencies in histone deacetylases (HDACs), which are essential
for trophoblast differentiation and are frequently downregulated in
preeclamptic placentas (106,107). Silencing of HDAC2 in
HTR-8/SVneo cells augments pyroptosis, and this effect is reduced
when PERK is knocked down (106). These data suggest that PERK
activation contributes to heightened trophoblast cell death in
placental disease. Multiple in vitro systems model ER stress
conditions that activate PERK signaling and influence trophoblast
survival. In BeWo cells, alternating hypoxia and reoxygenation at
1% O2 and ambient air increases phosphorylated eIF2α
levels and lowers cell numbers (108). Interleukin 1β, a
pro-inflammatory cytokine elevated in preeclampsia, promotes
apoptosis in BeWo cells via PERK signaling, whereas progesterone
counteracts this effect (101).
Inhibition of PERK similarly diminishes apoptosis in BeWo cells
exposed to the endocannabinoid 2 arachidonoylglycerol (109). Cadmium, an environmental
pollutant and carcinogen, suppresses 11β-hydroxysteroid
dehydrogenase type 2 expression in JEG-3 cells, while PERK
silencing or antioxidant treatment with melatonin or
N-acetylcysteine restores its expression (110,111). Collectively, these findings
indicate that PERK signaling governs trophoblast survival across
diverse ER stress contexts.
ATF6 signaling and dysregulation of
angiogenic factors
In early-onset preeclampsia with FGR, placental
tissues show higher ATF6α protein levels than those in healthy
controls (90,108). Whether ATF6α activation is also
increased remains unresolved, since neither the active form nor
nuclear localization has been assessed. A defining feature of
preeclampsia is reduced secretion of PlGF, a proangiogenic factor
essential for endothelial integrity. In preeclamptic placentas,
lower PlGF expression is associated with nuclear localization of
ATF4 and ATF6β, but not with nuclear localization of ATF6α or XBP1,
and nuclear ATF4 and ATF6β directly repress PlGF transcription
(112,113). In BeWo cells exposed to ER
stressors, such as thapsigargin or hypoxia followed by
reoxygenation, simultaneous silencing of ATF4 and ATF6β raises PlGF
expression (113). These
results suggest that ATF4 and ATF6β act as negative regulators of
PlGF, and that modulating ATF4/ATF6β signaling may help restore the
angiogenic balance in pregnancies with placental dysfunction.
ROS, mitochondrial dysfunction and metabolic
reprogramming
During pregnancy and the neonatal period, energy
requirements for both the mother and fetus rise markedly. Because
mitochondria drive cellular energy metabolism, FGR is often
accompanied by intensified mitochondrial stress (114). Mitochondria use oxygen as the
final electron acceptor during aerobic respiration, a process that
inevitably generates ROS (114). Excess ROS damage biomolecules,
and since the ETC is both a principal source and a target of ROS,
mitochondrial dysfunction commonly coexists with persistent
oxidative stress (114).
Evidence shows that FGR offspring present with elevated ROS,
abnormal antioxidant enzyme activity, increased lipid peroxidation
and impaired ATP synthesis (115), underscoring oxidative stress as
a central driver of metabolic disturbance.
The liver is densely populated with mitochondria
and is pivotal for systemic metabolism, which makes it particularly
susceptible to oxidative injury (Fig. 4) (116). Previous research has
demonstrated pronounced hepatic oxidative and mitochondrial stress
in FGR (116). In a spontaneous
FGR pig model, neonatal livers showed higher α1-acid glycoprotein
levels, indicating systemic oxidative stress, together with
increased ETC complex IV expression, compared with those in normal
birth weight littermates, consistent with ATP depletion from
excessive hydrolysis (116). In
a maternal caloric restriction rat model, cutting maternal energy
intake by 50% raised the levels of the lipid peroxidation marker
4HNE and lowered GSH levels in offspring at 3 weeks of age. When
offspring resumed normal diets after weaning, oxidative stress
markers normalized in adulthood, suggesting that prenatal
undernutrition alone can induce oxidative injury (117). Maternal protein restriction
models have further been used to clarify the role of catch-up
growth. Offspring exposed to a low-protein diet both in utero and
throughout postnatal life, did not undergo catch-up growth,
remained metabolically healthy in adulthood and generated less ROS
(118). By contrast, switching
to a normal diet at weaning in the LP2 group (offspring exposed to
a maternal low-protein diet during gestation and lactation,
switched to a normal-protein control diet at weaning) or at birth
in the LP3 group (offspring exposed to a maternal low-protein diet
during gestation only, with diets switched to a normal-protein
control diet from birth) triggered catch-up growth (118). Among these groups, only LP2
offspring developed hypercholesterolemia, impaired glucose
tolerance and altered drug metabolism at 4 months of age (119). These alterations coincided with
aerobic metabolic disruption, including increased hepatic protein
abundance of lactate dehydrogenase and phosphorylated pyruvate
dehydrogenase, decreased protein abundance of citrate synthase and
complex II, elevated levels of SOD1 and SOD2, reduced CAT, and
increased 4-hydroxynonenal as a marker of lipid peroxidation,
compared with those in control diet-fed offspring (119). The LP2 group also exhibited
upregulation of the ROS-promoting protein p66 Src homology 2
domain-containing transforming protein C, the 66-kDa isoform of the
Shc adaptor protein that promotes mitochondrial ROS production and
is closely linked to ER stress (119). Consistent findings have
indicated that catch-up growth after weaning raises hepatic oxygen
consumption, shifts antioxidant capacity and reduces expression of
mitochondrial DNA-encoded genes (120). Notably, the LP3 offspring do
not exhibit these metabolic and mitochondrial abnormalities
observed in LP2 offspring, suggesting that the timing of
nutritional recovery is a critical determinant of oxidative stress
and hepatic injury (119,120).
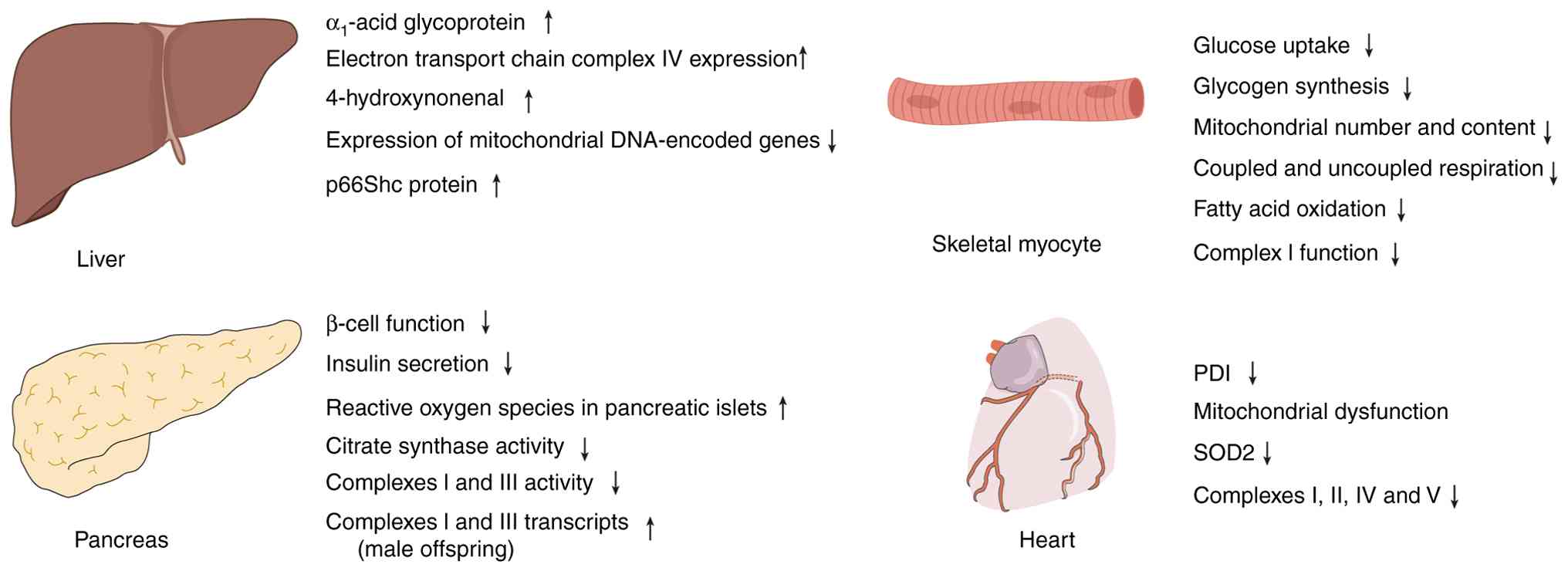 | Figure 4ROS accumulation, mitochondrial
dysfunction and metabolic reprogramming in FGR and maternal
nutritional models across multiple tissues. In the liver, the
spontaneous FGR pig model shows that neonatal piglets exhibit
elevated levels of α1-acid glycoprotein and electron
transport chain complex IV. In maternal caloric restriction
experiments in rats, a reduction in maternal energy intake led to
an increase in the lipid peroxidation marker 4-hydroxynonenal.
Post-weaning catch-up growth reduced the expression of
mitochondrial DNA-encoded genes. In the pancreas, FGR offspring
exhibit impaired β-cell function and insufficient insulin
secretion. Their islet ROS levels increase with age, accompanied by
decreased activities of citrate synthase and complexes I and III.
In skeletal muscle, early adult FGR offspring display reduced
insulin-stimulated glucose uptake and glycogen synthesis. Maternal
caloric restriction experiments similarly demonstrate that the
mitochondrial number and content are reduced, accompanied by
decreased coupled and uncoupled respiration, fatty acid oxidation,
and complex I function. In the heart, prenatal nicotine exposure
decreases cardiac PDI levels, and PDI deficiency further induces
mitochondrial dysfunction and oxidative injury, manifested as
reduced protein levels of SOD2 and mitochondrial complexes I, II,
IV and V. FGR, fetal growth restriction; PDI, protein disulfide
isomerase; ROS, reactive oxygen species; p66Shc, p66 Src homology 2
domain-containing transforming protein C; SOD2, superoxide
dismutase 2. |
In the pancreas, FGR offspring exhibit impaired
β-cell function and insufficient insulin secretion from birth
(121), a phenotype that
persists into adulthood (122).
Animal studies have further demonstrated age-dependent increases in
pancreatic islet ROS levels. For instance, in rats, ROS levels were
already elevated at 1 week of age and exceeded twice the control
levels by 15 weeks, accompanied by reduced citrate synthase
activity, impaired complex I and III function, and insufficient ATP
production (123). In maternal
protein restriction models using rats, 3-month-old male offspring
displayed elevated islet ROS levels and upregulation of complex I
and III transcripts (124),
suggesting that oxidative stress and mitochondrial dysfunction may
exhibit sex-specific differences.
Skeletal muscle metabolism is also markedly
impaired in FGR. In a rat model of uteroplacental insufficiency,
FGR offspring in early adulthood showed reduced insulin-stimulated
glucose uptake and glycogen synthesis in skeletal muscle, together
with decreased ATP production and impaired aerobic enzyme activity
(125). Pig models have
revealed that FGR piglets are more susceptible to diet-induced
mitochondrial damage in skeletal muscle when exposed to a high-fat
diet after birth (126).
Maternal caloric restriction experiments have demonstrated reduced
mitochondrial number and content in offspring at 10 weeks of age,
with concomitant reductions in coupled and uncoupled respiration,
fatty acid oxidation, and complex I function (127).
The heart is another organ that is highly
vulnerable to FGR-associated oxidative stress (128). Because mammalian hearts have
limited regenerative capacity after birth, mitochondria-dependent
oxidative stress may shorten the proliferative window (128). Prenatal nicotine exposure
reduces the levels of protein disulfide isomerase (PDI), a protein
essential for cardioprotection against ischemic injury (129). PDI deficiency exacerbates
mitochondrial dysfunction and oxidative damage, leading to reduced
levels of SOD2 and decreased expression of ETC complexes I, II, IV
and V in cardiac tissue (129).
Epigenetic regulation and placental
oxidative stress
Epigenetics refers to heritable modifications of
chromatin that regulate gene expression without altering the
underlying DNA sequence (130).
A typical example is DNA methylation, which involves the addition
of a methyl group to cytosine residues under the catalysis of DNA
methyltransferases (131). In
mammals, DNA methylation predominantly occurs at CpG sites, where
cytosine is followed by guanine in the 5'-3' direction (132). Methylation of gene-regulatory
regions, such as promoters, is generally associated with
transcriptional silencing (132). In addition, DNA methylation
serves key roles in X chromosome inactivation and genomic
imprinting (133). Regulatory
non-coding RNAs also contribute to transcriptional,
post-transcriptional and translational regulation, and thus, are
important components of epigenetic regulation (134,135).
These epigenetic mechanisms not only govern
fundamental molecular processes but also have direct relevance to
placental function and fetal growth (136). Current evidence indicates that
epigenetic alterations serve critical roles in the regulation of
fetal development. Genome-wide methylation analysis of placental
samples from 12 monochorionic twin pregnancies complicated by sFGR
revealed a global shift toward hypomethylation compared with normal
placentas, involving 5,625 hypomethylated and 452 hypermethylated
CpG sites, primarily located in CpG islands, gene bodies and
promoter regions (136).
Multi-omics analysis has further demonstrated activation of
oxidative stress pathways in sFGR placentas. RNA sequencing
identified 68 differentially expressed genes related to oxidative
stress, while DNA hypomethylation was enriched in hypoxia-related
pathways. Integrated analysis identified HK2 as a key upregulated
node, linked to hypoxia-induced metabolic stress and oxidative
injury, with strong predictive potential (area under the curve,
0.917), suggesting its pivotal role in hypoxia-driven
metabolic-oxidative pathways (137). An animal study also supports
this link. In a rat model of FGR induced by a high-sucrose,
low-copper diet, placentas exhibited reduced size, elevated
oxidative stress and global DNA hypomethylation, while copper
supplementation ameliorated oxidative stress and partially rescued
placental morphology (138). Hu
et al (139) reported
mitochondrial abnormalities in placental villi from human
monochorionic diamniotic twin pregnancies complicated by sFGR,
accompanied by elevated mitochondrial oxidative stress and
widespread hypomethylation of mitochondrial DNA. Epigenetic
profiling further showed aberrant upregulation of mitochondrial
long non-coding RNAs (lncRNAs/lncs), including lncND5, lncND6 and
lncCyt b, along with reduced expression of cytochrome c
oxidase I, a key component of respiratory chain complex IV,
suggesting that dysregulated mitochondrial lncRNAs may contribute
to respiratory dysfunction in sFGR (139). In trophoblasts isolated from
the placentas of female patients with preeclampsia, mono-, di- and
tri-methylation of histone H3 lysine 9, accompanied by reduced SOD
expression, indicated that intrauterine oxidative stress may impair
placental antioxidant defenses through H3K9 methylation and
contribute to preeclampsia and fetal programming (140).
MicroRNAs (miRNAs/miRs) represent another layer of
epigenetic regulation with strong implications for FGR.
miR-199a-5p, previously linked to cardiovascular disease (141), inhibits VEGFA expression in
endometrial mesenchymal stem cells (142), suppresses angiogenesis and
induces oxidative stress in tumor models (143). In placentas from human
monochorionic twin pregnancies complicated by sFGR, miR-199a-5p is
upregulated, and associated with impaired angiogenesis, elevated
oxidative stress and mitochondrial dysfunction (144). miR-199a also targets
hypoxia-inducible factor 1-α (HIF-1α) (145) and is markedly elevated in
severe preterm FGR (146).
HIF-1α induces miR-210-3p, which suppresses fibroblast growth
factor 1 and disrupts placental development, contributing to
adverse placental outcomes in selective intrauterine growth
restriction twin pregnancies (147). Other miRNAs also serve
essential roles in oxidative stress regulation. miR-144
downregulates Nrf2, impairing cellular tolerance to oxidative
stress (148), while miR-451
promotes antioxidant gene expression by inhibiting 14-3-3ζ, a
repressor of the transcription factor FoxO3 (149). Both miRNAs are dysregulated
under ischemic conditions and protect erythrocytes from oxidative
damage (150). Oxidative stress
induced by N-(4-hydroxyphenyl)-retinamide upregulates miR-16 and
miR-26b in ARPE-19 cells (151). Chronic oxidative stress in
human trabecular meshwork cells downregulates miR-29b, resulting in
extracellular matrix gene upregulation (152). Similarly, primary fibroblasts
exposed to H2O2 exhibit altered miR-155 and
miR-16 expression (152). These
findings are of particular relevance, as miR-155, miR-16 and
miR-29b regulate normal placental development: miR-155 suppresses
trophoblast proliferation and migration; miR-16 inhibits
trophoblast invasion, proliferation and angiogenesis; and miR-29b
reduces invasion and angiogenesis while promoting apoptosis
(153-155). Other miRNAs with known links to
oxidative stress include miR-204 and miR-1. miR-204 regulates
oxidative stress responses in human trabecular meshwork cells by
enhancing apoptosis, reducing cell viability and promoting
accumulation of oxidized proteins (156). miR-1 is upregulated by ROS in
ischemic myocardium (157) and
has also been implicated in preeclampsia (158). Furthermore, downregulation of
miR-21 is strongly associated with FGR (159). In placental cells, reduced
miR-21 expression decreases trophoblast invasion of the maternal
decidua, impairs migration and restricts growth, highlighting its
role in trophoblast function (160). miR-21 expression itself can be
induced by ROS (161).
Taken together, evidence from human studies, animal
models and in vitro experiments demonstrates that epigenetic
alterations occupy a central position in the pathogenesis of FGR
and are closely intertwined with placental oxidative stress
(136,138,140). Aberrations such as global DNA
hypomethylation, dysregulation of non-coding RNAs and miRNA
imbalances are frequently accompanied by elevated oxidative stress,
mitochondrial damage and impaired angiogenesis (136,139,144). These findings suggest that
oxidative stress may serve as a mechanistic bridge linking
epigenetic alterations to placental dysfunction (138,140). Mapping the epigenetic landscape
of FGR not only provides insights into its molecular pathology but
also offers promising directions for antioxidant-based
interventions aimed at improving placental function and optimizing
fetal outcomes (138,159).
Calcium signaling in placental vascular
endothelium, NO production and mechanisms of imbalance under
oxidative stress
In normal pregnancies, endothelial cells on the
fetal side of the placenta sense blood flow-induced shear stress
and, via the mechanosensitive channel Piezo1, promote
phosphorylation of endothelial NO synthase (eNOS) and enhancement
of NO signaling, thereby supporting placental vasodilation and
maintaining a relatively low-resistance perfusion environment
(162). In placentas from
pregnancies complicated by FGR, NO signaling and phosphorylation of
eNOS at Ser1177 are increased, and can be further augmented by
pharmacological activation of Piezo1, suggesting a compensatory
endothelial response aimed at enhancing the NO pathway in the
setting of altered placental blood flow (162). However, under persistent
oxidative stress, NO is readily scavenged by excessive superoxide
anions to form peroxynitrite, which reduces the effective
bioavailability of NO and prevents this compensatory upregulation
from fully restoring placental perfusion (163,164).
Abnormal intracellular calcium signaling is a key
link between oxidative stress and the imbalance of NO production.
In endothelial cells, persistent calcium oscillations induced by
stimuli such as ATP drive sustained NO generation, whereas in human
umbilical vein endothelial cells derived from preeclamptic
patients, both sustained calcium signaling and sustained NO
production are attenuated, indicating that disrupted calcium
signaling is a direct cause of insufficient NO generation (165). In human umbilical vein
endothelial cells obtained from pregnancies complicated by FGR and
gestational hypertension, ATP-induced intracellular calcium
responses show a prolonged time to peak and a reduced plateau
during the sustained phase. After depletion of ER calcium stores,
store-operated calcium entry driven by high extracellular calcium
is markedly enhanced, suggesting an adaptive remodeling of
calcium-buffering capacity and of the composition or functional
coupling of calcium channels (166). Sustained calcium signals
provide a critical temporal window for continuous release of
endothelial vasodilators. Thus, when the sustained calcium plateau
is blunted while compensatory calcium influx is paradoxically
enhanced, endothelial calcium homeostasis is more prone to
disruption, leading to associated functional impairment (166). Consistent with this, placental
endothelial cells in placentas from female patients with
preeclampsia exhibit marked S-glutathionylation of eNOS, a
modification associated with eNOS uncoupling, characterized by
reduced NO production and increased superoxide generation, thereby
providing molecular support for the concept that oxidative stress
reduces effective NO bioavailability (167). In trophoblast cells and mouse
models overexpressing an isoform of the transcription factor
storkhead box 1, which recapitulate key features of preeclampsia,
supplementation with tetrahydrobiopterin preserves NOS coupling,
and alleviates nitrosative and redox imbalance, as well as
excessive mitochondrial activation, suggesting that insufficient
cofactor supply may amplify NO dysregulation in the context of
oxidative stress (168).
Furthermore, in a rat model of intrauterine growth restriction
induced by maternal low-protein diet, offspring develop impaired
endothelium-dependent vasodilation accompanied by increased
arginase activity, reduced NO production and enhanced superoxide
generation. Administration of L-arginine or pharmacological
inhibition of arginase restored vasodilatory responses, indicating
that upregulated arginase competes with eNOS for L-arginine and
promotes eNOS uncoupling, thereby directly linking oxidative stress
to NO deficiency (169).
Pregnancy-associated increases in uterine blood
flow rely on localized calcium release in vascular smooth muscle
that activates large-conductance calcium-activated potassium (BKCa)
channels, generating spontaneous transient outward currents and
membrane hyperpolarization, which in turn inhibit voltage-dependent
calcium channel-mediated calcium entry, lower myogenic tone and
help maintain low-resistance perfusion (170). A mechanistic study has shown
that, in uterine arteries, pregnancy induces ten-eleven
translocation methylcytosine dioxygenase 1-mediated active DNA
demethylation of the BKCa channel β1-subunit gene, which in turn
upregulates β1-subunit expression and enhances BKCa channel
function, providing an epigenetic explanation for how the endocrine
milieu chronically tunes the efficiency of calcium
signaling-vasodilation coupling (171). When pregnancy is complicated by
hypoxia, a condition commonly associated with impaired placental
perfusion, gestational hypoxia blunts pregnancy-induced enhancement
of calcium release and spontaneous transient outward currents, and
is accompanied by activation of ER stress and oxidative stress
pathways, rendering uterine arteries more prone to a high-tone
state and thereby increasing the risk of preeclampsia and fetal
intrauterine growth restriction (172). In the placental circulation,
BKCa channel function is impaired in chorionic plate arteries from
preeclamptic pregnancies and is associated with increased placental
vascular resistance, indicating that dysregulation of the BKCa
channel system may consolidate oxidative stress and
calcium-dependent contractile dominance into a hemodynamic outcome
of low perfusion (173).
Furthermore, purinergic receptor-dependent pathways can form a
positive feedback loop between calcium signaling and oxidative
stress. For example, in human umbilical vein endothelial cells
exposed to high glucose, purinergic receptor P2X 4 (P2X4)
expression is upregulated, accompanied by increased intracellular
calcium levels, elevated ROS levels and reduced NO production.
Inhibition of P2X4 signaling attenuates inflammatory injury,
suggesting that P2X4 is a key coupling molecule that links calcium
influx to oxidative stress and contributes to reduced NO
bioavailability (174).
Antioxidant-based therapeutic strategies in
FGR
As aforementioned, oxidative stress is closely
involved in the pathogenesis and progression of FGR. Protecting the
placenta and fetus from oxidative damage has therefore emerged as a
promising therapeutic strategy. Table II summarizes the application of
antioxidants in the treatment of FGR.
| Table IIClinical and preclinical studies
investigating interventions targeting oxidative stress in FGR. |
Table II
Clinical and preclinical studies
investigating interventions targeting oxidative stress in FGR.
| Authors, year | Intervention |
Clinical/preclinical | Model | Key findings | (Refs.) |
|---|
| Alers et al,
2013 | Melatonin | Clinical
(NCT01695070) | 12 women with
severe early-onset FGR | Maternal melatonin
therapy was well-tolerated, increased fetal melatonin levels, and
reduced oxidative stress in the placenta of FGR fetuses. | (175) |
| Miller et
al, 2014 | Melatonin | Preclinical | Pregnant sheep with
FGR | Melatonin could
improve white matter and axonal injury in FGR lamb brains and was
accompanied by improvements in the behavioral capacities of FGR
lambs. | (178) |
| Asadi et al,
2022 | Pentoxifylline | Clinical
(IRCT2014031 7017034N9) | 40 pregnant women
with the diagnosis of severe early-onset FGR | Enhanced fetal
growth and serum antioxidant capacity, thereby contributing to
improved neonatal outcomes and reduced mortality. | (176) |
| Rumbold et
al, 2006 | Vitamins C and
E | Clinical
(ISRCTN00416244) | Nulliparous women
between 14 and 22 weeks of gestation | Supplementation
with vitamins C and E during pregnancy did not improve maternal or
neonatal outcomes. | (177) |
| Wang et al,
2024 | MitoQ | Preclinical | Wistar rats | Oral administration
of the mitochondria-targeted antioxidant MitoQ could effectively
protect against uterine artery vascular dysfunction and
remodeling. | (61) |
| Yang et al,
2021 | MitoQ | Preclinical | Reduced uterine
perfusion pressure mice | The efficacy of
mitochondria-targeted antioxidant therapy was highly
gestational-stage dependent: Potentially beneficial in late
pregnancy but harmful in early pregnancy; excessive suppression of
trophoblastic oxidative stress during early gestation may impair
placentation. | (179) |
| Stanley et
al, 2012 | Tempol | Preclinical | FGR mice | The antioxidant
tempol increased fetal weight and crown-rump length, and enhanced
uterine artery blood flow velocity. | (180) |
| Herrera et
al, 2017 | NAC | Preclinical | FGR guinea
pigs | Lowered placental
vascular resistance and restored fetal growth, while rescuing
eNOS-dependent vasodilation and normalizing endothelial eNOS
expression with reversal of Nos3 promoter CpG-170
hypomethylation. | (181) |
| Vega et al,
2016; Bourque et al, 2012 | Resveratrol | Preclinical | Wistar rats | Improved oxidative
stress markers in the mother, fetus and placenta, and reduced fetal
mortality in a prenatal hypoxia model. | (182,183) |
A pilot clinical study first confirmed the
feasibility and safety of maternal oral melatonin supplementation.
Treatment was well tolerated, increased fetal melatonin levels and
reduced oxidative stress in FGR placentas (175). Another pilot study suggested
that pentoxifylline may improve endothelial function and promote
vasodilation by lowering inflammation-mediated cytokine levels. In
women with early-onset FGR, pentoxifylline administration was
associated with increased fetal weight, enhanced serum antioxidant
capacity, improved neonatal outcomes and reduced mortality
(176). By contrast, a large
multicenter randomized controlled trial involving 1,877 nulliparous
women between 14 and 22 weeks of gestation evaluated vitamin C
(1,000 mg/day) and vitamin E (400 IU/day) supplementation. The
trial showed no significant difference between the intervention and
placebo groups in terms of the incidence of low birth weight or in
rates of infant death or severe adverse outcomes, suggesting that
routine vitamin C and E supplementation during pregnancy does not
improve maternal or fetal outcomes (177).
Findings from animal studies further support the
potential of antioxidant interventions. In a sheep model of FGR,
maternal melatonin supplementation from 0.7 gestation to term
reduced cerebral lipid peroxidation, improved white matter
myelination and axonal integrity in growth-restricted offspring,
and ameliorated neurobehavioral deficits (178). In rats, the
mitochondria-targeted antioxidant MitoQ was shown to cross the
placenta, improve uterine artery reactivity and prevent
hypoxia-induced vascular remodeling (61). However, in a reduced uterine
perfusion pressure mouse model of preeclampsia, in which mild
oxidative stress is required for trophoblast proliferation,
invasion and migration, administration of MitoQ in early pregnancy
may interfere with placental development and exacerbate
preeclampsia-like phenotypes (179). These findings highlight the
need for careful timing and dosing when considering antioxidant
therapies in pregnancy (61,179).
Other antioxidants have also shown therapeutic
benefits in experimental models. In a mouse model of FGR, tempol
increased fetal body weight and crown-rump length, and improved
uterine artery blood flow velocity (180). In guinea pigs, maternal oral
administration of NAC reduced placental vascular resistance,
restored fetal weight, and increased the fetal-to-placental weight
ratio at term. NAC also improved eNOS-dependent relaxation in fetal
aortas and umbilical arteries, normalized eNOS mRNA levels in
endothelial cells, and reversed hypomethylation of the nitric oxide
synthase 3 (Nos3) promoter at CpG-170, suggesting that NAC confers
vascular benefits at least in part through epigenetic modulation of
Nos3 (181). Similarly, in
Wistar rats, maternal resveratrol supplementation ameliorated
oxidative stress marker levels in mothers, fetuses and placentas,
and in a hypoxic pregnancy model, it reduced fetal mortality and
improved intrauterine survival (182,183).
Collectively, clinical and preclinical evidence
suggests that antioxidant-based therapies hold promise for
mitigating FGR by alleviating oxidative stress and improving
placental and fetal outcomes. Nevertheless, the results remain
heterogeneous, and the safety, timing and specificity of
antioxidant interventions require careful consideration before
translation into clinical practice.
Conclusions and future perspectives
Placental oxidative stress serves a central role in
the pathogenesis and progression of FGR. During early pregnancy,
moderate levels of oxidative stress facilitate trophoblast
invasion, spiral artery remodeling and angiogenesis. However, when
perfusion abnormalities and recurrent ischemia-reperfusion occur,
the oxidative burden rises sharply. ROS generated from NOX, XO and
mitochondria accumulate, triggering ER stress and the UPR. This
subsequently activates key signaling pathways, including the
PERK-eIF2α/ATF4, IRE1α-XBP1 and ATF6 signaling pathways. In
parallel, both placental and fetal tissues undergo metabolic
reprogramming, with mitochondrial dysfunction as the central
hallmark. Oxidative stress is further accompanied by marked
epigenetic alterations, such as global DNA hypomethylation,
aberrant H3K9 modifications and dysregulation of noncoding RNAs,
including miR-199a-5p, miR-210-3p and miR-21. Several antioxidants,
such as melatonin, pentoxifylline, NAC and tempol, show potential
in alleviating FGR, although the precise choice of molecular
targets and the optimal timing of intervention may be critical for
successful clinical translation.
Future research should aim to establish an
integrated biomarker system that combines oxidative stress markers
(such as MDA, 4HNE, 8-OHdG and d-ROMs), uterine artery Doppler
indices reflecting impaired uteroplacental perfusion (184), metabolic profiling and
epigenetic signatures to enable early risk stratification and
therapeutic monitoring. Clinically, antioxidant interventions must
strike a balance between preserving physiological ROS levels
required for placental development and preventing pathological
oxidative injury. Promising strategies may include
mitochondria-targeted therapies and selective inhibition of XO,
potentially in combination with pro-angiogenic or immunomodulatory
approaches. Optimizing treatment windows and dosing regimens,
together with long-term maternal-fetal follow-up, will be essential
to translate antioxidant-based interventions from experimental
evidence to a tangible clinical benefit.
Availability of data and materials
Not applicable.
Authors' contributions
DC, SY, FG and QS reviewed the literature and wrote
the manuscript. CW and KF critically revised the manuscript for
important intellectual content and approved the final manuscript
version to be published. Data authentication is not applicable. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present review was funded by the Science and Technology
Development Program of Jinan Municipal Health Commission (grant no.
2024203001), and The Medical Clinical and Technology Transfer Fund
of China International Medical Exchange Foundation (grant no.
Z-2014-08-2309-6).
References
|
1
|
No authors listed. Fetal growth
restriction: ACOG practice bulletin, number 227. Obstet Gynecol.
137:e16–e28. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Temming LA, Dicke JM, Stout MJ, Rampersad
RM, Macones GA, Tuuli MG and Cahill AG: Early second-trimester
fetal growth restriction and adverse perinatal outcomes. Obstet
Gynecol. 130:865–869. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Belkacemi L, Nelson DM, Desai M and Ross
MG: Maternal undernutrition influences placental-fetal development.
Biol Reprod. 83:325–331. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Di Martino DD, Avagliano L, Ferrazzi E,
Fusè F, Sterpi V, Parasiliti M, Stampalija T, Zullino S, Farina A,
Bulfamante GP, et al: Hypertensive disorders of pregnancy and fetal
growth restriction: clinical characteristics and placental lesions
and possible preventive nutritional targets. Nutrients.
14:32762022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pontes KFM and Araujo Júnior E:
Cytomegalovirus and pregnancy: Current evidence for clinical
practice. Rev Assoc Med Bras (1992). 70:e202405092024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu X, He S, Li Y, Guo D, Chen X, Liang B,
Wang M, Huang H and Xu L: Fetal genetic findings by chromosomal
microarray analysis and karyotyping for fetal growth restriction
without structural malformations at a territory referral center:
10-year experience. BMC Pregnancy Childbirth. 23:732023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baud O and Berkane N: Hormonal changes
associated with intra-uterine growth restriction: Impact on the
developing brain and future neurodevelopment. Front Endocrinol
(Lausanne). 10:1792019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fleisch MC and Hoehn T: Intrauterine fetal
death after multiple umbilical cord torsion-complication of a twin
pregnancy following assisted reproduction. J Assist Reprod Genet.
25:277–279. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schoots MH, Gordijn SJ, Scherjon SA, van
Goor H and Hillebrands JL: Oxidative stress in placental pathology.
Placenta. 69:153–161. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Grzeszczak K, Łanocha-Arendarczyk N,
Malinowski W, Ziętek P and Kosik-Bogacka D: Oxidative stress in
pregnancy. Biomolecules. 13:17682023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Myatt L and Cui X: Oxidative stress in the
placenta. Histochem Cell Biol. 122:369–382. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yuba T, Koyama Y, Kinishi Y, Uokawa R,
Ootaki C, Shimada S and Fujino Y: Analysis of maternal and fetal
oxidative stress during delivery with epidural analgesia. Reprod
Sci. 31:2753–2762. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sultana Z, Qiao Y, Maiti K and Smith R:
Involvement of oxidative stress in placental dysfunction, the
pathophysiology of fetal death and pregnancy disorders.
Reproduction. 166:R25–R38. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang Y, Jin H, Qiu Y, Liu Y, Wen L, Fu Y,
Qi H, Baker PN and Tong C: Reactive oxygen species are essential
for placental angiogenesis during early gestation. Oxid Med Cell
Longev. 2022:42909222022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chiarello DI, Abad C, Rojas D, Toledo F,
Vázquez CM, Mate A, Sobrevia L and Marín R: Oxidative stress:
Normal pregnancy versus preeclampsia. Biochim Biophys Acta Mol
Basis Dis. 1866:1653542020. View Article : Google Scholar
|
|
16
|
Jauniaux E, Poston L and Burton GJ:
Placental-related diseases of pregnancy: Involvement of oxidative
stress and implications in human evolution. Hum Reprod Update.
12:747–755. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brien M, Larose J, Greffard K, Julien P
and Bilodeau JF: Increased placental phospholipase A2
gene expression and free F2-isoprostane levels in
response to oxidative stress in preeclampsia. Placenta. 55:54–62.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McElwain CJ, Tuboly E, McCarthy FP and
McCarthy CM: Mechanisms of endothelial dysfunction in pre-eclampsia
and gestational diabetes mellitus: Windows into future
cardiometabolic health? Front Endocrinol (Lausanne). 11:6552020.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Biri A, Bozkurt N, Turp A, Kavutcu M,
Himmetoglu O and Durak I: Role of oxidative stress in intrauterine
growth restriction. Gynecol Obstet Invest. 64:187–192. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tasta O, Swiader A, Grazide MH, Rouahi M,
Parant O, Vayssière C, Bujold E, Salvayre R, Guerby P and
Negre-Salvayre A: A role for 4-hydroxy-2-nonenal in premature
placental senescence in preeclampsia and intrauterine growth
restriction. Free Radic Biol Med. 164:303–314. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fujimaki A, Watanabe K, Mori T, Kimura C,
Shinohara K and Wakatsuki A: Placental oxidative DNA damage and its
repair in preeclamptic women with fetal growth restriction.
Placenta. 32:367–372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kimura C, Watanabe K, Iwasaki A, Mori T,
Matsushita H, Shinohara K and Wakatsuki A: The severity of hypoxic
changes and oxidative DNA damage in the placenta of early-onset
preeclamptic women and fetal growth restriction. J Matern Fetal
Neonatal Med. 26:491–496. 2013. View Article : Google Scholar
|
|
23
|
Zhao S, Zhong S, Wang F, Wang H, Xu D and
Li G: Microcystin-LR exposure decreased the fetal weight of mice by
disturbance of placental development and ROS-mediated endoplasmic
reticulum stress in the placenta. Environ Pollut. 256:1133622020.
View Article : Google Scholar
|
|
24
|
Li L, Zhou L, Li W, Shi F, Feng X and
Zhuang J: Oxidative stress biomarkers in fetal growth restriction:
A systematic review and meta-analysis. Arch Gynecol Obstet.
312:1063–1084. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guerby P, Swiader A, Tasta O, Pont F,
Rodriguez F, Parant O, Vayssière C, Shibata T, Uchida K, Salvayre R
and Negre-Salvayre A: Modification of endothelial nitric oxide
synthase by 4-oxo-2(E)-nonenal(ONE) in preeclamptic placentas. Free
Radic Biol Med. 141:416–425. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Blok EL, Burger RJ, Bergeijk JEV,
Bourgonje AR, Goor HV, Ganzevoort W and Gordijn SJ: Oxidative
stress biomarkers for fetal growth restriction in umbilical cord
blood: A scoping review. Placenta. 154:88–109. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Toblli JE, Cao G, Oliveri L and Angerosa
M: Effects of iron deficiency anemia and its treatment with iron
polymaltose complex in pregnant rats, their fetuses and placentas:
Oxidative stress markers and pregnancy outcome. Placenta. 33:81–87.
2012. View Article : Google Scholar
|
|
28
|
Huang J, Zheng L, Wang F, Su Y, Kong H and
Xin H: Mangiferin ameliorates placental oxidative stress and
activates PI3K/Akt/mTOR pathway in mouse model of preeclampsia.
Arch Pharm Res. 43:233–241. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schoots MH, Bourgonje MF, Bourgonje AR,
Prins JR, van Hoorn EGM, Abdulle AE, Muller Kobold AC, van der
Heide M, Hillebrands JL, van Goor H and Gordijn SJ: Oxidative
stress biomarkers in fetal growth restriction with and without
preeclampsia. Placenta. 115:87–96. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Begum R, Thota S, Abdulkadir A, Kaur G,
Bagam P and Batra S: NADPH oxidase family proteins: Signaling
dynamics to disease management. Cell Mol Immunol. 19:660–686. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Matsubara S and Sato I: Enzyme
histochemically detectable NAD(P)H oxidase in human placental
trophoblasts: Normal, preeclamptic, and fetal growth
restriction-complicated pregnancy. Histochem Cell Biol. 116:1–7.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cheng G, Cao Z, Xu X, van Meir EG and
Lambeth JD: Homologs of gp91phox: Cloning and tissue expression of
Nox3, Nox4, and Nox5. Gene. 269:131–140. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hernandez I, Fournier T, Chissey A,
Therond P, Slama A, Beaudeux JL and Zerrad-Saadi A: NADPH oxidase
is the major source of placental superoxide in early pregnancy:
Association with MAPK pathway activation. Sci Rep. 9:139622019.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cui XL, Brockman D, Campos B and Myatt L:
Expression of NADPH oxidase isoform 1 (Nox1) in human placenta:
Involvement in preeclampsia. Placenta. 27:422–431. 2006. View Article : Google Scholar
|
|
35
|
Dechend R, Viedt C, Müller DN, Ugele B,
Brandes RP, Wallukat G, Park JK, Janke J, Barta P, Theuer J, et al:
AT1 receptor agonistic antibodies from preeclamptic patients
stimulate NADPH oxidase. Circulation. 107:1632–1639. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Poinsignon L, Chissey A, Ajjaji A,
Hernandez I, Vignaud ML, Ferecatu I, Fournier T, Beaudeux JL and
Zerrad-Saadi A: Placental cartography of NADPH oxidase (NOX) family
proteins: Involvement in the pathophysiology of preeclampsia. Arch
Biochem Biophys. 749:1097872023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pacher P, Nivorozhkin A and Szabó C:
Therapeutic effects of xanthine oxidase inhibitors: Renaissance
half a century after the discovery of allopurinol. Pharmacol Rev.
58:87–114. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Griffith JA, Schafner KJ, Garner KL,
DeVallance E, Lewis SE, Nurkiewicz TR, Kelley EE, Maxwell BA,
Goldsmith WT and Bowdridge EC: Maternal electronic cigarette
inhalation exposure during gestation: Impacts on prolactin and
xanthine oxidase. Cardiovasc Toxicol. 25:1095–1106. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Griffith JA, King RD, Dunn AC, Lewis SE,
Maxwell BA, Nurkiewicz TR, Goldsmith WT, Kelley EE and Bowdridge
EC: Maternal nano-titanium dioxide inhalation exposure alters
placental cyclooxygenase and oxidant balance in a sexually
dimorphic manner. Adv Redox Res. Apr 10–2024.Epub ahead of print.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Griffith JA, Dunn A, DeVallance E,
Schafner KJ, Engles KJ, Batchelor TP, Goldsmith WT, Wix K, Hussain
S, Bowdridge EC and Nurkiewicz TR: Maternal nano-titanium dioxide
inhalation alters fetoplacental outcomes in a sexually dimorphic
manner. Front Toxicol. 5:10961732024. View Article : Google Scholar
|
|
41
|
Liu S, Zhang H, Yan B, Zhao H, Wang Y, Gao
T and Liang H: Maternal high-fructose consumption provokes
placental oxidative stress resulting in asymmetrical fetal growth
restriction in rats. J Clin Biochem Nutr. 69:68–76. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shenhav S, Harel I, Solt I, Shenhav A,
Fytlovich S, Aharoni D, Rimler A, Anteby EY and Ovadia YS:
Fetoplacental unit involvement in uric acid production in women
with severe preeclampsia: A prospective case control pilot study. J
Matern Fetal Neonatal Med. 37:23993042024. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Asghar ZA, Thompson A, Chi M, Cusumano A,
Scheaffer S, Al-Hammadi N, Saben JL and Moley KH: Maternal fructose
drives placental uric acid production leading to adverse fetal
outcomes. Sci Rep. 6:250912016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bednarek-Jędrzejek M, Dzidek S, Tousty P,
Kwiatkowska E, Cymbaluk-Płoska A, Góra T, Czuba B, Torbé A and
Kwiatkowski S: Birth weight <3rd percentile prediction using
additional biochemical markers-the uric acid level and angiogenesis
markers (sFlt-1, PlGF)-an exploratory study. Int J Environ Res
Public Health. 19:150592022. View Article : Google Scholar
|
|
45
|
Ahn CS and Metallo CM: Mitochondria as
biosynthetic factories for cancer proliferation. Cancer Metab.
3:12015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Contreras L, Drago I, Zampese E and Pozzan
T: Mitochondria: The calcium connection. Biochim Biophys Acta.
1797:607–618. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Luongo TS, Lambert JP, Gross P, Nwokedi M,
Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E,
van Berlo JH, et al: The mitochondrial
Na+/Ca2+ exchanger is essential for
Ca2+ homeostasis and viability. Nature. 545:93–97. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tait SWG and Green DR: Mitochondrial
regulation of cell death. Cold Spring Harb Perspect Biol.
5:a0087062013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Collins Y, Chouchani ET, James AM, Menger
KE, Cochemé HM and Murphy MP: Mitochondrial redox signalling at a
glance. J Cell Sci. 125:801–806. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Turrens JF: Mitochondrial formation of
reactive oxygen species. J Physiol. 552:335–344. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kowaltowski AJ, de Souza-Pinto NC,
Castilho RF and Vercesi AE: Mitochondria and reactive oxygen
species. Free Radic Biol Med. 47:333–343. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Quinlan CL, Perevoschikova IV, Goncalves
RL, Hey-Mogensen M and Brand MD: The determination and analysis of
site-specific rates of mitochondrial reactive oxygen species
production. Methods Enzymol. 526:189–217. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Murphy MP: How mitochondria produce
reactive oxygen species. Biochem J. 417:1–13. 2009. View Article : Google Scholar
|
|
54
|
Fischbacher A, von Sonntag C and Schmidt
TC: Hydroxyl radical yields in the Fenton process under various pH,
ligand concentrations and hydrogen peroxide/Fe(II) ratios.
Chemosphere. 182:738–744. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Szabó C, Ischiropoulos H and Radi R:
Peroxynitrite: Biochemistry, pathophysiology and development of
therapeutics. Nat Rev Drug Discov. 6:662–680. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Mailloux RJ: Mitochondrial antioxidants
and the maintenance of cellular hydrogen peroxide levels. Oxid Med
Cell Longev. 2018:78572512018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Tong W, Allison BJ, Brain KL, Patey OV,
Niu Y, Botting KJ, Ford SG, Garrud TA, Wooding PFB, Lyu Q, et al:
Placental mitochondrial metabolic adaptation maintains cellular
energy balance in pregnancy complicated by gestational hypoxia. J
Physiol. Jan 27–2025.Epub ahead of print. View Article : Google Scholar
|
|
58
|
Guo Y, Huang C, Xu C, Qiu L and Yang F:
Dysfunction of ZNF554 promotes ROS-induced apoptosis and autophagy
in fetal growth restriction via the p62-Keap1-Nrf2 pathway.
Placenta. 143:34–44. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Dalton-O'Reilly J, Heazell AEP, Greenwood
S, Dilworth M and Desforges M: Sex-specific alterations in
placental mitochondria, oxidative damage and apoptosis in mice of
advanced maternal age. Reproduction. 170:e2501602025.PubMed/NCBI
|
|
60
|
O'Brien KA, Gu W, Houck JA, Holzner LMW,
Yung HW, Armstrong JL, Sowton AP, Baxter R, Darwin PM,
Toledo-Jaldin L, et al: Genomic selection signals in andean
highlanders reveal adaptive placental metabolic phenotypes that are
disrupted in preeclampsia. Hypertension. 81:319–329. 2024.
View Article : Google Scholar
|
|
61
|
Wang Z, Camm EJ, Nuzzo AM, Spiroski AM,
Skeffington KL, Ashmore TJ, Rolfo A, Todros T, Logan A, Ma J, et
al: In vivo mitochondria-targeted protection against uterine artery
vascular dysfunction and remodelling in rodent hypoxic pregnancy. J
Physiol. 602:1211–1225. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Li J, Dong X, Liu JY, Gao L, Zhang WW,
Huang YC, Wang Y, Wang H, Wei W and Xu DX: FUNDC1-mediated
mitophagy triggered by mitochondrial ROS is partially involved in
1-nitropyrene-evoked placental progesterone synthesis inhibition
and intrauterine growth retardation in mice. Sci Total Environ.
908:1683832024. View Article : Google Scholar
|
|
63
|
Feng H, Sun Z, Han B, Xia H, Chen L, Tian
C, Yan S, Shi Y, Yin J, Song W, et al: Miro2 sulfhydration by
CBS/H2S promotes human trophoblast invasion and
migration via regulating mitochondria dynamics. Cell Death Dis.
15:7762024. View Article : Google Scholar
|
|
64
|
Lee KM, Kim TH, Noh EJ, Han JW, Kim JS and
Lee SK: 25-Hydroxycholesterol induces oxidative stress, leading to
apoptosis and ferroptosis in extravillous trophoblasts. Chem Biol
Interact. 403:1112142024. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Chang EI, Stremming J, Knaub LA,
Wesolowski SR, Rozance PJ, Sucharov CC, Reusch JEB and Brown LD:
Mitochondrial respiration is lower in the intrauterine
growth-restricted fetal sheep heart. J Physiol. 602:2697–2715.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Dimasi CG, Darby JRT, Cho SKS, Saini BS,
Holman SL, Meakin AS, Wiese MD, Macgowan CK, Seed M and Morrison
JL: Reduced in utero substrate supply decreases mitochondrial
abundance and alters the expression of metabolic signalling
molecules in the fetal sheep heart. J Physiol. 602:5901–5922. 2024.
View Article : Google Scholar
|
|
67
|
Burton GJ, Woods AW, Jauniaux E and
Kingdom JC: Rheological and physiological consequences of
conversion of the maternal spiral arteries for uteroplacental blood
flow during human pregnancy. Placenta. 30:473–482. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Chouchani ET, Pell VR, Gaude E,
Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord
ENJ, Smith AC, et al: Ischaemic accumulation of succinate controls
reperfusion injury through mitochondrial ROS. Nature. 515:431–435.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Fukushima CT, Dancil IS, Clary H, Shah N,
Nadtochiy SM and Brookes PS: Reactive oxygen species generation by
reverse electron transfer at mitochondrial complex I under
simulated early reperfusion conditions. bioRxiv: Nov 21 2023. (Epub
ahead of print).doi: 2023.11.21.568136. 2023.
|
|
70
|
Many A and Roberts JM: Increased xanthine
oxidase during labour-implications for oxidative stress. Placenta.
18:725–726. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Rashdan NA and Lloyd PG: Fluid shear
stress upregulates placental growth factor in the vessel wall via
NADPH oxidase 4. Am J Physiol Heart Circ Physiol. 309:H1655–H1666.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Hernandez I, Chissey A, Guibourdenche J,
Atasoy R, Coumoul X, Fournier T, Beaudeux JL and Zerrad-Saadi A:
Human placental NADPH oxidase mediates sFlt-1 and PlGF secretion in
early pregnancy: Exploration of the TGF-β1/p38 MAPK pathways.
Antioxidants (Basel). 10:2812021. View Article : Google Scholar
|
|
73
|
Cindrova-Davies T, Spasic-Boskovic O,
Jauniaux E, Charnock-Jones DS and Burton GJ: Nuclear factor-kappa
B, p38, and stress-activated protein kinase mitogen-activated
protein kinase signaling pathways regulate proinflammatory
cytokines and apoptosis in human placental explants in response to
oxidative stress: Effects of antioxidant vitamins. Am J Pathol.
170:1511–1520. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yang Y, Pei X, Jin Y, Wang Y and Zhang C:
The roles of endoplasmic reticulum stress response in female
mammalian reproduction. Cell Tissue Res. 363:589–597. 2016.
View Article : Google Scholar
|
|
75
|
Wang M, Wey S, Zhang Y, Ye R and Lee AS:
Role of the unfolded protein response regulator GRP78/BiP in
development, cancer, and neurological disorders. Antioxid Redox
Signal. 11:2307–2316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Cui W, Li J, Ron D and Sha B: The
structure of the PERK kinase domain suggests the mechanism for its
activation. Acta Crystallogr D Biol Crystallogr. 67:423–428. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Cullinan SB and Diehl JA: PERK-dependent
activation of Nrf2 contributes to redox homeostasis and cell
survival following endoplasmic reticulum stress. J Biol Chem.
279:20108–20117. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Senft D and Ronai ZA: UPR, autophagy, and
mitochondria crosstalk underlies the ER stress response. Trends
Biochem Sci. 40:141–148. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Bailey D and O'Hare P: Transmembrane bZIP
transcription factors in ER stress signaling and the unfolded
protein response. Antioxid Redox Signal. 9:2305–2321. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Shamu CE and Walter P: Oligomerization and
phosphorylation of the Ire1p kinase during intracellular signaling
from the endoplasmic reticulum to the nucleus. EMBO J.
15:3028–3039. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Hassler J, Cao SS and Kaufman RJ: IRE1, a
double-edged sword in pre-miRNA slicing and cell death. Dev Cell.
23:921–923. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Upton JP, Wang L, Han D, Wang ES, Huskey
NE, Lim L, Truitt M, McManus MT, Ruggero D, Goga A, et al: IRE1α
cleaves select microRNAs during ER stress to derepress translation
of proapoptotic caspase-2. Science. 338:818–822. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Yoshida H, Matsui T, Yamamoto A, Okada T
and Mori K: XBP1 mRNA is induced by ATF6 and spliced by IRE1 in
response to ER stress to produce a highly active transcription
factor. Cell. 107:881–891. 2001. View Article : Google Scholar
|
|
84
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Nishitoh H, Matsuzawa A, Tobiume K,
Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A and Ichijo H: ASK1
is essential for endoplasmic reticulum stress-induced neuronal cell
death triggered by expanded polyglutamine repeats. Genes Dev.
16:1345–1355. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Novoa I, Zeng H, Harding HP and Ron D:
Feedback inhibition of the unfolded protein response by
GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol.
153:1011–1022. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Li G, Mongillo M, Chin KT, Harding H, Ron
D, Marks AR and Tabas I: Role of ERO1-alpha-mediated stimulation of
inositol 1,4,5-triphosphate receptor activity in endoplasmic
reticulum stress-induced apoptosis. J Cell Biol. 186:783–792. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Puthalakath H, O'Reilly LA, Gunn P, Lee L,
Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin
J, Motoyama N, et al: ER stress triggers apoptosis by activating
BH3-only protein Bim. Cell. 129:1337–1349. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Du L, He F, Kuang L, Tang W, Li Y and Chen
D: eNOS/iNOS and endoplasmic reticulum stress-induced apoptosis in
the placentas of patients with preeclampsia. J Hum Hypertens.
31:49–55. 2017. View Article : Google Scholar
|
|
90
|
Lian IA, Løset M, Mundal SB, Fenstad MH,
Johnson MP, Eide IP, Bjørge L, Freed KA, Moses EK and Austgulen R:
Increased endoplasmic reticulum stress in decidual tissue from
pregnancies complicated by fetal growth restriction with and
without pre-eclampsia. Placenta. 32:823–829. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Grasso E, Gori S, Soczewski E, Fernández
L, Gallino L, Vota D, Martínez G, Irigoyen M, Ruhlmann C, Lobo TF,
et al: Impact of the reticular stress and unfolded protein response
on the inflammatory response in endometrial stromal cells. Sci Rep.
8:122742018. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Cheng S, Huang Z, Jash S, Wu K, Saito S,
Nakashima A and Sharma S: Hypoxia-reoxygenation impairs
autophagy-lysosomal machinery in primary human trophoblasts
mimicking placental pathology of early-onset preeclampsia. Int J
Mol Sci. 23:56442022. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Li H, Zhao W, Wang L, Luo Q, Yin N, Lu X,
Hou Y, Cui J and Zhang H: Human placenta-derived mesenchymal stem
cells inhibit apoptosis of granulosa cells induced by IRE1α pathway
in autoimmune POF mice. Cell Biol Int. 43:899–909. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Gao XX, Lin S, Jiang PY, Ye MY, Chen W, Hu
CX and Chen YH: Gestational cholestasis induced intrauterine growth
restriction through triggering IRE1α-mediated apoptosis of
placental trophoblast cells. FASEB J. 36:e223882022. View Article : Google Scholar
|
|
95
|
Liong S and Lappas M: Endoplasmic
reticulum stress is increased in adipose tissue of women with
gestational diabetes. PLoS One. 10:e01226332015. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Shen WB, Wang B, Yao R, Goetzinger KR, Wu
S, Gao H and Yang P: Obesity impacts placental function through
activation of p-IRE1a-XBP1s signaling. Front Cell Dev Biol.
11:10233272023. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Sahoo PK, Krishnamoorthy C, Wood JR,
Hanson C, Anderson-Berry A, Mott JL and Natarajan SK: Palmitate
induces integrated stress response and lipoapoptosis in
trophoblasts. Cell Death Dis. 15:312024. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Rampersaud AM, Dunk CE, Lye SJ and Renaud
SJ: Palmitic acid induces inflammation in placental trophoblasts
and impairs their migration toward smooth muscle cells through
plasminogen activator inhibitor-1. Mol Hum Reprod. 26:850–865.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Huovinen M, Ietta F, Repo JK, Paulesu L
and Vähäkangas KH: The effect of ethanol and nicotine on ER stress
in human placental villous explants. Curr Res Toxicol.
3:1000812022. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Muthuraj PG, Sahoo PK, Kraus M, Bruett T,
Annamalai AS, Pattnaik A, Pattnaik AK, Byrareddy SN and Natarajan
SK: Zika virus infection induces endoplasmic reticulum stress and
apoptosis in placental trophoblasts. Cell Death Discov. 7:242021.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Liu L, Zhang Y, Wang Y, Peng W, Zhang N
and Ye Y: Progesterone inhibited endoplasmic reticulum stress
associated apoptosis induced by interleukin-1β via the
GRP78/PERK/CHOP pathway in BeWo cells. J Obstet Gynaecol Res.
44:463–473. 2018. View Article : Google Scholar
|
|
102
|
Yung HW, Calabrese S, Hynx D, Hemmings BA,
Cetin I, Charnock-Jones DS and Burton GJ: Evidence of placental
translation inhibition and endoplasmic reticulum stress in the
etiology of human intrauterine growth restriction. Am J Pathol.
173:451–462. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Thomson S, Waters KA, Hennessy A and
Machaalani R: The unfolded protein response and apoptotic
regulation in the human placenta due to maternal cigarette smoking
and pre-eclampsia. Reprod Toxicol. 105:120–127. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Gupta MB, Abu Shehab M, Nygard K, Biggar
K, Singal SS, Santoro N, Powell TL and Jansson T: IUGR Is
associated with marked hyperphosphorylation of decidual and
maternal plasma IGFBP-1. J Clin Endocrinol Metab. 104:408–422.
2019. View Article : Google Scholar :
|
|
105
|
Castro KR, Prado KM, Lorenzon AR, Hoshida
MS, Alves EA, Francisco RPV, Zugaib M, Marques ALX, Silva ECO,
Fonseca EJS, et al: Serum from preeclamptic women triggers
endoplasmic reticulum stress pathway and expression of angiogenic
factors in trophoblast cells. Front Physiol. 12:7996532022.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Liu J and Yang W: Mechanism of histone
deacetylase HDAC2 in FOXO3-mediated trophoblast pyroptosis in
preeclampsia. Funct Integr Genomics. 23:1522023. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Wang P, Zhao C, Zhou H, Huang X, Ying H,
Zhang S, Pan Y and Zhu H: Dysregulation of histone deacetylases
inhibits trophoblast growth during early placental development
partially through TFEB-dependent autophagy-lysosomal pathway. Int J
Mol Sci. 24:118992023. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Yung HW, Atkinson D, Campion-Smith T,
Olovsson M, Charnock-Jones DS and Burton GJ: Differential
activation of placental unfolded protein response pathways implies
heterogeneity in causation of early- and late-onset pre-eclampsia.
J Pathol. 234:262–276. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Almada M, Costa L, Fonseca B, Alves P,
Braga J, Gonçalves D, Teixeira N and Correia-da-Silva G: The
endocannabinoid 2-arachidonoylglycerol promotes endoplasmic
reticulum stress in placental cells. Reproduction. 160:171–180.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Shi XT, Zhu HL, Xiong YW, Liu WB, Zhou GX,
Cao XL, Yi SJ, Dai LM, Zhang C, Gao L, et al: Cadmium
down-regulates 11β-HSD2 expression and elevates active
glucocorticoid level via PERK/p-eIF2α pathway in placental
trophoblasts. Chemosphere. 254:1267852020. View Article : Google Scholar
|
|
111
|
Shi XT, Zhu HL, Xu XF, Xiong YW, Dai LM,
Zhou GX, Liu WB, Zhang YF, Xu DX and Wang H: Gestational cadmium
exposure impairs placental angiogenesis via activating GC/GR
signaling. Ecotoxicol Environ Saf. 224:1126322021. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Chau K, Hennessy A and Makris A: Placental
growth factor and pre-eclampsia. J Hum Hypertens. 31:782–786. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Mizuuchi M, Cindrova-Davies T, Olovsson M,
Charnock-Jones DS, Burton GJ and Yung HW: Placental endoplasmic
reticulum stress negatively regulates transcription of placental
growth factor via ATF4 and ATF6β: Implications for the
pathophysiology of human pregnancy complications. J Pathol.
238:550–561. 2016. View Article : Google Scholar :
|
|
114
|
Cadenas E and Davies KJ: Mitochondrial
free radical generation, oxidative stress, and aging. Free Radic
Biol Med. 29:222–230. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Rodríguez-Rodríguez P, Ramiro-Cortijo D,
Reyes-Hernández CG, López de Pablo AL, González MC and Arribas SM:
Implication of oxidative stress in fetal programming of
cardiovascular disease. Front Physiol. 9:6022018. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Wang J, Chen L, Li D, Yin Y, Wang X, Li P,
Dangott LJ, Hu W and Wu G: Intrauterine growth restriction affects
the proteomes of the small intestine, liver, and skeletal muscle in
newborn pigs. J Nutr. 138:60–66. 2008. View Article : Google Scholar
|
|
117
|
Devarajan A, Rajasekaran NS, Valburg C,
Ganapathy E, Bindra S and Freije WA: Maternal perinatal calorie
restriction temporally regulates the hepatic autophagy and redox
status in male rat. Free Radic Biol Med. 130:592–600. 2019.
View Article : Google Scholar
|
|
118
|
Sohi G, Marchand K, Revesz A, Arany E and
Hardy DB: Maternal protein restriction elevates cholesterol in
adult rat offspring due to repressive changes in histone
modifications at the cholesterol 7alpha-hydroxylase promoter. Mol
Endocrinol. 25:785–798. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Oke SL, Sohi G and Hardy DB: Perinatal
protein restriction with postnatal catch-up growth leads to
elevated p66Shc and mitochondrial dysfunction in the adult rat
liver. Reproduction. 159:27–39. 2020. View Article : Google Scholar
|
|
120
|
Theys N, Clippe A, Bouckenooghe T, Reusens
B and Remacle C: Early low protein diet aggravates unbalance
between antioxidant enzymes leading to islet dysfunction. PLoS One.
4:e61102009. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Setia S, Sridhar MG, Bhat V, Chaturvedula
L, Vinayagamoorti R and John M: Insulin sensitivity and insulin
secretion at birth in intrauterine growth retarded infants.
Pathology. 38:236–238. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Milovanovic I, Njuieyon F, Deghmoun S,
Chevenne D, Levy-Marchal C and Beltrand J: SGA children with
moderate catch-up growth are showing the impaired insulin secretion
at the age of 4. PLoS One. 9:e1003372014. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Simmons RA, Suponitsky-Kroyter I and Selak
MA: Progressive accumulation of mitochondrial DNA mutations and
decline in mitochondrial function lead to beta-cell failure. J Biol
Chem. 280:28785–28791. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Theys N, Bouckenooghe T, Ahn MT, Remacle C
and Reusens B: Maternal low-protein diet alters pancreatic islet
mitochondrial function in a sex-specific manner in the adult rat.
Am J Physiol Regul Integr Comp Physiol. 297:R1516–R1525. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Lane RH, Chandorkar AK, Flozak AS and
Simmons RA: Intrauterine growth retardation alters mitochondrial
gene expression and function in fetal and juvenile rat skeletal
muscle. Pediatr Res. 43:563–570. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Liu J, Chen D, Yao Y, Yu B, Mao X, He J,
Huang Z and Zheng P: Intrauterine growth retardation increases the
susceptibility of pigs to high-fat diet-induced mitochondrial
dysfunction in skeletal muscle. PLoS One. 7:e348352012. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Beauchamp B, Ghosh S, Dysart MW, Kanaan
GN, Chu A, Blais A, Rajamanickam K, Tsai EC, Patti ME and Harper
ME: Low birth weight is associated with adiposity, impaired
skeletal muscle energetics and weight loss resistance in mice. Int
J Obes (Lond). 39:702–711. 2015. View Article : Google Scholar :
|
|
128
|
Puente BN, Kimura W, Muralidhar SA, Moon
J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R,
Garcia JA, et al: The oxygen-rich postnatal environment induces
cardiomyocyte cell-cycle arrest through DNA damage response. Cell.
157:565–579. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Barra NG, Lisyansky M, Vanduzer TA, Raha
S, Holloway AC and Hardy DB: Maternal nicotine exposure leads to
decreased cardiac protein disulfide isomerase and impaired
mitochondrial function in male rat offspring. J Appl Toxicol.
37:1517–1526. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Dwi Putra SE, Neuber C, Reichetzeder C,
Hocher B and Kleuser B: Analysis of genomic DNA methylation levels
in human placenta using liquid chromatography-electrospray
ionization tandem mass spectrometry. Cell Physiol Biochem.
33:945–952. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Nelissen EC, van Montfoort AP, Dumoulin JC
and Evers JL: Epigenetics and the placenta. Hum Reprod Update.
17:397–417. 2011. View Article : Google Scholar
|
|
132
|
Curradi M, Izzo A, Badaracco G and
Landsberger N: Molecular mechanisms of gene silencing mediated by
DNA methylation. Mol Cell Biol. 22:3157–3173. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Handy DE, Castro R and Loscalzo J:
Epigenetic modifications: Basic mechanisms and role in
cardiovascular disease. Circulation. 123:2145–2156. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Saxena A and Carninci P: Long non-coding
RNA modifies chromatin: Epigenetic silencing by long non-coding
RNAs. Bioessays. 33:830–839. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Neve B, Jonckheere N, Vincent A and Van
Seuningen I: Epigenetic regulation by lncRNAs: An overview focused
on UCA1 in colorectal cancer. Cancers (Basel). 10:4402018.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Shi D, Zhou X, Cai L, Wei X, Zhang L, Sun
Q, Zhou F and Sun L: Placental DNA methylation analysis of
selective fetal growth restriction in monochorionic twins reveals
aberrant methylated CYP11A1 gene for fetal growth restriction.
FASEB J. 37:e232072023. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Bi Y, Yang J, Hu Y, Lin Y, Gao L, Lv J and
Wang Y: Placental epigenetic and transcriptional dysregulation in
type I selective fetal growth restriction. Placenta. 168:268–275.
2023. View Article : Google Scholar
|
|
138
|
Ergaz Z, Guillemin C, Neeman-Azulay M,
Weinstein-Fudim L, Stodgell CJ, Miller RK, Szyf M and Ornoy A:
Placental oxidative stress and decreased global DNA methylation are
corrected by copper in the Cohen diabetic rat. Toxicol Appl
Pharmacol. 276:220–230. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Hu Y, Lin Y, Yang J, Wang S, Gao L, Bi Y
and Wang Y: Mitochondrial dysfunction and oxidative stress in
selective fetal growth restriction. Placenta. 156:46–54. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Gu Y, Cooper D, Lewis DF, Zoorob D and
Wang Y: Oxidative stress contributes to hypermethylation of histone
H3 lysine 9 in placental trophoblasts from preeclamptic
pregnancies. Front Endocrinol (Lausanne). 15:13712202024.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Tian X, Yu C, Shi L, Li D, Chen X, Xia D,
Zhou J, Xu W, Ma C, Gu L and An Y: MicroRNA-199a-5p aggravates
primary hypertension by damaging vascular endothelial cells through
inhibition of autophagy and promotion of apoptosis. Exp Ther Med.
16:595–602. 2018.PubMed/NCBI
|
|
142
|
Hsu CY, Hsieh TH, Tsai CF, Tsai HP, Chen
HS, Chang Y, Chuang HY, Lee JN, Hsu YL and Tsai EM: miRNA-199a-5p
regulates VEGFA in endometrial mesenchymal stem cells and
contributes to the pathogenesis of endometriosis. J Pathol.
232:330–343. 2014. View Article : Google Scholar
|
|
143
|
Li B, He L, Zuo D, He W, Wang Y, Zhang Y,
Liu W and Yuan Y: Mutual regulation of MiR-199a-5p and HIF-1α
modulates the warburg effect in hepatocellular carcinoma. J Cancer.
8:940–949. 2017. View Article : Google Scholar
|
|
144
|
Meng M, Cheng YKY, Wu L, Chaemsaithong P,
Leung MBW, Chim SSC, Sahota DS, Li W, Poon LCY, Wang CC and Leung
TY: Whole genome miRNA profiling revealed miR-199a as potential
placental pathogenesis of selective fetal growth restriction in
monochorionic twin pregnancies. Placenta. 92:44–53. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Rane S, He M, Sayed D, Vashistha H,
Malhotra A, Sadoshima J, Vatner DE, Vatner SF and Abdellatif M:
Downregulation of miR-199a derepresses hypoxia-inducible
factor-1alpha and Sirtuin 1 and recapitulates hypoxia
preconditioning in cardiac myocytes. Circ Res. 104:879–886. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Whitehead CL, Teh WT, Walker SP, Leung C,
Larmour L and Tong S: Circulating MicroRNAs in maternal blood as
potential biomarkers for fetal hypoxia in-utero. PLoS One.
8:e784872013. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Li L, Huang X, He Z, Xiong Y and Fang Q:
miRNA-210-3p regulates trophoblast proliferation and invasiveness
through fibroblast growth factor 1 in selective intrauterine growth
restriction. J Cell Mol Med. 23:4422–4433. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Sangokoya C, Telen MJ and Chi JT: microRNA
miR-144 modulates oxidative stress tolerance and associates with
anemia severity in sickle cell disease. Blood. 116:4338–4348. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Yu D, dos Santos CO, Zhao G, Jiang J,
Amigo JD, Khandros E, Dore LC, Yao Y, D'Souza J, Zhang Z, et al:
miR-451 protects against erythroid oxidant stress by repressing
14-3-3zeta. Genes Dev. 24:1620–1633. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Wang X, Zhu H, Zhang X, Liu Y, Chen J,
Medvedovic M, Li H, Weiss MJ, Ren X and Fan GC: Loss of the
miR-144/451 cluster impairs ischaemic preconditioning-mediated
cardioprotection by targeting Rac-1. Cardiovasc Res. 94:379–390.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Kutty RK, Samuel W, Jaworski C, Duncan T,
Nagineni CN, Raghavachari N, Wiggert B and Redmond TM: MicroRNA
expression in human retinal pigment epithelial (ARPE-19) cells:
Increased expression of microRNA-9 by N-(4-hydroxyphenyl)
retinamide. Mol Vis. 16:1475–1486. 2010.PubMed/NCBI
|
|
152
|
Luna C, Li G, Qiu J, Epstein DL and
Gonzalez P: Role of miR-29b on the regulation of the extracellular
matrix in human trabecular meshwork cells under chronic oxidative
stress. Mol Vis. 15:2488–2497. 2009.PubMed/NCBI
|
|
153
|
Li P, Guo W, Du L, Zhao J, Wang Y, Liu L,
Hu Y and Hou Y: microRNA-29b contributes to pre-eclampsia through
its effects on apoptosis, invasion and angiogenesis of trophoblast
cells. Clin Sci (Lond). 124:27–40. 2013. View Article : Google Scholar
|
|
154
|
Wang Y, Fan H, Zhao G, Liu D, Du L, Wang
Z, Hu Y and Hou Y: miR-16 inhibits the proliferation and
angiogenesis-regulating potential of mesenchymal stem cells in
severe pre-eclampsia. FEBS J. 279:4510–4524. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Zhang Y, Diao Z, Su L, Sun H, Li R, Cui H
and Hu Y: MicroRNA-155 contributes to preeclampsia by
down-regulating CYR61. Am J Obstet Gynecol. 202:466.e1–e7. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Li G, Luna C, Qiu J, Epstein DL and
Gonzalez P: Role of miR-204 in the regulation of apoptosis,
endoplasmic reticulum stress response, and inflammation in human
trabecular meshwork cells. Invest Ophthalmol Vis Sci. 52:2999–3007.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Chen T, Ding G, Jin Z, Wagner MB and Yuan
Z: Insulin ameliorates miR-1-induced injury in H9c2 cells under
oxidative stress via Akt activation. Mol Cell Biochem. 369:167–174.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Zhu XM, Han T, Sargent IL, Yin GW and Yao
YQ: Differential expression profile of microRNAs in human placentas
from preeclamptic pregnancies vs normal pregnancies. Am J Obstet
Gynecol. 200:661.e1–e7. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Hu XQ and Zhang L: MicroRNAs in
uteroplacental vascular dysfunction. Cells. 8:13442019. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Maccani MA, Padbury JF and Marsit CJ:
miR-16 and miR-21 expression in the placenta is associated with
fetal growth. PLoS One. 6:e212102011. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Tu H, Sun H, Lin Y, Ding J, Nan K, Li Z,
Shen Q and Wei Y: Oxidative stress upregulates PDCD4 expression in
patients with gastric cancer via miR-21. Curr Pharm Des.
20:1917–1923. 2014. View Article : Google Scholar
|
|
162
|
Morley LC, Debant M, Gaunt HJ, Simpson NAB
and Beech DJ: Nitric oxide synthase phosphorylation in
fetoplacental endothelium is enhanced by agonism of Piezo1
mechanosensor in small for gestational age babies. Reprod Fertil.
4:e2201002023. View Article : Google Scholar :
|
|
163
|
Morley LC, Debant M, Walker JJ, Beech DJ
and Simpson NAB: Placental blood flow sensing and regulation in
fetal growth restriction. Placenta. 113:23–28. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Hu XQ, Song R and Zhang L: Effect of
oxidative stress on the estrogen-NOS-NO-KCa channel
pathway in uteroplacental dysfunction: Its implication in pregnancy
complications. Oxid Med Cell Longev. 2019:91942692019.
|
|
165
|
Krupp J, Boeldt DS, Yi FX, Grummer MA,
Bankowski Anaya HA, Shah DM and Bird IM: The loss of sustained
Ca(2+) signaling underlies suppressed endothelial nitric oxide
production in preeclamptic pregnancies: Implications for new
therapy. Am J Physiol Heart Circ Physiol. 305:H969–H979. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Cortés MP, Alonso C, Vinet R,
Valdivia-Cortés K, Muñoz-Sagredo L, Bahamondez-Canas TF and
Cárdenas AM: Ca2+ signals in human umbilical endothelial
cells derived from pregnancy with fetal growth restriction
associated with hypertensive disorder. Biomed Rep. 20:762024.
View Article : Google Scholar
|
|
167
|
Guerby P, Swiader A, Augé N, Parant O,
Vayssière C, Uchida K, Salvayre R and Negre-Salvayre A: High
glutathionylation of placental endothelial nitric oxide synthase in
preeclampsia. Redox Biol. 22:1011262019. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Chatre L, Ducat A, Spradley FT, Palei AC,
Chéreau C, Couderc B, Thomas KC, Wilson AR, Amaral LM, Gaillard I,
et al: Increased NOS coupling by the metabolite tetrahydrobiopterin
(BH4) reduces preeclampsia/IUGR consequences. Redox Biol.
55:1024062022. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Grandvuillemin I, Buffat C, Boubred F,
Lamy E, Fromonot J, Charpiot P, Simoncini S, Sabatier F,
Dignat-George F, Peyter AC, et al: Arginase upregulation and eNOS
uncoupling contribute to impaired endothelium-dependent
vasodilation in a rat model of intrauterine growth restriction. Am
J Physiol Regul Integr Comp Physiol. 315:R509–R520. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Hu XQ, Song R, Romero M, Dasgupta C, Huang
X, Holguin MA, Williams V, Xiao D, Wilson SM and Zhang L: Pregnancy
increases Ca2+ sparks/spontaneous transient outward
currents and reduces uterine arterial myogenic tone. Hypertension.
73:691–702. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Hu XQ, Dasgupta C, Chen M, Xiao D, Huang
X, Han L, Yang S, Xu Z and Zhang L: Pregnancy reprograms
large-conductance Ca2+-activated K+ channel
in uterine arteries: Roles of ten-eleven translocation
methylcytosine dioxygenase 1-mediated active demethylation.
Hypertension. 69:1181–1191. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Hu XQ, Song R, Romero M, Dasgupta C, Min
J, Hatcher D, Xiao D, Blood A, Wilson SM and Zhang L: Gestational
hypoxia inhibits pregnancy-induced upregulation of Ca2+
sparks and spontaneous transient outward currents in uterine
arteries via heightened endoplasmic reticulum/oxidative stress.
Hypertension. 76:930–942. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
He M, Li F, Yang M, Fan Y, Beejadhursing
R, Xie Y, Zhou Y and Deng D: Impairment of BKca channels in human
placental chorionic plate arteries is potentially relevant to the
development of preeclampsia. Hypertens Res. 41:126–134. 2018.
View Article : Google Scholar
|
|
174
|
Lv Q, Xue Y, Li G, Zou L, Zhang X, Ying M,
Wang S, Guo L, Gao Y, Li G, et al: Beneficial effects of evodiamine
on P2X(4)-mediated inflammatory injury of human umbilical vein
endothelial cells due to high glucose. Int Immunopharmacol.
28:1044–1049. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Alers NO, Jenkin G, Miller SL and Wallace
EM: Antenatal melatonin as an antioxidant in human pregnancies
complicated by fetal growth restriction-a phase I pilot clinical
trial: Study protocol. BMJ Open. 3:e0041412013. View Article : Google Scholar
|
|
176
|
Asadi N, Roozmeh S, Vafaei H, Asmarian N,
Jamshidzadeh A, Bazrafshan K, Kasraeian M, Faraji A, Shiravani Z,
Mokhtar Pour A, et al: Effectiveness of pentoxifylline in severe
early-onset fetal growth restriction: A randomized double-blinded
clinical trial. Taiwan J Obstet Gynecol. 61:612–619. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Rumbold AR, Crowther CA, Haslam RR, Dekker
GA and Robinson JS; ACTS Study Group: Vitamins C and E and the
risks of preeclampsia and perinatal complications. N Engl J Med.
354:1796–1806. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Miller SL, Yawno T, Alers NO,
Castillo-Melendez M, Supramaniam VG, VanZyl N, Sabaretnam T, Loose
JM, Drummond GR, Walker DW, et al: Antenatal antioxidant treatment
with melatonin to decrease newborn neurodevelopmental deficits and
brain injury caused by fetal growth restriction. J Pineal Res.
56:283–294. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Yang Y, Xu P, Zhu F, Liao J, Wu Y, Hu M,
Fu H, Qiao J, Lin L, Huang B, et al: The potent antioxidant MitoQ
protects against preeclampsia during late gestation but increases
the risk of preeclampsia when administered in early pregnancy.
Antioxid Redox Signal. 34:118–136. 2021. View Article : Google Scholar
|
|
180
|
Stanley JL, Andersson IJ, Hirt CJ, Moore
L, Dilworth MR, Chade AR, Sibley CP, Davidge ST and Baker PN:
Effect of the anti-oxidant tempol on fetal growth in a mouse model
of fetal growth restriction. Biol Reprod. 87(25): 1–8. 2012.
View Article : Google Scholar
|
|
181
|
Herrera EA, Cifuentes-Zúñiga F, Figueroa
E, Villanueva C, Hernández C, Alegría R, Arroyo-Jousse V, Peñaloza
E, Farías M, Uauy R, et al: N-Acetylcysteine, a glutathione
precursor, reverts vascular dysfunction and endothelial epigenetic
programming in intrauterine growth restricted guinea pigs. J
Physiol. 595:1077–1092. 2017. View Article : Google Scholar
|
|
182
|
Vega CC, Reyes-Castro LA,
Rodríguez-González GL, Bautista CJ, Vázquez-Martínez M, Larrea F,
Chamorro-Cevallos GA, Nathanielsz PW and Zambrano E: Resveratrol
partially prevents oxidative stress and metabolic dysfunction in
pregnant rats fed a low protein diet and their offspring. J
Physiol. 594:1483–1499. 2016. View Article : Google Scholar
|
|
183
|
Bourque SL, Dolinsky VW, Dyck JRB and
Davidge ST: Maternal resveratrol treatment during pregnancy
improves adverse fetal outcomes in a rat model of severe hypoxia.
Placenta. 33:449–452. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Pedroso MA, Palmer KR, Hodges RJ, Costa
FDS and Rolnik DL: Uterine artery doppler in screening for
preeclampsia and fetal growth restriction. Rev Bras Ginecol Obstet.
40:287–293. 2018. View Article : Google Scholar : PubMed/NCBI
|














