Introduction
Sea water drowning is a crucial public safety
problem and is the third leading cause of accidental fatality,
claiming ~372,000 lives annually worldwide (1,2).
In near-drowning patients, acute lung injury (ALI) or acute
respiratory distress syndrome (ARDS) is one of the most common
complications. ALI is a life-threatening clinical syndrome that
occurs in critically ill patients because of an uncontrolled
systemic inflammatory response. This response can either result
from direct injury to the lung or from indirect injury in the
setting of a systemic process following lipopolysaccharide (LPS)
exposure or drowning (3,4). However, the mechanisms through
which ALI is induced by drowning have not been well elucidated.
Severe inflammatory responses, when coupled with an
excessive oxidative stress response, are considered to be the main
causes of ALI (5,6). The onset of ALI is followed by a
strong inflammatory response in lung tissue. Inflammatory cells
such as alveolar macrophages (AMs) infiltrate lung tissue and
produce inflammatory cytokines (7). Also, the abnormal production of
reactive oxygen species (ROS) is considered to further worsen the
onset and development of lung injury (8). Our previous studies indicated that
endothelial semaphorin 7A promoted seawater aspiration-induced
acute lung injury through plexin C1 and β1 integrin (9). It was also shown that seawater
inhalation induced ALI ROS generation and the endoplasmic reticulum
stress pathway (10). This
suggested that severe inflammatory responses and ROS production
exited in drowning-induced ALI (drowning-ALI). Innate immune cells
located in the lung epithelium play an essential role by producing
pro-inflammatory factors to eliminate pathogens and releasing
anti-inflammatory factors to maintain lung homeostasis (11). Pulmonary macrophage is a critical
cell of the pneumonic innate immune system in lung injury,
suggesting that macrophages are activated and polarized in response
to LPS-induced lung injury (12). However, the effect of macrophages
in drowning-ALI is poorly understood.
SH3 domain-containing GRB2-like protein B1 (SH3GLB1)
or Bax-interacting Factor 1 (Bif 1), is an Endophilin B protein.
SH3GLB1 is expressed in most tissues, with high expression in the
heart and skeletal muscle (13).
Studies have reported that SH3GLB1 plays a key role in cancer
(14,15), Parkinson's disease (16), Alzheimer's disease (17) and cerebral ischemic injury
(18). However, whether SH3GLB1
is involved in maintaining normal cardiac function and myocardial
I/R injury remains unclear. Furthermore, the molecular mechanism
responsible for SH3GLB1 related cardiac regulation requires further
investigation.
Therefore, the objectives of this study were to
examine the effects of macrophages in drowning-ALI and to elucidate
the underlying molecular mechanism involved.
Materials and methods
Animals
All animal procedures followed the eighth edition
(2011) of the Guide for the Care and Use of Laboratory Animals
(https://www.ncbi.nlm.nih.gov/books/NBK54050/) and were
approved by the Fourth Military Medical University Animal Ethics
Committee (approval no. 20250088). A total of 20 SH3GLB1-deficient
mice on a C57BL/6J background and 20 of their wild-type littermates
were produced by Shanghai Model Organisms Center. Animals were
housed at 22±0.5°C and 60±5% relative humidity under a 12-h
light/dark cycle. Mice were allocated randomly and subsequent
histological and functional analyses were performed by
investigators unaware of genotype.
Drowningand LPS-induced ALI (LPS-ALI)
models
A total of 30 8-week-old pathogen-free, 15 male WT
C57BL/6 mice or 15 SH3GLB1 KO mice (20-22 g) were housed at
22±0.5°C and 60±5% relative humidity under a 12-h light/dark cycle.
Then they were used to establish an LPS-induced ALI model (19). Living conditions were as in the
previous section. In brief, mice are anesthetized with isoflurane
using an induction concentration of 5% for 2-5 min until the
animals lose consciousness. For maintenance, the concentration was
kept at 2.5% for 2-3 min, depending on the duration of seawater
perfusion. Then, 50 µl of 10 mg/kg LPS (cat. no. O55:B5;
MilliporeSigma) was slowly dripped into the lungs from the air pipe
to the distal end of the lung with a microinjector. To induce
drowning-related ALI, a seawater solution matching East China Sea
parameters (1,300 mmol/l osmolality; pH 8.2; SW 1.05; containing
6.518 g/l NaCl, 3.305 g/l MgSO4, 2.447 g/l
MgCl2, 1.141 g/l CaCl2, 0.725 g/l KCl, 0.202
g/l NaHCO3, 0.083 g/l NaBr) was slowly instilled at 4
ml/kg into the distal airways via a microinjector inserted through
the trachea (20). After 0.5, 1,
2 and 3 h of seawater inhalation, arterial blood from the right
common carotid artery was collected for blood gas analysis.
PaO2/FiO2≤300 mmHg (40 kPa) indicated
successful modelling.
Histopathological analysis
Paraffin sections of lungs (5 µg) were
stained with hematoxylin-eosin (room temperature; 1 min) and
evaluated independently by two pathologists unaware of group
assignment. Injury was graded 0-3 (absent to severe) for
interstitial/alveolar oedema, hemorrhage, septal thickening and
inflammatory-cell infiltration (21).
Single-cell preparation and single-cell
RNA-sequencing
Lungs were harvested and enzymatically dissociated
with 0.4 mg/ml collagenase A plus 0.4 mg/ml DNase to yield
single-cell suspensions; material from identical time points was
pooled. After Fc-blockade (anti-CD16/32, Invitrogen; cat. no.
MFCR00), cells were stained with anti-CD45 (BioLegend, Inc.; cat.
no. 480027) and CD45+ leukocytes were purified by
fluorescence-activated cell sorting for subsequent assays. Using
the 10X Genomics Chromium platform (Shanghai OE Biotech Co., Ltd.),
barcoded gel beads were loaded to saturation so that each gel
bead-in-emulsion (GEMs) contained one cell and one bead. After
lysis, poly-A RNA hybridized to the beads, which were then pooled
for reverse transcription; every cDNA received a 5′ UMI and a
cell-specific barcode. Libraries were checked on a Bioanalyzer
2,100 with an Agilent high-sensitivity DNA chip and quantified by
the Qubit HS DNA kit (Q33230; Thermo Fisher, Inc.) before 150-bp
paired-end sequencing on a NovaSeq 6,000 (Illumina, Inc.). Reads
were demultiplexed and aligned with Cell Ranger 4.0 (10X Genomics,
Inc.) under default settings. Gene Ontology (GO) functional
enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG)
pathway enrichment analyses of differentially expressed genes were
performed with ClusterProfiler (v4.2.0; Shanghai OE Biotech Co.,
Ltd.). The SRA data are accessible with the following link:
https://www.ncbi.nlm.nih.gov/sra/PRJNA1416051.
Flow cytometry analysis
After harvesting, cells were resuspended in BD
Pharmingen Stain Buffer (BD Biosciences; cat. no. 554656;) and
incubated for 30 min at 4°C with the following macrophage-focused
panel: APC-F4/80 (cat. no. 123115; BioLegend, Inc.; 5 µl),
Pacific Blue-Ly6G (cat. no. 127612; BioLegend, Inc.; 5 µl),
FITC-CD11b (cat. no. 101205; BioLegend, Inc.; 5 µl) and
PE-CD11c (cat. no. 117307; BioLegend, Inc.; 5 µl). Viability
was assessed with DAPI plus BD Horizon Fixable Viability Stain 780
(cat. no. 565388; 1:1,000) and 510 (cat. no. 564406; 1:1,000).
Following three washes in stain buffer, samples were acquired on an
ImageStream Mark II (Luminex) and analyzed with IDEAS v6.2 (Cytek
Biosciences, Inc.).
Transmission electron microscopy
(TEM)
Lung tissue was initially fixed overnight at 4°C in
4% glutaraldehyde prepared in PBS, then post-fixed for 1 h with 1%
osmium tetroxide. After dehydration the specimens were
resin-embedded and sectioned at 80 nm. Ultrastructural images were
recorded on a JEOL JEM-1230 transmission electron microscope (JEOL,
Ltd.) operating at 80 kV and mitochondrial cross-sectional areas
were measured with ImageJ 1.54p (National Institutes of
Health).
Reverse transcription-quantitative (RT-q)
PCR
RNA was extracted with TRIzol® (cat. no.
15596026; Thermo Fisher Scientific, Inc.). The purity of extracted
RNA was evaluated using the 260/280 ratio, with values between 1.8
and 2.0 indicating good purity. The RNA concentration ranged from
500 to 1,000 ng/ml. After gDNA removal (PrimeScript RT kit; cat.
no. RR047A; Takara Biotechnology Co., Ltd.), cDNA was synthesized
and analyzed by SYBR-based qPCR (SYBR Premix Ex Taq II; cat. no.
RR820L; Takara Biotechnology Co., Ltd. Transcript levels were
quantified by the 2-ΔΔCq method relative to GAPDH
(22). Primers (provided by
Beijing Tsingke Biotech Co., Ltd.): SH3GLB1, F 5′-GTG TGA GCG GAG
AGG CG-3′, R 5′-TCT TCT GTG AAC TGC ACG GC-3′; GAPDH, F: 5′-GAC ATG
CCG CCT GGA GAA AC-3′; R: 5′-AGC CCA GGA TGC CCT TTA GT-3′.
Western blot analysis
Lung samples and bone marrow-derived macrophages
(BMDMs) were homogenized in RIPA buffer. Proteins were separated by
SDS-PAGE, transferred onto PVDF membranes, blocked with 5% skimmed
milk for 1 h and probed with primary antibodies overnight at 4°C.
Primary antibodies: GAPDH (1:1,000; cat. no. ab8245; Abcam), HSP90
(1:1,000; cat. no. 13171-1-AP, Proteintech Group, Inc.), SH3GLB1
(1:1,000; cat. no. sc-374146, Santa Cruz Biotechnology, Inc.), LC3
(1:1,000; cat. no. 12741T; CST Biological Reagents Co., Ltd.), P62
(1:1,000; cat. no. ab109012, Abcam), Rab7 (1:1,000; cat. no.
A12308, ABclonal) and Parkin (1:1,000; cat. no. ab77924, Abcam).
After washing with 1X TBST (containing 0.1% Tween-20), membranes
were incubated with HRP-conjugated secondary antibodies (1:5,000,
cat. no. 7074S/7076S; CST Biological Reagents Co., Ltd.) and bands
were revealed using ECL-HRP substrate (cat. no. WBAVDCH01;
MilliporeSigma) and quantified in Image Lab (Bio-Rad Laboratories,
Inc.).
Isolation, culture and BMDM
treatments
Primary mouse BMDMs were isolated from 6to
11-week-old mice sacrificed under approved protocols (23). Bones (femur/tibia) were dissected
aseptically, cleaned of soft tissue and epiphyses removed. Marrow
cavities were rinsed with ice-cold DMEM (cat. no. 11995; Gibco;
Thermo Fisher Scientific, Inc.) using a 1 ml syringe. The flushed
cells were pelleted (300 × g, 5 min, room temperature), subjected
to red-cell lysis and filtered through a 40 µm nylon mesh
(Falcon; Corning Life Sciences) to obtain a single-cell suspension.
To drive differentiation, these bone-marrow progenitors were
maintained for 7 days at 37°C in complete DMEM (10% FBS, 1% P/S)
supplemented with 20 ng/ml mouse M-CSF (cat. no. 315-02-500;
PeproTech, Inc.). Differentiation was confirmed by flow-cytometric
detection of F4/80. For experiments, macrophages were exposed to
200 ng/ml LPS (cat. no. S7850; Selleck Chemicals) dissolved in 20%
artificial seawater for 24 h.
Bronchoalveolar lavage fluid (BALF)
collection
After sedating the mice, the airways were washed by
inserting a 14-G catheter into the trachea and gently instilling
and withdrawing 1 ml of PBS three times; the resulting BALF was
harvested for subsequent analyses (24).
ELISA assay
Post-ALI, carotid blood was withdrawn and
centrifuged (1,000 rpm; 10 min; 4°C) to obtain serum that was kept
at -80°C. In parallel, BALF was harvested. Cytokine concentrations
(TNF-α, IL-6, IL-1β; cat nos. RK04875, RK00008 and RK05253;
ABclonal Biotech Co., Ltd.) in both fluids were quantified with
commercial ELISA kits following the supplier's protocol.
Seahorse analysis
Mitochondrial oxygen consumption rate (OCR) was
monitored on an XF24 Extracellular Flux Analyzer (Agilent Seahorse;
Agilent Technologies, Inc.) (25). BMDMs were plated at
1.6×105 cells per XF24 well, transfected with siRNA for
48 h and treated as indicated. OCR was then recorded using the Mito
Stress Test with the following injector concentrations: oligomycin
0.6 µM, FCCP 0.75 µM, antimycin A 2 µM and
rotenone 1 µM. Basal and maximal respiration were extracted
with Seahorse Wave desktop software Wave Desktop v2.6.3.5 (Agilent
Seahorse; Agilent Technologies, Inc.); ATP-linked respiration and
spare respiratory capacity were calculated according to the kit's
algorithm.
Mitochondrial ROS assay
Mitochondrial ROS were visualized with MitoSOX Red
(cat. no. M36008; Invitrogen; Thermo Fisher Scientific, Inc.) per
the supplier's instructions. Fluorescence images were captured on
an FV3000 confocal microscope (Olympus Corporation) and quantified
by ImageJ v1.54p (National Institutes of Health) (26) In brief, the fluorescence of
exposed cells/fluorescence of control cells represented ROS
production and the average fluorescence intensity was analyzed by
ImageJ v1.54p (National Institutes of Health).
Mitochondrial membrane potential (Δψm)
measurements
Δψm was evaluated with the JC-1 assay (Beyotime
Biotechnology) (27). After 20
min of incubation at 37°C, healthy mitochondria (JC-1 aggregates)
emitted red light (570 nm), whereas depolarized mitochondria (JC-1
monomers) emitted green light (535 nm). Red/green fluorescence
ratios were acquired on an FV3000 confocal microscope (Olympus
Corporation) and used as a proxy for Δψm.
Coimmunoprecipitation (Co-IP)
BMDMs were rinsed three times with ice-cold PBS and
solubilized in IP buffer (25 mM Tris-HCl pH 7.4, 150 mM NaCl, 1%
NP-40, 1 mM EDTA, 5% glycerol; cat. no. 87787; Thermo Fisher
Scientific, Inc.) plus protease/phosphatase inhibitors (cat. no.
5872; CST Biological Reagents Co., Ltd.). Lysates were rotated
overnight at 4°C with 1 µg antibody, then with Protein A/G
magnetic beads for 4 h. After three washes with IP buffer,
bead-bound proteins were eluted, electrophoresed and transferred to
PVDF for blotting with anti-SH3GLB1 and anti-Rab7. A total of 40
µl of original lysate were retained as input/loading
control.
LC-MS/MS analysis
LC-MS/MS was contracted to Shanghai Baipu
Biotechnology Co., Ltd. Gel slices were reduced, alkylated,
acetone-precipitated and digested overnight with trypsin (Promega
Corporation; 1:50 w/w) at 37°C. Peptides were recovered by
centrifugation (16,000 × g; 15 min; 20°C), desalted on C18
StageTips and loaded onto a trap column in 0.1% FA. Separation used
a 300 nl min-¹ home-packed RP column coupled to a
Q-Exactive HF-X (Thermo Fisher Scientific, Inc.). A Top-20 DDA
method (350-1 800 m/z; 60,000 at 200 m/z for MS, 15,000 at 200 m/z
for HCD-MS/MS) generated raw files searched against the UniProt
mouse database with MaxQuant 1.6.1.0. Data are available via
ProteomeXchange with identifier PXD073650 (https://www.ebi.ac.uk/pride/archive/projects/PXD073650).
Cell transfection and adenoviral
infection
BMDMs were transfected with 20 nM siRNA (sense
5′-GCA CAG UGU UAC CAG UAU A, antisense 5′-UAU ACU GGU AAC ACU GUG
C) using Lipofectamine® RNAiMAX (cat. no. 13778075;
Invitrogen; Thermo Fisher Scientific, Inc.) for 6 h, then
maintained in fresh complete medium for 48 h at 37°C. For
overexpression, cells were exposed to adenovirus (MOI 50) for 6 h,
washed and cultured another 48 h at 37°C; efficiency was verified
by western blotting.
Immunofluorescence staining
To label mitochondria or lysosomes, live BMDMs
infected with SH3GLB1-expressing adenoviral vectors were stained
with the MitoTracker Red CMXRos probe (150 nM; cat. no. M7512;
Thermo Fisher Scientific, Inc.) or LysoTracker Red DND99 probe (100
nM; cat. no. T39855; TargetMol Chemicals Inc.) at 37°C for 20 min.
The images were acquired with a confocal microscope (FV3000;
Olympus Corporation).
Mitophagic flux analysis
A mitochondrion-targeting Keima (mKeima) adenovirus
(Shanghai GeneChem Co., Ltd.) was used to infect the cells for 6 h.
Their Keima fluorescence density was observed at 48 h after mKeima
infection by a Zeiss LSM780 confocal microscope (Zeiss AG).
Lysosomal mitochondrial degradation was evaluated with mKeima as a
marker for the evaluation of mitophagic flux. The pH sensitivity of
mKeima was used to determine if the mitochondria were in acidic
(561 nm, red) or neutral compartments (excitation 488 nm, green).
Pairs of images were collected in sequence at 488 nm (green) and
561 nm (red) wavelengths for ratiometric analysis of mKeima
fluorescence (28). The
fluorescence density of mKeima was calculated as the ratio of
561/488 nm fluorescence integrated densities in each x630 field by
ImageJ v1.54p (National Institutes of Health).
Pull-down assays
Recombinant GST-tagged proteins were incubated with
glutathione sepharose beads for 4 h at 4°C. After pre-binding, the
beads were incubated overnight at 4°C with purified interacting
proteins in binding buffer C (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM
EDTA, 6 mM sodium deoxycholate, 1% NP-40, 1 mM PMSF, plus protease
inhibitors). Beads were washed extensively with the same buffer and
bound proteins were eluted in 2X SDS sample buffer for immunoblot
analysis.
Statistical analysis
Continuous variables are presented as the means ±
Standard deviations. The normality of the data distribution was
examined with a Shapiro-Wilk normality test. Two-tailed Student's
t-tests and Mann-Whitney U tests were performed to see if there
were any significant differences between the two groups. A one-way
ANOVA was used to compare more than two variables with one
independent variable, followed by a Tukey multi-comparison test.
One-way ANOVA was used to evaluate the difference between time and
concentration, followed by Dunnett's multi-comparison test. Data
were analyzed using the GraphPad Prism Software 8.3.0 (Dotmatics)
and SPSS Statistics, v25 (IBM Corp.). P<0.05 was considered to
indicate a statistically significant difference.
Results
The regulation of macrophages in
drowning-ALI is similar to that in LPS-ALI
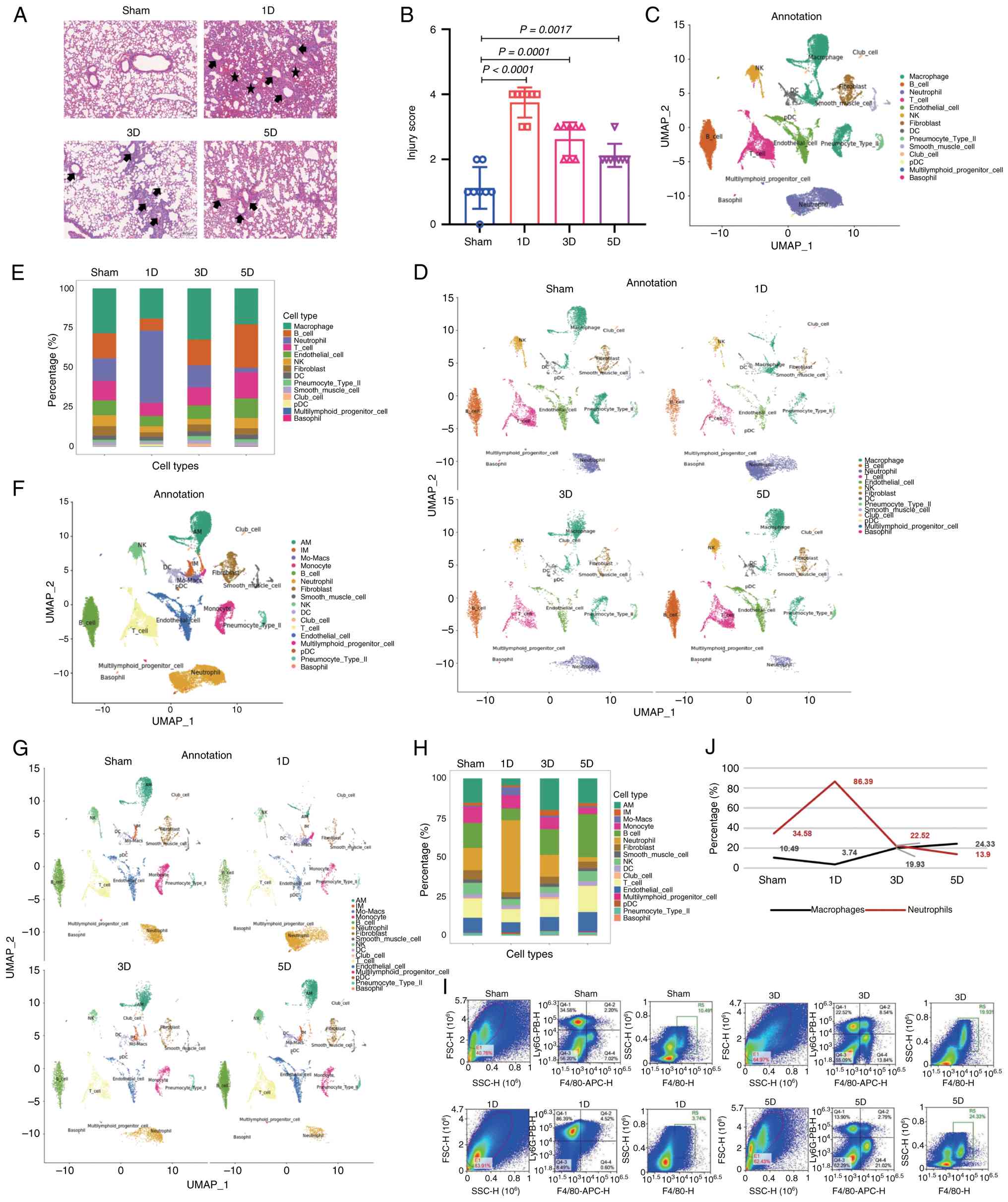
The mice were treated with sea water, followed by
hematoxylin and eosin staining. Compared with that in the normal
group (sham), on the first day, lung inflammation was markedly
aggravated, characterized by inflammation in the bronchi and
alveoli, inflammatory cell infiltration, thickening of the alveolar
septa and pulmonary oedema. From Day 1-3, the degree of lung
inflammation gradually decreased, with inflammation limited to the
peribronchial area. By Day 5, the inflammatory infiltration had
recovered (inflammation moved into the repair phase) (Fig. 1A). Moreover, scoring of the lung
injury revealed that the inflammation score was the highest on the
first day (Fig. 1B).
To dissect the contribution of immune cells to
drowning-ALI progression, we performed single-cell profiling. Lungs
were collected from sham mice and mice after sea water
administration for 1, 3 and 5 days. Then, the lung cells were
normalized and clustered and the clusters were annotated on the
basis of their specific gene expression markers: macrophages
(CD68+C5ar1+), dendritic cells
(CD11c+MHC-II+Dpp4+), T and
natural killer T (NKT) cells (CD3e+), B cells
(CD19+CD79a/b+), neutrophils
(CXCR2+C5ar1+) and NK cells
(NKg7+NCR1+Gzma+; Fig. 1C). It was found that on the first
day of seawater treatment, the macrophages in the lung tissue were
depleted and the proportion of neutrophils increased. On the third
day, the number of neutrophils began to decrease and the proportion
of macrophages increased. On the fifth day, the neutrophils were
depleted and the proportion of macrophages increased (Fig. 1D and E). Although macrophages are
implicated in the inflammatory process of ALI, little is known
about their role in drowning-ALI. To determine the regulation of
macrophages in drowning-ALI, the macrophages were classified and it
was found that, on the first day of seawater treatment, the
alveolar macrophages were depleted. On the third day, both alveolar
macrophages and mononuclear macrophages began to increase in
number. On the fifth day, the number of alveolar macrophages
increased (Fig. 1F-H). Alveolar
macrophages mostly originate from mononuclear macrophages in ALI.
Thus, single-cell sequencing trajectory analysis was performed and
it was found that after administration to sea water, alveolar
macrophages were derived mainly from mononuclear macrophages, which
was consistent with the source of alveolar macrophages in ALI
(Fig. 1I). Thus, to verify the
pattern of macrophages in seawater-induced acute lung injury, flow
cytometry analysis of F4/80 (marked macrophages) and Ly6G (marked
neutrophils) was conducted. The results revealed that on Day 1
after seawater treatment, the macrophages were depleted, whereas
the number of neutrophils sharply increased. On Days 3 and 5, the
number of macrophages increased, whereas the number of neutrophils
decreased. Furthermore, flow cytometry antibodies against CD11c
were used to identify alveolar macrophages and antibodies against
CD11b were used to identify monocyte-derived macrophages.
CD11c+ cells were markedly reduced on Day 1 after
seawater treatment and gradually increased on Days 3 and 5. This
finding indicates that on Day 1 after seawater treatment, the
alveolar macrophages were depleted but increased on Days 3 and 5
and mostly differentiated from monocyte-derived macrophages
(Fig. 1J), which was consistent
with the single-cell sequencing data. These data indicate that
immune cells, particularly macrophages, in drowning-ALI were
similar to those involved in LPS-induced ALI.
The mitochondrial protein SH3GLB1 is
highly expressed in alveolar macrophages in drowning-ALI
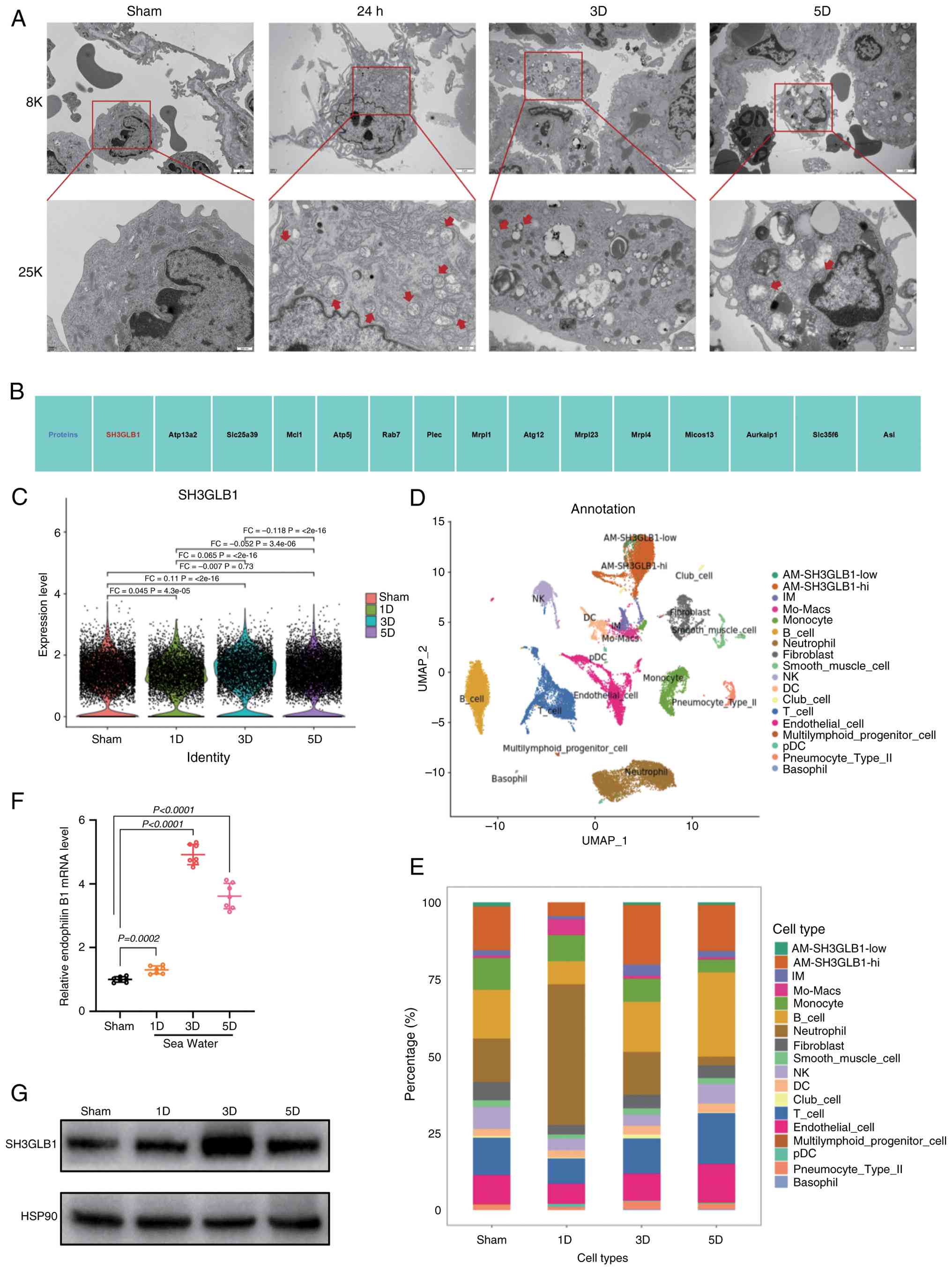
Mitochondria play essential roles in macrophage
inflammation during LPS-ALI. Transmission electron microscopy
revealed that sea water induced mitochondrial morphological
abnormalities, as indicated by greater swelling, a decreased number
of mitochondrial cristae and a reduced area of cristae (Fig. 2A). Next, single-cell sequencing
data was analyzed and the top 15 mitochondrial proteins identified,
among which SH3GLB1 had the highest score (Fig. 2B). Furthermore, it was found that
SH3GLB1 was highly expressed in alveolar macrophages and that its
expression was highest on the third day of seawater treatment
(Fig. 2C). On the basis of the
expression of SH3GLB1, the present study subsequently distinguished
between alveolar macrophages with high and low expression of
SH3GLB1 (Fig. 2D and E). It was
found that the alveolar macrophages with high SH3GLB1 expression
were depleted on the first day of seawater treatment, increased on
the third day and increased on the fifth day. This finding was
consistent with the changes in macrophages. To determine the effect
of sea water on SH3GLB1, western blotting and qPCR was used to
assess its expression. SH3GLB1 expression was elevated in alveolar
macrophages treated with seawater (Fig. 2F and G). These results indicated
a strong association between increased SH3GLB1 expression and sea
water-induced ALI.
SH3GLB1 deficiency protected against
inflammation in ALI
The present study subsequently performed Gene
Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG)
pathway enrichment analyses based on SH3GLB1 expression. The
results indicated that SH3GLB1high AMs showed an
enrichment in regulation of the inflammatory response and the
oxidative stress response (Fig.
3A-C). To assess the effects of SH3GLB1 on ALI, SH3GLB1 KO mice
were generated. To gain insight into the function of increased
SH3GLB1 in ALI, SH3GLB1 KO and control WT mice were subjected to
sea water treatment or a sham operation at 8 weeks of age. Notably,
SH3GLB1 deficiency markedly improved sea water-induced ALI and
LPS-ALI (Fig. 3D and E). The
present study next measured the concentration of inflammatory
factors in the BALF via ELISA and found that SH3GLB1 deficiency
markedly decreased the sea wateror LPS-induced expression of
inflammatory factors, including IL-1β, TNFα and IL-6 (Fig. 3F-H). In agreement with these
results, SH3GLB1 deficiency reduced the sea wateror LPS-induced
elevations in the serum concentrations of these inflammatory
factors (Fig. 3I-K). To
determine whether SH3GLB1 defects directly influence macrophage
inflammation, we isolated BMDMs and then infected them with
adenoviruses carrying siRNA targeting SH3GLB1 or a control
scrambled siRNA. Western blot and qPCR analyses revealed that the
siRNA effectively knocked down SH3GLB1 expression (Fig. 3L-M). As expected, ELISA analysis
demonstrated that SH3GLB1 knockdown in BMDMs markedly reduced the
secretion of inflammatory factors induced by LPS (Fig. 3N) and sea water (Fig. 3O). Taken together, the data
indicated that SH3GLB1 defects were critical for protection against
ALI.
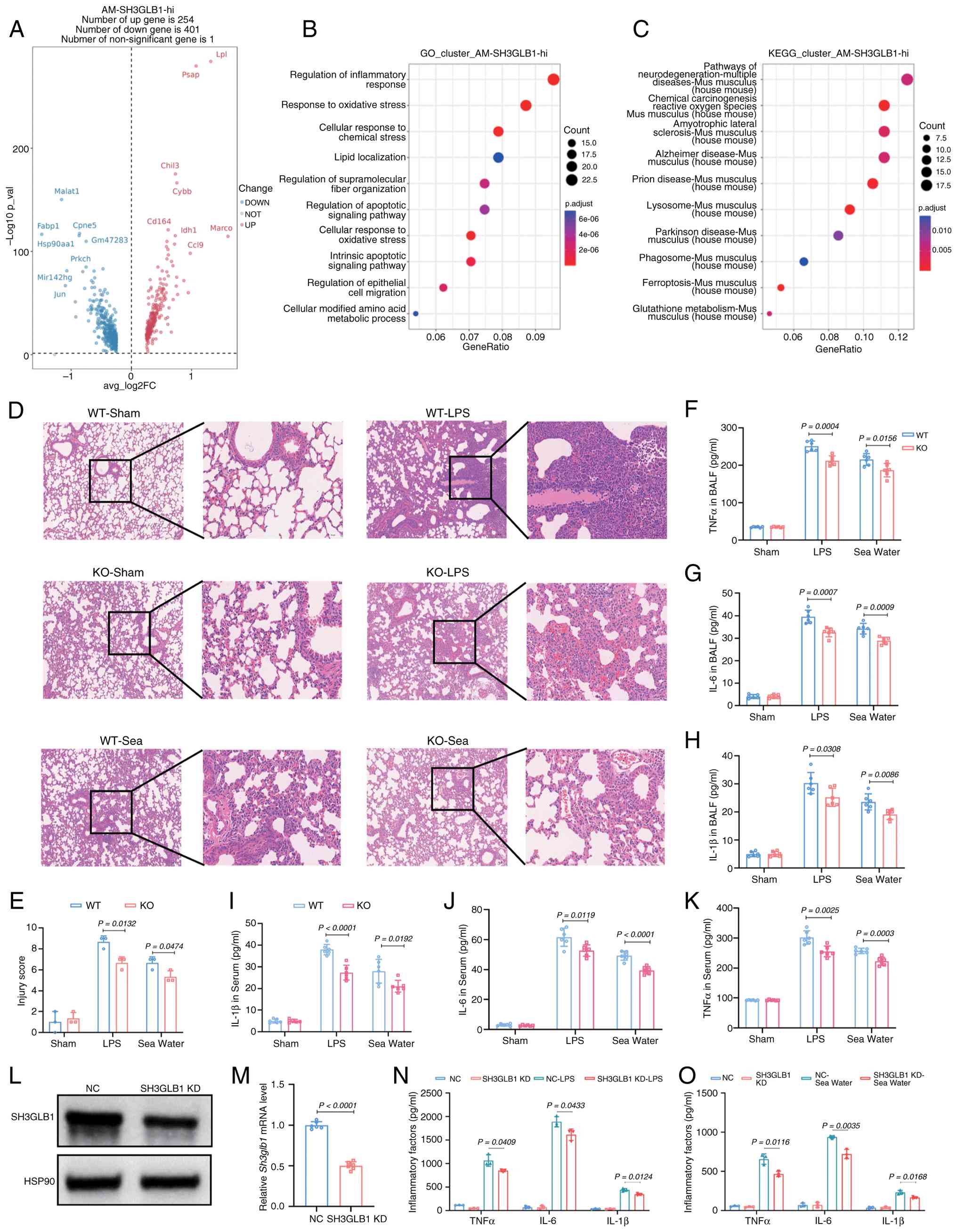 | Figure 3SH3GLB1 deficiency protects against
inflammation in ALI. (A) Gene expression of SH3GLB1high
AMs in cohort 1 (n=3) represented in a volcano plot, with the fold
change (log2) expressed as the level in sham versus sea water-ALI
subjects on the x-axis and the P-value on the y-axis (log10).
Significantly (P<0.05) regulated genes are marked in colors: red
indicates upregulated genes and blue indicates downregulated genes
in ALI. (B) Representative GO analysis of SH3GLB1high
AMs. (C) Representative KEGG analysis of SH3GLB1high
AMs. (D) Representative hematoxylin and eosin staining of lung
tissues (scale bars, 200 µm). (E) Representative severity
scores of lung injury (n=3). Representative ELISA results showing
the levels of inflammatory factors, including (F) TNFα, (G) IL-6
and (H) IL-1β, in the BALF (n=6). (I-K) Representative ELISA
analysis of the levels of inflammatory factors, including (I)
IL-1β, (J) IL-6 and (K) TNFα, in the serum (n=6). (L)
Representative western blots of SH3GLB1 protein levels in NC or
SH3GLB1-KD BMDMs. (M) Relative SH3GLB1 mRNA levels in (NC or
SH3GLB1-KD BMDMs. n=6/group. Representative ELISA analysis of the
levels of inflammatory factors, including IL-1β, IL-6 and TNFα, in
the supernatants of cells treated with (N) LPS or (O) seawater,
(n=3).The data are presented as the means ± SEMs; P<0.05 was
considered to indicate statistical significance. SH3GLB1, SH3
domain-containing GRB2-like protein B1; AMs, alveolar macrophages,
ALI, acute lung injury; GO, Gene Ontology; KEGG, Kyoto Encyclopedia
of Genes and Genomes; BALF, bronchoalveolar lavage fluid; BMDMs,
bone marrow-derived macrophages; KD, knockdown; WT, wild-type; KO,
knockout; NC, negative control; LPS, lipopolysaccharide. |
Reduced SH3GLB1 ameliorates mitochondrial
dysfunction in macrophages from ALI model mice
There is a close relationship between ALI and
mitochondrial homeostasis. Next, the present study examined the
mitochondrial morphology of SH3GLB1 KO and WT mice at 8 weeks old
by means of TEM. At 8 weeks old, no significant changes in
mitochondrial morphology were seen in the SH3GLB1 KO mice compared
with the control WT. However, it was found that SH3GLB1 KO
alleviated mitochondrial morphological abnormalities, as indicated
by greater swelling, a decreased number of mitochondrial cristae
and a reduced area of cristae induced by sea water (Fig. 4A).
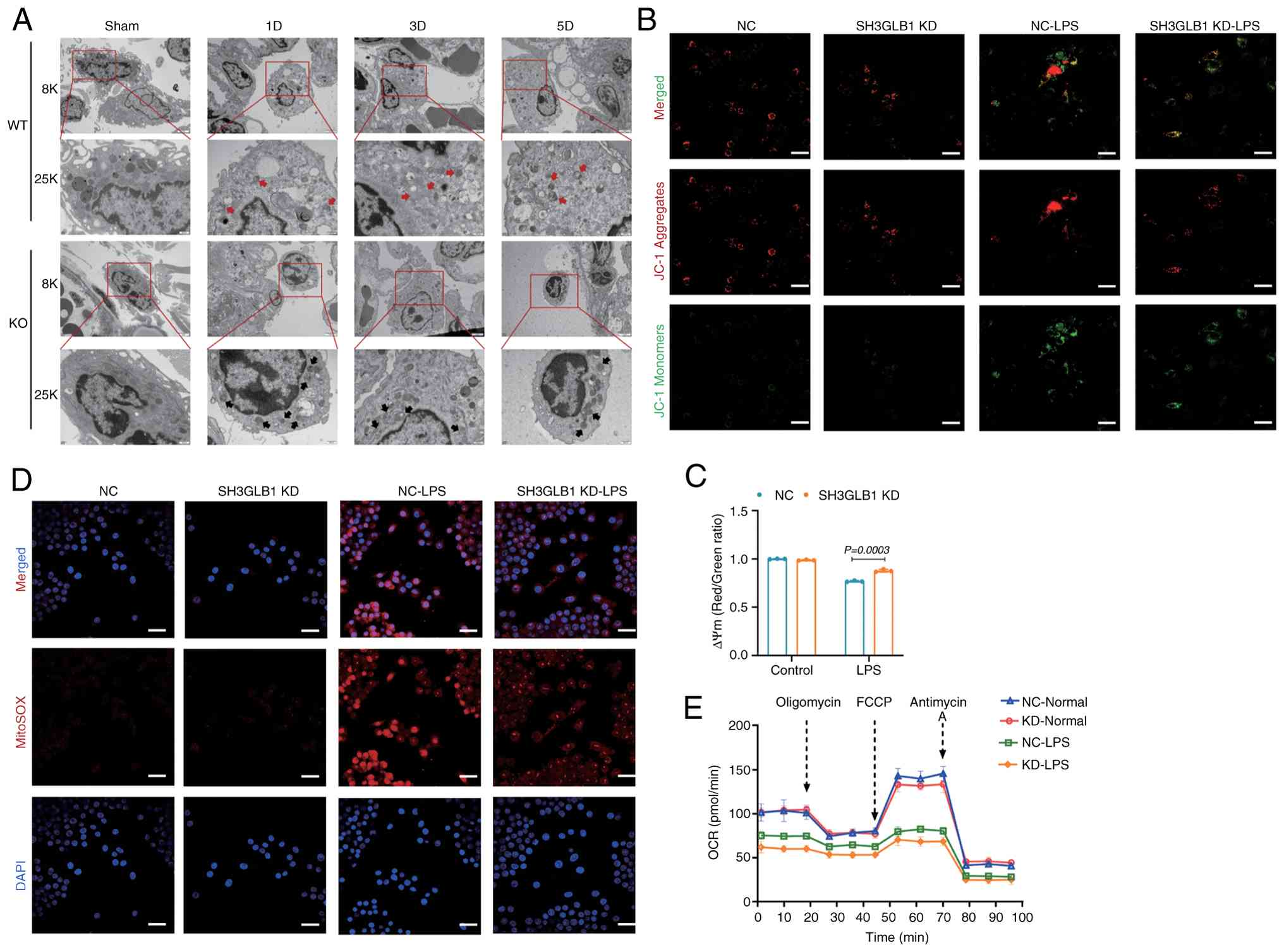 | Figure 4Reduced SH3GLB1 ameliorates
LPS-induced mitochondrial dysfunction in macrophages. (A)
Representative TEM images of macrophage mitochondria in the lung
(scale bars, 2 µm/500 nm). Representative (B) confocal
microscopy and (C) flow cytometry analysis of the mitochondrial
membrane potential by JC-1 staining in BMDMs. A high level of green
fluorescence (x-axis) represents a reduced mitochondrial membrane
potential (ΔΨm) and a high level of red fluorescence (y-axis)
represents a normal ΔΨm. The red fluorescence rate was analyzed
(n=3). (D) Representative confocal microscopy images of
mitochondrial ROS production (MitoSOX fluorescence; red) in NC or
SH3GLB1-KD BMDMs under normal or LPS conditions (scale bar, 50
µm). (E) Analysis of the OCR of mitochondrial respiratory
capacity, including basal respiration, ATP production, maximal
respiration and spare respiration, in NC or SH3GLB1-KD BMDMs under
normal or LPS conditions. The data are presented as the means ±
SEMs; P<0.05 was considered to indicate statistical
significance. SH3GLB1, SH3 domain-containing GRB2-like protein B1;
LPS, lipopolysaccharide; TEM, transmission electron microscopy;
BMDMs, bone marrow-derived macrophages; KD, knockdown; OCR, oxygen
consumption rate; NC, negative control. |
Given that SH3GLB1 deficiency improved BMDM
inflammation, it was next determined whether SH3GLB1 can directly
affect mitochondrial function in BMDMs. Therefore, the
mitochondrial membrane potential was tested using JC-1 staining and
it was found that SH3GLB1 knockdown markedly increased the
mitochondrial membrane potential in BMDMs under LPS conditions
(Fig. 4B and C). Additionally,
confocal microscopy images to were used assess mitochondrial ROS
levels and it was found that SH3GLB1 knockdown decreased
mitochondrial ROS production in BMDMs under LPS conditions
(Fig. 4D). Due to these profound
alterations in the structure of mitochondria and mitochondrial
oxidation, OCR was analyzed to assess mitochondrial respiration. As
expected, SH3GLB1 knockdown markedly enhanced mitochondrial
respiration in BMDMs under LPS, as shown by basal respiration, ATP
production, maximum respiration and reserve respiration (Fig. 4E). Taken together, these data
showed that the inactivation of SH3GLB1 increases the potential
loss of mitochondrial membrane potential, reduces the generation of
mitochondrial ROS and induced by LPS in mitochondrial respiratory
failure.
Restoration of SH3GLB1 expression
provokes ALI and mitochondrial dysfunction
To determine whether SH3GLB1 overexpression can
aggravate sea water-induced ALI or LPS-ALI, SH3GLB1 expression was
restored by inhalation of adenoviruses expressing the SH3GLB1-GFP
fusion protein into the mouse lung. Notably, the hematoxylin and
eosin results indicated that SH3GLB1 overexpression markedly
aggravated sea water-induced ALI or LPS-ALI (Fig. 5A and B). Furthermore, the present
study measured the concentrations of inflammatory factors in the
BALF and found that SH3GLB1 overexpression markedly increased the
sea wateror LPS-induced expression of inflammatory factors
(Fig. 5C and E). Similarly,
SH3GLB1 overexpression increased sea water or LPS-induced increases
in the serum concentrations of these inflammatory factors (Fig. 5F-H). Next, SH3GLB1 was
overexpressed in BMDMs via adenovirus infection. Western blot and
qPCR analyses revealed markedly increased expression of SH3GLB1 in
BMDMs (Fig. 5I and J). ELISAs
revealed that SH3GLB1 overexpression in BMDMs markedly promoted the
secretion of inflammatory factors induced by LPS (Fig. 5K) and sea water (Fig. 5L). Overall, the data revealed
that restoring SH3GLB1 expression in the lung exacerbated ALI.
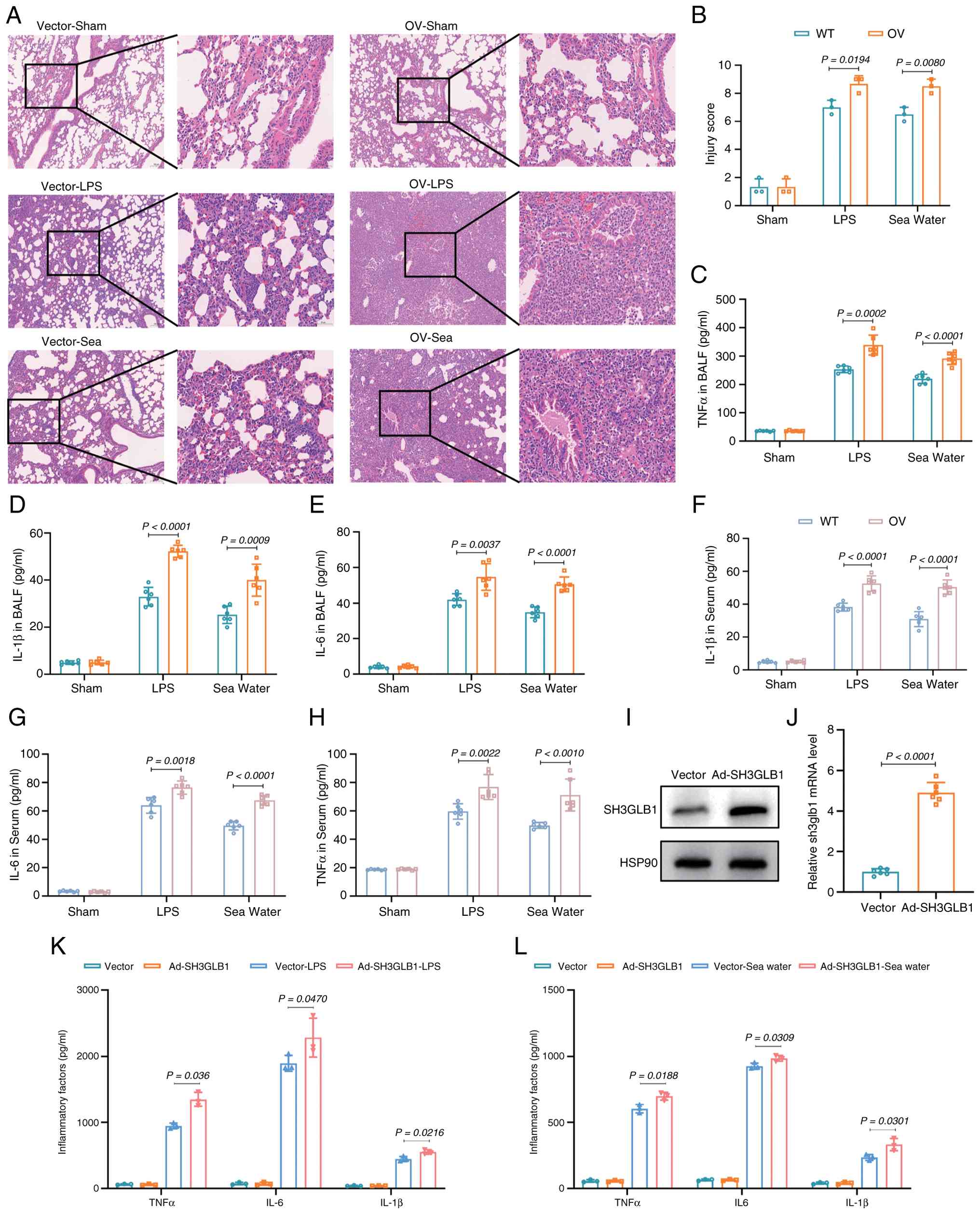 | Figure 5Restoration of SH3GLB1 expression
provokes ALI. (A) Representative hematoxylin and eosin staining of
lung tissues (scale bars, 100 µm). (B) Representative
severity scores of lung injury (n=3). Representative ELISA results
showing the levels of inflammatory factors, including (C) TNFα, (D)
IL-1β and (E) IL-6, in the BALF (n=6). Representative ELISA
analysis of the levels of inflammatory factors, including (F)
IL-1β, (G) IL-6 and (H) TNFα, in the serum (n=6). (I) Western blots
of SH3GLB1 expression. (J) Relative SH3GLB1 mRNA levels in vectoror
SH3GLB1-overexpressing BMDMs. n=6/group. Representative ELISA
analysis of the levels of inflammatory factors, including IL-1β,
IL-6 and TNFα, in the supernatants of cells treated with (K) LPS or
(L) sea water (n=3).The data are presented as the means ± SEMs;
P<0.05 was considered to indicate statistical significance.
SH3GLB1, SH3 domain-containing GRB2-like protein B1; ALI, acute
lung injury; BMDMs, bone marrow-derived macrophages; WT, wild-type;
OV; overexpression. |
Next, alveolar macrophages (AMs) mitochondrial
morphology was examined using TEM in SH3GLB1overexpressing and
control mice at 8 weeks of age. No significant alterations in
mitochondrial morphology were observed in SH3GLB1-overexpressing
mice compared with control mice. However, SH3GLB1 overexpression
provoked mitochondrial morphological abnormalities, as indicated by
increased swelling, an increased number of mitochondrial cristae
and an increased area of cristae induced by sea water or LPS
(Fig. 6A). The present study
also found that SH3GLB1 expression markedly decreased the
mitochondrial membrane potential in BMDMs under LPS conditions
(Fig. 6B and C). Additionally,
confocal microscopy images indicated that SH3GLB1 overexpression
increased mitochondrial ROS production in BMDMs under LPS
conditions (Fig. 6D). The
present study analyzed the mitochondrial OCR to assess the
mitochondrial respiratory capacity. SH3GLB1 overexpression markedly
reduced mitochondrial respiratory capacity, as indicated by basal
respiration, ATP production, maximal respiration and spare
respiration, in BMDMs under LPS conditions (Fig. 6E). Taken together, these data
demonstrated that SH3GLB1 overexpression facilitated mitochondrial
membrane potential loss and increased mitochondrial ROS production
and mitochondrial respiratory dysfunction induced by LPS.
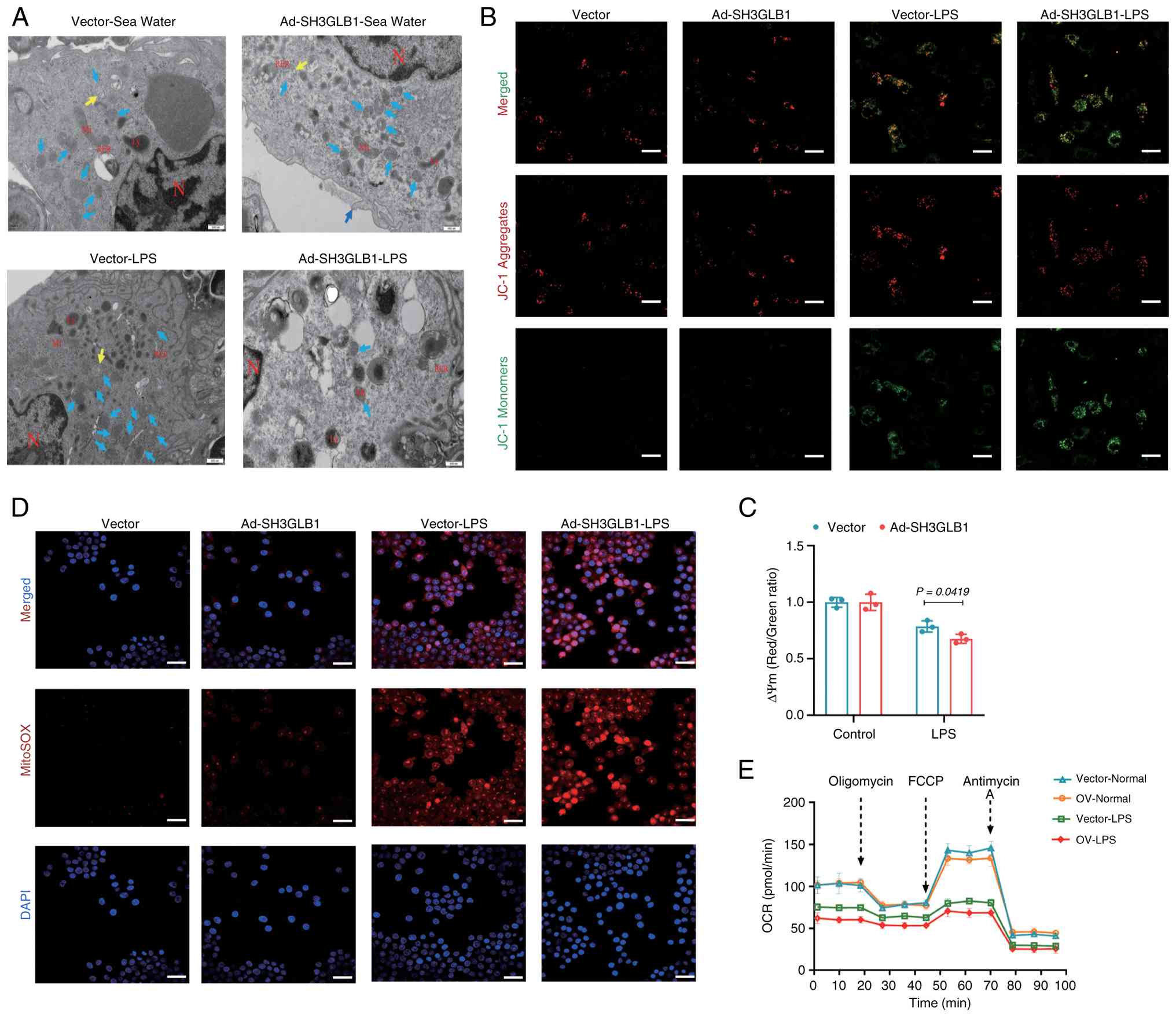 | Figure 6SH3GLB1 overexpression provoked
mitochondrial dysfunction in macrophages. (A) Representative TEM
images of macrophage mitochondria in the lung (scale bars, 2
µm/500 nm). Representative (B) confocal microscopy and (C)
flow cytometry analysis of the mitochondrial membrane potential by
JC-1 staining in BMDMs (n=3). (D) Representative confocal
microscopy images of mitochondrial ROS production in vectoror
SH3GLB1-OV BMDMs under normal or LPS conditions (scale bar, 50
µm). (E) Analysis of the OCR of mitochondrial respiratory
capacity, including basal respiration, ATP production, maximal
respiration and spare respiration, in vectoror
SH3GLB1-overexpressing BMDMs under normal or LPS conditions. The
data are presented as the means ± SEMs; P<0.05 was considered to
indicate statistical significance. SH3GLB1, SH3 domain-containing
GRB2-like protein B1; TEM, transmission electron microscopy; OCR,
oxygen consumption rate; BMDMs, bone marrow-derived
macrophages. |
SH3GLB1 interacts with the lysosomal
protein Rab7 to contribute to macrophage inflammation
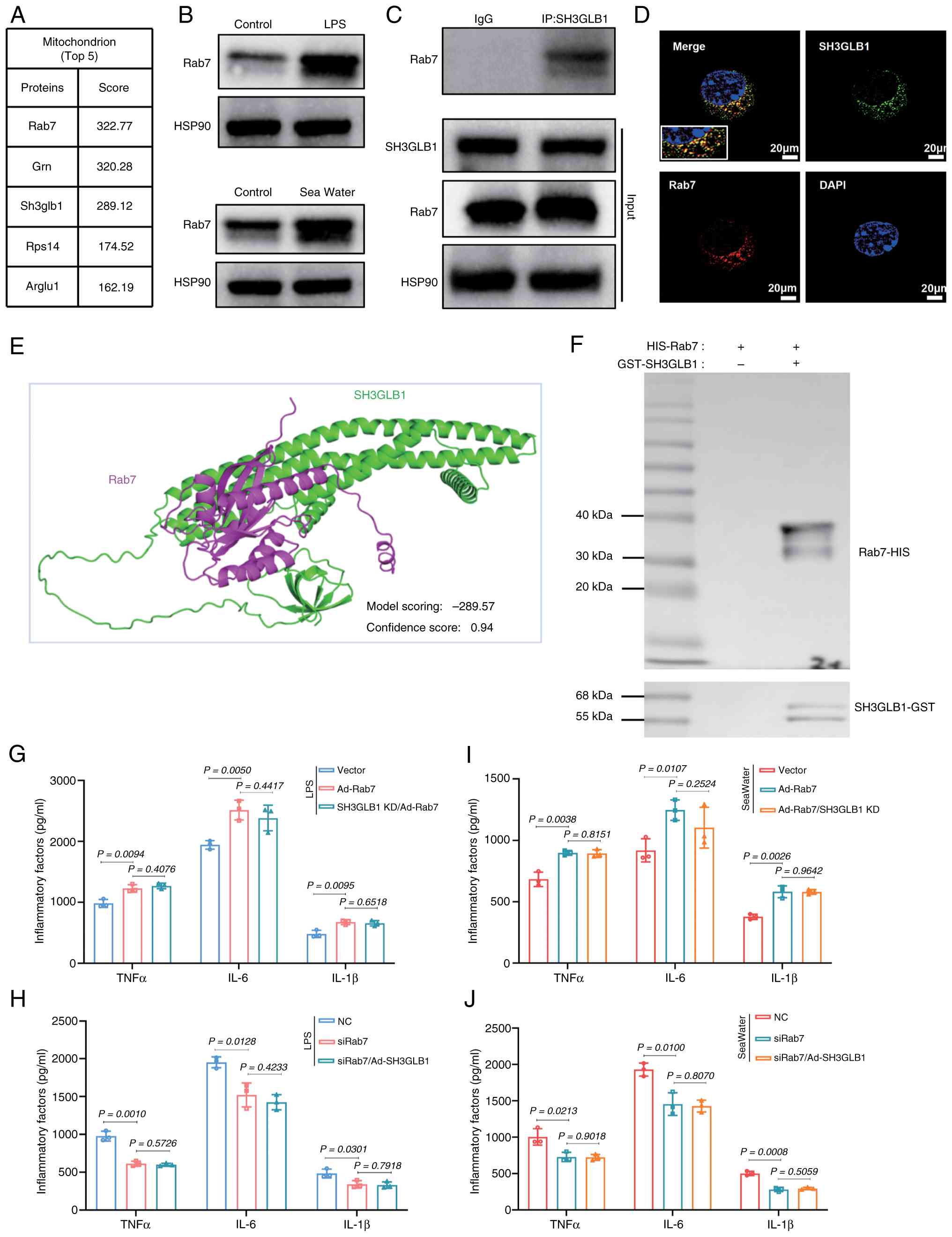
To understand the molecular mechanisms of SH3GLB1 in
macrophage function, IP-LC-MS/MS was employed to identify potential
endogenous SH3GLB1 binding partners with a specific antibody
against SH3GLB1 in BMDMs. The top 5 mitochondrial proteins were
identified, among which Rab7 had the highest score (Fig. 7A). LPS or sea water increased
Rab7 expression in BMDMs (Fig.
7B). Co-IP assays revealed that SH3GLB1 interacted with the
lysosomal protein Rab7 (Fig.
7C). Moreover, immunofluorescence staining of BMDMs was
performed and the colocalization of SH3GLB1 and Rab7 was observed
(Fig. 7D). The present study
subsequently conducted molecular docking analysis, which revealed a
docking score of -289.57, which is less than -1 and the confidence
score was 0.94 (a score higher than 0.7 indicated that Rab7 was
more likely to bind to SH3GLB1) (Fig. 7E). The results of the pull-down
experiments indicated that the purified Rab7 interacted with
SH3GLB1 in vitro (Fig.
7F). These results suggested the direct binding between SH3GLB1
and Rab7. To determine whether SH3GLB1 promotes inflammation
through Rab7, BMDMs were subjected to Rab7 overexpression or
SH3GLB1 knockdown. The results revealed that the overexpression of
Rab7 followed by the knockdown of SH3GLB1 failed to improve the
release of inflammatory factors in BMDMs induced by LPS or sea
water (Fig. 7G and I).
Similarly, Rab7 knockdown followed by SH3GLB1 overexpression failed
to change the release of inflammatory factors induced by LPS or sea
water (Fig. 7H and J). These
findings indicated that SH3GLB1 was required for Rab7 to contribute
to macrophage inflammation.
SH3GLB1 facilitates Rab7-mediated
mitophagy
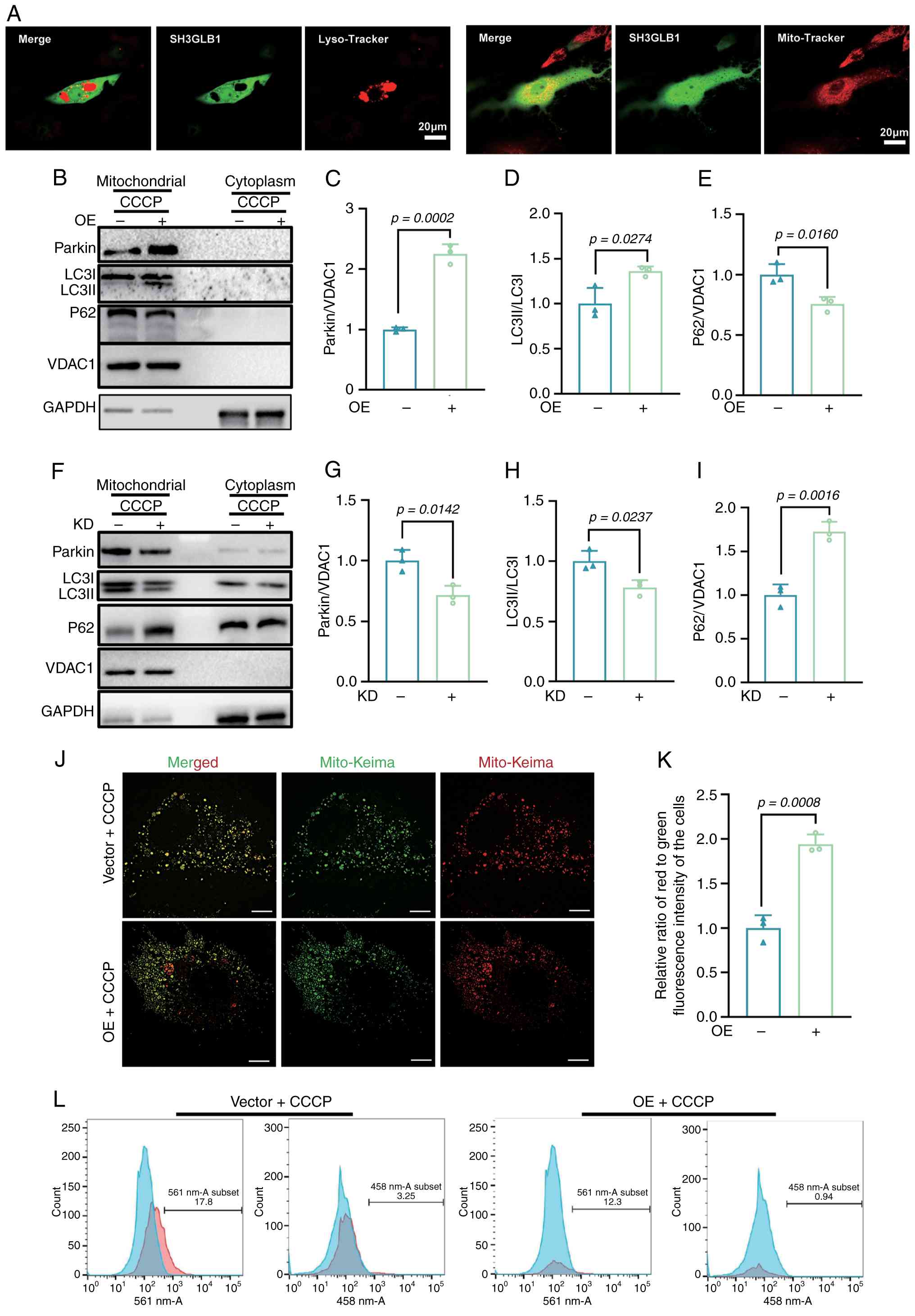
To clarify the localization of SH3GLB1 in BMDMs,
immunofluorescence staining was performed and the colocalization of
SH3GLB1 with lysosomes (labelled with LysoTracker) and mitochondria
(labelled with MitoTracker) was observed (Fig. 8A). Rab7 plays a crucial role in
the interaction between mitochondria and lysosomes, participating
in the formation and fusion of mitochondrial autophagosomes, as
well as in the contact and dissociation processes between
mitochondria and lysosomes. To investigate whether SH3GLB1 is
involved in Rab7-mediated mitophagy, cells overexpressing SH3GLB1
were treated with the mitochondrial autophagy inducer carbonyl
cyanide-chlorophenylhydrazone and the overexpression of SH3GLB1
markedly increased the expression of the mitophagy-related proteins
Parkin and LC3 while decreasing the expression of P62 (Fig. 8B-E). Additionally, mKeima
expressed in BMDMs was imaged by confocal microscopy and assayed by
flow cytometry. As shown in Fig. 8J
and L, the overexpression of SH3GLB1 markedly facilitated
mitophagy. However, SH3GLB1 knockdown decreased the expression of
the mitophagy-related proteins Parkin and LC3 while decreasing the
expression of P62 (Fig. 8F-I).
Taken together, these findings indicated that SH3GLB1 facilitated
adverse Rab7-mediated mitophagy.
Discussion
The present study demonstrated that the regulation
of macrophages in drowning-ALI was similar to that in LPS-ALI.
Specifically, SH3GLB1 was highly expressed in macrophages of
drowning-ALI and was related to inflammation. Furthermore, SH3GLB1
deletion ameliorated LPSor drowning-ALI. By contrast, the
restoration of SH3GLB1 expression provoked ALI. Mechanistically,
SH3GLB1 interacted with Rab7 to contribute to adverse mitophagy,
which resulted in mitochondrial dysfunction. These findings
suggested that SH3GLB1 may be a potential target for protection of
the lungs against ALI induced by LPS or drowning stimuli.
ALI is characterized by infiltration of a large
number of inflammatory cells, epithelial injury, pulmonary oedema,
increased microvascular permeability and diffuse alveolar damage
(29). Similarly, the present
study found that drowning-ALI was characterized by leukocyte
accumulation, pulmonary oedema, increased alveolar permeability and
diffuse alveolar damage. In particular, macrophages are implicated
in the inflammatory process in ALI (30). The present study performed
single-cell profiling of lung immune cells isolated from
drowning-ALI lungs. This demonstrated neutrophil infiltration in
the early stage of acute lung injury. As the inflammation
progressed, the neutrophils died and the number of recruited
macrophages increased. Over time, the recruited macrophages became
exhausted and tissue-resident macrophages became involved. This
process is similar to that of LPS-ALI. The results of the present
study indicated that macrophages were involved in the inflammatory
process of drowning-ALI.
Mitochondrial quality control modulates cell fate
and homeostasis and diminished mitochondrial quality control
results in mitochondrial dysfunction, increased ROS production,
reduced ATP production and often the induction of intrinsic
apoptosis (31). A study showed
that aberrant mitochondria contribute to ALI (32). The present study showed that
mitochondrial injury was implicated in drowning-ALI. It focused on
the mitochondrial protein SH3GLB1, a member of the endophilin
protein family (33), which is
located in the outer mitochondrial membrane (34). A previous study reported that
SH3GLB1 plays a key role in regulating mitochondrial injury and
cell death (14). The present
study found that SH3GLB1 was highly expressed in macrophages, which
is consistent with the pattern of macrophages in drowning-ALI.
Notably, macrophages with high SH3GLB1 expression were associated
with inflammation and the response to oxidative stress. The present
study found that SH3GLB1 deletion protected against mitochondrial
injury and oxidative stress induced by drowning or LPS. By
contrast, restoring SH3GLB1 expression in the lungs provoked
drowningor LPS-induced mitochondrial dysfunction. These findings
suggested for the first time that the mitochondrial protein SH3GLB1
participates in ALI.
To explore the mechanisms by which SH3GLB1 affects
mitochondria, the present study found that SH3GLB1 interacted with
Rab7, a small G protein of the Rab family that is located primarily
in late endosomes and lysosomes (35). Rab7 plays a significant role in
various inflammatory responses, particularly in regulating
intracellular transport and lysosomal function (36,37). Its functions in colitis, acute
pancreatitis and immune responses indicate that the regulation of
Rab7 is crucial for maintaining intracellular homeostasis and
alleviating inflammatory responses (38). The present study reported that
SH3GLB1 provoked inflammation by the regulation of Rab7 in ALI. It
has also been reported that Rab7 regulates transport from late
endosomes to lysosomes and is involved in the biogenesis and
degradation processes of lysosomes (39,40). Autophagy is an important form of
lysosomal degradation (41).
Among these processes, mitophagy plays a significant role in
inflammation through lysosomal degradation (42). A previous study showed that
autophagy-related key genes (including SH3GLB1) with diagnostic and
prognostic value in sepsis and discovered associations between key
genes and immune cell signatures (43). Sepsis induced ALI is a leading
cause of poor prognosis in clinical patients (44). The present study indicated that
the overexpression of SH3GLB1 led to the formation of
autophagosomes, which colocalized with mitochondria and lysosomes,
indicating that SH3GLB1 was involved in mitophagy. The results of
the present study revealed that increased SH3GLB1 promoted
mitophagy, whereas reduced SH3GLB1 inhibited mitophagy. This is
similar to a study by Takahashi et al (14). This may provide new directions
for the discovery of promising biomarkers for different factors
induced ALI.
Although the findings of the present study possess
some clinical relevance, there are several limitations. First,
ALI-induced damage, including apoptosis, has not been extensively
explored. Second, while the present study found that SH3GLB1
interacted with Rab7, it did not focus on further mechanisms.
Therefore, these limitations should be considered in future
studies.
In summary, the findings of the present study showed
that the regulation of macrophages in drowning-ALI was similar to
that in LPS-ALI. Specifically, SH3GLB1 was found to be related to
macrophage inflammation and mitochondrial homeostasis.
Mechanistically, SH3GLB1 was required for mitophagy regulated by
Rab7. These findings suggest that SH3GLB1 may be a potential target
for the protection of lungs against ALI induce by LPS or drowning
stimuli.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
HJN, JYD and FGJ conceived and designed the
research; HJN, JYD and YHG, WZ, JC, XG performed the experiments,
analyzed the data, interpreted the results of the experiments and
prepared the figures; HJN, JYD and FGJ drafted the manuscript; all
the authors contributed to editing and revision. FGJ and HJN
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All animal procedures followed the eighth edition
(2011) of the Guide for the Care and Use of Laboratory Animals
(https://www.ncbi.nlm.nih.gov/books/NBK54050/) and were
approved by the Fourth Military Medical University Animal Ethics
Committee (approval no. 20250088).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was financially supported by the Program for
National Science Funds of China (grant no. 82270084).
References
|
1
|
Jin F and Li C: Seawater-drowning-induced
acute lung injury: From molecular mechanisms to potential
treatments. Exp Ther Med. 13:2591–2598. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Qiu YB, Wan BB, Liu G, Wu YX, Chen D, Lu
MD, Chen JL, Yu RQ, Chen DZ and Pang QF: Nrf2 protects against
seawater drowning-induced acute lung injury via inhibiting
ferroptosis. Respir Res. 21:2322020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sun XQ, Wu C, Qiu YB, Wu YX, Chen JL,
Huang JF, Chen D and Pang QF: Heme oxygenase-1 attenuates seawater
drowninginduced acute lung injury through a reduction in
inflammation and oxidative stress. Int Immunopharmacol.
74:1056342019. View Article : Google Scholar
|
|
4
|
Zhang L, Kong D, Huang J, Wang Q and Shao
L: The therapeutic effect and the possible mechanism of
C-phycocyanin in lipopolysaccharide and seawater-induced acute lung
injury. Drug Des Devel Ther. 16:1025–1040. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jiang W, Ma C, Bai J and Du X: Macrophage
SAMSN1 protects against sepsis-induced acute lung injury in mice.
Redox Biol. 56:1024322022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jiang L, Yang D, Zhang Z, Xu L, Jiang Q,
Tong Y and Zheng L: Elucidating the role of Rhodiola rosea L. in
sepsis-induced acute lung injury via network pharmacology: Emphasis
on inflammatory response, oxidative stress, and the PI3K-AKT
pathway. Pharm Biol. 62:272–284. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hsieh PC, Wu YK, Yang MC, Su WL, Kuo CY
and Lan CC: Deciphering the role of damage-associated molecular
patterns and inflammatory responses in acute lung injury. Life Sci.
305:1207822022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jiang J, Huang K, Xu S, Garcia JGN, Wang C
and Cai H: Targeting NOX4 alleviates sepsis-induced acute lung
injury via attenuation of redox-sensitive activation of
CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction. Redox
Biol. 36:1016382020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang M, Yan X, Liu W, Sun R, Xie Y and
Jin F: Endothelial semaphorin 7A promotes seawater
aspiration-induced acute lung injury through plexin C1 and β1
integrin. Mol Med Rep. 16:4215–4221. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li PC, Wang BR, Li CC, Lu X, Qian WS, Li
YJ, Jin FG and Mu DG: Seawater inhalation induces acute lung injury
via ROS generation and the endoplasmic reticulum stress pathway.
Int J Mol Med. 41:2505–2516. 2018.PubMed/NCBI
|
|
11
|
Cao Z, Lis R, Ginsberg M, Chavez D, Shido
K, Rabbany SY, Fong GH, Sakmar TP, Rafii S and Ding BS: Targeting
of the pulmonary capillary vascular niche promotes lung alveolar
repair and ameliorates fibrosis. Nat Med. 22:154–162. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cheng P, Li S and Chen H: Macrophages in
lung injury, repair, and fibrosis. Cells. 10:4362021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cuddeback SM, Yamaguchi H, Komatsu K,
Miyashita T, Yamada M, Wu C, Singh S and Wang HG: Molecular cloning
and characterization of Bif-1. A novel Src homology 3
domain-containing protein that associates with Bax. J Biol Chem.
276:20559–20565. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Takahashi Y, Hori T, Cooper TK, Liao J,
Desai N, Serfass JM, Young MM, Park S, Izu Y and Wang HG: Bif-1
haploinsufficiency promotes chromosomal instability and accelerates
Myc-driven lymphomagenesis via suppression of mitophagy. Blood.
121:1622–1632. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Takahashi Y, Karbowski M, Yamaguchi H,
Kazi A, Wu J, Sebti SM, Youle RJ and Wang HG: Loss of Bif-1
suppresses Bax/Bak conformational change and mitochondrial
apoptosis. Mol Cell Biol. 25:9369–9382. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wong AS, Lee RH, Cheung AY, Yeung PK,
Chung SK, Cheung ZH and Ip NY: Cdk5-mediated phosphorylation of
endophilin B1 is required for induced autophagy in models of
Parkinson's disease. Nat Cell Biol. 13:568–579. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang DB, Kinoshita Y, Kinoshita C, Uo T,
Sopher BL, Cudaback E, Keene CD, Bilousova T, Gylys K, Case A, et
al: Loss of endophilin-B1 exacerbates Alzheimer's disease
pathology. Brain. 138(Pt 7): 2005–2019. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang DB, Uo T, Kinoshita C, Sopher BL, Lee
RJ, Murphy SP, Kinoshita Y, Garden GA, Wang HG and Morrison RS: Bax
interacting factor-1 promotes survival and mitochondrial elongation
in neurons. J Neurosci. 34:2674–2683. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou M, Meng L, He Q, Ren C and Li C:
Valsartan attenuates LPS-induced ALI by modulating NF-κB and MAPK
pathways. Front Pharmacol. 15:13210952024. View Article : Google Scholar
|
|
20
|
Ma L, Chen X, Wang R, Duan H, Wang L,
Liang L, Nan Y, Liu X, Liu A and Jin F:
3,5,4′-Tri-O-acetylresveratrol decreases seawater
inhalation-induced acute lung injury by interfering with the NF-κB
and i-NOS pathways. Int J Mol Med. 37:165–172. 2016. View Article : Google Scholar
|
|
21
|
McGuigan RM, Mullenix P, Norlund LL, Ward
D, Walts M and Azarow K: Acute lung injury using oleic acid in the
laboratory rat: Establishment of a working model and evidence
against free radicals in the acute phase. Curr Surg. 60:412–417.
2003. View Article : Google Scholar
|
|
22
|
Deng J, Chang X, Zhang X, Li C, Guo G,
Song H, Zheng Y, Zhang C, Yang B, Zhang C, et al: Endophilin B1 is
essential for maintaining cardiac function by regulating
mitocytosis. Cell Mol Life Sci. 82:1302025. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yan J, Zhang Y, Yu H, Zong Y, Wang D,
Zheng J, Jin L, Yu X, Liu C, Zhang Y, et al: GPSM1 impairs
metabolic homeostasis by controlling a pro-inflammatory pathway in
macrophages. Nat Commun. 13:72602022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Engels M, Bilgic E, Pinto A, Vasquez E,
Wollschläger L, Steinbrenner H, Kellermann K, Akhyari P,
Lichtenberg A and Boeken U: A cardiopulmonary bypass with deep
hypothermic circulatory arrest rat model for the investigation of
the systemic inflammation response and induced organ damage. J
Inflamm (Lond). 11:262014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang R, Ruan B, Wang R, Zhang X, Xing P,
Li C, Zhang Y, Chang X, Song H, Zhang S, et al: Cardiomyocyte βII
spectrin plays a critical role in maintaining cardiac function by
regulating mitochondrial respiratory function. Cardiovasc Res.
120:1312–1326. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Deng J, Zhang N, Chen F, Yang C, Ning H,
Xiao C, Sun K, Liu Y, Yang M, Hu T, et al: Irisin ameliorates high
glucose-induced cardiomyocytes injury via AMPK/mTOR signal pathway.
Cell Biol Int. 44:2315–2325. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qi B, He L, Zhao Y, Zhang L, He Y, Li J,
Li C, Zhang B, Huang Q, Xing J, et al: Akap1 deficiency exacerbates
diabetic cardiomyopathy in mice by NDUFS1-mediated mitochondrial
dysfunction and apoptosis. Diabetologia. 63:1072–1087. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Deng Z, He M, Hu H, Zhang W, Zhang Y, Ge
Y, Ma T, Wu J, Li L, Sun M, et al: Melatonin attenuates
sepsis-induced acute kidney injury by promoting mitophagy through
SIRT3-mediated TFAM deacetylation. Autophagy. 20:151–165. 2024.
View Article : Google Scholar :
|
|
29
|
Xia L, Zhang C, Lv N, Liang Z, Ma T, Cheng
H, Xia Y and Shi L: AdMSC-derived exosomes alleviate acute lung
injury via transferring mitochondrial component to improve
homeostasis of alveolar macrophages. Theranostics. 12:2928–2947.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fan EKY and Fan J: Regulation of alveolar
macrophage death in acute lung inflammation. Respir Res. 19:502018.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Larson-Casey JL, He C and Carter AB:
Mitochondrial quality control in pulmonary fibrosis. Redox Biol.
33:1014262020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Islam MN, Das SR, Emin MT, Wei M, Sun L,
Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S and
Bhattacharya J: Mitochondrial transfer from bone-marrow-derived
stromal cells to pulmonary alveoli protects against acute lung
injury. Nat Med. 18:759–765. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sparks AB, Hoffman NG, McConnell SJ,
Fowlkes DM and Kay BK: Cloning of ligand targets: Systematic
isolation of SH3 domain-containing proteins. Nat Biotechnol.
14:741–744. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li J, Barylko B, Eichorst JP, Mueller JD,
Albanesi JP and Chen Y: Association of endophilin B1 with
cytoplasmic vesicles. Biophys J. 111:565–576. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rodriguez-Furlan C, Borna R and Betz O:
RAB7 GTPases as coordinators of plant endomembrane traffic. Front
Plant Sci. 14:12409732023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bucci C, Bakke O and Progida C: Rab7b and
receptors trafficking. Commun Integr Biol. 3:401–404. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mousa SA, Shaqura M, Khalefa BI, Zöllner
C, Schaad L, Schneider J, Shippenberg TS, Richter JF, Hellweg R,
Shakibaei M and Schäfer M: Rab7 silencing prevents µ-opioid
receptor lysosomal targeting and rescues opioid responsiveness to
strengthen diabetic neuropathic pain therapy. Diabetes.
62:1308–1319. 2013. View Article : Google Scholar
|
|
38
|
Kelly C, Canning P, Buchanan PJ, Williams
MT, Brown V, Gruenert DC, Elborn JS, Ennis M and Schock BC:
Toll-like receptor 4 is not targeted to the lysosome in cystic
fibrosis airway epithelial cells. Am J Physiol Lung Cell Mol
Physiol. 304:L371–L382. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schleinitz A, Pöttgen LA, Keren-Kaplan T,
Pu J, Saftig P, Bonifacino JS, Haas A and Jeschke A: Consecutive
functions of small GTPases guide HOPS-mediated tethering of late
endosomes and lysosomes. Cell Rep. 42:1119692023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jin X, Wang K, Wang L, Liu W, Zhang C, Qiu
Y, Liu W, Zhang H, Zhang D, Yang Z, et al: RAB7 activity is
required for the regulation of mitophagy in oocyte meiosis and
oocyte quality control during ovarian aging. Autophagy. 18:643–660.
2022. View Article : Google Scholar :
|
|
41
|
Levine B and Kroemer G: Biological
functions of autophagy genes: A disease perspective. Cell.
176:11–42. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Picca A, Faitg J, Auwerx J, Ferrucci L and
D'Amico D: Mitophagy in human health, ageing and disease. Nat
Metab. 5:2047–2061. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang L, Zhou L, Li F, Chen X, Li T, Zou Z,
Zhi Y and He Z: Diagnostic and prognostic value of
autophagy-related key genes in sepsis and potential correlation
with immune cell signatures. Front Cell Dev Biol. 11:12183792023.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wu D, Spencer CB, Ortoga L, Zhang H and
Miao C: Histone lactylation-regulated METTL3 promotes ferroptosis
via m6A-modification on ACSL4 in sepsis-associated lung injury.
Redox Biol. 74:1031942024. View Article : Google Scholar : PubMed/NCBI
|