Introduction
Liver diseases are hepatic pathological changes
caused by numerous endogenous and exogenous pathogenic factors such
as drugs, chemical agents, viral infection, chronic alcohol
consumption and malnutrition. According to epidemiological
statistics, liver diseases account for two million deaths
world-wide annually and are gradually becoming a major challenge to
public health (1).
Comprehensively understanding the pathogenesis underlying liver
diseases and precisely clarifying specific therapeutic targets may
provide new diagnostic approaches and improve the prognosis.
Ferroptosis is a unique form of regulated cell death which is
morphologically, biochemically and genetically different from other
regulated cell deaths, such as apoptosis, necroptosis, autophagy
and pyroptosis. This process is driven by iron-dependent
phospholipid peroxidation, which relies on reactive oxygen species
(ROS), metal iron and phospholipid containing polyunsaturated fatty
acid chains (PUFA-PLs). The correlation of ferroptosis with the
pathogenesis of various diseases involving almost every organ in
the body has been identified recently (2). Particularly, the liver plays a
central role in cell metabolism and is the primary iron storage
organ, making it a preferential target of ferroptosis. Emerging
evidence supports the implication of ferroptosis in the occurrence
and progression of various liver diseases, including drug-induced
liver injury, liver ischemia-reperfusion injury, alcohol-associated
liver disease, non-alcoholic fatty liver disease (NAFLD) and
hepatocellular carcinoma (HCC) (3).
Autophagy is a tightly orchestrated intracellular
process in eukaryotic cells by which cytoplasmic materials are
conveyed to the lysosomal compartment for degradation and recycling
(4). To date, three major types
of autophagy have been defined: Macroautophagy, microautophagy and
chaperone-mediated autophagy (CMA). During macroautophagy, de
novo-synthesized double membrane-bound vesicle (referred to as
an autophagosome) engulfs cytosolic substrates and then fuses with
lysosomes to form an autolysosome. In microautophagy, a portion of
the cytoplasm is directly captured through invaginations or
protrusions of the lysosomal membrane.
CMA involves the identification of KFERQ-like motifs
present in the cytosolic proteins by heat shock cognate 71 kDa
protein cytosolic (Hsc70c), a chaperone protein, followed by the
direction to the lysosomal membrane receptor lysosomal-associated
membrane protein 2A and the translocation of cargo proteins to the
lysosomal lumen (5). Among these
three forms, macroautophagy is the best characterized and is
hereafter referred to as autophagy for simplicity. Autophagy occurs
at a basal level for the constitutive turnover of cytosolic
components to maintain normal cellular homeostasis. This sensitive
and highly inducible process drives cell response to diverse stress
conditions, such as nutrient deprivation, metabolic stress,
oxidative stress, endoplasmic reticulum (ER)-stress and microbial
infection (6). Thus, autophagy
is primarily regarded as a cytoprotective mechanism, although
prolonged constitutive defective or excessive autophagy can be
deleterious. Accordingly, this process has implications for
substantial human pathologies. Autophagy deregulation in either
liver cells or non-parenchymal cells contributes to numerous liver
diseases, including NAFLD, alcohol liver injury, drug-induced liver
injury and HCC (7).
Although ferroptosis was originally identified as a
type of autophagy-independent cell death, extensive evidence has
uncovered the crosstalk between autophagy and ferroptosis. The
present review outlined the research on the crosstalk between
ferroptosis and autophagy under the pathogenesis or treatment of
liver diseases, delineated the pivotal role of the crosstalk and
analyzed the involved molecular regulators or signal pathways,
hopefully providing new insights into liver diseases and effective
strategies for their prevention or amelioration.
Overview of ferroptosis
Cells undergoing ferroptosis typically round up,
detach and lose plasma membrane integrity. Alterations in
mitochondrial morphology and cristae structure are the
characteristic morphological features used as ferroptosis markers.
Obvious mitochondrial shrinkage with condensed membrane densities,
reduction or disappearance of mitochondrial cristae and rupture of
the outer mitochondrial membrane are observed in response to
ferroptosis activators (8).
However, the size and structural integrity of the nucleus are
retained, and nuclear condensation or chromatin margination is
rarely observed during ferroptosis (9). A number of genes regulate the
highly intricated ferroptotic process. For example, ferroptosis
induction may depend on the RAt Sarcoma virus (RAS),
which is the most common oncogene in cancers. Cancer cells
harboring RAS mutations are highly sensitive to ferroptosis
(10). Lung cancer cells
transfected with short hairpin RNAs targeting RAS exhibited
resistance to ferroptosis inducer erastin (11) and rhabdomyosarcoma cells
overexpressing RAS were markedly less susceptible to ferroptosis,
indicating the positive regulatory role of RAS in ferroptosis
(12). By contrast, heat
shock protein beta-1 (HspB1) was a highly expressed
following ferroptosis induction in cervical cancer cells,
osteosarcoma cells and prostate cancer cells. It negatively
regulates ferroptosis in vitro and in vivo:
ferroptosis is promoted by silencing HspB1 but inhibited by
upregulating HspB1 (13).
In biochemical aspects, one central event leading to
ferroptosis is aberrant iron homeostasis (14). Iron is an indispensable element
required by all living organisms, and the control of its levels is
a dynamic process involving its uptake, storage, utilization and
efflux. Serum ferric ion is bound by transferrin and subsequently
imported into cells via transferrin receptor (TFR1)-mediated
endocytosis. In the endosomes, ferric iron is reduced to ferrous
iron and then released into a labile iron pool in the cytoplasm.
Excess iron can be stored in ferritin, which is the primary iron
storage protein complex composed of ferritin light chain (FTL) and
ferritin heavy chain 1 (FTH1). Iron export requires the iron efflux
pump ferroportin-1 (FPN1), which produces ferric iron from ferrous
iron (15). Ferrous iron plays
numerous important functions in the regulation of multiple
biochemical processes. In addition to catalyzing ROS generation via
the Fenton reaction, iron is incorporated into several
ROS-generating enzymes and thus induces the other core event in
ferroptosis, as shown by the accumulation of lipid peroxidation
accompanied by depleted glutathione (GSH) and insufficient
glutathione peroxidase 4 (GPX4) (16). GSH is a tripeptide synthesized
from glutamate, cysteine and glycine and acts as a direct ROS
scavenger and a powerful antioxidant that limits oxidative damage
to cellular components. The rate-limiting precursor for GSH
synthesis is cysteine, which is imported into cells by system Xc-,
a cystine and glutamate antiporter in the plasma membrane
comprising xCT (solute carrier family 7 member 11) and solute
carrier family 3 member 2. GSH is a necessary cofactor of GPX4, an
antioxidant enzyme that utilizes reduced GSH to convert toxic lipid
peroxides to non-toxic phosphatidyl alcohols to confer resistance
to lipid peroxidation and subsequently prevent ferroptosis
(17). Therefore, an increase in
labile iron pool and lipid peroxidation are considered typical
presentations of ferroptosis and are used as markers of ferroptotic
cell death (Fig. 1).
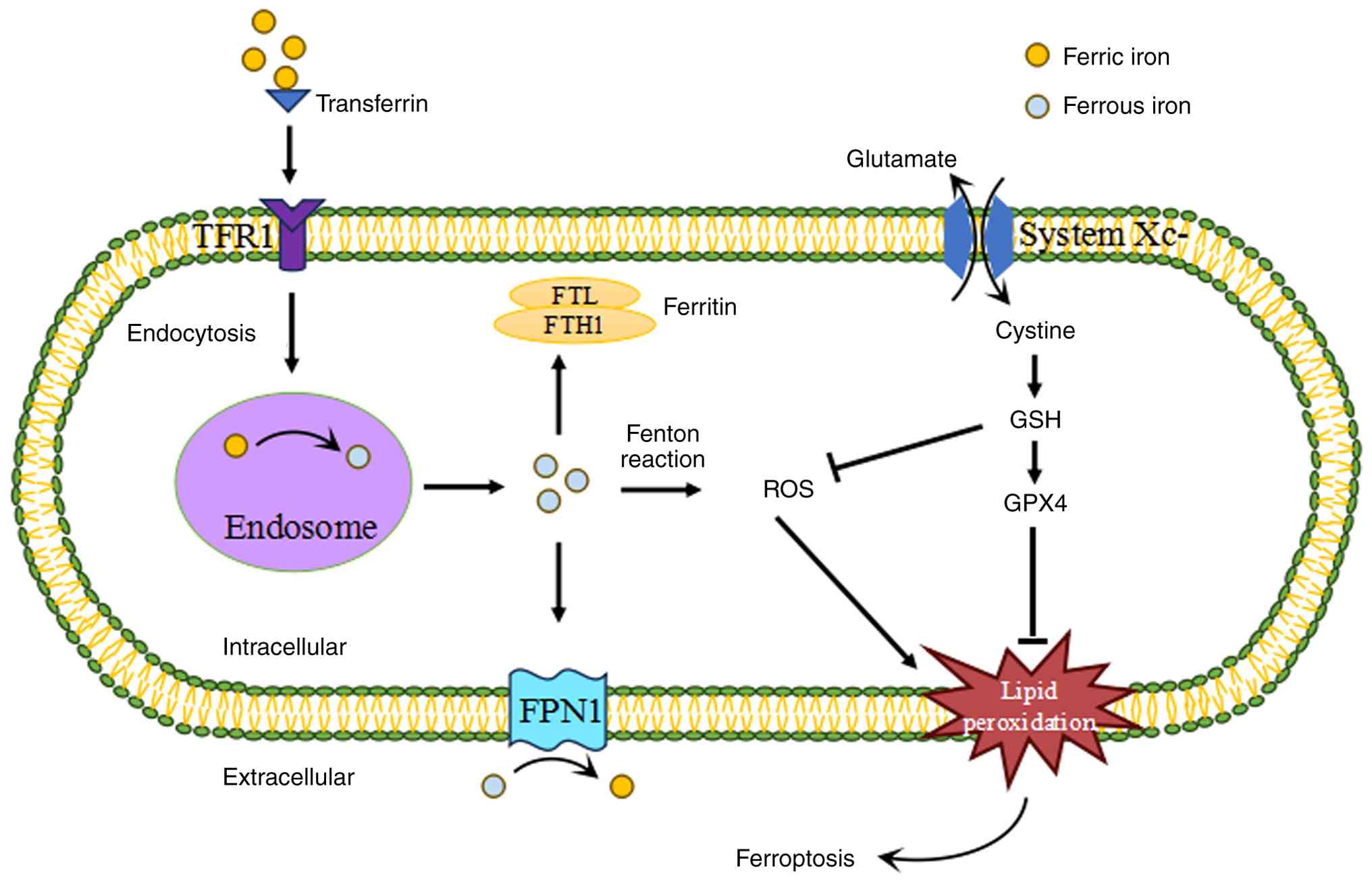 | Figure 1The process of ferroptosis. Cells
acquires transferrin-bound ferric iron via TFR1-mediated
endocytosis. Ferric iron is reduced to ferrous iron in the endosome
and then released into the cytoplasm. Excess iron can be stored by
ferritin, including FTL and FTH1. Iron export requires the iron
efflux pump FPN1, which produces ferric iron from ferrous iron.
System Xc- imports cysteine into cells with a 1:1 countertransport
of glutamate and then induces the generation of GSH. GSH is a major
ROS scavenger and a necessary cofactor of GPX4, an antioxidant
enzyme that converts the toxic lipid peroxides to non-toxic
phosphatidyl alcohols for preventing ferroptosis. Therefore,
ferroptosis is characterized by the iron overload and lipid
peroxidation. TFR1, transferrin receptor 1; FTL, ferritin light
chain; FTH1, ferritin heavy chain 1; FPN1, ferroportin-1; GSH,
glutathione; ROS, reactive oxygen species; GPX4, glutathione
peroxidase 4. |
Intra- and intercellular signaling events,
environmental stresses or small molecules can regulate ferroptosis
by directly or indirectly controlling iron accumulation and/or
lipid peroxidation (18). For
example, exogenous iron (such as ferric ammonium citrate and ferric
citrate) supplementation, increased iron uptake (such as TFR1
overexpression), decreased iron storage (such as ferritin
knockdown) and impaired iron efflux (such as FPN1 knockdown)
contribute to iron overload and enhance the sensitivity to
ferroptosis (19,20). By contrast, iron chelators such
as deferoxamine or desferrioxamine mesylate block ferroptosis via
the inhibition of iron overload (21). Blocking of system
Xc--mediated cystine import using excessive glutamate
results in GSH depletion and consequent ROS accumulation,
ultimately leading to a void in the antioxidant defenses and lipid
peroxidation that triggers ferroptosis (22). Ferroptosis can also be triggered
by downregulated GPX4 through genetic deletion, covalently
inactivating GPX4 (such as RSL3 and ML162) and promoting GPX4
degradation (such as FIN56), whereas overexpressing GPX4 or
pharmacologically blocking its degradation may enhance antioxidant
capacity and display potent protective effects against ferroptosis
(23). Lipid metabolism
regulates ferroptosis by controlling the peroxidizable levels of
PUFA-PLs, the most important lipids required for ferroptosis, and
the associated processes of phospholipid peroxidation. The
arachidonic acid-mediated depletion of PUFA-PLs severely
counteracts ferroptosis in a number of cell lines, and a similar
phenomenon is observed upon the pharmacological inhibition or
genetic inactivation of the acyl-coenzyme A synthetase long chain,
which is a requirement for the synthesis of PUFA-PLs. Furthermore,
lipid antioxidants such as ferrostatin-1 (Fer-1) or liproxstatin-1
prevent ferroptosis by inhibiting lipid peroxidation and are
commonly used as ferroptosis inhibitors (24).
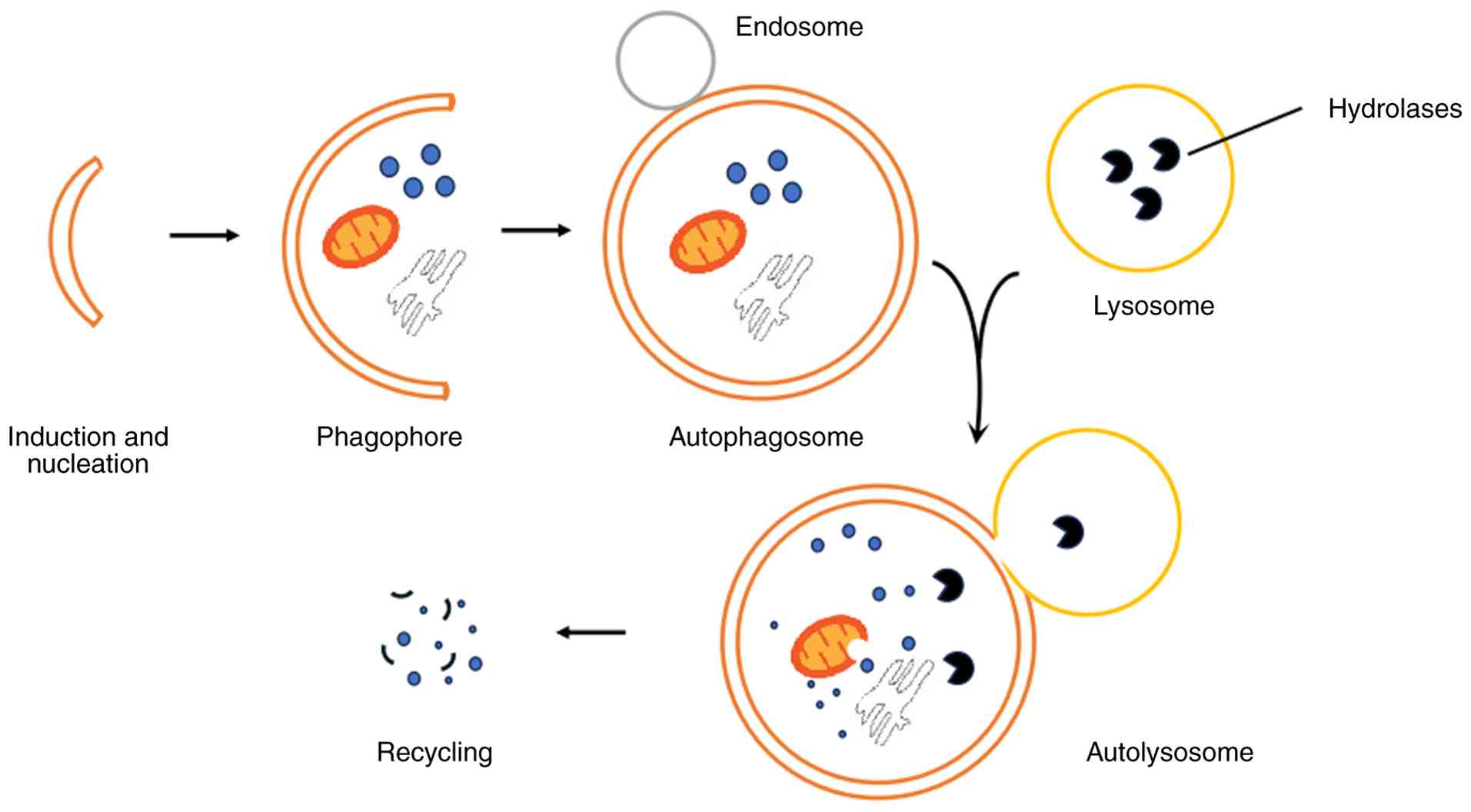
Overview of autophagy
As an evolutionarily conserved degradation system,
the complete autophagic process encompasses a series of consecutive
steps. Upon induction, the nucleation of the initial autophagosomal
vesicle (a very flat organelle similar to Golgi cisterna and
designated as phagophore or isolation membrane) occurs at multiple
sites throughout the cytoplasm. Subsequent to nucleation, the
phagophore expands to sequester its cargos via the addition of
membrane presumably derived from the ER, Golgi complex, plasma
membrane and mitochondria. The edges of the phagophore bend and
ultimately seal to generate a typically spherical, double-membraned
autophagosome with a diameter of 300-900 nm, depending on the
organisms and cargo types (6).
The autophagosome then moves to lysosome, and its outer membrane
fuses with the lysosome to form an autolysosome, where the inner
membrane of the autophagosome and its contents are degraded by
lysosomal hydrolases (5). Before
fusing with the lysosome, the autophagosome is hypothesized to
firstly fuse with early or late endosomes to become an amphisome
and acquire the necessary machineries for its subsequent fusion
with the lysosome (25).
Finally, the breakdown parts are exported back into the cytoplasm
by membrane permeases for reuse as building blocks of anabolic
processes or as an energy supplement (Fig. 2).
A specific family of genes called
autophagy-related genes (Atgs) constitutes the
multistage molecular machinery of autophagy. Originally identified
in yeast, >30 mammalian Atg orthologs have been found to
play critical roles in autophagy. Among these Atg proteins, the
essential subsets in autophagosome formation and maturation are
referred to as the core molecular machinery and grouped into the
following four categories based on their respective functions: The
UNC51-like kinase 1 kinase complex; two ubiquitin-like conjugation
systems; class III phosphatidylinositol 3-kinase (PI3KIII) complex;
and two transmembrane proteins, Atg9 and vacuole membrane protein
1(VPM1) (26,27). The ULK1 kinase complex is
composed of ULK1 and its regulatory subunit FIP200, Atg13 and
Atg101 and carries out the initiation of autophagy. Under nutrient
deprivation conditions, ULK1 is stimulated and Atg13, FIP200 and
ULK1 itself are phosphorylated, resulting in the recruitment of
other autophagy proteins for phagophore nucleation and assembly
(28). Two ubiquitin-like [Atg12
and Atg8/microtubule-associated protein light chain 3 (LC3)]
conjugation systems facilitate phagophore membrane elongation and
expansion. The first conjugation event involves the covalent
attachment of Atg12 to Atg5, which requires Atg7 and Atg10 acting
as E1 and E2-like enzymes, respectively. Atg12-Atg5 then interacts
noncovalently with Atg16 and oligomerizes to a large units called
Atg16L complex, which functions as an E3-like enzyme in the second
conjugation system to facilitate the conjugation of a single
phosphatidylethanolamine (PE) to the carboxyl terminus of LC3. For
this conjugation to occur, LC3 is initially cleaved by Atg4
protease. The proteolyzed LC3 (LC3I) is then processed by the same
E1-like enzyme Atg7 and are further transferred to the E2-like
enzyme Atg3. The Atg16 complex finally ligates LC3I to PE to form
LC3II, a lipidated form attached to autophagosome. Of note, the
distribution of cytosolic LC3 to autophagosomes and the amount of
LC3II are commonly regarded as markers for autophagy (29). The PI3KIII complex, which
comprises vacuolar protein sorting 34 (Vps34), Beclin1/Atg6 and
Vps15, preforms membrane modification involving
phosphatidylinositol phosphorylation to produce
phosphatidylinositol 3-phosphate. This molecule then serves as a
docking particle that promotes Atg protein complex formation at the
nucleation site, membrane enclosing and the cytoplasmic components
sequestration (26). The
transmembrane protein Atg9 localizes to autophagy-related
structures, namely, omegasomes, as well as the Golgi apparatus and
endosomes. Atg9 shuttles among these organelles and potentially
contributes to membrane transport to the forming autophagosome
(30). VPM1 is an ER- and Golgi
apparatus-associated membrane protein. By directly interacting with
Beclin1, VPM1 may bring PI3KIII components to the phagophore and
promote the autophagosome formation (31).
Autophagy regulation is extremely complicated and
involves multiple stimulatory or inhibitory factors or signal
pathways, such as PI3KI-protein kinase B (AKT)-mammalian target of
rapamycin complex 1 (mTORC1), adenosine monophosphate-dependent
protein kinase (AMPK)-mTORC1 pathway, B cell lymphoma 2
(Bcl2)-Beclin1 pathway and p53. mTOR is a conserved
serine/threonine protein kinase and exists in two distinct
complexes, mTORC1 and mTOR2, which are formed by binding with
multiple companion proteins and defined by the presence of the
companion proteins Raptor and Rictor, respectively. Particularly,
mTORC1 integrates various upstream signaling pathways to block or
induce autophagy. PI3KI can increase the membrane recruitment of
phosphoinositide-dependent kinase 1, which phosphorylates and
activates AKT. AKT further inhibits the downstream tuberous
sclerosis complex (TSC) and activates mTORC1, leading to autophagy
suppression. By contrast, AMPK can phosphorylate and activate TSC
and induce autophagy by inhibiting mTORC1 activity (32). Beclin1 is a mammalian autophagy
protein identified as a novel Bcl2-interacting protein. Increased
Beclin1 and Bcl2 interaction disturbs the PI3KIII complex
formation, thus inhibiting autophagy. By contrast, the disruption
of the Bcl2-Beclin1 complex upregulates autophagy (33). p53 may exert both pro- and
anti-autophagy functions depending on its compartmental
localization. Cytosolic p53 effectively represses autophagy,
whereas nuclear p53 stimulates autophagy through the
transcriptional repression of mTORC1 activity and the induction of
damage-regulated autophagy modulator expression (34). Furthermore, epigenetic
alterations, including DNA methylation, histone modification and
non-coding RNAs expression, not only modify Atgs but also
affect signaling genes, thus inhibiting or promoting of autophagy
(35) (Fig. 3). Of note, these regulatory
pathways may be interconnected and influence the dynamic autophagic
process at different steps.
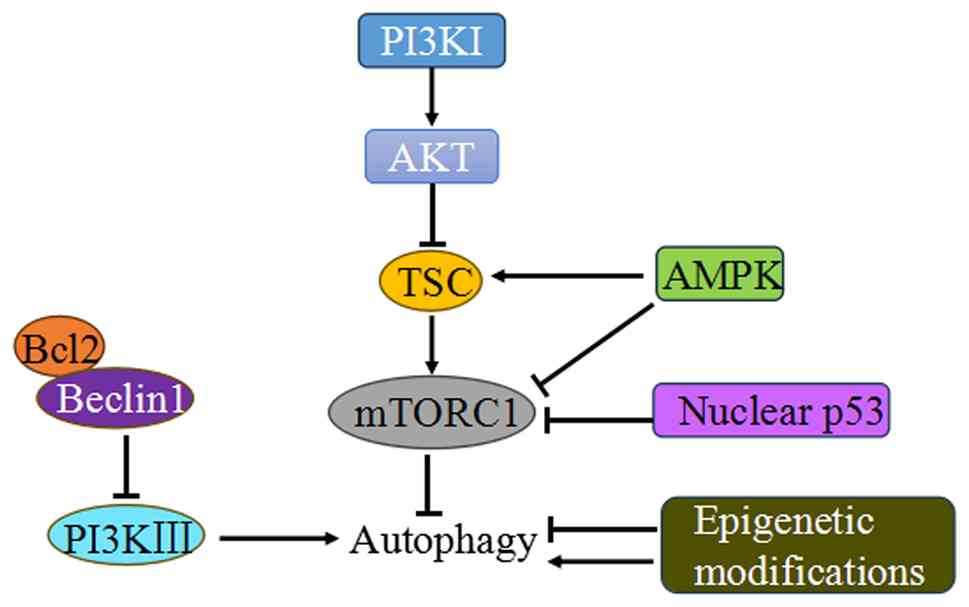 | Figure 3Major regulatory pathways of
autophagy. Autophagy regulation involves multiple stimulatory or
inhibitory factors or signal pathways, such as PI3KI-AKT-mTORC1,
AMPK-mTORC1, Bcl2-Beclin1 pathways, p53 and epigenetic modulations.
PI3KI, class I phosphatidylinositol 3-kinase; AKT, protein kinase
B; TSC, tuberous sclerosis complex; mTORC1, mammalian target of
rapamycin complex 1; AMPK, adenosine monophosphate-dependent
protein kinase; Bcl2, B cell lymphoma 2; PI3KIII, class III
phosphatidylinositol 3-kinase. |
Autophagy can be a bulk, nonselective, degradative
process, with random engulfment of cellular components. Under
certain circumstances, autophagy occurs in a selective manner
dependent on autophagy adaptor proteins, such as p62, neighbor of
BRCA1, optineurin and nuclear dot protein 52 KDa. These proteins
interact simultaneously with cargos and LC3 protein anchored in the
autophagosomal double membrane for autophagosome targeting.
Selective autophagy contributes to organelle quality control and
homeostasis regulation by degrading specific soluble proteins
(ferritinophagy), damaged and excess organelles (mitophagy,
lipophagy, lysophagy, ER-phagy, ribophagy, perophagy and
nucleophagy), aggregated proteins (aggrephagy) and invasive
bacteria (xenophagy) (36).
Crosstalk between autophagy and ferroptosis
in diverse liver diseases
Multiple Atgs, such as Atg5,
Atg7 and Beclin1, have been identified as potential
positive regulators of ferroptosis using RNAi screening methods.
The genetic and pharmacological inhibition of autophagy greatly
attenuates and delays ferroptosis, highlighting the vital
involvement of autophagy in the regulation of ferroptotic cell
death (37). Particularly,
selective autophagy including nuclear receptor coactivator 4
(NCOA4)-mediated ferritinophagy, Ras-related protein Rab7a
(Rab7)-mediated lipophagy, p62-mediated clockophagy and
Hsp90-mediated CMA, degrades ferritin, lipid droplets, aryl
hydrocarbon receptor nuclear translocator-like protein 1 and GPX4
to induce iron overload and/or lipid peroxidation, eventually
promoting ferroptosis (Fig. 4).
Conversely, ferroptosis induction activates autophagy by
stimulating autophagosomes formation, whereas ferroptosis
inhibition impairs autophagic degradation in a context-dependent
manner (38). Thus, the signal
pathways or essential molecules involved in these two processes may
either be shared or be interconnected (39). The crosstalk between autophagy
and ferroptosis has emerged as a vital factor in the occurrence,
development and therapeutic application of various liver diseases.
Studies have demonstrated that this crosstalk is an important
mechanism underlying drug- or toxin-induced liver injury (40-55), and is also closely associated
with liver fibrosis (23,56-65)
and HCC (66-81). Moreover, it functions in the
liver damage of metabolic syndrome (MS) (82), NAFLD (83), non-alcoholic steatohepatitis
(NASH) (84), acute liver injury
(ALI) (85-89), diabetic liver injury (90) and liver cell senescence (91). Further details supporting this
crosstalk as a pivotal mediator in these liver diseases are listed
in Table I).
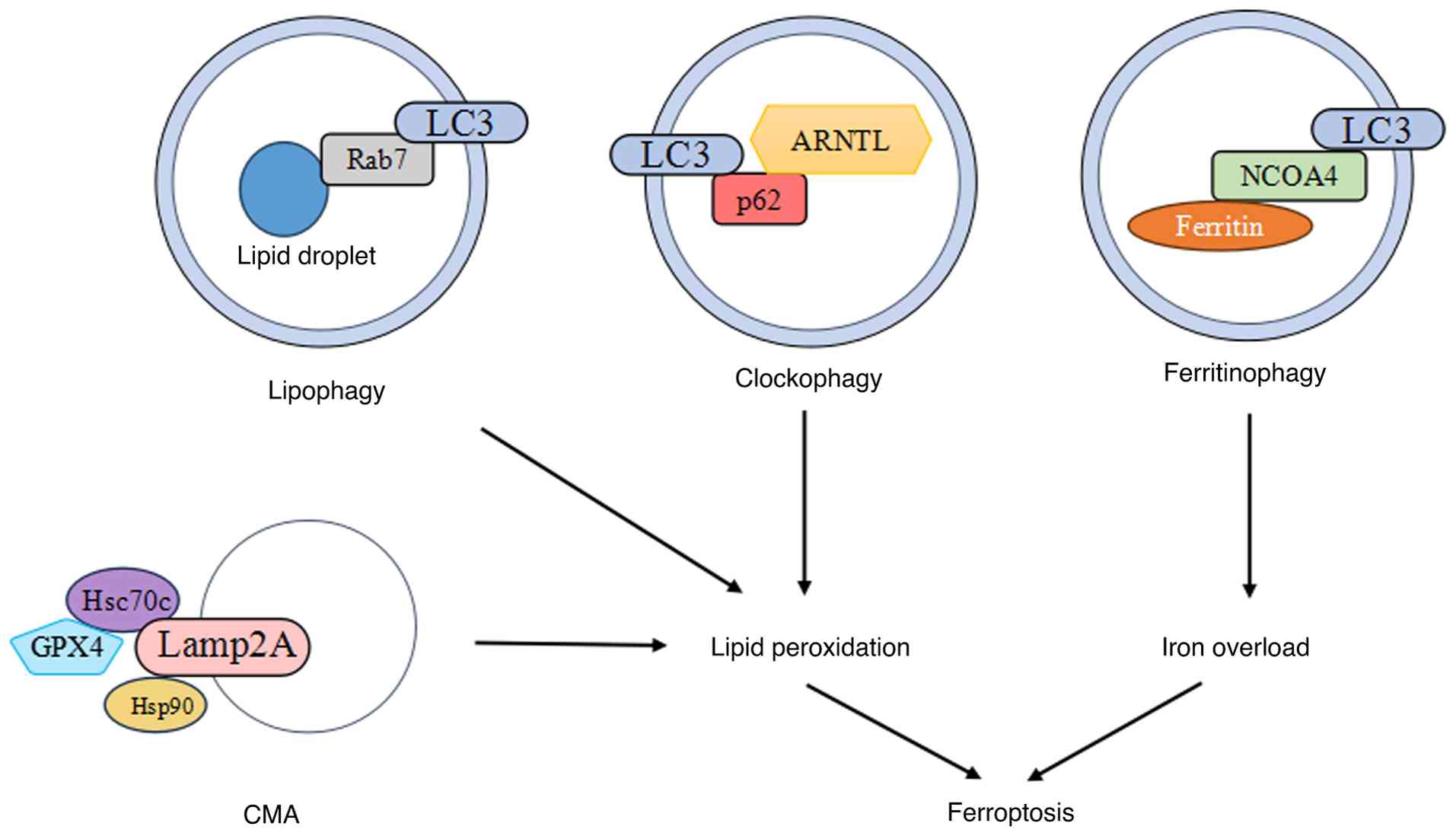 | Figure 4Selective autophagy promotes
ferroptosis. Rab7-mediated lipophagy, p62-mediated clockophagy and
Hsp90-mediated CMA promote lipid peroxidation in ferroptosis;
NCOA4-mediated ferritinophagy promotes iron accumulation in
ferroptosis. Rab7, Ras related protein Rab7a; LC3,
microtubule-associated protein light chain 3; ARNTL, aryl
hydrocarbon receptor nuclear translocator-like protein 1; Hsc70c,
heat shock cognate 71 KDa protein cytosolic; Lamp2,
lysosome-associated membrane protein 2; Hsp90, heat shock protein
90; GPX4, glutathione peroxidase 4; CMA, chaperone-mediated
autophagy; NCOA4, nuclear receptor coactivator 4. |
 | Table ICrosstalk between autophagy and
ferroptosis in diverse liver diseases. |
Table I
Crosstalk between autophagy and
ferroptosis in diverse liver diseases.
| Authors, year | Type of liver
diseases |
Autophagy/ferroptosis modulators | Experimental models
|
Autophagy/ferroptosis status | Related biological
effects | (Refs.) |
|---|
| In
vitro | In vivo |
|---|
| Zhou et al,
2022 | Drug-induced liver
injury | Rifampicin | HepG, AML12 | C57BL/6 mice |
Ferritinophagy↑; ferroptosis↑ | NCOA4, LC3II,
acidic vesicular organelles, FTH1-LC3 colocalization, iron content,
MDA↑; p62, FTH1, HspA8↓ | (40) |
| Wang et al,
2024 | | Methotrexate | HepG2, AML12 | C57BL/6 mice |
Ferritinophagy↑; ferroptosis↑ | NCOA4, LC3II,
autophagosomes, FTH1-LC3 colocalization, HMGB1, MDA↑; p62, FTH1, GSH, GPX4↓ | (41) |
| Liang et al,
2023 | | Toosendanin | HepG2 | Balb/c mice |
Ferritinophagy↑; ferroptosis↑ | NCOA4, TFR1,
ferrous ion, ROS, lipid peroxidation, PERK, p-PERK, p-eIF2α,
ATF4↑; FTH1, GPX4, FPN1↓ | (42) |
| Liu et al,
2025 | | Triptolide | HL7702 | C57BL/6J mice | Lipophagy↑; ferroptosis↑ | Rab7, LC3II,
LC3-Rab7 colocalization, iron content, mtROS, ROS, MDA, Ptgs2,
FFAs↑; p62, GSH, GPX4, lipid
droplets, MMP↓ | (43) |
| Wu et al,
2024 | | Acetaminophen | Mouse primary
hepatocytes | KM mice |
Ferritinophagy↑; ferroptosis↑ | LC3II, Iron
content, MDA, Keap1↑; p62,
FTH1, xCT, SLC3A2, GSH, Nrf2, HO-1↓ | (44) |
| Cai et al,
2022 | | Acetaminophen | HL7702 | C57BL/6 mice | Autophagy↓;
ferroptosis↑ | LC3II, p62, ROS,
xCT↑; FTH1, Nrf2, HO-1, GPX4↓;
mitochondrial shrinkage and cristae loss | (45) |
| Ren et al,
2025 | | Acetaminophen | AML12, HepG2 | C57BL/6J mice | Autophagy↓;
ferroptosis↑ | Foxo1, iron
content, MDA, ROS, Ptgs2, p62↑; LC3II, GSH, MMP, xCT, GPX4↓ | (46) |
| Huang et al,
2023 | Toxin-induced liver
injury | Acrylamide | HepG2 | C57BL/6J mice |
Ferritinophagy↑; ferroptosis↑ | NCOA4, LC3II,
autophagy flux, iron content, ROS, lipid ROS, MDA↑; p62, FTH1, GSH, GPX4, MMP,
Nrf2↓ | (47) |
| Zhong, et al
2024 | | Copper sulfate | | Broiler chicks |
Ferritinophagy↑; ferroptosis↑ | NCOA4, ACLS4, MDA,
Keap1, TXNIP, lipid peroxidation↑; GPX4, GSH, xCT, FSP1, Nrf2, SOD1,
TRX↓; mitochondrial shrinkage or cristae loss | (48) |
| He et al,
2022 | | Cadmium
chloride | AML12 | Balb/c mice |
Ferritinophagy↑; ferroptosis↑ | LC3II, iron
content, ferrous ion, MDA, lipid peroxidation, Ptgs2, GRP78,
p-PERK, p-elF2α, ATF4, CHOP↑;
NCOA4, p62, FTH1, GSH, GPX4↓ | (49) |
| Wei et al,
2022 | | Nickel
chloride | | ICR mice |
Ferritinophagy↑; ferroptosis↑ | Iron content,
mtROS, MDA, Ptgs2↑; NCOA4,
FTH1, GSH, GPX4, MMP, mitochondrial respiratory chain
complexes↓ | (50) |
| Yu et al,
2023 | | Sodium
arsenite | LMH | HY-line white
chickens |
Ferritinophagy↑; ferroptosis↑ | NCOA4, LC3II, Atg5,
Atg7, autophagosomes, iron content, lipid ROS, MDA, Ptgs2, p-AMPK
(Thr172), p-ULK1 (Ser555)↑;
p62, FTH1, GPX4, xCT, MMP, p-mTOR (Ser2448) ↓; mitochondrial
shrinkage and cristae loss | (51) |
| Xu et al,
2023 | | Sodium
fluoride | BRL3A | SD rats | Autophagy↓;
ferroptosis↑ | LC3II, p62, ferrous
ion, mtROS, MDA, ACSL4, TFR1, TOMM20↑; FTH1, autophagy flux, MMP, GSH,
GPX4↓ | (52) |
| Song et al,
2022 | | Ethanol- or
acetaldehyde | HepG2, HL7702 | |
Ferritinophagy↑; mitophagy↓; ferroptosis↑ | NCOA4,
ferritin-lysosome colocalization, iron content, ROS, MDA,
TFR1↑; p62, GSH, FTH1, PINK1,
Parkin↓ | (53) |
| Song et al,
2024 | | Aflatoxin B1 | HepG2 | C57BL/6 J mice |
Ferritinophagy↑; ferroptosis↑ | GPX4, T-AOC, CAT↓
Ferritin, FTH11, FTL↓; Beclin1, LC3II, Atg5, p-mTOR, p-AKT, p-PI3K,
p-ULK1↑ | (54) |
| Jiang et al,
2024 | | Deoxynivalenol | AML12 | C57BL/6 mice |
Ferritinophagy↑; mitophagy↑; ferroptosis↑ | Iron content, MDA,
Ptgs2, ACSL4, NCOA4, PINK1, Parkin, LC3II, p-JNK, p-JUN↑; NCOA4-FTH1 interaction↑; mitochondria-lysosomes
colocalization↑; xCT, GPX4,
CAT, SOD, FTH, p62, PDCD4↓ | (55) |
| Liang et al,
2023 | Liver fibrosis | Silica
nanoparticles | HL7702 | F344 rats |
Ferritinophagy↑; ferroptosis↑ | NCOA4, LC3II,
ferrous ion, MDA, Ptgs2↑;
GPX4, FTH1↓, mitochondrial membrane rupture | (56) |
| Bi et al,
2024 | | CCl4 | HepG2 | Friend Virus B
mice; liver tissues of patients with liver fibrosis | Mitophagy↑; ferroptosis↑ | MDA, iron content,
Pink1, Parkin, FUNDC1↑;
FUNDC1-GPX4 interaction↑; GSH,
MMP, xCT↓; mitochondrial shrinkage and cristae loss | (57) |
| Zhang et al,
2020 | | Sorafenib, erastin,
or RSL3 | HSC-LX2, HSC-T6,
primary mouse HSCs primary, human HSCs | BDL-induced liver
fibrosis model in C57BL/6 mice; human liver resection tissues with
liver cirrhosis complicated with HCC |
Ferritinophagy↑; ferroptosis↑ | NCOA4, LC3II, LC3
puncta, Atg16L1, autophagy flux, autophagic vesicles, iron content,
MDA, ACSL4, Ptgs2, xCT, SLC11A2, FBXW7, FBXW7-ZFP36
interaction↑; p62, FTH1, GSH,
GPX4, ZFP36↓ | (23) |
| Zhang et al,
2018 | | Erastin, buthionine
sulfoximine, and sorafenib | HSC-LX2, HSC-T6,
primary mouse HSCs, primary human HSCs | BDL-induced liver
fibrosis model in C57BL/6 mice; human liver resection tissues with
liver cirrhosis complicated with HCC |
Ferritinophagy↑; ferroptosis↑ | NCOA4, LC3II,
Beclin1, autophagic vesicles, iron content, lipid ROS, ROS, MDA,
GSH, ELAVL1, Ptgs2, ACSL4, xCT, SLC11A2↑; p62, FTH1↓ | (58) |
| Shen et al,
2021 | | Erastin, sorafenib
and RSL3 | HSC-LX2, HSC-T6,
primary mouse HSCs, primary human HSCs | CCl4-induced liver
fibrosis model in ICR mice; human liver resection tissues with
liver cirrhosis complicated with HCC |
Ferritinophagy↑; ferroptosis↑ | NCOA4, LC3II, LC3
puncta, Beclin1, autophagy flux, iron content, lipid ROS, MDA,
Ptgs2, METTL4, m6A RNA methylation↑; p62, FTH1, GSH, FTO↓ | (59) |
| Tan et al,
2022 | | MSC-ex | HSC-LX2 | CCl4 induced liver
fibrosis model in BALB/c mice |
Ferritinophagy↑; ferroptosis↑ | LC3II, Beclin1,
ferrous ion, ROS, lipid peroxidation↑; GSH, GPX4, MMP, xCT↓; mitochondrial
shrinkage | (60) |
| Kong et al,
2019 | | Artesunate | HSC-LX2, primary
mouse HSCs | CCl4 induced liver
fibrosis model in ICR mice |
Ferritinophagy↑; ferroptosis↑ | LC3II, LC3 puncta,
autophagic flux, autophagosomes ferrous ion, lipid ROS, MDA, lipid
peroxidation, Ptgs2↑; NCOA4,
FTH1, p62, GSH, GPX4↓; mitochondrial shrinkage | (61) |
| Zheng et al,
2022 | | Curcumol | HSC-T6 | |
Ferritinophagy↑; ferroptosis↑ | NCOA4, LC3II,
Beclin1, autophagy flux, iron content, ROS, ACSL4, Ptgs2↑; p62, FTH1, MMP, GPX4, xCT↓;
mitochondrial shrinkage and cristae loss | (62) |
| Zhang et al,
2021 | |
Dihydroartemisinin | Primary rat
HSCs | CCl4 induced liver
fibrosis model in SD rats |
Ferritinophagy↑; ferroptosis↑ | NCOA4, LC3II, Atg5,
iron content, lipid ROS, MDA, ACSL4, SLC11A2↑; GPX4, xCT, GSH↓; mitochondrial
shrinkage and cristae loss | (63) |
| Li et al,
2025 | | Artemether | HSC-LX2 | CCl4 induced liver
fibrosis model in ICR mice |
Ferritinophagy↑; ferroptosis↑ | NCOA4, LC3II, MDA,
ROS, iron content↑; FTH1, p62,
GSH↓ | (64) |
| Yi et al,
2021 | | Berberine | HSC-LX2,
HSC-T6 | Thioacetamide (TAA)
and CCl4-induced liver fibrosis model in C57BL/6 mice | Autophagy↓;
ferroptosis↑ | p62, ferrous ion,
ROS, MDA, 4-HNE, Ptgs2, FTH1 ubiquitination↑; LC3II, Atg5, Atg7, FTH1, GSH, GPX4,
autophagosome, autophagic flux↓; mitochondrial shrinkage | (65) |
| Liu et al,
2020 | HCC | Sorafenib | HepG2, Huh7 | |
Ferritinophagy↑; ferroptosis↑ | LC3II, autophagic
flux, autophagosome, ferrous ion, MDA, cIARS, cIARS-ALKBH5
interaction, BCl2-Beclin1 interaction↑; NCOA4, p62, FTH1, GSH ↓ | (66) |
| Bi et al,
2023 | | Lenvatinib;
erastin | HepG2, Huh7 | | ER-phagy↑; ferroptosis↑ | circFAM134B, LC3II,
ferrous ion, MDA, ROS↑;
FAM134B, REEP5, GSH, GPX4, xCT↓ | (67) |
| Liu et al,
2022 | | Sorafenib | HepG2, Huh7 | | ER-phagy↑; ferroptosis↑ | Autophagosomes
engulfed ER fragments, swelled and deformed ER, ferrous ion, lipid
ROS↑; FAM134B, Trap-α, REEP5,
GPX4↓ | (68) |
| Yang et al,
2023 | | Sorafenib | HepG2, Huh-7 | |
Ferritinophagy↑; ferroptosis↑ | NCOA4, ferrous ion,
lipid ROS, MDA, PTBP1↑; GSH,
FTH1↓ | (69) |
| Hu et al,
2022; Han et al, 2021 | | PNO1 | Hep3B, HLE | Xenografted human
HCC from BALB/c nude mice | Autophagy↑; ferroptosis↓ | LC3II, Beclin1,
Atg5, Atg7, autophagic flux, p-Erk, xCT, GSH↑; p62, lipid ROS, MDA↓ | (70,71) |
| Zhang et al,
2023 | | TSPO | HCCLM3,
MHCC97H | Xenografted human
HCC from BALB/c nude mice | Autophagy↓;
ferroptosis↓ | p62, GSH, GPX4,
Nrf2, HO-1, NQO1, GCLC, GCLM, PPAR-γ↑; LC3II, autophagy flux, ferrous ion,
ROS, lipid ROS, Keap1-Nrf2 interaction↓ | (72) |
| Wang et al,
2021 | | Erastin, sorafenib,
or sulfasalazine | HepG2 | Xenografted human
HCC from C57BL/6 mice |
Ferritinophagy↑; ferroptosis↑ | GFP-LC3 puncta,
ferrous ions, MDA, p-AMPK(Thr172) ↑; NCOA4, GSH, system Xc-activity,
BCAT2, SREBP1, SREBP1-BCAT2 interaction↓ | (73) |
| Wu et al,
2023 | | IGF1 | HCCLM3, Huh7 | Xenografted human
HCC from BALB/c nude mice | CMA↓;
ferroptosis↓ | GPX4, GSH, GPX4-CKB
interaction, AKT-CKB interaction, p-CKB (Thr133), p-GPX4
(Ser104)↑; lipid ROS,
GPX4-Hsc70c-Lamp2A interaction↓ | (74) |
| Li et al,
2024 | | Erastin, sorafenib,
RSL3 | HepG2, Huh7 | Xenografted human
HCC from BALB/c nude mice |
Ferritinophagy↑; ferroptosis↑ | ROS, iron content,
MDA, LC3II, Atg5, NCOA4, WTAP, YTHDC2↑; GSH FTH1, p62↓ | (75) |
| Cao et al,
2024 | | USP24 | HepG2, SMMC-7721,
Huh7, HCCLM3 | Xenografted human
HCC from BALB/c nude mice |
Ferritinophagy↑; ferroptosis↑ | NCOA4, ULK1, Atg5,
Beclin1, LC3II, autophagic vesicles, USP24-Beclin1 interaction,
iron content, TFR1, MDA↑;
Beclin1 polyubiquitination, p62, GPX4, FPN1, GSH↓ | (76) |
| Jiang et al,
2025 | | USP2 | Huh7, HepG2,
SMMC-7721, and SNU-449 | Xenografted human
HCC from BALB/c nude mice | Clockophagy↓;
ferroptosis↓ | ARNTL, USP2-ARNTL
interaction, GSH, HIF1α, xCT↑;
ARNTL polyubiquitination, ROS, MDA↓ | (77) |
| Xiu et al,
2022 | | Caryophyllene
oxide | HCCLM3, HUH7 | Xenografted human
HCC from BALB/c nude mice |
Ferritinophagy↑; ferroptosis↑ | NCOA4, LC3II,
LC3II-NCOA4 colocalization, lysosome, ferrous ions, ROS, lipid
peroxidation, MDA↑; GPX4,
Nrf2, HO-1, and NQO1, T-AOC, FTH1↓; mitochondrial shrinkage and
cristae loss | (78) |
| Xiu et al,
2023 | | Esculetin | HUH7, HCCLM3 | Xenografted human
HCC from BALB/c nude mice |
Ferritinophagy↑; ferroptosis↑ | NCOA4, LC3II,
autophagy flux, lysosome, ferrous ions, ROS, MDA, lipid
peroxidation↑; p62, FTH1,
GPX4, NFE2L2, HO-1, T-AOC, MMP↓; mitochondrial shrinkage and
cristae loss | (79) |
| Zhu et al,
2023 | | EChLESs | SNU-387, HUH-7,
Bel7402 | Xenografted human
HCC from BALB/c nude mice |
Ferritinophagy↑; ferroptosis↑ | NCOA4, LC3II,
autophagosomes, ferrous ions, mtROS, lipid peroxidation,
HIF-α↑; p62, FTH1, GSH, MMP,
p-mTOR (Ser2448), mTOR↓; mitochondrial shrinkage | (80) |
| Li et al,
2021 | | Artesunate combined
with sorafenib | HepG2, Huh7,
SNU-182, SNU-449 | Xenografted human
HCC from BALB/c nude mice |
Ferritinophagy↑; ferroptosis↑ | Ferrous ions, ROS,
mtROS, MDA, lipid peroxidation, cathepsin L, cathepsin B, lysosomal
activity↑; FTH1, FTL, TFR1,
GSH, MMP, ATP↓ | (81) |
| Cui et al,
2023 | MS liver
injury | High-fat and
high-fructose diet | | SD rats | Ferritinophagy↓;
ferroptosis↑ | FTH1, TFR1, Iron
content, MDA, lipid peroxidation↑; NCOA4, GPX4, SOD, xCT, FPN1,
NCOA4-FTH1 colocalization and interaction↓ | (82) |
| Liu et al,
2023 | NAFLD | Saturated fatty
acids | AML12 | C57BL/6J | Autophagy↓;
ferroptosis↑ | p62, lipid
droplets, MDA, lipid peroxidation, Keap1, Ptgs2, ACSL4,
p-mTOR↑; LC3II, Atg7,
autophagic flux, autophagosome, GSH, GPX4, Nrf2, xCT↓ | (83) |
| Honma et al,
2023 | NASH | Iron dextran | | HFC diet fed
SHRSP5/Dmcr rats | Ferritinophagy,
lipophagy↑;
ferroptosis↑ | NCOA4, Rab10,
nuclear TFEB, DNM2, ULK1, UVRAG, Atg14, Lamp2, TRPML1, CLN3, iron
content, 4-HNE, ALOX15, Ptgs2, calcineurin activity↑; GPX4↓; mitochondrial shrinkage | (84) |
| Li et al,
2023 | Acute liver
injury | Liensinine | RSL3-treated
alternatively activated macrophages | LPS/D-GalN-induced
liver injury model in C57BL/6 mice | Ferritinophagy↓;
ferroptosis↓ | LC3II, p62,
autophagosomes↑; ferrous ions,
ROS, lipid peroxidation, damaged mitochondria ↓; ferritin-Lamp1
colocalization, LC3-Lamp1 co localization↓ | (85) |
| Zhang et al,
2025 | | Quercetin | Mouse primary
hepatocytes |
LPS/γ-D-glutamyl-meso-diaminopimelic
acid-induced liver injury model in C57 mice | Ferritinophagy↓;
ferroptosis↓ | MDA, iron content,
lipid ROS, ACSL4, ALOX15, NCOA4, LC3-NCOA4 colocalization,
FTH1-Lamp1 colocalization, p-STAT3, IL-6↓; GPX4, GSH, xCT, SLC3A2,
FTH1↑; mitochondrial shrinkage
and cristae loss | (86) |
| Wang et al,
2022 | | YAP1 | LPS-treated HL7702
cells | CLP-induced liver
injury model in C57BL/6 mice | Ferritinophagy↓;
ferroptosis↓ | FTH1, xCT, GSH,
GPX4↑; NCOA4, LC3II, ROS,
lipid ROS, ferrous ions, MDA, ACSL4, SFXN1, LC3-ferritin co
localization, ferritin-Lamp1 colocalization, NCOA4-FTH1
interaction, damaged mitochondria↓ | (87) |
| Jia et al,
2022 | | SARS-CoV-2 | | SARS-CoV-2
patients' liver |
Ferritinophagy↑; ferroptosis↑ | NCOA4, LC3II, serum
ferritin, iron content, ROS, lipid peroxidation↑; FTH1↓ | (88) |
| Liu et al,
2023 | | Sulforaphane |
H2O2 treated HL7702
or BRL cells | CCL4-induced liver
injury model in SD rats | Autophagy↑; ferroptosis↓ | LC3II, autophagic
flux, Nrf2, xCT, GSH, GPX4, Beclin1-xCT interaction↑; p62, ferrous ion, ROS, lipid ROS,
damaged mitochondria↓ | (89) |
| Savic et al,
2024 | Diabetic liver
damage | Sulforaphane | |
Streptozotocin-induced diabetes model in
C57BL/6 mice | Ferritinophagy↓;
ferroptosis↓ | GSH, GPX4, p-Nrf2,
p-ACC, SOD, MnSOD, CAT, TrxR↑;
FTH1, FPN1, FTH1-LC3 colocalization, iron content, 4-HNE,
lipofuscin↓ | (90) |
| Wang et al,
2024 | Liver cell
senescence | DMC | Etoposide treated
HL77O2 cells | C57BL/6J mice |
Ferritinophagy↑; ferroptosis↑ | NCOA4, LC3II, FECH,
iron content, MDA, LPCAT3, POR, ALOX5↑; p62, ferritin↓; mitochondrial
shrinkage and cristae loss | (91) |
Crosstalk between autophagy and
ferroptosis in drug-induced liver injury
Drug-induced liver injury is one of the most
challenging liver diseases in clinical practice. It considerably
interrupts the drug therapy and increases treatment difficulty
(92). Increasing evidence
substantiates that the crosstalk between autophagy and ferroptosis
participates in the pathogenesis of drug-induced liver injury.
Rifampicin, a common chemotherapy agent for tuberculosis, not only
promotes ferroptosis, manifested by increasing lipid peroxidation
and intracellular iron content, but also markedly increases the
expressions of LC3II and NCOA4 while decreasing the expressions of
p62 and ferritin, thereby inducing NCOA4-mediated ferritinophagy
(40). NCOA4 is a specific
ferritinophagy receptor that recognize ferritin and binds to its
hub subunit FTH1 and interacts with LC3, transporting ferritin to
the autophagosome for lysosomal degradation and iron release
(93). In cell or mouse models,
blocking ferritinophagy via knockdown of NCOA4 or
Atg5, or with the treatment of 3-methyladenine (3-MA),
reduces ferritin degradation and iron overload, partially
alleviating ferroptosis and ultimately mitigating
rifampicin-induced cytotoxicity, liver steatosis and tissue injury.
Hsc70 is supposed to manipulate ferritinophagy by modulating the
ferritin degradation and ferroptosis sensitivity of liver cells.
Its expression decreases with the prolonged rifampicin treatment.
Hsc70 inducer treatment substantially inhibits NCOA4-mediated
ferritinophagy and ferroptosis, thereby alleviating the liver
injury caused by rifampicin (40).
Liver injury is a common adverse reaction to
methotrexate, a drug broadly employed for the treatment of
rheumatoid arthritis and various tumors treatment (94). Research has highlighted the
significant role of ferritinophagy-dependent ferroptosis in
methotrexate-induced hepatotoxicity. In liver cells, methotrexate
treatment upregulates LC3II and NCOA4, reduces p62 and FTH1 and
triggers ferroptosis. Additionally, NCOA4 knockdown clearly
inhibits FTH1 degradation, suppresses ferroptosis, and mitigates
methotrexate-induced cell death, indicating that NCOA4-mediated
ferritinophagy induced by methotrexate facilitates ferroptosis.
Mechanistically, the overexpression and cytoplasmic translocation
of high-mobility group box 1 (HMGB1), a damage-associated molecular
pattern molecule (95), is
responsible for the regulation of methotrexate-induced
ferritinophagy and ferroptosis. Depletion or pharmacological
inhibition of HMGB1 substantially alleviates methotrexate-induced
hepatotoxicity by diminishing ferritinophagy-mediated ferroptosis
(41).
Ferritinophagy-dependent ferroptosis also
contributes to the liver injury caused by toosendanin, which is a
natural compound extracted from traditional Chinese medicine,
Melia toosendan Sieb. et Zucc. with multiple bioactivities
(96). Toosendanin treatment
reduces liver cell viability in a concentration-dependent manner;
causes mouse liver injury; increases ROS, lipid peroxidation and
iron contents; and decreases GSH level and GPX4 expression, all of
which are consistent with the process of ferroptosis. Moreover, the
activation of protein kinase R-like endoplasmic reticulum kinase
(PERK)-eukaryotic initiation factor 2 α subunit (eIF2a)-activation
transcription factor 4 (ATF4) signaling pathway causes cellular
iron overload and enhanced sensitivity to ferroptosis by
upregulating the ATF3-mediated expression of NCOA4 and TFR1, which
are related to the impaired iron storage caused by ferritinophagy
induction and the increased iron uptake caused by the promotion of
iron importation, respectively (42).
Triptolide is a natural compound isolated from
Tripterygium wilfordii Hook. F. with severe liver injury
(97). Previous reports have
demonstrated triptolide-induced liver cells ferroptosis as a
biological process highly dependent on lipophagy. Triptolide
administration markedly elevates Rab7 and LC3II expressions,
reduces p62 expression and augments the colocalization of LC3 and
Rab7 proteins in human normal liver cells and mice livers,
supporting the activation of Rab7-mediated lipophagy. Triptolide
also increases the levels of malondialdehyde, iron and
prostaglandin endoperoxide synthase 2, depletes GSH and GPX4, and
causes significant mitochondria damage. Genetic or pharmacological
depletion of lipophagy reverses ferroptosis and attenuates liver
cell damage (43). Given the
lipolytic effect of lipophagy on lipid droplets (98), the activation of Rab7-mediated
lipophagy is hypothesized to promote the release of free fatty
acids, which subsequently impair mitochondrial function and amplify
oxidative stress. This cascade ultimately drives lipid
peroxidation, ferroptosis and liver injury development.
Acetaminophen (paracetamol) overdose is responsible
for the greatest proportion of drug-induced liver injury in Western
countries (99). The reactive
metabolite of acetaminophen induces GSH depletion and covalently
binds to mitochondrial proteins, initiating mitochondrial damage
and ROS overproduction and resulting in impaired antioxidant
capacity and lipid peroxidation (100). ROS generation from damage
mitochondria also trigger autophagy induction in mouse livers and
in primary cultured liver cells following acetaminophen treatment
(101). ROS-induced autophagy
is a critical factor that controls intracellular iron concentration
through ferritin degradation and TFR1 expression during ferroptosis
(38). In acetaminophen-induced
liver injury, excessive lipid peroxidation, GSH depletion, GPX4
suppression, increased LC3II expression, p62 degradation and
autophagosomes accumulation were observed in hepatic cells, which
are in accordance with the typical features of ferroptosis and
autophagy, respectively. Ferroptosis is responsible for
acetaminophen-induced liver injury, and ferroptosis inhibitor could
eliminate these ferroptosis characteristics and alleviate acute
hepatotoxicity. These studies collectively demonstrate the
involvement of autophagy and ferroptosis in acetaminophen-induced
liver injury and raise the possibility of a positive feedback loop
between autophagy and ferroptosis in liver cells in response to
acetaminophen (101,102). However, this relationship
between autophagy and ferroptosis during acetaminophen-induced
liver injury remains controversial. It has been reported that
acetaminophen injection led to a significant release of
intracellular iron and subsequent ROS accumulation by activating
ferritinophagy on the one hand and inhibiting the endogenous
nuclear factor E2-related factor 2 (Nrf2)/heme oxygenase-1 (HO-1)
antioxidant pathway on the other hand, which contributes to clear
intracellular ROS. The combined effect of both factors culminates
in liver cell ferroptosis and finally induces significant liver
injury (44). In contrast to a
cell death mechanism favoring ferroptosis, autophagy also plays a
critical protective role against acetaminophen-induced hepatic cell
death. Instead of exacerbating acetaminophen-induced liver injury,
the pharmacological induction of autophagy exhibits a protective
effect. The removal of damaged mitochondria by mitophagy and the
consequent reduction in ROS production could mediate this
protection (101). Another
study demonstrated that acetaminophen administration decreased the
expression levels of molecules related to the Nrf2/HO-1 antioxidant
pathway and increased the ROS content, resulting in impaired
antioxidant capacity and oxidative stress in mouse liver.
Simultaneously, weakened autophagy activity and enhanced
ferroptosis were observed (45).
Furthermore, it has been confirmed that the forehead box
transcription factor class O 1 (Foxo1) is a promoter for both the
suppression of autophagy and the induction of ferroptosis triggered
by acetaminophen. Notably, mice subjected to acetaminophen
treatment exhibit elevated levels of Foxo1. However,
hepatocyte-specific deletion of Foxo1 ameliorates liver injury by
stimulating autophagy and inhibiting ferroptosis (46).
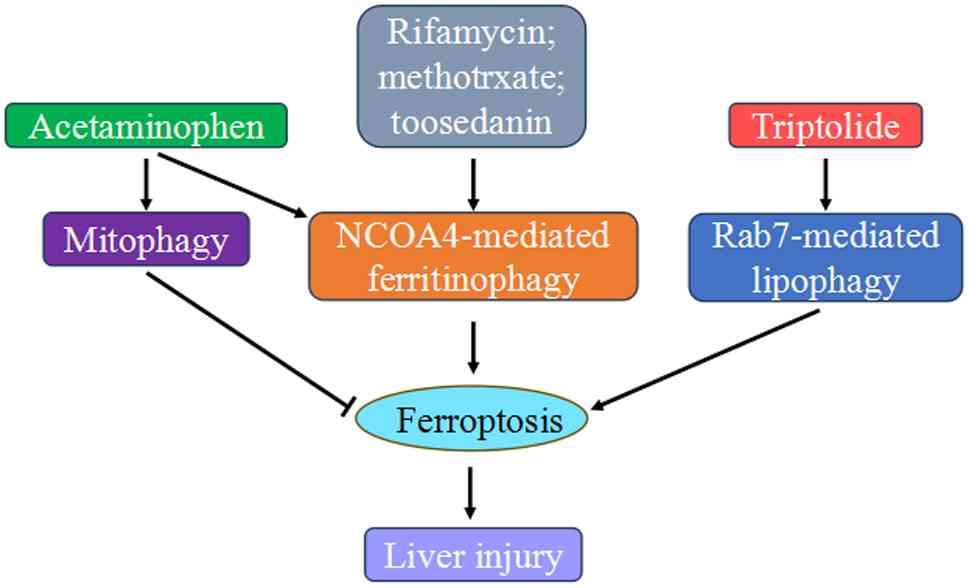
Taken together, drugs such as rifampicin,
methotrexate, toosendanin, and triptolide induce liver damage
through ferritinophagy- or lipophagy-dependent ferroptosis.
Acetaminophen overdose also involves both autophagy and
ferroptosis, but their relationship is a subject of debate, with
autophagy exerting both protective and detrimental effects
(Fig. 5).
Crosstalk between autophagy and
ferroptosis in toxin-induced liver injury
The liver is the main organ in the body for the
accumulation and detoxification of a number of toxins, making it
highly susceptible to their adverse effects. The crosstalk between
autophagy and ferroptosis is implicated in the molecular mechanisms
of liver injury induced by various exogenous toxins. Acrylamide is
a heat-induced toxic agent that widely exists in the environment,
and liver injury is a prevalent reported toxic effect of this
chemical (103). An
RNA-sequencing and bioinformatics analysis for evaluating and
comparing the overall gene expression pattern of liver cells showed
that exposure to acrylamide markedly activated oxidative stress
signaling pathways and upregulated autophagy and ferroptosis
pathways (47). Further
experiments confirmed that acrylamide decreased mitochondrial
membrane potential (MMP) and increased ROS production and Nrf2
levels, leading to GSH deactivation and GPX4 degradation and
inevitably inducing oxidative stress and ferroptosis (47,104). Acrylamide also induced
autophagy activation as indicated by LC3II elevation and p62
reduction (105). Moreover, ROS
scavenger and autophagy inhibition consistently and markedly
suppressed acrylamide-induced autophagy and ferroptosis. These
results suggest that acrylamide-induced ferroptosis is dependent on
oxidative stress-driven autophagy. Acrylamide treatment also
increases the NCOA4 level and decreases the FTH1 level, indicating
the activation of ferritinophagy. Notably, quercetin, a natural
antioxidant flavonoid, possesses the ability to counter
acrylamide-induced liver injury by targeting autophagy-dependent
ferroptosis. Quercetin effectively reduces ROS accumulation to
combat acrylamide-induced oxidative stress and autophagy and
eventually inhibits ferroptosis. Additionally, quercetin
specifically binds to NCOA4 protein to disrupt the NCOA4A-FTH1
interaction, impairs ferritinophagy, and consequently blocks
ferroptosis by preventing FTH1 degradation and reducing the amount
of bioavailable intracellular iron in acrylamide-exposed liver
cells (47).
Cadmium, nickel and copper are common harmful heavy
metals released by industrial manufacturing or naturally present in
the environment. They greatly endanger liver health. Excessive
intake leads to abnormal accumulation and homeostasis imbalance of
these metals in the liver, damaging its histomorphology and
interfering with its normal physiological functions (106). Autophagy and ferroptosis are
pivotal underlying mechanisms involved in the liver injury induced
by these metals and the crosstalk between these processes occur
during the pathogenesis of this condition. In chicken liver, copper
exposure inactivates the Nrf2/Kelch-like ECH-associated protein 1
(KEAP1) signaling pathway, reduces the expression of its downstream
gene targets such as xCT and GPX4, disrupts the
endogenous antioxidant GSH metabolism, promotes lipid peroxidation
and induces the typical morphological characteristics in
mitochondria associated with ferroptosis. Copper also markedly
augments the mRNA and protein expression levels of NCOA4.
Therefore, NCOA4-mediated ferritinophagy plays a critical role in
promoting copper induced ferroptosis in the liver (48). Similar patterns occur with nickel
or cadmium exposure, which enhances ferritin degradation through
NCOA4-mediated ferritinophagy and activates ferroptosis as
indicated by the occurrence of iron accumulation, lipid
peroxidation and impairment of the antioxidant system. The use of
ferroptosis inhibitors or autophagy inhibitors markedly promotes
ferroptosis resistance and reduces liver damage. These results
demonstrate that ferritinophagy as a trigger of ferroptosis is
closely involved in the cell death caused by nickel or cadmium
exposure (49,50). Mechanistically, mitochondria
damage, manifesting as mitochondrial ROS increase, MMP
depolarization and interference with mitochondrial respiratory
chain, is a significant factor for autophagy and ferroptosis
induction under nickel exposure (50). Regarding the toxic effect of
cadmium on the liver, the ER stress response is also activated
through the PERK-eIF2α-ATF4-C/EBP homologous protein signaling
pathway. ER stress activation not only initiates autophagy but also
exacerbates ferroptosis, indicating that ER stress participates in
the synergistic effect of ferritinophagy and ferroptosis (49).
Arsenic and fluoride are widely known as hazardous
nonmetallic pollutants. Their contamination poses severe threats to
livestock and humans and the liver is an important target organ of
their toxicology (107).
Chicken liver injury induced by long-term arsenic exposure is
inseparable from ferritinophagy-mediated ferroptosis, and the
mitochondria might play a crucial role in the process. Arsenic
exposure induced mitochondrial dysfunction and led to enhanced ROS
production, which triggered ferritinophagy via the AMPK/mTOR/ULK1
signaling pathway and markedly altered the expression levels of
ferroptosis-related proteins in chicken livers (51). Mitochondrial damage, increased
mitochondrial ROS generation and free iron-mediated ferroptosis are
typically observed in cell and animal models of fluoride-induced
liver injury (108). Increased
LC3II expression and p62 accumulation are also noted, indicating
the fusion of autophagosomes and lysosomes is inhibited and
autophagic degradation is hindered (52). Such outcomes might be attributed
to the excess iron, which exerts an inhibitory effect on the
autophagic process (109). The
autophagy activator rapamycin not only ameliorates fluoride-induced
autophagic flux blockage by correcting autophagic degradation
impairment but also inhibits fluoride-mediated ferroptosis by
reducing the iron content and suppressing lipid peroxidation.
Additionally, the ferroptosis inhibitor Fer-1 simultaneously
ameliorates ferroptosis and alleviates the impaired autophagic
degradation at the same time (52). Thus, a bidirectional regulation
exists between autophagy and ferroptosis in fluoride-induced liver
injury.
Dysregulated ferroptosis has been implicated in
alcohol liver injury, as evidenced by iron disorder and the
increased intracellular lipid peroxidation (53,110). The ferroptosis-promoting effect
of ethanol and its highly active metabolite acetaldehyde is highly
dependent on autophagy. On the one hand, ethanol or acetaldehyde
promotes ferritin degradation and free iron release via
NCOA4-mediated ferritinophagy, contributing to ferroptosis. On the
other hand, PTEN-induced putative kinase 1 (PINK1)/Parkin-mediated
mitophagy is arrested, leading to mitochondrial damage without
clearance in time and elevated ROS and further stimulating
ferroptosis. Ferroptosis inhibitor Fer-1 reverses elevated
ferritinophagy, implying that ferroptosis induces autophagy
activation in a feedback manner (53). Silibinin is the major active
constituent of milk thistle seed extract and has multiple targets.
It simultaneously reverses NCOA4-mediated ferritinophagy, restores
PINK1/Parkin-mediated mitophagy, modulates iron metabolism and
limits lipid peroxidation, thereby exhibiting hepatoprotective
properties in ethanol- or acetaldehyde-induced liver injury model
(53).
Aflatoxin B1 and deoxynivalenol, two extensively
prevalent mycotoxins, poses a considerable public health threat
owing to its high propensity to contaminate agricultural products
and induce liver damage upon consumption. One manner in which both
of them inflicts liver damage is through ferritinophagy-mediated
ferroptosis (54,55). At the molecular level, aflatoxin
B1 disturbs iron balance by affecting ferritinophagy, a process
that is reliant on the PI3K/AKT/mTOR/ULK1 pathway. The disruption
leads to iron accumulation, which in turn intensifies lipid
peroxidation and causes ferroptosis (54). The ability of deoxynivalenol to
regulate ferritinophagy is achieved partly by accelerating the
ubiquitination-mediated degradation of programmed cell death
protein 4, thereby upregulating NCOA4 expression through the c-Jun
N-terminal kinase-c-Jun axis. Moreover, deoxynivalenol has the
capacity to induce excessive mitophagy, and the administration of a
mitophagy activator further exacerbated the hepatic cell
ferroptosis-promoting effect of deoxynivalenol, suggesting that
mitophagy actually promotes ferroptosis, rather than exerting an
inhibitory effect (55).
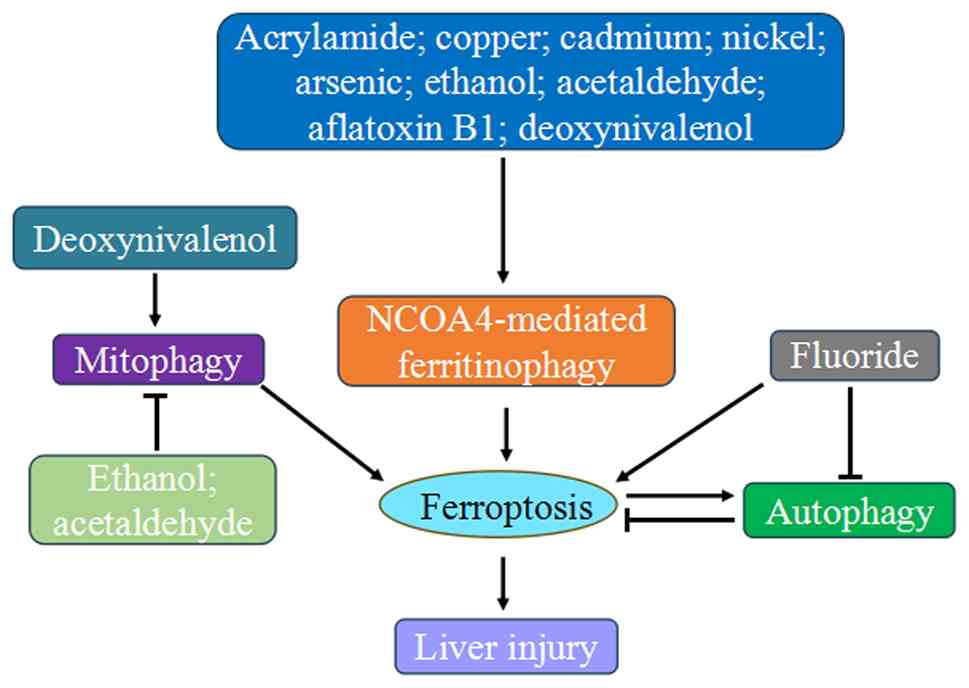
Overall, these findings provide substantial evidence
of a crosstalk between autophagy and ferroptosis in the liver
injury caused by various toxins, including acrylamide, heavy metal,
nonmetallic pollutants, alcohol, and mycotoxins. Mainly through
activating ferritinophagy, they disrupt iron homeostasis, leading
oxidative stress and lipid peroxidation, which trigger ferroptosis
(Fig. 6). Notably, substances
like quercetin and silibinin show promise in mitigating liver
damage by targeting these pathways.
Crosstalk between autophagy and
ferroptosis in liver fibrosis
Liver fibrosis is a reversible pathophysiological
condition mainly characterized by the death of normal liver cells
and the excess deposition of extracellular matrix. Activated
hepatic stellate cells (HSCs) are the most important producers of
extracellular matrix (111).
The crosstalk between autophagy and ferroptosis happens during the
induction or prevention of liver fibrosis. Silica nanoparticles
(SiNPs) are composed of silicon dioxide and have a number of
applications, including in biomedicine, food and chemical and
textile industries. Animal experiments on respiratory exposure to
SiNPs reveal that SiNPs accumulate primarily in the liver and
induce liver toxicity, manifested as ferritinophagy-mediated
ferroptosis and fibrosis (56,112). SiNPs administration causes
mitochondrial vacuolation and mitochondrial membrane rupture,
increases lipid peroxidation and iron accumulation and decreases
GPX4 levels, suggesting that SiNPs lead to ferroptosis in liver
cells. SiNPs exposure also evokes ferritinophagy, indicated by
NCOA4 and LC3II upregulation and FTH1 downregulation. NCOA4
knockdown can alleviate SiNP-triggered ferroptosis in liver cells,
indicating that NCOA4-mediated ferrinophagy is responsible for
ferroptosis (56). Mitophagy is
also reported to promote liver fibrosis through a
ferroptosis-dependent manner. FUN14 domain-containing protein 1
(FUNDC1), a mitophagy receptor highly expressed in the liver
tissues of patients suffering from liver fibrotic injury as well as
carbon tetrachloride-challenged mice, has been identified as a
culprit in eliciting liver cell ferroptosis. Through directly
interacting with GPX4, FUNDC1 facilitates the recruitment of GPX4
into mitochondria, where it undergoes degradation by mitophagy,
ultimately triggering ferroptosis (57). Parenchymal cell death further
activates HSCs, resulting in the upregulated expression of liver
fibrosis indicators, especially a-smooth muscle actin and collagen
I and II (56).
Targeted scavenging of activated HSCs and the
consequent blocking of the fibrogenic effect at the source have
been proposed as an effective therapeutic approach to reverse liver
fibrosis. Ferroptosis induction has been proven to be a new control
measure for HSCs activation. RNA binding proteins ZFP36 ring finger
protein (ZFP36) and ELAV like RNA binding protein 1(ELAVL1) plays a
pivotal role in triggering HSCs ferroptosis to alleviate liver
fibrosis caused by ferroptosis inducers such as sorafenib, erastin,
and RSL3. The activation of ferritinophagy is necessary for both
proteins to regulate ferroptosis in HSCs (23,58). Exposure to ferroptosis inducers
apparently increases the expression level of E3 ubiquitin ligase
F-box and WD repeat domain containing 7 (FBXW7) in HSCs. As shown
by immunocoprecipitation assay and ubiquitination assay, FBXW7
directly binds to ZFP36 and decreases ZFP36 protein expression by
ubiquitination-mediated proteasomal degradation. ZFP36
downregulation promotes ferritinophagy activation by stabilizing
Atg16L1 mRNA, which mediates ferroptosis by degrading FTH1
in a NCOA4-dependent manner (23). Contrary to the decrease in ZFP36,
ELAVL1 expression increases markedly through the inhibition of the
ubiquitin-proteasome pathway following ferroptosis inducers
treatment. ELAVL1 abrogates Beclin1 mRNA decay by binding to
the AU-rich elements within the 3'-untranslated region (UTR), and
in turn contributes to ferritinophagy activation and ferroptosis
induction in HSCs (58).
Additionally, the autophagy signaling pathway is involved in
N6-methyladenosine (m6A) modification-induced HSC
ferroptosis. RNA sequencing shows that ferroptosis inducers
markedly increases m6A modification in Beclin1
mRNA by upregulating methylase METTL4 and downregulating
demethylase FTO. m6A-binding protein YTHDF1 promotes
Beclin1 production by recognizing m6A binding
sites, thereby triggering ferritinophagy, and eventually leading to
ferroptosis (59). Moreover,
Beclin1 protein is enriched in mesenchymal stem cells-derived
exosomes (MSC-ex), which are the membrane vesicles encapsulating
MSCs-derived proteins, lipids, mRNAs and noncoding RNA (113). MSC-ex specifically targets HSCs
activation and promotes HSCs ferroptosis by delivering Beclin1
proteins into HSCs, which improves autophagy marker LC3II
expression and promotes iron release. Possibly MSC-ex induce
ferroptosis through exosomal Beclin1-induced ferritinophagy, and
Beclin1 can be a potential biofactor for alleviating liver fibrosis
(60).
Natural active products from traditional Chinese
medicines, such as artesunate, berberine, curcumol,
dihydroartemisinin and artemether, have made marked advances in the
prevention and treatment of liver fibrosis by inducing ferroptosis.
Studies have highlighted the importance of autophagy as an emerging
mechanism of these products in enhancing ferroptosis and proposed a
potential novel therapeutic strategy for liver fibrosis.
Artesunate, berberine, curcumol, dihydroartemisinin or artemether
treatment clearly promote ferroptosis to eliminate activated HSCs,
reduce the deposition of extracellular matrix, alleviate mouse
liver fibrosis and restore mouse liver function. In terms of
mechanism, artesunate, curcumol, dihydroartemisinin and artemether
mediate FTH1 degradation by increasing NCOA4, leading to
ferritinophagy activation, iron accumulation, lipid peroxidation
elevation, antioxidant capacity loss and ferroptosis occurrence. By
contrast, the interdiction of ferritinophagy completely abolishes
the induced ferroptosis and diminishes the efficacy of these
natural products against liver fibrosis (61-64). Regarding the anti-fibrosis effect
of berberine, the autophagy impairment caused by berberine is
incapable of eliminating the increased ROS production and
contributes to oxidative stress via a feed-forward loop, which
accelerates the ubiquitin-mediated degradation of ferritin and the
release of iron. Iron accumulation further amplifies ROS generation
through the Fenton reaction, and triggers ferroptosis through lipid
peroxidation and GSH depletion in activated HSCs, thereby providing
a brake on the fibrogenic response (65).
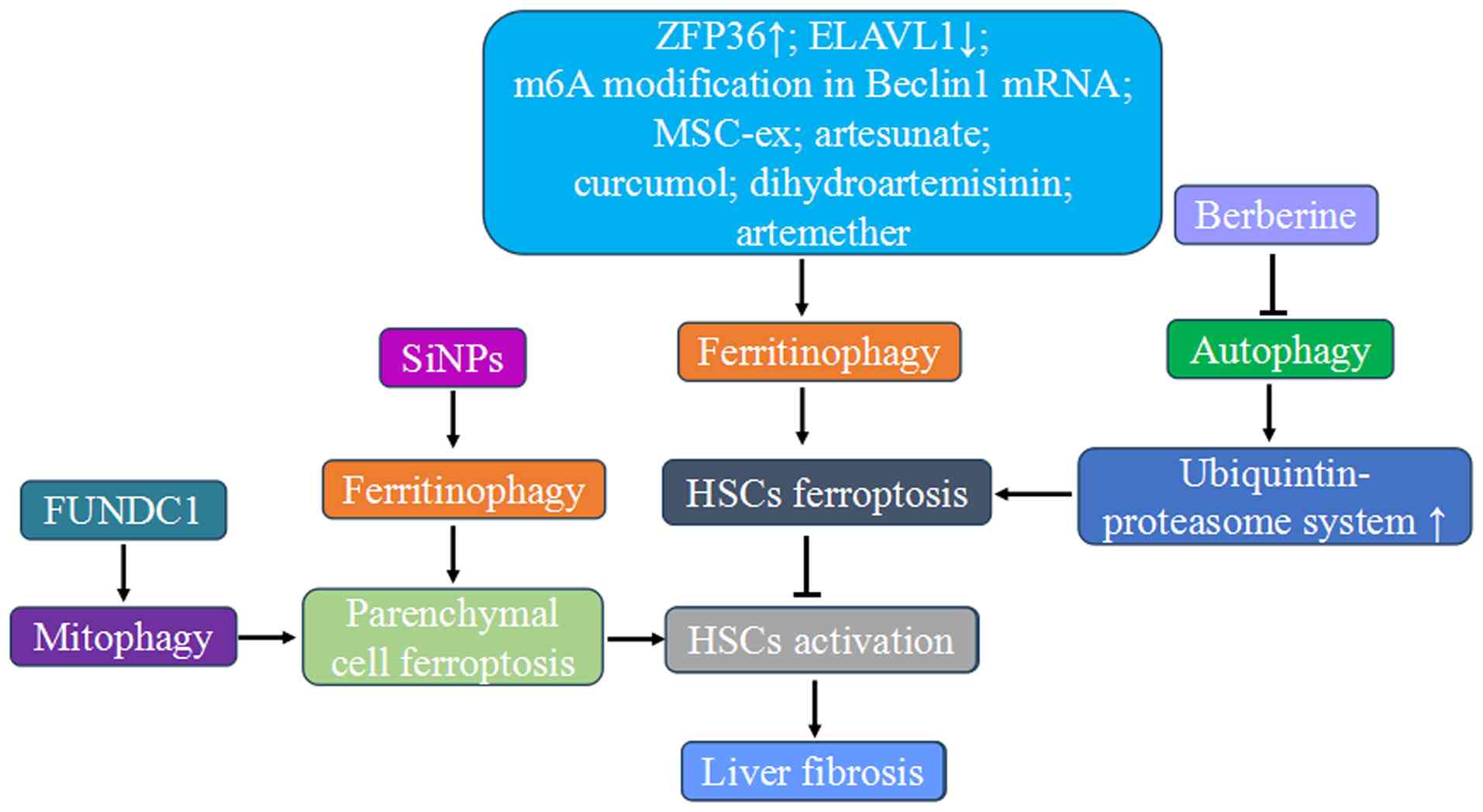
Thus, liver fibrosis involves liver parenchymal cell
ferroptosis and HSCs activation. Ferritinophagy and mitophagy can
promote this process. Targeting activated HSCs through ferroptosis
induction offers a therapeutic approach. Natural products from
traditional Chinese medicine have potential in preventing liver
fibrosis by promoting ferroptosis, with autophagy playing a key
role (Fig. 7).
Crosstalk between autophagy and
ferroptosis in HCC
HCC is among the most widely occurring tumors with
high incidence and mortality rates (66). Differentially expressed
ferritinophagy-related genes are markedly associated with patient
prognosis: individuals with high FTH1 and FTL expression levels
exhibit considerably poorer survival rates than individuals with
low expression levels (114).
Hence, ferritinophagy possibly regulates HCC by influencing
ferroptosis. Circular RNAs (circRNAs) and RNA-binding proteins
participates in the development and progression of HCC by
regulating autophagy-mediated ferroptosis. CircRNAs, a group of
endogenous non-coding RNAs with covalently closed-loop structures,
originate from the back-splicing events of primary mRNAs. By
binding with their corresponding microRNAs or proteins, circRNAs
participate in the multifaceted biological regulation of cellular
metabolism (115). A study has
found that circIARS, a novel circRNA derived from the IARS
gene, was the most highly expressed circRNA after ferroptosis
inducer sorafenib treatment in HCC cells (66). circIARS increased the
vulnerability to sorafenib and this effect partially depended on
repressing the biological role of the RNA binding protein AlkB
Homolog 5 (ALKBH5), a negative regulator of ferritinophagy in HCC
cells. Mechanistic identification revealed that circIARS physically
interacted with ALKBH5 and promoted phagophore nucleation and
ferritinophagy induction by suppressing the dissociation of
Bcl2-Beclin1 complex, resulting in the ferroptotic events by
increasing iron-binding ferritin turnover and iron release
(66). Another study
demonstrated the regulatory effect of the circRNA of the family
with sequence similarity 134, member B (circFAM134B) for
ER-phagy-mediated ferroptosis in HCC cells treated with
multi-targeted tyrosine kinase inhibitor lenvatinib. FAM134B is a
receptor protein for selectively recognizing and degrading ER
fragments and the expressions of both circFAM134B and FAM134B in
HCC cells were effectively induced by lenvatinib. circFAM134B acted
as a sponge that competitively bound to poly (A) binding protein
cytoplasmic 4, thereby destabilizing FAM134B mRNA by
facilitating nonsense-mediated mRNA decay and suppressing the
process of FAM134B-mediated ER-phagy. Moreover, circFAM134B is a
positive regulator of ferroptosis, and loss of circFAM134B markedly
reversed the ferroptosis phenotype in HCC cells. Concurrently, the
levels of FAM134B and its colocalization with LC3 markedly
increased after si-circFAM134B transfection. These results suggest
that targeting circFAM134B could markedly affect FAM134B-mediated
ER-phagy, thereby improving the ferroptosis sensitivity of HCC
cells to lenvatinib (67).
FAM134B-mediated ER-phagy also plays an important role in the
execution of sorafenib-induced ferroptosis in HCC cells. Sorafenib
effectively induced the direct interaction between PABPC1 and
FAM134B mRNA, promoting the translational activation of
FAM134B and inducing ER-phagy. FAM134B knockdown not only
blocked ER-phagy, but also improved the ferroptosis sensitivity of
HCC cells (68). Distinct from
other selective autophagy pathways that often promote ferroptosis
by degrading the cellular components critical for redox
homeostasis, ER-phagy inhibits ferroptosis by maintaining ER
integrity and modulating stress signaling (116). Moreover, upregulated
polypyrimidine tract-binding protein 1 (PTBP1), an RNA-binding
proteins, was observed in sorafenib-treated HCC cells. PTBP1
physically interacted with the 5'UTR of the NCOA4 mRNA
sequence and promoted the activation of ferritinophagy by
regulating NCOA4 translation, causing the enhanced
degradation of ferritin, accelerated accumulation of intracellular
iron and impaired ferroptosis resistance of HCC cells (69). Partner of NOB1 (PNO1) is another
RNA-binding protein that inhibits autophagy-mediated ferroptosis by
GSH metabolic reprogramming in HCC cells. By promoting autophagy
via the mitogen-activated protein kinases signaling pathway, PNO1
mainly affects the levels of intracellular glutamate, which
activates system Xc- to import additional cysteine. This highly
activated GSH biosynthesis inhibits lipid ROS generation and
finally counteracts ferroptosis. Thus, PNO1 is a bona fide
ferroptosis inhibitor. The combination of PNO1 inhibition with
ferroptosis-inducing drugs markedly strengthens ferroptosis
sensitivity (70,71).
Mitochondria damage has become a major factor in
oxidative stress and tumorigenesis. Mitophagy prevents the
accumulation of dysfunctional mitochondria to ensure the quantity
and quality of the mitochondrial population and maintain stable ROS
levels, which lead to oxidative stress (117). Mitophagy and ferroptosis are
associated with HCC prognosis. A mitophagy-related signature based
on a consensus clustering analysis has identified several
mitophagy-related genes closely related to the ferroptosis status
and progression of HCC. This signature also exhibits a predictive
effect on the prognosis of patients with HCC (118). Moreover, owing to its critical
role in maintaining cellular function, targeting mitochondria can
be a promising new strategy for HCC treatment. For example,
mitochondrial translocator protein (TSPO), a conserved
transmembrane protein primarily localized at the outer
mitochondrial membrane, is highly expressed in HCC and is markedly
associated with poor tumor differentiation, advanced stage, and
poor prognosis. Thus, TSPO serves as a putative oncogene and
represents a viable candidate for mitochondrial-directed
therapeutic strategies in HCC treatment (119). Gain- and loss-of-function
experiments present that TSPO upregulation inhibits ferroptosis in
HCC cells through the Nrf2-mediated upregulation of antioxidant
gene expression, thereby promoting HCC development. Additionally,
autophagy inhibition is involved in the underlying mechanisms of
TSPO action in HCC. TSPO directly interacts with the autophagy
receptor p62, thus interfering with the autophagy degradation of
p62. The excessively accumulated p62 competes with Nrf2 for binding
to the E3 ubiquitin ligase KEAP1 and disrupts the KEAP1-Nrf2
association, thus preventing Nrf2 from proteasomal degradation and
leading to Nrf2 stabilization. Ultimately, ferroptosis is inhibited
through the Nrf2-mediated activation of antioxidant gene
transcription (72).
Some metabolic enzymes controlling ferroptosis
sensitivity in HCC cells involve the autophagy pathway.
Branched-chain amino acid aminotransferase 2 (BCAT2) is a key
aminotransferase enzyme acting upon sulfur amino acids for the
synthesis of glutamate and positively regulates the function of
system Xc− (120).
It serves as a specific ferroptosis inhibitor in HCC because the
BCAT2-induced increase in intracellular glutamate levels boosts
system Xc− activity, enhances cystine uptake and
increases GSH levels to protect HCC cells against ferroptosis
(73). The ferritinophagy
induction mediated by ferroptosis inducers (erastin, sorafenib and
sulfasalazine) leads to rapid ROS accumulation because of the
increased cellular iron levels. It also induces AMPK
phosphorylation on threonine residue 172, which inhibits the
nuclear translocation of sterol response element binding protein 1
and consequently suppresses the transcription of its direct target
gene, BCAT2 and enhances the ferroptosis susceptibility of HCC
cells (73). Similar to BCAT2,
creatine kinase B (CKB) is a novel ferroptosis suppressor regulated
by CMA. This metabolic enzyme catalyzes the reversible transfer of
a phosphoryl group from ATP to creatine and is pivotal in
maintaining cellular energy balance (121). The activation of insulin-like
growth factor 1 receptor signaling, which frequently occurs in HCC,
enhances the interaction between AKT and CKB. Subsequently,
AKT-mediated CKB Thr133 phosphorylation reduces its binding to
creatine, thereby gained the ability to interact with GPX4 and
phosphorylated GPX4 at Ser104. This phosphorylation is adjacent to
the CMA target motif in GPX4 and counteracts CMA-mediated GPX4
degradation, which is dependent on its binding to Hsc70, therefore
greatly alleviating ferroptosis in HCC cells and exacerbating tumor
growth (74).
In HCC, the crosstalk between autophagy and
ferroptosis involves the coordination of the post-transcriptional
and post-translational modulation. For example, m6A
modification in HCC cells was clearly stimulated by ferroptosis
inducers and the elevated level of m6A writer, WT1 associated
protein (WTAP), served as the primary cause of this observed
increase. Atg5 mRNA is recognized as a downstream target of
WTAP-driven m6A modification, which is then recognized and bound by
YTH domain-containing protein 2. This process increases Atg5
translation and expression, subsequently initiating ferritinophagy
and resulting in ferroptosis activation in HCC (75). Moreover, other investigations
have emphasized that the importance of ubiquitination regulation as
a critical mechanism governing autophagy-dependent ferroptosis in
HCC. Ubiquitin-specific protease 24 (USP24) downregulation and USP2
upregulation were detected in HCC tissues, showing significant
correlation with altered autophagy-ferroptosis axis (76,77). USP24 maintains Beclin1 stability
by inhibiting its polyubiquitination, which prevents its
proteasomal degradation. This facilitates ferritin degradation via
ferritinophagy and subsequently increase the sensitivity of HCC
cells to ferroptosis (76).
Conversely, USP2 functions as a negative regulator of
clockophagy-induced ferroptosis. Mechanistically, USP2 interacts
with brain and muscle ARNT-like protein 1 (ARNTL), which promotes
its deubiquitination and weakens its interaction with p62. This
process reduces the autophagic degradation of ARNTL and stabilizes
this protein. Consequently, stabilized ARNTL directly activates
transcription of hypoxia inducible factor 1α (HIF1α) and xCT,
thereby reducing the susceptibility of HCC cells to ferroptosis
(77).
The poor clinical efficacy, serious adverse
reactions and side effects have greatly limited the drug therapy
for treating HCC. Compounds found in plants open new avenues for
anti-HCC medication development because of their unique chemical
structures and potent efficacy. It has been revealed that these
compounds may kill HCC cells in vitro and in vivo
through triggering ferritinophagy-mediated ferroptosis (78,79). A number of plants, such as
Senecio salignus, Chenopodium ambrosioides L., and
Cannabis sativa L., contain caryophyllene oxide, a natural
bicyclic sesquiterpene that possesses significant anti-HCC
activities by promoting ferritinophagy and ferroptosis (122). Caryophyllene oxide markedly
increases the expressions of NCOA4 and LC3II and the release of
free iron by degrading ferritin, leading to a
ferritinophagy-related phenomenon. When the HCC cells accumulate a
substantial amount of ROS produced by the Fenton reaction, it
causes oxidative stress and lipid peroxidation and induces
ferroptosis and finally affects the growth and proliferation of HCC
cells (78). A similar relation
between the inhibitory effects of esculetin on HCC and
ferritinophagy-mediated ferroptosis was validated. As a coumarin
derivative extracted from lemon leaves, Rehmannia glutinosa
(Rehmannia glutinosa var.), belladonna (Belladonna
baccifera Lam.) and other plants (123), esculetin specifically targets
NCOA4, FTH1 and LC3II to activate ferritinophagy. As a consequence
of iron deposition, increased ROS generation results in oxidative
stress and lipid peroxidation. Meanwhile, esculetin decreases GSH
contents by suppressing the Nrf2 signal pathway and inhibiting the
expression of its target antioxidant proteins (GPX4 and HO-1). This
activity further promotes ROS accumulation, thus triggering
ferroptosis in HCC cells (79).
Another study has demonstrated that the electrophilic
sesquiterpenes isolated from Eupatorium chinense L.
(EChLESs) suppressed HCC growth by enhancing
ferritinophagy-mediated ferroptosis. Following exposure to EChLESs,
ferroptosis was induced in HCC cells characterized by the crumpled
and broken mitochondria, elevated mitochondrial reactive oxygen
species (mtROS) levels, inactivated GSH-dependent antioxidant
defense system, increased intracellular iron levels and the
excessive lipid peroxidation. EChLESs elevated the mRNA expression
levels of NCOA4 by activating HIF1α and weakened the
degradation of NCOA4 protein at transcriptional and
post-transcriptional levels in HCC cells. NCOA4 knockdown
partly abolished ferroptosis and reversed the cell viability
inhibition caused by EChLESs, indicating that NCOA4-mediated
ferrinophagy constitutes an indispensable mechanism underlying
EChLESs-elicited ferroptosis (80). Sorafenib, a potent ferroptosis
inducer, is an effective first-line therapeutic drug for advanced
HCC. Unfortunately, drug resistance markedly limits its efficacy
(124). Artesunate is an ideal
compound that could increase sorafenib sensitivity in HCC
treatment. The synergistic effect of these agents involves
enhancing ferroptosis by activating lysosome function and promoting
ferritin degradation. Sorafenib directly acts on oxidative stress
via the impairment of mitochondrial functions and the inhibition of
GSH contents, whereas artesunate mainly activates lysosomes, as
evidenced by the increased cathepsin L and cathepsin B levels and
subsequent ferritin degradation. The convergence of oxidative
stress triggered by sorafenib and ferritinophagy mediated by
artesunate gives rise to uncontrolled lipid peroxidation,
ultimately culminating in ferroptosis (81).
Therefore, these results demonstrate the crosstalk
between autophagy and ferroptosis markedly influences HCC
progression and prognosis. Various factors, including circRNAs,
RNA-binding proteins, metabolic enzymes and post-transcriptional
and post-translational modifications, regulate this crosstalk.
Natural plant compounds enhance ferritinophagy-mediated ferroptosis
for HCC treatment. Combining these compounds with existing drugs
like sorafenib may overcome resistance and improve therapeutic
efficacy (Fig. 8).
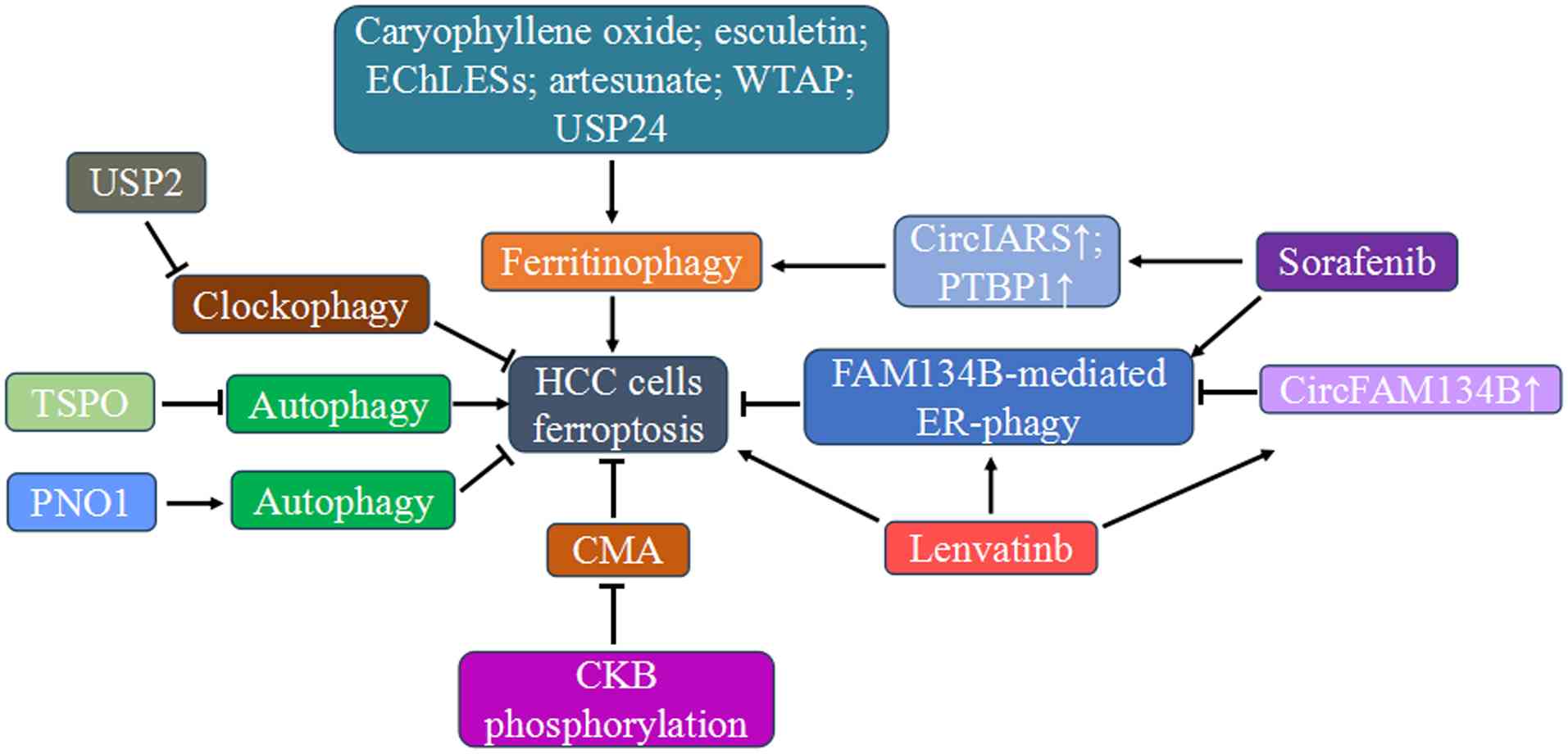 | Figure 8Crosstalk between autophagy and
ferroptosis in HCC. HCC, hepatocellular carcinoma; USP24,
ubiquitin-specific protease 24; WTAP, WT1 associated protein;
PTBP1, polypyrimidine tract-binding protein 1; EChLESs,
electrophilic sesquiterpenes isolated from Eupatorium
chinense L.; TSPO, mitochondrial translocator protein; PNO1,
partner of NOB1; CircFAM134B, circRNA of the family with sequence
similarity 134, member B; CMA, chaperone-mediated autophagy; CKB,
creatine kinase B. |
Crosstalk between autophagy and
ferroptosis in other liver diseases
MS is a pathological condition defined by a cluster
of metabolic disorders that increases the risk of cardiovascular
diseases and diabetes. Iron overload holds a crucial position in
the causation of liver damage in MS (125). In a liver injury rat model of
MS induced by a high-fat and high-fructose diet, the protein
expression levels of FTH1 and TFR1 were increased, whereas those of
NCOA4 and FPN1 and the NCOA4-FTH1 interaction were decreased in the
liver tissues compared with those in the control group. Moreover,
the ferroptosis-related proteins GPX4 and xCT were markedly
decreased, and oxidative stress was markedly induced in the liver
of MS rats. Therefore, decreased NCOA4 expression and impaired
binding of NCOA4 to FTH1 diminish the FTH1 degradation by
downregulating ferritinophagy, and decreased FPN1 expression
inhibits iron output and release to other tissues, both of which
effectively increase liver iron overload and, in turn, trigger
oxidative stress and ferroptosis (82). NAFLD is an abnormal liver
condition associated with MS. In high-fat diet-induced NAFLD murine
models, autophagosomes biogenesis was suppressed through both mTOR
activation and Atg7 downregulation, resulting in decreased
lipophagy that promoted lipid droplets accumulation and
lipotoxicity, such as lipid peroxidation, a key determinant of
ferroptosis (43). In addition
to enhanced lipid peroxidation, the suppression of autophagosome
formation, which alters the protein abundance or subcellular
distribution of p62, stabilizes KEAP1 by disturbing the balance
between the KEAP1-p62 and KEAP1-Nrf2 interaction and enhances
ferroptosis by counteracting the anti-ferroptotic functions of Nrf2
(83). NASH is a progressive
subtype of NAFLD, and iron overload is often observed in patients
with NASH (126). Iron overload
is followed by oxidative stress and the increased uptake of
intracellular calcium ions. The calcium ion-activated calcineurin
mediates the dephosphorylation of cytosol transcription factor EB
(TFEB) and promotes its subsequent migration into the nucleus. In
nuclear, TFEB upregulates the mRNA expression of autophagy- and
lysosome-related genes and simultaneously accelerates
NCOA4-mediated ferritinophagy and lipophagy. These iron
overload-mediated double effects on lipid and iron metabolic
dysregulation acts together to aggravate NASH via ferroptosis
(84).
ALI is an acute inflammatory liver disease induced
by various factors and accompanied by severe impairment of liver
function (127). Alternatively
activated macrophages (AAMs) exert a beneficial hepatoprotection in
ALI by producing anti-inflammatory factors, inhibiting the
necroptosis-necroinflammation axis and slowing down liver
inflammation progression. However, AAMs exhibit a high degree of
sensitivity towards ferroptosis, and the quantity of AAMs is below
the normal levels during ALI pathogenesis (128). Liensinine, a major
pharmacologically active ingredient isolated from lotus seeds,
possesses potential therapeutic effects against ALI. By inhibiting
ferroptosis while increasing the number of AAMs, liensinine
alleviated the lipopolysaccharide (LPS)/D-galactosamine-induced
pathological liver injury and the liver inflammatory response in a
mouse model. In vitro experiments revealed that liensinine
could alleviate ferritinophagy and inhibit ferrous iron release and
lipid peroxidation to boost AAMs resistance to ferroptosis,
potentially by blocking the autophagosome-lysosome fusion and
subsequently inhibiting the recruitment of ferritin into the
lysosomes for degradation (85).
Likewise, quercetin demonstrated a hepatoprotective effect against
LPS and γ-D-glutamyl-meso-di aminopimelic acid-induced ALI by
targeting the interplay between ferritinophagy and ferroptosis.
Specifically, quercetin suppressed interleukin-6/signal transducer
and activator of transcription 3-dependent ferritinophagy, thereby
attenuating liver cell ferroptosis and liver damage (86). Another study reported the
protective effect of Yes-associated protein 1 (YAP1) in
sepsis-induced ALI by regulating the process of
ferritinophagy-mediated ferroptosis (87). As a transcriptional coactivator
controlled by the Hippo signaling pathway (129), YAP1 deficiency markedly
exacerbated the oxidative damage, aggravated ferroptosis- and
ferritinophagy-associated morphological features and inflammatory
response in the septic liver tissue. Consistently, YAP1
overexpression restrained the ferrous iron synthesis by disrupting
the association between NCOA4 and ferritin and preventing
siderofexin 1-induced mitochondria iron accumulation, which
consequently suppressed mtROS generation and lipid peroxidation.
Eventually, the liver cells were protected from ferroptosis in this
sepsis-induced ALI model (87).
Ferritinophagy-mediated ferroptosis is also involved in the
pathophysiological mechanism behind the liver injury caused by
severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)
infection. Presumably, SARS-CoV-2 infection-induced cytokine
storms, which are characterized by the exuberant release of
inflammatory cytokines, stimulate liver cells to secrete ferritin
and encourage hyperferritinemia. Elevated ferritin levels can
initiate NCOA4-mediated ferritinophagy followed by the induction of
ferroptosis. With the death of hepatic cells, more ferritin is
further released. Therefore, the mutual stimulation of ferritin and
liver cells destruction establishes a vicious loop that
continuously aggravates liver injury (88). A further study demonstrated that
Nrf2-dependent autophagy activation is essential for preventing
ferroptosis in ALI under sulforaphane intervention. As a powerful
Nrf2 agonist found in cruciferous vegetables (130), Nrf2 activation induced by
sulforaphane treatment not only upregulates its downstream target
genes, including xCT, GPX4 and multiple Atgs,
but also disrupts the formation of Beclin1-xCT complex by promoting
autophagy. Consequently, the membrane localization of xCT and its
cysteine transport function for GSH synthesis are enhanced,
cooperatively generating anti-ferroptosis effects in carbon
tetrachloride-induced rat ALI model and
H2O2-induced liver cell injury model
(89).
Diabetes is accompanied by the development of
profound pathological alterations in the liver, and the loss of
liver cells resulting from ferroptosis contributes to diabetic
liver damage (131). Similar
anti-ferroptotic potential and hepatoprotective effect via the
activation of Nrf2 signaling pathways in the diabetic liver were
observed following the treatment with of sulforaphane, and the
occurrence of anti-ferroptotic phenotype within the diabetic liver
was associated with attenuated ferritinophagy levels. Sulforaphane
treatment of diabetic mice markedly restored Nrf2 expression,
increased the levels of its downstream target proteins related to
antioxidative defense, reversed the increase in iron accumulation
by suppressing the colocalization of FTH1 and LC3 for degradation
and by diminishing iron absorption from endosomes, and finally
prevented iron-induced lipid peroxidation and ferroptosis (90).
Cellular senescence is a state of permanent,
terminal cell cycle arrest resulting in the degeneration of
cellular structure and function (132). With the concomitant impairment
of ferrinophagy and ferroptosis, senescent cells alter cellular
iron acquisition and storage. The selective elimination of
senescent cells via ferroptosis improves physical function and
ameliorates age-related pathologies (133). 4,4'-Dimethoxychalcone (DMC) is
a natural flavonoid enriched in Angelica keiskei koidzumi
and has the ability to induce the ferroptosis of senescent liver
cells and alleviate senescence-associated phenotypes in old mouse
liver (91). The clearance of
senescent liver cells triggered by DMC is synergistically mediated
by the activation of ferritinophagy and the inhibition of
ferrochelatase (FECH), an essential enzyme for heme biosynthesis by
inserting a ferrous ion into protoporphyrin IX (134). By binding to FECH, DMC inhibits
the former's enzymatic activity, which likely contributes to free
iron accumulation. Meanwhile, the autophagic degradation of
ferritin after DMC treatment led to the accumulation of labile iron
pool and finally caused ferroptosis in senescent liver cells
(91).
In summary, the crosstalk between autophagy and
ferroptosis emerges as a key event in diverse liver diseases,
including those associated with MS, NAFLD, NASH, ALI, diabetic
liver damage and liver cell senescence. Interventions at this
pathway bring about significant opportunities for disease
management, as evidenced by compounds like liensinine, quercetin,
and sulforaphane.
Conclusion and future perspectives
A number of liver diseases have a tight connection
with the occurrence of autophagy and ferroptosis (3,7).
While recent reviews have expertly summarized the research
advancements on the autophagy-ferroptosis crosstalk within specific
conditions such as steatohepatitis and alcohol liver injury
(135,136), the scope is inherently confined
to single liver disease categories and may not capture the full
spectrum of how this crosstalk operates across the heterogeneous
landscape of liver pathologies. A comprehensive and systematic
investigation into the functional interplay and signaling pathways
linking ferroptosis and autophagy, particularly in relation to the
progression or potential therapeutic strategies for diverse liver
diseases, remains notably limited. To address this gap, the present
review systematically categorized liver diseases into four major
types, mainly including drug-induced liver injury, toxin-induced
liver injury, liver fibrosis and HCC. This novel classification
allows the present study to dissect the shared and distinct
mechanisms of autophagy-ferroptosis crosstalk across different
types of liver diseases, providing a comparative framework not
covered in previous reviews. Moreover, prior reviews acknowledge
the dual roles of autophagy in ferroptosis but lack a unified
framework to explain these contradictions. The present review
presented an interpretive framework to reconcile the contradictory
roles of autophagy in ferroptosis, highlighting critical
context-dependency (such as disease type, disease stage or cell
type) as a key determinant.
Undoubtedly, the extensive research aforementioned
has provided conclusive evidence that crosstalk between ferroptosis
and autophagy frequently occurs in liver pathological conditions.
Nevertheless, the intricate interplay between the mechanisms
underlying ferroptosis and autophagy has led to conflicting
conclusions in existing studies. In addition, the fact that some
studies employ inappropriate experimental methods or models may
exert an impact on the interpretation of experimental results. The
present study critically analyzed the conflicting findings and
evaluate methodological limitations in current researches, which
has remained unaddressed in prior reviews.
Analysis of conflicting findings
Dependent upon the available observation, autophagy
plays dual roles in the occurrence of ferroptosis (both promoting
and inhibiting). Ferroptosis sensitivity is positively correlated
with intracellular autophagy level in most cases. In drug-induced
liver injury (40-42,44), toxin-induced liver injury
(47-51,53-55), liver fibrosis (23,56,58-64) and HCC models (66,69,73,75,76,78-81), the activation of NCOA4-mediated
ferritinophagy facilitated ferroptosis by enhancing ferritin
decomposition. However, compromised ferritinophagy, iron
accumulation and increased ferroptosis levels were observed in MS
rat liver (82). Moreover,
autophagosome biogenesis downregulation and ferroptosis promotion
constitute the significant molecular mechanism underlying the
development of NAFLD (83).
Mitophagy also shows contradictory effects on ferroptosis (53,55,57). On the one hand, cells undergo
mitophagy to remove damaged mitochondria, thereby protecting cells
from oxidative stress and suppressing ferroptosis (53,101). On the other hand, mitophagy may
amplify lipid peroxidation and ferroptosis by promoting GPX4
degradation (55,57). Such discrepancy suggests that the
role of autophagy in ferroptosis is highly context-dependent. It
may stem from variations in pathological stages with altered basal
autophagy levels, which critically influence whether autophagy acts
as a pro- or anti-ferroptotic mechanism. During the early stages of
liver injury, autophagy likely plays a predominantly protective
role by eliminating damaged organelles (such as mitochondria via
mitophagy) and mitigating oxidative stress (such as GSH metabolic
reprogramming via autophagy) (70,71), thereby suppressing ferroptosis.
With the progression of liver diseases, cells are persistently
stimulated by external factors (such as oxidative stress,
pathogens) or internal factors (such as hormonal fluctuations or
iron overload), the basal autophagy levels changes, thereby
modifying the cell's susceptibility to ferroptosis. For example,
prolonged autophagy (such as ferritinophagy or lipophagy)
activation may shift toward intensifying ferroptosis by promoting
iron overload and/or lipid peroxidation (43,53,58,76), contributing to liver disease
progression. The regulation of autophagy-ferroptosis crosstalk may
also involve compensatory mechanisms. Take the MS live injury model
as an example: Simultaneous downregulation of ferritinophagy and
upregulation of ferroptosis were observed. This phenomenon reflects
that, under condition of iron overload, autophagy might undergo
compensatory inhibition to limit further iron release and
ferroptosis (82). Moreover,
various liver cell types may display unique reactions to autophagy
modulation. The diverse cellular pathways come into play when liver
cells with various phenotypes are stimulated, giving rise to
varying regulatory effects on ferroptosis. For example, liver cells
possibly depend more on autophagy for ferritin breakdown and iron
homeostasis, making them susceptible to ferroptosis upon autophagy
activation. Conversely, stellate cells may employ autophagy for
fibrotic purposes, with a weaker impact on ferroptosis (3).
In liver cells subjected to cadmium, nickel,
erastin, sorafenib, or sulfasalazine to induce ferritinophagy, a
decline in FTH1 expression was observed, accompanied by a parallel
downward trend in NCOA4 levels instead of the anticipated increase
(49,50,73). Given the NCOA4 abundance is
dually modulated by autophagy and the ubiquitin-proteasome system,
its downregulation might indicate a feedback mechanism involving
ubiquitin-dependent proteasomal degradation, which is promoted by
high iron levels and involves iron-dependent binding to the E3
ubiquitin ligase HERC2 (137).
However, the detailed molecular events underlying NCOA4 regulation
to determine its preferred interaction with HERC2 and mediate its
proteasomal degradation remain undefined. Besides, a highly
orchestrated network of transcription factor (such as ATF3 and
TFEB) (43,84), epigenetic effectors (such as
ELAVL1 and circIARS) (58,66) and signaling hubs (such as
PI3K/AKT and AMPK) (51,54) dictate the regulation of NCOA4
expression and the following ferritinophagy. Whether the relevant
pathways present in different liver diseases have a same role in
NCOA4 regulation and whether they have the same regulatory role in
the different pathological stages are concerns that deserve
sustained attention.
Limitations of experimental methods and
models
Current researches indeed exhibit some
methodological limitations. Using pharmacologic modulators such as
rapamycin, chloroquine, 3-MA and bafilomycin A1 (52,61,81,105), they have largely explored the
effect of autophagy on ferroptosis in certain experimental liver
disease animal and cellular models. However, these pharmacological
modulators have off-target effects (such as mTOR-independent roles
for rapamycin), complicating data interpretation. In the future,
specific genetic approaches to regulate the autophagy pathway
should be combined to interpretate these data. Measuring autophagy
and ferroptosis at one specific moment may fail to reflect the
ongoing and evolving changes in these processes, thereby
restricting the accuracy of result analysis. Dynamic time-course
experiments are required to elucidate the mechanistic interplay
between autophagy and ferroptosis at critical stages. Since
transmission electron microscopy (TEM) is universally acknowledged
as the gold-standard technique for the detection of autophagy and
ferroptosis, the lack of ultrastructural validation via TEM in
various studies remains a limitation (40-44,47,49,50,52,53,66,69,70-74,81,87,88,90). Incorporating this approach will
be critical to strengthen evidence for the functional role of the
autophagy-ferroptosis crosstalk in liver diseases.
A shared characteristic of these investigations is
their emphasis on the regulatory activities of ferritinophagy on
ferroptosis. By contrast, reports regarding other types of
autophagy that modulate ferroptosis are comparatively limited.
Notably, researchers appear to have neglected the reverse
regulation of ferroptosis on autophagy. Except for the mutual
regulation of autophagy and ferroptosis in fluoride-induced liver
injury (52), this phenomenon
has not been reported in the other previously discussed liver
diseases. The fact that there are more studies on the role of
autophagy in ferroptosis compared with the regulation of autophagy
by ferroptosis may be related to the easier observation of the
responses of ferroptosis when regulating autophagy, the clearer
regulatory pathways of autophagy on ferroptosis, and the greater
clinical application potential of regulating ferroptosis through
autophagy. Thus, whether other forms of selective autophagy occur
concurrently and play functional roles in ferroptosis modulation
and whether ferroptosis reciprocally regulates autophagy to form a
feedback loop remain to be further elucidated.
Future directions
The autophagy-ferroptosis axis has emerged as a
promising target across diverse liver diseases. Indeed, modulating
this axis can markedly alleviate drug- or toxin-induced
hepatotoxicity (40,41,47), reverse the progression of liver
fibrosis (61,64), effectively eliminate HCC cells
(78,79), or counteract drug resistance in
HCC cells (81), exhibiting a
broad range of beneficial effects. However, to translate these
mechanistic insights into clinical applications, addressing the
following testable questions and methodological challenges is
imperative.
First, the functional outcome of the
autophagy-ferroptosis crosstalk is highly dependent on the specific
pathological stage and the involved cell type, leading to a
paradigm where context defines therapeutic strategy. In drug- or
toxin-induced acute liver injury, early activation of autophagy may
prevent liver cells from undergoing ferroptosis, thereby limiting
the spread of injury. However, if the injury becomes chronic,
persistent autophagic stress may ultimately drive ferroptosis and
contribute to the progression from injury to fibrosis (40). In established liver fibrosis,
while protecting hepatocytes from ferroptosis remains crucial, a
parallel goal is to eliminate activated HSCs. In this context,
selectively enhancing autophagy in HSCs can trigger ferroptosis and
reverse fibrosis (61). By
contrast, activating autophagy to induce ferroptosis in HCC cells
could be an effective way to suppress tumor growth and overcome
chemoresistance (78,81). Therefore, a universal solution is
unlikely to be effective; instead, biomarker-guided, stage-specific
interventions are required. For example, in liver fibrosis,
inhibiting unrestrained autophagy-mediated ferroptosis in liver
cells may be crucial in protecting against liver injury and
fibrosis, while for stellate cells, promoting autophagy-mediated
ferroptosis to eliminate activated HSCs might be the more important
therapeutic approach. This raises a key question: How can we design
delivery systems to selectively induce pro-ferroptotic autophagy in
activated HSCs while ensuring that liver cells are shielded from
such effects? Nanoparticle-based drug delivery systems that are
decorated with ligands specific to liver cells or HSCs surface
markers offer a promising strategy to achieve this selective
modulation.
Second, the intricate integration of autophagy and
ferroptosis processes poses significant challenges for developing
targeted therapies. It is necessary to elucidate their regulatory
pathways in specific contexts. For instance, what are the key
molecular switches that determine whether autophagy inhibits or
promotes ferroptosis? Additionally, is it possible for the dual
effects of autophagy on ferroptosis to occur simultaneously and
could the simultaneous modulation of autophagy and ferroptosis
yield more favorable therapeutic outcomes on liver diseases
compared to targeting either one alone? Moreover, the role of
liver-resident cells such as Kupffer cells, endothelial cells and
stellate cells in modulating the response of liver cells to
autophagy and ferroptosis must be considered, since these cells may
modulate disease progression through paracrine signaling or direct
cell-cell interactions. To dissect these multi-layered
interactions, advanced omics technologies are indispensable.
Specifically, single-cell and spatial transcriptomics facilitates
the identification of core responsive cell types mediating the
autophagy-ferroptosis crosstalk and the signal transmission between
adjacent cells by high-resolution mapping of cellular heterogeneity
and spatial localization patterns in the liver microenvironment
(138). For instance, Kupffer
cells may modulate autophagy-ferroptosis crosstalk in neighboring
liver cells through the secretion of cytokines such as
interleukin-6 and tumor necrosis factor-a. These cytokines can
either activate or inhibit autophagy in liver cells, thereby
influencing ferroptosis sensitivity (85,86). In addition to Kupffer cells,
liver sinusoidal endothelial cells also play a role in the
regulatory network. They release extracellular vesicles containing
key autophagy factors, which can be taken up by liver cells and
thus affect the balance between autophagy and ferroptosis (139). Moreover, HSCs transmit
paracrine stimuli, such as certain cytokines and non-coding RNAs,
via exosomes. The levels of these stimuli and autophagy are
concomitantly or reciprocally regulated, which in turn alters the
ferroptosis status in liver cells (140). This integrative method also
enables the discovery of context-dependent regulators governing
this crosstalk or representative biomarkers linking autophagic and
ferroptotic pathways, thereby providing a rational basis for
developing precision medicine approaches targeting specific liver
pathologies, particular disease stages, and specific cells. Taking
liver fibrosis as an instance, the interaction between activated
HSCs and liver cells through autophagy-ferroptosis regulation can
promote the deposition of extracellular matrix (56). Specific blockage of the paracrine
signaling between HSCs and liver cells might restore the normal
autophagy-ferroptosis balance in liver cells, thereby slowing down
or reversing the progression of liver fibrosis.
Third, a wide range of compounds modulate autophagy
and/or ferroptosis, either inducing or inhibiting these processes
(141). Owing to the close
interconnection between autophagy and ferroptosis in regulating
cellular metabolism, their overlapping signaling pathways and
regulatory mechanisms are primarily manifested in the regulation of
iron metabolism, lipid peroxidation, the AMPK/mTOR pathway, as well
as the interactions among key regulatory proteins (37-39). These existing compound libraries
hold the potential to provide a wealth of candidates for the
identification of specific modulators of autophagy-ferroptosis
crosstalk. Additionally, they can bring considerable convenience
for accelerating the screening speed and improving the screening
efficiency. However, it must be pointed out that a number of these
substances that modulate autophagy and/or ferroptosis lack
sufficient specificity and whether other mechanisms are involved in
their actions and the cytotoxicity in non-diseased tissues remains
a concern (142). A critical
issue for future interventions is how to optimize the selectivity
and targeting specificity of autophagy and ferroptosis modulation
to pathological tissues while minimizing undesired deleterious
consequences on normal tissues. Moreover, several chemotherapy
drugs (such as sorafenib and lenvatinib) exhibit dual regulatory
effects on autophagy and ferroptosis in liver cells (58,76); however, their efficacy is
compromised by adverse reactions and drug resistance. Hence,
further screening for safe and effective therapeutic drugs with
higher specificity is necessary. Multiple studies have demonstrated
that bioactive phytochemicals isolated from traditional Chinese
medicine exert potent regulatory effects on autophagy-ferroptosis
crosstalk, thus presenting novel therapeutic options for liver
diseases treatment (61-65,78-81,86). However, their multiple-targets
and multiple-pathways mechanisms may contribute to the
translational gap between the preclinical findings and actual
clinical outcomes (43,97), highlighting the necessity for
optimized translational research designs. Organoids and
patient-derived xenografts are emerging as promising models that
can more accurately mimic the cellular heterogeneity and spatial
organization of human liver tissues (143). Advanced computational tools,
such as artificial intelligence-based drug screening and
bioinformatics tools, aid in finding novel therapeutic compounds
targeting specific components of the autophagy-ferroptosis axis.
They also predict the potential off-target effects and drug
resistance mechanisms, facilitating the optimization of therapeutic
regimens (144,145). Future work should integrate
advanced models with complementary high-performance tools to
overcome current limitations and unlock their therapeutic
promise.
Last, beyond the currently recognized liver
diseases, the crosstalk between autophagy and ferroptosis in
understudied liver diseases (such as cholestatic liver injury and
hepatic encephalopathy) has yet to be clarified. Future research
should explore whether dysregulation of this axis contribute to
diseases progression by dissecting specific regulatory
mechanisms.
In conclusion, the crosstalk between autophagy and
ferroptosis represents a crucial event in multiple liver diseases.
Further advances in unveiling the detailed signals and mechanisms
of these phenomena may offer a novel outlook for interpreting the
symptoms of these liver diseases and hold significant implications
for developing therapeutic strategies targeting the
autophagy-ferroptosis crosstalk in the management of liver
diseases.
Availability of data and materials
Not applicable.
Author's contributions
YW wrote and revised the manuscript. KZ, YS, ZL,
PL, YC, BH, HL and YQ revised the manuscript for important
intellectual content. JR conceived the present study. Data
authentication is not applicable. All authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
PUFA-PLs
|
phospholipid containing
polyunsaturated fatty acid chains
|
|
CMA
|
chaperone-mediated autophagy
|
|
Hsc70c
|
heat shock cognate 71 KDa protein
cytosolic
|
|
FTH1
|
ferritin heavy chain 1
|
|
LC3
|
microtubule-associated protein light
chain 3
|
|
NCOA4
|
nuclear receptor coactivator 4
|
|
Rab7
|
Ras-related protein Rab7a
|
|
HSCs
|
hepatic stellate cells
|
|
FUNDC1
|
FUN14 domain-containing protein 1
|
|
ELAVL1
|
ELAV like RNA binding protein 1
|
|
MSC-ex
|
mesenchymal stem cells-derived
exosomes
|
|
circFAM134B
|
circRNA of the family with sequence
similarity 134, member B
|
|
PTBP1
|
polypyrimidine tract-binding protein
1
|
|
TSPO
|
mitochondrial translocator
protein
|
|
BCAT2
|
branched-chain amino acid
aminotransferase 2
|
|
WTAP
|
WT1 associated protein
|
|
USP24
|
ubiquitin-specific protease 24
|
|
ARNTL
|
brain and muscle ARNT-like protein
1
|
|
EChLESs
|
electrophilic sesquiterpenes isolated
from Eupatorium chinense L.
|
|
TFEB
|
transcription factor EB
|
|
YAP1
|
Yes-associated protein 1
|
Acknowledgements
Not applicable.
Funding
The present study was supported by the Fundamental Research
Program of Shanxi Province (grants nos. 202103021224295 and
202303021222294), the Open Fund of Shanxi Key Laboratory of
Innovative Drug for the Treatment of Serious Diseases Basing on the
Chronic Inflammation, Shanxi University of Chinese Medicine (grant
no. SXInFDL2024-009), the Research Project Supported by Shanxi
Scholarship Council of China (grants nos. 2022-166 and 2024-134),
and Funding for the Discipline Construction of Pharmacology and
Toxicology of Traditional Chinese Medicine, Shanxi University of
Chinese Medicine (grant no. 2025XK36).
References
|
1
|
Wang C, Ma C, Gong L, Guo Y, Fu K, Zhang
Y, Zhou H and Li Y: Macrophage polarization and its role in liver
disease. Front Immunol. 12:8030372021. View Article : Google Scholar :
|
|
2
|
Wang X, Zhou Y, Min J and Wang F: Zooming
in and out of ferroptosis in human disease. Front Med. 17:173–206.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen J, Li X, Ge C, Min J and Wang F: The
multifaceted role of ferroptosis in liver disease. Cell Death
Differ. 29:467–480. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Klionsky DJ, Petroni G, Amaravadi RK,
Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cadwell K,
Cecconi F, Choi AMK, et al: Autophagy in major human diseases. EMBO
J. 40:e1088632021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Parzych KR and Klionsky DJ: An overview of
autophagy: morphology, mechanism, and regulation. Antioxid Redox
Signal. 20:460–473. 2014. View Article : Google Scholar
|
|
6
|
Zhang C, Wang W, Du C, Li H, Zhou K, Luan
Z, Chang Y, Liu S and Wei Y: Autophagy in the pharmacological
activities of celastrol (Review). Exp Ther Med. 25:2682023.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qian H, Chao X, Williams J, Fulte S, Li T,
Yang L and Ding WX: Autophagy in liver diseases: A review. Mol
Aspects Med. 82:1009732021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun
X, Kang R and Tang D: Ferroptosis: Process and function. Cell Death
Differ. 23:369–379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dolma S, Lessnick SL, Hahn WC and
Stockwell BR: Identification of genotype-selective antitumor agents
using synthetic lethal chemical screening in engineered human tumor
cells. Cancer Cell. 3:285–296. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Andreani C, Bartolacci C and Scaglioni PP:
Ferroptosis: A specific vulnerability of RAS-driven cancers? Front
Oncol. 12:9239152022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yagoda N, von Rechenberg M, Zaganjor E,
Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM,
Boniface JJ, et al: RAS-RAF-MEK-dependent oxidative cell death
involving voltage-dependent anion channels. Nature. 447:864–868.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schott C, Graab U, Cuvelier N, Hahn H and
Fulda S: Oncogenic RAS mutants confer resistance of RMS13
rhabdomyosarcoma cells to oxidative stress-induced ferroptotic cell
death. Front Oncol. 5:1312015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X,
Wang H, Cao L and Tang D: HSPB1 as a novel regulator of ferroptotic
cancer cell death. Oncogene. 34:5617–5625. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rochette L, Dogon G, Rigal E, Zeller M,
Cottin Y and Vergely C: Lipid peroxidation and iron metabolism: Two
corner stones in the homeostasis control of ferroptosis. Int J Mol
Sci. 24:4492022. View Article : Google Scholar
|
|
15
|
Chen X, Yu C, Kang R and Tang D: Iron
metabolism in ferroptosis. Front Cell Dev Biol. 8:5902262020.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang WS and Stockwell BR: Ferroptosis:
Death by lipid peroxidation. Trends Cell Biol. 26:165–176. 2016.
View Article : Google Scholar
|
|
17
|
Chen X, Li J, Kang R, Klionsky DJ and Tang
D: Ferroptosis: Machinery and regulation. Autophagy. 17:2054–2081.
2021. View Article : Google Scholar :
|
|
18
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu J, Kuang F, Kroemer G, Klionsky DJ,
Kang R and Tang D: Autophagy-dependent ferroptosis: machinery and
regulation. Cell Chem Biol. 27:420–435. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen HJ, Sugiyama M, Shimokawa F, Murakami
M, Hashimoto O, Matsui T and Funaba M: Response to iron overload in
cultured hepatocytes. Sci Rep. 10:211842020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yao X, Zhang Y, Hao J, Duan HQ, Zhao CX,
Sun C, Li B, Fan BY, Wang X, Liu WX, et al: Deferoxamine promotes
recovery of traumatic spinal cord injury by inhibiting ferroptosis.
Neural Regen Res. 14:532–541. 2019. View Article : Google Scholar :
|
|
22
|
Liu Y, Wang W, Li Y, Xiao Y, Cheng J and
Jia J: The 5-lipoxygenase inhibitor zileuton confers
neuroprotection against glutamate oxidative damage by inhibiting
ferroptosis. Biol Pharm Bull. 38:1234–1239. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Z, Guo M, Li Y, Shen M, Kong D, Shao
J, Ding H, Tan S, Chen A, Zhang F and Zheng S: RNA-binding protein
ZFP36/TTP protects against ferroptosis by regulating autophagy
signaling pathway in hepatic stellate cells. Autophagy.
16:1482–1505. 2020. View Article : Google Scholar :
|
|
24
|
Liang D, Minikes AM and Jiang X:
Ferroptosis at the intersection of lipid metabolism and cellular
signaling. Mol Cell. 82:2215–2227. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Klionsky DJ and Codogno P: The mechanism
and physiological function of macroautophagy. J Innate Immun.
5:427–433. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ariosa AR and Klionsky DJ: Autophagy core
machinery: Overcoming spatial barriers in neurons. J Mol Med
(Berl). 94:1217–1227. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang Z and Klionsky DJ: Mammalian
autophagy: Core molecular machinery and signaling regulation. Curr
Opin Cell Biol. 22:124–131. 2010. View Article : Google Scholar
|
|
28
|
Hosokawa N, Hara T, Kaizuka T, Kishi C,
Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et
al: Nutrient-dependent mTORC1 association with the
ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell.
20:1981–1991. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang S, Yazaki E, Sakamoto H, Yamamoto H
and Mizushima N: Evolutionary diversification of the
autophagy-related ubiquitin-like conjugation systems. Autophagy.
18:2969–2984. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Webber JL, Young AR and Tooze SA: Atg9
trafficking in mammalian cells. Autophagy. 3:54–56. 2007.
View Article : Google Scholar
|
|
31
|
Ropolo A, Grasso D, Pardo R, Sacchetti ML,
Archange C, Lo Re A, Seux M, Nowak J, Gonzalez CD, Iovanna JL and
Vaccaro MI: The pancreatitis-induced vacuole membrane protein 1
triggers autophagy in mammalian cells. J Biol Chem.
282:37124–37133. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Deleyto-Seldas N and Efeyan A: The
mTOR-autophagy axis and the control of metabolism. Front Cell Dev
Biol. 9:6557312021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu HD and Qin ZH: Beclin 1, Bcl-2 and
autophagy. Adv Exp Med Biol. 1206:109–126. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
White E: Autophagy and p53. Cold Spring
Harb Perspect Med. 6:a0261202016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hu LF: Epigenetic regulation of autophagy.
Adv Exp Med Biol. 1206:221–236. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vargas JNS, Hamasaki M, Kawabata T, Youle
RJ and Yoshimori T: The mechanisms and roles of selective autophagy
in mammals. Nat Rev Mol Cell Biol. 24:167–185. 2023. View Article : Google Scholar
|
|
37
|
Gao M, Monian P, Pan Q, Zhang W, Xiang J
and Jiang X: Ferroptosis is an autophagic cell death process. Cell
Res. 26:1021–1032. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Park E and Chung SW: ROS-mediated
autophagy increases intracellular iron levels and ferroptosis by
ferritin and transferrin receptor regulation. Cell Death Dis.
10:8222019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lv X, Tang W, Qin J, Wang W, Dong J and
Wei Y: The crosslinks between ferroptosis and autophagy in asthma.
Front Immunol. 14:11407912023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhou J, Tan Y, Hu L, Fu J, Li D, Chen J
and Long Y: Inhibition of HSPA8 by rifampicin contributes to
ferroptosis via enhancing autophagy. Liver Int. 42:2889–2899. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang C, Leng M, Ding C, Zhu X, Zhang Y,
Sun C and Lou P: Ferritinophagy-mediated ferroptosis facilitates
methotrexate-induced hepatotoxicity by high-mobility group box 1
(HMGB1). Liver Int. 44:691–705. 2024. View Article : Google Scholar
|
|
42
|
Liang Y, Chen S, Han S, Luo L, Shen F and
Huang Z: Toosendanin induced hepatotoxicity via triggering
PERK-eIF2α-ATF4 mediated ferroptosis. Toxicol Lett. 377:51–61.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu S, Su Y, Han B, Yin L, Li H, Wang Y,
Zhou K, Li P and Wei Y: Activation of Rab7-mediated lipophagy is
required for triptolide to induce ferroptosis in hepatic cells.
Food Chem Toxicol. 203:1155682025. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wu A, Li M, Chen Y, Zhang W, Li H, Chen J,
Gu K and Wang X: Multienzyme active manganese oxide alleviates
acute liver injury by mimicking redox regulatory system and
inhibiting ferroptosis. Adv Healthc Mater. 13:e23025562024.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cai X, Hua S, Deng J, Du Z, Zhang D, Liu
Z, Khan NU, Zhou M and Chen Z: Astaxanthin activated the Nrf2/HO-1
pathway to enhance autophagy and inhibit ferroptosis, ameliorating
acetaminophen-induced liver injury. ACS Appl Mater Interfaces.
14:42887–42903. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ren J, Wang Z, Zhang Y, Pei H, Wen R,
Zhang Z, Zhu S, Sun X, Yin B, Li S and Ma Y: Sesamin alleviates
drug-induced hepatotoxicity via autophagy enhancement and
ferroptosis inhibition. Redox Rep. 30:25888632025. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Huang T, Zhang K, Wang J, He K, Zhou X and
Nie S: Quercetin alleviates acrylamide-induced liver injury by
inhibiting autophagy-dependent ferroptosis. J Agric Food Chem.
71:7427–7439. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhong G, Li Y, Ma F, Huo Y, Liao J, Han Q,
Hu L and Tang Z: Copper exposure induced chicken hepatotoxicity:
Involvement of ferroptosis mediated by lipid peroxidation,
ferritinophagy, and inhibition of FSP1-CoQ10 and Nrf2/SLC7A11/GPX4
axis. Biol Trace Elem Res. 202:1711–1721. 2024. View Article : Google Scholar
|
|
49
|
He Z, Shen P, Feng L, Hao H, He Y, Fan G,
Liu Z, Zhu K, Wang Y, Zhang N, et al: Cadmium induces liver
dysfunction and ferroptosis through the endoplasmic
stress-ferritinophagy axis. Ecotoxicol Environ Saf. 245:1141232022.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wei L, Zuo Z, Yang Z, Yin H, Yang Y, Fang
J, Cui H, Du Z, Ouyang P, Chen X, et al: Mitochondria damage and
ferroptosis involved in Ni-induced hepatotoxicity in mice.
Toxicology. 466:1530682022. View Article : Google Scholar
|
|
51
|
Yu L, Lv Z, Li S, Jiang H, Han B, Zheng X,
Liu Y and Zhang Z: Chronic arsenic exposure induces ferroptosis via
enhancing ferritinophagy in chicken livers. Sci Total Environ.
890:1641722023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xu W, Hu Z, Zhang J, Tang Y, Xing H, Xu P,
Ma Y and Niu Q: Cross-talk between autophagy and ferroptosis
contributes to the liver injury induced by fluoride via the
mtROS-dependent pathway. Ecotoxicol Environ Saf. 250:1144902023.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Song XY, Liu PC, Liu WW, Zhou J, Hayashi
T, Mizuno K, Hattori S, Fujisaki H and Ikejima T: Silibinin
inhibits ethanol- or acetaldehyde-induced ferroptosis in liver cell
lines. Toxicol In Vitro. 82:1053882022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Song C, Wang Z, Cao J, Dong Y and Chen Y:
Hesperetin alleviates aflatoxin B1 induced liver toxicity in mice:
modulating lipid peroxidation and ferritin autophagy. Ecotoxicol
Environ Saf. 284:1168542024. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Jiang J, Zhou X, Chen H, Wang X, Ruan Y,
Liu X and Ma J: 18β-Glycyrrhetinic acid protects against
deoxynivalenol-induced liver injury via modulating ferritinophagy
and mitochondrial quality control. J Hazard Mater. 471:1343192024.
View Article : Google Scholar
|
|
56
|
Liang Q, Ma Y, Wang F, Sun M, Lin L, Li T,
Duan J and Sun Z: Ferritinophagy was involved in long-term SiNPs
exposure induced ferroptosis and liver fibrosis. Nanotoxicology.
17:157–175. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bi Y, Liu S, Qin X, Abudureyimu M, Wang L,
Zou R, Ajoolabady A, Zhang W, Peng H, Ren J and Zhang Y: FUNDC1
interacts with GPx4 to govern hepatic ferroptosis and fibrotic
injury through a mitophagy-dependent manner. J Adv Res. 55:45–60.
2024. View Article : Google Scholar :
|
|
58
|
Zhang Z, Yao Z, Wang L, Ding H, Shao J,
Chen A, Zhang F and Zheng S: Activation of ferritinophagy is
required for the RNA-binding protein ELAVL1/HuR to regulate
ferroptosis in hepatic stellate cells. Autophagy. 14:2083–2103.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Shen M, Li Y, Wang Y, Shao J, Zhang F, Yin
G, Chen A, Zhang Z and Zheng S: N (6)-methyladenosine modification
regulates ferroptosis through autophagy signaling pathway in
hepatic stellate cells. Redox Biol. 47:1021512021. View Article : Google Scholar
|
|
60
|
Tan Y, Huang Y, Mei R, Mao F, Yang D, Liu
J, Xu W, Qian H and Yan Y: HucMSC-derived exosomes delivered BECN1
induces ferroptosis of hepatic stellate cells via regulating the
xCT/GPX4 axis. Cell Death Dis. 13:3192022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kong Z, Liu R and Cheng Y: Artesunate
alleviates liver fibrosis by regulating ferroptosis signaling
pathway. Biomed Pharmacother. 109:2043–2053. 2019. View Article : Google Scholar
|
|
62
|
Zheng Y, Zhao T and Wang J, Jiang R, Huang
J, Li W and Wang J: Curcumol alleviates liver fibrosis through
inducing autophagy and ferroptosis in hepatic stellate cells. FASEB
J. 36:e226652022. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhang Z, Wang X, Wang Z, Zhang Z, Cao Y,
Wei Z, Shao J, Chen A, Zhang F and Zheng S: Dihydroartemisinin
alleviates hepatic fibrosis through inducing ferroptosis in hepatic
stellate cells. Biofactors. 47:801–818. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Li M, Sun Y, Wei Y, Li Y, Shao JJ, Guo M,
Zheng S and Zhang Z: Artemether relieves liver fibrosis by
triggering ferroptosis in hepatic stellate cells via
DHHC12-mediated S-palmitoylation of the BECN1 protein. Free Radic
Biol Med. 231:120–135. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yi J, Wu S, Tan S, Qin Y, Wang X, Jiang J,
Liu H and Wu B: Berberine alleviates liver fibrosis through
inducing ferrous redox to activate ROS-mediated hepatic stellate
cells ferroptosis. Cell Death Discov. 7:3742021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Liu Z, Wang Q, Wang X, Xu Z, Wei X and Li
J: Circular RNA cIARS regulates ferroptosis in HCC cells through
interacting with RNA binding protein ALKBH5. Cell Death Discov.
6:722020. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bi T, Lu Q, Pan X, Dong F, Hu Y, Xu Z, Xiu
P, Liu Z and Li J: circFAM134B is a key factor regulating
reticulophagy-mediated ferroptosis in hepatocellular carcinoma.
Cell Cycle. 22:1900–1920. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Liu Z, Ma C, Wang Q, Yang H, Lu Z, Bi T,
Xu Z, Li T, Zhang L, Zhang Y, et al: Targeting FAM134B-mediated
reticulophagy activates sorafenib-induced ferroptosis in
hepatocellular carcinoma. Biochem Biophys Res Commun. 589:247–253.
2022. View Article : Google Scholar
|
|
69
|
Yang H, Sun W, Bi T, Wang Q, Wang W, Xu Y,
Liu Z and Li J: The PTBP1-NCOA4 axis promotes ferroptosis in liver
cancer cells. Oncol Rep. 49:452023. View Article : Google Scholar
|
|
70
|
Hu X, He Y, Han Z, Liu W, Liu D, Zhang X,
Chen L, Qi L, Chen L, Luo Y, et al: PNO1 inhibits
autophagy-mediated ferroptosis by GSH metabolic reprogramming in
hepatocellular carcinoma. Cell Death Dis. 13:10102022. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Han Z, Liu D, Chen L, He Y, Tian X, Qi L,
Chen L, Luo Y, Chen Z, Hu X, et al: PNO1 regulates autophagy and
apoptosis of hepatocellular carcinoma via the MAPK signaling
pathway. Cell Death Dis. 12:5522021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhang D, Man D, Lu J, Jiang Y, Ding B, Su
R, Tong R, Chen J, Yang B, Zheng S, et al: Mitochondrial TSPO
promotes hepatocellular carcinoma progression through ferroptosis
inhibition and immune evasion. Adv Sci (Weinh). 10:e22066692023.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wang K, Zhang Z, Tsai HI, Liu Y, Gao J,
Wang M, Song L, Cao X, Xu Z, Chen H, et al: Branched-chain amino
acid aminotransferase 2 regulates ferroptotic cell death in cancer
cells. Cell Death Differ. 28:1222–1236. 2021. View Article : Google Scholar :
|
|
74
|
Wu K, Yan M, Liu T, Wang Z, Duan Y, Xia Y,
Ji G, Shen Y, Wang L, Li L, et al: Creatine kinase B suppresses
ferroptosis by phosphorylating GPX4 through a moonlighting
function. Nat Cell Biol. 25:714–725. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Li Y, Guo M, Qiu Y, Li M, Wu Y, Shen M,
Wang Y, Zhang F, Shao J, Xu X, et al: Autophagy activation is
required for N6-methyladenosine modification to regulate
ferroptosis in hepatocellular carcinoma. Redox Biol. 69:1029712024.
View Article : Google Scholar
|
|
76
|
Cao J, Wu S, Zhao S, Wang L, Wu Y, Song L,
Sun C, Liu Y, Liu Z, Zhu R, et al: USP24 promotes
autophagy-dependent ferroptosis in hepatocellular carcinoma by
reducing the K48-linked ubiquitination of Beclin1. Commun Biol.
7:12792024. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Jiang H, Wang X, Zhu Z, Song C, Li D, Yun
Y, Hui L, Bao L, O'Connor DP, Ma J and Xu G: DCAF7 recruits USP2 to
facilitate hepatocellular carcinoma progression by suppressing
clockophagy-induced ferroptosis. Cell Death Dis. 16:6542025.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Xiu Z, Zhu Y, Han J, Li Y, Yang X, Yang G,
Song G, Li S, Li Y, Cheng C, et al: Caryophyllene oxide induces
ferritinophagy by regulating the NCOA4/FTH1/LC3 pathway in
hepatocellular carcinoma. Front Pharmacol. 13:9309582022.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Xiu Z, Li Y, Fang J, Han J, Li S, Li Y,
Yang X, Song G, Li Y, Jin N, et al: Inhibitory effects of esculetin
on liver cancer through triggering NCOA4 pathway-mediation
ferritinophagy in vivo and in vitro. J Hepatocell Carcinoma.
10:611–629. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zhu ZH, Xu XT, Shen CJ, Yuan JT, Lou SY,
Ma XL, Chen X, Yang B and Zhao HJ: A novel sesquiterpene lactone
fraction from Eupatorium chinense L. suppresses hepatocellular
carcinoma growth by triggering ferritinophagy and mitochondrial
damage. Phytomedicine. 112:1546712023. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Li ZJ, Dai HQ, Huang XW, Feng J, Deng JH,
Wang ZX, Yang XM, Liu YJ, Wu Y, Chen PH, et al: Artesunate
synergizes with sorafenib to induce ferroptosis in hepatocellular
carcinoma. Acta Pharmacol Sin. 42:301–310. 2021. View Article : Google Scholar
|
|
82
|
Cui F, Mi H, Wang R, Du Y, Li F, Chang S,
Su Y, Liu A and Shi M: The effect of chronic intermittent hypobaric
hypoxia improving liver damage in metabolic syndrome rats through
ferritinophagy. Pflugers Arch. 475:1251–1263. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Liu P, Anandhan A, Chen J, Shakya A,
Dodson M, Ooi A, Chapman E, White E, Garcia JG and Zhang DD:
Decreased autophagosome biogenesis, reduced NRF2, and enhanced
ferroptotic cell death are underlying molecular mechanisms of
non-alcoholic fatty liver disease. Redox Biol. 59:1025702023.
View Article : Google Scholar
|
|
84
|
Honma K, Kirihara S, Nakayama H, Fukuoka
T, Ohara T, Kitamori K, Sato I, Hirohata S, Fujii M, Yamamoto S, et
al: Selective autophagy associated with iron overload aggravates
non-alcoholic steatohepatitis via ferroptosis. Exp Biol Med
(Maywood). 248:1112–1123. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Li J, Huang Q, Lv M, Ma W, Sun J, Zhong X,
Hu R, Ma M, Han Z, Zhang W, et al: Role of liensinine in
sensitivity of activated macrophages to ferroptosis and in acute
liver injury. Cell Death Discov. 9:1892023. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Zhang H, Shi H, Li X, Zhou S, Song X, Ma
N, Meng M, Chang G and Shen X: Quercetin alleviates
LPS/iE-DAP-induced liver injury by suppressing ferroptosis via
regulating ferritinophagy and intracellular iron efflux. Redox
Biol. 81:1035572025. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Wang J, Zhu Q, Li R, Zhang J, Ye X and Li
X: YAP1 protects against septic liver injury via ferroptosis
resistance. Cell Biosci. 12:1632022. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Jia FJ and Han J: Liver injury in
COVID-19: Holds ferritinophagy-mediated ferroptosis accountable.
World J Clin Cases. 10:13148–13156. 2022. View Article : Google Scholar
|
|
89
|
Liu J, Huang C, Liu J, Meng C, Gu Q, Du X,
Yan M, Yu Y, Liu F and Xia C: Nrf2 and its dependent autophagy
activation cooperatively counteract ferroptosis to alleviate acute
liver injury. Pharmacol Res. 187:1065632023. View Article : Google Scholar
|
|
90
|
Savic N, Markelic M, Stancic A, Velickovic
K, Grigorov I, Vucetic M, Martinovic V, Gudelj A and Otasevic V:
Sulforaphane prevents diabetes-induced hepatic ferroptosis by
activating Nrf2 signaling axis. Biofactors. 50:810–827. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Wang T, Yang C, Li Z, Li T, Zhang R, Zhao
Y, Cheng T, Zong Z, Ma Y, Zhang D and Deng H: Flavonoid
4,4'-dimethoxychalcone selectively eliminates senescent cells via
activating ferritinophagy. Redox Biol. 69:1030172024. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Björnsson HK and Björnsson ES:
Drug-induced liver injury: Pathogenesis, epidemiology, clinical
features, and practical management. Eur J Intern Med. 97:26–31.
2022. View Article : Google Scholar
|
|
93
|
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh
HJ III, Kang R and Tang D: Autophagy promotes ferroptosis by
degradation of ferritin. Autophagy. 12:1425–1428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Di Martino V, Verhoeven DW, Verhoeven F,
Aubin F, Avouac J, Vuitton L, Lioté F, Thévenot T and Wendling D:
Busting the myth of methotrexate chronic hepatotoxicity. Nat Rev
Rheumatol. 19:96–110. 2023. View Article : Google Scholar
|
|
95
|
Ni YA, Chen H, Nie H, Zheng B and Gong Q:
HMGB1: An overview of its roles in the pathogenesis of liver
disease. J Leukoc Biol. 110:987–998. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Zhuo Y, Zhang Y, Li M, Wu H, Gong S, Hu X,
Fu Y, Shen X, Sun B, Wu JL and Li N: Hepatotoxic evaluation of
toosendanin via biomarker quantification and pathway mapping of
large-scale chemical proteomics. Food Chem Toxicol. 153:1122572021.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Wei YM, Wang YH, Xue HQ, Luan ZH, Liu BW
and Ren JH: Triptolide, a potential autophagy modulator. Chin J
Integr Med. 25:233–240. 2019. View Article : Google Scholar
|
|
98
|
Bai Y, Meng L, Han L, Jia Y, Zhao Y, Gao
H, Kang R, Wang X, Tang D and Dai E: Lipid storage and lipophagy
regulates ferroptosis. Biochem Biophys Res Commun. 508:997–1003.
2019. View Article : Google Scholar
|
|
99
|
Jaeschke H and Ramachandran A:
Acetaminophen hepatotoxicity: Paradigm for understanding mechanisms
of drug-induced liver injury. Annu Rev Pathol. 19:453–478. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Lőrincz T, Jemnitz K, Kardon T, Mandl J
and Szarka A: Ferroptosis is involved in acetaminophen induced cell
death. Pathol Oncol Res. 21:1115–1121. 2015. View Article : Google Scholar
|
|
101
|
Ni HM, Bockus A, Boggess N, Jaeschke H and
Ding WX: Activation of autophagy protects against
acetaminophen-induced hepatotoxicity. Hepatology. 55:222–232. 2012.
View Article : Google Scholar
|
|
102
|
Yamada N, Karasawa T, Kimura H, Watanabe
S, Komada T, Kamata R, Sampilvanjil A, Ito J, Nakagawa K, Kuwata H,
et al: Ferroptosis driven by radical oxidation of n-6
polyunsaturated fatty acids mediates acetaminophen-induced acute
liver failure. Cell Death Dis. 11:1442020. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Zhang L, Yang L, Luo Y, Dong L and Chen F:
Acrylamide-induced hepatotoxicity through oxidative stress:
mechanisms and interventions. Antioxid Redox Signal. 38:1122–1137.
2023. View Article : Google Scholar
|
|
104
|
Tan X, Li L, Wang J, Zhao B, Pan J, Wang
L, Liu X, Liu X and Liu Z: Resveratrol prevents acrylamide-induced
mitochondrial dysfunction and inflammatory responses via targeting
circadian regulator Bmal1 and Cry1 in hepatocytes. J Agric Food
Chem. 67:8510–8519. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Erfan OS, Sonpol HMA and Abd El-Kader M:
Protective effect of rapamycin against acrylamide-induced
hepatotoxicity: The associations between autophagy, apoptosis, and
necroptosis. Anat Rec (Hoboken). 304:1984–1998. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Esfandiari M and Hakimzadeh MA: Assessment
of environmental pollution of heavy metals deposited on the leaves
of trees at Yazd bus terminals. Environ Sci Pollut Res Int.
29:32867–32881. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
González-Alfonso WL, Petrosyan P, Del Razo
LM, Sánchez-Peña LC, Tapia-Rodríguez M, Hernández-Muñoz R and
Gonsebatt ME: Chronic exposure to arsenic and fluoride starting at
gestation alters liver mitochondrial protein expression and induces
early onset of liver fibrosis in male mouse offspring. Biol Trace
Elem Res. 203:930–943. 2025. View Article : Google Scholar :
|
|
108
|
Wang HW, Liu J, Wei SS, Zhao WP, Zhu SQ
and Zhou BH: Mitochondrial respiratory chain damage and
mitochondrial fusion disorder are involved in liver dysfunction of
fluoride-induced mice. Chemosphere. 241:1250992020. View Article : Google Scholar
|
|
109
|
Jahng JWS, Alsaadi RM, Palanivel R, Song
E, Hipolito VEB, Sung HK, Botelho RJ, Russell RC and Sweeney G:
Iron overload inhibits late stage autophagic flux leading to
insulin resistance. EMBO Rep. 20:e479112019. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Liu CY, Wang M, Yu HM, Han FX, Wu QS, Cai
XJ, Kurihara H, Chen YX, Li YF and He RR: Ferroptosis is involved
in alcohol-induced cell death in vivo and in vitro. Biosci
Biotechnol Biochem. 84:1621–1628. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Horn P and Tacke F: Metabolic
reprogramming in liver fibrosis. Cell Metab. 36:1439–1455. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Huang Y, Li P, Zhao R, Zhao L, Liu J, Peng
S, Fu X, Wang X, Luo R, Wang R and Zhang Z: Silica nanoparticles:
Biomedical applications and toxicity. Biomed Pharmacother.
151:1130532022. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Liu J, Yan Z, Yang F, Huang Y, Yu Y, Zhou
L, Sun Z, Cui D and Yan Y: Exosomes derived from human umbilical
cord mesenchymal stem cells accelerate cutaneous wound healing by
enhancing angiogenesis through delivering angiopoietin-2. Stem Cell
Rev Rep. 17:305–317. 2021. View Article : Google Scholar
|
|
114
|
Wang G, Li J, Zhu L, Zhou Z, Ma Z, Zhang
H, Yang Y, Niu Q and Wang X: Identification of hepatocellular
carcinoma-related subtypes and development of a prognostic model: A
study based on ferritinophagy-related genes. Discov Oncol.
14:1472023. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Zhang Y, Luo J, Yang W and Ye WC: CircRNAs
in colorectal cancer: Potential biomarkers and therapeutic targets.
Cell Death Dis. 14:3532023. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Chen Y, Mi Y, Zhang X, Ma Q, Song Y, Zhang
L, Wang D, Xing J, Hou B, Li H, et al: Dihydroartemisinin-induced
unfolded protein response feedback attenuates ferroptosis via
PERK/ATF4/HSPA5 pathway in glioma cells. J Exp Clin Cancer Res.
38:4022019. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Vara-Perez M, Felipe-Abrio B and Agostinis
P: Mitophagy in cancer: A tale of adaptation. Cells. 8:4932019.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Liu C, Wu Z, Wang L, Yang Q and Huang J
and Huang J: A Mitophagy-related gene signature for subtype
identification and prognosis prediction of hepatocellular
carcinoma. Int J Mol Sci. 23:121232022. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Pan JH, Kang YQ, Li Q, Xing W, Chen YH,
Yan Y, Luo DX, Qiu Y, Yuan YF, Zeng WA and Chen DT: TSPO is a novel
biomarker for prognosis that regulates cell proliferation through
influencing mitochondrial functions in HCC. Heliyon. 9:e225902023.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Fu F, Lai Q, Hu J, Zhang L, Zhu X, Kou J,
Yu B and Li F: Ruscogenin alleviates myocardial ischemia-induced
ferroptosis through the activation of BCAT1/BCAT2. Antioxidants
(Basel). 11. pp. 5832022, View Article : Google Scholar
|
|
121
|
Rahbani JF, Roesler A, Hussain MF,
Samborska B, Dykstra CB, Tsai L, Jedrychowski MP, Vergnes L, Reue
K, Spiegelman BM and Kazak L: Creatine kinase B controls futile
creatine cycling in thermogenic fat. Nature. 590:480–485. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Fidyt K, Fiedorowicz A, Strządała L and
Szumny A: β-caryophyllene and β-caryophyllene oxide-natural
compounds of anticancer and analgesic properties. Cancer Med.
5:3007–3017. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Zhang L, Xie Q and Li X: Esculetin: A
review of its pharmacology and pharmacokinetics. Phytother Res.
36:279–298. 2022. View Article : Google Scholar
|
|
124
|
Tang W, Chen Z, Zhang W, Cheng Y, Zhang B,
Wu F, Wang Q, Wang S, Rong D, Reiter FP, et al: The mechanisms of
sorafenib resistance in hepatocellular carcinoma: Theoretical basis
and therapeutic aspects. Signal Transduct Target Ther. 5:872020.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Kim JE, Kim JS, Jo MJ, Cho E, Ahn SY, Kwon
YJ and Ko GJ: The roles and associated mechanisms of adipokines in
development of metabolic syndrome. Molecules. 27:3342022.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Sheka AC, Adeyi O, Thompson J, Hameed B,
Crawford PA and Ikramuddin S: Nonalcoholic steatohepatitis: A
review. JAMA. 323:1175–1183. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Woolbright BL and Jaeschke H: Sterile
inflammation in acute liver injury: Myth or mystery? Expert Rev
Gastroenterol Hepatol. 9:1027–1029. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Starkey Lewis P, Campana L, Aleksieva N,
Cartwright JA, Mackinnon A, O' Duibhir E, Kendall T, Vermeren M,
Thomson A, Gadd V, et al: Alternatively activated macrophages
promote resolution of necrosis following acute liver injury. J
Hepatol. 73:349–360. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Szulzewsky F, Holland EC and Vasioukhin V:
YAP1 and its fusion proteins in cancer initiation, progression and
therapeutic resistance. Dev Biol. 475:205–221. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Otoo RA and Allen AR: Sulforaphane's
multifaceted potential: From neuroprotection to anticancer action.
Molecules. 28:69022023. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Stancic A, Velickovic K, Markelic M,
Grigorov I, Saksida T, Savic N, Vucetic M, Martinovic V, Ivanovic A
and Otasevic V: Involvement of ferroptosis in diabetes-induced
liver pathology. Int J Mol Sci. 23:93092022. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Di Micco R, Krizhanovsky V, Baker D and
d'Adda di Fagagna F: Cellular senescence in ageing: from mechanisms
to therapeutic opportunities. Nat Rev Mol Cell Biol. 22:75–95.
2021. View Article : Google Scholar :
|
|
133
|
Masaldan S, Clatworthy SAS, Gamell C,
Meggyesy PM, Rigopoulos AT, Haupt S, Haupt Y, Denoyer D, Adlard PA,
Bush AI and Cater MA: Iron accumulation in senescent cells is
coupled with impaired ferritinophagy and inhibition of ferroptosis.
Redox Biol. 14:100–115. 2018. View Article : Google Scholar
|
|
134
|
Sishtla K, Lambert-Cheatham N, Lee B, Han
DH, Park J, Sardar Pasha SPB, Lee S, Kwon S, Muniyandi A, Park B,
et al: Small-molecule inhibitors of ferrochelatase are
antiangiogenic agents. Cell Chem Biol. 29:1010–1023.e14. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Zhao S, Guo Y and Yin X: Autophagy,
ferroptosis, apoptosis and pyroptosis in metabolic
dysfunction-associated steatotic liver disease. Front Biosci
(Landmark Ed). 29:302024. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Wang Y, Zhou X, Chen H and Li Z: Molecular
mechanisms of alcohol-associated liver disease-ferroptosis and
autophagy crosstalk. Mol Biol Rep. 52:3612025. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Mancias JD, Pontano Vaites L, Nissim S,
Biancur DE, Kim AJ, Wang X, Liu Y, Goessling W, Kimmelman AC and
Harper JW: Ferritinophagy via NCOA4 is required for erythropoiesis
and is regulated by iron dependent HERC2-mediated proteolysis.
Elife. 4. pp. e103082015, View Article : Google Scholar
|
|
138
|
Andrews TS, Nakib D, Perciani CT, Ma XZ,
Liu L, Winter E, Camat D, Chung SW, Lumanto P, Manuel J, et al:
Single-cell, single-nucleus, and spatial transcriptomics
characterization of the immunological landscape in the healthy and
PSC human liver. J Hepatol. 80:730–743. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Chen T, Zhang H, Shan W, Zhou J and You Y:
Liver sinusoidal endothelial cells in hepatic fibrosis:
Opportunities for future strategies. Biochem Biophys Res Commun.
766:1518812025. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Mastoridou EM, Goussia AC, Glantzounis GK,
Kanavaros P and Charchanti AV: Autophagy and exosomes:
Cross-regulated pathways playing major roles in hepatic stellate
cells activation and liver fibrosis. Front Physiol. 12:8013402022.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Ahsan A, Liu M, Zheng Y, Yan W, Pan L, Li
Y, Ma S, Zhang X, Cao M, Wu Z, et al: Natural compounds modulate
the autophagy with potential implication of stroke. Acta Pharm Sin
B. 11:1708–1720. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Wu Z, Zhu Y, Liu W, Balasubramanian B, Xu
X, Yao J and Lei X: Ferroptosis in liver disease: Natural active
compounds and therapeutic implication. Antioxidants (Basel). 13.
pp. 3522024, View Article : Google Scholar
|
|
143
|
Hou X, Du C, Lu L, Yuan S, Zhan M, You P
and Du H: Opportunities and challenges of patient-derived models in
cancer research: patient-derived xenografts, patient-derived
organoid and patient-derived cells. World J Surg Oncol. 20:372022.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Liu X, Zhang J, Wang X, Teng M, Wang G and
Zhou X: Application of artificial intelligence large language
models in drug target discovery. Front Pharmacol. 16:15973512025.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Ren J and Wu M: Identification of novel
therapeutic targets for MAFLD based on bioinformatics analysis
combined with Mendelian randomization. Int J Mol Sci. 26:31662025.
View Article : Google Scholar : PubMed/NCBI
|