Introduction
Diabetic nephropathy (DN), an important complication
of diabetes mellitus, is characterized by progressive deterioration
of kidney function and structural alterations within the kidney. It
is the primary etiologic factor of end-stage renal disease
worldwide (1). DN pathogenesis
is intricate and involves various cellular and molecular
mechanisms, including metabolic disturbances, hemodynamic
fluctuations, dysregulated mitophagy and endoplasmic reticulum
stress, accompanied by elevated reactive oxygen species (ROS) and
advanced glycation end products (AGEs), which alter numerous
pathways in cells, inducing an inflammatory response, fibrosis and
adverse pathological outcomes. Sustained exposure to elevated
glucose levels, in conjunction with hypertension, obesity and
dyslipidemia, is involved in the emergence and development of DN
(2). Glomerular basement
membrane thickening, mesangial expansion, interstitial fibrosis and
podocyte damage represent the principal histological
characteristics of DN (3,4).
However, the mechanisms underlying these pathological changes
remain poorly understood.
Recent studies have indicated that posttranslational
modifications (PTMs) may profoundly regulate protein function,
shape cellular phenotypes to adapt to environmental conditions and
modulate the occurrence and progression of DN (5-8).
PTMs are chemical modifications in proteins that occur after their
synthesis and can notably affect their activity, stability,
localization and interactions with other molecules. Examples of
PTMs include phosphorylation, glycosylation, acetylation and
ubiquitination. Studies have suggested that targeting specific PTMs
or the enzymes that mediate their modification may offer an
alternative way to modulate the disease process and potentially
decelerate the progression of DN (9,10). Lactate and lactate-mediated
lactylation are novel epigenetic modifications. Lactate and
lactylation serve as a bridge between metabolic reprogramming and
epigenetic modifications, and they have been found to participate
in the pathogenesis and progression of various diseases,
particularly cancer (such as gastric cancer, colorectal cancer,
breast cancer, lung cancer and melanoma), cardiovascular diseases
and metabolic disorders (11,12). DN, which is associated with
metabolic diseases, has attracted increasing attention due to its
roles in lactate and lactylation. However, current research on
lactylation in kidney diseases is limited, and the best of our
knowledge neither phenotypic analyses nor mechanistic studies have
been performed. Therefore, a systematic review and further
investigations into the cellular functions of lactylation in DN are
warranted.
Lactate accumulation within the kidney
Glucose metabolism serves as the principal mechanism
of energy metabolism within cells. In a quiescent state, cells
convert glucose into pyruvate, and under optimal oxygen conditions,
pyruvate is fully oxidized in the mitochondria to yield carbon
dioxide and water, producing substantial energy. In hypoxic
environments, pyruvate is converted into lactate, a reaction
catalyzed by lactate dehydrogenase (LDH). For a long time, lactate
has been considered a superfluous and detrimental metabolic
byproduct in mammals. However, the understanding of lactate has
progressively expanded (13).
Investigations have revealed that lactate carries out a substantial
role in the pathophysiological processes underlying disorders,
fostering a revised understanding of this metabolic product. At the
dawn of the 20th century, Warburg et al (14) reported that, under conditions of
ample oxygen, tumor cells can still preferentially generate lactate
via glycolysis after assimilating glucose, and this metabolic trait
can facilitate cancer cell proliferation and invasion. The latter
phenomenon is known as the classic 'Warburg effect' in lactate
metabolism and is intricately associated with multiple autoimmune
and inflammatory disorders, tumors and metabolic diseases.
The kidney is a high-energy-consuming organ that
performs important functions in maintaining body homeostasis. In a
healthy kidney, there is a net uptake of lactate via
gluconeogenesis, and the kidney has a higher net lactate clearance
rate when compared with skeletal muscle, heart and other tissues,
with only the liver clearing more lactate from the circulation
(15). Consequently, lactate may
play a key role in the initiation and progression of diabetic
nephropathy. A previous study suggests that urinary lactate
excretion is considerably increased by ~2.7-fold in patients with
DN (16). A notable association
was observed between urinary lactate levels and eGFR; in addition,
elevated lactate levels were shown to induce fibrosis and impair
mitochondrial function in DN (16,17). Azushima et al (18) suggested that dysregulated kidney
metabolism, leading to increased lactate biosynthesis, might be
involved in DN pathogenesis. Moreover, elevated urinary lactate
concentrations in individuals with DN have also been demonstrated
to be potential biomarkers for predicting the progression of kidney
diseases. Studies indicate that a higher urine lactate/creatinine
ratio reflects increased renal glycolysis and impaired
mitochondrial oxidative phosphorylation and is associated with
faster eGFR decline in type 2 diabetes and DN cohorts. Urinary
lactate is therefore considered a non-invasive metabolic stress
marker and may complement albuminuria and tubular injury markers
for risk stratification, but it does not specify which downstream
pathways or cell types are affected (17,19,20). However, the precise underlying
mechanism remains elusive.
Lactylation-related modifications have been
implicated as potential diagnostic and prognostic indicators in DN,
but they are still in the early biomarker-discovery phase rather
than in established routine clinical use. Mechanistic and lactylome
studies show that global lactylation and specific lactylated
proteins are upregulated in diabetic kidney tissue and associate
with podocyte injury, mitochondrial dysfunction, inflammation and
renal fibrosis in rodent and human DN models (21-23). Urinary lactate, as a proxy for
the lactate-lactylation axis, has been identified as a non-invasive
early diagnostic biomarker for DN, with meta- and cohort analyses
showing elevated urinary lactate precedes or predicts albuminuria
and kidney-function decline (18,24). Machine-learning and
lactylation-related gene/protein signatures built from omics data
are being tested as prognostic risk scores in other diseases (for
example, gastric cancer and lung adenocarcinoma) and analogous
approaches are now being explored in DN-related fibrotic and
inflammatory pathways, though DN-specific clinical-prognostic
models remain largely preclinical (24-27). In DN specifically, lactylation is
currently positioned as a pathophysiological biomarker and
therapeutic target rather than a fully validated, clinically
deployed diagnostic/prognostic test (22-24). Compared with conventional
biomarkers of DN (for example, albumin-to-creatinine ratio, serum
creatinine/eGFR and tubular proteins such as NGAL or KIM-1),
lactylation-linked measures offer several conceptual and
mechanistic advantages, even if the majority are still
investigational (20,21,28,29). Lactylation integrates metabolic
stress (glycolysis upregulation), inflammation and epigenetic
reprogramming into a single PTM layer, so alterations in lactylated
sites may appear before overt structural damage or sustained
proteinuria (21,23,24). This metabolic-epigenetic readout
may capture subclinical tubular and immune-cell activation earlier
than late-stage glomerular or tubular leakage markers (22,28). Lactylation targets specific
residues on key proteins, allowing site-specific biomarkers that
directly reflect distinct pathogenic processes (autophagy
suppression, mitochondrial dysfunction or inflammasome activation)
rather than generic tubular injury (21-23). This specificity may improve
stratification of DN phenotypes (predominantly inflammatory vs.
fibrotic) and improve targeted therapies. Lactylation is tightly
coupled to intracellular lactate concentration and glycolytic flux,
so it can rapidly reflect changes in glucose control, hypoxia or
drug effects (for example, SGLT2 inhibitors or GLP-1R agonists)
that traditional biomarkers change slowly (21,22,24). This dynamic range may be useful
for monitoring treatment response or detecting subclinical relapses
before conventional tests deteriorate. Lactylome and
lactylation-related gene/protein signatures can be combined with
transcriptomic, proteomic and metabolomic data to build composite
risk scores that outperform single-variable biomarkers in capturing
heterogeneous progression trajectories (25-27,30). Such models can be embedded into
decision-support tools for personalized prognostication (for
example, distinguishing slow- vs. fast-progressors) in DN.
Lactylation involves 'writers' (for example, p300/CBP), 'erasers'
[histone deactylases (HDACs) or sirtuins (SIRT)] and 'readers',
therefore, abnormal lactylation patterns can pinpoint druggable
nodes in DN, blurring the line between biomarker and therapeutic
target (21,23,24). This contrasts with traditional
biomarkers such as albuminuria, which are readouts of damage rather
than actionable molecular targets themselves. Nevertheless, there
are limitations to the use of lactylation as a biomarker of DN.
Indeed, the majority of lactylation data in DN are still
preclinical or small-scale human cohorts, and robust, standardized
assays for global or site-specific lactylation are not yet
available in routine pathology. In addition, lactylation-based
signatures require LC-MS/MS or advanced antibody-based platforms
(for example, pan-lactylation western blotting or
immunofluorescence), which are more complex and expensive compared
with current ELISA- or urine dipstick-based tests. Therefore,
lactylation-related modifications are emerging as promising
research-class diagnostic and prognostic indicators in DN, with key
advantages in mechanistic depth, pathway specificity and
therapeutic-target alignment; however, they are not yet substitutes
for traditional biomarkers in clinical practice and will require
larger validation studies and assay standardization before being
adopted in guidelines.
Biogenesis of lactate
The synthesis of lactase is predominantly
facilitated by multiple LDH isoenzymes with high renal expression
(31). LDH is a tetrameric
protein composed of LDHA and LDHB isoforms, which exhibit distinct
kinetic characteristics. Specifically, LDHA and LDHB demonstrate
greater affinities for pyruvate and lactate, respectively (32). In the kidney, LDHA has been
identified as being produced mainly in proximal segments, whereas
LDHB is localized in distal segments. Furthermore, a previous study
suggested cell-specific production of both LDH isoforms in response
to acute and chronic kidney disease (CKD). Hypoxia may increase
LDHA production, while LDHB levels remain relatively stable, and
CKD downregulates both isoforms (33). These discoveries suggest that
lactate cell-cell shuttling, identified in astrocytes, may also
carry out a key role in the kidney. The latter notion delineates a
function for lactate in the distribution of oxidative or
gluconeogenic substrates and in cellular signaling (34).
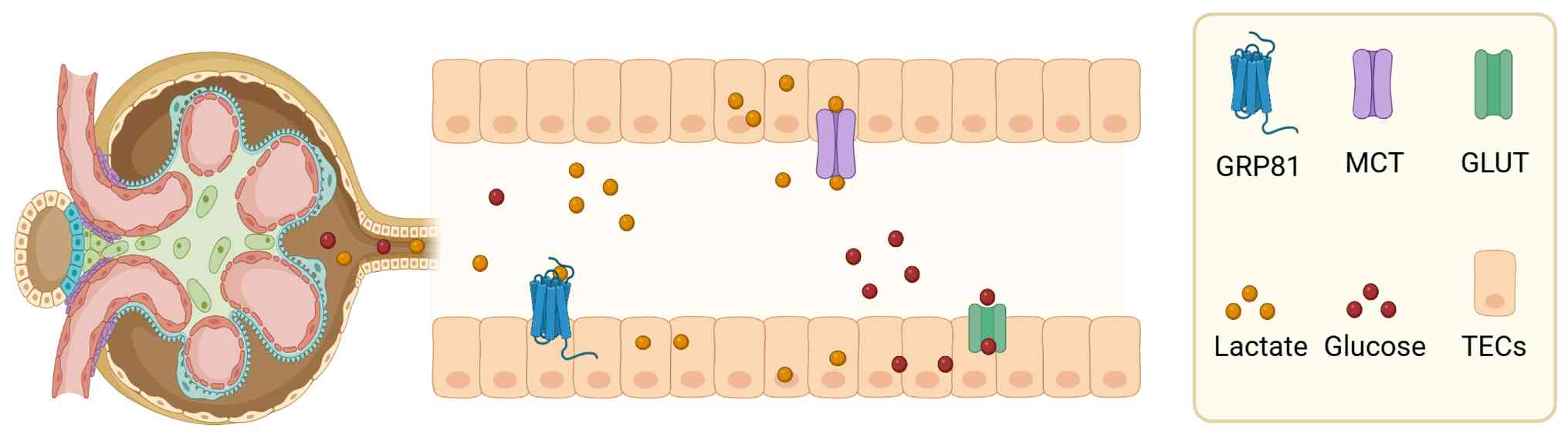
Lactate reabsorption in the kidney
Lactate is readily filtered through the glomerular
capillary. Consequently, the net reabsorption of lactate must
transpire along the nephron. Studies show that renal lactate
reabsorption in mammals is high (>95%) (35,36). Lactate is conveyed through two
main categories of transporters that are part of solute carrier
proteins and exhibit selectivity for monocarboxylates:
Proton-coupled monocarboxylate transporters (MCTs) and
sodium-coupled MCTs (SMCTs) (37,38). The MCT family comprises several
members that facilitate the cotransport of monocarboxylate anions
and protons, thereby balancing the amounts of substrate traversing
the plasma membrane in response to the combined concentration
gradients. Therefore, L-lactate and further MCT substrates traverse
from the production site to the utilization site. Proximal tubular
cells express proton-linked MCTs, including MCT1 and MCT2, and may
help transport lactate from the kidney to the bloodstream or
promote lactate or pyruvate uptake from the blood to achieve
gluconeogenesis (36). SMCTs
include SMCT1 and SMCT2. In mammalian kidneys, SMCT2 (a
low-affinity Na+-lactate cotransporter) controls the
majority of lactate reabsorption in the early segment of proximal
convoluted tubules (S1), whereas high-affinity SMCT1 in distal
proximal tubules (S2-S3) minimizes urinary lactate levels. A study
using the c/ebpδ null mouse model demonstrated that the
transcription factor C/EBPδ regulates the expression of slc5a8 and
slc5a12 by acting on their promoters. Consequently, the functional
double knock-out of these Na+/lactate co-transporters in
the kidney led to a 29-fold elevation in urine lactate levels,
suggesting that SMCTs account for the predominant lactate
reabsorption in the kidneys (39). In the DN, Zucker rats with
elevated plasma insulin levels showed elevated circulating lactate
levels and diminished muscle MCT4 and MCT1 content compared with
control animals (40). In
streptozotocin (STZ)-induced diabetes, resting plasma lactate
levels are increased (41), and
MCT1 and MCT4 densities and lactate transport are reduced (42).
In addition, lactate can enter the intracellular
space through GPR81 (or hydroxycarboxylic acid receptor 1), whose
activation downregulates cAMP and suppresses the protein kinase A
(PKA) pathway (43).
Furthermore, lactate-related GPR81 activation may signal via a
non-canonical, cAMP/PKA-independent pathway that involves the GPR81
adaptor protein β-arrestin under some conditions (44). In fact, GPR81 is expressed mainly
in fat tissue and, to a lesser degree, can also be detected in the
brain, kidney, liver, skeletal muscle and immune cells, although
its role in non-adipose tissues remains controversial (45).
In immune cells such as macrophages, lactate-induced
GPR81 activation triggers an anti-inflammatory signaling cascade.
Specifically, GPR81 activation recruits the adaptor protein
β-arrestin 2, which interacts with and inhibits the NLRP3
inflammasome or Toll-like receptor 4 (TLR4) signaling complexes,
thereby suppressing NF-kB activation and the subsequent release of
pro-inflammatory cytokines, including IL-1b and TNF-a (44,46). Furthermore, GPR81-mediated
signaling has been shown to inhibit the activity of yes-associated
protein, leading to the downregulation of pro-inflammatory
responses in macrophages following lipopolysaccharide (LPS)
stimulation (47). In models of
intestinal and hepatic injury, GPR81 activation has been
demonstrated to maintain tissue homeostasis and alleviate
inflammatory damage by inhibiting adenylyl cyclase to lower
intracellular cAMP levels or by modulating downstream kinase
activities (48).
The controversy over GPR81 in the kidney stems from
its lower expression levels compared with that in adipose tissue,
as well as its functional differences. While some studies have
shown that GPR81 activation has anti-inflammatory effects in
vitro in cultured macrophages and in vivo in intestinal,
synovial, muscle, myocardial and cancer tissues by suppressing
innate immunity (47-49), others have shown that GPR81 can
promote kidney fibrosis by inhibiting the cAMP/PKA pathway in renal
tubular cells (50,51). Lactate binding to GPR81 activates
a Gi-coupled signal that inhibits adenylyl cyclase, lowering
intracellular cAMP and thereby suppressing PKA/cyclic AMP response
element-binding protein (CREB) activity in renal stromal and
vascular cells. This loss of cAMP/PKA signaling removes an
important antifibrotic inhibitor system: CREB-driven Smad7
expression falls, TGF-β/Smad3 signaling is disinhibited and
RhoA/ROCK-dependent cytoskeletal remodeling and α-smooth muscle
actin (α-SMA) expression are favored, all of which promote
myofibroblast differentiation and extracellular matrix (ECM;
collagen, fibronectin) accumulation. At the same time, reduced
cAMP/PKA weakens anti-inflammatory tone, allowing increased
NF-κB-dependent cytokine production and leukocyte recruitment,
further stimulating fibroblast activation within the
tubulointerstitium. Because chronic kidney injury increases
glycolysis and lactate production, this creates a feed-forward loop
that drives progressive renal fibrosis: Hypoxia/glycolysis
activates lactate/GPR81 which inhibits cAMP/PKA resulting in
amplification of TGF-β signaling and inflammation (52-55). Nevertheless, these mechanisms
remain poorly understood, and additional studies are necessary to
determine the exact context-specific roles of GPR81 in the kidney
(Fig. 1).
Given the aforementioned findings, the present
review created an overview schematic diagram of lactate metabolism
in the DN (Fig. 2), from which
it can be observed that lactate metabolism in the kidney is key for
its pathophysiological effects on the kidney. An in-depth
exploration of lactate function in the kidney will help in
understanding the development of DN.
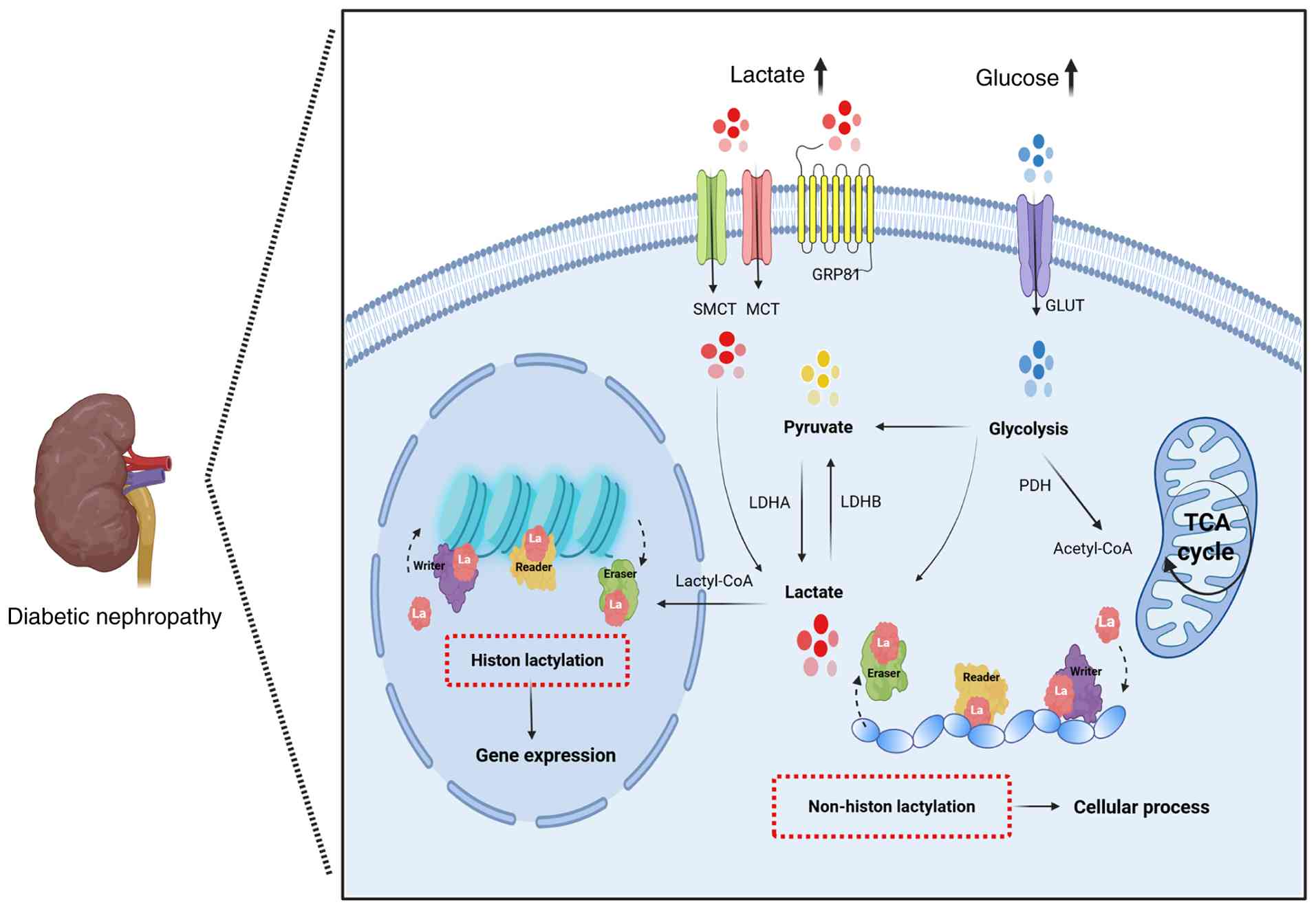 | Figure 2Overview of lactate metabolism and
mechanisms of histone or non-histone lactylation in diabetic
nephropathy. Intracellular lactate, mainly coming from glucose,
directly generates lactate via pyruvate through glycolysis.
Intracellular lactate undergoes rapid transport by SMCTs/MCTs.
Lactate in the microenvironment partially enters the circulation
and undergoes gluconeogenic metabolism in the kidney for
regenerating glucose. Lactate in the microenvironment may also be
involved in signaling pathways via GPR81 to regulate gene
expression. Lactate undergoes conversion into lactyl-CoA, which
transfers lactyl groups onto lysine moieties on histones and
non-histone proteins for lactylation, a process controlled by
epigenetic writers, readers and erasers, impacting gene expression
in cells epigenetically. MCTs, proton-coupled monocarboxylate
transporters; SMCTs, sodium-coupled monocarboxylate transporters;
LDHA, lactate dehydrogenase A; LDHB, lactate dehydrogenase B; PDH,
pyruvate dehydrogenase; La, lactylation residue. Figure created in
Biorender.com. |
Lactylation and DN
Discovery of lactylation
When cells undergo metabolic alterations, the
metabolic paradigm can shift from oxidative phosphorylation to
glycolysis, and the architecture and functionality of proteins
within the cells also undergo adaptive transformations. Proteins
can employ PTMs to modify their physicochemical properties by
adding chemical groups to amino acid residues, thereby altering
spatial configuration, increasing structural complexity and
governing the diversity and functionality of active proteins.
Common forms of PTMs include acetylation, ubiquitination,
phosphorylation and glycosylation, which carry out important roles
in the development of various diseases (56). Over the past 5 years, lactylation
has been considered a new PTM process. Indeed, lactate can serve as
a substrate for PTMs, thereby regulating the expression and
function of associated genes through lactylation (57).
In 2019, Zhang et al (58) first reported the histone
lactylation modification, laying the groundwork for subsequent
research. Their investigation revealed a mass deviation in lysine
residues of protein hydrolysis peptides, consistent with the mass
alteration induced by lactate binding to lysine residues. By
employing an array of experimental techniques, including western
blotting, isotope labeling and mass spectrometry-based
quantification, the latter authors validated the presence of
histone lactylation. In a different study, same research group also
reported that lactate is converted into lactyl-CoA and subsequently
transferred to histone lysine residues via lactate acyltransferase,
representing the classical process of lactylation modification
(59).
Regulation of lactylation
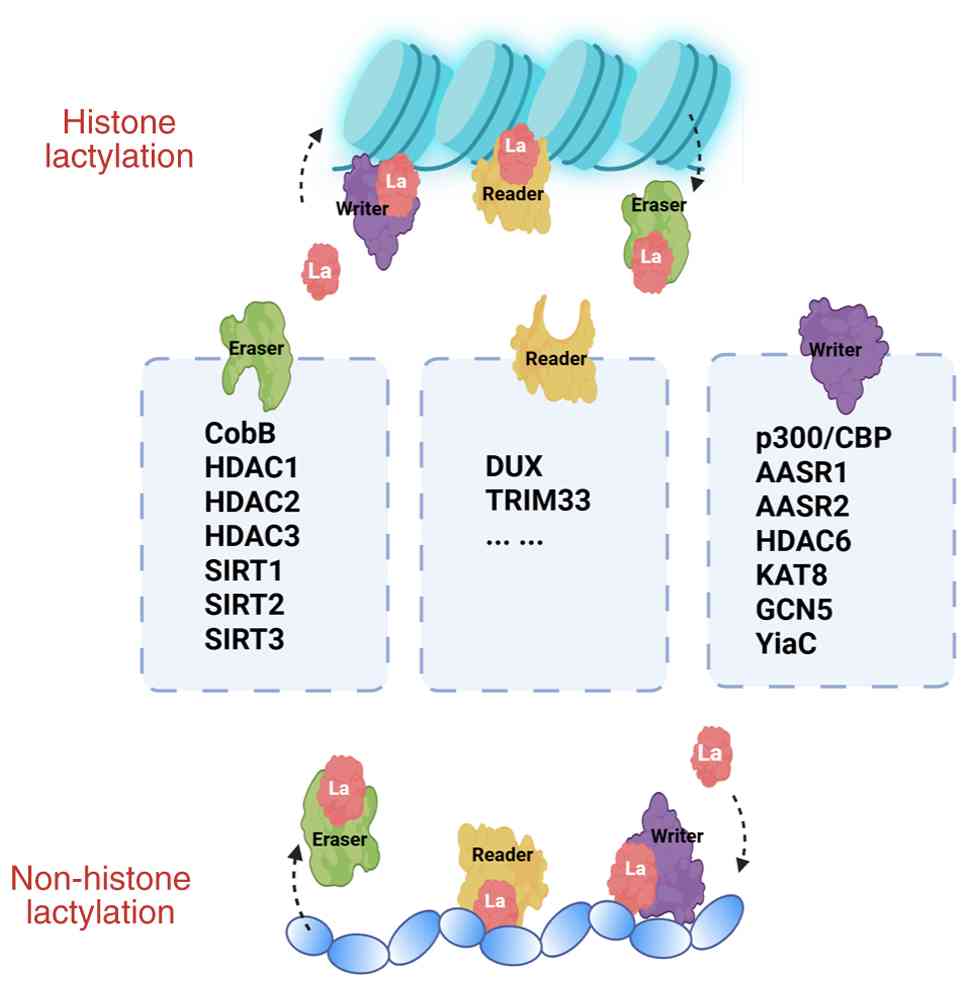
Enzymatic lactylation modifications are
predominantly regulated by histone acetyltransferases and HDACs.
Currently, several lactyl-CoA transferases, also called writers,
have been documented in mammals, including p300/CBP (60), KAT8 (61), general control of amino acid
synthesis 5 (GCN5) (62), the
GCN5-related N-acetyltransferase (GNAT) family protein YiaC and
mitochondrial alanyl-tRNA synthase 1/2 (AARS1/2) (51). Conversely, LDHs, also called
'erasers', include mainly the NAD-dependent protein deacetylases
CobB (63), HDAC1-3, SIRT1-3
(64). To the best of our
knowledge, studies on non-enzymatic lactylation modifications are
relatively rare, and further investigations are warranted (Fig. 3).
Previous studies have revealed that lysine
lactylation is regulated by both putative enzymatic (65) and non-enzymatic mechanisms
(66), but several key aspects
remain controversial. Clarifying these issues is essential for
accurate interpretation of lactate-driven epigenetic regulation in
DN. Current DN data clearly support increased protein lactylation
and both enzymatic and non-enzymatic lactylation routes in kidney
disease, but they do not yet directly demonstrate non-enzymatic
(D-lactoylglutathione-driven) lactylation in DN models. Available
data in kidney disease and DN show i) global increases in lysine
lactylation in db/db mice and other DN models and identify specific
lactylated targets such as ACSF2, and histone sites (H3K14la) that
drive mitochondrial dysfunction, EMT, inflammasome activation and
fibrosis, and ii) conceptual and experimental separation of
enzymatic lactylation (L-lactyl-CoA/p300-type) from non-enzymatic
D-lactylation via S-D-lactoylglutathione (LGSH), with the latter
firmly established biochemically in other systems and explicitly
described as the 'non-enzymatic lactylation' route (22,66,67). However, available articles to
date primarily infer that both mechanisms may operate under
high-lactate/oxidative stress rather than directly mapping
LGSH-dependent lactylation sites in DN tissue; thus, the presence
of non-enzymatic lactylation in DN should be framed as
mechanistically plausible and supported at the conceptual level,
but not yet conclusively demonstrated by DN-specific LGSH tracing
or stereospecific (L-vs. D-) lactyl-proteomics (23,68).
Enzymatic pathway (lactyl-CoA and
p300/AARS1 dependent)
Evidence suggests that intracellular L-lactate can
be converted to lactyl-CoA, which then serves as an acyl-donor for
lysine lactylation catalyzed by acyltransferases such as p300,
while AARS1 and other aminoacyl-tRNA synthetases have also been
reported to act as lactyltransferases that directly use lactate and
ATP (69). However, the very low
steady-state concentration of lactyl-CoA and the modest effect of
p300 knockdown on global lactylation levels have raised the issue
of whether p300 is the predominant physiological lactyltransferase
in vivo (67,70-73).
Non-enzymatic pathway
(high-lactate-driven)
The physiological relevance of non-enzymatic
lactylation remains a subject of intense debate. Although Gaffney
et al (66) provided
foundational evidence by demonstrating that lactyl-CoA and S-D-LGSH
can act as non-enzymatic acyl donors for lysine residues in
vitro, translating these findings to the diabetic kidney
remains contentious. As argued by Zhao et al (74) and Chen et al (67), the low intracellular abundance of
lactyl-CoA relative to acetyl-CoA challenges the kinetic viability
of spontaneous lactylation as a competitor to enzymatic acetylation
under pathophysiological conditions. Furthermore, the majority of
DN-related studies rely on pan-anti-Kla antibodies (75-77). Which lack the stereospecificity
required to distinguish between enzymatic L-lactylation (the
product of L-lactate-driven enzymatic pathways) and non-enzymatic
D-lactylation (the byproduct of the glyoxalase-linked non-enzymatic
route) (71,72). Consequently, the 'lactylome'
expansion observed in diabetic models, while associated with high
glycolytic flux, may be predominantly driven by the recruitment of
p300/CBP or other acting as acyltransferases (78). Thus, while the non-enzymatic
pathway is chemically plausible, its actual contribution to the
pathogenesis of DN remains inferred from correlative data rather
than confirmed by direct, site-specific mapping of D-lactylation
in vivo.
Potential impact on DN and 'metabolic
memory'
DN is characterized by long-term exposure of renal
cells, particularly podocytes and tubular epithelial cells, to a
high-glucose, high-lactate microenvironment, and has been
associated with elevated lactate to increased histone lactylation,
epithelial-mesenchymal transition (EMT), inflammation and fibrosis
in diabetic kidneys. Under such sustained conditions, non-enzymatic
lactylation of histone and non-histone proteins could serve as a
relatively slow-turnover 'molecular archive' of prior metabolic
states, thereby contributing to the phenomenon of metabolic memory
in DN, but this hypothesis has yet to be rigorously tested in
vivo and should be interpreted with caution (23,24,68,79).
Future studies combining quantitative metabolomics,
isotopic tracing and site-specific lactylation profiling in
diabetic kidneys are needed to disentangle enzymatic vs.
non-enzymatic (for example, acyltransferase-dependent lactoyl-CoA
pathways vs. glyoxalase-2-mediated lactoyl-glutathione chemistry)
sources of protein lactylation and to map their relative
contributions to DN progression. In parallel, integrating
flux-resolved lactate tracing with stoichiometric lactyl-proteomics
at defined stages of diabetic kidney injury will be essential to
associate specific lactylation events to upstream metabolic nodes,
clarify how 'writer' and 'eraser' activities are rewired in the
diabetic milieu, and identify targetable lactylation sites that
drive inflammation, fibrosis and mitochondrial dysfunction
(21,22,66).
Classification of lactylation
modifications
Lactylation modification primarily encompasses
histone and non-histone lactylation. Histone lactylation primarily
influences gene expression by altering chromosome architecture,
regulating transcription factor binding and altering promoter
accessibility. Non-histone lactylation predominantly governs
protein functions via steric hindrance, conformational alterations
and charge neutralization, thus impacting molecular interactions,
enzyme activity, subcellular localization and protein function
(80).
Although the enzymatic mechanisms are still being
investigated, the phenotypic outcomes driven by those enzymes are
already observed and well described in DN. Indeed, experimental and
translational work shows that histone and non-histone lactylation
are markedly upregulated in diabetic kidneys and associate lactate
accumulation to EMT, podocyte injury, endothelial-mesenchymal
transition and fibrosis. Lactylome and site-specific studies
demonstrate that specific lactylation events are tightly coupled to
defined pathogenic programs in DN, such as mitochondrial
dysfunction and profibrotic gene expression (21,23,24,68). Compared with conventional DN
biomarkers (albuminuria, eGFR, NAG or L-FABP), lactylation-based
readouts may offer several theoretical advantages. Pathway
specificity: Lactylation directly reports the activity of the
lactate-lactylation axis, integrating metabolic stress with
epigenetic and signaling changes, whereas albuminuria and
creatinine mainly reflect structural damage or filtration loss
(20,21,23,24). Cell-type and site resolution:
Histone or protein lactylation can be quantified at specific
residues and in defined renal compartments (podocytes, tubular
cells, endothelial cells), potentially distinguishing the
predominant pathogenic mechanisms in individual patients (21,23,24). Dynamic association with
treatment: Interventions that lower lactate or inhibit
LDH/p300/MCTs reduce histone lactylation and ameliorate DN
phenotypes in experimental models, suggesting that lactylation
levels might serve as pharmacodynamic markers of pathway-targeted
therapies (21,24,67,68).
Histone lactylation in DN
Histones, which are fundamental proteins that
associate with DNA and form the chromatin architecture, encompass
H2A, H2B, H3 and H4, also referred to as the core histones, which
are characterized by highly conserved amino acid sequences
(81). Initial evidence for
histone lactylation arises from high-performance liquid
chromatography-tandem mass spectrometry (HPLC-MS/MS) data, in which
lactylation of lysine residues was detected with a mass shift
identical to that resulting from the addition of a lactate group to
the ε-amino group of lysine, suggesting that lactylation
modification can transpire to the core histone H3 (58). Currently, numerous studies have
pinpointed lactylation sites on histones across various species;
the primary sites recognized to undergo lactylation include H3K9
(82), H3K14 (83), H3K18, H3K23 (84), H3K56 (85), H4K5 (86), H4K8 (87), H4K12 (88) and H4K16 (89), with H3K18 emerging as the most
thoroughly investigated lactylation site (90). Histone lactylation predominantly
enhances gene transcription, modulates cell necrosis and
proliferation, programs macrophages and initiates and progresses
tumors (60,91,92).
Studies assessing the implications of histone
lactylation in the onset and development of DN are rare.
Nevertheless, Zhang et al (77) reported that H3K14 lactylation may
be involved in DN through the regulation of EMT. The researchers
discovered that AGE-dependent induction of renal tubular epithelial
cells (RTECs) increased kidney lactate levels, and that reducing
lactate levels could notably hinder EMT progression and ameliorate
renal tubular fibrosis in DN. Mechanistically, lactate can
upregulate histone 3 lysine 14 lactylation (H3K14la) in DN. Further
ChIP-sequencing (seq) and RNA-seq data indicated that histone
lactylation is involved in EMT by upregulating Krüppel-like factor
5 (KLF5).
Furthermore, KLF5 binds the cdh1 promoter and
suppresses its transcription, thereby accelerating EMT in DN. In a
separate study on RTECs, it was found that H3 lysine 18 lactylation
(H3K18la) levels were notably elevated in the kidneys of DN mice.
Furthermore, the study revealed that a Kruppel-like zinc finger
protein transcription factor family member, Glis1 could bind to the
lactyltransferase KAT5 and markedly reduce the binding affinity
between histones and KAT5, thereby further decreasing H3K18
lactylation levels of H3K18 (93) (Fig. 4).
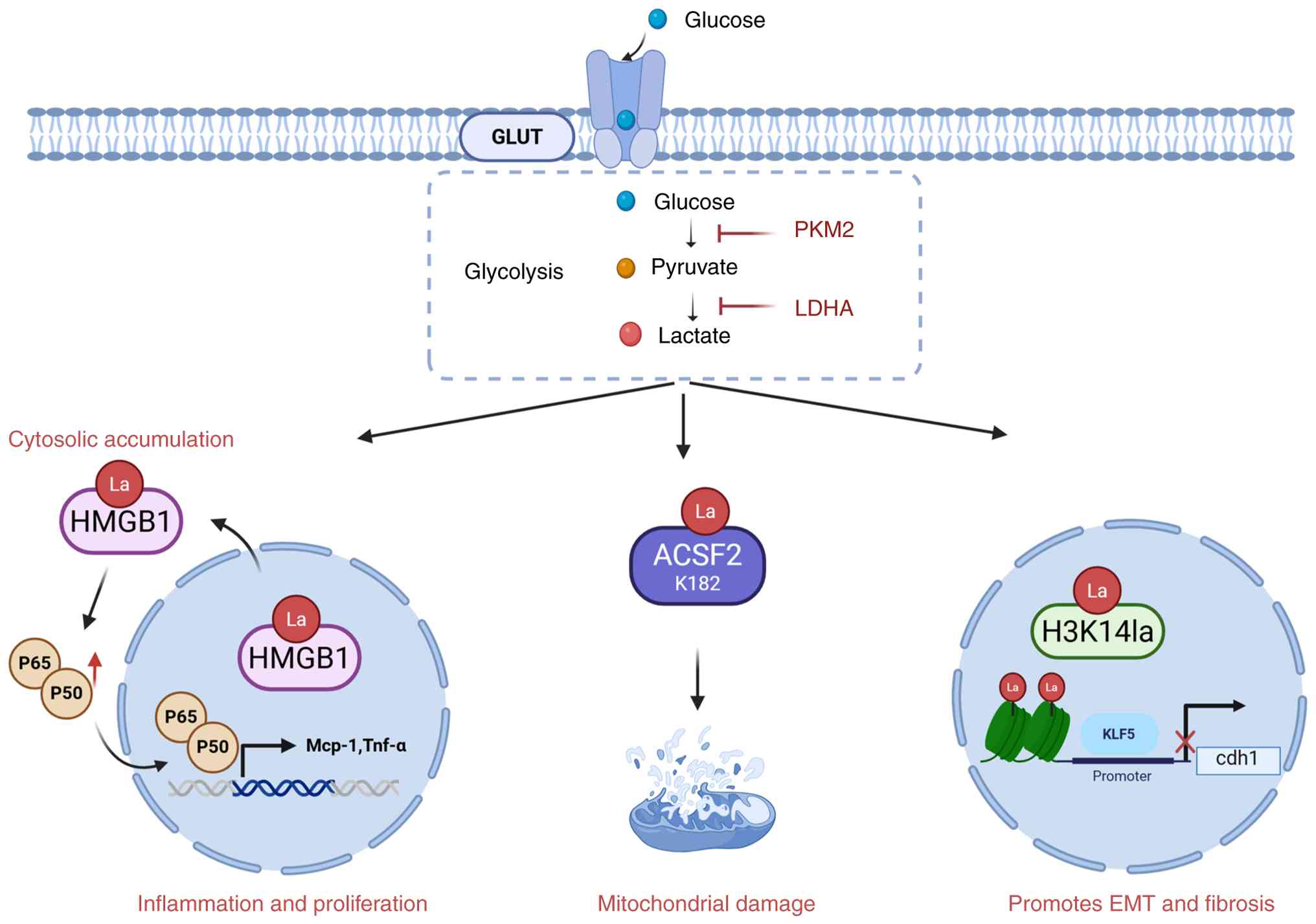 | Figure 4Lactylation facilitates disease
development and progression in diabetic nephropathy. Current
research on lactylation regulation in diabetic nephropathy has
shown that ACSF2, H3K14 and HMGB1 undergo lactylation in diabetic
nephropathy, which enhanced epithelial-mesenchymal transition,
mitochondrial damage and inflammation, thus exacerbating DN
progression. PKM2, pyruvate kinase M2; LDHA, lactate dehydrogenase
A; GLUT, glucose transporters; La, lactylation residue; ACSF2, acyl
CoA synthetase family member 2; HMGB1, High Mobility Group Box-1;
KLF5, Krüppel-like Factor 5; EMT, epithelial-mesenchymal
transition. Figure created in Biorender.com. |
Non-histone lactylation in DN
Non-histone lactylation represents another
pronounced regulatory site for lactylation; however, lysine
moieties on non-histone proteins more rarely accept lactoyl
substitutions than histones (60). A previous study revealed that
proteins with lactylation in the fungus Botrytis cinerea are
predominantly localized in the nucleus, mitochondria and cytoplasm
and participate in various cellular processes (94). In protozoan parasites, a diverse
array of non-histone lactylated proteins is also present, actively
engaging in processes such as splicing, cap binding, RNA export,
translation and degradation, the majority of which of which are
involved in glycolysis and can undergo lactylation (95). To date, research on non-histone
proteins regulated by lactylation in DN is scarce. The present
review focuses primarily on non-histone proteins confirmed to be
regulated by lactylation and summarizes the identified targets
implicated in DN progression (Table
I). Although lactylation of the majority of non-histone
proteins, such as p53, was first characterized in cancer and other
systems, their downstream signaling roles are highly relevant to
DN. For example, in podocytes exposed to a diabetic milieu, p53
activation is a well-established driver of apoptosis via
transcriptional upregulation of pro-apoptotic genes (for example,
Bax, PUMA and Noxa) and caspase activation (21,58,80,96-99).
 | Table IAn overview of associated non-histone
lactylation under investigation. |
Table I
An overview of associated non-histone
lactylation under investigation.
| Authors, year | Protein | Site | Disease | Molecule or
pathway | (Refs.) |
|---|
| Chen et al,
2024 | ACSF2 | K182 | Diabetic
nephropathy | Aggravated renal
tubule injury with excessive ROS accumulation, resulting in
mitochondrial damage. | (75) |
| Wang et al,
2022 | PKM2 | K62 | Wound healing | Suppresses its
tetramer-to-dimer transition, inducing its pyruvate kinase activity
and decreasing nuclear amounts to suppress inflammatory metabolic
adaptation. | (122) |
| Du et al,
2023 | HMGB1 | Pan | Liver
ischemia-reperfusion injury | Suppressed
macrophage chemotaxis and inflammatory activation. | (139) |
| Huang et al,
2024 | YY1 | K183 | Autoimmune
uveitis | Promoted microglial
dysfunction by increasing inflammatory cytokine secretion and
enhancing cell migration and proliferation. | (151) |
| Retinopathy of
prematurity | Increases the
transcriptional ability of YY1, directly upregulates FGF2 and
promotes angiogenesis. | (152) |
| Fan et al,
2022 | Snail | Pan | Myocardial
infarction | Disrupts
endothelial cell function and triggers mesenchymal-like function
after hypoxia via TGF-β/Smad2 pathway activation. | (162) |
| Wu et al,
2025 | | K9 | Idiopathic
pulmonary fibrosis | Increases ROS
production and mtDNA release by promoting peroxisome
proliferator-activated receptor γ coactivator 1α and PTEN-induced
kinase 1/PARKIN pathways | (304) |
| An et al,
2023 | Fis1 | K20 | Sepsis-induced
acute kidney injury | Promotes excessive
mitochondrial fission and then induces ATP depletion, mitochondrial
reactive oxygen species accumulation and mitochondrial
apoptosis. | (172) |
| Qiao et al,
2024 | Ezrin | K263 | Sepsis-associated
acute kidney injury | Interaction with
MYD88 and IRAK1, activating the NF-KB pathway, promotes
inflammatory responses. | (178) |
| Luo et al,
2022 | HIF-1α | Pan | Prostate
cancer | Promotes
angiogenesis and vasculogenic mimicry. | (187) |
| Li et al,
2025 | | K12 | Tumorigenesis | Increased
transcriptional activity by increased promoter occupancy and
upregulation of hypoxia-responsive related genes. | (302) |
| Cheng et al,
2024 | PFKP | K688 | Colorectal
cancer | Attenuated enzyme
activity formed a negative feedback loop in glycolysis and lactic
acid production. | (193) |
| Yan et al,
2024 | SOX9 | Pan | Non-small cell lung
cancer | Promote cell
stemness, migration and invasion via promoting glycolysis. | (200) |
| Wang et al,
2023 | Mecp2 | K271 | Cardiovascular
disease | Alters MAPK
signaling by regulating EGFR phosphorylation, thereby regulating
Vcam-1, Icam-1, Mcp-1, IL-1β, IL-6 and Enos in ECs, which in turn
inhibits atherosclerosis. | (206) |
| Li et al,
2024 | NEDD4 | K33 | APAP-induced liver
injury | Suppresses protein
interaction with Caspase-11. Restraining lactylation decreases
non-canonical pyroptosis in macrophages and alleviates liver
injury. | (215) |
| Huang et al,
2024 | TFEB | K91 | Cancer | Prevents the
interaction with E3 ubiquitin ligase WWP2, suppressing TFEB
ubiquitination and proteasome degradation, which enhances TFEB
activity and autophagy. | (229) |
| Zong et al,
2024 | P53 | K120/139 | Cancer | Lactylation of p53
reduces its liquid-liquid phase separation, DNA binding and
transcriptional activation, contributing to tumorigenesis. | (240) |
| Zhang et al,
2024 | AMPKα | Pan | Intervertebral disc
degeneration | Downregulates AMPKα
phosphorylation, enhances NP cell senescence and decreases
autophagy and ECM production through glycolysis. | (248) |
ACSF2
Acyl-CoA synthetase family member 2 (ACSF2) is a
non-histone protein that has been demonstrated to be subject to
lactylation and to exert functional effects during the development
of DN. ACSF2 catalyzes the first reaction of fatty acid metabolism
via the formation of a thioester with CoA (100). ACSF2 and other genes
controlling fatty acid oxidation can be induced by hepatocyte
nuclear factor 4 (HNF4), which affects adipocyte differentiation by
regulating fatty acid oxidation (101). A previous study reported that
ACSF2 was highly expressed in RTECs and localized mainly to
mitochondria. Proximal renal tubular cells have increased basal
metabolism and are enriched in mitochondria. Cells with elevated
metabolic rates prefer fatty acid oxidation for energy production,
as it yields more ATP than glucose oxidation (68). ACSF2 knockdown in HK2 cells
enhances hypoxia-reoxygenation (HR)-related mitophagy,
mitochondrial function restoration and reduction in mitochondrial
superoxide levels. The aforementioned evidence suggests that ACSF2
may have an important function in kidney disorders.
Further studies revealed that ASCF2 can be regulated
by lactylation in DN. Chen et al (75) reported markedly enhanced lysine
lactylation in the kidneys of both diabetic individuals and db/db
mice. Through lactylome analysis of kidney samples from db/db mice,
the authors detected 165 (356 lysine lactylation sites) upregulated
and 17 (22 lysine lactylation sites) downregulated proteins.
Subcellular localization analysis revealed that the majority of
proteins were lactylated in mitochondria (115 proteins, 269 sites).
Among these mitochondrion-localized proteins, ACSF2 and the
corresponding K182la lactylated modification impair mitochondrial
function in HK-2 cells exposed to high glucose, indicating that
targeting mitochondrial ACSF2 and lactylation might represent a
potential approach for DN (75)
(Fig. 4).
PKM2
Pyruvate kinase (PK) serves as a pivotal regulator
of glycolysis. PK can increase the rate of aerobic glycolysis by
catalyzing the conversion of phosphoenolpyruvate to pyruvate and
activating the pentose phosphate pathway. Competitive PK represents
one of the four distinct forms of this protein (M1, M2, L and R)
encoded by two genes (PKM and PKLR), with PKM2 comprising four
domains (A, B, C and N). PKM2 expression is apparent from the onset
of embryo development and continues over a lifetime in virtually
all tissues. Increased PKM2 activity reduces lactate production.
Previous research has demonstrated that oxaloacetate is present at
high levels and stimulates the Warburg effect via PKM2-mediated
pyruvate activation (102).
With reduced PKM2 activity, monomeric and dimeric PKM2 subtypes
undergo nuclear translocation, followed by interaction with hypoxia
inducible factor (HIF)-1α and regulation of various proglycolytic
enzymes (103). In the nucleus,
dimeric PKM2 functions as a histone kinase to increase c-Myc
expression, upregulating the expression of proglycolytic enzymes
for the induction of the Warburg effect (104).
Given the role of PKM2 in energy metabolism and the
substantial energy demand of the kidney, PKM2 plays a key role in
kidney disorders, particularly in DN. DN is characterized by a
chronic inflammatory response driven by macrophages, leading to
gradual deterioration of glomerular filtration barriers,
accompanied by glomerular hypertrophy, ECM buildup and induction of
the TGF-β pathway in RTECs, whose transformation is promoted
(95). In addition,
transcription factors such as NF-κB, STAT3 and HIF-1α are essential
for DN initiation and advancement (105). PKM2 regulates various adhesion
molecules, chemokines and inflammatory cytokines through the
aforementioned pathways. In the context of DN, PKM2 is
phosphorylated in glomerular endothelial cells, leading to its
isomerization and nuclear translocation to regulate inflammation
(106). These modulatory
functions increase STAT3 and NF-κB phosphorylation and upregulate
the expression of the adhesion molecule intercellular adhesion
molecule-1 (ICAM-1), facilitating the infiltration of inflammatory
cells and participating in DN progression (107). Qi et al (108) reported that PKM2 activation is
associated with reduced levels of toxic glucose metabolites and
preserved kidney function, while suppressing PKM2 phosphorylation
might diminish kidney inflammation, offering protection against DN
(108). Tetrameric PKM2
suppresses PKM2 phosphorylation, inhibits aberrant glycolytic
activity and downregulates ICAM-1, type I collagen a3 and TGF-β1 in
DN, in addition to preventing renal fibrosis due to elevated
albuminuria in diabetic patients (109,110). Tetrameric PKM2 also suppresses
macrophage adhesion and NF-κB and STAT3 pathway activation, further
alleviating kidney fibrosis (111,112). A decrease in tetrameric PKM2
levels in DN leads to diminished glucose metabolic flux and
enhances the accumulation of toxic glucose metabolites in
podocytes, confirming the potential of PKM2 in preventing or
alleviating severe diabetic microangiopathy (113,114). Since PKM2 is capable of
mitigating renal failure in diabetes, pharmacological strategies
that increase PKM2 expression may improve the prevention of DN
progression (115).
Indeed, research demonstrates that PTMs can modify
the structural and functional features of PKM2, playing a pivotal
role in endogenous allosteric regulation (116,117). For example, citrullination of
PKM2 R106 results in a reprogrammed interaction of PKM2 ligands for
increased activity (118),
whereas SUMOylation of PKM2 K270 induces a conformational shift of
PKM2 from the tetramer to the dimer, diminishing PK activity
(119). Furthermore, PKM2
acetylation at K433 increases its activity (120), whereas O-GlcNAcylation or K311
sacculation impedes PKM2 activity and results in an enhanced
Warburg effect (121). In
addition, PKM2 can undergo lactylation. For the first time, Wang
et al (122) identified
PKM2 as a lactylation substrate in proinflammatory macrophages.
They demonstrated that PKM2 lactylation enhances its PK activity
and diminishes its tetramer-to-dimer transition and nuclear
localization. Lactate reduces glycolysis by activating PKM2 to a
greater extent, thereby promoting macrophage phenotypic changes
along the inflammatory-reparative continuum. Increased PKM2
lactylation at K62 may directly convert PKM2 into a tetramer with
increased PK activity.
HMGB1
HMGB1, a cytoplasmic protein with an HMG-box domain,
serves as a quintessential representative of damage-associated
molecular patterns. It can translocate into the nuclear
compartment, where it binds to nucleosomes to regulate gene
transcription. HMGB1 resides within the nuclear compartment and
functions as a DNA-scaffolding protein (123,124). In addition, nuclear HMGB1 may
be secreted into the extracellular space, where it acts as a key
inducer of the inflammatory response by recruiting and activating
macrophages and other immune cells (125,126). Under typical conditions, HMGB1
primarily facilitates gene transcription, DNA repair and various
other biological processes (127). Upon release from immune or
damaged cells, it functions as an inflammatory modulator that binds
to membrane receptors, activating downstream intracellular
pathways, including the NF-kB-mediated proinflammatory response, in
the pathogenesis of kidney disease (128).
Multiple studies have elucidated function of HMGB1
in DN (129-136). Circulating and urinary HMGB1
levels are both high in patients with DN. Serum from mice with DN
induces HMGB1 expression, suggesting a potential association
between increased HMGB1 levels and DN (129). The increase in HMGB1 secretion
contributes to DN progression by modulating the TLR2/TLR4-NF-κB
pathway. In addition to HMGB1 upregulation in the presence of DN
serum, elevated apoptosis and reduced autophagy were observed in
MPC5, a mouse podocyte line. HMGB1 downregulation mitigated DN
serum-dependent podocyte apoptosis, indicating that HMGB1 may act
as an important risk factor for podocyte damage (130). In DN biopsy samples, both TLR4
and HMGB1 are overexpressed in tubular regions, suggesting that
HMGB1 is involved in the TLR4-mediated tubular inflammatory
response in DN (131). The
HMGB1 suppressor glycyrrhizic acid was shown to improve
inflammatory responses in STZ-induced DN rats by modulating
receptor for AGE (RAGE)/TLR4-associated ERK and p38 MAPK/NF-kB
induction (132). Notably,
HMGB1 was found to increase autophagy under conditions of oxidative
stress and other stimuli, particularly in cancer cells (133). The autophagy inducer rapamycin
(Rap) promotes histological and renal functions in an
ischemia/reperfusion rat model via the inhibition of HMGB1 release
(134). Previous data suggest
that renal tubular cells and podocytes are the primary sources of
secreted HMGB1 within the kidney (135). In addition, evidence indicates
that RAGE is the principal receptor for HMGB1, and suppression of
bone marrow-derived RAGE has been shown to enhance kidney function
in mice with experimental DN (136).
HMGB1 can also undergo PTM through lactylation.
HMGB1 secretion involves two phases: Nuclear-to-cytoplasmic
transport and exosomal release into the extracellular milieu. HMGB1
translocation from the nuclear compartment to the cytosol is
facilitated by various PTMs (137). Yang et al (138) first showed that HMGB1 may be
secreted into exosomes (exos) by macrophages during sepsis
following lactylation. In hepatocytes, reducing HMGB1 lactylation
and secretion, either by inhibiting lactate production or by using
a p300 suppressor, could blunt HMGB1 cytoplasmic accumulation and
subsequent exosomal secretion. It could inhibit macrophage
chemotaxis and improve the subsequent inflammatory response,
eventually protecting against liver ischemia-reperfusion (LI/R)
injury (139). Wu et al
(140) revealed that HMGB1 is
markedly upregulated in the kidneys of DN mice and in mesangial
cells treated with high glucose, with cytosolic HMGB1 increasing
the inflammatory response and proliferation in mesangial cells.
Mechanistically, they reported that cytoplasmic HMGB1 accumulation
is modulated by the nucleocytoplasmic translocation of
lactate-dependent HMGB1 acetylation and lactylation and further
induces NF-κB signaling via direct binding to IκBα, which helps
explain the regulation of HMGB1 lactylation (Fig. 4). In patients with acute kidney
injury (AKI) with acute decompensated heart failure, Zhu et
al (141) indicate that
lactate serves as an independent predictor. Combined application of
lactate and low-dose LPS to mice considerably provokes HMGB1
lactylation levels. Additionally, lactate-mediated HMGB1
lactylation is positively associated with circulating neutrophil
extracellular traps levels.
YY1
YY1, also referred to as δ, NF-E1, UCRBP and CF1,
is an important constituent of the GL1-Kruppel group of
ubiquitously expressed and evolutionarily conserved zinc-finger
transcription factors. The YY1 gene is located on human chromosome
14q32.2. The YY1 protein comprises 414 amino acid residues and has
a relative molecular weight of 65 kDa. Adjacent to the N2 terminus
of YY1, there is an acidic domain, followed by a sequence linking
12 guanosine nucleotides and a region abundant in Gly and Ala. The
four zinc fingers at its C-terminus engage with target promoters,
which typically harbor YY1 binding elements: CCAT and ACAT. YY1 is
a multifunctional protein with regulatory roles in normal
physiological processes, for example, developmental processes and
cell differentiation and division, and performs dual functions of
both repressing and activating transcription, resulting in its
designation as Yin-Yang 1 (142). Shi et al (143) first reported that YY1 is a
transcription factor that interacts with the adenoviral P5 promoter
and that its transcriptional repression activity can be converted
to transcriptional activation by adenoviral E1A. Reflective of this
dual functionality, YY1 can both inhibit and activate
transcription, contingent on the cotranscription factors it
recruits.
Mounting evidence suggests that YY1 is involved in
the pathogenesis of DN. Indeed, YY1 is important for the initiation
of renal fibrosis associated with DN. In addition, YY1 serves as a
notable modulator of α-SMA and epithelial-EMT-related proteins in
high glucose (HG)-induced DN. In both cultured HK-2 cells and
diabetic mouse models, HG markedly increased YY1 levels and nuclear
translocation through mTORC1/P70S6K signaling. Moreover, YY1
downregulation via short hairpin RNA substantially diminished
HG-dependent α-SMA expression and activity and inhibited
HG-triggered EMT, thereby alleviating kidney fibrosis in db/db mice
with DN. YY1 overexpression elevated creatinine, blood urea
nitrogen and urinary albumin levels; enhanced Masson and Sirius red
staining signals; and upregulated laminin and type IV collagen,
thereby promoting kidney fibrosis in a DN mouse model (144). Another investigation revealed
that YY1 is upregulated by diabetic hyperglycemia in DN mice.
Notably, YY1 is exclusively upregulated in mesangial cells, which
are important for ECM production and the subsequent development of
glomerulosclerosis in DN (145).
Additionally, the divergent functions of YY1 in
various diseases might be attributed to the recruitment of distinct
cofactors upon its stimulation, which subsequently modulates YY1
function to either suppress or activate transcription (146). Numerous studies have reported
that YY1 binds to a diverse array of transcription factors. YY1
forms a complex with nuclear factor erythroid 2-related factor 2,
binding to the TGFβ1 promoter and acting as a cofactor for YY1. YY1
directly binds to the TGFβ1 promoter and suppresses TGFβ1
expression transcriptionally in human kidney mesangial cells. In
mice, although YY1 levels are high in mesangial cells at the early
stages of DN lesions, they are reduced at later disease stages. YY1
silencing in the kidney exacerbates glomerulosclerosis, which is
mitigated by YY1 overexpression (147).
Furthermore, patients with higher YY1 expression
developed DN more slowly compared with those with lower YY1
expression. Additionally, the small-molecule compound eudesmin can
suppress TGFβ1 and other profibrotic proteins by upregulating YY1
in human kidney mesangial cells and by alleviating DN lesions in DN
mice through YY1 upregulation (148). The findings of Yang et
al (144) indicated that
mitochondrial dysfunction and YY1 upregulation in RTECs occurred
earlier than the onset of tubulointerstitial fibrosis (TIF) in DN.
Their data also demonstrated the generation of an mTOR-YY1
heterodimer triggered by HG-upregulated YY1, with nuclear
translocation impairing mitochondrial function in RTECs by
inactivating PGC-1α, leading to TIF in the early stages of DN via
EMT. These findings highlight YY1 as a new modulator of
mitochondrial function in RTECs, and a reduction in YY1 at the
early stages may serve as a promising preventive approach for
DN-related TIF.
Previous studies on epigenetic alterations in YY1
include phosphorylation at S118, which plays a role in
atherosclerosis (149), and
deacetylation, which is associated with renal fibrosis (150). Using a specific YY1-K183la
antibody (YY1-K183La), both animal and cell culture data indicated
that YY1 could also be subject to lactylation, with microglial
activation coexisting in autoimmune uveitis (AU). By modulating YY1
lactylation through interventions targeting lactate levels, YY1
mutations and p300 regulation, microglial activation was markedly
mitigated. Furthermore, CUT and Tag analysis demonstrated that YY1
lactylation enhances several inflammatory genes associated with
alterations in microglial functions, including migration,
proliferation and activation. In summary, these findings suggest
that YY1 lactylation is pivotal for microglial activation in AU.
For the first time, YY1 lactylation was shown to be a potential
pathogenic factor in AU (151).
Another investigation of retinal angiogenesis also demonstrated
that lactylation at K183 in YY1, a non-histone protein, enhances
YY1 transcriptional capacity, directly upregulating FGF2 expression
and promoting angiogenesis; the overexpression of p300, a
lactylation writer, resulted in increased YY1 lactylation, whereas
its inhibition via A485 led to reduced YY1 lactylation in both
animal and cell culture assays, suggesting that p300 may modulate
YY1 lactylation, thereby influencing the progression of retinal
angiogenesis (152).
Snail1
The Snail family is classified within the
superfamily of zinc finger proteins. In mammals, the Snail gene
family comprises three members: Snail1, Snail2 and Snail3 (153). Members of the Snail family
encode products with analogous structures, featuring conserved
carboxyl (DNA-binding) and variable amino (regulatory) terminal
domains, with the carboxyl terminal end incorporating 4-6 cysteine
(Cys)-histidine (His) zinc-finger motifs that establish
coordination bonds with Zn2+ ions, facilitating binding
to oligonucleotide sequences in the E-boxes of target gene
promoters and modulating their transcription (154). Snail1, which is located on
human chromosome 20q13.2, encompasses 264 amino acids (155). It is pivotal in EMT and
considerably influences several pathophysiological events
associated with EMT, for example, embryonic development, tumor
invasion and metastasis, wound healing and organ fibrosis. Previous
investigations have revealed that EMT is essential in renal
fibrosis and that Snail1 downregulates E-cadherin by interacting
with the CAGGTG sequence in the E-box of the E-cadherin promoter,
thereby initiating a key step in the EMT process (156).
In DN, renal fibrosis is always the endpoint event
that influences disease progression and prognosis, highlighting a
notable role for Snail1 in this mechanism. Snail1 substantially
affects EMT by suppressing E-cadherin expression while promoting
the upregulation of vimentin and fibronectin (FN) (157). The activation of Snail1
facilitates EMT in tubular epithelial cells and contributes to TIF
(158). Conversely, knockdown
of Snail1 reverses EMT in tubular epithelial cells and mitigates
the progression of diabetes-related TIF (159). Snail1 induces TIF and alters
E-cadherin and FN levels in tubular cells. Notably, K85- and
K146-linked ubiquitination of Snail1 destabilizes it and
facilitates its degradation via the ubiquitin-proteasome system
(UPS) (160). A mechanistic
study revealed that USP22, through its deubiquitinase activity,
deubiquitinates and stabilizes Snail1. In in vivo
experiments, disrupting USP22 could ameliorate kidney pathology and
enhance kidney function in a db/db mouse diabetes model by
downregulating Snail1, thereby inhibiting EMT and diminishing ECM
generation (161).
A study in the heart revealed that lactate could
accelerate the lactylation of Snail1, facilitating its nuclear
translocation and binding to the TGF-β promoter, thereby
upregulating TGF-β. Snail1 suppression could diminish
lactate-induced EMT following myocardial infarction (MI)/hypoxia
and improve cardiac function. These findings indicate that lactate
may not only serve as a key prognostic biomarker for heart attack
and heart failure but also affect the pathophysiological processes
of heart fibrosis via the promotion of EMT after MI. Lactate
administration increased Snail1 nuclear translocation in response
to hypoxia, with nuclear Snail1 interacting with the TGFβ1 gene for
TGF-β1 upregulation. Furthermore, Snail1 suppression inhibited EMT
and TGF-β/Smad2 activation associated with lactate upon hypoxia,
demonstrating a role for Snail1 in lactate-triggered
TGF-β/Smad2-dependent EMT. In animal studies, Snail1 knockdown
ameliorated heart function impairment and endothelial-mesenchymal
transition induced by lactate post-MI. The study also revealed that
lactate stimulates both Snail1 acetylation and lactylation in
response to hypoxia/MI. Treatment with the MCT suppressor,
α-cyano-4-hydroxycinnamate markedly alleviated Snail1 lactylation
induced by lactate, suggesting that lactate-associated Snail1
lactylation and nuclear translocation represent a notable mechanism
behind lactate-induced EMT via TGF-β/Smad2 signaling induction
after MI (162).
Mitochondrial fission 1 protein
(Fis1)
Mitochondria are highly dynamic organelles that
maintain homeostasis through continuous fusion and fission.
Disruption of fission-fusion equilibrium in mitochondria, for
example, exaggerated mitochondrial fission, modifies several
cellular events, including oxidative stress, apoptosis and
inflammatory responses, and contributes to the progression of
various diseases such as diabetic nephropathy, renal fibrosis,
cardiovascular diseases and neurodegenerative disorders,
constituting a hallmark of numerous pathologies (163,164). Fis1, which serves as a
mitochondrial outer membrane adaptor, can bind to the fission
executor dynamin-related protein 1 to facilitate mitochondrial
fission (165). Multiple
studies have validated increased Fis1 expression in tubular cells
from cases of DN (166).
Moreover, strong associations of Fis with Mfn1 and mitochondrial
breakdown in tubular cells were observed, indicating a pivotal
regulatory role for Fis in mitochondrial dynamics in kidney
tubules. Research on Chinese medicines has revealed that
formononetin can alleviate albuminuria and kidney histopathology by
reducing RTEC apoptosis and mitochondrial breakdown and, more
importantly, restoring the expression of Fis1 and
apoptosis-associated proteins, for example, Bax, Bcl-2 and
cleaved-caspase-3, in HK-2 cells exposed to high glucose (167). Additional results indicated
that another herbal compound, astragaloside II, could also
ameliorate albuminuria, kidney histopathology, podocyte foot
process effacement and podocyte apoptosis in a rat diabetes model,
which was partly related to restoring kidney levels of Fis1 and
autophagy-associated proteins (168).
Numerous reports have investigated the PTMs of
Fis1. A previous investigation revealed that phosphorylated Fis1
could mediate mitochondrial breakdown (169). Additionally, Fis1 can be
ubiquitinated by the E3 ubiquitin ligase Parkin, which targets Fis1
for proteasome-dependent degradation and dysregulation of this
process is associated with mitochondrial dysfunction and aging
(170). High levels of
acetyl-CoA induce the acetylation of Fis1, promoting its
ubiquitin-proteasomal degradation and thereby attenuating
mitochondrial fission (171).
Furthermore, An et al (172) reported that lactate could
induce an increase in Fis1 lactylation at K20 (Fis1 K20la) and
further promote aggravated mitochondrial fission and subsequent
functional impairment. The activation of PDHA1, a subunit of
pyruvate dehydrogenase (which catalyzes the conversion of pyruvate
to acetyl-CoA), can reduce Fis1 K20 lactylation, thereby
alleviating sepsis-associated AKI (SAKI). These findings emphasized
lactylation as a novel PTM of the non-histone Fis1.
Ezrin
Ezrin, a member of the ezrin-radixin-moesin family,
also known as p81, can be phosphorylated by the EGFR tyrosine
kinase (173). Ezrin is a
globular structural protein (molecular weight of 78 kDa) that
connects the cell membrane and actin filaments. It can associate
with several proteins, including CD44, CD43, L-selectin, ICAM-1,
ICAM-2, PSGL-1 and the death receptor CD95/Fas, thereby engaging in
cell adhesion and apoptosis (174). Ezrin is expressed in multiple
tissues, with elevated expression in the small intestine, stomach,
lung, pancreas and kidney (175). It functions as a linker between
the membrane and microfilaments, facilitating interactions between
the cytoplasmic membrane and the actin cytoskeleton while
preserving structural integrity, such as maintaining the integrity
of the cellular epithelium and mediating intracellular transduction
of extracellular mechanical signals. As a
membrane-microfilament-linking protein, ezrin can generate membrane
protrusions, regulating cellular remodeling, including cell
proliferation, deformation, migration and adhesion, thereby playing
key roles in sustaining cell shape and modulating cell movement.
Ezrin comprises 586 amino acids and contains phosphorylation sites,
including Tyr145, Tyr353 and Ser66.
Furthermore, Ezrin contains two cysteine residues,
which are considered to undergo nitration modification (176). Ezrin serves as a key
crosslinker protein within this complex, facilitates the
development of elongated projections of the podocyte slit diaphragm
and controls several cellular activities, for example, adhesion and
mobility. Disrupted ezrin/actin complexes have been detected in
animal models of podocyte injury, for example, in rats administered
puromycin aminonucleoside and the amount of podocyte ezrin is
inversely associated with the severity of proteinuria-related
kidney disease in pediatric patients (177).
Regarding lactylation, Qiao et al (178) partially clarified the
association between elevated blood lactate levels and the increased
incidence and unfavorable prognosis of SAKI. The increase in Ezrin
may further exacerbate inflammatory responses, primarily through
its interactions with MYD88 and IRAK1, thereby activating the NF-κB
pathway. Histone H3K18 lactylation (H3K18la) is increased in SAKI.
Moreover, this lactate-induced histone modification is enriched at
the promoter of Ras homolog gene family member A (RhoA) and is
positively associated with transcription. Rectification of altered
lactate levels reversed atypical histone lactylation at the RhoA
promoter. Investigation of the associated mechanisms revealed that
histone lactylation stimulated the RhoA/Rho-associated protein
kinase (ROCK)/Ezrin signaling pathway and activated NF-κB, the
inflammatory response and apoptotic cell death, exacerbating
impaired kidney function. Additionally, ezrin can undergo
lactylation as a non-histone protein. Mutation of the K263 site
(K263R) was found to counteract the regulatory effects of lactate
on ezrin-mediated renal injury.
HIF-1α
HIF-1 is a heterodimeric protein containing α and β
subunits, and the α and β subunits can operate as transcription
factors only following dimerization. HIF-1α represents the
functional subunit of HIF-1, while HIF-1β serves as the structural
subunit of HIF-1 and is expressed at relatively stable levels
within cells. Under normoxia, synthesis of the HIF-1α subunit
occurs concurrently with its degradation via the UPS, resulting in
minimal detection. However, under hypoxia, HIF-1 breakdown is
suppressed, promoting the accumulation and nuclear translocation of
HIF-1α, where it binds to HIF-1β for the generation of HIF-1
complexes (179). Under
hypoxia, HIF-1α upregulation stimulates the expression of numerous
target genes, regulating cellular proliferation, neovascularization
and remodeling, programmed cell death, energy metabolism and iron
transport.
There is tissue-specific expression of HIF-1α in
the kidneys. Elevated glucose levels enhance HIF-1α expression and
activity in human mesangial cells, thereby inducing
glomerulosclerosis. Conversely, high glucose can also diminish the
stability and function of HIF-1α in the proximal tubular HK-2 cell
line, exacerbating DN tubulointerstitial injury (180). Another perspective suggests
that under conditions of low oxygen and high glucose, high HIF-1α
expression in cells renders kidney tissue and cells more
susceptible to fibrosis (181).
Prolonged activation of HIF-1α in proximal tubules can promote TIF.
Inhibition of HIF-1α attenuates fibronectin expression associated
with low oxygen in high-glucose cells, thereby mitigating fibrosis
(182). Throughout DN
progression, elevated glucose alters HIF-1α stability and activity,
influencing vascular endothelial growth factor (VEGF) transcription
in renal cells, leading to decreased VEGF production and promoting
TIF (183). Another
investigation revealed that in DN, during the compensatory phase,
the upregulation of HIF-1α expression under hypoxic conditions can
also inhibit apoptosis and ROS formation through the induction of
mitochondrial autophagy, providing a protective effect (184). However, a sustained
high-glucose and low-oxygen environment disrupts HIF-1α stability
and inhibits mitochondrial autophagy (185). Impaired mitochondrial autophagy
further exacerbates mitochondrial damage, ROS imbalance and
disrupted intracellular homeostasis, thereby promoting the
progression of DN. Additionally, HIF-1α is implicated in
DN-associated inflammation, stimulating the production of various
cytokines involved in immune responses. Under high-glucose
conditions, high HIF-1α expression increases the levels of
inflammation- and fibrosis-associated cytokines, for example,
TGF-1β, endothelin and fibronectin, in mesangial cells,
exacerbating the incidence of interstitial fibrosis in DN (186).
Regarding the lactylation of HIF-1α in prostate
cancer (PCa), Luo et al (187) demonstrated that HIF-1α serves
as a transcriptional inducer of KIAA1199, which is involved in
pathways such as glycolysis, hypoxia and angiogenesis. Given that
lactylation has been identified as a key mechanism of
posttranscriptional regulation initiated by lactate, the potential
for lactate to stimulate HIF-1α lactylation in PCa cells was
further examined. Initially, in PCa cells treated with varying
lactate amounts for 72 h under normoxic conditions, increased
HIF-1α lactylation was observed. Following 72 h of culture with 10
mM lactate, substantial HIF-1α lactylation was noted in both the
PC-3 and DU145 cell lines. Additionally, immunofluorescence
revealed that lactate administration under normoxic conditions
enhances colocalization of lactylation and HIF-1α in the PC-3 cell
line, an effect reversed by MCT1 silencing. These findings indicate
that lactate augments HIF-1α expression through its
lactylation.
PFKP
Phosphofructokinase-1 (PFK-1) converts
fructose-6-phosphate into fructose-1,6-bisphosphate and is
modulated by fructose-2,6-bisphosphate(188). PFK-1 is the principal
rate-limiting enzyme in glycolysis and represents an important
juncture in this metabolic process. There are three variants of
PFK-1 in vertebrates, designated PFKM (muscle-specific), PFKL
(liver-specific) and PFKP (platelet-specific), based on their
discovery locations, with PFKP assumed to play a pivotal role in
the kidneys (189). PFKP, a
predominant PFK-1 isoform involved in tumor cell glycolysis, has
emerged as a promising anticancer target. The kidney ranks among
the human organs with the greatest metabolic rates, primarily
driven by tubular epithelial cells. The kidney needs to maintain
energy homeostasis, as disruptions in energy metabolism may cause
cellular dysfunction, cell death and multiple renal pathologies. In
diabetic patients, alterations in metabolic substrates and oxygen
delivery lead to hypoxia, enhanced glycolysis and lipid
accumulation in tubules, culminating in elevated amounts of ROS,
proinflammatory mediators and profibrotic molecules, as well as
increased PTEC apoptosis and kidney fibrosis. Lang et al
(190) reported that PFKP is
highly expressed and is markedly upregulated in glomerular tissues
from db/db mice. Similarly, PFKP is upregulated in samples from
patients with DN compared with control kidney tissue samples from
patients with cancer.
Furthermore, elevated ACR levels, increased
mortality, and more pronounced foot process fusion were observed in
db/db mice treated with CTZ, a PFKP suppressor (191). Additionally, the application of
FBP, a product of PFKP, ameliorated proteinuria and kidney damage
in a db/db mouse model. PFKP inhibition exacerbates diabetic kidney
injury and cytoskeletal remodeling in podocytes (192). These findings suggest that PFKP
may function as an endogenous protective factor in DN.
PFKP may also be modified by lactylation. Cheng
et al (193) noted that
lactate could induce PFKP lactylation, which diminishes its
enzymatic activity in the fetal human colon (FHC) cell line from
the colon of a 13-week-old embryo. They reported that the FHC cell
line uses a negative feedback mechanism in which lactate suppresses
PFKP in glycolysis, highlighting the importance of deregulated
lactate and PFKP lactylation in colorectal cancer development.
However, the mechanisms by which PFKP lactylation levels are
decreased following DCA administration and by which PFKP activity
is modulated by lactylation require further exploration.
SOX9
SOX9 is an essential transcription factor within
the SOX family. The name of the SOX family derives from its
homology to the SRY gene found on the male Y chromosome. It is
widely expressed across various developing organs, including the
kidney, cartilage, pancreas, liver, heart valves, testicles and
other organs (194). SOX9
primarily consists of four domains: An HMG box (N-terminal) that
binds to DNA, a dimerization domain (N-terminal) and two
transcription activation domains (C-terminal), one of which is rich
in proline (Pro)/glutamine (Gln)/alanine (Ala), whereas the other
transcription activation domain is enriched in Pro/Gln/serine
(Ser). SOX9 predominantly binds and activates target genes via the
HMG box, thereby fulfilling its role as a transcription factor. Its
activation domain is located at amino acids 402-509 at the
C-terminal end of SOX9 (195,196). Increasing evidence suggests
that SOX9 is pivotal in embryonic development, organ tissue
development and differentiation, and is extensively implicated in
the onset and progression of tumors affecting multiple organs,
including the kidney, prostate, lung, liver, pancreas, skin, breast
and ovary (197). In the
context of DN, a study by Kishi et al (198) revealed that SOX9 can prompt
mesangial cells to undergo chondrogenic phenotype transformation,
thereby promoting the progression of DN.
Furthermore, SOX9 overexpression led to increased
ectopic levels of proteoglycans and COL2 in mesangial cells. SOX9
partially colocalized with HIF-1α and BMP4 in diabetic glomeruli.
It may be because HIF-1α and BMP4 can upregulate SOX9 and induce
subsequent chondrogenic phenotype transformation in DN (198). Given that renal fibrosis is a
notable pathophysiological mechanism in DN, SOX9 also contributes
to it. Research by Li et al (199) revealed that SOX9 silencing
alleviated TGF-β induced renal fibrosis. When renal fibroblasts are
stimulated with TGF-β in vitro, the TGF-β/Smad complex binds
to the conserved enhancer region of SOX9, considerably upregulating
SOX9 and thereby promoting cellular fibrosis.
In addition, SOX9 expression in the tumor
microenvironment can be controlled by non-coding RNAs, RNA
methylation and PTMs (for example, phosphorylation and
lactylation). In non-small cell lung cancer, Yan et al
(200) reported that hypoxia
enhances SOX9 expression and lactylation and that these effects are
reversed by suppressing glycolysis. In addition, SOX9 silencing
blunted the malignant features of these cells. In tumor-bearing
mice, SOX9 overexpression promoted tumor growth, which was
attenuated by glycolysis suppression.
Mecp2
Methyl-CpG binding protein 2 (MeCP2) represents an
important DNA methylation binding protein belonging to the
methyl-CpG binding domain (MBD) family. Presently, the majority of
fundamental studies on MeCP2 have focused on neurological diseases,
for example, Rett syndrome and MeCP2 duplication syndrome (201). The biological function of MeCP2
primarily lies in its capacity as a DNA methylation-binding
protein. Previous investigations have established that MeCP2
possesses six structural domains, the most important of which are
the MBD and the transcriptional repressor domain. These two
structural domains predominantly facilitate the interaction of
MeCP2 with methylated DNA sites and recruit additional factors to
form transcriptional repression complexes, thereby executing
transcriptional repression functions (202). MeCP2 can also recruit histone
deacetylase complexes. MeCP2 modulates gene expression
posttranslationally by inhibiting nuclear miRNA processing. Studies
of Mecp2 PTMs revealed that phosphorylated MeCP2 (p-MeCP2) directly
interacts with DiGeorge syndrome critical region 8 (DGCR8), a vital
factor in nuclear micro RNA processing and disrupts the
Drosha-DGCR8 complex (203).
Conversely, homeodomain interacting protein kinase 2 (HIPK2), which
regulates gene expression by phosphorylating transcription factors,
phosphorylates MeCP2 at Ser80, with p-MeCP2 produced by HIPK2
contributing to apoptotic cell death (204). p-MeCP2, HIPK2 and NOX4 are
upregulated in animal and cell models of diabetes. Notably, the
mechanism outlined by these studies may be relevant to DN and other
renal disorders in humans, as renal Mecp2 is upregulated in
clinical CKD, lupus nephritis, focal segmental glomerulosclerosis,
IgA nephropathy and DN (205).
Lactylation is considered a novel form of PTM of
Mecp2. Given that exercise generates the lactate used in
lactylation, Wang et al (206) first explored the fundamental
role of exercise in atherosclerotic cardiovascular disease (ASCVD)
via Mecp2 k271 lactylation. They demonstrated that exercise
markedly elevated Mecp2k271la levels in an ASCVD mouse model,
resulting in diminished inflammation in endothelial cells and
suppressed ASCVD progression. In the inflammatory state,
Mecp2k271la suppresses Ereg expression, which modulates Egfr/MAPK
signaling, thereby upregulating IL-1β, IL-6, Icam-1, Vcam-1 and
Mcp-1 and downregulating Enos in endothelial cells, thereby
mitigating plaque progression in ASCVD. Specifically, increased
Mecp2k271la expression decreased ASCVD by downregulating adhesion
(Vcam-1, Icam-1 and Mcp-1) and proinflammatory (IL-1β and IL-6)
factors, while increasing Enos in ECs.
Neuronal precursor cell expressed,
developmentally downregulated 4 (NEDD4)
NEDD4 represents a variant of E3 ubiquitin ligase
that can bind to lysine residues associated with the upstream
ubiquitin-activating enzyme E1 and conjugating enzyme E2, resulting
in the ubiquitination of its substrate proteins. NEDD4 enzymes are
broadly expressed in yeast and mammalian cells (207). NEDD4 can exert antiapoptotic
effects through various mechanisms. Research has demonstrated that
NEDD4 can specifically interact with PTEN to facilitate its
degradation by the neural proteasome system, thereby counteracting
apoptosis (208). Additional
findings indicate that NEDD4 activates PI3K/Akt signaling to
diminish apoptosis in myocardial cells triggered by
ischemia/reperfusion (209).
Furthermore, NEDD4 expressed in vascular endothelial cells degrades
VEGFR2, which is involved in endocytosis (210). In the kidney, NEDD4L is
predominantly located in proximal tubules and is markedly
upregulated in mice afflicted with DN. Co-immunoprecipitation
experiments demonstrated that NEDD4L overtly interacts with
Ca/calmodulin-dependent protein kinase β (CaMKKβ). Furthermore,
NEDD4L suppression under high-glucose conditions inhibited the
rapid degradation of CaMKKβ. Cell culture assays demonstrated that
abnormal NEDD4L expression adversely affects CaMKKβ protein
stability. Inhibition of NEDD4L results in a reduction in urinary
protein excretion, TIF, oxidative stress and impaired mitochondrial
function. Additional cell culture assays revealed that small
interfering-Nedd4L curtails mitochondrial fusion and ROS
accumulation and that these effects are counteracted by si-CaMKKβ
(211). Another investigation
indicated that mice lacking NEDD4L, particularly in renal tubular
cells, had mild kidney disease because of ENaC upregulation
(212), since NEDD4L
potentially contributes to renal regulation of potassium (213) and sodium (214) balance.
Li et al (215) also reported that lactates
modulate NEDD4 K33 lactylation, thereby inhibiting the interaction
between Caspase-11 and NEDD4. Furthermore, inhibiting lactylation
diminishes non-canonical pyroptosis in macrophages and alleviates
hepatic damage. Their research connects lactate to the intricate
modulation of non-canonical inflammasomes and establishes a
foundation for targeting lactylation to address
caspase-11-associated non-canonical pyroptosis and
acetaminophen-dependent liver damage. Given that LPS enhances
macrophage histone lactylation, the authors examined whether LPS
affects NEDD4 lactylation. As anticipated, LPS could increase NEDD4
lactylation and synergize with exogenous lactate intake.
Transcription factor EB (TFEB)
TFEB is a constituent of the
microphthalmia-associated transcription factor/transcription factor
E family, is widely expressed across various cell types, and
regulates numerous cellular biological functions (216). The TFEB protein comprises 476
amino acid residues, including an acidic activation domain, a
helix-loop-helix leucine zipper, a glutamate-rich domain and
sequences rich in serine. Previous evidence revealed that TFEB can
undergo phosphorylation at serine residue 142 (S142), serine
residue 211 (S211) and the terminal serine-rich domain. It
initiates transcription of corresponding genes by recognizing E-box
and M-box sequences and engaging in various pathophysiological
processes (217). Recent
investigations have shown that TFEB can directly interact with the
coordinated lysosomal expression and regulation network in the
promoters of lysosome-related genes. Overexpression of TFEB induces
expression of these genes, thereby augmenting lysosome biosynthesis
and function (218). TFEB also
interacts with the promoter regions of autophagy-related genes,
regulating autophagy by facilitating autophagosome generation,
autophagosome-lysosome fusion and the degradation of autophagic
substrates (219). Furthermore,
TFEB is important for modulating lipid metabolism, organelle
autophagy, the immune response, inflammation and carcinogenesis
(220).
In DN progression, TFEB plays a key role. Yuan
et al (221) reported
that mesenchymal stem cells (MSCs) enhance lysosomal function by
activating TFEB, augment autophagy and mitigate DN by promoting the
M2 macrophage phenotype. Previous findings revealed that MSCs can
bolster the anti-inflammatory response in M2 macrophages;
downregulate TNF-α, monocyte chemotactic protein and IL-1β in DN
mice; safeguard against kidney damage; increase the protein
expression of tissue protease B, TFEB, LC3II and Beclin1 in
macrophages; decrease the protein expression of P62; and increase
lysosomal autophagy. Additional studies have also indicated that
MSCs ameliorate lysosomal dysfunction in high-glucose-induced
RAW264.7 cells, increase autophagic flux and alleviate cellular
inflammation triggered by high glucose (222). Research by Zhao et al
(223) revealed a notable
reduction in the number of autophagic vesicles in podocytes of
patients of DN, accompanied by diminished autophagy. In mice with
late-stage diabetes AGEs activate mTOR, inhibiting TFEB
translocation into the nucleus, thereby further downregulating
lysosome-related genes and autophagy-related genes, obstructing
autophagosome and autolysosome formation and impeding autophagic
flux, thus contributing to the progression of DN.
Current evidence indicates that TFEB activation is
predominantly regulated by PTMs, such as phosphorylation and
acetylation (224,225). mTORC1-related phosphorylation
plays a fundamental role by sequestering TFEB in the cytosol, while
inactivating mTORC1 triggers TFEB dephosphorylation and
translocation into the nucleus to initiate the transcription of
target genes (226). A previous
study also reported that the transcription of TFEB itself might be
controlled by a positive feedback loop (227) and that STIP1 homology and U-box
containing protein 1 (STUB1) directs TFEB for UPS-dependent
breakdown (228). Huang et
al (229) demonstrated that
lactate covalently modifies TFEB, resulting in stable lactylation.
Mechanistically, lactylation at K91 prevents TFEB interaction with
the E3 ubiquitin ligase WWP2, thereby inhibiting TFEB
ubiquitination and proteasomal degradation, increasing TFEB
activity and enhancing autophagy. Using specific antibodies
targeting lactylated K91, augmented TFEB lactylation was detected
in human pancreatic cancer specimens. These findings indicate that
lactylation represents a new regulatory mechanism for TFEB and that
TFEB lactylation may associate with increased autophagy in
fast-growing cells, including malignant cells.
P53
P53 is a key tumor suppressor gene located at
17p13.1 on the human chromosome and encodes the 43.7-kDa p53
protein. p53 is involved in and regulates multiple
pathophysiological processes, for example, controlling the cell
cycle in tissues, facilitating apoptosis, preserving genetic
stability and suppressing tumor angiogenesis (230-232). Furthermore, increasing evidence
indicates that p53 contributes to and modulates the physiological
and pathological mechanisms underlying kidney disease. Under
physiological conditions, renal p53 protein levels remain low and
are primarily maintained at a minimal level through ubiquitination
and degradation pathways (233). However, when cells encounter
stress conditions such as elevated glucose, the p53 protein can
rapidly accumulate and affect downstream targets, producing
corresponding biological effects (234). Increased p53 levels and
phosphorylation are observed in both tubular cells and podocytes in
chronic DN. Nuclear p53 content is markedly elevated in DN and IgA
nephropathy cases compared with control cases with minimal change
disease, confirming p53 activation in the context of progressive
kidney damage in humans (235).
STZ-induced diabetic mice, along with db/db mice, showed elevated
kidney levels of p53, BAX and Puma, as well as increased apoptotic
death of epithelial cells (236). Mitigating oxidative stress
through catalase overexpression reduced p53 levels and alleviated
diabetic kidney damage (237).
Human kidney allograft rejection is associated with tubular damage,
ongoing fibrosis and induction of p53 and p21. Indeed, p53 and p21
levels in allografts are positively associated with creatinine
levels and negatively associated with the eGFR (238), underscoring the clinical
importance of p53 hyperactivation in transplantation failure.
However, the extent to which p53 contributes to the progression of
human nephropathies remains to be explored, as extensive p53
activation has been observed in various in vivo models of
kidney disease. The aggravation of I/R injury in diabetes also
results in a p53-mediated increase in serum creatinine levels,
increased tubular damage and increased apoptotic cell death.
DN-related AKI, triggered by both diabetes and ischemia, is also
alleviated by pifithrin-α administration and PTC p53 ablation
(234). By contrast, global p53
knockout mice develop more severe AKI than their wild-type
counterparts after I/R due to heightened inflammation, indicating
that p53 may, under specific conditions, also protect the kidney
(239).
In a recently published study, proteomics revealed
multiple AARS1 targets, for example, p53 with lactylated K120 and
K139 (DNA binding domains). The use of p53 variants harboring
constitutively lactylated lysine moieties demonstrated that AARS1
lactylation of p53 suppresses its liquid-liquid phase separation,
DNA binding and transcriptional activation. AARS1 levels and p53
lactylation were markedly associated with poor survival in patients
with breast cancer harboring wild-type p53. β-alanine disrupts the
interaction between lactate and AARS1, reduces p53 lactylation and
alleviates tumorigenesis in model animals, indicating that AARS1 is
involved in tumorigenesis by associating metabolic events to
proteome changes in tumor cells (240).
AMPKα
Adenosine monophosphate-activated protein kinase
(AMPK) is a heterotrimer consisting of one catalytic (α) and two
regulatory (β and γ) subunits. AMPK-α1 and α2 have tissue-specific
distributions: α1 is predominantly expressed in the kidney, liver,
lung, heart and brain, whereas α2 is primarily expressed in the
heart, liver, skeletal muscle and brain neuronal cells. The α
subunit of AMPK is the essential catalytic component that contains
a conventional serine/threonine kinase domain. Furthermore, the
central region of the α subunit also functions as an autoinhibitory
domain that interacts with the kinase domain in the absence of a
notable activator of the AMPK signaling pathway, AMP, thereby
hindering activation of the active site (241). In the context of metabolic
stress, hypoxia, ischemia and similar conditions, AMPK is activated
as the concentration of AMP rises and the AMP/ATP ratio increases,
thereby increasing ATP consumption. Activating agents for AMPK
include liver kinase B1 (LKB1), CaMKKβ and TGF-β-activating kinase
(242).
AMPKα is prominently expressed in various kidney
cell types, for example, mesangial cells, glomerular endothelial
cells and podocytes. Impaired AMPKα signaling, podocyte damage,
metabolic dysregulation, insulin resistance and reduced adiponectin
levels are notably associated with DN initiation and progression.
AMPKα serves as a pivotal modulator of energy metabolism in cells.
AMPKα activation can trigger the activation of insulin receptor
tyrosine kinases, inducing the phosphorylation of downstream
effector proteins, further phosphorylation, and the activation of
Akt, ultimately facilitating GLUT4 transport to the plasma
membrane, which considerably enhances glucose uptake in skeletal
muscles, improves insulin sensitivity, augments fatty acid
oxidation and modulates gene transcription (243). Presently, AMPKα is considered a
beneficial regulator of insulin sensitivity. Knockout of the AMPKα
gene can induce insulin resistance in mice, whereas AMPKα
overexpression or targeted activation of AMPKα exerts
insulin-sensitizing effects, reducing podocyte injury, diminishing
proteinuria and deferring the onset and progression of DN (244). Moreover, AMPKα is also an
upstream regulatory factor of SIRT1, elevating the AMP/ATP ratio,
particularly the NAD+/NADH ratio, to boost SIRT1 activity or
increasing nicotinamide phosphoribosyl transferase to increase NAD+
levels, further enhancing SIRT1 activity (245). SIRT1 can also deacetylate the
upstream kinase LKB1 of AMPKα, promoting LKB1 translocation to the
cytoplasm and activating AMPKα. The interplay between AMPKα and
SIRT1 is underscored by these findings, suggesting that they confer
protective effects in damaged kidneys by affecting the renal
filtration barrier and alleviating renal pathology. Furthermore,
AMPKα activators can restore Thr172 phosphorylation of the AMPKα
subunit, decrease mTORC1 phosphorylation induced by elevated
glucose levels, reduce podocyte apoptosis and reduce proteinuria
(246). Research on podocytes
in DN has demonstrated that upregulating AMPKα expression can
increase the levels of the Nephrin and Podocin proteins within
cells, effectively ameliorating podocyte injury (247).
A recent investigation demonstrated that glutamine
concentrations are decreased in human and aged Sprague-Dawley rat
nucleus pulposus (NP) tissues with severe degeneration, whereas
lactate levels and lactylation are increased. Exogenous glutamine
curtails glycolysis and increases lactate levels, downregulating
AMPKα lactylation and increasing AMPKα phosphorylation.
Furthermore, glutamine administration mitigated senescence in NP
cells and promoted autophagy and ECM production by suppressing
glycolysis and AMPKα lactylation; impaired glycolysis also
suppressed lactylation. These findings suggest that glutamine may
prevent intervertebral disc degeneration by suppressing glycolysis
and AMPKα lactylation, thereby fostering autophagy and curtailing
NP cell senescence (248).
Limitations
Several non-histone lactylation events discussed in
the present review have been characterized in non-DN contexts, and
their relevance to DN is mechanistically plausible but still
requires direct validation. For instance, in pro-inflammatory
macrophages, lactate-induced PKM2 K62 lactylation stabilizes the
tetrameric (high-activity) form, reduces nuclear translocation and
attenuates the Warburg effect as a negative-feedback 'metabolic
brake'. Because podocytes and mesangial cells in DN also display
tumor-like aerobic glycolysis, it is tempting to speculate that
PKM2 lactylation might similarly restrain excessive glycolytic flux
and toxic metabolite accumulation in early DN, a concept that
aligns with the current interest in PKM2 tetramerizing agonists;
however, no study has yet directly mapped PKM2 K62la in diabetic
kidneys, making this a hypothesis that warrants targeted metabolic
and lactyl-proteomic validation in DN models (70,78,122,249).
Likewise, YY1 K183 lactylation in microglia has
been shown to enhance its DNA binding and transcriptional
activation of FGF2, thereby promoting angiogenesis under hypoxia,
and this effect is driven by p300 and can be blocked by the p300
inhibitor A-485. Given that YY1 is upregulated in DN mesangial
cells and acts as an important co-factor for TGF-β-induced
profibrotic gene expression, high lactate levels in the diabetic
microenvironment may reasonably be hypothesized to amplify TGF-β
signaling through YY1 K183 lactylation, increasing mesangial
'sensitivity' to fibrogenic cues and fueling a
metabolism-epigenetics vicious cycle; however, direct evidence for
YY1 lactylation in DN kidneys is still lacking and should be a
focus of future work (152,250,251).
In SAKI, lactate-driven H3K18 and Ezrin K263
lactylation promote RhoA/ROCK/Ezrin signaling, NF-κB activation,
inflammation and renal dysfunction, with Ezrin K263la enhancing
Ezrin-MYD88 interaction. Because Ezrin is a core linker between the
podocyte slit diaphragm and the actin cytoskeleton, it is plausible
that chronic lactate elevation in DN could induce similar Ezrin
lactylation, simultaneously promoting NF-κB-mediated inflammation
and destabilizing cytoskeletal anchoring, thereby contributing to
foot process effacement and proteinuria; this mechanism has not yet
been directly demonstrated in DN and represents an important
hypothesis connecting 'inflammation-to-structure' damage at the
molecular level (24,178,251,252).
Finally, recent work identifies AARS1 as a
lactate-responsive lactyltransferase that modifies p53 at K120 and
K139, impairing its DNA binding, liquid-liquid phase separation and
transcriptional activation of canonical target genes, and
associating with poor outcomes in patients with breast cancer with
wild-type p53. Because p53 hyperactivation is a major driver of
podocyte apoptosis and tubular cell senescence in DN,
AARS1-mediated p53 lactylation raises the possibility of a
stage-dependent 'protective vs. pathogenic' switch: Early in DN,
elevated lactate might transiently dampen p53 transcriptional
activity and delay cell death, whereas in advanced disease,
disruption of lactate handling or dominance of other PTMs (for
example, acetylation and phosphorylation) could override this
brake, leading to overt apoptosis and fibrosis. This dynamic model
is conceptually attractive but remains speculative until p53
K120/K139 lactylation is quantified across DN stages in animal
models and in human kidney tissue (240,253,254). Overall, while extrapolating
non-histone lactylation data from macrophage, microglial,
angiogenesis and sepsis-induced AKI models provides a coherent
mechanistic framework for DN, these assumptions must be explicitly
labeled as hypotheses and systematically tested in diabetic kidney
models using site-specific lactyl-proteomics and functional
assays.
Prospect of lactylation as a therapeutic
target for DN
Drugs targeting lactylation
Given that previous studies have shown that
lactylation can contribute to the development of DN by regulating
multiple key signaling pathways, for example, ROS, inflammation,
mitochondrial dysfunction, autophagy, apoptosis and ferroptosis,
effective therapeutic strategies based on lactylation regulation
are promising but currently lacking. Owing to the incomplete
elucidation of the roles of lactate and lactylation in DN, this
therapeutic approach may attract increasing interest. With respect
to therapeutic applications, a series of drugs have been shown to
affect lactylation, focusing mainly on lactate production or
inhibition.
First, LDHA drives pyruvate conversion to lactate,
and LDHA inhibitors such as oxamate, (R)-GNE-140, and fargesin
effectively inhibit lactylation. Oxamate has been studied in depth
for its mechanism and value in lactylation and has been shown to
inhibit LDHA activity and, subsequently, affect histone Kla
expression, particularly H3K18la (255). Oxamate is a classical isosteric
and isoelectronic analogue of pyruvate that mimics the carboxyl and
carbonyl groups of α-keto acid, allowing it to bind in the LDHA
active site and competitively interfere with the conversion of
pyruvate to lactate. Structure-activity studies indicate that
maintaining the pyruvate-like α-keto-carboxylate pharmacophore is
key for LDHA binding, while prodrug strategies, such as
esterification of the carboxyl group, can improve membrane
permeability and may enhance uptake across RTECs before
intracellular hydrolysis regenerates the active inhibitor (256-259). Oxamate is a polar pyruvate
analogue with poor oral bioavailability and requires relatively
high systemic doses (often administered intraperitoneally or
intravenously in animals) to achieve meaningful LDH inhibition
in vivo. Newer LDH inhibitors (such as N-hydroxyindole LDH-A
inhibitors, NCI-006, GNE-140 and pyrazole-based LDH inhibitors)
with improved potency and lipophilicity show improved tissue
penetration but still display relatively short half-lives and are
primarily cleared renally and hepatically, which is relevant in DN
with impaired kidney function (257,260-265). Preclinical studies show that
oxamate can suppress whole-body glycolytic flux, lower ATP
production and increase ROS, which is beneficial against tumors but
may compromise energy homeostasis in high-demand tissues such as
the kidney, heart and brain. At high concentrations, oxamate and
related compounds may inhibit other glycolytic enzymes (for
example, PK or enolase), raising concerns regarding systemic
fatigue, hypoglycemia-like symptoms and worsening of ischemic
injury in DN, especially in patients with already reduced renal
reserve (260,261,266,267).
Second, lactate transporters are important for
increasing lactate transport; MCT suppressors, for example,
AZD-3965, α-cyano-4-hydroxycinnamate, lonidamine, AR-C155858, the
MCT1/4 inhibitor syrosingopine, MCT1 inhibitors (meplazumab,
phloretin and thalidomide) and derivatives (lenalidomide and
pomalidomide), can also be used to alter lactate levels, regulating
lactylation (268). In
addition, drugs that target SMCT are scarce. Common drugs used in
the clinic may exert suppressive effects on MCT/SMCT, for example,
the non-steroidal anti-inflammatory drugs ibuprofen and salicylic
acid (269,270). Some aforementioned drugs are
being examined in ongoing clinical trials. AZD3965 is a potent,
nanomolar inhibitor that selectively targets MCT1 and competes with
endogenous monocarboxylates (such as lactate) at the
substrate-binding site, locking the transporter in a non-productive
conformation and thereby blocking bidirectional lactate transport
across the plasma membrane. The compound contains an acidic or
pseudo-acidic scaffold that forms hydrogen bonds and ionic
interactions in the central cavity of MCT1, a feature identified as
a key SAR element for high-affinity binding and effective
inhibition of lactate flux (271-274). AZD3965 is an oral, selective
MCT1 inhibitor with dose-dependent target engagement demonstrated
in phase I oncology trials, and its pharmacokinetic profile
supports once-daily dosing at a maximum tolerated dose of ~20 mg in
adults. It achieves substantial inhibition of lactate transport in
high-MCT1-expressing tissues, including tumor, heart and retina,
which is central to both its efficacy and toxicity (275-277). The main dose-limiting
toxicities of AZD3965 are on-target, reversible ocular events
(retinal functional changes) and potential cardiac effects in
tissues with high MCT1 expression, alongside common low-grade
symptoms such as nausea and fatigue. In DN, where cardiometabolic
comorbidities and microvascular eye disease are prevalent, MCT1/4
inhibition might aggravate retinal or cardiac stress if not
carefully monitored, although kidney-focused delivery or
intermittent dosing could leverage renal benefits (reduced lactate
influx and lactylation) while minimizing systemic risks (24,276,277).
Additionally, studies on other targets have yielded
encouraging findings (262,278-285). Since p300/CBP regulates
lactylation as a writer, its inhibitors have therapeutic potential;
C646, a small-molecule inhibitor designed to inhibit p300 HAT
activity, was experimentally validated for targeting histone
lactylation (278). A-485 is a
highly selective, competitive small-molecule inhibitor that targets
the catalytic domain of p300/CBP by occupying the acyl-CoA (such as
acetyl-CoA or lactyl-CoA) binding pocket and structurally mimicking
key interactions of the CoA thioester scaffold. By blocking the
CoA-binding channel, A-485 prevents transfer of acyl groups,
including lactyl moieties, to lysine residues on histone and
non-histone substrates, thereby suppressing lactylation-dependent
transcriptional programs (21,262). A-485 is an orally bioavailable,
small-molecule CBP/p300 HAT inhibitor with moderate plasma
half-life and good tissue distribution in rodents, achieving
sustained suppression of histone acetylation at tolerable doses.
Because p300/CBP is ubiquitously expressed, systemic exposure to
A-485 affects multiple organs, including bone, liver, the immune
system and potentially the kidney (279-283). In preclinical models, A-485 and
related CBP/p300 HAT inhibitors can cause hematologic changes
(transient leukocytosis), alter osteoclast/osteoblast balance and
modulate inflammatory signaling, reflecting broad epigenetic
reprogramming. For DN, this implies that while p300 inhibition
might reduce pathogenic histone and non-histone lactylation in
renal cells, long-term systemic blockade could interfere with
normal repair, immune responses and bone metabolism, necessitating
kidney-targeted delivery or carefully titrated dosing regimens
(279-281,283). HDAC suppressors enhance histone
lactylation. For example, mouse P19 Embryonal Carcinoma
cells-derived Neural Stem/Progenitor Cells treated with the HDACi
MS275 (entinostat) presented increased global histone Kla levels;
in addition (284), apicidin, a
specific suppressor of HDAC1-3, also increased histone Kla levels
(30). As SIRTs act as
dehydrogenases, strategies for the use of epigenetic small
molecules to upregulate sirtuins at the gene and protein levels
represent promising options; currently available SIRT-activating
compounds include piceatannol, resveratrol, SRT1720, SRT2104,
ADTL-SA1215, honokiol and UBCS039 (285). However, it is still necessary
to explore whether these inhibitors affect lactylation and related
diseases in future studies. The majority of the aforementioned
drugs have been studied in oncology and have been shown to exert
antitumor effects; thus, drugs targeting lactylation might provide
new therapeutic options for DN in the future.
Other therapeutic methods targeting
lactylation
The regulation of lactate, histone lactylation and
non-histone protein lactylation can be epigenetically modulated by
microRNAs (miRNAs). miRNAs are 18-25 nucleotide-long non-coding
RNAs that regulate target genes posttranscriptionally by generating
incomplete complementary base pairs at the 3' untranslated regions
(3' UTRs) of target mRNAs (286). Further evidence showed that the
virus-encoded miR-N20 targets HIF-1α to suppress host glycolysis,
leading to downregulation of H3K18la and H4K12la (287). In addition, exos, a current
research focus in DN, have not been extensively investigated
because of the notable role lactylation may carry out in their
function. MSCs are highly valued for their multipotency, paracrine
functions, immunomodulatory properties and tissue repair
capabilities. In particular, the exos produced by MSCs (MSC-Exos)
contain high amounts of bioactive compounds and promote
intercellular communication, with key roles in multiple
pathophysiological events. The evidence suggests that MSC-Exos
effectively treat DN by improving tissue repair, inhibiting
fibrosis and reducing the inflammatory response. Recent findings
highlight the potential use of MSC-Exos, underscoring their wide
therapeutic applicability in DN (288). Whether MSC-Exos can be
administered and retreated by extracting exosomes from urine to
alleviate DN progression via lactylation also deserves further
examination.
Impact of infectious agents on lactate
metabolism and DN
In the context of the COVID-19 pandemic,
individuals with diabetes and obesity face a disproportionately
higher risk of severe COVID-19, intensive care unit admission and
AKI, and DN or obesity plus diabetes markedly increase
COVID-19-related mortality (289-294). These observations support the
classification of patients with COVID-19 who have underlying
obesity or diabetes as a global high-risk group that warrants
intensified renal monitoring and preventive strategies. SARS-CoV-2
infection is characterized by profound systemic inflammation and
tissue hypoxia, which together enhance aerobic glycolysis,
upregulate LDH expression, and lead to excessive lactate
accumulation and lactic acidosis. Experimental and translational
studies indicate that elevated intracellular lactate can promote
histone and non-histone lactylation, thereby reprogramming immune
and stromal cells and contributing to organ fibrosis in various
inflammatory settings (295-297). It is therefore plausible that,
in patients with pre-existing diabetes or obesity, COVID-19-induced
lactate overload may exacerbate renal inflammation and fibrosis
through lactylation-mediated pathways, further accelerating the
progression of DN and long-term kidney dysfunction (67,74,290,298-302). These data highlight the urgency
of elucidating lactate-lactylation signaling in high-risk
populations with COVID-19, which may uncover novel targets to
mitigate post-infectious renal fibrosis in patients with metabolic
comorbidities.
Beyond SARS-CoV-2, emerging evidence suggests that
a broad spectrum of infectious agents, including both bacteria and
other viruses, can perturb host lactate metabolism via metabolic
reprogramming, potentially exacerbating the progression of DN.
During bacterial infections or sepsis, pathogen-associated
molecular patterns such as LPS induce a 'Warburg-like' metabolic
shift in both immune and renal cells, leading to excessive lactate
accumulation (58,138). This hyperlactatemic
microenvironment triggers both histone and non-histone lactylation.
For instance, in SAKI, lactate-driven lactylation of H3K18 and
Ezrin has been shown to activate the RhoA/ROCK pathway, amplifying
inflammatory responses and destabilizing the podocyte cytoskeleton
(178). Similarly, other viral
infections, such as influenza A, have been reported to upregulate
LDH activity, where lactate-mediated signaling can suppress type I
interferon production, thereby impairing host immunity and
magnifying tissue damage (303). Given that patients with DN are
highly susceptible to recurrent infections, these infection-induced
alterations in the lactylation landscape may serve as a 'second
hit' that accelerates the deterioration of renal function.
Conclusion and prospects
Lactylation is associated with metabolic
reprogramming, suggesting that dynamic changes in lactylation are
linked to nutritional supply, oxygen levels and energy demand and
can affect biological processes important in the pathogenesis of
metabolic disorders. Whereas diabetes is a typical metabolic
disorder, several aspects of lactylation modification, especially
diabetic complications, remain unexplored. Non-histone lactylation,
which is distinct from other PTMs, is still in its early stages of
research but has already shown great potential in biomedical
research and clinical applications. Although several DN-related
non-histone proteins that can be lactylated and participate in
numerous aspects of DN pathophysiology have been identified
(Fig. 5), extensive research is
imperative to elucidate the specific functions and regulatory
mechanisms involved in DN. In addition, with rapid advances in
sequencing techniques, several comprehensive databases for
integrating and predicting PTM sites have been developed by
collating data from high-throughput sequencing and experimental
verification sources, providing comprehensive information and
research tools for the study of protein PTMs. However, to the best
of our knowledge, there are currently no available lactylation
databases. Therefore, making full use of existing technologies,
such as high-throughput sequencing, proteomics and bioinformatics
tools, to systematically study lactylation is important for
revealing its roles in cellular signal transduction, metabolic
regulation, and disease pathogenesis. Such tools would help improve
understanding of the importance of lactylation and reveal
lactylation targeting as a new treatment approach for different
stages of various diseases.
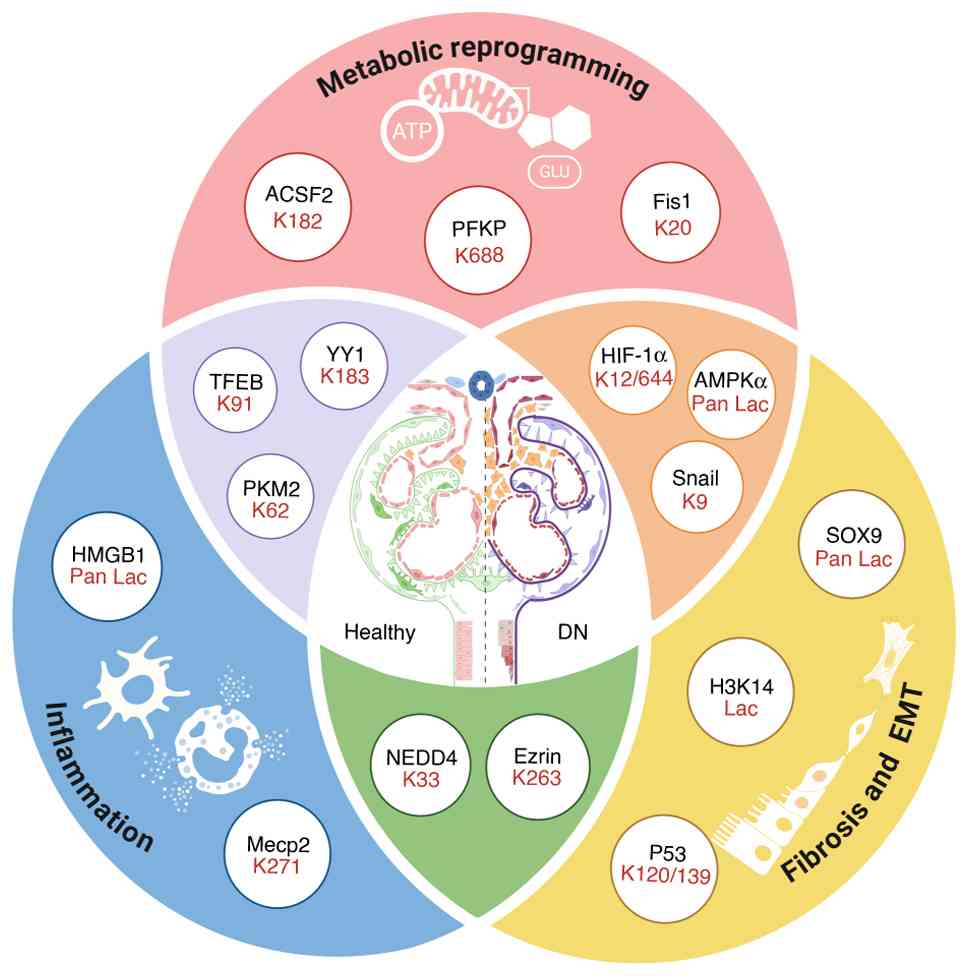 | Figure 5Possible mechanism of lactylation in
diabetic nephropathy pathogenesis. The diagram illustrates the
categorization of lactylated proteins based on their primary
pathological impact in DN. Metabolic Reprogramming: Lactylation of
ACSF2, PFKP, HIF-1α, Snail, AMPKα, Fis1, YY1, TFEB and PKM2.
Inflammation: Lactylation of HMGB1, Mecp2, YY1, TFEB, PKM2, NEDD4
and Ezrin. Fibrosis and EMT: Lactylation of H3K14, P53, SOX9,
NEDD4, Ezrin, HIF-1α, Snail and AMPKα. ACSF2, Acyl-CoA synthetase
family member 2; PKM2, pyruvate kinase M2; HMGB1, high-mobility
group box 1; YY1, yin yang 1; Fis1, fission 1; HIF-1α,
hypoxia-inducible factor-1α; PFKP, platelet isoform of
phosphofructokinase 1; Sox9, sex determining region Y (SRY)-related
HMG-box gene 9; Mecp2, methylated CpG-binding protein 2; NEDD4,
neuronally expressed developmentally downregulated 4; TFEB,
transcription factor EB; AMPKα, adenosine monophosphate-activated
protein kinase α; DN, diabetic nephropathy; ROS, reactive oxygen
species; EMT, epithelial-mesenchymal transition. Figure created in
Biorender.com. |
However, while targeting the lactate-lactylation
axis holds therapeutic promise, several critical hurdles must be
overcome before clinical translation. First, current evidence is
predominantly derived from preclinical models; therefore, robust,
large-scale clinical trials are urgently required to validate the
safety and efficacy of lactylation-modulating agents specifically
in patients with DN. Secondly, a major challenge in clinical
studies is the lack of molecular specificity. Numerous 'writers'
(for example, p300/CBP) and 'erasers' (for example, HDACs) of
lactylation also regulate other post-translational modifications,
such as acetylation. Consequently, systemic inhibition of these
enzymes may lead to broad epigenetic off-target effects,
potentially disrupting normal cellular homeostasis. Thirdly, as
lactate serves as a vital energy substrate for high-demand organs
such as the heart and brain, systemic interference with lactate
metabolism could compromise metabolic homeostasis, leading to
adverse effects such as systemic fatigue or organ dysfunction.
Furthermore, dose-limiting toxicities observed in Phase I trials of
MCT1 inhibitors (for example, AZD3965), underscore the necessity
for developing kidney-targeted delivery systems to minimize
systemic risks and improve the therapeutic index.
In conclusion, the present review summarizes
lactylation and key regulatory molecules during the progression of
DN, with a focus on the relationship between lactylated non-histone
proteins and DN. Currently, few drugs target lactylation for the
treatment of metabolic diseases, especially DN. The limited
availability of potential treatments is related to the small
numbers of druggable targets and drugs identified to date, as well
as the incomplete understanding of lactylation or its regulatory
mechanisms. Once these issues are effectively addressed, the use of
lactylation as a target for DN treatment would be feasible.
Availability of data and materials
Not applicable.
Authors' contributions
YYX was responsible for conceptualization, writing
the original draft and visualization. YYX and YHX were responsible
for literature collection and curation. NSW, DKG and YHX were
responsible for supervision, writing, reviewing and editing. Data
authentication is not applicable. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present review was funded by the National Key R&D
Program of China (grant no. 2019YFE0110500), Science and Technology
Development Fund of Macau (grant no. 0055/2019/AMJ, 0022/2024/RIB1
and 006/2023/SKL), Shanghai Sixth People's Hospital East
Outstanding Young Fund Project (grant no. 2019006) and Shanghai
Sixth People's Hospital Clinic Research Project (grant no.
ynhg202103).
References
|
1
|
Li MJ, Liu HY, Zhang YQ, Li SR, Zhang JH
and Li R: Global burden of chronic kidney disease and its
attributable risk factors (1990-2021): An analysis based on the
global burden of disease study. Front Endocrinol (Lausanne).
16:15632462025. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
de Sa JR, Rangel EB, Canani LH, Bauer AC,
Escott GM, Zelmanovitz T, Bertoluci MC and Silveiro SP: The
2021-2022 position of Brazilian Diabetes Society on diabetic kidney
disease (DKD) management: An evidence-based guideline to clinical
practice. Screening and treatment of hyperglycemia, arterial
hypertension, and dyslipidemia in the patient with DKD. Diabetol
Metab Syndr. 14:812022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sanghavi SF, Roark T, Zelnick LR, Najafian
B, Andeen NK, Alpers CE, Pichler R, Ayers E and de Boer IH:
Histopathologic and clinical features in patients with diabetes and
kidney disease. Kidney360. 1:1217–1225. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barutta F, Bellini S and Gruden G:
Mechanisms of podocyte injury and implications for diabetic
nephropathy. Clin Sci (Lond). 136:493–520. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang M, Huang Z, Li X, He P, Sun H, Peng Y
and Fan Q: Apabetalone, a BET protein inhibitor, inhibits kidney
damage in diabetes by preventing pyroptosis via modulating the
P300/H3K27ac/PLK1 axis. Pharmacol Res. 207:1073062024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen Q, Xie C, Tang K, Luo M, Zhang Z, Jin
Y, Liu Y, Zhou L and Kong Y: The E3 ligase Trim63 promotes podocyte
injury and proteinuria by targeting PPARalpha to inhibit fatty acid
oxidation. Free Radic Biol Med. 209:40–54. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Song S, Hu T, Shi X, Jin Y, Liu S, Li X,
Zou W and Wang C: ER Stress-perturbed intracellular protein
O-GlcNAcylation aggravates podocyte injury in diabetes nephropathy.
Int J Mol Sci. 24:176032023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
He M, Wang Z, Miao Z, Zhao Y, Wei L, Zhang
L, Yin R, Wang Y and Yang L: Post-translational modifications in
diabetic kidney disease (Review). Int J Mol Med. 57:882026.
View Article : Google Scholar
|
|
9
|
Gu W, Wang X, Zhao H, Geng J, Li X, Zheng
K, Guan Y, Hou X, Wang C and Song G: Resveratrol ameliorates
diabetic kidney injury by reducing lipotoxicity and modulates
expression of components of the junctional adhesion
molecule-like/sirtuin 1 lipid metabolism pathway. Eur J Pharmacol.
918:1747762022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qi B, Chen Y, Chai S, Lu X and Kang L:
O-linked beta-N-acetylglucosamine (O-GlcNAc) modification: Emerging
pathogenesis and a therapeutic target of diabetic nephropathy.
Diabet Med. 42:e154362025. View Article : Google Scholar
|
|
11
|
Zhu W, Guo S, Sun J, Zhao Y and Liu C:
Lactate and lactylation in cardiovascular diseases: current
progress and future perspectives. Metabolism. 158:1559572024.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu B, Liu Y, Li N and Geng Q: Lactate and
lactylation in macrophage metabolic reprogramming: Current progress
and outstanding issues. Front Immunol. 15:13957862024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li X, Yang Y, Zhang B, Lin X, Fu X, An Y,
Zou Y, Wang JX, Wang Z and Yu T: Lactate metabolism in human health
and disease. Signal Transduct Target Ther. 7:3052022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Warburg O, Wind F and Negelein E: The
metabolism of tumors in the body. J Gen Physiol. 8:519–530. 1927.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dimitriadis GD, Maratou E, Kountouri A,
Board M and Lambadiari V: Regulation of postabsorptive and
post-prandial glucose metabolism by Insulin-Dependent and
Insulin-independent mechanisms: An Integrative Approach. Nutrients.
13:1592021. View Article : Google Scholar
|
|
16
|
Lee DY, Kim JY, Ahn E, Hyeon JS, Kim GH,
Park KJ, Jung Y, Lee YJ, Son MK, Kim SW, et al: Associations
between local acidosis induced by renal LDHA and renal fibrosis and
mitochondrial abnormalities in patients with diabetic kidney
disease. Transl Res. 249:88–109. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Darshi M, Kugathasan L, Maity S, Sridhar
VS, Fernandez R, Limonte CP, Grajeda BI, Saliba A, Zhang G, Drel
VR, et al: Glycolytic lactate in diabetic kidney disease. JCI
Insight. 9:e1688252024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Azushima K, Kovalik JP, Yamaji T, Ching J,
Chng TW, Guo J, Liu JJ, Nguyen M, Sakban RB, George SE, et al:
Abnormal lactate metabolism is linked to albuminuria and kidney
injury in diabetic nephropathy. Kidney Int. 104:1135–1149. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kugathasan L, Darshi M, Srinivasan Sridhar
V, Drel V, Tumova J, Liu JJ, Wang J, Perkins BA, Lytvyn Y,
Natarajan L, et al: 12-OR: Urine lactate is a disease biomarker for
diabetic kidney disease. Diabetes. 72:12–OR. 2023. View Article : Google Scholar
|
|
20
|
Chen Y, Liu X, Shengbu M, Shi Q, Jiaqiu S
and Lai X: Biomarkers: New advances in diabetic nephropathy.
Natural Product Communications. 20:1934578X2513217582025.
View Article : Google Scholar
|
|
21
|
Xu Y, Li X, Mao Z and Xue C: Protein
lactylation in kidney diseases. Front Cell Dev Biol.
13:15331752025. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cheng Y and Guo L: Lactate metabolism and
lactylation in kidney diseases: Insights into mechanisms and
therapeutic opportunities. Ren Fail. 47:24697462025. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou M, Liu L, Sun Y and Wang X:
Lactylation in diabetes mellitus and its complications: Mechanisms
of action and therapeutic potential-recent advances. Front
Endocrinol (Lausanne). 16:17106452025. View Article : Google Scholar
|
|
24
|
Wei X, Long M, Yu J and Du Y: The
lactate-lactylation axis in renal fibrosis: Potential mechanisms in
diabetic kidney disease. Ann Med. 57:25873262025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gao M, Wang M, Zhou S, Hou J, He W, Shu Y
and Wang X: Machine learning-based prognostic model of
lactylation-related genes for predicting prognosis and immune
infiltration in patients with lung adenocarcinoma. Cancer Cell Int.
24:4002024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fu C, Jia Z, Fu Y, Zhang Y, Wu Y, Cao X,
Cao D and Jiang J: The clinical prognostic value of
lactylation-regulated proteins in gastric cancer. J Proteome Res.
25:446–459. 2026. View Article : Google Scholar :
|
|
27
|
Yang X, Li G, Jiang R, Yang Y, Zhu H,
Huang L, Li T, Zhou J and Liu Z: Lactylation-related gene
signatures for prognosis and treatment response prediction in
radiation-resistant non-small cell lung cancer. Discover Oncol.
16:17872025. View Article : Google Scholar
|
|
28
|
Wu Q, Qiu X and Chen H: Advancements in
early biomarkers of acute kidney injury: From traditional
indicators to a paradigm shift in lactate metabolism. Ann Med.
58:26123832026. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Concepción M, Quiroz J, Suarez J, Paz J,
Roseboom P, Ildefonso S, Cribilleros D, Zavaleta F, Coronado J and
Concepción L: Novel Biomarkers for the diagnosis of diabetic
nephropathy. Caspian J Intern Med. 15:382–391. 2024.PubMed/NCBI
|
|
30
|
Cai D, Yuan X, Cai DQ, Li A, Yang S, Yang
W, Duan J, Zhuo W, Min J, Peng L and Wei J: Integrative analysis of
lactylation-related genes and establishment of a novel prognostic
signature for hepatocellular carcinoma. J Cancer Res Clin Oncol.
149:11517–11530. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kemp E, Jacobsen IA and Amtrup F: LDH
release into perfusates of preserved kidneys. Scand J Urol Nephrol.
10:142–146. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Valvona CJ, Fillmore HL, Nunn PB and
Pilkington GJ: The regulation and function of lactate dehydrogenase
a: Therapeutic potential in brain tumor. Brain Pathol. 26:3–17.
2016. View Article : Google Scholar
|
|
33
|
Osis G, Traylor AM, Black LM, Spangler D,
George JF, Zarjou A, Verlander JW and Agarwal A: Expression of
lactate dehydrogenase A and B isoforms in the mouse kidney. Am J
Physiol Renal Physiol. 320:F706–F718. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brooks GA: Cell-cell and intracellular
lactate shuttles. J Physiol. 587:5591–5600. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Becker HM, Klier M and Deitmer JW:
Nonenzymatic augmentation of lactate transport via monocarboxylate
transporter isoform 4 by carbonic anhydrase II. J Membr Biol.
234:125–135. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Iwanaga T and Kishimoto A: Cellular
distributions of monocarboxylate transporters: A review. Biomed
Res. 36:279–301. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Halestrap AP: Monocarboxylic acid
transport. Compr Physiol. 3:1611–1643. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Becker HM, Mohebbi N, Perna A, Ganapathy
V, Capasso G and Wagner CA: Localization of members of MCT
monocarboxylate transporter family Slc16 in the kidney and
regulation during metabolic acidosis. Am J Physiol Renal Physiol.
299:F141–F154. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Thangaraju M, Ananth S, Martin PM, Roon P,
Smith SB, Sterneck E, Prasad PD and Ganapathy V: c/ebpdelta Null
mouse as a model for the double knock-out of slc5a8 and slc5a12 in
kidney. J Biol Chem. 281:26769–26773. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Py G, Lambert K, Perez-Martin A, Raynaud
E, Prefaut C and Mercier J: Impaired sarcolemmal vesicle lactate
uptake and skeletal muscle MCT1 and MCT4 expression in obese Zucker
rats. Am J Physiol Endocrinol Metab. 281:E1308–F1315. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Py G, Lambert K, Milhavet O, Eydoux N,
Prefaut C and Mercier J: Effects of streptozotocin-induced diabetes
on markers of skeletal muscle metabolism and monocarboxylate
transporter 1 to monocarboxylate transporter 4 transporters.
Metabolism. 51:807–813. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Enoki T, Yoshida Y, Hatta H and Bonen A:
Exercise training alleviates MCT1 and MCT4 reductions in heart and
skeletal muscles of STZ-induced diabetic rats. J Appl Physiol
(1985). 94:2433–2438. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ahmed K, Tunaru S and Offermanns S:
GPR109A, GPR109B and GPR81, a family of hydroxy-carboxylic acid
receptors. Trends Pharmacol Sci. 30:557–562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hoque R, Farooq A, Ghani A, Gorelick F and
Mehal WZ: Lactate reduces liver and pancreatic injury in Toll-like
receptor- and inflammasome-mediated inflammation via GPR81-mediated
suppression of innate immunity. Gastroenterology. 146:1763–1774.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jones NK, Stewart K, Czopek A, Menzies RI,
Thomson A, Moran CM, Cairns C, Conway BR, Denby L, Livingstone DEW,
et al: Endothelin-1 mediates the systemic and renal hemodynamic
effects of GPR81 activation. Hypertension. 75:1213–1222. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Brown TP and Ganapathy V: Lactate/GPR81
signaling and proton motive force in cancer: Role in angiogenesis,
immune escape, nutrition, and Warburg phenomenon. Pharmacol Ther.
206:1074512020. View Article : Google Scholar
|
|
47
|
Yang K, Xu J, Fan M, Tu F, Wang X, Ha T,
Williams DL and Li C: Lactate suppresses macrophage
pro-inflammatory response to LPS stimulation by inhibition of YAP
and NF-κB Activation via GPR81-mediated signaling. Front Immunol.
11:5879132020. View Article : Google Scholar
|
|
48
|
Ranganathan P, Shanmugam A, Swafford D,
Suryawanshi A, Bhattacharjee P, Hussein MS, Koni PA, Prasad PD,
Kurago ZB, Thangaraju M, et al: GPR81, a Cell-surface receptor for
lactate, regulates intestinal homeostasis and protects mice from
experimental colitis. J Immunol. 200:1781–1789. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Llibre A, Kucuk S, Gope A, Certo M and
Mauro C: Lactate: A key regulator of the immune response. Immunity.
58:535–554. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Guo S, Ye M, Zhu W and Liu C: From fuel to
epigenetic signal: Lactate-lactylation axis orchestrates diabetic
complications. Pharmacol Res. 222:1080522025. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liu J, Zhao F and Qu Y: Lactylation: A
novel post-translational modification with clinical implications in
CNS diseases. Biomolecules. 14:11752024. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ishihara S, Hata K, Hirose K, Okui T,
Toyosawa S, Uzawa N, Nishimura R and Yoneda T: The lactate sensor
GPR81 regulates glycolysis and tumor growth of breast cancer. Sci
Rep. 12:62612022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chen S, Zhou L, Sun J, Qu Y and Chen M:
The role of cAMP-PKA pathway in Lactate-induced intramuscular
triglyceride accumulation and mitochondria content increase in
mice. Front Physiol. 12:7091352021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang H, Yang M, Liu X, Fan J and Wang C: G
protein-coupled receptor-mediated renal fibrosis: A key focus on
kidney disease drug development. Front Pharmacol. 16:16458882025.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhi Y, Fan K, Liu S, Hu K, Zan X, Lin L,
Yang Y, Gong X, Chen K, Tang L, et al: Deletion of GPR81 activates
CREB/Smad7 pathway and alleviates liver fibrosis in mice. Mol Med.
30:992024. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wu X, Xu M, Geng M, Chen S, Little PJ, Xu
S and Weng J: Targeting protein modifications in metabolic
diseases: Molecular mechanisms and targeted therapies. Signal
Transduct Target Ther. 8:2202023. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Liu Y, Guo X, Hu X, Zhou S, Yang Q, Feng P
and Zeng L: Lysine lactylation in diseases: Beyond histone
lactylation. Cell Death Dis. 17:552025. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang D, Tang Z, Huang H, Zhou G, Cui C,
Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al: Metabolic
regulation of gene expression by histone lactylation. Nature.
574:575–580. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Moreno-Yruela C, Zhang D, Wei W, Bæk M,
Liu W, Gao J, Danková D, Nielsen AL, Bolding JE, Yang L, et al:
Class I histone deacetylases (HDAC1-3) are histone lysine
delactylases. Sci Adv. 8:eabi66962022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cui H, Xie N, Banerjee S, Ge J, Jiang D,
Dey T, Matthews QL, Liu RM and Liu G: Lung myofibroblasts promote
macrophage profibrotic activity through lactate-induced histone
lactylation. Am J Respir Cell Mol Biol. 64:115–125. 2021.
View Article : Google Scholar
|
|
61
|
Xie B, Zhang M, Li J, Cui J, Zhang P, Liu
F, Wu Y, Deng W, Ma J, Li X, et al: KAT8-catalyzed lactylation
promotes eEF1A2-mediated protein synthesis and colorectal
carcinogenesis. Proc Natl Acad Sci USA. 121:e23141281212024.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wang N, Wang W, Wang X, Mang G, Chen J,
Yan X, Tong Z, Yang Q, Wang M, Chen L, et al: Histone lactylation
boosts reparative gene activation post-myocardial infarction. Circ
Res. 131:893–908. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Dong H, Zhang J, Zhang H, Han Y, Lu C,
Chen C, Tan X, Wang S, Bai X, Zhai G, et al: YiaC and CobB regulate
lysine lactylation in Escherichia coli. Nat Commun. 13:66282022.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wang J, Wang Z, Wang Q, Li X and Guo Y:
Ubiquitous protein lactylation in health and diseases. Cell Mol
Biol Lett. 29:232024. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zong Z, Ren J, Yang B, Zhang L and Zhou F:
Emerging roles of lysine lactyltransferases and lactylation. Nat
Cell Biol. 27:563–574. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Gaffney DO, Jennings EQ, Anderson CC,
Marentette JO, Shi T, Schou Oxvig AM, Streeter MD, Johannsen M,
Spiegel DA, Chapman E, et al: Non-enzymatic Lysine lactoylation of
glycolytic enzymes. Cell Chem Biol. 27:206–213.e6. 2020. View Article : Google Scholar
|
|
67
|
Chen X, Yuan Y, Zhou F, Li L, Pu J, Zeng Y
and Jiang X: Lactylation: From homeostasis to pathological
implications and therapeutic strategies. MedComm (2020).
6:e702262025. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zheng T, Gu YP, Wang JM, Huang TT, Gou LS
and Liu YW: Lactate-triggered histone lactylation contributes to
podocyte epithelial-mesenchymal transition in diabetic nephropathy
in mice. Chem Biol Interact. 408:1114182025. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Li H, Liu C, Li R, Zhou L, Ran Y, Yang Q,
Huang H, Lu H, Song H, Yang B, et al: AARS1 and AARS2 sense
L-lactate to regulate cGAS as global lysine lactyltransferases.
Nature. 634:1229–1237. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Che X, Zhang Y, Chen X, Xie G, Li J, Xu C,
Zhang C, Zhu Y and Yang X: The lactylation-macrophage interplay:
Implications for gastrointestinal disease therapeutics. Front
Immunol. 16:16081152025. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Wan L, Zhang H, Liu J, He Q, Zhao J, Pan
C, Zheng K and Tang Y: Lactylation and human disease. Expert Rev
Mol Med. 27:e102025. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Lu Z, Zheng X, Shi M, Yin Y, Liang Y, Zou
Z, Ding C, He Y, Zhou Y and Li X: Lactylation: The emerging
frontier in post-translational modification. Front Genet.
15:14232132024. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wang T, Ye Z, Li Z, Jing DS, Fan GX, Liu
MQ, Zhuo QF, Ji SR, Yu XJ, Xu XW and Qin Y: Lactate-induced protein
lactylation: A bridge between epigenetics and metabolic
reprogramming in cancer. Cell Prolif. 56:e134782023. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhao L, Qi H, Lv H, Liu W, Zhang R and
Yang A: Lactylation in health and disease: Physiological or
pathological? Theranostics. 15:1787–1821. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Chen J, Feng Q, Qiao Y, Pan S, Liang L,
Liu Y, Zhang X, Liu D and Liu Z and Liu Z: ACSF2 and lysine
lactylation contribute to renal tubule injury in diabetes.
Diabetologia. 67:1429–1443. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Hong J, Xu H, Yu L, Yu Z, Chen X, Meng Z,
Zhu J, Li J and Zhu M: AARS1-mediated lactylation of H3K18 and
STAT1 promotes ferroptosis in diabetic nephropathy. Cell Death
Differ. Sep 23–2025. View Article : Google Scholar : Epub ahead of
print. PubMed/NCBI
|
|
77
|
Zhang X, Chen J, Lin R, Huang Y, Wang Z,
Xu S, Wang L, Chen F, Zhang J, Pan K and Yin Z: Lactate drives
epithelial-mesenchymal transition in diabetic kidney disease via
the H3K14la/KLF5 pathway. Redox Biol. 75:1032462024. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Qian J, Huang C, Wang M, Liu Y, Zhao Y, Li
M, Zhang X, Gao X, Zhang Y, Wang Y, et al: Nuclear translocation of
metabolic enzyme PKM2 participates in high glucose-promoted HCC
metastasis by strengthening immunosuppressive environment. Redox
Biol. 71:1031032024. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Song M, Liu B, Wang H and Sun W: Lactate
metabolism and lactylation modification: New opportunities and
challenges in cardiovascular disease. MedComm (2020). 6:e702692025.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Peng X and Du J: Histone and non-histone
lactylation: Molecular mechanisms, biological functions, diseases,
and therapeutic targets. Mol Biomed. 6:382025. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Tessarz P and Kouzarides T: Histone core
modifications regulating nucleosome structure and dynamics. Nat Rev
Mol Cell Biol. 15:703–708. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Dai W, Wu G, Liu K, Chen Q, Tao J, Liu H
and Shen M: Lactate promotes myogenesis via activating H3K9
lactylation-dependent up-regulation of Neu2 expression. J Cachexia
Sarcopenia Muscle. 14:2851–2865. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Sun T, Zhang JN, Lan T, Shi L, Hu L, Yan
L, Wei C, Hei L, Wu W, Luo Z, et al: H3K14 lactylation exacerbates
neuronal ferroptosis by inhibiting calcium efflux following
intracerebral hemorrhagic stroke. Cell Death Dis. 16:5532025.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Zhang J, Yang Y, Chen X, Chen H, Kuang E,
Deng M and Wang F: Histone lactylation dynamics are associated with
impaired maternal-to-zygotic transition in vitro-cultured goat
embryos. Theriogenology. 256:1178592026. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Jiang Z, Xiong N, Yan R, Li ST, Liu H, Mao
Q, Sun Y, Shen S, Ye L, Gao P, et al: PDHX acetylation facilitates
tumor progression by disrupting PDC assembly and activating
lactylation-mediated gene expression. Protein Cell. 16:49–63. 2025.
View Article : Google Scholar :
|
|
86
|
Yang Y, Wen J, Lou S, Han Y, Pan Y, Zhong
Y, He Q, Zhang Y, Mo X, Ma J and Shen N: DNAJC12 downregulation
induces neuroblastoma progression via increased histone H4K5
lactylation. J Mol Cell Biol. 16:mjae0562025. View Article : Google Scholar :
|
|
87
|
Guan B, Zhou M, Dai W, Zhang J, Xie B, Mi
Y, Zhang X, Wei P, Liu Y, Li S, et al: Peritumoral colonic
epithelial cell-derived GDF15 sustains colorectal cancer via
regulation of glycolysis and histone lactylation. Nat Aging.
5:2449–2465. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Yang Y, Song L, Yu L, Zhang J and Zhang B:
H4K12 lactylation potentiates mitochondrial oxidative stress via
the Foxo1 pathway in diabetes-induced cognitive impairment. J Adv
Res. 78:391–407. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Jiao Q, Ren Y, Teng X, Feng M, Liu X, Cai
Y, Hu T, Wang M and Wang Y: Positive feedback between histone H4K16
lactylation and glycolysis promotes MAFLD progression. Hepatol Int.
Dec 2–2025. View Article : Google Scholar : Epub ahead of
print. PubMed/NCBI
|
|
90
|
Galle E, Wong CW, Ghosh A, Desgeorges T,
Melrose K, Hinte LC, Castellano-Castillo D, Engl M, de Sousa JA,
Ruiz-Ojeda FJ, et al: H3K18 lactylation marks tissue-specific
active enhancers. Genome Biol. 23:2072022. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Pan L, Feng F, Wu J, Fan S, Han J, Wang S,
Yang L, Liu W, Wang C and Xu K: Demethylzeylasteral targets lactate
by inhibiting histone lactylation to suppress the tumorigenicity of
liver cancer stem cells. Pharmacol Res. 181:1062702022. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Pan RY, He L, Zhang J, Liu X, Liao Y, Gao
J, Liao Y, Yan Y, Li Q, Zhou X, et al: Positive feedback regulation
of microglial glucose metabolism by histone H4 lysine 12
lactylation in Alzheimer's disease. Cell Metab. 34:634–648.e6.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Chen J, He J, Wang X, Bai L, Yang X, Chen
J, He Y and Chen K: Glis1 inhibits RTEC cellular senescence and
renal fibrosis by downregulating histone lactylation in DKD. Life
Sci. 361:1232932025. View Article : Google Scholar
|
|
94
|
Gao M, Zhang N and Liang W: Systematic
analysis of lysine lactylation in the plant fungal pathogen
botrytis cinerea. Front Microbiol. 11:5947432020. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Zhao W, Yu H, Liu X, Wang T, Yao Y, Zhou
Q, Zheng X and Tan F: Systematic identification of the lysine
lactylation in the protozoan parasite Toxoplasma gondii. Parasit
Vectors. 15:1802022. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Dong H, Sun Y, Nie L, Cui A, Zhao P, Leung
WK and Wang Q: Metabolic memory: Mechanisms and diseases. Signal
Transduct Target Ther. 9:382024. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Erekat NS: Programmed cell death in
diabetic nephropathy: A review of apoptosis, autophagy, and
necroptosis. Med Sci Monit. 28:e9377662022. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Yu H, Zhu T, Ma D, Cheng X, Wang S and Yao
Y: The role of nonhistone lactylation in disease. Heliyon.
10:e362962024. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Tang S, Sun Y, Sun W, Kang X, Zhao X,
Jiang L, Gao Q, An X, Ji H and Lian F: Programmed cell death in
diabetic kidney disease: Mechanisms and therapeutic targeting. J
Inflamm Res. 18:13001–13037. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Watkins PA, Maiguel D, Jia Z and Pevsner
J: Evidence for 26 distinct acyl-coenzyme A synthetase genes in the
human genome. J Lipid Res. 48:2736–2750. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Perera RJ, Marcusson EG, Koo S, Kang X,
Kim Y, White N and Dean NM: Identification of novel PPARgamma
target genes in primary human adipocytes. Gene. 369:90–99. 2006.
View Article : Google Scholar
|
|
102
|
Wiese EK, Hitosugi S, Loa ST, Sreedhar A,
Andres-Beck LG, Kurmi K, Pang YP, Karnitz LM, Gonsalves WI and
Hitosugi T: Enzymatic activation of pyruvate kinase increases
cytosolic oxaloacetate to inhibit the Warburg effect. Nat Metab.
3:954–968. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Palsson-McDermott EM, Curtis AM, Goel G,
Lauterbach MAR, Sheedy FJ, Gleeson LE, van den Bosch MWM, Quinn SR,
Domingo-Fernandez R, Johnston DGW, et al: Pyruvate kinase M2
regulates Hif-1α activity and IL-1β induction and is a critical
determinant of the warburg effect in LPS-activated macrophages.
Cell Metab. 21:3472015. View Article : Google Scholar
|
|
104
|
Yang W, Xia Y, Hawke D, Li X, Liang J,
Xing D, Aldape K, Hunter T, Alfred Yung WK and Lu Z: PKM2
phosphorylates histone H3 and promotes gene transcription and
tumorigenesis. Cell. 150:685–696. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Ouyang X, Han SN, Zhang JY, Dioletis E,
Nemeth BT, Pacher P, Feng D, Bataller R, Cabezas J, Stärkel P, et
al: Digoxin suppresses pyruvate kinase M2-promoted HIF-1α
transactivation in steatohepatitis. Cell Metab. 27:11562018.
View Article : Google Scholar
|
|
106
|
Kocak MZ, Aktas G, Atak BM, Duman TT, Yis
OM, Erkus E and Savli H: Is Neuregulin-4 a predictive marker of
microvascular complications in type 2 diabetes mellitus? Eur J Clin
Invest. 50:e132062020. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Amdur RL, Feldman HI, Gupta J, Yang W,
Kanetsky P, Shlipak M, Rahman M, Lash JP, Townsend RR, Ojo A, et
al: Inflammation and progression of CKD: The CRIC study. Clin J Am
Soc Nephrol. 11:1546–1556. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Qi W, Keenan HA, Li Q, Ishikado A, Kannt
A, Sadowski T, Yorek MA, Wu IH, Lockhart S, Coppey LJ, et al:
Pyruvate kinase M2 activation may protect against the progression
of diabetic glomerular pathology and mitochondrial dysfunction. Nat
Med. 23:753–762. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Li L, Tang L, Yang X, Chen R, Zhang Z,
Leng Y and Chen AF: Gene regulatory effect of pyruvate kinase M2 is
involved in renal inflammation in type 2 diabetic nephropathy. Exp
Clin Endocrinol Diabetes. 128:599–606. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Gordin D, Shah H, Shinjo T, St-Louis R, Qi
W, Park K, Paniagua SM, Pober DM, Wu IH, Bahnam V, et al:
Characterization of glycolytic enzymes and pyruvate kinase M2 in
type 1 and 2 diabetic nephropathy. Diabetes Care. 42:1263–1273.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Liu H, Takagaki Y, Kumagai A, Kanasaki K
and Koya D: The PKM2 activator TEPP-46 suppresses kidney fibrosis
via inhibition of the EMT program and aberrant glycolysis
associated with suppression of HIF-1alpha accumulation. J Diabetes
Investig. 12:697–709. 2021. View Article : Google Scholar :
|
|
112
|
Zhang Z, Deng X, Liu Y, Liu Y, Sun L and
Chen F: Correction to: PKM2, function and expression and
regulation. Cell Biosci. 9:592019. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Angiari S, Runtsch MC, Sutton CE,
Palsson-McDermott EM, Kelly B, Rana N, Kane H, Papadopoulou G,
Pearce EL, Mills KHG, et al: Pharmacological activation of pyruvate
kinase M2 inhibits CD4+ T cell pathogenicity and
suppresses autoimmunity. Cell Metab. 31:391–405.e8. 2020.
View Article : Google Scholar
|
|
114
|
Sweetwyne MT, Gruenwald A, Niranjan T,
Nishinakamura R, Strobl LJ and Susztak K: Notch1 and Notch2 in
podocytes play differential roles during diabetic nephropathy
development. Diabetes. 64:4099–4111. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Yokoyama M, Tanuma N, Shibuya R, Shiroki
T, Abue M, Yamamoto K, Miura K, Yamaguchi K, Sato I, Tamai K and
Satoh K: Pyruvate kinase type M2 contributes to the development of
pancreatic ductal adenocarcinoma by regulating the production of
metabolites and reactive oxygen species. Int J Oncol. 52:881–891.
2018.PubMed/NCBI
|
|
116
|
Zheng S, Liu Q, Liu T and Lu X:
Posttranslational modification of pyruvate kinase type M2 (PKM2):
Novel regulation of its biological roles to be further discovered.
J Physiol Biochem. 77:355–363. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Prakasam G, Iqbal MA, Bamezai RNK and
Mazurek S: Posttranslational modifications of pyruvate kinase M2:
Tweaks that benefit cancer. Front Oncol. 8:222018. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Coassolo S, Davidson G, Negroni L, Gambi
G, Daujat S, Romier C and Davidson I: Citrullination of pyruvate
kinase M2 by PADI1 and PADI3 regulates glycolysis and cancer cell
proliferation. Nat Commun. 12:17182021. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Xia L, Jiang Y, Zhang XH, Wang XR, Wei R,
Qin K and Lu Y: SUMOylation disassembles the tetrameric pyruvate
kinase M2 to block myeloid differentiation of leukemia cells. Cell
Death Dis. 12:1012021. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Das Gupta K, Shakespear MR, Curson JEB,
Murthy AMV, Iyer A, Hodson MP, Ramnath D, Tillu VA, von Pein JB,
Reid RC, et al: Class IIa histone deacetylases drive Toll-like
receptor-inducible glycolysis and macrophage inflammatory responses
via pyruvate kinase M2. Cell Rep. 30:2712–2728.e8. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Singh JP, Qian K, Lee JS, Zhou J, Han X,
Zhang B, Ong Q, Ni W, Jiang M, Ruan HB, et al: O-GlcNAcase targets
pyruvate kinase M2 to regulate tumor growth. Oncogene. 39:560–573.
2020. View Article : Google Scholar :
|
|
122
|
Wang J, Yang P, Yu T, Gao M, Liu D, Zhang
J, Lu C, Chen X, Zhang X and Liu Y: Lactylation of PKM2 suppresses
inflammatory metabolic adaptation in Pro-inflammatory macrophages.
Int J Biol Sci. 18:6210–6225. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Sosa RA, Terry AQ, Kaldas FM, Jin YP,
Rossetti M, Ito T, Li F, Ahn RS, Naini BV, Groysberg VM, et al:
Disulfide High-mobility group box 1 drives ischemia-reperfusion
injury in human liver transplantation. Hepatology. 73:1158–1175.
2021. View Article : Google Scholar
|
|
124
|
Chen R, Kang R and Tang D: The mechanism
of HMGB1 secretion and release. Exp Mol Med. 54:91–102. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Yang H, Zeng Q, Silverman HA, Gunasekaran
M, George SJ, Devarajan A, Addorisio ME, Li J, Tsaava T, Shah V, et
al: HMGB1 released from nociceptors mediates inflammation. Proc
Natl Acad Sci USA. 118:e21020341182021. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Venereau E, Casalgrandi M, Schiraldi M,
Antoine DJ, Cattaneo A, De Marchis F, Liu J, Antonelli A, Preti A,
Raeli L, et al: Mutually exclusive redox forms of HMGB1 promote
cell recruitment or proinflammatory cytokine release. J Exp Med.
209:1519–1528. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Tang D, Kang R, Livesey KM, Kroemer G,
Billiar TR, Van Houten B, Zeh HJ III and Lotze MT: High-mobility
group box 1 is essential for mitochondrial quality control. Cell
Metab. 13:701–711. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Park JS, Gamboni-Robertson F, He Q,
Svetkauskaite D, Kim JY, Strassheim D, Sohn JW, Yamada S, Maruyama
I, Banerjee A, et al: High mobility group box 1 protein interacts
with multiple Toll-like receptors. Am J Physiol Cell Physiol.
290:C917–C924. 2006. View Article : Google Scholar
|
|
129
|
Kim J, Sohn E, Kim CS, Jo K and Kim JS:
The role of high-mobility group box-1 protein in the development of
diabetic nephropathy. Am J Nephrol. 33:524–529. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Jin J, Gong J, Zhao L, Zhang H, He Q and
Jiang X: Inhibition of high mobility group box 1 (HMGB1) attenuates
podocyte apoptosis and epithelial-mesenchymal transition by
regulating autophagy flux. J Diabetes. 11:826–836. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Lin M, Yiu WH, Wu HJ, Chan LY, Leung JC,
Au WS, Chan KW, Lai KN and Tang SC: Toll-like receptor 4 promotes
tubular inflammation in diabetic nephropathy. J Am Soc Nephrol.
23:86–102. 2012. View Article : Google Scholar
|
|
132
|
Zhang H, Zhang R, Chen J, Shi M, Li W and
Zhang X: High mobility group box1 inhibitor glycyrrhizic acid
attenuates kidney injury in streptozotocin-induced diabetic rats.
Kidney Blood Press Res. 42:894–904. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Kang R, Livesey KM, Zeh HJ III, Lotze MT
and Tang D: HMGB1 as an autophagy sensor in oxidative stress.
Autophagy. 7:904–906. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Ling H, Chen H, Wei M, Meng X, Yu Y and
Xie K: The effect of autophagy on inflammation cytokines in renal
ischemia/reperfusion injury. Inflammation. 39:347–356. 2016.
View Article : Google Scholar
|
|
135
|
Zhao ZB, Marschner JA, Iwakura T, Li C,
Motrapu M, Kuang M, Popper B, Linkermann A, Klocke J, Enghard P, et
al: Tubular epithelial cell HMGB1 promotes AKI-CKD transition by
sensitizing cycling tubular cells to oxidative stress: A rationale
for targeting HMGB1 during AKI recovery. J Am Soc Nephrol.
34:394–411. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Tesch G, Sourris KC, Summers SA, McCarthy
D, Ward MS, Borg DJ, Gallo LA, Fotheringham AK, Pettit AR, Yap FY,
et al: Deletion of bone-marrow-derived receptor for AGEs (RAGE)
improves renal function in an experimental mouse model of diabetes.
Diabetologia. 57:1977–1985. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Liu Y, Lu T, Zhang C, Xu J, Xue Z,
Busuttil RW, Xu N, Xia Q, Kupiec-Weglinski JW and Ji H: Activation
of YAP attenuates hepatic damage and fibrosis in liver
ischemia-reperfusion injury. J Hepatol. 71:719–730. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Yang K, Fan M, Wang X, Xu J, Wang Y, Tu F,
Gill PS, Ha T, Liu L, Williams DL and Li C: Lactate promotes
macrophage HMGB1 lactylation, acetylation, and exosomal release in
polymicrobial sepsis. Cell Death Differ. 29:133–146. 2022.
View Article : Google Scholar :
|
|
139
|
Du S, Zhang X, Jia Y, Peng P, Kong Q,
Jiang S, Li Y, Li C, Ding Z and Liu L: Hepatocyte HSPA12A inhibits
macrophage chemotaxis and activation to attenuate liver
ischemia/reperfusion injury via suppressing glycolysis-mediated
HMGB1 lactylation and secretion of hepatocytes. Theranostics.
13:3856–3871. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Wu K, Zha H, Wu T, Liu H, Peng R, Lin Z,
Lv D, Liao X, Sun Y and Zhang Z: Cytosolic Hmgb1 accumulation in
mesangial cells aggravates diabetic kidney disease progression via
NFκB signaling pathway. Cell Mol Life Sci. 81:4082024. View Article : Google Scholar
|
|
141
|
Zhu L, Zheng Q, Liu X, Ding H, Ma M, Bao
J, Cai Y and Cao C: HMGB1 lactylation drives neutrophil
extracellular trap formation in lactate-induced acute kidney
injury. Front Immunol. 15:14755432024. View Article : Google Scholar
|
|
142
|
Lee JS, See RH, Galvin KM, Wang J and Shi
Y: Functional interactions between YY1 and adenovirus E1A. Nucleic
Acids Res. 23:925–931. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Shi Y, Seto E, Chang LS and Shenk T:
Transcriptional repression by YY1, a human GLI-Kruppel-related
protein, and relief of repression by adenovirus E1A protein. Cell.
67:377–388. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Yang T, Shu F, Yang H, Heng C, Zhou Y,
Chen Y, Qian X, Du L, Zhu X, Lu Q and Yin X: YY1: A novel
therapeutic target for diabetic nephropathy orchestrated renal
fibrosis. Metabolism. 96:33–45. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Jha JC, Banal C, Okabe J, Gray SP, Hettige
T, Chow BSM, Thallas-Bonke V, De Vos L, Holterman CE, Coughlan MT,
et al: NADPH oxidase Nox5 accelerates renal injury in diabetic
nephropathy. Diabetes. 66:2691–2703. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Gordon S, Akopyan G, Garban H and Bonavida
B: Transcription factor YY1: Structure, function, and therapeutic
implications in cancer biology. Oncogene. 25:1125–1142. 2006.
View Article : Google Scholar
|
|
147
|
Deng Z, Wan M, Cao P, Rao A, Cramer SD and
Sui G: Yin Yang 1 regulates the transcriptional activity of
androgen receptor. Oncogene. 28:3746–3757. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Gao P, Li L, Yang L, Gui D, Zhang J, Han
J, Wang J, Wang N, Lu J, Chen S, et al: Yin Yang 1 protein
ameliorates diabetic nephropathy pathology through transcriptional
repression of TGFbeta1. Sci Transl Med. 11:eaaw20502019. View Article : Google Scholar
|
|
149
|
Wei SY, Shih YT, Wu HY, Wang WL, Lee PL,
Lee CI, Lin CY, Chen YJ, Chien S and Chiu JJ: Endothelial Yin Yang
1 phosphorylation at S118 induces atherosclerosis under flow. Circ
Res. 129:1158–1174. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Du L, Qian X, Li Y, Li XZ, He LL, Xu L,
Liu YQ, Li CC, Ma P, Shu FL, et al: Sirt1 inhibits renal tubular
cell epithelial-mesenchymal transition through YY1 deacetylation in
diabetic nephropathy. Acta Pharmacol Sin. 42:242–251. 2021.
View Article : Google Scholar
|
|
151
|
Huang J, Wang X, Li N, Fan W, Li X, Zhou
Q, Liu J, Li W, Zhang Z, Liu X, et al: YY1 lactylation aggravates
autoimmune uveitis by enhancing microglial functions via
inflammatory genes. Adv Sci (Weinh). 11:e23080312024. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Wang X, Fan W, Li N, Ma Y, Yao M, Wang G,
He S, Li W, Tan J, Lu Q and Hou S: YY1 lactylation in microglia
promotes angiogenesis through transcription activation-mediated
upregulation of FGF2. Genome Biol. 24:872023. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Yoshino J, Monkawa T, Tsuji M, Inukai M,
Itoh H and Hayashi M: Snail1 is involved in the renal
epithelial-mesenchymal transition. Biochem Biophys Res Commun.
362:63–68. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Katoh M and Katoh M: Comparative genomics
on SNAI1, SNAI2, and SNAI3 orthologs. Oncol Rep. 14:1083–1086.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Kaufhold S and Bonavida B: Central role of
Snail1 in the regulation of EMT and resistance in cancer: A target
for therapeutic intervention. J Exp Clin Cancer Res. 33:622014.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Barrallo-Gimeno A and Nieto MA: The Snail
genes as inducers of cell movement and survival: Implications in
development and cancer. Development. 132:3151–3161. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Grande MT, Sanchez-Laorden B, Lopez-Blau
C, De Frutos CA, Boutet A, Arévalo M, Rowe RG, Weiss SJ,
López-Novoa JM and Nieto MA: Snail1-induced partial
epithelial-to-mesenchymal transition drives renal fibrosis in mice
and can be targeted to reverse established disease. Nat Med.
21:989–997. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Li SS, Sun Q, Hua MR, Suo P, Chen JR, Yu
XY and Zhao YY: Targeting the Wnt/β-catenin signaling pathway as a
potential therapeutic strategy in renal tubulointerstitial
fibrosis. Front Pharmacol. 12:7198802021. View Article : Google Scholar
|
|
159
|
Zhao Y, Yin Z, Li H, Fan J, Yang S, Chen C
and Wang DW: MiR-30c protects diabetic nephropathy by suppressing
epithelial-to-mesenchymal transition in db/db mice. Aging Cell.
16:387–400. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Sala L, Franco-Valls H, Stanisavljevic J,
Curto J, Vergés J, Peña R, Duch P, Alcaraz J, García de Herreros A
and Baulida J: Abrogation of myofibroblast activities in metastasis
and fibrosis by methyltransferase inhibition. Int J Cancer.
145:3064–3077. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Zhao X, He X, Wei W and Huang K: USP22
aggravated diabetic renal tubulointerstitial fibrosis progression
through deubiquitinating and stabilizing Snail1. Eur J Pharmacol.
947:1756712023. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Fan M, Yang K, Wang X, Chen L, Gill PS, Ha
T, Liu L, Lewis NH, Williams DL and Li C: Lactate promotes
endothelial-to-mesenchymal transition via Snail1 lactylation after
myocardial infarction. Sci Adv. 9:eadc94652023. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Mao RW, He SP, Lan JG and Zhu WZ: Honokiol
ameliorates cisplatin-induced acute kidney injury via inhibition of
mitochondrial fission. Br J Pharmacol. 179:3886–3904. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Kleele T, Rey T, Winter J, Zaganelli S,
Mahecic D, Perreten Lambert H, Ruberto FP, Nemir M, Wai T,
Pedrazzini T and Manley S: Distinct fission signatures predict
mitochondrial degradation or biogenesis. Nature. 593:435–439. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Haileselassie B, Mukherjee R, Joshi AU,
Napier BA, Massis LM, Ostberg NP, Queliconi BB, Monack D, Bernstein
D and Mochly-Rosen D: Drp1/Fis1 interaction mediates mitochondrial
dysfunction in septic cardiomyopathy. J Mol Cell Cardiol.
130:160–169. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Zhan M, Usman IM, Sun L and Kanwar YS:
Disruption of renal tubular mitochondrial quality control by
Myo-inositol oxygenase in diabetic kidney disease. J Am Soc
Nephrol. 26:1304–1321. 2015. View Article : Google Scholar
|
|
167
|
Huang Q, Chen H, Yin K, Shen Y, Lin K, Guo
X, Zhang X, Wang N, Xin W, Xu Y and Gui D: Formononetin attenuates
renal tubular injury and mitochondrial damage in diabetic
nephropathy partly via regulating Sirt1/PGC-1α pathway. Front
Pharmacol. 13:9012342022. View Article : Google Scholar
|
|
168
|
Su J, Gao C, Xie L, Fan Y, Shen Y, Huang
Q, Wang N, Xu Y, Yang N and Gui D: Astragaloside II ameliorated
podocyte injury and mitochondrial dysfunction in
streptozotocin-induced diabetic rats. Front Pharmacol.
12:6384222021. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Wang S, Zhu H, Li R, Mui D, Toan S, Chang
X and Zhou H: DNA-PKcs interacts with and phosphorylates Fis1 to
induce mitochondrial fragmentation in tubular cells during acute
kidney injury. Sci Signal. 15:eabh11212022. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Bragoszewski P, Turek M and Chacinska A:
Control of mitochondrial biogenesis and function by the
ubiquitin-proteasome system. Open Biol. 7:1700072017. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Wang L, Zhang T, Wang L, Cai Y, Zhong X,
He X, Hu L, Tian S, Wu M, Hui L, et al: Fatty acid synthesis is
critical for stem cell pluripotency via promoting mitochondrial
fission. EMBO J. 36:1330–1347. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
An S, Yao Y, Hu H, Wu J, Li J, Li L, Wu J,
Sun M, Deng Z, Zhang Y, et al: PDHA1 hyperacetylation-mediated
lactate over-production promotes sepsis-induced acute kidney injury
via Fis1 lactylation. Cell Death Dis. 14:4572023. View Article : Google Scholar
|
|
173
|
Roberts RE and Hallett MB: Neutrophil cell
shape change: Mechanism and signalling during cell spreading and
phagocytosis. Int J Mol Sci. 20:13832019. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Wehbi VL and Tasken K: Molecular
mechanisms for cAMP-mediated immunoregulation in T cells-role of
anchored protein kinase A signaling units. Front Immunol.
7:2222016. View Article : Google Scholar
|
|
175
|
Garcia-Ortiz A and Serrador JM: ERM
proteins at the crossroad of leukocyte polarization, migration and
intercellular Adhesion. Int J Mol Sci. 21:15022020. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Zhang X, Li G, Guo Y, Song Y, Chen L, Ruan
Q, Wang Y, Sun L, Hu Y, Zhou J, et al: Regulation of ezrin tension
by S-nitrosylation mediates non-small cell lung cancer invasion and
metastasis. Theranostics. 9:2555–2571. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
McRobert EA and Bach LA: Ezrin contributes
to impaired podocyte migration and adhesion caused by advanced
glycation end products. Nephrology (Carlton). 21:13–20. 2016.
View Article : Google Scholar
|
|
178
|
Qiao J, Tan Y, Liu H, Yang B, Zhang Q, Liu
Q, Sun W, Li Z, Wang Q, Feng W, et al: Histone H3K18 and ezrin
lactylation promote renal dysfunction in sepsis-associated acute
kidney injury. Adv Sci (Weinh). 11:e23072162024. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Catrina SB and Zheng X: Hypoxia and
hypoxia-inducible factors in diabetes and its complications.
Diabetologia. 64:709–716. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Yamazaki T, Mimura I, Tanaka T and Nangaku
M: Treatment of diabetic kidney disease: Current and future.
Diabetes Metab J. 45:11–26. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Feng X, Wang S, Sun Z, Dong H, Yu H, Huang
M and Gao X: Ferroptosis enhanced diabetic renal tubular injury via
HIF-1α/HO-1 pathway in db/db mice. Front Endocrinol (Lausanne).
12:6263902021. View Article : Google Scholar
|
|
182
|
Mei S, Li L, Zhou X, Xue C, Livingston MJ,
Wei Q, Dai B, Mao Z, Mei C and Dong Z: Susceptibility of renal
fibrosis in diabetes: Role of hypoxia inducible factor-1. FASEB J.
36:e224772022. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Dou L and Jourde-Chiche N: Endothelial
toxicity of high glucose and its by-Products in diabetic kidney
disease. Toxins (Basel). 11:5782019. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Fu ZJ, Wang ZY, Xu L, Chen XH, Li XX, Liao
WT, Ma HK, Jiang MD, Xu TT, Xu J, et al: HIF-1alpha-BNIP3-mediated
mitophagy in tubular cells protects against renal
ischemia/reperfusion injury. Redox Biol. 36:1016712020. View Article : Google Scholar
|
|
185
|
Sulkshane P, Ram J, Thakur A, Reis N,
Kleifeld O and Glickman MH: Ubiquitination and receptor-mediated
mitophagy converge to eliminate oxidation-damaged mitochondria
during hypoxia. Redox Biol. 45:1020472021. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Liu C, Yang M, Li L, Luo S, Yang J, Li C,
Liu H and Sun L: A Glimpse of inflammation and Anti-inflammation
therapy in diabetic kidney disease. Front Physiol. 13:9095692022.
View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Luo Y, Yang Z, Yu Y and Zhang P: HIF1α
lactylation enhances KIAA1199 transcription to promote angiogenesis
and vasculogenic mimicry in prostate cancer. Int J Biol Macromol.
222:2225–2243. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Yi W, Clark PM, Mason DE, Keenan MC, Hill
C, Goddard WA III, Peters EC, Driggers EM and Hsieh-Wilson LC:
Phosphofructokinase 1 glycosylation regulates cell growth and
metabolism. Science. 337:975–980. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Fernandes PM, Kinkead J, McNae I, Michels
PAM and Walkinshaw MD: Biochemical and transcript level differences
between the three human phosphofructokinases show optimisation of
each isoform for specific metabolic niches. Biochem J.
477:4425–4441. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Lang L, Chemmalakuzhy R, Shay C and Teng
Y: PFKP signaling at a glance: An emerging mediator of cancer cell
metabolism. Adv Exp Med Biol. 1134:243–258. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Meira DD, Marinho-Carvalho MM, Teixeira
CA, Veiga VF, Da Poian AT, Holandino C, de Freitas MS and
Sola-Penna M: Clotrimazole decreases human breast cancer cells
viability through alterations in cytoskeleton-associated glycolytic
enzymes. Mol Genet Metab. 84:354–362. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Zhang Z, Liang W, Luo Q, Hu H, Yang K, Hu
J, Chen Z, Zhu J, Feng J, Zhu Z, et al: PFKP activation ameliorates
foot process fusion in podocytes in diabetic kidney disease. Front
Endocrinol (Lausanne). 12:7970252021. View Article : Google Scholar
|
|
193
|
Cheng Z, Huang H, Li M and Chen Y:
Proteomic analysis identifies PFKP lactylation in SW480 colon
cancer cells. iScience. 27:1086452024. View Article : Google Scholar
|
|
194
|
Pritchett J, Athwal V, Roberts N, Hanley
NA and Hanley KP: Understanding the role of SOX9 in acquired
diseases: Lessons from development. Trends Mol Med. 17:166–174.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Symon A and Harley V: SOX9: A genomic view
of tissue specific expression and action. Int J Biochem Cell Biol.
87:18–22. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Lefebvre V and Dvir-Ginzberg M: SOX9 and
the many facets of its regulation in the chondrocyte lineage.
Connect Tissue Res. 58:2–14. 2017. View Article : Google Scholar
|
|
197
|
Grimm D, Bauer J, Wise P, Krüger M,
Simonsen U, Wehland M, Infanger M and Corydon TJ: The role of SOX
family members in solid tumours and metastasis. Semin Cancer Biol.
67:122–153. 2020. View Article : Google Scholar
|
|
198
|
Kishi S, Abe H, Akiyama H, Tominaga T,
Murakami T, Mima A, Nagai K, Kishi F, Matsuura M, Matsubara T, et
al: SOX9 protein induces a chondrogenic phenotype of mesangial
cells and contributes to advanced diabetic nephropathy. J Biol
Chem. 286:32162–32169. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Li H, Cai H, Deng J, Tu X, Sun Y, Huang Z,
Ding Z, Dong L, Chen J, Zang Y and Zhang J: TGF-β-mediated
upregulation of Sox9 in fibroblast promotes renal fibrosis. Biochim
Biophys Acta Mol Basis Dis. 1864:520–532. 2018. View Article : Google Scholar
|
|
200
|
Yan F, Teng Y, Li X, Zhong Y, Li C, Yan F
and He X: Hypoxia promotes non-small cell lung cancer cell
stemness, migration, and invasion via promoting glycolysis by
lactylation of SOX9. Cancer Biol Ther. 25:23041612024. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Baubec T, Ivanek R, Lienert F and
Schubeler D: Methylation-dependent and -independent genomic
targeting principles of the MBD protein family. Cell. 153:480–492.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Clouaire T and Stancheva I: Methyl-CpG
binding proteins: Specialized transcriptional repressors or
structural components of chromatin? Cell Mol Life Sci.
65:1509–1522. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Cheng TL, Wang Z, Liao Q, Zhu Y, Zhou WH,
Xu W and Qiu Z: MeCP2 suppresses nuclear microRNA processing and
dendritic growth by regulating the DGCR8/Drosha complex. Dev Cell.
28:547–560. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Partscht P, Simon A, Chen NP, Erhardt S
and Schiebel E: The HIPK2/CDC14B-MeCP2 axis enhances the spindle
assembly checkpoint block by promoting cyclin B translation. Sci
Adv. 9:eadd69822023. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Oh HJ, Kato M, Deshpande S, Zhang E, Das
S, Lanting L, Wang M and Natarajan R: Inhibition of the processing
of miR-25 by HIPK2-Phosphorylated-MeCP2 induces NOX4 in early
diabetic nephropathy. Sci Rep. 6:387892016. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Wang Y, Chen L, Zhang M, Li X, Yang X,
Huang T, Ban Y, Li Y, Li Q, Zheng Y, et al: Exercise-induced
endothelial Mecp2 lactylation suppresses atherosclerosis via the
Ereg/MAPK signalling pathway. Atherosclerosis. 375:45–58. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Ingham RJ, Gish G and Pawson T: The Nedd4
family of E3 ubiquitin ligases: Functional diversity within a
common modular architecture. Oncogene. 23:1972–1984. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Wang X, Trotman LC, Koppie T, Alimonti A,
Chen Z, Gao Z, Wang J, Erdjument-Bromage H, Tempst P, Cordon-Cardo
C, et al: NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN.
Cell. 128:129–139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Fujio Y, Nguyen T, Wencker D, Kitsis RN
and Walsh K: Akt promotes survival of cardiomyocytes in vitro and
protects against ischemia-reperfusion injury in mouse heart.
Circulation. 101:660–667. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Murdaca J, Treins C, Monthouel-Kartmann
MN, Pontier-Bres R, Kumar S, Van Obberghen E and Giorgetti-Peraldi
S: Grb10 prevents Nedd4-mediated vascular endothelial growth factor
receptor-2 degradation. J Biol Chem. 279:26754–26761. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Han F, Wu S, Dong Y, Liu Y, Sun B and Chen
L: Aberrant expression of NEDD4L disrupts mitochondrial homeostasis
by downregulating CaMKKβ in diabetic kidney disease. J Transl Med.
22:4652024. View Article : Google Scholar
|
|
212
|
Henshall TL, Manning JA, Alfassy OS, Goel
P, Boase NA, Kawabe H and Kumar S: Deletion of Nedd4-2 results in
progressive kidney disease in mice. Cell Death Differ.
24:2150–2160. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Al-Qusairi L, Basquin D, Roy A, Rajaram
RD, Maillard MP, Subramanya AR and Staub O: Renal tubular
Ubiquitin-Protein ligase NEDD4-2 is required for renal adaptation
during long-term potassium depletion. J Am Soc Nephrol.
28:2431–2442. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Wu P, Su XT, Gao ZX, Zhang DD, Duan XP,
Xiao Y, Staub O, Wang WH and Lin DH: Renal tubule Nedd4-2
deficiency stimulates Kir4.1/Kir5.1 and Thiazide-sensitive NaCl
cotransporter in distal convoluted tubule. J Am Soc Nephrol.
31:1226–1242. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Li Q, Zhang F, Wang H, Tong Y, Fu Y, Wu K,
Li J, Wang C, Wang Z, Jia Y, et al: NEDD4 lactylation promotes APAP
induced liver injury through Caspase11 dependent non-canonical
pyroptosis. Int J Biol Sci. 20:1413–1435. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Aksan I and Goding CR: Targeting the
microphthalmia basic helix-loop-helix-leucine zipper transcription
factor to a subset of E-box elements in vitro and in vivo. Mol Cell
Biol. 18:6930–6938. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Settembre C, Di Malta C, Polito VA, Garcia
Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D,
Colella P, et al: TFEB links autophagy to lysosomal biogenesis.
Science. 332:1429–1433. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Song JX, Sun YR, Peluso I, Zeng Y, Yu X,
Lu JH, Xu Z, Wang MZ, Liu LF, Huang YY, et al: A novel curcumin
analog binds to and activates TFEB in vitro and in vivo independent
of MTOR inhibition. Autophagy. 12:1372–1389. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Kim SH, Kim G, Han DH, Lee M, Kim I, Kim
B, Kim KH, Song YM, Yoo JE, Wang HJ, et al: Ezetimibe ameliorates
steatohepatitis via AMP activated protein kinase-TFEB-mediated
activation of autophagy and NLRP3 inflammasome inhibition.
Autophagy. 13:1767–1781. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Settembre C, Zoncu R, Medina DL, Vetrini
F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, et
al: A lysosome-to-nucleus signalling mechanism senses and regulates
the lysosome via mTOR and TFEB. EMBO J. 31:1095–1108. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Yuan Y, Li L, Zhu L, Liu F, Tang X, Liao
G, Liu J, Cheng J, Chen Y and Lu Y: Mesenchymal stem cells elicit
macrophages into M2 phenotype via improving transcription factor
EB-mediated autophagy to alleviate diabetic nephropathy. Stem
Cells. 38:639–652. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Rask-Madsen C and King GL: Diabetes:
Podocytes lose their footing. Nature. 468:42–44. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Zhao X, Chen Y, Tan X, Zhang L, Zhang H,
Li Z, Liu S, Li R, Lin T, Liao R, et al: Advanced glycation
end-products suppress autophagic flux in podocytes by activating
mammalian target of rapamycin and inhibiting nuclear translocation
of transcription factor EB. J Pathol. 245:235–248. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Jacinto E: TFEBulous control of traffic by
mTOR. EMBO J. 30:3215–3216. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Li T, Yin L, Kang X, Xue W, Wang N, Zhang
J, Yuan P, Lin L and Li Y: TFEB acetylation promotes lysosome
biogenesis and ameliorates Alzheimer's disease-relevant phenotypes
in mice. J Biol Chem. 298:1026492022. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Martina JA and Puertollano R: Protein
phosphatase 2A stimulates activation of TFEB and TFE3 transcription
factors in response to oxidative stress. J Biol Chem.
293:12525–12534. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Settembre C, De Cegli R, Mansueto G, Saha
PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch
TJ, et al: TFEB controls cellular lipid metabolism through a
starvation-induced autoregulatory loop. Nat Cell Biol. 15:647–658.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Sha Y, Rao L, Settembre C, Ballabio A and
Eissa NT: STUB1 regulates TFEB-induced autophagy-lysosome pathway.
EMBO J. 36:2544–2552. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Huang Y, Luo G, Peng K, Song Y, Wang Y,
Zhang H, Li J, Qiu X, Pu M, Liu X, et al: Lactylation stabilizes
TFEB to elevate autophagy and lysosomal activity. J Cell Biol.
223:e2023080992024. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Qu D, Jiang M, Huang D, Zhang H, Feng L,
Chen Y, Zhu X, Wang S and Han J: Synergistic effects of the
enhancements to mitochondrial ROS, p53 activation and apoptosis
generated by aspartame and potassium sorbate in HepG2 cells.
Molecules. 24:4572019. View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Kruiswijk F, Labuschagne CF and Vousden
KH: p53 in survival, death and metabolic health: A lifeguard with a
licence to kill. Nat Rev Mol Cell Biol. 16:393–405. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
232
|
Saldana-Meyer R and Recillas-Targa F:
Transcriptional and epigenetic regulation of the p53 tumor
suppressor gene. Epigenetics. 6:1068–1077. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Mulay SR, Thomasova D, Ryu M and Anders
HJ: MDM2 (murine double minute-2) links inflammation and tubular
cell healing during acute kidney injury in mice. Kidney Int.
81:1199–1211. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Peng J, Li X, Zhang D, Chen JK, Su Y,
Smith SB and Dong Z: Hyperglycemia, p53, and mitochondrial pathway
of apoptosis are involved in the susceptibility of diabetic models
to ischemic acute kidney injury. Kidney Int. 87:137–150. 2015.
View Article : Google Scholar :
|
|
235
|
Yang R, Xu X, Li H, Chen J, Xiang X, Dong
Z and Zhang D: p53 induces miR199a-3p to suppress SOCS7 for STAT3
activation and renal fibrosis in UUO. Sci Rep. 7:434092017.
View Article : Google Scholar : PubMed/NCBI
|
|
236
|
Brezniceanu ML, Liu F, Wei CC, Chénier I,
Godin N, Zhang SL, Filep JG, Ingelfinger JR and Chan JS:
Attenuation of interstitial fibrosis and tubular apoptosis in db/db
transgenic mice overexpressing catalase in renal proximal tubular
cells. Diabetes. 57:451–459. 2008. View Article : Google Scholar
|
|
237
|
Brezniceanu ML, Liu F, Wei CC, Tran S,
Sachetelli S, Zhang SL, Guo DF, Filep JG, Ingelfinger JR and Chan
JS: Catalase overexpression attenuates angiotensinogen expression
and apoptosis in diabetic mice. Kidney Int. 71:912–923. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
238
|
Qi R, Wang J, Jiang Y, Qiu Y, Xu M, Rong R
and Zhu T: Snai1-induced partial epithelial-mesenchymal transition
orchestrates p53-p21-mediated G2/M arrest in the progression of
renal fibrosis via NF-κB-mediated inflammation. Cell Death Dis.
12:442021. View Article : Google Scholar
|
|
239
|
Sutton TA, Hato T, Mai E, Yoshimoto M,
Kuehl S, Anderson M, Mang H, Plotkin Z, Chan RJ and Dagher PC: p53
is renoprotective after ischemic kidney injury by reducing
inflammation. J Am Soc Nephrol. 24:113–124. 2013. View Article : Google Scholar :
|
|
240
|
Zong Z, Xie F, Wang S, Wu X, Zhang Z, Yang
B and Zhou F: Alanyl-tRNA synthetase, AARS1, is a lactate sensor
and lactyltransferase that lactylates p53 and contributes to
tumorigenesis. Cell. 187:2375–2392.e33. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
241
|
Russo GL, Russo M and Ungaro P:
AMP-activated protein kinase: A target for old drugs against
diabetes and cancer. Biochem Pharmacol. 86:339–350. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
242
|
Momcilovic M, Hong SP and Carlson M:
Mammalian TAK1 activates Snf1 protein kinase in yeast and
phosphorylates AMP-activated protein kinase in vitro. J Biol Chem.
281:25336–25343. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
243
|
Rogacka D, Piwkowska A, Audzeyenka I,
Angielski S and Jankowski M: Involvement of the AMPK-PTEN pathway
in insulin resistance induced by high glucose in cultured rat
podocytes. Int J Biochem Cell Biol. 51:120–130. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
244
|
Jørgensen SB, Viollet B, Andreelli F,
Frøsig C, Birk JB, Schjerling P, Vaulont S, Richter EA and
Wojtaszewski JF: Knockout of the alpha2 but not alpha1
5'-AMP-activated protein kinase isoform abolishes
5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not
contraction-induced glucose uptake in skeletal muscle. J Biol Chem.
279:1070–1079. 2004. View Article : Google Scholar
|
|
245
|
Canto C, Gerhart-Hines Z, Feige JN,
Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P and Auwerx
J: AMPK regulates energy expenditure by modulating NAD+ metabolism
and SIRT1 activity. Nature. 458:1056–1060. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
246
|
Godel M, Hartleben B, Herbach N, Liu S,
Zschiedrich S, Lu S, Debreczeni-Mór A, Lindenmeyer MT, Rastaldi MP
and Hartleben G: Role of mTOR in podocyte function and diabetic
nephropathy in humans and mice. J Clin Invest. 121:2197–2209. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
247
|
Guo H, Wang Y, Zhang X, Zang Y, Zhang Y,
Wang L, Wang H, Wang Y, Cao A and Peng W: Astragaloside IV protects
against podocyte injury via SERCA2-dependent ER stress reduction
and AMPKα-regulated autophagy induction in streptozotocin-induced
diabetic nephropathy. Sci Rep. 7:68522017. View Article : Google Scholar
|
|
248
|
Zhang Y, Huang Z, Han W, Wu J, Li S, Qin
T, Zhang C, Shi M, Han S, Gao B, et al: Glutamine suppresses
senescence and promotes autophagy through glycolysis
inhibition-mediated AMPKα lactylation in intervertebral disc
degeneration. Commun Biol. 7:3252024. View Article : Google Scholar
|
|
249
|
Hou X, Hong Z, Zeng H, Zhang C, Zhang P,
Ma D and Han Z: Lactylation in cancer biology: Unlocking new
avenues for research and therapy. Cancer Commun (Lond).
45:1367–1406. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
250
|
Wu Z, Peng Y, Chen W, Xia F, Song T and Ke
Q: Lactylation-driven transcriptional activation of FBXO33 promotes
gallbladder cancer metastasis by regulating p53 polyubiquitination.
Cell Death Dis. 16:1442025. View Article : Google Scholar : PubMed/NCBI
|
|
251
|
Feng L, Feng YY, Ren Q, Fu P and Ma L:
Mesangial cells in diabetic kidney disease: From mechanisms to
therapeutic implications. Int J Biol Sci. 21:4762–4781. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
252
|
Zhang S, Luo M, Lu Z and Shi Q: Lactate
and lactylation in sepsis-associated acute kidney injury: Clinical
evidence from the MIMIC-IV database and mechanistic insights. Front
Med (Lausanne). 12:17081452025. View Article : Google Scholar : PubMed/NCBI
|
|
253
|
Zhang D, Liang C, Wu C, Hawanga M, Wan S,
Xu L, Zhang X, Liu Y, Hu F, Wang M, et al: Nonhistone lactylation:
A hub for tumour metabolic reprogramming and epigenetic regulation.
J Transl Med. 23:9012025. View Article : Google Scholar : PubMed/NCBI
|
|
254
|
Kung CP and Murphy ME: The role of the p53
tumor suppressor in metabolism and diabetes. J Endocrinol.
231:R61–R75. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
255
|
Sun T, Liu B, Li Y, Wu J, Cao Y, Yang S,
Tan H, Cai L, Zhang S, Qi X, et al: Oxamate enhances the efficacy
of CAR-T therapy against glioblastoma via suppressing
ectonucleotidases and CCR8 lactylation. J Exp Clin Cancer Res.
42:2532023. View Article : Google Scholar : PubMed/NCBI
|
|
256
|
Marlier JF, Cleland WW and Zeczycki TN:
Oxamate is an alternative substrate for pyruvate carboxylase from
Rhizobium etli. Biochemistry. 52:2888–2894. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
257
|
Altinoz MA and Ozpinar A: Oxamate
targeting aggressive cancers with special emphasis to brain tumors.
Biomed Pharmacother. 147:1126862022. View Article : Google Scholar : PubMed/NCBI
|
|
258
|
Ye W, Zheng Y, Zhang S, Yan L, Cheng H and
Wu M: Oxamate improves glycemic control and insulin sensitivity via
inhibition of tissue lactate production in db/db Mice. PLoS One.
11:e01503032016. View Article : Google Scholar : PubMed/NCBI
|
|
259
|
Friberg A, Rehwinkel H, Nguyen D, Pütter
V, Quanz M, Weiske J, Eberspächer U, Heisler I and Langer G:
Structural evidence for isoform-selective allosteric inhibition of
lactate dehydrogenase A. ACS Omega. 5:13034–13041. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
260
|
Hollenberg AM, Smith CO, Shum LC, Awad H
and Eliseev RA: Lactate dehydrogenase inhibition with oxamate
exerts bone anabolic effect. J Bone Miner Res. 35:2432–2443. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
261
|
Qiao T, Xiong Y, Feng Y, Guo W, Zhou Y,
Zhao J, Jiang T, Shi C and Han Y: Inhibition of LDH-A by oxamate
enhances the efficacy of Anti-PD-1 treatment in an NSCLC humanized
mouse model. Front Oncol. 11:6323642021. View Article : Google Scholar : PubMed/NCBI
|
|
262
|
Shibata S, Sogabe S, Miwa M, Fujimoto T,
Takakura N, Naotsuka A, Kitamura S, Kawamoto T and Soga T:
Identification of the first highly selective inhibitor of human
lactate dehydrogenase B. Sci Rep. 11:213532021. View Article : Google Scholar : PubMed/NCBI
|
|
263
|
Rai G, Brimacombe KR, Mott BT, Urban DJ,
Hu X, Yang SM, Lee TD, Cheff DM, Kouznetsova J, Benavides GA, et
al: Discovery and optimization of potent, Cell-active
pyrazole-based inhibitors of lactate dehydrogenase (LDH). J Med
Chem. 60:9184–9204. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
264
|
Han JH, Lee EJ, Park W, Ha KT and Chung
HS: Natural compounds as lactate dehydrogenase inhibitors:
Potential therapeutics for lactate dehydrogenase inhibitors-related
diseases. Front Pharmacol. 14:12750002023. View Article : Google Scholar : PubMed/NCBI
|
|
265
|
El Hassouni B, Franczak M, Capula M, Vonk
CM, Gomez VM, Smolenski RT, Granchi C, Peters GJ, Minutolo F and
Giovannetti E: Lactate dehydrogenase A inhibition by small
molecular entities: Steps in the right direction. Oncoscience.
7:76–80. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
266
|
Hwang D, Kim T, Kyun S, Jang I, Park HY,
Kim SW, Han JS, So JM, Lee CH, Park J, et al: Oxamate suppresses
whole-body energy metabolism at rest and during exercise in mice by
inhibiting fat oxidation and altering lactate dynamics. Phys Act
Nutr. 29:26–34. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
267
|
Zhai X, Yang Y, Wan J, Zhu R and Wu Y:
Inhibition of LDH-A by oxamate induces G2/M arrest, apoptosis and
increases radiosensitivity in nasopharyngeal carcinoma cells. Oncol
Rep. 30:2983–2991. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
268
|
Ouyang J, Wang H and Huang J: The role of
lactate in cardiovascular diseases. Cell Commun Signal. 21:3172023.
View Article : Google Scholar : PubMed/NCBI
|
|
269
|
Cui D and Morris ME: The drug of abuse
gamma-hydroxybutyrate is a substrate for sodium-coupled
monocarboxylate transporter (SMCT) 1 (SLC5A8): Characterization of
SMCT-mediated uptake and inhibition. Drug Metab Dispos.
37:1404–1410. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
270
|
Emoto A, Ushigome F, Koyabu N, Kajiya H,
Okabe K, Satoh S, Tsukimori K, Nakano H, Ohtani H and Sawada Yl:
H(+)-linked transport of salicylic acid, an NSAID, in the human
trophoblast cell line BeWo. Am J Physiol Cell Physiol.
282:C1064–C1075. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
271
|
Richard AN, Natalie B, Helen B, Sikka A,
Thomas H, Phillips N, Nakjang S, Miwa S, Crossland R, Rand V, et
al: Inhibition of monocarboxyate transporter 1 by AZD3965 as a
novel therapeutic approach for diffuse large B-cell lymphoma and
Burkitt lymphoma. Haematologica. 102:1247–1257. 2017. View Article : Google Scholar
|
|
272
|
Bola BM, Chadwick AL, Michopoulos F,
Blount KG, Telfer BA, Williams KJ, Smith PD, Critchlow SE and
Stratford IJ: Inhibition of monocarboxylate transporter-1 (MCT1) by
AZD3965 enhances radiosensitivity by reducing lactate transport.
Mol Cancer Ther. 13:2805–2816. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
273
|
Beloueche-Babari M, Wantuch S, Casals
Galobart T, Koniordou M, Parkes HG, Arunan V, Chung YL, Eykyn TR,
Smith PD and Leach MO: MCT1 Inhibitor AZD3965 increases
mitochondrial metabolism, facilitating combination therapy and
noninvasive magnetic resonance spectroscopy. Cancer Res.
77:5913–5924. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
274
|
Xu Z and Wang X, Cheng H, Li J, Zhang X
and Wang X: The role of MCT1 in tumor progression and targeted
therapy: A comprehensive review. Front Immunol. 16:16104662025.
View Article : Google Scholar : PubMed/NCBI
|
|
275
|
Halford SER, Jones P, Wedge S, Hirschberg
S, Katugampola S, Veal G, Payne G and Plummer ER: A first-in-human
first-in-class (FIC) trial of the monocarboxylate transporter 1
(MCT1) inhibitor AZD3965 in patients with advanced solid tumours. J
Clin Oncol. 35:2516. 2017. View Article : Google Scholar
|
|
276
|
Halford S, Veal GJ, Wedge SR, Payne GS,
Bacon CM, Sloan P, Dragoni I, Heinzmann K, Potter S, Salisbury BM,
et al: A Phase I Dose-escalation Study of AZD3965, an oral
monocarboxylate transporter 1 inhibitor, in patients with advanced
cancer. Clin Cancer Res. 29:1429–1439. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
277
|
Huang T, Feng Q, Wang Z, Li W, Sun Z,
Wilhelm J, Huang G, Vo T, Sumer BD and Gao J: Tumor-Targeted
inhibition of monocarboxylate transporter 1 improves T-cell
immunotherapy of solid tumors. Adv Healthc Mater. 10:20005492021.
View Article : Google Scholar
|
|
278
|
Wu G, Pan Y, Chen M, Liu Z, Li C, Sheng Y,
Li H, Shen M and Liu H: Lactylation drives hCG-triggered
luteinization in hypoxic granulosa cells. Int J Biol Macromol.
280:1355802024. View Article : Google Scholar : PubMed/NCBI
|
|
279
|
Leng S, Huang W, Chen Y, Yang Y, Feng D,
Liu W, Gao T, Ren Y, Huo M, Zhang J, et al: SIRT1 coordinates with
the CRL4B complex to regulate pancreatic cancer stem cells to
promote tumorigenesis. Cell Death Differ. 28:3329–3343. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
280
|
Peng J, Li J, Huang J, Xu P, Huang H, Liu
Y, Yu L, Yang Y, Zhou B, Jiang H, et al: p300/CBP inhibitor A-485
alleviates acute liver injury by regulating macrophage activation
and polarization. Theranostics. 9:8344–8361. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
281
|
Jaschke NP, Breining D, Hofmann M, Pählig
S, Baschant U, Oertel R, Traikov S, Grinenko T, Saettini F, Biondi
A, et al: Small-molecule CBP/p300 histone acetyltransferase
inhibition mobilizes leukocytes from the bone marrow via the
endocrine stress response. Immunity. 57:364–378.e9. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
282
|
Ji C, Xu W, Ding H, Chen Z, Shi C, Han J,
Yu L, Qiao N, Zhang Y, Cao X, et al: The p300 inhibitor A-485
exerts antitumor activity in growth hormone pituitary adenoma. J
Clin Endocrinol Metab. 107:e2291–e2300. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
283
|
Ansari MSZ, Stagni V, Iuzzolino A, Rotili
D, Mai A, Del Bufalo D, Lavia P, Degrassi F and Trisciuoglio D:
Pharmacological targeting of CBP/p300 drives a redox/autophagy axis
leading to senescence-induced growth arrest in non-small cell lung
cancer cells. Cancer Gene Therapy. 30:124–136. 2023. View Article : Google Scholar :
|
|
284
|
Dai SK, Liu PP, Li X, Jiao LF, Teng ZQ and
Liu CM: Dynamic profiling and functional interpretation of histone
lysine crotonylation and lactylation during neural development.
Development. 149:dev2000492022. View Article : Google Scholar : PubMed/NCBI
|
|
285
|
Bursch KL, Goetz CJ and Smith BC: Current
trends in sirtuin activator and inhibitor development. Molecules.
29:11852024. View Article : Google Scholar : PubMed/NCBI
|
|
286
|
Yu S, Li Y, Lu X, Han Z, Li C, Yuan X and
Guo D: The regulatory role of miRNA and lncRNA on autophagy in
diabetic nephropathy. Cell Signal. 118:1111442024. View Article : Google Scholar : PubMed/NCBI
|
|
287
|
Zhang Y and Zhang X: Virus-induced histone
lactylation promotes virus infection in crustacean. Adv Sci
(Weinh). 11:e24010172024. View Article : Google Scholar : PubMed/NCBI
|
|
288
|
Liu L, Chen Y, Li X, Wang J and Yang L:
Therapeutic potential: The role of mesenchymal stem cells from
diverse sources and their derived exosomes in diabetic nephropathy.
Biomed Pharmacother. 175:1166722024. View Article : Google Scholar : PubMed/NCBI
|
|
289
|
van Son J, Oussaada SM, Şekercan A, Beudel
M, Dongelmans DA, van Assen S, Eland IA, Moeniralam HS, Dormans
TPJ, van Kalkeren CAJ, et al: Overweight and obesity are associated
with acute kidney injury and acute respiratory distress syndrome,
but not with increased mortality in hospitalized COVID-19 patients:
A retrospective cohort study. Front Endocrinol (Lausanne).
12:7477322021. View Article : Google Scholar :
|
|
290
|
Schiller M, Solger K, Leipold S, Kerl HU
and Kick W: Diabetes-associated nephropathy and obesity influence
COVID-19 outcome in type 2 diabetes patients. J Community Hosp
Intern Med Perspect. 11:590–596. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
291
|
Gao M, Piernas C, Astbury NM,
Hippisley-Cox J, O'Rahilly S, Aveyard P and Jebb SA: Associations
between body-mass index and COVID-19 severity in 6•9 million people
in England: A prospective, community-based, cohort study. Lancet
Diabetes Endocrinol. 9:350–359. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
292
|
Sörling A, Nordberg P, Hofmann R, Häbel H
and Svensson P: Association between CKD, obesity, cardiometabolic
risk factors, and severe COVID-19 outcomes. Kidney Int Rep.
8:775–784. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
293
|
Martín-Del-Campo F, Ruvalcaba-Contreras N,
Velázquez-Vidaurri AL, Cueto-Manzano AM, Rojas-Campos E,
Cortés-Sanabria L, Espinel-Bermúdez MC, Hernández-González SO,
Nava-Zavala AH, Fuentes-Orozco C, et al: Morbid obesity is
associated with mortality and acute kidney injury in hospitalized
patients with COVID-19. Clin Nutr ESPEN. 45:200–205. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
294
|
Al-Sabah S, Al-Haddad M, Al-Youha S, Jamal
M and Almazeedi S: COVID-19: Impact of obesity and diabetes on
disease severity. Clin Obes. 10:e124142020. View Article : Google Scholar : PubMed/NCBI
|
|
295
|
Li LN, Li WW, Xiao LS and Lai WN:
Lactylation signature identifies liver fibrosis phenotypes and
traces fibrotic progression to hepatocellular carcinoma. Front
Immunol. 15:14333932024. View Article : Google Scholar : PubMed/NCBI
|
|
296
|
Wang P, Xie D, Xiao T, Cheng C, Wang D,
Sun J, Wu M, Yang Y, Zhang A and Liu Q: H3K18 lactylation promotes
the progression of arsenite-related idiopathic pulmonary fibrosis
via YTHDF1/m6A/NREP. J Hazard Mater. 461:1325822024. View Article : Google Scholar
|
|
297
|
Liang H, Xu L and Yang Y: Lactate and
lactylation: Novel perspectives on fibrosis pathogenesis and
therapeutic directions. J Transl Med. 23:7052025. View Article : Google Scholar : PubMed/NCBI
|
|
298
|
Nechipurenko YD, Semyonov DA, Lavrinenko
IA, Lagutkin DA, Generalov EA, Zaitceva AY, Matveeva OV and Yegorov
YE: The role of acidosis in the pathogenesis of severe forms of
COVID-19. Biology (Basel). 10:8522021.PubMed/NCBI
|
|
299
|
Wang X, Li Y, Qiang G, Wang K, Dai J,
McCann M, Munoz MD, Gil V, Yu Y, Li S, et al: Secreted EMC10 is
upregulated in human obesity and its neutralizing antibody prevents
diet-induced obesity in mice. Nat Commun. 13:73232022. View Article : Google Scholar : PubMed/NCBI
|
|
300
|
Gupta GS: The lactate and the lactate
dehydrogenase in inflammatory diseases and major risk factors in
COVID-19 patients. Inflammation. 45:2091–2123. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
301
|
Bao C, Ma Q, Ying X, Wang F, Hou Y, Wang
D, Zhu L, Huang J and He C: Histone lactylation in macrophage
biology and disease: From plasticity regulation to therapeutic
implications. eBioMedicine. 111:1055022025. View Article : Google Scholar :
|
|
302
|
Li C, Fu C, Zhou W, Li H, Liu Z, Wu G, He
T, Shen M and Liu H: Lactylation modification of HIF-1α enhances
its stability by blocking VHL recognition. Cell Commun Signal.
23:3642025. View Article : Google Scholar
|
|
303
|
Thyrsted J, Storgaard J, Blay-Cadanet J,
Heinz A, Thielke AL, Crotta S, de Paoli F, Olagnier D, Wack A,
Hiller K, et al: Influenza A induces lactate formation to inhibit
type I IFN in primary human airway epithelium. iScience.
24:1033002021. View Article : Google Scholar : PubMed/NCBI
|
|
304
|
Wu J, Li Y, Yang Y, Chen R, Wang H, Xie K,
Lan J, Guo M, Zhang Y and Li X: Wogonoside inhibits fibroblast
activation and pulmonary fibrosis by dual regulation of Snail1
lactylation and PGC1α/PINK1-mediated mitophagy. Phytomedicine.
147:1572402025. View Article : Google Scholar
|