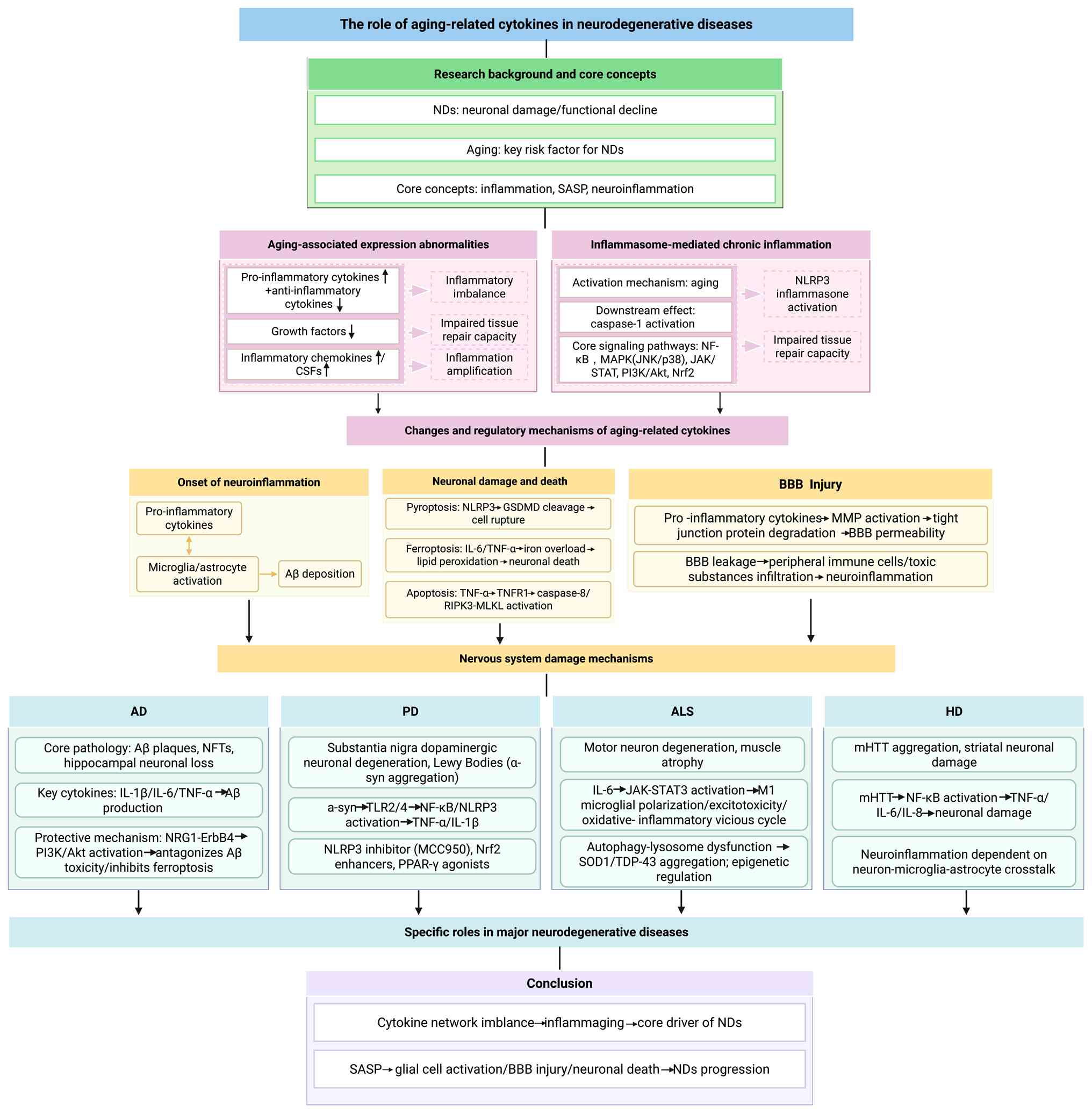
Aging is a multifaceted process characterized by the
progressive decline of biological function, accompanied by chronic
inflammation and disruptions in cellular homeostasis. This process
is associated with the pathogenesis of neurodegenerative disease
(ND). Throughout aging, immune system dysfunction results in an
imbalance between pro- and anti-inflammatory factors, creating a
persistent inflammatory microenvironment that exacerbates neuronal
damage and synaptic dysfunction (1). Cytokines released by the
senescence-associated secretory phenotype (SASP) serve a key role
in mediating neuroinflammation and neuronal damage (1). Key aging-associated cytokines
include IL-6, tumor necrosis factor-α (TNF-α), transforming growth
factor-β (TGF-β) and chemokines such as chemokine (C-C motif)
ligand 2 (CCL2) and C-X-C motif chemokine ligand 10 (CXCL10)
(2). These factors intensify
neuroinflammatory responses by activating microglia and astrocytes,
which promote β-amyloid (Aβ) deposition, τ hyperphosphorylation and
α-synuclein (α-syn) aggregation, ultimately resulting in neuronal
apoptosis and synaptic dysfunction (2).
IL-6 and TNF-α perpetuate chronic neuroinflammation
through the NF-κB signaling pathway, as evidenced in
lipopolysaccharide (LPS)-stimulated murine microglial cell lines
and LPS-hyperresponsive TNFAIP3/A20-deficient murine
neuroinflammation models (3,4).
By contrast, TGF-β may serve a dual role, being involved in immune
regulation and potentially contributing to fibrosis, as observed in
both in vitro fibroblast cultures and murine models of
tissue injury (3,5). Therapeutic strategies targeting
aging-associated cytokines, including neutralizing antibodies,
receptor antagonists or gene editing techniques, have demonstrated
potential in delaying disease progression primarily in mouse models
of obesity, type 2 diabetes and aging; however, their safety and
efficacy in human subjects require further validation (6,7).
The present study aimed to review the pathological mechanisms and
therapeutic prospects of age-related cytokines in ND, providing a
theoretical foundation for the development of novel intervention
strategies (Fig. 1).
Cytokines are soluble proteins or glycoproteins with
low molecular weight, typically ranging from 6 to 70 kDa, that
facilitate intercellular signal transmission and serve crucial
roles in regulating physiological processes such as immune
responses, cell proliferation, differentiation, metabolism,
apoptosis and tissue repair (8).
These molecules are secreted by immune cells, including
macrophages, lymphocytes and mast cells, as well as by non-immune
cells such as endothelial cells, fibroblasts, astrocytes, microglia
and other stromal cells (8).
The cytokine family is diverse and can be
categorized based on structural characteristics and functional
roles. Structurally, cytokines are classified into families,
including TNF, IL, IFN, colony-stimulating factor (CSF), TGF,
chemokines and GF (9).
Furthermore, cytokines are categorized into pro- and
anti-inflammatory factors based on their primary biological
effects. This classification is not definitive as certain
cytokines, including TGF-β1, IL-6 and IL-10, may display both pro-
and anti-inflammatory characteristics depending on the specific
microenvironment (9).
Pro-inflammatory cytokines, including members of the
IL-1 family, TNF-α, IL-6, IL-8, IL-12, IL-17, IL-18, IFN-γ and
resistin, are pivotal in initiating inflammatory responses.
However, the persistent presence or upregulation of these cytokines
can result in chronic inflammation, which is associated with aging
and various age-related diseases, such as cardiovascular disease,
diabetes and Alzheimer's disease (AD). Conversely,
anti-inflammatory cytokines, such as IL-4, IL-10, TGF-β, IL-13 and
IL-1 receptor antagonist (IL-1RA), typically decline during the
aging process, promoting the development of chronic low-grade
inflammation and accelerating the aging process (10).
Beyond the classical pro- and anti-inflammatory
dichotomy, other families of cytokines and signaling molecules play
key roles in intercellular communication and tissue homeostasis.
GF, such as nerve GF (NGF), vascular endothelial GF (VEGF),
insulin-like GF 1 (IGF-1), neuregulin (NRG), and fibroblast GF, are
typically not categorized as either anti-inflammatory or
pro-inflammatory (11). As the
aging process advances, the levels of GF generally diminish,
leading to a marked decrease in the capacity for tissue repair and
regeneration. Furthermore, chemokines and CSFs do not directly
amplify or inhibit the inflammatory response but primarily modulate
immune cell functions (12).
Based on their function, chemokines are classified
into inflammatory and homeostatic categories. Inflammatory
chemokines enhance the inflammatory response by recruiting immune
cells to sites of inflammation and include molecules such as
monocyte chemoattractant protein-1 (MCP-1), fractalkine and
macrophage inflammatory protein-1 (13). homeostatic chemokines serve a key
role in regulating the migration of immune cells, attenuating
immune responses, maintaining immune homeostasis and facilitating
tissue repair. Key chemokines in this category include CXCL12,
CCL18 and CXCL13 (14).
During the aging process, there is a marked
elevation in the levels of pro-inflammatory factors, including
IL-6, TNF-α, and IL-1β. These factors contribute to the maintenance
of chronic low-grade inflammation by facilitating the activation of
immune cells and perpetuating the cytokine cascade (15). For example, IL-6 overexpression
in collagen-induced arthritis mice and immunoglobulin heavy chain
enhancer (Eµ)-IL-6 transgenic mice promotes inflammation via
nuclear factor IL-6-driven transcription (16). Moreover, elevated serum IL-6 in
humans is associated with cardiovascular disease and type 2
diabetes (17). TNF-α, primarily
acting via NF-κB, contributes to aging-associated pathologies such
as muscle atrophy and neurodegeneration. This is supported by
studies in LPS-stimulated primary murine macrophages and C57BL/6
mice, where TNF/TNF receptor 1 (TNFR1) ablation alleviates
lethality caused by NF-κB pathway deficiency (18,19). Inflammatory chemokines are also
upregulated with age, promoting monocyte, macrophage and T cell
recruitment to inflamed sites (20). Their continuous elevation
sustains chronic inflammation and worsens age-associated diseases
(13).
Under physiological conditions, anti-inflammatory
factors are crucial for maintaining immune system balance and
homeostasis by mitigating excessive immune responses. During aging,
the expression of key anti-inflammatory cytokines such as IL-10 and
IL-1RA is typically diminished (21). This compromises the immune system
capacity to resolve inflammation. Specifically, lower serum IL-10
levels, as observed in a cross-sectional study of 193 adults aged
>60 years, impair the suppression of excessive immune responses
and are associated with features of metabolic syndrome (22). Similarly, decreased IL-1RA
expression leads to heightened activity of the pro-inflammatory
cytokines IL-1α and IL-1β (23,24). By contrast, TGF-β undergoes a
functional shift rather than a quantitative decline. Although TGF-β
serves crucial roles in tissue repair and immune regulation, its
signaling becomes dysregulated with age (25). This results in overactivation of
the TGF-β pathway, which is associated with pro-fibrotic responses
and contributes to tissue senescence and pathological fibrosis,
thereby exacerbating chronic inflammation (26).
Throughout the aging process, GF levels typically
decrease, which directly impacts tissue repair, cellular
regeneration and immune system functionality (27). To clarify their distinct roles in
the aging-associated decline, major GFs can be categorized into two
functional groups based on their primary physiological actions:
Neurotrophic and angiogenic factors, which support neuronal
survival and vascular health, and metabolic and repair-associated
factors, which regulate tissue maintenance and regeneration.
In chronic low-grade inflammation, as a compensatory
mechanism for the declining immune system, levels of G-CSF and
GM-CSF rise. This elevation leads to increased production of immune
cells, such as neutrophils and macrophages, thereby enhancing
immune efficacy. These changes increase the susceptibility of
elderly individuals to inflammatory phenomena (40).
Inflammatory senescence is a hallmark of aging,
characterized by prolonged immune system activation and a
persistent increase in pro-inflammatory factors (41). One of the key mechanisms
underlying inflammatory aging is the activation of inflammasomes,
which initiate a sustained immune response by promoting the release
of pro-inflammatory cytokines such as IL-1β and IL-18 (42). This persistent pro-inflammatory
response not only exacerbates tissue damage but also contributes to
a decline in immune function (41). Chronic activation of
inflammasomes accelerates the aging process and is closely
associated with the onset of age-related diseases such as AD,
atherosclerosis, and type 2 diabetes (43). Therefore, the continuous
activation of inflammasomes is a key driver of age-related immune
decline and disease progression.
As aging advances, the intracellular levels of
reactive oxygen species (ROS) increase. ROS not only directly
damage cell components but also function as signaling molecules to
activate the NOD-like receptor protein 3 (NLRP3) inflammasome. In
murine bone marrow-derived macrophages, ATP-induced oxidative
stress upregulates NLRP3 expression and promotes its cytoplasmic
aggregation, thereby activating Caspase-1 and driving IL-1β/IL-18
secretion (44). During aging,
mitochondrial function declines, and the damage signals released by
mitochondria, such as cytochrome C, activate the NLRP3 inflammasome
(45). Concurrently, the
increase in mitochondrial peroxides augments ROS production,
thereby promoting inflammasome activation (46). Senescent cells typically exhibit
damage or leakage of the cell membrane, resulting in the release of
intracellular endogenous harmful molecules, such as ATP and uric
acid crystals, into the extracellular environment. These molecules
serve as danger signals, activating the NLRP3 inflammasome in
immune cells and triggering an immune response (47).
During the aging process, the activation of
inflammasomes not only influences the immune response through
intrinsic mechanisms but also intensifies the inflammatory response
by engaging cellular signaling pathways, such as the NF-κB, MAPK,
JAK/STAT, phosphoinositide 3-kinase (PI3K)/Akt and Nrf2) signaling
pathways.
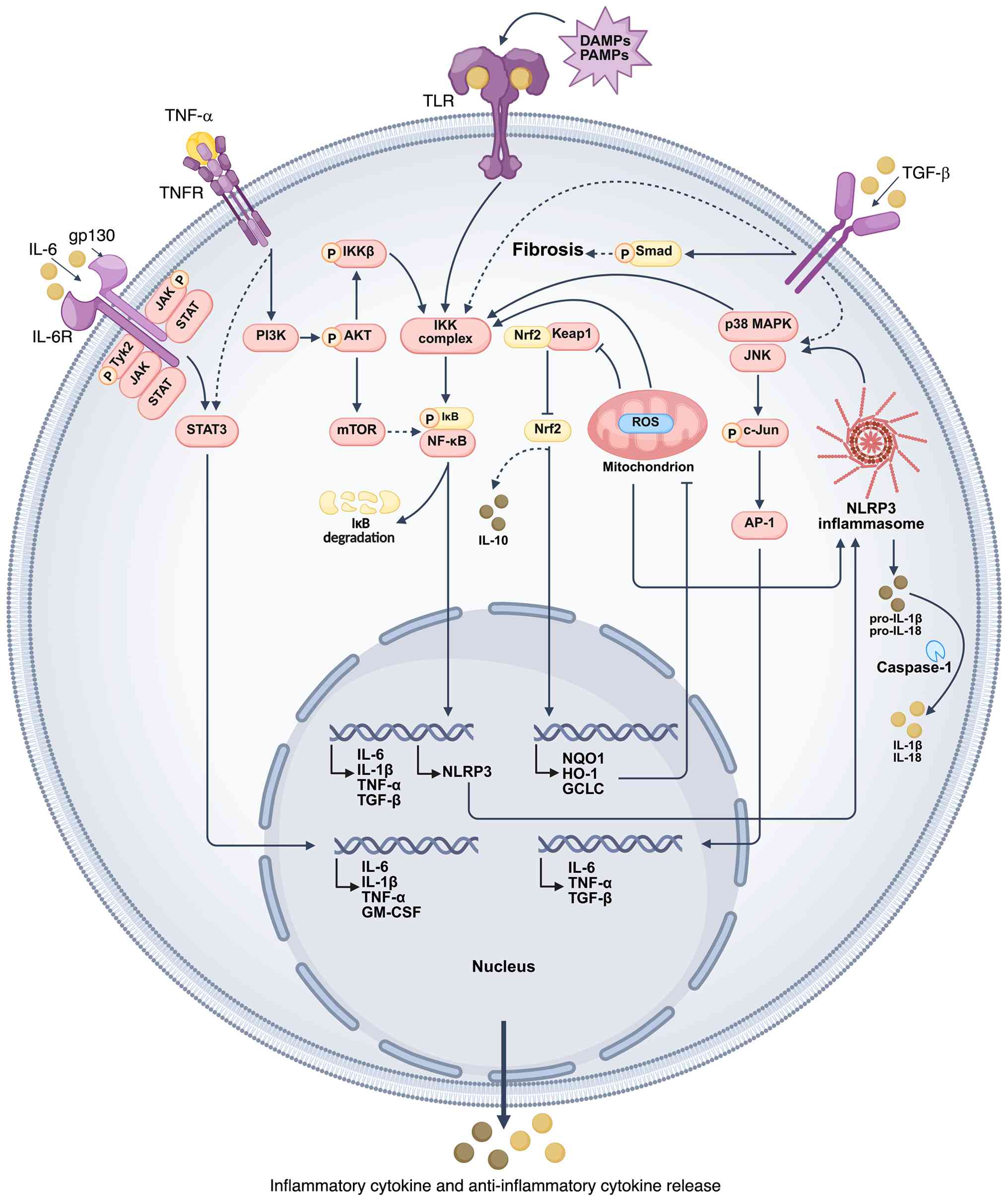
NF-κB serves as a key transcription factor that
governs physiological processes such as immune response, cell
proliferation, survival and aging (48). Inflammasomes initiate a cascade
of reactions via the activation of NLRP3, thereby facilitating the
upregulation of pro-inflammatory cytokines, including IL-1β, IL-6
and TNF-α. This process is predominantly mediated by the regulatory
function of the NF-κB signaling pathway (49,50). The activation of NLRP3 results in
degradation of IκB, an inhibitor of NF-κB, thereby alleviating the
suppression of NF-κB. This facilitates translocation of NF-κB into
the nucleus, where it initiates the transcription of genes
associated with inflammation (51). Prolonged activation of NF-κB
sustains the pro-inflammatory response in immune cells and may
contribute to the progression of aging-associated diseases.
Consistent with this, NF-κB activation has been identified as a key
mediator of synaptic repair in early-stage AD mouse models
(52,53).
MAPK signaling pathway is a key pathway in cell
response to external stimuli, involving cell proliferation,
differentiation, survival and death (54). NLRP3 inflammasome activates JNK
and p38 MAPK through stress signals such as ROS, enhances the
secretion of IL-6, TNF-α and other pro-inflammatory factors and
maintains and aggravates chronic low-grade inflammation (55). This ROS-MAPK-NLRP3 axis has been
validated in an 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
(MPTP)-induced Parkinson's disease (PD) mouse model, where its
inhibition alleviates neuroinflammation and motor deficit (56). Overactivation of MAPK signaling
increases cellular oxidative stress, promotes the aging process and
serves a key role in aging-associated diseases, such as PD
(57).
The JAK/STAT signaling pathway serves a key role in
cytokine signal transduction, primarily regulating cell
proliferation, differentiation, immune responses and senescence
(58). In aging, the JAK/STAT
pathway is activated by pro-inflammatory cytokines, such as IL-6
and TNF-α, thereby amplifying the immune cell response to
inflammation, perpetuating chronic inflammation and accelerating
the aging process (59).
Dysregulation of this pathway is associated with immune system
disorders, particularly in aging-associated diseases such as NDs,
cardiovascular disease and diabetes. Metabolic stressors can
exacerbate inflammation by augmenting JAK/STAT signaling, directly
linking this pathway to the pathophysiology of multiple age-related
conditions, such as rheumatoid arthritis, atherosclerosis, and PD
(60,61).
The PI3K/Akt signaling pathway is key to the
regulation of cell survival, proliferation and metabolism and it
plays a crucial role in cellular adaptation to stress. While the
PI3K/Akt pathway typically exerts an anti-inflammatory effect by
inhibiting pro-inflammatory pathways, its prolonged activation may
contribute to chronic low-grade inflammation during aging (62). Concurrently, the NLRP3
inflammasome can activate the PI3K/Akt pathway via pro-inflammatory
factors, such as IL-6, which are upregulated by the NF-κB signaling
pathway, thereby enhancing cytokine secretion and perpetuating
chronic low-grade inflammation (63). Excessive activation of the
PI3K/Akt pathway may result in the persistence of inflammatory
responses, contributing to age-associated metabolic disorders and
diminished immune function. In BV-2 murine microglial cells and a
transient middle cerebral artery occlusion/reperfusion (tMCAO/R)
mouse model of cerebral ischemia-reperfusion, modulation of this
pathway directly regulates microglial phenotype and autophagic
activity (64).
Nrf2 serves as a key transcription factor in cell
antioxidant defense mechanisms, primarily sustaining redox
homeostasis via the regulation of antioxidant enzyme expression,
including heme oxygenase-1 (HO-1) and NAD(P)H quinone dehydrogenase
1 (65). Throughout aging,
oxidative stress accumulation stimulates Nrf2 activation, thereby
augmenting the cell antioxidant capacity (66). In the context of chronic
low-grade inflammation, Nrf2 mitigates the accumulation of ROS and
lipid peroxidation by modulating the expression of antioxidant
genes, which in turn attenuates the activation of NLRP3
inflammasomes (67).
Concurrently, Nrf2 suppresses excessive immune responses by
upregulating anti-inflammatory factors, such as IL-10 (68). Conversely, during aging, the
regulation of Nrf2 becomes impaired. This impairment is driven by
upstream factors such as the upregulation of glycogen synthase
kinase-3β and leads to a functional decline compromising the
cellular responsiveness to oxidative stress, as demonstrated in
aged C57BL/6 mouse models of hepatic ischemia-reperfusion injury
and senescent L02 hepatocytes (69,70) (Fig. 2).
TGF-β is a pleiotropic cytokine involved in immune
regulation, cell proliferation and aging (71). During aging, TGF-β signaling
becomes dysregulated, contributing to chronic low-grade
inflammation and fibrosis (72).
Within the inflammatory network, TGF-β positively modulatse immune
responses via Smad-dependent pathways (73). Moreover, it acts with NF-κB and
MAPK signaling to promote tissue remodeling. Activation of the
NLRP3 inflammasome may exacerbate fibrosis by upregulating TGF-β
expression (74).
Neuroinflammation has been reported to result in the
activation of inflammatory cells within the brain, primarily
microglia and astrocytes (75).
Concurrently, factors such as immune senescence, mitochondrial
dysfunction, autophagy and dysfunction of the ubiquitin-proteasome
system contribute to a sustained state of chronic inflammation
(76). In this activated state,
inflammatory cells release cytokines, which are implicated in the
pathogenesis of various types of ND (77), including AD and PD, by inducing
neuronal synaptic dysfunction and excitotoxicity.
Under physiological conditions, microglia exhibit
phagocytic activity, facilitating the removal of damaged neurons
and promoting tissue repair. Concurrently, astrocytes contribute to
neuroprotection by clearing debris from the cerebrospinal fluid.
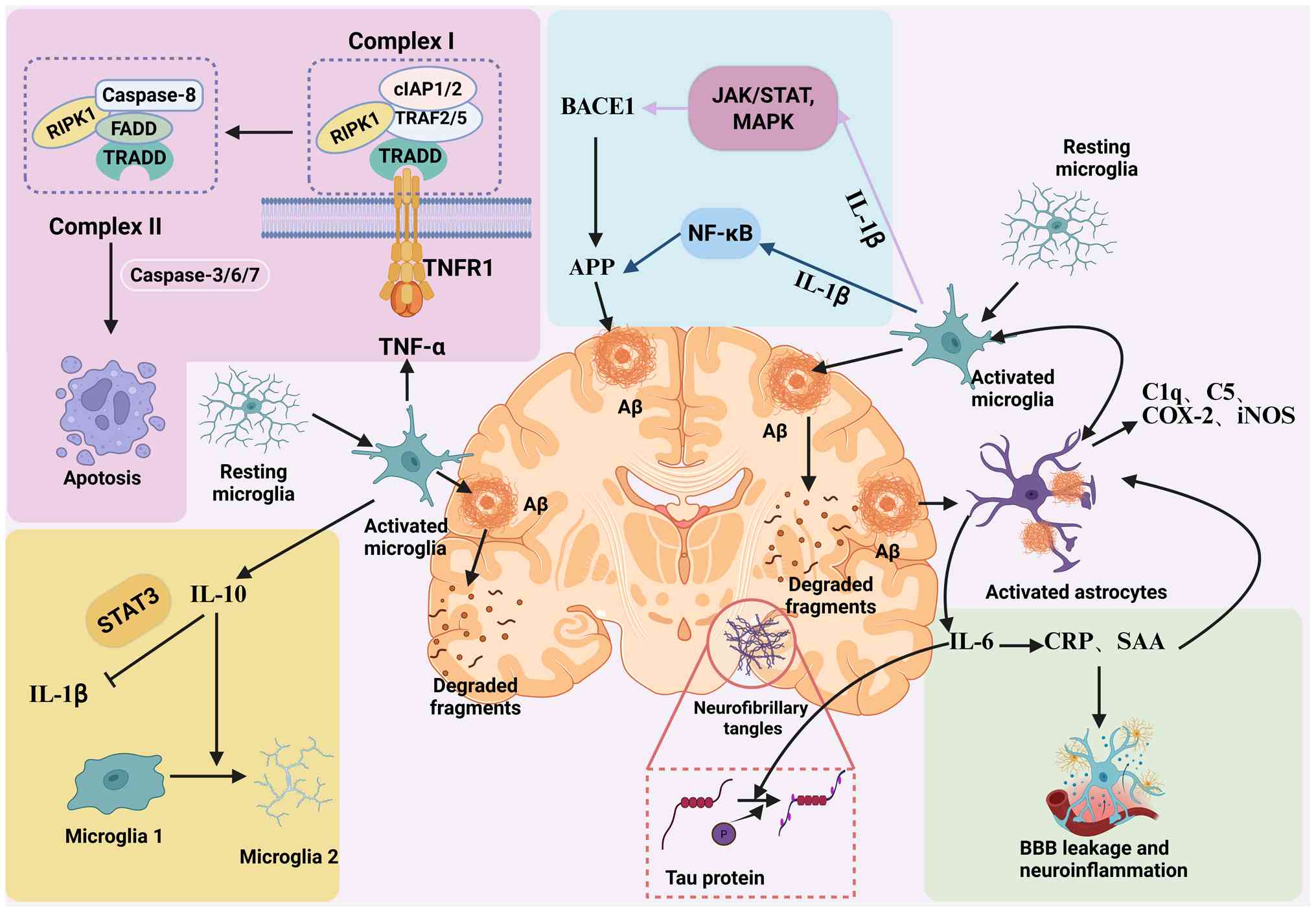
During neuroinflammation, IL-1 secreted by activated microglia and
astrocytes engages MAPK signaling, leading to upregulation of
β-site amyloid precursor protein cleaving enzyme 1 (BACE1) and
enhanced Aβ formation (78).
IL-1 also promotes τ hyperphosphorylation, contributing to
neurofibrillary tangle (NFT) pathology, as evidenced by elevated
p38 MAPK expression in IL-1β-infused rat brain (79). τ protein is key for the growth
and development of neuronal axons, serving as an essential molecule
for the assembly and stabilization of the microtubule cytoskeleton
(80). In AD,
hyperphosphorylated τ proteins are major components of paired
helical filaments, establishing a connection to NFTs (81). Furthermore, IL-4 has been shown
to impede Aβ clearance, resulting in increased Aβ deposition and
amyloid plaque formation (82-84). This is evidenced by experiments
in 4-month-old amyloid precursor protein (APP) transgenic TgCRND8
mice with pre-existing plaques (82). Overexpression of murine IL-4 in
the hippocampus via adeno-associated virus 2/1 chimeric vector
(AAV2/1) notably aggravates cerebral Aβ deposition and plaque
burden (82,85).
In addition to their pro-inflammatory role,
microglia and astrocytes produce immunosuppressive factors to limit
inflammation (86). For example,
IL-10 is upregulated in the rat cerebral cortex following LPS
injection, with increased mRNA at 8 and protein expression at 24 h
post-injection (87). IL-10 has
been demonstrated to induce the expression of anti-inflammatory
microRNAs, which negatively regulate toll-like receptor (TLR)
signaling pathways and modify the stability of inflammatory
cytokine mRNA (88).
There are two primary categories of cell death:
Accidental cell death (ACD) and programmed cell death (PCD). ACD
occurs as a reaction to unforeseen injurious stimuli, such as
necrosis (89). By contrast, PCD
is an orderly process of self-extinction initiated by gene
regulation. PCD occurs in a spatially and temporally constrained
manner during normal neuronal development, facilitating the
establishment of neural structures and shaping the central nervous
system (CNS) (90,91). In the pathogenesis of nervous
system disease, anomalies in the signaling cascades of PCD,
including apoptosis, ferroptosis, autophagy, pyroptosis and
necroptosis, are evident (92,93).
Pyroptosis is characterized by cell rupture and the
release of cell contents, which leads to abnormal microglial
activation, intense inflammation and the promotion of ND (94). Pyroptosis is primarily induced by
inflammasome activation (NLRP3) triggered by pathological factors
such as mitochondrial dysfunction, ROS accumulation or Aβ during
aging. Upon activation, Caspase-1 cleaves gasdermin D, whose
N-terminal fragment forms membrane pores, leading to cell swelling,
rupture and release of pro-inflammatory cytokines (95,96). Caspase-1 cleaves pro-IL-1β and
pro-IL-18 into their mature forms, amplifying inflammation
(96). Damage-associated
molecular patterns (DAMPs) released by pyrocytes initiate
inflammatory responses in adjacent glial cells via interaction with
TLR4 or purinergic ligand-gated ion channel 7 receptors,
potentially leading to further neuronal damage (97).
Iron-dependent cell death, also known as
ferroptosis, represents a distinct form of PCD characterized by
iron accumulation in cells (98). This process leads to neuronal
damage and cell death through iron-mediated lipid peroxidation.
Aging is associated with disruptions in cell iron homeostasis,
resulting in iron overload and the generation of excessive ROS via
the Fenton reaction (98,99).
These ROS oxidize lipids, compromising the integrity of the cell
membrane (100). Excessive ROS
activate NF-κB, leading to the formation of inflammasomes and the
release of pro-inflammatory cytokines such as IL-6, TNF-α and
IL-1β, which contribute to neuroinflammation (101). Hepcidin, a peptide hormone
involved in iron homeostasis, inhibits cell iron efflux by
interacting with ferroportin 1 (102). Hepcidin levels are associated
with IL-6, which promotes hepcidin expression, thereby increasing
intracellular iron levels. Ferritin, an iron storage protein, is
upregulated during inflammation via the IL-6/STAT3 pathway
(103). Additionally, IL-1β,
IL-6 and TNF-α can indirectly induce ferritin synthesis by
enhancing hepcidin transcription.
Necrosis was initially identified as an alternative
pathway to the death receptor pathway (104). In cell death due to ischemia,
physical injury, oxidative stress or pathogen infection, the
integrity of the cell membrane is compromised, leading to the
release of DAMPs and the activation of immune cells, such as
microglia and astrocytes, via pattern recognition receptors. This
triggers downstream inflammatory signaling pathways (105). DAMPs trigger the formation of
the NLRP3 inflammasome complex through ROS-dependent pathways and
promote the maturation and secretion of pro-inflammatory cytokines
such as IL-1β and IL-18 via caspase-1-dependent mechanisms
(106). High mobility group box
1 (HMGB1), a nuclear protein, is released into the extracellular
environment during ACD (107).
While HMGB1 has limited direct pro-inflammatory effects, it
indirectly enhances the release of pro-inflammatory cytokines by
recruiting inflammatory cells such as microglia (108). Under the influence of
microglia-derived IL-1α, TNF-α and complement component 1, q
subcomponent, astrocytes transform into A1-reactive astrocytes
(109). These A1-reactive
astrocytes produce CXCL10, which facilitates immune cell
infiltration and enables immune cells to cross the blood-brain
barrier (BBB) into the CNS, exacerbating neuroinflammation
(110).
The BBB serves as a key protective mechanism within
the CNS, comprising endothelial cells, pericytes, astrocytic
end-feet and a basement membrane. Under physiological conditions,
the BBB is notably impermeable; however, in pathological states,
the release of vasoactive substances, cytokines and chemical
mediators enhances its permeability, thereby compromising its
barrier function (111). Damage
to the BBB may be associated with the onset of age-associated ND
and the deterioration of cognitive function (111-113).
The activation, migration and cytokine release by
some immune cells can compromise the integrity of the BBB.
Compromised BBB permits the entry of peripheral fibrinogen into the
brain, which activates microglia and promotes neuroinflammation
(114). During aging, the
activation of microglia and astrocytes upregulates MMP activity,
resulting in the degradation of tight junction proteins and
increased BBB permeability (115). Furthermore, BBB damage
facilitates the entry of neurotoxic substances, including plasma
protein, inflammatory cells and toxins, into the brain parenchyma,
thereby inducing neuroinflammation and neuronal damage (116). In conclusion, aging compromises
the integrity of the BBB via endothelial damage and
neuroinflammation. This leads to BBB leakage, which exacerbates
inflammation and neurodegeneration in the brain, thereby
establishing a deleterious feedback loop.
AD is a prevalent neurodegenerative disorder among
the elderly, current estimates suggest that 44 million people live
with dementia worldwide at present (117). This is predicted to more than
triple by 2050 as the population ages. Primarily characterized by
cognitive decline, memory impairment, language disturbance and
behavioral alteration. The hallmark pathological features of AD
include the deposition of Aβ, primarily in the cerebral cortex and
hippocampal regions, leading to the formation of amyloid plaques,
abnormal phosphorylation of τ protein, resulting in NFTs that
compromise neuronal structure and function (118), and neuroinflammation, with
extensive research indicating that the inflammatory response is a
predominant mechanism in the pathogenesis of AD (119,120).
Microglia and astrocytes are capable of initiating
an inflammatory response following an injury to the CNS. Aβ rapidly
activates microglia, resulting in alteration to their morphological
and phenotypical characteristics, which promote phagocytosis and
induce localized immune cells (121). However, sustained microglial
activation and unresolved inflammation within the brain are
detrimental to neurons and synapses, promoting chronic
dysregulation of glial cells and contributing to the deterioration
of brain structure and function (122). Aβ42 has been shown to induce
microglial phagocytosis of viable neurons, resulting in early
synaptic loss in AD (123).
Following exposure to Aβ, microglia secrete a range of
pro-inflammatory cytokines and chemokines, including IL-6, IL-1β,
TNF-α, macrophage inflammatory protein-1α and MCP-1 (124). This secretion leads to the
recruitment and activation of astrocytes and peripheral immune
cells.
Pro-inflammatory cytokines such as IL-1β, IL-6, and
TNF-α play a predominant role in exacerbating AD pathology. They
not only maintain the inflammatory environment but also directly
interact with and aggravate Aβ plaques and NFTs (84). IL-1β is elevated in the brain of
patients with AD, particularly in association with diffuse plaques,
and is predominantly produced by activated microglia during early
disease stages (125). In
vitro, IL-1β treatment of human neuroblastoma SH-SY5Y cells
enhances APP transcription via NF-κB activation, leading to
increased APP synthesis (125).
These findings are supported by APP/presenilin 1 (PS1) transgenic
mouse models, where IL-1β overexpression is associated with
elevated cortical APP levels and accelerated amyloid deposition
(125,126). IL-1β also enhances BACE1 and
γ-secretase activity via MAPK and JAK/STAT signaling, contributing
to Aβ overproduction (127).
Neuroinflammation upregulates BACE1 activity while
downregulating the expression of Aβ-degrading enzymes. Inflammatory
processes lead to the downregulation of Aβ-degrading enzymes,
including insulin-degrading enzyme and neutral endopeptidase,
contributing to the accumulation of Aβ in the brain. These effects
create a self-reinforcing cycle that accelerates plaque formation
(128,129). Moreover, the intermediate form
of APP activates microglia, resulting in the excessive secretion of
IL-1β, which stimulates astrocytes and promotes the release of
pro-inflammatory substances (130).
TNF-α binding to TNFR1 recruits TNFR1-associated
death domain protein, TNFR-associated factor 2/5,
receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and
cellular inhibitor of apoptosis protein 1/2 (cIAP1/2) to form
complex I, leading to canonical NF-κB activation andinflammation
(134). When NF-κB activation
is blocked (such as by cIAP1/2 inhibition), Caspase-8 is activated,
initiating apoptosis (135). If
Caspase-8 is suppressed, RIPK1 deubiquitinates and associates with
Fas-associated death domain protein (FADD) and receptor-interacting
serine/threonine-protein kinase 3 to form complex II, which
phosphorylates mixed lineage kinase domain-like pseudokinase and
triggers necroptosis, a pro-inflammatory cell death pathway
(136,137).
IL-10 suppresses NLRP3 inflammasome activity via
STAT3 signaling, decreasing IL-1β release and promoting microglial
M2 polarization (138).
However, in AD, IL-10 expression is typically decreased, which
diminishes its inhibitory effect on excessive inflammatory
responses. The impaired anti-inflammatory response fuels chronic
neuroinflammation and pathological progression.
As the intercellular signaling proteins that serve
as ligands for receptor tyrosine kinases within the ErbB receptor
family, NRGs and their corresponding receptors are key to organ
development and maintenance, as well as the pathogenesis of NDs
(139). Notably, NRG1 may exert
a protective effect by modulating the pathological progression of
AD. Compared with age-matched normal controls, the immunoreactivity
intensities of ErbB4 and phosphorylated ErbB4 are elevated in the
neurons of the CA1-2 transition zone in AD brains (36). Furthermore, ErbB4 expression is
increased in the medial neurons of the basolateral amygdala cortex
and the superior frontal gyrus neurons of patients with AD
(36). In the cerebral cortex
and hippocampus of APP/PS1 double transgenic mice, ErbB4
immunoreactivity is significantly higher than that in age-matched
wild-type controls (36). In
APP/PS1 mice, ErbB4 immunoreactivity is elevated in the cerebral
cortex and hippocampus compared with wild-type controls (36). These reports suggest that
aberrant alterations in ErbB4 may contribute to the pathological
progression of AD, while NRG1 exhibits neuroprotective effects
against neurotoxicity induced by the Swedish amyloid precursor
(36).
NRG1 mitigates the neurotoxic effects associated
with the expression of APP C-terminal fragment in SH-SY5Y human
neuroblastoma cells, reducing ROS accumulation and mitochondrial
membrane potential loss via ErbB4 signaling (140). Another study examined the
downstream signaling pathway of NRG1, highlighting its role in
counteracting Aβ42-induced neurotoxicity (141). The findings indicate that
inhibiting the activation of the PI3K/Akt pathway negates NRG1
ability to prevent Aβ42-induced lactate dehydrogenase release,
increases the number of TUNEL-positive cells and elevates ROS
accumulation in primary cortical neurons (142). These findings confirm
NRG1/PI3K/Akt signaling as a viable therapeutic target for
Aβ-induced neurotoxicity in AD (Fig.
3).
Our previous study demonstrated that recombinant
Neuregulin-1β treatment alleviates LPS-induced neuroinflammation by
reducing the number of microglial cells and astrocytes as well as
the expression of IL-1β (143).
Moreover, our group recently reported that targeted activation of
ErbB4 using the small-molecule agonist
4-bromo-1-hydroxy-2-naphthoic acid (E4A), an NRG1 mimic, exerts
neuroprotective effects in multiple disease-related models
(144-146). Specifically, E4A alleviates
neuronal damage in D-galactose-induced senescence (144), ameliorates cognitive deficit in
APP/PS1 mice via dedicator of cytokinesis 3 (DOCK3) signaling
(145) and mitigates
neuroinflammation in a polystyrene microplastic exposure model
(146). These findings not only
corroborate the key role of the NRG1-ErbB4 axis in neuronal
protection and the regulation of neuroinflammation but also offer
direct experimental evidence in support of the cytokine-centered
approach for treating AD and other ND.
PD is a neurodegenerative disorder marked by
progressive extrapyramidal dysfunction. The primary pathological
characteristics of PD include the degeneration of dopaminergic
(DAergic) neurons in the substantia nigra (SN), leading to
decreased levels of dopamine and the accumulation of α-syn within
the cytoplasm and axons of DAergic neurons, forming Lewy bodies
(147). Imamura et al
(148) identified the
infiltration of activated microglial cells in the SN of brain of
patients with PD postmortem. These activated microglial cells
secrete pro-inflammatory cytokines, such as TNF-α, IL-1β and IL-6,
and express class II major histocompatibility complex (MHC)
molecules. These activated microglial cells contribute to neuronal
damage in patients with PD (148). Furthermore, neuroimaging
studies employing radioactive tracers specific to microglial cell
activation have revealed the presence of persistent
neuroinflammation in PD (149,150).
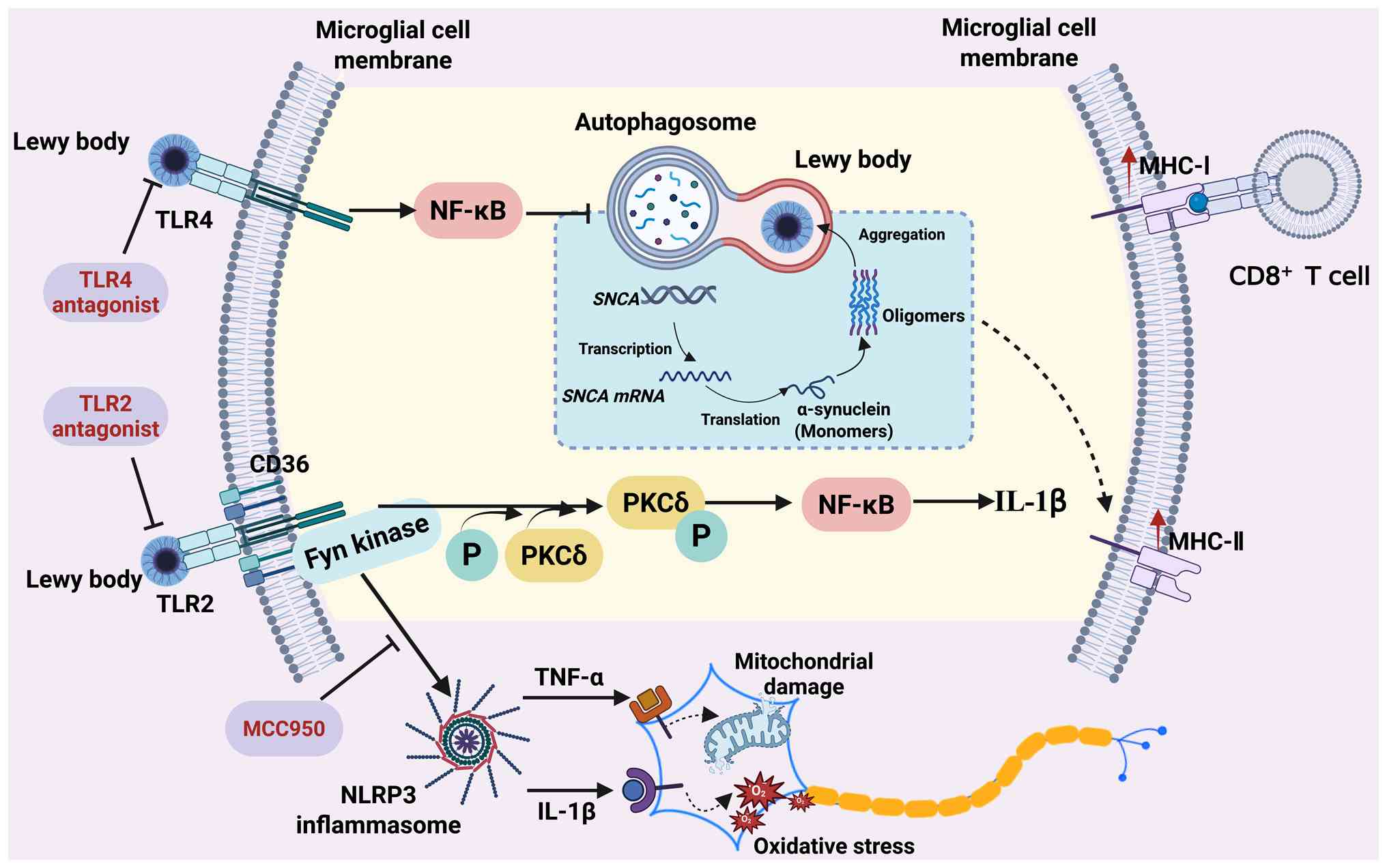
The aggregation of abnormal and insoluble α-syn is
key in the pathogenesis of PD (151). Misfolded α-syn serves as a DAMP
to dysregulate TLR2/TLR4-myeloid differentiation primary response
protein 88-NF-κB signaling in microglia, inducing TNF-α and IL-1β
production. Treatment of BV2 mouse or primary microglia with
aggregated α-syn upregulates the production of TNF-α, IL-1β, MCP-1
and IFN-γ (152). Additionally,
α-syn binding to TLR2 triggers NLRP3 inflammasome activation,
promoting IL-1β maturation and release (153). These IL-1β-driven
neuroinflammatory responses contribute to DAergic neuron
degeneration in PD. Furthermore, the knockout of TLR2 decreases the
uptake of α-syn by mouse microglia (154) (Fig. 4).
TNF-α is upregulated in the mouse striatum prior to
DAergic neuron degeneration, implicating it in early PD
pathogenesis. Genetic ablation of TNF receptors or pharmacological
inhibition of TNF-α (using thalidomide) attenuates MPTP-induced
neuronal loss (162,163). A cohort study demonstrated an
association between early anti-TNF therapy and decreased PD
incidence (164). α-syn binding
to TLR2 activates the NLRP3 inflammasome. In patients with PD,
NLRP3 colocalizes with microglia in the SN (165). The NLRP3 inhibitor MCC950
attenuates inflammasome activation and mitigates motor deficit,
nigrostriatal degeneration and α-syn aggregation in mouse models
(165,166). Nrf2 activation by dimethyl
fumarate decreases ROS production in neurons of SNCA (p.A53T)
transgenic mice and protects against MPTP- and α-syn-induced
DAergic neuron damage (167,168). Peroxisome
proliferator-activated receptor (PPAR)-γ agonists, such as
pioglitazone and rosiglitazone, alleviate MPTP-induced inflammation
and protect nigrostriatal function in mice and monkeys;
pioglitazone also decreases glial cell activation and prevents
DAergic neuron loss in MPTP-treated mice (162,169). Collectively, these findings
support targeting TNF-α, NLRP3, Nrf2 and PPAR-γ as therapeutic
strategies to impede PD progression.
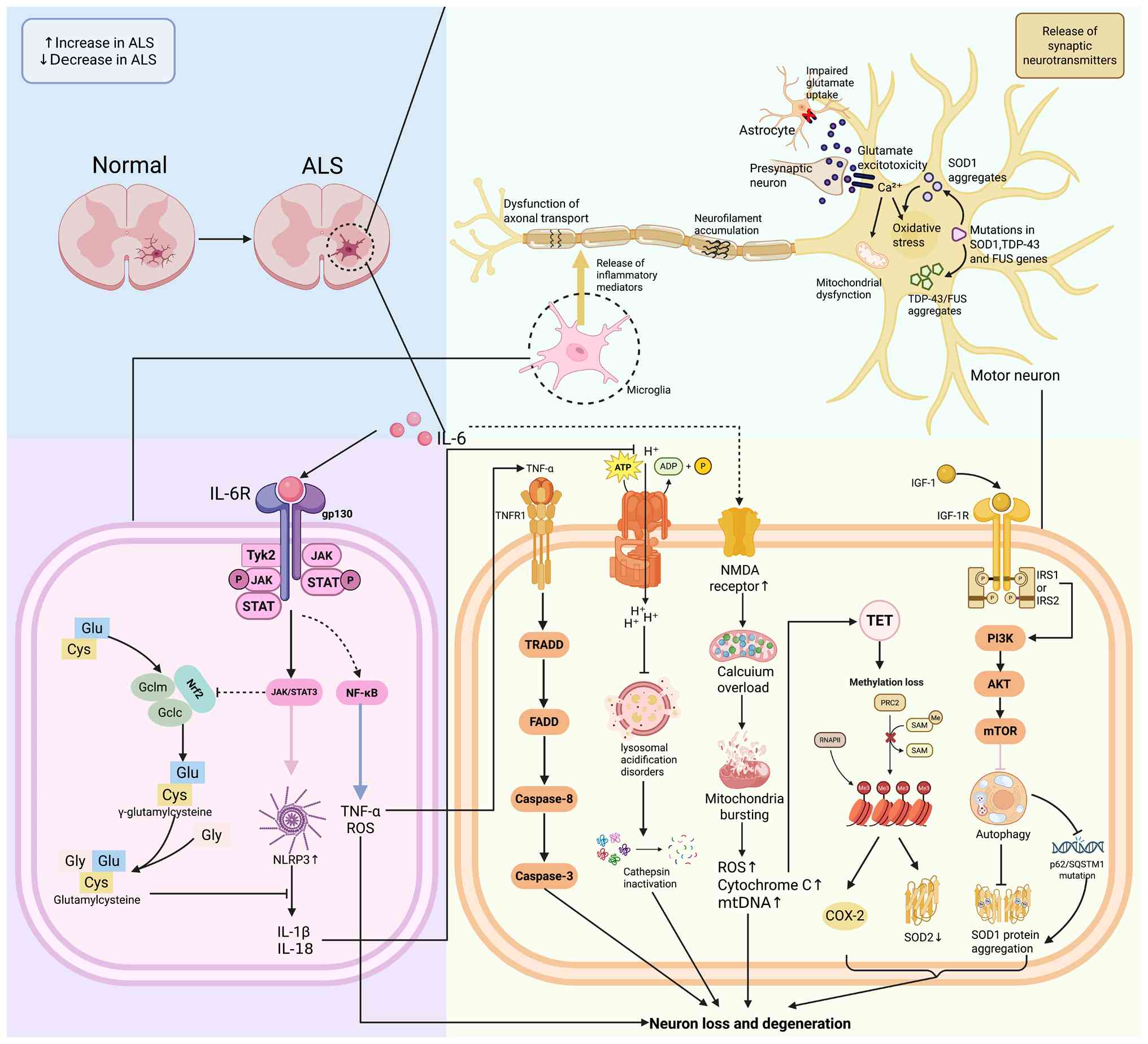
ALS is a neurodegenerative disorder marked by the
progressive degeneration of motor neurons, leading to symptoms such
as muscle weakness, atrophy and impaired motor function (163). The pathology of ALS involves
the degeneration of both upper and lower motor neurons within the
spinal cord and cerebral cortex, culminating in the inability to
control muscle movements (164). The resultant motor neuron death
precipitates muscle atrophy and weakness, leading to mortality,
often due to respiratory failure caused by paralysis of the
respiratory muscles (170).
Neuroinflammation is a prevalent pathological feature in ALS,
regardless of the presence of genetic mutations, and is
characterized by the infiltration of activated microglia and
astrocytes. These activated glial cells produce pro-inflammatory
cytokines, which are upregulated in postmortem tissue of patients
with ALS (171).
Aging cells release pro-inflammatory factors such as
IL-6, IL-1β and TNF-α through the SASP, which activate glial cells
and initiate chronic CNS inflammation (172). In vivo studies
demonstrate that systemic administration of recombinant murine IL-6
in C57BL/6 mice promotes microglial polarization toward the M1
phenotype, characterized by elevated CD16/32 expression, and
exacerbates neuroinflammation (173-175). IL-6 is a potential therapeutic
target for ALS. In superoxide dismutase 1 (SOD1)G93A
transgenic mice, IL-6 promotes microglial M1 polarization via
JAK2/STAT3 signaling, resulting in TNF-α and ROS release that
directly damages spinal motor neurons (176,177). IL-6 also downregulates
glutamate transporter 1, causing excitotoxicity, and impairs the
Nrf2 antioxidant pathway, resulting in mitochondrial ROS
accumulation and NLRP3 inflammasome activation (178,179). These IL-6-driven processes
collectively establish an oxidative-inflammatory cycle that
accelerates motor neuron death.
Paralleling the persistent elevation of
inflammatory cytokines, a concurrent failure in cell waste disposal
mechanisms contributes to proteotoxicity and disease progression in
ALS. With advancing age, autophagic function declines, resulting in
the inefficient degradation of aberrant proteins, including
transactive response DNA-binding protein 43 (TDP-43) and SOD1
aggregates (180). In the
SOD1G93A transgenic mouse model of ALS, these autophagic
deficits contribute to motor neuron degeneration. In individuals
with ALS, persistent activation of the mechanistic target of
rapamycin pathway inhibits autophagy initiation, while mutations in
p62/sequestosome-1 compromise substrate recognition (181,182). Additionally, lysosomal
acidification disorder, characterized by increased pH, leads to
cathepsin inactivation, thereby exacerbating protein toxicity
(183).
The inflammatory state in ALS is strengthened and
regulated by persistent changes at the epigenetic level.
Age-associated DNA methylation loss and abnormal histone
modifications upregulate pro-inflammatory genes, while suppressing
neuroprotective genes (184).
For example, microRNA-155 enhances microglial JAK/STAT signaling by
inhibiting suppressor of cytokine signaling 1, whereas the
downregulation of microRNA-146a disrupts NF-κB pathway regulation,
creating a positive feedback loop that drives ALS progression
(185). Pro-inflammatory
factors, including IL-1α, TNF-α and complement component 1, q
subcomponent, A chain, are released by microglia activated by
neuroinflammation, inducing neurotoxicity via alterations in
astrocyte activity (109).
TNF-α activates the Caspase-8/FADD signaling pathway via TNFR1,
while IL-6 upregulates the pro-apoptotic protein Bax via the
JAK/STAT3 pathway, both of which are implicated in the pathogenesis
of ALS (186) (Fig. 5).
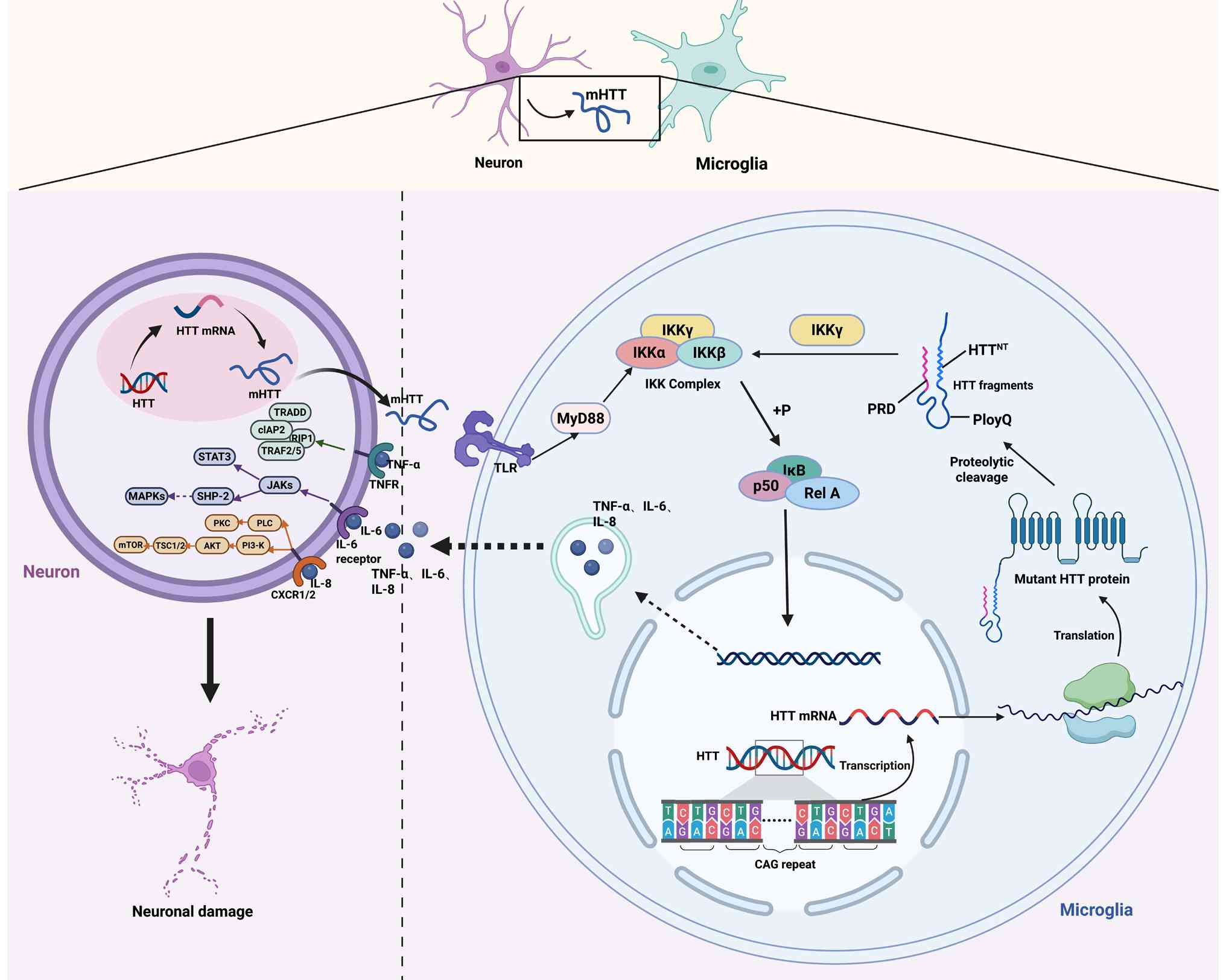
HD is a deleterious autosomal dominant hereditary
neurological disorder, characterized by mood disturbance, weight
loss, movement abnormality and dementia (187). The gene responsible for HD was
first cloned in 1993, and the highly conserved protein it encodes,
whose function remains unclear, is huntingtin (HTT) (187,188). In individuals with HD, the
polymorphic trinucleotide repeat sequence CAGn at the 5'end of the
gene undergoes expansion beyond the normal repeat threshold,
leading to the translation of an extended polyglutamine tract
within the protein (187). The
proteolytic cleavage of the mutant HTT protein plays a critical
role in the pathogenesis of HD (189). Studies utilizing in vivo
models, such as R6/2 HD mice, demonstrate that these aberrant HTT
fragments initiate a complex cascade of compensatory and
deleterious molecular processes, including neuroinflammation
(190,191). Such processes result in
atrophy, fragility and damage to nerve cells, rendering them
susceptible to stressors, such as excitotoxic stress, oxidative
damage, pro-apoptotic signals, energy depletion, impaired
proteolysis and neurophysiological defects. Collectively, these
factors may contribute to neuronal death (189).
Compared with other CNS disorders, the involvement
of microglia in HD remains insufficiently investigated (192). Singhrao et al (193) demonstrated microglial
impairment in patients with HD (193). The aforementioned study
observed an increased number of microglial cells in the caudate
nucleus and putamen, associated with elevated expression levels of
complement factors. Sapp et al (194) investigated microglial
morphological alterations associated with HD and identified
structurally activated microglia in the cortex, globus pallidus and
neostriatum. Notably, in the cortex and striatum, the aggregation
of thymosin β-4-reactive microglia intensifies in parallel with the
progression of neuropathological grade (194). Another study reported
microglial accumulation in HD tissue and the R6/2 mouse model of
the striatum (192).
Accumulated HTT expression induces transcriptional
alterations in neuronal cells, and it is possible that microglial
transcription is similarly impacted (192). Björkqvist et al
(195) suggested that
postmortem tissue from patients with HD exhibit distinct
inflammatory characteristics. Specifically, inflammatory molecules
such as TNF-α and IL-1β are markedly elevated in the striatum,
whereas MMP-9, IL-6 and IL-8 show increased expression in the
cortex and cerebellar regions (192). This contrasts with the
neuroinflammatory profiles observed in other types of ND, such as
PD or AD, which typically involve the upregulation of a broader
range of inflammatory molecules (196). The presence of inflammatory
regulators in the striatum may signify ongoing pathological
processes, while the dysregulation of molecules such as MMP-9, IL-6
and IL-8 suggests a more generalized role of mutant HTT protein
expression (197). Furthermore,
studies have demonstrated that monocytes expressing mutant HTT
protein in patients with HD exhibit increased secretion of IL-6
(198-200). By contrast with other
neurological disorders, including AD and multiple sclerosis, the
involvement of peripheral immune cells, such as neutrophils and
lymphocytes, in HD has not been thoroughly explored (201). Furthermore, studies have
indicated that significant infiltration of T cells is not observed
in postmortem tissue of individuals with HD (202,203). Consequently, neuroinflammation
in HD is predominantly sustained by the interactions among neurons,
microglia and astrocytes (192)
(Fig. 6).
Cytokines associated with aging are key to the
pathogenesis of ND. Throughout the aging process, there is a
persistent elevation of pro-inflammatory factors, including IL-6,
TNF-α and IL-1β, coupled with a decline in anti-inflammatory
factors such as IL-10 and TGF-β. This shift results in
inflammaging, which exacerbates chronic neuroinflammation (204,205). This imbalance facilitates the
deposition of Aβ, abnormal phosphorylation of τ protein and
aggregation of α-syn through the activation of microglia and
astrocytes, leading to neuronal apoptosis, synaptic dysfunction and
impairment of the BBB (206).
Excessive activation of the NLRP3 inflammasome
intensifies the inflammatory cascade, promoting oxidative stress
and mitochondrial dysfunction. This creates an
inflammation-oxidation feedback loop that accelerates
neurodegenerative damage (207). Additionally, cytokines released
by the SASP, through the recruitment of immune cells and the
dysregulation of signaling pathways such as NF-κB and JAK/STAT,
represent a common mechanism underlying diseases such as AD and PD
(208,209) (Table I). While interventions targeting
cytokines, such as IL-1β neutralizing antibodies and NLRP3
inhibitors in mouse autoinflammatory disease models and
inflammatory arthritis models have demonstrated potential in
delaying disease progression, their safety and efficacy in humans
require further validation (210,211). Future research should
investigate the precise regulation of the cytokine network to
achieve a balance between neuroinflammation and neuroprotection.
Targeting senescence-associated cytokines offers a novel
therapeutic avenue for ND.
Not applicable.
XD, XR, YW and WZ conceived the study. XD, XR and
YW wrote the manuscript. WZ edited the manuscript. Data
authentication is not applicable. All authors have read and
approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by Jiangsu Province Shuangchuang
Talent Plan (grant no. JSSCRC 2021533).
|
1
|
Banerjee P, Kotla S, Reddy Velatooru L,
Abe RJ, Davis EA, Cooke JP, Schadler K, Deswal A, Herrmann J, Lin
SH, et al: Senescence-associated secretory phenotype as a hinge
between cardiovascular diseases and cancer. Front Cardiovasc Med.
8:7639302021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Calsolaro V and Edison P:
Neuroinflammation in Alzheimer's disease: Current evidence and
future directions. Alzheimers Dement. 12:719–732. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang Q, Lenardo MJ and Baltimore D: 30
years of NF-κB: A blossoming of relevance to human pathobiology.
Cell. 168:37–57. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vande Walle L, Van Opdenbosch N, Jacques
P, Fossoul A, Verheugen E, Vogel P, Beyaert R, Elewaut D,
Kanneganti TD, van Loo G and Lamkanfi M: Negative regulation of the
NLRP3 inflammasome by A20 protects against arthritis. Nature.
512:69–73. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Derynck R, Turley SJ and Akhurst RJ: TGFβ
biology in cancer progression and immunotherapy. Nat Rev Clin
Oncol. 18:9–34. 2021. View Article : Google Scholar
|
|
6
|
Franceschi C, Garagnani P, Parini P,
Giuliani C and Santoro A: Inflammaging: A new immune-metabolic
viewpoint for age-related diseases. Nat Rev Endocrinol. 14:576–590.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
López-Otín C, Blasco MA, Partridge L,
Serrano M and Kroemer G: The hallmarks of aging. Cell.
153:1194–1217. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu C, Chu D, Kalantar-Zadeh K, George J,
Young HA and Liu G: Cytokines: From clinical significance to
quantification. Adv Sci (Weinh). 8:e20044332021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kaminska P, Tempes A, Scholz E and Malik
AR: Cytokines on the way to secretion. Cytokine Growth Factor Rev.
79:52–65. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Morrisette-Thomas V, Cohen AA, Fülöp T,
Riesco É, Legault V, Li Q, Milot E, Dusseault-Bélanger F and
Ferrucci L: Inflamm-aging does not simply reflect increases in
pro-inflammatory markers. Mech Ageing Dev. 139:49–57. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cotman CW, Berchtold NC and Christie LA:
Exercise builds brain health: Key roles of growth factor cascades
and inflammation. Trends Neurosci. 30:464–472. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wakefield PE, James WD, Samlaska CP and
Meltzer MS: Colony-stimulating factors. J Am Acad Dermatol.
23:903–912. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lei W, Jia L, Wang Z, Liang Z, Zhao A, Liu
Y, Tian Y, Zhao L, Chen Y, Shi G, et al: CC chemokines family in
fibrosis and aging: From mechanisms to therapy. Ageing Res Rev.
87:1019002023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rot A and von Andrian UH: Chemokines in
innate and adaptive host defense: Basic chemokinese grammar for
immune cells. Annu Rev Immunol. 22:891–928. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ajoolabady A, Pratico D, Tang D, Zhou S,
Franceschi C and Ren J: Immunosenescence and inflammaging:
Mechanisms and role in diseases. Ageing Res Rev. 101:1025402024.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hirano T, Akira S, Taga T and Kishimoto T:
Biological and clinical aspects of interleukin 6. Immunol Today.
11:443–449. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jones SA and Jenkins BJ: Recent insights
into targeting the IL-6 cytokine family in inflammatory diseases
and cancer. Nat Rev Immunol. 18:773–789. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Frankola KA, Greig NH, Luo W and Tweedie
D: Targeting TNF-α to elucidate and ameliorate neuroinflammation in
neurodegenerative diseases. CNS Neurol Disord Drug Targets.
10:391–403. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hayden MS and Ghosh S: Regulation of NF-κB
by TNF family cytokines. Semin Immunol. 26:253–266. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mukaida N, Harada A and Matsushima K:
Interleukin-8 (IL-8) and monocyte chemotactic and activating factor
(MCAF/MCP-1), chemokines essentially involved in inflammatory and
immune reactions. Cytokine Growth Factor Rev. 9:9–23. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Opal SM and DePalo VA: Anti-inflammatory
cytokines. Chest. 117:1162–1172. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Freitas RS, de Souza Silva CM, Ferreira
Fratelli C, Ramos de Lima L, Morato Stival M, Schwerz Funghetto S,
Rodrigues da Silva IC and Vieira de Andrade R: IL-10 and IL-1β
serum levels, genetic variants, and metabolic syndrome: Insights
into older Adults' clinical characteristics. Nutrients.
16:12412024. View Article : Google Scholar
|
|
23
|
Tahtinen S, Tong AJ, Himmels P, Oh J,
Paler-Martinez A, Kim L, Wichner S, Oei Y, McCarron MJ, Freund EC,
et al: IL-1 and IL-1ra are key regulators of the inflammatory
response to RNA vaccines. Nat Immunol. 23:532–542. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dinarello CA: Immunological and
inflammatory functions of the interleukin-1 family. Annu Rev
Immunol. 27:519–550. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tominaga K and Suzuki HI: TGF-β signaling
in cellular senescence and Aging-related pathology. Int J Mol Sci.
20:50022019. View Article : Google Scholar
|
|
26
|
Peng D, Fu M, Wang M, Wei Y and Wei X:
Targeting TGF-β signal transduction for fibrosis and cancer
therapy. Mol Cancer. 21:1042022. View Article : Google Scholar
|
|
27
|
Hage C and Salvatori R: Growth hormone and
aging. Endocrinol Metab Clin North Am. 52:245–257. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Perovic M, Tesic V, Mladenovic Djordjevic
A, Smiljanic K, Loncarevic-Vasiljkovic N, Ruzdijic S and Kanazir S:
BDNF transcripts, proBDNF and proNGF, in the cortex and hippocampus
throughout the life span of the rat. Age (Dordr). 35:2057–2070.
2013. View Article : Google Scholar
|
|
29
|
Covaceuszach S, Capsoni S, Ugolini G,
Spirito F, Vignone D and Cattaneo A: Development of a non invasive
NGF-based therapy for Alzheimer's disease. Curr Alzheimer Res.
6:158–170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Grunewald M, Kumar S, Sharife H, Volinsky
E, Gileles-Hillel A, Licht T, Permyakova A, Hinden L, Azar S,
Friedmann Y, et al: Counteracting age-related VEGF signaling
insufficiency promotes healthy aging and extends life span.
Science. 373:eabc84792021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ferrara N, Gerber HP and LeCouter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Maggio M, Ble A, Ceda GP and Metter EJ:
Decline in insulin-like growth factor-I levels across adult life
span in two large population studies. J Gerontol A Biol Sci Med
Sci. 61:182–183. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Junnila RK, List EO, Berryman DE, Murrey
JW and Kopchick JJ: The GH/IGF-1 axis in ageing and longevity. Nat
Rev Endocrinol. 9:366–376. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Holbro T and Hynes NE: ErbB receptors:
Directing key signaling networks throughout life. Annu Rev
Pharmacol Toxicol. 44:195–217. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ou GY, Lin WW and Zhao WJ: Neuregulins in
neurodegenerative diseases. Front Aging Neurosci. 13:6624742021.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Woo RS, Lee JH, Yu HN, Song DY and Baik
TK: Expression of ErbB4 in the neurons of Alzheimer's disease brain
and APP/PS1 mice, a model of Alzheimer's disease. Anat Cell Biol.
44:116–127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lira-Junior R, Åkerman S, Gustafsson A,
Klinge B and Boström EA: Colony stimulating factor-1 in saliva in
relation to age, smoking, and oral and systemic diseases. Sci Rep.
7:72802017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pixley FJ and Stanley ER: CSF-1 regulation
of the wandering macrophage: Complexity in action. Trends Cell
Biol. 14:628–638. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin EY, Nguyen AV, Russell RG and Pollard
JW: Colony-stimulating factor 1 promotes progression of mammary
tumors to malignancy. J Exp Med. 193:727–740. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang N, Fang Y, Hou Y, Cheng D, Dressler
EV, Wang H, Wang J, Wang G, Li Y, Liu H, et al: Senescent cells
promote breast cancer cells motility by secreting GM-CSF and bFGF
that activate the JNK signaling pathway. Cell Commun Signal.
22:4782024. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Franceschi C, Bonafè M, Valensin S,
Olivieri F, De Luca M, Ottaviani E and De Benedictis G:
Inflamm-aging. An evolutionary perspective on immunosenescence. Ann
N Y Acad Sci. 908:244–254. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xu J and Núñez G: The NLRP3 inflammasome:
Activation and regulation. Trends Biochem Sci. 48:331–344. 2023.
View Article : Google Scholar
|
|
43
|
Latz E, Xiao TS and Stutz A: Activation
and regulation of the inflammasomes. Nat Rev Immunol. 13:397–411.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tschopp J and Schroder K: NLRP3
inflammasome activation: The convergence of multiple signalling
pathways on ROS production? Nat Rev Immunol. 10:210–215. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang C and Youle RJ: The role of
mitochondria in apoptosis*. Annu Rev Genet. 43:95–118. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Groß CJ, Mishra R, Schneider KS, Médard G,
Wettmarshausen J, Dittlein DC, Shi H, Gorka O, Koenig PA, Fromm S,
et al: K+ Efflux-independent NLRP3 inflammasome activation by small
molecules targeting mitochondria. Immunity. 45:761–73. 2016.
View Article : Google Scholar
|
|
47
|
Di Virgilio F, Dal Ben D, Sarti AC,
Giuliani AL and Falzoni S: The P2X7 Receptor in Infection and
Inflammation. Immunity. 47:15–31. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Oeckinghaus A, Hayden MS and Ghosh S:
Crosstalk in NF-κB signaling pathways. Nat Immunol. 12:695–708.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Qiao Y, Wang P, Qi J, Zhang L and Gao C:
TLR-induced NF-κB activation regulates NLRP3 expression in murine
macrophages. FEBS Lett. 586:1022–1026. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liu C, Zhao Y and Zhao WJ: Positive Effect
of 6-Gingerol on functional plasticity of microglia in a rat model
of LPS-induced depression. J Neuroimmune Pharmacol. 19:202024.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Karin M and Ben-Neriah Y: Phosphorylation
meets ubiquitination: The control of NF-[kappa]B activity. Annu Rev
Immunol. 18:621–663. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sun E, Motolani A, Campos L and Lu T: The
Pivotal Role of NF-µB in the Pathogenesis and Therapeutics of
Alzheimer's Disease. Int J Mol Sci. 23:89722022. View Article : Google Scholar
|
|
53
|
Li X, Yan S, Li M, Liu R, Lu Q, Lu M, Bai
F and Shen QD: Piezoelectric nanoparticle-driven rhythmic
ultrasound neuromodulation for treatment of early-stage Alzheimer's
disease. Biomaterials. 328:1239052026. View Article : Google Scholar
|
|
54
|
Kharitidi D, Manteghi S and Pause A:
Pseudophosphatases: Methods of analysis and physiological
functions. Methods. 65:207–218. 2014. View Article : Google Scholar
|
|
55
|
O'Brien WT, Pham L, Symons GF, Monif M,
Shultz SR and McDonald SJ: The NLRP3 inflammasome in traumatic
brain injury: Potential as a biomarker and therapeutic target. J
Neuroinflammation. 17:1042020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yang S, Han J, Xue M, Zhang Y, Yan A, Gao
X, He J, Sun X, Fu S, Liu D and Huang B: Coixol ameliorates
dopaminergic neurodegeneration by inhibiting neuroinflammation and
protecting mitochondrial function. Front Pharmacol. 16:16579102025.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zaman B, Mostafa I, Hassan T, Ahmed S,
Esha NJI, Chowdhury FA, Bosu T, Chowdhury HN, Mallick A, Islam MS,
et al: Tolperisone hydrochloride improves motor functions in
Parkinson's disease via MMP-9 inhibition and by downregulating p38
MAPK and ERK1/2 signaling cascade. Biomed Pharmacother.
174:1164382024. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Owen KL, Brockwell NK and Parker BS:
JAK-STAT Signaling: A Double-edged sword of immune regulation and
cancer progression. Cancers (Basel). 11:20022019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
O'Shea JJ and Murray PJ: Cytokine
signaling modules in inflammatory responses. Immunity. 28:477–487.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hu X, Li J, Fu M, Zhao X and Wang W: The
JAK/STAT signaling pathway: From bench to clinic. Signal Transduct
Target Ther. 6:4022021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Doshi NK, Pesaresi T, Pagadala T, Dion W,
Zhang Y, David NL, Amorim T, Wang W, Kumar GVN, Zhu B, et al:
Branched chain amino acids prime metabolic inflammation. Mol Metab.
104:1023082026. View Article : Google Scholar :
|
|
62
|
Sun K, Luo J, Guo J, Yao X, Jing X and Guo
F: The PI3K/AKT/mTOR signaling pathway in osteoarthritis: A
narrative review. Osteoarthritis Cartilage. 28:400–409. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lee YJ, Heo JS, Suh HN, Lee MY and Han HJ:
Interleukin-6 stimulates alpha-MG uptake in renal proximal tubule
cells: Involvement of STAT3, PI3K/Akt, MAPKs, and NF-kappaB. Am J
Physiol Renal Physiol. 293:F1036–F1046. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wang H, Li X, Liu Z, Lu R, Li X and Zhang
X: ANXA1 regulates microglia polarization and autophagy via
PI3K/Akt/mTOR pathway to reduce inflammatory injury after cerebral
ischemia-reperfusion. Cell Signal. 139:1123362026. View Article : Google Scholar
|
|
65
|
Zhou J, Zheng Q and Chen Z: The Nrf2
pathway in liver diseases. Front Cell Dev Biol. 10:8262042022.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Motohashi H and Yamamoto M: Nrf2-Keap1
defines a physiologically important stress response mechanism.
Trends Mol Med. 10:549–557. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wang D, Wu J, Le S, Wang H, Luo J, Li R,
Chen X, Song Y, Wu L, Ye P, et al: Oltipraz, the activator of
nuclear factor erythroid 2-related factor 2 (Nrf2), protects
against the formation of BAPN-induced aneurysms and dissection of
the thoracic aorta in mice by inhibiting activation of the
ROS-mediated NLRP3 inflammasome. Eur J Pharmacol. 936:1753612022.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wang L and He C: Nrf2-mediated
anti-inflammatory polarization of macrophages as therapeutic
targets for osteoarthritis. Front Immunol. 13:9671932022.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Shih PH and Yen GC: Differential
expressions of antioxidant status in aging rats: The role of
transcriptional factor Nrf2 and MAPK signaling pathway.
Biogerontology. 8:71–80. 2007. View Article : Google Scholar
|
|
70
|
Wu Y, Zhang Y, Zhang S, Yang H and Ci X:
Aging-associated GSK3β overexpression exacerbates hepatic
ischemia-reperfusion injury through Nrf2 deficiency-induced
hepatocyte ferroptosis. Life Sci. 388:1242142026. View Article : Google Scholar
|
|
71
|
Deng Z, Fan T, Xiao C, Tian H, Zheng Y, Li
C and He J: TGF-β signaling in health, disease, and therapeutics.
Signal Transduct Target Ther. 9:612024. View Article : Google Scholar
|
|
72
|
Cáceres FT, Gaspari TA, Samuel CS and
Pinar AA: Serelaxin inhibits the profibrotic TGF-β1/IL-1β axis by
targeting TLR-4 and the NLRP3 inflammasome in cardiac
myofibroblasts. FASEB J. 33:14717–14733. 2019. View Article : Google Scholar
|
|
73
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Feng J, Liu H, Jiang K, Gong X, Huang R,
Zhou C, Mao J, Chen Y, Xu H, Zhang X, et al: Enhanced oxidative
stress aggravates BLM-induced pulmonary fibrosis by promoting
cellular senescence through enhancing NLRP3 activation. Life Sci.
358:1231282024. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Mohammad ZB, Yudin SCY, Goldberg BJ, Serra
KL and Klegeris A: Exploring neuroglial signaling: Diversity of
molecules implicated in microglia-to-astrocyte neuroimmune
communication. Rev Neurosci. 36:91–117. 2025. View Article : Google Scholar :
|
|
76
|
Hegde AN, Smith SG, Duke LM, Pourquoi A
and Vaz S: Perturbations of ubiquitin-proteasome-mediated
proteolysis in aging and Alzheimer's disease. Front Aging Neurosci.
11:3242019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Yang G, Xu X, Gao W, Wang X, Zhao Y and Xu
Y: Microglia-orchestrated neuroinflammation and synaptic
remodeling: Roles of pro-inflammatory cytokines and receptors in
neurodegeneration. Front Cell Neurosci. 19:17006922025. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Sawikr Y, Yarla NS, Peluso I, Kamal MA,
Aliev G and Bishayee A: Neuroinflammation in Alzheimer's disease:
The preventive and therapeutic potential of polyphenolic
nutraceuticals. Adv Protein Chem Struct Biol. 108:33–57. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Sheng JG, Jones RA, Zhou XQ, McGinness JM,
Van Eldik LJ, Mrak RE and Griffin WS: Interleukin-1 promotion of
MAPK-p38 overexpression in experimental animals and in Alzheimer's
disease: Potential significance for tau protein phosphorylation.
Neurochem Int. 39:341–348. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Nizynski B, Dzwolak W and Nieznanski K:
Amyloidogenesis of Tau protein. Protein Sci. 26:2126–2150. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Braak H, Braak E and Strothjohann M:
Abnormally phosphorylated tau protein related to the formation of
neurofibrillary tangles and neuropil threads in the cerebral cortex
of sheep and goat. Neurosci Lett. 171:1–4. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Chakrabarty P, Tianbai L, Herring A,
Ceballos-Diaz C, Das P and Golde TE: Hippocampal expression of
murine IL-4 results in exacerbation of amyloid deposition. Mol
Neurodegener. 7:362012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Koller EJ, McFarland KN, Angelle C, Howard
J, Ryu D, Dillon KD, Erquizi A, Beheray M, De La Cruz EG, Cruz PE,
et al: Antagonizing Il10 and Il4 signaling via intracerebral decoy
receptor expression attenuates Aβ accumulation. Acta Neuropathol
Commun. 13:512025. View Article : Google Scholar
|
|
84
|
Zheng C, Zhou XW and Wang JZ: The dual
roles of cytokines in Alzheimer's disease: Update on interleukins,
TNF-α, TGF-β and IFN-γ. Transl Neurodegener. 5:72016. View Article : Google Scholar
|
|
85
|
Bagyinszky E, Giau VV, Shim K, Suk K, An
SSA and Kim S: Role of inflammatory molecules in the Alzheimer's
disease progression and diagnosis. J Neurol Sci. 376:242–254. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Burmeister AR and Marriott I: The
Interleukin-10 family of cytokines and their role in the CNS. Front
Cell Neurosci. 12:4582018. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Park KW, Lee HG, Jin BK and Lee YB:
Interleukin-10 endogenously expressed in microglia prevents
lipopolysaccharide-induced neurodegeneration in the rat cerebral
cortex in vivo. Exp Mol Med. 39:812–819. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Mosser DM and Zhang X: Interleukin-10: New
perspectives on an old cytokine. Immunol Rev. 226:205–218. 2008.
View Article : Google Scholar
|
|
89
|
Peng F, Liao M, Qin R, Zhu S, Peng C, Fu
L, Chen Y and Han B: Regulated cell death (RCD) in cancer: Key
pathways and targeted therapies. Signal Transduct Target Ther.
7:2862022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Moujalled D, Strasser A and Liddell JR:
Molecular mechanisms of cell death in neurological diseases. Cell
Death Differ. 28:2029–2044. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Fricker M, Tolkovsky AM, Borutaite V,
Coleman M and Brown GC: Neuronal cell death. Physiol Rev.
98:813–880. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Fuchs Y and Steller H: Programmed cell
death in animal development and disease. Cell. 147:742–758. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Buss RR, Sun W and Oppenheim RW: Adaptive
roles of programmed cell death during nervous system development.
Annu Rev Neurosci. 29:1–35. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Wang Y, Lv MN and Zhao WJ: Research on
ferroptosis as a therapeutic target for the treatment of
neurodegenerative diseases. Ageing Res Rev. 91:1020352023.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Shi J, Zhao Y, Wang K, Shi X, Wang Y,
Huang H, Zhuang Y, Cai T, Wang F and Shao F: Cleavage of GSDMD by
inflammatory caspases determines pyroptotic cell death. Nature.
526:660–665. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Vasudevan SO, Behl B and Rathinam VA:
Pyroptosis-induced inflammation and tissue damage. Semin Immunol.
69:1017812023. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Franchi L, Eigenbrod T, Muñoz-Planillo R
and Nuñez G: The inflammasome: A caspase-1-activation platform that
regulates immune responses and disease pathogenesis. Nat Immunol.
10:241–247. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun
X, Kang R and Tang D: Ferroptosis: Process and function. Cell Death
Differ. 23:369–379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Lee J and Hyun DH: The Interplay between
intracellular iron homeostasis and neuroinflammation in
neurodegenerative diseases. Antioxidants (Basel). 12:9182023.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Nemeth E, Tuttle MS, Powelson J, Vaughn
MB, Donovan A, Ward DM, Ganz T and Kaplan J: Hepcidin regulates
cellular iron efflux by binding to ferroportin and inducing its
internalization. Science. 306:2090–2093. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Nairz M and Weiss G: Iron in infection and
immunity. Mol Aspects Med. 75:1008642020. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Degterev A, Huang Z, Boyce M, Li Y, Jagtap
P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar
|
|
105
|
Takeuchi O and Akira S: Pattern
recognition receptors and inflammation. Cell. 140:805–820. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Schroder K and Tschopp J: The
inflammasomes. Cell. 140:821–832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Scaffidi P, Misteli T and Bianchi MEL:
Release of chromatin protein HMGB1 by necrotic cells triggers
inflammation. Nature. 418:191–195. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Park JS, Svetkauskaite D, He Q, Kim JY,
Strassheim D, Ishizaka A and Abraham E: Involvement of toll-like
receptors 2 and 4 in cellular activation by high mobility group box
1 protein. J Biol Chem. 279:7370–7377. 2004. View Article : Google Scholar
|
|
109
|
Liddelow SA, Guttenplan KA, Clarke LE,
Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS,
Peterson TC, et al: Neurotoxic reactive astrocytes are induced by
activated microglia. Nature. 541:481–487. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Lawrence JM, Schardien K, Wigdahl B and
Nonnemacher MR: Roles of neuropathology-associated reactive
astrocytes: A systematic review. Acta Neuropathol Commun.
11:422023. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Senatorov VV Jr, Friedman AR, Milikovsky
DZ, Ofer J, Saar-Ashkenazy R, Charbash A, Jahan N, Chin G, Mihaly
E, Lin JM, et al: Blood-brain barrier dysfunction in aging induces
hyperactivation of TGFβ signaling and chronic yet reversible neural
dysfunction. Sci Transl Med. 11:eaaw82832019. View Article : Google Scholar
|
|
112
|
Montagne A, Barnes SR, Sweeney MD,
Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua
L, et al: Blood-brain barrier breakdown in the aging human
hippocampus. Neuron. 85:296–302. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Nation DA, Sweeney MD, Montagne A, Sagare
AP, D'Orazio LM, Pachicano M, Sepehrband F, Nelson AR, Buennagel
DP, Harrington MG, et al: Blood-brain barrier breakdown is an early
biomarker of human cognitive dysfunction. Nat Med. 25:270–276.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Ryu JK, Rafalski VA, Meyer-Franke A, Adams
RA, Poda SB, Rios Coronado PE, Pedersen LØ, Menon V, Baeten KM,
Sikorski SL, et al: Fibrin-targeting immunotherapy protects against
neuroinflammation and neurodegeneration. Nat Immunol. 19:1212–1223.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Banks WA, Gray AM, Erickson MA, Salameh
TS, Damodarasamy M, Sheibani N, Meabon JS, Wing EE, Morofuji Y,
Cook DG and Reed MJ: Lipopolysaccharide-induced blood-brain barrier
disruption: Roles of cyclooxygenase, oxidative stress,
neuroinflammation, and elements of the neurovascular unit. J
Neuroinflammation. 12:2232015. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Davalos D, Ryu JK, Merlini M, Baeten KM,
Le Moan N, Petersen MA, Deerinck TJ, Smirnoff DS, Bedard C,
Hakozaki H, et al: Fibrinogen-induced perivascular microglial
clustering is required for the development of axonal damage in
neuroinflammation. Nat Commun. 3:12272012. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Lane CA, Hardy J and Schott JM:
Alzheimer's disease. Eur J Neurol. 25:59–70. 2018. View Article : Google Scholar
|
|
118
|
Scheltens P, De Strooper B, Kivipelto M,
Holstege H, Chételat G, Teunissen CE, Cummings J and van der Flier
WM: Alzheimer's disease. Lancet. 397:1577–1590. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Jagust W: Imaging the evolution and
pathophysiology of Alzheimer disease. Nat Rev Neurosci. 19:687–700.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Chen Y and Yu Y: Tau and neuroinflammation
in Alzheimer's disease: Interplay mechanisms and clinical
translation. J Neuroinflammation. 20:1652023. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Meraz-Ríos MA, Toral-Rios D,
Franco-Bocanegra D, Villeda-Hernández J and Campos-Peña V:
Inflammatory process in Alzheimer's disease. Front Integr Neurosci.
7:592013. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Frank-Cannon TC, Alto LT, McAlpine FE and
Tansey MG: Does neuroinflammation fan the flame in
neurodegenerative diseases? Mol Neurodegener. 4:472009. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Blennow K and Zetterberg H: Biomarkers for
Alzheimer's disease: Current status and prospects for the future. J
Intern Med. 284:643–663. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Lue LF, Kuo YM, Roher AE, Brachova L, Shen
Y, Sue L, Beach T, Kurth JH, Rydel RE and Rogers J: Soluble amyloid
beta peptide concentration as a predictor of synaptic change in
Alzheimer's disease. Am J Pathol. 155:853–862. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Ng A, Tam WW, Zhang MW, Ho CS, Husain SF,
McIntyre RS and Ho RC: IL-1β, IL-6, TNF-α and CRP in elderly
patients with depression or Alzheimer's disease: Systematic review
and meta-analysis. Sci Rep. 8:120502018. View Article : Google Scholar
|
|
126
|
Lopez-Rodriguez AB, Hennessy E, Murray CL,
Nazmi A, Delaney HJ, Healy D, Fagan SG, Rooney M, Stewart E, Lewis
A, et al: Acute systemic inflammation exacerbates neuroinflammation
in Alzheimer's disease: IL-1β drives amplified responses in primed
astrocytes and neuronal network dysfunction. Alzheimers Dement.
17:1735–1755. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Liao YF, Wang BJ, Cheng HT, Kuo LH and
Wolfe MS: Tumor necrosis factor-alpha, interleukin-1beta, and
interferon-gamma stimulate gamma-secretase-mediated cleavage of
amyloid precursor protein through a JNK-dependent MAPK pathway. J
Biol Chem. 279:49523–49532. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Millot P, San C, Bennana E, Porte B,
Vignal N, Hugon J, Paquet C, Hosten B and Mouton-Liger F: STAT3
inhibition protects against neuroinflammation and BACE1
upregulation induced by systemic inflammation. Immunol Lett.
228:129–134. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Wang YR, Zeng XQ, Wang J, Fowler CJ, Li
QX, Bu XL, Doecke J, Maruff P, Martins RN, Rowe CC, et al:
Autoantibodies to BACE1 promote Aβ accumulation and
neurodegeneration in Alzheimer's disease. Acta Neuropathol.
148:572024. View Article : Google Scholar
|
|
130
|
Pawluk H, Woźniak A, Grześk G,
Kołodziejska R, Kozakiewicz M, Kopkowska E, Grzechowiak E and
Kozera G: The role of selected Pro-inflammatory cytokines in
pathogenesis of ischemic stroke. Clin Interv Aging. 15:469–484.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Peng W, Lu W, Jiang X, Xiong C, Chai H,
Cai L and Lan Z: Current progress on Neuroinflammation-mediated
postoperative cognitive dysfunction: An update. Curr Mol Med.
23:1077–1086. 2023. View Article : Google Scholar
|
|
132
|
Shan C and Zhang C and Zhang C: The role
of IL-6 in neurodegenerative disorders. Neurochem Res. 49:834–846.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Sochocka M, Diniz BS and Leszek J:
Inflammatory response in the CNS: Friend or Foe? Mol Neurobiol.
54:8071–8089. 2017. View Article : Google Scholar :
|
|
134
|
Bouwmeester T, Bauch A, Ruffner H, Angrand
PO, Bergamini G, Croughton K, Cruciat C, Eberhard D, Gagneur J,
Ghidelli S, et al: A physical and functional map of the human
TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol.
6:97–105. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Schorn F, Werthenbach JP, Hoffmann M,
Daoud M, Stachelscheid J, Schiffmann LM, Hildebrandt X, Lyu SI,
Peltzer N, Quaas A, et al: cIAPs control RIPK1 kinase
activity-dependent and-independent cell death and tissue
inflammation. EMBO J. 42:e1136142023. View Article : Google Scholar
|
|
136
|
Newton K, Wickliffe KE, Dugger DL,
Maltzman A, Roose-Girma M, Dohse M, Kőműves L, Webster JD and Dixit
VM: Cleavage of RIPK1 by caspase-8 is crucial for limiting
apoptosis and necroptosis. Nature. 574:428–431. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Wang L, Du F and Wang X: TNF-alpha induces
two distinct caspase-8 activation pathways. Cell. 133:693–703.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Liu H, Zhou YC and Song W: Involvement of
IL-10R/STAT3 pathway in amyloid β clearance by microlgia in
Alzheimer's disease. Int Immunopharmacol. 101:1082632021.
View Article : Google Scholar
|
|
139
|
Gallucci M, Limbucci N, Catalucci A and
Caulo M: Neurodegenerative diseases. Radiol Clin North Am.
46:799–817. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Ryu J, Yu HN, Cho H, Kim HS, Baik TK, Lee
SJ and Woo RS: Neuregulin-1 exerts protective effects against
neurotoxicities induced by C-terminal fragments of APP via ErbB4
receptor. J Pharmacol Sci. 119:73–81. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Baik TK, Kim YJ, Kang SM, Song DY, Min SS
and Woo RS: Blocking the phosphatidylinositol 3-kinase pathway
inhibits neuregulin-1-mediated rescue of neurotoxicity induced by
Aβ1-42. J Pharm Pharmacol. 68:1021–1029. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Woo RS, Lee JH, Kim HS, Baek CH, Song DY,
Suh YH and Baik TK: Neuregulin-1 protects against neurotoxicities
induced by Swedish amyloid precursor protein via the ErbB4
receptor. Neuroscience. 202:413–423. 2012. View Article : Google Scholar
|
|
143
|
Jiang Q, Chen S, Hu C, Huang P, Shen H and
Zhao W: Neuregulin-1 (Nrg1) signaling has a preventive role and is
altered in the frontal cortex under the pathological conditions of
Alzheimer's disease. Mol Med Rep. 14:2614–2624. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Liu C, Zhao Y, Dao JJ, Zhang W, Liu J, Ma
YK, Qiao CM, Cui C, Chen SX, Shen YQ and Zhao WJ: Targeted ErbB4
receptor activation ameliorates neuronal deficits via DOCK3
signaling in a transgenic mouse AD model. Neurotherapeutics.
22:e007392025. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Dao JJ, Zhang W, Liu C, Li Q, Qiao CM, Cui
C, Shen YQ, Chen SX and Zhao WJ: Targeted ErbB4 receptor activation
prevents D-galactose-induced neuronal senescence via inhibiting
ferroptosis pathway. Front Pharmacol. 16:15286042025. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Liu C, Zhao Y, Zhang W, Dao JJ, Li Q,
Huang J, Li ZF, Ma YK, Qiao CM, Cui C, et al: Targeted activation
of ErbB4 receptor ameliorates neuronal deficits and
neuroinflammation in a food-borne polystyrene microplastic exposed
mouse model. J Neuroinflammation. 22:862025. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Koeglsperger T, Rumpf SL, Schließer P,
Struebing FL, Brendel M, Levin J, Trenkwalder C, Höglinger GU and
Herms J: Neuropathology of incidental Lewy body & prodromal
Parkinson's disease. Mol Neurodegener. 18:322023. View Article : Google Scholar
|
|
148
|
Imamura K, Hishikawa N, Sawada M, Nagatsu
T, Yoshida M and Hashizume Y: Distribution of major
histocompatibility complex class II-positive microglia and cytokine
profile of Parkinson's disease brains. Acta Neuropathol.
106:518–526. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Pajares M, I Rojo A, Manda G, Boscá L and
Cuadrado A: Inflammation in Parkinson's disease: Mechanisms and
therapeutic implications. Cells. 9:16872020. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Isik S, Yeman Kiyak B, Akbayir R, Seyhali
R and Arpaci T: Microglia mediated neuroinflammation in Parkinson's
disease. Cells. 12:10122023. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Bennett MC: The role of alpha-synuclein in
neurodegenerative diseases. Pharmacol Ther. 105:311–331. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Yi-Bin W, Xiang L, Bing Y, Qi Z, Fei-Tong
J, Minghong W, Xiangxiang Z, Le K, Yan L, Ping S, et al: Inhibition
of the CEBPβ-NFκB interaction by nanocarrier-packaged Carnosic acid
ameliorates glia-mediated neuroinflammation and improves cognitive
function in an Alzheimer's disease model. Cell Death Dis.
13:3182022. View Article : Google Scholar
|
|
153
|
Singh J, Habean ML and Panicker N:
Inflammasome assembly in neurodegenerative diseases. Trends
Neurosci. 46:814–831. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Kim C, Ho DH, Suk JE, You S, Michael S,
Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ and Lee SJ:
Neuron-released oligomeric α-synuclein is an endogenous agonist of
TLR2 for paracrine activation of microglia. Nat Commun. 4:15622013.
View Article : Google Scholar
|
|
155
|
Choi I, Zhang Y, Seegobin SP, Pruvost M,
Wang Q, Purtell K, Zhang B and Yue Z: Microglia clear
neuron-released α-synuclein via selective autophagy and prevent
neurodegeneration. Nat Commun. 11:13862020. View Article : Google Scholar
|
|
156
|
Fellner L, Irschick R, Schanda K, Reindl
M, Klimaschewski L, Poewe W, Wenning GK and Stefanova N: Toll-like
receptor 4 is required for α-synuclein dependent activation of
microglia and astroglia. Glia. 61:349–360. 2013. View Article : Google Scholar :
|
|
157
|
Cebrián C, Zucca FA, Mauri P, Steinbeck
JA, Studer L, Scherzer CR, Kanter E, Budhu S, Mandelbaum J,
Vonsattel JP, et al: MHC-I expression renders catecholaminergic
neurons susceptible to T-cell-mediated degeneration. Nat Commun.
5:36332014. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Chu Y, Dodiya H, Aebischer P, Olanow CW
and Kordower JH: Alterations in lysosomal and proteasomal markers
in Parkinson's disease: Relationship to alpha-synuclein inclusions.
Neurobiol Dis. 35:385–398. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Cole TA, Zhao H, Collier TJ, Sandoval I,
Sortwell CE, Steece-Collier K, Daley BF, Booms A, Lipton J, Welch
M, et al: α-Synuclein antisense oligonucleotides as a
disease-modifying therapy for Parkinson's disease. JCI Insight.
6:e1356332021. View Article : Google Scholar
|
|
160
|
Reynolds AD, Glanzer JG, Kadiu I,
Ricardo-Dukelow M, Chaudhuri A, Ciborowski P, Cerny R, Gelman B,
Thomas MP, Mosley RL and Gendelman HE: Nitrated
alpha-synuclein-activated microglial profiling for Parkinson's
disease. J Neurochem. 104:1504–1525. 2008. View Article : Google Scholar
|
|
161
|
Reynolds AD, Stone DK, Hutter JA, Benner
EJ, Mosley RL and Gendelman HE: Regulatory T cells attenuate Th17
cell-mediated nigrostriatal dopaminergic neurodegeneration in a
model of Parkinson's disease. J Immunol. 184:2261–2271. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Chaturvedi RK and Beal MF: PPAR: A
therapeutic target in Parkinson's disease. J Neurochem.
106:506–518. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Rowland LP and Shneider NA: Amyotrophic
lateral sclerosis. N Engl J Med. 344:1688–1700. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Feldman EL, Goutman SA, Petri S, Mazzini
L, Savelieff MG, Shaw PJ and Sobue G: Amyotrophic lateral
sclerosis. Lancet. 400:1363–1380. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Gordon R, Albornoz EA, Christie DC,
Langley MR, Kumar V, Mantovani S, Robertson AAB, Butler MS, Rowe
DB, O'Neill LA, et al: Inflammasome inhibition prevents α-synuclein
pathology and dopaminergic neurodegeneration in mice. Sci Transl
Med. 10:eaah40662018. View Article : Google Scholar
|
|
166
|
Chen J, Mao K, Yu H, Wen Y, She H, Zhang
H, Liu L, Li M, Li W and Zou F: p38-TFEB pathways promote microglia
activation through inhibiting CMA-mediated NLRP3 degradation in
Parkinson's disease. J Neuroinflammation. 18:2952021. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Brandes MS, Zweig JA, Tang A and Gray NE:
NRF2 activation ameliorates oxidative stress and improves
mitochondrial function and synaptic plasticity, and in A53T
α-Synuclein hippocampal neurons. Antioxidants (Basel). 11:262021.
View Article : Google Scholar
|
|
168
|
Campolo M, Casili G, Biundo F, Crupi R,
Cordaro M, Cuzzocrea S and Esposito E: The neuroprotective effect
of dimethyl fumarate in an MPTP-Mouse model of Parkinson's disease:
Involvement of reactive oxygen Species/Nuclear Factor-κB/Nuclear
transcription factor related to NF-E2. Antioxid Redox Signal.
27:453–471. 2017. View Article : Google Scholar :
|
|
169
|
Dehmer T, Heneka MT, Sastre M, Dichgans J
and Schulz JB: Protection by pioglitazone in the MPTP model of
Parkinson's disease correlates with I kappa B alpha induction and
block of NF kappa B and iNOS activation. J Neurochem. 88:494–501.
2004. View Article : Google Scholar
|
|
170
|
Wijesekera LC and Leigh PN: Amyotrophic
lateral sclerosis. Orphanet J Rare Dis. 4:32009. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Calma AD, Pavey N, Menon P and Vucic S:
Neuroinflammation in amyotrophic lateral sclerosis: Pathogenic
insights and therapeutic implications. Curr Opin Neurol.
37:585–592. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Ungerleider K, Beck J, Lissa D, Turnquist
C, Horikawa I, Harris BT and Harris CC: Astrocyte senescence and
SASP in neurodegeneration: Tau Joins the loop. Cell Cycle.
20:752–764. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Guerrero AR, Uchida K, Nakajima H,
Watanabe S, Nakamura M, Johnson WE and Baba H: Blockade of
interleukin-6 signaling inhibits the classic pathway and promotes
an alternative pathway of macrophage activation after spinal cord
injury in mice. J Neuroinflammation. 9:402012. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Garbuzova-Davis S, Ehrhart J, Sanberg PR
and Borlongan CV: Potential role of Humoral IL-6 cytokine in
mediating Pro-inflammatory endothelial cell response in amyotrophic
lateral sclerosis. Int J Mol Sci. 19:4232018. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Serizawa K, Tomizawa-Shinohara H, Magi M,
Yogo K and Matsumoto Y: Anti-IL-6 receptor antibody improves pain
symptoms in mice with experimental autoimmune encephalomyelitis. J
Neuroimmunol. 319:71–79. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Atta AA, Ibrahim WW, Mohamed AF and
Abdelkader NF: Microglia polarization in nociplastic pain:
Mechanisms and perspectives. Inflammopharmacology. 31:1053–1067.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Khan AW, Farooq M, Hwang MJ, Haseeb M and
Choi S: Autoimmune Neuroinflammatory diseases: Role of
interleukins. Int J Mol Sci. 24:79602023. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Li SJ, Liu W, Wang JL, Zhang Y, Zhao DJ,
Wang TJ and Li YY: The role of TNF-α, IL-6, IL-10, and GDNF in
neuronal apoptosis in neonatal rat with hypoxic-ischemic
encephalopathy. Eur Rev Med Pharmacol Sci. 18:905–909. 2014.
|
|
179
|
IL6 receptor 358Ala variant and
trans-signaling are disease modifiers in amyotrophic lateral
sclerosis. Neurol Neuroimmunol Neuroinflamm. 7:e6502019.
|
|
180
|
Zhang W, Xiao D, Mao Q and Xia H: Role of
neuroinflammation in neurodegeneration development. Signal
Transduct Target Ther. 8:2672023. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Labrador L, Rodriguez L, Beltran S,
Hernandez F, Gomez L, Ojeda P, Bergmann C, Calegaro-Nassif M, Kerr
B, Medinas DB, et al: Overexpression of autophagy enhancer
PACER/RUBCNL in neurons accelerates disease in the SOD1G93A ALS
mouse model. Biol Res. 57:862024. View Article : Google Scholar
|
|
182
|
Chu CT: Mechanisms of selective autophagy
and mitophagy: Implications for neurodegenerative diseases.
Neurobiol Dis. 122:23–34. 2019. View Article : Google Scholar :
|
|
183
|
Huang J, Yu Y, Pang D, Li C, Wei Q, Cheng
Y, Cui Y, Ou R and Shang H: Lnc-HIBADH-4 regulates
Autophagy-lysosome pathway in amyotrophic lateral sclerosis by
targeting Cathepsin D. Mol Neurobiol. 61:4768–4782. 2024.
View Article : Google Scholar :
|
|
184
|
Martínez-Iglesias O, Naidoo V, Carrera I,
Corzo L and Cacabelos R: Nosustrophine: An epinutraceutical
bioproduct with effects on DNA methylation, histone acetylation and
sirtuin expression in Alzheimer's disease. Pharmaceutics.
14:24472022. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Giagnorio E, Malacarne C, Cavalcante P,
Scandiffio L, Cattaneo M, Pensato V, Gellera C, Riva N, Quattrini
A, Dalla Bella E, et al: MiR-146a in ALS: Contribution to early
peripheral nerve degeneration and relevance as disease biomarker.
Int J Mol Sci. 24:46102023. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Madaro L, Passafaro M, Sala D, Etxaniz U,
Lugarini F, Proietti D, Alfonsi MV, Nicoletti C, Gatto S, De Bardi
M, et al: Denervation-activated STAT3-IL-6 signalling in
fibro-adipogenic progenitors promotes myofibres atrophy and
fibrosis. Nat Cell Biol. 20:917–927. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Hersch SM and Rosas HD: Neuroprotection
for Huntington's disease: Ready, set, slow. Neurotherapeutics.
5:226–236. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
No authors listed. A novel gene containing
a trinucleotide repeat that is expanded and unstable on
Huntington's disease chromosomes. The Huntington's disease
collaborative research group. Cell. 72:971–983. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Wellington CL, Brinkman RR, O'Kusky JR and
Hayden MR: Toward understanding the molecular pathology of
Huntington's disease. Brain Pathol. 7:979–1002. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Etxeberria-Rekalde E,
Alzola-Aldamizetxebarria S, Flunkert S, Hable I, Daurer M, Neddens
J and Hutter-Paier B: Quantification of Huntington's disease
related markers in the R6/2 mouse model. Front Mol Neurosci.
13:6172292020. View Article : Google Scholar
|
|
191
|
Paldino E, Migliorato G and Fusco FR:
Neuroimmune pathways involvement in neurodegeneration of R6/2 mouse
model of Huntington's disease. Front Cell Neurosci. 18:13600662024.
View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Möller T: Neuroinflammation in
Huntington's disease. J Neural Transm (Vienna). 117:1001–1008.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Singhrao SK, Neal JW, Morgan BP and Gasque
P: Increased complement biosynthesis by microglia and complement
activation on neurons in Huntington's disease. Exp Neurol.
159:362–376. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Sapp E, Kegel KB, Aronin N, Hashikawa T,
Uchiyama Y, Tohyama K, Bhide PG, Vonsattel JP and DiFiglia M: Early
and progressive accumulation of reactive microglia in the
Huntington disease brain. J Neuropathol Exp Neurol. 60:161–172.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Björkqvist M, Wild EJ, Thiele J,
Silvestroni A, Andre R, Lahiri N, Raibon E, Lee RV, Benn CL, Soulet
D, et al: A novel pathogenic pathway of immune activation
detectable before clinical onset in Huntington's disease. J Exp
Med. 205:1869–1877. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Hirsch EC, Vyas S and Hunot S:
Neuroinflammation in Parkinson's disease. Parkinsonism Relat
Disord. 18(Suppl 1): S210–S212. 2012. View Article : Google Scholar
|
|
197
|
Fung JN, Lee JD, Adam R, O'Sullivan JD and
Woodruff TM: Peripheral and central elevation of IL-8 in patients
with Huntington's disease. Mol Immunol. 179:84–93. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Eide S, Misztal M and Feng ZP:
Interleukin-6 as a marker of Huntington's disease progression:
Systematic review and meta-analysis. Brain Behav Immun Health.
30:1006352023. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Träger U, Magnusson A, Lahiri Swales N,
Wild E, North J, Lowdell M and Björkqvist M and Björkqvist M:
JAK/STAT signalling in Huntington's disease immune cells. PLoS
Curr. 5:ecurrents.hd.5791c897b5c3bebeed93b1d1da0c06482013.
|
|
200
|
Kwan W, Magnusson A, Chou A, Adame A,
Carson MJ, Kohsaka S, Masliah E, Möller T, Ransohoff R, Tabrizi SJ,
et al: Bone marrow transplantation confers modest benefits in mouse
models of Huntington's disease. J Neurosci. 32:133–142. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Gómez-Jaramillo L, Cano-Cano F,
González-Montelongo MDC, Campos-Caro A, Aguilar-Diosdado M and
Arroba AI: A new perspective on Huntington's disease: How a
neurological disorder influences the peripheral tissues. Int J Mol
Sci. 23:60892022. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Crotti A and Glass CK: The choreography of
neuroinflammation in Huntington's disease. Trends Immunol.
36:364–373. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Rocha NP, Ribeiro FM, Furr-Stimming E and
Teixeira AL: Neuroimmunology of Huntington's disease: Revisiting
evidence from human studies. Mediators Inflamm. 2016:86531322016.
View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Franceschi C, Capri M, Monti D, Giunta S,
Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M,
et al: Inflammaging and anti-inflammaging: A systemic perspective
on aging and longevity emerged from studies in humans. Mech Ageing
Dev. 128:92–105. 2007. View Article : Google Scholar
|
|
205
|
Ouyang W, Rutz S, Crellin NK, Valdez PA
and Hymowitz SG: Regulation and functions of the IL-10 family of
cytokines in inflammation and disease. Annu Rev Immunol. 29:71–109.
2011. View Article : Google Scholar
|
|
206
|
O'Neil SM, Hans EE, Jiang S, Wangler LM
and Godbout JP: Astrocyte immunosenescence and deficits in
interleukin 10 signaling in the aged brain disrupt the regulation
of microglia following innate immune activation. Glia. 70:913–934.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Heneka MT, Kummer MP, Stutz A, Delekate A,
Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, et
al: NLRP3 is activated in Alzheimer's disease and contributes to
pathology in APP/PS1 mice. Nature. 493:674–678. 2013. View Article : Google Scholar :
|
|
208
|
Roxburgh CS and McMillan DC: Therapeutics
targeting innate immune/inflammatory responses through the
interleukin-6/JAK/STAT signal transduction pathway in patients with
cancer. Transl Res. 167:61–66. 2016. View Article : Google Scholar
|
|
209
|
Halle A, Hornung V, Petzold GC, Stewart
CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ and
Golenbock DT: The NALP3 inflammasome is involved in the innate
immune response to amyloid-beta. Nat Immunol. 9:857–865. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Dinarello CA: Interleukin-1 in the
pathogenesis and treatment of inflammatory diseases. Blood.
117:3720–3732. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Ising C, Venegas C, Zhang S, Scheiblich H,
Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM,
Tejera D, et al: NLRP3 inflammasome activation drives tau
pathology. Nature. 575:669–673. 2019. View Article : Google Scholar : PubMed/NCBI
|