Introduction
Ubiquitination is a key mechanism of
post-translational modification, accomplished through a cascade
reaction involving E1, E2 and E3 enzymes. E1 activates ubiquitin
and transfers it to E2, while E3 determines substrate specificity
and catalyzes ubiquitin chain formation (1). This process marks target proteins
for degradation by the proteasome, constituting the
ubiquitin-proteasome system (UPS) (2). The UPS regulates physiological
processes such as the cell cycle, inflammation and DNA repair, and
its dysregulation is closely associated with various diseases,
including cancer and neurodegenerative disorders (3,4).
Among the enzymes involved, E3 ubiquitin ligases act as direct
'molecular switches' that interact with target proteins and have
thus emerged as important therapeutic targets (2,5).
E3 ligases can be classified into four major families, including
RING, HECT, RBR and CRL complexes (6-8).
HECT-type E3 ligases possess a unique C-terminal HECT domain that
mediates ubiquitin transfer via a cysteine-dependent thioester
intermediate, conferring broader substrate selectivity (9,10). Consequently, HECT-type E3s are
likely to participate in ubiquitination across a wide spectrum of
diseases. Several studies have highlighted the critical functions
of HECT-type E3 ubiquitin ligases in regulating the molecular
networks underlying disease pathogenesis and progression (6,11,12).
By regulating protein ubiquitination, HECT-type E3
ligases play pivotal roles in protein stability, signal
transduction and quality control within the protein synthesis
pathway. These regulatory functions include the following: i)
direct regulation of structural/functional protein synthesis,
whereby certain HECT family members directly bind and ubiquitinate
target proteins that affect liver function (for example, SMURF2
promotes the degradation of TGF-β receptor I via ubiquitination,
thereby inhibiting the TGF-β/Smad signaling pathway and influencing
fibrosis and tumor progression) (13,14); ii) regulation of ribosomal
protein homeostasis and synthesis efficiency, whereby HECT-type E3s
indirectly modulate protein synthesis by maintaining ribosomal
protein stability (for example, HUWE1 ubiquitinates the ribosomal
protein RPL7 as part of the ribosomal quality control program,
impacting ribosome pool integrity and consequently affecting
protein translation) (15); and
iii) synthetic reprogramming under stress conditions, whereby,
under pathological conditions, such as endoplasmic reticulum stress
(ERS), the unfolded protein response promotes the degradation of
misfolded proteins: HECT-type E3s are involved in this adaptive
regulation of protein synthesis, with examples including the
interaction of E6AP with HSP70 and the roles of SMURF1 with IRAK2
and WFS1 in these processes (16-18). The liver, as a central metabolic
organ, synthesizes key functional proteins such as albumin,
coagulation factors (for example, fibrinogen, Factors II, VII, IX,
X), and apolipoproteins, all of which are essential for substance
metabolism, bile secretion, coagulation and immune regulation.
These functions rely on the precise regulation of protein
homeostasis.
HECT-type E3 ubiquitin ligases are key regulators
involved in multiple hepatic functions and disease processes, where
they are involved in mediating the homeostatic balance between
protein synthesis and degradation. In the field of liver diseases,
although the mechanisms of an increasing number of HECT-type E3
ligases are being elucidated, only those associated with lipid
droplet formation have been systematically summarized to date
(19). Through comprehensive
literature review, it was identified that HECT-type E3 ubiquitin
ligases exhibit a remarkable 'dual role' across the spectrum of
liver pathologies, wherein identical enzymes can exert
diametrically opposite functions depending on the disease stage,
cellular context, and prevailing signaling environment. This
functional versatility arises from several interconnected
mechanisms: Substrate selection shifts as the disease progresses,
with distinct target proteins becoming available or prioritized at
different pathological phases; the cellular context dictates both
the expression of potential substrates and the availability of
accessory proteins that direct ligase activity; and dynamic
signaling cues, including post-translational modifications,
metabolic pressures, and fine-tuned ligase-substrate interactions.
Consequently, the same ligase may suppress pathological initiation,
promote disease progression, or exert protective effects in one
cell type while driving pathogenesis in another. This
context-dependent functional duality reflects the complexity and
spatiotemporal specificity of HECT family regulatory networks.
Therefore, in the present review, the current knowledge of the
roles of HECT-type E3 ubiquitin ligases in liver pathology was
summarized, with particular emphasis on how identical enzymes
orchestrate opposing effects across different disease stages,
cellular contexts, and signaling environments. This perspective
provides crucial insights into liver disease pathogenesis and
reveals both challenges and opportunities for therapeutic
intervention.
Structure and classification of HECT-type E3
ubiquitin ligases
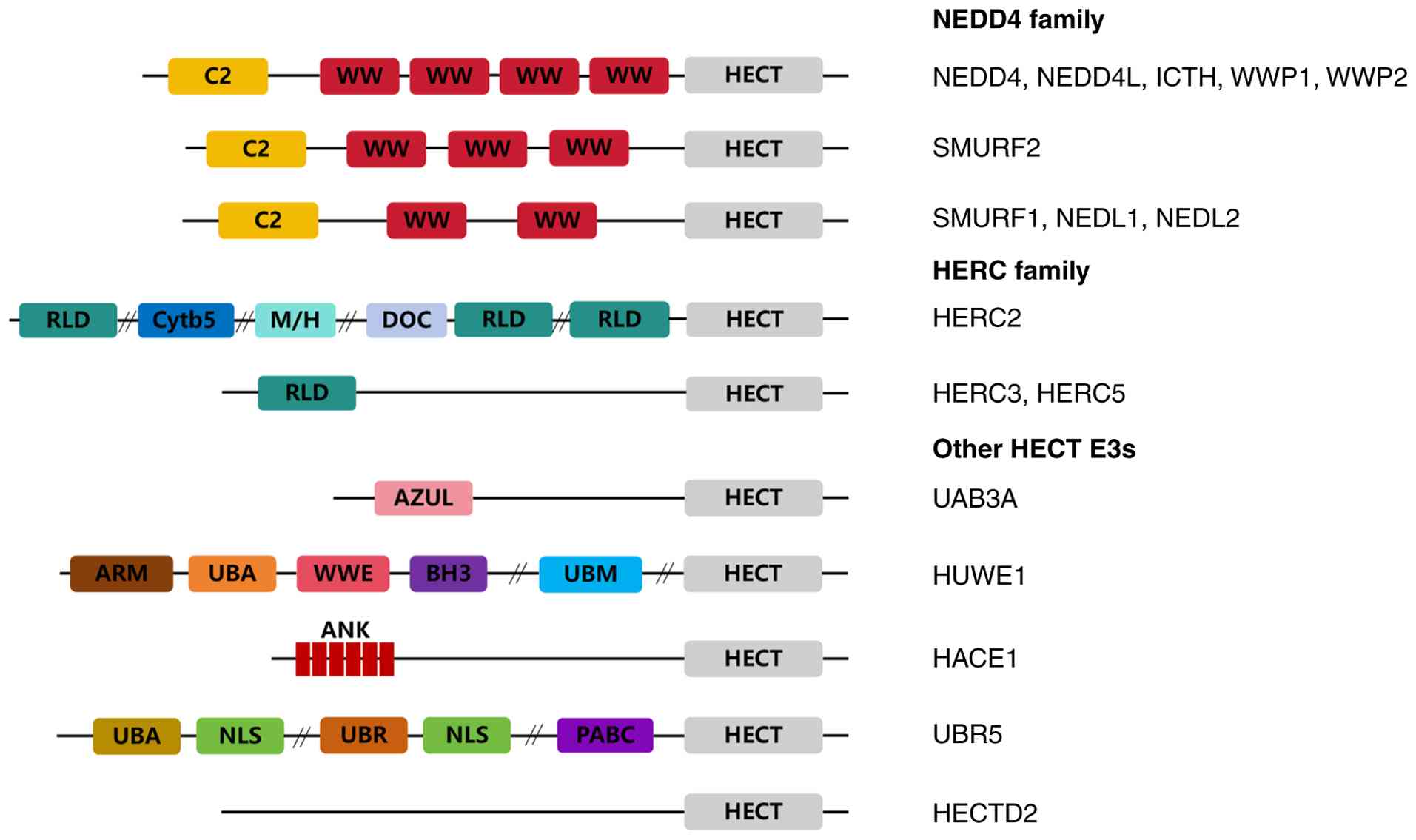
The HECT-type E3 family comprises 28 members,
characterized by a core HECT domain consisting of an N-lobe that
binds to the E2 enzyme and a C-lobe containing the catalytic
cysteine (Cys) residue (9,20,21). The E2 transfers ubiquitin to the
catalytic cysteine within the C-lobe, forming a thioester
intermediate; subsequent conformational changes facilitate the
transfer of ubiquitin to lysine residues or non-canonical sites on
the substrate (22,23). Accessory domains (for example,
WW, C2 and RCC1) are responsible for substrate recognition and
subcellular localization, leading to the classification of
HECT-type E3s into NEDD4, HERC and other subfamilies (Fig. 1) (24,25).
NEDD4 subfamily
The NEDD4 subfamily, also referred to as
C2-WW-HECT-type E3s, typically contains an N-terminal C2 domain
(mediating Ca2+-dependent membrane targeting), two to
four WW domains (recognizing PPxY motifs or phosphorylated
sequences), and a C-terminal HECT domain (4,26,27). The C2 domain is a
Ca2+-dependent phospholipid-binding motif that mediates
membrane association and recognition of membrane-associated
substrates (4,26). WW domains mediate protein-protein
interactions by recognizing PPxY, LPxY, or related sequences, as
well as phosphorylated Ser/Thr-Pro motifs (4,27). Essentially, the C2 and WW domains
determine the specificity of target protein binding for NEDD4
family members. Known NEDD4 subfamily E3s include NEDD4, NEDD4L,
ITCH, SMURF1, SMURF2, WWP1, WWP2, HECW1 and HECW2 (28). The NEDD4 family represents the
most extensively studied group of HECT-type E3s, which regulate
processes such as plasma membrane protein stability and misfolded
protein degradation, and are widely implicated in the mechanisms of
liver diseases (29).
HERC subfamily
HERC subfamily members contain an N-terminal
RCC1-like domain (involved in regulating GTPases) and a C-terminal
HECT domain. They are subdivided into members such as HERC1-6 based
on variations in their intermediate domains. The conserved RCC1
domain mediates GTPase regulation, contributing to pleiotropic
roles in cellular homeostasis (30). Their diverse intermediate domains
(for example, zinc fingers, ISG15-binding domains) confer distinct
functions to individual members, including roles in DNA repair,
antiviral immunity and metabolic regulation, potentially exerting
protective or repair functions in pathological processes such as
liver injury and hepatitis (30,31).
Other HECT members
The other group of HECT members includes 13
HECT-type E3s (for example, HECTD1-4, E6AP/UBE3A, HUWE1, TRIP12,
UBR5, UBE3B, UBE3C, HACE1, G2E3 and AREL1), all of which possess a
C-terminal HECT domain but often contain N-terminal or intermediate
domains that are unstructured or lack defining features, precluding
their classification into the aforementioned subfamilies (32,33). These E3s have been reported to
participate in various disease processes by regulating energy
metabolism, ERS, oxidative stress and cell death (34,35).
Role of HECT-type E3 ubiquitin ligases in
metabolic dysfunction-associated steatotic liver disease
(MASLD)/metabolic dysfunction-associated steatohepatitis
(MASH)
MASLD, formerly known as non-alcoholic fatty liver
disease, is associated with systemic metabolic dysregulation and
has a global prevalence of up to 38%. The MASLD nomenclature
accurately reflects the pathophysiology and systemic metabolic
implications of this common liver condition (36-39). The core mechanisms of MASLD
involve multiple factors, including lipotoxicity, insulin
resistance, pro-inflammatory diets and gut dysbiosis, leading to
hepatic steatosis combined with cardiovascular risks associated
with metabolic abnormalities. These processes can drive hepatic
inflammation and the development of systemic low-grade
inflammation, with its inflammatory subtype termed MASH (36). Undoubtedly, the intrinsic
functions of HECT-type E3 ligases play significant roles in
regulating the molecular networks underlying these pathogenic
mechanisms.
Role of HECT-type E3 in mediating lipid
metabolism in MASLD/MASH
Dysregulation of lipid homeostasis is a central
pathological event in patients with MASLD. Several HECT-type E3
ligases directly modulate key transcriptional regulators and
enzymes within lipid metabolic pathways.
The roles of proteins from the NEDD4 subfamily in
the pathogenesis of MASLD/MASH have been extensively reported.
Research on NEDD4 within this subfamily in MASLD remains relatively
limited; current evidence indicates that it is upregulated under
conditions of elevated hepatic free fatty acids, where it regulates
high-density lipoprotein metabolism by ubiquitinating the scavenger
receptor SR-BI (40).
Conversely, a study in geese reported opposing findings, showing
increased NEDD4 expression in fatty liver that appeared to suppress
hepatic steatosis-related injury via ubiquitination of PTEN and
IGF-1R (41), suggesting
potential species-specific functional differences. NEDD4L has been
more extensively studied, revealing a dual role: Wherein reduced
AT-rich interactive domain 2 expression in MASLD livers leads to
NEDD4L transcriptional downregulation, impairing JAK2
ubiquitination and subsequent degradation, which results in
hyperactivation of the JAK-STAT pathway and ultimately exacerbates
hepatic steatosis (42),
indicating that the normal function of NEDD4L suppresses lipid
accumulation. However, the markedly elevated NEDD4L levels observed
in MASH serve to promote the K48-linked ubiquitination and
degradation of lysosomal-associated protein transmembrane 5
(LAPTM5), activating the cell division cycle 42 (CDC42)-MAPK axis
and paradoxically worsening lipid accumulation (43), while NEDD4L-mediated degradation
of TMBIM1 has also been confirmed to aggravate steatosis (44). The role of ITCH in lipid
metabolism is similarly complex. ITCH knockout mice fed a high-fat
diet exhibit reduced hepatic lipid accumulation and inhibited
atherosclerosis formation, suggesting that ITCH promotes lipid
accumulation, Previous studies indicated that ITCH knockout mice
fed a high-fat diet exhibit reduced hepatic lipid accumulation and
suppressed atherosclerosis formation. This is associated with ITCH
enhancing LDL uptake by mediating the ubiquitination of proteins
such as sirtuin 6 (SIRT 6), Sterol regulatory element binding
protein 2 (SREBP 2) and liver kinase B1, thereby influencing lipid
accumulation. Clinical samples similarly reveal a negative
correlation between human ITCH protein abundance and hepatic
steatosis severity (45-48). Additionally, ITCH binds to the
PPAY motif in Spartin through its WW domain; this interaction
recruits ITCH from the cytoplasm to the surface of lipid droplets,
leading to the ubiquitination of surface proteins. This process may
impair lipid droplet degradation in hepatocytes (49), thereby promoting or exacerbating
the onset and progression of MASLD. The regulatory mechanism of
SMURF1 in lipid metabolism is particularly unique. Although
considered a protective factor that is upregulated in human and
mouse MASLD, SMURF1 deficiency alleviates hepatic steatosis. This
occurs because SMURF1 interacts with SREBP-1c not to ubiquitinate
and degrade it, but to occupy the binding site for its primary E3
ligase, FBW7A, thereby inhibiting SREBP-1c ubiquitination and
degradation, which markedly exacerbates hepatic steatosis (50,51). However, another study observed
that SMURF1 knockout leads to spontaneous hepatic steatosis and
increased susceptibility to HFD-induced MASLD, associated with a
sharp increase in PPARγ expression. Research on alcoholic
steatohepatitis also suggests that SMURF1 deficiency restricts
lipid degradation, thereby exacerbating hepatic lipotoxicity
(52,53). These findings indicate that
SMURF1 exerts multiple regulatory effects on lipid metabolism,
though it is generally considered a protective E3 ligase in MASLD.
SMURF2 participates in lipid metabolism by regulating C/EBPβ
stability; under high-fat conditions, PRMT1 suppresses SMURF2
expression, thereby inhibiting the ubiquitination and degradation
of C/EBPβ, which promotes preadipocyte proliferation and enhances
lipid droplet formation induced by PPARγ pathway activation
(54). Furthermore, WWP1
knockout mitigates hepatic lipid accumulation by reducing AKT
phosphorylation levels (55).
A limited number of studies have reported that other
HECT-type E3 ligases outside the NEDD4 subfamily are associated
with lipid accumulation in MASLD/MASH. HUWE1, a prominent member of
the other HECT family members, promotes lipid accumulation through
multiple pathways: it directly binds and ubiquitinates PPARα for
degradation, suppressing the fatty acid metabolism-related
LXR/RXR/PPARα axis, a process modulated by PAQR3 and PAQR9 through
competitive binding and significantly influenced by MCT1 via
lactate metabolism (56-59); additionally, HUWE1 is linked to
mTORC1 regulation, mediating WIPI2 ubiquitination and degradation
(60), with both mechanisms
collectively exacerbating hepatic steatosis. Meanwhile, UBE3A
overexpression significantly exacerbates hepatic steatosis in MASLD
by mediating ubiquitination of PDHA1 and ACAT1, reprogramming
hepatic energy metabolism to promote cholesterol and triglyceride
accumulation in the liver (61).
These findings demonstrate that HECT-type E3 ubiquitin ligases
exert precise and complex regulatory effects on lipid accumulation
in MASLD by targeting multiple key lipid metabolism pathways,
including SREBP, PPAR, JAK-STAT and mTOR.
HECT-type E3 in mediating inflammation in
MASLD/MASH
The inflammatory response is a key driver of the
progression from MASLD to MASH, and HECT-type E3 ubiquitin ligases
play an important role in shaping the hepatic inflammatory
microenvironment by regulating various inflammation-related
signaling pathways. Members of the NEDD4 subfamily are involved in
the regulation of inflammation. NEDD4 is upregulated under
conditions of elevated hepatic free fatty acids and exacerbates
liver inflammation and promotes atherosclerosis by ubiquitinating
SR-BI and affecting high-density lipoprotein metabolism (40). NEDD4L exhibits significant dual
effects in inflammatory regulation: Downregulated NEDD4L expression
in MASH leads to impaired ubiquitination of thioredoxin interacting
protein (Txnip), consequently enhancing ERS and promoting
hepatocyte apoptosis (62);
however, another study found markedly elevated NEDD4L levels in
MASH that worsen inflammatory responses through LAPTM5 degradation
and activation of the CDC42-MAPK axis (43). The association of ITCH with
inflammation is equally complex. ITCH knockout mice fed a high-fat
diet exhibit suppressed macrophage polarization and show protection
against body weight gain and insulin resistance (45); however, further investigation
revealed that ITCH knockout leads to elevated branched-chain amino
acid (BCAA) levels, which promote inflammatory responses (48). This contradiction between
macroscopic protective effects and microscopic pro-inflammatory
effects highlights the complexity of ITCH within the global
metabolic-immune network. SMURF1 indirectly participates in
MASLD-related inflammation and fibrosis by regulating the TGF-β
pathway (50-52,63). Notably, SMURF1 and SMURF2 also
play a role in hepatic stellate cell (HSC) activation during liver
fibrosis by interacting with talin1 (TLN1) and mediating its
ubiquitination and degradation, thereby regulating the TLN1/FAK
signaling axis and subsequently influencing HSC activation status
and downstream inflammatory responses in models of hepatic
ischemia-reperfusion injury associated with liver transplantation
(64). SMURF2-mediated
ubiquitination of AXIN1 attenuates AXIN1-induced Smad7
phosphorylation, inhibiting TGF-β-mTORC1 pathway activation and
delaying MASLD-hepatocellular carcinoma (HCC) progression (63). TRIP12, an important member of the
other HECT family members, exerts pro-inflammatory effects
indirectly through the gut-liver axis: TRIP12 expression is
upregulated in the intestines of MASLD and MASH mice, where it
promotes K48-linked ubiquitination and degradation of nuclear
transcription factor EB, leading to intestinal barrier disruption,
increased permeability and worsened MASH severity (65). Additionally, in a proteomic
analysis of alcohol-induced hepatic steatosis, TRIP12 was
identified as the most significantly altered protein (66), suggesting that it may also
participate in local hepatic pathological processes. Although
direct studies of HERC subfamily members in MASLD/MASH are limited,
their RCC1-like domain-mediated GTPase regulation and known
functions in DNA repair, antiviral immunity and metabolic
regulation suggest that they may exert protective or reparative
functions in pathological processes such as liver injury and
hepatitis (30,31). While other HECT members,
including HECTD1-4, UBR5 and HACE1, have not been extensively
studied in MASLD/MASH, their roles in regulating energy metabolism,
ERS, oxidative stress, and cell death in other disease models
provide important clues for future exploration of these E3 ligases
in the context of MASLD inflammation (34,35).
Mechanistic synthesis
Synthesizing the aforementioned findings, HECT-type
E3 ubiquitin ligases exhibit a distinct dual role in the
pathogenesis of MASLD/MASH, wherein the same E3 ligase can both
promote and inhibit disease progression. This functional duality
reflects the complexity and spatiotemporal specificity of the
regulatory networks involving HECT family members. First, members
of the same NEDD4 subfamily target different substrates in distinct
cellular environments or disease stages, producing diametrically
opposite effects. NEDD4L serves as a paradigmatic example: In MASH,
NEDD4L downregulation impairs degradation of Txnip and JAK2,
activating ERS and the JAK-STAT pathway respectively, thereby
promoting apoptosis and steatosis (42,62); however, in advanced MASH,
upregulated NEDD4L recognizes the PY motif of LAPTM5 through its WW
domain, leading to the ubiquitination and degradation of LAPTM5,
and subsequently activates the CDC42-MAPK axis, exacerbating lipid
accumulation and inflammation (Fig.
2) (43). Such
substrate-switching mechanisms may be determined by cell type,
subcellular localization, post-translational modifications, or
interactions with accessory proteins, enabling NEDD4L to play both
disease-promoting and disease-suppressing roles at different
disease stages.
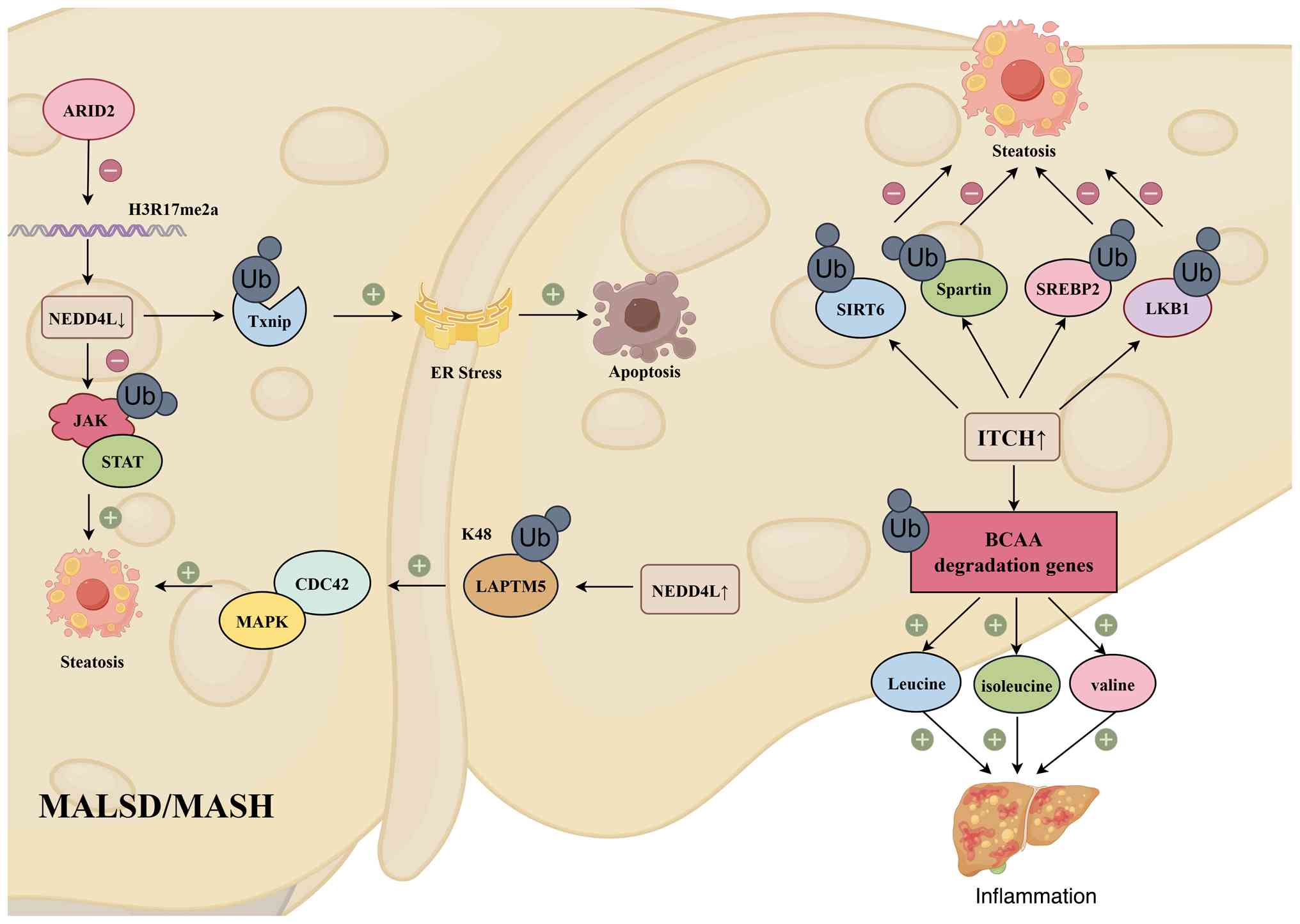 | Figure 2Role of HECT-type E3 ubiquitin
ligases involved in MASLD/MASH. NEDD4L and ITCH exhibit a classic
dual-role effect in the course of MASLD/MASH. NEDD4L may mediate
steatosis through specific pathways regardless of whether it is up-
or downregulated. Meanwhile, the inherent ITCH upregulation in
MASLD/MASH simultaneously exerts both steatosis-suppressing and
inflammation-promoting effects. ARID2, AT-rich interactive domain
2; TxNIP, thioredoxin interacting protein; LAPTM5,
lysosomal-associated protein transmembrane domain 5; CDC42, cell
division cycle 42 protein; SIRT 6, sirtuin 6; SREBP2, sterol
regulatory element-binding protein 2; LKB1, liver kinase B1; BCAA,
branched-chain amino acid. MASLD, metabolic dysfunction-associated
steatotic liver disease; MASH, metabolic dysfunction-associated
steatohepatitis. |
This E3 ligase can exert functions contrary to its
classical enzymatic activity through non-canonical mechanisms. The
regulatory mechanisms of SMURF1 provide multiple illustrations.
Although considered a protective E3 ligase, SMURF1 stabilizes
SREBP-1c by occupying the binding site for its primary E3 ligase
FBW7A, rather than through classical ubiquitin-mediated
degradation, a 'steric hindrance' mechanism that paradoxically
promotes steatosis (50,51); simultaneously, SMURF1 exerts
protective effects by suppressing PPARγ, while in HSCs, SMURF1
performs protective functions by inhibiting HSC activation and
inflammatory responses through ubiquitination and degradation of
TLN1. These findings indicate that the effect of SMURF1 ultimately
depends on the dynamic balance of different substrates in specific
cell types and under specific metabolic stresses (52,64).
ITCH also plays a 'dual role' in MASLD/MASH,
revealing the holistic and complex nature of HECT-type E3 ligase
function. This is demonstrated by the marked hepatoprotective
effect observed in systemic ITCH knockout mice (45), while human clinical samples show
a negative correlation between ITCH protein abundance and hepatic
steatosis severity (46).
Paradoxically, ITCH knockout-induced alterations in BCAA profiles
promote inflammation (Fig. 2)
(48). This suggests that ITCH
simultaneously suppresses lipid accumulation and exerts
pro-inflammatory effects within the MASLD/MASH environment, making
it difficult to determine whether ITCH possesses therapeutic
potential in MASLD/MASH.
These findings suggest that a single E3 ligase may
exert opposing effects across the disease spectrum. Specific E3s,
notably NEDD4, NEDD4L, and ITCH, engage diverse substrates across
different cell types, simultaneously influencing pro-disease and
protective outcomes. Their ultimate functional output is determined
by the integration of dominant microenvironmental cues, cellular
metabolic status and tissue-specific stress responses. This
pervasive duality indicates that HECT-type E3 ligases are not mere
linear promoters or suppressors of disease but function as dynamic,
environmentally responsive molecular hubs. Their activity is
precisely calibrated by a confluence of factors, including disease
stage, cellular origin, metabolic pressure and local tissue
microenvironment. In conclusion, HECT-type E3 ubiquitin ligases act
as complex regulators of MASLD/MASH, with functional duality
arising from substrate diversity, spatiotemporal expression,
non-canonical catalytic activities and trans-organ regulatory
networks. Defining a single ligase simply as protective or
pathogenic is incomplete.
Role of HECT-type E3 ubiquitin ligases in
liver fibrosis
Liver fibrosis is a wound-healing response to
chronic liver injury, characterized by excessive deposition of
extracellular matrix (ECM), which represents a common pathological
outcome of chronic liver diseases. The central events driving
fibrosis include HSC activation and disruption of ECM metabolism
triggered by inflammation and oxidative stress. Current research
has confirmed the involvement of HECT-type E3 ubiquitin ligases in
the pathogenesis of liver fibrosis (67). However, unlike MASLD/MASH, the
role of most HECT-type E3 ubiquitin ligases in liver fibrosis lacks
bidirectional effects, which may be attributed to the limited
number of studies conducted.
A carbon tetrachloride (CCl4)-induced
liver fibrosis model established by Song et al (68) revealed that NEDD4-2 in HSC
promoted the ubiquitination and degradation of the hepatoprotective
factor tyrosine kinase receptor B. This enhanced TGF-β-mediated
Smad2/3 phosphorylation, thereby exacerbating fibrotic injury
(68). Furthermore,
phosphorylated (p-) NEDD4L levels were increased in liver fibrosis,
and p-NEDD4L promoted HSC activation, contributing to fibrosis
aggravation (69). Thus,
NEDD4/NEDD4L appear to promote liver fibrosis, although further
research is needed to confirm these roles and elucidate the
underlying molecular mechanisms. In contrast to NEDD4/NEDD4L, WWP2,
which belongs to the same NEDD4 subfamily, has been demonstrated to
be a distinct antifibrotic gene. Research on WWP2 emerged from a
pharmacological study that demonstrated that costunolide inhibits
cholestatic and CCl4-induced fibrosis. This effect is
based on costunolide promoting the interaction between WWP2 and
Notch3, leading to UPS-mediated degradation of Notch3 (70). A few studies exist regarding the
HERC subfamily's role in fibrosis. Because of its unique
ISG15-binding domain, HERC5 mediates ISGylation, a ubiquitin-like
modification. HERC5 was shown to mediate P53 ISGylation, inhibit
HSC activation, and alleviate fibrosis, a process regulated by
upstream miR-145 and zinc-finger proteins (71). Another study in a murine fibrosis
model using single-cell RNA-sequencing revealed that HERC6 was
predominantly enriched in inflammatory neutrophils and promoted
liver injury. This effect was suppressed by the CCR2/CCR5 dual
antagonist cenicriviroc (72),
suggesting that CCR family proteins act as upstream regulators of
HERC6 and provide initial insights into its cell-specific
localization. Among other HECT members, Kim et al (73) reported that E6AP expression is
upregulated in fibrosis, where it ubiquitinates MAPK pathway
proteins, inhibiting HSC activation and exerting an antifibrotic
effect. It is worth noting that the HECT-type E3 ubiquitin ligase
identified in the aforementioned study has not been demonstrated to
simultaneously promote and inhibit the progression of liver
fibrosis during its disease course. Therefore, it may be a
therapeutic target for the treatment of liver fibrosis.
Unlike the aforementioned genes, the SMURF proteins
are distinguished by their specificity in regulating Smad family
proteins. Numerous studies have focused on the role of SMURF1 and
SMURF2 in liver fibrosis. One study indicated that SMURF1
ubiquitinates Smad1/5 (74), but
its relevance in liver fibrosis has not been explored. However,
SMURF2, a member of the NEDD4 family, simultaneously exhibits both
a promoting effect on liver fibrosis and resistance to fibrosis.
Two separate studies reported that SMURF2 binds to and
ubiquitinates Smad7, promoting Smad2/3 phosphorylation and
ultimately worsening fibrosis (75,76). However, Cai et al
(77) found that SMURF2 was
upregulated in both cholestatic and CCl4-induced
cirrhosis models and exerted antifibrotic effects through both
ubiquitination-dependent and miRNA-mediated mechanisms. Although
the role of SMURF2 in fibrosis remains controversial, two
pharmacological studies targeting antifibrotic therapy have
identified potential SMURF2 activators. Jiang et al
(78) synthesized an isothiazole
derivative, J-1155, which exhibited high selectivity for SMURF2,
upregulating its expression and inhibiting fibrosis. A similar
effect was observed with curcumin in treating fibrosis (79). These studies provide valuable
agonist tools for future SMURF2 research. In summary, current
research on HECT-type E3s in fibrosis primarily focuses on SMURF2.
However, whether this upregulation during fibrosis is protective or
detrimental remains debatable and warrants further
investigation.
Mechanistic synthesis
The involvement of HECT-type E3 ligases in liver
fibrosis demonstrates their functional diversity and
context-dependent activities. First, distinct family members can
exert opposing effects within the same pathological process. It
remains challenging to develop targeted therapies by focusing on a
specific domain of NEDD4 or a particular HERC subfamily member. For
instance, SMURF1/2 exhibits potentially opposing effects on
fibrosis compared with NEDD4/4L and WWP2. HERC5/HERC6 also show
opposing actions. Second, a single E3 ligase can exhibit functional
contradictions, as exemplified by SMURF2, which has been reported
to be both profibrotic and antifibrotic, depending on the
experimental model and mechanistic context. Collectively, these
factors contribute to failure of domain-targeted therapies.
Role of HECT-type E3 ubiquitin ligases in
viral hepatitis
Viral hepatitis poses a significant public health
threat and is a leading cause of mortality worldwide (80). The five main types are hepatitis
A (HAV), B (HBV), C (HCV), D (HDV) and E (HEV) viruses. HECT-type
E3 ligases play dual roles in viral hepatitis by regulating both
host antiviral immunity and the viral life cycle (81). However, these studies have
primarily focused on the more common HBV and HCV, with only a few
studies on HAV. Thus, strategies targeting specific HECT-type E3s
may offer novel avenues for antiviral therapy. The following
sections detail these cutting-edge findings, organized by virus
type.
HECT-type E3 ubiquitin ligases in
HBV
Multiple HECT-type E3 ligases have been implicated
in HBV infection and exhibit complex and often opposing functions
via distinct mechanisms. Several HECT-type E3 ligases promote HBV
replication and release through various strategies. ITCH expression
is decreased in HBV-positive human cells, leading to suppressed
ubiquitination of Notch1 and subsequent aberrant activation of the
Notch pathway, which promotes transcription of covalently closed
circular DNA (cccDNA) and enhances HBV replication (82). HERC5 is hijacked by the HBV X
protein (HBx), which directly binds HERC5 for ISGylation
modification, conferring enhanced replication capability and
interferon-α resistance to HBV (83). UBR5 mediates K29-linked
ubiquitination of the HBV core protein, which may be involved in
regulating HBV transcription, RNA encapsidation, reverse
transcription and viral release, although the functional
consequences require further elucidation (84).
Conversely, other HECT-type E3 proteins exert
protective effects by targeting viral proteins for degradation.
NEDD4 directly induces proteasomal degradation of HBx via
K48-linked ubiquitination, suggesting an antitumor role in
HBV-associated HCC (85).
However, NEDD4 exhibits functional duality as it also interacts
with and ubiquitinates γ2-Adaptin, promoting HBV nucleocapsid
assembly and disrupting the multivesicular body (MVB) pathway, with
NEDD4 knockdown significantly reducing HBV release (86-88). Thus, NEDD4 simultaneously
facilitates viral production while suppressing viral oncoprotein
stability. E6AP forms a p53/E6AP/HBx ternary complex that leads to
HBx ubiquitination and degradation, and all-trans retinoic acid
activates this pathway to suppress HBV replication (89,90).
HECT-type E3 ubiquitin ligases in
HCV
HCV infection involves multiple HECT-type E3 ligases
acting via diverse mechanisms that collectively influence viral
replication, particle release and disease progression. Several
HECT-type E3s promote HCV propagation. ITCH plays a proviral role
where increased reactive oxygen species (ROS) induce ITCH
phosphorylation, leading to ubiquitination of the ATPase VPS4A, a
protein necessary for MVB formation, thereby promoting HCV particle
release (91). HERC5's role
remains controversial, with some studies reporting that HERC5
specifically promotes NS5A ISGylation, thereby enhancing HCV
replication (92), while others
demonstrate that the inhibitory effect of ISG15 on HCV RNA
replication does not require HERC5 binding (93).
By contrast, other HECT-type E3s exert anti-HCV
effects. E6AP represents the most extensively studied protective
factor, initially characterized as promoting HCV pathogenesis
through nonstructural protein 5B (NS5B)-recruited ubiquitination of
retinoblastoma tumor suppressor protein (pRb) leading to
chromosomal instability (94),
but subsequently established as an anti-HCV replication factor
through direct binding and ubiquitination of the viral nucleocapsid
core protein (95,96). The HCV core protein suppresses
E6AP expression via DNA promoter methylation to evade
ubiquitin-proteasome system-mediated degradation, while p53
upregulation enhances E6AP expression and recruits E6AP to form a
ternary complex with the HCV core, promoting its ubiquitination and
degradation (97-99). All-trans retinoic acid activates
E6AP by inhibiting its methylation, thereby suppressing HCV
replication (100).
SMURF2 has dual functions in HCV infection. It
enhances ubiquitination and degradation of the viral protease
NS3-4A, inhibiting TGF-β-induced Smad2/3 phosphorylation and
alleviating HCV-associated fibrosis (101). However, the SMURF2/NS3-4A
complex also interacts with quinolinate phosphoribosyl-transferase
(QPRT), leading to QPRT degradation, which lowers the
NAD+/NADH ratio, potentially shifting lipid metabolism
to favor HCV replication (102).
HECT-type E3 ubiquitin ligases in
HAV
Current evidence for HECT-type E3 involvement in HAV
infection primarily focuses on a single mechanism. ITCH facilitates
HAV release, and its catalytic activity is essential for the
efficient release of quasi-enveloped virions (eHAV). During acute
infection, eHAV is released into the blood via the MVB pathway,
which involves an interaction between the pX domain and the ESCRT
complex and the HECT domain of ITCH, resulting in inhibition of
viral activity and release (103).
Mechanistic synthesis
Synthesizing the findings across different hepatitis
viruses reveals that HECT-type E3 ubiquitin ligases exhibit a
striking dual role in viral hepatitis, wherein the same E3 ligase
can both promote and inhibit viral replication, host pathogenesis,
or antiviral responses, depending on the viral context, cellular
environment, and target substrate specificity. This functional
duality profoundly reflects the evolutionary arms race between host
antiviral defense and viral evasion strategies.
The same HECT-type E3 ligases can exert opposing
effects on the same virus by targeting different viral or host
proteins. NEDD4 in HBV infection provides a paradigmatic example:
While NEDD4 promotes HBV nucleocapsid assembly and viral release
through γ2-Adaptin ubiquitination and HBc modification (86-88), it simultaneously induces
proteasomal degradation of HBx via K48-linked ubiquitination,
potentially suppressing HBV-associated hepatocarcinogenesis
(85). This substrate-specific
duality positions NEDD4 as both a facilitator of viral production
and a suppressor of viral oncoprotein stability, suggesting that
its net effect may depend on the relative abundance and
accessibility of different substrates during different stages of
infection.
The same E3 ligase plays different roles in distinct
hepatitis viruses, reflecting virus-specific coevolutionary
adaptations. ITCH exemplifies this virus-dependent functional
diversity: In HAV infection, ITCH facilitates eHAV release through
ESCRT pathway interactions (103); in HCV infection, ROS-induced
ITCH phosphorylation promotes VPS4A ubiquitination and viral
particle release (91); whereas
in HBV infection, decreased ITCH expression leads to Notch1
stabilization and enhanced cccDNA transcription (82). This virus-specific engagement
suggests that therapeutic targeting of ITCH would require careful
consideration of viral context.
E6AP demonstrates how the same E3 ligase can be
reinterpreted from pathogenic to protective as mechanistic
understanding deepens. Initially characterized as a pro-HCV factor
through NS5B-mediated pRb ubiquitination, promoting chromosomal
instability (94), E6AP was
subsequently identified as an anti-HCV factor through direct HCV
core ubiquitination and degradation (95,96). The discovery of the
p53/E6AP/viral core axis operating in both HCV and HBV infections
unified these observations, establishing E6AP as a host restriction
factor whose activity is subverted by viral proteins through
epigenetic suppression (97-99).
HERC5 exemplifies the controversy arising from the
context-dependent mechanisms. Conflicting studies on HERC5's role
in HCV replication and whether it inhibits or promotes viral RNA
synthesis through NS5A ISGylation (92,93) may reflect differences in
experimental systems, viral strains, or the balance between the
direct antiviral effects and proviral consequences of
ISGylation.
In conclusion, HECT-type E3 ubiquitin ligases
function as critical nodes at the host-virus interface during viral
hepatitis, with their dual roles arising from substrate diversity,
virus-specific adaptations and the complex interplay between viral
life cycle requirements and host antiviral defenses. However, owing
to the presence of the aforementioned factors, therapeutic
approaches that simply target a specific enzyme or HECT domain
cannot be developed. This limitation has constrained the
advancement of viral hepatitis treatments.
Role of HECT-type E3 ubiquitin ligases in
HCC
HCC is a highly prevalent malignancy with a high
mortality rate worldwide. Its development stems from the
accumulation of genetic and epigenetic alterations caused by
chronic liver injury, which involves the dysregulation of complex
signaling pathways. The diverse roles of HECT-type E3 ligases in
HCC have been partially summarized. For instance, NEDD4 and SMURF1
can promote tumor proliferation, invasion and metastasis by
degrading tumor suppressors (for example, PTEN) or stabilizing
oncoproteins (for example, c-Myc) via ubiquitination. Conversely,
specific HECT enzymes (for example, HACE1) exert tumor-suppressive
functions, such as inhibition of the Rac1 signaling pathway, and
their loss or mutation is associated with HCC progression (104). Furthermore, the HECT family
participates in regulating DNA damage repair, inflammatory
microenvironment and metabolic reprogramming, potentially driving
HCC malignant transformation indirectly by affecting genomic
stability or immune evasion (Fig.
3) (105). Based on these
findings, this section provides a detailed and updated description
of the dual roles of HECT-type E3 ligases in HCC pathogenesis.
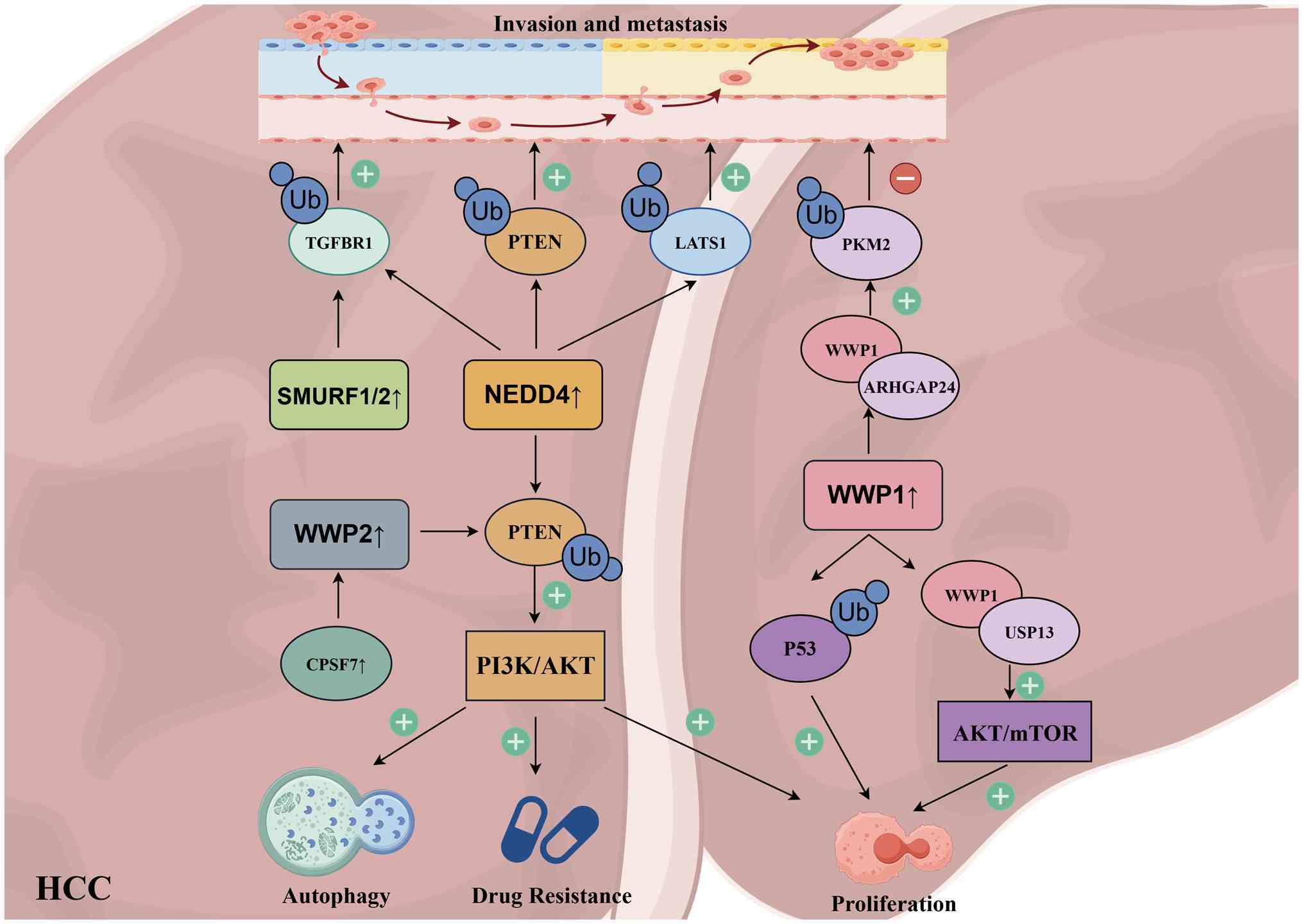 | Figure 3Role of HECT-type E3 ubiquitin
ligases involved in HCC. In HCC, PTEN, as a classic tumor
suppressor gene, can undergo ubiquitination and degradation by
multiple HECT-type E3 ubiquitin ligases. Among these, NEDD4 holds
the greatest translational potential as a HECT-type E3 ubiquitin
ligase target due to its ability to simultaneously activate
multiple signaling pathways and exert oncogenic effects. WWP1,
however, exhibits a classic 'dual role' effect in HCC. It can both
suppress tumor growth and potentially promote tumor invasion and
metastasis. HCC, hepatocellular carcinoma; TGFBR1, TGF-β type I
receptor; LATS1, large tumor suppressor kinase 1; PKM2, pyruvate
kinase M2; ARHGAP24, RhoGAP 24; CPSF7, cleavage and polyadenylation
specificity factor 7; USP13, ubiquitin specific peptidase 13. |
HECT-type E3 ubiquitin ligases in
proliferation
In studies investigating the function of HECT-type
E3 ligases in HCC, multiple studies have confirmed NEDD4 as an
oncogene that mediates the ubiquitination and degradation of the
classical tumor suppressor PTEN, accelerating HCC progression
(106-110). Two clinical studies found that
patients with HCC and high NEDD4 expression had shorter survival
times, and subsequent experimental validation indicated that NEDD4
upregulation suppresses PTEN expression, thereby promoting PI3K-AKT
phosphorylation (106,107). This finding was corroborated by
Zhou et al (108), who
also suggested that the NEDD4/PTEN/PI3K/AKT axis is modulated by
the interaction between craniofacial development protein 1 and
NEDD4 (108). Beyond PTEN, some
research shows that NEDD4 also promotes HCC invasion and metastasis
by ubiquitinating large tumor suppressor kinase 1 (LATS1) in the
HIPPO pathway, and TGF-β type I receptor (TGFBR1) in the TGF-β
pathway (109,110). Therefore, NEDD4 is currently
the most promising HECT-type E3 enzyme to serve as a novel
therapeutic target for HCC.
In studies of other HECT-type E3 ligases, HECTD1/2
appear to promote HCC proliferation, although they have been
reported to do so through distinct mechanisms. HECTD1 targets
growth factor receptor-bound protein 2 (GRB2), and its degradation
inhibits MAPK pathway-mediated angiogenesis, although this
ubiquitination is competitively inhibited by elevated Golgi protein
73 levels in HCC (111). HECTD2
is highly expressed in HCC and promotes proliferation through KEAP1
ubiquitination and subsequent antioxidant response activation
(112). Liu et al
(113) suggested that E6AP
interacts with G protein-coupled receptor 26 (GPR26) through its
HECT domain. By altering the conformation of this domain, E6AP
induces ubiquitination at the K286 site of GPR26, thereby promoting
tumor proliferation (113).
UBR5 interacts with the tumor suppressor esophageal cancer-related
gene 4 (ECRG4). ECRG4 overexpression conversely downregulates UBR5
expression, leading to increased p21 levels and anti-proliferative
effects (114). Wang et
al (115) found that
targeting and inhibiting UBR5 with Echinacoside promoted apoptosis
and inhibited glycolysis in HepG2 cells (115). Leboeuf et al (116) demonstrated that simultaneous
small interfering RNA silencing of UBR1, UBR2, UBR4 and UBR5
inhibited the Arg/N-degron pathway, reducing HCC cell proliferation
while increasing spontaneous apoptosis (116). HACE1 was initially found to be
downregulated in HCC tissues, and patients with low HACE1
expression had lower overall survival rates (117). Mechanistic studies revealed
that HACE1 mediates the ubiquitination of SREBP cleavage-activating
protein (SCAP), thereby inhibiting SREBP2-mediated cholesterol
biosynthesis, which is crucial for membrane synthesis during rapid
proliferation. However, this process is antagonized by the
ZDHHC3-mediated S-acylation of SCAP at Cys264 (118). Additionally, LINC00161 recruits
EZH2, leading to HACE1 methylation and promotion of HCC progression
(119). Therefore, HACE1 is
currently recognized as a tumor suppressor that inhibits HCC
proliferation. These HECT-type E3 ligases have been extensively
studied for their roles in regulating HCC proliferation; however,
no significant bidirectional effects have been observed.
Consequently, they represent promising novel therapeutic targets
for HCC diagnosis and treatment, although they currently lack the
level of recognition attained by NEDD4.
The HECT-type E3 ligases described below have
demonstrated dual functionality in current research, representing a
significantly controversial aspect of HCC proliferation studies,
with findings potentially exhibiting both promoting and inhibitory
effects. Focus was first addressed on studies of other HECT-type E3
ubiquitin ligases within the NEDD4 subfamily, excluding NEDD4
itself, that regulates HCC proliferation. The role of NEDD4L in HCC
proliferation remains controversial. Lee et al (120) demonstrated that NEDD4L
expression is significantly upregulated upon activation of the
canonical cancer-related Wnt/β-catenin pathway. Conversely, Zhao
et al (121) reported
that downregulation of NEDD4L in HCC tissues correlates with poor
prognosis. The authors proposed that NEDD4L binds to ERK1/2 not to
ubiquitinate it but rather to promote its phosphorylation and
activation, thereby inducing apoptosis and inhibiting HCC cell
proliferation (121). Another
study also characterized NEDD4L as a tumor suppressor, attributing
this role to its ability to ubiquitinate cyclin-dependent kinase 5
(CDK5). The same study indicated that ubiquitin-specific peptidase
1 (USP1) acts as a deubiquitinating enzyme, counteracting NEDD4L's
effect on CDK5 (122). Research
on ITCH in HCC proliferation is relatively limited but suggests
tumor-suppressive functions. Ge et al (123) found that ITCH ubiquitinates
transforming growth factor β-activated kinase 1 (TAK1). TAK1
degradation leads to inhibition of the MAPK and NF-κB pathways,
which are critical for cell proliferation and survival. The
glycosyltransferase Rab-like GTPase activating protein and GRAM
domain-containing protein 4 (GRAMD4) recruits ITCH, thereby
promoting TAK1 degradation and inhibiting HCC progression (123). Additionally, ITCH has been
linked to programmed cell death ligand 1 (PD-L1) regulation through
GLI1 ubiquitination, although this primarily affects immune evasion
rather than direct proliferation control (124). Conflicting reports exist
regarding the role of WWP1 in HCC proliferation. Cheng et al
(125) identified WWP1 as an
oncogenic factor and found that its expression was elevated in
tissues of patients with HCC and positively correlated with
alpha-fetoprotein levels. WWP1 knockout led to HCC cell apoptosis
and inhibited proliferation via the upregulation of caspase-3 and
P53, although the involvement of ubiquitination was not clarified
(125). By contrast, another
study reported a tumor-suppressive role for WWP1, associated with
its binding to and degradation of pyruvate kinase M2 (PKM2),
inhibiting HCC cell proliferation. However, this interaction
requires RhoGAP 24 (ARHGAP24) as a scaffolding protein (126). Furthermore, in HCC, WWP1 can be
influenced by the deubiquitinating enzyme USP13, leading to
activation of AKT and mTOR phosphorylation and accelerated HCC
proliferation (127). Similar
to NEDD4, WWP2 is also a key regulator of PTEN, cleavage and
polyadenylation specificity factor 7 is upregulated, inducing the
upregulation of WWP2 and promoting the ubiquitination and
degradation of PTEN (128).
However, the current body of research on WWP2 remains insufficient,
and its potential as a therapeutic target warrants further
investigation.
Research on the role of the HERC subfamily in HCC
proliferation, although limited, suggests pro-tumorigenic
functions. HERC2 is significantly upregulated in primary
hepatocytes under inflammatory stimulation, promoting STAT3
activation during the inflammation-to-cancer transition and
enhancing cancer stemness (129). HERC4 is elevated in HCC
tissues, enhancing cell proliferation through the suppression of
Salvador 1 (SAV1) (130,131).
HERC5 is also upregulated in HCC, promoting cancer cell
proliferation and influencing apoptosis. Wang et al
(132) synthesized a
disubstituted quinazoline derivative, HZ-6d, which binds to HERC5,
inhibits its expression, and activates P53, effectively delaying
HCC growth in vivo (132). Interestingly, integrated omics
and clinical data analyses identified HERC5 as a prognostic marker
for post-liver transplant recurrence in patients with HCC, with low
expression associated with poor prognosis because of immune escape
mechanisms (133).
HUWE1, TRIP12 and UBE3B/C regulate HCC
proliferation. HUWE1 mediates p53 degradation and participates in
the regulation of HCC cell apoptosis, which is modulated by the
deubiquitinating enzyme USP7 (134). Similar to HUWE1, TRIP12 is also
regulated by USP7-mediated de-ubiquitination. Overexpression of
USP7 blocks TRIP12-mediated ubiquitination and degradation of
p14ARF, promoting HCC proliferation (135). A large-scale population
sequencing study found that UBE3C overexpression in HCC tissues was
significantly associated with reduced survival, and that UBE3C
promoted HCC progression by regulating epithelial-mesenchymal
transition (EMT), which indirectly affects proliferation (136). Tao et al (137) also described UBE3C upregulation
in HCC and its regulation by the upstream miR-542-3p. Although
these genes have also been shown to potentially regulate HCC
proliferation, the current body of research remains insufficient.
Therefore, it remains unclear whether they exhibit bidirectional
effects.
HECT-type E3 ubiquitin ligases in
invasion and metastasis
HECT-type E3 ubiquitin ligases play critical roles
in HCC invasion and metastasis by regulating cell migration,
adhesion, EMT and ECM remodeling. NEDD4 promotes HCC cell invasion
and metastasis via multiple mechanisms. Beyond its role in
proliferation, research shows that NEDD4 ubiquitinates LATS1 in the
HIPPO pathway and TGFBR1 in the TGF-β pathway, thereby relieving
the tumor-suppressive effects of these pathways and enhancing the
migratory and invasive capacity of HCC cells (109,110). The HIPPO pathway is a key
regulator of organ size and metastasis, and its inactivation by
NEDD4-mediated LATS1 degradation promotes nuclear translocation of
YAP/TAZ, driving EMT and metastasis. Similarly, TGFBR1
ubiquitination by NEDD4 modulates TGF-β signaling, which has dual
roles in HCC but often promotes metastasis in advanced stages.
SMURF1 and SMURF2 predominantly exert inhibitory
effects on invasion and metastasis in HCC, particularly through
negative regulation of the TGF-β signaling pathway, which is a
major driver of EMT and metastasis. Liu et al (138) demonstrated that in HCC,
SMURF1/2 binds to TGFBR1, inducing its ubiquitination and
degradation, which suppresses the TGF-β pathway and its
pro-metastatic effects. TGF-β Receptor associated protein 1
(TGFBRAP1) competes with SMURF1/2 for the TGFBR1 binding site,
thereby enhancing cancer stemness and metastatic potential.
Similarly, Niemann-Pick disease, type C1 protein exerts an effect
analogous to TGFBRAP1 (138,139). SMURF1 can also suppress TGF-β
pathway activation by binding to and ubiquitinating TGFBR2
(140). Beyond TGF-β signaling,
SMURF1 ubiquitinates ultra-violet radiation resistance associated,
which inhibits HCC proliferation and metastasis by promoting
autophagosome maturation and lysosomal degradation of EGFR, a key
driver of metastatic signaling (141). Signal transducer and activator
of transcription 1, which can have tumor-suppressive functions in
HCC, is also targeted for degradation by SMURF1, but this process
depends on the presence of SIRT 7. In addition, hsa_circ_0000235
participates in SMURF1's regulation of HSP60 in a manner similar to
that of SIRT 7 (142,143). These findings collectively
support the metastasis-suppressive role of SMURF1 in HCC.
Therefore, the aforementioned studies conclusively demonstrate the
roles of NEDD4 and SMURF1/2 in the regulation of HCC invasion and
metastasis. Notably, NEDD4 has emerged as a well-defined target
that promotes proliferation, invasion and metastasis and has
significant translational potential.
The functions of HECT-type E3 enzymes remain
controversial because of limited studies and their bidirectional
activity. ITCH indirectly inhibits HCC invasion and metastasis
through TAK1 ubiquitination and degradation, which leads to
inhibition of the MAPK and NF-κB pathways, both of which can
promote EMT and metastasis. GRAMD4 recruits ITCH to promote this
process, thereby suppressing HCC progression (123). Additionally, the ITCH/GLI1 axis
may indirectly influence HCC metastatic potential by regulating
PD-L1 expression and subsequent immune evasion; however, its direct
effects on cell migration remain to be explored (124). The role of WWP1 in HCC invasion
and metastasis is unclear. One study reported that WWP1, through
binding to and degradation of PKM2, inhibits HCC cell migration and
invasion, requiring ARHGAP24 as a scaffolding protein (126). However, the oncogenic functions
of WWP1 described in other studies suggest context-dependent
effects on metastasis, which require further investigation. HERC4,
through suppression of SAV1, may influence HCC metastasis, as SAV1
is a core component of the HIPPO pathway that regulates YAP/TAZ
activity and subsequent EMT (130,131). UBE3C overexpression promotes
HCC progression by regulating EMT in HCC cells, directly
implicating it in metastatic spread (136). UBR5 influences HCC cell
migration through the regulation of the Arg/N-degron pathway, as
simultaneous silencing of UBR1, UBR2, UBR4 and UBR5 reduces HCC
cell migration (116).
HECT-type E3 ubiquitin ligases in drug
resistance
HECT-type E3 ubiquitin ligases significantly
contribute to therapeutic resistance in HCC by affecting the
responses to multiple classes of anticancer drugs, including
targeted therapies and chemotherapy. NEDD4 plays an important role
in drug resistance by regulating the PTEN/PI3K/AKT axis. Inhibition
of NEDD4/PTEN axis-mediated autophagy can enhance the sensitivity
of HCC animal models and cell lines to sorafenib, a first-line
multi-kinase inhibitor used in advanced HCC (107,108,144). Autophagy is a cellular survival
mechanism that promotes resistance to therapy, and NEDD4-mediated
PTEN degradation activates AKT, which in turn promotes autophagy
and survival under stress conditions. In the context of the FOLFOX
(5-fluorouracil, oxaliplatin and leucovorin) chemotherapy regimen,
glycolysis-enhanced histone lactylation forms an H3K14la/NEDD4/PTEN
axis regulating multidrug resistance in HCC (145). This epigenetic mechanism links
metabolic reprogramming to drug resistance through NEDD4
upregulation. Notably, this study indicated that NEDD4L is not
involved in PTEN regulation in this context (146).
The following enzymes may be underrepresented
because of the limited number of studies on drug resistance.
SMURF1/2 may indirectly influence drug resistance through their
regulation of TGF-β signaling. TGFBRAP1 competes with SMURF1/2 for
TGFBR1 binding, thereby enhancing cancer stemness and HCC
resistance to regorafenib, another multi-kinase inhibitor used in
second-line treatment for HCC (138). HECTD2 is highly expressed in
lenvatinib-resistant HCC cell lines, patient tissues,
patient-derived organoids and xenografts. Lenvatinib is another
multi-kinase inhibitor used as a first-line treatment for advanced
HCC. HECTD2 promotes ubiquitination of KEAP1, a negative regulator
of antioxidant response. KEAP1 degradation leads to the activation
of NRF2 and the subsequent antioxidant response, which reduces
oxidative stress-induced cell death and decreases sensitivity to
lenvatinib in HCC cells (112).
WWP2 contributes to doxorubicin resistance in HCC through its
regulation of PTEN. METTL3-mediated m6A modification of WWP2
increases its stability, leading to enhanced PTEN ubiquitination
and AKT activation, which counteracts the antitumor effects of
doxorubicin, an anthracycline antibiotic used in HCC chemotherapy
(147). UBE3B is associated
with lenvatinib resistance in HCC. Lobeline, an m6A antagonist,
downregulates UBE3B expression, partially reversing lenvatinib
resistance. This suggests that UBE3B expression, which is
potentially regulated by m6A modifications, contributes to the
resistant phenotype (148).
Mechanistic synthesis
Synthesizing the aforementioned findings, HECT-type
E3 ubiquitin ligases exhibit a dual-role effect in the pathogenesis
of HCC across all aspects of tumor biology, including
proliferation, invasion, metastasis and drug resistance. Different
members of the same subfamily, and even the same E3 ligase in
different cellular contexts or targeting different substrates, can
either promote or suppress HCC progression. This functional duality
profoundly reflects the intricate regulation of complex signaling
networks in HCC and the functional diversity of the HECT family
members.
NEDD4 acts as a definitive oncogene by degrading
PTEN to activate PI3K-AKT signaling and promote proliferation
(107,108). It also enhances invasion and
metastasis by ubiquitinating LATS1 and TGFBR1 (109,110) and contributes to drug
resistance through autophagy regulation and epigenetic mechanisms
(107,108,144). Although it is now widely
recognized that inhibiting NEDD4 significantly suppresses tumor
proliferation, invasion and metastasis while improving drug
resistance, no studies have described its dual role in HCC
treatment, demonstrating its immense translational potential.
Unfortunately, no clinical trials have confirmed whether this
effect can be effectively applied to HCC therapy.
Unlike NEDD4, the following genes fully demonstrate
the complexity of HECT-type E3 ubiquitin ligases in HCC
progression. WWP1 illustrates how the same E3 ligase may exhibit
contradictory roles owing to context-dependent interactions and
substrate availability. WWP1 has been reported both as an oncogene
promoting proliferation through caspase-3 and P53 regulation
(125), and as a tumor
suppressor that inhibits migration and invasion through PKM2
degradation in an ARHGAP24-dependent manner (126). This functional switch may
depend on the presence of scaffolding proteins such as ARHGAP24,
which direct WWP1 to specific substrates, and on the cellular
context that determines which substrates are available or relevant.
The involvement of USP13 in the regulation of WWP1 stability and
subsequent AKT/mTOR activation further complicates its functional
characterization (127). Other
HECT members exhibit specialized roles in specific aspects of HCC
biology. HECTD1 regulates angiogenesis through GRB2 modulation
(111). HECTD2 promotes
proliferation and lenvatinib resistance through KEAP1
ubiquitination and NRF2 activation (112). E6AP enhances tumor activity
through GPR26 degradation (113). UBR5 affects proliferation,
apoptosis, glycolysis and migration through multiple mechanisms
(115,116). HACE1 acts as a tumor suppressor
by regulating cholesterol metabolism and immune evasion through
SCAP ubiquitination, with its activity antagonized by S-acylation
(118). HUWE1 and TRIP12
regulate apoptosis through the p53/p14ARF axis and are modulated by
USP7 (134,135). UBE3C promotes EMT and
metastasis (136), while UBE3B
contributes to lenvatinib resistance (148). This functional specialization
reflects the diverse roles of different HECT members within the
complex signaling network of HCC.
From structural and functional perspectives, the
domain architecture of HECT-type E3 ligases determines their
substrate recognition specificity and functional outcomes in HCC.
The WW domains of NEDD4 and WWP1/2 mediate the recognition of
substrates containing PPxY motifs, such as PTEN, enabling these
ligases to regulate the PI3K-AKT pathway. The C2 and WW domains of
SMURF1/2 enable interaction with membrane-associated proteins such
as TGF-β receptors, allowing precise regulation of TGF-β signaling.
The RCC1-like domains of the HERC family confer the ability to
regulate GTPase-related signaling and respond to inflammatory
stimuli. Through their diverse N-terminal domains, other HECT
members participate in the regulation of fundamental biological
processes including metabolism (HACE1), stress responses (HECTD2)
and cell death (HUWE1 and TRIP12). This structure-function
relationship explains the molecular basis of the protective or
pathogenic roles of different HECT members in HCC. In conclusion,
HECT-type E3 ubiquitin ligases function as critical regulatory
nodes in HCC, influencing tumor proliferation, invasion,
metastasis, and drug resistance through diverse mechanisms. Their
functional duality arises from substrate diversity,
context-dependent interactions, scaffolding protein involvement,
post-translational modifications and tumor microenvironment.
Limitations and conclusion
Although numerous studies have highlighted the
potential of HECT-type E3 ubiquitin ligases as therapeutic targets
in liver diseases, significant limitations remain. First, research
coverage is uneven. The majority of evidence focuses on the NEDD4
subfamily (for example, NEDD4 and SMURF1/2) and HUWE1, whereas
functional studies on the HERC subfamily (for example, HERC5/6) and
unclassified members (for example, HECTD2/3 and UBE3C) are scarce,
potentially leading to the oversight of critical regulatory nodes.
Second, our mechanistic understanding is often incomplete. The
precise characterization of ubiquitination sites and linkage types
(for example, K48 vs. K63) is frequently lacking, and some
conclusions rely heavily on single animal models without validation
in human tissues. However, further comparative analyses were
impeded by several limitations. Third, the majority of studies on
MASLD lack clear definitions of disease staging, thereby precluding
a stratified comparison between early- and late-stage disease.
Moreover, liver fibrosis and cirrhosis are frequently conflated in
the literature, obscuring distinctions critical for mechanistic
interpretation. Forth, numerous studies fail to specify the precise
cell types in which HECT-type E3 ubiquitin ligases exert their
functions; for example, whether their actions are localized to
hepatocytes or HSC. Collectively, these gaps constrain a more
nuanced understanding of the potentially divergent roles of
different HECT-type E3 ligases across the continuum of disease
progression.
Most critically, the evidence supporting clinical
translation remains limited. While NEDD4 has emerged as a promising
oncogenic target in HCC, with multiple clinical correlation studies
supporting its prognostic value, most HECT-type E3 ligases lack
validation in human patient cohorts. Even for NEDD4, therapeutic
translation remains hindered by the absence of selective
small-molecule inhibitors that have advanced to clinical trials.
For other family members such as SMURF1/2, ITCH and NEDD4L, the
current findings are predominantly derived from preclinical models,
with human data limited to correlative expression analyses that
cannot establish causality. Although SMURF2 agonists (for example,
J-1155) and E6AP activators (for example, all-trans retinoic acid)
show promise in preclinical models, their selectivity, safety and
cross-species efficacy require further validation. Furthermore,
targeted therapies may cause off-target effects because of the dual
role nature of these enzymes (for example, profibrotic yet
anti-carcinogenic actions). Furthermore, evidence supporting its
clinical translation is weak. Finally, functional contradictions
are evident: For instance, NEDD4L can suppress ER stress but also
activate pro-inflammatory pathways in MASLD; SMURF2 may promote
TGF-β/Smad signaling in fibrosis while exerting protective effects
via miRNA mechanisms. These paradoxes suggest highly
context-dependent regulation, potentially fine-tuned by
microenvironmental metabolic pressure, immune status, or epigenetic
modifications. However, most current studies have focused on single
disease models or isolated molecular mechanisms, lacking systematic
analysis across disease stages and multi-organ interactions, which
hampers a comprehensive understanding of their functional
duality.
Future investigations should integrate multi-omics
technologies (for example, single-cell sequencing and spatial
transcriptomics) to delineate the dynamic expression and substrate
profiles of HECT-type E3s across different liver disease stages and
cellular subpopulations. Structurally, techniques such as
cryo-electron microscopy and AI-based prediction can elucidate
structure-function relationships, facilitating the design of
allosteric modulators to circumvent functional contradictions. For
clinical translation, patient-derived organoids or humanized mouse
models are essential for validating the conservation of regulatory
networks, along with the exploration of biomarkers for prognostic
stratification. Greater attention should also be directed towards
understudied members that may influence disease progression by
modulating the immune microenvironment or metabolic
reprogramming.
HECT-type E3 ubiquitin ligases function as critical
molecular hubs in the pathogenesis of diverse liver diseases and
exhibit a remarkable dual role that profoundly influences disease
initiation, progression and therapeutic responses. This functional
duality manifests across the entire spectrum of hepatic
pathologies, wherein the same ligase can both promote and inhibit
disease progression through context-dependent substrate selection,
spatiotemporally specific expression patterns, and the integration
of diverse microenvironmental cues. Upon analyzing the underlying
reasons for this trend, it is considered that the 'dual-role'
nature of HECT-type E3 ubiquitin ligases may offer a plausible
explanation. Most of these genes do not consistently exhibit
uniform effects across multiple pathological features throughout
disease progression, which may lead to suboptimal outcomes in
clinical studies and subsequent stagnation in translational
development. As a result, advancing from fundamental research to
clinical application remains particularly challenging. In metabolic
dysfunction-associated liver disease, these enzymes simultaneously
modulate lipid homeostasis and inflammatory responses through
opposing actions on key metabolic regulators and signaling
networks. During fibrogenesis, individual family members
demonstrate both profibrotic and antifibrotic activities depending
on the cellular context and experimental models. In viral
hepatitis, HECT-type E3 ligases occupy a central position at the
host-virus interface, either restricting replication through direct
viral protein degradation or facilitating propagation by hijacking
the host cellular machinery. Within HCC, these ligases exert
pleiotropic effects, including proliferation, invasion, metastasis
and drug resistance, and function as either oncogenic drivers or
tumor suppressors through diverse substrate interactions.
Although the specific mechanisms underlying this
'dual role' effect cannot be fully elucidated due to variations in
the depth of previous studies, it is proposed that analyzing
differences in disease stage, tissue specificity and heterogeneity
among experimental models can partially explain the conflicting
findings regarding HECT-type E3 ubiquitin ligases in liver
diseases. Stage-specific substrate switching accounts for numerous
contradictory observations, as exemplified by NEDD4L in MASLD/MASH:
Its downregulation in early stages impairs Txnip and JAK2
degradation, thereby exacerbating hepatic steatosis; conversely,
its upregulation in late stages targets LAPTM5, activating the
pro-inflammatory CDC42-MAPK signaling pathway. Cellular specificity
within the liver microenvironment further reinforces functional
duality: SMURF1 protects hepatocytes from steatosis via PPARγ
regulation yet promotes HSC activation through TLN1 ubiquitination;
similarly, ITCH knockout confers systemic metabolic protection
while simultaneously elevating pro-inflammatory BCAA levels in
specific cellular compartments. Heterogeneity in experimental
models, including interspecies differences, disease induction
methods (for example, CCl4 vs. cholestatic models), and
temporal analysis points, has generated conflicting reports
regarding the roles of SMURF2 in fibrosis and HERC5 in HCV
replication. The innovative contribution of the present review lies
in integrating these disparate observations into a unified
framework that positions HECT-type E3s as dynamic,
context-dependent molecular hubs rather than linear
disease-promoting factors. Unlike previous reviews that merely
catalog individual ligase functions in isolation, paradigm-shifting
connections were establish by elucidating how substrate switching
across disease stages generates functional duality, revealing how
scaffold proteins (for example, ARHGAP24 directing WWP1 activity)
dictate context-specific outcomes, and integrating
microenvironmental signals, such as metabolic stress, inflammatory
cues and viral hijacking, as critical determinants of ligase
behavior. This framework transforms apparent contradictions from
obstacles into opportunities, providing a mechanistic roadmap for
developing stage-specific and cell-type-selective therapeutic
strategies. By targeting auxiliary domains rather than conserved
catalytic sites, this approach holds promise for inhibiting
pathological activity while preserving beneficial functions.
Given the highly context-dependent nature of
HECT-type E3 ligase activity, current therapeutic strategies aimed
at directly targeting the conserved HECT domain to treat common
chronic liver diseases remain inadvisable. The structural
similarity of catalytic domains across family members raises
significant concerns regarding off-target effects, whereas the dual
roles of individual ligases across different disease stages and
cell types complicate predictable therapeutic outcomes. Future
efforts should focus on elucidating upstream regulatory mechanisms,
developing strategies to modulate specific protein-protein
interactions involving accessory domains, and identifying
context-specific biomarkers that can predict functional
directionality. Only through such refined approaches can the
tremendous therapeutic potential of HECT-type E3 ligases be safely
and effectively harnessed.
Future efforts should therefore prioritize the
following: i) Expanding clinical correlation studies with
functional validation in patient-derived models; ii) elucidating
upstream regulatory mechanisms that govern context-specific
activities; iii) developing strategies to modulate specific
protein-protein interactions involving accessory domains rather
than the conserved HECT domain; and iv) identifying
context-specific biomarkers that can predict functional
directionality. Only through such refined approaches can the
tremendous therapeutic potential of HECT-type E3 ligases be safely
and effectively harnessed, moving beyond correlative observations
toward mechanism-based interventions. The path to clinical
translation requires patience and rigor; however, the central
position of these enzymes in liver disease networks offers
unprecedented opportunities for eventual therapeutic
innovation.
Availability of data and materials
Not applicable.
Authors' contributions
TXL and HC read and analyzed the references and
wrote the manuscript. PZ and CZY performed literature review. ZFS
conceptualized the study and provided written ideas and financial
support. All authors read and approved the final version of the
manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant nos. 81974040 and 82270412).
References
|
1
|
Cruz Walma DA, Chen Z, Bullock AN and
Yamada KM: Ubiquitin ligases: Guardians of mammalian development.
Nat Rev Mol Cell Biol. 23:350–367. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Buetow L and Huang DT: Structural insights
into the catalysis and regulation of E3 ubiquitin ligases. Nat Rev
Mol Cell Biol. 17:626–642. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sheng X, Xia Z, Yang H and Hu R: The
ubiquitin codes in cellular stress responses. Protein Cell.
15:157–190. 2024. View Article : Google Scholar :
|
|
4
|
Sampson C, Wang Q, Otkur W, Zhao H, Lu Y,
Liu X and Piao HL: The roles of E3 ubiquitin ligases in cancer
progression and targeted therapy. Clin Transl Med. 13:e12042023.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Berndsen CE and Wolberger C: New insights
into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol.
21:301–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ciechanover A: The unravelling of the
ubiquitin system. Nat Rev Mol Cell Biol. 16:322–324. 2015.
View Article : Google Scholar : PubMed/NCBI
Morreale FE and Walden H: Types of
ubiquitin ligases. Cell. 165:248–248.e1. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Asmamaw MD, Liu Y, Zheng YC, Shi XJ and
Liu HM: Skp2 in the ubiquitin-proteasome system: A comprehensive
review. Med Res Rev. 40:1920–1949. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bernassola F, Chillemi G and Melino G:
HECT-type E3 ubiquitin ligases in cancer. Trends Biochem Sci.
44:1057–1075. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Metzger MB, Hristova VA and Weissman AM:
HECT and RING finger families of E3 ubiquitin ligases at a glance.
J Cell Sci. 125:531–537. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shah SS and Kumar S: Adaptors as the
regulators of HECT ubiquitin ligases. Cell Death Differ.
28:455–472. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Goto J, Otaki Y, Watanabe T and Watanabe
M: The role of HECT-Type E3 ligase in the development of cardiac
disease. Int J Mol Sci. 22:60652021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Filali-Mouncef Y, Hunter C, Roccio F,
Zagkou S, Dupont N, Primard C, Proikas-Cezanne T and Reggiori F:
The ménage à trois of autophagy, lipid droplets and liver disease.
Autophagy. 18:50–72. 2022. View Article : Google Scholar :
|
|
13
|
You Y, Gao C, Wu J, Qu H, Xiao Y, Kang Z,
Li J and Hong J: Enhanced expression of ARK5 in hepatic stellate
cell and hepatocyte synergistically promote liver fibrosis. Int J
Mol Sci. 23:130842022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Maghames CM, Lobato-Gil S, Perrin A,
Trauchessec H, Rodriguez MS, Urbach S, Marin P and Xirodimas DP:
NEDDylation promotes nuclear protein aggregation and protects the
ubiquitin proteasome system upon proteotoxic stress. Nat Commun.
9:43762018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mishra A, Godavarthi SK, Maheshwari M,
Goswami A and Jana NR: The ubiquitin ligase E6-AP is induced and
recruited to aggresomes in response to proteasome inhibition and
may be involved in the ubiquitination of Hsp70-bound misfolded
proteins. J Biol Chem. 284:10537–10545. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu J, Chen Y, Huang Q, Liu W, Ji X, Hu F,
Zhu Y, Zhang L and Dong G: IRAK2 counterbalances oncogenic Smurf1
in colon cancer cells by dictating ER stress. Cell Signal.
48:69–80. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo X, Shen S, Song S, He S, Cui Y, Xing
G, Wang J, Yin Y, Fan L, He F and Zhang L: The E3 ligase Smurf1
regulates Wolfram syndrome protein stability at the endoplasmic
reticulum. J Biol Chem. 286:18037–18047. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rinella ME, Lazarus JV, Ratziu V, Francque
SM, Sanyal AJ, Kanwal F, Romero D, Abdelmalek MF, Anstee QM, Arab
JP, et al: A multisociety Delphi consensus statement on new fatty
liver disease nomenclature. Hepatology. 78:1966–1986. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rotin D and Kumar S: Physiological
functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell
Biol. 10:398–409. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huibregtse JM, Scheffner M, Beaudenon S
and Howley PM: A family of proteins structurally and functionally
related to the E6-AP ubiquitin-protein ligase. Proc Natl Acad Sci
USA. 92:2563–2567. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang L, Kinnucan E, Wang G, Beaudenon S,
Howley PM, Huibregtse JM and Pavletich NP: Structure of an
E6AP-UbcH7 complex: Insights into ubiquitination by the E2-E3
enzyme cascade. Science. 286:1321–1326. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maspero E, Mari S, Valentini E, Musacchio
A, Fish A, Pasqualato S and Polo S: Structure of the HECT:Ubiquitin
complex and its role in ubiquitin chain elongation. EMBO Rep.
12:342–349. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Scheffner M and Kumar S: Mammalian HECT
ubiquitin-protein ligases: Biological and pathophysiological
aspects. Biochim Biophys Acta. 1843:61–74. 2024. View Article : Google Scholar
|
|
24
|
Zheng N and Shabek N: Ubiquitin ligases:
Structure, function, and regulation. Annu Rev Biochem. 86:129–157.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang X, Trotman LC, Koppie T, Alimonti A,
Chen Z, Gao Z, Wang J, Erdjument-Bromage H, Tempst P, Cordon-Cardo
C, et al: NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN.
Cell. 128:129–139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Staub O, Dho S, Henry P, Correa J,
Ishikawa T, McGlade J and Rotin D: WW domains of Nedd4 bind to the
proline-rich PY motifs in the epithelial Na+ channel deleted in
Liddle's syndrome. EMBO J. 15:2371–2380. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Song MS and Pandolfi PP: The HECT family
of E3 ubiquitin ligases and PTEN. Semin Cancer Biol. 85:43–51.
2022. View Article : Google Scholar
|
|
28
|
Shearwin-Whyatt L, Dalton HE, Foot N and
Kumar S: Regulation of functional diversity within the Nedd4 family
by accessory and adaptor proteins. Bioessays. 28:617–628. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sala-Gaston J, Costa-Sastre L, Pedrazza L,
Martinez-Martinez A, Ventura F and Rosa JL: Regulation of MAPK
signaling pathways by the large HERC ubiquitin ligases. Int J Mol
Sci. 24:49062023. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
García-Cano J, Martinez-Martinez A,
Sala-Gaston J, Pedrazza L and Rosa JL: HERCing: Structural and
functional relevance of the large HERC ubiquitin ligases. Front
Physiol. 10:10142019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pérez-Villegas EM, Ruiz R, Bachiller S,
Ventura F, Armengol JA and Rosa JL: The HERC proteins and the
nervous system. Semin Cell Dev Biol. 132:5–15. 2022. View Article : Google Scholar
|
|
32
|
Kao SH, Wu HT and Wu KJ: Ubiquitination by
HUWE1 in tumorigenesis and beyond. J Biomed Sci. 25:672018.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Qi L, Xu X and Qi X: The giant E3 ligase
HUWE1 is linked to tumorigenesis, spermatogenesis, intellectual
disability, and inflammatory diseases. Front Cell Infect Microbiol.
12:9059062022. View Article : Google Scholar :
|
|
34
|
Wenmaekers S, Viergever BJ, Kumar G,
Kranenburg O, Black PC, Daugaard M and Meijer RP: A potential role
for HUWE1 in modulating cisplatin sensitivity. Cells. 10:12622021.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yuan B, Liu J, Cao J, Yu Y, Zhang H, Wang
F, Zhu Y, Xiao M, Liu S, Ye Y, et al: PTPN3 acts as a tumor
suppressor and boosts TGF-β signaling independent of its
phosphatase activity. EMBO J. 38:e999452019. View Article : Google Scholar
|
|
36
|
Hagström H, Vessby J, Ekstedt M and Shang
Y: 99% of patients with NAFLD meet MASLD criteria and natural
history is therefore identical. J Hepatol. 80:e76–e77. 2024.
View Article : Google Scholar
|
|
37
|
Wong VW, Ekstedt M, Wong GL and Hagström
H: Changing epidemiology, global trends and implications for
outcomes of NAFLD. J Hepatol. 79:842–852. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
European Association for the Study of the
Liver (EASL); European Association for the Study of Diabetes
(EASD); European Association for the Study of Diabetes (EASD);
European Association for the Study of Obesity (EASO):
EASL-EASD-EASO clinical practice guidelines on the management of
metabolic dysfunction-associated steatotic liver disease (MASLD). J
Hepatol. 81:492–542. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Targher G, Byrne CD and Tilg H: MASLD: A
systemic metabolic disorder with cardiovascular and malignant
complications. Gut. 73:691–702. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hu S, Zhu Y, Zhao X, Li R, Shao G, Gong D,
Hu C, Liu H, Xu K, Liu C, et al: Hepatocytic lipocalin-2 controls
HDL metabolism and atherosclerosis via Nedd4-1-SR-BI axis in mice.
Dev Cell. 58:2326–2337.e5. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yan C, Zhao M, Li S, Liu T, Xu C, Liu L,
Geng T and Gong D: Increase of E3 ubiquitin ligase NEDD4 expression
leads to degradation of its target proteins PTEN/IGF1R during the
formation of goose fatty liver. J Anim Sci. 98:skaa2702020.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cao HJ, Jiang H, Ding K, Qiu XS, Ma N,
Zhang FK, Wang YK, Zheng QW, Xia J, Ni QZ, et al: ARID2 mitigates
hepatic steatosis via promoting the ubiquitination of JAK2. Cell
Death Differ. 30:383–396. 2023. View Article : Google Scholar
|
|
43
|
Jiang L, Zhao J, Yang Q, Li M, Liu H, Xiao
X, Tian S, Hu S, Liu Z, Yang P, et al: Lysosomal-associated protein
transmembrane 5 ameliorates non-alcoholic steatohepatitis by
promoting the degradation of CDC42 in mice. Nat Commun.
14:26542023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhao GN, Zhang P, Gong J, Zhang XJ, Wang
PX, Yin M, Jiang Z, Shen LJ, Ji YX, Tong J, et al: Tmbim1 is a
multivesicular body regulator that protects against non-alcoholic
fatty liver disease in mice and monkeys by targeting the lysosomal
degradation of Tlr4. Nat Med. 23:742–752. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Marino A, Menghini R, Fabrizi M,
Casagrande V, Mavilio M, Stoehr R, Candi E, Mauriello A,
Moreno-Navarrete JM, Gómez-Serrano M, et al: ITCH deficiency
protects from diet-induced obesity. Diabetes. 63:550–561. 2014.
View Article : Google Scholar
|
|
46
|
Stöhr R, Mavilio M, Marino A, Casagrande
V, Kappel B, Möllmann J, Menghini R, Melino G and Federici M: ITCH
modulates SIRT6 and SREBP2 to influence lipid metabolism and
atherosclerosis in ApoE null mice. Sci Rep. 5:90232015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Huang H, Peng B, Chen Q, Wang Y and Li R:
Long non-coding RNA nuclear-enriched abundant transcript 1 (NEAT1)
facilitates foam cell formation and atherosclerosis progression
through the miR-17-5p/Itchy E3 ubiquitin protein ligase
(ITCH)/liver kinase B1 (LKB1) axis. Circ J. 88:1697–1708. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Menghini R, Hoyles L, Cardellini M,
Casagrande V, Marino A, Gentileschi P, Davato F, Mavilio M, Arisi
I, Mauriello A, et al: ITCH E3 ubiquitin ligase downregulation
compromises hepatic degradation of branched-chain amino acids. Mol
Metab. 59:1014542022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hooper C, Puttamadappa SS, Loring Z,
Shekhtman A and Bakowska JC: Spartin activates
atrophin-1-interacting protein 4 (AIP4) E3 ubiquitin ligase and
promotes ubiquitination of adipophilin on lipid droplets. BMC Biol.
8:722010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lin W, Zhang X, Zhang C, Li L, Zhang J,
Xie P, Zhan Y and An W: Deletion of Smurf1 attenuates liver
steatosis via stabilization of p53. Lab Invest. 102:1075–1087.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang X, Zhan Y, Lin W, Zhao F, Guo C,
Chen Y, Du M, Li D, Zhang L, An W, et al: Smurf1 aggravates
non-alcoholic fatty liver disease by stabilizing SREBP-1c in an E3
activity-independent manner. FASEB J. 34:7631–7643. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhu K, Tang Y, Xu X, Dang H, Tang LY, Wang
X, Wang XW and Zhang YE: Non-proteolytic ubiquitin modification of
PPARγ by Smurf1 protects the liver from steatosis. PLoS Biol.
16:e30000912018. View Article : Google Scholar
|
|
53
|
Petrasek J, Erhartova D and Levine B:
Protective effect of SMAD-specific E3 ubiquitin protein ligase 1 in
alcoholic steatohepatitis in mice. Hepatol Commun. 3:1450–1458.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhu Q, Wang D, Liang F, Tong X, Liang Z,
Wang X, Chen Y and Mo D: Protein arginine methyltransferase PRMT1
promotes adipogenesis by modulating transcription factors C/EBPβ
and PPARγ. J Biol Chem. 298:1023092022. View Article : Google Scholar
|
|
55
|
Nozaki Y, Kobayashi M, Wakasawa H, Hoshino
S, Suwa F, Ose Y, Tagawa R and Higami Y: Systemic depletion of WWP1
improves insulin sensitivity and lowers triglyceride content in the
liver of obese mice. FEBS Open Bio. 13:1086–1094. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Feng WW, Bang S, Takacs EM, Day C,
Crawford KJ, Al-Sheyab R, Almufarrej DB, Wells W, Ilchenko S,
Kasumov T, et al: Hepatic Huwe1 loss protects mice from
non-alcoholic fatty liver disease through lipid metabolic rewiring.
iScience. 26:1084052023. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhao Z, Xu D, Wang Z, Wang L, Han R, Wang
Z, Liao L and Chen Y: Hepatic PPARα function is controlled by
polyubiquitination and proteasome-mediated degradation through the
coordinated actions of PAQR3 and HUWE1. Hepatology. 68:289–303.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lin Y, Chen L, You X, Li Z, Li C and Chen
Y: PAQR9 regulates hepatic ketogenesis and fatty acid oxidation
during fasting by modulating protein stability of PPARα. Mol Metab.
53:1013312021. View Article : Google Scholar
|
|
59
|
Luo X, Li Z, Chen L, Zhang X, Zhu X, Wang
Z and Chen Y: Monocarboxylate transporter 1 in the liver modulates
high-fat diet-induced obesity and hepatic steatosis in mice.
Metabolism. 143:1555372023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wan W, You Z, Zhou L, Xu Y, Peng C, Zhou
T, Yi C, Shi Y and Liu W: mTORC1-regulated and HUWE1-mediated WIPI2
degradation controls autophagy flux. Mol Cell. 72:303–315.e6. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Peng K, Wang S, Liu R, Zhou L, Jeong GH,
Jeong IH, Liu X, Kiyokawa H, Xue B, Zhao B, et al: Effects of UBE3A
on cell and liver metabolism through the ubiquitination of PDHA1
and ACAT1. Biochemistry. 62:1274–1286. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Guo Q, Xin M, Lu Q, Feng D, Yang V, Peng
LF, Whelan KA, Hu W, Wu S, Yang X, et al: A novel NEDD4L-TXNIP-CHOP
axis in the pathogenesis of nonalcoholic steatohepatitis.
Theranostics. 13:2210–2225. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Li Z, Zhao J, Wu Y, Fan S, Yuan H, Xia J,
Hu L, Yang J, Liu J, Wu X, et al: TRAF2 decrease promotes the
TGF-β-mTORC1 signal in MAFLD-HCC through enhancing AXIN1-mediated
Smad7 degradation. FASEB J. 38:e234912024. View Article : Google Scholar
|
|
64
|
Li T, Liu R, Cao H, Deng S, Wang G, Wang
X, Zhao P, Li X, Zhu J, Shao S, et al: Epithelial membrane protein
1 drives hepatic stellate cell activation via the TLN1/FAK cascade
in MASLD donor liver transplantation. Mol Biomed. 6:1162025.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Liu D, Chen L, Wang Z, Li Z, Liu L and
Peng L: Ubiquitination of TFEB increased intestinal permeability to
aggravate metabolic dysfunction-associated steatohepatitis.
Hepatology. 82:1534–1550. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhang Y, Zhan C, Chen G and Sun J:
Label-free quantitative proteomics and bioinformatics analyses of
alcoholic liver disease in a chronic and binge mouse model. Mol Med
Rep. 18:2079–2087. 2018.PubMed/NCBI
|
|
67
|
Horn P and Tacke F: Metabolic
reprogramming in liver fibrosis. Cell Metab. 36:1439–1455. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Song Y, Wei J, Li R, Fu R, Han P, Wang H,
Zhang G, Li S, Chen S, Liu Z, et al: Tyrosine kinase receptor B
attenuates liver fibrosis by inhibiting TGF-β/SMAD signaling.
Hepatology. 78:1433–1447. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Chen C, Bi Y, Chen B and He S: Nedd4L
signaling contributes to carbon tetrachloride-induced liver
fibrosis in female mice and is associated with enteric
dysbacteriosis. Gastroenterol Rep (Oxf). 13:goaf0222025. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ge MX, Liu HT, Zhang N, Niu WX, Lu ZN, Bao
YY, Huang R, Yu DK, Shao RG and He HW: Costunolide represses
hepatic fibrosis through WW domain-containing protein 2-mediated
Notch3 degradation. Br J Pharmacol. 177:372–387. 2020. View Article : Google Scholar
|
|
71
|
Shen J, Wang Z, Liu M, Zhu YJ, Zheng L,
Wang LL, Cheng JL, Liu TT, Zhang GD, Yang TY, et al:
LincRNA-ROR/miR-145/ZEB2 regulates liver fibrosis by modulating
HERC5-mediated p53 ISGylation. FASEB J. 37:e229362023. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Guo Y, Zhao C, Dai W, Wang B, Lai E, Xiao
Y, Tang C, Huang Z and Gao J: C-C motif chemokine receptor 2
inhibition reduces liver fibrosis by restoring the immune cell
landscape. Int J Biol Sci. 19:2572–2587. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Kim JY, Kim KM, Yang JH, Cho SS, Kim SJ,
Park SJ, Ahn SG, Lee GH, Yang JW, Lim SC, et al: Induction of E6AP
by microRNA-302c dysregulation inhibits TGF-β-dependent
fibrogenesis in hepatic stellate cells. Sci Rep. 10:4442020.
View Article : Google Scholar
|
|
74
|
Zhang N, Yang P, Li Y, Ouyang Q, Hou F,
Zhu G, Zhang B, Huang J, Jia J and Xu A: Serum iron overload
activates the SMAD pathway and hepcidin expression of hepatocytes
via SMURF1. J Clin Transl Hepatol. 12:227–235. 2024.PubMed/NCBI
|
|
75
|
Lee H, Yu DM, Bahn MS, Kwon YJ, Um MJ,
Yoon SY, Kim KT, Lee MW, Jo SJ, Lee S, et al: Hepatocyte-specific
prominin-1 protects against liver injury-induced fibrosis by
stabilizing SMAD7. Exp Mol Med. 54:1277–1289. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Cai Y, Zhou CH, Fu D and Shen XZ:
Overexpression of Smad ubiquitin regulatory factor 2 suppresses
transforming growth factor-β mediated liver fibrosis. J Dig Dis.
13:327–334. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Cai Y, Huang G, Ma L, Dong L, Chen S, Shen
X and Zhang S, Xue R, Sun D and Zhang S: Smurf2, an E3 ubiquitin
ligase, interacts with PDE4B and attenuates liver fibrosis through
miR-132 mediated CTGF inhibition. Biochim Biophys Acta Mol Cell
Res. 1865:297–308. 2018. View Article : Google Scholar
|
|
78
|
Jiang XL, Liu C, Zhan ZY, Lan XQ, Wu YL,
Nan JX, Jin CH and Lian LH: Thiazole isomers as potential ALK5
inhibitors alleviate P2X7R-mediated inflammation during liver
fibrosis. Int Immunopharmacol. 153:1144722025. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Kong D, Zhang Z, Chen L, Huang W, Zhang F,
Wang L, Wang Y, Cao P and Zheng S: Curcumin blunts
epithelial-mesenchymal transition of hepatocytes to alleviate
hepatic fibrosis through regulating oxidative stress and autophagy.
Redox Biol. 36:1016002020. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Danpanichkul P, Suparan K, Kaeosri C,
Jatupornpakdee P, Attia AM, Suenghataiphorn T, Thongpiya J,
Sukphutanan B, Huang DQ, Noureddin M, et al: Global trend of
MASH-associated liver cancer: A systematic analysis from the global
burden of disease 2021. Clin Gastroenterol Hepatol. 23:1346–1355.
2025. View Article : Google Scholar
|
|
81
|
Cao X, Chen Y, Chen Y and Jiang M: The
role of tripartite motif family proteins in chronic liver diseases:
Molecular mechanisms and therapeutic potential. Biomolecules.
14:10382024. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wang Z, Kawaguchi K, Honda M, Hashimoto S,
Shirasaki T, Okada H, Orita N, Shimakami T, Yamashita T, Sakai Y,
et al: Notch signaling facilitates hepatitis B virus covalently
closed circular DNA transcription via cAMP response element-binding
protein with E3 ubiquitin ligase-modulation. Sci Rep. 9:16212019.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Bawono RG, Abe T, Qu M, Kuroki D, Deng L,
Matsui C, Ryo A, Suzuki T, Matsuura Y, Sugiyama M, et al: HERC5 E3
ligase mediates ISGylation of hepatitis B virus X protein to
promote viral replication. J Gen Virol. 102:2021. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Langerová H, Lubyová B, Zábranský A,
Hubálek M, Glendová K, Aillot L, Hodek J, Strunin D, Janovec V,
Hirsch I and Weber J: Hepatitis B core protein is
post-translationally modified through K29-linked ubiquitination.
Cells. 9:25472020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Wan T, Lei Z, Tu B, Wang T, Wang J and
Huang F: NEDD4 induces K48-linked degradative ubiquitination of
hepatitis B virus x protein and inhibits HBV-associated HCC
progression. Front Oncol. 11:6251692021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Rost M, Mann S, Lambert C, Döring T, Thomé
N and Prange R: Gamma-adaptin, a novel ubiquitin-interacting
adaptor, and Nedd4 ubiquitin ligase control hepatitis B virus
maturation. J Biol Chem. 281:29297–29308. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Lambert C, Döring T and Prange R:
Hepatitis B virus maturation is sensitive to functional inhibition
of ESCRT-III, Vps4, and gamma 2-adaptin. J Virol. 81:9050–9060.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Zheng Y, Wang M, Li S, Bu Y, Xu Z, Zhu G,
Wu C, Zhao K, Li A, Chen Q, et al: Hepatitis B virus hijacks TSG101
to facilitate egress via multiple vesicle bodies. PLoS Pathog.
19:e10113822023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Lim HY, Han J, Yoon H and Jang KL: Tumor
suppressor p53 inhibits hepatitis B virus replication by
downregulating HBx via E6AP-mediated proteasomal degradation in
human hepatocellular carcinoma cell lines. Viruses. 14:23132022.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Han J and Jang KL: All-trans retinoic acid
downregulates HBx levels via E6-associated protein-mediated
proteasomal degradation to suppress hepatitis B virus replication.
PLoS One. 19:e03053502024. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Deng L, Liang Y, Ariffianto A, Matsui C,
Abe T, Muramatsu M, Wakita T, Maki M, Shibata H and Shoji I:
Hepatitis C virus-induced ROS/JNK signaling pathway activates the
E3 ubiquitin ligase itch to promote the release of HCV particles
via polyubiquitylation of VPS4A. J Virol. 96:e01811212022.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Domingues P, Bamford CGG, Boutell C and
McLauchlan J: Inhibition of hepatitis C virus RNA replication by
ISG15 does not require its conjugation to protein substrates by the
HERC5 E3 ligase. J Gen Virol. 96:3236–3242. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Abe T, Minami N, Bawono RG, Matsui C, Deng
L, Fukuhara T, Matsuura Y and Shoji I: ISGylation of hepatitis C
virus NS5A protein promotes viral RNA replication via recruitment
of cyclophilin A. J Virol. 94:e00532–20. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Munakata T, Liang Y, Kim S, McGivern DR,
Huibregtse J, Nomoto A and Lemon SM: Hepatitis C virus induces
E6AP-dependent degradation of the retinoblastoma protein. PLoS
Pathog. 3:1335–1347. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Shirakura M, Murakami K, Ichimura T,
Suzuki R, Shimoji T, Fukuda K, Abe K, Sato S, Fukasawa M, Yamakawa
Y, et al: E6AP ubiquitin ligase mediates ubiquitylation and
degradation of hepatitis C virus core protein. J Virol.
81:1174–1185. 2007. View Article : Google Scholar :
|
|
96
|
Moriishi K, Shoji I, Mori Y, Suzuki R,
Suzuki T, Kataoka C and Matsuura Y: Involvement of PA28gamma in the
propagation of hepatitis C virus. Hepatology. 52:411–420. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Kwak J, Shim JH, Tiwari I and Jang KL:
Hepatitis C virus core protein inhibits E6AP expression via DNA
methylation to escape from ubiquitin-dependent proteasomal
degradation. Cancer Lett. 380:59–68. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Yoon H and Jang KL: Hydrogen peroxide
inhibits hepatitis C virus replication by downregulating hepatitis
C virus core levels through e6-associated protein-mediated
proteasomal degradation. Cells. 13:622023. View Article : Google Scholar
|
|
99
|
Park JM, Yoon H, Jeong Y and Jang KL:
Tumour suppressor p53 inhibits hepatitis C virus replication by
inducing E6AP-mediated proteasomal degradation of the viral core
protein. FEBS Lett. 596:2525–2537. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Lee HK, Yoon H and Jang KL: All-trans
retinoic acid inhibits HCV replication by downregulating core
levels via E6AP-mediated proteasomal degradation. Biochem Biophys
Res Commun. 594:15–21. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Verga-Gérard A, Porcherot M,
Meyniel-Schicklin L, André P, Lotteau V and Perrin-Cocon L:
Hepatitis C virus/human interactome identifies SMURF2 and the viral
protease as critical elements for the control of TGF-β signaling.
FASEB J. 27:4027–4040. 2013. View Article : Google Scholar
|
|
102
|
Wang Z, Gao Y, Zhang C, Hu H, Guo D, Xu Y,
Xu Q, Zhang W, Deng S, Lv P, et al: Quinolinate
phosphoribosyltransferase is an antiviral host factor against
hepatitis C virus infection. Sci Rep. 7:58762017. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Shirasaki T, González-López O, McKnight
KL, Xie L, Shiota T, Chen X, Feng H and Lemon SM: Nonlytic
quasi-enveloped hepatovirus release is facilitated by pX protein
interaction with the E3 ubiquitin ligase ITCH. J Virol.
96:e01195222022. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Wang H, Li Q, Tang Q, Shi G, Wu G, Mao X,
Wu C, Zhang L, Liu J, Li J and Li B: Role and therapeutic potential
of E3s in the tumor microenvironment of hepatocellular carcinoma.
Front Immunol. 15:14837212024. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Miao Y, Wang S, Zhang J, Liu H, Zhang C,
Jin S and Bai D: Strategic advancement of E3 ubiquitin ligase in
the management of hepatocellular carcinoma. Med Oncol. 41:1782024.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Hang X, Zhu S, Di H, Wu Z, Chu K, Wang J,
Xin H, Yu G, Peng H, Miao X and Xu W: NEDD4 depletion inhibits
hepatocellular carcinoma growth via targeting PTEN. Cell Physiol
Biochem. 39:768–779. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Huang ZJ, Zhu JJ, Yang XY and Biskup E:
NEDD4 promotes cell growth and migration via PTEN/PI3K/AKT
signaling in hepatocellular carcinoma. Oncol Lett. 14:2649–2656.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Zhou Y, Qiu J, Liu S, Wang P, Ma D, Zhang
G, Cao Y, Hu L, Wang Z, Wu J and Jiang C: CFDP1 promotes
hepatocellular carcinoma progression through activating
NEDD4/PTEN/PI3K/AKT signaling pathway. Cancer Med. 12:425–444.
2023. View Article : Google Scholar :
|
|
109
|
Zheng H, Ke X, Li D and Wang Q, Wang J,
Liu X, Deng M, Deng X, Xue Y, Zhu Y and Wang Q: NEDD4 promotes cell
growth and motility in hepatocellular carcinoma. Cell Cycle.
17:728–738. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Li K, Niu Y, Yuan Y, Qiu J, Shi Y, Zhong
C, Qiu Z, Li K, Lin Z, Huang Z, et al: Insufficient ablation
induces E3-ligase Nedd4 to promote hepatocellular carcinoma
progression by tuning TGF-β signaling. Oncogene. 41:3197–3209.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Liu Y, Hu X, Zhou S, Sun T, Shen F and
Zeng L: Golgi protein 73 promotes angiogenesis in hepatocellular
carcinoma. Research (Wash D C). 7:04252024.PubMed/NCBI
|
|
112
|
Dong R, Fei Y, He Y, Gao P, Zhang B, Zhu
M, Wang Z, Wu L, Wu S, Wang X, et al: Lactylation-driven HECTD2
limits the response of hepatocellular carcinoma to lenvatinib. Adv
Sci (Weinh). 12:e24125592025. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Liu F, Yang W, Hu M, Zhang Y, Sun B, Yang
H, Brosius J and Deng C: Constitutive activity of GPR26 regulated
by ubiquitin-dependent degradation and its antitumor role. FEBS J.
288:4655–4682. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Wu Y, Xiang Q, Lv X, Xiang X, Feng Z, Tian
S, Tang J, Xiang T and Gong J: C2orf40 inhibits hepatocellular
carcinoma through interaction with UBR5. J Gastroenterol Hepatol.
36:2581–2591. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Wang M, Ma X, Wang G, Song Y, Zhang M, Mai
Z, Zhou B, Ye Y and Xia W: Targeting UBR5 in hepatocellular
carcinoma cells and precise treatment via echinacoside
nanodelivery. Cell Mol Biol Lett. 27:922022. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Leboeuf D, Abakumova T, Prikazchikova T,
Rhym L, Anderson DG, Zatsepin TS and Piatkov KI: Downregulation of
the Arg/N-degron pathway sensitizes cancer cells to chemotherapy in
vivo. Mol Ther. 28:1092–1104. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Gao ZF, Wu YN, Bai ZT, Zhang L, Zhou Q and
Li X: Tumor-suppressive role of HACE1 in hepatocellular carcinoma
and its clinical significance. Oncol Rep. 36:3427–3435. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Wu M, Zhou X, Zhou X, Wang G, Zeng Y, Li
J, Prochownik EV, Wang F and Li Y: ZDHHC3-mediated SCAP S-acylation
promotes cholesterol biosynthesis and tumor immune escape in
hepatocellular carcinoma. Cell Rep. 43:1149622024. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Zhang Y, Chen S, You L, He Z, Xu P and
Huang W: LINC00161 upregulated by M2-like tumor-associated
macrophages promotes hepatocellular carcinoma progression by
methylating HACE1 promoters. Cytotechnology. 76:777–793. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Lee HS, Park MH, Yang SJ, Park KC, Kim NS,
Kim YS, Kim DI, Yoo HS, Choi EJ and Yeom YI: Novel candidate
targets of Wnt/beta-catenin signaling in hepatoma cells. Life Sci.
80:690–698. 2007. View Article : Google Scholar
|
|
121
|
Zhao F, Gong X, Liu A, Lv X, Hu B and
Zhang H: Downregulation of Nedd4L predicts poor prognosis, promotes
tumor growth and inhibits MAPK/ERK signal pathway in hepatocellular
carcinoma. Biochem Biophys Res Commun. 495:1136–1143. 2018.
View Article : Google Scholar
|
|
122
|
Bian S, Ni W, Zhou L, Tong Y, Dai C, Zhao
X, Qiang Y, Gao J, Xiao Y, Liu W, et al: Ubiquitin-specific
protease 1 facilitates hepatocellular carcinoma progression by
modulating mitochondrial fission and metabolic reprogramming via
cyclin-dependent kinase 5 stabilization. Cell Death Differ.
31:1202–1218. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Ge QY, Chen J, Li GX, Tan XL, Song J, Ning
D, Mo J, Du PC, Liu QM, Liang HF, et al: GRAMD4 inhibits tumour
metastasis by recruiting the E3 ligase ITCH to target TAK1 for
degradation in hepatocellular carcinoma. Clin Transl Med.
11:e6352021. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Chen S, Zhou B, Huang W, Li Q, Yu Y, Kuang
X, Huang H, Wang W and Xie P: The deubiquitinating enzyme USP44
suppresses hepatocellular carcinoma progression by inhibiting
Hedgehog signaling and PDL1 expression. Cell Death Dis. 14:8302023.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Cheng Q, Cao X, Yuan F, Li G and Tong T:
Knockdown of WWP1 inhibits growth and induces apoptosis in hepatoma
carcinoma cells through the activation of caspase3 and p53. Biochem
Biophys Res Commun. 448:248–254. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Yang W, Wang B, Yu Q, Liu T, Li T, Tian T,
Jin A, Ding L, Chen W, Wang H, et al: ARHGAP24 represses β-catenin
transactivation-induced invasiveness in hepatocellular carcinoma
mainly by acting as a GTPase-independent scaffold. Theranostics.
12:6189–6206. 2022. View Article : Google Scholar
|
|
127
|
Zhu Q, Yuan Z, Huo Q, Lu Q, Wu Q, Guo J,
Fu W, Lu Y, Zhong L, Shang W, et al: YY1 induced USP13
transcriptional activation drives the malignant progression of
hepatocellular carcinoma by deubiquitinating WWP1. Cell Mol Biol
Lett. 30:562025. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Fang S, Zhang D, Weng W, Lv X, Zheng L,
Chen M, Fan X, Mao J, Mao C, Ye Y, et al: CPSF7 regulates liver
cancer growth and metastasis by facilitating WWP2-FL and targeting
the WWP2/PTEN/AKT signaling pathway. Biochim Biophys Acta Mol Cell
Res. 1867:1186242020. View Article : Google Scholar
|
|
129
|
Liu Y, Xu Q, Deng F, Zheng Z, Luo J, Wang
P, Zhou J, Lu X, Zhang L, Chen Z, et al: HERC2 promotes
inflammation-driven cancer stemness and immune evasion in
hepatocellular carcinoma by activating STAT3 pathway. J Exp Clin
Cancer Res. 42:382023. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Zheng Y, Li J, Pan C, Zhou G, Zhuge L, Jin
L and Fang P: HERC4 is overexpressed in hepatocellular carcinoma
and contributes to the proliferation and migration of
hepatocellular carcinoma cells. DNA Cell Biol. 36:490–500. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Huang F, Tang X, Sun T, Wang G, Ru Q and
Zheng Y: SAV1, regulated by HERC4, inhibits the proliferation,
migration, and invasion of hepatocellular carcinoma. Transl Cancer
Res. 10:349–360. 2021. View Article : Google Scholar
|
|
132
|
Wang Y, Ding Q, Xu T, Li CY, Zhou DD and
Zhang L: HZ-6d targeted HERC5 to regulate p53 ISGylation in human
hepatocellular carcinoma. Toxicol Appl Pharmacol. 334:180–191.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Xue F, Higgs BW, Huang J, Morehouse C, Zhu
W, Yao X, Brohawn P, Xiao Z, Sebastian Y, Liu Z, et al: HERC5 is a
prognostic biomarker for post-liver transplant recurrent human
hepatocellular carcinoma. J Transl Med. 13:3792015. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Yi J, Gong X, Yin XY, Wang L, Hou JX, Chen
J, Xie B, Chen G, Wang LN, Wang XY, et al: Parthenolide and arsenic
trioxide co-trigger autophagy-accompanied apoptosis in
hepatocellular carcinoma cells. Front Oncol. 12:9885282022.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Cai JB, Shi GM, Dong ZR, Ke AW, Ma HH, Gao
Q, Shen ZZ, Huang XY, Chen H, Yu DD, et al: Ubiquitin-specific
protease 7 accelerates p14(ARF) degradation by deubiquitinating
thyroid hormone receptor-interacting protein 12 and promotes
hepatocellular carcinoma progression. Hepatology. 61:1603–1614.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Jiang JH, Liu YF, Ke AW, Gu FM, Yu Y, Dai
Z, Gao Q, Shi GM, Liao BY, Xie YH, et al: Clinical significance of
the ubiquitin ligase UBE3C in hepatocellular carcinoma revealed by
exome sequencing. Hepatology. 59:2216–2227. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Tao J, Liu Z, Wang Y, Wang L, Yao B, Li Q,
Wang C, Tu K and Liu Q: MiR-542-3p inhibits metastasis and
epithelial-mesenchymal transition of hepatocellular carcinoma by
targeting UBE3C. Biomed Pharmacother. 93:420–428. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Liu K, Tian F, Chen X, Liu B, Tian S, Hou
Y, Wang L, Han M, Peng S, Tan Y, et al: Stabilization of TGF-β
receptor 1 by a receptor-associated adaptor dictates feedback
activation of the TGF-β signaling pathway to maintain liver cancer
stemness and drug resistance. Adv Sci (Weinh). 11:e24023272024.
View Article : Google Scholar
|
|
139
|
Li S, Yan L, Li C, Lou L, Cui F, Yang X,
He F and Jiang Y: NPC1 controls TGFBR1 stability in a cholesterol
transport-independent manner and promotes hepatocellular carcinoma
progression. Nat Commun. 16:4392025. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Ning J, Ye Y, Bu D, Zhao G, Song T, Liu P,
Yu W, Wang H, Li H, Ren X, et al: Imbalance of TGF-β1/BMP-7
pathways induced by M2-polarized macrophages promotes
hepatocellular carcinoma aggressiveness. Mol Ther. 29:2067–2087.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Feng X, Jia Y, Zhang Y, Ma F, Zhu Y, Hong
X, Zhou Q, He R, Zhang H, Jin J, et al: Ubiquitination of UVRAG by
SMURF1 promotes autophagosome maturation and inhibits
hepatocellular carcinoma growth. Autophagy. 15:1130–1149. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Dong L, Wei X, Yu L, Li Y and Chen L:
Inhibition of SIRT7 promotes STAT1 activation and STAT1-dependent
signaling in hepatocellular carcinoma. Cell Signal. 114:1110052024.
View Article : Google Scholar
|
|
143
|
Yang L, Tan W, Wang M, Wei Y, Xie Z, Wang
Q, Zhang Z, Zhuang H, Ma X, Wang B, et al: circCCNY enhances
lenvatinib sensitivity and suppresses immune evasion in
hepatocellular carcinoma by serving as a scaffold for SMURF1
mediated HSP60 degradation. Cancer Lett. 612:2174702025. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Zhang W, Du D, Lu H, Zhang D, Liu L, Li J,
Chen Z, Yu X, Ye M, Wang W, et al: FAT10 mediates the
sorafenib-resistance of hepatocellular carcinoma cells by
stabilizing E3 ligase NEDD4 to enhance PTEN/AKT pathway-induced
autophagy. Am J Cancer Res. 14:1523–1544. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Zeng Y, Jiang H, Chen Z, Xu J, Zhang X,
Cai W, Zeng X, Ma P, Lin R, Yu H, et al: Histone lactylation
promotes multidrug resistance in hepatocellular carcinoma by
forming a positive feedback loop with PTEN. Cell Death Dis.
16:592025. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Tong HV, Hoan NX, Binh MT, Quyen DT, Meyer
CG, Hang DTT, Hang DTD, Son HA, Van Luong H, Thuan ND, et al:
Upregulation of enzymes involved in ISGylation and ubiquitination
in patients with hepatocellular carcinoma. Int J Med Sci.
17:347–353. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Qin Y, Wang CJ, Ye HL, Ye GX, Wang S, Pan
DB, Wang J, Shen HJ and Xu SQ: WWP2 overexpression inhibits the
antitumor effects of doxorubicin in hepatocellular carcinoma. Cell
Biol Int. 46:1682–1692. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Zhao L, Ma H, Jiang Y, Li Y, Qiao L, Chen
Y, Jiang X, Wang L, Wang S and Fan X: Identification of an m6A
natural inhibitor, lobeline, that reverses lenvatinib resistance in
hepatocellular tumors. J Nat Prod. 87:1983–1993. 2024. View Article : Google Scholar : PubMed/NCBI
|