Introduction
According to the 11th edition of the International
Diabetes Federation Diabetes Atlas, the global prevalence of
diabetes is projected to increase substantially (1). Diabetic kidney disease (DKD), a
microvascular complication of diabetes, occurs in ~40% of
individuals with diabetes and represents a predominant cause of
end-stage renal disease (2).
Despite its clinical importance, the pathogenesis of DKD remains
incompletely understood, with contributing factors including
hyperglycaemia, inflammatory injury, metabolic dysregulation,
oxidative stress and haemodynamic alterations (3-5).
Current therapeutic strategies in clinical practice are limited to
glycemic, lipid and blood pressure management, as no effective
disease-modifying treatments are available (6). Consequently, early detection of DKD
is important, as timely intervention can notably mitigate
associated morbidity and mortality.
Recent advances have highlighted the pivotal role of
metabolic reprogramming and epigenetic modifications in the
progression of DKD (7-9). Within the hyperglycemic and hypoxic
microenvironment characteristic of DKD, renal cells undergo
considerable metabolic alterations, preferentially utilizing
glycolysis for energy production and generating excessive lactate
even under normoxic conditions, a phenomenon analogous to the
'Warburg effect' observed in cancer cells (10-12). Once regarded merely as a
metabolic byproduct of hypoxia, lactate is now recognized to
perform essential physiological functions (13,14). Accumulating evidence suggests
that lactate serves as a key signalling molecule, modulating gene
expression and cellular functions through a novel epigenetic
mechanism involving posttranslational modification (PTM) known as
lactylation (9,15-17).
Emerging evidence implicates cell death pathways,
particularly autophagy and ferroptosis, in the pathogenesis of DKD
(18-20). Notably, lactylation has been
identified as a potential regulatory mechanism in cell death
processes (21). Renal fibrosis,
a hallmark pathological feature of advanced kidney disease with
various etiologies, carries out a pivotal role in driving the
progression of DKD to renal functional decline (22,23). In addition to the
well-established TGF-β signalling pathway (24,25), previous investigations have
revealed substantial contributions of epigenetic modifications to
the pathogenesis of renal fibrosis (26-28). However, the precise regulatory
mechanisms of lactylation in diabetic kidney fibrosis, particularly
its integration with dysregulated ferroptosis, autophagy, and
fibrotic processes, remain poorly understood. Elucidating these
mechanisms could provide key insights into the transition from
metabolic dysregulation to irreversible organ damage in DKD. While
individual studies have implicated lactylation in fibrotic or
inflammatory pathways (29,30), a cohesive framework integrating
ferroptosis and autophagy remains to be established (31).
Based on current evidence, the present review
proposes the concept that lactylation is a key modulator of cell
death decisions in DKD. The present review synthesizes current
evidence on the complex metabolic-epigenetic interplay between
ferroptosis and autophagy, to explore how their concurrent and
synergistic interactions may contribute to fibrotic remodeling and
DKD progression. The present review aims to propose novel targeted
therapeutic strategies and foster interdisciplinary research at the
nexus of metabolic and epigenetic regulation.
The glycolytic-lactate axis: A metabolic
signature of the DKD microenvironment
Glycolysis in DKD
Local tissue oxidative stress has been identified as
a key factor in the development and progression of DKD. Multiple
pathways generating reactive oxygen species, including glycolysis
and the polyol pathway, have been implicated in the pathogenesis of
DKD (32). Glycolysis, a
fundamental metabolic pathway conserved across organisms, generates
pyruvate and two ATP molecules through the catabolism of glucose.
This pathway represents the initial stage of glucose catabolism,
comprising ten enzymatic reactions. While the majority of these
reactions are reversible, the three steps catalyzed by hexokinase
(HK), phosphofructokinase-1 (PFK1) and pyruvate kinase (PK) are
irreversible, with the activity of these key enzymes playing a key
role in regulating the overall rate of glycolytic flux (33,34). The transport of glucose across
the cell membrane, facilitated by glucose transporter proteins, is
a key regulatory step that governs cellular glucose uptake
(35).
Within the cytoplasmic compartment, HK catalyzes the
phosphorylation of glucose to glucose-6-phosphate, representing
both the initial committed step and the first rate-limiting
reaction in the glycolytic pathway (36). Among the four HK isoforms (HK1,
HK2, HK3 and HK4), HK2 exhibits superior glycolytic promotion
efficiency, with its renal activity being markedly diminished in
diabetic animal models, suggesting its potential therapeutic
relevance in DKD (37,38). PFK1 governs the second
rate-limiting step, facilitating the conversion of
fructose-6-phosphate to fructose-1,6-bisphosphate, thereby serving
as a key regulatory node in glycolytic flux (39). PK, the terminal rate-limiting
enzyme, catalyzes the transformation of phosphoenolpyruvate to
pyruvate, influencing both glycolytic flux and energy metabolism
through pyruvate fate determination (40). The mammalian PK system comprises
four isoforms: Erythrocyte PK, liver PK and muscle isoforms PKM1
and PKM2 (41). Emerging
evidence suggests that PKM2 is a potential biomarker for early DKD
detection and a potential therapeutic target (42,43). Elevated plasma lactate levels, a
glycolytic byproduct, are observed in diabetic patients (44), with lactate dehydrogenase (LDH)
isoforms mediating the reversible conversion between pyruvate and
lactate: LDHA catalyzes pyruvate-to-lactate conversion, and LDHB
facilitates the reverse reaction (45,46). This conserved energy pathway is
precisely regulated by its rate-limiting enzymes (HK, PFK1 and
PKM2), with LDHA-mediated pyruvate-to-lactate conversion
representing the terminal regulatory hub.
The lactate shuttle mechanism and its
signaling role
Lactate, a pivotal metabolite generated through the
Warburg effect, has dual functions as both an energy substrate and
a metabolic byproduct (47). The
lactate shuttle theory elucidates the mechanisms by which lactate
operates within biological systems. This theory, which involves
both intercellular and intracellular lactate shuttling, delineates
the comprehensive process of lactate transmembrane transport
(48). The intercellular lactate
shuttle was initially proposed and systematically articulated by
Brooks in 1985 (49). This
concept posits that lactate, which functions as a metabolic
intermediate, traverses the interstitium and vascular system,
supplying carbon sources for gluconeogenesis and bio-oxidation,
thereby fostering the progressive recognition of the novel
biological roles of lactate (14,48). Brooks further advanced the
intracellular lactate shuttle theory in 1998 (50), which posits that lactate produced
in the cytosol via glycolysis or glycogenolysis can directly enter
the mitochondria of the same cell for oxidation, bypassing the need
for prior conversion to pyruvate in the cytosol. This theory is
underpinned by studies on LDH enzyme kinetics (51,52). Brooks also introduced the
mitochondrial lactate oxidation complex model, which reinforces
this theoretical framework (14,48). Nevertheless, the intracellular
lactate shuttle theory remains a subject of debate and warrants
further in-depth investigation (53-55).
The transport of lactate across cell membranes is
facilitated by the monocarboxylate transporter (MCT) protein family
(56). The MCT family,
classified within the solute carrier family 16 (SLC16), carries out
a key role in human pathophysiology by regulating the bidirectional
transport of key metabolites in fundamental metabolic processes
(57). The MCT family comprises
14 proteins, each exhibiting distinct tissue distributions that
reflect their specific roles in various metabolic and physiological
contexts. Among these, MCT1, MCT2 and MCT4 are widely expressed and
catalyze the coupled, bidirectional transport of protons and
monocarboxylates, including lactate (58). Under normal physiological
conditions, the coordinated activity of MCT1, MCT2 and MCT4
collectively maintains systemic lactate homeostasis. Specifically,
MCT1 and MCT2 primarily mediate lactate influx into cells, whereas
MCT4 is predominantly responsible for lactate efflux (59,60).
Lactate, which is traditionally regarded as a
metabolic byproduct of anaerobic glycolysis, has emerged as a
multi-functional signalling molecule that carries out pivotal roles
in both physiological and pathological processes (13,14,61). Mechanistically, lactate serves as
a substrate for gluconeogenesis, which is mediated by LDHB, thereby
contributing to energy homeostasis (62). Furthermore, lactate functions as
a key regulator of the cellular redox balance by modulating the
intracellular ratio of reduced to oxidized nicotinamide adenine
dinucleotide (NADH/NAD+), thereby stabilizing the redox
state through its involvement in alternative metabolic pathways
(63). Additionally, lactate
accumulation has been shown to facilitate intracellular fatty acid
synthesis, a key process for maintaining cellular membrane
integrity, signal transduction and energy storage (14,64). Notably, lactate has been
identified as a regulator of gene expression through lactylation, a
novel epigenetic modification mechanism (15). These findings collectively
challenge the conventional view of lactate as a metabolic waste
product, instead positioning it as a central signalling molecule
within the lactate shuttle framework, with the capacity to modulate
gene expression networks. Notably, lactate has been demonstrated to
activate TGF-β through a pH-dependent mechanism, thereby promoting
fibrotic processes (65).
Concurrently, lactate induces a mild reactive oxygen species burst
that triggers nuclear factor erythroid 2-related factor 2
activation, enhancing antioxidant defenses and cell survival
(66,67). These dual signaling roles, which
are intricately linked to lactate metabolism and transport, are
summarized in Fig. 1 and will be
further discussed in the context of DKD pathogenesis in subsequent
sections.
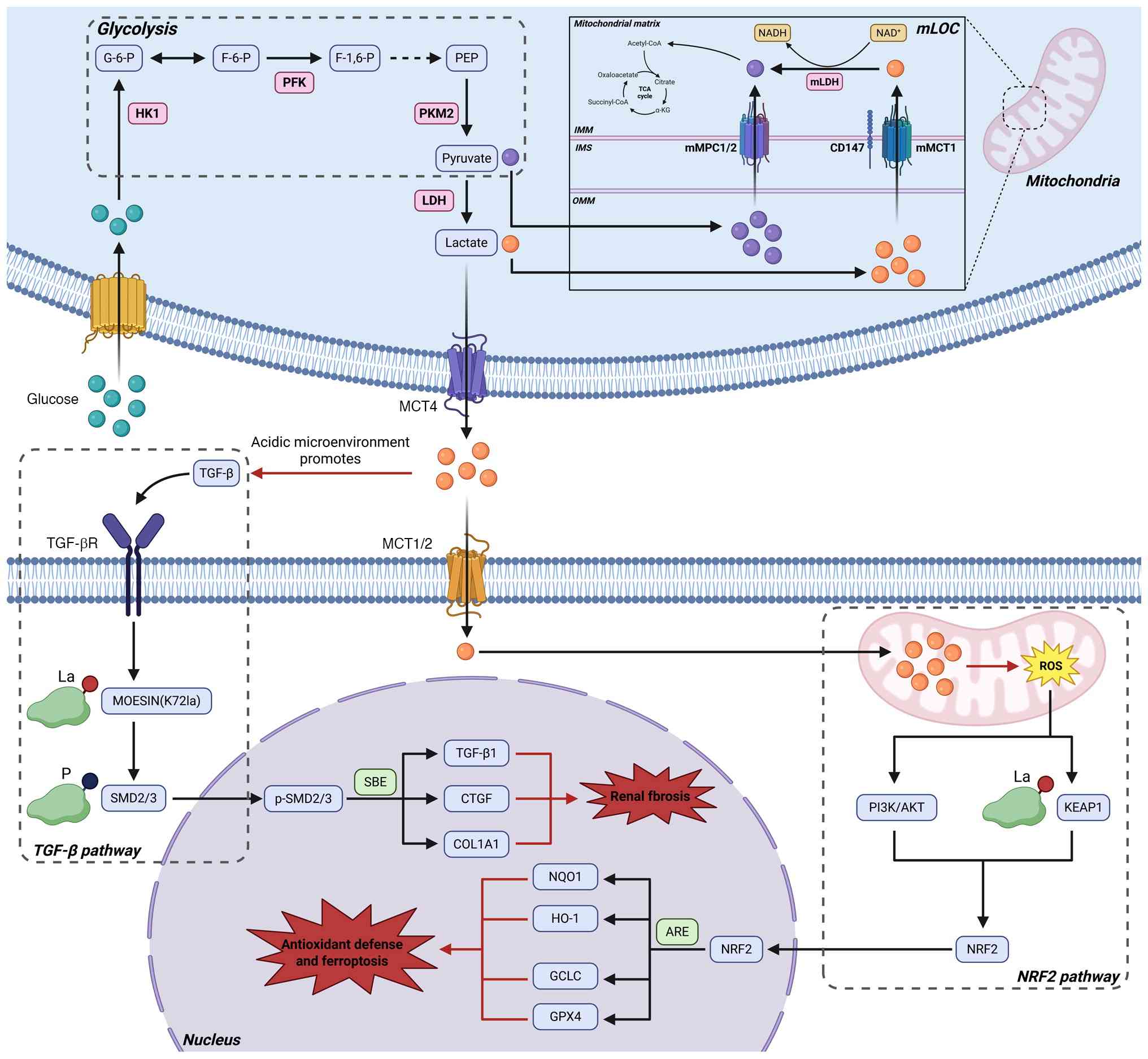 | Figure 1Lactate shuttling and its dual
signaling roles in DKD. This schematic integrates intercellular
lactate transport (mediated by MCT1/MCT4), mitochondrial lactate
oxidation complex (mLOC)-mediated intracellular lactate metabolism,
along with lactate-induced activation of pro-fibrotic TGF-β
signaling via a pH-dependent mechanism and the activation of the
antioxidant NRF2 response through a mild ROS burst. HK1: Hexokinase
1; G-6-P, Glucose-6-phosphate; F-6-P, Fructose-6-phosphate; PFK,
Phosphofructokinase; F-1,6-P, Fructose-1,6-bisphosphate; PEP,
Phosphoenolpyruvate; PKM2, Pyruvate kinase M2; LDH, Lactate
dehydrogenase; mLOC, Mitochondrial lactate oxidation complex; IMM,
Inner mitochondrial membrane; IMS, Intermembrane space; OMM, Outer
mitochondrial membrane; NAD+, Nicotinamide adenine
dinucleotide; NADH, Nicotinamide adenine dinucleotide hydrogen;
mLDH, Mitochondrial lactate dehydrogenase; TCA cycle, Tricarboxylic
acid cycle; α-KG, α-ketoglutarate; mMPC1/2, Mitochondrial pyruvate
carrier 1/2; CD147, Cluster of Differentiation 147; mMCT1,
Mitochondrial monocarboxylate transporter 1; MCT1/2,
Monocarboxylate transporter 1/2; MCT4, Monocarboxylate transporter
4; TGF-β, Transforming growth factor β; TGF-βR, Transforming growth
factor β receptor; La, Lactylation; P, Phosphorylation; MOESIN,
Membrane-organizing extension spike protein; SMD2/3, Mothers
against decapentaplegic homolog 2/3; p-SMD2/3, Phosphorylated
mothers Against decapentaplegic homolog 2/3; SBE, SMAD binding
element; CTGF, Connective tissue growth factor; COL1A1, Collagen
type 1 α chain; ROS, Reactive oxygen species; PI3K,
Phosphoinositide 3-kinase; AKT, AKT serine/threonine kinase; KEAP1,
Kelch-like ECH-associated protein 1; NRF2, Nuclear factor erythroid
2-related factor 2; ARE, Antioxidant Response Element; NQO1,
NADP-quinone oxidoreductase; HO-1, Heme oxygenase 1; GCLC,
Glutamate-cysteine ligase catalytic subunit; GPX4, Glutathione
peroxidase 4. |
Dysregulated glycolysis and lactate
accumulation in DKD
Metabolic reprogramming is increasingly recognized
as a key feature of DKD, primarily driven by enhanced glycolysis
and subsequent lactate accumulation. The sustained renal hypoxia
observed during the initial stages of the pathological progression
carries out a particularly key role in this process. Clinical
investigations have established that elevated lactate
concentrations and diminished redox potential are closely
associated with this hypoxic state, a phenomenon analogous to, yet
distinct from, the Warburg effect observed in malignant cells
(11,68).
Substantial evidence indicates that under diabetic
conditions, renal cells exhibit increased dependence on glycolysis
despite sufficient oxygen availability, resulting in notable
lactate production and accumulation. Srivastava et al
(69) demonstrated that sirtuin
(SIRT) 3 deficiency is associated with aberrant glycolytic
activation in diabetic renal fibrosis, characterized by the
upregulation of key glycolytic enzymes (HK2 and PKM2), elevated
lactate levels and the activation of hypoxia-inducible factor 1α
(HIF-1α), which subsequently perpetuates dysregulated glycolysis.
These findings are corroborated by Liu et al (70) Park et al (71) and Jain et al (72). These findings collectively
establish that disrupted PKM2 tetramer-dimer ratios and HIF-1α
accumulation under hyperglycemic conditions promote pathological
glycolytic activation and accelerate renal fibrotic progression.
Consequently, these findings suggest that therapeutic targeting of
aberrant glycolysis warrants further investigation as a potential
strategy for DKD intervention.
HIF-1, a heterodimeric transcription factor
comprising an oxygen-sensitive α-subunit and a constitutively
expressed β-subunit, is ubiquitously expressed in hypoxic cellular
environments (73). The
pathogenesis of renal injury in DKD is driven primarily by chronic
exposure to hyperglycemia and hypoxia, with HIF-1α serving as a
central mediator of the adaptive hypoxic response (74). HIF-1α exerts its regulatory
functions through binding to hypoxia-response elements
(5'-RCGTG-3') within the promoter regions of target genes, thereby
modulating critical processes, including glycolysis and
angiogenesis (75). Experimental
evidence has demonstrated that dysregulated HIF-1α activation in
DKD promotes renal interstitial fibrosis and is associated with
pathological structural alterations and proteinuria (76,77). Furthermore, HIF-1α orchestrates
the downregulation of oxidative phosphorylation through
hypoxia-mediated signalling pathways while simultaneously enhancing
the expression of key glycolytic enzymes (HK, PFK, PKM2 and LDHA),
thereby augmenting glycolytic flux (78,79). These mechanistic insights
position HIF-1α as a potential therapeutic target for DKD
management.
A substantial body of clinical evidence has reported
an association between the activation of glycolytic pathways and
the pathogenesis of DKD. In a large-scale cohort study involving
4,888 patients, Tang et al (80) observed a positive association
between LDH levels and DKD incidence in patients with type 2
diabetes mellitus (T2DM). Furthermore, Lee et al (51) in both human and animal models of
DKD, disease progression is mechanistically associated with
LDHA-mediated lactic acidosis under hypoxic conditions, which
subsequently induces fibrotic changes and mitochondrial
dysfunction. Through comprehensive integration of clinical data,
multiomics analyses and in vivo/in vitro experimental
models, Darshi et al (81) reported that lactate functions not
only as a biomarker for progressive renal functional decline but
also as a pivotal pathogenic metabolite in DKD development. These
findings are corroborated by multiple animal studies (44,82), which collectively highlight the
key regulatory role of LDH activity in glycemic homeostasis and DKD
progression.
The intracellular accumulation of lactate disrupts
renal acid-base homeostasis, and persistent metabolic acidosis can
lead to progressive renal damage and irreversible nephron injury
(83). Renal tubular epithelial
cells play an important role in systemic pH regulation through
precise modulation of proton secretion (84). TGF-β, a well-established central
mediator of fibrotic processes, is implicated in of DKD
pathogenesis (85). In pulmonary
fibrosis models, lactate has been reported to activate TGF-β in a
pH-dependent manner, subsequently promoting fibrogenesis and
upregulating HIF-1α expression, thereby establishing a positive
feedback loop that enhances lactate production (65,86,87). While this specific mechanism has
not been directly demonstrated in renal fibrosis, emerging evidence
suggests a robust interplay between the lactate and TGF-β
signalling pathways in the kidney: Lactate promotes renal fibrosis
through activation of the transient receptor potential vanilloid 4
channel and the TGF-β/Smad2/3 signalling axis (88), with lactate accumulation
exacerbating renal fibrosis in DKD animal models (77,89,90). These findings suggest that
lactate and TGF-β may act synergistically to drive fibrotic
processes in the kidney, although the precise molecular mechanisms
warrant further experimental investigation.
The DKD microenvironment is shaped by three
interconnected pathological factors: Hyperglycemia, hypoxia and
acidosis. These elements form a self-amplifying feedforward cycle
that drives glycolytic flux and lactate accumulation. Within the
distinct metabolic landscape of DKD, lactic acid emerges as a key
metabolic byproduct, serving as both a pathogenic signaling
molecule and a precursor for lactylation. A key question is how
this persistent metabolic stress translates into sustained
pathological gene expression and cellular fate determination. It is
plausible that lactylation may act as a regulator in this process,
potentially associating enhanced glycolytic flux with stable
epigenetic modifications and PTMs. Elucidating the intricate
mechanisms underlying these processes may provide novel insights
into the pathogenesis of DKD and inform innovative therapeutic
strategies (Fig. 2) (25,91).
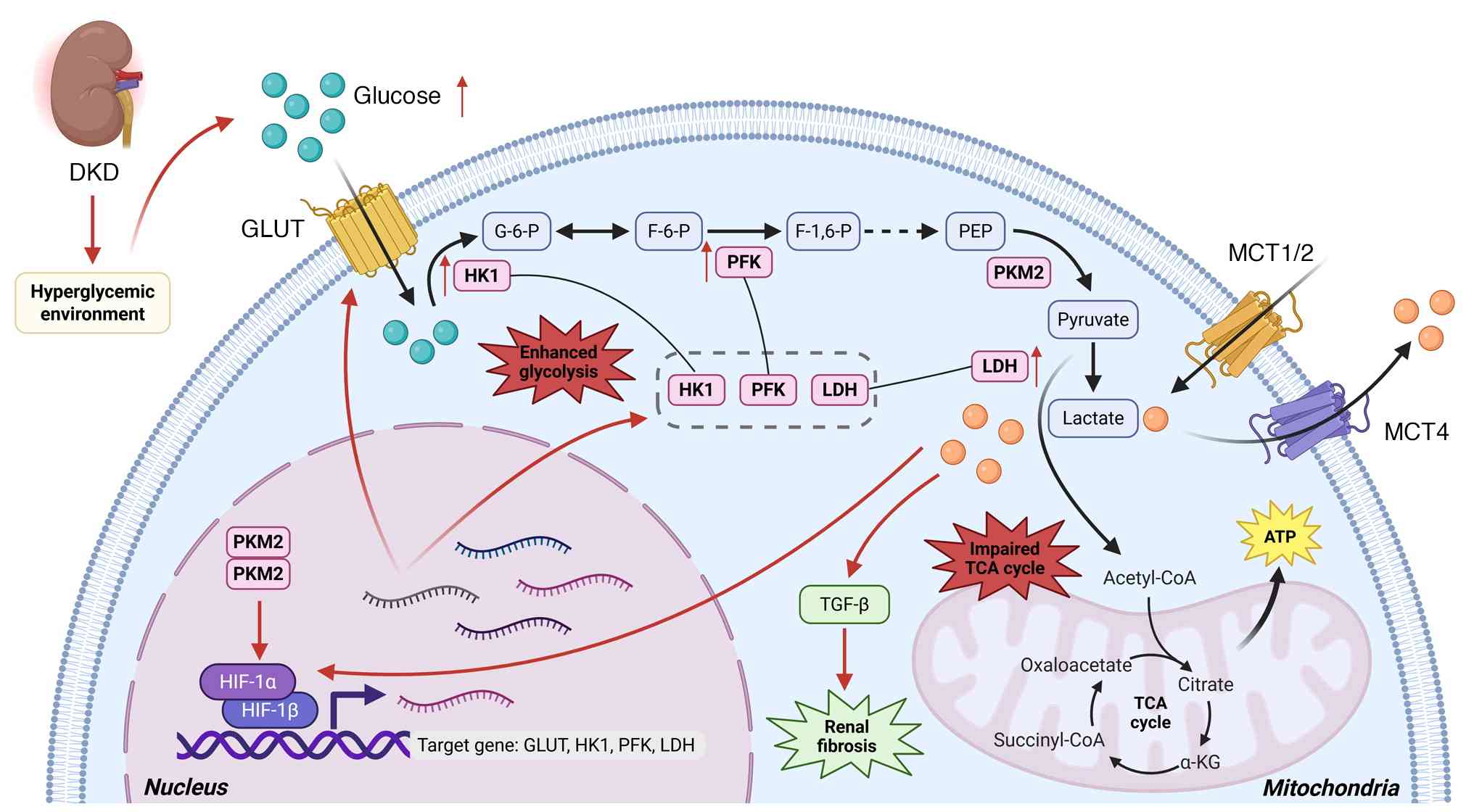 | Figure 2Hyperglycemia-induced metabolic
reprogramming fosters a microenvironment conducive to lactylation
in DKD. In diabetic kidneys, the synergistic effects of
hyperglycemia and hypoxia induce excessive glycolysis, resulting in
aberrant lactate accumulation. Under diabetic conditions, impaired
lactate homeostasis leads to both intracellular and extracellular
lactate accumulation. This lactate surplus triggers the activation
of TGF-β signaling pathways, thereby promoting fibrotic processes,
while simultaneously stabilizing HIF-1α in the nucleus, which
upregulates glycolytic enzyme expression and establishes a
self-perpetuating glycolytic loop. Furthermore, the diversion of
pyruvate from mitochondrial oxidative phosphorylation to lactate
production exacerbates energy deficits and mitochondrial
dysfunction. DKD, Diabetic kidney disease; GLUT, Glucose
transporter; HK1, Hexokinase 1; G-6-P, Glucose-6-phosphate; F-6-P,
Fructose-6-phosphate; PFK, Phosphofructokinase; F-1,6-P,
Fructose-1,6-bisphosphate; PEP, Fructose-1,6-bisphosphate; PKM2,
Pyruvate kinase M2; LDH, Lactate dehydrogenase; MCT1/2,
Monocarboxylate transporter 1/2; MCT4, Monocarboxylate transporter
4; HIF-1α, Hypoxia-inducible factor 1-α; HIF-1β, Hypoxia-inducible
factor 1-β; ATP, Adenosine triphosphate; TGF-β, Transforming growth
factor β; TCA cycle, Tricarboxylic acid cycle; α-KG,
α-ketoglutarate. |
Molecular mechanisms of lactylation in
DKD
In 2019, a research team led by Yingming et
al (15) at the University
of Chicago first reported a novel PTM termed lysine lactylation
(Kla). This modification involves the transfer of lactyl groups to
specific amino acid residues, particularly lysine, on target
proteins, thereby modulating the expression of associated genes and
proteins (14,47). This seminal discovery opened new
avenues for lactylation research, positioning lactate as a key
mediator in epigenetic regulatory pathways (92). The distinctive metabolic milieu
characteristic of DKD provides an optimal biochemical environment
for lactylation modifications. The next section will discuss the
mechanistic pathways through which metabolic stress in DKD may be
linked to functional epigenetic and post-translational signaling
networks.
Enzymatic and non-enzymatic lactylation
modifications
Lactylation modifications can be classified into two
distinct mechanisms: Enzymatic and non-enzymatic processes. While
lactate serves as the primary substrate in both pathways, its
stereochemical configuration differs between the two mechanisms.
Specifically, L-lactate is utilized in enzymatic lactylation,
whereas D-lactate is implicated in nonenzymatic lactylation
(93).
Enzymatic lactylation modification
Enzymatic lactylation constitutes a reversible,
enzyme-regulated dynamic process, which is orchestrated by three
principal functional components: Writers, erasers and readers
(94). The dysregulation of this
intricate regulatory system, particularly within the distinct
metabolic milieu of DKD, is postulated to constitute a fundamental
mechanistic pathway underlying the pathogenesis of pathological
hyperlactylation.
Writers
The enzymatic addition of lactyl groups to specific
protein residues is mediated by a class of enzymes referred to as
'writers' (95). Current
evidence suggests that this catalytic activity may occur through at
least two distinct biochemical pathways: The L-lactyl-CoA pathway
and the lactate-AMP pathway (96). DKD microenvironment displays
features that may favor the activation of these pathways. Chronic
hyperglycemic conditions coupled with hypoxia can increase lactate
production, thereby providing a substantial substrate pool for
these enzymatic reactions.
In the L-lactyl-CoA pathway, lactate is
enzymatically converted into the high-energy donor lactyl-CoA by
acetyl-CoA synthetase 2 (ACSS2) and GTP-specific succinyl-CoA
synthetase (GTPSCS) within the nucleus, which serve as substrates
for subsequent lactylation modifications. Zhu et al
(97) reported that ACSS2
facilitates histone lactylation, tumor growth and immune evasion
through its interaction with lysine acetyltransferase 2A (KAT2A),
thereby identifying ACSS2 and KAT2A as previously unrecognized
lactyl-CoA synthetases and lactyltransferases, respectively. In a
separate study, Liu et al (98) showed that GTPSCS translocates to
the nucleus and cooperates with p300 to increase histone
lactylation without concurrent succinylation, establishing GTPSCS
as the first enzyme that catalyzes lactyl-CoA synthesis for
epigenetic histone lactylation. Collectively, these findings
suggest that both ACSS2 and GTPSCS may functional 'lactyl-CoA
synthetases' in the conversion of lactate to lactyl-CoA.
While the complete repertoire of lactylation writers
has not been fully elucidated, current evidence points to
functional overlap with enzyme families that mediate other PTMs,
including histone acetyltransferases (HATs), such as CBP/p300,
GCN5, KAT5, KAT7 (HBO1) and KAT8, as well as other modifiers, such
as α-tubulin acetyltransferase 1 and histone deacetylase (HDAC) 6
(99). CBP and p300,
ubiquitously expressed paralogues essential for animal development,
function as transcriptional coactivators for numerous transcription
factors, including nuclear receptors (100). These proteins contain multiple
core domains, such as the HAT domain, bromodomain and ZZ-type zinc
finger domain, and regions rich in cysteine and histidine residues
[encompassing the plant homeodomain and really interesting new gene
(RING) domains], which facilitate acyl group transfer to diverse
substrates (101). Notably,
p300 was the first protein identified to exhibit lactylation writer
activity (15). CBP/p300
mediates the L-lactyl-CoA pathway by utilizing its bromodomain to
recognize lactyl-CoA and catalyze the transfer of the lactyl group
to lysine residues on target proteins, thereby influencing
chromatin remodelling and the transcription of pro-oncogenic genes.
This process requires lactyl-CoA as a high-energy donor substrate
rather than free lactate directly (102,103). Notably, lactyl-CoA originates
from lactate, which is itself produced from pyruvate through LDH
catalysis, linking this pathway and glycolytic flux. The activation
of CBP/p300 has been associated with reactive oxygen species
production, inflammation and extracellular matrix (ECM) protein
synthesis in DKD. The hyperglycemic and hypoxic conditions
characteristic of DKD further amplify lactate generation,
potentially intensifying these pathological processes (104,105). Elevated reactive oxygen species
levels and inflammation are known contributors to renal fibrosis
(106). To the best of our
knowledge, while no studies have explicitly demonstrated
CBP/p300-mediated lactylation in DKD, based on parallels with
acetylations, it is plausible that this process is likely to occur
under DKD-specific conditions. If confirmed, this would position
CBP/p300 as a promising therapeutic target for DKD
intervention.
The lactate-AMP pathway involves aminoacyl-tRNA
synthetases 1 and 2 (AARS1/2) in lactylation. AARS1 is
predominantly localized in the cytoplasm, whereas AARS2 is
primarily located in the mitochondria. These enzymes employ an
ATP-dependent mechanism to directly convert lactate and ATP into a
lactyl-AMP intermediate, which subsequently transfers the lactyl
group to lysine residues on substrate proteins, resulting in
lactylation (107-109). In human embryonic kidney cells,
the knockdown of AARS1 expression suppresses lactylation, whereas
its overexpression increases lactylation levels (107,110), suggesting that AARS enzymes may
function as lactyltransferases in this process. This regulatory
mechanism may also be operative in the kidney, potentially offering
new insights for DKD prevention and treatment. The discovery of the
lactate-AMP pathway, in contrast to the first pathway, points to a
potentially more direct cellular response to intracellular lactate
levels. In the context of DKD, renal cells experience chronic
lactate overload, which may induce persistent activation of
AARS1/2, resulting in extensive lactylation of both cytoplasmic and
mitochondrial proteins. This process could directly disrupt
cellular functions, thereby exacerbating the mitochondrial
dysfunction and metabolic inflexibility characteristic of DKD.
Further studies investigating whether the expression or activity of
AARS1/2 is modulated in renal cells could provide valuable insights
and potentially identify novel therapeutic targets.
Erasers
The elimination of lactylation modifications plays a
pivotal role in dynamic cellular signalling and is regulated by
specific enzymatic erasers (95). To date, two primary classes of
erasers have been reported in lactylation regulation: Class I
HDAC1-3 and SIRT deacetylases 1-3 (111,112). These erasers are considered to
function in opposition to lactylation writers to maintain metabolic
homeostasis. It has been hypothesized that in DKD, dysregulation of
epigenetic modifications, characterized by diminished eraser
activity in conjunction with heightened writer function, may
contribute to a pathological state of lactylation.
In vitro studies have demonstrated that class
I HDAC1-3 considerably reduce lysine lactylation levels on
histones, with HDAC1 and HDAC3 exhibiting distinct site-specific
activity in histone delactylation (113-115). Additionally, Du et al
(116) established through
overexpression and knockout experiments that SIRT1 and SIRT3 serve
as potent erasers in mammalian cells, modulating lactylation across
both histone and non-histone protein substrates. Notably, SIRT2 has
been shown to carry out a key role in non-histone protein
delactylation, specifically by removing lactylation modifications
from METTL16, thereby influencing cuproptosis regulation (117).
The activity of these erasers appears to be context
dependent and potentially influenced by factors such as expression
levels, PTMs, subcellular localization and the availability of
cofactors and coenzymes (118).
It remains unclear whether such regulatory mechanisms operate
within the metabolically dysregulated environment of DKD to prevent
excessive lactylation accumulation. HDACs and SIRTs have been the
subject of extensive investigation in the context of renal
pathology. Current evidence implicates HDACs in DKD pathogenesis,
particularly through their association with pathological processes,
including fibrosis, oxidative stress and inflammation (119). Daude et al (120) demonstrated that reduced HDAC2
activity attenuates renal injury in diabetic model rats. While HDAC
inhibitors have demonstrated some efficacy in improving renal
function in DKD animal models, clinical trials have identified
notable toxic and off-target effects, highlighting the need for the
development of agents with improved safety profiles (121). Among the SIRT family members,
SIRT1 is predominantly expressed in the nucleus and widely
expressed in renal tubular cells and podocytes, SIRT2 is uniquely
expressed in the cytoplasm and SIRT3, which is localized to the
mitochondrial matrix, serves as a key regulator of organelle
acetylation (122,123). SIRT1 is the most extensively
studied member of this family, with the SIRT family collectively
participating in diverse biological processes, including the
oxidative stress response, cell cycle regulation, metabolism and
apoptosis (124). Current
research indicates that SIRT1-3 contribute to renal homeostasis
maintenance, with the upregulation of SIRT1, SIRT2 and SIRT3
demonstrating protective effects against renal injury and fibrosis
in DKD (125-128). However, the potential role of
HDAC1-3 and SIRT1-3 in lactylation erasure within the context of
DKD remains unexplored. If their delactylase activity is preserved
in the DKD microenvironment, this could imply novel functional
roles for these enzymes and may advance the understanding of DKD
pathophysiology.
Readers
The biological consequences of lactylation are
mediated by specialized 'reader' proteins that specifically
recognize and interpret lactylated amino acid residues,
subsequently recruiting downstream effector complexes to modulate
gene expression or protein function (96). Although research on
enzyme-catalyzed lactylation readers is still in its nascent
stages, considerable progress has been made. For instance, Hu et
al (129) identified Brg1
as a reader of H3K18la, demonstrating its accumulation at promoter
regions of genes associated with pluripotency and epithelial
junctions, thereby playing a pivotal role in induced pluripotent
stem cell reprogramming. Additionally, recent studies have revealed
that TRIM33β functions as a reader of H3K18la, regulating the TGF-β
signalling pathway (130),
whereas DPF2, a reader of H3K14la, is important for chromatin
remodelling and gene expression modulation in cancer (131).
In the context of DKD, it remains an open question
whether specific epigenetic readers are recruited by lactylation to
drive fibrotic progression. Notably, the reader protein TRIM33β,
which recognizes H3K18la and modulates TGF-β signaling, represents
a promising candidate. Within the unique microenvironment of DKD,
lactate-induced H3K18la could facilitate the recruitment of TRIM33β
to the promoters of profibrotic genes, potentially activating their
transcriptional. If confirmed, this would establish a mechanistic
association between glycolytic flux and fibrotic pathogenesis. The
identification and functional characterization of such DKD-specific
epigenetic readers are key to elucidating the molecular mechanisms
by which lactylation signaling contributes to disease
progression.
In summary, enzymatic lactylation serves as an
active regulatory interface that associates metabolic processes
with cellular functions. In DKD, the accumulation of excess
substrates (lactic acid) and activated writers, coupled with
impaired erasers, leads to the saturation of cellular environments
with lactylation markers. These markers are subsequently recognized
by specific reader proteins, which initiate downstream signaling
cascades promoting fibrosis and programmed cell death. The
dysregulation of these three key molecular components suggests a
shift of lactylation from a dynamic regulatory mechanism toward a
contributor to pathology, thereby influencing cellular fate
decisions and fibrotic remodeling in DKD.
Non-enzymatic lactylation
modification
Non-enzymatic lactylation, which is distinct from
its enzymatic counterpart, occurs independently of enzymatic
catalysis in metabolic regulation. This process is predominantly
mediated by D-lactate, which undergoes lactylation through a
nucleophilic substitution reaction between S,D-lactoylglutathione
(LGSH) and lysine residues (132). LGSH is synthesized by
glyoxalase 1 (GLO1), which catalyzes the reaction between
methylglyoxal (MGO), a byproduct of glycolysis and glutathione
(GSH) (133). MGO, a
well-characterized metabolic byproduct of glycolysis, is generated
from glycolytic intermediates such as dihydroxyacetone phosphate
and glyceraldehyde 3-phosphate (134). Under physiological conditions,
MGO is detoxified by the GLO system, which comprises GLO1 and GLO2.
GLO1 converts MGO into stable LGSH, whereas GLO2 hydrolyses LGSH to
yield D-lactate and regenerate GSH (135).
In DKD, the endogenous protective mechanisms may
become compromised. Chronic hyperglycemia markedly elevates the
generation of glycolytic intermediates, resulting in the
pathological accumulation of MGO. MGO serves as a key precursor for
the formation of advanced glycation end products, which are
strongly implicated in the pathogenesis and progression of DKD
(136,137). A causal relationship between
GLO1 and DKD has been established, particularly in tissues such as
the skin and salivary glands, raising the possibility of its
utility as a DKD biomarker (138). Consequently, impaired MGO
clearance resulting from glycolytic overflow or dysfunctional GLO
activity promotes the diversion of MGO into the non-enzymatic
lactylation pathway, leading to the production of D-lactate and
LGSH.
Non-enzymatic lactylation predominantly targets
lysine residues, whose reactive ε-amino groups exhibit increased
susceptibility to modification under conditions of metabolic
stress. Elevated lactate concentrations, particularly within the
acidic pH environment associated with DKD-induced metabolic
acidosis, can facilitate the protonation of carboxyl groups of
lactate. This protonation markedly enhances the reactivity of
lactate, enabling a direct nucleophilic attack on the ε-amino
groups of lysine to form stable amide bonds, thereby generating
Kla. This 'mass effect'-driven mechanism differs from the tightly
regulated process of enzymatic lactylation and has been proposed as
a potential indicator of metabolic dysregulation (93,135,139).
In summary, the DKD microenvironment may be subject
to both enzymatic and non-enzymatic lactylation modifications,
which may transition from regulatory mechanisms to contributors to
pathogenesis. These modification pathways may function in a
coordinated manner: Non-enzymatic lactylation induces widespread,
stochastic protein damage, establishing the foundational
pathological conditions. Within this framework, signal-driven
enzymatic lactylation may contribute to specific pathological
programs, including the transcriptional activation of fibrotic
genes and the direct suppression of autophagy and ferroptosis. This
synergistic interplay facilitates the efficient conversion of
high-throughput glycolytic states into the precise execution of
cellular damage and death pathways, potentially contributing to
fibrotic remodeling (Fig.
3).
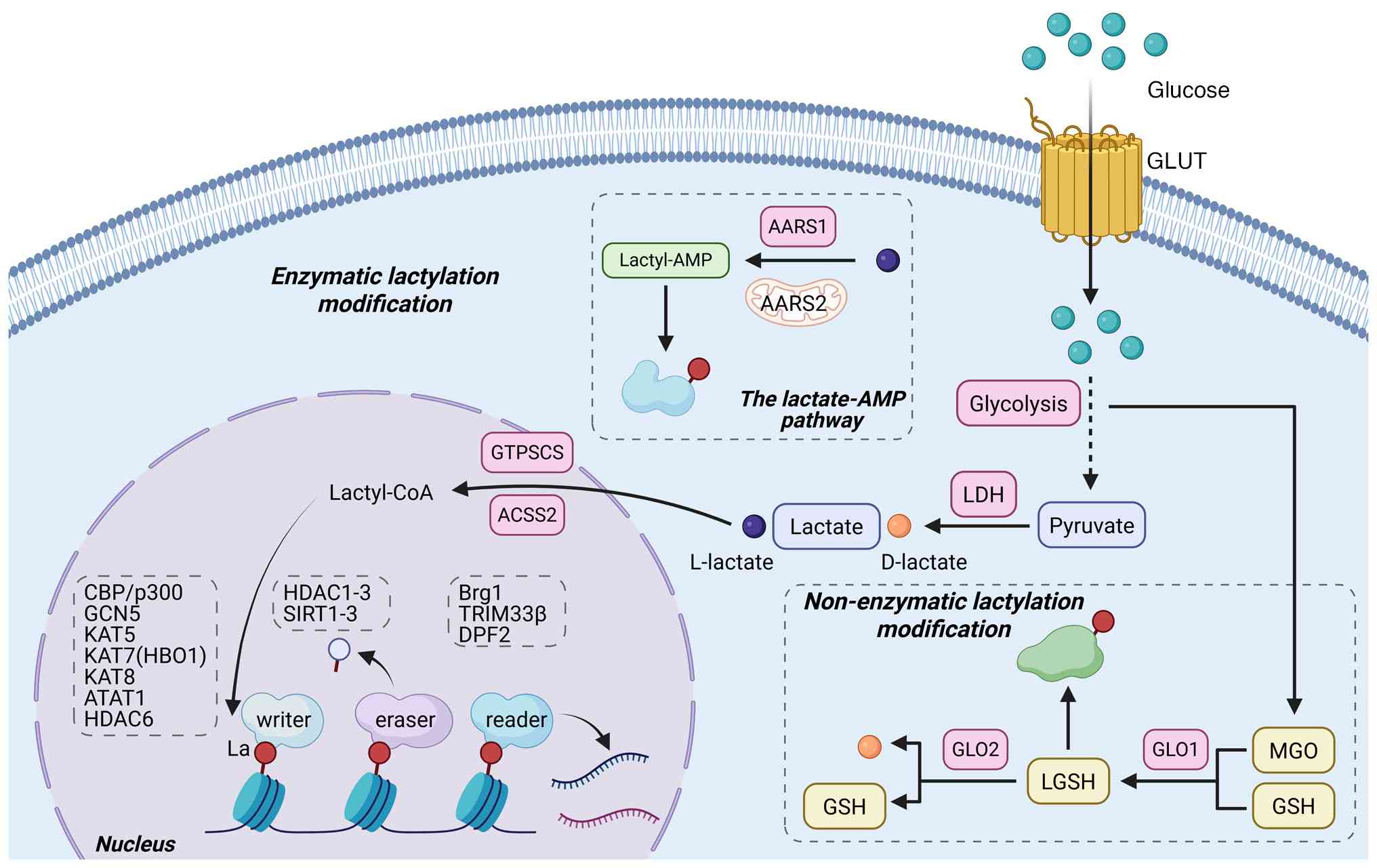 | Figure 3Molecular mechanisms of lactylation,
enzymatic and non-enzymatic pathways. Lysine lactylation is
mediated through two distinct biochemical pathways, the enzymatic
and non-enzymatic mechanisms. The enzymatic pathway is
characterized by a highly regulated process involving three
functional protein classes, writers catalyze the addition of
lactate groups, erasers facilitate their removal and readers
recognize the modification to propagate downstream signaling. By
contrast, the non-enzymatic pathway directly modifies lysine
residues through D-lactate derived from the glycolytic MGO pathway
in a stochastic, concentration-dependent manner, independent of
enzymatic catalysis. This non-enzymatic modification serves as a
molecular indicator of severe metabolic dysregulation. GLUT,
Glucose transporter; AARS1, Alanyl-tRNA synthetase 1; AARS2,
Alanyl-tRNA synthetase 2; Lactyl-AMP, Lactoyl adenosine
monophosphate; LDH, Lactate dehydrogenase; GTPSCS, GTP-specific
succinyl-coA synthetase; ACSS2, Acyl-coA synthetase short-chain
family member 2; Lactyle-CoA, Lactyl-coenzyme A; La, Lactylation;
CBP, CREB-binding protein; p300, E1A binding protein p300; GCN5,
General control nonderepressible 5; KAT5, Lysine acetyltransferase
5; KAT7 (HBO1), Lysine acetyltransferase 7; KAT8, Lysine
acetyltransferase 8; ATAT1, α-tubulin N-acetyltransferase 1; HDAC6,
Histone deacetylase 6; HDAC1-3, Histone deacetylase 1-3; SIRT1-3,
Sirtuin 1-3; Brg1, Brahma-related gene 1; TRIM33β, Tripartite
motif-containing protein 33 β; DRF2, D4-related factor 2; LGSH,
Lactoylglutathione; GLO2, Glyoxalase 2; GSH, Glutathione; GLO1,
Glyoxalase 1; MGO, Methylglyoxal. |
Histone and non-histone lactylation:
Functions and implications
Lactylation modifications can be classified into two
distinct categories on the basis of their protein targets: Histone
lactylation and non-histone lactylation. Histone lactylation
predominantly involves the modification of lysine residues on
histones by L-lactate, whereas non-histone lactylation primarily
targets glycolytic enzymes through D-lactate, thereby regulating
glycolysis via a negative feedback mechanism (47). Although these two forms of
lactylation operate through distinct mechanisms, they may together
link metabolic signals into functional outputs, potentially playing
synergistic roles in the pathogenesis of DKD.
Histone lactylation
Histones, as the most extensively characterized and
earliest identified lactylated proteins, have served as
prototypical models for investigating the functional mechanisms of
Kla. The process of histone lactylation entails the covalent
attachment of a lactyl group to lysine residues, which directly
modulates chromatin architecture and transcriptional regulation
(12,15,140). This PTM has been proposed as a
molecular link between aberrant lactate accumulation in DKD and the
transcriptional activation of genes implicated in fibrotic
processes and oxidative stress responses.
In DKD, renal fibrosis is primarily driven by the
activation of myofibroblasts and excessive accumulation of ECM,
processes associated with various fibrotic and growth factors
(141). Metabolic dysregulation
and chronic hyperglycemia-induced inflammatory damage are
intricately linked, operating both independently and
synergistically to exacerbate renal injury (142,143). Emerging evidence suggests that
histone lactylation is an epigenetic mechanism that links
hyperlactate signaling in DKD to sustained activation of fibrotic
and inflammatory gene programs. It is plausible that in renal
cells, hyperglycemia-induced glycolytic flux and subsequent lactate
accumulation may lead to histone lactylation, which localizes to
the promoters of key pathogenic genes, potentially contributing to
the initiation and persistence of fibrotic states.
Consistent with this hypothesis, experimental
findings have identified specific histone lactylation sites that
may be implicated in DKD. Recent investigations have identified
multiple lactylation sites on histones H3 and H4, specifically at
H3K9, H3K14, H3K18, H3K27, H3K56, H4K8, and H4K12 (140,144). Notably, lactylation at H3K14
and H3K18 has been implicated in the pathological progression of
DKD. Zhang et al (7)
demonstrated that H3K14la promotes epithelial-mesenchymal
transition (EMT) by activating the fibrotic transcription factor
KLF5 in renal tubular epithelial cells. Similarly, Hu et al
(142) reported that H3K18la
facilitates renal fibrosis and endothelial-mesenchymal transition
(EndoMT) by modulating insulin-like growth factor-binding
protein 5 (IGFBP5). Importantly, H3K18la activates the
NLRP3 inflammasome, establishing a direct molecular association
between lactate accumulation, chronic inflammation and tissue
remodeling in DKD (142). These
findings suggest that histone lactylation may represent a
regulatory mechanism contributing to fibrosis in DKD. While the
roles of other lactylation sites in DKD have not been fully
investigated, their potential involvement in the diabetic milieu
through alternative PTMs (Table
I) (7,145-159) raises the possibility of complex
epigenetic crosstalk.
 | Table IHistone lactylation in DKD and other
histone modifications in the context of DM. |
Table I
Histone lactylation in DKD and other
histone modifications in the context of DM.
| Histone site | Lactylation in
DKD? | Known mechanism in
DKD (via lactylation) | Known mechanism in
DM (via other PTMs) | (Refs.) |
|---|
| H3K9 | No | Not yet
reported | Sodium butyrate
activates p300, increases H3K9bu, suppresses expression of renal
injury-related genes, and ameliorates nephropathy. | (145) |
| In response to
hyperglycaemia, TGF-β1 induces the nuclear redistribution of the
repressive histone mark H3K9me3, promoting fibroblast activation
and thereby contributing to DKD. | (146) |
| ACSS2 promotes
podocyte injury in diabetic kidney disease by catalysing H3K9ac,
which activates the Raptor/mTORC1 pathway and subsequently
suppresses podocyte autophagy. | (147) |
| Exendin-4
alleviates diabetic kidney disease by activating SIRT1, which
reduces H3K9ac levels at the Txnip promoter and thereby
downregulates Txnip expression. | (148) |
| SIRT2
downregulation exacerbates oxidative stress and inflammation in
diabetic osteoarthritis by compromising H3K9 deacetylation. | (149) |
| H3K14 | Yes | H3K14la activates
KLF5 expression, promoting EMT in renal tubular cells. | Downregulation of
SIRT2 exacerbates diabetic osteoarthritis by promoting H3K14
deacetylation, which in turn amplifies oxidative stress and
inflammatory responses. | (7,149) |
| H3K18 | Yes | H3K18la facilitates
IGFBP5-mediated renal fibrosis and EndoMT. Also implicated
in NLRP3 inflammasome activation. | The elevation of
H3K18cr represents a key mechanism through which NaCr mediates its
renoprotective effects. | (141,150) |
| H3K27 | No | Not yet
reported | EZH2 and JMJD3
cooperatively drive fibroblast activation and renal fibrosis in DKD
via the coordinated regulation of H3K27me3 and pro-fibrotic
signalling. | (146) |
| Advanced glycation
end products AGEs may alter the H3K27me3 landscape in podocytes,
contributing to DKD progression. | (151) |
| H3K56 | No | Not yet
reported | SIRT6-mediated
β-catenin signalling deacetylates it, thereby preventing renal
fibrosis. | (152) |
| In diabetic
retinopathy, the hyperglycaemic milieu induces SIRT6
downregulation, which relieves the repression of HIF-1α and is
accompanied by increased H3K56ac, thereby driving the upregulation
of VEGF gene expression. | (153) |
| Deficiency of SIRT6
in pancreatic β-cells elevates H3K56ac, contributing to glucose
intolerance and impaired glucose-stimulated insulin secretion in
mice. | (154) |
| In diabetic OA,
hyperglycaemia suppresses SIRT2 and increases H3K56ac, contributing
to diabetic oxidative stress and inflammatory responses. | (149) |
| Extensive H3K56
acetylation in human adipocytes is associated with the KEGG T2DM
pathway. | (155) |
| H3K56
deacetylation, mediated by SIRT1, is involved in oxidative stress
processes in diabetes. | (156) |
| H4K8 | No | Not yet
reported | Activation of NF-κB
and TGF-β signalling, along with increased PCAF/CBP expression,
elevates H4K8ac levels, upregulating ANP, BNP and NEP expression in
diabetic hearts and kidneys. | (157) |
| In diabetic
cardiomyopathy, pharmacological reversal of H4K8ac helps alleviate
the inflammatory (NF-κB/MCP-1), pro-fibrotic (TGF-β/Smad7), and
apoptotic (PARP/Caspase-3) cascades. pro-fibrotic (TGF-β/Smad7),
and apoptotic (PARP/Caspase-3) cascades. | (158) |
| H4K12 | No | Not yet
reported | In diabetic mice,
the upregulation of HDAC2 and HDAC3 leads to reduced levels of
histone H4K12ac, which consequently suppresses the expression of
memory-associated proteins (BDNF, SYP, PSD-95) and leads to
cognitive dysfunction. | (159) |
Histone lactylation has been proposed as an
epigenetic mechanism that may link glycolytic flux to sustained
pathological gene activation in DKD. However, several key questions
remain unresolved. First, the functional roles of other histone
lactylation sites in DKD require further investigation, as they
could be involved in additional fibrotic and inflammatory gene
programs. Second, the existence of cell-specific lactylation
patterns remains unclear. Finally, the interplay between histone
lactylation and other PTMs in reshaping chromatin landscapes merits
further study to understand its potential role in disease
progression. Determining whether histone lactylation plays a
primary role or amplifies pre-existing fibrotic signals will be
important for understanding its role in DKD pathogenesis.
Non-histone lactylation
Protein lactylation, which extends beyond histones,
involves a diverse array of non-histone proteins, including
metabolic enzymes, transcription factors, and signalling proteins.
This post-translational modification exerts rapid and direct
regulatory effects on protein activity, stability and molecular
interactions, thereby facilitating precise and dynamic cellular
responses to metabolic fluctuations (140,160,161). In the context of DKD,
non-histone protein lactylation has been implicated in cellular
injury, directly disrupting fundamental cellular processes and
contributing to disease pathogenesis.
Emerging evidence underscores the pivotal role of
non-histone lactylation in the pathogenesis of DKD. A key mechanism
involves the suppression of protective autophagy, wherein
lactylation of lysine 970 in Lysyl-tRNA synthetase 1 (LARS1)
activates the mTORC1 signaling pathway, thereby inhibiting
autophagy and contributing to podocyte injury (162). Furthermore, non-histone
lactylation exacerbates metabolic dysregulation and oxidative
stress. Specifically, lactylation of lysine 182 in acyl-CoA
synthetase family member 2 compromises its enzymatic activity,
aggravating mitochondrial dysfunction and impairing HK-2 cell
viability under hyperglycemic conditions (163). Additionally, lactylation has
been associated with apoptotic pathways, as demonstrated by the
lactylation of lysine 206 in E3 ubiquitin ligase TRIM65. This
modification attenuates its catalytic activity, resulting in the
accumulation of ferroportin 2 (IREB2) and phosphodiesterase 4,
which subsequently induces ferroptosis in renal tubular epithelial
cells and exacerbates aberrant glycolysis (164). Collectively, these findings
suggest that non-histone lactylation may represent a regulatory
mechanism that can influence the pathological and physiological
functions of key metabolic and signaling nodes in DKD.
Emerging evidence indicates that non-histone
lactylation modifications may be involved in modulating the
activity of key glycolytic enzymes, with potential implications for
disease pathogenesis. Specifically, mannose has been demonstrated
to inhibit lactylation at lysine 433 of the PKM2 protein while
facilitating its nuclear translocation, resulting in the
suppression of both lactate production and glycolytic flux
(165). In the context of DKD,
lactylation of PKM2 may establish a positive feedback mechanism
that amplifies glycolytic activity and lactate accumulation,
thereby exacerbating metabolic dysregulation. Furthermore,
lactylation modifications of key signaling molecules, including
HIF-1α and mTOR, have been shown to enhance their stability and
functional activity, contributing to the reinforcement of
pathological conditions (162,166).
In summary, non-histone lactylation has emerged as
an important effector in mediating metabolic injury in DKD.
Distinct from the regulatory role of histone lactylation, which
orchestrates pathological processes through epigenetic modulation
of gene expression, non-histone lactylation exerts direct
functional impacts on cellular mechanisms. Specifically, it has
been associated with cellular dysfunction through effects on
autophagy, mitochondrial function and ferroptosis pathways. These
mechanisms, in conjunction with fibrotic reprogramming, may
collectively contribute to the progressive renal tissue damage
observed in advanced stages of DKD.
Interplay between lactylation and other
PTMs
PTMs form a complex and interconnected regulatory
network that contributes to the regulation of cellular metabolism
and function. Lactylation, a recently identified PTM (29,167,168), is an integral component of this
network and interacts with other PTMs through mechanisms of
competition, cooperation and functional collaboration, with
potential implications for cell fate (93,99). Within the unique pathological
microenvironment of DKD, aberrant lactate accumulation may disrupt
the homeostatic balance among PTMs, leading to a comprehensive
'reprogramming' of the modification landscape. This molecular
reprogramming has been associated with disease-specific alterations
in both gene expression profiles and protein functional states,
potentially influencing key cellular fate decisions.
Competitive interactions can occur when multiple
PTMs target identical amino acid residues. Lactylation specifically
targets the ε-amino group of lysine (Kla), but this site can also
be modified by other modifications, including acetylation (169), crotonylation (170), phosphoglycerolation (171) and ubiquitination (172). The interplay between
lactylation and acetylation has been implicated in the pathogenesis
of DKD. HDAC1-3, which function as 'erasers' of lactylation,
demonstrate dual enzymatic activities as both de-lactylases and
de-acetylases (173). Their
dysregulation in DKD may create a molecular milieu that favors PTM
imbalances. Pyruvate, a key metabolic intermediate, undergoes
conversion into either acetyl-CoA or lactyl-CoA through specific
acyltransferases, mediated by enzymatic or non-enzymatic reactions
(174). While acetyl-CoA serves
as the acyl donor for acetylation, lactyl-CoA functions as the
substrate for lactylation. In the hyperlactatemic environment
characteristic of DKD, the increased lactyl-CoA to acetyl-CoA ratio
may function as a molecular switch, preferentially promoting
lactylation over acetylation. This metabolic shift potentially
redirects transcriptional regulation from homeostatic maintenance
toward the activation of pro-inflammatory and pro-fibrotic gene
networks, thereby influencing cellular fate decisions between
pathological activation and programmed cell death.
Dynamic interactions between cooperation and
synergy are considered to be important in cellular processes
(175-177). Proteins exhibit collaborative
behavior through PTMs, wherein distinct modification mechanisms may
synergistically influence functional outcomes. Phosphorylation
represents a prominent example of such cooperative interactions. In
the context of DKD, TGF-β-mediated Smad phosphorylation constitutes
the central mechanism underlying fibrotic progression (178). Cappelli et al (179) elucidated that phosphorylated
Smad2/3 recruits the transcriptional coactivator p300, establishing
a molecular platform for histone lactylation at TGF-β target genes,
thereby potentiating their transcriptional activation. This
cooperative interplay may initiate a signaling cascade that
exacerbates fibrotic programming. Moreover, functional synergy can
occur when multiple PTMs collectively modulate protein stability.
As the principal regulator of hypoxia response in DKD, HIF-1α
lactylation has been demonstrated to impede Von Hippel-Lindau
recognition, consequently inhibiting ubiquitin-mediated proteasomal
degradation and augmenting HIF-1α stability (166). This mechanism could contribute
to a robust positive feedback loop: HIF-1α can promote glycolytic
flux and lactate production, which in turn stabilizes HIF-1α
through lactylation, further amplifying glycolytic activity and
creating a self-perpetuating cycle that contributes to metabolic
dysregulation and its associated pathological consequences
(65,78,79).
In conclusion, the interplay between lactylation
and other PTMs is not only of biochemical interest but also
represents a regulatory mechanism that may be dysregulated in DKD.
The competition for lysine residues can reshape the epigenetic
landscape, while the cooperation with phosphorylation could enhance
pro-fibrotic signaling pathways. Additionally, the synergistic
stabilization of pivotal factors such as HIF-1α can entrench cells
in a pathological metabolic state. This complex regulatory network
may help explain how the initial metabolic perturbations induced by
hyperglycemia and hypoxia could lead to a robust and coordinated
pathological program, linking the intricate interplay among
ferroptosis, autophagy and fibrosis that underlies the progression
of DKD. Future investigations should be aimed at delineating these
interaction networks within relevant renal cell populations to help
identify key therapeutic targets for intervention.
Comparative insights: Lactylation in
cancer vs. DKD
Lactylation, a previously identified PTM (14,15), was initially discovered in
oncology research, revealing how the Warburg effect drives tumor
progression and immune evasion through lactate. In both cancer and
DKD, lactate accumulation can drive histone and non-histone
lactylation; however, due to differences in the microenvironment
and cellular lineages, the functional outcomes and regulatory
contexts differ substantially. In tumors, lactylation is generally
associated with promoting cell proliferation and immune suppression
(180). In contrast, in DKD,
lactylation is primarily linked to fibrotic remodeling, metabolic
dysfunction, and the modulation of cell death pathways (181,182).
A potential distinction may lie in the concept of
the 'lactate clock', referring to the duration and intensity of
lactate exposure. In cancer, intermittent hypoxia and lactate
shuttling between glycolytic and oxidative zones contribute to a
dynamically fluctuating lactylation landscape, which may facilitate
tumor cell adaptation to microenvironmental changes. In comparison,
DKD is characterized by persistent hyperglycemia and chronic
hypoxia, leading to sustained lactate overload. This results in a
relatively static and pathologically fixed lactylation state, which
might explain why lactylation in DKD is more frequently associated
with irreversible tissue damage and fibrosis rather than adaptive
growth (182,183).
From a molecular perspective, common 'writers' and
'erasers' appear to exert context-dependent regulatory roles in
different disease settings. For example, p300/CBP, as canonical
lactylation writers, primarily promote oncogene expression in
cancer (184), whereas in DKD,
they amplify pro-fibrotic and pro-inflammatory transcriptional
programs (181). Specifically,
p300-mediated H4K12 lactylation has been shown to suppress
ferroptosis by upregulating GCLC in colorectal cancer stem cells,
thereby promoting chemotherapy resistance (185). In the context of DKD, however,
the same p300/H4K12la axis has been implicated in renal injury
through the regulation of AKR1B1 (30). Similarly, HDAC1-3 and SIRT1-3
function as lactylation erasers in both diseases, yet their
regulatory targets and biological effects appear distinct:
Inhibition of these erasers is often associated with suppressed
tumor growth in cancer (180),
whereas their activation in DKD has been associated with
renoprotective effects through the restoration of autophagic flux
and attenuation of fibrosis (181).
These comparative observations suggest that
therapeutic strategies targeting the lactylation axis may need to
account for disease-specific contexts. Thus, while lactylation
represents a conserved metabolic-epigenetic regulatory mechanism,
its pathological consequences in cancer vs. DKD appear to be
substantially shaped by the distinct microenvironment and cellular
background (186). Elucidating
these differences may not only deepen the understanding of DKD
pathogenesis but also provide a rationale for the development of
context-selective intervention strategies.
Deciphering the regulatory nexus of
lactylation in cell death pathways and fibrotic tissue
remodeling
Ferroptosis and lactylation
Ferroptosis, an iron-dependent, regulated cell
death mechanism mediated by lipid peroxidation, has been
increasingly recognized as a notable contributor to the
pathogenesis of DKD. The distinctive DKD microenvironment, marked
by hyperglycemia, hypoxia and oxidative stress, fosters conditions
conducive to ferroptosis through the inhibition of the
cystine/glutamate antiporter (system Xc-), depletion of GSH and
subsequent accumulation of cytotoxic lipid peroxides in renal cells
(187-189). Recent studies have suggested
that lactylation may serve as a metabolic regulator that influences
the activation of this cell death pathway (190,191).
The role of glycolytic flux and lactate in this
regulatory mechanism has been explored in recent studies. Yang
et al (164)
demonstrated that lactate promotes ferroptosis through lactylation
at K206 of TRIM65, which inhibits its E3 ubiquitin ligase activity,
thereby suppressing the degradation of IREB2. This results in IREB2
accumulation, dysregulated iron metabolism, enhanced lipid
peroxidation and ultimately ferroptosis in renal tubular epithelial
cells, exacerbating kidney injury. IREB2 modulates intracellular
free iron levels by regulating the mRNA expression of the
transferrin receptor (192).
These findings suggest a potential connection wherein lactylation
may be involved in the modulation of pro-ferroptotic signaling
pathways in a context-dependent manner (31,193). In addition to its regulatory
role in iron homeostasis, lactylation may also affect the
antioxidant defense system. Specifically, lactylation of GPX4, a
pivotal enzyme responsible for the detoxification of lipid
peroxides, has been experimentally validated to facilitate
ferroptosis in models of cardiac injury (194,195). This evidence strongly suggests
that in DKD, GPX4 lactylation likely suppresses its enzymatic
activity, thereby compromising cellular capacity to eliminate lipid
peroxides and consequently enabling the progression of
ferroptosis.
Furthermore, lactylation may facilitate ferroptosis
induction through the modulation of systemic metabolic homeostasis.
In DKD, the characteristic glycolytic shift accompanied by lactate
accumulation has been associated with the suppression of the
pentose phosphate pathway, a key cellular source of NADPH. This
metabolic perturbation compromises the regeneration of reduced GSH,
thereby attenuating the GPX4-mediated antioxidant defense system
and establishing a cellular milieu conducive to ferroptosis
(196,197). Moreover, the elevated lactate
environment is associated with a reduction in the
NAD+/NADH ratio, which may disrupt cellular redox
homeostasis and contribute to the oxidative stress conditions that
can lead to ferroptotic cell death (198).
In conclusion, lactylation emerges as a key
regulatory mechanism in DKD-associated ferroptosis, potentially
affecting key executioner proteins such as TRIM65 and potentially
GPX4, while also influencing cellular antioxidant defenses through
metabolic reprogramming. Several key questions remain to be
addressed: What is the complete repertoire of ferroptosis-related
proteins undergoing lactylation across diverse renal cell
populations? How does the interplay between lactylation and other
PTMs influence ferroptotic susceptibility? Furthermore, elucidating
the integration of the lactylation-ferroptosis axis with other cell
death pathways will be essential for understanding the complex
regulatory networks governing cell fate determination in diabetic
nephropathy.
Autophagy and lactylation
Autophagy, a fundamental cellular process essential
for maintaining homeostasis, demonstrates a complex dualistic role
in DKD. Dysregulation of autophagy, manifesting either as excessive
activation or, more frequently, as impaired function, has been
extensively implicated in the pathogenesis of renal injury
(199,200). Under hyperglycemic conditions,
activation of energy-sensing pathways such as AMP-activated protein
kinase can stimulate autophagy; however, the prevailing metabolic
milieu in DKD paradoxically results in autophagic suppression.
Furthermore, the phosphorylation of various oxidases enhances key
metabolic processes, including glycolysis, glucose transport and
fatty acid metabolism, thereby exerting additional influence on
cellular homeostasis (201).
Notably, the glycolysis-lactylation axis has recently emerged as a
key regulatory mechanism that may affect autophagic flux,
potentially influencing cell fate decisions.
The relationship between lactylation and autophagy
has been explored through several studies. Sun et al
(202) demonstrated that
lactate serves as a key metabolic signal connecting glycolysis to
autophagy. The suppression of autophagy by lactylation has been
linked to the mTORC1 signaling pathway. Experimental evidence
derived from DKD models indicates that lactylation of LARS1 at K970
may contribute to mTORC1 activation, suggesting a potential
regulatory role in autophagic flux. This activation has been
associated with inhibition of autophagosome formation and
disruption of autophagic flux, ultimately resulting in podocyte
injury and apoptosis (162,203). This mechanistic pathway
provides an example of how a specific lactylation event may
influence a key regulatory node and affect a cytoprotective
cellular process.
Beyond mTORC1 signaling, lactylation is poised to
directly target the core autophagic machinery. Key proteins
involved in autophagosome formation and degradation, including LC3,
ATG5 and p62, have been identified as potential lactylation
substrates. Specifically, lactylation of LC3 may interfere with its
lipidation process or receptor binding capacity, while lactylation
of p62 could impair its functional activity or degradation,
potentially leading to its pathological accumulation and subsequent
disruption of selective autophagy. Although direct evidence linking
these mechanisms to DKD remains limited, studies in other
biological contexts offer relevant insights. Notably, lactylation
of RUBCN at K33 under high-lactate conditions has been reported to
enhance LC3-associated phagocytosis, illustrating the ability of
lactate to directly modify and functionally regulate
autophagy-related proteins (204).
Lactylation exerts deleterious effects on
mitophagy, a key process for the selective elimination of damaged
mitochondria. The impairment of mitophagy exacerbates mitochondrial
dysfunction, potentially amplifying oxidative stress and
establishing a self-perpetuating cycle of cellular damage. This
phenomenon is evidenced by studies in acute kidney injury models,
where lactylation of aldehyde dehydrogenase 2 has been associated
with reduced mitophagy and mitochondrial dysfunction (95,182,205). A comparable mechanism is
postulated to occur in the energetically compromised
microenvironment of DKD, where lactylation-induced mitophagy
failure would further deplete cells of functional mitochondria,
potentially impairing the energy-demanding autophagy process
itself.
In conclusion, the glycolysis-lactate axis has been
implicated in multifaceted regulatory effects on autophagy in DKD
through three principal mechanisms: Modulation of key signaling
pathways, regulation of core autophagy-related proteins and
selective elimination of damaged organelles via mitophagy. This
coordinated dysregulation compromises essential cellular defense
mechanisms, thereby increasing renal cell susceptibility to
secondary injuries. The dual role of lactate in both suppressing
autophagy and promoting ferroptosis may represent a regulatory node
in determining cellular fate, collectively contributing to the
progression of renal injury. Future investigations aimed at
comprehensive profiling of the lactate-modified autophagy proteome
in DKD and precise characterization of the regulatory mechanisms
underlying these modifications could provide valuable insights.
Lactylation as an epigenetic contributor
to renal fibrosis
The early phases of DKD are characterized by mild
renal inflammation, which progresses to renal fibrosis and
sclerosis, culminating in end-stage renal disease (206). Renal fibrosis is characterized
by the activation and proliferation of myofibroblasts and excessive
ECM deposition (141), with
TGF-β serving as a pivotal profibrotic mediator (85). Consequently, protein lactylation,
driven by glycolytic flux, has been proposed as a key epigenetic
mechanism. This process may contribute to the conversion of
metabolic signals into a sustained fibrotic program across diverse
renal cell populations, potentially serving as a metabolic mediator
for the establishment of pathological memory.
Lactylation has been associated with direct
regulatory effects on fibrotic phenotypes in renal cells through
distinct molecular mechanisms across different cell types. In renal
tubular epithelial cells, H3K14la functions as a key mediator of
myofibroblast differentiation via EMT, facilitating ECM production.
This process is mechanistically associated with the KLF5
gene activation, which drives mesenchymal matrix secretion and
contributes to tubulointerstitial fibrosis (7). In glomerular endothelial cells,
hyperglycemic conditions upregulate IGFBP5, enhancing
glycolytic flux and subsequent lactic acid production. The
resulting H3K18la binds to the promoter regions of NLRP3 and IL-1β,
initiating pyroptosis and promoting EndoMT (142). Furthermore, in podocytes,
lactylation targets non-histone proteins, specifically modifying
K970 in the LARS1 protein, which activates mTORC1 signaling,
impairs autophagy flux and exacerbates podocyte injury and
detachment, ultimately contributing to glomerulosclerosis (162). Collectively, these findings
suggest that lactylation may represent an epigenetic mechanism that
can influence transcriptional networks and cellular functions in
renal cells, associating transient metabolic stress with persistent
fibrotic pathology.
The fibrotic cascade in DKD is intricately
associated with chronic inflammation, where macrophages have been
recognized as central mediators in this process (142,207). Lactate metabolism has been
implicated in macrophage polarization, with H4K12la associated with
M2 phenotype acquisition. This polarization has been associated
with activation of genes involved in glycolysis and oxidative
phosphorylation, potentially contributing to renal fibrosis
(208,209). Additionally, H3K18la in
macrophages upregulates Tgfb1 expression, which in turn
activates the TGF-β1/Smad3 signaling pathway. This activation
drives macrophage-to-myofibroblast transition, potentially
contributing to fibrotic progression (89). Collectively, lactate-mediated
immune modulation may contribute to a renal microenvironment
characterized by both immunosuppressive and pro-fibrotic
features.
Lactation has also been implicated as a key
function in intercellular signaling mechanisms. The metabolic
reprogramming of renal tubule epithelial cells and podocytes has
been associated with both autonomous fibrotic changes and increased
lactate efflux into the extracellular milieu. This extracellular
lactate may function as a paracrine signaling molecule that can be
taken up by adjacent cells, including fibroblasts and macrophages.
Within these recipient cells, lactate-mediated lactylation, such as
H4K12la in (myo)fibroblasts, has been associated with NF-κB pathway
activation and enhanced pro-inflammatory and pro-fibrotic gene
expression (210). This process
may contribute to a feedback loop: M2 macrophages secrete TGF-β,
which can enhance glycolytic activity and lactate production in
renal tubule cells, potentially perpetuating the cycle.
In conclusion, lactylation has been implicated as a
key regulatory mechanism contributing to fibrotic processes across
diverse renal cell populations. This phenomenon involves direct
cellular reprogramming, immune microenvironment remodeling and the
modulation of metabolic-epigenetic crosstalk. As summarized in
Table II (89,142,162-164,208-212), lactylation targets in various
renal cell types have been associated with fundamental pathological
pathways, including ferroptosis, autophagy suppression and
inflammatory activation, potentially contributing to fibrotic
progression.
| Table IILactylation targets, functional
outcomes, and signalling pathways across different renal cell
types. |
Table II
Lactylation targets, functional
outcomes, and signalling pathways across different renal cell
types.
| Cell type | Lactylation
target | Functional
outcome | Involved
pro-fibrotic pathway/gene | (Refs.) |
|---|
| Tubular epithelial
cells | ACSF2(K182la) | Mitochondrial ROS
accumulation and dysfunction in HK-2 cells under hyperglycaemic
conditions. | Therapeutic
targeting of mitosis and ferroptosis | (163) |
| TRIM65 | TRIM65 confers
protection against ferroptosis and suppresses excessive glycolysis
by mediating the ubiquitination and degradation of IREB2 and
PDK4. | Ferroptosis | (164) |
| Tsc1 | Activated mTORC1
signalling, triggered by Tsc1 loss, acts as a master driver of this
metabolic switch, ultimately leading to aberrant tubular epithelial
cell proliferation, cyst formation, and renal fibrosis. | mTORC1 | (214) |
| Podocytes | LARS1 | mTORC1 activation
drives DKD progression by inhibiting autophagic activity, a process
that accelerates podocyte apoptosis and injury. | mTORC1,
autophagy | (162) |
| Glomerular
endothelial cells | H3K18la | Promotes NLRP3
inflammasome-induced EndoMT and renal fibrosis, accelerating the
progression of DKD. |
EndoMT/IGFBP5 | (141) |
| Macrophages | H4K12la | Enhances HIF-1α
transcription and augments PA-induced inflammatory responses in
macrophages. | Slc16a3 | (212) |
| Stimulates
macrophage M2 polarization and exacerbates renal tissue
fibrosis. | TMAO, TGF-β/Smad,
Wnt/β-catenin | (208,209) |
| H3K18la | Activates
Tgfb1 expression, and the subsequent TGF-β1 activates the
Smad3 pathway to drive the MMT process. |
Tgfb1/TGF-β1/Smad3 | (89) |
| (Myo)
fibroblasts | H4K12la | Lactate, produced
by PFKFB3-driven glycolysis, mediates H4K12la to activate the
transcription of NF-κB pathway genes, including Ikbkb,
Rela and Relb. | PFKFB3/NF-κB | (210) |
The elucidation of lactylation mechanisms presents
both a considerable scientific challenge and a therapeutic
opportunity. As depicted in Fig.
4, the intricate lactylation networks highlight the need for
highly targeted intervention strategies. Future research should
prioritize the development of cell-specific delivery systems to
precisely modulate pathogenic lactylation events while preserving
physiological homeostasis. Furthermore, elucidating the temporal
dynamics of these modifications during DKD progression and
investigating their interplay with other PTMs are essential.
Current evidence suggests that targeted disruption of this
coordinated lactylation network may represent a promising
therapeutic approach to attenuate the progressive fibrotic cascade
in DKD.
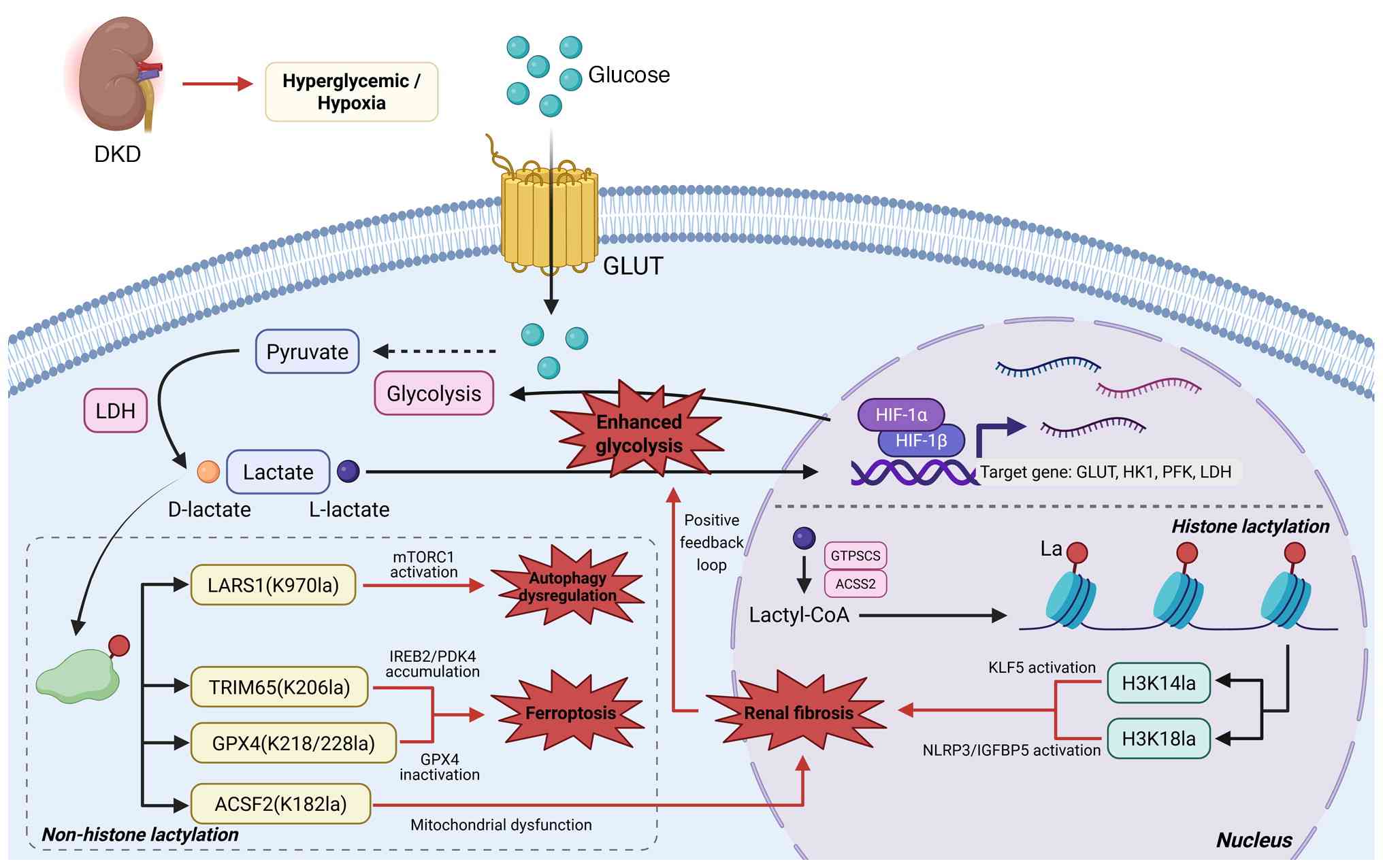 | Figure 4The glycolytic-lactate axis
orchestrates fibrotic remodeling in DKD through
lactylation-mediated modulation of ferroptosis and autophagy.
Elevated lactate concentrations induce cellular dysfunction through
non-histone lactylation, suppress cytoprotective autophagy and
initiate ferroptosis, thereby activating key pathways associated
with cellular damage and death. Concurrently, lactylation drives
renal fibrosis via a dual regulatory mechanism, histone
modification-mediated activation of profibrotic gene expression and
signaling protein alteration-induced functional impairment. The
resultant fibrotic tissue remodeling exacerbates hypoxic conditions
and inflammatory responses, establishing a self-perpetuating cycle
that progressively amplifies disease pathogenesis. DKD, Diabetic
kidney disease; GLUT, Glucose transporter; LDH, Lactate
dehydrogenase; LARS1, Mechanistic target of rapamycin complex 1;
K970la, Lysine 970 lactylation; mTORC1, Mechanistic target of
rapamycin complex 1; TRIM65, Tripartite motif-containing 65;
K206la, Lysine 206 lactylation; IREB2, Iron-responsive
element-binding protein 2; PDK4, Pyruvate dehydrogenase kinase 4;
GPX4, Glutathione peroxidase 4; K218/228la, Lysine 218/228
lactylation; ACSF2, Acyl-coA synthetase family member 2; K182la,
Lysine 182 lactylation; HIF-1α, Hypoxia-inducible factor 1-α;
HIF-1β, Hypoxia-inducible factor 1-β; HK1, Hexokinase 1; PFK,
Phosphofructokinase; GTPSCS, GTP-specific succinyl-coA synthetase;
ACSS2, Acyl-coA synthetase short-chain family member 2;
Lactyle-CoA, Lactyl-coenzyme A; La, Lactylation; H3K14la, Histone
H3 lysine 14 lactylation; H3K18la, Histone H3 lysine 18
lactylation; KLF5, Krüppel-like factor 5; NLRP3, NLR family pyrin
domain containing 3; IGFBP5, Insulin-like growth factor-binding
protein 5. |
Clinical translational potential of
glycolysis and lactylation in DKD
The rapidly advancing field of DKD research
underscores the importance of therapeutic interventions that target
fundamental pathogenic mechanisms rather than merely addressing
symptomatic manifestations. While current standard-of-care
approaches, primarily focused on glycemic, lipid and blood pressure
regulation, have shown limited efficacy in halting disease
progression, they highlight a need for additional therapeutic
options. The identification of the glycolysis-lactylation axis as a
key contributor to DKD pathogenesis has opened a new avenue for
therapeutic intervention.
Therapeutic strategies targeting the glycolysis,
lactylation axis can be categorized into three distinct
intervention levels based on their mechanism of action: i)
Inhibition of lactate generation (upstream metabolic enzymes), ii)
blockade of lactate transport (MCT inhibitors) and iii) direct
modulation of the lactylation machinery (writers, erasers and
readers). A summary of representative agents targeting each level
is provided in Table III
(70,72,104,105,125-128,130,213-228). This section discusses the
potential of targeting the glycolysis-lactylation axis, reviews
strategies for modulating lactylation modifications and considers
translational challenges, providing a perspective on emerging
therapeutic strategies for DKD.
| Table IIISummary of therapeutic agents
targeting the glycolysis-lactylation axis in DKD. |
Table III
Summary of therapeutic agents
targeting the glycolysis-lactylation axis in DKD.
| Intervention
level | Mechanism of
action | Agent/compound | Key
description | (Refs.) |
|---|
| Inhibition of
lactate generation (upstream metabolic intervention) | PKM2 activator | TEPP-46 | Promotes PKM2
tetramer formation, inhibits its nuclear pro-glycolytic function,
restores metabolic homeostasis. | (70,213) |
| | Suppresses
apoptosis and fibrosis. | (214) |
| | Reduces nuclear
PKM2 translocation, downstream LDHA and GLUT1 transcription and
lactate production. | (215) |
| 15n | Novel
imidazopyrimidine derivative with a similar mechanism to TEPP-46,
attenuates renal fibrosis. | (72) |
| PKM2 inhibitor | LncRNA
ARAP1 | Promotes aberrant
glycolysis and fibrosis in DKD via the EGFR/PKM2/HIF-1α
pathway. | (216,217) |
| LDHA inhibitor | Oxamate | Directly inhibits
LDHA activity, reduces lactate production and protein
lactylation. | (218) |
| FX11 | Directly blocks
pyruvate-to-lactate conversion, reduces lactate concentration and
downstream lactylation levels. | |
| HIF-1α
inhibitor | LW6 | Inhibits central
hypoxic signaling, downregulates glycolytic enzyme expression,
fundamentally reduces lactate synthesis. | (219-222) |
| Blockade of lactate
transport (intercellular communication intervention) | MCT4 inhibitor | VB124 | Inhibits lactate
efflux, disrupts intercellular lactate shuttling, potentially
attenuates fibrotic signaling in recipient cells such as
fibroblasts. | (223) |
| Syrosingopine | Inhibits MCT4,
restores adaptive kidney repair. | (223) |
| AZD3965 | Primarily studied
in oncology; may provide insights into pathological cellular
communication in DKD. | (224) |
| Direct modulation
of lactylation machinery (epigenetic intervention) | CBP/p300
inhibitor | A-485 (HAT
inhibitor) | Inhibits
lactylation-writing enzymes; may delay disease progression by
directly suppressing pro-fibrotic lactylation marks. | (104,105,225) |
| CPI644 (bromodomain
inhibitor)
NEO2734 (Dual bromodomain inhibitor)
CCS1477 (PROTAC inhibitor) | | |
| AARS1
inhibitor | β-alanine | Inhibits novel
lactylation writer, ameliorates renal injury and fibrosis. | (226) |
| SIRT1 agonist | Resveratrol | Enhances
delactylase activity, removes pathological modifications, improves
renal injury and fibrosis. | (125-128, 227,228) |
| SIRT3 agonist | SRT2104 Picroside
Shennanone Metformin | | |
| Regulation of
lactylation recognition (signal transduction intervention) | Reader protein
inhibitor | TRIM33β
inhibitor | Interferes with
reader protein recognition of lactylation sites, blocks downstream
pro-fibrotic signal transduction (for example, TGF-β) | (130) |
Targeting the glycolytic-lactate
axis
Inhibitors of key glycolytic
enzymes
Accumulating evidence indicates that dysregulation
of PKM2 is associated with the pathogenesis and progression of DKD
(229). PKM2 exists in a
dynamic equilibrium between its highly active tetrameric form and
less active dimeric state, with the latter serving as a nuclear
transcription co-activator. In the context of DKD, the shift toward
the dimeric form facilitates PKM2 nuclear translocation, where it
interacts with HIF-1α to upregulate glycolytic gene expression,
thereby perpetuating a metabolic regulatory feedback loop (70,230,231). Pharmacological activation of
PKM2 using compounds such as TEPP-46 promotes tetramer formation,
which inhibits its nuclear function, restores metabolic
homeostasis, attenuates fibrotic progression and preserves renal
function (213). Notably, Jian
et al (72) identified a
novel imidazopyrimidine derivative, 15n, which exhibits mechanisms
of action analogous to TEPP-46 and demonstrates efficacy in
mitigating renal fibrosis in preclinical models. This therapeutic
strategy exemplifies a targeted approach to modulating underlying
metabolic dysregulation in DKD.
Direct inhibition of lactate production has been
proposed as a therapeutic approach. Pharmacological inhibitors of
LDHA, such as oxamate and FX11, have been reported to reduce
intracellular lactate concentrations and attenuate protein
lactylation (218). This
strategy is of interest given the evidence associating LDHA
activity with DKD pathogenesis (44,51,80,81). By targeting LDHA, this approach
may influence lactate accumulation, metabolic acidosis and the
formation of a lactate-enriched microenvironment. These strategies
for inhibiting lactate production that represent distinct
intervention points within the upstream metabolic cascade (Table III). However, the systemic
administration of these metabolic enzyme inhibitors presents
challenges, as they could affect energy homeostasis in non-target
tissues, underscoring the need for kidney-specific delivery
strategies.
Lactate transporter inhibitors
The functional expression of MCT1 and MCT4 is
essential for lactate transport, facilitated by their association
with the ancillary protein Basigin (CD147) (232-236). In DKD, glomerular cells
exhibiting a glycolytic phenotype predominantly utilize MCT4 for
lactate efflux. Elevated MCT4 expression levels have been observed
in patients with T2DM, suggesting a potential role in disease
pathogenesis (237).
Pharmacological inhibition of MCT4 may disrupt intracellular
lactate efflux, potentially leading to autotoxicity and impairment
of intercellular lactate shuttles, thereby attenuating fibrotic
signaling in recipient cells, including fibroblasts and
macrophages. While MCT4 inhibitors such as VB124 and AZD3965 have
been primarily studied in oncological contexts (223,224), their application in DKD could
provide key insights into their therapeutic potential by targeting
pathological lactate-mediated cellular communication.
Notably, MCT4 inhibition represents a distinct
intervention strategy targeting lactate transport rather than its
generation, disrupting intercellular lactate shuttling and
potentially attenuating fibrotic signaling in recipient cells such
as fibroblasts and macrophages (Table III). Preclinical evidence in
kidney injury models has demonstrated that the MCT4 inhibitor
syrosingopine can restore adaptive kidney repair by suppressing
proinflammatory endothelial activation, supporting the therapeutic
relevance of targeting lactate transport in renal disease (226).
HIF-1α inhibitors
As a central regulator of hypoxic responses and a
key modulator of glycolytic flux in DKD, HIF-1α emerges as a
promising therapeutic target. Pharmacological inhibition of HIF-1α,
exemplified by the compound LW6, has been demonstrated to
facilitate its proteasomal degradation, attenuate EMT, normalize
aberrant glycolysis and mitigate renal fibrotic processes (219-221). These inhibitory agents have
been associated with renoprotective effects, potentially through
transcriptional downregulation of key glycolytic enzymes and
reducing lactic acid production (222). Nevertheless, the therapeutic
application of HIF-1α inhibitors is challenging due to their
pleiotropic physiological functions, particularly in angiogenesis
and erythropoiesis (238,239). Future research aimed at
developing kidney-selective or microenvironment-responsive HIF-1α
modulators could help reduce off-target effects and improve
therapeutic efficacy in DKD.
Targeting lactylation modifications
Targeting writers
The inhibition of writers, particularly CBP/p300,
emerges as a promising therapeutic strategy for mitigating
pathogenic lactogenic processes. Current research has identified
four distinct classes of CBP/p300 inhibitors: HAT domain inhibitors
(for example, A-485), bromodomain inhibitors (for example, CPI644),
dual inhibitors targeting both CBP/p300 and BRD4 bromodomains (for
example, NEO2734) and proteolysis-targeting chimera inhibitors (for
example, CCS1477) (225).
Mechanistically, CBP/p300 activation has been shown to upregulate
NADPH oxidase activity, fibrotic pathways and inflammatory
responses in diabetic nephropathy (104,105), suggesting that its inhibition
could potentially attenuate disease progression through direct
suppression of fibrotic lactogenic markers. Although these
compounds have been primarily studied in oncology, their
application in DKD models warrants investigation as a potential
therapeutic approach. Concurrently, targeting AARS1/2 within the
lactate-AMP signaling pathway could offer an alternative approach
to influencing lactylation in response to intracellular lactate
levels. Recent studies have identified that AARS1 promotes
glycolytic reprogramming in DKD by lactylating Akt and p65, and its
pharmacological inhibition with β-alanine has been shown to
ameliorate renal injury and fibrosis in preclinical models
(240-242). This approach represents a
distinct epigenetic intervention strategy, directly targeting the
lactylation machinery rather than upstream lactate
availability.
Targeting erasers
The augmentation of eraser activity represents a
complementary therapeutic approach in disease management. SIRT1-3,
which exhibit deacetylase activity, have been reported to be
downregulated in DKD. Activation of these SIRTs has been
empirically validated to ameliorate renal injury and fibrosis
(125-128). SIRT1 agonists, including the
naturally occurring compound resveratrol and the synthetic molecule
SRT2104, have been investigated for their therapeutic potential.
However, the clinical application of resveratrol is constrained by
its limited bioavailability and off-target effects (227). Similarly, SIRT3 agonists such
as picroside, shennanone and metformin have been shown to confer
renal protection, potentially mediated through the restoration of
deacetylation functions (228).
The enhancement of eraser activity may offer the distinct advantage
of modulating pathological modifications while preserving
physiological signaling pathways.
Targeting readers
The targeted inhibition of reader proteins that
specifically recognize lactylation marks offers a novel therapeutic
strategy characterized by high specificity, potentially reducing
off-target effects on other PTM pathways. For example,
small-molecule inhibitors that selectively interfere with the
interaction between the H3K18la reader TRIM33β and its lactylated
histone substrate might allow modulation of its role in TGF-β
signaling and fibrotic processes (130). This approach could achieve
higher specificity by interfering with signal interpretation rather
than modification deposition. Nevertheless, this research area
remains in its early stages of exploration. A pivotal research
objective is the systematic identification and functional
validation of pathogenic reader proteins specifically implicated in
DKD, which would facilitate the development of a new class of
therapeutic agents targeting the terminal step of
lactylation-mediated signal transduction.
Therapeutic modulation of lactylation poses
considerable challenges, primarily arising from its intricate
biological mechanisms. The extensive structural and functional
similarities between lactylation-regulating enzymes and those
involved in other acylation processes (for example, acetylation)
may increase the potential for off-target effects. Non-selective
inhibition could inadvertently affect key physiological pathways.
Therefore, the development of highly specific inhibitors capable of
distinguishing Kla from Kac, coupled with the advancement of
kidney-targeted drug delivery systems, is considered an essential
prerequisite for successful clinical translation. Future progress
in this field will likely require achieving a level of precision
that corresponds to the complexity of the PTM network.
Challenges and future directions
The clinical translation of therapeutics targeting
the glycolysis-lactylation axis encounters substantial challenges
that warrant careful consideration. A primary obstacle is achieving
pharmacological specificity, as systemic inhibition of core
metabolic or epigenetic pathways could lead to off-target effects
and toxicity, highlighting the need for kidney-selective drug
delivery platforms. Furthermore, the functional roles of specific
lactylation modifications in the pathogenesis of DKD remain
incompletely characterized, highlighting the need for advanced
spatial multi-omics approaches and cell-type-specific genetic tools
to identify the most biologically relevant and therapeutically
actionable targets. Additionally, while monotherapeutic strategies
targeting this axis show potential, the complex, multifactorial
pathophysiology of DKD may require rationally designed combination
therapies that simultaneously modulate fibrosis, inflammation and
metabolic dysregulation to achieve synergistic therapeutic efficacy
and mitigate the emergence of treatment resistance.
Conclusion and perspectives
The present review synthesizes emerging evidence
suggesting that glycolysis-driven lactylation may represent a key
mechanism in DKD, though direct causal evidence remains limited and
further experimental validation is required to establish definitive
mechanistic links. The present review discussed how the
hyperglycemic and hypoxic microenvironment can increase glycolytic
flux, leading to lactate accumulation that may serve as a substrate
for both enzymatic and non-enzymatic lactylation. This modification
has been proposed to function as a regulatory bridge, linking
metabolic states to epigenetic and post-translational signals.
Specifically, how lactylation has been implicated in key
pathological processes, affecting ferroptosis, influencing
autophagic flux and contributing to pro-fibrotic gene programs
across diverse renal cell types was explored. The intricate
interplay between lactylation and other PTMs may further contribute
to its impact, forming a complex regulatory network that could
stabilize the pathological phenotype.
Looking ahead, this field holds considerable
potential for advancing the understanding of DKD. Future research
aimed at understanding the cell-specific and temporal dynamics of
lactylation in the kidney could be important for defining its roles
in disease initiation vs. progression. The identification of novel
pathogenic 'reader' proteins and the exploration of crosstalk
between lactylation and other metabolic signaling pathways may
represent promising avenues for discovery. Additionally, while
therapeutic targeting presents challenges, the continued
development of highly specific agents and advanced delivery systems
may offer potential for applying these mechanistic insights to
therapeutic strategies.
In summary, lactylation represents an intersection
of metabolic and epigenetic regulation in DKD. Despite the
complexities and unresolved questions, further exploration of this
pathway may not only advance the understanding of diabetic kidney
injury but also reveal new therapeutic opportunities for managing
DKD.
Availability of data and materials
Not applicable.
Authors' contributions
All authors read and approved the final manuscript.
TZ conceptualized and directed the research project, authored the
initial manuscript draft and designed the graphical
representations. XZ, YX, YL, SX and XY systematically gathered and
organized scientific literature and associated datasets pertaining
to diabetes and lactylation from publicly accessible repositories.
TL and LL conducted comprehensive reviews of the manuscript,
providing critical feedback and substantive revisions. Data
authentication not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent of publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present review was supported by the Natural Science
Foundation of the Xinjiang Uygur Autonomous Region (grant no.
2024D01C128).
References
|
1
|
Genitsaridi I, Salpea P, Salim A, Sajjadi
SF, Tomic D, James S, Thirunavukkarasu S, Issaka A, Chen L, Basit
A, et al: 11th edition of the IDF diabetes atlas: Global, regional,
and national diabetes prevalence estimates for 2024 and projections
for 2050. Lancet Diabetes Endocrinol. 14:149–156. 2026. View Article : Google Scholar
|
|
2
|
Alicic RZ, Rooney MT and Tuttle KR:
Diabetic kidney disease: Challenges, progress, and possibilities.
Clin J Am Soc Nephrol. 12:2032–2045. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhou T, Fang YL, Tian TT and Wang GX:
Pathological mechanism of immune disorders in diabetic kidney
disease and intervention strategies. World J Diabetes.
15:1111–1121. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Naaman SC and Bakris GL: Diabetic
nephropathy: Update on pillars of therapy slowing progression.
Diabetes Care. 46:1574–1586. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jung CY and Yoo TH: Pathophysiologic
mechanisms and potential biomarkers in diabetic kidney disease.
Diabetes Metab J. 46:181–197. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang C, Chen DF, Yin YH, Liu X, Liu Y, Li
X, Kong C and Zhao Q: Multi-target regulation of cellular
senescence by traditional Chinese medicine: A novel strategy to
preventing diabetic kidney disease. Ren Fail. 47:25112752025.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang X, Chen J, Lin R, Huang Y, Wang Z,
Xu S, Wang L, Chen F, Zhang J, Pan K and Yin Z: Lactate drives
epithelial-mesenchymal transition in diabetic kidney disease via
the H3K14la/KLF5 pathway. Redox Biol. 75:1032462024. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Maekawa H, Zhou YL, Aoi Y, Fain ME,
Kaminski DS, Kong H, Sebo ZL, Chakrabarty RP, Howard BC, Andersen
G, et al: SGLT2 inhibition protects kidney function by
SAM-dependent epigenetic repression of inflammatory genes under
metabolic stress. J Clin Invest. 135:e1889332025. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cannito S, Giardino I, D'Apolito M,
Ranaldi A, Scaltrito F, Pettoello-Mantovani M and Piscazzi A: From
metabolic to epigenetic memory: The impact of hyperglycemia-induced
epigenetic signature on kidney disease progression and
complications. Genes (Basel). 16:14422025. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zlacká J and Zeman M: Glycolysis under
circadian control. Int J Mol Sci. 22:136662021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yadav D, Yadav A, Bhattacharya S, Dagar A,
Kumar V and Rani R: GLUT and HK: Two primary and essential key
players in tumor glycolysis. Semin Cancer Biol. 100:17–27. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu HW, Huang H and Zhao YM: Interplay
between metabolic reprogramming and post-translational
modifications: From glycolysis to lactylation. Front Immunol.
14:12112212023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ferguson BS, Rogatzki MJ, Goodwin ML, Kane
DA, Rightmire Z and Gladden LB: Lactate metabolism: Historical
context, prior misinterpretations, and current understanding. Eur J
Appl Physiol. 118:691–728. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li XL, Yang YY, Zhang B, Lin X, Fu X, An
Y, Zou Y, Wang JX, Wang Z and Yu T: Lactate metabolism in human
health and disease. Signal Transduct Target Ther. 7:3052022.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang D, Tang ZY, Huang H, Zhou G, Cui C,
Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al: Metabolic
regulation of gene expression by histone lactylation. Nature.
574:575–580. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hughes E, Wang XX, Sabol L, Barton K,
Hegde S, Myakala K, Krawczyk E, Rosenberg A and Levi M: Role of
nuclear receptors, lipid metabolism, and mitochondrial function in
the pathogenesis of diabetic kidney disease. Am J Physiol Renal
Physiol. 329:F510–F547. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang C, Xue S, Ren P, Han S, Zhou Y, Si
Y, Han X, Zhang X, Zhang Y, Chen N, et al: Advances in the
epigenetic mechanisms of diabetic nephropathy pathogenesis.
Diabetes Metab Syndr Obes. 18:2629–2639. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen X, Tsvetkov AS, Shen HM, Isidoro C,
Ktistakis NT, Linkermann A, Koopman WJH, Simon HU, Galluzzi L, Luo
S, et al: International consensus guidelines for the definition,
detection, and interpretation of autophagy-dependent ferroptosis.
Autophagy. 20:1213–1246. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Qiu S, Xie D, Guo S, Wang Z, Cai Y, Wang
X, Hu Z, Wang S, Lin C, Yao H, et al: MAPK14/SLC7A11/GPX4 axis
dysregulation drives podocyte ferroptosis via mediating
glycerophospholipid metabolism. Cell Death Discov. 12:1472026.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang SS, Sun YT, Sun WJ, Kang X, Zhao X,
Jiang L, Gao Q, An X, Ji H and Lian F: Programmed cell death in
diabetic kidney disease: Mechanisms and therapeutic targeting. J
Inflamm Res. 18:13001–13037. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang WL, Shan GY, Bi GS, Hu Z, Yi Y, Zeng
D, Lin Z and Zhan C: Lactylation and regulated cell death. Biochim
Biophys Acta Mol Cell Res. 1872:1199272025. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ma H, Lee S, Yang Y, Bedi P and Chou SY:
Pentoxifylline protects against loss of function and renal
interstitial fibrosis in chronic experimental partial ureteral
obstruction. Pathophysiology. 25:419–425. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zeng LF, Xiao Y and Sun L: A Glimpse of
the mechanisms related to renal fibrosis in diabetic nephropathy.
In: Renal Fibrosis: Mechanisms and Therapies. Advances in
Experimental Medicine and Biology. Liu BC, Lan HY and Lv LL: 1165.
Springer; Singapore: pp. 49–79. 2019, View Article : Google Scholar
|
|
24
|
Li L, Zhang LM, Li Z, Wang Q, Zhang Q, Li
S and Ji J: TGFβ1 accelerated the progression of diabetic
nephropathy via up-regulating BRD4/Notch1/YAP signaling induced
fibrosis and proliferation in fibroblasts. Sci Rep. 15:253382025.
View Article : Google Scholar
|
|
25
|
Al-Masri S, Coelho JN and Thomas L:
Molecular pathways and emerging therapeutic targets in the
pathogenesis of diabetic kidney disease. Front Physiol.
17:17470532026. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fontecha-Barriuso M, Martin-Sanchez D,
Ruiz-Andres O, Poveda J, Sanchez-Niño MD, Valiño-Rivas L,
Ruiz-Ortega M, Ortiz A and Sanz AB: Targeting epigenetic DNA and
histone modifications to treat kidney disease. Nephrol Dial
Transplant. 33:1875–1886. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu YJ, Liu ZH, Liu LJ, Su Y, Chen J, Qing
B, Tang C, Zhang Z, Ji R, Cao X, et al: Methionine restriction
alleviates kidney fibrosis through epigenetic repression of the
TGF-β-Smad3-Hoxc8/P-TEFb axis. Nat Commun. 17:13012025. View Article : Google Scholar
|
|
28
|
Wei QQ, Xiao X, Huo E, Guo C, Zhou X, Hu
X, Dong C, Shi H and Dong Z: Hypermethylation and suppression of
microRNA219a-2 activates the ALDH1L2/GSH/PAI-1 pathway for
fibronectin degradation in renal fibrosis. Mol Ther. 33:249–262.
2025. View Article : Google Scholar
|
|
29
|
Liang H, Xu L and Yang Y: Lactate and
lactylation: Novel perspectives on fibrosis pathogenesis and
therapeutic directions. J Transl Med. 23:7052025. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fan Q: Mechanism of histone H4K12
lactylation regulating AKR1B1 promoting kidney injury in diabetes
and treatment effect of fecal microbiota transplantation. J Am Soc
Nephrol. 36: View Article : Google Scholar : 2025.
|
|
31
|
Chen C, Lin A, Zhao J, Lin X, Ye Q, Guo J,
Liu J and Xiang A: Beyond metabolism: Exploring the regulatory and
therapeutic implications of lactate and lactylation in
cancer-regulated cell death. Cell Death Dis. 17:1842026. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Forbes JM, Coughlan MT and Cooper ME:
Oxidative stress as a major culprit in kidney disease in diabetes.
Diabetes. 57:1446–1454. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Akram M: Mini-review on glycolysis and
cancer. J Cancer Educ. 28:454–457. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wei L, Zhang B, Tu Y and Liu A: Research
progress on glycolysis mechanism of psoriasis. Psoriasis (Auckl).
14:195–206. 2024.
|
|
35
|
Yu Q, Guo M, Zeng W, Zeng M, Zhang X,
Zhang Y, Zhang W, Jiang X and Yu B: Interactions between NLRP3
inflammasome and glycolysis in macrophages: New insights into
chronic inflammation pathogenesis. Immun Inflamm Dis. 10:e5812022.
View Article : Google Scholar :
|
|
36
|
Pająk B, Zieliński R and Priebe W: The
impact of glycolysis and its inhibitors on the immune response to
inflammation and autoimmunity. Molecules. 29:12982024. View Article : Google Scholar
|
|
37
|
Feng J, Li J, Wu L, Yu Q, Ji J, Wu J, Dai
W and Guo C: Emerging roles and the regulation of aerobic
glycolysis in hepatocellular carcinoma. J Exp Clin Cancer Res.
39:1262020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sun W, Wang Y, Miao X, Wang Y, Zhang L,
Xin Y, Zheng S, Epstein PN, Fu Y and Cai L: Renal improvement by
zinc in diabetic mice is associated with glucose metabolism
signaling mediated by metallothionein and Akt, but not Akt2. Free
Radic Biol Med. 68:22–34. 2014. View Article : Google Scholar
|
|
39
|
Chen S, Zou Y, Song C, Cao K, Cai K, Wu Y,
Zhang Z, Geng D, Sun W, Ouyang N, et al: The role of glycolytic
metabolic pathways in cardiovascular disease and potential
therapeutic approaches. Basic Res Cardiol. 118:482023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gómez-Milán E, Cardenete G and
Sánchez-Muros MJ: Annual variations in the specific activity of
fructose 1,6-bisphosphatase, alanine aminotransferase and pyruvate
kinase in the Sparus aurata liver. Comp Biochem Physiol B Biochem
Mol Biol. 147:49–55. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dong G, Mao Q, Xia W, Xu Y, Wang J, Xu L
and Jiang F: PKM2 and cancer: The function of PKM2 beyond
glycolysis. Oncol Lett. 11:1980–1986. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Park YS, Han JH, Park JH, Choi JS, Kim SH
and Kim HS: Pyruvate kinase M2: A new biomarker for the early
detection of diabetes-induced nephropathy. Int J Mol Sci.
24:26832023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chun N, Wyatt CM and He JC: Identification
of a protective proteomic signature and a potential therapeutic
target in diabetic nephropathy. Kidney Int. 92:780–781. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Irena A, Klaudia G, Maria S, Tomasz K,
Patrycja R, Dorota R and Agnieszka P: Role of lactate dehydrogenase
A in the regulation of podocyte metabolism and glucose uptake under
hyperglycemic conditions. Sci Rep. 15:141622025. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Forkasiewicz A, Dorociak M, Stach K,
Szelachowski P, Tabola R and Augoff K: The usefulness of lactate
dehydrogenase measurements in current oncological practice. Cell
Mol Biol Lett. 25:352020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Claps G, Faouzi S, Quidville V, Chehade F,
Shen S, Vagner S and Robert C: The multiple roles of LDH in cancer.
Nat Rev Clin Oncol. 19:749–762. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sun C, Li J, Dong L, Mou Y, Zhang B and
Song X: Lactylation: A novel epigenetic regulator of cellular
senescence. Aging Dis. 17:890–906. 2025.PubMed/NCBI
|
|
48
|
Brooks GA: The science and translation of
lactate shuttle theory. Cell Metab. 27:757–785. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Brooks GA: Lactate shuttles in nature.
Biochem Soc Trans. 30:258–264. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Brooks GA: Mammalian fuel utilization
during sustained exercise. Comp Biochem Physiol B Biochem Mol Biol.
120:89–107. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lee DY, Kim JY, Ahn E, Hyeon JS, Kim GH,
Park KJ, Jung Y, Lee YJ, Son MK, Kim SW, et al: Associations
between local acidosis induced by renal LDHA and renal fibrosis and
mitochondrial abnormalities in patients with diabetic kidney
disease. Transl Res. 249:88–109. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Reeves WB, Wang W and Li K: Renal
epithelial lactate dehydrogenase A is essential for ischemic AKI. J
Am Soc Nephrol. 36: View Article : Google Scholar : 2025.
|
|
53
|
Hashimoto T and Brooks GA: Mitochondrial
lactate oxidation complex and an adaptive role for lactate
production. Med Sci Sports Exerc. 40:486–494. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sahlin K, Fernström M, Svensson M and
Tonkonogi M: No evidence of an intracellular lactate shuttle in rat
skeletal muscle. J Physiol. 541:569–574. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Halestrap AP: Monocarboxylic acid
transport. Compr Physiol. 3:1611–1643. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Enerson BE and Drewes LR: Molecular
features, regulation, and function of monocarboxylate transporters:
Implications for drug delivery. J Pharm Sci. 92:1531–1544. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bågenholm V, Nordlin KP, Pasquadibisceglie
A, Belinskiy A, Holm CM, Hotiana HA, Gotfryd K, Delemotte L,
Nour-Eldin HH, Pedersen PA and Gourdon P: Cryo-EM structure of the
human monocarboxylate transporter 10. Structure. 33:891–902.e4.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Halestrap AP: The SLC16 gene
family-structure, role and regulation in health and disease. Mol
Aspects Med. 34:337–349. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Sun S, Li H, Chen J and Qian Q: Lactic
acid: No longer an inert and end-product of glycolysis. Physiology
(Bethesda). 32:453–463. 2017.PubMed/NCBI
|
|
60
|
Contreras-Baeza Y, Sandoval PY, Alarcón R,
Galaz A, Cortés-Molina F, Alegría K, Baeza-Lehnert F, Arce-Molina
R, Guequén A, Flores CA, et al: Monocarboxylate transporter 4
(MCT4) is a high affinity transporter capable of exporting lactate
in high-lactate microenvironments. J Biol Chem. 294:20135–20147.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Rabinowitz JD and Enerbäck S: Lactate: The
ugly duckling of energy metabolism. Nat Metab. 2:566–571. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Brooks GA: Lactate as a fulcrum of
metabolism. Redox Biol. 35:1014542020. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Liu X, Zhang Y, Zhuang L, Olszewski K and
Gan B: NADPH debt drives redox bankruptcy: SLC7A11/xCT-mediated
cystine uptake as a double-edged sword in cellular redox
regulation. Genes Dis. 8:731–745. 2020. View Article : Google Scholar
|
|
64
|
Hoy AJ, Nagarajan SR and Butler LM: Tumour
fatty acid metabolism in the context of therapy resistance and
obesity. Nat Rev Cancer. 21:753–766. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kottmann RM, Kulkarni AA, Smolnycki KA,
Lyda E, Dahanayake T, Salibi R, Honnons S, Jones C, Isern NG, Hu
JZ, et al: Lactic acid is elevated in idiopathic pulmonary fibrosis
and induces myofibroblast differentiation via pH-dependent
activation of transforming growth factor-β. Am J Respir Crit Care
Med. 186:740–751. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Tauffenberger A, Fiumelli H, Almustafa S
and Magistretti PJ: Lactate and pyruvate promote oxidative stress
resistance through hormetic ROS signaling. Cell Death Dis.
10:6532019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zhu JF, Guo DP, Lv HN, Liang ZY, Song J
and Zeng W: Histone lactylation-mediated up-regulation of IGF2BP2
enhances ferroptosis resistance via Nrf2 in colorectal cancer. Clin
Transl Med. 15:e705512025. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Blantz RC: Phenotypic characteristics of
diabetic kidney involvement. Kidney Int. 86:7–9. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Srivastava SP, Li J, Kitada M, Fujita H,
Yamada Y, Goodwin JE, Kanasaki K and Koya D: SIRT3 deficiency leads
to induction of abnormal glycolysis in diabetic kidney with
fibrosis. Cell Death Dis. 9:9972018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Liu H, Takagaki Y, Kumagai A, Kanasaki K
and Koya D: The PKM2 activator TEPP-46 suppresses kidney fibrosis
via inhibition of the EMT program and aberrant glycolysis
associated with suppression of HIF-1α accumulation. J Diabetes
Investig. 12:697–709. 2021. View Article : Google Scholar :
|
|
71
|
Park J, Joo YS, Nam BY, Kim G, Park JT,
Yoo TH, Kang SW and Han SH: Pyruvate kinase M2 activation maintains
mitochondrial metabolism by regulating the interaction between
HIF-1α and PGC-1α in diabetic kidney disease. Mol Med. 31:2662025.
View Article : Google Scholar
|
|
72
|
Jain M, Sayyed AA, Gondaliya P, Kapoor S,
Chatterjee DR, Das R, Kalia K, Khairnar A and Shard A:
Imidazopyrimidine-based pyruvate kinase M2 activator halts diabetic
nephropathy progression via modulating epithelial-to-mesenchymal
transition and fibrosis. Chem Biol Interact. 420:1116732025.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Semenza GL and Wang GL: A nuclear factor
induced by hypoxia via de novo protein synthesis binds to the human
erythropoietin gene enhancer at a site required for transcriptional
activation. Mol Cell Biol. 12:5447–5454. 1992.PubMed/NCBI
|
|
74
|
Guan Z, Jin X, Guan Z, Liu S, Tao K and
Luo L: The gut microbiota metabolite capsiate regulate SLC2A1
expression by targeting HIF-1α to inhibit knee
osteoarthritis-induced ferroptosis. Aging Cell. 22:e138072023.
View Article : Google Scholar
|
|
75
|
Wang M, Mao C, Ouyang L, Liu Y, Lai W, Liu
N, Shi Y, Chen L, Xiao D, Yu F, et al: Long noncoding RNA LINC00336
inhibits ferroptosis in lung cancer by functioning as a competing
endogenous RNA. Cell Death Differ. 26:2329–2343. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Yang R, Liu W, Zhou Y, Cheng B, Liu S, Wu
R, Liu Y and Li J: Modulating HIF-1α/HIF-2α homeostasis with
Shen-Qi-Huo-Xue formula alleviates tubular ferroptosis and
epithelial-mesenchymal transition in diabetic kidney disease. J
Ethnopharmacol. 343:1194782025. View Article : Google Scholar
|
|
77
|
Wusiman R, Haimiti S, Abuduaini H, Yang M,
Wang Y, Gu M, Sailike A and Gao L: Increased SUMO-activating enzyme
subunit 1 promotes glycolysis and fibrotic phenotype of diabetic
nephropathy. Biochem Pharmacol. 237:1169202025. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Gong N, Wang W, Fu Y, Zheng X, Guo X, Chen
Y, Chen Y, Zheng S and Cai G: The crucial role of metabolic
reprogramming in driving macrophage conversion in kidney disease.
Cell Mol Biol Lett. 30:722025. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Semenza GL: HIF-1: Upstream and downstream
of cancer metabolism. Curr Opin Genet Dev. 20:51–56. 2010.
View Article : Google Scholar
|
|
80
|
Tang L, Yang Q, Ma R, Zhou P, Peng C, Xie
C, Liang Q, Wu T, Gao W, Yu H, et al: Association between lactate
dehydrogenase and the risk of diabetic kidney disease in patients
with type 2 diabetes. Front Endocrinol (Lausanne). 15:13699682024.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Darshi M, Kugathasan L, Maity S, Sridhar
VS, Fernandez R, Limonte CP, Grajeda BI, Saliba A, Zhang G, Drel
VR, et al: Glycolytic lactate in diabetic kidney disease. JCI
Insight. 9:e1688252024. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Azushima K, Kovalik JP, Yamaji T, Ching J,
Chng TW, Guo J, Liu JJ, Nguyen M, Sakban RB, George SE, et al:
Abnormal lactate metabolism is linked to albuminuria and kidney
injury in diabetic nephropathy. Kidney Int. 104:1135–1149. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Wesson DE, Buysse JM and Bushinsky DA:
Mechanisms of metabolic acidosis-induced kidney injury in chronic
kidney disease. J Am Soc Nephrol. 31:469–482. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Brown D and Wagner CA: Molecular
mechanisms of acid-base sensing by the kidney. J Am Soc Nephrol.
23:774–780. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Han YS, Yoon YM, Go G, Lee JH and Lee SH:
Melatonin protects human renal proximal tubule epithelial cells
against high glucose-mediated fibrosis via the cellular prion
protein-TGF-β-Smad signaling axis. Int J Med Sci. 17:1235–1245.
2020. View Article : Google Scholar
|
|
86
|
Kumari S, Singh K, Khadia M, Kumar R,
Bansal V and Mishra A: Exploring the influence of metabolic changes
in fibrotic lung diseases. Pulm Circ. 15:e701632025. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Liu Z, Liu W, Wei H, Ping Y, Yu Z, Dong Z,
Ren J, Zhang S and Liu S: Elevated lactate production exacerbates
PM2.5-induced pulmonary fibrosis by stabilizing TGF-β1. J Adv Res.
S2090-1232(25)00587-9. 2025.Epub ahead of print. View Article : Google Scholar
|
|
88
|
Zhao BY, Xu YP, Chen YL, Cai Y, Gong Z, Li
D, Kuang H, Liu X, Zhou H, Liu G and Yin Y: Activation of TRPV4 by
lactate as a critical mediator of renal fibrosis in spontaneously
hypertensive rats after moderate- and high-intensity exercise.
Front Physiol. 13:9270782022. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Xiang T, Wang X, Huang S, Zhou K, Fei S,
Zhou B, Yue K, Li Q, Xue S, Dai Y, et al: Inhibition of PKM2 by
shikonin impedes TGF-β1 expression by repressing histone
lactylation to alleviate renal fibrosis. Phytomedicine.
136:1563242025. View Article : Google Scholar
|
|
90
|
Wei X, Long M, Yu J and Du Y: The
lactate-lactylation axis in renal fibrosis: Potential mechanisms in
diabetic kidney disease. Ann Med. 57:25873262025. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Efiong EE, Maedler K, Effa E, Osuagwu UL,
Peters E, Ikebiuro JO, Soremekun C, Ihediwa U, Niu J, Fuchs M, et
al: Decoding diabetic kidney disease: A comprehensive review of
interconnected pathways, molecular mediators, and therapeutic
insights. Diabetol Metab Syndr. 17:1922025. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Xiong Y, Zhou J, Wang JR and Huang H: How
lactate and lactylation shape the immunity system in
atherosclerosis (review). Int J Mol Med. 56:1632025. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Deng D, Luo Y, Hong Y, Ren X, Zu X and
Feng J: Lactylation: A new direction for tumor-targeted therapy.
Biochim Biophys Acta Rev Cancer. 1880:1893992025. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Gao X, Pang C, Fan Z, Wang Y, Duan Y and
Zhan H: Regulation of newly identified lysine lactylation in
cancer. Cancer Lett. 587:2166802024. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Ma F and Yu W: The roles of lactate and
lactylation in diseases related to mitochondrial dysfunction. Int J
Mol Sci. 26:72492025. View Article : Google Scholar
|
|
96
|
Wang D, Rong H, Ma K and Peng J:
Lactylation in tumor: Mechanisms and therapeutic potentials. Front
Immunol. 16:16095962025. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Zhu R, Ye X, Lu X, Xiao L, Yuan M, Zhao H,
Guo D, Meng Y, Han H, Luo S, et al: ACSS2 acts as a lactyl-CoA
synthetase and couples KAT2A to function as a lactyltransferase for
histone lactylation and tumor immune evasion. Cell Metab.
37:361–376.e7. 2025. View Article : Google Scholar
|
|
98
|
Liu R, Ren X, Park YE, Feng H, Sheng X,
Song X, AminiTabrizi R, Shah H, Li L, Zhang Y, et al: Nuclear
GTPSCS functions as a lactyl-CoA synthetase to promote histone
lactylation and gliomagenesis. Cell Metab. 37:377–394.e9. 2025.
View Article : Google Scholar
|
|
99
|
Tan Q, Liu M and Tao X: Targeting
lactylation: From metabolic reprogramming to precision therapeutics
in liver diseases. Biomolecules. 15:11782025. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH,
Lee JE, Wang C, Brindle PK, Dent SYR and Ge K: Distinct roles of
GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in
nuclear receptor transactivation. EMBO J. 30:249–262. 2011.
View Article : Google Scholar
|
|
101
|
Li Y, Cao Q, Hu Y, He B, Cao T, Tang Y,
Zhou XP, Lan XP and Liu SQ: Advances in the interaction of
glycolytic reprogramming with lactylation. Biomed Pharmacother.
177:1169822024. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Chen Y, Wu J, Zhai L, Zhang T, Yin H, Gao
H, Zhao F, Wang Z, Yang X, Jin M, et al: Metabolic regulation of
homologous recombination repair by MRE11 lactylation. Cell.
187:294–311.e21. 2024. View Article : Google Scholar
|
|
103
|
He Y, Song T, Ning J, Wang Z, Yin Z, Jiang
P, Yuan Q, Yu W and Cheng F: Lactylation in cancer: Mechanisms in
tumour biology and therapeutic potentials. Clin Transl Med.
14:e700702024. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Lazar AG, Vlad ML, Manea A, Simionescu M
and Manea SA: Activated histone acetyltransferase p300/CBP-related
signalling pathways mediate up-regulation of NADPH oxidase,
inflammation, and fibrosis in diabetic kidney. Antioxidants
(Basel). 10:13562021. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Chung S, Kim S, Son M, Kim M, Koh ES, Shin
SJ, Park CW and Kim HS: Inhibition of p300/CBP-associated factor
attenuates renal tubulointerstitial fibrosis through modulation of
NF-kB and Nrf2. Int J Mol Sci. 20:15542019. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Miguel V, Tituaña J, Herrero JI, Herrero
L, Serra D, Cuevas P, Barbas C, Puyol DR, Márquez-Expósito L,
Ruiz-Ortega M, et al: Renal tubule Cpt1a overexpression protects
from kidney fibrosis by restoring mitochondrial homeostasis. J Clin
Invest. 131:e1406952021. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Zong Z, Xie F, Wang S, Wu X, Zhang Z, Yang
B and Zhou F: Alanyl-tRNA synthetase, AARS1, is a lactate sensor
and lactyltransferase that lactylates p53 and contributes to
tumorigenesis. Cell. 187:2375–2392.e33. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Ju J, Zhang H, Lin M, Yan Z, An L, Cao Z,
Geng D, Yue J, Tang Y, Tian L, et al: The alanyl-tRNA synthetase
AARS1 moonlights as a lactyltransferase to promote YAP signaling in
gastric cancer. J Clin Invest. 134:e1745872024. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Mao Y, Zhang J, Zhou Q, He X, Zheng Z, Wei
Y, Zhou K, Lin Y, Yu H, Zhang H, et al: Hypoxia induces
mitochondrial protein lactylation to limit oxidative
phosphorylation. Cell Res. 34:13–30. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Li H, Liu C, Li R, Zhou L, Ran Y, Yang Q,
Huang H, Lu H, Song H, Yang B, et al: AARS1 and AARS2 sense
L-lactate to regulate cGAS as global lysine lactyltransferases.
Nature. 634:1229–1237. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Dai SK, Liu PP, Li X, Jiao LF, Teng ZQ and
Liu CM: Dynamic profiling and functional interpretation of histone
lysine crotonylation and lactylation during neural development.
Development. 149:dev2000492022. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Chen C, Zhang YG, Zang Y, Fan Z, Han Y,
Bai X, Wang A, Zhang J, Wang J and Zhang K: SIRT3 functions as an
eraser of histone H3K9 lactylation to modulate transcription for
inhibiting the progression of esophageal cancer. Mol Cell
Proteomics. 24:1009732025. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Moreno-Yruela C, Zhang D, Wei W, Bæk M,
Liu W, Gao J, Danková D, Nielsen AL, Bolding JE, Yang L, et al:
Class I histone deacetylases (HDAC1-3) are histone lysine
delactylases. Sci Adv. 8:eabi66962022. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Gonzatti MB, Hintzen JCJ, Sharma I, Najar
MA, Tsusaka T, Marcinkiewicz MM, Da Silva Crispim CV, Snyder NW,
Burslem GM and Goldberg EL: Class I histone deacetylases catalyze
lysine lactylation. J Biol Chem. 301:1106022025. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Chen Z, Zhong M, Lin Y, Zhang W, Zhu Y,
Chen L, Huang Z, Luo K, Lu Z, Huang Z and Yan Y: METTL7B-induced
histone lactylation prevents heart failure by ameliorating cardiac
remodelling. J Mol Cell Cardiol. 202:64–80. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Du RH, Gao YM, Yan C, Ren X, Qi S, Liu G,
Guo X, Song X, Wang H, Rao J, et al: Sirtuin 1/sirtuin 3 are robust
lysine delactylases and sirtuin 1-mediated delactylation regulates
glycolysis. iScience. 27:1109112024. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Sun L, Zhang Y, Yang B, Sun S, Zhang P,
Luo Z, Feng T, Cui Z, Zhu T, Li Y, et al: Lactylation of METTL16
promotes cuproptosis via m6A-modification on FDX1 mRNA
in gastric cancer. Nat Commun. 14:65232023. View Article : Google Scholar
|
|
118
|
Sun S, Xu Z, He L, Shen Y, Yan Y, Lv X,
Zhu X, Li W, Tian WY, Zheng Y, et al: Metabolic regulation of
cytoskeleton functions by HDAC6-catalyzed α-tubulin lactylation.
Nat Commun. 15:83772024. View Article : Google Scholar
|
|
119
|
Dewanjee S, Vallamkondu J, Kalra RS,
Chakraborty P, Gangopadhyay M, Sahu R, Medala V, John A, Reddy PH,
De Feo V and Kandimalla R: The emerging role of HDACs: Pathology
and therapeutic targets in diabetes mellitus. Cells. 10:13402021.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Daude RB, Bhadane R and Shah JS:
Alpha-cyperone mitigates renal ischemic injury via modulation of
HDAC-2 expression in diabetes: Insights from molecular dynamics
simulations and experimental evaluation. Eur J Pharmacol.
975:1766432024. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Lechner S, Malgapo MIP, Grätz C, Steimbach
RR, Baron A, Rüther P, Nadal S, Stumpf C, Loos C, Ku X, et al:
Target deconvolution of HDAC pharmacopoeia reveals MBLAC2 as common
off-target. Nat Chem Biol. 18:812–820. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Wang Y, He J, Liao M, Hu M, Li W, Ouyang
H, Wang X, Ye T, Zhang Y and Ouyang L: An overview of Sirtuins as
potential therapeutic target: Structure, function and modulators.
Eur J Med Chem. 161:48–77. 2019. View Article : Google Scholar
|
|
123
|
Morigi M, Perico L and Benigni A: Sirtuins
in renal health and disease. J Am Soc Nephrol. 29:1799–1809. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Covarrubias AJ, Perrone R, Grozio A and
Verdin E: NAD+ metabolism and its roles in cellular
processes during ageing. Nat Rev Mol Cell Biol. 22:119–141. 2021.
View Article : Google Scholar :
|
|
125
|
Zhao X, He S, Luo X, Zhang X, Jiang X,
Liang Y, Tang T, Qi K, Wang Y, Zhang Y and Li P: Co-treatments of
vitamin D and mesenchymal stem cells effectively alleviate the
diabetic kidney disease through attenuating the SIRT1-mediated
pathways. Stem Cell Res Ther. 16:4512025. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Zhang R, Chang R, Wang H, Chen J, Lu C,
Fan K, Zhang Y, Li L, Yan S and Dong H: Untargeted metabolomic and
proteomic analysis implicates SIRT2 as a novel therapeutic target
for diabetic nephropathy. Sci Rep. 15:42362025. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Yang S, Yang G, Wang X, Xiang J, Kang L
and Liang Z: SIRT2 alleviated renal fibrosis by deacetylating SMAD2
and SMAD3 in renal tubular epithelial cells. Cell Death Dis.
14:6462023. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Wang LF, Li Q, Le Zhao J, Wen K, Zhang YT,
Zhao QH, Ding Q, Li JH, Guan XH, Xiao YF, et al: CD38 deficiency
prevents diabetic nephropathy by inhibiting lipid accumulation and
oxidative stress through activation of the SIRT3 pathway. Biochem
Cell Biol. 103:1–12. 2025.
|
|
129
|
Hu X, Huang X, Yang Y, Sun Y, Zhao Y,
Zhang Z, Qiu D, Wu Y, Wu G and Lei L: Dux activates
metabolism-lactylation-MET network during early iPSC reprogramming
with Brg1 as the histone lactylation reader. Nucleic Acids Res.
52:5529–5548. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Nuñez R, Sidlowski PFW, Steen EA,
Wynia-Smith SL, Sprague DJ, Keyes RF and Smith BC: The TRIM33
bromodomain recognizes histone lysine lactylation. ACS Chem Biol.
19:2418–2428. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Zhai G, Niu Z, Jiang Z, Zhao F, Wang S,
Chen C, Zheng W, Wang A, Zang Y, Han Y and Zhang K: DPF2 reads
histone lactylation to drive transcription and tumorigenesis. Proc
Natl Acad Sci USA. 121:e24214961212024. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Liu S, Ma Q, Zeng C, Li H, Su J, Song Z,
Yan R, Zhao Z, Tian S and Huang W: Crosstalk between lactylation
and RNA modifications in tumorigenesis: Mechanisms and therapeutic
implications. Biomark Res. 13:1102025. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Anaya-Sanchez A, Feng Y, Berude JC and
Portnoy DA: Detoxification of methylglyoxal by the glyoxalase
system is required for glutathione availability and virulence
activation in Listeria monocytogenes. PLoS Pathog. 17:e10098192021.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Ding L, Hou Y, Liu J, Wang X, Wang Z, Ding
W and Zhao K: Circulating concentrations of advanced glycation end
products, carboxymethyl lysine and methylglyoxal are associated
with renal function in individuals with diabetes. J Ren Nutr.
34:154–160. 2024. View Article : Google Scholar
|
|
135
|
Gaffney DO, Jennings EQ, Anderson CC,
Marentette JO, Shi T, Schou Oxvig AM, Streeter MD, Johannsen M,
Spiegel DA, Chapman E, et al: Non-enzymatic lysine lactoylation of
glycolytic enzymes. Cell Chem Biol. 27:206–213.e6. 2020. View Article : Google Scholar
|
|
136
|
Liu L, Zhang S, Xu J, Cao Y, Cui D, Liu C,
Shen B, Wu Y and Zhang Q: Mass-spectrometry-based quantitative
proteomic analysis reveals that methylglyoxal and carnosine
influence oxidative stress and RNA-processing associated proteins
in renal proximal tubule epithelial cells. Mol Biol Rep.
52:1002025. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Huang X, Zhou H, Tan T and Luo Y:
Dose-dependent scavenging of methylglyoxal by naringenin in
diabetic mice. ACS Omega. 10:18615–18621. 2025.PubMed/NCBI
|
|
138
|
Adeshara K, Dahlstrom EH, Lehto M, Groop
PH and Sandholm N; On Behalf of The Finn Diane Study Group: 7-OR:
Causal associations between glyoxalase 1 expression and kidney
disease in type 1 diabetes. Diabetes. 73(Suppl 1): 7–OR. 2024.
View Article : Google Scholar
|
|
139
|
Gong H, Zhong H, Cheng L, Li LP and Zhang
DK: Post-translational protein lactylation modification in health
and diseases: A double-edged sword. J Transl Med. 22:412024.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Jiang X, Yang Y, Li X, Li T, Yu T and Fu
X: Lactylation: An innovative approach to disease control. Aging
Dis. 16:2130–2150. 2024.PubMed/NCBI
|
|
141
|
Singh J, Jain A, Bhamra R, Rathi V and
Dhingra AK: The mechanistic role of different mediators in the
pathophysiology of nephropathy: A review. Curr Drug Targets.
24:104–117. 2023. View Article : Google Scholar
|
|
142
|
Hu X, Chen W, Yang M, Li M, Li X and
Ouyang S: IGFBP5 promotes EndoMT and renal fibrosis through H3K18
lactylation in diabetic nephropathy. Cell Mol Life Sci. 82:2152025.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Elendu C, John Okah M, Fiemotongha KDJ,
Adeyemo BI, Bassey BN, Omeludike EK and Obidigbo B: Comprehensive
advancements in the prevention and treatment of diabetic
nephropathy: A narrative review. Medicine (Baltimore).
102:e353972023. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Xu Y, Zhang L, Shang D and Xiang H:
Lactylation: From molecular insights to disease relevance.
Biomolecules. 15:8102025. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Zhou T, Xu H, Cheng X, He Y, Ren Q, Li D,
Xie Y, Gao C, Zhang Y, Sun X, et al: Sodium butyrate attenuates
diabetic kidney disease partially via histone butyrylation
modification. Mediators Inflamm. 2022:76433222022. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Qu P, Li L, Jin Q, Liu D, Qiao Y, Zhang Y,
Sun Q, Ran S, Li Z, Liu T and Peng L: Histone methylation
modification and diabetic kidney disease: Potential molecular
mechanisms and therapeutic approaches (review). Int J Mol Med.
54:1042024. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Lu J, Li XQ, Chen PP, Zhang JX, Liu L,
Wang GH, Liu XQ, Jiang TT, Wang MY, Liu WT, et al: Activation of
acetyl-CoA synthetase 2 mediates kidney injury in diabetic
nephropathy. JCI Insight. 8:e1658172023. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Wang MJ, Cai X, Liang RY, Zhang EM, Liang
XQ, Liang H, Fu C, Zhou AD, Shi Y, Xu F and Cai MY: SIRT1-dependent
deacetylation of Txnip H3K9ac is critical for exenatide-improved
diabetic kidney disease. Biomed Pharmacother. 167:1155152023.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Qu ZA, Ma XJ, Huang SB, Hao XR, Li DM,
Feng KY and Wang WM: SIRT2 inhibits oxidative stress and
inflammatory response in diabetic osteoarthritis. Eur Rev Med
Pharmacol Sci. 24:2855–2864. 2020.PubMed/NCBI
|
|
150
|
He Y, Xie Y, Zhou T, Li D, Cheng X, Yang
P, Luo C, Liu Y, Guo M, Wan Q, et al: Sodium crotonate alleviates
diabetic kidney disease partially via the histone crotonylation
pathway. Inflammation. 48:254–275. 2025. View Article : Google Scholar
|
|
151
|
Liebisch M and Wolf G: AGE-induced
suppression of EZH2 mediates injury of podocytes by reducing
H3K27me3. Am J Nephrol. 51:676–692. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Cai J, Liu Z, Huang X, Shu S, Hu X, Zheng
M, Tang C, Liu Y, Chen G, Sun L, et al: The deacetylase sirtuin 6
protects against kidney fibrosis by epigenetically blocking
β-catenin target gene expression. Kidney Int. 97:106–118. 2020.
View Article : Google Scholar
|
|
153
|
Zorrilla-Zubilete MA, Yeste A, Quintana
FJ, Toiber D, Mostoslavsky R and Silberman DM: Epigenetic control
of early neurodegenerative events in diabetic retinopathy by the
histone deacetylase SIRT6. J Neurochem. 144:128–138. 2018.
View Article : Google Scholar
|
|
154
|
Qin K, Zhang N, Zhang Z, Nipper M, Zhu Z,
Leighton J, Xu K, Musi N and Wang P: SIRT6-mediated transcriptional
suppression of Txnip is critical for pancreatic beta cell function
and survival in mice. Diabetologia. 61:906–918. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Lo KA, Bauchmann MK, Baumann AP, Donahue
CJ, Thiede MA, Hayes LS, des Etages SA and Fraenkel E: Genome-wide
profiling of H3K56 acetylation and transcription factor binding
sites in human adipocytes. PLoS One. 6:e197782011. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Guo Q, Li X, Han H, Li C, Liu S, Gao W and
Sun G: Histone lysine methylation in TGF-β1 mediated p21 gene
expression in rat mesangial cells. Biomed Res Int.
2016:69272342016. View Article : Google Scholar
|
|
157
|
Malek V, Sharma N and Gaikwad AB: Histone
acetylation regulates natriuretic peptides and neprilysin gene
expressions in diabetic cardiomyopathy and nephropathy. Curr Mol
Pharmacol. 12:61–71. 2019. View Article : Google Scholar
|
|
158
|
Malek V and Gaikwad AB: Telmisartan and
thiorphan combination treatment attenuates fibrosis and apoptosis
in preventing diabetic cardiomyopathy. Cardiovasc Res. 115:373–384.
2019. View Article : Google Scholar
|
|
159
|
Aggarwal A, Yadav B, Sharma N, Kaur R and
Rishi V: Disruption of histone acetylation homeostasis triggers
cognitive dysfunction in experimental diabetes. Neurochem Int.
170:1055922023. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Wang J, Yang P, Yu T, Gao M, Liu D, Zhang
J, Lu C, Chen X, Zhang X and Liu Y: Lactylation of PKM2 suppresses
inflammatory metabolic adaptation in pro-inflammatory macrophages.
Int J Biol Sci. 18:6210–6225. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Li Q, Lin G, Zhang K, Liu X, Li Z, Bing X,
Nie Z, Jin S, Guo J and Min X: Hypoxia exposure induces lactylation
of Axin1 protein to promote glycolysis of esophageal carcinoma
cells. Biochem Pharmacol. 226:1164152024. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Fan Z, Zhang Y, Yuan L, Gao Y, Tian X,
Tian J, Wan J, Li B, Wang X, Wang S, et al: LARS1 lactylation
inhibits autophagy by activating mTORC1 to promote podocytes injury
in diabetic kidney disease. Cell Signal. 134:1119552025. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Chen J, Feng Q, Qiao Y, Pan S, Liang L,
Liu Y, Zhang X, Liu D and Liu Z and Liu Z: ACSF2 and lysine
lactylation contribute to renal tubule injury in diabetes.
Diabetologia. 67:1429–1443. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Yang G, Liu X, Li Y, Li L, Xiang J, Liang
Z, Jiang M and Yang S: TRIM65 as a key regulator of ferroptosis and
glycolysis in lactate-driven renal tubular injury and diabetic
kidney disease. Cell Rep. 44:1160912025. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Jin H, Wu P, Lv C, Zhang S, Zhang Y, Li C,
Gao R, Shan G, Bi H, Chang H, et al: Mannose inhibits PKM2
lactylation to induce pyroptosis in bladder cancer and activate
antitumor immune responses. Commun Biol. 8:6892025. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Li C, Fu C, Zhou W, Li H, Liu Z, Wu G, He
T, Shen M and Liu H: Lactylation modification of HIF-1α enhances
its stability by blocking VHL recognition. Cell Commun Signal.
23:3642025. View Article : Google Scholar
|
|
167
|
Yan X, Zhang G, Wei H, Yang L and Jiang X:
Lysine lactylation: Regulatory mechanisms, role in health and
disease, and its therapeutic potential. Drug Discov Today.
30:1044202025. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Zhang W, Shan G, Bi G, Hu Z, Yi Y, Zeng D,
Lin Z and Zhan C: Lactylation and regulated cell death. Biochim
Biophys Acta Mol Cell Res. 1872:1199272025. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Yu Y, Zhao F, Yue Y, Zhao Y and Zhou DX:
Lysine acetylation of histone acetyltransferase adaptor protein
ADA2 is a mechanism of metabolic control of chromatin modification
in plants. Nat Plants. 10:439–452. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Wang X, Qu Y, Li Z and Xia Q: Histone
crotonylation in tumors (review). Mol Clin Oncol. 22:392025.
View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Mikkat S, Kreutzer M and Patenge N: Lysine
phoshoglycerylation is widespread in bacteria and overlaps with
acylation. Microorganisms. 12:15562024. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Gui W, Davidson GA and Zhuang Z: Chemical
methods for protein site-specific ubiquitination. RSC Chem Biol.
2:450–467. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Latham T, Mackay L, Sproul D, Karim M,
Culley J, Harrison DJ, Hayward L, Langridge-Smith P, Gilbert N and
Ramsahoye BH: Lactate, a product of glycolytic metabolism, inhibits
histone deacetylase activity and promotes changes in gene
expression. Nucleic Acids Res. 40:4794–4803. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Ma Q, Hu T, Lin Y, Li J, Huang J, Zhang Q,
Gong T, Zhou X, Lei L, Zou J, et al: Crosstalk between lysine
lactylation and acetylation regulates lactate dehydrogenase in
streptococcus mutans. Genomics Proteomics Bioinformatics.
qzaf0732025.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Chu F, Sharma S, Ginsberg SD and Chiosis
G: PTMs as molecular encoders: Reprogramming chaperones into
epichaperomes for network control in disease. Trends Biochem Sci.
50:892–905. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Beltrao P, Bork P, Krogan NJ and van Noort
V: Evolution and functional cross-talk of protein
post-translational modifications. Mol Syst Biol. 9:7142013.
View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Cheng Z, Cheng Z, Zhang Y and Zhang S:
'Intrinsic disorder-protein modification-LLPS-tumor' regulatory
axis: From regulatory mechanisms to precision medicine. Biochim
Biophys Acta Rev Cancer. 1880:1892422025. View Article : Google Scholar
|
|
178
|
Loboda A, Sobczak M, Jozkowicz A and Dulak
J: TGF-β1/Smads and miR-21 in Renal Fibrosis and Inflammation.
Mediators Inflamm. 2016:83192832016. View Article : Google Scholar
|
|
179
|
Cappelli C, Tellez A, Jara C, Alarcón S,
Torres A, Mendoza P, Podestá L, Flores C, Quezada C, Oyarzún C and
San Martín R: The TGF-β profibrotic cascade targets
ecto-5'-nucleotidase gene in proximal tubule epithelial cells and
is a traceable marker of progressive diabetic kidney disease.
Biochim Biophys Acta Mol Basis Dis. 1866:1657962020. View Article : Google Scholar
|
|
180
|
Liu Z, Li A, Ma Z, Wang J, Chen X, Wang Z
and Fu R: Lactate metabolism and protein lactylation in cancer. Mol
Biomed. 7:152026. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Guo S, Ye M, Zhu W and Liu C: From fuel to
epigenetic signal: Lactate-lactylation axis orchestrates diabetic
complications. Pharmacol Res. 222:1080522025. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Cheng Y and Guo L: Lactate metabolism and
lactylation in kidney diseases: Insights into mechanisms and
therapeutic opportunities. Ren Fail. 47:24697462025. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Fang Y, Li Z, Yang L, Li W, Wang Y, Kong
Z, Miao J, Chen Y, Bian Y and Zeng L: Emerging roles of lactate in
acute and chronic inflammation. Cell Commun Signal. 22:2762024.
View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Chen C, Wang J, Zhu X, Zhang S, Yuan X, Hu
J, Liu C, Liu L, Zhang Z and Li J: Lactylation as a metabolic
epigenetic modification: Mechanistic insights and regulatory
pathways from cells to organs and diseases. Metabolism.
169:1562892025. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Deng J, Li Y, Yin L, Liu S, Li Y, Liao W,
Mu L, Luo X and Qin J: Histone lactylation enhances GCLC expression
and thus promotes chemoresistance of colorectal cancer stem cells
through inhibiting ferroptosis. Cell Death Dis. 16:1932025.
View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Wang X, Tang J, Zhang S, Chen X, Zhang J
and Fu Y: The dual faces of lactylation: From post-translational
modification to disease regulation. J Pharm Anal. 1015892026.
View Article : Google Scholar
|
|
187
|
Liu S, Yao S, Yang H, Liu S and Wang Y:
Autophagy: Regulator of cell death. Cell Death Dis. 14:6482023.
View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Shan XM, Chen CW, Zou DW, Gao YB, Ba YY,
He JX, Zhu ZY and Liang JJ: Suppression of ferroptosis through the
SLC7A11/glutathione/glutathione peroxidase 4 axis contributes to
the therapeutic action of the Tangshenning formula on diabetic
renal tubular injury. Chin Med. 19:1512024. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Wang Y, Zhang Q, Lv S, Wang X, Liu Q, Liu
X, Zhang Y and Liu G: PHGDH alleviates DKD by regulating
YB1/SLC7A11-mediated ferroptosis in podocytes. Transl Res.
282:1–13. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Xiang J, Yang G, Li L, Liao T, Li Y, Liu
X, Kang L, Wang X, Yang S and Liang Z: Lactate orchestrates the
TGFβ pathway and ferroptosis nexus in organ fibrosis via USP2
lactylation. Commun Biol. 8:18552025. View Article : Google Scholar
|
|
191
|
Yu H, Yin H, Mei Y, Bai X, Zhang C, Feng
Z, Li M, Chen H, Liang S, Gou X, et al: Nucleophosmin 1 lactylation
in graft kidney induces ferroptotic trigger waves that exacerbate
delayed graft function. Nat Commun. 17:2772025. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Wang Y, Yue Q, Song X, Du W and Liu R:
Hypoxia/reoxygenation-induced glycolysis mediates myocardial
ischemia-reperfusion injury through promoting the lactylation of
GPX4. J Cardiovasc Transl Res. 18:762–774. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Gan Y, Zhang J, Fu X, Wang Y, Zhao C, Dai
Y, Yan H, Liu Q, Sun W and Liu L: Lactylation-mitochondria axis in
chronic kidney disease: Metabolic reprogramming, epigenetic
dysregulation, and therapeutic potential. Mol Cell Biochem.
481:1533–1545. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Yu X, Guo Q, Zhang H, Wang X, Han Y and
Yang Z: Hypoxia-inducible factor-1α can reverse the Adriamycin
resistance of breast cancer adjuvant chemotherapy by upregulating
transferrin receptor and activating ferroptosis. FASEB J.
38:e238762024. View Article : Google Scholar
|
|
195
|
Hu W, Liang K, Zhu H, Zhao C, Hu H and Yin
S: Ferroptosis and its role in chronic diseases. Cells.
11:20402022. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
TeSlaa T, Ralser M, Fan J and Rabinowitz
JD: The pentose phosphate pathway in health and disease. Nat Metab.
5:1275–1289. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Sheng YC, Huang JN, Wu WL, Wan XR, Wang J,
Qin ZH and Wang Y: TIGAR plays neuroprotective roles in
MPP+/MPTP-induced Parkinson's disease by alleviating
ferroptosis. Eur J Pharmacol. 995:1774302025. View Article : Google Scholar
|
|
198
|
Quinn WJ III, Jiao J, TeSlaa T, Stadanlick
J, Wang Z, Wang L, Akimova T, Angelin A, Schäfer PM, Cully MD, et
al: Lactate limits T cell proliferation via the NAD(H) redox state.
Cell Rep. 33:1085002020. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Yu J, Liu Y, Li H and Zhang P:
Pathophysiology of diabetic kidney disease and autophagy: A review.
Medicine (Baltimore). 102:e339652023. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Li QR, Xu HY, Ma RT, Ma YY and Chen MJ:
Targeting autophagy: A promising therapeutic strategy for diabetes
mellitus and diabetic nephropathy. Diabetes Ther. 15:2153–2182.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Egan DF, Shackelford DB, Mihaylova MM,
Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor
R, et al: Phosphorylation of ULK1 (hATG1) by AMP-activated protein
kinase connects energy sensing to mitophagy. Science. 331:456–461.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Sun W, Jia M, Feng Y and Cheng X: Lactate
is a bridge linking glycolysis and autophagy through lactylation.
Autophagy. 19:3240–3241. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Panwar V, Singh A, Bhatt M, Tonk RK,
Azizov S, Raza AS, Sengupta S, Kumar D and Garg M: Multifaceted
role of mTOR (mammalian target of rapamycin) signaling pathway in
human health and disease. Signal Transduct Target Ther. 8:3752023.
View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Sun L, Wu S, Wang H, Zhang T, Zhang M, Bai
X, Zhang X, Li B, Zhang C, Li Y, et al: PDCD6 regulates lactate
metabolism to modulate LC3-associated phagocytosis and
antibacterial defense. Nat Commun. 15:101572024. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Li J, Shi X, Xu J, Wang K, Hou F, Luan X
and Chen L: Aldehyde dehydrogenase 2 lactylation aggravates
mitochondrial dysfunction by disrupting PHB2 mediated mitophagy in
acute kidney injury. Adv Sci (Weinh). 12:e24119432025. View Article : Google Scholar :
|
|
206
|
Wang M, Zhang Q, Lou S, Jin L, Wu G, Wu W,
Tang Q, Wang Y, Long X, Huang P, et al: Inhibition of MD2 by
natural product-drived JM-9 attenuates renal inflammation and
diabetic nephropathy in mice. Biomed Pharmacother. 168:1156602023.
View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Wei J, Xu Z and Yan X: The role of the
macrophage-to-myofibroblast transition in renal fibrosis. Front
Immunol. 13:9343772022. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Tang Y, Li Y, Yang X, Lu T, Wang X, Li Z,
Liu J and Wang J: Intestinal metabolite TMAO promotes CKD
progression by stimulating macrophage M2 polarization through
histone H4 lysine 12 lactylation. Cell Death Differ. 33:314–326.
2026. View Article : Google Scholar :
|
|
209
|
Hu M, Yao Z, Xu L, Peng M, Deng G, Liu L,
Jiang X and Cai X: M2 macrophage polarization in systemic sclerosis
fibrosis: Pathogenic mechanisms and therapeutic effects. Heliyon.
9:e162062023. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Wang Y, Li H, Jiang S, Fu D, Lu X, Lu M,
Li Y, Luo D, Wu K, Xu Y, et al: The glycolytic enzyme PFKFB3 drives
kidney fibrosis through promoting histone lactylation-mediated
NF-κB family activation. Kidney Int. 106:226–240. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Cao H, Luo J, Zhang Y, Mao X, Wen P, Ding
H, Xu J, Sun Q, He W, Dai C, et al: Tuberous sclerosis 1 (Tsc1)
mediated mTORC1 activation promotes glycolysis in tubular
epithelial cells in kidney fibrosis. Kidney Int. 98:686–698. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Ma XM, Geng K, Wang P, Jiang Z, Law BYK
and Xu Y: MCT4-dependent lactate transport: A novel mechanism for
cardiac energy metabolism injury and inflammation in type 2
diabetes mellitus. Cardiovasc Diabetol. 23:962024. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Wang Z, Yu J, Hao D, Liu X and Wang X:
Transcriptomic signatures responding to PKM2 activator TEPP-46 in
the hyperglycemic human renal proximal epithelial tubular cells.
Front Endocrinol (Lausanne). 13:9653792022. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Ishihara SI, Kayes MI, Makino H, Matsuda
H, Kumagai A, Hayashi Y, Ferdaus SA, Kawakita E, Koya D and
Kanasaki K: The PKM2 activator TEPP-46 suppresses cellular
senescence in hydrogen peroxide-induced proximal tubular cells and
kidney fibrosis in CD-1db/db mice. J Diabetes Investig.
16:598–607. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Chen Y, Bai X, Chen J, Huang M, Hong Q,
Ouyang Q, Sun X, Zhang Y, Liu J, Wang X, et al: Pyruvate kinase M2
regulates kidney fibrosis through pericyte glycolysis during the
progression from acute kidney injury to chronic kidney disease.
Cell Prolif. 57:e135482024. View Article : Google Scholar :
|
|
216
|
Sachan R, Kundu A, Dey P, Son JY, Kim KS,
Lee DE, Kim HR, Park JH, Lee SH, Kim JH, et al: Dendropanax
morbifera protects against renal fibrosis in streptozotocin-induced
diabetic rats. Antioxidants (Basel). 9:842020. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Li X, Ma TK, Wang M, Zhang XD, Liu TY, Liu
Y, Huang ZH, Zhu YH, Zhang S, Yin L, et al: YY1-induced
upregulation of LncRNA-ARAP1-AS2 and ARAP1 promotes diabetic kidney
fibrosis via aberrant glycolysis associated with EGFR/PKM2/HIF-1α
pathway. Front Pharmacol. 14:10693482023. View Article : Google Scholar
|
|
218
|
Peng J, Jiang Z, Song J, Chen J, Fu Z,
Zhang H, Zhen J, Tuerdi M, Luo M, Wu J and Sun T: Identification of
lactylation-related hub genes as novel therapeutic and diagnostic
targets for thoracic aortic dissection. Cell Signal.
134:1119442025. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Jiang T, Dong Y, Wang L, Wu T, Yu X, Xiao
Y and Zhong T: Genistein mitigates renal fibrosis in diabetic
nephropathy by suppressing epithelial-mesenchymal transition and
aberrant glycolysis through downregulating the expression of
HIF-1α. Food Biosci. 71:1071722025. View Article : Google Scholar
|
|
220
|
Yang H, Liu M, Song S, Xu Q, Lee J, Sun J,
Xue S, Sun X and Che C: HIF-1α promotes inflammatory responses in
aspergillus fumigatus keratitis by activating pyroptosis through
caspase-8/GSDMD pathway. Invest Ophthalmol Vis Sci. 66:322025.
View Article : Google Scholar
|
|
221
|
Zhou L, Zhang H, Liu L, Zhang F, Wang L,
Zheng P, Mao Z, Zhu X, Zi G, Chen L, et al: Intermittent hypoxia
aggravates asthma inflammation via NLRP3/IL-1β-dependent pyroptosis
mediated by HIF-1α signalling pathway. Chin Med J (Engl).
138:1714–1729. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Jia Y, Chen J, Zheng Z, Tao Y, Zhang S,
Zou M, Yang Y, Xue M, Hu F, Li Y, et al: Tubular epithelial
cell-derived extracellular vesicles induce macrophage glycolysis by
stabilizing HIF-1α in diabetic kidney disease. Mol Med. 28:952022.
View Article : Google Scholar
|
|
223
|
Singh M, Afonso J, Sharma D, Gupta R and
Kumar V, Rani R, Baltazar F and Kumar V: Targeting monocarboxylate
transporters (MCTs) in cancer: How close are we to the clinics?
Semin Cancer Biol. 90:1–14. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Silva A, Antunes B, Batista A,
Pinto-Ribeiro F, Baltazar F and Afonso J: In vivo anticancer
activity of AZD3965: A systematic review. Molecules. 27:1812021.
View Article : Google Scholar
|
|
225
|
Wu X, Zhang X, Tang S and Wang Y: The
important role of the histone acetyltransferases p300/CBP in cancer
and the promising anticancer effects of p300/CBP inhibitors. Cell
Biol Toxicol. 41:322025. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Tiwari R, Sharma R, Rajendran G, Borkowski
GS, An SY, Schonfeld M, O'Sullivan J, Schipma MJ, Zhou Y, Courbon
G, et al: Postischemic inactivation of HIF prolyl hydroxylases in
endothelium promotes maladaptive kidney repair by inducing
glycolysis. J Clin Invest. 135:e1762072024. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Rakshe PS and Gaikwad AB: Diabetic kidney
disease: Exploring mechanistic depths and the future of
pharmacological intervention. Naunyn Schmiedebergs Arch Pharmacol.
399:1625–1638. 2026. View Article : Google Scholar
|
|
228
|
Chen Y, Wang Y, Yu D, Zhao H and Li P:
Sirtuin 3 in diabetic kidney disease: Mechanisms and
pharmacotherapy. Ren Fail. 47:25439272025. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Chen DQ, Han J, Liu H, Feng K and Li P:
Targeting pyruvate kinase M2 for the treatment of kidney disease.
Front Pharmacol. 15:13762522024. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Alquraishi M, Puckett DL, Alani DS,
Humidat AS, Frankel VD, Donohoe DR, Whelan J and Bettaieb A:
Pyruvate kinase M2: A simple molecule with complex functions. Free
Radic Biol Med. 143:176–192. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Wang Y, Liu J, Jin X, Zhang D, Li D, Hao
F, Feng Y, Gu S, Meng F, Tian M, et al: O-GlcNAcylation
destabilizes the active tetrameric PKM2 to promote the Warburg
effect. Proc Natl Acad Sci USA. 114:13732–13737. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
232
|
Kirk P, Wilson MC, Heddle C, Brown MH,
Barclay AN and Halestrap AP: CD147 is tightly associated with
lactate transporters MCT1 and MCT4 and facilitates their cell
surface expression. EMBO J. 19:3896–3904. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Payen VL, Mina E, Van Hée VF, Porporato PE
and Sonveaux P: Monocarboxylate transporters in cancer. Mol Metab.
33:48–66. 2020. View Article : Google Scholar :
|
|
234
|
Bonen A: The expression of lactate
transporters (MCT1 and MCT4) in heart and muscle. Eur J Appl
Physiol. 86:6–11. 2001. View Article : Google Scholar
|
|
235
|
Li S, Nguyen TT and Bonanno JA: CD147
required for corneal endothelial lactate transport. Invest
Ophthalmol Vis Sci. 55:4673–4681. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
236
|
Schneiderhan W, Scheler M, Holzmann KH,
Marx M, Gschwend JE, Bucholz M, Gress TM, Seufferlein T, Adler G
and Oswald F: CD147 silencing inhibits lactate transport and
reduces malignant potential of pancreatic cancer cells in in vivo
and in vitro models. Gut. 58:1391–1398. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
237
|
Lokman FE, Seman NA, Ismail AA, Yaacob NA,
Mustafa N, Khir AS, Hussein Z and Wan Mohamud WN: Gene expression
profiling in ethnic Malays with type 2 diabetes mellitus, with and
without diabetic nephropathy. J Nephrol. 24:778–789. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
238
|
Qiu F, Fan S, Diao Y, Liu J, Li B, Li K
and Zhang W: The mechanism of chebulae fructus immaturus promote
diabetic wound healing based on network pharmacology and
experimental verification. J Ethnopharmacol. 322:1175792024.
View Article : Google Scholar
|
|
239
|
Semenza GL: HIF-1: Mediator of
physiological and pathophysiological responses to hypoxia. J Appl
Physiol (1985). 88:1474–1480. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
240
|
Yang L, Guo D, Wu K, Li Y, Xi Y, Qin W,
Chen X, Zhou C and Tang J: Lactate metabolism: The String-puller
for the development of pancreatic cancer. Biology (Basel).
14:12132025.PubMed/NCBI
|
|
241
|
Tian L, Cheng S and Li X and Li X:
AARS1-mediated lactylation of Akt/p65 drives their activation and
glycolytic reprogramming in diabetic kidney disease. J Am Soc
Nephrol. 36: View Article : Google Scholar : 2025.
|
|
242
|
Hu K, Yu Z, Yuan Y, Yi J, Li J, Zhu M,
Meng Z, Liu Y and Cao D: Lactate/AARS1/H3K18la/LDHA positive
feedback loop triggers ferroptosis, which participates in diabetic
nephropathy via the modulation of ACSL4 transcription. Acta Biochim
Biophys Sin (Shanghai). Oct 30–2025.Epub ahead of print. View Article : Google Scholar
|














