Introduction
Despite current treatments, ischemic heart disease
is the primary cause of global morbidity and mortality (1). Although thrombolysis or
revascularization can effectively mitigate myocardial ischemic
injury, the subsequent reperfusion can cause severe damage. This
reperfusion-induced injury can account for up to 50% of the final
myocardial infarction area and contribute to adverse events such as
arrhythmia and cardiac arrest (2). The pathogenic mechanisms of
ischemia-reperfusion (I/R) injury include myocardial inflammation,
apoptosis, calcium overload, excessive production of reactive
oxygen species (ROS), and disturbances in energy metabolism
(3). However, therapeutic
targets must be identified and applied to facilitate recovery.
Therefore, exploring the mechanisms of myocardial I/R injury (MIRI)
and developing preventive and therapeutic measures are of clinical
significance.
Mitochondria are the energy supply and metabolic
hubs of cells and have been a major focus of cardiovascular
research (4). Studies indicate
that mitochondrial dysfunction is one of the key mechanisms by
which mitochondria are implicated in I/R injury (5). The ATP balance of cardiomyocytes is
typically maintained by mitochondria through oxidative
phosphorylation (OXPHOS). However, during ischemia, hypoxia shifts
energy production from OXPHOS to glycolysis, impairing ATP
synthesis. Furthermore, an abrupt influx of oxygen results in a
surge of ROS. ROS disrupt mitochondrial structure, damage
mitochondrial membranes, respiratory chai and mitochondrial DNA
(6). In contrast to conventional
perspectives, emerging evidence suggests that mitochondrial
dynamics are central to metabolic regulation. Mitofusin proteins
1/2 (MFN1/2) dynamically remodel mitochondrial architecture to form
interconnected networks for content sharing and segregate damaged
components by fusion with dynamin-related protein 1 (DRP1)
(7,8). This dynamic equilibrium critically
regulates energy efficiency and oxidative stress tolerance.
Furthermore, PINK1/Parkin pathway-mediated impaired mitophagy is
directly associated with Parkinson's disease and diabetes (9).
Mitochondria also act as signaling hubs by releasing
ROS, calcium ions and metabolites, including acetyl-CoA and
α-ketoglutarate (α-KG), to regulate nuclear epigenetic
modifications and gene expression (10). For instance,
mitochondrial-derived ROS and metabolites can activate oncogenic
pathways or disrupt insulin signaling, connecting mitochondrial
dysfunction to tumor progression and metabolic disorders (11). Therefore, mitochondrial research
holds significant potential. A thorough investigation of the
mechanisms regulating mitochondrial homeostasis is conducive to the
development of more effective therapeutic strategies. Studies have
demonstrated that mitochondrial function can induce abnormal flux
changes in glycolysis and reprogram cell metabolism (11). Recently, it has been indicated
that changes in glycolytic activity can regulate mitochondrial
physiology through negative feedback mechanisms (12). However, the molecular mechanisms
and physiological consequences of this bidirectional dialogue
remain largely unexplored.
In addition to its role as a glycolytic end-product,
lactate is now recognized as an important mediator and a substrate
for a novel post-translational modification: Lactylation (13). This epigenetic mark, formed when
lactate is covalently attached to lysine residues, is driven by
intracellular lactate levels and regulated by 'writers' (for
instance, P300/CBP) and 'erasers' [such as histone deacetylase
(HDAC)]. In lactylation, lactate molecules bind covalently to
lysine residues in proteins via ester bonds. The main regulatory
factors of lactylation are typically associated with increased
intracellular lactate concentrations. Furthermore,
acylation-modifying enzymes, such as acyltransferase P300/CBP and
delactase HDAC, play important roles. Previous studies have
revealed that lactylation regulates the progression of numerous
diseases, including tumors, sepsis and immune diseases (14,15). Recent research indicates that
dexmedetomidine can regulate myocardial I/R-induced ferroptosis
through MDH2 K241 lactylation (16). However, there are few studies on
lactylation in MIRI, and the underlying mechanisms remain
unexplored.
The present study aimed to investigate the mechanism
and role of lactylation in MIRI regulation. It was observed that
increased lactylation levels exacerbated mitochondrial and cardiac
dysfunction following MIRI. Mechanistically, it was confirmed that
IDH2 lactylation was upregulated after MIRI. This upregulation led
to decreased enzymatic activity and α-KG-regulated AMPK
phosphorylation. These findings improve understanding of the role
of lactate in mitochondrial function and lay the groundwork for
developing novel targeted therapies to improve MIRI.
Materials and methods
Animal experiment
The ethics approval (approval no.
AHGDMU-LAC-B-202301-0004) for animal research was obtained from the
Experimental Animal Ethics Committee of Guangdong Medical
University (animal facility license, SYXK 2022-0286; Zhanjiang,
China). C57BL/6 mice were purchased from SPF Biotechnology Co.,
Ltd. Mice were housed under SPF conditions in a controlled
environment. The animal facility maintained a temperature of 22±2°C
and relative humidity of 50±10%, with a 12/12-h light/dark cycle
(lights on at 7:00 a.m.). Ventilation was set at 15 air changes per
hour, and all animals had ad libitum access to sterilized
food and autoclaved water. All procedures complied with the
institutional guidelines for animal care and use. A total of 110
male adult mice, weighing 25±5 g, were raised to 8-weeks old before
receiving the experimental treatment. They were then fed normally
for 1 week to reduce stress. A total of 100 neonatal mice were
placed in a thermostatic incubator on postnatal day 2 to prepare
for the isolation of primary cardiomyocytes.
I/R model
After successful intubation, the mice were connected
with an anesthesia ventilator. Then, the intercostal space was
fully expanded to expose the surgical field. An 8-0 suture needle
was employed to ligate the threading of the left anterior
descending coronary artery, and the left heart apex was white and
the ECG showed MI. After 45 min of ischemia elapsed, the heart was
re-exposed. The ligature was removed, and the ECG returned to
normal, signifying successful reperfusion. Subsequently, the chest
wall was sutured in layers, and resuscitation was carried out on a
warming pad.
Anesthesia procedures
General anesthesia was achieved with isoflurane for
all surgical interventions. Anesthesia was induced in an induction
chamber with 5% isoflurane delivered in 100% oxygen at a flow rate
of 1.5 l/min. Following the loss of righting and pedal reflexes,
the mouse was secured in the supine position on a heated surgical
platform equipped with continuous temperature maintenance. After
orotracheal intubation, anesthesia was maintained by connecting the
endotracheal tube to the gas delivery system, delivering 1.5-2.0%
isoflurane in 100% oxygen at a constant flow rate of 0.9 l/min
throughout the procedure.
Administration methods
Following myocardial I/R, mice were treated with
either 400 mg/kg lactate (cat. no. L1750; MilliporeSigma) via
intraperitoneal (i.p.) injection, or with 50 mg/kg
3-(1H-1,2,3-Triazol-4-yl) pyridine (3-TYP; cat. no. HY-108331;
MedChemExpress) i.p. 24 h prior to I/R. For genetic manipulation,
8-week-old mice received a tail vein (i.v.) injection of AAV9 virus
(2×10¹¹ GC/mouse); surgical procedures were performed 14 days
post-injection. Evans Blue-TTC staining: The areas of myocardial
infarction and area at risk (AAR) after I/R were determined and
evaluated using TTC (cat. no. T8877-5G; MilliporeSigma) and Evans
blue stain (cat. no. E2129; MilliporeSigma) after 3 days of I/R
modelling of mouse hearts. For the procedure, once the mice were
anesthetized and intubated, a thin tube was inserted into the
ascending aorta. Then, the aorta was perfused retrogradely with
1.5% Evans blue dye for 1 min. Subsequently, the entire heart was
frozen for 10 min at -80°C and sliced into 2 mm slices. After
rewarming and setting, the slices were placed in 1.5% TTC solution
and incubated at 37°C for 10 min, followed by incubation in 10%
formalin for 1 h. Finally, the slices were scanned, images were
captured and analyzed.
Mouse echocardiography
Mice were depilated with a razor and anaesthetized
with isoflurane. Mice were immobilized on a laboratory bench and
cardiac function was assessed by echocardiography (Vevo 3100;
Fujifilm Visual Sonics). The procedure was as follows: B-mode and
M-mode images were acquired in parasternal short-axis views with an
MS-400 ultrasound probe, and left ventricular end-systolic
diameter, left ventricular end-diastolic diameter, left ventricular
ejection fraction (LVEF%) and left ventricular fractional
shortening (LVFS%) were examined and calculated for measurements in
three consecutive cardiac cycles. EF% and FS% were calculated, and
each group was replicated in 5 mice.
Animal euthanasia
According to the AVMA Guidelines for the Euthanasia
of Animals (2020 Edition), at the end of the experimental protocol,
all animals were humanely euthanized. Adult mice were deeply
anesthetized with an i.p. injection of pentobarbital sodium (150
mg/kg body weight). Following confirmation of complete loss of
pedal and righting reflexes, euthanasia was ensured by cervical
dislocation prior to tissue collection. Neonatal mice were
anesthetized with 2% isoflurane, and after confirming loss of
consciousness and absence of reflexes, they were euthanized by
cervical dislocation, followed by tissue collection (17).
Humane endpoints
Throughout the study, animals were monitored daily
for signs of distress. The following humane endpoints were
established: i) weight loss exceeding 20% of baseline body weight;
ii) inability to ambulate or access food and water; iii) signs of
severe respiratory distress; iv) persistent hypothermia; and v)
moribund condition. Any animal meeting any of these criteria was
immediately euthanized by cervical dislocation under deep
isoflurane anesthesia to minimize suffering.
Cell culture and treatment
Primary cardiomyocytes
Briefly, ventricles from 2-day-old C57BL/6 neonatal
mice (euthanized by cervical dislocation under 2% isoflurane
anesthesia for 2 min) were excised, minced, and digested with 0.1%
trypsin (cat. no. T1300; Beijing Solarbio Science & Technology
Co., Ltd.) at 4°C for 14 h, followed by two 15-min digestions with
0.1 mg/ml type II collagenase (cat. no. 9001-12-1; Beijing Solarbio
Science & Technology Co., Ltd.) in PBS containing 5 mg/ml BSA
at 37°C under constant stirring. The cell suspension was collected,
neutralized with two volumes of DMEM/10% fetal bovine serum (FBS;
cat. no. 10099141C; Gibco; Thermo Fisher Scientific, Inc.) and
centrifuged at 500 × g for 3 min. Cells were resuspended in DMEM
supplemented with 10% FBS and 1% penicillin-streptomycin and plated
onto 100-mm dishes for 2 h at 37°C to allow fibroblast attachment
(18,19). Non-adherent cardiomyocytes were
collected, reseeded onto pre-coated dishes, and cultured at 37°C in
5% CO2.
Cell culture
Primary mouse cardiomyocytes and HL-1 cells (Cell
Bank; Chinese Academy of Sciences) were cultured in high-glucose
DMEM supplemented with 10% FBS at 37°C in a 5% CO2
humidified incubator.
Plasmids
Before transfection, 6-well plates were inoculated
with 5×104 cells/well in 3 ml of media and cultured for
24 h. When the cell confluence reached 40-50%, the cells were
transfected with IDH2-WT and IDH2-K275R plasmid (2,000 ng/well)
using lipofectamine 3000 reagent (cat. no. L3000015; Invitrogen;
Thermo Fisher Scientific, Inc.) following the manufacturer's
instructions (Table SI). After
8-10 h of incubation, cells' culture medium was replaced with
normal medium, then these cells were used for further
experiments.
Lentivirus transfection
ShRNAs (Table
SII) were purchased from Shanghai Genechem Co., Ltd. Lentiviral
particles were produced using a second-generation SIN packaging
system. The three plasmids (GV-based transfer vector, pHelper1.0,
and pHelper2.0) were co-transfected into 293T cells (Shanghai
GeneChem Co., Ltd.). After a 6-h incubation at 37°C, the medium was
replaced. Viral supernatants were collected at 48 and 72 h
post-transfection, pooled, and concentrated by ultracentrifugation
(120,000 × g, 2 h, 4°C) using a Beckman ultracentrifuge (Beckman
Coulter, Inc.). The concentrated virus was resuspended in PBS and
stored at -80°C. The optimal multiplicity of infection (MOI) was
predetermined as 100 in 96-well plates. For stable transfection,
cells at 60% confluence were infected with LV-sh-IDH2 lentivirus
(5×105 TU/ml) in infection solution P, followed by
medium replacement at regular intervals. At 80-90% confluence,
cells were expanded and selected with 3 µg/ml puromycin for
48 h. Stable clones were subsequently harvested and cryopreserved
in liquid nitrogen or at -80°C.
Hypoxia/reoxygenation (H/R) model
When the cell growth confluence was ~70-80%, the H/R
model was established. The cells were washed twice with PBS. After
that, an appropriate quantity of DMEM (without serum and
antibiotics) was added. The parameters were preset in order to
incubate the cells in a hypoxia incubator (37°C, 95% N2,
5% CO2, 1% O2) for 12 h. Complete culture
medium was added, and the specimens were put into a regular
constant temperature cell incubator for 3 h. These were then
employed for the subsequent experiments.
Cell treatment
Experimental cells were pretreated with
dichloroacetate (DCA; cat. no. B7174; APeXBIO Technology LLC) or
sodium lactate (NaLac; cat. no. L7022; MilliporeSigma) for 24 h,
both of the drug concentrations were 20 mmol/l. STO-609 (cat. no.
S8274; Selleck Chemicals) was used at the concentration of 10
µmol/l for the inhibition of AMPK signaling pathway in
vitro, which is a selective and cell-permeable inhibitor of
CaM-KK2.
Lactate measurement
The lactate of blood, cell and myocardial tissue
samples was measured using lactate assay kit (cat. no. BC2235;
Beijing Solarbio Science & Technology Co., Ltd.). According to
the instructions of the kit, the established standard curve was
used to determine the OD value at the wavelength of 450 nm, and the
lactate concentration was calculated accordingly.
Western blotting
Protein lysates were extracted from cardiac tissue
and experimental cells. Protein concentrations were determined
using the bicinchoninic acid (BCA) assay (cat. no. P0399S; Beyotime
Institute of Biotechnology). Equal amounts of protein (30 µg
per lane) were separated by electrophoresis on 10%
SDS-polyacrylamide gels at 4°C and subsequently transferred onto
PVDF membranes at 4°C. Following 1-h blocking with 5% non-fat milk
at 25°C, the membranes were incubated overnight with the following
primary antibodies at 4°C: phosphorylated (p-)AMPK (1:2,000; cat.
no. 50081; Proteintech Group, Inc.), IDH2 (1:2,000; cat. no. D8E3B;
Proteintech Group, Inc.), DRP1 (1:2,000; cat. no. 26187-1-AP;
Proteintech Group, Inc.), MnSOD2 (1:2,000; cat. no. 66474-1-Ig;
Proteintech Group, Inc.), AMPK (1:2,000; cat. no. 10929-2-AP; Cell
Signaling Technology, Inc.), Cytochrome C (1:2,000; cat. no.
PTM5351; PTM Biolab, Inc.; http://www.ptm-biolab.com.cn/index.html), Tubulin
(1:4,000; cat. no. 11224-1-AP; Cell Signaling Technology, Inc.) and
Pan-Kla (1:2,000; cat. no. PTM-1401; PTM Biolab, Inc.).
Subsequently, the horseradish peroxidase-conjugated secondary
antibody (1:5,000; cat. nos. SA00001-9 and SA00012-6; Proteintech
Group, Inc.) was incubated for 1 h at ambient temperature.
Visualization of the blots was achieved using enhanced
chemiluminescence (ECL; cat. no. WBKLS0500 MilliporeSigma).
Densitometric analysis was performed using ImageJ software (version
1.53t; National Institutes of Health).
Creatine kinase (CK)-MB
The CK-MB kit (cat. no. A032-1-1; Nanjing Jiancheng
Bioengineering Institute) was used to reflect myocardial injury.
After adding chromogen, the samples were incubated for 10 min at
37°C. After washing, and absorbance values were measured at 450
nm.
Immunofluorescence staining
Cells or tissue sections were fixed with 4%
paraformaldehyde for 15 min at room temperature, permeabilized with
0.1% Triton X-100 (cat. no. P0096; Beyotime Institute of
Biotechnology) for 10 min and blocked with 5% BSA in PBS for 1 h at
room temperature to reduce non-specific binding. Samples were then
incubated overnight at 4°C with the following primary antibodies:
Pan-Kla (cat. no. PTM-1401; PTM Biolab, Inc.) and cTNT (cat. no.
Ab005550; Beyotime Institute of Biotechnology) diluted 1:200 in
blocking buffer. After washing three times with PBS, samples were
incubated with the appropriate Alexa Fluor 488-conjugated goat
anti-mouse IgG (H+L) (cat. no. A0428) and Alexa Fluor
594-conjugated goat anti-rabbit IgG (H+L) (cat. no. A0516; both
from Beyotime Institute of Biotechnology) at a dilution of 1:500
for 1 h at room temperature in the dark. Nuclei were counterstained
with DAPI (cat. no. 4083S; Cell Signaling Technology, Inc.; diluted
to 5×10−4 mg/ml) for 5 min. Slides were mounted with
antifade mounting medium and visualized using a fluorescence
microscope (Olympus Corporation). Images were captured and
processed using ImageJ software.
TUNEL staining
Apoptosis was assessed by TUNEL assay (cat. no.
C1090; Beyotime Institute of Biotechnology). Fresh frozen
myocardial sections (5 µm) were fixed with 4%
paraformaldehyde (cat. no. P0099; Beyotime Institute of
Biotechnology) for 30 min at room temperature, permeabilized with
0.1% Triton X-100 for 10 min, and incubated with TUNEL reaction
solution for 2 h. After washing, sections were mounted and imaged
by fluorescence microscopy (Olympus Corporation). At least five
randomly selected fields of view per section were captured at ×200
magnification for quantitative analysis.
ROS detection
DHE staining
Intracellular ROS levels were measured by DHE
staining reagent (cat. no. S0063; Beyotime Institute of
Biotechnology). The cells were incubated for 20 min at 37°C in a
serum-free medium supplemented with 10 µM DHE fluorescent
probe. For animal experiments, 5-µm fresh-frozen slices of
mouse hearts were prepared immediately after the experimental
treatment; 100 µl of DHE working solution at a concentration
of 20 µM was used for each slice, and the slices were
incubated in a wet box and protected from light for 30 min at 37°C.
Fluorescence microscopy was utilized to examine and analyze the red
fluorescence intensity of DHE.
MitoSOX staining
Mitochondrial ROS was determined by MitoSOX staining
reagent (cat. no. MT14; Dojindo Laboratories, Inc.). Cells were
added with the 10 µmol/l MitoSOX Deep Red working solution,
and the sample was incubated for 30 min in a 5% CO2
incubator at 37°C. Following two washes with Hank's Balanced Salt
Solution (cat. no. C0219; Beyotime Institute of Biotechnology), the
fluorescence intensity was measured using a fluorescence
microscope.
Mouse 8-OHdG production assay
The content of mouse 8-OHdG was determined by ELISA
reagent (cat. no. E-EL-0028; Elabscience Biotechnology Co., Ltd.).
The fresh cell homogenate was added to the microplate for detection
according to the instructions of the manufacturer, and the OD value
of the reaction products at 450 nm was detected by a microplate
reader and calculated in accordance with the measured standard
curve.
ATP measurement, mitochondrial membrane
potential detection and NADPH/NADP+ ratio assay
The ATP content was determined using an assay kit
(cat. no. S0026; Beyotime Institute of Biotechnology) according to
the manufacturer's instructions. Mitochondrial membrane potential
was measured with a JC-1 kit (cat. no. C2003S; Beyotime Institute
of Biotechnology). High levels of JC-1 monomers indicate a decrease
in the mitochondrial membrane potential. After incubation according
to the manufacturer's instructions, JC-1 monomers were measured
using a BD FACSCanto II flow cytometer, and the data were analyzed
with FlowJo software (version 10.8; BD Biosciences). The level of
NADPH/NADP+ was measured using a kit (cat. no. S0179;
Beyotime Institute of Biotechnology).
Co-immunoprecipitation (Co-IP)
Cells at 70-80% confluence were lysed in RIPA buffer
at 4°C for 20-30 min, and debris was removed by centrifugation
(10,000 × g, 10 min, 4°C). Supernatants containing 50-100 µg
protein were incubated with primary antibody overnight at 4°C,
followed by incubation with 30 µl Protein A/G agarose beads
for 4 h at 4°C with gentle agitation. Beads were washed 3-5 times
with PBS to remove no-specific binding, and bound proteins were
eluted by boiling in SDS-PAGE loading buffer at 95°C for 5-10 min.
The eluate was collected by magnetic separation for subsequent
analysis.
Transmission electron microscopy
(TEM)
Samples were fixed with 2.5% glutaraldehyde in 0.1 M
phosphate buffer (pH 7.4) for 2 h at 4°C, followed by post-fixation
with 1% osmium tetroxide for 1 h at 4°C, after dehydration, finally
prepared as serial sections (80 nm) by an ultramicrotome and
stained with uranyl acetate for 15 min at room temperature and lead
citrate for 10 min at room temperature. After being rinsed and
vacuum-dried at room temperature, the ultrastructure of the
samples. Semi-quantitative analysis, using ImageJ software (version
1.53t; National Institutes of Health) to measure the size.
Oxygen consumption rate (OCR) assay
To measure the OCR, a 24-well XFe plate (Agilent
Technologies, Inc.) was utilized. Cells were seeded at a density of
1×104 per well and left to settle naturally for 1 h.
Next, the cell plates were incubated overnight. Once the cell
confluence reached 70-80%, the H/R model of cell culture was
created. Thereafter, 0.5 µM rotenone, 1 µM FCCP and 2
µM oligomycin were added to each well according to the
provided instructions. Subsequently, OCR was determined using a
Seahorse XF analyzer.
IDH2 enzyme activity and α-KG content
detection
An IDH activity assay kit (cat. no. S0526S; Beyotime
Institute of Biotechnology) was used for enzyme activity detection.
In accordance with the manufacturer's guidelines, fresh samples
were diluted with buffer. These diluted samples were then
incorporated into the NAD+ reaction system, and the
absorbance change at 450 nm was gauged using a microplate reader. A
test kit (cat. no. ADS-F-S001; Jiangsu ADS Biotechnology Co., Ltd.)
was used to detect α-KG, and the corresponding reagent and sample
were added to the fresh sample supernatant after centrifugation
(3,000 × g, 10 min, 4°C). The mixture was vortexed evenly and then
incubated at 37°C in the dark for 10 min. The absorbance (OD) of
each well at 450 nm wavelength was measured.
Omics sequencing and analysis
Proteomics
Heart tissue proteins were extracted, digested with
trypsin, and lactylated peptides were enriched using anti-pan-Kla
antibody beads. Enriched peptides were analyzed by liquid
chromatography-mass spectrometry (LC-MS/MS) on a timsTOF Pro mass
spectrometer (Bruker Daltonics; Bruker Corporation) operating in
positive ionization mode with a capillary voltage of 1.60 kV. The
instrument was operated in PASEF (parallel accumulation-serial
fragmentation) mode. Precursors with charge states 0 to 5 were
selected for fragmentation, and 10 PASEF-MS/MS scans were acquired
per cycle with a dynamic exclusion of 30 s. Precursors and
fragments were analyzed at the TOF detector over a mass range of
100-1,700 m/z. The nitrogen gas temperature was set to 180°C, the
nebulizer pressure to 1.4 bar, and the dry gas flow rate to 3.0
l/min. Raw data were searched against the UniProt mouse database
using MaxQuant with FDR <1%. Bioinformatics analyzes included
functional annotation [Gene Ontology, Kyoto Encyclopedia of Genes
and Genomes (KEGG), subcellular localization], differential
expression screening (fold change >1.5 or <0.67, P<0.05),
enrichment analysis by Fisher's exact test, and protein-protein
interaction network construction using STRING database (https://string-db.org/).
Metabolomics
Heart tissue metabolites were extracted and analyzed
by UPLC-Q Exactive HF-X MS. Raw data were processed using Compound
Discoverer 3.1 with metabolite identification against mzCloud and
KEGG databases (https://www.kegg.jp/kegg/pathway.html). Metabolites
with RSD >0.5 in QC samples were excluded. Missing values were
imputed by KNN, followed by sum normalization and Pareto scaling.
OPLS-DA was performed to identify differential metabolites (VIP
>1, fold change >1.5 or <0.67, P<0.05). Pathway
enrichment was conducted using KEGG with Fisher's exact test
(P<0.05).
Molecular docking
PDB structure files of IDH2 were downloaded from the
UniProt database (https://www.uniprot.org/), and SDF structure files of
isocitrate were obtained from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/). GROMACS
software (version 2020.4; GROMACS development team; https://www.gromacs.org) was used to simulate the
molecular dynamics of the modified IDH2 protein structure. PyMOL
2.1 software (version 2.1; Schrödinger, LLC; https://pymol.org) was used to visualize the spatial
conformation of the amino acid side chain of the simulated IDH2
K275la (Tables SIII and
SIV).
Quantification and statistical
analyses
All quantitative data were obtained from independent
experiments with triplicate repeats, and expressed as the mean ±
SD. Two-tailed unpaired Student's t-test between two groups and
one-way ANOVA followed by Bonferroni test for multiple comparison
were performed for statistical analysis using SPSS 15.0 software
(SPSS, Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
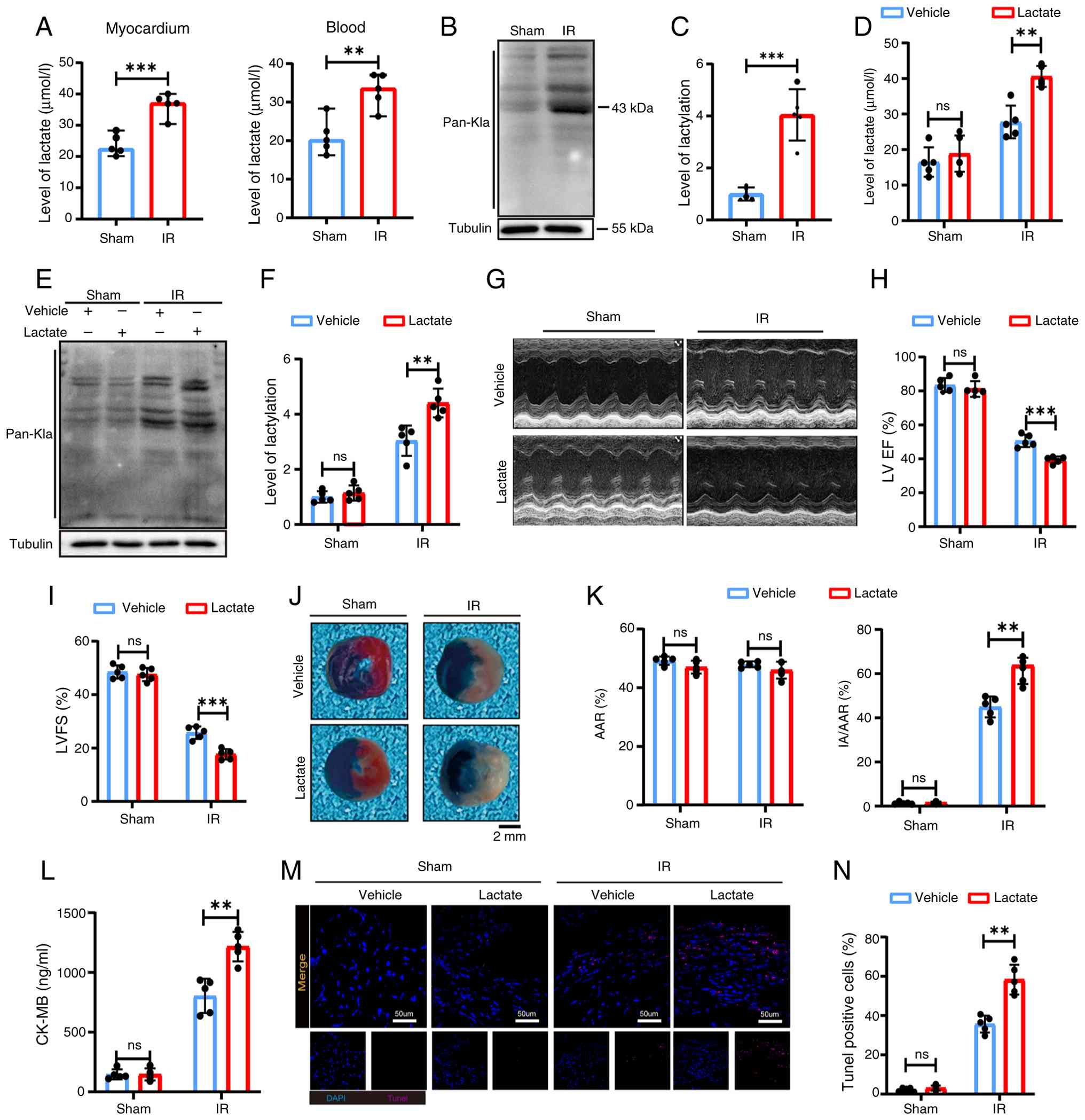
Elevated lactate and lactylation levels
are associated with MIRI
To investigate the changes in lactate and
lactylation during MIRI, a mouse model of MIRI was developed.
Lactate levels were significantly elevated in both peripheral blood
and myocardial tissue (Fig. 1A),
with myocardium experiencing a peak at 6 h post-I/R, followed by a
gradual decline (Fig. S1A).
Quantification using pan-antibody staining confirmed a significant
increase in lactate levels in the myocardial tissue (Fig. 1B and C).
Subsequently, exogenous lactate was administered to
assess its effects on MIRI. The introduction of exogenous lactate
increased myocardial lactate accumulation compared with the I/R
group (Fig. 1D), along with a
significant upregulation of lactylation (Fig. 1E and F). The I/R + lactate group
exhibited a significant decline in cardiac function and increased
injury compared with the I/R + vehicle group. These results were
demonstrated by decreases in LVFS% and LVEF% (Fig. 1G-I) and increased myocardial
infarct size (Fig. 1J and
K).
Furthermore, exogenous lactate administration led to
higher CK-MB levels (Fig. 1L)
and increased cardiomyocyte apoptosis, as indicated by TUNEL
staining (Fig. 1M and N)
compared with the I/R group. These findings suggest that
lactylation levels are elevated in MIRI, which may contribute to
enhanced myocardial injury and adverse outcomes.
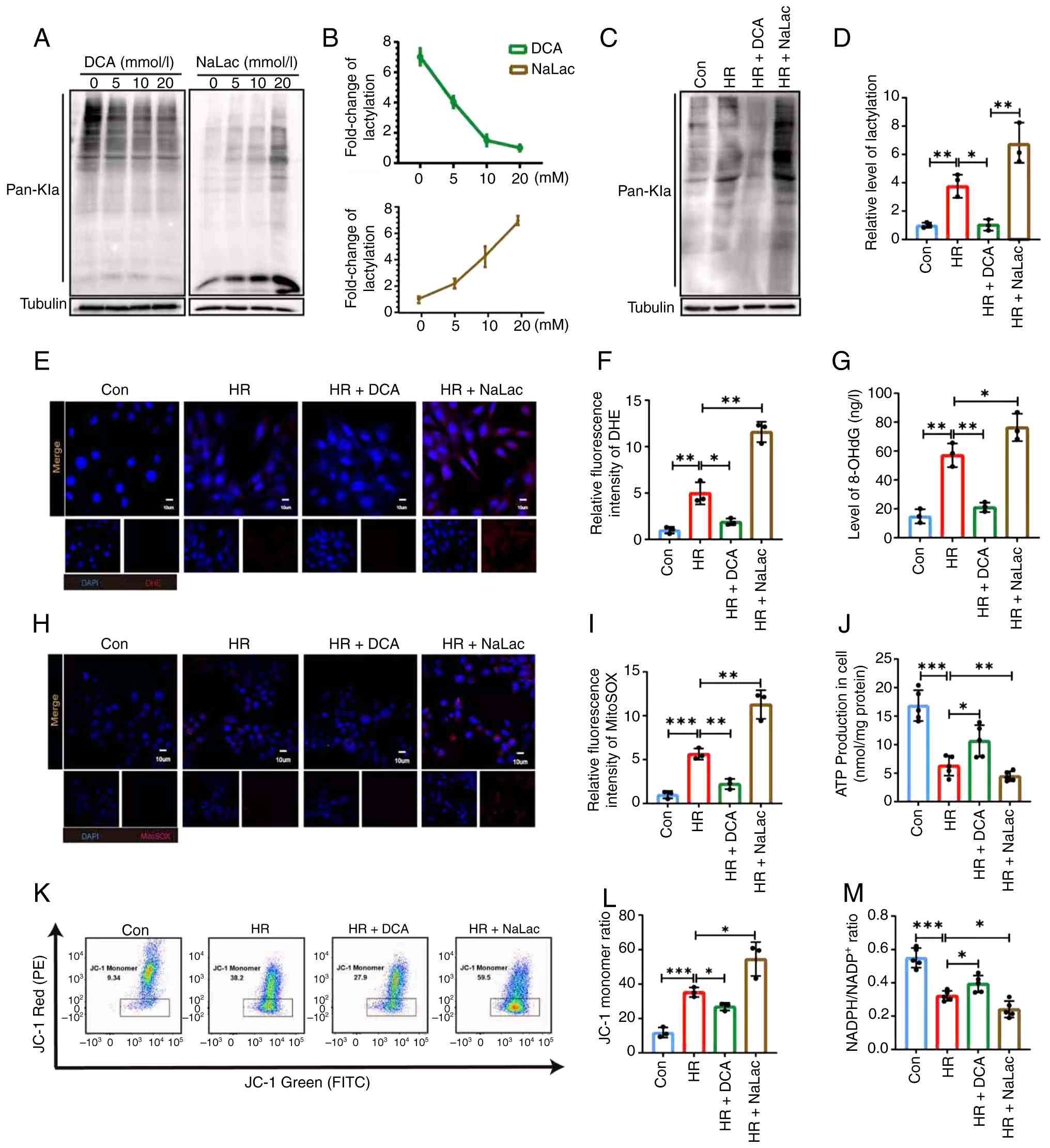
Myocardial mitochondrial dysfunction is
affected by lactylation in vitro
To further investigate the effect of lactylation on
MIRI, HL-1 cells and primary cardiomyocytes were pretreated with
NaLac or DCA, a lactate dehydrogenase inhibitor, as aforementioned.
A dose-dependent reduction in cell lactylation was observed
following treatment with varying concentrations of DCA. Conversely,
cells treated with NaLac exhibited a gradient increase in
lactylation (Fig. 2A and B).
Pan-modified antibody detection indicated that lactylation levels
were higher in the H/R group than in the Control (Con) group.
However, DCA administration decreased lactylation levels compared
with the HR group, and NaLac treatment further increased them
(Fig. 2C and D).
Immunofluorescence staining revealed similar trends in and Pan-Kla
levels (Fig. S1B and C).
Given the established link between cellular lactate
levels and mitochondrial dysfunction in disease, it was
investigated whether lactylation is involved in H/R-induced
mitochondrial dysfunction in cardiomyocytes. First, DHE and MitoSOX
staining demonstrated that inhibiting lactate production decreased
H/R-induced mitochondrial ROS production in cardiomyocytes, whereas
the addition of exogenous NaLac further promoted intracellular ROS
production (Fig. 2E, F, H and
I). Similarly, H/R increased the mitochondrial damage marker
8-OHdG (Fig. 2G), reduced ATP
production (Fig. 2J), decreased
mitochondrial membrane potential (Fig. 2K-M), and lowered NADPH levels.
These manifestations of mitochondrial dysfunction were further
aggravated by NaLac treatment compared with the HR group. However,
the addition of DCA ameliorated the H/R-induced mitochondrial
dysfunction phenotypes. Collectively, these findings indicate that
myocardial mitochondrial dysfunction is affected by lactylation
in vitro.
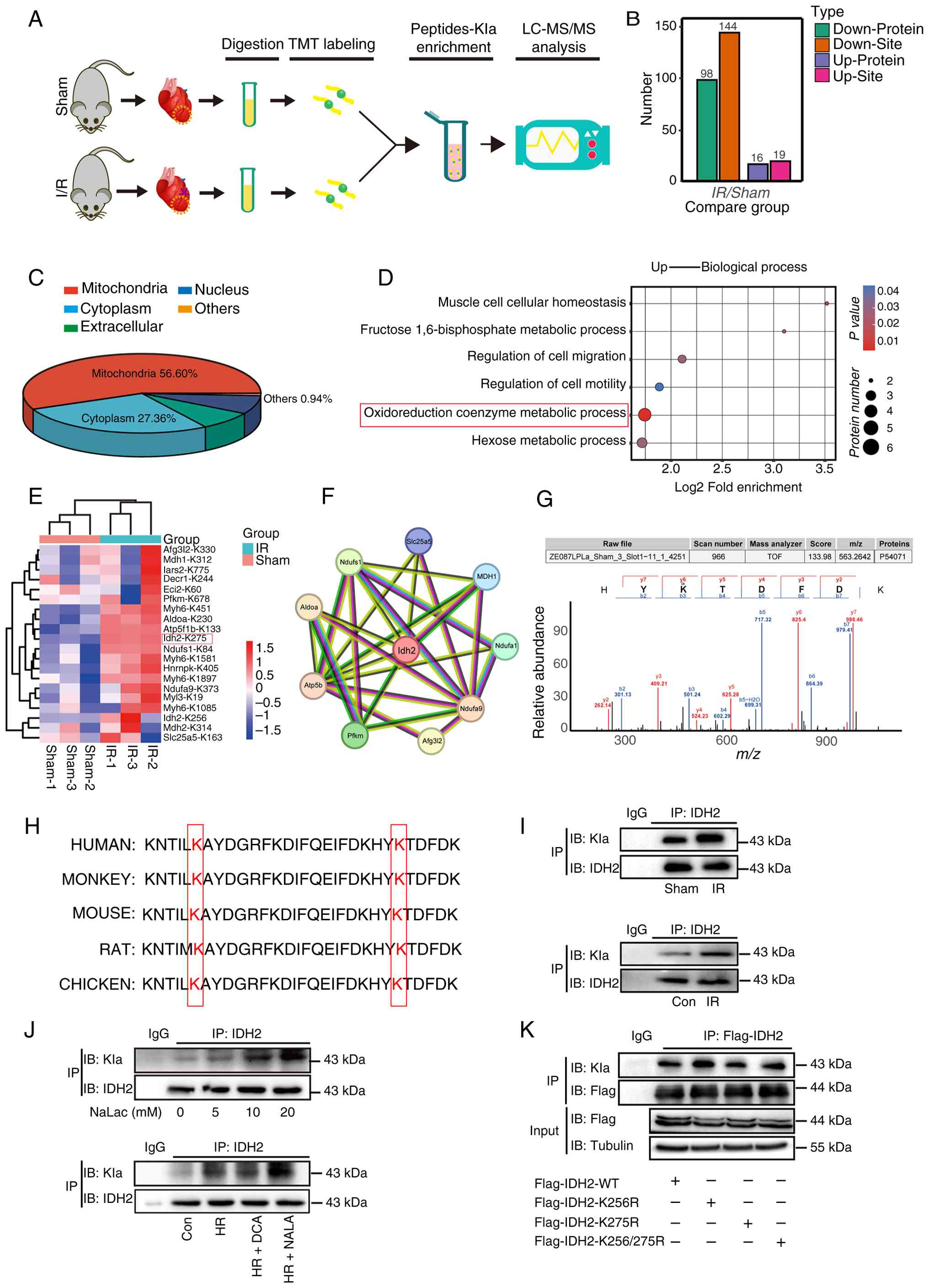
IDH2 K275 lactylation levels are elevated
in I/R myocardium
The lactylome proteomics analysis was performed to
identify specific lactylation targets in myocardial I/R (Fig. 3A). LC-MS analysis revealed that
98 proteins exhibited decreased lactylation and 16 exhibited
increased lactylation in the I/R group compared with the
sham-operated group. Furthermore, 144 sites exhibited reduced
lactylation and 19 exhibited increased lactylation (Fig. 3B). Functional analysis of the
lactylated proteins revealed predominant mitochondrial localization
(Fig. 3C). An enrichment
analysis of the genes associated with increased levels of
lactylation revealed entries related to the oxidoreduction coenzyme
metabolic process and upregulation of the redox homeostasis
biological process (Fig.
3D).
Based on the top 20 altered sites (Fig. 3E), a protein-protein interaction
network constructed using the STRING database identified IDH2 as a
key hub protein (Fig. 3F).
Collision-induced dissociation analysis demonstrated a
characteristic tandem MS spectrum at the lysine (K) site of IDH2
(Fig. 3G). Genetic conservation
analysis of the IDH2 lysine sites across multiple species using the
UniProt website revealed that the IDH2 K275 site was conserved
across species (Fig. 3H). IDH2
is an important rate-limiting enzyme in the tricarboxylic acid
(TCA) cycle. It catalyzes the oxidative decarboxylation of
isocitrate to provide energy and support metabolic processes. The
results of non-targeted metabolomics also indicated a decrease in
the concentration of its catalytic product, α-KG (Fig. S2A). Animal and cellular I/R
models exhibited significantly elevated IDH2 lactylation in Co-IP
assays (Figs. 3I and S2B). This lactylation was
pharmacologically modulated: DCA decreased it, and NaLac increased
it in a concentration-dependent manner, without affecting total
IDH2 expression (Figs. 3J and
S2C). Lactylome analysis
indicated elevated lactylation at K256 and K275. Therefore, the
deacylating mutant plasmids K256R and K275R were used to identify
the major lactylation sites in IDH2. Co-IP assay revealed a more
significant reduction in lactylation in K275R than in K256R
(Figs. 3K and S2D), suggesting that K275 plays a
predominant role in IDH2 lactylation. Therefore, it is proposed
that the primary factor contributing to the impairment of protein
function and enzyme activity in MIRI is the lactylation of IDH2,
rather than changes in its protein expression. This, in turn,
affects a series of downstream mitochondrial functions. These
findings indicate that IDH2 K275 may be a key regulatory site for
MIRI.
K275 mutant effectively attenuates
mitochondrial dysfunction in H/R cardiomyocytes
HL-1 cell lines with stable IDH2 knockout were
established using lentivirus to investigate whether IDH2 K275la
affects mitochondrial homeostasis (Fig. S3A and B) and subsequently
transfected with an IDH2 K275 mutant plasmid for rescue
experiments. Co-IP results indicated that the K275R mutant group in
the H/R model exhibited no IDH2 lactylation compared with the
wild-type (WT) group (Fig. 4A and
B). In the HR group, ATP content and the NADPH/NADP+
ratio decreased, whereas 8-OHdG levels increased. However, the
markers associated with mitochondrial function were significantly
improved in the K275 mutant group (Fig. 4C-E). The OCR assay results
revealed impaired maximum mitochondrial levels and basal
respiratory rates in the HR-WT group compared with the Con group.
However, these findings were reversed in the K275 mutation
(Fig. 4F and G). DHE and MitoSOX
fluorescence staining indicated that mitochondrial ROS levels were
significantly higher in the HR + WT group than in the Con group.
Conversely, the K275 mutant decreased mitochondrial ROS production
(Fig. 4H-K). The mutant group
consistently stabilized the H/R-induced loss of mitochondrial
membrane potential, as indicated by an increased JC-1 monomer ratio
(Fig. 4L and M). Western blots
confirmed the reversals in the effects on mitochondrial dynamin
DRP1, MnSOD2, and mitochondrial apoptosis Cytochrome C (Cyt C) in
the HR-WT treated group with a K275 mutant plasmid (Fig. 4N and O). These findings indicate
that lactylation at the K275 site of IDH2 is essential in
regulating H/R-induced mitochondrial dysfunction in
cardiomyocytes.
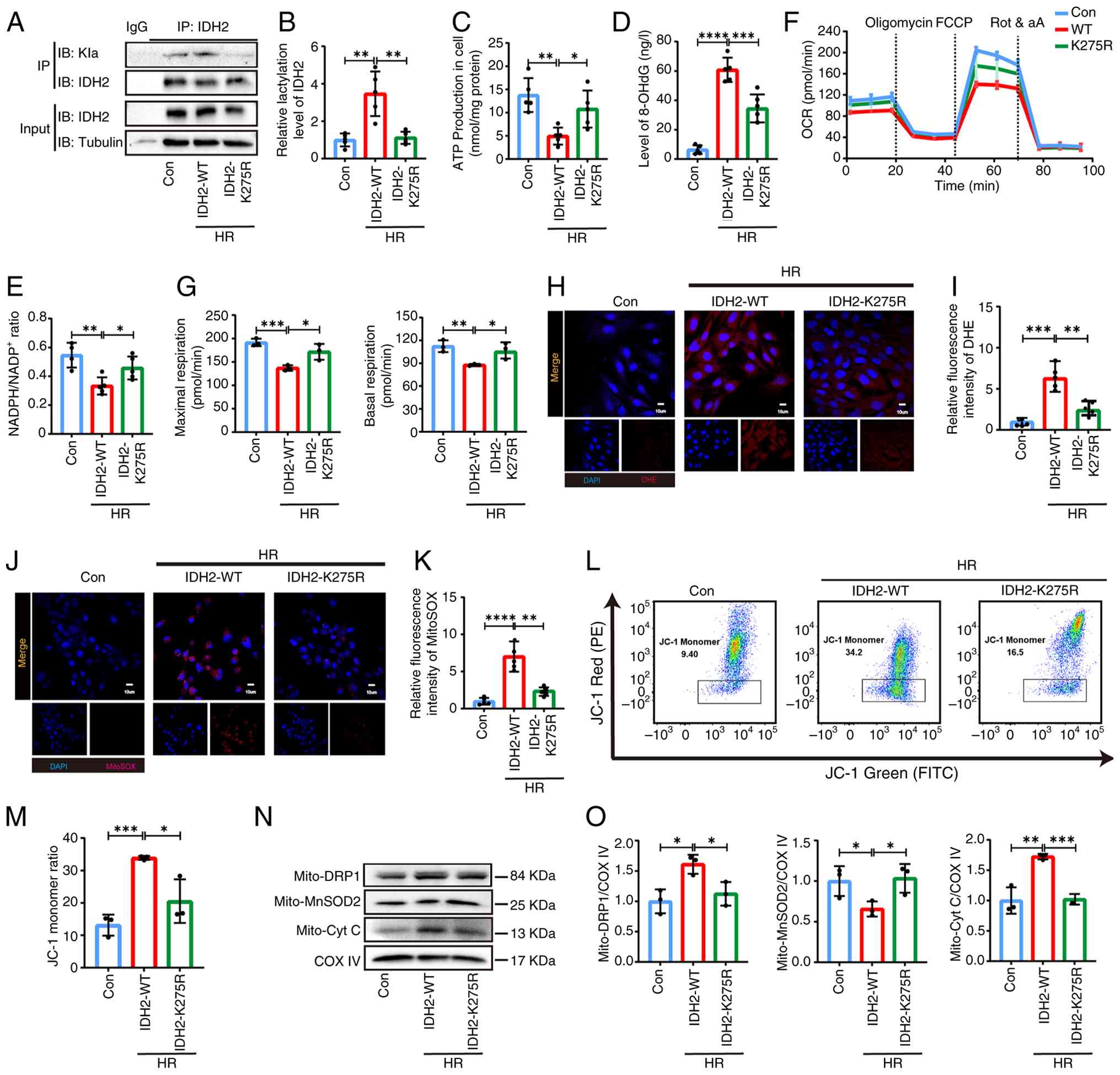 | Figure 4K275 mutant effectively attenuates
mitochondrial dysfunction in H/R cardiomyocytes. (A and B) IP
experiment for the lactylation level of IDH2 in IDH2-WT and IDH2
K275R plasmids and quantitative analysis (n=5). (C) Detection of
intracellular ATP production after plasmid transfection (n=5). (D)
8-OHdG levels in cardiomyocytes (n=5). (E) Intracellular
NADPH/NADP+ levels (n=5). (F and G) Oxygen consumption
rate measured by mitochondrial stress test in HL-1 cells and
quantitative respiration function (n=3). (H and I) DHE staining
assay for reactive oxygen species level in cells and quantitative
analysis (n=5). (J and K) The mitochondrial oxidative stress level
of cardiomyocytes detected by MitoSOX staining (n=5). (L and M)
Mitochondrial membrane potential in HL-1 cardiomyocytes detected by
JC-1 staining flow cytometry and quantitative analysis (n=3). (N
and O) The protein levels of DRP1, MnSOD2 and Cyt C protein
expression detected by western blot (n=3). These data were
representative results of three repetitions at least. Data are
expressed as the mean ± SD *P<0.05,
**P<0.01 and ***P<0.001. H/R,
hypoxia/reoxygenation; IDH2, isocitrate dehydrogenase 2; WT,
wild-type; IP, immunoprecipitation; 8-OHdG,
8-hydroxy-2′-deoxyguanosine; DRP1, dynamin-related protein 1;
MnSOD2, manganese superoxide dismutase 2; Cyt C, cytochrome C. |
IDH2 K275la reduces IDH2 activity and
exacerbates mitochondrial dysfunction in cardiomyocytes by
inhibiting the phosphorylation of AMPK
It was previously reported that IDH2 product α-KG
activates AMPK via upstream kinases (20). It was hypothesized that
IDH2-α-KG-AMPK axis regulates mitochondrial function (Fig. 5A). IDH2 catalytic activity was
examined at varying NaLac concentrations. IDH2 enzymatic activity
was inversely correlated with NaLac concentration (Fig. 5B). Correspondingly, α-KG levels
also decreased with increases in NaLac concentration. This downward
trend was consistent with that of IDH2 (Fig. 5C). Subsequently, it was observed
that IDH2 catalytic activity and α-KG content decreased in the
HR-WT group compared with the Con group and reversed by IDH2 K275R
(Fig. 5D and E).
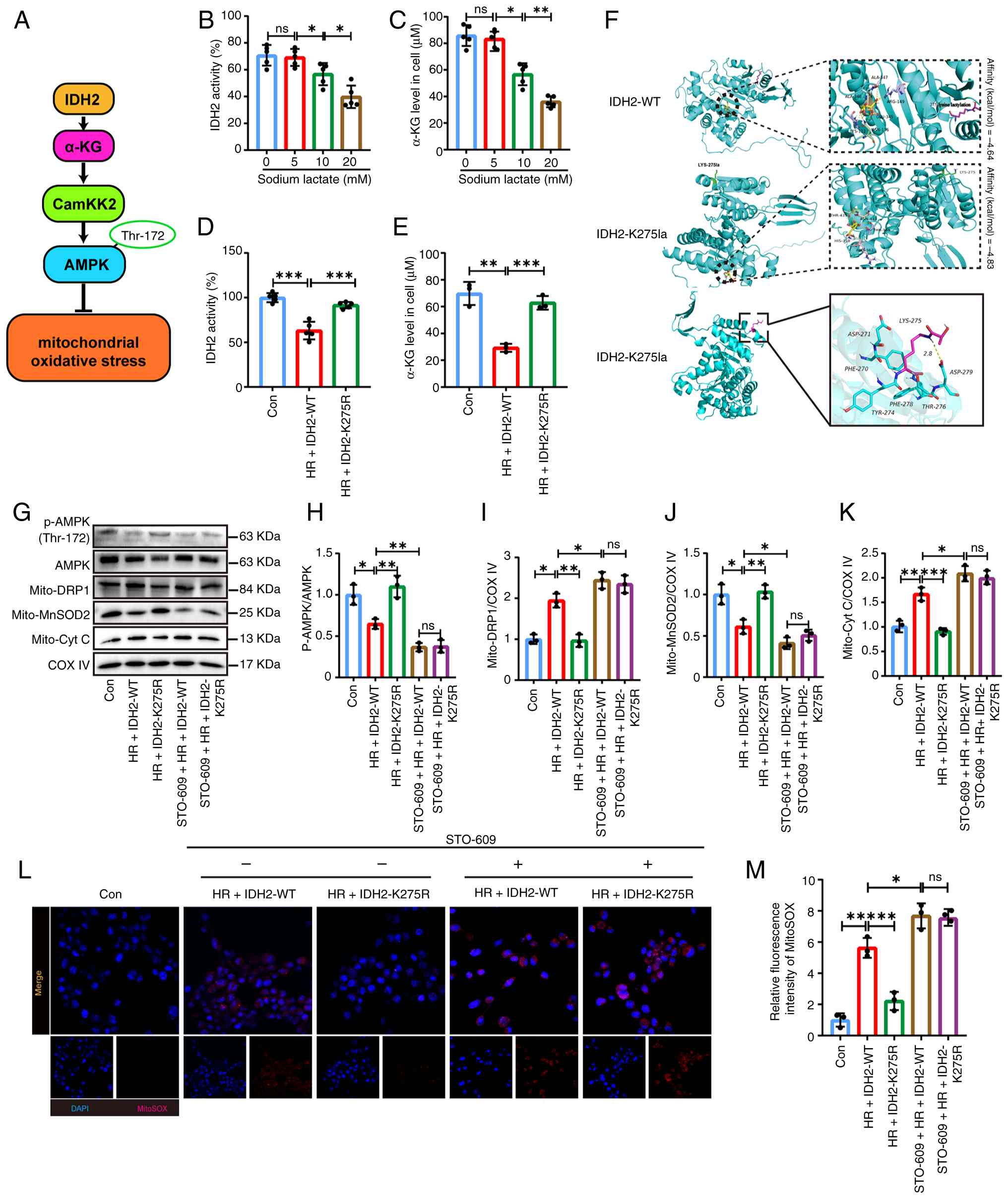 | Figure 5IDH2 K275la reduces IDH2 activity and
exacerbates mitochondrial dysfunction in cardiomyocytes by
inhibiting the phosphorylation of AMPK. (A) The mechanism diagram
of the conjecture. (B) Detection of IDH2 activity under 0, 5, 10,
20 mM sodium lactate gradient (n=5). (C) Detection of α-KG
production under sodium lactate gradient (n=5). (D) Detection of
IDH2 activity by comparing between WT and K275R mutant (n=5). (E)
α-KG production between WT and K275R mutant detected by α-KG assay
kit (n=3). (F) Molecular dynamics simulation of IDH2 structure
before and after lactylation using GROMACS software, and
visualization of the lactylated IDH2 tertiary structure using PyMOL
2.1. (G-K) Protein levels of p-AMPK, AMPK, DRP1, MnSOD2 and Cyt C
detected by western blotting after STO-609 treatment, and their
semi-quantitative analyzes (n=3). (L and M) Representative images
of MitoSOX staining for STO-609 treatment group (n=3). These data
were representative results of three repetitions at least. Data are
expressed as the mean ± SD. *P<0.05,
**P<0.01 and ***P<0.001. IDH2,
isocitrate dehydrogenase 2; AMPK, adenosine
5′-monophosphate-activated protein kinase; α-KG, α-ketoglutaric
acid; WT, wild-type; p-, phosphorylated; DRP1, dynamin-related
protein 1; MnSOD2, manganese superoxide dismutase 2; Cyt C,
cytochrome C; H/R, hypoxia/reoxygenation; ns, not significant. |
Molecular dynamics simulations of the modified IDH2
protein structure were performed using GROMACS to clarify the
specific molecular structural changes upon lactylated IDH2 binding
to isocitric acid. The average docking binding energy scores of
IDH2 with its substrate increased after lactylation. The binding
affinity of the modified IDH2 and isocitric acid was reduced
according to the principle of the lowest binding energy and the
highest binding affinity. Furthermore, visualization of the IDH2
tertiary structure following lactylation revealed that IDH2 K275la
may affect local stability. This was accomplished by impeding the
formation of the salt bridge between Lys275 and ASP279 in the
protein structure. Consequently, the spatial conformation of the
amino acid side chain was readily altered, preventing IDH2 from
binding to its substrate and thereby reducing its catalytic
activity (Fig. 5F).
The AMPK phosphorylation inhibitor, STO-609, was
used to determine whether this pathway mediates K275la effects.
Western blot analysis demonstrated that the K275 mutation increased
p-AMPK levels, along with beneficial changes in MnSOD2, DRP1 and
Cyt C. As expected, STO-609 treatment abolished the increased AMPK
phosphorylation and the mitochondrial protective effects of the
K275 mutation (Fig. 5G-K).
However, mitochondrial ROS production was significantly increased
after STO-609 treatment compared with the HR + IDH2 + K275R group
(Fig. 5L and M).
Furthermore, cells were exposed to α-KG to validate
its mediation of mitochondrial dysfunction. Western blot analysis
revealed that α-KG treatment in the H/R model restored AMPK
phosphorylation levels compared with the HR group. Treatment of
myocytes with α-KG reduced the expression of the mitochondrial
kinetic protein DRP1 and increased the levels of the antioxidant
protein MnSOD2 (Fig. S3C and
D). These results indicate that IDH2 K275la downregulates IDH2
catalytic activity and exacerbates mitochondrial dysfunction in
cardiomyocytes by inhibiting the phosphorylation-activation of the
α-KG/AMPK pathway.
Inhibition of IDH2 K275la alleviates MIRI
and partially eliminates the aggravating effect of lactate on
cardiac tissue
To validate the in vivo role of IDH2 K275,
mice were administered AAV9 carrying IDH2-WT or IDH2-K275R via the
tail vein 14 days before I/R surgery (Fig. 6A). A green fluorescent protein
(GFP) tag demonstrated successful infection with GFP-tagged viruses
(Fig. 6B), and Co-IP experiments
confirmed that the virus reduced IDH2 lactylation in the myocardium
compared with the WT group (Fig.
6C). Subsequently, Evans blue-TTC staining revealed a smaller
myocardial infarct area in K275R mice (Fig. 6D and E). TEM revealed a large
number of irregularly accumulated heart mitochondria, swollen
mitochondria, and destroyed mitochondrial ridge structures, while
mitochondrial swelling improved, the structure recovered, and the
arrangement became regular in the mutant group (Fig. 6F). The K275 mutant group
demonstrated improved cardiac function (LVEF% and LVFS%), increased
NADPH/NADP+ ratio, and decreased markers of myocardial
damage and ROS production compared with the I/R group. Notably, the
exacerbation of these parameters by exogenous NaLac was partially
reversed in the K275R mice (Fig.
6G-M). These results suggest that inhibiting IDH2-K275la
alleviated MIRI and partially eliminated the disease-aggravating
effect of lactate on cardiac tissue.
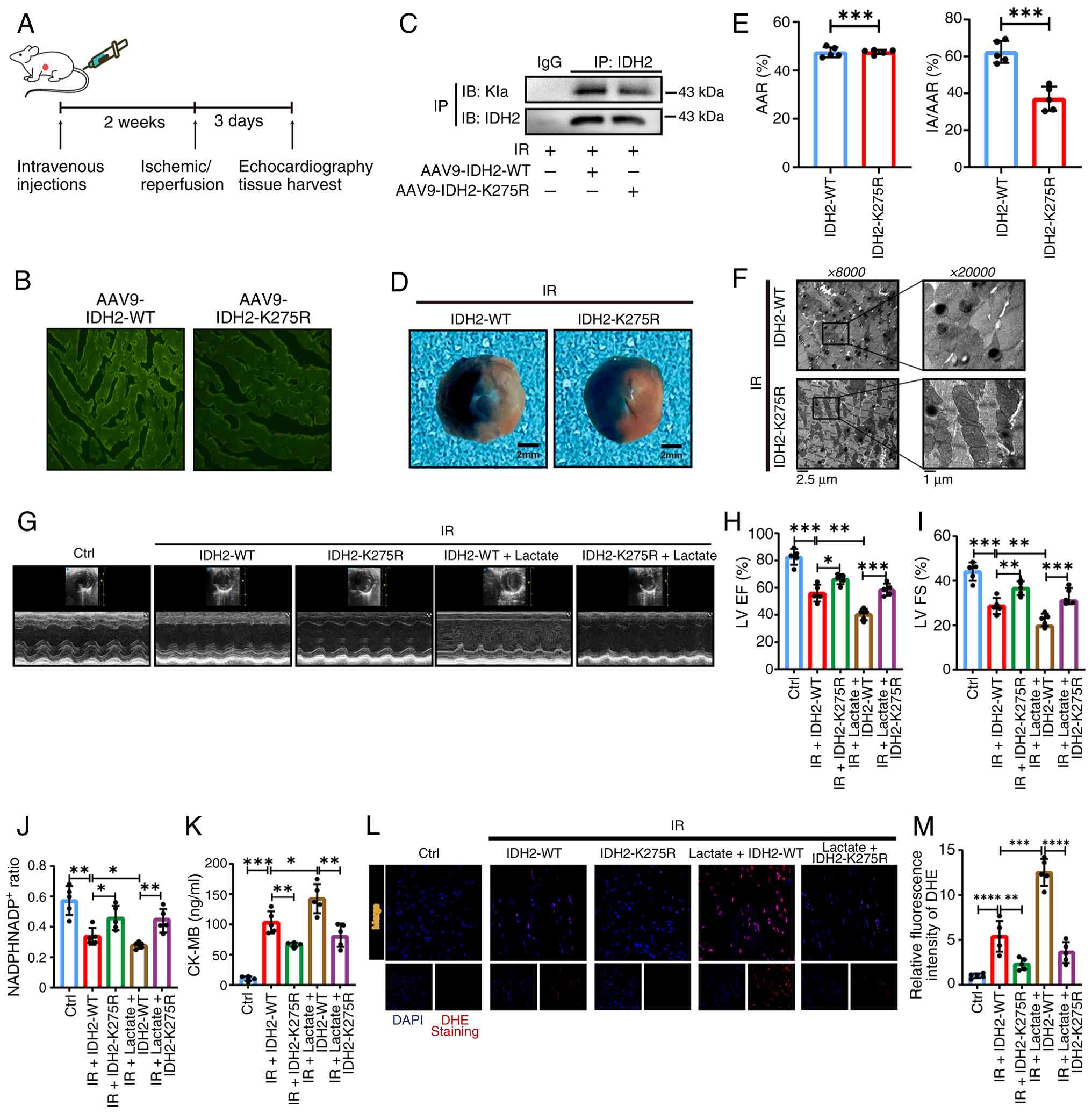 | Figure 6Inhibition of IDH2 K275la alleviates
myocardial ischemia-reperfusion injury and partially eliminates the
aggravating effect of lactate on cardiac injury. (A) Schematic
diagram of virus injection method and experimental scheme. (B) The
efficiency of virus infection in mouse heart observed by
fluorescence microscopy (n=5). (C) IP assay for detecting the IDH2
lactylation after cardiac virus infection (n=5). (D and E) Evans
blue-TTC staining showing ischemic area of myocardial infarction
(scale, 2 mm; n=5). (F) Electron microscopy of mitochondria in
mouse left ventricular myocardium (scale, 1 µm; n=5). (G-I)
Echocardiography to evaluate cardiac function and quantitative
analysis of LVEF% and LVFS% (n=5). (J) NADPH/NADP+ ratio
in mouse myocardial tissue (n=5). (K) CK-MB content in myocardial
tissue of mice (n=5). (L and M) The reactive oxygen species
production level detected by DHE staining (n=5). These data were
representative results of three repetitions at least. Data are
expressed as the mean ± SD. *P<0.05,
**P<0.01 and ***P<0.001. IDH2,
isocitrate dehydrogenase 2; IP, immunoprecipitation; LVEF, left
ventricular ejection fraction; LVFS, left ventricular fractional
shortening; CK, creatine kinase; WT, wild-type; I/R,
ischemia/reperfusion; IA, myocardial infarction; AAR, area at
risk. |
SIRT3 regulates myocardial oxidative
injury by delactylating IDH2 in MIRI
Lactylation is a newly identified form of acylation
modification that may share comparable regulatory enzymes with
other lysine acylation modifications. It has been recently
suggested that Sirtuins (SIRTs) may regulate lactylation (21). RCSB and UniProt databases were
used to analyze the molecular docking of several known
acylation-modifying SIRT enzymes with IDH2 and to explore the
regulatory enzymes that affect IDH2 lactylation. SIRT3, 4, and 6
exhibited strong binding affinity for IDH2 (Fig. 7A). Co-IP identified SIRT3 as the
most potent inhibitor of IDH2 lactylation, whose expression was
decreased in the I/R model (Fig. 7B
and C). Previous studies have demonstrated that SIRT3 levels
are reduced in I/R injury, and that SIRT3 overexpression promotes
recovery of cardiac function and reduces injury markers in
reperfusion injury (22). 3-TYP,
a specific SIRT3 inhibitor, was used to investigate whether SIRT3
regulates IDH2 lactylation in vivo. The findings of the
present study revealed that 3-TYP did not affect the SIRT3
expression. Rather, it inhibited the delactylation function of
SIRT3 and increased IDH2 lactylation. Conversely, the effect of
3-TYP was nullified in mice treated with the K275 mutant (Fig. 7D and E). 3-TYP treatment
downregulated α-KG content compared with the WT group, an effect
reversed in the K275 mutation group (Fig. 7F). 3-TYP administration further
reduced AMPK phosphorylation, increased the expression of
mitochondrial function-related proteins DRP1 and Cyt C, decreased
MnSOD2, downregulated the proportion of reduced coenzyme, elevated
ROS production, and increased CK-MB compared with the I/R + WT
group. Conversely, the IDH2 mutant reversed the previously
described inhibition of AMPK phosphorylation and mitochondrial
dysfunction and increased the 3-TYP-induced damage markers
(Fig. 7G-K). These findings
indicate that SIRT3 is the upstream regulatory enzyme of IDH2
acylation and that IDH2 K275 is a critical regulatory site for
SIRT3-mediated myocardial injury in MIRI.
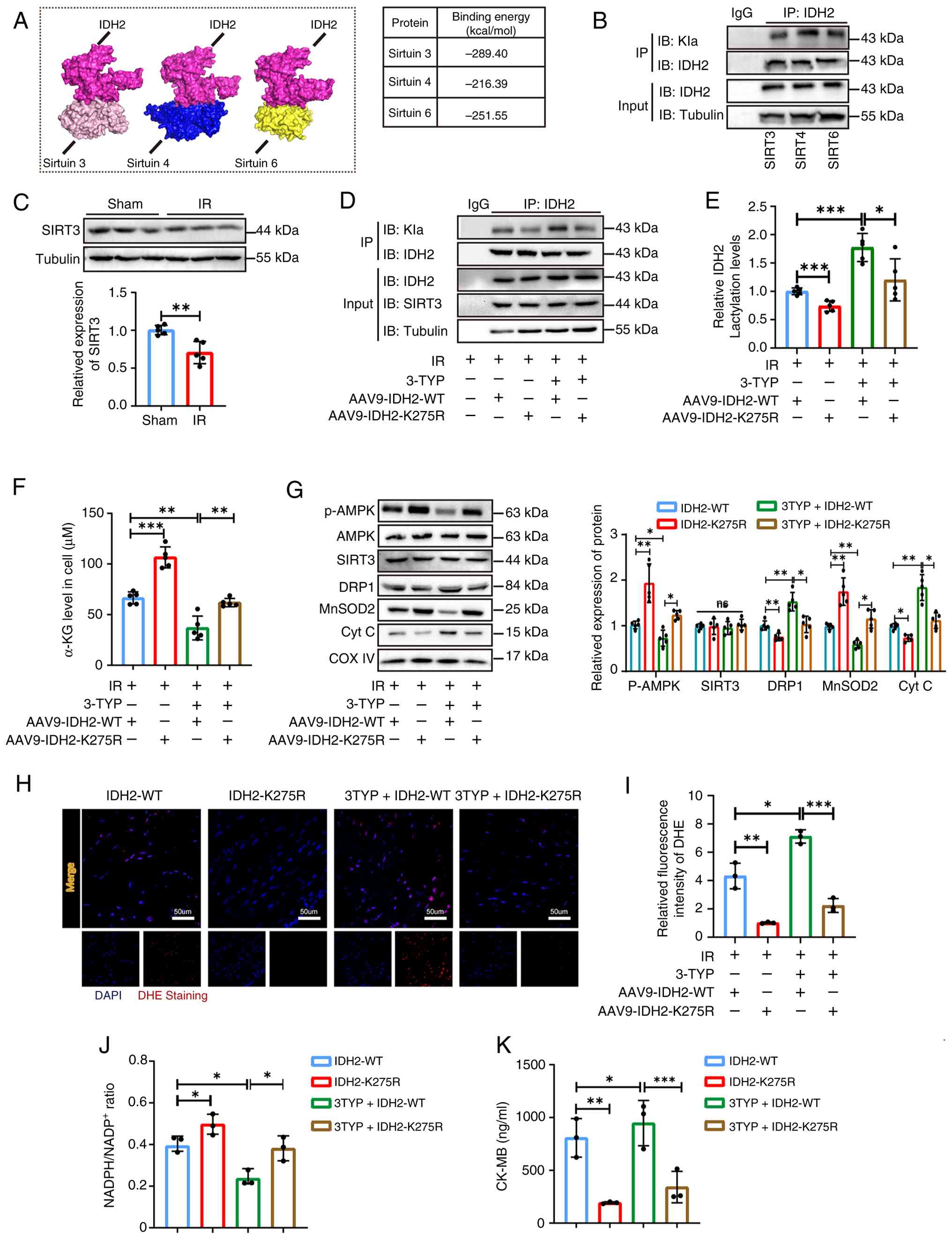 | Figure 7SIRT3 regulates myocardial oxidative
injury by delactylating IDH2 in MIRI. (A) IDH2 docked with SIRT3,
SIRT4, and SIRT6 molecules, respectively. (B) The level of IDH2
lactylation detected by IP assay after overexpressing SIRT3, SIRT4,
SIRT6 (n=5). (C) SIRT3 protein expression levels in MIRI detected
by western blot (n=5). (D and E) The level of IDH2 lactylation
detected by IP experiment and semi-quantitative analyses (n=5). (F)
α-KG production under 3-TYP treatment (n=5). (G) Protein levels of
p-AMPK, AMPK, DRP1, MnSOD2 and Cyt C assayed by western blotting
after 3-TYP treatment, and their semi-quantitative analyses (n=5).
(H and I) Representative images of DHE staining for 3-TYP treatment
group (n=5). (J and K) The NADPH/NADP+ ratio and CK-MB
levels in the 3-TYP treatment group (n=5). These data were
representative results of three repetitions at least. Data are
expressed as the mean ± SD. *P<0.05,
**P<0.01 and ***P<0.001. IDH2,
isocitrate dehydrogenase 2; MIRI, myocardial ischemia-reperfusion
injury; IP, immunoprecipitation; 3-TYP, 3-(1H-1,2,3-Triazol-4-yl)
pyridine; α-KG, α-ketoglutaric acid; p-, phosphorylated; AMPK,
adenosine 5′-monophosphate-activated protein kinase; DRP1,
dynamin-related protein 1; MnSOD2, manganese superoxide dismutase
2; Cyt C, cytochrome C; CK, creatine kinase; WT, wild-type; I/R,
ischemia/reperfusion. |
Discussion
The present study demonstrated that
hyper-lactylation, triggered by increased lactate levels,
contributes to the early aggravation of I/R injury in mice. The
findings of the present study reveal that IDH2 K275 lactylation
impairs its catalytic activity, reduces α-KG production, and alters
activation of the AMPK signaling pathway, thereby leading to
mitochondrial dysfunction. The present study is the first, to the
best of our knowledge, to demonstrate that IDH2 can be modified by
lactylation in addition to acetylation and identifies K275 as a
novel functional site in I/R hearts. Furthermore, by exploring the
relationship between lactylation and mitochondrial function, it was
identified, for the first time, that IDH2 lactylation can regulate
its activity, thereby affecting mitochondrial function. These
findings advance the understanding of the physiological role of
lactylation, provide a novel perspective on MIRI pathogenesis, and
suggest potential therapeutic strategies.
Lactate, previously considered a metabolic waste
product, is now recognized as essential for cellular functions. For
instance, lactate can be transported to cells, absorbed, used,
oxidized, and metabolized in the mitochondria to provide energy for
cells (23), indicating that
lactate can be used as a metabolic fuel in emergencies.
Furthermore, lactate can eliminate and suppress immune cells in the
immune microenvironment (24).
In 2019, lactate was found to exert its function through histone
lactylation, providing new insights into its role. Subsequent
research has demonstrated that lactylation is prevalent among
non-histone proteins across various cell types and plays
significant roles in energy metabolism, cell signaling,
transcriptional regulation and organ dysfunction (25,26). For instance, lactylation plays a
key regulatory role by activating the ubiquitin-proteasome system,
leading to the retention of erythrocyte mitochondria and
contributing to lupus (27).
Furthermore, in cardiovascular disease, the downregulation of α-MHC
lactylation may result in structural and functional impairments of
the heart and exacerbate heart failure (28). Regarding immune regulation, PKM2
lactylation suppresses the inflammatory metabolic adaptation of
pro-inflammatory macrophages (29). The present findings are
consistent with this expanding understanding. A
lactate-concentration-dependent increase in lactylation was
observed in the I/R model, and lactate supplementation exacerbated
myocardial dysfunction. DCA ameliorated H/R-induced mitochondrial
dysfunction in vitro, whereas NaLac exacerbated this effect.
However, the mechanism by which lactate contributes to the
occurrence and progression of I/R myocardial injury remains unclear
and warrants further study.
IDH2, a mitochondrial isoform of isocitrate
dehydrogenase, is a key enzyme in the TCA cycle. It contributes to
energy production and cellular redox balance by catalyzing the
oxidative decarboxylation of isocitrate to α-KG and producing NADPH
(30,32). IDH2 helps defend against
oxidative stress by maintaining cellular NADPH pools (31). Its dysfunction, associated with
elevated ROS and mitochondrial impairment, is implicated in cancer,
neurodegenerative diseases and aging (33-35). For instance, IDH2 deficiency in
mice exacerbates mitochondrial oxidative stress in the liver and
accelerates age-associated phenotypes (36). In cardiac pathology, IDH2-null
mice are more susceptible to pressure overload-induced hypertrophy,
leading to cardiomyocyte metabolic dysregulation and oxidative
stress (37). Notably, the
oncometabolite 2-HG is produced by gain-of-function mutations in
IDH2 (for instance, R172), which drive oncogenesis in AML and
gliomas (38). This has rendered
mutant IDH2 a promising therapeutic target, with inhibitors such as
enasidenib and vorasidenib developed for clinical use (39,40). Targeting protein sites with drugs
disrupts cellular metabolism and inhibits tumor cell proliferation.
This suggests that the precise targeting of key protein
modification sites is effective for disease treatment. In the
present study, IDH2 K275 was mutated in cells to reduce
lactylation. This mutation improved mitochondrial function in
cardiomyocytes. Furthermore, the present study demonstrated that
IDH2 mutation can alleviate I/R-induced heart injury in animal
experiments. These findings provide a practical approach for
treating MIRI and may represent a novel therapeutic target.
However, further investigation is required to determine the factors
regulating lactylation.
Protein lactylation is regulated by enzymes that
often govern other lysine acylation such as p300/CBP and HDACs. It
has been previously reported that SIRT1 and SIRT3 are 'erasers' of
histone and non-histone Kla, with distinct regulatory mechanisms
and substrate specificities (41). Previous studies have demonstrated
that SIRT3, an NAD+-dependent deacetylase, is vital for
regulating cellular metabolism, stress reaction and organismal
longevity (42,43). SIRT3 plays a key role in
maintaining mitochondrial homeostasis. It regulates the deacylation
of mitochondrial proteins and affects the activity of enzymes
involved in important metabolic pathways, including fatty acid
oxidation, the TCA cycle and OXPHOS (44,45). SIRT3 affects mitochondrial
dynamics and biogenesis, and controls redox homeostasis. These
processes are crucial for sustaining mitochondrial morphology,
function and distribution. Studies have demonstrated that SIRT3 can
regulate PGC-1-α, a protein implicated in neurodegenerative and
cardiovascular diseases (46,47), increasing mitochondrial
biogenesis and ensuring an adequate supply of functional
mitochondria. Furthermore, SIRT3 is involved in mitophagy by
regulating the expression and activity of PINK1/Parkin (48). SIRT3, a crucial cellular
deacylation enzyme, reverses various lysine acylations, including
acetylation, crotonylation, β-hydroxybutyrylation and
propionylation (49,50). In terms of mitochondrial
oxidative stress, SIRT3 can directly deacetylate and activate SOD2,
thereby enhancing mitochondrial ROS scavenging and protecting them
from oxidative damage. The present findings indicate that SIRT3
binds to IDH2 K275, acting as a regulator of IDH2 lactylation and
IDH2 activity in MIRI, thereby affecting mitochondrial function.
Significant downregulation of SIRT3 was found in MIRI, consistent
with other studies. IDH2 K275 mutation can reverse mitochondrial
dysfunction and reduce ROS production after SIRT3 inhibition. The
downregulation of SIRT3 may be attributed to oxygen and nutrient
deficiencies during the ischemic phase, leading to metabolic shifts
that favor lactylation. However, whether SIRT3 knockdown initiates
the enhanced lactylation of IDH2 during IR remains unclear. Whether
corresponding molecular drugs that act on IDH2-K275 and effectively
inhibit its lactylation are available remains a key focus of our
subsequent research and clinical translation. It has been recently
demonstrated that SIRT3 can be activated by salvianolic acid B,
which directly deacetylates and activates superoxide dismutase and
inhibits NLRP3 inflammasome activation by regulating mitochondrial
ROS levels, thereby alleviating the inflammatory response
accompanied by oxidative stress, which may affect IDH2 lactylation
(51). Although salvianolic acid
B has been reported to regulate histone lactylation, it is unknown
whether it can regulate non-histone proteins. Follow-up studies are
being conducted, and relevant experiments are being designed to
validate our conjecture regarding salvianolic acid B and expect new
findings.
The present study has several limitations. First,
the regulation of SIRT3-mediated IDH2 lactylation was verified
in vivo but not validated in cellular studies. Second, the
precise mechanisms driving lactate elevation in MIRI have not been
explored. Third, the present study did not explore the
overexpression of SIRT3 in cellular or mouse models to determine
whether it can prevent I/R-induced increase in IDH2 lactylation.
Future studies should consider introducing overexpression vectors
for verification. Moreover, the downstream consequences of reduced
α-KG levels beyond AMPK inhibition require further investigation.
Furthermore, the current lactylome data revealed other
significantly modified proteins (for instance, ALDOA and NDUFS1)
involved in metabolism and mitochondrial electron transport, whose
roles in MIRI merit further study. From a therapeutic perspective,
modern drug discovery increasingly focuses on small molecules that
directly modulate specific PTMs (such as phosphorylation,
acetylation and ubiquitination). Therefore, identifying a
small-molecule compound that selectively targets the IDH2 K275 site
to inhibit lactylation represents a promising strategy for clinical
translation.
As illustrated in the mechanistic diagram (Fig. 8), sufficient evidence is provided
that lactate elevations inhibit mitochondrial IDH2 function by
modulating lactate levels in IDH2 K275 after myocardial I/R, which
affects the production of the downstream metabolite α-KG, thereby
affecting the activation of the AMPK pathway. The downregulation
levels of SIRT3 in MIRI limit IDH2 K275 delactylation and further
aggravated mitochondrial dysfunction in cardiomyocytes. Thus,
future research should investigate diagnosis and treatment methods
using this novel target.
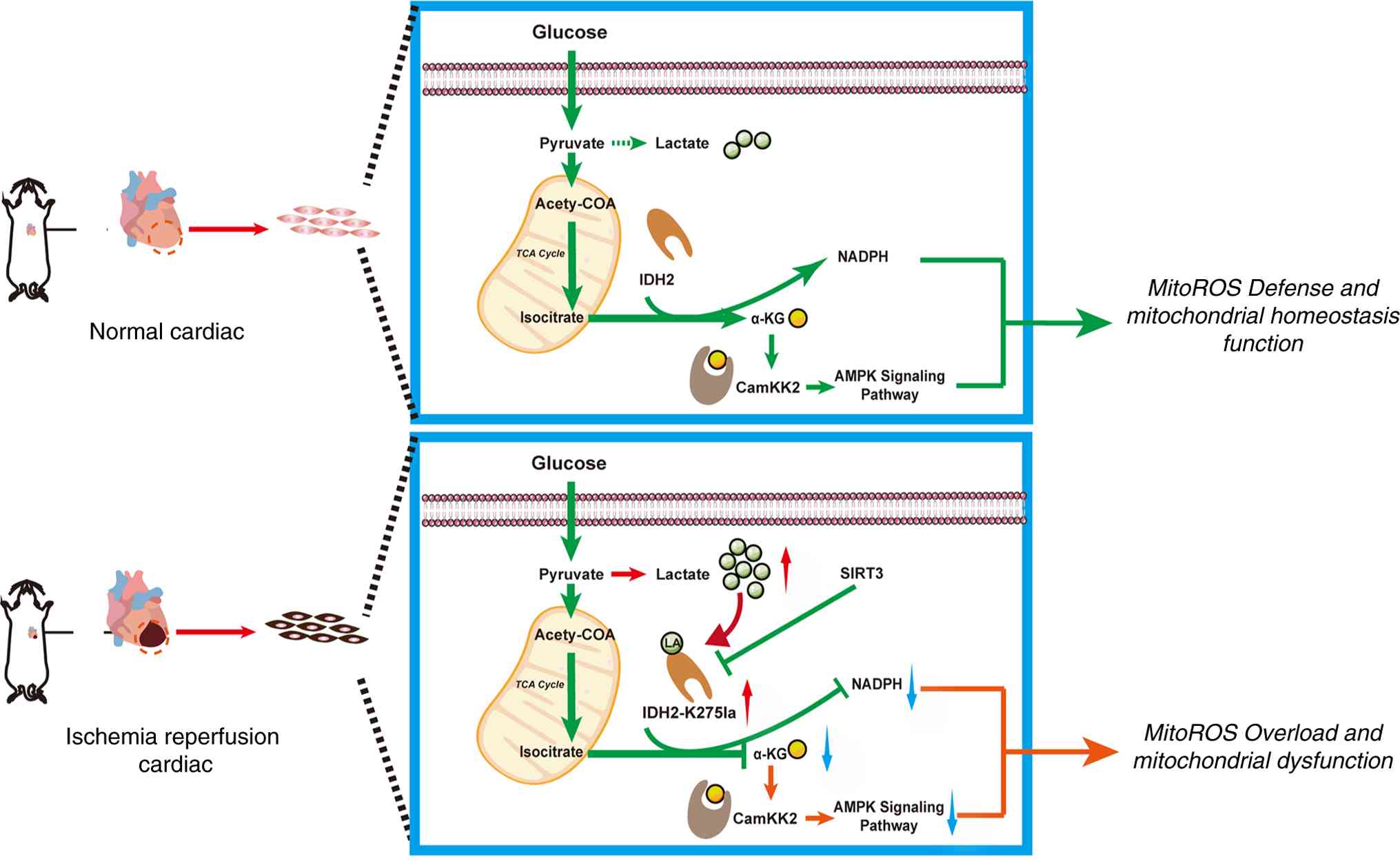 | Figure 8Schematic model of IDH2 lactylation
in MIRI pathogenesis. Under physiological conditions, cardiac
lactate does not accumulate excessively, and IDH2 lactylation
remains at a basal level. This preserves IDH2 enzymatic activity,
allowing it to maintain mitochondrial functional homeostasis and
protect against oxidative stress. However, during MIRI, elevated
intracellular lactate drives hyper-lactylation of IDH2. This
aberrant modification impairs IDH2 function, disrupting downstream
α-KG production and AMPK pathway activation. Consequently,
mitochondrial dysfunction ensues, leading to myocardial damage and
loss of cardiac function. Importantly, delactylating the IDH2-K275
site to reduce its lactylation can restore IDH2 activity, preserve
mitochondrial homeostasis, and ultimately mitigate cardiac injury.
IDH2, isocitrate dehydrogenase 2; myocardial ischemia-reperfusion
injury; α-KG, α-ketoglutaric acid; AMPK, adenosine
5′-monophosphate-activated protein kinase; ROS, reactive oxygen
species; TCA, tricarboxylic acid. |
Supplementary Data
Availability of data and materials
The data generated in the present study are included
in the figures and/or tables of this article.
The data generated in the present study may be found
in the ProteomeXchange under accession number PXD076346 or at the
following URL: https://proteomecentral.proteomex-change.org/ui?view=datasets&search=PXD076346;
and in the National Genomics Data Center under accession number
OMIX015583 or at the following URL: https://ngdc.cncb.ac.cn/search/specific?db=omix&q=OMIX015583.
Authors' contributions
LQZ led the project. CSW and SMS designed and
conceived the study. CSW, JDL, LX and SMS drafted the manuscript.
LX and CSW performed data analysis. LX and CSW wrote, reviewed and
edited the manuscript. SYCh, SYCa, SML and JNC fed the experimental
mice. CSW, SYCh and JDL performed the animal experiments. ZQY and
KD performed cell cultures. SYCa, LX and LQZ revised the
manuscript. CSW, JDL and XDY performed the functional experiments.
CSW and LQZ confirm the authenticity of all the raw data. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The ethics approval (approval no.
AHGDMU-LAC-B-202301-0004) for animal research was obtained from the
Experimental Animal Ethics Committee of Guangdong Medical
University (Zhanjiang, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
MIRI
|
myocardial ischemia-reperfusion
injury
|
|
PTM
|
post-translation modification
|
|
DCA
|
dichloroacetic acid
|
|
NaLac
|
sodium lactate
|
|
8-OHdG
|
8-hydroxy-2′-deoxyguanosine
|
|
DHE
|
dihydroethidium
|
|
IDH2
|
isocitrate dehydrogenase 2
|
|
DRP1
|
dynamin-related protein 1
|
|
MnSOD2
|
manganese superoxide dismutase 2
|
|
Cyt C
|
cytochrome C
|
|
SIRT3
|
sirtuin 3
|
|
α-KG
|
α-ketoglutaric acid
|
|
AAV9
|
adeno-associated virus 9
|
|
ROS
|
reactive oxygen species
|
|
NADPH
|
nicotinamide adenine dinucleotide
phosphate
|
|
AMPK
|
adenosine 5′-monophosphate-activated
protein kinase
|
|
CK-MB
|
creatine kinase-MB
|
|
3-TYP
|
3-(1H-1,2,3-Triazol-4-yl)
pyridine
|
|
LVEF
|
left ventricular ejection
fraction
|
|
LVFS
|
left ventricular fractional
shortening
|
Acknowledgments
The authors would like to thank the experimental
facility of the Affiliated Hospital of Guangdong Medical University
for help in the use of confocal microscopy and flow cytometry; Dr
Li Xu (Affiliated Second Hospital of Guangdong Medical University,
Zhanjiang, China) and Professor Jing Tang (Affiliated Hospital of
Guangdong Medical University, Zhanjiang, China) for support and
discussion during the present study.
Funding
The present study was supported by the Natural Science
Foundation of Guangdong (grant no. 2022A1515012103), the Natural
Science Foundation of Guangdong (grant no. 2024A1515013119) and the
National Natural Science Foundation of China (grant no.
82370281).
References
|
1
|
Benjamin EJ, Blaha MJ, Chiuve SE, Cushman
M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C,
et al: Heart disease and stroke Statistics-2017 update: A report
from the American heart association. Circulation. 135:e146–e603.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Heusch G: Myocardial ischaemia-reperfusion
injury and cardioprotection in perspective. Nat Rev Cardiol.
17:773–789. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chan DC: Mitochondrial dynamics and its
involvement in disease. Annu Rev Pathol. 15:235–259. 2020.
View Article : Google Scholar
|
|
5
|
Dambrova M, Zuurbier CJ, Borutaite V,
Liepinsh E and Makrecka-Kuka M: Energy substrate metabolism and
mitochondrial oxidative stress in cardiac ischemia/reperfusion
injury. Free Radic Biol Med. 165:24–37. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ruiz-Meana M, Fernandez-Sanz C and
Garcia-Dorado D: The SR-mitochondria interaction: A new player in
cardiac pathophysiology. Cardiovasc Res. 88:30–39. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang Y and Patti GJ: The Warburg effect: A
signature of mitochondrial overload. Trends Cell Biol.
33:1014–1020. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Quinn WJ III, Jiao J, TeSlaa T, Stadanlick
J, Wang Z, Wang L, Akimova T, Angelin A, Schäfer PM, Cully MD, et
al: Lactate limits T cell proliferation via the NAD(H) redox state.
Cell Rep. 33:1085002020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bohn T, Rapp S, Luther N, Klein M, Bruehl
TJ, Kojima N, Aranda Lopez P, Hahlbrock J, Muth S, Endo S, et al:
Tumor immunoevasion via acidosis-dependent induction of regulatory
tumor-associated macrophages. Nat Immunol. 19:1319–1329. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang X, Lu Y, Hang J, Zhang J, Zhang T,
Huo Y, Liu J, Lai S, Luo D, Wang L, et al: Lactate-modulated
immunosuppression of Myeloid-derived suppressor cells contributes
to the radioresistance of pancreatic cancer. Cancer Immunol Res.
8:1440–1451. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vaupel P, Schmidberger H and Mayer A: The
Warburg effect: Essential part of metabolic reprogramming and
central contributor to cancer progression. Int J Radiat Biol.
95:912–919. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mao Y, Zhang J, Zhou Q, He X, Zheng Z, Wei
Y, Zhou K, Lin Y, Yu H, Zhang H, et al: Hypoxia induces
mitochondrial protein lactylation to limit oxidative
phosphorylation. Cell Res. 34:13–30. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang D, Tang Z, Huang H, Zhou G, Cui C,
Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al: Metabolic
regulation of gene expression by histone lactylation. Nature.
574:575–580. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiong J, He J, Zhu J, Pan J, Liao W, Ye H,
Wang H, Song Y, Du Y, Cui B, et al: Lactylation-driven
METTL3-mediated RNA m6A modification promotes immunosuppression of
tumor-infiltrating myeloid cells. Mol Cell. 82:1660–1677.e10. 2022.
View Article : Google Scholar
|
|
15
|
Yang K, Fan M, Wang X, Xu J, Wang Y, Tu F,
Gill PS, Ha T, Liu L, Williams DL and Li C: Lactate promotes
macrophage HMGB1 lactylation, acetylation, and exosomal release in
polymicrobial sepsis. Cell Death Differ. 29:133–146. 2022.
View Article : Google Scholar :
|
|
16
|
She H, Hu Y, Zhao G, Du Y, Wu Y, Chen W,
Li Y, Wang Y, Tan L, Zhou Y, et al: Dexmedetomidine ameliorates
myocardial Ischemia-reperfusion injury by inhibiting MDH2
lactylation via regulating metabolic reprogramming. Adv Sci
(Weinh). 11:e24094992024. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Leary S, Underwood W, Anthony R, Cartner
S, Grandin T, Greenacre C, Gwaltney-Brant S, Ann McCrackin M, Meyer
R, Miller D, et al: AVMA Guidelines for the Euthanasia of Animals:
2020 Edition. Schaumburg, IL: American Veterinary Medical
Association; 2020
|
|
18
|
Ehler E, Moore-Morris T and Lange S:
Isolation and culture of neonatal mouse cardiomyocytes. J Vis Exp.
501542013.PubMed/NCBI
|
|
19
|
Rigaud VOC, Hoy RC, Kurian J, Zarka C,
Behanan M, Brosious I, Pennise J, Patel T, Wang T, Johnson J, et
al: RNA-Binding Protein LIN28a regulates new myocyte formation in
the heart through long noncoding RNA-H19. Circulation. 147:324–337.
2023. View Article : Google Scholar
|
|
20
|
Jin L, Chun J, Pan C, Kumar A, Zhang G, Ha
Y, Li D, Alesi GN, Kang Y, Zhou L, et al: The PLAG1-GDH1 axis
promotes anoikis resistance and tumor metastasis through
CamKK2-AMPK signaling in LKB1-Deficient lung cancer. Mol Cell.
69:87–99.e87. 2018. View Article : Google Scholar
|
|
21
|
Sun L, Zhang Y, Yang B, Sun S, Zhang P,
Luo Z, Feng T, Cui Z, Zhu T, Li Y, et al: Lactylation of METTL16
promotes cuproptosis via m6A-modification on FDX1 mRNA in gastric
cancer. Nat Commun. 14:65232023. View Article : Google Scholar
|
|
22
|
Ma L, Shi H, Li Y, Gao W, Guo J, Zhu J,
Dong Z, Sun A, Zou Y and Ge J: Hypertrophic preconditioning
attenuates myocardial ischemia/reperfusion injury through the
deacetylation of isocitrate dehydrogenase 2. Sci Bull (Beijing).
66:2099–2114. 2021. View Article : Google Scholar
|
|
23
|
Ørn S and van Hall G: Does a normal
peripheral lactate value always indicate an aerobic tissue
metabolism? Eur J Heart Fail. 19:1034–1035. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bhattacharya B, Mohd Omar MF and Soong R:
The Warburg effect and drug resistance. Br J Pharmacol.
173:970–979. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li W, Zhou C, Yu L, Hou Z, Liu H, Kong L,
Xu Y, He J, Lan J, Ou Q, et al: Tumor-derived lactate promotes
resistance to bevacizumab treatment by facilitating autophagy
enhancer protein RUBCNL expression through histone H3 lysine 18
lactylation (H3K18la) in colorectal cancer. Autophagy. 20:114–130.
2024. View Article : Google Scholar :
|
|
26
|
Li F, Si W, Xia L, Yin D, Wei T, Tao M,
Cui X, Yang J, Hong T and Wei R: Positive feedback regulation
between glycolysis and histone lactylation drives oncogenesis in
pancreatic ductal adenocarcinoma. Mol Cancer. 23:902024. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhu Z, Huang C, Chen J, Wan L, Zhang C and
Wang J: Lactylation at the crossroads of immune metabolism and
epigenetic regulation: Revealing its role in rheumatic immune
diseases. J Transl Med. 24:252025. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang N, Zhang Y, Xu J, Wang P, Wu B, Lu
S, Lu X, You S, Huang X, Li M, et al: α-myosin heavy chain
lactylation maintains sarcomeric structure and function and
alleviates the development of heart failure. Cell Res. 33:679–698.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang J, Yang P, Yu T, Gao M, Liu D, Zhang
J, Lu C, Chen X, Zhang X and Liu Y: Lactylation of PKM2 suppresses
inflammatory metabolic adaptation in Pro-inflammatory macrophages.
Int J Biol Sci. 18:6210–6225. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang H, Xiong Q, He G, Tang J, Sun L,
Cheng S, Ke M, Chen S, Hu Y, Feng J, et al: Hepatic IDH2 regulates
glycolysis and gluconeogenesis. Metabolism. 143:1555592023.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang GF, Jensen MV, Gray SM, El K, Wang
Y, Lu D, Becker TC, Campbell JE and Newgard CB: Reductive TCA cycle
metabolism fuels glutamine- and glucose-stimulated insulin
secretion. Cell Metab. 33:804–817.e5. 2021. View Article : Google Scholar :
|
|
32
|
Kim H, Lee JH and Park JW: IDH2 deficiency
exacerbates acetaminophen hepatotoxicity in mice via mitochondrial
dysfunction-induced apoptosis. Biochim Biophys Acta Mol Basis Dis.
1865:2333–2341. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Barnabas GD, Lee JS, Shami T, Harel M,
Beck L, Selitrennik M, Jerby-Arnon L, Erez N, Ruppin E and Geiger
T: Serine biosynthesis is a metabolic vulnerability in IDH2-driven
breast cancer progression. Cancer Res. 81:1443–1456. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu X and Gong Y: Isocitrate dehydrogenase
inhibitors in acute myeloid leukemia. Biomark Res. 7:222019.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Si X, Shao M, Teng X, Huang Y, Meng Y, Wu
L, Wei J, Liu L, Gu T, Song J, et al: Mitochondrial isocitrate
dehydrogenase impedes CAR T cell function by restraining
antioxidant metabolism and histone acetylation. Cell Metab.
36:176–192.e10. 2024. View Article : Google Scholar
|
|
36
|
Han SJ, Choi HS, Kim JI, Park JW and Park
KM: IDH2 deficiency increases the liver susceptibility to
ischemia-reperfusion injury via increased mitochondrial oxidative
injury. Redox Biol. 14:142–153. 2018. View Article : Google Scholar
|
|
37
|
Noh MR, Kong MJ, Han SJ and Park KM:
Isocitrate dehydrogenase 2 deficiency aggravates prolonged high-fat
diet intake-induced hypertension. Redox Biol. 34:1015482020.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim
SH, Ito S, Yang C, Wang P, Xiao MT, et al: Oncometabolite
2-hydroxyglutarate is a competitive inhibitor of
α-ketoglutarate-dependent dioxygenases. Cancer Cell. 19:17–30.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Stein EM, DiNardo CD, Pollyea DA, Fathi
AT, Roboz GJ, Altman JK, Stone RM, DeAngelo DJ, Levine RL, Flinn
IW, et al: Enasidenib in mutant IDH2 relapsed or refractory acute
myeloid leukemia. Blood. 130:722–731. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mellinghoff IK, van den Bent MJ,
Blumenthal DT, Touat M, Peters KB, Clarke J, Mendez J, Yust-Katz S,
Welsh L, Mason WP, et al: Vorasidenib in IDH1- or IDH2-Mutant
Low-Grade Glioma. N Engl J Med. 389:589–601. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Du R, Gao Y, Yan C, Ren X, Qi S, Liu G,
Guo X, Song X, Wang H, Rao J, et al: Sirtuin 1/sirtuin 3 are robust
lysine delactylases and sirtuin 1-mediated delactylation regulates
glycolysis. iScience. 27:1109112024. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Morigi M, Perico L and Benigni A: Sirtuins
in renal health and disease. J Am Soc Nephrol. 29:1799–1809. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gong Y, Tang N, Liu P, Sun Y, Lu S, Liu W,
Tan L, Song C, Qiu X, Liao Y, et al: Newcastle disease virus
degrades SIRT3 via PINK1-PRKN-dependent mitophagy to reprogram
energy metabolism in infected cells. Autophagy. 18:1503–1521. 2022.
View Article : Google Scholar :
|
|
44
|
Kim TS, Jin YB, Kim YS, Sun Y, Lu S, Liu
W, Tan L, Song C, Qiu X, Liao Y, et al: SIRT3 promotes
antimycobacterial defenses by coordinating mitochondrial and
autophagic functions. Autophagy. 15:1356–1375. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li Z, Hu O, Xu S, Lin C, Yu W, Ma D, Lu J
and Liu P: The SIRT3-ATAD3A axis regulates MAM dynamics and
mitochondrial calcium homeostasis in cardiac hypertrophy. Int J
Biol Sci. 20:831–847. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ding Y, Yang H, Wang Y, Chen J, Ji Z and
Sun H: Sirtuin 3 is required for osteogenic differentiation through
maintenance of PGC-1α-SOD2-mediated regulation of mitochondrial
function. Int J Biol Sci. 13:254–264. 2017. View Article : Google Scholar
|
|
47
|
Gao J, Zhang K, Wang Y, Guo R, Liu H, Jia
C, Sun X, Wu C, Wang W, Du J and Chen J: A machine learning-driven
study indicates emodin improves cardiac hypertrophy by modulation
of mitochondrial SIRT3 signaling. Pharmacol Res. 155:1047392020.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Long D, Deng Z, Zhao X, Xu Y, Li W, Mo X,
Zhong Y, Li M, He A, Zhang Z, et al: m7G-modified mt-tRF3b-LeuTAA
regulates mitophagy and metabolic reprogramming via SUMOylation of
SIRT3 in chondrocytes. Biomaterials. 314:1229032025. View Article : Google Scholar
|
|
49
|
Li R, Yan L, Sun X and Zheng W: A bicyclic
pentapeptide-based highly potent and selective pan-SIRT1/2/3
inhibitor harboring Nε-thioacetyl-lysine. Bioorg Med Chem.
28:1153562020. View Article : Google Scholar
|
|
50
|
Zhang X, Cao R, Niu J, Yang S, Ma H, Zhao
S and Li H: Molecular basis for hierarchical histone
de-β-hydroxybutyrylation by SIRT3. Cell Discov. 5:352019.
View Article : Google Scholar
|
|
51
|
Wei XH, Chen J, Wu XF, Zhang Q, Xia GY,
Chu XY, Xia H, Lin S and Shang HC: Salvianolic acid B alleviated
myocardial ischemia-reperfusion injury via modulating
SIRT3-mediated crosstalk between mitochondrial ROS and NLRP3.
Phytomedicine. 136:1562602025. View Article : Google Scholar
|