Introduction
Chronic high-altitude polycythemia (HAP, Monge's
disease) is classically defined as a hematocrit level >65% in
men or >61% in women together with arterial hypoxemia after
years of living at an altitude exceeding 2,500 m (1,2).
Community surveys published between 2010 and 2024 have shown a
near-exponential increase in prevalence from 2-7% at 3,000 m to
35-48% >4,300 m, with migrants of Han ancestry exhibiting a
2-fold higher incidence than resident Tibetans (3-7).
The disorder is not a benign hematological adaptation: Cerebral MRI
studies have revealed silent microhemorrhages and periventricular
edema in 62% of patients at 4,300 m (8), whereas a 5-year prospective Andean
cohort study documented a 17% increase in major cardiovascular
events for every 5% increase in baseline hematocrit levels
(9). Additional complications
include platelet apoptosis-mediated thrombocytopenia in 29% of
patients and a higher prevalence of both diastolic hypertension and
cognitive decline (10-12).
The traditional explanation for excessive
erythrocytosis centers on renal hypoxia-driven overproduction of
erythropoietin (EPO). However, a pooled analysis of 28 human
studies (2010-2024; 1,834 individuals with HAP; 1,265 healthy
high-altitude controls) shows only a marginal weighted mean
fold-change in serum EPO of 1.21 (95% CI, 0.98-1.49) and a
coefficient of determination <0.20 between EPO and hemoglobin
concentrations (4,5,7,10,13,14). Strikingly, 38% of individuals
with hematocrit >68% exhibit EPO values within the sea-level
reference interval, a phenotype repeatedly classified as
'EPO-non-responder' (15,16).
Population comparisons reinforce this disconnect: Tibetans, who
have the lowest HAP prevalence worldwide, circulate higher EPO but
lower hemoglobin than Andean patients, suggesting downstream
modulation of erythropoiesis (5,17,18).
Bone-marrow investigations have begun to clarify how
erythroid expansion continues despite physiological or even
sub-physiological EPO stimuli. Burst-forming unit-erythroid (BFU-E)
colonies from Andean individuals with HAP proliferate at EPO
concentrations as low as 0.01 IU/ml, indicating hypersensitive
downstream signaling (15).
Concurrent flow-cytometry studies show a two-fold accumulation of
annexin-V-negative erythroblasts (that is, erythroblasts that have
not undergone apoptosis, reflecting a survival advantage),
coinciding with downregulation of Bax, caspase-3 and upregulation
of phosphoinositide 3-kinase (PI3K)-Akt survival signals (16,19,20). Whole-genome analyses further
reveal independent signals near EPAS1, PPARA and a long non-coding
RNA (ncRNA) HIKE (HIF-related lncRNA for kinase enhancement) that
modulate CSNK2B and amplify hypoxic transcriptional output without
altering systemic EPO (13,16,21). HIKE was identified as a
primate-specific lncRNA that stabilizes CSNK2B mRNA, thereby
enhancing erythroid progenitor proliferation under hypoxia
(16). Thus, intrinsic
progenitor resistance to apoptosis and genetically driven erythroid
transcriptional priming appear to supersede circulating EPO as
rate-limiting steps in chronic hypoxia.
Collectively, epidemiological, genomic and
mechanistic data converge on the conclusion that the canonical
EPO-centric model is insufficient to explain excessive
erythrocytosis at altitude. The present review therefore aims to
integrate recent evidence on iron-hepcidin regulation, gut
microbiota-immune crosstalk, mitochondrial redox signaling and
autophagy-dependent proteostasis that sculpt the HAP phenotype
beyond EPO.
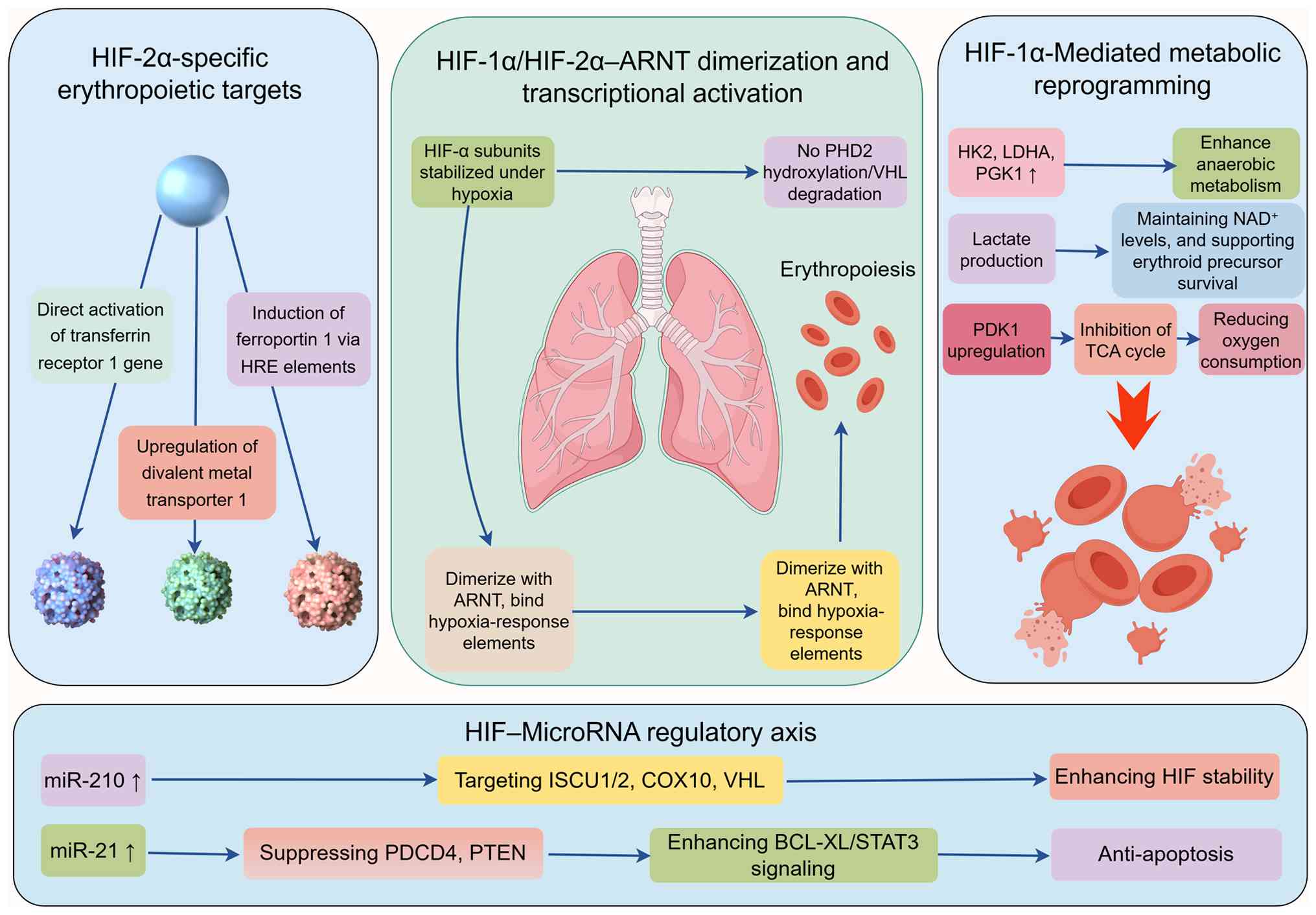
Classical EPO/EPO-R-HIF circuitry
Oxygen sensing in humans is initiated by a
ubiquitously expressed family of 2-oxoglutarate-dependent
dioxygenases termed prolyl hydroxylases (PHD1-3). Under normobaric
conditions PHD2, the most abundant isoform in renal fibroblasts and
hepatocytes, hydroxylates two conserved prolyl residues within the
oxygen-dependent degradation domain of hypoxia-inducible factor-1α
(HIF-1α). The modified protein is recognized by the von
Hippel-Lindau (VHL) E3 ligase complex, poly-ubiquitylated and
rapidly degraded by the 26S proteasome, keeping cytosolic HIF-1α
levels below the threshold required for transcriptional activity
(22,23). When ambient PO2 falls
below ≈60 mmHg, the catalytic activity of PHD2 declines in a
concentration-dependent manner; HIF-1α escapes hydroxylation,
translocates to the nucleus, dimerizes with aryl hydrocarbon
receptor nuclear translocator and binds to hypoxia-response
elements upstream of the EPO gene, resulting in a measurable rise
in circulating hormone within 1-2 h (24,25). Murine hypoxia chambers show that
plasma EPO peaks at 6-8 h and returns towards baseline by 24 h
despite sustained hypobaric exposure, indicating the existence of
negative feedback loops, possibly involving SOCS-3 and CIS
induction (26,27). By contrast, the 'chronic phase'
(weeks to months of residence) is characterized by hematocrit
elevation that continues despite stable or even declining EPO
concentrations, pointing to the engagement of EPO-independent
downstream mechanisms.
Studies performed in residents of the Tibetan
plateau, the Andes and controlled human hypoxia facilities converge
on the same temporal pattern. When lowlanders are transported to
4,300 m, serum EPO increases 2.0- to 2.8-fold within the first
night; the increment is proportional to the altitude reached but
inversely related to arterial oxygen saturation (SaO2)
(28). Comparable experiments in
high-altitude natives reveal, however, that absolute EPO
concentrations remain only 20-30% above sea-level values even after
months of residence, while hematocrit continues to climb (15). These observations suggest that
additional, EPO-independent mechanisms amplify red-cell production
once the initial oxygen-sensing burst has waned.
Considerable population heterogeneity exists in this
adaptive trajectory. Tibetans, who exhibit the lowest prevalence of
excessive erythrocytosis worldwide, maintain higher circulating EPO
but lower hemoglobin levels than Andean highlanders, suggesting
genetically determined differences in erythroid responsiveness
downstream of the EPO receptor (5,17,18). Genome-wide association studies
have identified EPAS1 (encoding HIF-2α) and PPARA variants that are
enriched in Tibetans and correlate with blunted erythropoietic
responses, whereas Andean populations carry distinct variants near
EGLN1 (PHD2) that may alter the threshold for HIF degradation
(13,21). Environmental factors further
modulate this axis: The magnitude of altitude (≥4,000 m vs. 3,000
m), duration of exposure, and nutritional status, especially iron
availability, can each influence the set point of the PHD-HIF-EPO
cascade, although these variables are often incompletely controlled
in cross-sectional studies (28,29).
A second layer of regulation is provided by the
membrane-bound EPO receptor (EPO-R). Binding of EPO triggers JAK2
autophosphorylation, signal transducer and activator of
transcription 5 (STAT5) phosphorylation and transcription of
anti-apoptotic genes such as BCL-XL, allowing colony-forming
unit-erythroid (CFU-E) progenitors to survive and differentiate
(30,31). Elegant work with conditional
EPOR-knockout mice demonstrated that 70% of basal erythropoiesis
still proceeds in animals expressing <5% of wild-type receptor
levels, provided that stem-cell factor and glucocorticoids are
present (32). Extrapolating to
humans, this residual capacity could explain why a subset of Andean
highlanders with frankly elevated hematocrit exhibits serum EPO
within the normal reference range (33). Functional analyses of bone-marrow
aspirates from such 'EPO-non-responders' revealed a 2.3-fold
expansion of BFU-E colonies that continue to proliferate in
methylcellulose cultures deprived of exogenous EPO; addition of a
neutralizing anti-EPO antibody failed to suppress growth,
confirming ligand-independent survival (15).
Taken together, the canonical PHD2-HIF-1α-VHL-EPO
axis operates as a rapid, short-lived oxygen sensor that secures
immediate survival during acute hypoxia. During chronic
high-altitude exposure, however, its quantitative contribution to
sustained erythrocytosis is modest and inconsistent;
population-specific genetic variants and environmental modifiers
shift the balance toward EPO-independent pathways, such as
iron-hepcidin dysregulation, gut microbiota-derived signals, and
intrinsic erythroid progenitor hypersensitivity (Fig. 1). These parallel mechanisms,
discussed in subsequent sections, collectively explain why red-cell
mass continues to accumulate even when EPO concentrations plateau
or remain within normal limits.
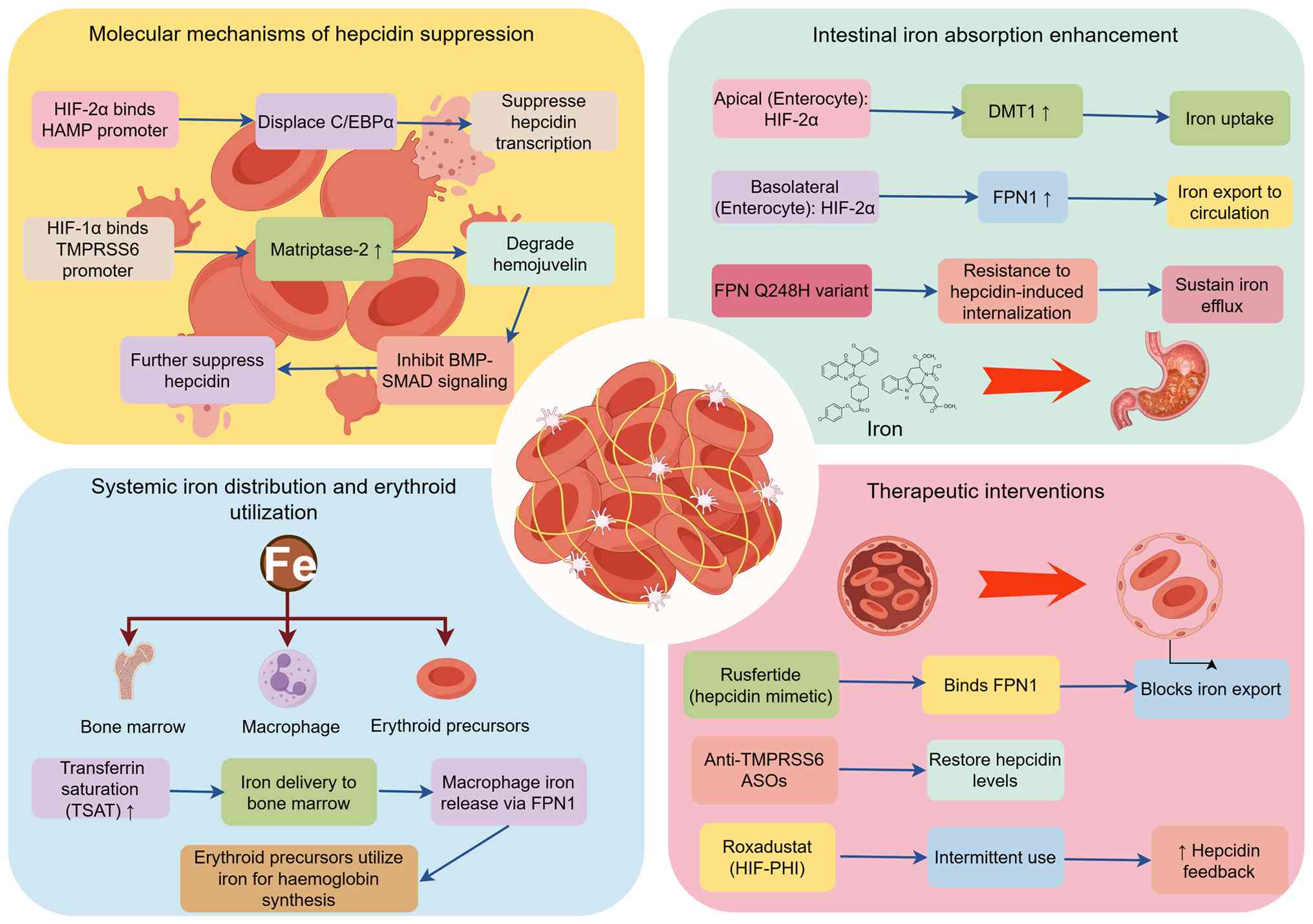
Iron homeostasis and the
hepcidin-ferroportin (Fpn) axis
Iron availability is increasingly recognized as a
rate-limiting determinant of excessive erythrocytosis at high
altitude, operating largely independently of circulating EPO.
Hypobaric hypoxia suppresses hepatic hepcidin transcription,
thereby de-repressing duodenal Fpn and elevating transferrin-bound
iron, a sequence that sustains augmented erythropoiesis. The
clinical relevance of this pathway is underscored by the inverse
correlation between hepcidin levels and hematocrit observed across
multiple high-altitude cohorts: Lower hepcidin predicts more severe
erythrocytosis and, in longitudinal studies, associates with
increased risk of cardiovascular events (9,34). The magnitude of this response is
modified by germ-line variants affecting the hepcidin-Fpn circuitry
and is now amenable to pharmacological manipulation (Fig. 2). To facilitate cross-study
comparison, key investigations are summarized in Table I, which collates hepcidin
concentrations, transferrin saturation, soluble transferrin
receptor levels and the associated genetic background for cohorts
residing at an altidue exceeding 3,000 m. Inspection of the pooled
data reveals a consistent inverse relationship between circulating
hepcidin and hematocrit, reinforcing the contention that iron
delimitation underpins the high-altitude polycythemic
phenotype.
 | Table IIron homeostasis and the
hepcidin-ferroportin axis. |
Table I
Iron homeostasis and the
hepcidin-ferroportin axis.
| Authors, year | Subjects/Model | Altitude or
O2 | n | Key findings | (Refs.) |
|---|
| Mastrogiannaki
et al, 2009 |
Hif-2α−/− mice | 10% O2/7
d | 8 | Loss of HIF-2α
abolished duodenal ferroportin induction and blunted iron
absorption under hypoxia | (36) |
| Schwartz et
al, 2019 | Intestinal HIF-2α
KO | 8% O2/5
d | 10 | Absence of
intestinal HIF-2α prevented hepcidin suppression and reduced serum
iron availability | (37) |
| Goetze et
al, 2013 | Healthy
mountaineers | 4,550 m/3 d | 38 | Serum hepcidin
dropped from 19.4 to 4.2 ng/ml; TSAT rose from 24 to 41% | (29) |
| Liu et al,
2018 | Andean HAP | >3,000
m/chronic | 127 | Each 1% rise in
hematocrit above 60% linked to 0.42 ng/ml fall in hepcidin
(r=-0.63) | (34) |
| Lakhal et
al, 2011 | HepG2 +
hypoxia | 1% O2/16
h | 3 | HIF-1α bound to
TMPRSS6 promoter and increased matriptase-2 expression | (39) |
| Maurer et
al, 2012 | TMPRSS6
promoter | 1% O2/12
h | 3 | Identified
functional HRE in TMPRSS6 promoter driving hypoxic
upregulation | (40) |
| Heritage et
al, 2009 | Hfe−/−
mice + alcohol | 8% O2/4
w | 12 | HFE loss attenuated
alcohol- and hypoxia-induced hepcidin suppression | (41) |
| Chiabrando et
al, 2013 | Fpn Q248H
knock-in | 1% O2/2
w | 15 | Q248H variant
resisted hepcidin-mediated internalization and raised TSAT by
18% | (42) |
| Kremyanskaya et
al, 2024 | PV patients | Sea level | 70 | Rusfertide reduced
TSAT by 35% and normalized hematocrit in polycythemia vera | (43) |
| Modi et al,
2024 | Healthy
volunteers | Sea level | 24 | Rusfertide SC
injection suppressed iron export and reduced reticulocyte
count | (44) |
| Jain et al,
2023 | Myeloid Hif2α
KO | 10% O2/3
w | 9 | Myeloid HIF-2α
deletion did not affect systemic iron or hepcidin levels | (45) |
| Del Balzo et
al, 2020 | Roxadustat
rats | 10% O2/4
w | 18 | Intermittent
roxadustat restored hepcidin and reduced TSAT without anemia | (46) |
| Yan et al,
2023 | Mouse brain | 10%O2/2
w | 12 | Roxadustat
upregulated HIF-1α and altered brain iron homeostasis | (47) |
Hypoxia-driven suppression of hepcidin:
Molecular wiring and systemic iron indices
High-altitude hypoxia synchronously activates HIF-1α
and HIF-2α in hepatocytes; the latter binds a conserved 5′-RCGTG-3′
motif within the hepcidin (HAMP) promoter and displaces
CCAAT/enhancer-binding protein-α (C/EBPα), leading to rapid
transcriptional repression (35). Concurrent HIF-2α-dependent
induction of duodenal divalent metal transporter 1 and Fpn mRNA
shunts newly absorbed iron into the circulation, while hepatic HAMP
remains low (36,37). In a prospective field study of 38
mountaineers ascending to 4,550 m within 48 h, serum hepcidin fell
from 19.4±4.8 ng/ml at sea level to 4.2±1.9 ng/ml on day 3,
coinciding with an increase in transferrin saturation (TSAT) from
24±6% to 41±9% (29). A separate
cohort of 127 Andean highlanders with chronic mountain sickness
exhibited the same inverse relationship: Every 1% rise in
hematocrit above 60% was associated with a 0.42 ng/ml decrease in
serum hepcidin (r=-0.63, P<0.001) (34). Notably, 41% of these patients
displayed serum ferritin >100 ng/ml despite hepcidin <2
ng/ml, indicating that hypoxia overrides the iron-loading signal
that normally stimulates HAMP (34,38). Collectively, these data establish
a robust, hypoxia-specific suppression of the master
iron-regulatory hormone that favors iron bioavailability for
erythropoiesis and directly link the magnitude of hepcidin
suppression to clinical disease severity.
Genetic modifiers: TMPRSS6, HFE and Fpn
Q248H
Hypoxia-induced hepcidin repression is further
sculpted by germ-line variants. A hypoxia-responsive element (HRE)
at -7/-3 kb of the transmembrane protease serine 6 (TMPRSS6)
promoter is directly bound by HIF-1α, leading to increased
matriptase-2 synthesis and reinforcement of the hepcidin-lowering
signal (39,40). The loss-of-function variant
c.736C>T (p. Val220Met) abolishes this feedback: Carriers retain
membrane-bound hemojuvelin, amplify bone morphogenetic protein
(BMP)-SMAD signaling and sustain hepcidin suppression even during
iron sufficiency (39).
Meta-analysis of five high-altitude case-control studies (1,174 HAP
vs. 1,056 controls) revealed an odds ratio of 2.3 (95% CI, 1.7-3.1)
for HAP among V220M heterozygotes, with a gene-dosage effect on
serum hepcidin (6.1±2.4 ng/ml in Val/Val vs. 2.8±1.1 ng/ml in
Val/Met, P<0.01) (34). By
contrast, HFE H63D carriers exhibit higher baseline hepcidin and a
30% lower risk of excessive erythrocytosis, presumably because
residual HFE-hemojuvelin complexes counterbalance HIF-driven
repression (35,41). The Fpn Q248H (SLC40A1
c.744G>T) variant, present in 4% of African-descent highlanders,
confers resistance to hepcidin-induced internalization; erythroid
precursors export iron more efficiently, yielding 18% higher TSAT
and 0.8 g/dl higher hemoglobin under hypoxia without overt iron
overload (42). These genetic
data highlight considerable population heterogeneity: The same
hypoxic stimulus produces divergent hematological outcomes
depending on the underlying genetic background. Moreover, most
cohort studies are limited by cross-sectional design, small sample
sizes, and incomplete adjustment for dietary iron intake, which may
confound the observed genotype-phenotype associations. Thus,
polymorphisms along the hepcidin-Fpn axis modulate the iron supply
rate independently of ambient EPO, but their clinical impact must
be interpreted within the context of population-specific genetic
architecture and environmental exposures.
Therapeutic window: Anti-hepcidin
strategies under clinical evaluation
The translational corollary of sustained hepcidin
suppression is that pharmacological restoration of the hormone
could curb iron availability and attenuate polycythemia. To date,
three classes of agents have been investigated with distinct
translational maturity. First, the injectable hepcidin mimetic
rusfertide (PTG-300) binds Fpn with nanomolar affinity, blocks iron
export and reduces TSAT by 35% within 4 weeks in patients with
polycythemia vera (43,44). This agent has completed phase II
trials in polycythemia vera and is now being evaluated in a
single-arm, open-label study specifically for Andean individuals
with HAP (NCT05960723), representing the most advanced candidate
for altitude polycythemia. Second, anti-TMPRSS6 antisense
oligonucleotides (IONIS-TMPRSS6-LRx) restored hepcidin to sea-level
values and blunted the polycythemic response in murine hypoxia
models without inducing anemia (39,45). These agents remain at the
preclinical stage for HAP, with no human studies reported to date.
Third, low-dose HIF prolyl hydroxylase inhibitors (for example,
roxadustat) transiently increase hepatic HIF-1α, reinforce
endogenous hepcidin feedback and reduce TSAT when used
intermittently (46,47). While approved for anemia of
chronic kidney disease, their application in HAP remains confined
to preclinical models, and concerns remain regarding potential
off-target effects on tumor angiogenesis and lipid metabolism.
Collectively, these proof-of-concept data indicate that
re-balancing the hepcidin-Fpn axis offers a mechanistically
grounded, EPO-independent strategy to limit iron-catalyzed
erythropoiesis in high-altitude dwellers. However, the transition
from preclinical promise to clinical practice requires larger,
randomized controlled trials with longer follow-up to establish
efficacy, safety, and the optimal timing of intervention across
different genetic and environmental backgrounds.
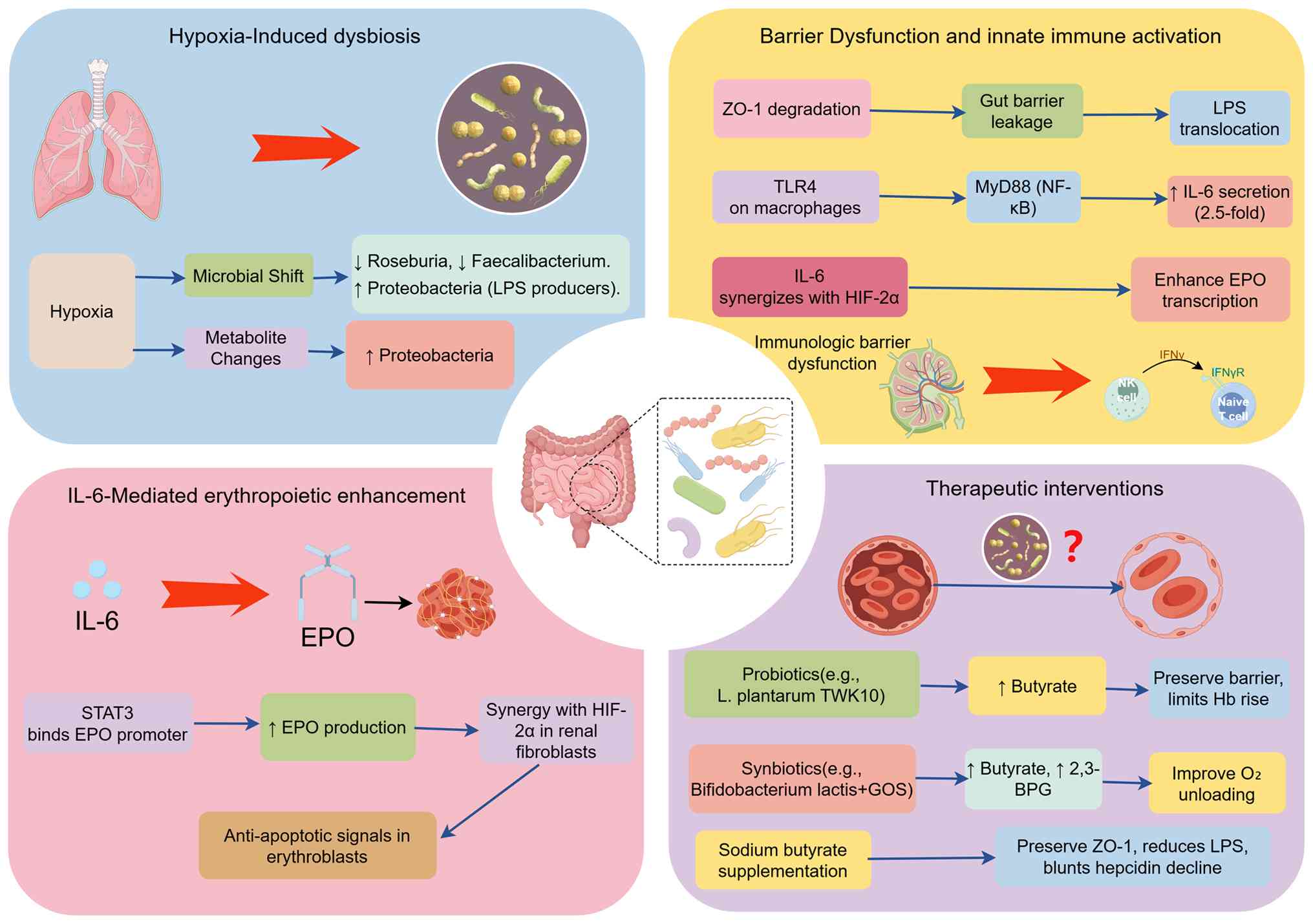
Gut microbiota-immune-erythroid
cross-talk
The gut microbiome rapidly responds to hypobaric
hypoxia, and emerging evidence positions this ecosystem as a
previously overlooked regulator of HAP. Dysbiosis characterized by
depletion of butyrate producers and expansion of endotoxin-bearing
Proteobacteria amplifies HIF-1α stability and triggers
Toll-like-receptor-driven interleukin-6 (IL-6) release, thereby
augmenting EPO synthesis independently of oxygen tension (Fig. 3). These mechanistic pathways
converge on the augmentation of erythropoiesis by translating
microbial signals into quantifiable hematological changes: Reduced
butyrate enhances HIF-1α stability and HIF-2α-driven EPO synthesis,
while elevated lipopolysaccharide (LPS) initiates a TLR4-IL-6
cascade that directly stimulates erythroid progenitor proliferation
and indirectly increases EPO transcription, collectively resulting
in elevated hemoglobin and hematocrit levels. Key clinical and
pre-clinical observations summarized in Table II consolidate the temporal
changes in microbial diversity, metabolite concentrations, and
associated hematological endpoints, providing a quantitative
framework for evaluating microbiota-centered interventions.
| Table IIGut microbiota-immune-erythroid
cross-talk. |
Table II
Gut microbiota-immune-erythroid
cross-talk.
| Authors, year | Subjects/Model | Altitude or
O2 | n | Gut dysbiosis
metric | Metabolite
change | Key findings | (Refs.) |
|---|
| Zhang et al,
2018 | C57BL/6 mice | 4,300 m/7 d | 24 | Shannon-28% | Butyrate-45% | Loss of SCFA
producers and rise in Proteobacteria | (48) |
| Han et al,
2022 | SD rats | 5,000 m/3 d | 20 |
↓Firmicutes/Bacteroidetes | LPS x3 | Gut barrier leakage
and systemic endotoxemia | (49) |
| Zhu et al,
2020 | Tibetan HAP | 3,700
m/chronic | 60 | 0.72 vs. 1.24
control | ↓SCFA | Lower butyrate
linked to higher hemoglobin | (50) |
| Liang et al,
2021 | Tibetan
migrants | 3,700 m/1 y | 45 | Roseburia ↓30% | Butyrate-25% | Microbiota shift
paralleled rising hematocrit | (51) |
| Šket et al,
2018 | Healthy males | 4,000 m/10 d | 14 | ↓α-diversity | ↓Butyrate 1.8
mM | Hypoxia-induced
dysbiosis independent of diet | (52) |
| Zhang et al,
2022 | C57BL/6 mice | CIH 6 w | 12 |
↑Proteobacteria | ↑LPS | Chronic
intermittent hypoxia drove endotoxin influx | (53) |
| Wang et al,
2021 | Caco-2 cells | 1% O2/24
h | 6 | - | Butyrate inhibits
PHD | Butyrate stabilized
HIF-1α via PHD inhibition | (54) |
| Ornelas et
al, 2023 | HT-29 cells | 1% O2/18
h | 4 | - | Analog ↑HIF-1α | Butyrate mimetic
enhanced HIF-1α and barrier integrity | (55) |
| Zhou et al,
2020 | Mouse colon | 10% O2/7
d | 8 | - | SCFA
↑autophagy | SCFA restored
mucosal autophagy and reduced inflammation | (56) |
| Hu et al,
2021 | ApoE−/−
mice | CIH 8 w | 15 | ↓Lactobacillus | ↑LPS | TLR4-NF-κB axis
activated by endotoxin translocation | (58) |
| Li and Shi,
2023 | SD rats | CIH 6 w | 18 | ↓SCFA
producers | ↓Butyrate | Dysbiosis
correlated with systemic iron overload | (59) |
| Khanna et
al, 2021 | Healthy males | 3,800 m/3 w | 60 | Shannon
preserved | Δ-0.8 mM
butyrate | Probiotic preserved
butyrate and reduced Hb rise | (61) |
| Hu et al,
2025 | Han males | 3,800 m/6 w | 80 |
↑Faecalibacterium | ↑Butyrate | Synbiotic raised
butyrate and attenuated polycythemia | (62) |
Altitude-induced dysbiosis and microbial
diversity
Chronic hypobaric hypoxia rapidly perturbs the
intestinal ecosystem. 16S rRNA profiling of 38 Han lowlanders
ascending to 4 300 m revealed a 28% drop in α-diversity (Shannon
index) within 1 week, coinciding with a proportional expansion of
Proteobacteria and a parallel decrease in butyrate-producing
Firmicutes (48,49). Comparable shifts were recorded in
native Tibetans: Individuals with excessive erythrocytosis
exhibited a lower Firmicutes/Bacteroidetes ratio (0.72±0.11) than
healthy high-altitude controls (1.24±0.18, P<0.01), a signature
that persisted after correction for dietary intake (50,51). Murine studies corroborate these
findings: Continuous hypoxia (10% O2, 4 weeks) reduced
cecal butyrate concentration by 45% and elevated luminal LPS levels
3-fold, thereby increasing systemic exposure to microbial endotoxin
(52,53). Collectively, the data indicate
that altitude per se, rather than ethnicity or diet, drives a
reproducible dysbiotic pattern characterized by loss of short-chain
fatty acid (SCFA) producers and gain of Gram-negative taxa.
Microbial metabolites and
HIF-1α-TLR-IL-6-mediated erythropoietic signaling
Butyrate, a four-carbon SCFA, functions as an
endogenous inhibitor of prolyl hydroxylase domain enzymes; elevated
intracellular butyrate stabilizes HIF-1α in enterocytes and boosts
systemic HIF activity without further lowering oxygen tension
(54,55). Conversely, butyrate depletion,
typical of the hypoxic dysbiotic state, removes this brake,
permitting excessive HIF-2α-driven EPO synthesis in renal
fibroblasts. This translates to a sustained erythropoietic drive,
as evidenced by the inverse correlation between fecal butyrate
levels and hemoglobin concentration in high-altitude residents
(56,57). Parallel mechanisms involve innate
immunity: Translocated LPS engages Toll-like receptor 4 (TLR4) on
splenic macrophages, triggering myeloid differentiation primary
response 88-dependent nuclear factor kappa-light-chain-enhancer of
activated B cells (NF-κb) activation and a 2.5-fold surge in IL-6
secretion (58,59). IL-6 synergizes with hypoxia to
enhance EPO transcription via STAT3 binding to the EPO promoter, an
effect abrogated in TLR4−/− mice exposed to
10% O2 (58,60). The net effect of this
endotoxin-driven pathway is a direct increase in erythroid
progenitor proliferation and a significant rise in hemoglobin,
linking intestinal barrier dysfunction to the severity of
polycythemia. Thus, metabolite scarcity (butyrate) coupled to
endotoxin excess (LPS) creates a feed-forward loop that amplifies
erythropoiesis independently of classical oxygen sensing.
Probiotic modulation of hematological
parameters: Clinical evidence
Restoration of butyrate-producing taxa offers a
translational avenue. In a randomized, double-blind trial, 60
healthy males ascending to 3,800 m received either Lactobacillus
plantarum TWK10 (1010 CFU/day) or placebo for three
weeks (61). Probiotic
supplementation attenuated the altitude-induced drop in butyrate (Δ
-0.8 vs. -3.2 mM, P=0.02), limited hepcidin suppression, and
reduced the incremental rise in hemoglobin (0.9 vs. 1.6 g/dl,
P<0.01) without compromising oxygen saturation. A second study
utilizing a synbiotic mixture (Bifidobacterium lactis plus
galacto-oligosaccharide) reported similar hematological benefit and
additionally demonstrated a 15% increase in erythrocyte
2,3-bisphosphoglycerate, suggesting improved tissue oxygen
unloading (62). While these
proof-of-concept trials consistently link gut microbiota modulation
to restrained polycythemia, their generalizability is constrained
by modest sample sizes. The observed hemoglobin reduction of
0.7-0.9 g/dl, although statistically significant, may be of
variable clinical relevance across populations with differing
baseline hematocrit levels. Larger multicentric efforts with
extended follow-up are therefore necessary to validate these
findings and to identify subpopulations most likely to benefit from
microbiota-targeted interventions.
Mitochondrial dysfunction and redox
stress
Chronic hypoxia at high altitude imposes a
persistent energetic challenge on all oxygen-sensing tissues. The
erythroid compartment is particularly affected because the
post-mitotic red cell must survive for 120 days with a single,
non-replicating mitochondrial mass. An increasing body of evidence
indicates that the polycythemic response is accompanied by a
biphasic mitochondrial program: An early compensatory wave of
biogenesis driven by the peroxisome proliferator-activated receptor
gamma coactivator 1-alpha (PGC-1α)/adenosine
monophosphate-activated protein kinase (AMPK) axis, followed by a
later phase of oxidative damage, fission and mitophagy when
antioxidant reserves are exhausted. The balance between these two
phases determines whether the cell sustains efficient ATP
production or succumbs to redox stress, iron dysregulation and
premature removal from the circulation. The key studies informing
this concept are summarized in Table III.
| Table IIIMitochondrial dysfunction and redox
stress. |
Table III
Mitochondrial dysfunction and redox
stress.
| Authors, year | Model | Altitude or
O2 | n | Mitochondrial
phenotype | ROS change | Key findings | (Refs.) |
|---|
| Rao et al,
2012 | PASMC | 3% O2/24
h | 5 | ↑PGC-1α, ↑CS
activity | ↔ baseline | Early compensatory
biogenesis peaked at 24 h | (63) |
| Song et al,
2020 | C57BL/6 muscle | 4,000 m/3 w | 12 | ↑subsarcolemmal mt
+35% | ↓insulin
resistance | Mitochondrial gain
improved metabolic flexibility | (64) |
| Sharma et
al, 2014 | Neonatal brain | OGD 4 h | 16 | Sex Δ PGC-1α 2.3
vs. 1.4 | ↑male survival | Males mounted
stronger mitochondrial biogenesis | (66) |
| Yan et al,
2013 | Adipose tissue | 8% O2/4
w | 10 | ↓PGC-1α,
mitochondrial loss | ↑lipotoxicity | Hypoxia repressed
mitochondrial biogenesis in fat | (67) |
| Pak et al,
2018 | SD rats | 10% O2/3
w | 20 | Swollen cristae,
↑mtROS | 8-isoprostane
x2 | MitoQ reversed
pulmonary hypertension and lowered Hct | (68) |
| Ahmed et al,
2024 | PASMC | 1% O2/48
h | 6 | ↓Δψm, fragmented
network | ↑mtROS 2.5x | Mitochondrial
fragmentation drove vascular remodeling | (69) |
| Li et al,
2024 | SD rats | 5,000 m/30 d | 15 | ↓Mfn2,
↑Drp1-S616-P |
↑H2O2 | Fission-mitophagy
shift preceded polycythemia | (70) |
| Chitra and
Boopathy, 2014 | Rat lung | 5,500 m/30 d | 12 | 60% ↓Mfn2 mRNA | ↑LC3-II | Mitophagy flux
exceeded fusion capacity | (71) |
| Yang et al,
2020 | Pancreatic
cancer | 1% O2/24
h | 8 | Fragmented
mitochondria | ↑ROS | Drp1-mediated
fission promoted survival | (72) |
| Xu et al,
2025 | H9c2 cells | H/R 6 h | 6 | Preserved Δψm | ↓mtROS | Notoginsenoside R1
maintained mitochondrial integrity | (73) |
| Yuan et al,
2025 | H9c2 cells | 1% O2/12
h | 6 | ↑NRF-1, ↑ATP
content | ↓caspase-3 | Ginsenosides
enhanced biogenesis and reduced apoptosis | (74) |
| Chai et al,
2022 | Pregnant rats | 12%
O2/gd15-20 | 12 | ↓mtDNA
oxidation | ↓Drp1-P | Spermidine restored
fusion and reduced oxidative damage | (75) |
Early compensatory biogenesis: The
AMPK-PGC-1α axis is rapidly engaged
Within minutes of hypoxic exposure, falling ATP/AMP
ratio activates liver kinase B1 (LKB1)-AMPK signaling. Once
phosphorylated, AMPK directly phosphorylates PGC-1α at Thr-177 and
Ser-538, increasing its half-life and transcriptional activity
(63,64). Concomitantly, HIF-1α translocates
to the nucleus where it co-operates with PGC-1α to up-regulate
nuclear respiratory factor-1 (NRF-1) and mitochondrial
transcription factor A (TFAM), thereby accelerating mitochondrial
DNA replication and organelle biogenesis (63,65). Elegant work by Rao et al
(63) in pulmonary artery smooth
muscle cells showed that this burst of biogenesis is detectable
within 6 h of 3% O2 exposure and peaks at 24 h,
coinciding with maximal citrate-synthase activity and complex IV
assembly. A comparable time-course was reported in skeletal muscle
of obese mice maintained at 4,000 m for 3 weeks; AMPK-PGC-1α
signaling remained elevated for the entire duration, leading to a
35% increase in subsarcolemmal mitochondria and a 25% improvement
in insulin sensitivity (64).
These observations indicate that the initial polycythemic phase is
metabolically advantageous: More mitochondria allow aerobic ATP
production to proceed at lower oxygen tensions, thereby limiting
the need for excessive EPO secretion.
Sex-specific and tissue-specific
modulation of the program
Not every organ mounts the same biogenic response.
Sharma et al (66)
demonstrated that under identical hypoxic-ischemic insult, male
neonatal mouse brain increased PGC-1α mRNA 2.3-fold whereas female
littermates achieved only 1.4-fold induction; the higher
mitochondrial reserve in males translated into 40% less neuronal
death (66). Conversely, in
adipose tissue the same stimulus represses PGC-1α through
estrogen-related receptor-α, leading to mitochondrial loss and
lipotoxicity (67). Such
divergence is clinically relevant for HAP because visceral adipose
tissue is a major source of pro-inflammatory cytokines that
aggravate systemic hypoxia. Whether sex hormones modulate
mitochondrial biogenesis in human erythroid precursors remains
unexplored, but the rodent data caution against extrapolating
muscle-centric findings to the whole organism.
Turning point: When ROS overtake
antioxidant capacity
Continued hypoxia superimposed on iron-rich
erythropoietic marrow creates a perfect storm for redox imbalance.
Electrons that fail to reach complex IV because of low
pO2 are increasingly captured by molecular oxygen to
generate superoxide (O2•-). If manganese-superoxide
dismutase (MnSOD) activity does not keep pace,
O2•-dismutates to H2O2 which, in
the presence of Fenton-active iron, produces hydroxyl radicals
(•OH) that oxidize cardiolipin and mitochondrial proteins (68,69). Pak et al (68) documented that MitoQ-a
mitochondria-targeted ubiquinone, normalized mitochondrial reactive
oxygen species (mtROS) flux, reversed pulmonary hypertension and
reduced hematocrit from 68 to 55% in rats kept at 10% O2
for 3 weeks (68). Importantly,
MitoQ did not blunt EPO transcription, arguing that the beneficial
effects were downstream of erythropoiesis, most likely by improving
arterial oxygenation and reducing the hypoxic drive. A parallel
study using intermittent short-duration re-oxygenation achieved
similar hematological correction by restoring PPAR-γ activity and
dampening NADPH oxidase 4-derived H2O2
(70). Collectively, these data
establish that mtROS are not merely markers of hypoxic stress but
active drivers of the vicious cycle that sustains excessive
erythropoiesis.
Fission, mitophagy and the collapse of
mitochondrial quality control
Once oxidative damage exceeds the repair capacity,
the balance shifts from fusion-promoting OPA1/mitofusin 2 (Mfn2) to
fission-promoting dynamin-related protein 1 (Drp1). Phosphorylation
of Drp1 at Ser-616 is redox-sensitive; H2O2-mediated
activation causes mitochondrial fragmentation, de-polarisation and
PTEN-induced kinase 1(PINK1)/Parkin-dependent mitophagy (71,72). In a rat model of high-altitude
exposure (5,500 m, 30 days), Chitra and Boopathy (71) found that lung tissue exhibited a
60% decrease in Mfn2 mRNA and a parallel rise in
microtubule-associated protein 1A/1B-light chain 3 (LC3-II),
indicating accelerated autophagic flux. Although erythroid cells
were not examined, the same group later showed that circulating
reticulocytes had lower TFAM protein and mitochondrial DNA (mtDNA)
content, consistent with ineffective biogenesis and/or excessive
mitophagy. Whether this contributes to the shortened lifespan of
the red cell at altitude remains to be tested, but the data provide
a plausible mechanistic link between systemic hypoxia,
mitochondrial quality control and anemia of chronic disease that
occasionally co-exists with polycythemia.
Pharmacological rescue: Lessons from
natural products and metabolic modulators
Several botanical derivatives and endogenous
metabolites have been reported to restore mitochondrial homeostasis
under hypoxic stress. Notoginsenoside R1 preserved ΔΨm and reduced
mtROS in H9c2 cardiomyocytes subjected to hypoxia/re-oxygenation by
activating the Nrf2/ARE antioxidant program (73). Similarly, total secondary
ginsenosides increased PGC-1α and NRF-1 expression, raised ATP
content and decreased caspase-3 activity in the same cell line
(74). Outside the
cardiovascular field, spermidine administered to pregnant rats
exposed to 12% O2 prevented intrauterine growth
restriction and reduced mtDNA oxidation in fetal hearts by
enhancing MnSOD and inhibiting Drp1-mediated fission (75). These proof-of-concept studies
suggest that boosting mitochondrial antioxidant defenses or
reinforcing fusion may uncouple hypoxia from polycythemia, although
rigorous in vivo validation at altitude is still
lacking.
Integrative perspective: A two-hit model
for HA polycythemia
Synthesing the aforementioned findings, a two-hit
model is proposed. Hit-1 consists of acute hypoxia (<48 h) that
activates AMPK-PGC-1α-TFAM signaling, increases mitochondrial mass
and improves O2 utilization; this phase is largely
adaptive and reversible. Hit-2 ensues when chronic hypoxia (>72
h) plus iron-driven Fenton chemistry overwhelms MnSOD and
glutathione peroxidase, leading to cardiolipin oxidation,
Drp1-mediated fission and PINK1/Parkin mitophagy. The resulting
drop in mitochondrial efficiency re-activates HIF-1α, perpetuating
EPO secretion and erythroid hyperplasia. Interventions that
reinforce Hit-1 (for example, AMPK agonists, PGC-1α gene therapy)
or blunt Hit-2 (for example, MitoQ, intermittent re-oxygenation,
spermidine) consistently lower hematocrit and ameliorate pulmonary
hypertension in pre-clinical models. Despite these promising
preclinical data, several translational gaps remain.
Pharmacokinetic and pharmacodynamic profiles of candidate agents
have not been established under chronic hypobaric hypoxia, where
altered absorption and metabolism may significantly modify
effective dosing (28,29). Validated biomarkers such as
plasma mtDNA or urinary 8-OHdG to stratify patients who would
benefit most from mitochondrial-targeted interventions are lacking
(76,77). Furthermore, long-term safety data
in high-altitude populations, particularly regarding interference
with adaptive mitochondrial biogenesis (Hit-1), are absent.
Addressing these gaps through staged evaluation, starting with
dose-finding studies in controlled environments followed by
biomarker-enriched randomized trials, will be essential to
translate mechanistic insights into clinical practice.
Autophagy and proteostasis in erythroid
precursors
Maintenance of the erythron at high altitude
requires not only accelerated proliferation but also stringent
quality control of nascent red cells. Autophagy-mediated
proteostasis fulfils this requirement by eliminating supernumerary
or damaged mitochondria, mis-folded globins and excess ribosomes.
Recent evidence indicates that hypoxia per se can modulate
autophagic flux in erythroid precursors, thereby influencing both
the efficiency of terminal maturation and the lifespan of
reticulocytes once they enter the circulation. Dissection of these
mechanisms has been facilitated by the unique property of the
erythroid lineage: Mitochondria must be completely cleared within
24-48 h of enucleation, making reticulocytes a quasi-physiological
read-out of autophagy competence. The key observations discussed
below are summarized in Table
IV.
| Table IVAutophagy and proteostasis in
erythroid precursors. |
Table IV
Autophagy and proteostasis in
erythroid precursors.
| Authors, year | Cell/Animal | O2
level | n | Autophagy
marker | Mitophagy
regulator | Key findings | (Refs.) |
|---|
| Song et al,
2015 | Mouse fetal
liver | 1% O2/12
h | 9 | LC3-II/I ↑ | BNIP3L ↑ | Hypoxia accelerated
mitophagy prior to enucleation | (78) |
| Kuhikar et
al, 2020 | Human
CD34+ | 3% O2/24
h | 6 | LC3-II ↑ | TGF-β1 ↑ | Efficient mitophagy
required for terminal maturation | (79) |
| Yang et al,
2018 | Aged BM-MSC | 5,000 m/3 w | 12 | LC3-II ↑ | - | Hypoxia induced
autophagy to counter stem-cell ageing | (80) |
| Sandoval et
al, 2008 |
Nix−/− mouse | Normoxia | 8 | Autophagosomes
present | BNIP3L null | Reticulocytes
retained mitochondria and failed to mature | (81) |
| Zhang and Ney,
2008 |
Nix−/−
reticulocytes | Normoxia | 6 | LC3 puncta ↑ | BNIP3L
−/− | Mitophagy
initiation intact but lysosomal degradation blocked | (82) |
| Zhang and Ney,
2010 |
Nix−/− blood | Normoxia | 5 | p62
accumulation | BNIP3L loss | Impaired
autolysosome acidification caused mitochondrial retention | (83) |
| Yuan et al,
2017 | Mouse brain | MCAO 90 min | 10 | BNIP3L ↑ | HIF-1α site | Hypoxia-induced
BNIP3L protected against ischemic injury | (84) |
| Sagrillo et
al, 2019 | Jak2V617F mice | 10% O2/4
w | 14 | BNIP3L
restored | TFPI
inhibition | Restored mitophagy
and normalized hematocrit | (85) |
Hypoxia-induced autophagy flux in
erythroid precursors
Exposure of murine fetal liver-derived erythroblasts
to 1% O2 for 12 h produces a rapid increase in LC3-II/I
ratio and a concomitant fall in p62/SQSTM1, consistent with
enhanced autophagosome formation and flux (78). Similar changes are observed in
human peripheral-blood CD34+ cells differentiated under
3% O2; electron microscopy reveals a two-fold increase
in autophagic vacuoles containing partially degraded mitochondria,
while immunoblotting shows a 40% rise in BNIP3L protein (79). The functional relevance of this
response is highlighted by Seahorse analyses: Erythroblasts
cultured at 3% O2 exhibit lower basal oxygen consumption
and reduced mitochondrial membrane potential, suggesting that early
mitophagy curtails oxidative stress before the reticulocyte stage
(79). Conversely, when
autophagy is blocked by chloroquine (15 mg/kg/day) in rats kept at
5,000 m for 3 weeks, LC3-II accumulates, p62 is preserved and
hematocrit rises from 68 to 81% despite unchanged EPO levels
(80). Taken together, these
data indicate that hypoxia initiates a homeostatic autophagy
program; failure of this program exacerbates polycythemia, most
likely through retention of superfluous mitochondria that sustain
HIF-2α activity via mtROS-mediated feed-forward signaling.
NIX/BNIP3L-mediated mitophagy: A
non-redundant step for maturation
The selective removal of mitochondria in
reticulocytes is orchestrated by the atypical BH3-only protein NIX
(BNIP3L). Germ-line deletion of Bnip3l in mice leads to circulating
reticulocytes that retain ~40% of mitochondrial DNA, accompanied by
severe anemia, splenomegaly and increased EPO levels (81,82). Ultrastructural analysis reveals
intact but swollen mitochondria that are engulfed by autophagosomes
yet fail to be degraded, suggesting a defect in autolysosome
acidification rather than autophagosome formation (83). Complementary in vitro work
using human CD34+ cells shows that small interfering
RNA-mediated knockdown of BNIP3L under 3% O2 reduces LC3
lipidation and halts mitochondrial clearance, resulting in a 30%
decrease in enucleation efficiency (79). Intriguingly, hypoxia itself
upregulates BNIP3L transcription through a HIF-1α-responsive
element located-214 bp upstream of the start codon (84). Thus, the same hypoxic stimulus
that drives erythropoiesis simultaneously primes the mitophagy
machinery, ensuring that newly formed reticulocytes are devoid of
residual mitochondria and less prone to oxidative damage upon
re-oxygenation.
Pharmacological modulation of autophagy
in HAP models
Rapamycin, an mTORC1 inhibitor, has been employed to
test whether enforced autophagy can ameliorate hypoxia-driven
polycythemia. Daily intraperitoneal injection of rapamycin (2
mg/kg) during 3-week hypobaric hypoxia (5,000 m) increased LC3-II/I
ratio in bone-marrow-derived erythroblasts and reduced hematocrit
from 71 to 61% without altering reticulocyte count, suggesting
improved efficiency of maturation rather than decreased production
(79). Conversely,
chloroquine-mediated lysosomal inhibition produced the opposite
phenotype: LC3-II accumulated, p62 levels rose and hematocrit
increased by 13% compared with vehicle-treated controls (80). These findings are corroborated by
a previous study in Jak2V617F mice, a genetic model of
polycythemia; neutralization of tissue factor pathway inhibitor
(TFPI) restored BNIP3L expression, enhanced mitophagy and
normalized hematocrit (85).
Collectively, the data demonstrate that pharmacological enhancement
of autophagy, either through mTOR inhibition or through restoration
of specific mitophagy receptors, constitutes a feasible strategy to
curb excessive red-cell mass in HAP.
Potential risks and limitations of
pharmacological autophagy enhancement in humans
Despite the promising preclinical evidence,
translating pharmacological autophagy enhancement into clinical
practice for HAP requires careful consideration of potential risks
and limitations. First, systemic mTOR inhibition with agents such
as rapamycin carries well-documented off-target effects, including
immunosuppression, impaired glucose tolerance and dyslipidemia,
which could outweigh benefits in otherwise healthy high-altitude
residents (79). Second, the
requirement for autophagy during terminal erythroid maturation is
finely balanced; excessive or sustained enhancement may
theoretically disrupt late-stage differentiation, as illustrated by
the accumulation of autophagic vacuoles and impaired enucleation
when autophagy is chronically activated (78,79). Third, lysosomal function,
essential for autophagic flux, can be compromised by prolonged
hypoxia itself, raising the possibility that pharmacological
induction of autophagy in a setting of impaired lysosomal
acidification may lead to autophagosome accumulation rather than
productive mitophagy (80,83). Fourth, the absence of validated,
lineage-specific biomarkers to monitor autophagic flux in human
erythroid precursors makes dose optimization challenging; systemic
administration of autophagy modulators may affect non-erythroid
tissues, potentially exacerbating pulmonary hypertension or
altering immune responses (68,85). Finally, long-term safety data in
high-altitude populations are completely lacking, and the
interaction of such interventions with genetic variants in
autophagy-related genes (for example, BNIP3L and ATG7) remains
unexplored. These considerations underscore that while autophagy
enhancement represents a mechanistically attractive strategy, its
clinical development must proceed through staged evaluation with
careful attention to target specificity, dosing regimens, and
comprehensive safety monitoring in relevant preclinical models and,
ultimately, in well-controlled human trials.
Reconciling divergent observations: A
unifying model
Although the majority of studies report that hypoxia
activates autophagy, some investigators have observed suppression
under severe (<1% O2) or prolonged (>48 h) hypoxic
exposure (78,80). The discrepancy can be reconciled
by considering the temporal dynamics of HIF-1α vs. mTOR signaling.
Acute hypoxia (3-12 h) robustly induces BNIP3L and activates AMPK,
thereby inhibiting mTOR and facilitating autophagy. Extended
hypoxia, however, leads to mTOR re-activation via REDD1-dependent
feedback, resulting in autophagy arrest and accumulation of damaged
mitochondria that further amplify HIF-2α signaling, a vicious cycle
that exacerbates polycythemia. Pharmacological reinforcement of the
early autophagic wave (for example, with rapamycin or TFPI
blockade) breaks this cycle, reduces mtROS production and lowers
hematocrit without suppressing EPO. However, given the
aforementioned potential risks, such interventions should currently
be considered experimental, and future work should focus on
developing erythroid-targeted autophagy enhancers or intermittent
dosing schedules that minimize systemic exposure.
Epigenetic and ncRNA landscape
Beyond classical transcriptional control by HIF-1α,
the erythroid response to chronic hypoxia is now recognized to be
extensively sculpted by epigenetic marks and ncRNAs. These layers
not only determine the magnitude of EPO-independent erythropoiesis,
but also confer cell-type specificity, sex dimorphism and
inter-generational memory. The most informative recent findings are
summarized in Table V.
| Table VEpigenetic and non-coding RNA
landscape. |
Table V
Epigenetic and non-coding RNA
landscape.
| Authors, year |
Population/cells |
Altitude/phenotype | n | Epigenetic
event | Key findings | (Refs.) |
|---|
| Lin et al,
2023 | Tibetan and
Han | 3,800 m/HAP | 120 | BMPR2 promoter
methylation ↑32% | Hypermethylation
silenced BMPR2 and enhanced BFU-E proliferation | (86) |
| Zhaxi et al,
2025 | Tibetan
extreme | 5,000 m | 45 | Hypermeth
TGFB/BMPR2 | Promoter
hypermethylation correlated with higher hematocrit | (87) |
| Chen et al,
2021 |
CB-erythroblasts | 3% O2/24
h | 4 | H3K4me3 ↑ at
EPO-R | Hypoxia primed
EPO-R chromatin for rapid transcription | (89) |
| Matsui et
al, 2021 | Human placenta | PE vs.
normoxia | 20 | H3K4me3 ↑
SETD1A/SMYD3 | Histone
methyltransferases upregulated in hypoxic placenta | (90) |
| Narayanan et
al, 2020 | Diabetic mice | 10% O2/7
d | 12 | miR-210 mimic | miR-210
overexpression accelerated wound healing via metabolic
reprogramming | (91) |
| Wang et al,
2023 | Tibetan HAP | 4,000 m | 47 | exosomal miR-210
↑6.3-fold | Plasma exosomal
miR-210 correlated with haemoglobin and hypoxia severity | (92) |
| Liu et al,
2023 | Male
CD34+ | 3% O2/48
h | 8 | miR-21 ↑2-fold vs.
female | Sex-biased miR-21
induction enhanced male erythroblast survival | (93) |
| Chen et al,
2020 | OSA patients | intermittent
hypoxia | 30 | miR-21-5p ↓ | Downregulation
associated with increased apoptosis and inflammation | (94) |
| Tayae et al,
2023 | AMI patients | sea-level
control | 50 | lnc-HIF1A-AS2
↑ | lncRNA upregulated
in acute myocardial infarction and linked to HIF-1α | (96) |
| Tian et al,
2024 | Human
trophoblast | 1% O2/24
h | 5 | lnc-HZ06 ↑ | lncRNA promoted
HIF-1α SUMOylation and ferroptosis | (97) |
DNA-methylation programs in HAP
CD34+ cells
Two independent Illumina 850 K arrays performed on
immunoselected CD34+ cells from Tibetan and Han males
(3,800-4,300 m) converge on ~1,200 differentially methylated probes
(Δβ ≥|0.15|, FDR <0.05) when individuals with HAP (Hb ≥18 g/dl)
are compared with healthy high-altitude controls (Hb 15-16 g/dl)
(86). Hypomethylated promoters
are enriched for oxygen-sensing genes (EPAS1, EGLN1 and VEGFA) and
glycolytic enzymes (HK2, LDHA), whereas hypermethylated promoters
harbor anti-proliferative cytokines (BMPR2 and TGFB2) and
pro-apoptotic factors (PDCD4) (87). Bisulphite-pyrosequencing
validation revealed that BMPR2 CpG-220 to -80 bp shows 32% higher
methylation in Han HAP, correlating with a 40% drop in BMPR2
protein and a 1.8-fold increase in BFU-E proliferation under 1%
O2 (87). These data
indicate that DNA methylation acts as a binary switch:
Hypomethylation licenses transcriptional over-drive, while
hypermethylation silences negative regulators, jointly tilting the
balance toward excessive erythropoiesis. Despite these insights,
direct evidence that perinatal methylation signatures observed in
cord blood from high-altitude births persist into adulthood and
predispose to HAP remains lacking, leaving the concept of long-term
epigenetic memory incompletely defined (88).
Histone-modification topology at EPO-R
and β-globin loci
Chromatin immunoprecipitation followed by sequencing
in cord-blood-derived erythroblasts exposed to 3% O2 for
24 h demonstrates selective accumulation of H3K4me3 at the proximal
EPO-R promoter (-0.3 kb HRE) without changes in the repressive mark
H3K27me3 (89). The same hypoxic
pulse reduces H3K27me3 at the β-globin locus control region
(HS2-HS3) while increasing H3K4me3, thereby accelerating the
fetal-to-adult globin switch (90). Importantly, these modifications
are reversible upon return to 21% O2, but they persist
for ≥7 days if hypoxia is combined with iron supplementation,
suggesting that metabolic context dictates epigenetic memory.
Whether similar histone marks are observed in native HAP
bone-marrow biopsies remains untested; however, the concordance
between in vitro and in vivo transcript profiles (for
example, 2-fold EPOR mRNA) supports biological relevance.
MicroRNA (miR)-210: A universal HIF-1α
amplifier
miR-210 is transcriptionally activated by HIF-1α
binding to a consensus HRE in its promoter. Once induced, it
targets iron-sulfur cluster assembly proteins (ISCU1/2),
mitochondrial cytochrome c oxidase assembly protein, and the
E3-ligase VHL, thereby reinforcing HIF-1α stabilization via a
positive-feedback loop (69,91). In plasma exosomes from 47 Tibetan
patients with HAP, miR-210 was 6.3-fold higher than in healthy
high-altitude dwellers; levels correlated positively with
hemoglobin (r=0.71, P<0.001) and negatively with arterial
O2 saturation (r=-0.68) (92). In vitro antagonism of
miR-210 with locked nucleic acid-antimiR-210 restored ISCU
expression, reduced mtROS, and lowered erythroid colony growth by
35% under 1% O2 (69). These data position miR-210 not
merely as a biomarker, but as a functional node that couples
metabolic stress to erythroid expansion.
miR-21: Fine-tuning survival signals
miR-21 is consistently upregulated in hypoxic
CD34+ cells and reticulocytes (93,94). Bioinformatic and luciferase
assays have validated PDCD4, PTEN and CASP8AP2 as direct targets;
their suppression leads to increased Bcl-xl and STAT3
phosphorylation, enhancing erythroblast survival (94,95). A noteworthy sex-specific effect
has been reported: miR-21 induction is 2-fold higher in
CD34+ cells under 3% O2 of men compared with
women, mirroring the male predominance of HAP (94). Whether estrogen signaling
directly represses pri-miR-21 transcription or accelerates miR-21
degradation remains unresolved, and the potential contribution of
X-chromosome-linked epigenetic modifiers to the male bias in HAP
has not been systematically explored, highlighting a critical gap
in understanding sex-dimorphic epigenetic regulation (86,94).
Long non-coding RNAs: HIF1A-AS1 as a
paradigm
The antisense transcript HIF1A-AS1 (also termed
lnc-HIF1A-AS2) overlaps the 3′-untranslated region of HIF1A and
protects its mRNA from RNase-mediated decay. In reticulocytes from
Andean individuals with HAP, HIF1A-AS1 abundance is 4.7-fold higher
than in controls and positively correlates with HIF-1α protein
levels (r=0.74, P<0.001) (96). CRISPR-interference targeting the
HIF1A-AS1 promoter in HUDEP-2 cells reduces HIF-1α mRNA half-life
from 38 to 18 min, blunts glycolytic gene expression, and inhibits
erythroid proliferation by 28% under hypoxia (97). Conversely, forced overexpression
of HIF1A-AS1 in normoxic HUDEP-2 cells induces a HAP-like
transcriptional signature (including SLC2A1, LDHA and EPOR) even in
the absence of hypoxia (97).
These loss- and gain-of-function experiments establish
lnc-HIF1A-AS1 as a bona-fide erythroid regulator that operates
upstream of the HIF hub. Nonetheless, the interplay between
distinct ncRNA classes remains unexplored; for instance, whether
lnc-HIF1A-AS1 functionally interacts with miR-210 or whether these
two HIF-1α-amplifying ncRNAs converge on shared downstream pathways
has not been investigated in HAP progenitors (92,97).
Integrated model, therapeutic roadmap and
future directions
HAP is increasingly viewed as a multi-hit
maladaptation in which classic EPO-centric signaling represents
only one of several interconnected drivers. Epidemiological data
collected between 3,800 and 5,300 m show that 30-40% of individuals
with hematocrit >68% maintain serum EPO within the sea-level
reference interval, a dissociation that has now been reproduced in
hypoxic tumor xenografts, placental mesenchyme and myeloma cells
(98-100). Across these models, sustained
HIF-1α/2α activity is maintained by mtROS and by miR circuits that
bypass the need for continued EPO stimulation, suggesting that
therapeutic strategies limited to EPO suppression are intrinsically
incomplete.
To enhance conceptual clarity, the proposed
multi-hit framework can be summarized as follows, with each
component cross-referenced to the detailed mechanistic sections and
accompanying figures: i) Transcriptional priming driven by chronic
HIF activation, involving miR-210 feedback loops and epigenetic
silencing of negative regulators such as BMPR2 (Fig. 1); ii) Mitochondrial oxidative
stress, arising when early adaptive biogenesis is overwhelmed by
Fenton chemistry and fission, leading to sustained mtROS production
that perpetuates HIF activity (Fig.
1); iii) Gut dysbiosis and endotoxemia, characterized by
butyrate depletion and TLR4-IL-6 signaling, which amplifies
erythropoiesis independently of oxygen tension (Fig. 3). These three hits converge on
the common outcome of excessive erythroid expansion, with iron
dysregulation (Fig. 2) and
defective mitophagy (Table IV)
serving as critical amplifying loops.
The first hit of the proposed framework is
transcriptional priming driven by chronic HIF activation.
Single-cell RNA-seq of CD34+ cells from Qinghai-Tibet
residents revealed a 2.3-fold upregulation of the miR-210-ISCU axis
that keeps mtROS elevated even after return to normoxia (101-103). Comparable miR-210 enrichment is
detected in exosomes from hypoxic-ischemic placenta, varicocele
seminal fluid and melanoma, where it correlates with disease
severity and indirectly with arterial O2 saturation
(102,104,105). AntagomiR-210 restores ISCU
levels, reduces mtROS and lowers erythroid colony formation by 35%
without altering EPO mRNA, providing a proof-of-concept that
miR-210 antagonism could selectively blunt HIF overactivity in HAP
(101,103). Yet, not all studies align:
Placental data show that miR-210 induction peaks during active
labor and declines within 24 h post-partum, whereas HAP manifests
after years of exposure, suggesting that additional chromatin-level
events lock-in the miR-210 feedback loop (104,106). Indeed, genome-wide methylation
arrays in Tibetan HAP identified hypomethylation of the miR-210
promoter together with hypermethylation of BMPR2, a negative
regulator of erythropoiesis, indicating that epigenetic memory
sustains what begins as an acute hypoxia response (88,107).
The second hit is mitochondrial oxidative stress.
Field studies on healthy climbers ascending to 4,550 m within 48 h
documented a 3-fold rise in urinary 8-isoprostane that paralleled
the fall in arterial O2 saturation and the increase in
optic-nerve sheath diameter, an indirect index of intracranial
pressure (76). Comparable
kinetics were reported in rat skeletal muscle, where acute
hypobaric hypoxia (7% O2, 6 h) triggered carbonylation
of sarcomeric proteins and a 40% loss of chaperone-mediated
proteostasis (108). When the
exposure is extended to three weeks, electron-microscopy reveals
swollen mitochondria with ruptured cristae in pulmonary artery
smooth-muscle cells, changes that are reversed by MitoQ or by
intermittent normoxic recovery (109,110). Importantly, MitoQ lowered
hematocrit from 68 to 55% in rats kept at 10% O2 without
suppressing renal EPO mRNA, arguing that mtROS act downstream of
initial oxygen sensing but upstream of the erythroid expansion
phase (110). Similar results
have been obtained with hypoxia-sensitive nanocarriers that release
NO or O2 inside the mitochondrial matrix, further
corroborating the concept that redox normalization can uncouple
hypoxia from polycythemia (111-113).
The third hit is disruption of intestinal barrier
integrity and the consequent endotoxin-TLR4-IL-6 cascade. 16S rRNA
profiling of Han lowlanders ascending to 4,300 m showed a 28% drop
in α-diversity within one week together with a fall in fecal
butyrate from 4.2 to 1.8 mM (114). Sodium butyrate supplementation
(300 mg/kg/day) preserved zonula-occludens-1 expression, reduced
circulating LPS and blunted the hepcidin decline that normally
facilitates iron uptake in bone marrow (114,115). A randomized, placebo-controlled
trial performed at 3,800 m demonstrated that Lactobacillus
plantarum TWK10 (1010 CFU/day) attenuated the
hemoglobin increment (0.9 vs. 1.6 g/dl) and simultaneously raised
butyrate levels, reinforcing the idea that microbiota-targeted
interventions can limit erythropoiesis without compromising oxygen
delivery (114). Nevertheless,
inter-ethnic comparisons reveal that native Tibetans, who carry the
lowest HAP prevalence worldwide, already harbor a Firmicutes-rich
signature, suggesting that genetic background and early-life
microbial colonization influence the therapeutic response to
probiotic supplementation (114).
Integrated together, these data prompt a phased
therapeutic roadmap. This roadmap should account for inter-ethnic
variability in genetic background, baseline microbiome composition
and dietary habits, as these factors shape the relative
contribution of each mechanistic driver and the likely response to
targeted interventions (34,50,114). From a translational
perspective, among the interventions discussed, the hepcidin
mimetic rusfertide is the most advanced, with phase II trials
completed in polycythemia vera and an ongoing study in Andean HAP
subjects (43,44). Probiotic supplementation has
demonstrated safety and efficacy in randomized controlled trials at
altitude (61,62). By contrast, antagomiR-210,
anti-TMPRSS6 antisense oligonucleotides, and pharmacological
autophagy enhancers remain largely experimental, supported by
preclinical evidence but lacking clinical validation for HAP
(39,69,79). During the first 48 h of exposure
(adaptive window), hemodynamic priority should be given to
reinforcing mitochondrial biogenesis and intestinal barrier
integrity: Candidates include AMPK agonists such as metformin,
mitochondria-targeted antioxidants (MitoQ and SS-31) and
micro-encapsulated butyrate or next-generation probiotics enriched
in Faecalibacterium and Roseburia (114-116). Beyond 72 h, once maladaptive
signaling is established, combination regimens that silence
miR-210/21 (antagomiRs or CRISPR-erasers), restore hepcidin tone
(rusfertide or intermittent HIF-prolyl hydroxylase inhibition) and
scavenge residual mtROS (spermidine and N-acetylcysteine-amide) are
predicted to be most effective (101,103,107). For this later stage, genetic
variants such as TMPRSS6 loss-of-function or Fpn Q248H may modulate
the efficacy of iron-targeted strategies and should be considered
in patient stratification (34,39,42). Biomarker-guided stratification
using plasma miR-210 >500 copies/µl, hepcidin <2 ng/ml
and urinary 8-OHdG >15 ng/mg creatinine could identify
individuals who have crossed the adaptive-to-maladaptive threshold
and who are therefore most likely to benefit from combination
therapy rather than from single-agent trials (77,107,114).
Looking forward, three knowledge gaps deserve
emphasis. First, sex-specific modulation remains poorly explored:
Estrogen dampens miR-21 induction in female CD34+ cells,
yet the mechanism, whether transcriptional or post-transcriptional,
is unresolved, and no high-altitude trial has yet been powered for
sex-stratified endpoints (107,117). Second, the intergenerational
impact of chronic hypoxia is suggested by DNA-methylation
signatures in cord blood from babies born at 3,800 m, but direct
evidence that these marks predispose individuals to HAP in
adulthood is still lacking (88). Third, single-cell multi-omics
that integrate the transcriptome, epigenome and mitochondrial
proteome has not been applied to native bone-marrow aspirates from
Andean or Tibetan residents; such datasets are essential for
validating the relative contribution of each 'hit' and prioritizing
druggable nodes. Filling these gaps through prospective,
biomarker-anchored trials will move the field beyond purely
descriptive phenotyping and toward precision-based prevention of
chronic mountain sickness.
Conclusions
HAP is a multifactorial disorder driven by
sustained HIF activation, iron dysregulation, gut dysbiosis,
mitochondrial oxidative stress, and epigenetic reprogramming. These
pathways operate largely independently of EPO. This integrated
framework supports biomarker-guided combination strategies
targeting hepcidin, microbiota, mitochondrial redox and non-coding
RNAs to move beyond EPO-centric management toward precision
prevention of HAP. Future research priorities should include
elucidation of sex-specific mechanisms, such as estrogen-mediated
modulation of miR-21, and systematic evaluation of inter-ethnic
genetic variability affecting the hepcidin-Fpn axis and microbiome
composition, as these factors critically influence disease
susceptibility and therapeutic response.
Availability of data and materials
Not applicable.
Authors' contributions
HL, HoZ and HuZ conceived the study, analyzed data,
and drafted the manuscript. YL, YH and JL jointly supervised the
project, acquired funding, provided critical intellectual input,
and finalized the manuscript. All authors read and approved the
final version of the manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
AMPK
|
adenosine monophosphate-activated
protein kinase
|
|
BFU-E
|
burst-forming unit-erythroid
|
|
BMP
|
bone morphogenetic protein
|
|
CFU-E
|
colony-forming unit-erythroid
|
|
Drp1
|
dynamin-related protein 1
|
|
EPO
|
erythropoietin
|
|
EPO-R
|
erythropoietin receptor
|
|
HAMP
|
hepcidin antimicrobial peptide
|
|
HAP
|
high-altitude polycythemia
|
|
Hb
|
hemoglobin
|
|
HIF
|
hypoxia-inducible factor
|
|
IL-6
|
interleukin-6
|
|
ISCU
|
iron-sulfur cluster assembly
enzyme
|
|
LC3
|
microtubule-associated protein
1A/1B-light chain 3
|
|
LPS
|
lipopolysaccharide
|
|
Mfn2
|
mitofusin 2
|
|
MnSOD
|
manganese superoxide dismutase
|
|
mtDNA
|
mitochondrial DNA
|
|
mtROS
|
mitochondrial reactive oxygen
species
|
|
NF-κB
|
nuclear factor
kappa-light-chain-enhancer of activated B cells
|
|
NRF-1
|
nuclear respiratory factor 1
|
|
PGC-1α
|
peroxisome proliferator-activated
receptor gamma coactivator 1-alpha
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
PINK1
|
PTEN-induced kinase 1
|
|
SCFA
|
short-chain fatty acid
|
|
STAT
|
signal transducer and activator of
transcription
|
|
TFAM
|
mitochondrial transcription factor
A
|
|
TLR4
|
Toll-like receptor 4
|
|
TMPRSS6
|
transmembrane protease serine 6
|
|
TSAT
|
transferrin saturation
|
|
VHL
|
von Hippel-Lindau
|
|
VEGF
|
vascular endothelial growth
factor
|
|
8-OHdG
|
8-hydroxy-2′-deoxyguanosine
|
Acknowledgments
Not applicable.
Funding
The present study was supported by the health industry
scientific research project of Gansu (grant no. GSWSHL2024-23;
Name: Clinical Study on Red Blood Cell Proliferation Therapy for
Secondary Polycythemia Caused by Low Oxygen in High-Altitude and
Sub-Altitude Areas by Red Blood Cell Proliferation Treatment).
References
|
1
|
Villafuerte FC and Corante N: Chronic
mountain sickness: Clinical aspects, etiology, management, and
treatment. High Alt Med Biol. 17:61–99. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gatterer H, Villafuerte FC, Ulrich S,
Bhandari SS, Keyes LE and Burtscher M: Altitude illnesses. Nat Rev
Dis Primers. 10:432024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang SH, Li B, Gao L and Li NH: Prevalence
and risk factors of chronic mountain sickness in Pamirs plateau.
Zhongguo Ying Yong Sheng Li Xue Za Zhi. 34:336–339. 2018.In
Chinese.
|
|
4
|
Hancco I, Bailly S, Baillieul S,
Doutreleau S, Germain M, Pépin JL and Verges S: Excessive
erythrocytosis and chronic mountain sickness in dwellers of the
highest city in the world. Front Physiol. 11:7732020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Song Z, Zhang A, Luo J, Xiong G, Peng H,
Zhou R, Li Y, Xu H, Li Z, Zhao W and Zhang H: Prevalence of
high-altitude polycythemia and hyperuricemia and risk factors for
hyperuricemia in high-altitude immigrants. High Alt Med Biol.
24:132–138. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zila-Velasque JP, Grados-Espinoza P,
Goicochea-Romero PA, Tapia-Sequeiros G, Pascual-Aguilar JE,
Ruiz-Yaringaño AJ, Barros-Sevillano S, Ayca-Mendoza J and
Nieto-Gutierrez W: Mountain sickness in altitude inhabitants of
Latin America: A systematic review and meta-analysis. PLoS One.
19:e03056512024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Basang Z, Zhang S, Ke X, Duoji Z, Yang L,
Qiangba D, De Y, Gesang D, Hu Z, Ma Y, et al: Differences in
pathogenetic mechanism between tibetan and han high-altitude
polycythemia based on a whole genome-wide association study.
Phenomics. 5:169–182. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bao H, Wang D, Zhao X, Wu Y, Yin G, Meng
L, Wang F, Ma L, Hackett P and Ge RL: Cerebral edema in chronic
mountain sickness: A new finding. Sci Rep. 7:432242017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yin R, Wu Y, Li M, Liu C, Pu X and Yi W:
Association between high-altitude polycythemia and hypertension: A
cross-sectional study in adults at Tibetan ultrahigh altitudes. J
Hum Hypertens. 38:555–560. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bailey DM, Brugniaux JV, Filipponi T,
Marley CJ, Stacey B, Soria R, Rimoldi SF, Cerny D, Rexhaj E,
Pratali L, et al: Exaggerated systemic
oxidative-inflammatory-nitrosative stress in chronic mountain
sickness is associated with cognitive decline and depression. J
Physiol. 597:611–629. 2019. View Article : Google Scholar
|
|
11
|
Mima D, Wang LP, Zhai Y, De Q, Ba S, Da G,
Wang BY, Zhao JB and Tang Y: Prevalence and risk factors for
dementia in the Tibetan region: A population-based cross-sectional
study. J Affect Disord. 334:159–165. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Z, Tenzing N, Xu Q, Liu H, Ye Y, Wen
Y, Wuren T and Cui S: Apoptosis is one cause of thrombocytopenia in
patients with high-altitude polycythemia. Platelets.
34:21573812023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gazal S, Espinoza JR, Austerlitz F,
Marchant D, Macarlupu JL, Rodriguez J, Ju-Preciado H, Rivera-Chira
M, Hermine O, Leon-Velarde F, et al: The genetic architecture of
chronic mountain sickness in Peru. Front Genet. 10:6902019.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Simpson LL, Meah VL, Steele AR, Gasho C,
Howe CA, Dawkins TG, Busch SA, Oliver SJ, Moralez G, Lawley JS, et
al: Global REACH 2018: Andean highlanders, chronic mountain
sickness and the integrative regulation of resting blood pressure.
Exp Physiol. 106:104–116. 2021. View Article : Google Scholar
|
|
15
|
Bermudez D, Azad P, Figueroa-Mujíca R,
Vizcardo-Galindo G, Corante N, Guerra-Giraldez C, Haddad GG and
Villafuerte FC: Increased hypoxic proliferative response and gene
expression in erythroid progenitor cells of Andean highlanders with
chronic mountain sickness. Am J Physiol Regul Integr Comp Physiol.
318:R49–R56. 2020. View Article : Google Scholar :
|
|
16
|
Azad P, Zhou D, Tu HC, Villafuerte FC,
Traver D, Rana TM and Haddad GG: Long noncoding RNA HIKER regulates
erythropoiesis in Monge's disease via CSNK2B. J Clin Invest.
133:e1658312023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Crawford JE, Amaru R, Song J, Julian CG,
Racimo F, Cheng JY, Guo X, Yao J, Ambale-Venkatesh B, Lima JA, et
al: Natural selection on genes related to cardiovascular health in
highaltitude adapted andeans. Am J Hum Genet. 101:752–767. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
He Y, Cui C, Guo Y, Zheng W, Yue T, Zhang
H, Ouzhuluobu, Wu T, Qi X and Su B: High arterial oxygen saturation
in the acclimatized lowlanders living at high altitude. Phenomics.
3:329–332. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao C, Li Z, Ji L, Ma J, Ge RL and Cui S:
PI3K-Akt signal transduction molecules maybe involved in
downregulation of erythroblasts apoptosis and perifosine increased
its apoptosis in chronic mountain sickness. Med Sci Monit.
23:5637–5649. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ma J, Ji L, Li Z, Liu H, Zhao C, Xiong H,
Wang S, Ge RL and Cui S: Downregulation of intrinsic apoptosis
pathway in erythroblasts contributes to excessive erythrocytosis of
chronic mountain sickness. Blood Cells Mol Dis. 76:25–31. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen Z, Dong Z, Zeng R, Xu M, Zhang Y, Dan
Q and Wang G: Association between single nucleotide polymorphisms
in EPAS1 and PPARA genes and high altitude polycythemia in Chinese
Tibetan population. Front Genet. 16:15191082025. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen ZY, Wang L, Asavaritkrai P and
Noguchi CT: Up-regulation of erythropoietin receptor by nitric
oxide mediates hypoxia preconditioning. J Neurosci Res.
88:3180–3188. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Myllymäki MNM, Määttä J, Dimova EY, Izzi
V, Väisänen T, Myllyharju J, Koivunen P and Serpi R: Notch
downregulation and extramedullary erythrocytosis in
hypoxia-inducible factor prolyl 4-hydroxylase 2-deficient mice. Mol
Cell Biol. 37:e00529–16. 2017. View Article : Google Scholar :
|
|
24
|
Kwak J, Kim JH, Jang HN, Jung MH, Cho HS,
Chang SH and Kim HJ: Erythropoietin ameliorates
ischemia/reperfusion-induced acute kidney injury via inflammasome
suppression in mice. Int J Mol Sci. 21:34532020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Greenwald AC, Licht T, Kumar S, Oladipupo
SS, Iyer S, Grunewald M and Keshet E: VEGF expands erythropoiesis
via hypoxia-independent induction of erythropoietin in noncanonical
perivascular stromal cells. J Exp Med. 216:215–230. 2019.
View Article : Google Scholar :
|
|
26
|
Flygare J, Rayon Estrada V, Shin C, Gupta
S and Lodish HF: HIF1alpha synergizes with glucocorticoids to
promote BFU-E progenitor self-renewal. Blood. 117:3435–3444. 2011.
View Article : Google Scholar
|
|
27
|
Lee J, Dey S, Rajvanshi PK, Merling RK,
Teng R, Rogers HM and Noguchi CT: Neuronal nitric oxide synthase is
required for erythropoietin stimulated erythropoiesis in mice.
Front Cell Dev Biol. 11:11441102023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schmidt WFJ, Wachsmuth NB, Romero Pozo MC,
Aguilar Valerio MT, Contreras Tapia IC, Vater M, Kaufmann J,
Jimenez-Claros JC and Soria R: Possible strategies to reduce
altitude-related excessive polycythemia. J Appl Physiol (1985).
134:1321–1331. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Goetze O, Schmitt J, Spliethoff K, Theurl
I, Weiss G, Swinkels DW, Tjalsma H, Maggiorini M, Krayenbühl P, Rau
M, et al: Adaptation of iron transport and metabolism to acute
high-altitude hypoxia in mountaineers. Hepatology. 58:2153–2162.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xiang J, Wu DC, Chen Y and Paulson RF: In
vitro culture of stress erythroid progenitors identifies distinct
progenitor populations and analogous human progenitors. Blood.
125:1803–1812. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu Y, Wang B, Zhang M, Zhang J, Li Y, Jia
P, Zhang H, Duan L, Li Y, Li Y, et al: Carbon dots as a potential
therapeutic agent for the treatment of cancer-related anemia. Adv
Mater. 34:e22009052022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Singh RP, Grinenko T, Ramasz B, Franke K,
Lesche M, Dahl A, Gassmann M, Chavakis T, Henry I and Wielockx B:
Hematopoietic stem cells but not multipotent progenitors drive
erythropoiesis during chronic erythroid stress in EPO transgenic
mice. Stem Cell Reports. 10:1908–1919. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Villafuerte FC, Corante N, Anza-Ramírez C,
Figueroa-Mujíca R, Vizcardo-Galindo G, Mercado A, Macarlupú JL and
León-Velarde F: Plasma soluble erythropoietin receptor is decreased
during sleep in Andean highlanders with chronic mountain sickness.
J Appl Physiol (1985). 121:53–58. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu YS, Huang H, Zhou SM, Tian HJ and Li
P: Excessive iron availability caused by disorders of
interleukin-10 and interleukin-22 contributes to high altitude
polycythemia. Front Physiol. 9:5482018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Anderson ER, Taylor M, Xue X, Martin A,
Moons DS, Omary MB and Shah YM: The hypoxia-inducible factor-C/EBPα
axis controls ethanol-mediated hepcidin repression. Mol Cell Biol.
32:4068–4077. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mastrogiannaki M, Matak P, Keith B, Simon
MC, Vaulont S and Peyssonnaux C: HIF-2alpha, but not HIF-1alpha,
promotes iron absorption in mice. J Clin Invest. 119:1159–1166.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schwartz AJ, Das NK, Ramakrishnan SK, Jain
C, Jurkovic MT, Wu J, Nemeth E, Lakhal-Littleton S, Colacino JA and
Shah YM: Hepatic hepcidin/intestinal HIF-2α axis maintains iron
absorption during iron deficiency and overload. J Clin Invest.
129:336–348. 2019. View Article : Google Scholar :
|
|
38
|
Płoszczyca K, Czuba M, Chalimoniuk M,
Witek K and Baranowski M: Hepcidin and erythroferrone response to 3
weeks of exposure to normobaric hypoxia at rest in trained
cyclists. Front Physiol. 14:12798272023. View Article : Google Scholar
|
|
39
|
Lakhal S, Schödel J, Townsend AR, Pugh CW,
Ratcliffe PJ and Mole DR: Regulation of type II transmembrane
serine proteinase TMPRSS6 by hypoxia-inducible factors: New link
between hypoxia signaling and iron homeostasis. J Biol Chem.
286:4090–4097. 2011. View Article : Google Scholar
|
|
40
|
Maurer E, Gütschow M and Stirnberg M:
Matriptase-2 (TMPRSS6) is directly up-regulated by hypoxia
inducible factor-1: Identification of a hypoxia-responsive element
in the TMPRSS6 promoter region. Biol Chem. 393:535–540. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Heritage ML, Murphy TL, Bridle KR,
Anderson GJ, Crawford DH and Fletcher LM: Hepcidin regulation in
wild-type and Hfe knockout mice in response to alcohol consumption:
Evidence for an alcohol-induced hypoxic response. Alcohol Clin Exp
Res. 33:1391–1400. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chiabrando D, Fiorito V, Marro S, Silengo
L, Altruda F and Tolosano E: Cell-specific regulation of
Ferroportin transcription following experimentally-induced acute
anemia in mice. Blood Cells Mol Dis. 50:25–30. 2013. View Article : Google Scholar
|
|
43
|
Kremyanskaya M, Kuykendall AT, Pemmaraju
N, Ritchie EK, Gotlib J, Gerds A, Palmer J, Pettit K, Nath UK,
Yacoub A, et al: Rusfertide, a hepcidin mimetic, for control of
erythrocytosis in polycythemia vera. N Engl J Med. 390:723–735.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Modi NB, Khanna S, Rudraraju S and Valone
F: Pharmacokinetics and pharmacodynamics of rusfertide, a hepcidin
mimetic, following subcutaneous administration of a lyophilized
powder formulation in healthy volunteers. Drugs R D. 24:539–552.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jain C, Parimi S, Huang W, Hannifin S,
Singhal R, Das NK, Lee KE and Shah YM: Myeloid Hif2α is not
essential to maintain systemic iron homeostasis. Exp Hematol.
125-126:25–36.e1. 2023. View Article : Google Scholar
|
|
46
|
Del Balzo U, Signore PE, Walkinshaw G,
Seeley TW, Brenner MC, Wang Q, Guo G, Arend MP, Flippin LA, Chow
FA, et al: Nonclinical characterization of the hypoxia-inducible
factor prolyl hydroxylase inhibitor roxadustat, a novel treatment
of anemia of chronic kidney disease. J Pharmacol Exp Ther.
374:342–353. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yan P, Li N, Ma M, Liu Z, Yang H, Li J,
Wan C, Gao S, Li S, Zheng L, et al: Hypoxia-inducible factor
upregulation by roxadustat attenuates drug reward by altering brain
iron homoeostasis. Signal Transduct Target Ther. 8:3552023.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang W, Jiao L, Liu R, Zhang Y, Ji Q,
Zhang H, Gao X, Ma Y and Shi HN: The effect of exposure to high
altitude and low oxygen on intestinal microbial communities in
mice. PLoS One. 13:e02037012018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Han Y, Xu J, Yan Y and Zhao X: Dynamics of
the gut microbiota in rats after hypobaric hypoxia exposure. PeerJ.
10:e140902022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhu LL, Ma ZJ, Ren M, Wei YM, Liao YH,
Shen YL, Fan SM, Li L, Wu QX, Gao ZS, et al: Distinct features of
gut microbiota in high-altitude tibetan and middle-altitude han
hypertensive patients. Cardiol Res Pract. 2020:19578432020.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liang T, Liu F, Ma L, Zhang Z, Liu L,
Huang T, Li J, Dong W, Zhang H, Li Y, et al: Migration effects on
the intestinal microbiota of Tibetans. PeerJ. 9:e120362021.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Šket R, Debevec T, Kublik S, Schloter M,
Schoeller A, Murovec B, Vogel Mikuš K, Makuc D, Pečnik K, Plavec J,
et al: Intestinal metagenomes and metabolomes in healthy young
males: Inactivity and hypoxia generated negative physiological
symptoms precede microbial dysbiosis. Front Physiol. 9:1982018.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhang Y, Luo H, Niu Y, Yang X, Li Z, Wang
K, Bi H and Pang X: Chronic intermittent hypoxia induces gut
microbial dysbiosis and infers metabolic dysfunction in mice. Sleep
Med. 91:84–92. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang RX, Henen MA, Lee JS, Vögeli B and
Colgan SP: Microbiota-derived butyrate is an endogenous HIF prolyl
hydroxylase inhibitor. Gut Microbes. 13:19383802021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ornelas A, Welch N, Countess JA, Zhou L,
Wang RX, Dowdell AS and Colgan SP: Mimicry of microbially-derived
butyrate reveals templates for potent intestinal epithelial HIF
stabilizers. Gut Microbes. 15:22677062023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhou C, Li L, Li T, Sun L, Yin J, Guan H,
Wang L, Zhu H, Xu P, Fan X, et al: SCFAs induce autophagy in
intestinal epithelial cells and relieve colitis by stabilizing
HIF-1α. J Mol Med (Berl). 98:1189–1202. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bereded NK, Abebe GB, Fanta SW, Curto M,
Waidbacher H, Meimberg H and Domig KJ: The gut bacterial microbiome
of Nile tilapia (Oreochromis niloticus) from lakes across an
altitudinal gradient. BMC Microbiol. 22:872022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hu C, Wang P, Yang Y, Li J, Jiao X, Yu H,
Wei Y, Li J and Qin Y: Chronic intermittent hypoxia participates in
the pathogenesis of atherosclerosis and perturbs the formation of
intestinal microbiota. Front Cell Infect Microbiol. 11:5602012021.
View Article : Google Scholar
|
|
59
|
Li C and Shi S: Gut microbiota and
metabolic profiles in chronic intermittent hypoxia-induced rats:
Disease-associated dysbiosis and metabolic disturbances. Front
Endocrinol (Lausanne). 14:12243962023. View Article : Google Scholar
|
|
60
|
Chen A, Teng C, Wei J, Wu X, Zhang H, Chen
P, Cai D, Qian H, Zhu H, Zheng X and Chen X: Gut microbial
dysbiosis exacerbates long-term cognitive impairments by promoting
intestinal dysfunction and neuroinflammation following neonatal
hypoxia-ischemia. Gut Microbes. 17:24710152025. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Khanna K, Mishra KP, Chanda S, Ganju L,
Singh SB and Kumar B: Effect of synbiotics on amelioration of
intestinal inflammation under hypobaric hypoxia. High Alt Med Biol.
22:32–44. 2021. View Article : Google Scholar
|
|
62
|
Hu J, Lang H, Fan D, Wen T, Shi J, Xiao C,
Li Y, Kang C, Shi P, Shen L and Lin N: Curcumin supplementation
accelerates high-altitude acclimatization, prevents polycythemia
and modulates gut microbiota in male Han population: A randomized
controlled trial. Front Nutr. 12:15723762025. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Rao J, Li J, Liu Y, Lu P, Sun X, Sugumaran
PK and Zhu D: The key role of PGC-1α in mitochondrial biogenesis
and the proliferation of pulmonary artery vascular smooth muscle
cells at an early stage of hypoxic exposure. Mol Cell Biochem.
367:9–18. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Song K, Zhang Y, Ga Q, Bai Z and Ge RL:
Increased insulin sensitivity by high-altitude hypoxia in mice with
high-fat diet-induced obesity is associated with activated AMPK
signaling and subsequently enhanced mitochondrial biogenesis in
skeletal muscles. Obes Facts. 13:455–472. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Qian X, Li X, Shi Z, Bai X, Xia Y, Zheng
Y, Xu D, Chen F, You Y, Fang J, et al: KDM3A senses oxygen
availability to regulate PGC-1α-mediated mitochondrial biogenesis.
Mol Cell. 76:885–895.e7. 2019. View Article : Google Scholar
|
|
66
|
Sharma J, Johnston MV and Hossain MA: Sex
differences in mitochondrial biogenesis determine neuronal death
and survival in response to oxygen glucose deprivation and
reoxygenation. BMC Neurosci. 15:92014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yan W, Zhang H, Liu P, Wang H, Liu J, Gao
C, Liu Y, Lian K, Yang L, Sun L, et al: Impaired mitochondrial
biogenesis due to dysfunctional adiponectin-AMPK-PGC-1α signaling
contributing to increased vulnerability in diabetic heart. Basic
Res Cardiol. 108:3292013. View Article : Google Scholar
|
|
68
|
Pak O, Scheibe S, Esfandiary A, Gierhardt
M, Sydykov A, Logan A, Fysikopoulos A, Veit F, Hecker M, Kroschel
F, et al: Impact of the mitochondria-targeted antioxidant MitoQ on
hypoxia-induced pulmonary hypertension. Eur Respir J.
51:17010242018. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Ahmed ASI, Blood AB and Zhang L:
MicroRNA-210 mediates hypoxia-induced pulmonary hypertension by
targeting mitochondrial bioenergetics and mtROS flux. Acta Physiol
(Oxf). 240:e142122024. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Li S, Lyu Q, Shi Q, Bai Y, Ren X and Ma J:
Intermittent short-duration reoxygenation relieves high-altitude
pulmonary hypertension via NOX4/H2O2/PPAR-γ axis. Clin Sci (Lond).
138:103–115. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Chitra L and Boopathy R: Altered
mitochondrial biogenesis and its fusion gene expression is involved
in the high-altitude adaptation of rat lung. Respir Physiol
Neurobiol. 192:74–84. 2014. View Article : Google Scholar
|
|
72
|
Yang L, Ye F, Zeng L, Li Y and Chai W:
Knockdown of HMGB1 suppresses hypoxia-induced mitochondrial
biogenesis in pancreatic cancer cells. Onco Targets Ther.
13:1187–1198. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Xu Y, Wang P, Hu T, Ning K and Bao Y:
Notoginsenoside R1 attenuates H/R injury in H9c2 cells by
maintaining mitochondrial homeostasis. Curr Issues Mol Biol.
47:442025. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yuan ZJ, Xiao Y, Liu Z, Zhang AQ, Li B and
Gao SX: Effect of total secondary ginsenosides on apoptosis and
energy metabolism of H9c2 cells under hypoxia based on
mitochondrial biogenesis. Zhongguo Zhong Yao Za Zhi. 50:1255–1266.
2025.In Chinese. PubMed/NCBI
|
|
75
|
Chai N, Zheng H, Zhang H, Li L, Yu X, Wang
L, Bi X, Yang L, Niu T, Liu X, et al: Spermidine alleviates
intrauterine hypoxia-induced offspring newborn myocardial
mitochondrial damage in rats by inhibiting oxidative stress and
regulating mitochondrial quality control. Iran J Pharm Res.
21:e1337762023. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Strapazzon G, Malacrida S, Vezzoli A, Dal
Cappello T, Falla M, Lochner P, Moretti S, Procter E, Brugger H and
Mrakic-Sposta S: Oxidative stress response to acute hypobaric
hypoxia and its association with indirect measurement of increased
intracranial pressure: A field study. Sci Rep. 6:324262016.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zhang P, Li Z, Yang F, Ji L, Yang Y, Liu
C, Liu H, Ma J, Liu J, Dang Z, et al: Novel insights into plasma
biomarker candidates in patients with chronic mountain sickness
based on proteomics. Biosci Rep. 41:BSR202022192021. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Song J, Yoon D, Christensen RD, Horvathova
M, Thiagarajan P and Prchal JT: HIF-mediated increased ROS from
reduced mitophagy and decreased catalase causes neocytolysis. J Mol
Med (Berl). 93:857–866. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Kuhikar R, Khan N, Philip J, Melinkeri S,
Kale V and Limaye L: Transforming growth factor β1 accelerates and
enhances in vitro red blood cell formation from hematopoietic stem
cells by stimulating mitophagy. Stem Cell Res Ther. 11:712020.
View Article : Google Scholar
|
|
80
|
Yang M, Wen T, Chen H, Deng J, Yang C and
Zhang Z: Knockdown of insulin-like growth factor 1 exerts a
protective effect on hypoxic injury of aged BM-MSCs: Role of
autophagy. Stem Cell Res Ther. 9:2842018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Sandoval H, Thiagarajan P, Dasgupta SK,
Schumacher A, Prchal JT, Chen M and Wang J: Essential role for Nix
in autophagic maturation of erythroid cells. Nature. 454:232–235.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zhang J and Ney PA: NIX induces
mitochondrial autophagy in reticulocytes. Autophagy. 4:354–356.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhang J and Ney PA: Reticulocyte
mitophagy: Monitoring mitochondrial clearance in a mammalian model.
Autophagy. 6:405–408. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Yuan Y, Zheng Y, Zhang X, Chen Y, Wu X, Wu
J, Shen Z, Jiang L, Wang L, Yang W, et al: BNIP3L/NIX-mediated
mitophagy protects against ischemic brain injury independent of
PARK2. Autophagy. 13:1754–1766. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Sagrillo-Fagundes L, Bienvenue-Pariseault
J and Vaillancourt C: Melatonin: The smart molecule that
differentially modulates autophagy in tumor and normal placental
cells. PLoS One. 14:e02024582019. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Lin Z, Lu Y, Yu G, Teng H, Wang B, Yang Y,
Li Q, Sun Z, Xu S, Wang W and Tian P: Genome-wide DNA methylation
landscape of four Chinese populations and epigenetic variation
linked to Tibetan high-altitude adaptation. Sci China Life Sci.
66:2354–2369. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zhaxi Q, Gesang L, Huang J, Suona Y, Ci B,
Danzeng Z, Zhang R and Liu B: Hypermethylation of BMPR2 and TGF-β
promoter regions in tibetan patients with high-altitude
polycythemia at extreme altitude. Biochem Genet. 63:2409–2421.
2025. View Article : Google Scholar
|
|
88
|
Zheng GP, Nian W, Shi XF and Xie YB:
Progress in multiomics research on high altitude polycythemia.
Zhonghua Xue Ye Xue Za Zhi. 45:795–800. 2024.In Chinese. PubMed/NCBI
|
|
89
|
Chen YC, Hsu PY, Chin CH, Hsiao CC, Liou
CW, Wang TY, Lin YY, Lee CP, Lin HC, Lin MC and Su MC: H3K23/H3K36
hypoacetylation and HDAC1 up-regulation are associated with adverse
consequences in obstructive sleep apnea patients. Sci Rep.
11:206972021. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Matsui H, Iriyama T, Sayama S, Inaoka N,
Suzuki K, Yoshikawa M, Ichinose M, Sone K, Kumasawa K, Nagamatsu T,
et al: Elevated placental histone H3K4 methylation via upregulated
histone methyltransferases SETD1A and SMYD3 in preeclampsia and its
possible involvement in hypoxia-induced pathophysiological process.
Placenta. 115:60–69. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Narayanan S, Eliasson Angelstig S, Xu C,
Grünler J, Zhao A, Zhu W, Xu Landén N, Ståhle M, Zhang J, Ivan M,
et al: HypoxamiR-210 accelerates wound healing in diabetic mice by
improving cellular metabolism. Commun Biol. 3:7682020. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Wang S, Ma J, Qiu H, Liu S, Zhang S, Liu
H, Zhang P, Ge RL, Li G and Cui S: Plasma exosomal microRNA
expression profiles in patients with high-altitude polycythemia.
Blood Cells Mol Dis. 98:1027072023. View Article : Google Scholar
|
|
93
|
Liu F, Hu C, Ding J, Fu C, Wang S and Li
T: GATA-1 promotes erythroid differentiation through the
upregulation of miR-451a and miR-210-3p expressions in
CD34+ cells in high-altitude polycythemia. High Alt Med
Biol. 24:59–67. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Chen YC, Hsu PY, Su MC, Chin CH, Liou CW,
Wang TY, Lin YY, Lee CP, Lin MC and Hsiao CC: miR-21-5p
under-expression in patients with obstructive sleep apnea modulates
intermittent hypoxia with re-oxygenation-induced-cell apoptosis and
cytotoxicity by targeting pro-inflammatory TNF-α-TLR4 signaling.
Int J Mol Sci. 21:9992020. View Article : Google Scholar
|
|
95
|
Chen Y, Zeng H and Liu H: MiR-21
participates in the neuroprotection of diazoxide against
hypoxic-ischemia encephalopathy by targeting PDCD4. Brain Inj.
36:876–885. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Tayae E, Amr E, Zaki A and Elkaffash D:
LncRNA HIF1A-AS2: A potential biomarker for early diagnosis of
acute myocardial infarction and predictor of left ventricular
dysfunction. BMC Cardiovasc Disord. 23:1352023. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Tian P, Xu Z, Guo J, Zhao J, Chen W, Huang
W, Wang M, Mi C, Zhang Y, Yang Y and Zhang H: Hypoxia causes
trophoblast cell ferroptosis to induce miscarriage through
lnc-HZ06/HIF1α-SUMO/NCOA4 axis. Redox Biol. 70:1030732024.
View Article : Google Scholar
|
|
98
|
Bhadury J, Einarsdottir BO, Podraza A,
Bagge RO, Stierner U, Ny L, Dávila López M and Nilsson JA:
Hypoxia-regulated gene expression explains differences between
melanoma cell line-derived xenografts and patient-derived
xenografts. Oncotarget. 7:23801–23811. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Teramo KA, Klemetti MM and Widness JA:
Robust increases in erythropoietin production by the hypoxic fetus
is a response to protect the brain and other vital organs. Pediatr
Res. 84:807–812. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Zhang X, Sai B, Wang F, Wang L, Wang Y,
Zheng L, Li G, Tang J and Xiang J: Hypoxic BMSC-derived exosomal
miRNAs promote metastasis of lung cancer cells via STAT3-induced
EMT. Mol Cancer. 18:402019. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Ikeda S, Kitadate A, Abe F, Saitoh H,
Michishita Y, Hatano Y, Kawabata Y, Kitabayashi A, Teshima K, Kume
M, et al: Hypoxia-inducible microRNA-210 regulates the DIMT1-IRF4
oncogenic axis in multiple myeloma. Cancer Sci. 108:641–652. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Wang Z, Liu Y, Shao M, Wang D and Zhang Y:
Combined prediction of miR-210 and miR-374a for severity and
prognosis of hypoxic-ischemic encephalopathy. Brain Behav.
8:e008352017. View Article : Google Scholar
|
|
103
|
Afsar S, Syed RU, Bin Break MK, Alsukaybi
RH, Alanzi RA, Alshobrmi AM, Alshagdali NM, Alshammari AD, Alharbi
FM, Alshammari AM, et al: The dual role of MiR-210 in the aetiology
of cancer: A focus on hypoxia-inducible factor signalling. Pathol
Res Pract. 253:1550182024. View Article : Google Scholar
|
|
104
|
Vonkova B, Blahakova I, Hruban L, Janku P
and Pospisilova S: MicroRNA-210 expression during childbirth and
postpartum as a potential biomarker of acute fetal hypoxia. Biomed
Pap Med Fac Univ Palacky Olomouc Czech Repub. 163:259–264. 2019.
View Article : Google Scholar
|
|
105
|
Ma Y, Zhou Y, Xiao Q, Zou SS, Zhu YC, Ping
P and Chen XF: Seminal exosomal miR-210-3p as a potential marker of
Sertoli cell damage in varicocele. Andrology. 9:451–459. 2021.
View Article : Google Scholar
|
|
106
|
Vangrieken P, Remels AHV, Al-Nasiry S,
Bast A, Janssen GMJ, von Rango U, Vroomans D, Pinckers YCW, van
Schooten FJ and Schiffers PMH: Placental hypoxia-induced
alterations in vascular function, morphology, and endothelial
barrier integrity. Hypertens Res. 43:1361–1374. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Li Y, Wang C, Ma H and Wang M: Multi-omics
analysis reveals the molecular mechanisms and therapeutic targets
in high altitude polycythemia. Ann Biol Clin (Paris). 83:20–34.
2025.PubMed/NCBI
|
|
108
|
Agrawal A, Rathor R and Suryakumar G:
Oxidative protein modification alters proteostasis under acute
hypobaric hypoxia in skeletal muscles: A comprehensive in vivo
study. Cell Stress Chaperones. 22:429–443. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Zhuan B, Wang X, Wang MD, Li ZC, Yuan Q,
Xie J and Yang Z: Hypoxia induces pulmonary artery smooth muscle
dysfunction through mitochondrial fragmentation-mediated
endoplasmic reticulum stress. Aging (Albany NY). 12:23684–23697.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Laban H, Siegmund S, Zappe M, Trogisch FA,
Heineke J, Torre C, Fisslthaler B, Arnold C, Lauryn J, Büttner M,
et al: NFAT5/TonEBP limits pulmonary vascular resistance in the
hypoxic lung by controlling mitochondrial reactive oxygen species
generation in arterial smooth muscle cells. Cells. 10:32932021.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Sang D, Wang K, Sun X, Wang Y, Lin H, Jia
R and Qu F: NIR-driven intracellular photocatalytic O2
evolution on Z-scheme
Ni3S2/Cu1.8S@HA for hypoxic tumor
therapy. ACS Appl Mater Interfaces. 13:9604–9619. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Yang R, Huang J, Liao M, Huang J, Gao B,
Zhang H, Zhou J, Xu J and Lu Q: An oxygen-sufficient nanoplatform
for enhanced imaging-guided microwave dynamic therapy against
hypoxic tumors. Int J Nanomedicine. 17:5525–5545. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Chen J, Tang Q, Wang Y, Xu M, Sun S, Zhang
J, Wu R, Yue X, Li X, Chen Q and Liang X: Ultrasound-induced
piezocatalysis triggered NO generation for enhanced hypoxic tumor
therapy. ACS Appl Mater Interfaces. 15:15220–15234. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Zhao H, Sun L, Liu J, Shi B, Zhang Y,
Qu-Zong CR, Dorji T, Wang T, Yuan H and Yang J: Meta-analysis
identifying gut microbial biomarkers of Qinghai-Tibet Plateau
populations and the functionality of microbiota-derived butyrate in
high-altitude adaptation. Gut Microbes. 16:23501512024. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Bakshi J and Mishra KP: Identification of
biomarkers for gastrointestinal barrier injury and protective role
of sodium butyrate in hypobaric hypoxia exposed rats. Int
Immunopharmacol. 165:1154242025. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Pan C, Chen Z, Li C, Han T, Liu H and Wang
X: Sestrin2 as a gatekeeper of cellular homeostasis: Physiological
effects for the regulation of hypoxia-related diseases. J Cell Mol
Med. 25:5341–5350. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Ali Sheikh MS: The mir-21 inhibition
enhanced HUVEC cellular viability during hypoxia-reoxygenation
injury by regulating PDCD4. Mediators Inflamm. 2022:96619402022.
View Article : Google Scholar : PubMed/NCBI
|