DNA damage and repair, as the core mechanism for
maintaining genomic stability, are critical for safeguarding the
integrity of genetic material and sustaining cellular homeostasis
and organismal health. DNA damage can be caused by a combination of
endogenous factors (such as oxidative stress or replication errors)
or exogenous factors (such as ultraviolet rays or chemical toxins)
(1,2), with major types of DNA damage
including single-strand breaks (SSBs), double-strand breaks (DSBs),
base modifications, cross-links and large insertions/deletions
(3). Failure to promptly repair
these damages may lead to genomic instability and abnormal cellular
function, and may even drive pathological processes such as cancer,
neurodegenerative diseases and autoimmune diseases (1,4-6).
In response to damage, cells have evolved several
repair mechanisms. Base excision repair (BER) specifically
recognizes and excises oxidatively damaged bases (such as
8-oxoguanine) through DNA glycosylases [such as 8-oxoguanine DNA
glycosylase 1 (OGG1)] (7,8).
Nucleotide excision repair (NER) removes large-scale damage.
Homologous recombination (HR) and non-homologous end joining (NHEJ)
repair DSB through precision repair or direct joining,
respectively. On one hand, RNA molecules can directly participate
in repair regulation, with their own chemical modifications (such
as 5-methylcytosine) can promote repair efficiency; furthermore,
transcripts or damage-associated RNAs can serve as repair
templates, guiding precise repair (9,10). On the other hand,
post-translational modifications of the repair proteins themselves,
such as phosphorylation and acetylation, precisely control their
localization, activity and function, forming the dynamic regulatory
core of the repair network (11). Notably, DNA repair defects are
not only closely associated with genomic instability and cancer
development but also contribute to immune system abnormalities. For
instance, defects in the KU complex, a core component of the
DNA-dependent protein kinase holoenzyme, can trigger autoimmune
response linked to T cell senescence (12,13). Therefore, the present review
summarizes the research on the mechanism of DNA repair-immune axis
dysregulation, the development of related diseases and the future
direction of targeted therapy.
Autoimmune diseases, such as systemic lupus
erythematosus (SLE) and rheumatoid arthritis (RA), are
fundamentally characterized by the immune system losing tolerance
to self-antigens, leading to the production of autoantibodies
and/or autoreactive T cells, thereby causing chronic inflammation
and tissue damage in specific organs or throughout multiple systems
of the body. Their pathogenesis involves genetic predisposition,
epigenetic regulatory abnormalities and immune homeostasis
imbalances (14-16). Previous studies have confirmed
that DNA damage and repair abnormalities exacerbate autoimmune
disease progression through multiple mechanisms. Uncleared
extracellular DNA (for example, microsomal-bound DNA) can be
recognized as a danger signal activating the cyclic GMP-AMP
synthase (cGAS)-stimulator of interferon genes (STING) pathway and
promoting type I interferon secretion to drive lupus-like
autoimmunity (17-19). Oxidative stress modulates DNA
methylation to affect immune cell differentiation, further
promoting the activation of autoreactive T cell (15,20). Meanwhile, sustained accumulation
of DNA damage can trigger chronic low-grade inflammation,
accelerating autoimmune processes (21).
Despite significant progress in the field, key
challenges remain. First, the causal relationship between DNA
damage and autoimmunity has not been fully elucidated. Whether
oxidative damage is the initial trigger or a secondary consequence
of chronic inflammation requires verification using dynamic
spatiotemporal tracking techniques (22). Second, the regulation of immune
cell repair pathways exhibits high heterogeneity. For instance,
single-cell RNA sequencing analysis have revealed that cGAS-STING
pathway serves a unique role in maintaining the antitumor functions
of CD8+ T cells (23), suggesting that different immune
cells may differentially regulate DNA damage response (DDR)
pathways through mechanisms such as epigenetic reprogramming.
Third, the immune checkpoint protein programmed death ligand 1
(PD-L1) serves a complex role in the cross-regulation between the
DDR and innate immunity. On one hand, PD-L1 expression is induced
by DNA damage signals [such as the ataxia telangiectasia mutated
(ATM)/ataxia telangiectasia and Rad3 related (ATR) pathways] and
may affect genomic stability by, for example, stabilizing key
repair complexes (24,25). On the other hand, PD-L1
negatively regulates the cGAS-STING pathway, providing a novel
direction for immune cell-specific targeting strategies (26).
Novel therapeutic strategies targeting the
dysregulation of the DNA repair-immune axis have shown promising
potential. STING agonists combined with poly adenosine diphosphate
ribose polymerase (PARP) inhibitors synergistically enhance DNA
damage-induced immunogenic death while remodeling the immune
microenvironment through epigenetic regulation (27,28). Nanomedicines targeting the
cGAS-STING-TANK-binding kinase 1 (TBK1) axis selectively inhibit
autoreactive T cells, achieving remission in experimental
autoimmune encephalomyelitis models (29,30). Radiotherapy activates the
cGAS-STING pathway by inducing DNA damage and releasing it into the
cytoplasm, and preclinical studies have confirmed that this key
antitumor immune activation mechanism can significantly enhance the
efficacy of immune checkpoint inhibitors (31-33). DNA damage markers provide
assessment tools, while repair pathway modulation demonstrates
therapeutic potential in preclinical models (34-37). The present review systematically
summarizes the central role of the DDR in autoimmunity and
constructs a conceptual framework for targeting the DDR-immune
axis, providing a theoretical basis for the development of novel
therapeutic strategies.
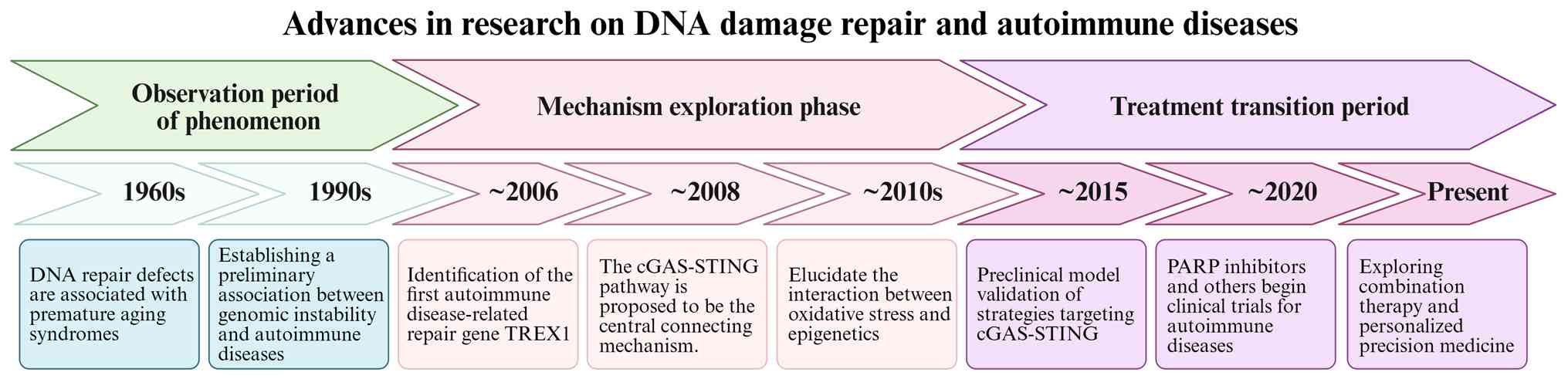
The trajectory of research on DNA damage and repair
in autoimmune diseases clearly demonstrates an evolutionary path
from phenomenological association to mechanistic dissection, and
ultimately towards targeted therapy, as presented in Fig. 1. Early studies (1960s-1990s)
established a preliminary link between DNA repair defects and human
diseases by observing syndromes such as xeroderma pigmentosum, and
also revealed evidence of genomic instability in patients with SLE
and other autoimmune conditions (38-41).
Entering the 21st century, mechanistic research
achieved a breakthrough in progress. The seminal discovery in 2006
that loss-of-function mutations in the three prime repair
exonuclease 1 (TREX1) genes cause a severe type I interferonopathy,
linked aberrant accumulation of cytoplasmic DNA to autoimmunity for
the first time (Fig. 1)
(42). The period from 2008 to
2013 yielded a pivotal discovery with the identification of the
cGAS-STING pathway as the cytosolic DNA sensor, which provided the
core molecular explanation for this association (Fig. 1) (43). This established the central
paradigm that unrepaired DNA triggers cGAS-STING activation,
initiating a cascade of events, a type I interferon response that
drives autoimmunity (44).
Concurrent research also revealed the synergistic role of oxidative
damage and epigenetic reprogramming in disrupting immune
homeostasis (15,20).
In recent years, research has entered a
translational phase. Preclinical models have demonstrated the
potential of targeting the cGAS-STING axis and enhancing
deoxyribonuclease 1-like 3 (DNase1L3) activity for the treatment of
autoimmune diseases (19,45,46).
In the mouse models of hereditary and induced lupus erythematosus,
a bifunctional enzyme (DNASE1/dnase1l3) was found to significantly
prevent the onset and death in mice with autoimmune diseases, which
directly proved the therapeutic potential of enhancing the activity
of DNase1L3 (19). In addition,
a class of covalent small molecule inhibitors (such as C-178 and
C-176) can directly target and inhibit sting protein to block type
I interferon response, which provides key preclinical validation
for the treatment of autoimmune diseases through drug inhibition of
cGAS-STING pathway (45).
Furthermore, the exploration of cell-free DNA (cfDNA) as a
biomarker for precision medicine strategies marks the advancement
of the field towards a new era of individualized targeted therapy
(47,48).
DNA damage is a major source of genomic instability,
comprising endogenous damage [including replication errors and
reactive oxygen species (ROS)-induced oxidative damage] and
exogenous damage (including UV radiation and chemical mutagens)
(49). At the molecular level,
damage includes SSBs, DSBs, base modifications, cross-links and
large insertions/deletions (3)
(Table I). These damages are
handled by specific repair pathways. For example, BER mainly
repairs oxidative base damage, while HR or NHEJ specifically deals
with DSBs. It is worth noting that these repair mechanisms are
closely related to autoimmune diseases. For example, BER defects
are related to SLE and RA, while the elimination of cytoplasmic DNA
is closely related to autoimmune syndrome characterized by type I
interferon reaction, which systematically explains the potential
pathways of DNA damage and repair imbalance involved in autoimmune
pathogenesis at the molecular level. Eukaryotes maintain genome
stability through five pathways: BER, NER, mismatch repair, HR and
NHEJ (3). Notably, ultraviolet
light-induced pyrimidine dimers are repaired primarily via the NER,
whereas radiation-induced DSBs are processed by either HR or NHEJ
(50), with distinct
spatiotemporal specificities: HR utilizes sister chromatids for DSB
repair exclusively in the S/G2 phase, whereas NHEJ
directly ligates breaks throughout the cell cycle (11,51) (Fig. 2). The precise execution of these
pathways is regulated by multi-level coordination:
Post-translational modifications precisely regulate repair
(52); RNA-mediated mechanisms
have expanded the traditional paradigms (for example, human DNA
polymerase θ can utilize RNA templates to direct the repair of DSB)
(10,53); and tissue-specific differences
markedly influence pathway choice (for example, neurons prefer NHEJ
due to absent HR factors, including BRCA1/2) (11,54). Endogenous DNA damage is closely
linked to the metabolic microenvironment. For instance, ROS
generated during oxidative stress induce base lesions that are
predominantly repaired by the BER pathway. Conversely, replication
stress can lead to stalled replication forks, whose restart and
stabilization depend on HR (Fig.
2) (55-57). The chromatin status affects
damage susceptibility and repair. Transcription factor-binding
regions are more vulnerable, while compact heterochromatin
inherently impedes the access of repair machinery (58). Separately, impaired DNA repair
itself is a direct driver of disease. For example,
FANCD2/FANCI-associated nuclease 1 deletion in chronic kidney
disease leads to tubular DNA damage accumulation and fibrosis,
highlighting how a deficiency in a specific repair factor can cause
pathogenic microenvironment-repair imbalance (59,60).
The DDR network coordinates repair through dynamic
signaling. Core sensors ATM/ATR kinases activate phosphorylation
cascades upon detecting DSBs or replication stress (61). Repair proteins form dynamic foci
[such as phosphorylated histone H2AX (γH2AX)] whose composition and
function changes with aging, increasing error-prone repair
(Fig. 2) (62). Epigenetic regulation via
chromatin remodeling complexes promotes repair factor recruitment
(58). Beyond chromatin, a
RNA-centric regulatory layer has emerged, where RNA-binding
proteins and long non-coding RNAs are increasingly recognized for
their role in fine-tuning DSB repair pathway choice and efficiency
(63,64). Critically, DNA damage repair is
intricately linked to cell fate decisions. Unfinished repair
activates the p53 pathway to induce cell cycle arrest or apoptosis
(65), while adult stem cells
employ continuous repair for rapid damage response during migration
(66). Damage-released DNA
fragments activate the cGAS-STING pathway, converting genomic
instability into antitumor immune responses, and the circadian
protein neuronal PAS domain protein 2 enhances HR repair by
stabilizing H2AX mRNA, suggesting circadian regulation of repair
efficiency (67-69). These mechanisms collectively
establish the pivotal role of DNA repair in cellular
homeostasis.
Technological innovations have markedly advanced
this field. High-resolution damage mapping achieves
single-nucleotide level precision (70). In addition, CRISPR-based
functional screening reveals cross-regulation between DNA repair
and MAPK/ERK signaling (71,72). Furthermore, stochastic kinetic
models quantify the intrinsic efficiency limitations of repair
pathways, such as the competition between repair and lesion bypass
in transcription-coupled repair (73). Additionally, emerging
technologies such as single-cell sequencing and nanoradiostatic
sensitizers are overcoming traditional research bottlenecks.
Single-cell sequencing resolves the critical issue of cellular
heterogeneity by mapping distinct DNA damage responses and repair
capacities across individual cells within complex tissues.
Nanoradiostatic sensitizers overcome therapeutic resistance by
irreversibly disrupting key DNA repair processes, thereby
transforming reparable lesions into lethal and immunogenic damage
(74). These tools not only
deepen the understanding of basic repair mechanisms but also
provide novel strategies for translating repair-related research
into clinical applications.
The interplay between the DDR and immune system
functionality is pivotal in maintaining immune homeostasis, while
the dysregulation of the DNA repair-immune axis contributes to
autoimmune diseases, chronic inflammation and immunosenescence
(75,76). This section explores how DNA
repair defects across immune cell lineages drive autoimmune
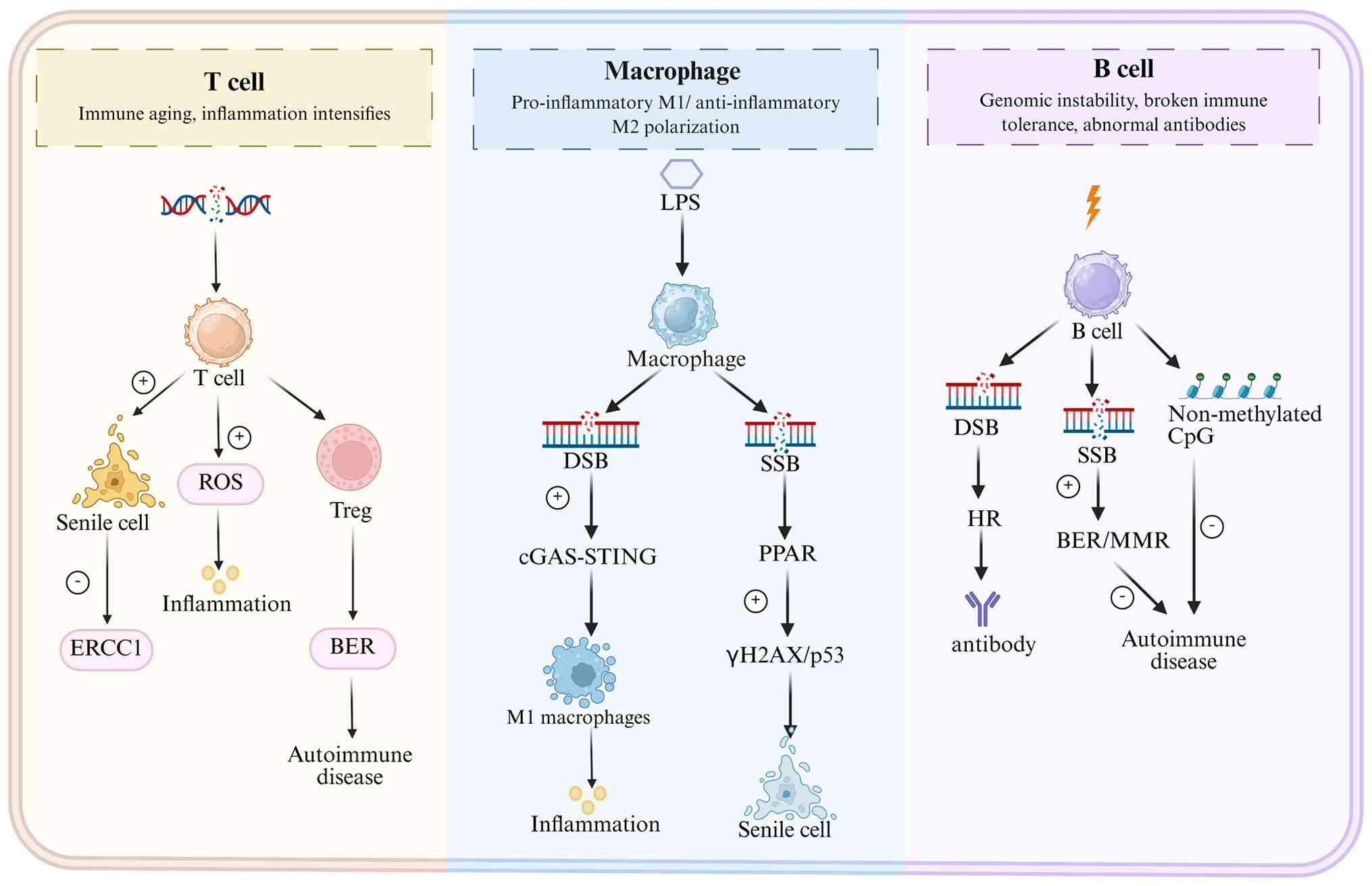
pathogenesis by disrupting immune homeostasis (Fig. 3).
T cells are the core of adaptive immunity, and T
cell function is markedly impacted by the accumulation of DNA
damage. Studies have demonstrated that the dynamic balance between
DNA damage and repair not only regulates T cell activation,
differentiation and homeostasis maintenance (77), but is also closely associated
with autoimmune diseases and immune senescence (78-80). During aging, T cells develop
genomic instability due to decreased DNA repair capacity,
manifesting as reduced proliferative potential and weakened
effector functions, ultimately leading to immune senescence
(81,82).
In autoimmune conditions, activated T cells
accumulate ROS-induced oxidative DNA damage, creating a vicious
cycle that further exacerbates inflammatory responses. In
autoimmune hepatitis, for instance, CD4+ T cells exhibit
markedly elevated 8-oxoguanine levels, which are positively
associated with hepatic inflammation severity (12). Notably, regulatory T cells
(Tregs), critical for immune tolerance maintenance, also exhibit
DNA repair capacity-dependent function. Defects in BER pathway
components in Tregs impair Foxp3 stability, driving their
conversion to pro-inflammatory T helper 17 (Th17) cells and thereby
disrupting autoimmune homeostasis (83-85) (Fig. 3).
As important effector cells of innate immunity, the
functional state of macrophages (for example, pro-inflammatory
M1-type or anti-inflammatory M2-type polarization) is closely
related to the dynamic balance of DNA damage and repair mechanisms.
During chronic inflammation and aging, the sustained DDR induces
macrophage conversion to a pro-inflammatory phenotype (M1), and
promotes the formation of an inflammatory microenvironment through
the secretion of pro-inflammatory factors such as IL-1β and TNF-α
(Fig. 3) (90). Genotoxic stresses in the
microenvironment (including ROS, chemotherapeutic agents or
radiation) can induce DNA damage in macrophages, which subsequently
regulates their immune phenotype and function through activation of
the cGAS-STING pathway or PARP-dependent repair pathways (91). Studies have demonstrated that
mtDNA release drives M1 polarization of macrophages via the
cGAS-STING pathway, while PARP-1 overactivation promotes
inflammatory factor production by modulating NF-κB signaling
(92,93). However, long-term DNA damage
accumulation also leads to macrophage dysfunction, characterized by
impaired phagocytic capacity and compromised antigen presentation,
which contributes to tumor progression or senescence-associated
inflammation (94,95) (Fig. 3). In the tumor microenvironment,
macrophages frequently exhibiting DNA damage promote immune escape
by secreting immunosuppressive factors such as IL-10 and TGF-β,
whereas during aging, persistent DNA damage in macrophages alters
their secretory phenotype, fostering chronic inflammatory
conditions in tissues (Fig. 3).
These findings reveal the central role of the DNA damage repair
balance in regulating macrophage function, providing novel
perspectives for the treatment of related diseases.
B cell activation and antibody production are
strictly dependent on the maintenance of genomic stability
(Fig. 3). The maintenance of B
cell function depends on some specific DNA repair pathways, such as
BER and HR. Impairment here markedly increases genomic instability
in B cells, causing errors in somatic hypermutation and class
switch recombination processes, ultimately resulting in aberrant
antibody production or breakdown of immune tolerance (96). Toll-like receptor 9 (TLR9)
recognizes pathogen DNA to drive B cell activation (97,98); however, the aberrant accumulation
of self-nucleic acids due to nuclease deficiencies (for example, in
TREX1) can be misrecognized by intracellular sensors such as TLR9
or cGAS-STING, driving inappropriate B cell activation and a type I
interferon response that is central to the pathogenesis of systemic
lupus erythematosus (99). In
the tumor microenvironment, B cells mediate antitumor immunity via
antigen presentation; however, their function is modulated by
damage-associated molecular patterns (DAMPs) released from tumor
cells. These DAMPs alter B-cell activation status, antigen
presentation efficiency and costimulatory molecule expression
(100). Notably, emerging
evidence suggests that in pathological contexts such as chronic
infection or cancer, persistent DNA damage stress within the tumor
microenvironment can skew B cell differentiation. This may
contribute to the expansion of regulatory B cells (Bregs) or
otherwise dysfunctional B cells that promote immune escape by
inducing immunosuppressive factor secretion (including IL-10 and
TGF-β secretion) (101). A
recent study has further revealed that specific DNA repair
pathways, such as NHEJ, serve essential roles in B cell antibody
diversity generation and affinity maturation (102). For example, during early
development, NHEJ facilitates V(D)J recombination to assemble the
primary B cell receptor repertoire; following activation, it
ensures the fidelity of class-switch recombination to alter
antibody class (102). This
provides novel perspectives for understanding the central position
of DNA repair mechanisms in adaptive immunity.
Innate immune cells [such as natural killer (NK)
cells and dendritic cells] can sense DNA damage signals through
nucleic acid sensors. For example, tumor-derived cfDNA or self-DNA
released from cells can be taken up by antigen-presenting cells
such as dendritic cells (DCs) (33). Activation of the cGAS-STING
pathway in these DCs promotes their maturation and the production
of cytokines (for example, type I interferons), which, in turn, can
enhance NK cell activation and drive adaptive antitumor immunity
(33,91,103,104). However, chronic DNA damage may
lead to hyperactivation of intrinsic immunity, triggering
autoinflammation or tissue damage (105). In addition, certain proteins
involved in the DNA damage response exhibit immunomodulatory
functions. For example, DNA repair factors (such as Growth arrest
and DNA damage-inducible 45 family of proteins) have been shown to
be involved in DNA repair and to regulate inflammatory responses in
various cell types, playing an important immunoregulatory role in a
variety of inflammatory and autoimmune diseases (106,107).
DNA damage drives inflammatory responses through
multiple mechanisms. Acute damage (such as that caused by infection
or radiotherapy) activates both the cGAS-STING and NF-κB pathways,
promoting the release of pro-inflammatory factors such as IL-6 and
TNF-α, thereby enhancing antitumor or antiviral immunity (94,103). Specifically, cGAS recognizes
cytoplasmic DNA to form dimers that catalyze cyclic GMP-AMP
production, subsequently activating the STING protein and
initiating type I interferon responses (108). However, chronic damage (such as
that caused by aging or metabolic stress) leads to a persistent
inflammatory microenvironment that promotes tumor progression or
aging-related diseases (90,109,110). For instance, senescent cells
mainly create a persistent pro-inflammatory microenvironment via
the senescence-associated secretory phenotype, which involves the
secretion of a large number of chemokines (such as CCL2, CXCL1 and
CXCL8). These factors actively recruit immune cells such as
monocytes and neutrophils. These cells, together with the senescent
cells themselves, release various inflammatory mediators, including
IL-1, IL-6 and MMPs, thereby establishing a chronic low-grade
inflammatory state (90,111). In addition, previous
foundational studies, along with more recent work, have shown that
DNA repair enzymes (such as mutT homologs) can maintain genomic
stability by preventing the incorporation of oxidatively damaged
nucleotides into DNA, offering a novel perspective for treating
autoimmune diseases by targeting genomic instability (100,112).
DNA damage is a key driver of immune senescence, a
process characterized by T cell dysfunction and chronic
inflammation (81,113). Senescent T cells exhibit
telomere shortening, mitochondrial dysfunction and sustained
activation of the DDR signaling pathway (81,114). Specifically, telomeres are
shortened by 50-100 base pairs annually on average, leading to
upregulated expression of the cell cycle arrest-related protein
p21. Concurrently, the mitochondrial membrane potential decreases,
ATP production diminishes and mtDNA release occurs (114). Impaired DNA repair capacity,
exemplified by excision repair cross-complementation group 1
(ERCC1) deficiency, accelerates hematopoietic senescence and
compromises immune cell reconstitution (115,116). Hematopoietic stem cells with
ERCC1 deficiency exhibit markedly reduced self-renewal capacity and
a tendency to differentiate into myeloid cells rather than lymphoid
cells, which disrupts immune homeostasis (116,117). Notably, an elevated frequency
of mutations in DNA repair genes in the elderly population is
associated with increased infiltration of CD8+ T cells
and M1 macrophages, suggesting a complex association between the
DDR and age-related immune remodeling (118). The frequency of somatic
mutations in repair genes (such as TP53 and ATM) in peripheral
blood cells of individuals >70 years of age is markedly higher
than that of young adults (119,120). This genomic instability,
combined with inflammatory immune cell infiltration, constitutes
the molecular foundation of immune senescence. Interventional
strategies, such as senolytics (senescent cell-targeting agents),
delay immune senescence via senescent cell elimination or DNA
repair enhancement ('genoprotection') (81,95,121). For instance, the combination of
dasatinib and quercetin specifically clears senescent immune cells
and restores thymic function (122).
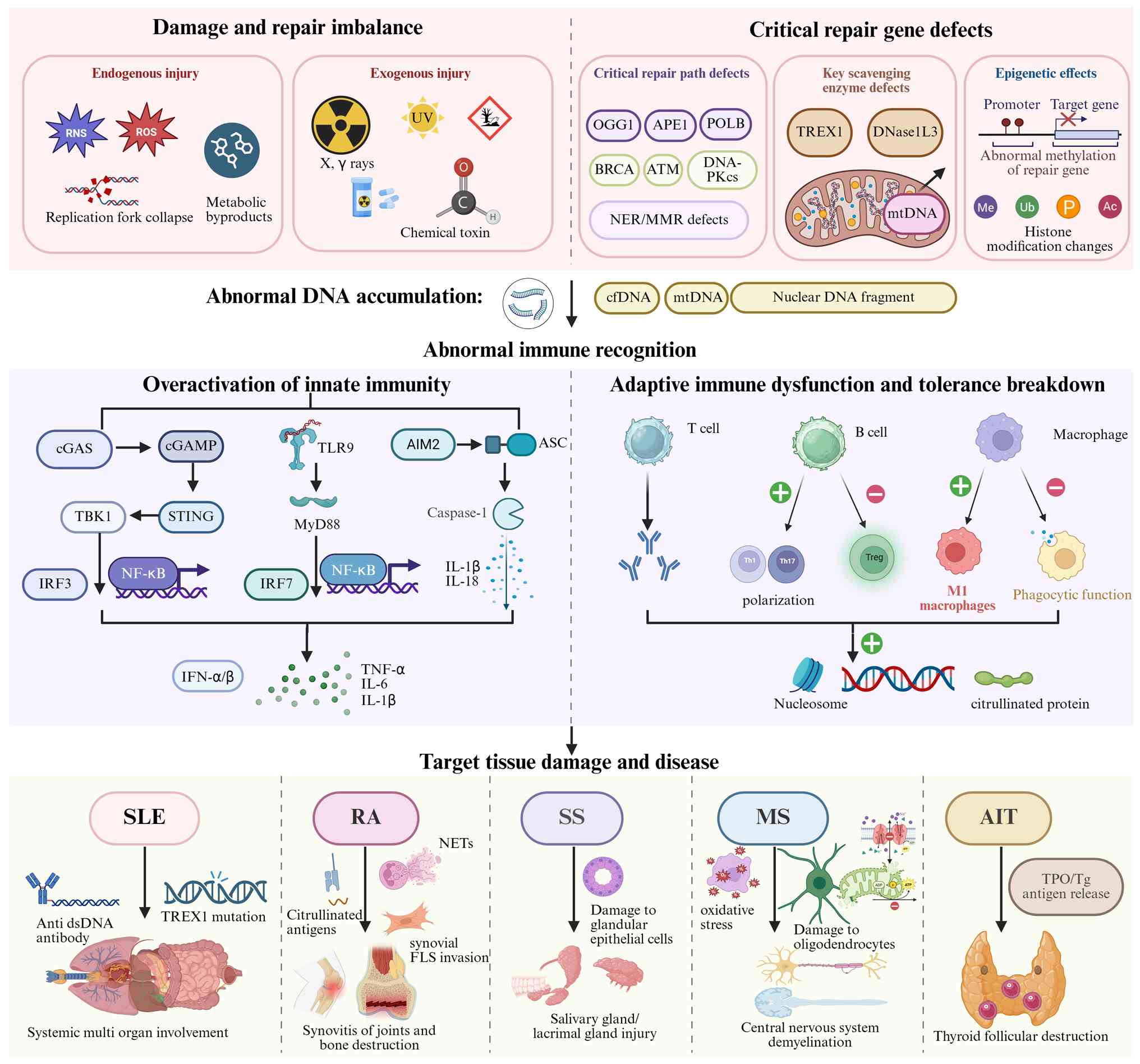
The accumulation of DNA damage and deficiencies in
repair pathways are now recognized as fundamental drivers in the
pathogenesis of various autoimmune diseases. While the clinical
manifestations and target organs differ, a common thread linking
conditions such as Sjögren's syndrome (SS), RA, SLE and multiple
sclerosis (MS) is the breakdown of immune tolerance triggered by
genomic instability (42,123,124),
The main core mechanisms involved in various autoimmune diseases
include the sustained accumulation of oxidative DNA damage,
functional defects in specific repair genes, resulting abnormal
accumulation of cytoplasmic DNA, and ultimately excessive type I
interference response and chronic inflammation triggered by innate
immune sensing pathways such as cGAS STING. These links are
intertwined, forming a 'damage inflammation' cycle that
collectively destroys immune tolerance (Fig. 4).
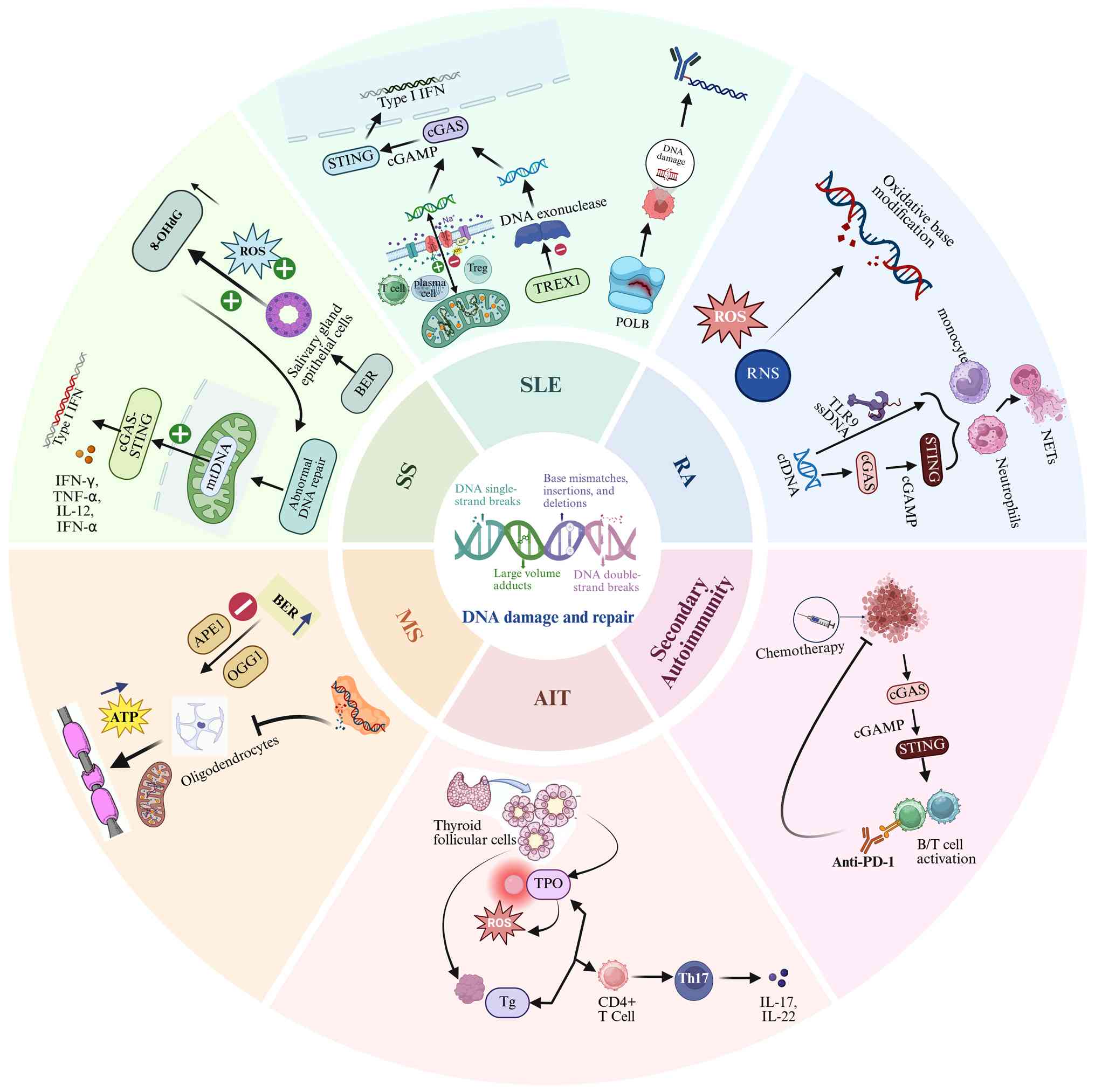
SS is a chronic autoimmune disease characterized by
lymphocytic infiltration of exocrine glands and autoantibody
production. Salivary gland epithelial cells from patients with SS
exhibit increased levels of oxidative DNA damage markers, such as
8-oxoguanine, and show evidence of an activated DDR, including
γH2AX (Fig. 4) (85,125,126). This intrinsic genomic stress is
considered to contribute to glandular dysfunction and the release
of immunostimulatory nucleic acids. The impaired DNA repair
capacity observed in patients with SS may accelerate disease
progression. Specifically, dysfunction of the BER pathway can lead
to ineffective repair of oxidative DNA damage, and unrepaired SSBs
may progress into more cytotoxic DSBs (127,128). Furthermore, defective
O6-methylguanine-DNA methyltransferase repair in
high-risk patients with SS and a tendency to develop lymphoma
further exacerbates genomic instability (129).
Defects in DNA clearance or abnormal accumulation of
DNA are key drivers of inflammation in SS (130). Concomitantly, cells exhibit
inherent defects in DNA repair capacity. This inability to
effectively resolve DNA damage leads to the accumulation of lesions
and triggers a compensatory hyperactive DNA damage response,
manifesting as enhanced p53 phosphorylation and G1 phase
cell cycle arrest (125,131).
Mechanistic studies have revealed that unrepaired DNA fragments
accumulating in the cytoplasm activate innate immune signaling
pathways, particularly the cGAS-STING pathway, leading to excessive
production of inflammatory factors. This triggers a type I
interferon response, subsequently impairing salivary and lacrimal
gland function (Figs. 4 and
5) (132,133). Furthermore, cytokines such as
IFN-γ, TNF-α, IL-12 and IFN-α can continuously activate
autoreactive T/B cells (128).
Currently, strategies focusing on enhancing DNA
repair, reducing oxidative stress or inhibiting downstream
signaling are being investigated in preclinical models. For
instance, key drugs that inhibit the cGAS-STING pathway, such as
rapamycin, have been demonstrated to significantly alleviate
pathological damage to the submandibular glands and improve
salivary gland function in SS model mice by restricting excessive
activation of this pathway (134). Similarly, drugs targeting the
TLR pathway, such as inamodine, can mitigate glandular damage and
inflammation in experimental SS by inhibiting the activation of
NLRP3 inflammasomes (135).
The core pathological features of RA include chronic
synovial inflammation, abnormal proliferation and enhanced
invasiveness of RA synovial fibroblasts (RA-FLS), and eventual
destruction of articular cartilage and bone (Fig. 4) (136). In the RA synovial
microenvironment, activated immune cells, including macrophages and
T cells, excessively produce and accumulate ROS, which contributes
to oxidative stress and perpetuates inflammation (137,138). The persistent inflammation and
oxidative stress microenvironment in synovium is a key factor
inducing local cellular DNA damage, and the two can form a mutually
exacerbating vicious cycle (139). Studies have shown that the
abundant pro-inflammatory factors in RA synovium upregulate AIM2
protein by activating NF-κB and other pathways. After recognizing
the cytoplasmic dsDNA, the AIM2 inflammasome recruits the adaptor
protein ASC through its PYD domain, which then recruits
pro-caspase-1 and assembles it into an activation platform,
eventually leading to the activation of Caspase-1 (139).
Clinical studies have found that patients with RA
not only have increased DNA damage, but also have inherent defects
in their DNA repair ability. Compared with healthy controls, the
levels of endogenous DNA SSB and DSBs in peripheral blood
mononuclear cells of patients with active RA were significantly
increased (140). A 2024 study
further revealed that the repair efficiency of RA patients' cells
for DNA DSBs was significantly lower after induced damage, and this
low repair efficiency was related to the single nucleotide
polymorphisms of specific genes (such as Rad51) in the HR repair
pathway (141). It has been
demonstrated that the levels of 8-hydroxy-2′-deoxyguanosine
(8-OHdG), a marker of oxidative DNA damage, in the synovial fluid
of patients with RA were significantly higher than those in the
non-arthritis control group (142). Furthermore, NADPH oxidase
4-mediated oxidative stress in RA-FLS markedly enhances their
migration and invasion capabilities, forming a critical pathogenic
axis (143). In RA, metabolic
and functional abnormalities of immune cells may lead to genomic
and mitochondrial instability. For example, elevated levels of
mtDNA have been detected in the synovial fluid of patients with RA,
suggesting that it may act as a DAMP to drive inflammation
(142). This discovery reveals
a key pathological mechanism in RA: DNA repair defects and cellular
energy metabolism crisis jointly lead to pathological death of
immune cells, such as pyroptosis, while released molecules such as
mtDNA act as DAMPs to continuously activate the immune system
(107). This mechanism closely
links DNA repair deficiency, cellular energy metabolism
dysregulation and pathological death of immune cells, which
provides a key explanation for the persistence of chronic
inflammation and immune dysregulation in RA. In addition, cfDNA is
a key ligand that activates cGAS-STING and TLR9 pathways in RA and
drives inflammation (144).
Notably, neutrophil extracellular traps are web-like structures
composed of decondensed chromatin and antimicrobial proteins
released by activated neutrophils. They contain a large amount of
cfDNA (including mtDNA) and citrullinated autoantigen. These
substances, as a new wave of cfDNA, aggravate inflammation again
through the TLR9/cGAS-STING pathway (Fig. 5) (145). A study has shown that the DNA
methylation group of peripheral blood mononuclear cells in patients
with RA showed significant changes and increased variability
compared with the healthy control group, indicating that the
systemic inflammatory state is directly related to epigenetic
regulation (146).
These findings provide novel perspectives for
precision diagnosis and treatment of RA. Monitoring DNA damage
markers such as 8-OHdG and cfDNA not only helps assess disease
activity but also predicts treatment response (142,147). Intervention strategies
targeting the DDR-inflammation axis, such as modulating epigenetic
modifications or using biologics that can indirectly ameliorate DNA
repair capacity, may represent novel approaches to break the
'damage-inflammation' vicious cycle (47,148).
The pathogenesis of SLE is closely linked to
autoantigen exposure resulting from defective DNA repair (150). TREX1 gene mutations represent
an important genetic factor in familial lupus (151,152). By impairing the function of the
encoded 3′-5′ DNA exonuclease, the mutation leads to massive
accumulation of undegraded self-DNA in the cytoplasm (42,153). Furthermore, mitochondrial
dysfunction serves a pivotal role in the pathogenesis of SLE
(154). In patients with active
SLE, the mitochondrial membrane potential exhibits marked changes,
leading to substantial release of mtDNA into the cytoplasm and
activation of type I interferon signaling via the cGAS-STING
pathway (155) (Fig. 5). For example, the
hypermetabolism of T cells and plasma cells may lead to temporary
or persistent elevation of the membrane potential (156,157), while the dysregulation of
regulatory B cells may lead to reduction of the membrane potential
due to metabolic defects (158). Furthermore, abnormal function
of DNA polymerase β promotes autoimmunity through a distinct
mechanism (159). One study
using mouse models demonstrated that inefficient mutations that
reduce polymerase activity impair DNA repair accuracy, producing
immunogenic aberrant DNA fragments, which can lead to lupus-like
autoimmune diseases (160).
Furthermore, impaired clearance of apoptotic cells induced by DNA
damage may lead to sustained exposure to nuclear antigens and
exacerbate immune complex deposition (161). Macrophages from patients with
SLE exhibit markedly reduced phagocytic capacity for apoptotic
cells, which is directly associated with disease activity (Fig. 4) (162). Nucleosome fragments released
from improperly cleared apoptotic cells can serve as autoantigens
to activate autoreactive B cells to produce autoantibodies,
establishing a vicious cycle. These findings provide novel targets
for the treatment of SLE (161,162). In recent years, small molecule
inhibitors targeting the cGAS-STING pathway (such as disulfiram
targeting ring finger protein 115, and cordycepin promoting STING
degradation) have shown promising efficacy in lupus mouse models,
markedly alleviating autoimmune symptoms (163,164). Meanwhile, assessing γH2AX,
oxidative damage products, or specific plasma free DNA not only
helps assess disease activity but may also provide an important
basis for early intervention strategies (165,166).
In MS, a clinical study has confirmed significant
oxidative DNA damage and impaired repair function in patients with
MS (124). Research indicates
increased DNA damage and delayed repair in peripheral blood
lymphocytes of patients with MS, alongside downregulated expression
of key BER enzymes (such as OGG1 and APE1). These molecular
alterations in DNA integrity and repair capacity are implicated in
the pathogenesis of MS and correlate with disease activity
(Fig. 5) (124). Genetic polymorphisms further
associate repair capacity with disease risk (124). This molecular damage has direct
clinical relevance: The levels of oxidative DNA markers (such as
8-OHdG) in patient body fluids are positively associated with the
severity of core symptoms, including neurological dysfunction,
fatigue and cognitive impairment (Fig. 5) (167). Under chronic inflammatory
conditions, persistent oxidative stress leads to the accumulation
of 8-oxoguanine in DNA. During the attempt to repair this damage,
the enzymatic activity of OGG1 itself triggers pro-inflammatory
signaling pathways, thereby exacerbating the inflammatory response.
OGG1 is a key enzyme for repairing oxidative DNA damage
(particularly 8-oxoguanine). An animal study has demonstrated that
inhibiting OGG1 activity can ameliorate symptoms of MS (Fig. 5) (85). Notably, epigenetic analyses have
revealed abnormal hypermethylation in the promoter regions of
specific BER genes within MS lesions, which may represent an
important factor contributing to persistent repair dysfunction
(168). In addition to nuclear
DNA damage, oligodendrocytes in patients with active MS exhibit
mitochondrial dysfunction, including markedly reduced membrane
potential and ATP production (Fig.
4) (169). Research has
demonstrated that the anti-inflammatory effects of small-molecule
inhibitors in MS models, coupled with the effective promotion of
remyelination by the epigenetic silencing inhibitor ESI1 in animal
models of demyelination, together provide a preclinical theoretical
foundation for intervening in MS by modulating DNA repair processes
(85,168).
The pathogenesis of AIT is closely related to
oxidative damage in thyroid follicular cells. Thyroid peroxidase
(TPO) generates ROS during hormone synthesis, and its dysfunction
can exacerbate the oxidative stress state (Fig. 5) (170,171). Excessive ROS can induce
oxidative DNA damage, including 8-OHdG. A study has shown that in
the thyroid tissue of patients with AIT and in experimental models,
the levels of such DNA damage markers are markedly elevated,
accompanied by inhibited DNA repair capacity (172). Persistent DNA damage can
promote apoptosis of follicular cells, leading to the release of
self-antigens such as TPO and thyroglobulin within the cells,
thereby disrupting immune tolerance (Fig. 4) (171,172). Additionally, in patients with
AIT, autophagosome formation-related genes, such as
autophagy-related 101 and beclin 1, have been found to be
hypomethylated, and this is associated with environmental iodine
levels (173). In thyroiditis
animal models, intervening in specific DNA repair mechanisms has
been shown to alleviate thyroiditis (172). For example, overexpression of
the DNA repair protein mutT homolog 1 (MTH1) can reduce
inflammation and damage induced by high iodine levels. High iodine
can induce DNA damage and inflammation in thyroid cells by
inhibiting MTH1, and increasing the infiltration of Th17 cells in
the thyroid (Fig. 5) (172).
The DDR in cancer therapy is a double-edged sword,
as it is associated with both therapeutic efficacy and the risk of
autoimmunity. Chemotherapy-induced DNA damage and tumor cell death
can lead to the release of self-antigens and cytoplasmic nucleic
acids (such as cytosolic chromatin fragments), which in turn
activate innate immune pathways such as the cGAS-STING pathway
(174,175). While this drives antitumor
immunity, it also provides a theoretical basis for breaking
self-tolerance and triggering treatment-related autoimmune
phenomena (176,177). In addition, intrinsic defects
in DDR genes (such as BRCA1/2 mutations) in tumor cells exacerbate
genomic instability and increase tumor immunogenicity (178). While this may enhance the
efficacy of immune checkpoint inhibitors, it could also elevate the
risk of autoimmune reactions due to aberrant immune activation
(178). Therefore, imbalance of
the DDR constitutes a key hub linking the therapeutic effects of
cancer treatment to autoimmune side effects. A recent study has
suggested that precise regulation of the DDR through strategies may
enhance antitumor immunity while reducing toxicity to normal
lymphocytes, offering a novel direction for optimizing treatment
strategies (179).
Building upon the elucidated connections between
DNA repair defects and autoimmune pathogenesis, targeting the DDR
has emerged as a promising therapeutic frontier. The integration of
mechanistic insights with clinical transformation may reveal novel
inhibitor strategies, combination therapies and precision medicine
approaches (Table II). However,
this path has notable challenges, including balancing efficacy with
genomic safety risks and navigating tissue-specific repair
dependencies.
PARP inhibitors have achieved significant efficacy
in the treatment of breast and ovarian cancer by inhibiting DNA
SSBR and inducing a synthetic lethal effect (Table II) (180-182). The repurposing of PARP
inhibitors in autoimmune diseases are still at the preclinical
exploration stage, and they may exert potential effects by
modulating DNA repair and inflammatory pathways (183,184). For example, in EAE model,
targeting DNA damage repair related protein PARP-1 can play a
therapeutic role by regulating immune cell migration and
inflammatory response (183).
In addition, a human phase I clinical trial marked the first step
in the transformation of a new PARP family inhibitor from
preclinical to clinical (184).
It is well-established that patients with SLE exhibit defective DNA
damage repair. This impairment contributes to the release of
self-antigens (such as cfDNA) and the activation of autoimmune
responses (39,149).
Although, theoretically, PARP inhibitors are
considered to have therapeutic potential in various autoimmune
diseases due to their anti-inflammatory properties and effects on
DNA repair, their preclinical exploration in areas such as SLE and
MS is still in its early stages (185,186). In addition, long-term use of
PARP inhibitors may exacerbate genomic instability. Studies have
shown that their prolonged use increases the risk of secondary
cancers; for instance, the risk of developing myelodysplastic
syndrome/acute myeloid leukemia is significantly higher in patients
with ovarian cancer (4-12%) (187). Consequently, rigorous
validation of their long-term safety and applicability across
heterogeneous patient cohorts remains imperative. Notably, the
application of PARP inhibitors in autoimmunity remains
controversial with critical safety considerations (188). PARP inhibition alone fails to
disrupt the persistent 'damage-inflammation' feedback loop in the
inflammatory microenvironment (189). Considering the complexity of
the inflammatory microenvironment in RA, theoretically, PARP
inhibitors alone may struggle to completely block the progression
of the disease (190).
Excessive PARP suppression may also impair DNA repair in immune
cells. The secondary malignancy risk is a major safety concern
(191). Long-term inhibition
accumulates unrepaired DNA lesions and increases lymphoma or
leukemia incidence, especially in BRCA wild-type individuals
(191,192). Another prominent safety issue
is hematotoxicity. PARP inhibitors induce myelosuppression,
including neutropenia and thrombocytopenia, which may worsen immune
deficiency in autoimmune patients (193). To navigate this risk-benefit
landscape, future strategies require rigorous validation.
Intermittent dosing regimens and nanoparticle-mediated targeted
delivery may minimize off-target effects on normal tissues
(194,195). Besides, integrating cfDNA
damage markers with imaging for long-term safety monitoring is
crucial for the early warning and monitoring of autoimmune
diseases. Preclinical validation using humanized autoimmune models
will further improve translational accuracy (183).
Inhibitors of ATM/ATR kinase, a central regulator
of the DDR, enhance sensitivity to radiotherapy or chemotherapy by
blocking cell cycle checkpoints (196,197) (Table II). Emerging studies have found
that ATM/ATR inhibitors also promote remodeling of the tumor immune
microenvironment through activation of the cGAS-STING pathway
(198-200). For example, inhibition of ATR
leads to accumulation of unrepaired cytoplasmic DNA, activates
cGAS-STING pathway and induces type I interferon secretion, which
enhances T cell infiltration and antitumor immunity (200,201). Emerging evidence from
preclinical models supports the combination of ATM/ATR inhibitors
and immune checkpoint inhibitors (for example, anti-PD-L1) as a
promising strategy for synergistic antitumor activity (202,203); however, this strategy
necessitates a balance between immune activation and carcinogenic
risks arising from unresolved DNA damage accumulation (204).
The antioxidant NAC attenuates oxidative DNA damage
by ROS and replenishes the levels of intracellular glutathione, a
key antioxidant involved in DNA repair (Table II) (205). Accumulating evidence indicates
that NAC monotherapy does not reduce the efficacy of chemotherapy;
instead, it protects normal tissues from treatment-induced
oxidative injury while preserving the cytotoxic effect of
chemotherapeutic agents on tumor cells (206-208). Preclinical and clinical study
has indicated that immunotherapeutic agents (such as immune
checkpoint inhibitors) reverse the immunosuppressive tumor
microenvironment and enhance T cell-dependent elimination of
residual tumor cells (209).
However, the generalizability of this strategy across different
cancer types, the optimal administration timing (for example,
concurrent vs. sequential with antitumor agents) and dosage
regimens remain to be validated in large-scale clinical trials.
Damage fragments (such as oxidized base
modifications or DSB markers) in cfDNA can be used as biomarkers
for real-time monitoring of the DNA repair status (210). For example, persistent high
levels of the DNA damage marker γH2AX were detected after
radiotherapy suggest insufficient DNA damage repair and may guide
the timing of combination therapy with ATM/ATR inhibitor (197,202). In addition, tumor-specific
cfDNA mutation profiles (for example, BRCA1/2 deletion) can predict
PARP inhibitor efficacy, and immunotherapy responders often exhibit
upregulated expression levels of STING pathway-related genes in
cfDNA (26,202). Future development of highly
sensitive assays and integrated multi-omics modeling is required to
improve predictive accuracy.
DNA repair inhibitors face several challenges. PARP
or ATM/ATR inhibitors may increase genomic instability in normal
tissues due to off-target effects, with long-term PARP inhibitor
use potentially inducing hematopoietic malignancies and ATR
inhibitors exhibiting hepatotoxicity in mouse models (196,204,211,212). Furthermore, inhibition of
specific repair pathways may force cells to rely on error-prone
alternative mechanisms (such as microhomology-mediated
end-joining), increasing oncogenic mutation risks (213). Significant differences exist in
DNA repair capacity and dependency across tissues: Hematopoietic
stem cells highly depend on NHEJ, whereas intestinal epithelial
cells preferentially use HR (214), leading to notable efficacy and
toxicity variations of the same inhibitor in different organs.
Strategies addressing these limitations include developing
tissue-selective delivery systems and intermittent dosing regimens
(194,215). Future research should integrate
single-cell sequencing with organoid models to elucidate
tissue-specific DNA repair dynamic networks across spatiotemporal
dimensions, thereby designing adaptive therapeutic strategies that
can adjust according to repair status or cell cycle.
Notwithstanding the substantial advances in
delineating the DDR-immune axis, critical gaps remain in
mechanistic clarity and translational validation. First, the causal
temporality between DDR defects and immune activation is still
unclear. A study has shown that loss-of-function mutations of
TREX1, an endoplasmic reticulum-associated exonuclease, can trigger
uncontrolled cGAS-STING activation, which is associated with
autoinflammatory diseases, including SLE (216). However, loss-of-function
mutations in TREX1 are clearly pathogenic in monogenic diseases
such as Aicardi-Goutières Syndrome, but their contribution to
complex autoimmune diseases such as SLE still requires more
population genetic studies. Second, the role of the cGAS-STING
pathway in autoimmunity is contradictory and appears to be dual
(152). On one hand,
overactivation of this pathway is closely related to type I
interferon-driven autoimmunity. On the other hand, animal models
suggest that a simple genetic defect (such as TREX1 deficiency) may
be insufficient to induce full tissue inflammation, often requiring
an environmental 'second hit', indicating that its pathogenicity
may be conditional rather than absolutely causative (217). Third, there is currently a lack
of clinically validated, standardized DDR-derived biomarkers that
can be used for disease stratification or efficacy prediction.
Although DDR-related autoantibodies (such as anti-PARP zinc finger
domain antibodies) are common in SLE and SS (39,218), biomarkers such as specific
damage features of cfDNA, including oxidative modifications, have
not yet been prospectively validated as reliable predictive tools,
partly due to the lack of standardized detection methods.
A prioritized research agenda should address these
gaps. Preclinically, using conditional knockout models of specific
cells such as synovial cells, the cell-specific role of TREX1-cGAS
crosstalk in RA and other diseases was precisely validated
(219). In human cohorts,
research suggests that specific forms of cfDNA, such as oxidatively
modified DNA, may be associated with disease activity (220), providing a basis for its use as
a biomarker for monitoring disease activity. A preclinical study
suggests that PARP inhibitors may exert therapeutic effects through
anti-inflammatory mechanisms, but their inhibitory effects on DNA
repair could pose long-term genomic risks (221). Therefore, exploring dosing
regimens that can distinguish between their anti-inflammatory
effects and genotoxic effects (such as low-dose or intermittent
administration) is a key direction for future research.
Research on the link between the DDR and
autoimmunity has unveiled a complex interplay between genomic
instability and immune homeostasis disruption. Unrepaired DNA
damage, such as oxidative base lesions or DSBs, can drive
autoimmune responses. The core mechanism is that unrepaired DNA
damage triggers a type I interferon storm through pathways such as
cGAS/STING, while defects in repair genes such as TREX1 lead to
exposure of self-nucleic acid antigens, both of which together
break immune tolerance and drive self-immunity. Furthermore,
epigenetic disorders caused by oxidative stress further exacerbate
the functional imbalance of immune cells such as Th17/Treg.
Therapeutic strategies targeting DDR pathways show significant
potential. Nanomedicines designed to block the cGAS-STING-TBK1 axis
can specifically inhibit autoreactive T-cell activation. DNase1L3
analogs help clear free DNA, reducing the autoantigen load and
alleviating lupus-like phenotypes in preclinical models.
Additionally, combining PARP inhibitors with immune checkpoint
blockade represents a promising translational approach by
synergistically inducing immunogenic cell death and remodeling the
immune microenvironment.
In conclusion, future research should leverage
single-cell multi-omics to decipher repair heterogeneity among
immune cell subsets and construct dynamic DNA damage maps to reveal
the spatiotemporal relationship between lesion accumulation and
inflammatory signaling. In addition, the development of novel
intervention strategies that can distinguish between
anti-inflammatory and genotoxic effects (such as intermittent
administration and tissue selective delivery systems) is expected
to balance efficacy and safety while avoiding systemic toxicity.
These advancements are poised to overcome the limitations of
conventional therapies and advance the field of autoimmune disease
treatment into the realm of personalized precision medicine.
Not applicable.
KW was primarily responsible for the writing,
review and revision of this manuscript. MW, QQX and HF compiled the
references and developed the tables and figures. YC, TSZ and XY
conceived the structure of the manuscript. LW, JWY and HS drafted
the initial manuscript. XF-L and JL participated in the review and
the critical revision of the manuscript for important intellectual
content. All authors read and approved the final version of the
manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
This study was supported by the Project for Excellent Young in
Colleges and Universities of Anhui Province (grant no.
2023AH030120), Postgraduate Innovation Research and Practice
Program of Anhui Medical University (grant nos. YJS20250083 and
YJS20250078) and Anhui Medical University Scientific Research
Institution Construction and Promotion Plan Fund (grant nos.
2024xkjT001 and 2025xkjT001).
|
1
|
Owiti NA, Nagel ZD and Engelward BP:
Fluorescence Sheds Light on DNA Damage, DNA repair, and mutations.
Trends Cancer. 7:240–248. 2021. View Article : Google Scholar
|
|
2
|
Hasan A, Rizvi SF, Parveen S and Mir SS:
Molecular chaperones in DNA repair mechanisms: Role in genomic
instability and proteostasis in cancer. Life Sci. 306:1208522022.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chatterjee N and Walker GC: Mechanisms of
DNA damage, repair, and mutagenesis. Environ Mol Mutagen.
58:235–263. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wu L, Sowers JR, Zhang Y and Ren J:
Targeting DNA damage response in cardiovascular diseases: from
pathophysiology to therapeutic implications. Cardiovasc Res.
119:691–709. 2023. View Article : Google Scholar
|
|
5
|
Wen J, Wang Y, Yuan M, Huang Z, Zou Q, Pu
Y, Zhao B and Cai Z: Role of mismatch repair in aging. Int J Biol
Sci. 17:3923–3935. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Klapp V, Álvarez-Abril B, Leuzzi G,
Kroemer G, Ciccia A and Galluzzi L: The DNA damage response and
inflammation in cancer. Cancer Discov. 13:1521–1545. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fan L, Liu W, Yang B, Zhang Y, Liu X, Wu
X, Ning B, Peng Y, Bai J and Guo L: A highly sensitive method for
simultaneous detection of hAAG and UDG activity based on
multifunctional dsDNA probes mediated exponential rolling circle
amplification. Talanta. 232:1224292021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pao PC, Patnaik D, Watson LA, Gao F, Pan
L, Wang J, Adaikkan C, Penney J, Cam HP, Huang WC, et al: HDAC1
modulates OGG1-initiated oxidative DNA damage repair in the aging
brain and Alzheimer's disease. Nat Commun. 11:24842020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen H, Yang H, Zhu X, Yadav T, Ouyang J,
Truesdell SS, Tan J, Wang Y, Duan M, Wei L, et al: m(5)C
modification of mRNA serves a DNA damage code to promote homologous
recombination. Nat Commun. 11:28342020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tsegay PS, Hernandez D, Qu F, Olatunji M,
Mamun Y, Chapagain P and Liu Y: RNA-guided DNA base damage repair
via DNA polymerase-mediated nick translation. Nucleic Acids Res.
51:166–181. 2023. View Article : Google Scholar :
|
|
11
|
Hustedt N and Durocher D: The control of
DNA repair by the cell cycle. Nat Cell Biol. 19:1–9. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y, Fu Z, Li X, Liang Y, Pei S, Hao S,
Zhu Q, Yu T, Pei Y, Yuan J, et al: Cytoplasmic DNA sensing by KU
complex in aged CD4(+) T cell potentiates T cell activation and
aging-related autoimmune inflammation. Immunity. 54:632–647.e9.
2021. View Article : Google Scholar
|
|
13
|
Felgentreff K, Baumann U, Klemann C,
Schuetz C, Viemann D, Wetzke M, Pannicke U, von Hardenberg S, Auber
B, Debatin KM, et al: Biomarkers of DNA damage response enable flow
cytometry-based diagnostic to identify inborn DNA repair defects in
primary immunodeficiencies. J Clin Immunol. 42:286–298. 2022.
View Article : Google Scholar :
|
|
14
|
Thatte AS, Billingsley MM, Weissman D,
Melamed JR and Mitchell MJ: Emerging strategies for nanomedicine in
autoimmunity. Adv Drug Deliv Rev. 207:1151942024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mu S, Wang W, Liu Q, Ke N, Li H, Sun F,
Zhang J and Zhu Z: Autoimmune disease: A view of epigenetics and
therapeutic targeting. Front Immunol. 15:14827282024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reynolds JA and Putterman C: Progress and
unmet needs in understanding fundamental mechanisms of
autoimmunity. J Autoimmun. 137:1029992023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
McCord JJ, Engavale M, Masoumzadeh E,
Villarreal J, Mapp B, Latham MP, Keyel PA and Sutton RB: Structural
features of Dnase1L3 responsible for serum antigen clearance.
Commun Biol. 5:8252022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Klein B and Günther C: Type I interferon
induction in cutaneous DNA damage Syndromes. Front Immunol.
12:7157232021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stabach PR, Sims D, Gomez-Bañuelos E,
Zehentmeier S, Dammen-Brower K, Bernhisel A, Kujawski S, Lopez SG,
Petri M, Goldman DW, et al: A dual-acting DNASE1/DNASE1L3 biologic
prevents autoimmunity and death in genetic and induced lupus
models. JCI Insight. 9:e1770032024. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zheng X and Sawalha AH: The role of
oxidative stress in epigenetic changes underlying autoimmunity.
Antioxid Redox Signal. 36:423–440. 2022. View Article : Google Scholar
|
|
21
|
Schmitz CRR, Maurmann RM, Guma FTCR, Bauer
ME and Barbé-Tuana FM: cGAS-STING pathway as a potential trigger of
immunosenescence and inflammaging. Front Immunol. 14:11326532023.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hopfner KP and Hornung V: Molecular
mechanisms and cellular functions of cGAS-STING signalling. Nat Rev
Mol Cell Biol. 21:501–521. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li W, Lu L, Lu J, Wang X, Yang C, Jin J,
Wu L, Hong X, Li F, Cao D, et al: cGAS-STING-mediated DNA sensing
maintains CD8(+) T cell stemness and promotes antitumor T cell
therapy. Sci Transl Med. 12:eaay90132020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Smith J, Tho LM, Xu N and Gillespie DA:
The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and
cancer. Adv Cancer Res. 108:73–112. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang W, Jin J, Wang Y, Fang L, Min L,
Wang X, Ding L, Weng L, Xiao T, Zhou T and Wang P: PD-L1 regulates
genomic stability via interaction with cohesin-SA1 in the nucleus.
Signal Transduct Target Ther. 6:812021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xue Z, Zheng S, Linghu D, Liu B, Yang Y,
Chen MK, Huang H, Song J, Li H, Wang J, et al: PD-L1 deficiency
sensitizes tumor cells to DNA-PK inhibition and enhances cGAS-STING
activation. Am J Cancer Res. 12:2363–2375. 2022.PubMed/NCBI
|
|
27
|
Cao L, Tian H, Fang M, Xu Z, Tang D, Chen
J, Yin J, Xiao H, Shang K, Han H and Li X: Activating cGAS-STING
pathway with ROS-responsive nanoparticles delivering a hybrid
prodrug for enhanced chemo-immunotherapy. Biomaterials.
290:1218562022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mahin J, Xu X, Li L and Zhang C:
cGAS/STING in skin melanoma: From molecular mechanisms to
therapeutics. Cell Commun Signal. 22:5532024. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang M, Zou Y, Zhou X and Zhou J:
Inhibitory targeting cGAS-STING-TBK1 axis: Emerging strategies for
autoimmune diseases therapy. Front Immunol. 13:9541292022.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lemos H, Mohamed E, Ou R, McCardle C,
Zheng X, McGuire K, Homer NZM, Mole DJ, Huang L and Mellor AL:
Co-treatments to Boost IDO activity and inhibit production of
downstream catabolites induce durable suppression of experimental
autoimmune encephalomyelitis. Front Immunol. 11:12562020.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu Z, Li Q, Zhu K, Zheng S, Hu H, Hou M,
Qi L, Chen S, Xu Y, Zhao B and Yan C: Cancer Radiosensitization
Nanoagent to Activate cGAS-STING pathway for molecular imaging
guided synergistic radio/chemo/immunotherapy. Adv Healthc Mater.
13:e23036262024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guo X, Tu P, Wang X, Du C, Jiang W, Qiu X,
Wang J, Chen L, Chen Y and Ren J: Decomposable nanoagonists enable
NIR-Elicited cGAS-STING activation for tandem-amplified
photodynamic-metalloimmunotherapy. Adv Mater. 36:e23130292024.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Deng L, Liang H, Xu M, Yang X, Burnette B,
Arina A, Li XD, Mauceri H, Beckett M, Darga T, et al:
STING-Dependent Cytosolic DNA sensing promotes radiation-induced
type I interferon-dependent antitumor immunity in immunogenic
tumors. Immunity. 41:843–852. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tumurkhuu G, Chen S, Montano EN, Ercan
Laguna D, De Los Santos G, Yu JM, Lane M, Yamashita M, Markman JL,
Blanco LP, et al: Oxidative DNA damage accelerates skin
inflammation in pristane-induced lupus model. Front Immunol.
11:5547252020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Meza-Sosa KF, Miao R, Navarro F, Zhang Z,
Zhang Y, Hu JJ, Hartford CCR, Li XL, Pedraza-Alva G, Pérez-Martínez
L, et al: SPARCLE, a p53-induced lncRNA, controls apoptosis after
genotoxic stress by promoting PARP-1 cleavage. Mol Cell.
82:785–802.e10. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yedidia-Aryeh L and Goldberg M: The
interplay between the cellular response to DNA double-strand breaks
and estrogen. Cells. 11:30972022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Puentes LN, Makvandi M and Mach RH:
Molecular imaging: PARP-1 and beyond. J Nucl Med. 62:765–770. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cleaver JE: Defective repair replication
of DNA in xeroderma pigmentosum. 1968. DNA Repair (Amst).
3:183–187. 2004.PubMed/NCBI
|
|
39
|
Mireles-Canales MP, González-Chávez SA,
Quiñonez-Flores CM, León-López EA and Pacheco-Tena C: DNA damage
and deficiencies in the mechanisms of its repair: Implications in
the pathogenesis of systemic lupus erythematosus. J Immunol Res.
2018:82143792018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cleaver JE: Cancer in xeroderma
pigmentosum and related disorders of DNA repair. Nat Rev Cancer.
5:564–573. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nath SK, Kilpatrick J and Harley JB:
Genetics of human systemic lupus erythematosus: The emerging
picture. Curr Opin Immunol. 16:794–800. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Crow YJ, Hayward BE, Parmar R, Robins P,
Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, et
al: Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1
cause Aicardi-Goutières syndrome at the AGS1 locus. Nat Genet.
38:917–920. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ishikawa H and Barber GN: STING is an
endoplasmic reticulum adaptor that facilitates innate immune
signalling. Nature. 455:674–678. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sun L, Wu J, Du F, Chen X and Chen ZJ:
Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates
the type I interferon pathway. Science. 339:786–791. 2013.
View Article : Google Scholar :
|
|
45
|
Haag SM, Gulen MF, Reymond L, Gibelin A,
Abrami L, Decout A, Heymann M, van der Goot FG, Turcatti G,
Behrendt R and Ablasser A: Targeting STING with covalent
small-molecule inhibitors. Nature. 559:269–273. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Knight JS, Subramanian V, O'Dell AA,
Yalavarthi S, Zhao W, Smith CK, Hodgin JB, Thompson PR and Kaplan
MJ: Peptidylarginine deiminase inhibition disrupts NET formation
and protects against kidney, skin and vascular disease in
lupus-prone MRL/lpr mice. Ann Rheum Dis. 74:2199–2206. 2015.
View Article : Google Scholar :
|
|
47
|
Adams C, Nair N, Plant D, Verstappen SMM,
Quach HL, Quach DL, Carvidi A, Nititham J, Nakamura M, Graf J, et
al: Identification of cell-specific differential DNA methylation
associated with methotrexate treatment response in rheumatoid
arthritis. Arthritis Rheumatol. 75:1088–1097. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mah LJ, El-Osta A and Karagiannis TC:
gammaH2AX: A sensitive molecular marker of DNA damage and repair.
Leukemia. 24:679–686. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ciccia A and Elledge SJ: The DNA damage
response: Making it safe to play with knives. Mol Cell. 40:179–204.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Marteijn JA, Lans H, Vermeulen W and
Hoeijmakers JH: Understanding nucleotide excision repair and its
roles in cancer and ageing. Nat Rev Mol Cell Biol. 15:465–481.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chang HHY, Pannunzio NR, Adachi N and
Lieber MR: Non-homologous DNA end joining and alternative pathways
to double-strand break repair. Nat Rev Mol Cell Biol. 18:495–506.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ceccaldi R and Cejka P: Mechanisms and
regulation of DNA end resection in the maintenance of genome
stability. Nat Rev Mol Cell Biol. 26:586–599. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chandramouly G, Zhao J, McDevitt S,
Rusanov T, Hoang T, Borisonnik N, Treddinick T, Lopezcolorado FW,
Kent T, Siddique LA, et al: Polθ reverse transcribes RNA and
promotes RNA-templated DNA repair. Sci Adv. 7:eabf17712021.
View Article : Google Scholar
|
|
54
|
Madabhushi R, Pan L and Tsai LH: DNA
damage and its links to neurodegeneration. Neuron. 83:266–282.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wallace SS: Base excision repair: A
critical player in many games. DNA Repair (Amst). 19:14–26. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Berti M, Cortez D and Lopes M: The
plasticity of DNA replication forks in response to clinically
relevant genotoxic stress. Nat Rev Mol Cell Biol. 21:633–651. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Whitaker AM, Schaich MA, Smith MR, Flynn
TS and Freudenthal BD: Base excision repair of oxidative DNA
damage: from mechanism to disease. Front Biosci (Landmark Ed).
22:1493–1522. 2017. View
Article : Google Scholar : PubMed/NCBI
|
|
58
|
Clouaire T and Legube G: A snapshot on the
cis chromatin response to DNA double-strand breaks. Trends Genet.
35:330–345. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Airik M, Phua YL, Huynh AB, McCourt BT,
Rush BM, Tan RJ, Vockley J, Murray SL, Dorman A, Conlon PJ and
Airik R: Persistent DNA damage underlies tubular cell
polyploidization and progression to chronic kidney disease in
kidneys deficient in the DNA repair protein FAN1. Kidney Int.
102:1042–1056. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhou W, Otto EA, Cluckey A, Airik R, Hurd
TW, Chaki M, Diaz K, Lach FP, Bennett GR, Gee HY, et al: FAN1
mutations cause karyomegalic interstitial nephritis, linking
chronic kidney failure to defective DNA damage repair. Nat Genet.
44:910–915. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Blackford AN and Jackson SP: ATM, ATR, and
DNA-PK: The trinity at the heart of the DNA damage response. Mol
Cell. 66:801–817. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sharma N, Coticchio G, Borini A, Tachibana
K, Nasmyth KA and Schuh M: Changes in DNA repair compartments and
cohesin loss promote DNA damage accumulation in aged oocytes. Curr
Biol. 34:5131–5148.e6. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zou S, Gou X and Wen K: Advances in the
role of long non-coding RNAs and RNA-binding proteins in regulating
DNA damage repair in cancer cells. Int J Mol Med. 52:932023.
View Article : Google Scholar
|
|
64
|
Michelini F, Pitchiaya S, Vitelli V,
Sharma S, Gioia U, Pessina F, Cabrini M, Wang Y, Capozzo I,
Iannelli F, et al: Damage-induced lncRNAs control the DNA damage
response through interaction with DDRNAs at individual
double-strand breaks. Nat Cell Biol. 19:1400–1411. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kastenhuber ER and Lowe SW: Putting p53 in
Context. Cell. 170:1062–1078. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Sahu S, Sridhar D, Abnave P, Kosaka N,
Dattani A, Thompson JM, Hill MA and Aboobaker A: Ongoing repair of
migration-coupled DNA damage allows planarian adult stem cells to
reach wound sites. Elife. 10:e637792021. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Motwani M, Pesiridis S and Fitzgerald KA:
DNA sensing by the cGAS-STING pathway in health and disease. Nat
Rev Genet. 20:657–674. 2019. View Article : Google Scholar
|
|
68
|
Kang TH and Sancar A: Circadian regulation
of DNA excision repair: Implications for chrono-chemotherapy. Cell
Cycle. 8:1665–1667. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Sulli G, Lam MTY and Panda S: Interplay
between circadian clock and cancer: New frontiers for cancer
treatment. Trends Cancer. 5:475–494. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Mingard C, Wu J, McKeague M and Sturla SJ:
Next-generation DNA damage sequencing. Chem Soc Rev. 49:7354–7377.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Awwad SW, Serrano-Benitez A, Thomas JC,
Gupta V and Jackson SP: Revolutionizing DNA repair research and
cancer therapy with CRISPR-Cas screens. Nat Rev Mol Cell Biol.
24:477–494. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Su D, Feng X, Colic M, Wang Y, Zhang C,
Wang C, Tang M, Hart T and Chen J: CRISPR/CAS9-based DNA damage
response screens reveal gene-drug interactions. DNA Repair (Amst).
87:1028032020. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Nicholson MD, Anderson CJ, Odom DT, Aitken
SJ and Taylor MS: DNA lesion bypass and the stochastic dynamics of
transcription-coupled repair. Proc Natl Acad Sci USA.
121:e24038711212024. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yang H, Lin P, Zhang B, Li F and Ling D: A
Nucleophilicity-Engineered DNA ligation blockade
nanoradiosensitizer induces irreversible DNA damage to overcome
cancer radioresistance. Adv Mater. 36:e24100312024. View Article : Google Scholar
|
|
75
|
Crow YJ and Manel N: Aicardi-Goutières
syndrome and the type I interferonopathies. Nat Rev Immunol.
15:429–440. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Glück S, Guey B, Gulen MF, Wolter K, Kang
TW, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L and Ablasser A:
Innate immune sensing of cytosolic chromatin fragments through cGAS
promotes senescence. Nat Cell Biol. 19:1061–1070. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zha S, Bassing CH, Sanda T, Brush JW,
Patel H, Goff PH, Murphy MM, Tepsuporn S, Gatti RA, Look AT and Alt
FW: ATM-deficient thymic lymphoma is associated with aberrant tcrd
rearrangement and gene amplification. J Exp Med. 207:1369–1380.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Grieves JL, Fye JM, Harvey S, Grayson JM,
Hollis T and Perrino FW: Exonuclease TREX1 degrades double-stranded
DNA to prevent spontaneous lupus-like inflammatory disease. Proc
Natl Acad Sci USA. 112:5117–5122. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Yousefzadeh MJ, Flores RR, Zhu Y,
Schmiechen ZC, Brooks RW, Trussoni CE, Cui Y, Angelini L, Lee KA,
McGowan SJ, et al: An aged immune system drives senescence and
ageing of solid organs. Nature. 594:100–105. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ahn J, Xia T, Konno H, Konno K, Ruiz P and
Barber GN: Inflammation-driven carcinogenesis is mediated through
STING. Nat Commun. 5:51662014. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Kell L, Simon AK, Alsaleh G and Cox LS:
The central role of DNA damage in immunosenescence. Front Aging.
4:12021522023. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Desdín-Micó G, Soto-Heredero G, Aranda JF,
Oller J, Carrasco E, Gabandé-Rodríguez E, Blanco EM, Alfranca A,
Cussó L, Desco M, et al: T cells with dysfunctional mitochondria
induce multimorbidity and premature senescence. Science.
368:1371–1376. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Dong Y, Yang C and Pan F:
Post-translational regulations of foxp3 in treg cells and their
therapeutic applications. Front Immunol. 12:6261722021. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Pieren DKJ, Smits NAM, Imholz S, Nagarajah
B, van Oostrom CT, Brandt RMC, Vermeij WP, Dollé MET and Guichelaar
T: Compromised DNA repair promotes the accumulation of regulatory T
cells with an aging-related phenotype and responsiveness. Front
Aging. 2:6671932021. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Karsten S: Targeting the DNA repair
enzymes MTH1 and OGG1 as a novel approach to treat inflammatory
diseases. Basic Clin Pharmacol Toxicol. 131:95–103. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Zhou Y and Mouw KW: DNA repair deficiency
and the immune microenvironment: A pathways perspective. DNA Repair
(Amst). 133:1035942024. View Article : Google Scholar
|
|
87
|
Schietinger A, Philip M, Krisnawan VE,
Chiu EY, Delrow JJ, Basom RS, Lauer P, Brockstedt DG, Knoblaugh SE,
Hämmerling GJ, et al: Tumor-specific T cell dysfunction is a
dynamic antigen-driven differentiation program initiated early
during tumorigenesis. Immunity. 45:389–401. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Vardhana SA, Hwee MA, Berisa M, Wells DK,
Yost KE, King B, Smith M, Herrera PS, Chang HY, Satpathy AT, et al:
Impaired mitochondrial oxidative phosphorylation limits the
self-renewal of T cells exposed to persistent antigen. Nat Immunol.
21:1022–1033. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
McKinney EF, Lyons PA, Carr EJ, Hollis JL,
Jayne DR, Willcocks LC, Koukoulaki M, Brazma A, Jovanovic V, Kemeny
DM, et al: A CD8+ T cell transcription signature predicts prognosis
in autoimmune disease. Nat Med. 16:586–591, 1p following 591. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Undi RB, Filiberti A, Ali N and Huycke MM:
Cellular carcinogenesis: Role of polarized macrophages in cancer
initiation. Cancers (Basel). 14:28112022. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Uchihara Y, Permata TBM, Sato H and
Shibata A: Modulation of immune responses by DNA damage signaling.
DNA Repair (Amst). 104:1031352021. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
West AP, Khoury-Hanold W, Staron M, Tal
MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff
DA, et al: Mitochondrial DNA stress primes the antiviral innate
immune response. Nature. 520:553–557. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Andrabi SA, Umanah GK, Chang C, Stevens
DA, Karuppagounder SS, Gagné JP, Poirier GG, Dawson VL and Dawson
TM: Poly(ADP-ribose) polymerase-dependent energy depletion occurs
through inhibition of glycolysis. Proc Natl Acad Sci USA.
111:10209–10214. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Shi C, Qin K, Lin A, Jiang A, Cheng Q, Liu
Z, Zhang J and Luo P: The role of DNA damage repair (DDR) system in
response to immune checkpoint inhibitor (ICI) therapy. J Exp Clin
Cancer Res. 41:2682022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Barros EM, McIntosh SA and Savage KI: The
DNA damage induced immune response: Implications for cancer
therapy. DNA Repair (Amst). 120:1034092022. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Caracciolo D, Riillo C, Arbitrio M, Di
Martino MT, Tagliaferri P and Tassone P: Error-prone DNA repair
pathways as determinants of immunotherapy activity: an emerging
scenario for cancer treatment. Int J Cancer. 147:2658–2668. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Willaume S, Rass E, Fontanilla-Ramirez P,
Moussa A, Wanschoor P and Bertrand P: A link between replicative
stress, lamin proteins, and inflammation. Genes (Basel).
12:5522021. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Marshak-Rothstein A: Toll-like receptors
in systemic autoimmune disease. Nat Rev Immunol. 6:823–835. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Crow YJ: Type I interferonopathies: A
novel set of inborn errors of immunity. Ann N Y Acad Sci.
1238:91–98. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Chen Y, Hua X, Huang B, Karsten S, You Z,
Li B, Li Y, Li Y, Liang J, Zhang J, et al: MutT homolog 1 inhibitor
karonudib attenuates autoimmune hepatitis by inhibiting DNA repair
in activated T cells. Hepatol Commun. 6:1016–1031. 2022. View Article : Google Scholar :
|
|
101
|
Shen P and Fillatreau S:
Antibody-independent functions of B cells: A focus on cytokines.
Nat Rev Immunol. 15:441–451. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Alt FW, Zhang Y, Meng FL, Guo C and Schwer
B: Mechanisms of programmed DNA lesions and genomic instability in
the immune system. Cell. 152:417–429. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Zouali M: B cells at the cross-roads of
autoimmune diseases and auto-inflammatory Syndromes. Cells.
11:40252022. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Marcus A, Mao AJ, Lensink-Vasan M, Wang L,
Vance RE and Raulet DH: Tumor-Derived cGAMP Triggers a
STING-mediated interferon response in non-tumor cells to activate
the NK cell response. Immunity. 49:754–763.e4. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Cheong A and Nagel ZD: Human variation in
DNA repair, immune function, and cancer risk. Front Immunol.
13:8995742022. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Liu H, Jia S, Guo K and Li R: INK4
cyclin-dependent kinase inhibitors as potential prognostic
biomarkers and therapeutic targets in hepatocellular carcinoma.
Biosci Rep. 42:BSR202210822022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Ma Y, Hossen MM, Huang JJ, Yin Z, Du J, Ye
Z, Zeng M and Huang Z: Growth arrest and DNA damage-inducible 45: A
new player on inflammatory diseases. Front Immunol. 16:15130692025.
View Article : Google Scholar :
|
|
108
|
Hou P, Zhu H, Sun X, Zhang N, Wang S,
Zheng X, Wang X, Feng Y, Zhang F, Li X, et al: PBLD orchestrates
the STING-Mediated antiviral immune response and autoimmune
diseases. Adv Sci (Weinh). 13:e145122026. View Article : Google Scholar :
|
|
109
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
López-Otín C, Blasco MA, Partridge L,
Serrano M and Kroemer G: The hallmarks of aging. Cell.
153:1194–1217. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Faget DV, Ren Q and Stewart SA: Unmasking
senescence: Context-dependent effects of SASP in cancer. Nat Rev
Cancer. 19:439–453. 2019. View Article : Google Scholar
|
|
112
|
Gad H, Koolmeister T, Jemth AS, Eshtad S,
Jacques SA, Ström CE, Svensson LM, Schultz N, Lundbäck T,
Einarsdottir BO, et al: MTH1 inhibition eradicates cancer by
preventing sanitation of the dNTP pool. Nature. 508:215–221. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
López-Otín C, Blasco MA, Partridge L,
Serrano M and Kroemer G: Hallmarks of aging: An expanding universe.
Cell. 186:243–278. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Goronzy JJ and Weyand CM: Successful and
Maladaptive T Cell Aging. Immunity. 46:364–378. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Cimprich KA, Li GM, Demaria S, Gekara NO,
Zha S and Chen Q: The crosstalk between DNA repair and immune
responses. Mol Cell. 83:3582–3587. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Kim DE, Dollé MET, Vermeij WP, Gyenis A,
Vogel K, Hoeijmakers JHJ, Wiley CD, Davalos AR, Hasty P, Desprez PY
and Campisi J: Deficiency in the DNA repair protein ERCC1 triggers
a link between senescence and apoptosis in human fibroblasts and
mouse skin. Aging Cell. 19:e130722020. View Article : Google Scholar :
|
|
117
|
Rossi DJ, Bryder D, Seita J, Nussenzweig
A, Hoeijmakers J and Weissman IL: Deficiencies in DNA damage repair
limit the function of haematopoietic stem cells with age. Nature.
447:725–729. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Cunha ALS and Perazzio SF: Effects of
immune exhaustion and senescence of innate immunity in autoimmune
disorders. Braz J Med Biol Res. 57:e132252024. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Jaiswal S, Fontanillas P, Flannick J,
Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N,
Chavez A, et al: Age-related clonal hematopoiesis associated with
adverse outcomes. N Engl J Med. 371:2488–2498. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Genovese G, Kähler AK, Handsaker RE,
Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM,
Fromer M, et al: Clonal hematopoiesis and blood-cancer risk
inferred from blood DNA sequence. N Engl J Med. 371:2477–2487.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Chang J, Wang Y, Shao L, Laberge RM,
Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W,
et al: Clearance of senescent cells by ABT263 rejuvenates aged
hematopoietic stem cells in mice. Nat Med. 22:78–83. 2016.
View Article : Google Scholar
|
|
122
|
Xu M, Pirtskhalava T, Farr JN, Weigand BM,
Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser
DG, et al: Senolytics improve physical function and increase
lifespan in old age. Nat Med. 24:1246–1256. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Galita G, Sarnik J, Zajac G, Brzezinska O,
Budlewski T, Poplawska M, Przybyłowska-Sygut K, Makowska JS and
Poplawski T: The association between inefficient repair of
oxidative DNA lesions and common polymorphisms of the key base
excision repair genes as well as their expression levels in
patients with rheumatoid arthritis. Arch Med Sci. 21:1010–1017.
2023.PubMed/NCBI
|
|
124
|
Filipek B, Macieja A, Binda A,
Szelenberger R, Gorniak L, Miller E, Swiderek-Matysiak M, Stasiolek
M, Majsterek I and Poplawski T: Oxidative DNA damage and repair
dynamics in multiple sclerosis: Insights from comet assay kinetics,
base excision repair gene expression, and genotype analysis.
Biomolecules. 15:7562025. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Ryo K, Yamada H, Nakagawa Y, Tai Y, Obara
K, Inoue H, Mishima K and Saito I: Possible involvement of
oxidative stress in salivary gland of patients with Sjogren's
syndrome. Pathobiology. 73:252–260. 2006. View Article : Google Scholar
|
|
126
|
Pagano G, Castello G and Pallardó FV:
Sjøgren's syndrome-associated oxidative stress and mitochondrial
dysfunction: prospects for chemoprevention trials. Free Radic Res.
47:71–73. 2013. View Article : Google Scholar
|
|
127
|
Caldecott KW: XRCC1 protein; Form and
function. DNA Repair (Amst). 81:1026642019. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
An Q, Zhao J, Zhu X, Yang B, Wu Z, Su Y,
Zhang L, Xu K and Ma D: Exploiting the role of T cells in the
pathogenesis of Sjögren's Syndrome for therapeutic treatment. Front
Immunol. 13:9958952022. View Article : Google Scholar
|
|
129
|
Guo K, Major G, Foster H, Bassendine M,
Collier J, Ross D and Griffiths I: Defective repair of
O6-methylguanine-DNA in primary Sjögren's syndrome patients
predisposed to lymphoma. Ann Rheum Dis. 54:229–232. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Vijayraghavan S, Blouin T, McCollum J,
Porcher L, Virard F, Zavadil J, Feghali-Bostwick C and Saini N:
Widespread mutagenesis and chromosomal instability shape somatic
genomes in systemic sclerosis. Nat Commun. 15:88892024. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Henriksson G, Brant M, Sallmyr A,
Fukushima S, Manthorpe R and Bredberg A: Enhanced DNA
damage-induced p53 peptide phosphorylation and cell-cycle arrest in
Sjögren's syndrome cells. Eur J Clin Invest. 32:458–465. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Ka Y, Tan T, Fan Y, Liu W, Wang A, Wang W,
Yuzhen G, Zhang J, Yao X, Lin X and Wu Y: STING pathways and
Sjögren's syndrome: Exploration from mechanism to treatment. Front
Immunol. 16:16490462025. View Article : Google Scholar
|
|
133
|
Goddard AM, Cho MG, Lerner LM and Gupta
GP: Mechanisms of immune sensing of DNA damage. J Mol Biol.
436:1684242024. View Article : Google Scholar
|
|
134
|
Zhu W and Wang Y, Guan Y, Lu Y, Li Y, Sun
L and Wang Y: Rapamycin can alleviate the submandibular gland
pathology of Sjögren's syndrome by limiting the activation of
cGAS-STING signaling pathway. Inflammopharmacology. 32:1113–1131.
2024. View Article : Google Scholar
|
|
135
|
Zhang Q, Yang XR and Deng Y: Iguratimod
alleviates experimental Sjögren's Syndrome by inhibiting NLRP3
inflammasome activation. Cell Biochem Biophys. 82:2275–2283. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Zheng L, Gu M, Li X, Hu X, Chen C, Kang Y,
Pan B, Chen W, Xian G, Wu X, et al: ITGA5(+) synovial fibroblasts
orchestrate proinflammatory niche formation by remodelling the
local immune microenvironment in rheumatoid arthritis. Ann Rheum
Dis. 84:232–252. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Lin W, Shen P, Song Y, Huang Y and Tu S:
Reactive oxygen species in autoimmune cells: Function,
differentiation, and metabolism. Front Immunol. 12:6350212021.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Zhang R, Lin X, Lin R, Chen Z, Miao C,
Wang Y, Deng X, Lin J, Lin S, Weng S and Chen M: Effectively
alleviate rheumatoid arthritis via maintaining redox balance,
inducing macrophage repolarization and restoring homeostasis of
fibroblast-like synoviocytes by metformin-derived carbon dots. J
Nanobiotechnology. 23:582025. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Xu C, Jing W, Liu C, Yuan B, Zhang X, Liu
L, Zhang F, Chen P, Liu Q, Wang H and Du X: Cytoplasmic DNA and
AIM2 inflammasome in RA: where they come from and where they go?
Front Immunol. 15:13433252024. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Souliotis VL, Vlachogiannis NI, Pappa M,
Argyriou A and Sfikakis PP: DNA damage accumulation, defective
chromatin organization and deficient DNA repair capacity in
patients with rheumatoid arthritis. Clin Immunol. 203:28–36. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Galita G, Sarnik J, Brzezinska O,
Budlewski T, Poplawska M, Sakowski S, Dudek G, Majsterek I,
Makowska J and Poplawski T: The association between inefficient
repair of DNA double-strand breaks and common polymorphisms of the
HRR and NHEJ repair genes in patients with rheumatoid arthritis.
Int J Mol Sci. 25:26192024. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Hajizadeh S, DeGroot J, TeKoppele JM,
Tarkowski A and Collins LV: Extracellular mitochondrial DNA and
oxidatively damaged DNA in synovial fluid of patients with
rheumatoid arthritis. Arthritis Res Ther. 5:R234–R240. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Lee HR, Yoo SJ, Kim J, Yoo IS, Park CK and
Kang SW: The effect of nicotinamide adenine dinucleotide phosphate
oxidase 4 on migration and invasion of fibroblast-like synoviocytes
in rheumatoid arthritis. Arthritis Res Ther. 22:1162020. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Zhu Q and Zhou H: The role of cGAS-STING
signaling in rheumatoid arthritis: from pathogenesis to therapeutic
targets. Front Immunol. 15:14660232024. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Corsiero E, Pratesi F, Prediletto E,
Bombardieri M and Migliorini P: NETosis as source of autoantigens
in rheumatoid arthritis. Front Immunol. 7:4852016. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Rodríguez-Ubreva J, de la Calle-Fabregat
C, Li T, Ciudad L, Ballestar ML, Català-Moll F, Morante-Palacios O,
Garcia-Gomez A, Celis R, Humby F, et al: Inflammatory cytokines
shape a changing DNA methylome in monocytes mirroring disease
activity in rheumatoid arthritis. Ann Rheum Dis. 78:1505–1516.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Dong C, Liu Y, Sun C, Liang H, Dai L, Shen
J, Wei S, Guo S, Leong KW, Chen Y, et al: Identification of
specific joint-inflammatogenic cell-free DNA molecules from
synovial fluids of patients with rheumatoid arthritis. Front
Immunol. 11:6622020. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Nair N, Plant D, Verstappen SM, Isaacs JD,
Morgan AW, Hyrich KL, Barton A and Wilson AG; MATURA investigators:
Differential DNA methylation correlates with response to
methotrexate in rheumatoid arthritis. Rheumatology (Oxford).
59:1364–1371. 2020. View Article : Google Scholar :
|
|
149
|
Lee HT, Lin CS, Lee CS, Tsai CY and Wei
YH: Increased 8-hydroxy-2′-deoxyguanosine in plasma and decreased
mRNA expression of human 8-oxoguanine DNA glycosylase 1,
anti-oxidant enzymes, mitochondrial biogenesis-related proteins and
glycolytic enzymes in leucocytes in patients with systemic lupus
erythematosus. Clin Exp Immunol. 176:66–77. 2014. View Article : Google Scholar
|
|
150
|
Bai Y, Tong Y, Liu Y and Hu H: Self-dsDNA
in the pathogenesis of systemic lupus erythematosus. Clin Exp
Immunol. 191:1–10. 2018. View Article : Google Scholar
|
|
151
|
Fredi M, Bianchi M, Andreoli L, Greco G,
Olivieri I, Orcesi S, Fazzi E, Cereda C and Tincani A: Typing TREX1
gene in patients with systemic lupus erythematosus. Reumatismo.
67:1–7. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Xu S, Xiao N, Du H, Zhou X, Huang M, Feng
S, Hu S, Zhang X, Zhang S, Cui D, et al: The SLE-associated
TREX1-P212fs mutation disrupts ER association leading to type I
interferonopathy. FASEB J. 38:e702132024. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Stetson DB, Ko JS, Heidmann T and
Medzhitov R: Trex1 prevents cell-intrinsic initiation of
autoimmunity. Cell. 134:587–598. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Poznyak AV, Orekhov NA, Churov AV,
Starodubtseva IA, Beloyartsev DF, Kovyanova TI, Sukhorukov VN and
Orekhov AN: Mitochondrial dysfunction in systemic lupus
erythematosus: Insights and therapeutic potential. Diseases.
12:2262024. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Sprenger HG, MacVicar T, Bahat A, Fiedler
KU, Hermans S, Ehrentraut D, Ried K, Milenkovic D, Bonekamp N,
Larsson NG, et al: Cellular pyrimidine imbalance triggers
mitochondrial DNA-dependent innate immunity. Nat Metab. 3:636–650.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Perl A, Nagy G, Gergely P, Puskas F, Qian
Y and Banki K: Apoptosis and mitochondrial dysfunction in
lymphocytes of patients with systemic lupus erythematosus. Methods
Mol Med. 102:87–114. 2004.PubMed/NCBI
|
|
157
|
Sumikawa MH, Iwata S, Zhang M, Miyata H,
Ueno M, Todoroki Y, Nagayasu A, Kanda R, Sonomoto K, Torimoto K, et
al: An enhanced mitochondrial function through glutamine metabolism
in plasmablast differentiation in systemic lupus erythematosus.
Rheumatology (Oxford). 61:3049–3059. 2022. View Article : Google Scholar
|
|
158
|
Bradford HF, McDonnell TCR, Stewart A,
Skelton A, Ng J, Baig Z, Fraternali F, Dunn-Walters D, Isenberg DA,
Khan AR, et al: Thioredoxin is a metabolic rheostat controlling
regulatory B cells. Nat Immunol. 25:873–885. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Paluri SL, Burak M, Senejani AG, Levinson
M, Rahim T, Clairmont K, Kashgarian M, Alvarado-Cruz I, Meas R,
Cardó-Vila M, et al: DNA glycosylase deficiency leads to decreased
severity of lupus in the Polb-Y265C mouse model. DNA Repair (Amst).
105:1031522021. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Senejani AG, Liu Y, Kidane D, Maher SE,
Zeiss CJ, Park HJ, Kashgarian M, McNiff JM, Zelterman D, Bothwell
AL and Sweasy JB: Mutation of POLB causes lupus in mice. Cell Rep.
6:1–8. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Ramirez-Ortiz ZG, Pendergraft WF III,
Prasad A, Byrne MH, Iram T, Blanchette CJ, Luster AD, Hacohen N, El
Khoury J and Means TK: The scavenger receptor SCARF1 mediates the
clearance of apoptotic cells and prevents autoimmunity. Nat
Immunol. 14:917–926. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Muñoz LE, Lauber K, Schiller M, Manfredi
AA and Herrmann M: The role of defective clearance of apoptotic
cells in systemic autoimmunity. Nat Rev Rheumatol. 6:280–289. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Yang D, Peng N, Zhang H, Qiu Z, Xu L and
Pan M: Cordycepin ameliorates autoimmunity by promoting STING
degradation via autophagy pathway. Br J Pharmacol. 182:1546–1560.
2025. View Article : Google Scholar
|
|
164
|
Zhang ZD, Shi CR, Li FX, Gan H, Wei Y,
Zhang Q, Shuai X, Chen M, Lin YL, Xiong TC, et al: Disulfiram
ameliorates STING/MITA-dependent inflammation and autoimmunity by
targeting RNF115. Cell Mol Immunol. 21:275–291. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Peng Y, Tao H, Liu D, Tang D, Wen C, Wu M,
Xu T, Wang G, Zheng X and Dai Y: Comprehensive analysis of eccDNA
characteristics and associated genes expression in peripheral blood
of ASLE and ISLE patients. Epigenetics. 20:24779032025. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Micheli C, Parma A, Tani C, Di Bello D,
Falaschi A, Chiaramonte A, Testi S, Mosca M and Scarpato R: UCTD
and SLE patients show increased levels of oxidative and DNA damage
together with an altered kinetics of DSB repair. Mutagenesis.
36:429–436. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Piña r-Morales R, Durán R, Bautista-Ga
rcía A, García-Mansilla MJ, Aliaga-Gaspar P, Vives-Montero F and
Barrero-Hernández FJ: The impact of oxidative stress on symptoms
associated with multiple sclerosis. Sci Rep. 15:229832025.
View Article : Google Scholar
|
|
168
|
Liu X, Xin DE, Zhong X, Zhao C, Li Z,
Zhang L, Dourson AJ, Lee L, Mishra S, Bayat AE, et al:
Small-molecule-induced epigenetic rejuvenation promotes SREBP
condensation and overcomes barriers to CNS myelin regeneration.
Cell. 187:2465–2484.e22. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
López-Muguruza E and Matute C: Alterations
of oligodendrocyte and myelin energy metabolism in multiple
sclerosis. Int J Mol Sci. 24:129122023. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Blanchin S, Estienne V, Durand-Gorde JM,
Carayon P and Ruf J: Complement activation by direct C4 binding to
thyroperoxidase in Hashimoto's thyroiditis. Endocrinology.
144:5422–5429. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Marique L, Van Regemorter V, Gérard AC,
Craps J, Senou M, Marbaix E, Rahier J, Daumerie C, Mourad M,
Lengelé B, et al: The expression of dual oxidase, thyroid
peroxidase, and caveolin-1 differs according to the type of immune
response (TH1/TH2) involved in thyroid autoimmune disorders. J Clin
Endocrinol Metab. 99:1722–1732. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Li F, Wu Y, Chen L, Hu L, Zhu F and He Q:
High iodine induces DNA damage in autoimmune thyroiditis partially
by inhibiting the DNA repair protein MTH1. Cell Immunol.
344:1039482019. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Ren B, Chen Y, Liu J, Zhou Z, He Y, Wan S,
Chen Y, Wu X, Du M, Gao H, et al: DNA methylation of genes that
mediate autophagosome formation contributes to iodine-induced
autoimmune thyroiditis: A population-based study conducted at
regions with different iodine levels in China. Int J Hyg Environ
Health. 265:1145372025. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Mackenzie KJ, Carroll P, Martin CA, Murina
O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch
A, et al: cGAS surveillance of micronuclei links genome instability
to innate immunity. Nature. 548:461–465. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Kroemer G, Galluzzi L, Kepp O and Zitvogel
L: Immunogenic cell death in cancer therapy. Annu Rev Immunol.
31:51–72. 2013. View Article : Google Scholar
|
|
176
|
Zhang D and Zhang B: cGAS/STING signaling
pathway in gynecological malignancies: From molecular mechanisms to
therapeutic values. Front Immunol. 16:15257362025. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Miller KN, Li B, Pierce-Hoffman HR, Patel
S, Lei X, Rajesh A, Teneche MG, Havas AP, Gandhi A, Macip CC, et
al: p53 enhances DNA repair and suppresses cytoplasmic chromatin
fragments and inflammation in senescent cells. Nat Commun.
16:22292025. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Liu X, Wang S, Lv H, Chen E, Yan L and Yu
J: Advances in the relationship of immune checkpoint inhibitors and
DNA damage repair. Curr Res Transl Med. 73:1034942025.PubMed/NCBI
|
|
179
|
Jiao X, Liu J, Wu Y, Zhong Q, Zhu L, Wang
L, Li H, Xiang M, Zhao X, Zhao G, et al: Sequential treatment with
PARPi and WEE1i enhances antitumor immune responses in preclinical
models of ovarian cancer. Sci Transl Med. 17:eadu69892025.
View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Clarke TL and Mostoslavsky R: DNA repair
as a shared hallmark in cancer and ageing. Mol Oncol. 16:3352–3379.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Kulkarni S, Brownlie J, Jeyapalan JN,
Mongan NP, Rakha EA and Madhusudan S: Evolving DNA repair synthetic
lethality targets in cancer. Biosci Rep. 42:BSR202217132022.
View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Farmer H, McCabe N, Lord CJ, Tutt AN,
Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I,
Knights C, et al: Targeting the DNA repair defect in BRCA mutant
cells as a therapeutic strategy. Nature. 434:917–921. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Cavone L, Aldinucci A, Ballerini C,
Biagioli T, Moroni F and Chiarugi A: PARP-1 inhibition prevents CNS
migration of dendritic cells during EAE, suppressing the
encephalitogenic response and relapse severity. Mult Scler.
17:794–807. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Polasek TM, Cole A, Bozón V, Manyak E,
Novak J, Yang B, Johnston BA, Parasuraman S, Paneliya KJ and Schuck
V: First-in-human phase 1 study to evaluate the clinical
pharmacology properties of RBN-3143, a novel inhibitor of
mono-adenosine diphosphate ribosyltransferase-PARP14. Clin
Pharmacol Drug Dev. 14:493–504. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Meira M, Sievers C, Hoffmann F, Bodmer H,
Derfuss T, Kuhle J, Haghikia A, Kappos L and Lindberg RL: PARP-1
deregulation in multiple sclerosis. Mult Scler J Exp Transl Clin.
5:20552173198946042019.
|
|
186
|
Franzese O and Graziani G: Role of PARP
inhibitors in cancer immunotherapy: Potential friends to immune
activating molecules and foes to immune checkpoints. Cancers
(Basel). 14:56332022. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Asare A, Corvigno S, Yao J, Zhao L,
Fleming ND, Celestino J, Hajek RA, Kim MS, Legarreta AF, Lu KH, et
al: Landscape of clonal hematopoiesis prior to PARP inhibitor
treatment in patients with ovarian cancer. Cancer Res. 84:5070.
2024. View Article : Google Scholar
|
|
188
|
Shuai Q, Bai X, Li G, Wang L, Chen J and
Chen L: Hematopoietic adverse events associated with PARP
inhibitors: A FAERS database study. Expert Opin Drug Saf. 1–11.
2024.Epub ahead of print.
|
|
189
|
Kondratska O, Grushka N, Pavlovych S,
Krasutska N, Tsyhankov S and Yanchii R: Effects of Poly
(ADP-ribose) polymerase inhibition on DNA integrity and gene
expression in ovarian follicular cells in mice with endotoxemia.
Iran Biomed J. 26:44–52. 2022.
|
|
190
|
Firestein GS and McInnes IB:
Immunopathogenesis of rheumatoid arthritis. Immunity. 46:183–196.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Rose M, Burgess JT, O'Byrne K, Richard DJ
and Bolderson E: PARP Inhibitors: Clinical relevance, mechanisms of
action and tumor resistance. Front Cell Dev Biol. 8:5646012020.
View Article : Google Scholar :
|
|
192
|
Morice PM, Leary A, Dolladille C, Chrétien
B, Poulain L, González-Martín A, Moore K, O'Reilly EM, Ray-Coquard
I and Alexandre J: Myelodysplastic syndrome and acute myeloid
leukaemia in patients treated with PARP inhibitors: A safety
meta-analysis of randomised controlled trials and a retrospective
study of the WHO pharmacovigilance database. Lancet Haematol.
8:e122–e134. 2021. View Article : Google Scholar
|
|
193
|
Xu Z, Vandenberg CJ, Lieschke E, Di Rago
L, Scott CL and Majewski IJ: CHK2 inhibition provides a strategy to
suppress hematologic toxicity from PARP Inhibitors. Mol Cancer Res.
19:1350–1360. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Cecchini M, Walther Z, Wei W, Hafez N,
Pilat MJ, Boerner SA, Durecki DE, Eder JP, Schalper KA, Chen AP and
LoRusso P: NCI 7977: A phase I dose-escalation study of
intermittent oral ABT-888 (Veliparib) plus intravenous irinotecan
administered in patients with advanced solid tumors. Cancer Res
Commun. 3:1113–1117. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Gralewska P, Gajek A, Marczak A and
Rogalska A: Targeted nanocarrier-based drug delivery strategies for
improving the therapeutic efficacy of PARP Inhibitors against
ovarian cancer. Int J Mol Sci. 25:83042024. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Sriramulu S, Thoidingjam S, Brown SL,
Siddiqui F, Movsas B and Nyati S: Molecular targets that sensitize
cancer to radiation killing: From the bench to the bedside. Biomed
Pharmacother. 158:1141262023. View Article : Google Scholar
|
|
197
|
Zhang J, Si J, Gan L, Zhou R, Guo M and
Zhang H: Harnessing the targeting potential of differential
radiobiological effects of photon versus particle radiation for
cancer treatment. J Cell Physiol. 236:1695–1711. 2021. View Article : Google Scholar
|
|
198
|
Zeng Q, Liu M, Wang Z, Zhou R and Ai K:
Enhancing radiotherapy-induced anti-tumor immunity via
nanoparticle-mediated STING agonist synergy. Mol Cancer.
24:1762025. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Yue B, Gao W, Lovell JF, Jin H and Huang
J: The cGAS-STING pathway in cancer immunity: dual roles,
therapeutic strategies, and clinical challenges. Essays Biochem.
69:EBC202530062025. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Taniguchi H, Chakraborty S, Takahashi N,
Banerjee A, Caeser R, Zhan YA, Tischfield SE, Chow A, Nguyen EM,
Villalonga ÁQ, et al: ATR inhibition activates cancer cell
cGAS/STING-interferon signaling and promotes antitumor immunity in
small-cell lung cancer. Sci Adv. 10:eado46182024. View Article : Google Scholar :
|
|
201
|
Qiu Y, Hu X, Zeng X and Wang H: Triple
kill: DDR inhibitors, radiotherapy and immunotherapy leave cancer
cells with no escape. Acta Biochim Biophys Sin (Shanghai).
54:1569–1576. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Xie D, Jiang B, Wang S, Wang Q and Wu G:
The mechanism and clinical application of DNA damage repair
inhibitors combined with immune checkpoint inhibitors in the
treatment of urologic cancer. Front Cell Dev Biol. 11:12004662023.
View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Hu M, Zhou M, Bao X, Pan D, Jiao M, Liu X,
Li F and Li CY: ATM inhibition enhances cancer immunotherapy by
promoting mtDNA leakage and cGAS/STING activation. J Clin Invest.
131:e1393332021. View Article : Google Scholar :
|
|
204
|
Lutfi N, Galindo-Campos MA and Yélamos J:
Impact of DNA damage response-targeted therapies on the immune
response to tumours. Cancers (Basel). 13:60082021. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Pedre B, Barayeu U, Ezeriņa D and Dick TP:
The mechanism of action of N-acetylcysteine (NAC): The emerging
role of H(2)S and sulfane sulfur species. Pharmacol Ther.
228:1079162021. View Article : Google Scholar
|
|
206
|
Muldoon LL, Wu YJ, Pagel MA and Neuwelt
EA: N-acetylcysteine chemoprotection without decreased cisplatin
antitumor efficacy in pediatric tumor models. J Neurooncol.
121:433–440. 2015. View Article : Google Scholar
|
|
207
|
Zavala-Valencia AC, Velasco-Hidalgo L,
Martínez-Avalos A, Castillejos-López M and Torres-Espíndola LM:
Effect of N-Acetylcysteine on cisplatin toxicity: A review of the
literature. Biologics. 18:7–19. 2024.PubMed/NCBI
|
|
208
|
Sancho-Martínez SM, Prieto-García L,
Prieto M, Fuentes-Calvo I, López-Novoa JM, Morales AI,
Martínez-Salgado C and López-Hernández FJ: N-acetylcysteine
transforms necrosis into apoptosis and affords tailored protection
from cisplatin cytotoxicity. Toxicol Appl Pharmacol. 349:83–93.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Topalian SL, Hodi FS, Brahmer JR,
Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD,
Sosman JA, Atkins MB, et al: Safety, activity, and immune
correlates of anti-PD-1 antibody in cancer. N Engl J Med.
366:2443–2454. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Heitzer E, Ulz P and Geigl JB: Circulating
tumor DNA as a liquid biopsy for cancer. Clin Chem. 61:112–123.
2015. View Article : Google Scholar
|
|
211
|
Ramachandran A and Jaeschke H: Oxidative
stress and acute hepatic injury. Curr Opin Toxicol. 7:17–21. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Wengner AM, Siemeister G, Lücking U,
Lefranc J, Wortmann L, Lienau P, Bader B, Bömer U, Moosmayer D,
Eberspächer U, et al: The novel ATR inhibitor BAY 1895344 Is
efficacious as monotherapy and combined with DNA damage-inducing or
repair-compromising therapies in preclinical cancer models. Mol
Cancer Ther. 19:26–38. 2020. View Article : Google Scholar
|
|
213
|
Zerio CJ, Bai Y, Sosa-Alvarado BA, Guzi T
and Lander GC: Human polymerase θ helicase positions DNA
microhomologies for double-strand break repair. Nat Struct Mol
Biol. 32:1061–1068. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Lodovichi S, Cervelli T, Pellicioli A and
Galli A: Inhibition of DNA repair in cancer therapy: Toward a
multi-target approach. Int J Mol Sci. 21:66842020. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Luo Z, Huang Y, Chen S, Zhang B, Huang H,
Dabiri S, Chen Y, Zhang A, Andreas AR and Li S: Delivery of PARP
inhibitors through 2HG-incorporated liposomes for synergistically
targeting DNA repair in cancer. Cancer Lett. 604:2172682024.
View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Rice GI, Rodero MP and Crow YJ: Human
disease phenotypes associated with mutations in TREX1. J Clin
Immunol. 35:235–243. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Gall A, Treuting P, Elkon KB, Loo YM, Gale
M Jr, Barber GN and Stetson DB: Autoimmunity initiates in
nonhematopoietic cells and progresses via lymphocytes in an
interferon-dependent autoimmune disease. Immunity. 36:120–131.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Espinosa A, Hennig J, Ambrosi A,
Anandapadmanaban M, Abelius MS, Sheng Y, Nyberg F, Arrowsmith CH,
Sunnerhagen M and Wahren-Herlenius M: Anti-Ro52 autoantibodies from
patients with Sjögren's syndrome inhibit the Ro52 E3 ligase
activity by blocking the E3/E2 interface. J Biol Chem.
286:36478–36491. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Luo WD, Wang YP, Lv J, Liu Y, Qu YQ, Xu
XF, Yang LJ, Lin ZC, Wang LN, Chen RH, et al: Age-related self-DNA
accumulation may accelerate arthritis in rats and in human
rheumatoid arthritis. Nat Commun. 14:43942023. View Article : Google Scholar :
|
|
220
|
Lee HT, Lin CS, Pan SC, Chen WS, Tsai CY
and Wei YH: The role of plasma cell-free mitochondrial DNA and
nuclear DNA in systemic lupus erythematosus. Front Biosci (Landmark
Ed). 27:3332022. View Article : Google Scholar
|
|
221
|
Alvarado-Cruz I, Mahmoud M, Khan M, Zhao
S, Oeck S, Meas R, Clairmont K, Quintana V, Zhu Y, Porciuncula A,
et al: Differential immunomodulatory effect of PARP inhibition in
BRCA1 deficient and competent tumor cells. Biochem Pharmacol.
184:1143592021. View Article : Google Scholar
|