Introduction of ferroptosis
Musculoskeletal disorders, including tendinopathy,
sarcopenia, osteoarthritis and osteoporosis, represent a major and
growing global health burden, with osteoarthritis affecting 32.5
million adults in the United States, osteoporosis affecting
8.4-17.7% of middle-aged and elderly adults and nearly half of
skeletal muscle mass being lost by the age of 80 years due to
sarcopenia (1,2), particularly in aging populations
(3). These conditions are
characterized by progressive tissue degeneration, impaired
regenerative capacity and limited therapeutic options capable of
restoring structural and functional integrity. Despite advances in
understanding inflammatory signaling and mechanical stress-related
injury, several aspects of musculoskeletal degeneration remain
insufficiently explained at the level of regulated cell death and
metabolic vulnerability (4,5).
This gap suggests that additional mechanisms may underlie the
progressive loss of tissue homeostasis observed in these disorders.
Previous studies have identified ferroptosis, an iron-dependent
form of regulated cell death driven by lipid peroxidation, as a
potential contributor to tissue degeneration in multiple organ
systems (6,7). Increasing evidence indicates that
ferroptosis may intersect with iron dysregulation, oxidative stress
and aging-associated metabolic alterations, features that are
highly relevant to musculoskeletal tissues (8-11). These findings raise the
possibility that ferroptosis represents a previously
underappreciated mechanism in musculoskeletal decline (Fig. 1).
Ferroptosis, first identified in 2012 (7), is a regulated form of
iron-dependent cell death characterized by excessive lipid
peroxidation and failure of antioxidant defense systems. It is
driven by intracellular iron accumulation, depletion of glutathione
(GSH), inactivation of GSH peroxidase 4 (GPX4) and subsequent
oxidative damage to membrane phospholipids (11-14). Unlike necrosis or apoptosis,
ferroptosis is associated with cellular metabolic state and redox
homeostasis, forming a self-amplifying cycle among iron overload,
reactive oxygen species (ROS) generation and lipid peroxidation
(12,15). At the molecular level,
dysregulation of iron-handling proteins, including transferrin
receptor (TFRC), ferritin and ferroportin, contributes to free iron
accumulation and promotes Fenton chemistry-mediated ROS production
(16-23). Lipid peroxidation of
polyunsaturated fatty acids, particularly in cellular membranes,
represents a hallmark event of ferroptosis, while GPX4 and the
system the cystine/glutamate antiporter (Xc−/) GSH axis
serve as central antioxidant defenses that counteract this process
(24-28). Mitochondrial dysfunction and
oxidative stress further amplify ferroptotic signaling, as altered
membrane potential, ROS overproduction and ferritinophagy-mediated
iron release accelerate lipid damage and cell death (Fig. 2) (29-33).
Musculoskeletal tissues possess unique metabolic and
structural characteristics that may render them particularly
susceptible to ferroptotic injury (4). Tendons and cartilage are relatively
avascular and rely heavily on tightly regulated redox balance,
while skeletal muscle exhibits high metabolic demand and
mitochondrial activity (34).
Moreover, aging-associated alterations in iron homeostasis, stem
cell function and antioxidant capacity may further amplify
ferroptotic vulnerability across these tissues (34). These features suggest that
ferroptosis could represent a convergent mechanism underlying
degeneration and impaired regeneration in the musculoskeletal
system.
The present review aims to provide a comprehensive
and disease-oriented synthesis of current evidence regarding
ferroptosis in musculoskeletal disorders. The fundamental molecular
mechanisms of ferroptosis is first summarized followed by
examination of its involvement in major musculoskeletal conditions,
including tendinopathy, sarcopenia, osteoarthritis and
osteoporosis. The present review further discuss aging-related
modulation, experimental modeling strategies and emerging
therapeutic perspectives to clarify the potential translational
significance of targeting ferroptosis in musculoskeletal
disease.
Application of ferroptosis in the
musculoskeletal system
Musculoskeletal tissues exhibit distinct metabolic
and structural characteristics that may influence their
susceptibility to ferroptotic injury. Skeletal muscle is highly
metabolically active and relies on robust mitochondrial function to
sustain contractile activity. This high oxidative demand renders
muscle fibers and muscle stem cells particularly sensitive to redox
imbalance and iron-mediated lipid peroxidation (35-38). Notably, activation of ferroptosis
in muscle stem cells has been shown to impair regenerative capacity
by disrupting intracellular iron homeostasis and antioxidant
defenses, leading to excessive lipid peroxidation and mitochondrial
dysfunction. These alterations compromise satellite cell viability
and functional maintenance, thereby contributing to accelerated
muscle aging (39). Tendons, by
contrast, are relatively avascular connective tissues with limited
intrinsic antioxidant capacity (40,41). Their restricted blood supply may
compromise redox buffering under stress conditions, potentially
increasing vulnerability to iron accumulation and lipid
peroxidation. Aging-associated dysfunction of tendon-derived stem
cells further contributes to impaired repair, and emerging research
suggests that ferroptosis may participate in this degenerative
process (9,42,43). However, the interaction between
cellular senescence, iron metabolism and ferroptosis remains
complex and may vary depending on ferritinophagy status and
intracellular iron handling (44). Bone tissue undergoes continuous
remodeling through tightly regulated interactions between
osteoblasts, osteoclasts and osteocytes (45). This dynamic process involves
substantial metabolic activity and iron flux. A recent study have
identified ferroptosis as a potential mechanism contributing to
osteocyte loss and cortical bone degeneration during aging
(46). Collectively, these
tissue-specific characteristics suggest that ferroptosis may
represent a convergent mechanism associating metabolic stress, iron
dysregulation and impaired regeneration across the musculoskeletal
system.
To provide a comparative overview of ferroptosis
involvement across musculoskeletal tissues and diseases, the
present review systematically summarizes key ferroptosis-related
biomarkers reported in tendinopathy, muscle degeneration,
sarcopenia, osteoporosis and other degenerative conditions,
highlighting shared molecular signatures and conserved regulatory
pathways (Table I).
 | Table IImportant biomarker of ferroptosis in
musculoskeletal system related diseases. |
Table I
Important biomarker of ferroptosis in
musculoskeletal system related diseases.
| Diseases | Biomarkers | (Refs.) |
|---|
| Tendinopathy | GPX4, SLC7A11,
FSP1 | (49) |
| Tendinopathy | ACSL4, GPX4,
TfR1 | (42) |
| Tendinopathy | GPX4, SLC7A11 | (43) |
| Aged tendon | GPX4, ACSL4 | (9) |
| Aged skeletal
muscle | TfR1, SLC39A14,
GPX4, FTH | (39) |
| Sarcopenia | SLC7A11, GPX4 | (57) |
| Muscle wasting | ACSL4, GPX4 | (57) |
| Muscle aging | FTH, GPX4, SLC7A11,
FTL | (56) |
| Muscle aging | ACSL4, GPX4 | (61) |
| Osteoporosis | GPX4, ACSL4 | (101) |
| Osteoporosis | TFR1, FPN, GPX4,
SLC7A11 | (120) |
| Postmenopausal
osteoporosis | GPX4, SLC7A11 | (102) |
| Intervertebral disc
degeneration | ACSL4, GPX4,
FTH | (115,123) |
| Intervertebral disc
degeneration | ACSL4, GPX4 | (113) |
| Osteoarthritis | FTH, GPX4 | (122) |
The occurrence of ferroptosis in
tendinopathy
Tendinopathy is commonly defined as unexplained
tendon pain and may be associated with tears, inflammatory entheses
or chronic degenerative lesions. It is a prevalent sports injury,
with varying incidence rates across athletic populations: The rate
of achilles tendon rupture reaches 52% in former elite runners, 24%
in competitive athletes and 18% in athletes under 45 years of age;
patellar tendinopathy affects 45% of volleyball players and 32% of
basketball players (47), and
chronic tendinopathy is often observed alongside more severe
conditions, such as tendon damage or rupture (48), although causal relationships
remain to be fully established. In 2022, a study suggested a
potential association between ferroptosis and tendinopathy. In
particular, Wu et al (49) reported that treatment with
Farrerol (FA) was associated with a reduction in
collagenase-induced tendinopathy, potentially involving modulation
of ferroptosis. The study observed that tendinopathy is associated
with iron accumulation in tendon cells and that FA treatment
coincided with reduced lipid peroxidation, increased GSH levels and
decreased iron accumulation, consistent with reduced ferroptosis.
However, the study also observed that FA treatment was associated
with reduced inflammatory cell response and infiltration (49), suggesting a potential association
between ferroptosis and inflammation. Differential gene expression
analysis from RNA sequencing of rotator cuff injury samples treated
with the anti-inflammatory agents celecoxib and lactoferrin showed
an association with ferroptosis (50). Subsequent analysis of
differential genes in rotator cuff injury identified key
ferroptosis-related genes, such as Acsl3, Cybb, Ascl4, Flt3, and
Sat1 and further confirmed that ferroptosis is indeed
interconnected with inflammation in rotator cuff tears. This
finding indicated a possible association between ferroptosis
inhibition and reduced inflammatory markers (51), though causality remains to be
determined. Ferroptosis is also frequently associated with
metabolic diseases, with diabetes being a typical example. A Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway analysis of
differential genes in rotator cuff tear samples from diabetic and
non-diabetic patients revealed an association with the ferroptosis
signaling pathway (52). In a
study on the db/db mouse model of diabetes, changes in the cellular
microenvironment were found to make the mice more prone to
tendinopathy, which was associated with ferroptosis (42). In the study by Wang et al
(42), YAP, a key transcription
factor in the Hippo signaling pathway known to be involved in
various types of cancer (53),
was found to associate with ACSL4 expression. Both YAP and ACSL4
levels were elevated in the diabetic microenvironment, coinciding
with increased markers of ferroptosis, suggesting a potential
regulatory association between YAP, ACSL4 and ferroptosis. The
study observed that ferroptosis inhibitors were associated with
reduced signs of diabetes-induced tendinopathy (42). The occurrence of tendinopathy is
accompanied by the aging of tendon stem cells, which is triggered
by oxidative stress. Gao et al (43) reported that hydrogen
peroxide-induced oxidative stress in tendon stem cells activates
the cGAS-STING signaling pathway in parallel with mitochondrial
damage, mitophagy dysregulation and ferroptosis-related molecular
alterations, ultimately contributing to tendinopathy-associated
pathological phenotypes. Mechanistically, these findings support
the cGAS-STING pathway as a stress-responsive damage-sensing
signaling axis activated under oxidative stress conditions, rather
than as a primary upstream driver of ferroptosis initiation. Within
this framework, cGAS-STING is more appropriately positioned as a
downstream modulatory and signal-amplifying node within the
oxidative stress-mitochondrial dysfunction-ferroptosis regulatory
network in tendon stem cells. Its activation may enhance
inflammatory signaling and ferroptosis susceptibility through
feedback regulation and pathway cross-talk, thereby contributing to
degenerative progression. Further studies are warranted to clarify
its hierarchical positioning and regulatory interactions in tendon
degeneration. Another study on tendon stem cell aging and
ferroptosis found that ferroptosis could promote tendon stem cell
aging, leading to tendon degeneration and aging, while also
affecting tendon-bone healing and mechanical strength (9). Therefore, tendon stem cell aging
may be triggered by ferroptosis, which occurs concurrently with
tendinopathy. Additionally, a bioinformatics analysis of sequencing
data from patients with frozen shoulder revealed an association
with ferroptosis. Ferroptosis influences the pathological processes
of frozen shoulder and Il-6, Hmox1 and Tlr4 genes have been
identified as potential therapeutic targets for this condition
(54), as supported by
transcriptomic and computational analyses conducted using
established bioinformatics frameworks.
Current research associating ferroptosis to tendon
stem cell aging and impaired regeneration is primarily derived from
indirect indicators, including alterations in molecular expression
profiles, increased oxidative stress levels, dysregulated iron
metabolism and activation of ferroptosis-related signaling
pathways. These findings mainly support associations and
mechanistic inferences, while direct causal validation at the level
of cell lineage and fate determination remains lacking. Future
studies integrating lineage tracing models, single-cell
transcriptomic analyses and spatial mapping of ferroptosis-specific
markers in human tendon tissues will be essential to systematically
validate the causal role of ferroptosis in tendon stem cell aging
and regenerative decline. Such approaches will enable mechanistic
dissection at the levels of cell fate determination, cellular
subpopulation heterogeneity and tissue microenvironmental
regulation, thereby establishing a more mechanistically robust and
translationally relevant theoretical framework. Collectively, these
studies indicate that ferroptosis is deeply integrated into the
pathophysiological processes of tendinopathy through coordinated
interactions among oxidative stress, inflammatory signaling,
metabolic dysregulation and tendon stem cell aging (9,42,49). These interconnected mechanisms
converge on iron accumulation, lipid peroxidation and membrane
damage, ultimately driving ferroptotic cell death and tendon
degeneration. the present review integrates these multi-axis
regulatory pathways into a unified mechanistic framework,
illustrating how mitochondrial damage-associated inflammatory
signaling, mechanical stress-induced oxidative injury and metabolic
reprogramming-driven lipid peroxidation collectively contribute to
ferroptosis in tendon pathology (Fig. 3).
The occurrence of ferroptosis in
muscle-related diseases
Iron metabolism plays a key role in muscle diseases,
and the occurrence of ferroptosis can lead to muscle-related
disorders (55-58). It is possible that ferroptosis
not only promotes muscle aging but also contributes to muscle
atrophy. Sarcopenia, which is a clinical manifestation of this
process, can further lead to muscle wasting, and both conditions
are associated with the occurrence of ferroptosis (Fig. 4).
 | Figure 4Role of ferroptosis in muscle-related
diseases. The diagram summarizes key mechanisms involved in
skeletal muscle aging, sarcopenia and muscle atrophy, including
iron dysregulation, mitochondrial dysfunction and impaired
antioxidant defense. Ferroptosis contributes to reduced muscle
regeneration, stem cell dysfunction and loss of muscle mass and
function. KEAP1, Kelch-like ECH-associated protein 1; NRF2, nuclear
factor erythroid 2-related factor 2; GPX4, glutathione peroxidase
4; TFR, transferrin receptor; MSTN, myostatin; SLC7A11, solute
carrier family 7 member 11; p53, tumor protein p53; p16,
cyclin-dependent kinase inhibitor 2A; p21, cyclin-dependent kinase
inhibitor 1A. |
Skeletal muscle aging
Skeletal muscle aging is a physiological phenomenon
that is often accompanied by the onset of sarcopenia and other
related diseases such as osteoporosis and osteoarthritis (59). Different subtypes of muscle stem
cells contribute to distinct aging phenotypes (60). Compared with young mice, aged
mice exhibit markedly reduced expression of GPX4 in skeletal
muscle. Moreover, defects in cystathionine γ-lyase (CSE) exacerbate
GPX4 reduction, thereby inducing ferroptosis in skeletal muscle
(61). As aging progresses, the
expression of TFRC declines in both skeletal muscle and satellite
cells. The loss of TFRC triggers ferroptosis, impairing skeletal
muscle regeneration (39). In a
study utilizing Tfr1-specific knockout mice, Tfr1 depletion
resulted in an ~60% irreversible reduction in satellite cell
population, along with defects in proliferation and differentiation
capacity, further promoting ferroptosis (39). The C2C12 cell line, derived from
murine satellite cells, is frequently used as an in vitro
model for muscle differentiation and regeneration (62,63). When appropriately stimulated,
these myoblasts can differentiate into mature myofibers (64). Liu et al (55) discovered that ferroptosis in
C2C12 myoblasts accelerates skeletal muscle aging, with a notable
reduction in the redox-active metabolite taurine, leading to iron
metabolism dysregulation. This suggests that aging and ferroptosis
may form a vicious cycle that mutually reinforces each other,
although the underlying mechanisms require further investigation.
Grevendonk et al (65)
found that regular exercise training enhances physical activity
levels and considerably counteracts aging, potentially through
mechanisms associated with mitochondrial function decline, although
ferroptosis was not explicitly addressed. However, Wang et
al (56) demonstrated that
lifelong aerobic exercise mitigates oxidative stress in aging
skeletal muscle by activating the Keap1/nuclear factor erythroid
2-related factor 2 (Nrf2) signaling pathway, thereby enhancing
antioxidant enzyme activity. Conversely, detraining inhibits this
pathway, resulting in increased oxidative stress and ferroptosis,
ultimately compromising muscle mass and function, leading to muscle
atrophy. These effects are particularly pronounced during the aging
process. Furthermore, skeletal muscle dysfunction may also arise
from various pathological conditions. For instance, chronic
obstructive pulmonary disease (COPD), which commonly develops with
aging, contributes to sarcopenia and skeletal muscle impairment
(66). In a cigarette
smoke-induced COPD mouse model, Myostatin expression was
upregulated and induced ferroptosis in skeletal muscle cells via
hypoxia inducible factor (HIF) 2 α (67). Therefore, the investigation of
ferroptosis in skeletal muscle should not be limited to primary
ferroptotic events within muscle tissue but should also consider
ferroptosis induced by aging, systemic diseases and other
physiological conditions. Collectively, research suggests that
skeletal muscle aging is accompanied by increased susceptibility to
ferroptosis, characterized by reduced GPX4 expression, altered iron
handling (including TFRC decline) and heightened oxidative stress
(39). Both intrinsic aging
processes and external stressors, such as chronic disease and
impaired antioxidant signaling, appear to converge on redox
imbalance and mitochondrial dysfunction (68). Disruption of iron homeostasis and
antioxidant defense may impair satellite cell maintenance and
regenerative capacity, thereby contributing to age-related muscle
decline (39). However, the
majority of existing data are derived from animal models and in
vitro systems, and direct validation in human aging muscle
remains limited. Further studies are required to clarify whether
ferroptosis represents a primary driver of muscle aging or a
downstream consequence of systemic metabolic stress (39,68).
Sarcopenia
Sarcopenia is a systemic syndrome characterized by
muscle atrophy, which is associated with an age-related decline in
muscle mass and function. It is recognized as one of the leading
causes of disability in the elderly (69), while also increasing the risk of
falls and mortality (70). In a
differential gene expression analysis comparing sarcopenic and
normal muscle tissues, 46 ferroptosis-related genes were identified
and found to be functionally enriched in key biological processes
and KEGG signaling pathways, particularly those related to steroid
hormone and glucocorticoid responses, carbon metabolism,
ferroptosis and the glyoxylate metabolic pathway in sarcopenia. A
total of 11 hub genes, Cdkn1a, Cs, DLD, Foxo1, Hspb1, Ldha, Mdh2
and Ywhaz, demonstrated high sensitivity and specificity for
diagnosing sarcopenia, as derived from transcriptomic and
network-based bioinformatics analyses conducted using established
analytical frameworks (71). In
a study by Huang et al (57) on ferroptosis in sarcopenia, the
SAMP8 mouse model of accelerated aging was used, the study found
that aged SAMP8 mice exhibited notable reductions in muscle mass
and fiber density, along with an increased expression of
ferroptosis-related markers in skeletal muscle. Moreover, iron
overload was shown to induce ferroptosis, impairing the
differentiation of C2C12 myoblasts into myotubes, reducing cell
viability and promoting the accumulation of lipid peroxidation
products. Mechanistically, this process was mediated by the
p53-SLC7A11 signaling pathway (57). SLC7A11, a member of the solute
carrier family 7, functions as an amino acid transporter involved
in GSH biosynthesis (72). Iron
overload upregulates p53 expression, which in turn inhibits SLC7A11
protein levels, leading to the accumulation of lipid peroxides and
the induction of ferroptosis in muscle cells (57). Sarcopenia is frequently
associated with various comorbidities. For instance, COPD is often
accompanied by sarcopenia. A bioinformatics analysis identified
SAA1 as a potential hub gene in COPD-associated sarcopenia, while
the NF-κB signaling pathway was implicated in the underlying
mechanism. Additionally, oxidative phosphorylation and ferroptosis
were suggested to play key roles in the development of this
comorbidity, based on published transcriptomic and computational
analyses using standardized bioinformatics pipelines (73). Furthermore, studies have
indicated that patients with multiple sclerosis may develop
sarcopenia due to gut microbiota dysbiosis caused by hemodialysis
(74). This suggests a
considerable role for gut microbiota in the progression of
sarcopenia. Recent research has also demonstrated that temperature
fluctuations may influence gut microbiota, leading to increased
serum levels of homocitrulline, which exacerbates ferroptosis and
results in mitochondrial dysfunction in muscle cells (75,76). Notably, activation of the Nrf2
pathway was shown to mitigate heat exposure-induced
ferroptosis-associated muscle atrophy (75). Additionally, a study by Liu et
al (77) proposed that
temperature fluctuations may impact muscle function via the
gut-muscle axis, thereby contributing to sarcopenia progression.
Taken together, emerging data indicate that ferroptosis may
participate in the pathophysiology of sarcopenia through
coordinated alterations in iron metabolism, p53-SLC7A11 signaling
and mitochondrial oxidative stress. Transcriptomic analyses
consistently identify ferroptosis-related gene enrichment in
sarcopenic muscle, although these findings largely reflect
associative bioinformatics evidence rather than direct functional
validation. Moreover, comorbid conditions such as COPD, gut
microbiota dysbiosis and environmental stress may amplify
ferroptotic susceptibility, suggesting that sarcopenia represents a
multifactorial state in which ferroptosis intersects with
inflammatory and metabolic signaling. Future studies integrating
human tissue validation and mechanistic intervention models are
necessary to determine whether ferroptosis is a primary driver or a
secondary consequence of muscle degeneration.
Skeletal muscle atrophy
Skeletal muscle atrophy is a systemic pathological
condition that may result from multiple factors (77). The occurrence of muscle atrophy
is characterized by a reduction in skeletal muscle size and
thinning of muscle fibers (78).
It can be broadly classified into two types: neurogenic and
myogenic (79). Regardless of
the type, muscle atrophy is invariably associated with a decline in
muscle strength and mass, ultimately affecting patients' quality of
life. Muscle atrophy is not an independent disease but rather a
pathological condition that accompanies the progression of various
diseases. Aging (80),
immobilization (81), cancer
(82), metabolic disorders
(83) and neurodegenerative
diseases have all been shown to contribute to muscle atrophy, often
accompanied by the loss of collagen fibrin (84). Additionally, immobilization due
to pain or postoperative recovery can lead to muscle atrophy
(85). Following nerve injury,
the control and nutritional support provided by nerves to muscles
become impaired, further inducing muscle atrophy (86). Given that muscle atrophy
frequently accompanies the onset and prognosis of various diseases,
Ni et al (58)
established a chronic kidney disease (CKD) mouse model and observed
skeletal muscle dysfunction and atrophy, which was found to be
associated with ferroptosis. In CKD, skeletal muscle atrophy, a
common complication, has been found to be mitigated by caffeic
acid, a natural phenolic compound, through inhibition of the
TLR4/MYD88/NF-κB signaling pathway and ferroptosis (87). Moreover, Shenshuai Yingyang
Jiaonang, a traditional Chinese herbal medicine, has been
demonstrated to alleviate CKD-induced muscle atrophy by activating
the HIF-1α/SLC7A11 pathway to inhibit ferroptosis (88). In patients with cancer undergoing
cisplatin chemotherapy, although cisplatin effectively targets
tumor cells, it has severe side effects, including muscle atrophy
(89), which compromises muscle
function. Programmed cell death protein 1 (PD-1), a membrane
protein expressed on immune cells, plays a key role in immune
responses and autoimmunity. Knockout of PD-1 induces considerable
skeletal muscle atrophy and following chemotherapy, PD-1-deficient
mice exhibit exacerbated muscle atrophy accompanied by inflammation
and oxidative stress. Notably, genes related to ferroptosis and
autophagy were notably upregulated in these models (90). Inflammation and oxidative stress
are recognized as major risk factors for muscle atrophy. In a study
by Sheng et al (91),
sepsis was found to induce muscle atrophy and weakness. Microarray
dataset analysis revealed notable enrichment of genes related to
muscle function, ferroptosis and the p53 signaling pathway, based
on published transcriptomic analyses using standardized
bioinformatics pipelines. Additionally, targeting the inhibition of
STAT6 was shown to ameliorate mitochondrial dysfunction,
ferroptosis and CHI3L1-mediated satellite cell damage, thereby
improving muscle atrophy and weakness. Furthermore, treatment with
dihydromyricetin was found to restore muscle mass and function. The
underlying mechanism primarily involved inhibiting excessive E3
ubiquitin-protein ligase activity, reducing malondialdehyde (MDA)
levels and restoring superoxide dismutase and GPX activity. This
led to a reduction in oxidative stress and ferroptosis, ultimately
improving muscle atrophy (89).
As muscle atrophy is a common complication in various diseases,
research on its association with ferroptosis remains limited. While
emerging evidence suggests a potential association between
ferroptosis and muscle atrophy, further investigations are needed
to elucidate their precise. Overall, available evidence suggests
that ferroptosis may contribute to skeletal muscle atrophy across
diverse pathological contexts, including CKD, chemotherapy, sepsis
and inflammatory states. A recurring theme involves oxidative
stress amplification, dysregulation of antioxidant defense systems
and activation of ferroptosis-related signaling pathways. However,
the several studies rely on transcriptomic enrichment analyses or
pharmacological intervention models, and the temporal relationship
between ferroptosis activation and muscle fiber loss remains
incompletely defined (92-94). Clarifying whether ferroptosis
acts as an initiating event or a downstream amplifier of muscle
atrophy will be key for determining its therapeutic relevance.
Bone-related disorders and
ferroptosis
Osteoporosis, osteoarthritis and lumbar IVD
represent three interrelated degenerative skeletal disorders, all
of which are closely linked to aging, altered mechanical loading
and hormonal changes. Osteoporosis has been shown to accelerate the
progression of both osteoarthritis and IVD. Conversely, the reduced
mobility and mechanical unloading resulting from joint and disc
degeneration can further exacerbate bone loss and promote the
development of osteoporosis (Fig.
5).
 | Figure 5Ferroptosis in skeletal system
diseases. The figure illustrates the involvement of ferroptosis in
osteoporosis, osteoarthritis and intervertebral disc degeneration.
Iron accumulation, oxidative stress and GPX4 inhibition promote
osteocyte loss, chondrocyte dysfunction and extracellular matrix
degradation, contributing to disease progression. ROS, reactive
oxygen species; GPX4, glutathione peroxidase 4. MMP, mitochondrial
membrane potential; Piezo1, Piezo-type mechanosensitive ion channel
component 1; AMPK, AMP-activated protein kinase; NCOA4, nuclear
receptor coactivator 4. |
Osteoporosis
Osteoporosis is a major risk factor for fractures,
particularly in elderly individuals. It is characterized by reduced
bone mineral density resulting from alterations in bone
microarchitecture, rendering bones susceptible to fractures even
under relatively low-impact forces. Although aging and decreased
levels of sex hormones are well-known contributors to osteoporosis,
it may also occur secondary to various pathological conditions
(95-97). Thus, osteoporosis is not limited
solely to the elderly population (98). Ferroptosis, a form of regulated
cell death driven by iron-dependent lipid peroxidation, has been
identified as a key mechanism underlying osteoblast impairment in
osteoporosis (99). Smoking is a
recognized risk factor for osteoporosis and has been associated
with ferroptosis-related bone homeostasis disruption. Jing et
al (100) demonstrated that
smoking increases ROS levels in bone marrow mesenchymal stem cells
(BMSCs), leading to activation of the AMPK signaling pathway. This,
in turn, promotes NCOA4-mediated ferritinophagy, resulting in
elevated intracellular iron levels and lipid peroxidation,
ultimately triggering ferroptosis and contributing to osteoporotic
pathology. Ferroptosis in BMSCs may impair their osteogenic
differentiation potential and simultaneously enhance osteoclast
activation, further increasing the risk of osteoporosis. Enhanced
osteoclastogenesis and bone resorption, processes frequently
observed in osteoporosis, have also been associated with
ferroptotic mechanisms (101).
During aging, activating ATF3 promotes iron uptake by upregulating
TFR1 and inhibits cystine import through suppression of SLC7A11,
leading to iron overload and lipid peroxidation. This culminates in
ferroptosis of osteocytes, cortical bone loss and ultimately
contributes to osteoporosis development (46). Keap1 has been shown to modulate
osteoblast ferroptosis through activation of the Nrf2/SLC7A11/GPX4
signaling axis, providing a potential therapeutic avenue for
osteoporosis (99). Estrogen
deficiency, particularly postmenopause, promotes iron accumulation
in bone tissue and induces ferroptosis in osteocytes, making
ferroptosis a promising therapeutic target for postmenopausal
osteoporosis (102). Moreover,
osteoporosis is a notable complication in patients with diabetes.
Recent studies have revealed that elevated levels of ferroptosis in
diabetic conditions impair the osteogenic commitment and
differentiation of BMSCs, leading to marked skeletal abnormalities
(103-105). Collectively, ferroptosis has
been implicated to varying extents in osteoporosis of diverse
etiologies, highlighting its potential as a mechanistic focus for
future research into osteoporosis pathogenesis. Current research
indicates that ferroptosis participates in osteoporosis across
diverse etiologies, including aging, estrogen deficiency, smoking
exposure and diabetes-associated metabolic dysfunction. A recurring
mechanistic pattern involves iron accumulation, ferritinophagy
activation, oxidative stress amplification and impairment of
osteoblast or osteocyte viability. However, the majority of studies
are based on animal models or in vitro BMSC systems, and the
temporal relationship between ferroptosis activation and structural
bone loss remains to be fully established (106,107). Whether ferroptosis represents a
primary pathogenic driver or a secondary response to metabolic and
hormonal imbalance warrants further investigation.
IVDD
IVDD is one of the most prevalent degenerative
conditions affecting the spine and is considered a major
contributor to various lumbar disorders, with a prevalence of 71%
in men and 77% in women <50 years of age and >90% in
individuals aged 50 years and older (108). Among these, lumbar disc
herniation (LDH) represents a typical manifestation (109). The senescence of
fibrochondrocytes within the intervertebral disc and the consequent
reduction in proteoglycan synthesis led to disc dehydration and
collapse. This structural deterioration increases the mechanical
stress on the annulus fibrosus, eventually causing the nucleus
pulposus to herniate through annular fissures, thereby initiating
LDH (110). Excessive
mechanical loading is widely recognized as a key pathogenic factor
in IVDD (111,112). Such loading activates the
mechanosensitive ion channel Piezo1, located on the plasma and
endoplasmic reticulum membranes, resulting in elevated
intracellular Ca2+ levels. This, in turn, induces
ferroptosis and endoplasmic reticulum stress (113). Xiang et al (114) further demonstrated an
association between IVDD and ferroptosis, identifying key oxidative
stress-related differentially expressed genes, including HMOX1,
KEAP1, MAPK1, HSPA5, TXNRD1, IL6, PPARA, JUN, HIF1A and DUSP1. The
principal signaling pathways implicated were the TNF signaling
pathway, HIF-1 signaling pathway, NOD-like receptor signaling
pathway and IL-17 signaling pathway. Additionally, a downregulation
of sirtuin (SIRT) 3 expression has been observed during IVDD
progression. An in vivo knockout study revealed that loss of
SIRT3 exacerbates disc degeneration and pain symptoms (115). SIRT3 is considered to mitigate
mitochondrial autophagy via the PINK1/Parkin signaling pathway and
is closely associated with ubiquitination processes (115). While IVD is an age-related
physiological process, proactive strategies to delay or prevent
disc degeneration may be effective in reducing the incidence and
progression of related spinal disorders (116). Taken together, available
studies suggest that ferroptosis may contribute to IVD through
mechanisms involving mechanical stress-induced calcium influx,
oxidative stress-related gene dysregulation and mitochondrial
dysfunction (113). Activation
of Piezo1 signaling and alterations in SIRT3 expression appear to
represent important regulatory nodes linking mechanical loading to
redox imbalance (117).
Nevertheless, current evidence is largely derived from
transcriptomic analyses and experimental models, and direct
clinical validation in human disc tissue remains limited (118). Further studies are required to
clarify the causal contribution of ferroptosis to disc structural
failure and pain progression.
Osteoarthritis (OA)
The pathogenesis of OA initially manifests at the
level of articular cartilage. Early cartilage matrix degradation,
particularly of collagen, prompts the proliferation and clustering
of chondrocytes, leading to the formation of osteophytes (119). This is accompanied by the
production of pro-inflammatory mediators and progressive cartilage
erosion, ultimately resulting in joint destruction and the onset of
OA (98). Iron homeostasis plays
a pivotal role in maintaining the health of articular cartilage.
Excessive iron accumulation induces oxidative stress and
ferroptosis in chondrocytes, thereby contributing to the
development of arthritis (120). Ferroptosis not only impairs
chondrocyte function but also activates inflammatory signaling
pathways, creating a vicious cycle that accelerates joint
inflammation. Single-cell RNA sequencing comparing healthy
cartilage and osteoarthritic cartilage revealed a considerable
increase in both inflammatory and fibrocartilage-like chondrocyte
populations in OA tissues, with enrichment of genes involved in
ferroptosis pathways (121),
based on published transcriptomic analyses using standardized
bioinformatics pipelines. Furthermore, a study by Xie et al
(122) demonstrated that the
antioxidant compound JP4-039 attenuates OA progression by promoting
PINK1/Parkin-dependent mitophagy, thereby protecting chondrocytes
from ferroptotic cell death. Iron overload catalyzes the generation
of ROS through mitochondrial dysfunction, which inhibits type II
collagen expression and promotes the expression of matrix-degrading
enzymes such as MMPs and ADAMTS. This enzymatic activity leads to
the degradation of collagen and proteoglycans, compromising the
structural integrity of cartilage (120). Excessive mechanical loading is
also an important risk factor for OA, wherein the mechanosensitive
ion channel Piezo1 contributes to disease progression by modulating
ferroptosis in chondrocytes (5).
Therefore, early therapeutic intervention targeting ferroptosis
holds promise for the effective treatment of OA (123). Overall, current data support a
contributory role of ferroptosis in osteoarthritis, particularly
through iron-induced oxidative stress, mitochondrial dysfunction
and inflammatory amplification in chondrocytes. Enrichment of
ferroptosis-related genes in single-cell transcriptomic analyses
and protective effects of ferroptosis-targeting interventions
suggest mechanistic relevance. However, much of the evidence
remains associative and the extent to which ferroptosis precedes or
follows cartilage matrix degradation is not yet fully defined.
Clarifying the sequence and cell-type specificity of ferroptotic
events will be key for translating these findings into therapeutic
strategies.
Aging is characterized by progressive dysregulation
of both iron metabolism and antioxidant defenses, which together
create a permissive environment for ferroptotic susceptibility in
musculoskeletal tissues. Age-related increases in intracellular
iron content have been observed in skeletal muscle and bone, which
can promote oxidative damage through enhanced Fenton chemistry and
lipid peroxidation, thereby impairing mitochondrial function and
cellular homeostasis (124).
Such iron accumulation is accompanied by a decline in GSH synthesis
and activity of GPX4 and other endogenous antioxidant systems,
weakening cellular capacity to neutralize ROS and lipid peroxides.
In addition, age-associated impairment of iron export and storage
mechanisms, such as reduced expression of ferroportin and altered
ferritin dynamics, may further contribute to iron retention and
redox imbalance. Together, these age-related changes in iron
handling and antioxidant capacity may sensitize muscle cells,
tendon-derived cells, chondrocytes and osteocytes to ferroptotic
injury, thus intersecting with and potentially accelerating
musculoskeletal decline (125).
Disease-oriented modeling and
pharmacological modulation of ferroptosis in musculoskeletal
systems
In vitro ferroptosis models are not merely
experimental tools but represent essential platforms for
understanding disease-specific mechanisms and evaluating
therapeutic strategies in musculoskeletal disorders. In the context
of tendon degeneration, muscle atrophy, cartilage degeneration and
bone remodeling, ferroptosis-related cellular models provide key
insights into how iron metabolism dysregulation, oxidative stress
and lipid peroxidation contribute to tissue dysfunction,
regenerative failure and progressive degeneration (126). Therefore, these models are
discussed not as isolated experimental systems, but as
disease-oriented platforms that integrate mechanistic investigation
with translational relevance in musculoskeletal pathology.
At the level of pathological microenvironment
modeling, ferroptosis can be induced by mimicking iron overload and
oxidative stress conditions characteristic of degenerative
musculoskeletal tissues. In the study conducted by Huang et
al (57), ferric ammonium
citrate was used to treat undifferentiated C2C12 myoblasts,
differentiated C2C12 myotubes and cells undergoing differentiation,
successfully inducing ferroptosis, thereby providing a model that
reflects iron accumulation-driven cellular injury in muscle
degeneration. Similarly, hydrogen peroxide
(H2O2) is frequently used in oxidative stress
and aging models relevant to musculoskeletal disorders.
Importantly, studies have demonstrated that ferrostatin-1 (Fer-1)
can effectively reverse cell death induced by
H2O2, suggesting that
H2O2-induced injury partially involves
ferroptotic mechanisms (43).
These models simulate disease-relevant microenvironmental stressors
and provide a biologically meaningful context for studying
ferroptosis in musculoskeletal degeneration.
At the level of molecular mechanism-oriented
modeling, classical ferroptosis inducers target core regulatory
nodes of ferroptotic signaling. Erastin, a commonly used
ferroptosis inducer, inhibits the Xc− system, leading to
reduced cystine uptake, depletion of intracellular GSH, impairment
of antioxidant defenses and subsequent lipid peroxidation-driven
cell death. In addition, erastin can induce ROS generation and
activate p53 signaling, which further suppresses SLC7A11
expression, reinforcing Xc− system inhibition and
amplifying ferroptotic vulnerability (27). RSL3 represents another
mechanistically distinct ferroptosis inducer. Although
traditionally regarded as a GPX4 inhibitor, Dorian et al
(127) demonstrated that RSL3
induces ferroptosis primarily through potent inhibition of
thioredoxin reductase 1 (TXNRD1), thereby revealing an alternative
redox regulatory axis in ferroptotic control. Together, these
models enable precise dissection of ferroptosis-regulatory networks
that are highly relevant to redox imbalance and metabolic
dysregulation in musculoskeletal diseases.
At the level of pharmacological modulation,
ferroptosis inhibitors provide essential tools for evaluating the
therapeutic potential of ferroptosis-targeted interventions. Fer-1
is widely employed as a canonical ferroptosis inhibitor and acts by
scavenging phospholipid hydroperoxyl radicals, thereby interrupting
lipid peroxidation chain reactions and stabilizing membrane
integrity. Comparative analyses by Miotto et al (128) demonstrated that although both
Fer-1 and Trolox exhibit chain-breaking antioxidant activity, Fer-1
shows superior efficacy in inhibiting lipid peroxidation. Unlike
conventional antioxidants that are consumed during redox reactions,
Fer-1 forms a catalytic-like redox cycle through complex formation
with Fe2+, enabling sustained ferroptosis inhibition
(118). Consistently, Rachid
et al (129) reported
that Fer-1 prevents ferroptotic cell death by protecting membrane
lipids without interfering with mitochondrial ROS production or
lysosomal membrane permeability, supporting its mechanistic
specificity in ferroptosis modulation.
Importantly, the sensitivity of different cell types
to ferroptosis inducers and inhibitors varies substantially, and
their molecular targets are not universally conserved across
tissues. This heterogeneity highlights the necessity of disease-
and tissue-specific selection of experimental ferroptosis models
and pharmacological agents, particularly in the musculoskeletal
system, where cellular phenotypes, metabolic profiles and
vascularization levels differ markedly among muscle, tendon,
cartilage and bone tissues.
Collectively, these in vitro ferroptosis
models should not be viewed as isolated experimental systems, but
as disease-oriented platforms that bridge mechanistic research and
translational application. While current ferroptosis inducers and
inhibitors are indispensable for mechanistic dissection, the
majority remain research-grade tools rather than clinically
applicable agents. Their translational limitations include limited
target specificity, poor bioavailability, lack of tissue-selective
delivery and restricted applicability in low-vascularized
musculoskeletal tissues such as tendons and cartilage. These
challenges underscore the necessity of integrating disease-specific
modeling with pharmacological optimization and targeted delivery
strategies, thereby transforming experimental ferroptosis models
into translational platforms for musculoskeletal therapy
development (Table II).
| Table IIFerroptosis related model (in
vitro). |
Table II
Ferroptosis related model (in
vitro).
A, Inducers
|
|---|
| Authors, year | Cells | Compound | (Refs.) |
|---|
| Wu et al,
2022 | Tenocytes | RSL3 | (49) |
| Gao et al,
2024 | TSPCs (tendon stem
cells) |
H2O2 | (43) |
| Huang et al,
2021 | C2C12 myoblasts,
C2C12 myotubes | Ferric citrate,
erastin | (57) |
| Ni et al,
2023 | C2C12 myoblasts,
C2C12 myotubes | Indolyl
sulfate | (58) |
| Wang et al,
2021 | C2C12
myoblasts | RSL3 | (61) |
| Liu et al,
2022 | BMSCs | Erastin, RSL3 | (138) |
| Jiang et al,
2024 | Ocy454 cell
line | FAC | (102) |
| Wan et al,
2023 | Chondrocyte | Erastin | (135) |
| Xie et al,
2025 | Chondrocytes | Tert-Butyl
Hydroperoxide, TBHP | (122) |
|
| B, Inhibitors |
|
| Authors, year | Cells | Compound | (Refs.) |
|
| Wang et al,
2023 | Tendon-derived stem
cells | Ferrostatin-1
(Fer-1) | (42) |
| Gao et al,
2024 | Tendon
stem/progenitor cells | Fer-1 | (43) |
| Huang et al,
2021 | C2C12 myoblasts,
C2C12 myotubes | Fer-1,
Deferoxamine | (57) |
| Ni et al,
2023 | C2C12 myoblasts,
C2C12 myotubes | Lobetyolin | (58) |
| Zhang et al,
2022 | C2C12 myotubes | UAMC-3203 | (67) |
| Jing et al,
2023 | Bone marrow
mesenchymal stem cells | Fer-1, DFO | (100) |
| Jiang et al,
2024 | Ocy454 cell
line | Lip-1, Fer-1 | (102) |
While pharmacological inducers and inhibitors
provide valuable tools for manipulating ferroptosis and exploring
its therapeutic potential, these compounds primarily target
downstream execution pathways, such as lipid peroxidation and
antioxidant defense systems. However, ferroptosis is also tightly
regulated at the post-translational level through dynamic
modifications of key proteins involved in iron metabolism, redox
balance and mitochondrial function. Understanding how
post-translational modifications (PTMs) modulate
ferroptosis-related signaling networks offers a deeper mechanistic
perspective beyond compound-based intervention and provides insight
into endogenous regulatory complexity. Therefore, the following
section focuses on the role of PTMs in fine-tuning ferroptotic
processes in musculoskeletal contexts.
Ferroptosis and post-translational
modifications
PTMs represent a decisive regulatory layer that
integrates upstream stress signals and determines ferroptotic cell
fate through dynamic modulation of key proteins involved in iron
metabolism, lipid peroxidation and antioxidant defense. Unlike
transcriptional regulation, which alters gene expression at the
mRNA level, PTMs provide rapid, reversible and spatially restricted
control over protein stability, enzymatic activity and subcellular
localization. In musculoskeletal tissues, where mechanical loading,
metabolic imbalance, inflammatory signaling and aging-related
oxidative stress frequently intersect, PTM-mediated fine-tuning may
function as a key checkpoint that dictates ferroptosis
susceptibility. Therefore, PTMs should not be viewed merely as
molecular decorations, but as core regulatory nodes that connect
upstream pathological stimuli with downstream ferroptotic execution
mechanisms (Fig. 6).
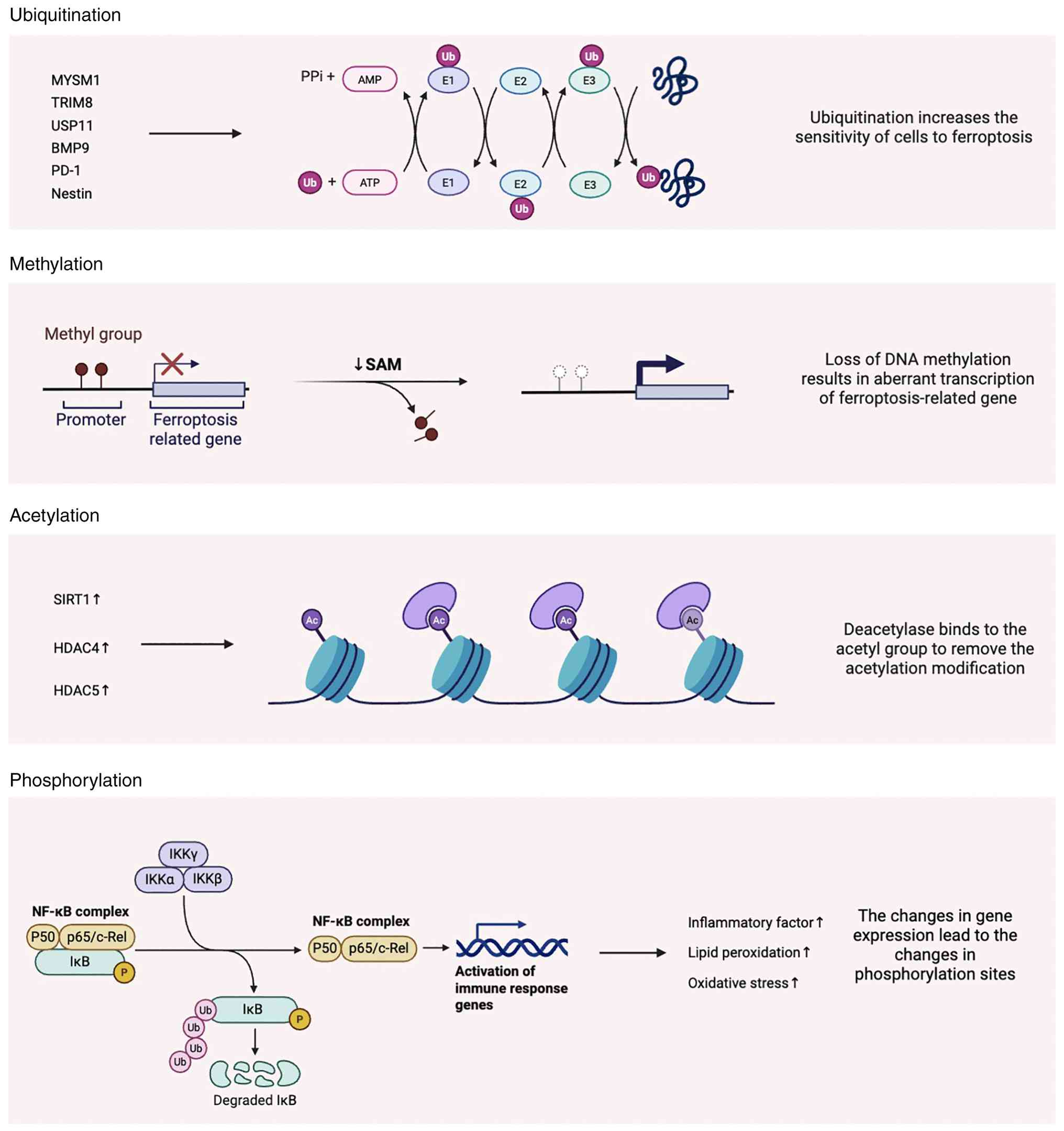 | Figure 6Post-translational modifications
regulating ferroptosis in musculoskeletal disorders. The diagram
highlights key post translation modifications, including
ubiquitination, phosphorylation, methylation and acetylation, which
modulate the stability, activity and localization of
ferroptosis-related proteins. These modifications act as regulatory
nodes linking upstream stress signals to ferroptotic cell death.
MYSM1, Myb-like, SWIRM and MPN domain-containing protein 1; TRIM8,
tripartite motif containing 8; USP11, ubiquitin specific peptidase
11; BMP9, bone morphogenetic protein 9; PD-1, programmed cell death
protein 1; Nestin, neuroepithelial stem cell protein; SIRT1,
sirtuin 1; HDAC4, histone deacetylase 4; HDAC5, histone deacetylase
5; p50, nuclear factor NF-kappa-B p50 subunit; p65, transcription
factor p65. |
Ubiquitination
Ubiquitination is a key post-translational
modification that regulates protein synthesis, stability and
degradation, thereby influencing cellular sensitivity to
ferroptosis. This process plays a vital role in maintaining bone
cell function and skeletal metabolic homeostasis. MYSM1, a histone
deubiquitinase, has been shown to promote osteogenic
differentiation, its deficiency leads to increased osteoclast
number and activity, disrupts hematopoietic stem cell (HSC)
function and heightens HSC susceptibility to ferroptosis,
contributing to bone marrow failure syndromes. Mechanistically,
MYSM1 deficiency reduces the translational efficiency of GPX4, a
key ferroptosis suppressor and may also impair the recruitment of
essential transcription factors such as Gata2 and Runx1 (130). Ubiquitination also contributes
to ferroptosis regulation in the context of arthritis. For example,
P21 modulates the mono-ubiquitination level of GPX4, indirectly
affecting chondrocyte sensitivity to ferroptosis (131). TRIM8, via its RING domain,
facilitates the ubiquitination and subsequent degradation of
YTHDF2, thereby influencing chondrocyte ferroptosis and modulating
arthritis progression (132).
In studies on IVDD, Zhu et al (115) reported that the deubiquitinase
USP11 alleviates oxidative stress-induced ferroptosis through
deubiquitination mechanisms. Similarly, Huang et al
(105) found that BMP9
alleviates iron accumulation-induced osteoporosis by upregulating
USP10, which removes K48-linked polyubiquitin chains from FOXO1.
This action prevents excessive cytoplasmic ubiquitination of FOXO1
and promotes GPX4 expression, thereby reducing ferroptosis.
Aberrant expression of E3 ubiquitin ligases is closely associated
with ferroptosis in musculoskeletal diseases (89), with PD-1 playing a regulatory
role in this context (90).
SMURF, identified as the E3 ligase for FTH1, activates ferroptosis
signaling and inhibits myoblast differentiation into myotubes
(133). Nestin, a cytoskeletal
protein, has been recognized as a key regulator of the
ferroptosis-autophagy crosstalk in skeletal muscle atrophy. It
interacts with MAP1LC3B (LC3B) and catalyzes its polyubiquitination
at lysine-51, reducing LC3B availability for autophagy and thereby
suppressing ferroptotic cell death (134). Moreover, the transcription
factor Nrf2, a master regulator of antioxidant responses, is itself
regulated by Keap1-mediated ubiquitin-dependent degradation. This
regulatory axis represents a promising direction for future
investigations into ferroptosis-related musculoskeletal
pathologies. Collectively, ubiquitination acts as a protein
stability checkpoint that determines the turnover of key
ferroptosis regulators such as GPX4, FTH1 and transcription
factors. By controlling degradation dynamics, ubiquitination
establishes a threshold for ferroptotic activation in
musculoskeletal cells.
Phosphorylation
Phosphorylation, a post-translational modification
in biological systems, wherein phosphate groups are covalently
attached to proteins or other biomolecules under enzymatic
catalysis. This modification profoundly influences protein
activity, subcellular localization and interaction networks,
thereby modulating numerous cellular pathways. In a study by Xue
et al (101), inhibition
of I-κB and p65 phosphorylation was shown to suppress activation of
the NF-κB signaling pathway, indicating that phosphorylation plays
a key regulatory role in inflammatory responses. Similarly, Chen
et al (9) demonstrated
that phosphorylation of AMPK is essential for regulating cellular
energy metabolism, antioxidant defense and regeneration. By
modulating the AMPK/Nrf2/GPX4 signaling axis, AMPK phosphorylation
delays the senescence of TSPCs, inhibits ferroptosis and enhances
tendon regeneration. Phosphorylation also plays a pivotal role in
ferroptosis associated with IVDD. For instance, PDK4 phosphorylates
the E1α subunit of the pyruvate dehydrogenase complex, thereby
inhibiting pyruvate decarboxylation and regulating cellular energy
metabolism and lipid biosynthesis (123). Wan et al (135) found that induction of AMPKα
phosphorylation in chondrocytes enhances its stability and activity
and that activation of the AMPK/Nrf2/HO-1 pathway attenuates
ferroptosis, offering a potential therapeutic strategy for
arthritis. Similarly, Zhou et al (136) reported that suppression of
PI3K/Akt phosphorylation mitigates ferroptosis by enhancing
antioxidant defenses through upregulation of GSH and GPX4, thereby
preventing lipid peroxidation and relieving joint pain. In another
study, Hyeon et al (137) demonstrated that donepezil
alleviates skeletal muscle insulin resistance in obese mice by
inhibiting inflammation and ferroptosis via the AMPK/FGF21 pathway.
This effect was mainly attributed to suppression of NF-κB
phosphorylation and I-κB degradation, leading to reduced expression
of pro-inflammatory markers. Moreover, mutations in genes encoding
phosphorylation-related enzymes or phospho-sites can disrupt
phosphorylation dynamics, resulting in aberrant signaling and
contributing to disease pathogenesis in musculoskeletal tissues.
Taken together, phosphorylation functions as a rapid signal
transduction mechanism that links metabolic and inflammatory
stimuli to ferroptosis-related antioxidant pathways, thereby
modulating cellular susceptibility to ferroptotic stress.
Methylation
Methylation refers to the enzymatic transfer of a
methyl group from a donor molecule to specific substrates, a
process typically catalyzed by methyltransferases. This epigenetic
and epitranscriptomic modification plays a key role in gene
expression regulation, RNA stability and cellular function,
including the modulation of ferroptosis in musculoskeletal tissues.
In a study by Liu (138),
NSUN5, a member of the NOP2/Sun RNA methyltransferase family, was
shown to play a pivotal role in regulating ferroptosis in BMSCs
through the NSUN5-FTH1/FTL axis. Specifically, NSUN5 catalyzes
5-methylcytosine (m5C) modifications on the mRNA of FTH1 and FTL,
and NSUN5 depletion results in a reduction of these modifications,
subsequently leading to decreased expression of FTH1 and FTL,
thereby promoting ferroptosis. Nrf2, a key regulator of antioxidant
defense, also plays a central role in the modulation of ferroptosis
in bone cells. Beyond its involvement in ubiquitination pathways,
Nrf2 has been found to regulate DNA methylation of the receptor
activator of nuclear factor κB ligand (RANKL) promoter via DNA
methyltransferase 3a (Dnmt3a), thus influencing RANKL expression
and osteoclastogenesis (102).
Moreover, methyltransferase-like 3 (METTL3), an RNA
N6-methyladenosine (m6A) methyltransferase, has emerged as a key
modulator in chondrocyte ferroptosis. Depletion of METTL3 was shown
to alleviate ferroptosis and cartilage damage, whereas its
overexpression enhanced m6A methylation of HMGB1 mRNA, promoting
ferroptotic responses (139).
In models of diabetic osteoporosis induced by high glucose and high
fat, METTL3 was found to activate the ASK1-p38 MAPK signaling
pathway, thereby promoting ferroptosis in osteoblasts. Knockdown of
METTL3 in MC3T3-E1 osteoblasts not only inhibited ASK1-p38
activation but also attenuated ferroptosis, highlighting METTL3 as
a potential molecular target in diabetic bone loss (140). Furthermore, methylation
directly impacts the expression and activity of
ferroptosis-regulating enzymes. Inhibition of GPX4 methylation has
been shown to mitigate ferroptosis in chondrocytes during arthritis
progression (141). In
ovariectomized mouse models of osteoporosis, reduced GPX4
expression was accompanied by hypermethylation of its promoter and
elevated levels of DNA methyltransferases DNMT1, DNMT3a and DNMT3b
(142). Although methylation
has been widely studied in cancer and neurological diseases, its
role in musculoskeletal disorders and ferroptosis is an emerging
area of interest and warrants further exploration. Overall,
methylation introduces an additional epigenetic and
epitranscriptomic layer of regulation, influencing ferroptosis
through long-term modulation of gene expression and mRNA stability,
particularly in aging and metabolic disorders.
Acetylation
Acetylation is a form of post-translational protein
modification involving the addition of an acetyl group to specific
amino acid residues, most commonly lysine (143). This modification can
considerably impact protein function, localization and
interactions, thereby influencing diverse cellular processes,
including ferroptosis. Recent studies have highlighted the
regulatory role of acetylation in ferroptosis within
musculoskeletal diseases (144,145). Overexpression of SIRT1, a class
III histone deacetylase, promotes the expression of FTL by
deacetylating it at lysine 181 (K181), which in turn suppresses
ferroptosis in chondrocytes and attenuates the progression of
arthritis (144,146). Histone deacetylases HDAC4 and
HDAC5 also contribute to ferroptosis regulation by reducing
acetylation of the tumor suppressor protein p53 at lysine 120
(K120). This deacetylation inhibits the activation of apoptotic and
ferroptotic pathways, thereby preserving skeletal muscle cell
viability and function under lipotoxic stress conditions (147). As such, acetylation-based
regulatory mechanisms represent a promising avenue for future
research in understanding and potentially targeting ferroptosis in
musculoskeletal pathology. Acetylation-dependent regulation
highlights the importance of metabolic-epigenetic crosstalk in
ferroptosis control, suggesting that cellular acetyl-CoA
availability and deacetylase activity may shape ferroptotic
outcomes in musculoskeletal pathology.
Emerging PTMs such as SUMOylation, O-GlcNAcylation,
lactylation and succinylation have recently been implicated in
redox regulation and metabolic stress adaptation in other disease
contexts. Although research associating these modifications to
ferroptosis in musculoskeletal tissues remains limited, their
established roles in regulating iron metabolism, mitochondrial
function and antioxidant signaling suggest potential relevance.
Future multi-omics and proteomics approaches may uncover additional
PTM-mediated regulatory circuits that fine-tune ferroptotic
susceptibility.
Therapeutic approaches for ferroptosis in
muscular system disorders
Interorgan communication
Guan et al (148) demonstrated that the gut
microbiota-derived metabolite capsiate (CAT) plays a regulatory
role in ferroptosis induced by OA. Specifically, CAT targets HIF-1α
to activate the expression of SLC2A1, thereby inhibiting
ferroptosis and attenuating OA progression. This finding highlights
the interorgan communication between the gut and the joints.
Metabolites produced by the gut microbiota can reach joint tissues
via the circulatory system or other pathways, thereby modulating
the occurrence of ferroptosis in joint-resident cells. Despite its
potential significance, research on interorgan communication in the
context of ferroptosis and musculoskeletal diseases remains
limited, particularly regarding the modulatory effects of gut
microbial metabolites on musculoskeletal health.
Exosome therapy
Extracellular vesicles, also known as exosomes
(EXOs), have been increasingly recognized as a promising tool for
mitigating tendon aging, with potential applications in tissue
regeneration and repair (9), and
the purified exosomes can also improve the biomechanics of the
rotator cuff (149).
Platelet-derived EXOs exhibit anti-inflammatory properties and
promote stem cell proliferation. In the study by Jin et al
(150), it was found that
systemic administration of bio-nanoparticles derived from human
exfoliated deciduous teeth stem cell-derived EXOs (SHED-EXOs)
effectively attenuated tendon degeneration in a naturally aging rat
model and localized delivery of microspheres loaded with SHED-EXOs
not only markedly reduced the accumulation of senescent cells and
ectopic ossification but also restored the regenerative and
reparative capacity of tendons in aged rats.
Pharmacological therapy
Celecoxib, a non-steroidal anti-inflammatory drug,
has been reported to exert ferroptosis-inhibitory effects (151). When combined with lactoferrin,
celecoxib enhances tendon repair. Zhang et al (50) conducted gene identification and
bioinformatics analysis to explore the underlying mechanisms and
identified Slc40a1, Tfrc and Cryab as ferroptosis-related genes key
for regulating tendon injury repair (50), as indicated by published
bioinformatics analyses. Lobetyolin, an active compound in
traditional Chinese medicine, has been found to alleviate skeletal
muscle damage by activating the Hedgehog signaling pathway,
particularly the transcription factor Gli1, thereby upregulating a
series of ferroptosis-inhibitory factors (58). Taurine, a redox-specific
metabolite (152), has been
shown to prevent ferroptosis by markedly increasing intracellular
GSH levels, reducing MDA and ROS production and restoring the
vitality of impaired myogenic differentiation. Taurine is also
essential for glycerophospholipid metabolism, playing a key role in
cell membrane repair. Additionally, it enhances mitochondrial
bioenergetics, increasing the energy reserves produced by muscle
satellite cells (55). Aconine
is a natural compound that suppresses osteoclast activity and
reduces bone resorption. It exerts its effects by inhibiting the
NF-κB signaling pathway and modulating the expression of GPX4 and
ACSL4, thereby regulating ferroptosis in osteoclasts (101). Biotin A (BCA), a naturally
occurring isoflavonoid isolated from Astragalus membranaceus,
exhibits anti-inflammatory and antioxidant properties. It directly
reduces intracellular iron levels by inhibiting TfR1 and promoting
FPN expression. In addition, BCA targets the Nrf2/system
Xc−/GPX4 signaling pathway to scavenge free radicals and
prevent lipid peroxidation (120). Selenium supplementation has
been shown to alleviate endoplasmic reticulum stress by
upregulating selenoprotein K (SelK) expression and reducing
intracellular free Ca2+ concentrations. Through both the
selenium-GPX4 and selenium-SelK axes, selenium mitigates
ferroptosis induced by mechanical overload and contributes to the
stabilization of the extracellular matrix (113).
Biomaterials and other approaches
Intramuscular injection of lentivirus expressing
Tfr1 has been shown to mitigate Tfr1 deficiency-induced iron
accumulation, thereby promoting skeletal muscle regeneration
(39). Fer-1, a widely used
ferroptosis inhibitor, upregulates CSE, thereby modulating the
CSE/H2S signaling pathway, scavenging ROS and preventing
lipid peroxidation. Hydrogen sulfide (H2S), a novel
gaseous signaling molecule derived from CSE, is widely distributed
across tissues and organs and plays a regulatory role in oxidative
stress and lipid metabolism (61). Ferroptosis exhibits a dual role
in cancer therapy. Zhang et al (153) designed a self-amplifying
nanodrug (RCH NPs) using human serum albumin as a carrier to
program the dual nature of ferroptosis. Within this system, ferric
porphyrin-induced ferroptosis is coupled with celecoxib-mediated
disruption of inflammation-related immune suppression, while
roscovitine genetically blocks IFN-γ-induced PD-L1 upregulation.
This strategy enhances the therapeutic potential of ferroptosis in
tumor treatment. Regular physical exercise (65) and newly developed brain-muscle
communication interventions that suppress detrimental central
signals while stabilizing peripheral muscles to prevent muscle
aging (154) may indirectly
regulate ferroptosis, providing new directions for future research.
In the study by Gao et al (155), a ROS-responsive injectable
hydrogel, PVA-tsPBA@SLC7A11 modRNA, was developed to mitigate IVDD.
Upon local injection into the degenerated disc, ROS-induced
cleavage of the PVA-tsPBA hydrogel facilitated the release of
encapsulated SLC7A11 modRNA, thereby suppressing ferroptosis in
nucleus pulposus cells and ameliorating IVDD. In a separate study,
Cit-AuNRs@Anti-TRPV1, a near-infrared (NIR)-activated nanoplatform,
was shown to protect chondrocytes against ferroptosis under NIR
irradiation and attenuate the progression of arthritis, offering a
promising therapeutic strategy for joint diseases (156).
Future perspectives
Ferroptosis has emerged as a key pathogenic
mechanism across a spectrum of musculoskeletal disorders, including
skeletal muscle aging and atrophy, tendinopathy, osteoarthritis,
IVDD and osteoporosis. Despite increasing research associating iron
dysregulation, oxidative stress amplification and mitochondrial
dysfunction to musculoskeletal tissue degeneration, several
fundamental questions remain unresolved.
First, the temporal sequence of ferroptotic
activation during tissue degeneration requires further
clarification. It remains unclear whether ferroptosis functions as
a primary driver of stem/progenitor cell dysfunction and
extracellular matrix degradation or as a secondary amplifier of
inflammatory and metabolic stress. Second, the interplay between
ferroptosis and other cellular regulatory pathways, such as
autophagy, warrants deeper investigation. For example, autophagy
deficiency has been shown to mitigate erastin-induced ferroptosis
(16), highlighting the
importance of autophagy-dependent regulatory networks that may also
be relevant in musculoskeletal tissues.
In addition, emerging evidence suggests potential
crosstalk between ferroptosis and other forms of metal-associated
regulated cell death. Xue et al (157) demonstrated that Cu2+
can enhance cellular susceptibility to ferroptosis by directly
binding to GPX4 and promoting its ubiquitination and degradation.
Although these findings were not limited to musculoskeletal models,
they raise the possibility that metal homeostasis beyond iron may
influence ferroptotic vulnerability in musculoskeletal
pathology.
Future research should prioritize tissue-specific
and stage-specific investigation of ferroptosis within the
musculoskeletal system. Particular attention should be given to the
interaction between mechanical loading, metabolic stress and
ferroptotic signaling, as well as to the development of targeted
therapeutic strategies for low-vascularized tissues such as tendon
and cartilage. Integrating multi-omics approaches with functional
validation in human tissues will be essential to translate
ferroptosis-based interventions into clinically relevant
therapies.
Overall, maintaining a focused mechanistic
framework centered on musculoskeletal diseases will be critical for
advancing both basic understanding and therapeutic innovation in
this field.
Availability of data and materials
Not applicable.
Authors' contributions
WG was responsible for the writing, original draft
of the manuscript. TX, FL and LY contributed to the writing, review
and editing of the manuscript. FL responsible for preparation of
figures. HZ and YY provided supervision and conceptualization of
the present study. GW contributed to the conceptualization of the
research and the acquisition of funding.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by research grants from the
Beijing Natural Science Foundation (grant no. 7262054) and the
National Natural Science Foundation of China (grant no. NSFC
82171190). Additionally, Dr Wang's research has received funding
through the Entrepreneurship and Innovation Program of Jiangsu
Province (grant no. KYCX22_3383). Support was also received from
the Shanghai Pujiang Program (grant no. 24PJD089) to Dr Haifeng
Zhang.
References
|
1
|
Joy EA, Briesacher M and Wiegand B:
Musculoskeletal failure. Am J Lifestyle Med. 18:826–829. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Leser JM, Harriot A, Buck HV, Ward CW and
Stains JP: Aging, osteo-sarcopenia, and musculoskeletal
mechano-transduction. Front Rehabil Sci. 2:7828482021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Safiri S, Kolahi AA, Cross M, Hill C,
Smith E, Carson-Chahhoud K, Mansournia MA, Almasi-Hashiani A,
Ashrafi-Asgarabad A, Kaufman J, et al: Prevalence, deaths, and
disability-adjusted life years due to musculoskeletal disorders for
195 countries and territories 1990-2017. Arthritis Rheumatol.
73:702–714. 2021. View Article : Google Scholar
|
|
4
|
Xiang W, Zhang T, Li B, Li S, Zhang B,
Fang S, Chen L, Gong Y, Huang B, Feng D, et al: Senescent
macrophages induce ferroptosis in skeletal muscle and accelerate
osteoarthritis-related muscle atrophy. Nat Aging. 5:1295–1316.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang S, Li W, Zhang P, Wang Z, Ma X, Liu
C, Vasilev K, Zhang L, Zhou X, Liu L, et al: Mechanical overloading
induces GPX4-regulated chondrocyte ferroptosis in osteoarthritis
via Piezo1 channel facilitated calcium influx. J Adv Res. 41:63–75.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cui W, Liu D, Gu W and Chu B:
Peroxisome-driven ether-linked phospholipids biosynthesis is
essential for ferroptosis. Cell Death Differ. 28:2536–2551. 2021.
View Article : Google Scholar :
|
|
7
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fan Y, Chen Z, Wang H, Jiang M, Lu H, Wei
Y, Hu Y, Mo L, Liu Y, Zhou C, et al: Isovitexin targets SIRT3 to
prevent steroid-induced osteonecrosis of the femoral head by
modulating mitophagy-mediated ferroptosis. Bone Res. 13:182025.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen D, Tang Q, Song W and He Y:
Platelet-derived exosomes alleviate tendon stem/progenitor cell
senescence and ferroptosis by regulating AMPK/Nrf2/GPX4 signaling
and improve tendon-bone junction regeneration in rats. J Orthop
Surg Res. 19:3822024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lu X, Sharko OL, Shmanai VV, Shchepinov MS
and Markworth JF: Deuterated polyunsaturated fatty acids alleviate
in vitro skeletal muscle dysfunction induced by oxidative stress.
Free Radic Biol Med. 248:222–237. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bertrand RL: Iron accumulation,
glutathione depletion, and lipid peroxidation must occur
simultaneously during ferroptosis and are mutually amplifying
events. Med Hypotheses. 101:69–74. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Riegman M, Sagie L, Galed C, Levin T,
Steinberg N, Dixon SJ, Wiesner U, Bradbury MS, Niethammer P,
Zaritsky A and Overholtzer M: Ferroptosis occurs through an osmotic
mechanism and propagates independently of cell rupture. Nat Cell
Biol. 22:1042–1048. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brown CW, Amante JJ, Chhoy P, Elaimy AL,
Liu H, Zhu LJ, Baer CE, Dixon SJ and Mercurio AM: Prominin2 drives
ferroptosis resistance by stimulating iron export. Dev Cell.
51:575–586.e4. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen GH, Song CC, Pantopoulos K, Wei XL,
Zheng H and Luo Z: Mitochondrial oxidative stress mediated
Fe-induced ferroptosis via the NRF2-ARE pathway. Free Radic Biol
Med. 180:95–107. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li Y, Zeng X, Lu D, Yin M, Shan M and Gao
Y: Erastin induces ferroptosis via ferroportin-mediated iron
accumulation in endometriosis. Hum Reprod. 36:951–964. 2021.
View Article : Google Scholar
|
|
16
|
Park E and Chung SW: ROS-mediated
autophagy increases intracellular iron levels and ferroptosis by
ferritin and transferrin receptor regulation. Cell Death Dis.
10:8222019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu Y, Jiang L, Wang H, Shen Z, Cheng Q,
Zhang P, Wang J, Wu Q, Fang X, Duan L, et al: Hepatic transferrin
plays a role in systemic iron homeostasis and liver ferroptosis.
Blood. 136:726–739. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gao M, Monian P, Quadri N, Ramasamy R and
Jiang X: Glutaminolysis and transferrin regulate ferroptosis. Mol
Cell. 59:298–308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tian Y, Lu J, Hao X, Li H, Zhang G, Liu X,
Li X, Zhao C, Kuang W, Chen D and Zhu M: FTH1 inhibits ferroptosis
through ferritinophagy in the 6-OHDA model of Parkinson's disease.
Neurotherapeutics. 17:1796–1812. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang X, Ding Y, Sun L, Shi M, Zhang P,
Huang Z, Wang J, He A, Wang J, Wei J, et al: Ferritin light chain
deficiency-induced ferroptosis is involved in preeclampsia
pathophysiology by disturbing uterine spiral artery remodelling.
Redox Biol. 58:1025552022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim H, Villareal LB, Liu Z, Haneef M,
Falcon DM, Martin DR, Lee HJ, Dame MK, Attili D, Chen Y, et al:
Transferrin receptor-mediated iron uptake promotes colon
tumorigenesis. Adv Sci (Weinh). 10:e22076932023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Feng H, Schorpp K, Jin J, Yozwiak CE,
Hoffstrom BG, Decker AM, Rajbhandari P, Stokes ME, Bender HG, Csuka
JM, et al: Transferrin receptor is a specific ferroptosis marker.
Cell Rep. 30:3411–3423.e7. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Henning Y, Blind US, Larafa S, Matschke J
and Fandrey J: Hypoxia aggravates ferroptosis in RPE cells by
promoting the Fenton reaction. Cell Death Dis. 13:6622022.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Borchert A, Kalms J, Roth SR, Rademacher
M, Schmidt A, Holzhutter HG, Kuhn H and Scheerer P: Crystal
structure and functional characterization of
selenocysteine-containing glutathione peroxidase 4 suggests an
alternative mechanism of peroxide. Biochim Biophys Acta Mol Cell
Biol Lipids. 1863:1095–1107. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fang X, Wang H, Han D, Xie E, Yang X, Wei
J, Gu S, Gao F, Zhu N, Yin X, et al: Ferroptosis as a target for
protection against cardiomyopathy. Proc Natl Acad Sci USA.
116:2672–2680. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Conrad M and Friedmann Angeli JP:
Glutathione peroxidase 4 (Gpx4) and ferroptosis: What's so special
about it? Mol Cell Oncol. 2:e9950472015. View Article : Google Scholar
|
|
27
|
Dixon SJ, Patel DN, Welsch M, Skouta R,
Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS
and Stockwell BR: Pharmacological inhibition of cystine-glutamate
exchange induces endoplasmic reticulum stress and ferroptosis.
Elife. 3:e025232014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ingold I, Berndt C, Schmitt S, Doll S,
Poschmann G, Buday K, Roveri A, Peng X, Porto Freitas F, Seibt T,
et al: Selenium utilization by GPX4 is required to prevent
hydroperoxide-induced ferroptosis. Cell. 172:409–422.e21. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gao M, Yi J, Zhu J, Minikes AM, Monian P,
Thompson CB and Jiang X: Role of mitochondria in ferroptosis. Mol
Cell. 73:354–363.e3. 2019. View Article : Google Scholar
|
|
30
|
Friedmann Angeli JP, Schneider M, Proneth
B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch
A, Eggenhofer E, et al: Inactivation of the ferroptosis regulator
Gpx4 triggers acute renal failure in mice. Nat Cell Biol.
16:1180–1191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian
D, Liu D, Zhang F, Ning S, Yao J and Tian X: Ischemia-induced ACSL4
activation contributes to ferroptosis-mediated tissue injury in
intestinal ischemia/reperfusion. Cell Death Differ. 26:2284–2299.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh
HJ III, Kang R and Tang D: Autophagy promotes ferroptosis by
degradation of ferritin. Autophagy. 12:1425–1428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Qiu B, Zandkarimi F, Bezjian CT, Reznik E,
Soni RK, Gu W, Jiang X and Stockwell BR: Phospholipids with two
polyunsaturated fatty acyl tails promote ferroptosis. Cell.
187:1177–1190.e18. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Blanc RS, Shah N, Hachmer S, Salama NAS,
Meng FW, Mousaei A, Puri G, Hwang JH, Wacker EE, Yang BA, et al:
Epigenetic erosion of H4K20me1 induced by inflammation drives aged
stem cell ferroptosis. Nat Aging. 5:1491–1509. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Campbell NJ and Maani CV: Histology
muscle. StatPearls [Internet] Treasure Island (FL): StatPearls
Publishing; 2026
|
|
36
|
Braithwaite JP and Al Khalili Y:
Physiology muscle myocyte. StatPearls [Internet] Treasure Island
(FL): StatPearls Publishing; 2026
|
|
37
|
Kuang S, Kuroda K, Le Grand F and Rudnicki
MA: Asymmetric self-renewal and commitment of satellite stem cells
in muscle. Cell. 129:999–1010. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
García-Prat L, Muñoz-Cánoves P and
Martinez-Vicente M: Dysfunctional autophagy is a driver of muscle
stem cell functional decline with aging. Autophagy. 12:612–613.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ding H, Chen S, Pan X, Dai X, Pan G, Li Z,
Mai X, Tian Y, Zhang S, Liu B, et al: Transferrin receptor 1
ablation in satellite cells impedes skeletal muscle regeneration
through activation of ferroptosis. J Cachexia Sarcopenia Muscle.
12:746–768. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Thorpe CT, Peffers MJ, Simpson D,
Halliwell E, Screen HR and Clegg PD: Anatomical heterogeneity of
tendon: Fascicular and interfascicular tendon compartments have
distinct proteomic composition. Sci Rep. 6:204552016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bi Y, Ehirchiou D, Kilts TM, Inkson CA,
Embree MC, Sonoyama W, Li L, Leet AI, Seo BM, Zhang L, et al:
Identification of tendon stem/progenitor cells and the role of the
extracellular matrix in their niche. Nat Med. 13:1219–1227. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang G, Wang S, Ouyang X, Wang H, Li X,
Yao Z, Chen S and Fan C: Glycolipotoxicity conferred tendinopathy
through ferroptosis dictation of tendon-derived stem cells by YAP
activation. IUBMB Life. 75:1003–1016. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gao Y, Sun W, Wang J, Zhao D, Tian H, Qiu
Y, Ji S, Wang S, Fu Q, Zhang F, et al: Oxidative stress induces
ferroptosis in tendon stem cells by regulating mitophagy through
cGAS-STING pathway. Int Immunopharmacol. 138:1126522024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Masaldan S, Clatworthy SAS, Gamell C,
Meggyesy PM, Rigopoulos AT, Haupt S, Haupt Y, Denoyer D, Adlard PA,
Bush AI and Cater MA: Iron accumulation in senescent cells is
coupled with impaired ferritinophagy and inhibition of ferroptosis.
Redox Biol. 14:100–115. 2018. View Article : Google Scholar
|
|
45
|
Kamel-ElSayed SA, Nezwek TA and Varacallo
MA: Physiology. Bone. StatPearls [Internet] Treasure Island (FL):
StatPearls Publishing; 2026
|
|
46
|
Yin Y, Chen GJ, Yang C, Wang JJ, Peng JF,
Huang XF, Tang QM and Chen LL: Osteocyte ferroptosis induced by
ATF3/TFR1 contributes to cortical bone loss during ageing. Cell
Prolif. 57:e136572024. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cushman DM, Stokes D, Vu L, Corcoran B,
Fredericson M, Eby SF and Teramoto M: Ultrasound as a predictor of
time-loss injury for the patellar tendon, Achilles tendon and
plantar fascia in division I collegiate athletes. Br J Sports Med.
59:241–248. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Charnoff J, Ponnarasu S, Sina RE and Naqvi
U: Tendinosis. StatPearls [Internet] Treasure Island (FL):
StatPearls Publishing; 2026
|
|
49
|
Wu Y, Qian J, Li K, Li W, Yin W and Jiang
H: Farrerol alleviates collagenase-induced tendinopathy by
inhibiting ferroptosis in rats. J Cell Mol Med. 26:3483–3494. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang Y, Shi L, Wang F, Wang L, Min N, Wen
L and Xue Q: Screening for
autophagy/hypoxia/ferroptosis/pyroptosis-related genes of tendon
injury and repair in a rat model after celecoxib and lactoferrin
treatment. J Orthop Surg Res. 18:3832023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tong Z, Li H, Jin Y, Sheng L, Ying M, Liu
Q, Wang C and Teng C: Mechanisms of ferroptosis with immune
infiltration and inflammatory response in rotator cuff injury.
Genomics. 115:1106452023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yuan Z, Zhu X, Dai Y, Shi L, Feng Z, Li Z,
Diao N, Guo A, Yin H and Ma L: Analysis of differentially expressed
genes in torn rotator cuff tendon tissues in diabetic patients
through RNA-sequencing. BMC Musculoskelet Disord. 25:312024.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Khalil AA, Smits D, Haughton PD, Koorman
T, Jansen KA, Verhagen MP, van der Net M, van Zwieten K, Enserink
L, Jansen L, et al: A YAP-centered mechanotransduction loop drives
collective breast cancer cell invasion. Nat Commun. 15:48662024.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang H, Zhou J, Liu Z, Wang K and Jiang
H: Bioinformatics analysis of ferroptosis in frozen shoulder. BMC
Med Genomics. 17:2342024. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Liu X, Zhou Y, Qi Z, Huang C and Lin D:
Taurine alleviates ferroptosis-induced metabolic impairments in
C2C12 myoblasts by stabilizing the labile iron pool and improving
redox homeostasis. J Proteome Res. 23:3444–3459. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang ZZ, Xu HC, Zhou HX, Zhang CK, Li BM,
He JH, Ni PS, Yu XM, Liu YQ and Li FH: Long-term detraining
reverses the improvement of lifelong exercise on skeletal muscle
ferroptosis and inflammation in aging rats: Fiber-type dependence
of the Keap1/Nrf2 pathway. Biogerontology. 24:753–769. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Huang Y, Wu B, Shen D, Chen J, Yu Z and
Chen C: Ferroptosis in a sarcopenia model of senescence accelerated
mouse prone 8 (SAMP8). Int J Biol Sci. 17:151–162. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ni SH, Zhang XJ, OuYang XL, Ye TC, Li J,
Li Y, Sun SN, Han XW, Long WJ, Wang LJ, et al: Lobetyolin
alleviates ferroptosis of skeletal muscle in 5/6 nephrectomized
mice via activation of hedgehog-GLI1 signaling. Phytomedicine.
115:1548072023. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Liu C, Zheng D, Zhang R, Li H, Tong X, Wu
Y, Zhang G, Wang S, Chen H, Ren Z, et al: Transcriptional diversity
in response to aging across skeletal muscles. Aging Cell.
24:e701642025. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kedlian VR, Wang Y, Liu T, Chen X, Bolt L,
Tudor C, Shen Z, Fasouli ES, Prigmore E, Kleshchevnikov V, et al:
Human skeletal muscle aging atlas. Nat Aging. 4:727–744. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wang Y, Yu R, Wu L and Yang G: Hydrogen
sulfide guards myoblasts from ferroptosis by inhibiting ALOX12
acetylation. Cell Signal. 78:1098702021. View Article : Google Scholar
|
|
62
|
Nguyen NUN, Hsu CC, Ali SR and Wang HV:
Actin-organizing protein palladin modulates C2C12 cell fate
determination. Biochem Biophys Rep. 39:1017622024.PubMed/NCBI
|
|
63
|
Sato S, Hanai T, Kanamoto T, Kawano F,
Hikida M, Yokoi H, Take Y, Magome T, Ebina K, Mae T, et al:
Vibration acceleration enhances proliferation, migration, and
maturation of C2C12 cells and promotes regeneration of muscle
injury in male rats. Physiol Rep. 12:e159052024. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yaffe D and Saxel O: Serial passaging and
differentiation of myogenic cells isolated from dystrophic mouse
muscle. Nature. 270:725–727. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Grevendonk L, Connell NJ, McCrum C, Fealy
CE, Bilet L, Bruls YMH, Mevenkamp J, Schrauwen-Hinderling VB,
Jörgensen JA, Moonen-Kornips E, et al: Impact of aging and exercise
on skeletal muscle mitochondrial capacity, energy metabolism, and
physical function. Nat Commun. 12:47732021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kaluźniak-Szymanowska A, Talarska D, Tobis
S, Styszyński A, Cofta S, Wieczorowska-Tobis K and Deskur-Śmielecka
E: Body compositions phenotypes of older adults with COPD. Front
Nutr. 11:14491892024. View Article : Google Scholar
|
|
67
|
Zhang L, Li D, Chang C and Sun Y:
Myostatin/HIF2α-mediated ferroptosis is involved in skeletal muscle
dysfunction in chronic obstructive pulmonary disease. Int J Chron
Obstruct Pulmon Dis. 17:2383–2399. 2022. View Article : Google Scholar
|
|
68
|
Ji F, Rheem H, Lee H, Eom M and Kim JH:
Multilevel regulation of skeletal muscle ferroptosis in aging: Sex-
and exercise-dependent effects on histological, molecular, and
genetic markers. Geroscience. Jan 10–2026.Epub ahead of print.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Lauretani F, Russo CR, Bandinelli S,
Bartali B, Cavazzini C, Di Iorio A, Corsi AM, Rantanen T, Guralnik
JM and Ferrucci L: Age-associated changes in skeletal muscles and
their effect on mobility: An operational diagnosis of sarcopenia. J
Appl Physiol (1985). 95:1851–1860. 2003. View Article : Google Scholar
|
|
70
|
Kirk B, Cawthon PM, Arai H, Ávila-Funes
JA, Barazzoni R, Bhasin S, Binder EF, Bruyere O, Cederholm T, Chen
LK, et al: The conceptual definition of sarcopenia: Delphi
consensus from the global leadership initiative in sarcopenia
(GLIS). Age Ageing. 53:afae0522024. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Chen Y, Zhang Y, Zhang S and Ren H:
Molecular insights into sarcopenia: Ferroptosis-related genes as
diagnostic and therapeutic targets. J Biomol Struct Dyn.
43:7989–8007. 2025. View Article : Google Scholar
|
|
72
|
Yan Y, Teng H, Hang Q, Kondiparthi L, Lei
G, Horbath A, Liu X, Mao C, Wu S, Zhuang L, et al: SLC7A11
expression level dictates differential responses to oxidative
stress in cancer cells. Nat Commun. 14:36732023. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wang W, Ren W, Zhu L, Hu Y and Ye C:
Identification of genes and key pathways underlying the
pathophysiological association between sarcopenia and chronic
obstructive pulmonary disease. Exp Gerontol. 187:1123732024.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Tang J and Zhang H, Yin L, Zhou Q and
Zhang H: The gut microbiota from maintenance hemodialysis patients
with sarcopenia influences muscle function in mice. Front Cell
Infect Microbiol. 13:12259912023. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Guo YF, Liu ZY, Zhou M, Kuang WH, Liu Y,
Huang Y, Yin P and Xia ZY: Heat exposure promotes sarcopenia via
gut microbiota-derived metabolites. Aging Cell. 29:e143702025.
View Article : Google Scholar
|
|
76
|
Liu Y, Guo Y, Liu Z, Feng X, Zhou R, He Y,
Zhou H, Peng H and Huang Y: Augmented temperature fluctuation
aggravates muscular atrophy through the gut microbiota. Nat Commun.
14:34942023. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Hesketh GE: Progressive muscular atrophy.
Physiotherapy. 33:57–60. 1947.PubMed/NCBI
|
|
78
|
Bean LA, Thomas C, Villa JF, Fitt AJ,
Javier AJS, Agrawal A, Whitney H, Dos Santos GN, White KE, Huot JR
and Welc SS: Klotho deficiency promotes skeletal muscle weakness
and is associated with impaired motor unit connectivity. Int J Mol
Sci. 26:79862025. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Bao S and Lei Y: Motor unit activity and
synaptic inputs to motoneurons in the caudal part of the injured
spinal cord. J Neurophysiol. 131:187–197. 2024. View Article : Google Scholar
|
|
80
|
Wang HH, Zhang Y, Qu TQ, Sang XQ, Li YX,
Ren FZ, Wen PC and Sun YN: Nobiletin improves D-galactose-induced
aging mice skeletal muscle atrophy by regulating protein
homeostasis. Nutrients. 15:18012023. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Yokogawa H, Higashida K and Nakai N: Iron
homeostasis disruption and lipid peroxidation in skeletal muscle
during short-term immobilization. FEBS Open Bio. Mar 19–2026.Epub
ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Bhatt BJ, Ghosh S, Mazurak V, Brun AQ,
Bathe O, Baracos VE and Damaraju S: Molecular subtypes of human
skeletal muscle in cancer cachexia. Nature. 646:973–982. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Ma X, Rao Z, Jin Z, Lu Y, Sun Z and Zheng
L: Resistance exercise counteracts skeletal muscle atrophy in T2DM
mice by upregulating FGF21 and activating PI3K/Akt pathway.
Biomolecules. 16:32025. View Article : Google Scholar
|
|
84
|
Cohen S, Brault JJ, Gygi SP, Glass DJ,
Valenzuela DM, Gartner C, Latres E and Goldberg AL: During muscle
atrophy, thick, but not thin, filament components are degraded by
MuRF1-dependent ubiquitylation. J Cell Biol. 185:1083–1095. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Lee J, Lee SH, Kim H and Chung SW: Effect
of electrical muscle stimulation on the improvement of deltoid
muscle atrophy in a rat shoulder immobilization model. J Orthop
Res. 42:2634–2645. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Liu Z, Zeng X, Bian W, Li H, Tegeleqi B,
Gao Z and Liu J: Exosomes from muscle-derived stem cells repair
peripheral nerve injury by inhibiting ferroptosis via the
Keap1-Nrf2-Ho-1 axis. J Cell Biochem. 125:e306142024. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
He J, He Z, Wang H, Zhang C, Pei T, Yan S,
Yan Y, Wang F, Chen Y, Yuan N, et al: Caffeic acid alleviates
skeletal muscle atrophy in 5/6 nephrectomy rats through the
TLR4/MYD88/NF-kB pathway. Biomed Pharmacother. 174:1165562024.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Ju L, Diao J, Zhang J, Dai F, Zhou H, Han
Z, Hu R, Pei T, Wang F, He Z, et al: Shenshuai Yingyang Jiaonang
ameliorates chronic kidney disease-associated muscle atrophy in
rats by inhibiting ferroptosis mediated by the HIF-1α/SLC7A11
pathway. Heliyon. 10:e290932024. View Article : Google Scholar
|
|
89
|
You L: Dihydromyricetin inhibits
ferroptosis to attenuate cisplatin-induced muscle atrophy. Physiol
Res. 73:405–413. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Liu X, Xu M, Yu Y, Chen Y, Weng X and Zhu
L: PD-1 alleviates cisplatin-induced muscle atrophy by regulating
inflammation and oxidative stress. Antioxidants (Basel).
11:18392022. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Sheng Z, Yu Z, Wang M, Zhou R, Chen S, Yu
X and Li F: Targeting STAT6 to mitigate sepsis-induced muscle
atrophy and weakness: Modulation of mitochondrial dysfunction,
ferroptosis, and CHI3L1-Mediated satellite cell loss. Biochem
Biophys Rep. 37:1016082023.
|
|
92
|
Tran L, Xie B, Assaf E, Ferrari R, Pipinos
II, Casale GP, Mota Alvidrez RI, Watkins S and Sachdev U:
Transcriptomic profiling identifies ferroptosis-related gene
signatures in ischemic muscle satellite cells affected by
peripheral artery disease-brief report. Arterioscler Thromb Vasc
Biol. 43:2023–2029. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Maimaiti Y, Abulitifu M, Ajimu Z, Su T,
Zhang Z, Yu Z and Xu H: FOXO regulation of TXNIP induces
ferroptosis in satellite cells by inhibiting glutathione
metabolism, promoting sarcopenia. Cell Mol Life Sci. 82:812025.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Co HKC, Wu CC, Lee YC and Chen SH:
Emergence of large-scale cell death through ferroptotic trigger
waves. Nature. 631:654–662. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Wáng YXJ, Xiao BH, Leung JCS, Griffith JF,
Aparisi Gómez MP, Bazzocchi A, Diacinti D, Chan WP, Guermazi A and
Kwok TCY: The observation that older men suffer from hip fracture
at DXA T-scores higher than older women and a proposal of a new low
BMD category, osteofrailia, for predicting fracture risk in older
men. Skeletal Radiol. 54:925–936. 2025. View Article : Google Scholar :
|
|
96
|
Florez H, Carrasco JL, Barberá M,
Hernández-Rodríguez J, Muxi A, Mocritcaia A, Prieto-González S, Cid
MC, Monegal A, Guañabens N and Peris P: Risk factors for
glucocorticoid induced osteoporosis in young adults. Front
Endocrinol (Lausanne). 16:15289622025. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Sornay-Rendu E, Duboeuf F and Chapurlat
RD: Postmenopausal women with normal BMD who have fractures have
deteriorated bone microarchitecture: A prospective analysis from
The OFELY study. Bone. 182:1170722024. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Porter JL and Varacallo MA:
Osteoporosis(archived). StatPearls [Internet] Treasure Island (FL):
StatPearls Publishing; 2026
|
|
99
|
Deng X, Lin B, Wang F, Xu P and Wang N:
Mangiferin attenuates osteoporosis by inhibiting osteoblastic
ferroptosis through Keap1/Nrf2/SLC7A11/GPX4 pathway. Phytomedicine.
124:1552822024. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Jing Z, Li Y, Zhang H, Chen T, Yu J, Xu X,
Zou Y, Wang X, Xiang K, Gong X, et al: Tobacco toxins induce
osteoporosis through ferroptosis. Redox Biol. 67:1029222023.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Xue C, Luo H, Wang L, Deng Q, Kui W, Da W,
Chen L, Liu S, Xue Y, Yang J, et al: Aconine attenuates
osteoclast-mediated bone resorption and ferroptosis to improve
osteoporosis via inhibiting NF-κB signaling. Front Endocrinol
(Lausanne). 14:12345632023. View Article : Google Scholar
|
|
102
|
Jiang Z, Qi G, He X, Yu Y, Cao Y, Zhang C,
Zou W and Yuan H: Ferroptosis in osteocytes as a target for
protection against post-menopausal osteoporosis. Adv Sci (Weinh).
11:e23073882024. View Article : Google Scholar
|
|
103
|
Xu CY, Xu C, Xu YN, Du SQ, Dai ZH, Jin SQ,
Zheng G, Xie CL and Fang WL: Poliumoside protects against type 2
diabetes-related osteoporosis by suppressing ferroptosis via
activation of the Nrf2/GPX4 pathway. Phytomedicine. 125:1553422024.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Li Y, Cai Z, Ma W, Bai L, Luo E and Lin Y:
A DNA tetrahedron-based ferroptosis-suppressing nanoparticle:
Superior delivery of curcumin and alleviation of diabetic
osteoporosis. Bone Res. 12:142024. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Huang Y, Zhang J, Zhu Y, Zhao R, Xie Z, Qu
X, Duan Y, Li N, Tang D and Luo X: BMP9 alleviates iron
accumulation-induced osteoporosis via the USP10/FOXO1/GPX4 axis. J
Adv Res. 79:953–972. 2026. View Article : Google Scholar :
|
|
106
|
Zhang Y, Cong Y, Du J, Guo D, Huang J, Pan
J, Liang Y, Zhang J, Ye Z, Liu Y and Zhou Y: Lif-deficiency promote
systemic Iron metabolism disorders and increases the susceptibility
of osteoblasts to ferroptosis. Bone. 189:1172662024. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Tang X, Yang M, Liu Y, Zhang H, Hong X,
Deng M, Liu P, Sun Q, Tu X and Shao G: Inhibition of ferroptosis
rescues BMP osteogenic differentiation impaired by iron overload in
the osteoporotic microenvironment. Cell Signal. 136:1121162025.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Teraguchi M, Yoshimura N, Hashizume H,
Muraki S, Yamada H, Minamide A, Oka H, Ishimoto Y, Nagata K,
Kagotani R, et al: Prevalence and distribution of intervertebral
disc degeneration over the entire spine in a population-based
cohort: The Wakayama spine study. Osteoarthritis Cartilage.
22:104–110. 2014. View Article : Google Scholar
|
|
109
|
Li W, Djuric N, Mink C and
Vleggeert-Lankamp CLA: Inflammation and macrophage polarization are
associated with Modic change type in lumbar radiculopathy. Brain
Spine. 5:1042492025. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Al Qaraghli MI and De Jesus O: Lumbar disc
herniation. StatPearls [Internet]. StatPearls Publishing; Treasure
Island, FL: 2026
|
|
111
|
Du J, Dong H, Huang M, Silberschmidt VV,
Meng L and Miao J: Regional variations of mechanical responses of
IVD to 7 different motions: An in vivo study combined with FEA and
DFIS. J Mech Behav Biomed Mater. 160:1067852024. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Guo J, Ding Q and Sun L: Association
between sarcopenia and intervertebral disc degeneration: A
bidirectional two-sample Mendelian randomization. J Back
Musculoskelet Rehabil. 38:902–913. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Jia C, Xiang Z, Zhang P, Liu L, Zhu X, Yu
R, Liu Z, Wang S, Liu K, Wang Z, et al: Selenium-SelK-GPX4 axis
protects nucleus pulposus cells against mechanical
overloading-induced ferroptosis and attenuates senescence of
intervertebral disc. Cell Mol Life Sci. 81:492024. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Xiang Q, Zhao Y and Li W: Identification
and validation of ferroptosis-related gene signature in
intervertebral disc degeneration. Front Endocrinol (Lausanne).
14:10897962023. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Zhu J, Sun R, Sun K, Yan C, Jiang J, Kong
F and Shi J: The deubiquitinase USP11 ameliorates intervertebral
disc degeneration by regulating oxidative stress-induced
ferroptosis via deubiquitinating and stabilizing Sirt3. Redox Biol.
62:1027072023. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Yao B, Cai Y, Wan L, Deng J, Zhao L, Wang
W and Han Z: BACH1 promotes intervertebral disc degeneration by
regulating HMOX1/GPX4 to mediate oxidative stress, ferroptosis, and
lipid metabolism in nucleus pulposus cells. J Gene Med.
25:e34882023. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Xiang Z, Zhang P, Jia C, Xu R, Cao D, Xu
Z, Lu T, Liu J, Wang X, Qiu C, et al: Piezo1 channel exaggerates
ferroptosis of nucleus pulposus cells by mediating mechanical
stress-induced iron influx. Bone Res. 12:202024. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Zou M, Wu S, Wang J, Xue W, Sun X, Liu L,
Yin P and Huang D: Bioinformatics analysis reveals hub genes linked
to programmed cell death in intervertebral disc degeneration. Appl
Biochem Biotechnol. 197:4475–4493. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Takemoto M, Sugishita Y, Takahashi-Suzuki
Y, Fujiya H, Niki H and Yudoh K: Repetitive compressive loading
downregulates mitochondria function and upregulates the cartilage
matrix degrading enzyme MMP-13 through the coactivation of
NAD-dependent sirtuin 1 and Runx2 in osteoarthritic chondrocytes.
Int J Mol Sci. 26:49672025. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
He Q, Yang J, Pan Z, Zhang G, Chen B, Li
S, Xiao J, Tan F, Wang Z, Chen P and Wang H: Biochanin A protects
against iron overload associated knee osteoarthritis via regulating
iron levels and NRF2/System xc-/GPX4 axis. Biomed Pharmacother.
157:1139152023. View Article : Google Scholar
|
|
121
|
Li H, Jiang X, Xiao Y, Zhang Y, Zhang W,
Doherty M, Nestor J, Li C, Ye J, Sha T, et al: Combining
single-cell RNA sequencing and population-based studies reveals
hand osteoarthritis-associated chondrocyte subpopulations and
pathways. Bone Res. 11:582023. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Xie Y, Lv Z, Li W, Lin J, Sun W, Guo H,
Jin X, Liu Y, Jiang R, Fei Y, et al: JP4-039 protects chondrocytes
from ferroptosis to attenuate osteoarthritis progression by
promoting Pink1/Parkindependent mitophagy. J Orthop Translat.
51:132–144. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Chen W, Yang W, Meng B, Wang X, Duan H, Xu
Q and Li H: Validation and the role of PDK4 relevant to ferroptosis
in degenerative lumbar disc disease. J Orthop Surg Res. 20:302025.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Seo AY, Xu J, Servais S, Hofer T, Marzetti
E, Wohlgemuth SE, Knutson MD, Chung HY and Leeuwenburgh C:
Mitochondrial iron accumulation with age and functional
consequences. Aging Cell. 7:706–716. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Chen X, Zhang Y, Zhao Y, Zhou M, Cheng P,
Yang H, Shu Y, Duan L, Wu Y, Sun Y, et al: SLC38A1 protects against
aging-related oxidative stress and lipid peroxidation in C2C12
myoblasts: Implication of a ferroptosis-related regulator for
skeletal muscle aging. Exp Gerontol. 210:1128802025. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Qiao C, Sun Z, Muhammad F, Gong W, Lin J,
Lv J, Cheng X, Fei Y, Xu N, Xie Y, et al: Ruthenium oxide nanozyme
for tendinopathy treatment via countering ferroptosis. Adv Healthc
Mater. 14:e25010352025. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Cheff DM, Huang C, Scholzen KC, Gencheva
R, Ronzetti MH, Cheng Q, Hall MD and Arnér ESJ: The ferroptosis
inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4
but of TXNRD1. Redox Biol. 62:1027032023. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Miotto G, Rossetto M, Di Paolo ML, Orian
L, Venerando R, Roveri A, Vučković AM, Bosello Travain V, Zaccarin
M, Zennaro L, et al: Insight into the mechanism of ferroptosis
inhibition by ferrostatin-1. Redox Biol. 28:1013282020. View Article : Google Scholar
|
|
129
|
Skouta R, Dixon SJ, Wang J, Dunn DE, Orman
M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A and
Stockwell BR: Ferrostatins inhibit oxidative lipid damage and cell
death in diverse disease models. J Am Chem Soc. 136:4551–4556.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Zhao J, Jia Y, Mahmut D, Deik AA,
Jeanfavre S, Clish CB and Sankaran VG: Human hematopoietic stem
cell vulnerability to ferroptosis. Cell. 186:732–747.e16. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Zheng Z, Shang X, Sun K, Hou Y, Zhang X,
Xu J, Liu H, Ruan Z, Hou L, Guo Z, et al: P21 resists ferroptosis
in osteoarthritic chondrocytes by regulating GPX4 protein
stability. Free Radic Biol Med. 212:336–348. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Liu R, Xiao Y, Zhang G, Han P, Lin Z and
Song H: TRIM8 enhances chondrocyte ferroptosis by inhibiting
YTHDF2-m6A mediated SREBF2 mRNA degradation to promote OA
progression. Int Immunopharmacol. 152:1144412025. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Xiong X, Li W, Yu C, Qiu M, Zhang Z, Hu C,
Zhu S, Yang L, Pen H, Song X, et al: SMURF1-induced ubiquitination
of FTH1 disrupts iron homeostasis and suppresses myogenesis. Int J
Mol Sci. 26:13902025. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Han S, Zhao X, Yu C, Cui C, Zhang Y, Zhu
Q, Qiu M, Yang C and Yin H: Nestin regulates autophagy-dependent
ferroptosis mediated skeletal muscle atrophy by ubiquitinating MAP
1LC3B. J Cachexia Sarcopenia Muscle. 16:e137792025. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Wan Y, Shen K, Yu H and Fan W: Baicalein
limits osteoarthritis development by inhibiting chondrocyte
ferroptosis. Free Radic Biol Med. 196:108–120. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Zhou X, Pan Y, Li J, Zhuang R, Tong P and
Xia H: Notopterol mitigates osteoarthritis progression and relieves
pain in mice by inhibiting PI3K/Akt/GPX4-mediated ferroptosis. Int
Immunopharmacol. 151:1143232025. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Gwon HJ, Cho W, Choi SW, Lim DS,
Tanriverdi EÇ, Abd El-Aty AM, Jeong JH and Jung TW: Donepezil
improves skeletal muscle insulin resistance in obese mice via the
AMPK/FGF21-mediated suppression of inflammation and ferroptosis.
Arch Pharm Res. 47:940–953. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Liu J, Ren Z, Yang L, Zhu L, Li Y, Bie C,
Liu H, Ji Y, Chen D, Zhu M and Kuang W: The NSUN5-FTH1/FTL pathway
mediates ferroptosis in bone marrow-derived mesenchymal stem cells.
Cell Death Discov. 8:992022. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Bao T, Liao T, Cai X, Lu B, Dai G, Pei S,
Zhang Y, Li Y and Xu B: METTL3 mediated ferroptosis in chondrocytes
and promoted pain in KOA via HMGB1 m6A modification. Cell Biol Int.
48:1755–1765. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Lin Y, Shen X, Ke Y, Lan C, Chen X, Liang
B, Zhang Y and Yan S: Activation of osteoblast ferroptosis via the
METTL3/ASK1-p38 signaling pathway in high glucose and high fat
(HGHF)-induced diabetic bone loss. FASEB J. 36:e221472022.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Shang J, Xiong C, Jiang W, Yu Z, Zhang J,
Zhang Y, Han L, Miao K, Yu C, Huang Y and Zhou X: Gossypol acetic
acid alleviates the ferroptosis of chondrocytes in osteoarthritis
by inhibiting GPX4 methylation. Curr Med Chem. 32:2422–2439. 2025.
View Article : Google Scholar
|
|
142
|
Ruan B, Dong J, Wei F, Huang Z, Yang B,
Zhang L, Li C, Dong H, Cao W, Wang H and Wang Y: DNMT
aberration-incurred GPX4 suppression prompts osteoblast ferroptosis
and osteoporosis. Bone Res. 12:682024. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Romanick SS, Godoy L, Lopez A, Matsumura
A, Boc K, Stewart TJ, Baker JE and Ferguson BS: Skeletal muscle
alpha actin acetylation enhances myosin binding and increases
calcium sensitivity. Biophys Rep (N Y). 5:1002262025.PubMed/NCBI
|
|
144
|
Xiong X, Huang H, Wang N, Zhou K and Song
X: Sirt1 overexpression inhibits chondrocyte ferroptosis via Ftl
deacetylation to suppress the development of osteoarthritis. J Bone
Miner Metab. 43:203–215. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Zhang Z, Zhang N, Li M, Ma X and Qiu Y:
Sappanone a alleviates osteoarthritis progression by inhibiting
chondrocyte ferroptosis via activating the SIRT1/Nrf2 signaling
pathway. Naunyn Schmiedebergs Arch Pharmacol. 397:8759–8770. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Bai L, Wang T, Zheng J, Bian C, Zhang Y
and Fan Y: SIRT1 mediated by baicalein and GFI1 promotes osteogenic
differentiation and ameliorates osteoporosis by inhibiting
ferroptosis in bone marrow mesenchymal stem cells. J Biochem Mol
Toxicol. 39:e703682025. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Martin SD, Connor T, Sanigorski A, McEwen
KA, Henstridge DC, Nijagal B, De Souza D, Tull DL, Meikle PJ,
Kowalski GM, et al: Class IIa HDACs inhibit cell death pathways and
protect muscle integrity in response to lipotoxicity. Cell Death
Dis. 14:7872023. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Guan Z, Jin X, Guan Z, Liu S, Tao K and
Luo L: The gut microbiota metabolite capsiate regulate SLC2A1
expression by targeting HIF-1α to inhibit knee
osteoarthritis-induced ferroptosis. Aging Cell. 22:e138072023.
View Article : Google Scholar
|
|
149
|
Ren Y, Zhang S, Wang Y, Jacobson DS,
Reisdorf RL, Kuroiwa T, Behfar A, Moran SL, Steinmann SP and Zhao
C: Effects of purified exosome product on rotator cuff tendon-bone
healing in vitro and in vivo. Biomaterials. 276:1210192021.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Jin S, Wang Y, Wu X, Li Z, Zhu L, Niu Y,
Zhou Y and Liu Y: Young exosome bio-nanoparticles restore
aging-impaired tendon stem/progenitor cell function and reparative
capacity. Adv Mater. 35:e22116022023. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Li Y, Ma M, Wang X, Li J, Fang Z, Li J,
Yang B, Lu Y, Xu X and Li Y: Celecoxib alleviates the DSS-induced
ulcerative colitis in mice by enhancing intestinal barrier
function, inhibiting ferroptosis and suppressing apoptosis.
Immunopharmacol Immunotoxicol. 46:240–254. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Liu C, He P, Guo Y, Tian Q, Wang J, Wang
G, Zhang Z and Li M: Taurine attenuates neuronal ferroptosis by
regulating GABAB/AKT/GSK3β/β-catenin pathway after
subarachnoid hemorrhage. Free Radic Biol Med. 193:795–807. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Zhang M, Qin X, Zhao Z, Du Q, Li Q, Jiang
Y and Luan Y: A self-amplifying nanodrug to manipulate the
Janus-faced nature of ferroptosis for tumor therapy. Nanoscale
Horiz. 7:198–210. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Kumar A, Vaca-Dempere M, Mortimer T,
Deryagin O, Smith JG, Petrus P, Koronowski KB, Greco CM, Segalés J,
Andrés E, et al: Brain-muscle communication prevents muscle aging
by maintaining daily physiology. Science. 384:563–572. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Gao T, Xu G, Ma T, Lu X, Chen K, Luo H,
Chen G, Song J, Ma X, Fu W, et al: ROS-responsive injectable
hydrogel loaded with SLC7A11-modRNA inhibits ferroptosis and
mitigates intervertebral disc degeneration in rats. Adv Healthc
Mater. 13:e24011032024. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Li W, Lv Z, Wang P, Xie Y, Sun W, Guo H,
Jin X, Liu Y, Jiang R, Fei Y, et al: Near infrared responsive gold
nanorods attenuate osteoarthritis progression by targeting TRPV1.
Adv Sci (Weinh). 11:e23076832024. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Xue Q, Yan D, Chen X, Li X, Kang R,
Klionsky DJ, Kroemer G, Chen X, Tang D and Liu J: Copper-dependent
autophagic degradation of GPX4 drives ferroptosis. Autophagy.
19:1982–1996. 2023. View Article : Google Scholar : PubMed/NCBI
|

















