Definition and types of autophagy
Autophagy, a word derived from the Greek words
'auto' and '-phagy', is a highly conserved physiological process in
which eukaryotic cells degrade abnormal proteins and damaged
organelles through the lysosomal system. In 1967, it was formally
proposed by Professor Christian de Duve, winner of the Nobel Prize
in Physiology or Medicine, while studying the effect of glucagon on
rat liver lysosomes (1). After
nearly 60 years of in-depth research, autophagy has been confirmed
to be involved in the occurrence and development of a variety of
diseases, such as cancer, cardiovascular diseases, metabolic
diseases, and neurological diseases (2-5).
Autophagy can be divided into three main types based
on the mechanism of substrate transport to lysosomes:
Macroautophagy, microautophagy, and chaperone-mediated autophagy
(CMA). Macroautophagy is a highly conserved mechanism of lysosomal
degradation. Unlike microautophagy or CMA, macroautophagy requires
the formation of double-layer membrane vesicles, which can isolate
intracellular components such as proteins, macromolecular
complexes, and organelles, and even invading pathogens (6). Microautophagy is a conserved
degradation pathway in which cytoplasmic components are directly
engulfed and degraded by the lysosomal system. In yeast, this
process is accomplished by direct invagination of the vacuolar
membrane, whereas in mammalian cells, it is mediated by
endolysosomes (7). The
degradation substrates of microautophagy include soluble proteins
and various organelles, such as peroxisomes, mitochondria, and even
some nuclear structures (8).
Finally, CMA is a particular protein degradation pathway. In this
process, the cytoplasmic chaperone protein heat shock protein
family A (Hsp 70) member 8, also know as heat shock cognate 70
(Hsc70) recognizes the pentapeptide motif (similar to the KFERQ
sequence) in the substrate protein to form a substrate/chaperone
protein complex, which binds to lysosome-associated membrane
protein 2A (LAMP2A) on the lysosomal surface, triggering LAMP2A
polymerization to create a transport complex. After the substrate
is unfolded, the complex mediates the internalization of the
substrate into the lysosome for degradation (9,10). However, with the continuous
advancement of autophagy research, Sahu et al (11) demonstrated that proteins
containing KFERQ-like motifs can also be degraded via an
alternative pathway, endosomal microautophagy (eMI). eMI operates
independently of LAMP2A, and the delivery of its cargo proteins to
late endosomes (LEs) is primarily driven by the endosomal sorting
complex required for transport (ESCRT) machinery mediated by
vacuolar protein sorting (VPS)4 and tumor susceptibility gene 101,
thereby facilitating the conversion of LEs into multivesicular
bodies (MVBs). The process is initiated by the recognition and
binding of cytosolic KFERQ-motif-containing proteins by Hsc70,
followed by electrostatic interaction between the cationic domain
of Hsc70 and the endosomal membrane, which induces localized
membrane invagination. Subsequently, the cargo proteins are
transported to lysosomes for degradation (12,13). Owing to its highly selective
degradation mechanism, eMI has emerged as a promising avenue with
potential applications in targeted cancer therapy (14).
The origin of the autophagosomal membrane is a
central, long-debated question in autophagy research. Currently,
three main models have been proposed. These involve membrane
sources from the endoplasmic reticulum (ER), mitochondria, and the
Golgi apparatus. In recent years, ER-mitochondria contact sites
have gained significant attention. Wilhelm Bernhard first observed
these structures in rat hepatocytes using electron microscopy as
early as 1952 (15). Research
has shown that under starvation conditions, key components of the
class III phosphatidylinositol-3-kinase (PI3KC3) complex [including
autophagy-related protein 14-like protein (ATG14L), VPS34, Beclin
1, and VPS15] become enriched in mitochondria-associated membranes
and may be recruited to ER-mitochondria contact sites during
autophagy induction. At the same time, autophagy related (ATG)5, a
marker of autophagosome formation, localizes to these contact sites
until the autophagosome is formed (16). Walker et al (17) proposed the omegasome model for
starved cells. This model features specific ER subdomains enriched
in phosphatidylinositol-3-phosphate (PI3P; also known as PtdIns3P),
called 'omegasomes' (17). These
cup-shaped structures protrude from the ER membrane and serve as
platforms for autophagosome formation. Double FYVE-containing
protein 1 binds to PI3P through its FYVE domain and simultaneously
localizes to the ER/Golgi membrane with its ER-targeting sequence
(18). Autophagy-related
proteins such as ATG5 then get recruited to these PI3P-rich
regions. These regions create a local environment favorable for the
curvature and expansion of the phagophore, or isolation membrane.
The Golgi apparatus is also a significant source of autophagosomal
membranes in mammals. Locke and Collins (19) initially observed this phenomenon
in 1965 in developing fat body cells of invertebrates. Later
research confirmed the involvement of the Golgi complex in early
autophagy (20). During
telophase, when the Golgi apparatus reassembles, Golgi-derived
membranes appear around autophagosomes, suggesting these supply
membrane material for autophagosome expansion. ATG9, the only known
transmembrane ATG protein, is also associated with the Golgi and
may help deliver membranes to autophagosome formation sites
(20). Of note, these models are
not mutually exclusive. They may play dominant roles in different
cell types or under distinct physiological conditions. The modes of
autophagosome formation can also be species-specific. In yeast,
phagophore membranes form at a specific cytoplasmic site, the
pre-autophagosomal structure, but such a structure is not evident
in mammalian cells (21). In
mammals, phagophore membranes mostly originate from the ER
(22), gradually forming through
interactions with other cytoplasmic membranes, such as the
trans-Golgi network and LEs (18). Under certain conditions,
membranes may also be derived from the nuclear envelope (23). Notably, the scarcity of
transmembrane proteins in autophagosomal membranes means mammalian
cells might still form autophagosomes through de novo lipid
synthesis. Despite progress in understanding membrane sources,
fully clarifying the formation mechanism of this organelle demands
further exploration. Key questions remain regarding autophagosome
formation. It is unclear whether basal and stimulus-induced
autophagosomal membranes are identical, whether the membrane source
depends on the type of stimulus, whether ER-contact is essential
for autophagosome formation, and whether formation dynamics differ
among membrane sources. Most of these aspects still require
clarification.
Formation of autophagosomes
Autophagy is a process that consists of a series of
molecular events that ultimately lead to the formation of
autophagosomes. Autophagosomes engulf cytoplasmic material and
ultimately fuse with lysosomes to degrade their contents. Each step
is regulated by a series of ATG proteins, many of which are
organized into large complexes that are recruited in a specific
order and whose activity is directly or indirectly regulated by
metabolic signaling pathways. Next, the complexes formed during
autophagy and the roles of the molecules within these complexes
during autophagy are briefly introduced (24) (Fig. 1).
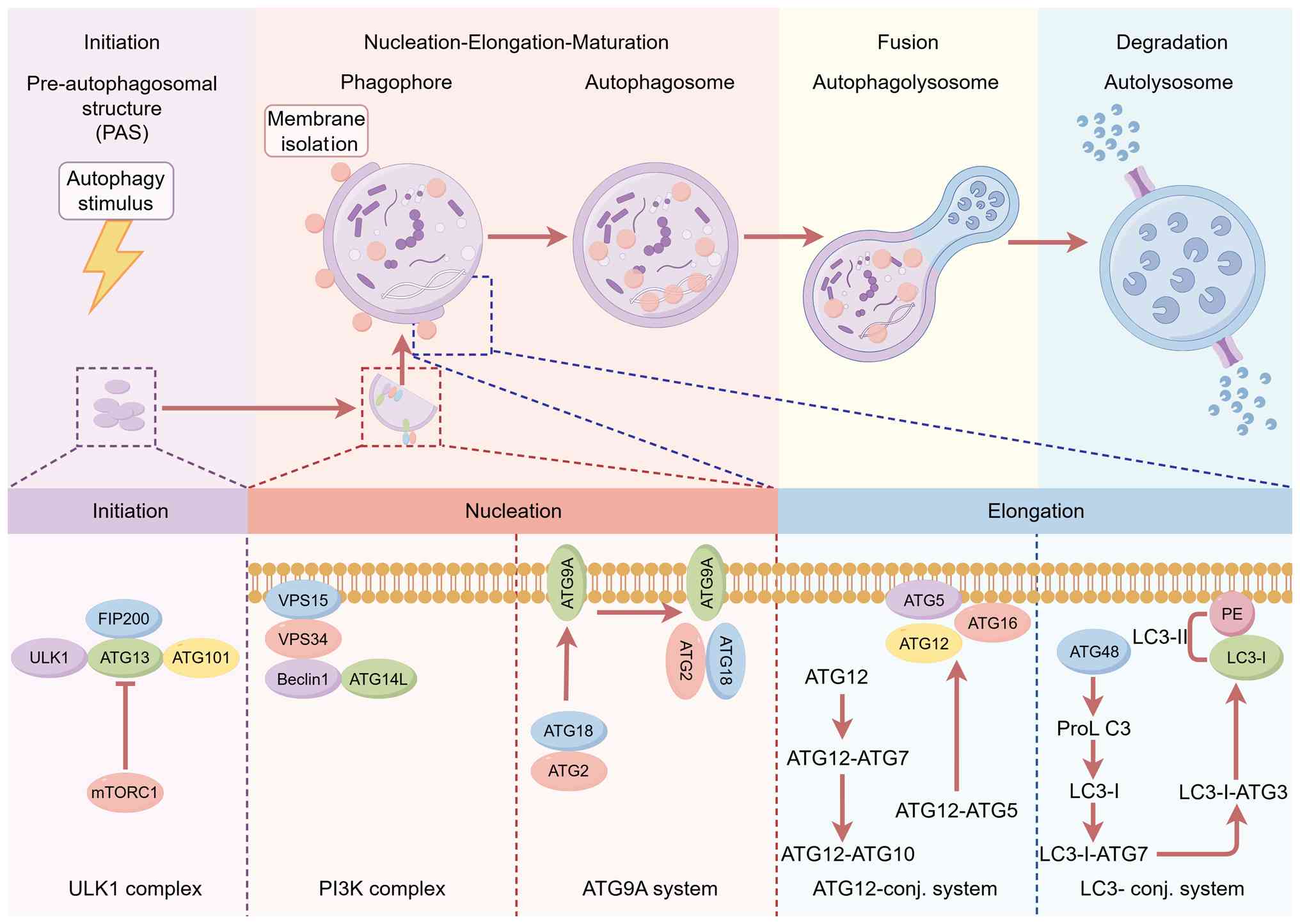 | Figure 1Autophagosome complex during the
process of autophagy. FIP200, focal adhesion kinase family
interacting protein of 200 kDa; ULK, Unc-51 like autophagy
activating kinase; ATG, autophagy related; mTORC1, mechanistic
target of rapamycin complex 1; VPS, vacuolar protein sorting; LC3,
microtubule-associated protein 1A/1B light chain 3B; PE,
phosphatidylethanolamine; SQSTM1, sequestosome 1; PI3K,
phosphatidylinositol-3-kinase; HOPS, homotypic fusion and vacuole
protein sorting. |
Autophagy initiation
First, autophagy initiation is associated with the
Unc-51 like autophagy activating kinase (ULK) complex, which
consists of ULK1, ATG13, RB1-inducible coiled-coil 1/focal adhesion
kinase family interacting protein of 200 kDa (FIP200), and ATG101,
and its activity is negatively regulated by mechanistic target of
rapamycin complex 1 (mTORC1) and positively regulated by
AMP-activated protein kinase (AMPK) (25). Activation of this complex
triggers membrane nucleation. As one of the key components of the
complex, ULK1 is a serine/threonine protein kinase. The N-terminus
of ULK1 contains a protein kinase domain, and the C-terminus is
responsible for interacting with the C-terminus of ATG13, which is
necessary for initiating autophagy (26). ATG101 is entirely composed of the
Hop1, Rev7, Mad2 (HORMA) domain. It forms a heterodimer with ATG13
through the HORMA domain at its N-terminus, thereby preventing
proteasomal degradation of ATG13 and enhancing the interaction
between ATG13 and ULK1 (27,28). ATG13 is located on the autophagic
isolation membrane and plays a role in connecting ULK1 with the
scaffold subunit FIP200 (29).
FIP200 is a predicted large coiled-coil protein involved in
scaffolding, and FIP200 is crucial for maintaining ULK1 kinase
activity. In the absence of FIP200, autophagy is severely inhibited
(30). Chu et al
(31) found that in a cellular
model investigating the regulation of autophagic initiation, linear
ubiquitin chain assembly complex (LUBAC) and OTU deubiquitinase
with linear linkage specificity (OTULIN) coordinately regulate
autophagy initiation and autophagosome maturation by mediating
linear ubiquitination and stabilization of ATG13. In
OTULIN-knockdown cells, hyperubiquitinated ATG13 is recruited to
phagophores and undergoes sustained expansion, thereby impairing
autophagosome maturation (31).
Thus, LUBAC and OTULIN are recognized as ubiquitin enzymes that
regulate the ubiquitination and protein levels of ATG13 to govern
autophagic initiation and maturation.
Membrane nucleation and autophagosome
formation
Secondly, membrane nucleation requires the
participation of PI3KC3-complex I (CI). The PI3KC3-CI complex is
composed of the catalytic subunit VPS34/PIK3C3,
VPS15/phosphoinositide-3-kinase regulatory subunit 4 (PIK3R4),
Beclin 1 (BECN1), and ATG14L. The complex adopts a V-shaped model,
where PIK3C3 and PIK3R4 form the right arm of the V shape and
possess catalytic functions, while BECN1 and ATG14 form the left
arm of the V shape and have regulatory functions. PIK3C3 contains a
C2 domain, a helical domain, and a kinase domain. The C2 domain of
PIK3C3 directly contacts the WD40 domain of PIK3R4, and this
binding is crucial for the kinase activity of PIK3C3 and
autophagosome formation (32,33). Beclin 1 serves as the core
component of the PI3KC3-CI complex. The coiled-coil domain of
Beclin 1 functions to assemble complexes containing distinct ATG14L
and UVRAG, thereby regulating the activity of VPS34 (34). When the ULK1 initiation complex
is activated in response to changes in the energy status of the
body, ULK1 binds to and phosphorylates Ser19 of ATG14, Ser249 of
VPS34, and Ser15 (as well as other sites) of Beclin 1, leading to
the activation of PI3KC3-CI and the production of PI3P (35,36). PI3P, as a marker on the isolation
membrane, is essential for recruiting specific autophagic factors
and membrane remodeling (37,38). A previous study has demonstrated
that in a mouse model of colitis, nuclear receptor binding factor 2
(NRBF2) functions as a regulatory subunit of the ATG14-Beclin
1-VPS34 complex to restrict intestinal inflammation and apoptosis
through positive regulation of macroautophagy/autophagy.
NRBF2-deficient mice exhibited exacerbated colitis symptoms
following dextran sulfate sodium challenge, revealing a novel
mechanism by which NRBF2 modulates the autophagic complex to limit
intestinal inflammation (39).
Nevertheless, whether NRBF2-mediated regulation of autophagy exerts
similar effects in other models of inflammatory disease remains to
be further investigated.
Autophagosome elongation
After the PI3KC3-CI complex is activated, the
extension of phagosomes and the formation of autophagosomes are
accomplished through two ubiquitin-like protein binding systems,
the ATG12 and ATG8 systems (40). In the ATG12 dimeric system, ATG7
and ATG10 couple ATG12 with ATG5. Subsequently, the ATG12-ATG5
complex binds to ATG16L1 and moves to the phagosome, forming the
ATG12-ATG5-ATG16L1 ubiquitin-like protein complex (41,42). The PI3P-binding protein WIPI2
recruits the ATG12-ATG5-ATG16L1 complex to the phagosome, promoting
the transition from phagosome initiation to phagosome expansion
(43). In the ATG8 system, the
homolog of microtubule-associated protein 1A/1B light chain 3B
(LC3) generated by the proteolysis of ATG4 from ATG8 is then
converted to LC3I. Subsequently, the E1 activating enzyme ATG7, the
E2 binding enzyme ATG3, and the E3 connecting enzyme
ATG12-ATG5-ATG16L1 activate LC3I and promote its interaction with
phosphatidylethanolamine, forming lipidated ATG8/LC3-II (44-47), coordinating the extension of the
phagosome. When the extended membrane closes around its cargo,
ESCRT, including VPS37A, charged MVB protein 2A, and AAA ATPase
VPS4, participates in the regulation of autophagosome closure
(48-50). Additionally, it has been found
that the ER-localized autophagy protein EPG-3/vacuole membrane
protein 1 and transmembrane protein 41B also play a key role in
controlling the maturation of phagosomes and autophagosome closure
(51-53).
Liu et al (54) demonstrated that hypoxia-inducible
factor-1α at the translational level (HITT) binds to the key
autophagy protein ATG5, thereby preventing the formation of the
ATG12-ATG5-ATG16L1 complex and suppressing autophagosome
biogenesis. This sensitized colorectal cancer cells to
PI-103-induced cell death both in vitro and in vivo
(in a nude mouse model). The study identified HITT as a novel
RNA-based regulator of autophagy (54). However, the structural basis
underlying the HITT-ATG5 interaction and its upstream regulatory
signals remain to be elucidated.
Autophagosome fusion with lysosome
The final step of autophagy is the fusion of the the
autophagosome with the lysosome, resulting in the degradation of
autophagic cargo by lysosomal hydrolases. A series of factors
regulates this process. The roles of the homotypic fusion and
protein sorting (HOPS) complex and the soluble NSF attachment
protein receptor (SNARE) complex in autophagic fusion are examined
(55,56). The HOPS complex is the core
complex for autophagosome-lysosome fusion and consists of six
subunits, including VPS11, VPS16, VPS18, VPS33A, VPS39, and VPS41
(57). In yeast, HOPS binds to
the vacuolar Rab7-like GTPase Ypt7 through its subunits Vps41 and
Vps39, and supports autophagosome-lysosome fusion by promoting
SNARE assembly. VPS41 is a molecular ruler for membrane fusion
mediated by HOPS (58).
Additionally, the HOPS complex promotes autophagosome-lysosome
fusion through interaction with (syntaxin 17) STX17. When VPS33A,
VPS16, or VPS39 are knocked out, autophagic flux is blocked,
leading to the accumulation of STX17 and LC3-positive
autophagosomes (59). The SNARE
complex is a major participant in controlling membrane-mediated
transport through vesicle fusion and has been proven to be the core
mechanism for autophagosome-lysosome fusion (60). SNAREs can be classified into
Q-SNAREs and R-SNAREs, and Q-SNARE proteins are further divided
into Q-a, Q-b, and Q-c SNARE proteins based on their amino acid
sequences in the SNARE domain (61). Q-SNARE and R-SNARE proteins
achieve membrane fusion by forming trans-SNARE complexes. For
example, the QaSNARE protein STX17 is located in the autophagosome,
while the R-SNARE protein vesicle-associated membrane protein 8 is
located in the lysosome/LE. By recruiting Qbc-SNARE protein
synaptic-associated protein 29 (SNAP29), they jointly form a
trans-SNARE complex to achieve the fusion of the autophagosome and
the lysosome (62). Using the
inhibitor SM15, SNAP29 O-GlcNAcylation can be enhanced, inhibiting
the formation of SNARE complexes and blocking the fusion of
autophagosomes and lysosomes (63).
Regulation of autophagy by nutrients
Autophagy is highly sensitive to changes in the
nutritional environment, cellular metabolism, energy status,
hypoxia, oxidative stress, DNA damage, protein aggregation, and the
presence of intracellular pathogens. Autophagy can be induced by
the deficiency of essential nutrients (such as glucose and amino
acids) or specific metabolites (such as fatty acids and ammonia).
Therefore, a thorough understanding of the mutual regulation
between autophagy and macronutrient concentrations is crucial for
formulating strategies to combat obesity.
Amino acids
Amino acids, as the fundamental building blocks of
proteins, play a crucial role in maintaining the environmental
nutrient levels necessary for life. When the supply of amino acids
is insufficient, proteins respond to cellular nutrient fluctuations
by activating the ubiquitin-proteasome degradation and autophagy
mechanisms (64). Conversely,
when there is an accumulation of specific amino acids in cells, the
accumulated amino acids induce autophagic dysfunction in cells and
promote disease progression (65). Two evolutionarily conserved
nutrient pathways, namely the mTORC1 pathway and the general
control non-derepressible 2 (GCN2) pathway, play key roles in amino
acid sensing (66,67).
mTORC1 senses amino acids through signal
transduction. Under conditions of sufficient amino acids, amino
acids activate Rag guanosine triphosphate (GTP)ases, promoting the
translocation of mTORC1 to the lysosomal surface and regulating
autophagy (68,69). Sciarretta et al (70) found that mice with high-fat diet
(HFD)-induced obesity and metabolic syndrome exhibited dysregulated
cardiac activation of Ras homolog enriched in brain and mTORC1
during ischemia. These HFD mice displayed suppressed cardiac
autophagy, which further exacerbated myocardial ischemic injury.
Pharmacological and genetic inhibition of mTORC1 restored autophagy
and abolished the increase in myocardial infarct size observed in
HFD mice (70). Under conditions
of amino acid starvation, Nowosad et al (71) discovered that a fraction of p27
is recruited to lysosomes, where it interacts with late
endosomal/lysosomal adaptor, MAPK and mTOR activator 1 (LAMTOR1).
The binding of p27 to LAMTOR1 prevents Ragulator complex assembly
and mTORC1 activation, thereby promoting autophagy. Conversely,
p27-deficient cells exhibited elevated mTORC1 signaling, along with
impaired lysosomal activity and autophagy. This phenotype is
associated with the cytoplasmic sequestration of transcription
factor EB (TFEB), which prevents the induction of lysosomal genes
required for lysosomal function (71). Additionally, Wong et al
(72) also found that during
amino acid starvation, autophagy can be induced by promoting the
dephosphorylation of ULK1. This occurs through the stimulation of
the dissociation of protein phosphatase 2A from its inhibitor,
Alpha4, which increases the phosphatase activity of ULK1 (72).
For the control of the GCN2 pathway, GCN2 is a
serine/threonine protein kinase with a high affinity for all
uncharged tRNAs. It senses the intracellular amino acid levels by
binding to uncharged tRNAs (73). When the intracellular amino acid
levels are low, GCN2 binds to specific uncharged tRNAs, triggering
the homodimerization and autophosphorylation activation of GCN2.
The activated GCN2 phosphorylates the eukaryotic translation
initiation factor 2α at the Ser51 site, and this phosphorylation
can upregulate the activation transcription factor 4 (ATF4) and
C/EBP homologous protein, thereby inducing autophagy by enhancing
the transcription of autophagy-related proteins (74). Disruptions in intracellular amino
acid levels occur frequently in obesity-related metabolic diseases,
and in these cases, autophagy dysregulation often occurs. However,
the exact mechanism of 'resetting and/or reactivation of mTOR'
remains unclear. Future research should further investigate the
regulatory mechanism by which changes in amino acid levels affect
the upstream molecules of mTOR (Fig.
2).
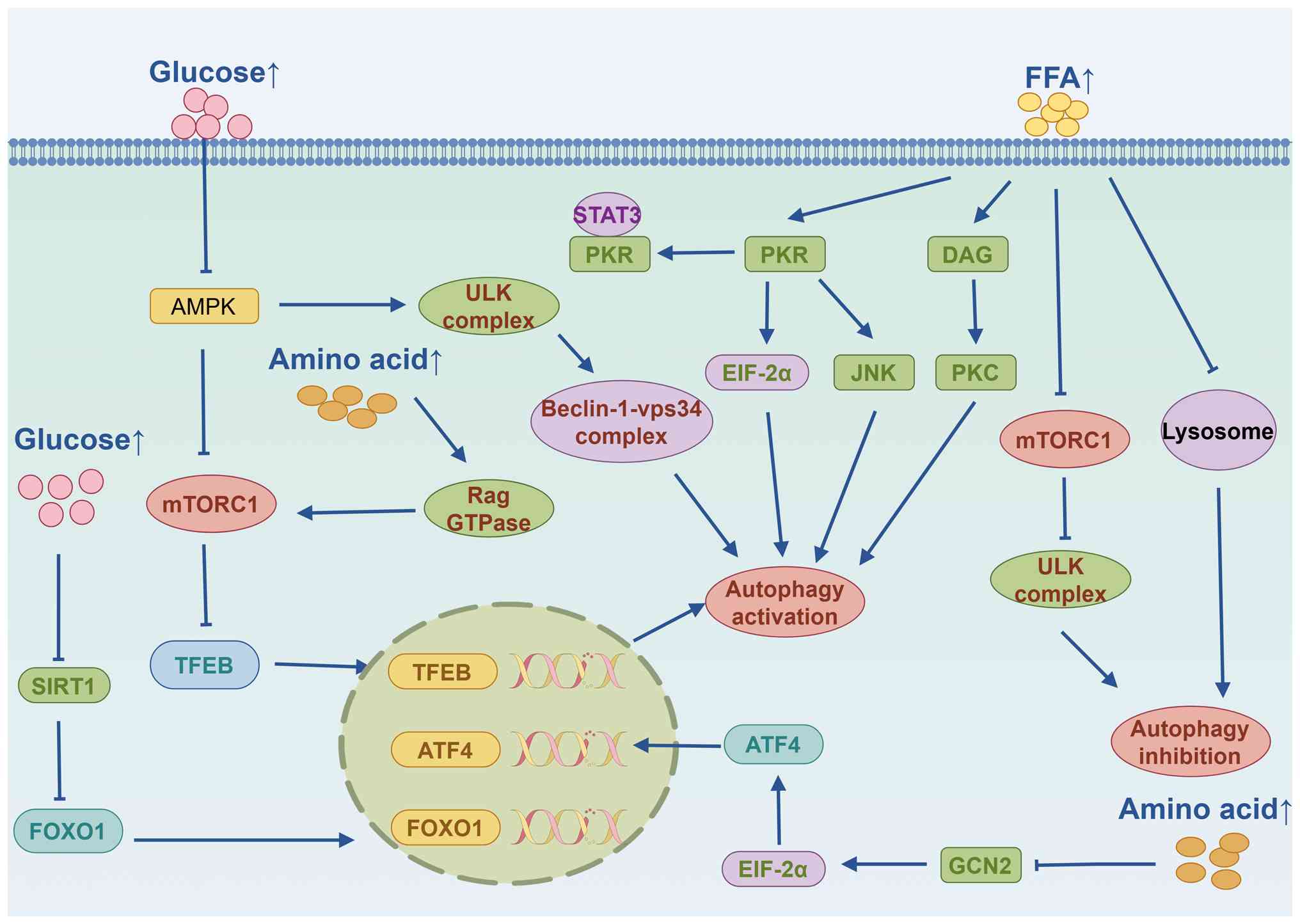 | Figure 2Regulation of autophagy by glucose,
amino acids, and fatty acids. FFA, free fatty acids; PKR, portein
kinase R; DAG, diacylglycerol; PKC, protein kinase C; EIF-2α,
eukaryotic translation initiation factor-2α; VPS, vacuolar protein
sorting; AMPK, AMP-activated protein kinase; mTORC1, mechanistic
target of rapamycin complex 1; SIRT1, sirtuin 1; TFEB,
transcription factor EB; ATF4, activation transcription factor 4;
FOXO1, forkhead box O1; GCN2, general control non-derepressible 2;
ULK, Unc-51 like autophagy activating kinase. |
Glucose
As the primary energy source, glucose plays a
central role in the metabolic processes throughout the body.
Glucose not only generates energy through oxidative phosphorylation
but also provides intermediate biomolecules, such as fatty acids,
amino acids, and nucleotides, for biosynthesis according to the
needs of cells. Glucose can affect the level of autophagy by
regulating the secretion of glucagon and insulin. Glucagon can
enhance autophagy, while insulin signaling inhibits autophagy
(75,76).
High glucose (HG) inhibits autophagy through the
AMPK pathway. For instance, Wang et al (77) found that when chondrocytes were
treated with different HG concentrations, the AMPK signaling
pathway and autophagy were significantly inhibited. At the same
time, apoptosis-related and senescence-related markers were
significantly upregulated. 5-Aminoimidazole-4-carboxamide
ribonucleotide (AICAR) reversed these phenomena. This research
indicates that HG can inhibit autophagy through the AMPK signaling
pathway, leading to chondrocyte apoptosis and senescence (77). During glucose deficiency, ATP
levels decrease, promoting the accumulation of AMP and the
activation of AMPK. AMPK can inhibit mTORC1 activation of autophagy
by stimulating the tuberous sclerosis complex (TSC)1-TSC2 (also
known as hamartin-tuberin) complex or directly through the
phosphorylation of the ULK1 complex (78). In agreement with this, Karabiyik
et al (79) demonstrated
that under glucose deprivation, AMPK activates ULK1, leading to
phosphorylation of the lipid kinase PI3P-phospahte 5-kinase, type
III (PIKFYVE) at Ser1548. The activated PIKFYVE subsequently
promotes the formation of phosphatidylinositol-5-phosphate
(PtdIns5P)-containing autophagosomes, thereby driving autophagy
upregulation. This novel finding that ULK1 orchestrates a
non-canonical autophagic pathway, PtdIns5P-dependent autophagy, not
only expands our understanding of autophagy but also unveils
potential therapeutic strategies for human diseases (80). Another key signaling molecule
involved in glucose-mediated autophagy regulation is the forkhead
box protein O (FOXO) family transcription factors.
NAD+-dependent sirtuins may deacetylate FOXO, thereby
promoting autophagy (81,82).
Future development of novel autophagy regulation strategies
targeting the glucose-sensing pathway will provide potential
therapeutic approaches for metabolic diseases related to obesity,
such as diabetes, obesity, and cancer (Fig. 2).
Lipids
Lipids are a diverse group of nutrients that include
fatty acids and cholesterol. They are characterized by their
hydrophobic carbon backbone and are used for energy storage,
membrane biosynthesis, and other cellular processes (83). Although increased lipid intake
and lipid storage disorders (such as in the state of obesity) can
lead to abnormal autophagy function of cells, compared to the
interaction between autophagy and amino acids or glucose, the
interaction mechanism between cell autophagy and lipids has not
been fully elucidated. However, research has shown that saturated
fatty acids and unsaturated fatty acids have a certain duality in
regulating autophagy (84).
Excessive intake of saturated fatty acids can lead
to the hypertrophy of fat cells and induce autophagy. Through
research using an in vitro model of 3T3-L1 adipocytes in
mice, it was found that high concentrations of palmitic acid (PA)
would induce autophagy through ER stress. Using 4-benzoylbenzoic
acid to block the ER stress pathway significantly reduced the level
of LC3-II, and autophagy levels were inhibited (85). Additionally, in the study on the
lipid toxicity effect of saturated fatty acids on brain astrocytes,
it was discovered that after treating primary astrocytes with PA,
the level of LC3-II (a marker of autophagosomes) increased. By
contrast, the flux of LC3-II in astrocytes decreased, and the
autophagy level of astrocytes was blocked (86). Compared with saturated fatty
acids, unsaturated fatty acid oleic acid (OA) can also trigger
autophagy, but the underlying molecular mechanism is different. The
research by Niso-Santano et al (87) showed that OA may induce autophagy
through a non-classical pathway. In this pathway, the LC3 protein
related to autophagosomes (LC3-II) binds to the multi-protein
PI3KIII class/VPS34 complex containing Beclin 1, thereby driving
the autophagic process of the Golgi apparatus (87). Furthermore, in a mouse model of
non-alcoholic fatty liver disease (NAFLD), it was demonstrated that
PA induces autophagic impairment by reducing the nuclear
translocation of TFEB and suppressing Cathepsin B activity. By
contrast, OA markedly alleviated PA-induced apoptosis and
autophagic dysfunction in hepatocytes, and improved hepatic glucose
and lipid metabolism, indicating that OA-driven autophagy
represents a key protective mechanism against NAFLD (88). Therefore, clarifying the
regulatory mechanism of fatty acid metabolism on autophagy will
provide novel intervention strategies for the treatment of
obesity-related metabolic diseases (Fig. 2).
Tissue specificity and model differences
in nutrient-mediated autophagy
The regulation of autophagy by nutrients exhibits
significant tissue specificity, with notable differences in results
among various research models. This phenomenon is particularly
prominent in the sensing mechanisms of core nutrients such as amino
acids, glucose, and lipids. Regarding the regulation by amino
acids, a study in a mouse model of diabetic cardiomyopathy found
that the deficiency of branched-chain amino acids could activate
autophagy in cardiac fibroblasts through the AMPK-ULK1 signaling
pathway, thereby exacerbating the cardiomyopathy phenotype in
diabetic mice (89). However,
Fermo et al (90) found
in a rat cerebral cortex study that the supplementation of
branched-chain amino acids could significantly increase the levels
of autophagy markers Beclin 1, ATG7, and ATG5 in the cerebral
cortex, enhance the expression of ATG12 in the striatum, and
increase the levels of ATG5 and LC3-I/II in the hippocampus,
thereby promoting autophagy in brain regions (90). These findings suggest that the
regulation of autophagy by amino acids has clear tissue dependence.
In terms of glucose regulation, Li et al (91) found that high blood sugar levels
in colorectal cancer (CRC) cells could inhibit autophagy by
activating the PI3K/AKT/mTOR pathway, thereby promoting the
proliferation and migration of CRC cells (91). However, in the in vitro
culture model of MC3T3-E1 osteoblast precursor cells, high blood
sugar significantly induced autophagy through the reactive oxygen
species (ROS)-AKT-mTOR axis, affecting the apoptosis and
proliferation of cells (92),
which exhibited a different regulatory pattern compared with the
tumor cell model. In lipid regulation research, Hossain et
al (93) found that free
fatty acids (FFAs) could induce the upregulation of DEAD-box
helicase 3 expression in HepG2 cells, thereby increasing miR-141,
inhibiting sirtuin 1 (SIRT1) expression, and reducing the levels of
autophagy-related proteins, ultimately decreasing autophagic flux
(93). In a cell model of
alcohol-related liver disease, FFA could downregulate LAMP2
expression through ATF4-mediated endoplasmic reticulum stress,
damaging autophagic flux, while overexpression of LAMP2 could
restore autophagy function and alleviate alcohol-induced liver
injury (94). In summary, the
tissue specificity and model differences in the nutrient-autophagy
regulatory axis essentially stem from the precise regulatory
network formed by biological systems during evolution to maintain
metabolic homeostasis in different tissues under specific
physiological conditions. Future research should address the
limitations of single-cell models and tissue types by establishing
a comparative analysis framework that spans multiple tissues and
models, thereby enabling a systematic investigation of dynamic
changes in nutrient-sensing pathways under various physiological
and pathological conditions. In-depth exploration in this direction
will not only help clarify the heterogeneous pathogenesis of
metabolic diseases but also provide a theoretical basis for the
formulation of precise nutritional intervention strategies, that
is, to design differentiated nutritional intervention plans based
on the autophagy status of different tissues and disease stages to
achieve precise regulation of autophagic activity.
Autophagy and obesity-related metabolic
diseases
Given the complex interplay between nutrient
metabolism and the autophagic process, it is reasonable to
hypothesize that autophagy plays a critical role in the
pathogenesis of obesity-related metabolic diseases. The
pathological state induced by obesity is typically characterized by
abnormal accumulation of lipid droplets, formation of protein
aggregates, and mitochondrial damage, all of which are primary
degradation substrates of the autophagic pathway. Consequently,
impaired autophagy may accelerate the progression of
obesity-related pathological alterations in multiple tissues. Based
on this, the following discussion will further explore the
intrinsic relationship between autophagy and obesity-related
metabolic diseases, aiming to provide insights for future research
and the development of therapeutic strategies.
Type 2 diabetes (T2D)
T2D is mainly characterized by dysfunction of
pancreatic β cells and insulin resistance. Obesity is considered a
contributing factor to type 2 diabetes mellitus (T2DM), and the
prevalence of T2DM increases with the increase in obesity
prevalence (95). A recent study
showed that autophagy plays a crucial role in maintaining the
typical structure and function of pancreatic β cells and in
mitigating insulin resistance in T2D (96). The mechanisms of autophagy in T2D
are next examined.
Firstly, basal autophagy has been proven to be
crucial in maintaining the typical structure and function of
pancreatic β cells and in mitigating insulin resistance. Under
conditions of insulin resistance induced by T2D or HFD, autophagic
vacuole formation in early-stage β cells is markedly upregulated to
clear the accumulation of various toxic intracellular aggregates.
This increase in autophagic activity at an early stage serves as a
distinctive protective defense mechanism (97). Furthermore, a study using an
animal model investigating autophagic activity in pancreatic β
cells revealed that ATG7-deficient mice exhibit impaired glucose
tolerance and reduced serum insulin levels, along with β-cell
morphological abnormalities including mitochondrial swelling, ER
distension, and vacuolar changes (98). Additionally, under conditions of
metabolic stress, Martino et al (99) established a tiered experimental
model comprising INS-1E cells, isolated rat islets, and isolated
human islets, and demonstrated that palmitate, as a representative
FFA, consistently activates autophagic pathways across species,
whereas HG treatment alone does not elicit this effect. Further
investigations revealed that inhibition of autophagy exacerbates
FFA-induced β-cell death (99).
Collectively, these findings indicate that autophagy is an
essential mechanism for maintaining β-cell structure and function,
and that its dysfunction may lead to organellar metabolic
abnormalities, impaired insulin secretion, and the onset of
hyperglycemia. Furthermore, in a HFD-induced T2D model, Meng et
al (100) observed in
zebrafish that a lipotoxic environment induces autophagic deficits,
disrupts insulin signaling, and leads to abnormal accumulation of
preproinsulin, thereby compromising cellular homeostasis and
promoting the development of insulin resistance. Concurrently, Budi
et al (101) reported in
a C57BL/6 mouse model fed a HFD that chronic high-fat feeding not
only elicits insulin resistance but also results in late-stage
autophagy inhibition, ER stress, and adipocyte apoptosis. The
suppression of autophagy may indirectly contribute to the
progression of insulin resistance by exacerbating ER stress and
apoptotic cell death (101).
Therefore, basal autophagy of pancreatic β cells appears to be an
adaptive response and is crucial for the homeostasis of pancreatic
β cells.
On the other hand, some studies have also reported
the adverse effects of autophagy in T2D. Excessive activation of
autophagy is associated with autophagic apoptosis and necrosis of
pancreatic β cells. For instance, in pancreatic tissues isolated
from rats, it was found that with age, the expression of autophagy
markers LAMP2 and LC3b and the apoptotic index significantly
increased in aged rats. Excessive activation of autophagy induced
autophagic apoptosis of pancreatic β cells (102). Additionally, a case-control
study of human pancreatic samples from patients with T2D and
healthy controls showed that autophagosomes and autophagic vesicles
were correlated with the level of β-cell death, and metformin
improved the autophagic changes in diabetic β cells and delayed
β-cell death (103). The known
autophagy inhibitor mTOR plays a crucial role in preventing
pancreatic β-cell apoptosis induced by oxidative stress and
autophagy-mediated cell death. It limits insulin resistance and the
development of T2D caused by pancreatic β-cell death (104). This indicates that autophagy
has a detrimental effect on pancreatic β cells, leading to
accelerated cell death and the development of T2D. However,
numerous unanswered questions remain regarding the specific
mechanisms that regulate pancreatic β-cell autophagy. Therefore,
further elucidation of the mechanism of pancreatic β-cell autophagy
dysregulation may provide novel therapeutic interventions for
diabetes.
NAFLD
NAFLD is the most common liver disease worldwide and
can progress to more severe non-alcoholic steatohepatitis (NASH),
liver cirrhosis, and hepatocellular carcinoma. Notably, 70-80% of
obese and diabetic patients will develop NAFLD (105). The pathogenesis of NAFLD
involves multiple concurrent events. During this process, defective
organelles, such as lipid droplets, mitochondria, ER, and
peroxisomes, accumulate in the cells and require degradation
systems such as autophagy to remove these harmful cellular
components. Previous research has demonstrated that autophagy
disorders are closely related to NAFLD (106).
NAFLD is characterized by the irregular deposition
of triglycerides in liver cells, mainly because the amount of fatty
acids entering the cells through intake or synthesis exceeds the
amount of fatty acids oxidized or excreted by the cells. Autophagy
dysfunction is considered an important marker for the aggravation
of fatty degeneration in NAFLD (107). In a HFD-induced NAFLD mouse
model, it was found that the autophagic activity in the liver was
downregulated in ATG7 gene-diminished autophagy-deficient mice,
accompanied by an increase in fatty degeneration (108). It is notable that even under a
regular diet, specific liver knockout of ATG7 in mice leads to the
occurrence of hepatic fatty degeneration, indicating that autophagy
plays a crucial role in preventing fat accumulation in liver cells
(109). Consistent with these
research findings, it was found that sequestosome 1 (SQSTM1)
induces the phosphorylation of ULK1 by promoting the interaction
between AMPK and ULK1, thereby inducing macroautophagy/autophagy.
The activity of the non-classical KEAP1-NFE2L2 pathway mediated by
SQSTM1 conferred protection to the liver, protecting it from the
effects of lipid toxicity in the liver of SQSTM1-knockout mice
(110). Additionally, TFEB is a
major regulator of lysosomal biogenesis and autophagy. Long-term
feeding of HFD leads to a decrease in the expression of TFEB
specifically in liver cells, resulting in impaired lipolysis
metabolism in the liver and hepatic fatty degeneration.
Overexpression of adenovirus TFEB restores the autophagy flow and
improves liver fatty degeneration in similar animal models
(111). However, Cao et
al (112) discovered that
in a mouse model of NAFLD and in in vitro cell culture
systems, M2-type macrophages promote hepatic stellate cell (HSC)
autophagy by secreting prostaglandin E2, which binds to the EP4
receptor on the HSC surface. This process subsequently exacerbates
HSC activation, extracellular matrix deposition, and liver
fibrosis, thereby promoting disease progression (112). In the white adipose tissue of
mice fed a HFD, the levels of Rubicon, a negative regulator of
autophagy, were decreased, accompanied by increased autophagic
flux. Adipocyte-specific ATG7-knockout mice exhibited reduced serum
FFA levels and alleviated HFD-induced lipotoxicity, hepatic
inflammation, and fibrosis. These findings suggest that autophagy
inhibition in white adipose tissue ameliorates liver pathology in
NAFLD through adipose-liver crosstalk (113). In summary, autophagy plays an
dual role in hepatic lipid metabolism, and autophagy
dysfunction-induced lipid metabolism abnormalities may be a
potential cause of NAFLD.
It is worth noting that the lack of autophagy in the
liver can also lead to liver damage and increased inflammation.
Research has found that Rubicon, a negative regulator of
autophagosome-lysosome fusion that interacts with Beclin 1, is
overexpressed in the liver tissues of mice fed with HFD, resulting
in ER stress, cell apoptosis, and increased lipid accumulation in
liver cells. At the same time, specific knockout of Rubicon shows a
significant reduction in liver steatosis and damage (114). Moreover, further research has
shown that autophagy can block the damage caused by
lipopolysaccharide (LPS) to the liver of mice. Specifically, liver
cell-specific autophagy knockout mice exhibit increased liver
damage and mortality due to high-dose LPS (115), suggesting the role of autophagy
in protecting against liver damage and inflammatory responses.
Consistent with this, as autophagy decreases, IL-1β induces
cytotoxicity and pro-inflammatory reactions in liver cells, leading
to the occurrence of liver damage. Therefore, therapies that
increase autophagy may be effective methods for treating liver
diseases such as NAFLD (116).
Collectively, during the progression of NAFLD, autophagy may
initially facilitate lipid clearance at early stages, yet
subsequently contribute to the amplification of cell death or
inflammatory signaling. However, whether regulatory molecules such
as Rubicon consistently assume the same functional role across
distinct disease phases, from simple steatosis to steatohepatitis,
remains to be further elucidated. Moreover, while most research has
focused on the effects of autophagy deficiency or enhancement,
relatively less attention has been paid to autophagic flux,
selective autophagy, and the functional shifts of autophagy-related
proteins within the pathological microenvironment. Future research
should systematically dissect the dual-edged role of autophagy in
the evolution of liver disease and its underlying regulatory
networks, thereby providing a more precise theoretical foundation
for the development of phase-adapted therapeutic strategies for
liver disorders.
Atherosclerosis
Atherosclerosis is a progressive and complex disease
characterized by excessive lipid accumulation in the arterial
intima. Abnormal lipid metabolism, insulin resistance, inflammatory
response, endothelial dysfunction, imbalance of adipocytokines, and
activation of inflammasomes, among other mechanisms, may play a
crucial role in the relationship between obesity and
atherosclerosis (117). Among
the various cell types involved in the formation of atherosclerotic
plaques, autophagy is dysregulated in macrophages, vascular smooth
muscle cells (VSMCs), and endothelial cells, which are of vital
importance for the initiation and development of atherosclerosis.
The changes in autophagy levels in these three types of cells are
next examined (118).
Monocyte-derived macrophages play a crucial role
throughout the entire pathological process of atherosclerosis, from
its onset, progression, to eventual plaque rupture (119). Research has shown that the
autophagy process of macrophages plays a critical protective role
in atherosclerosis, and inhibiting macrophage autophagy can
activate the mechanism of plaque instability and trigger necrotic
reactions. However, inducing macrophage autophagy through mTORC1
can effectively stabilize atherosclerotic plaques (120). Consistent with this, in a mouse
model of atherosclerotic plaque, it was found that macrophages had
impaired autophagic function, with decreased ATG14 expression.
Overexpressing ATG14 in macrophages enhanced the fusion of
autophagosomes and lysosomes, promoted lipid degradation, and
reduced oxidized low-density lipoprotein (oxLDL)-induced cell death
and inflammatory responses (121). Additionally, Li et al
(122) revealed in a mouse
model of atherosclerosis that the level of dosage-sensitive sex
reversal, adrenal hypoplasia critical region, on chromosome X, gene
1 (Dax1) in atherosclerotic plaques was elevated, and Dax1
inhibited autophagy by interacting with TFEB, leading to lipid
accumulation in macrophages and inflammation. By reducing the level
of Dax1 in mice using 2′-deoxycytidine, autophagy was enhanced, and
the progression of atherosclerosis was inhibited (122). However, regulatory molecules
such as mTORC1, ATG14, and Dax1 may exert dynamic or even opposing
functions at different stages of atherosclerosis. Future research
should integrate temporally controlled models with cell-specific
intervention strategies to systematically dissect the mechanistic
roles of autophagy across distinct pathological phases of
atherosclerosis. Such efforts will provide a more precise
theoretical foundation for the development of stage-adapted
therapeutic targets. Therefore, further exploration of the specific
mechanisms regulating macrophage autophagy may yield promising
therapeutic targets for treating atherosclerosis. Secondly, as the
main component of the vascular wall, VSMCs play a key role in the
development of atherosclerosis by converting from a contractile
phenotype to a synthetic phenotype or a macrophage-like phenotype
(123). Regular autophagic
activity is associated with the survival and stability of SMCs, but
excessive autophagy leads to SMC death and plaque instability. A
previous study has shown that in the early stage of
atherosclerosis, when SMCs are exposed to medium concentrations of
oxLDL (10-40 µg/ml), autophagy is activated as a protective
mechanism; while in the late stage of atherosclerosis, when exposed
to high concentrations of oxLDL (≥60 µg/ml), the protective
effect of autophagy weakens and instead exacerbates
autophagy-induced cell death (124). Liu et al (125) found in a mouse model of
atherosclerosis that Paeonol can induce autophagy in VSMCs by
activating the PI3KC3/Beclin 1 signaling pathway, inhibit VSMC
apoptosis, and alleviate the progression of atherosclerosis.
However, although pharmacological interventions such as Paeonol
have demonstrated protective effects under specific conditions,
whether these effects remain consistent across different
pathological stages and distinct cellular states requires
systematic validation. Future research should further elucidate the
spatiotemporally specific mechanisms governing the dynamic
regulatory network of autophagy in SMCs, thereby providing a more
precise theoretical foundation for stage-adapted therapeutic
strategies in atherosclerosis. Finally, the flat cells located on
the inner side of the vascular wall, vascular endothelial cells,
have a key function in maintaining homeostasis. Vascular
endothelial dysfunction is a key step in the formation of
atherosclerosis (126). In mice
models with excessive lipids, endothelial autophagy was revealed to
play a vital role in limiting lipid accumulation in the vascular
wall and maintaining lipid homeostasis. The absence of endothelial
autophagy significantly increased the burden of atherosclerosis
(127). Consistent with this,
Ding et al (128) found
that after oxLDL treatment, there was a decrease in v-Myb avian
myeloblastosis viral oncogene homolog-like 1 expression in
endothelial cells, leading to a reduction in pleckstrin homology
domain containing, family M member 1 expression, inhibition of
endothelial autophagy, and ultimately induction of endothelial cell
apoptosis, promoting the formation of atherosclerotic plaques
(128). Therefore, stimulating
endothelial cell autophagy may be particularly beneficial for the
treatment of atherosclerotic vascular diseases.
Therapies for autophagy
Lifestyle changes and exercise
Changes in lifestyle, such as calorie restriction,
weight loss, and physical exercise, can reverse metabolic rigidity,
meaning the reduced ability to adjust substrate oxidation based on
available substrates, as well as obesity, and insulin resistance.
Dietary restriction has been proven to be beneficial for weight
gain caused by excessive nutrient intake and complications related
to obesity (129,130). Gao et al (131) found that in obese and T2DM
animal models (db/db mice and mice fed a HFD), moderate (40%)
calorie restriction reversed β-cell dysfunction and insulin
resistance in obese mice induced by diet, and restored glucose
homeostasis, these changes being related to the upregulation of
autophagy in β-cells (131).
Moreover, in obesity-induced diabetic mouse models, it was found
that modified forms of calorie restriction, namely intermittent
fasting, could restore the autophagy flux in the pancreas and
improve glucose tolerance by enhancing glucose-stimulated insulin
secretion, β-cell survival, and pancreatic regeneration markers
such as neurogenin 3 nuclear expression, but could not rescue
β-cell death (132). In
addition, Roux-en-Y gastric bypass (RYGB) surgery was demonstrated
to rapidly reduce liver lipid toxicity in T2DM obese rats and
improve insulin sensitivity. The mechanism is related to RYGB
improving metabolic parameters, increasing plasma glucagon-like
peptide-1 (GLP-1), activating autophagy, and reducing liver fat
load (133). A clinical trial
conducted by Guevara-Cruz et al (134) demonstrated that modifications
in caloric intake, such as caloric restriction, or adjustments in
meal timing, exemplified by intermittent fasting, play a
significant role in stimulating both cellular autophagy and
mitophagy, thereby facilitating the elimination of aged and
dysfunctional mitochondria (134). Future research should
systematically evaluate the safety profile of autophagy-based
interventions and explore personalized dietary restriction
strategies tailored to the specific metabolic phenotypes of
patients.
It has long been recognized that exercise is a
powerful physiological stimulus for various metabolic adaptations,
exerting significant impacts on health and diseases. Exercise
training promotes autophagy and improves maximal oxygen uptake and
is considered a proper physiological stimulus for promoting
metabolic adaptations beneficial to health (135). For example, Xiang et al
(136) found that, compared
with metformin, exercise alone was more effective in inhibiting the
ubiquitin-proteasome system, increasing the level of autophagy, and
alleviating skeletal muscle atrophy. This is mainly achieved by
promoting the synthesis and degradation of autophagy through the
AMPK/ULK1 pathway (136). This
study indicates that exercise-induced autophagy plays a crucial
role in the related complications of T2D. In addition, exercise is
the first-line treatment and an important preventive measure for
patients with NAFLD. In a non-alcoholic fatty liver mouse model
with a HFD, it was found that 12-week swimming exercise prevented
hepatic lipid accumulation and alleviated hepatocyte injury in HFD
mice. This was mainly related to the fact that exercise
significantly reduced the expression of the lipid metabolism gene
fatty acid binding protein 1, restored lysosomal function
(including lysosomal proteolysis and maintenance of lysosomal
acidification), and increased autophagic flux (137). Consistent with this result, 15
weeks of moderate treadmill exercise in mice improved the
histological phenotype of NAFLD, including hepatic steatosis,
inflammation, and ballooning degeneration. It prevented HFD-induced
hepatic fat deposition and liver injury. This was mainly related to
the inhibition of abnormal lipid droplet expansion and the
enhancement of lysosomal clearance of lipid droplets during
lipophagy (138). In addition,
physical exercise is also considered a potential form of
'beneficial medicine' for treating atherosclerosis. Li et al
(139) found that long-term
swimming exercise could reduce weight gain in ApoE mice, improve
arterial structural disorder, reduce the burden of atherosclerotic
lesions, decrease the serum concentrations of total cholesterol,
total triglycerides, soluble intercellular adhesion molecule-1,
MMP-9, and IL-6, and increase the expression of autophagy markers
LC3 and Beclin 1 (139).
Moreover, regular exercise could enhance IL-1 signaling, stimulate
endothelial cell autophagy, maintain the homeostasis of arterial
endothelial cells in ApoE-deficient mice, and protect arteries from
vascular diseases (140).
Therefore, the benefits of exercise may be related to the autophagy
mechanism, which is the basis for its positive effects on
obesity-related metabolic diseases.
Pharmacological intervention
Given the crucial role of autophagy in the
development of various human diseases, numerous studies have been
conducted to develop drugs targeting autophagy. To date, the
research on drugs targeting autophagy regulation has mainly focused
on cancer, neurodegenerative diseases, and infectious diseases.
Although no clinical trials using autophagy-targeting drugs for
treating metabolic diseases have been carried out yet, the latest
research indicates that existing or under-development drugs or
compounds related to metabolic diseases may exert therapeutic
effects by regulating the autophagy process. The roles of autophagy
activators and inhibitors in obesity-related metabolic diseases are
next summarized.
Autophagy activators
The activation of autophagy is mainly achieved by
activating AMPK and/or inhibiting the PIK3CA/AKT/MTOR signaling
pathway. AMPK, as the primary energy sensor in cells, responds to
changes in the ATP-to-AMP ratio within cells. The activation of
AMPK provides a powerful target for triggering autophagy (141). AICAR is considered one of the
most commonly used pharmacological regulators of AMPK activity, and
it can cause allosteric activation of AMPK (142). For mice with T2D induced by a
HFD, AICAR treatment led to weight loss, reduction in abdominal fat
volume and quality, and improvement in the pathological morphology
of internal organs, indicating that AICAR has certain therapeutic
potential in treating T2D (143). Additionally, it was found that
the use of AICAR could prevent or reverse diabetic peripheral
neuropathy in both T2D and T1D mouse models, which was related to
increased AMPK phosphorylation and activation of mitochondrial
autophagy (144). Secondly,
metformin, as a first-line anti-diabetic drug, was shown to
effectively alleviate glucose and lipid metabolism disorders, renal
function damage in diabetic rats, oxidative stress in glomerular
cells cultured under HG conditions, and enhanced autophagy levels,
protecting the pathological process of diabetic nephropathy
(145). Concurrently, You et
al (146) also found that
metformin, by activating the AMPK-mTOR pathway, induces autophagy
and has a specific improvement effect on the development of
atherosclerosis (146). In
addition, thiazolidinedione drugs are widely used in clinical
practice to improve insulin sensitivity. It was found that
rosiglitazone could induce phosphorylation of AMPK and reduce
phosphorylation of p70S6 kinase (p70S6K). Inhibition of AMPK
impaired autophagy activation and exacerbated apoptosis induced by
palmitate, suggesting that rosiglitazone-induced autophagy
contributes to its protective function during palmitate treatment
of β cells (147). Finally,
resveratrol is a well-known polyphenolic compound with
anti-obesity, antitumor, anti-diabetic, antioxidant, and anti-aging
effects (148). In a mouse
model of hepatic steatosis, resveratrol induced autophagy through
the cAMP-PRKA-AMPK-SIRT1 signaling pathway, partially improving
hepatic steatosis in mice (149). Moreover, Xu et al
(150) found that resveratrol
activates AMPK and JNK1, thereby inhibiting mTOR and its downstream
effectors p70S6K1 and eukaryotic translation initiation factor
4E-binding protein 1, and disrupting the Beclin 1-Bcl-2 complex,
inducing myocardial cell autophagy and reducing myocardial cell
death to protect myocardial cells (150). Based on the aforementioned, it
is considered that preclinical studies of autophagy activators are
promising; however, further clinical research is still required to
evaluate the efficacy and safety of these treatments.
The PIK3CA/AKT/mTOR signaling pathway is the most
extensively studied autophagy-inhibiting pathway. Small molecule
compounds that inhibit the AKT, mTOR, and PIK3CA complexes within
this pathway can be used as potent autophagy inducers (151). There are two direct strategies
for targeting mTOR activity. Firstly, rapamycin, as an mTOR
inhibitor, modulates the activity of mTOR kinase through allosteric
regulation and activates autophagy (152). For example, in the microglia
and astrocytes of mice fed with HFDs, it was found that
neuroinflammation increased, autophagy and brain-derived
neurotrophic factor (BDNF) levels decreased, AMPK phosphorylation
was inhibited, mTOR phosphorylation was induced, and mice exhibited
depressive and anxiety-like behaviors. After treatment with the
mTOR inhibitor rapamycin, autophagy and BDNF levels increased, and
depressive and anxiety-like behaviors were also improved to a
certain extent (153).
Consistent with this, in the mouse model of diabetes nephropathy
caused by obesity, it was found that the treatment with mTOR
inhibitors improved the autophagy dysfunction of proximal renal
tubular epithelial cells mediated by obesity and restored autophagy
in the proximal renal tubules, improving the renal outcomes of
obese patients (154). In
addition to rapamycin, the mTOR inhibitor eptifibatide is expected
to become a new therapeutic drug for NASH or diseases related to
macrophage autophagy dysregulation. It was found that in
LPS-induced macrophages, eptifibatide improved liver autophagy and
NASH by inhibiting the ROS/p38/Nrf2 axis and the PI3K/AKT/mTOR
pathway and enhancing macrophage autophagy, further inhibiting the
progression of NAFLD (155).
Secondly, small molecules such as Torin1/2, PP242, and AZD8055 that
target the ATP-binding site of mTOR inhibit its kinase activity
through ATP-competitive inhibition (156,157). However, this strategy is mainly
applied in antitumor treatment, and the therapeutic prospects for
obesity-related metabolic diseases need to be further explored in
the future. Currently, AMPK pathway modulators, notably metformin,
represent the strategy with the lowest risk and the most clearly
defined path for clinical translation. Preclinical evidence has
robustly supported the role of autophagy as a therapeutic target in
metabolic diseases. Future frameworks for translational medicine
should prioritize the following directions: The development of
tissue-specific or cell type-specific autophagy-modulating
strategies to minimize systemic toxicity; in-depth investigations
into the stage-dependent functions of autophagy to identify
biomarkers capable of guiding therapeutic windows; and, upon
establishing preclinical safety, the prioritization of clinical
trials using metformin for specific metabolic complications,
alongside early-phase translational studies of agents such as
AICAR, in order to address critical gaps in human evidence.
Autophagy inhibitors
In addition to the therapeutic potential of
autophagy inducers for treating metabolic abnormalities associated
with obesity, inhibiting autophagy may also provide therapeutic
benefits for obesity and its metabolic diseases. Strategies for
inhibiting autophagy typically target the later stages of
autophagy, including the inhibition of autophagosome formation (the
initiation and maturation processes) and lysosomal degradation.
For the initiation of autophagy and the formation
of autophagosomes, small molecules and natural products targeting
the ULK1 complex and PI3KC3-C1 are two primary methods for
inhibiting the initiation of autophagy, which leads to the blockage
of autophagosome biogenesis. The ULK inhibitors MRT67307 and
MRT68921 were selected from a series of closely related analogs
generated during the initial TANK-binding kinase 1 screening
process and are currently mainly used for antitumor treatment
(158). Another early
identified ULK inhibitor, SBI-0206965, also exhibits potent
inhibition of AMPK or ULK1 signal transduction and cellular
function (159). However,
preclinical and clinical evidence regarding their application in
metabolic diseases such as obesity and T2D remains largely absent,
and their efficacy, specificity, and safety in the context of
metabolic disorders are entirely unknown. Additionally, a pan-PI3K
inhibitor, 3-MA, is widely used as an effective experimental tool
to block the early stage of autophagy. Research has found that 3-MA
can temporarily block the PI3K complex (class III) that promotes
autophagy, while continuously inhibiting the PI3K complex (class I)
of autophagy (160). These
kinase inhibitors have great potential in combating obesity-related
metabolic diseases. However, it should be particularly noted that
because these kinase inhibitors typically have broad kinase
activity inhibition, they may cause adverse effects and even
produce effects opposite to those intended.
Autophagy is a one-way ticket for lysosomal
degradation. Inhibiting lysosomal activity can significantly block
autophagic degradation. Lysosomal cavity alkalizing agents such as
chloroquine (CQ) and hydroxychloroquine (HCQ) exert their effects
by damaging lysosomal function and inhibiting autophagy. Mauthe
et al (161) discovered
that CQ can induce severe disorder in the autophagic independence
of the Golgi and endolysosomal systems, inhibit the interaction
between autophagosomes and lysosomes, and mediate autophagy
impairment (161).
Additionally, batroxobin A1, as an autophagy inhibitor, was shown
to block the vacuolar H-ATPase (V-ATPase) enzyme, which is
responsible for the acidification of lysosomes and other
intracellular organelles (162). For instance, Hirao et al
(163) found that batroxobin
blocked V-ATPase, inhibited gluconeogenic enzymes and mitochondrial
electron transfer enzymes in the kidneys of T2D rats, reduced
glucose levels in renal cytoplasm, and improved plasma glucose
levels in patients with T2D (163). In conclusion, drugs targeting
lysosomal function, such as CQ and HCQ, hold the greatest potential
for rapid clinical translation due to their established clinical
availability and preliminary evidence of metabolic benefits.
However, their well-known systemic side effects necessitate
rigorous monitoring. By contrast, upstream targets such as ULK1
offer greater mechanistic precision, yet their pleiotropic effects
and the current absence of metabolic disease-specific data render
the path to clinical application considerably more protracted. To
establish a robust translational medicine framework, future
research must prioritize: i) The development of tissue-specific
delivery strategies; ii) validation in more physiologically
relevant chronic disease models; and iii) the conduct of rigorous
early-phase clinical trials to delineate the safety windows and
preliminary efficacy of distinct target inhibitors in patients with
metabolic disorders.
Finally, in constructing a therapeutic framework
targeting autophagy for metabolic diseases, it is essential to
recognize the heterogeneity of metabolic risk across human
populations. A typical illustration of this is the 'obesity
paradox,' wherein a higher body mass index (BMI) is sometimes
associated with lower mortality in specific patient groups with
established cardiovascular conditions such as heart failure or
coronary artery disease (164).
This phenomenon underscores that BMI or body weight classification
alone is insufficient to accurately reflect underlying metabolic
status, regional fat distribution (such as visceral vs.
subcutaneous adipose tissue), systemic inflammatory tone, or the
extent of organ-specific steatosis. Future therapeutic strategies
must incorporate more refined stratification parameters, including
insulin sensitivity, hepatic fat content, specific autophagic flux
biomarkers, and cardiovascular functional status, to guide patient
segmentation and individualized intervention.
Conclusion and prospects
Autophagy, a highly conserved intracellular
degradation system, plays a central role in maintaining cellular
metabolic homeostasis. The present review systematically examined
the basic concepts, classification, molecular mechanisms of
autophagy, the regulation of autophagy by nutritional factors, and
its pivotal role in obesity-related metabolic diseases such as T2D,
NAFLD, and atherosclerosis. Research indicates that autophagy
exerts a dual regulatory effect on pancreatic β-cell function,
hepatic lipid metabolism, inflammatory responses, and vascular
homeostasis: Moderate activation helps clear damaged organelles and
abnormal proteins, thereby maintaining normal cellular function,
whereas either excessive or insufficient autophagy may exacerbate
metabolic disorders and tissue damage. Under metabolic stress
conditions such as obesity, autophagy function is often
dysregulated, further leading to lipid accumulation, insulin
resistance, and worsened inflammation. Currently, lifestyle
interventions and pharmacological approaches have demonstrated the
potential to improve metabolic abnormalities by modulating
autophagy pathways, suggesting that targeting autophagy may emerge
as a novel therapeutic strategy for metabolic diseases. However,
despite extensive attention on autophagy in the metabolic field,
several key issues remain to be elucidated: First, the mechanisms
of autophagy vary across different tissues, necessitating the use
of cell-specific gene-editing models to deeply dissect its
regulatory networks; second, how nutrients (such as amino acids,
glucose, and lipids) precisely regulate upstream signaling pathways
of autophagy requires further exploration; third, most autophagy
modulators are still in the preclinical research stage, and their
specificity, safety, and long-term efficacy need systematic
validation. Developing tissue-specific autophagy intervention
strategies will be an important future research direction.
In summary, this review systematically integrated
the basic knowledge and cutting-edge progress of autophagy in the
field of metabolism, clarified its potential as a novel target for
the treatment of metabolic diseases, and elucidated the core
challenges in current research at three levels: Tissue-specific
mechanisms, precise nutritional regulation, and clinical
translation bottlenecks. It provided a clear roadmap for
researchers and clinical practitioners to shift from basic
understanding to precise intervention, ultimately offering novel
perspectives and potential intervention methods for the prevention
and treatment of obesity and related metabolic diseases.
Availability of data and materials
Not applicable.
Authors' contributions
CL and XQ conceived the review. CL designed the
scope and structure of the review. XQ performed structured
literature searches. PL, DL, QG and CY critically synthesized and
interpreted findings. YT revised major sections of the manuscript.
All authors read and approved the final manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present review was supported by grants from the National
Natural Science Foundation of China (grant nos. 82370849, 82270839,
82200947 and 82400928), the Natural Science Foundation of Shandong
Province (grant no. ZR2023MH008), and the Shandong Provincial
Natural Science Foundation Youth Fund (grant no. ZR2024QH375).
References
|
1
|
Deter RL and De Duve C: Influence of
glucagon, an inducer of cellular autophagy, on some physical
properties of rat liver lysosomes. J Cell Biol. 33:437–449. 1967.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Matsuura A, Tsukada M, Wada Y and Ohsumi
Y: Apg1p, a novel protein kinase required for the autophagic
process in Saccharomyces cerevisiae. Gene. 192:245–250. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mizushima N, Noda T, Yoshimori T, Tanaka
Y, Ishii T, George MD, Klionsky DJ, Ohsumi M and Ohsumi Y: A
protein conjugation system essential for autophagy. Nature.
395:395–398. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Deretic V: Autophagy in inflammation,
infection, and immunometabolism. Immunity. 54:437–453. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maestri A, Garagnani P, Pedrelli M,
Hagberg CE, Parini P and Ehrenborg E: Lipid droplets, autophagy,
and ageing: A cell-specific tale. Ageing Res Rev. 94:1021942024.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Griffey CJ and Yamamoto A: Macroautophagy
in CNS health and disease. Nat Rev Neurosci. 23:411–427. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li WW, Li J and Bao JK: Microautophagy:
Lesser-known self-eating. Cell Mol Life Sci. 69:1125–1136. 2012.
View Article : Google Scholar
|
|
8
|
Schuck S: Microautophagy-distinct
molecular mechanisms handle cargoes of many sizes. J Cell Sci.
133:jcs2463222020. View Article : Google Scholar
|
|
9
|
Tekirdag K and Cuervo AM:
Chaperone-mediated autophagy and endosomal microautophagy: Joint by
a chaperone. J Biol Chem. 293:5414–5424. 2018. View Article : Google Scholar
|
|
10
|
Bourdenx M, Gavathiotis E and Cuervo AM:
Chaperone-mediated autophagy: A gatekeeper of neuronal
proteostasis. Autophagy. 17:2040–2042. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sahu R, Kaushik S, Clement CC, Cannizzo
ES, Scharf B, Follenzi A, Potolicchio I, Nieves E, Cuervo AM and
Santambrogio L: Microautophagy of cytosolic proteins by late
endosomes. Dev Cell. 20:131–139. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Saftig P and Klumperman J: Lysosome
biogenesis and lysosomal membrane proteins: Trafficking meets
function. Nat Rev Mol Cell Biol. 10:623–635. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bauer PO, Goswami A, Wong HK, Okuno M,
Kurosawa M, Yamada M, Miyazaki H, Matsumoto G, Kino Y, Nagai Y and
Nukina N: Harnessing chaperone-mediated autophagy for the selective
degradation of mutant huntingtin protein. Nat Biotechnol.
28:256–263. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lei K, Li J, Tu Z, Gong C, Liu J, Luo M,
Ai W, Wu L, Li Y, Zhou Z, et al: Endosome-microautophagy targeting
chimera (eMIATAC) for targeted proteins degradation and enhance
CAR-T cell anti-tumor therapy. Theranostics. 14:4481–4498. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bernhard W, Haguenau F, Gautier A and
Oberling C: Submicroscopical structure of cytoplasmic basophils in
the liver, pancreas and salivary gland; study of ultrafine slices
by electron microscope. Z Zellforsch Mikrosk Anat. 37:281–300.
1952.In Undetermined language. View Article : Google Scholar
|
|
16
|
Hamasaki M, Furuta N, Matsuda A, Nezu A,
Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et
al: Autophagosomes form at ER-mitochondria contact sites. Nature.
495:389–393. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Walker S, Chandra P, Manifava M, Axe E and
Ktistakis NT: Making autophagosomes: Localized synthesis of
phosphatidylinositol 3-phosphate holds the clue. Autophagy.
4:1093–1096. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Axe EL, Walker SA, Manifava M, Chandra P,
Roderick HL, Habermann A, Griffiths G and Ktistakis NT:
Autophagosome formation from membrane compartments enriched in
phosphatidylinositol 3-phosphate and dynamically connected to the
endoplasmic reticulum. J Cell Biol. 182:685–701. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Locke M and Collins JV: The structure and
formation of protein granules in the fat body of an insect. J Cell
Biol. 26:857–884. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Coutts AS and La Thangue NB: Regulation of
actin nucleation and autophagosome formation. Cell Mol Life Sci.
73:3249–3263. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Klionsky DJ: Autophagy: From phenomenology
to molecular understanding in less than a decade. Nat Rev Mol Cell
Biol. 8:931–937. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hayashi-Nishino M, Fujita N, Noda T,
Yamaguchi A, Yoshimori T and Yamamoto A: A subdomain of the
endoplasmic reticulu forms a cradle for autophagosome formation.
Nat Cell Biol. 11:1433–1437. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
English L, Chemali M, Duron J, Rondeau C,
Laplante A, Gingras D, Alexander D, Leib D, Norbury C, Lippé R and
Desjardins M: Autophagy enhances the presentation of endogenous
viral antigens on MHC class I molecules during HSV-1 infection. Nat
Immunol. 10:480–487. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Park JM, Lee DH and Kim DH: Redefining the
role of AMPK in autophagy and the energy stress response. Nat
Commun. 14:29942023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zachari M and Ganley IG: The mammalian
ULK1 complex and autophagy initiation. Essays Biochem. 61:585–596.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Noda NN and Fujioka Y: Atg1 family kinases
in autophagy initiation. Cell Mol Life Sci. 72:3083–3096. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qi S, Kim DJ, Stjepanovic G and Hurley JH:
Structure of the human Atg13-Atg101 HORMA heterodimer: An
interaction hub within the ULK1 complex. Structure. 23:1848–1857.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mercer CA, Kaliappan A and Dennis PB: A
novel, human Atg13 binding protein, Atg101, interacts with ULK1 and
is essential for macroautophagy. Autophagy. 5:649–662. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hosokawa N, Hara T, Kaizuka T, Kishi C,
Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et
al: Nutrient-dependent mTORC1 association with the
ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell.
20:1981–1991. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hara T, Takamura A, Kishi C, Iemura S,
Natsume T, Guan JL and Mizushima N: FIP200, a ULK-interacting
protein, is required for autophagosome formation in mammalian
cells. J Cell Biol. 181:497–510. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chu Y, Kang Y, Yan C, Yang C, Zhang T, Huo
H and Liu Y: LUBAC and OTULIN regulate autophagy initiation and
maturation by mediating the linear ubiquitination and the
stabilization of ATG13. Autophagy. 17:1684–1699. 2021. View Article : Google Scholar :
|
|
32
|
Ohashi Y, Tremel S and Williams RL: VPS34
complexes from a structural perspective. J Lipid Res. 60:229–241.
2019. View Article : Google Scholar :
|
|
33
|
Rostislavleva K, Soler N, Ohashi Y, Zhang
L, Pardon E, Burke JE, Masson GR, Johnson C, Steyaert J, Ktistakis
NT and Williams RL: Structure and flexibility of the endosomal
Vps34 complex reveals the basis of its function on membranes.
Science. 350:aac73652015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li X, He L, Che KH, Funderburk SF, Pan L,
Pan N, Zhang M, Yue Z and Zhao Y: Imperfect interface of Beclin1
coiled-coil domain regulates homodimer and heterodimer formation
with Atg14L and UVRAG. Nat Commun. 3:6622012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lamb CA, Yoshimori T and Tooze SA: The
autophagosome: Origins unknown, biogenesis complex. Nat Rev Mol
Cell Biol. 14:759–774. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Funderburk SF, Wang QJ and Yue Z: The
Beclin 1-VPS34 complex-at the crossroads of autophagy and beyond.
Trends Cell Biol. 20:355–362. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ktistakis NT and Tooze SA: Digesting the
expanding mechanisms of autophagy. Trends Cell Biol. 26:624–635.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nascimbeni AC, Codogno P and Morel E:
Phosphatidylinositol-3-phosphate in the regulation of autophagy
membrane dynamics. FEBS J. 284:1267–1278. 2017. View Article : Google Scholar
|
|
39
|
Wu MY, Liu L, Wang EJ, Xiao HT, Cai CZ,
Wang J, Su H, Wang Y, Tan J, Zhang Z, et al: PI3KC3 complex subunit
NRBF2 is required for apoptotic cell clearance to restrict
intestinal inflammation. Autophagy. 17:1096–1111. 2021. View Article : Google Scholar :
|
|
40
|
Nakatogawa H: Two ubiquitin-like
conjugation systems that mediate membrane formation during
autophagy. Essays Biochem. 55:39–50. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Popelka H, Reinhart EF, Metur SP, Leary
KA, Ragusa MJ and Klionsky DJ: Membrane binding and
homodimerization of Atg16 via two distinct protein regions is
essential for autophagy in yeast. J Mol Biol. 433:1668092021.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lystad AH, Carlsson SR and Simonsen A:
Toward the function of mammalian ATG12-ATG5-ATG16L1 complex in
autophagy and related processes. Autophagy. 15:1485–1486. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Campisi D, Hawkins N, Bonjour K and
Wollert T: The role of WIPI2, ATG16L1 and ATG12-ATG5 in selective
and nonselective autophagy. J Mol Biol. 437:1691382025. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Martens S and Fracchiolla D: Activation
and targeting of ATG8 protein lipidation. Cell Discov. 6:232020.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nakatogawa H, Ichimura Y and Ohsumi Y:
Atg8, a ubiquitin-like protein required for autophagosome
formation, mediates membrane tethering and hemifusion. Cell.
130:165–178. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Abreu S, Kriegenburg F, Gómez-Sánchez R,
Mari M, Sánchez-Wandelmer J, Skytte Rasmussen M, Soares Guimarães
R, Zens B, Schuschnig M, Hardenberg R, et al: Conserved Atg8
recognition sites mediate Atg4 association with autophagosomal
membranes and Atg8 deconjugation. EMBO Rep. 18:765–780. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Walczak M and Martens S: Dissecting the
role of the Atg12-Atg5-Atg16 complex during autophagosome
formation. Autophagy. 9:424–425. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yamamoto YH and Noda T: Autophagosome
formation in relation to the endoplasmic reticulum. J Biomed Sci.
27:972020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Takahashi Y, He H, Tang Z, Hattori T, Liu
Y, Young MM, Serfass JM, Chen L, Gebru M, Chen C, et al: An
autophagy assay reveals the ESCRT-III component CHMP2A as a
regulator of phagophore closure. Nat Commun. 9:28552018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Takahashi Y, Liang X, Hattori T, Tang Z,
He H, Chen H, Liu X, Abraham T, Imamura-Kawasawa Y, Buchkovich NJ,
et al: VPS37A directs ESCRT recruitment for phagophore closure. J
Cell Biol. 218:3336–3354. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Moretti F, Bergman P, Dodgson S, Marcellin
D, Claerr I, Goodwin JM, DeJesus R, Kang Z, Antczak C, Begue D, et
al: TMEM41B is a novel regulator of autophagy and lipid
mobilization. EMBO Rep. 19:e458892018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Morita K, Hama Y, Izume T, Tamura N, Ueno
T, Yamashita Y, Sakamaki Y, Mimura K, Morishita H, Shihoya W, et
al: Genome-wide CRISPR screen identifies TMEM41B as a gene required
for autophagosome formation. J Cell Biol. 217:3817–3828. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhao YG and Zhang H: The ER-localized
autophagy protein EPG-3/VMP1 regulates ER contacts with other
organelles by modulating ATP2A/SERCA activity. Autophagy.
14:362–363. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Liu H, Wang X, Li B, Xiang Z, Zhao Y, Lu
M, Lin Q, Zheng S, Guan T, Zhang Y and Hu Y: LncRNA HITT inhibits
autophagy by attenuating ATG12-ATG5-ATG16L1 complex formation.
Cancer Lett. 616:2175322025. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhang S, Li L, Liu X and Zhong Q: The
hookup model of the HOPS complex in autophagosome-lysosome fusion.
Autophagy. 20:714–715. 2024. View Article : Google Scholar :
|
|
56
|
Tian X, Teng J and Chen J: New insights
regarding SNARE proteins in autophagosome-lysosome fusion.
Autophagy. 17:2680–2688. 2021. View Article : Google Scholar :
|
|
57
|
van der Kant R, Jonker CTH, Wijdeven RH,
Bakker J, Janssen L, Klumperman J and Neefjes J: Characterization
of the mammalian CORVET and HOPS complexes and their modular
restructuring for endosome specificity. J Biol Chem.
290:30280–30290. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
König C, Shvarev D, Gao J, Haar E, Susan
N, Auffarth K, Langemeyer L, Moeller A and Ungermann C: Vps41
functions as a molecular ruler for HOPS tethering complex-mediated
membrane fusion. J Cell Sci. 138:jcs2637882025. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Jiang P, Nishimura T, Sakamaki Y, Itakura
E, Hatta T, Natsume T and Mizushima N: The HOPS complex mediates
autophagosome-lysosome fusion through interaction with syntaxin 17.
Mol Biol Cell. 25:1327–1337. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Nair U and Klionsky DJ: Autophagosome
biogenesis requires SNAREs. Autophagy. 7:1570–1572. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Fasshauer D, Sutton RB, Brunger AT and
Jahn R: Conserved structural features of the synaptic fusion
complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc Natl
Acad Sci USA. 95:15781–15786. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Itakura E, Kishi-Itakura C and Mizushima
N: The hairpin-type tail-anchored SNARE syntaxin 17 targets to
autophagosomes for fusion with endosomes/lysosomes. Cell.
151:1256–1269. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Pellegrini FR, De Martino S, Fianco G,
Ventura I, Valente D, Fiore M, Trisciuoglio D and Degrassi F:
Blockage of autophagosome-lysosome fusion through SNAP29
O-GlcNAcylation promotes apoptosis via ROS production. Autophagy.
19:2078–2093. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhang N and Zhao Y: Other molecular
mechanisms regulating autophagy. Adv Exp Med Biol. 1206:261–271.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yang Y, Shi G, Ge Y, Huang S, Cui N, Tan
L, Liu R and Yang X: Accumulated BCAAs and BCKAs contribute to the
HFD-induced deterioration of Alzheimer's disease via a
dysfunctional TREM2-related reduction in microglial β-amyloid
clearance. J Neuroinflammation. 21:3272024. View Article : Google Scholar
|
|
66
|
Fernandes SA, Angelidaki DD, Nüchel J, Pan
J, Gollwitzer P, Elkis Y, Artoni F, Wilhelm S, Kovacevic-Sarmiento
M and Demetriades C: Spatial and functional separation of mTORC1
signalling in response to different amino acid sources. Nat Cell
Biol. 26:1918–1933. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Masson GR: Towards a model of GCN2
activation. Biochem Soc Trans. 47:1481–1488. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zoncu R, Bar-Peled L, Efeyan A, Wang S,
Sancak Y and Sabatini DM: mTORC1 senses lysosomal amino acids
through an inside-out mechanism that requires the vacuolar
H(+)-ATPase. Science. 334:678–683. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Bar-Peled L, Schweitzer LD, Zoncu R and
Sabatini DM: Ragulator is a GEF for the rag GTPases that signal
amino acid levels to mTORC1. Cell. 150:1196–1208. 2012. View Article : Google Scholar
|
|
70
|
Sciarretta S, Zhai P, Shao D, Maejima Y,
Robbins J, Volpe M, Condorelli G and Sadoshima J: Rheb is a
critical regulator of autophagy during myocardial ischemia:
Pathophysiological implications in obesity and metabolic syndrome.
Circulation. 125:1134–1146. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Nowosad A, Jeannot P, Callot C, Creff J,
Perchey RT, Joffre C, Codogno P, Manenti S and Besson A: p27
controls Ragulator and mTOR activity in amino acid-deprived cells
to regulate the autophagy-lysosomal pathway and coordinate cell
cycle and cell growth. Nat Cell Biol. 22:1076–1090. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Wong PM, Feng Y, Wang J, Shi R and Jiang
X: Regulation of autophagy by coordinated action of mTORC1 and
protein phosphatase 2A. Nat Commun. 6:80482015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Dong J, Qiu H, Garcia-Barrio M, Anderson J
and Hinnebusch AG: Uncharged tRNA activates GCN2 by displacing the
protein kinase moiety from a bipartite tRNA-binding domain. Mol
Cell. 6:269–279. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
B'chir W, Maurin AC, Carraro V, Averous J,
Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P and Bruhat
A: The eIF2α/ATF4 pathway is essential for stress-induced autophagy
gene expression. Nucleic Acids Res. 41:7683–7699. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Schworer CM and Mortimore GE:
Glucagon-induced autophagy and proteolysis in rat liver: Mediation
by selective deprivation of intracellular amino acids. Proc Natl
Acad Sci USA. 76:3169–3173. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kanazawa T, Taneike I, Akaishi R,
Yoshizawa F, Furuya N, Fujimura S and Kadowaki M: Amino acids and
insulin control autophagic proteolysis through different signaling
pathways in relation to mTOR in isolated rat hepatocytes. J Biol
Chem. 279:8452–8459. 2004. View Article : Google Scholar
|
|
77
|
Wang B, Shi Y, Chen J, Shao Z, Ni L, Lin
Y, Wu Y, Tian N, Zhou Y, Sun L, et al: High glucose suppresses
autophagy through the AMPK pathway while it induces autophagy via
oxidative stress in chondrocytes. Cell Death Dis. 12:5062021.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Kim KH and Lee MS: Autophagy-a key player
in cellular and body metabolism. Nat Rev Endocrinol. 10:322–337.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Karabiyik C, Vicinanza M, Son SM and
Rubinsztein DC: Glucose starvation induces autophagy via
ULK1-mediated activation of PIKfyve in an AMPK-dependent manner.
Dev Cell. 56:1961–1975.e5. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yang Y and Klionsky DJ: An
AMPK-ULK1-PIKFYVE signaling axis for PtdIns5P-dependent autophagy
regulation upon glucose starvation. Autophagy. 17:2663–2664. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
James HA, O'Neill BT and Nair KS: Insulin
regulation of proteostasis and clinical implications. Cell Metab.
26:310–323. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ng F and Tang BL: Sirtuins' modulation of
autophagy. J Cell Physiol. 228:2262–2270. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Onal G, Kutlu O, Gozuacik D and Dokmeci
Emre S: Lipid droplets in health and disease. Lipids Health Dis.
16:1282017. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Ciesielska K and Gajewska M: Fatty acids
as potent modulators of autophagy activity in white adipose tissue.
Biomolecules. 13:2552023. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Yin J, Wang Y, Gu L, Fan N, Ma Y and Peng
Y: Palmitate induces endoplasmic reticulum stress and autophagy in
mature adipocytes: Implications for apoptosis and inflammation. Int
J Mol Med. 35:932–940. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Ortiz-Rodriguez A, Acaz-Fonseca E, Boya P,
Arevalo MA and Garcia-Segura LM: Lipotoxic effects of palmitic acid
on astrocytes are associated with autophagy impairment. Mol
Neurobiol. 56:1665–1680. 2019. View Article : Google Scholar
|
|
87
|
Niso-Santano M, Malik SA, Pietrocola F,
Bravo-San Pedro JM, Mariño G, Cianfanelli V, Ben-Younès A, Troncoso
R, Markaki M, Sica V, et al: Unsaturated fatty acids induce
non-canonical autophagy. EMBO J. 34:1025–1041. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Liu X, Li X, Su S, Yuan Y, Liu W, Zhu M,
Zheng Q, Zeng X, Fu F, Lu Y and Chen Y: Oleic acid improves hepatic
lipotoxicity injury by alleviating autophagy dysfunction. Exp Cell
Res. 429:1136552023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Zhou ZY, Song K, Liu ZY, Ke YF, Shi Y, Cai
K, Zhao R, Sun X, Tao H and Zhao JY: Branched-chain amino acids
deficiency promotes diabetic cardiomyopathy by activating autophagy
of cardiac fibroblasts. Theranostics. 14:7333–7348. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Fermo KT, da Silva Lemos I, Farias HR,
Rosso MP, Effting PS, Leipnitz G and Streck EL: Branched-chain
amino acids (BCAA) administration increases autophagy and the
autophagic pathway in brain tissue of rats submitted to a maple
syrup urine disease (MSUD) protocol. Metab Brain Dis. 38:287–293.
2023. View Article : Google Scholar
|
|
91
|
Li F, Wan X, Li Z and Zhou L: High glucose
inhibits autophagy and promotes the proliferation and metastasis of
colorectal cancer through the PI3K/AKT/mTOR pathway. Cancer Med.
13:e73822024. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Wang X, Feng Z, Li J, Chen L and Tang W:
High glucose induces autophagy of MC3T3-E1 cells via ROS-AKT-mTOR
axis. Mol Cell Endocrinol. 429:62–72. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Hossain MM, Mishra AK, Yadav AK, Ismail M,
Sata TN, Sah AK, Banik A, Sharma G and Venugopal SK: Free fatty
acid-induced DDX3 inhibits autophagy via miR-141 upregulation in
diet-induced MASLD mice model system. Ann Hepatol. 30:1017582025.
View Article : Google Scholar
|
|
94
|
Guo W, Zhong W, Hao L, Dong H, Sun X, Yue
R, Li T and Zhou Z: Fatty acids inhibit LAMP2-mediated autophagy
flux via activating ER stress pathway in alcohol-related liver
disease. Cell Mol Gastroenterol Hepatol. 12:1599–1615. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Piché ME, Tchernof A and Després JP:
Obesity phenotypes, diabetes, and cardiovascular diseases. Circ
Res. 126:1477–1500. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Al-Kuraishy HM, Jabir MS, Al-Gareeb AI,
Klionsky DJ and Albuhadily AK: Dysregulation of pancreatic β-cell
autophagy and the risk of type 2 diabetes. Autophagy. 20:2361–2372.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Yao D, GangYi Y and QiNan W: Autophagic
dysfunction of β cell dysfunction in type 2 diabetes, a
double-edged sword. Genes Dis. 8:438–447. 2020. View Article : Google Scholar
|
|
98
|
Jung HS, Chung KW, Won Kim J, Kim J,
Komatsu M, Tanaka K, Nguyen YH, Kang TM, Yoon KH, Kim JW, et al:
Loss of autophagy diminishes pancreatic beta cell mass and function
with resultant hyperglycemia. Cell Metab. 8:318–324. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Martino L, Masini M, Novelli M, Beffy P,
Bugliani M, Marselli L, Masiello P, Marchetti P and De Tata V:
Palmitate activates autophagy in INS-1E β-cells and in isolated rat
and human pancreatic islets. PLoS One. 7:e361882012. View Article : Google Scholar
|
|
100
|
Meng XH, Chen B and Zhang JP:
Intracellular insulin and impaired autophagy in a zebrafish model
and a cell model of type 2 diabetes. Int J Biol Sci. 13:985–995.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Budi YP, Li YH, Huang C, Wang ME, Lin YC,
Jong DS, Chiu CH and Jiang YF: The role of autophagy in high-fat
diet-induced insulin resistance of adipose tissues in mice. PeerJ.
10:e138672022. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Wang S, Sun QQ, Xiang B and Li XJ:
Pancreatic islet cell autophagy during aging in rats. Clin Invest
Med. 36:E72–E80. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Masini M, Bugliani M, Lupi R, del Guerra
S, Boggi U, Filipponi F, Marselli L, Masiello P and Marchetti P:
Autophagy in human type 2 diabetes pancreatic beta cells.
Diabetologia. 52:1083–1086. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Maiese K: mTOR: Driving apoptosis and
autophagy for neurocardiac complications of diabetes mellitus.
World J Diabetes. 6:217–224. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Younossi ZM, Golabi P, de Avila L, Paik
JM, Srishord M, Fukui N, Qiu Y, Burns L, Afendy A and Nader F: The
global epidemiology of NAFLD and NASH in patients with type 2
diabetes: A systematic review and meta-analysis. J Hepatol.
1:793–801. 2019. View Article : Google Scholar
|
|
106
|
Filali-Mouncef Y, Hunter C, Roccio F,
Zagkou S, Dupont N, Primard C, Proikas-Cezanne T and Reggiori F:
The ménage à trois of autophagy, lipid droplets and liver disease.
Autophagy. 18:50–72. 2022. View Article : Google Scholar :
|
|
107
|
Lian CY, Zhai ZZ, Li ZF and Wang L: High
fat diet-triggered non-alcoholic fatty liver disease: A review of
proposed mechanisms. Chem Biol Interact. 330:1091992020. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Byun S, Seok S, Kim YC, Zhang Y, Yau P,
Iwamori N, Xu HE, Ma J, Kemper B and Kemper JK: Fasting-induced
FGF21 signaling activates hepatic autophagy and lipid degradation
via JMJD3 histone demethylase. Nat Commun. 11:8072020. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Lee YA, Noon LA, Akat KM, Ybanez MD, Lee
TF, Berres ML, Fujiwara N, Goossens N, Chou HI, Parvin-Nejad FP, et
al: Autophagy is a gatekeeper of hepatic differentiation and
carcinogenesis by controlling the degradation of Yap. Nat Commun.
9:49622018. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Lee DH, Park JS, Lee YS, Han J, Lee DK,
Kwon SW, Han DH, Lee YH and Bae SH: SQSTM1/p62 activates
NFE2L2/NRF2 via ULK1-mediated autophagic KEAP1 degradation and
protects mouse liver from lipotoxicity. Autophagy. 16:1949–1973.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Settembre C, De Cegli R, Mansueto G, Saha
PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch
TJ, et al: TFEB controls cellular lipid metabolism through a
starvation-induced autoregulatory loop. Nat Cell Biol. 15:647–658.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Cao Y, Mai W, Li R, Deng S, Li L, Zhou Y,
Qin Q, Zhang Y, Zhou X, Han M, et al: Macrophages evoke autophagy
of hepatic stellate cells to promote liver fibrosis in NAFLD mice
via the PGE2/EP4 pathway. Cell Mol Life Sci. 79:3032022. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Sakane S, Hikita H, Shirai K, Myojin Y,
Sasaki Y, Kudo S, Fukumoto K, Mizutani N, Tahata Y, Makino Y, et
al: White adipose tissue autophagy and adipose-liver crosstalk
exacerbate nonalcoholic fatty liver disease in mice. Cell Mol
Gastroenterol Hepatol. 12:1683–1699. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Tanaka S, Hikita H, Tatsumi T, Sakamori R,
Nozaki Y, Sakane S, Shiode Y, Nakabori T, Saito Y, Hiramatsu N, et
al: Rubicon inhibits autophagy and accelerates hepatocyte apoptosis
and lipid accumulation in nonalcoholic fatty liver disease in mice.
Hepatology. 64:1994–2014. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Lalazar G, Ilyas G, Malik SA, Liu K, Zhao
E, Amir M, Lin Y, Tanaka KE and Czaja MJ: Autophagy confers
resistance to lipopolysaccharide-induced mouse hepatocyte injury.
Am J Physiol Gastrointest Liver Physiol. 311:G377–G386. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Shen Y, Malik SA, Amir M, Kumar P,
Cingolani F, Wen J, Liu Y, Zhao E, Farris AB, Raeman R and Czaja
MJ: Decreased hepatocyte autophagy leads to synergistic IL-1β and
TNF mouse liver injury and inflammation. Hepatology. 72:595–608.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Lovren F, Teoh H and Verma S: Obesity and
atherosclerosis: Mechanistic insights. Can J Cardiol. 31:177–183.
2015.PubMed/NCBI
|
|
118
|
Shan R, Liu N, Yan Y and Liu B: Apoptosis,
autophagy and atherosclerosis: Relationships and the role of Hsp27.
Pharmacol Res. 166:1051692021. View Article : Google Scholar
|
|
119
|
Xue S, Su Z and Liu D: Immunometabolism
and immune response regulate macrophage function in
atherosclerosis. Ageing Res Rev. 90:1019932023. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Tian F, Yu BL and Hu JR: mTOR mediates the
cross-talk of macrophage polarization and autophagy in
atherosclerosis. Int J Cardiol. 177:144–145. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Zhang H, Ge S, Ni B, He K, Zhu P, Wu X and
Shao Y: Augmenting ATG14 alleviates atherosclerosis and inhibits
inflammation via promotion of autophagosome-lysosome fusion in
macrophages. Autophagy. 17:4218–4230. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Li F, Wang Y, Chen S, Liu J, Wu X, Maimati
Y, Ding F, Wang X, Shen Y, Chen Q, et al: Nuclear receptor Dax1
promotes atherosclerosis by lipid transport inhibition and
autophagy suppression in macrophages. Eur Heart J. 46:4933–4949.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Miano JM, Fisher EA and Majesky MW: Fate
and state of vascular smooth muscle cells in atherosclerosis.
Circulation. 143:2110–2116. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Ding Z, Wang X, Schnackenberg L, Khaidakov
M, Liu S, Singla S, Dai Y and Mehta JL: Regulation of autophagy and
apoptosis in response to ox-LDL in vascular smooth muscle cells,
and the modulatory effects of the microRNA hsa-let-7 g. Int J
Cardiol. 168:1378–1385. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Liu Y, Song A, Wu H, Sun Y and Dai M:
Paeonol inhibits apoptosis of vascular smooth muscle cells via
up-regulation of autophagy by activating class III PI3K/Beclin-1
signaling pathway. Life Sci. 264:1187142021. View Article : Google Scholar
|
|
126
|
Gimbrone MA Jr and García-Cardeña G:
Endothelial cell dysfunction and the pathobiology of
atherosclerosis. Circ Res. 118:620–636. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Torisu K, Singh KK, Torisu T, Lovren F,
Liu J, Pan Y, Quan A, Ramadan A, Al-Omran M, Pankova N, et al:
Intact endothelial autophagy is required to maintain vascular lipid
homeostasis. Aging Cell. 15:187–191. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Ding SA, Liu H, Zheng R, Ge Y, Fu Z, Mei J
and Tang M: Downregulation of MYBL1 in endothelial cells
contributes to atherosclerosis by repressing PLEKHM1-inducing
autophagy. Cell Biol Toxicol. 40:402024. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Smith NJ, Caldwell JL, van der Merwe M,
Sharma S, Butawan M, Puppa M and Bloomer RJ: A comparison of
dietary and caloric restriction models on body composition,
physical performance, and metabolic health in young mice.
Nutrients. 11:3502019. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Webb VL and Wadden TA: Intensive lifestyle
intervention for obesity: Principles, practices, and results.
Gastroenterology. 152:1752–1764. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Gao X, Yan D, Zhao Y, Tao H and Zhou Y:
Moderate calorie restriction to achieve normal weight reverses
β-cell dysfunction in diet-induced obese mice: involvement of
autophagy. Nutr Metab (Lond). 12:342015. View Article : Google Scholar
|
|
132
|
Liu H, Javaheri A, Godar RJ, Murphy J, Ma
X, Rohatgi N, Mahadevan J, Hyrc K, Saftig P, Marshall C, et al:
Intermittent fasting preserves beta-cell mass in obesity-induced
diabetes via the autophagy-lysosome pathway. Autophagy.
13:1952–1968. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Ma N, Ma R, Tang K, Li X and He B:
Roux-en-Y gastric bypass in obese diabetic rats promotes autophagy
to improve lipid metabolism through mTOR/p70S6K signaling pathway.
J Diabetes Res. 2020:43265492020. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Guevara-Cruz M, Hernández-Gómez KG,
Condado-Huerta C, González-Salazar LE, Peña-Flores AK,
Pichardo-Ontiveros E, Serralde-Zúñiga AE, Sánchez-Tapia M, Maya O,
Medina-Vera I, et al: Intermittent fasting, calorie restriction,
and a ketogenic diet improve mitochondrial function by reducing
lipopolysaccharide signaling in monocytes during obesity: A
randomized clinical trial. Clin Nutr. 43:1914–1928. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Kitzman DW, Brubaker P, Morgan T,
Haykowsky M, Hundley G, Kraus WE, Eggebeen J and Nicklas BJ: Effect
of caloric restriction or aerobic exercise training on peak oxygen
consumption and quality of life in obese older patients with heart
failure with preserved ejection fraction: A randomized clinical
trial. JAMA. 315:36–46. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Xiang M, Yuan X, Zhang N, Zhang L, Liu Y,
Liu J, Gao Y, Xu Y, Sun W, Tang Q, et al: Effects of exercise,
metformin, and combination treatments on type 2 diabetic
mellitus-induced muscle atrophy in db/db mice: Crosstalk between
autophagy and the proteasome. J Physiol Biochem. 80:235–247. 2024.
View Article : Google Scholar
|
|
137
|
Pi H, Liu M, Xi Y, Chen M, Tian L, Xie J,
Chen M, Wang Z, Yang M, Yu Z, et al: Long-term exercise prevents
hepatic steatosis: A novel role of FABP1 in regulation of
autophagylysosomal machinery. FASEB J. 33:11870–11883. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Yang Y, Li X, Liu Z, Ruan X, Wang H, Zhang
Q, Cao L, Song L, Chen Y and Sun Y: Moderate treadmill exercise
alleviates NAFLD by regulating the biogenesis and autophagy of
lipid droplet. Nutrients. 14:49102022. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Li Y, Sun D, Zheng Y and Cheng Y: Swimming
exercise activates aortic autophagy and limits atherosclerosis in
ApoE−/− mice. Obes Res Clin Pract. 14:264–270. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Okutsu M, Yamada M, Tokizawa K, Marui S,
Suzuki K, Lira VA and Nagashima K: Regular exercise stimulates
endothelium autophagy via IL-1 signaling in ApoE deficient mice.
FASEB J. 35:e216982021. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Steinberg GR and Kemp BE: AMPK in health
and disease. Physiol Rev. 89:1025–1078. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Višnjić D, Lalić H, Dembitz V, Tomić B and
Smoljo T: AICAr, a widely used AMPK activator with important
AMPK-independent effects: A systematic review. Cells. 10:10952021.
View Article : Google Scholar
|
|
143
|
Tukhovskaya EA, Shaykhutdinova ER,
Pakhomova IA, Slashcheva GA, Goryacheva NA, Sadovnikova ES,
Rasskazova EA, Kazakov VA, Dyachenko IA, Frolova AA, et al: AICAR
improves outcomes of metabolic syndrome and type 2 diabetes induced
by high-fat diet in C57Bl/6 male mice. Int J Mol Sci. 23:157192022.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Chandrasekaran K, Choi J, Salimian M,
Hedayat AF and Russell JW: Administration of AICAR, an AMPK
activator, prevents and reverses diabetic polyneuropathy (DPN) by
regulating mitophagy. Int J Mol Sci. 26:802024. View Article : Google Scholar
|
|
145
|
Ren H, Shao Y, Wu C, Ma X, Lv C and Wang
Q: Metformin alleviates oxidative stress and enhances autophagy in
diabetic kidney disease via AMPK/SIRT1-FoxO1 pathway. Mol Cell
Endocrinol. 500:1106282020. View Article : Google Scholar
|
|
146
|
You G, Long X, Song F, Huang J, Tian M,
Xiao Y, Deng S and Wu Q: Metformin activates the AMPK-mTOR pathway
by modulating lncRNA TUG1 to induce autophagy and inhibit
atherosclerosis. Drug Des Devel Ther. 14:457–468. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Wu J, Wu JJ, Yang LJ, Wei LX and Zou DJ:
Rosiglitazone protects against palmitate-induced pancreatic
beta-cell death by activation of autophagy via 5′-AMP-activated
protein kinase modulation. Endocrine. 44:87–98. 2013. View Article : Google Scholar
|
|
148
|
Zhang LX, Li CX, Kakar MU, Khan MS, Wu PF,
Amir RM, Dai DF, Naveed M, Li QY, Saeed M, et al: Resveratrol (RV):
A pharmacological review and call for further research. Biomed
Pharmacother. 143:1121642021. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Zhang Y, Chen ML, Zhou Y, Yi L, Gao YX,
Ran L, Chen SH, Zhang T, Zhou X, Zou D, et al: Resveratrol improves
hepatic steatosis by inducing autophagy through the cAMP signaling
pathway. Mol Nutr Food Res. 59:1443–1457. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Xu K, Liu XF, Ke ZQ, Yao Q, Guo S and Liu
C: Resveratrol modulates apoptosis and autophagy induced by high
glucose and palmitate in cardiac cells. Cell Physiol Biochem.
46:2031–2040. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Levine B and Kroemer G: Biological
functions of autophagy genes: A disease perspective. Cell.
176:11–42. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Kim YC and Guan KL: mTOR: A pharmacologic
target for autophagy regulation. J Clin Invest. 125:25–32. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Li Y, Cheng Y, Zhou Y, Du H, Zhang C, Zhao
Z, Chen Y, Zhou Z, Mei J, Wu W and Chen M: High fat diet-induced
obesity leads to depressive and anxiety-like behaviors in mice via
AMPK/mTOR-mediated autophagy. Exp Neurol. 348:1139492022.
View Article : Google Scholar
|
|
154
|
Yamahara K, Kume S, Koya D, Tanaka Y,
Morita Y, Chin-Kanasaki M, Araki H, Isshiki K, Araki S, Haneda M,
et al: Obesity-mediated autophagy insufficiency exacerbates
proteinuria-induced tubulointerstitial lesions. J Am Soc Nephrol.
24:1769–1781. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Liu B, Deng X, Jiang Q, Li G, Zhang J,
Zhang N, Xin S and Xu K: Scoparone improves hepatic inflammation
and autophagy in mice with nonalcoholic steatohepatitis by
regulating the ROS/P38/Nrf2 axis and PI3K/AKT/mTOR pathway in
macrophages. Biomed Pharmacother. 125:1098952020. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Zhou J, Tan SH, Nicolas V, Bauvy C, Yang
ND, Zhang J, Xue Y, Codogno P and Shen HM: Activation of lysosomal
function in the course of autophagy via mTORC1 suppression and
autophagosome-lysosome fusion. Cell Res. 23:508–523. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Liu Q, Xu C, Kirubakaran S, Zhang X, Hur
W, Liu Y, Kwiatkowski NP, Wang J, Westover KD, Gao P, et al:
Characterization of Torin2, an ATP-competitive inhibitor of mTOR,
ATM, and ATR. Cancer Res. 73:2574–2586. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Petherick KJ, Conway OJL, Mpamhanga C,
Osborne SA, Kamal A, Saxty B and Ganley IG: Pharmacological
inhibition of ULK1 kinase blocks mammalian target of rapamycin
(mTOR)-dependent autophagy. J Biol Chem. 290:11376–11383. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Ahwazi D, Neopane K, Markby GR, Kopietz F,
Ovens AJ, Dall M, Hassing AS, Gräsle P, Alshuweishi Y, Treebak JT,
et al: Investigation of the specificity and mechanism of action of
the ULK1/AMPK inhibitor SBI-0206965. Biochem J. 478:2977–2997.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Wu YT, Tan HL, Shui G, Bauvy C, Huang Q,
Wenk MR, Ong CN, Codogno P and Shen HM: Dual role of
3-methyladenine in modulation of autophagy via different temporal
patterns of inhibition on class I and III phosphoinositide
3-kinase. J Biol Chem. 285:10850–10861. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr
M, Hijlkema KJ, Coppes RP, Engedal N, Mari M and Reggiori F:
Chloroquine inhibits autophagic flux by decreasing
autophagosome-lysosome fusion. Autophagy. 14:1435–1455. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Wang R, Wang J, Hassan A, Lee CH, Xie XS
and Li X: Molecular basis of V-ATPase inhibition by bafilomycin A1.
Nat Commun. 12:17822021. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Hirao J, Tojo A, Hatakeyama S, Satonaka H
and Ishimitsu T: V-ATPase blockade reduces renal gluconeogenesis
and improves insulin secretion in type 2 diabetic rats. Hypertens
Res. 43:1079–1088. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Şaylık F, Çınar T and Hayıroğlu Mİ: Effect
of the obesity paradox on mortality in patients with acute coronary
syndrome: A comprehensive meta-analysis of the literature. Balkan
Med J. 40:93–103. 2023. View Article : Google Scholar : PubMed/NCBI
|














