According to the International Diabetes Federation,
~588.7 million individuals globally are living with diabetes in
2024, representing a prevalence of 11.1% (1). This figure is projected to rise
substantially, reaching 852.5 million cases and a prevalence of
13.0% by 2050. The burden of diabetes is most pronounced in
middle-income countries, which account for 452.9 million cases in
2024, followed by high-income countries with 114.1 million and
low-income countries with 21.8 million cases (2). DFUs represent one of the most
debilitating complications of diabetes mellitus, affecting 15-25%
of diabetic patients during their lifetime and contributing to
>85% of non-traumatic lower limb amputations (3). Globally, DFUs impact ~18.6 million
individuals annually, with lifetime ulcer risk ranging from 19 to
34%, recurrence rates reaching 65% at 3-5 years and 5-year
mortality rates of 50-70% following initial ulceration (4). The burden of DFUs extends beyond
individual morbidity to encompass substantial healthcare costs,
with annual expenditures ranging from $9 to 13 billion in the
United States alone (5). Despite
advances in multidisciplinary care and evidence-based
interventions, amputation rates associated with DFUs continue to
rise in some regions, particularly among young and racial/ethnic
minority populations (4).
The pathophysiology of DFUs involves a complex
interplay of peripheral neuropathy, peripheral arterial disease
(PAD), chronic inflammation and impaired tissue regeneration
(6). Peripheral neuropathy,
affecting ~60% of diabetic patients, contributes to sensory loss
and abnormal foot mechanics that increase ulcer risk (7,8).
Vascular insufficiency secondary to PAD further compounds healing
impairment by limiting oxygen and nutrient delivery to affected
tissues (9). The diabetic
microenvironment is characterized by persistent hyperglycemia,
advanced glycation end-product (AGE) accumulation, oxidative stress
and immune dysregulation, factors that collectively sustain a
pro-inflammatory state dominated by cytokines such as TNF-α, IL-1β
and IL-6, which impair angiogenesis and delay repair (3).
While considerable attention has been directed
toward soft-tissue pathology in DFUs, bone involvement remains
frequently overlooked despite mounting evidence of its important
role in wound healing and ulcer recurrence (7,10). In diabetic patients, the skeleton
undergoes profound metabolic and structural alterations including
cortical thinning, reduced bone mineral density, impaired bone
marrow stromal cell function and dysregulated bone remodeling
(11). These changes not only
increase susceptibility to foot deformities and mechanical stress
but also compromise the capacity of the bone to support overlying
soft-tissue repair. Beyond endogenous repair mechanisms, bone
regeneration research has increasingly explored biomimetic
scaffolds and immunomodulatory fiber-based approaches (12-14).
Emerging evidence has fundamentally redefined the
understanding of bone from a passive structural scaffold to a
multifunctional endocrine organ (15-17). Bone cells, osteoblasts,
osteocytes and osteoclasts, secrete a constellation of bioactive
molecules termed 'osteokines' that exert systemic effects on
distant organs through endocrine, paracrine and autocrine
mechanisms (15). These
bone-derived factors, including OCN, sclerostin, FGF-23, receptor
activator of nuclear factor-κB ligand/osteoprotegerin (RANKL/OPG)
and lipocalin-2 (LCN2), regulate energy metabolism, vascular
biology, immune function and tissue repair capacity (18). OCN, in its undercarboxylated
form, stimulates insulin secretion from pancreatic β-cells and
enhances insulin sensitivity in peripheral tissues (19,20). Sclerostin, secreted predominantly
by osteocytes, inhibits Wnt/β-catenin signaling and suppresses bone
formation (21). The endocrine
dimension of bone extends beyond mineral homeostasis to influence
wound healing processes through modulation of angiogenesis,
macrophage polarization and extracellular matrix (ECM) remodeling
(22).
The concept of 'bone-origin repair' defined as the
targeted recruitment of intrinsic reparative programs within the
bone microenvironment through deliberate and controlled local bone
stimulation, represents an emerging therapeutic framework that may
be applicable to carefully selected cases of DFU (23). Building on principles derived
from bone marrow stimulation techniques (microfracture, drilling),
periosteal distraction and cortical transverse transport, this
approach aims to harness the regenerative and endocrine capacity of
the skeleton to secondarily support soft-tissue healing in
bone-adjacent ulcers (24).
However, this concept remains exploratory and requires clearly
defined clinical boundaries to prevent conceptual overreach and
inappropriate application in settings of active osteomyelitis,
structural instability or advanced Charcot neuroarthropathy
(25). The present review
synthesizes current understanding of bone pathology in diabetic
foot disease, examines the molecular and cellular mechanisms
underlying callus formation and its impairment in diabetes,
explores the endocrine functions of bone relevant to wound healing
and discusses the translational potential of bone-targeted
strategies as adjunctive therapies in diabetic wound management. By
integrating insights from osteoimmunology, vascular biology and
regenerative medicine, the present review aims to provide a
comprehensive framework for understanding and potentially
exploiting the bone-wound axis in clinical practice.
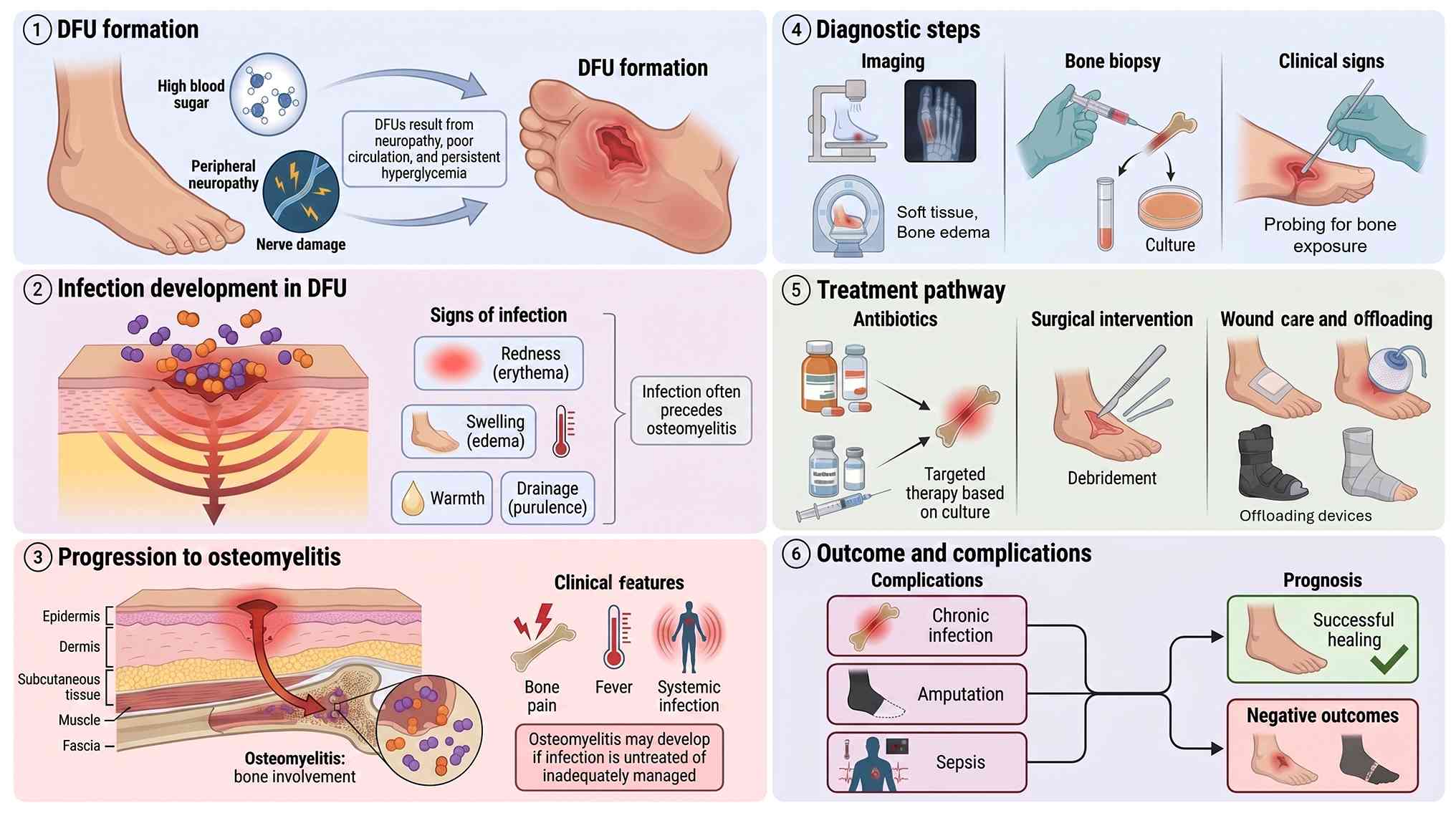
DFUs are open wounds that occur in individuals with
type 1 or type 2 diabetes, commonly in the setting of peripheral
neuropathy, poor glycemic control and PAD (26). These ulcers represent one of the
most frequent and severe complications of diabetes, affecting ~1/3
of patients during their lifetime. Globally, ~18.6 million
individuals are affected each year including ~1.6 million cases in
the United States and~50% of DFUs become infected, with ~20% of
infected cases progressing to partial or total foot amputation
(27).
The development of DFUs is driven by factors such as
neuropathy, vascular insufficiency, trauma and impaired immunity
(28). Neuropathy contributes to
sensory loss and abnormal foot mechanics, increasing ulcer risk
(29) and affects ~60% of
diabetic patients (8). Vascular
disease further impairs circulation and delays healing (30). Foot deformities and visual
impairment, particularly in older adults, elevate the likelihood of
unnoticed injuries (31). These
risks are exacerbated by hyperglycemia and prolonged diabetes
duration (32). In this setting,
chronic hyperglycemia serves as the upstream metabolic abnormality
linking vascular dysfunction and immune dysregulation in the
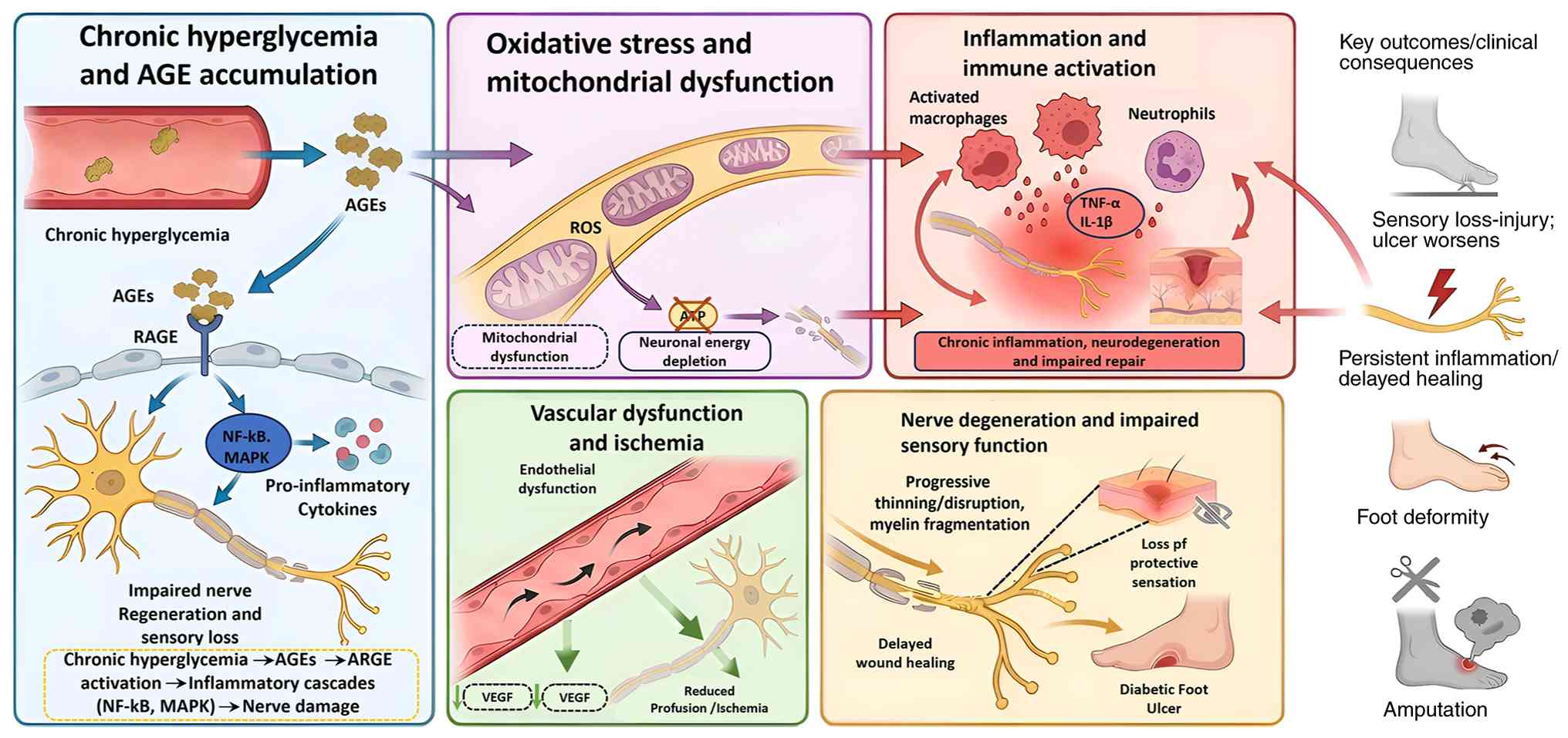
diabetic wound microenvironment (33-35).
Chronic hyperglycemia activates alternative
metabolic pathways, such as the polyol and hexosamine pathways,
which contribute to the overproduction of reactive oxygen species
and the formation of AGE products, both of which are key mediators
of cellular damage in diabetes (36). AGEs accumulate in hyperglycemic
environments through non-enzymatic reactions between reducing
sugars and proteins, lipids or nucleic acids (37). These AGEs exert their
pathological effects primarily through interaction with the
receptor for AGE (RAGE), a multiligand pattern recognition receptor
expressed on endothelial cells, macrophages and fibroblasts
(38). Binding of AGEs to RAGE
triggers intracellular signaling cascades, including NF-κB, MAPK
and JAK/STAT pathways (39,40). These cascades upregulate
pro-inflammatory cytokines such as TNF-α and IL-6, promote
oxidative stress via NADPH oxidase activation and suppress
reparative cell behaviors (41-43).
Metabolic injury then converges with vascular and
immune dysfunction. In chronic wounds such as DFUs, persistent
inflammatory signaling impairs fibroblast migration, disrupts ECM
turnover and compromises angiogenesis (44). At the same time, AGE-related
redox imbalance and inflammatory activation sustain chronic
inflammation and oxidative stress (45,46), while diabetic wound healing is
further impaired through AGE-RAGE-associated mechanisms (40,47). Rather than acting sequentially,
these metabolic, vascular and immune abnormalities reinforce one
another: Metabolic stress damages endothelial and stromal function,
vascular insufficiency worsens tissue hypoxia and repair failure,
and chronic inflammation further compromises microvascular
integrity and tissue regeneration. Together, these processes create
a wound environment that predisposes to ulceration and delays
healing (Fig. 1).
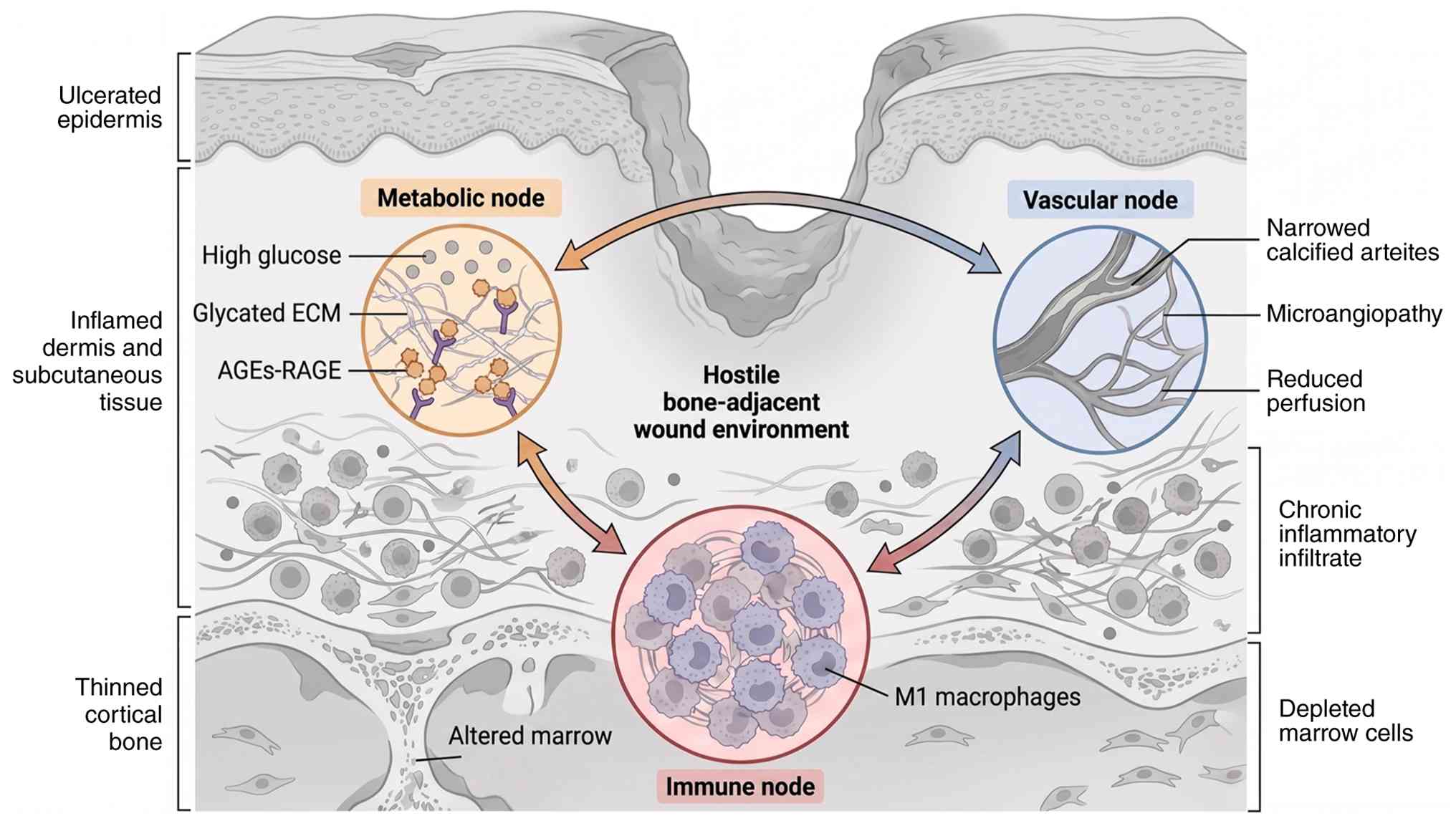
Bone involvement in DFUs is frequently overlooked,
yet it plays an important role in maintaining the structural
integrity of the foot and supporting the regenerative processes of
the wound bed. In individuals with diabetes, both metabolic and
mechanical factors compromise the bone-soft tissue unit, creating
an environment that fosters chronic wound formation and hinders
effective healing (48,49).
Cortical thinning in DFU is a key pathological
feature driven by a complex interplay of metabolic, neurological
and vascular factors (50).
Hyperglycemia negatively affects bone health by impairing the
function of osteoblasts and osteocytes, the primary cells involved
in bone formation and maintenance (51). This metabolic disturbance
disrupts normal bone metabolism by decreasing the production of ECM
components and hindering mineralization (52). Beyond reducing bone formation and
increasing fragility, it accelerates the apoptosis and senescence
of osteoblasts, further contributing to bone loss (53). By perturbing the delicate balance
between bone-forming osteoblasts and bone-resorbing osteoclasts,
diabetes impairs bone remodeling and contributes to cortical bone
thinning and reduced bone mineral density (54).
Neuropathy, a prevalent complication in DFU, further
exacerbates cortical thinning (58). These sensory and motor deficits
lead to abnormal mechanical loading of the foot, driven by altered
gait mechanics and loss of protective sensation, which ultimately
impairs bone remodeling responses (59). Beyond mechanical stress,
compromised neural signaling disrupts the intricate neurovascular
regulation of local blood flow and nutrient delivery, hindering the
bone's ability to repair and maintain its structural integrity
(60). Similarly, autonomic
dysfunction can dysregulate circulatory responses, diminishing the
supply of oxygen and nutrients essential for maintaining skeletal
health (61).
PAD, a frequent comorbidity in DFU, substantially
exacerbates cortical thinning by compromising the local blood
supply (62). Reduced
vascularity limits the delivery of oxygen, growth factors and
nutrients essential for osteoblast function and bone matrix
synthesis (63). The resulting
chronic ischemia impairs bone repair processes and promotes a
catabolic environment within bone, thereby accelerating cortical
loss (64). Collectively, the
interplay of hyperglycemia, AGEs, neuropathy and PAD creates a
vicious cycle that progressively depletes cortical bone mass,
increasing bone fragility and the risk of further complications in
patients with DFU (Fig. 2).
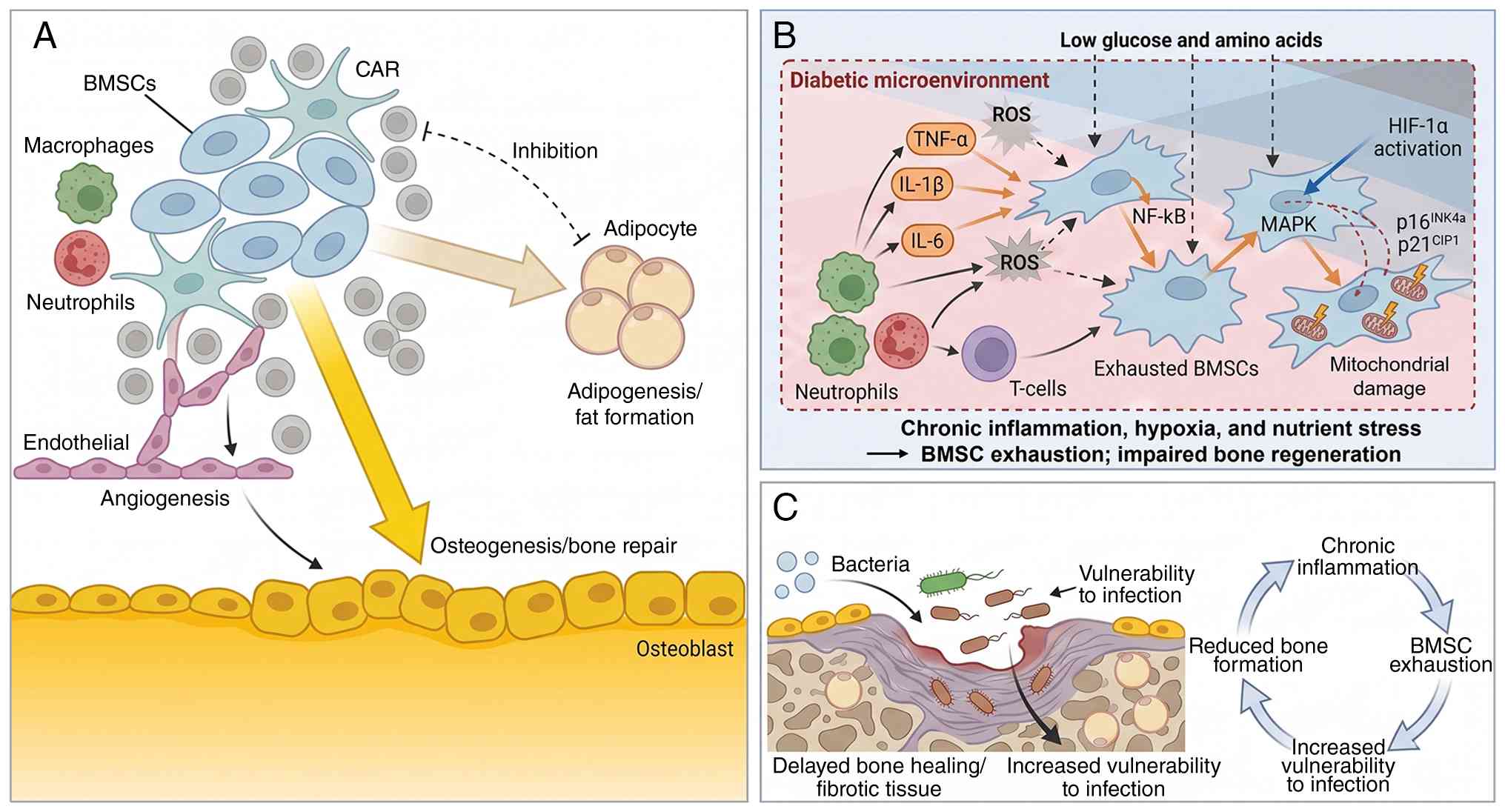
Beyond impaired remodeling, diabetes perturbs the
bone marrow stem/progenitor niche, reducing the availability and
fitness of osteogenic progenitors. Osteogenesis is severely
hindered in diabetic individuals by osteoblast dysfunction and the
exhaustion of bone marrow stromal cells (BMSCs), both of which are
key for bone regeneration (65).
In the context of DFU, this BMSC exhaustion contributes to impaired
repair and delayed wound healing (66-68), further compromising overall bone
health (69). Aging further
compounds this issue, driving a lineage shift whereby BMSCs
preferentially differentiate into adipocytes rather than
osteoblasts, a hallmark of age-related bone loss (70,71). Key to this balance are
CXCL12-abundant reticular (CAR) cells, which regulate the
transition between osteogenesis and adipogenesis (72). Disruptions in CAR-hematopoietic
cell communication promote marrow adipogenesis through the direct
conversion of CXCL12+ preadipocyte-like cells into lipid-laden
marrow adipocytes (72). This
suggests a cell-nonautonomous mechanism in which CXCL12-dependent
physical coupling with hematopoietic cells helps prevent premature
adipocytic differentiation.
The chronic inflammation, oxidative stress and
compromised microenvironment surrounding the ulcer create a hostile
setting for BMSCs, which are essential for bone regeneration
(73). Pro-inflammatory
cytokines, including TNF-α, IL-1β, IL-6 and IL-8, secreted by
immune cells such as macrophages and neutrophils, activate
inflammatory pathways such as NF-κB and MAPK, thereby promoting
BMSC senescence (74-76). This impairs the ability of BMSCs
to proliferate and differentiate into osteoblasts, while the
upregulation of cell-cycle inhibitors p16INK4a and p21CIP1 serves
as a marker of cellular aging (77). Simultaneously, oxidative stress
generated by immune-cell activity produces reactive oxygen species,
including O2−, H2O2, and
OH•. These reactive oxygen species damage cellular
components, particularly mitochondria (78-80) and further impair BMSC function
(81,82). Dysregulation of Nrf2, a key
regulator of antioxidant defenses, exacerbates this oxidative
damage and further limits the regenerative capacity of these cells
(83).
The DFU microenvironment is further compromised by
hypoxia and nutrient deprivation (3). While poor vascularization initially
activates hypoxia-inducible factor-1α(HIF-1α) signaling (84,85), prolonged oxygen deficiency
eventually impairs BMSC metabolism (86). These local deficits are
compounded by systemic metabolic disturbances that reduce the
availability of essential glucose and amino acids, depriving BMSCs
of the substrates required for repair (87). Together with chronic inflammation
and oxidative stress, these stressors establish a feedback loop in
which BMSC exhaustion diminishes the regenerative potential of bone
tissue. Consequently, bone repair becomes less efficient, further
weakening the capacity of diabetic bone to support local healing.
Ultimately, BMSC exhaustion in DFUs impairs bone regeneration and
may increase vulnerability to further complications, including
infection (Fig. 3).
Diabetic foot osteomyelitis (DFO) represents a
severe complication affecting 20-68% of patients with diabetic foot
ulcers, considerably increasing the risk of lower-extremity
amputation (88). While
neuropathy and vascular insufficiency create the general conditions
for ulcer formation (89),
impaired wound healing and microbial persistence, diabetes-related
changes in bone may further increase susceptibility to
osteomyelitis (90). Cortical
bone deterioration, driven by intracortical remodeling, can thin
the cortex from within through cavitation and coalescing pores,
increasing cortical porosity and compromising bone strength and
structural integrity (91-93). Consistent with this, regions with
lower cortical thickness and higher cortical porosity represent
structurally and mechanically weaker sites within bone (94). In parallel, marrow stromal
dysfunction may impair the local supportive capacity of the marrow
microenvironment (95-98). In this context, BMSC
exhaustion/senescence is associated with impaired self-renewal and
reduced osteogenic differentiation, thereby diminishing osteogenic
repair capacity (99). Once DFU
extends into deep soft tissue adjacent to bone (100), these bone-specific
abnormalities may leave the subjacent bone less able to withstand
and recover from contiguous bacterial insult, thereby facilitating
progression to established infection (100).
The progression from superficial ulceration to bone
infection follows a predictable cascade driven by the unique
pathophysiological environment of the diabetic foot. Superficial
DFUs typically begin as neuropathic ulcerations that, when left
untreated or inadequately managed, provide a direct pathway for
bacterial invasion (101). Loss
of protective sensation due to peripheral neuropathy allows
repetitive microtrauma to go unnoticed, while impaired immune
function and chronic hyperglycemia create conditions favorable for
bacterial proliferation and biofilm formation (3).
Bacteria penetrate compromised soft tissues,
initially colonizing the wound bed before extending into
subcutaneous tissues and eventually reaching the underlying bone
(102). This progression is
accelerated by PAD, which reduces perfusion and oxygen delivery
required for effective immune cell function (103). In addition, the polymicrobial
nature of the majority of DFIs, commonly involving Staphylococcus
aureus, Pseudomonas aeruginosa and anaerobic species, further
complicates the infectious process (104,105). Once bacteria establish
infection within bone tissue, they form resilient biofilms that
resist both host immune responses and antibiotic penetration,
leading to chronic, difficult to treat osteomyelitis (106,107). Diagnosis of DFO remains
challenging because systemic inflammatory markers may be absent or
minimal in diabetic patients despite severe bone infection
(108). A positive probe to
bone test in the setting of a chronic ulcer is highly suggestive of
underlying osteomyelitis and often necessitates aggressive
intervention, including surgical debridement and prolonged
antimicrobial therapy, to prevent progression to major amputation
(109). This progression
highlights the importance of early intervention, revascularization
and appropriate infection control as part of the wound healing
strategy (Fig. 4). It also
defines an important practical boundary for bone-origin repair,
since suspected osteomyelitis should first undergo appropriate
diagnostic evaluation, confirmed osteomyelitis should be treated
before bone-targeted intervention is considered (102) and any subsequent reconstruction
should follow only after infection control has been achieved
(110).
In bone-adjacent DFUs, the underlying skeleton
should be viewed not merely as a passive structural substrate, but
as a compartment with inherent regenerative and signaling capacity
(111). In the present review,
bone-origin repair refers to a hypothesis-driven, localized
strategy in which controlled stimulation of the cortical,
subcortical unit beneath or near a DFU is intended to trigger a
confined, callus-like response that may secondarily support
soft-tissue healing (112). At
present, this remains an emerging conceptual framework rather than
a validated clinical strategy. It is not proposed as a blanket
indication for bone-targeted intervention, but as a potential
adjunctive strategy to be applied under carefully defined clinical
conditions.
At the clinical level, these conditions are best
understood as provisional candidate scenarios rather than
established indications. The concept is most plausible in chronic,
bone-adjacent DFUs in which sensory loss and abnormal foot
mechanics indicate a strong neuropathic/mechanical component
(113,114), especially when ulceration
develops over deformity or other sites of repetitive stress
(31) and the underlying
bone-soft tissue unit is likely to be structurally compromised by
cortical thinning or impaired bone support (115,116). By contrast, ulcers dominated by
active osteomyelitis, critical ischemia, severe structural
instability or advanced Charcot neuroarthropathy should not be
considered candidates for bone-targeted stimulation until those
conditions have been appropriately excluded or treated (6,117,118).
A key factor in the clinical translation of
bone-origin repair is patient stratification by ulcer phenotype.
Conceptually, this framework is most plausible in neuropathic or
neuropathic-predominant DFUs, where sensory loss, deformity and
repetitive mechanical pressure are central drivers of tissue
breakdown (31). In these
settings, the underlying bone-soft tissue unit is exposed to
chronic stress, leading to structural vulnerabilities that may be
more relevant to a bone-targeted reparative concept (59,119). Conversely, in ischemic or
neuro-ischemic ulcers, limited macrovascular and microvascular
perfusion likely constrains both the local callus response and any
downstream soft-tissue benefit (120). Consequently, vascular
optimization remains a prerequisite, and severe ischemia represents
a poor candidate state for primary bone-targeted stimulation
(121). DFUs with mixed
neuropathic and ischemic features require the greatest caution, as
the coexistence of abnormal mechanical loading and perfusion
deficits makes both mechanistic attribution and therapeutic
outcomes more difficult to predict (6).
Mechanistically, the proposed core of bone-origin
repair is that controlled local bone stimulation activates
intrinsic reparative programs within the bone microenvironment
(122). Procedures such as
micro-drilling or osteoperforation analogs create localized
cortical or subchondral injuries intended to recruit progenitor
populations, initiate a callus-like healing cascade, and modulate
local vascular and immune signaling (123,124). In principle, this confined
response could enhance regional perfusion, mobilize reparative
mesenchymal cells and alter the periosteal microenvironment in ways
that secondarily support the metabolic and regenerative
requirements of the overlying ulcer bed (125,126). This hypothesis is biologically
grounded in the capacity of bone injury responses to recruit MSCs
(127,128), osteoprogenitors (129) and associated reparative
cellular programs (130,131).
However, the evidence level must be clearly
distinguished: At present, direct evidence that inducing a
peri-ulcer callus causes DFU healing is lacking. Tibial cortex
transverse transport (TTT) is a corticotomy-based distraction
procedure (132) and the
currently cited efficacy evidence derives from clinical studies
(133-135) in severe or recalcitrant DFUs
rather than from mechanism-specific trials (136). Consequently, clinical
experience with TTT provides only indirect support for the broader
principle that targeted manipulation of bone biology can influence
distal soft-tissue outcomes. In these studies, TTT has been
associated with improved healing, limb salvage, ankle-brachial
index, skin temperature and distal microvascular density (137,138). These observations are
consistent with bone-driven neovascular and reparative signaling,
but they do not establish peri-ulcer callus formation itself as the
proven causal mechanism of wound closure.
Clarifying the boundaries of bone-origin repair is
essential to prevent conceptual overreach. First, it is not a
proposal for blanket osteogenesis under all DFUs. Several ulcers
overlie weakened, deformed or infected bone (for example, Charcot
neuroarthropathy, osteopenic metatarsals and DFO). In such
settings, indiscriminate drilling or corticotomy risks fracture,
destabilization or spread of infection (139-141). Any bone-targeted maneuver must
be contingent on prior evaluation and, where necessary, treatment
of osteomyelitis and structural instability. Second, bone-origin
repair is not synonymous with unchecked or ectopic ossification.
The aim is to induce a confined, self-limited callus response
within cortical and subcortical bone, not to generate heterotopic
bone in fascia, tendons or dermis, which would likely exacerbate
pressure concentrations and impair joint mobility (142,143). Lessons from distraction
osteogenesis highlight that while sustained mechanical tension can
robustly stimulate osteogenesis and angiogenesis, it also carries
risks of infection, fracture and aberrant mineralization if poorly
controlled (144,145). Third, bone-origin repair is not
a substitute for foundational DFU care. Revascularization,
infection control (including management of DFO), pressure
off-loading and meticulous wound debridement remain primary
determinants of outcome (146,147). The promising results of TTT and
similar procedures have been achieved in the setting of optimized
conventional management, indicating that bone-targeted approaches
should be understood as adjunctive strategies layered onto standard
care rather than replacements for it (137). Finally, bone-origin repair is
not a single, fixed protocol; the depth, spatial distribution and
timing of bone stimulation are likely to require patient-specific
titration based on bone quality, vascular status and ulcer
characteristics.
Bone-origin repair, if translated into clinical
practice, should be layered onto standard DFU management rather
than introduced in parallel without prior optimization of vascular,
infectious and mechanical factors (148,149). Current DFU management is based
on systematic assessment of perfusion, infection and mechanical
loading, followed by regular wound evaluation, serial debridement,
appropriate moist dressings and pressure off-loading. When PAD is
present, revascularization should be performed when indicated,
whereas adjunctive therapies such as negative pressure wound
therapy are generally considered only after standard care has been
optimized and wound healing remains inadequate (149). In practical terms,
revascularization and infection assessment come first: Ischemic or
neuro-ischemic ulcers should undergo prompt vascular evaluation and
revascularization when indicated before elective bone-targeted
stimulation is considered (150,151), while clinically uninfected
ulcers should not receive antibiotics solely to promote healing and
suspected or confirmed infection should undergo guideline-based
antimicrobial and, when necessary, surgical management before any
bone procedure is contemplated (152,153). Local wound care should continue
throughout, including regular inspection, debridement and
appropriate dressings; negative pressure wound therapy may be
considered mainly for post-operative wounds or selected cases, but
not as a substitute for infection control or vascular optimization
(154). Mechanical protection
also remains essential, with ongoing off-loading or protected
weight-bearing determined by ulcer location, foot stability and
procedural extent; any progression in loading after a bone-targeted
intervention should be individualized and deferred until both
soft-tissue and structural conditions are clinically stable
(155,156).
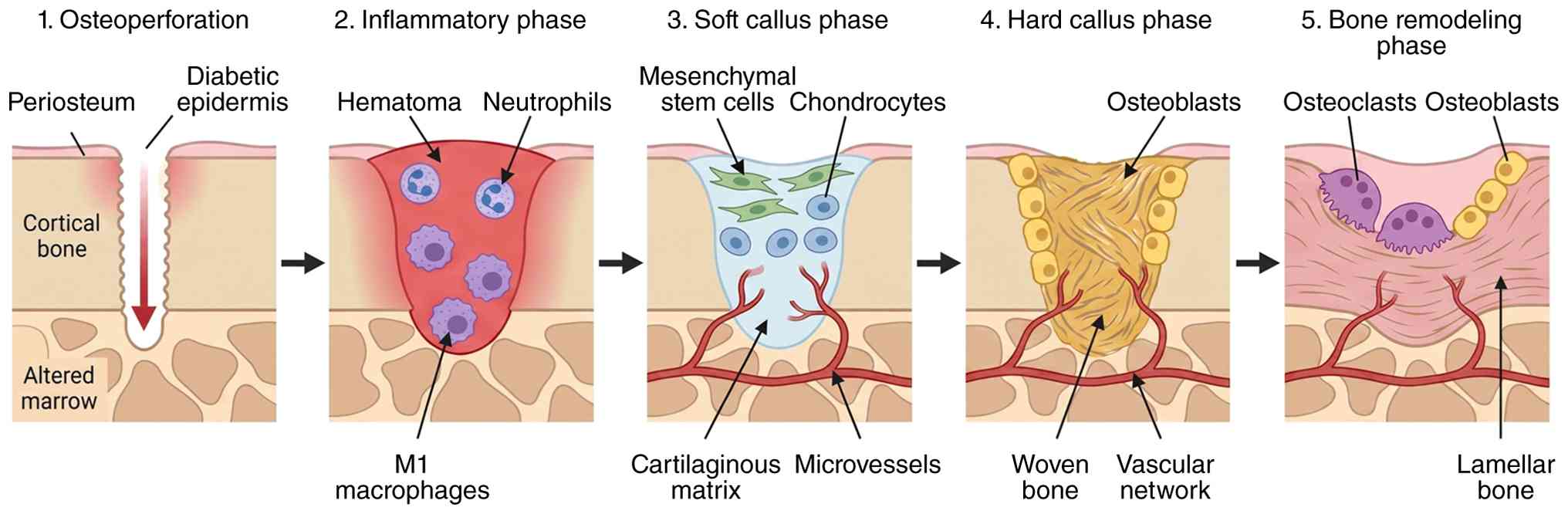
Callus formation is the central tissue response
through which mechanically competent bone is restored after injury
(157). This process represents
a sophisticated orchestration of cellular recruitment, phenotypic
switching and matrix deposition that recapitulates developmental
ossification programs (157,158). Therefore, understanding callus
biology at molecular and cellular resolution is important for
identifying how diabetes disrupts fracture healing and for
developing targeted interventions to restore bone-origin repair
capacity in diabetic foot ulcers.
Following a controlled cortical or subcortical
injury, callus formation proceeds through overlapping phases of
inflammation, soft callus formation, hard callus maturation and
remodeling. Each phase is defined by a characteristic 'cellular
cast' (159). Early on,
platelets and neutrophils dominate the hematoma, providing
hemostasis and releasing a first wave of chemokines and growth
factors. Monocytes subsequently differentiate into macrophages,
which are key regulators of the switch from inflammation to repair
(160); their phenotype
transitions from predominantly pro-inflammatory to pro-reparative
as healing progresses and that disruption of this switch leads to
delayed or abnormal callus formation (160,161).
Mesenchymal stromal cells (MSCs) are recruited from
periosteum, endosteum and bone marrow and migrate into the
hematoma, where they proliferate and differentiate along
chondrogenic and osteogenic lineages (128). After the early inflammatory
events, fracture repair progresses into the soft-callus stage
(162). This stage usually
begins a few days following the fracture. During this time, MSCs
migrate to the injury site and differentiate into chondrocytes and
osteoblasts. These newly formed cells generate a fibrocartilaginous
callus, which plays a vital role in stabilizing the fractured bone.
The soft callus functions as a temporary support structure,
connecting the broken bone fragments and creating a foundation for
subsequent bone repair (163).
As the healing process advances, the soft callus is slowly
converted into a hard callus during the stage referred to as hard
callus formation (Fig. 5)
(164). During this stage,
osteoblasts deposit osteoid, which subsequently mineralizes to
create a woven bone scaffold. At the same time, osteoclasts resorb
both cartilage and immature bone, contributing to callus remodeling
and restoration of normal bone architecture (163).
The developing callus becomes highly vascularized,
with endothelial cells and pericytes working together to form and
stabilize new blood vessels. Recent studies have also identified a
specialized subset of endothelial cells, known as 'type H' cells,
which are associated with osteoprogenitor cells and may serve as
key regulators of angiocrine signaling during bone repair (165,166). In addition to these classical
bone and vascular cells, immune cell subsets, including
macrophages, T cells and innate lymphoid populations, contribute to
callus quality (167).
Macrophages play pivotal roles throughout healing, with M1
macrophages dominating early inflammation and M2 macrophages
promoting resolution and osteogenesis (168). Osteoimmunology studies indicate
that macrophages regulate both osteoblast and osteoclast activity
throughout bone repair, and that sustained pro-inflammatory
macrophage phenotypes or defective efferocytosis can impair callus
maturation (161,167,169). These osteal macrophages are
located adjacent to osteoblasts and regulate bone formation through
efferocytosis and paracrine signaling (170). Taken together, these
observations suggest that immune and stromal cell behavior is
likely to be an important determinant of callus quality in any
bone-targeted reparative response.
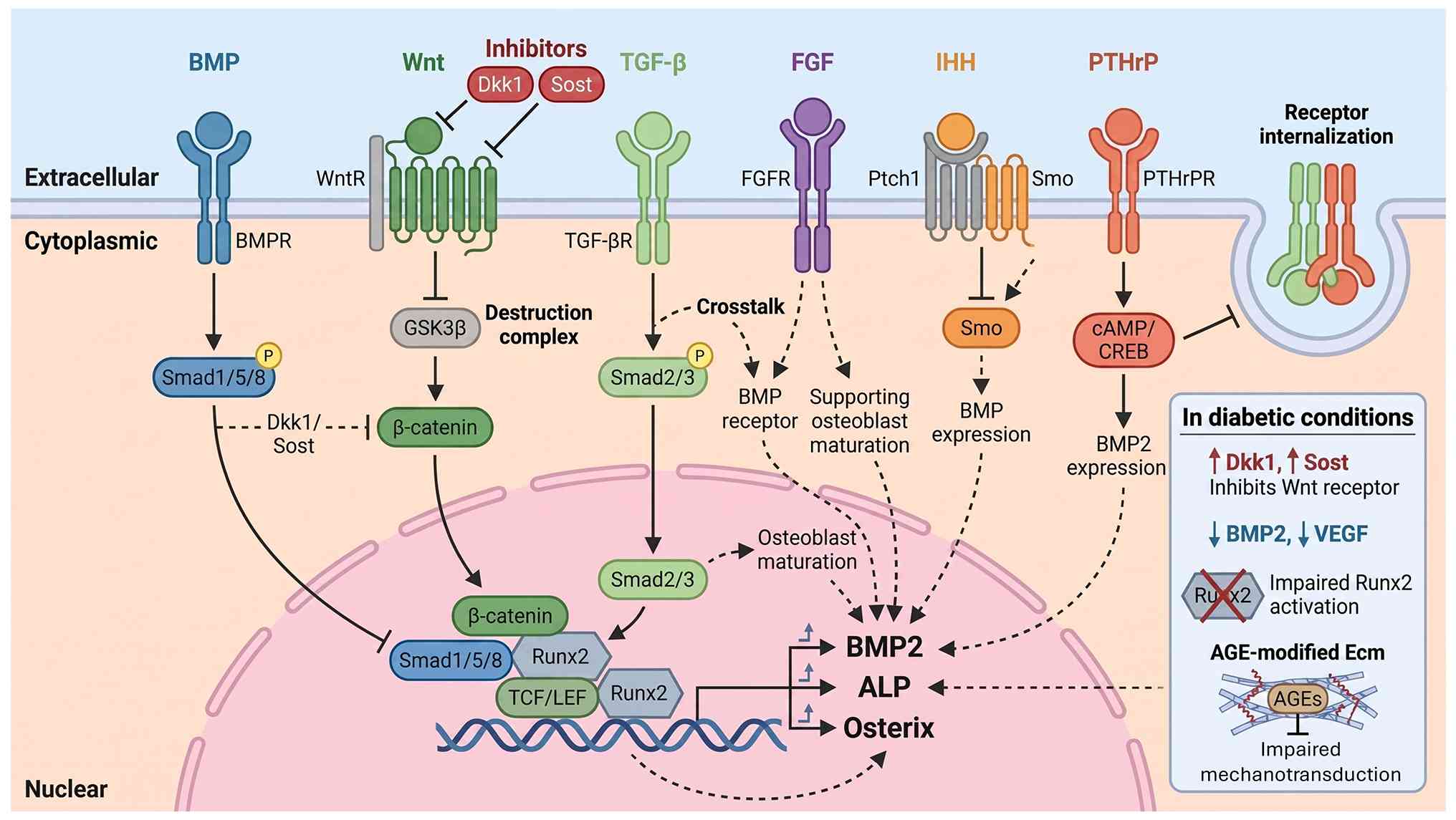
Callus formation is orchestrated by an integrated
network of signaling pathways. Members of the TGF-β and bone
morphogenetic protein (BMP) families are among the earliest and
most prominent (171). They
drive MSC recruitment, proliferation and differentiation, and
regulate the balance between chondrogenesis and osteogenesis;
canonical Smad-dependent TGF-β/BMP signaling is essential for
normal bone repair and perturbation of these pathways has been
linked to non-union and impaired callus quality (172,173).
The Wnt/β-catenin pathway is another major
regulator, promoting osteoblast differentiation and maturation
while suppressing excessive chondrogenesis (174). Human fracture-callus studies
have shown active Wnt signaling in normally healing callus and
altered Wnt activity in non-union tissue, underscoring its
relevance to callus competence (175,176). Hedgehog and Notch pathways
interact with both BMP/TGF-β and Wnt networks, fine-tuning
progenitor cell fate and the timing of endochondral ossification.
These pathways do not operate in isolation; rather, they form a
densely interconnected 'signal grammar' in which changes in one
pathway propagate through others, affecting cell recruitment,
proliferation and differentiation (177).
The HIF-VEGF axis couples angiogenesis with
osteogenesis through oxygen-sensing mechanisms. HIF-1 regulates
bone homeostasis and angiogenesis by controlling VEGF expression,
with the HIF-1/RANKL/Notch1 pathway bidirectionally regulating
macrophage differentiation into osteoclasts under different
conditions (178). VEGF serves
as the master regulator of vascular development and is required for
effective angiogenic-osteogenic coupling, with dose-dependent
effects on mesenchymal progenitor differentiation and angiocrine
functions of endothelium (179). Notch signaling exhibits
stage-specific functions, with Notch1-4 receptors controlling
osteoblast differentiation, matrix mineralization and osteoclast
recruitment through crosstalk with Wnt/β-catenin, BMP and RANKL/OPG
pathways (180).
The ECM adds a further layer of regulation: Its
composition, stiffness and degree of cross-linking influence
integrin signaling, mechanotransduction and the way cells interpret
growth-factor cues. Glycation-related changes and altered collagen
cross-linking can therefore have direct consequences for callus
architecture and mechanics, beyond their effects on bone cells per
se (181,182). ECM stiffness regulates MSC
differentiation through mechanotransduction pathways, while
glycation-induced matrix modifications alter mechanical properties
and cellular interactions (183). The bone ECM guides specific
tissue formation at implantation sites through osteoinductive,
osteoconductive and osteogenic signals (Fig. 6) (183).
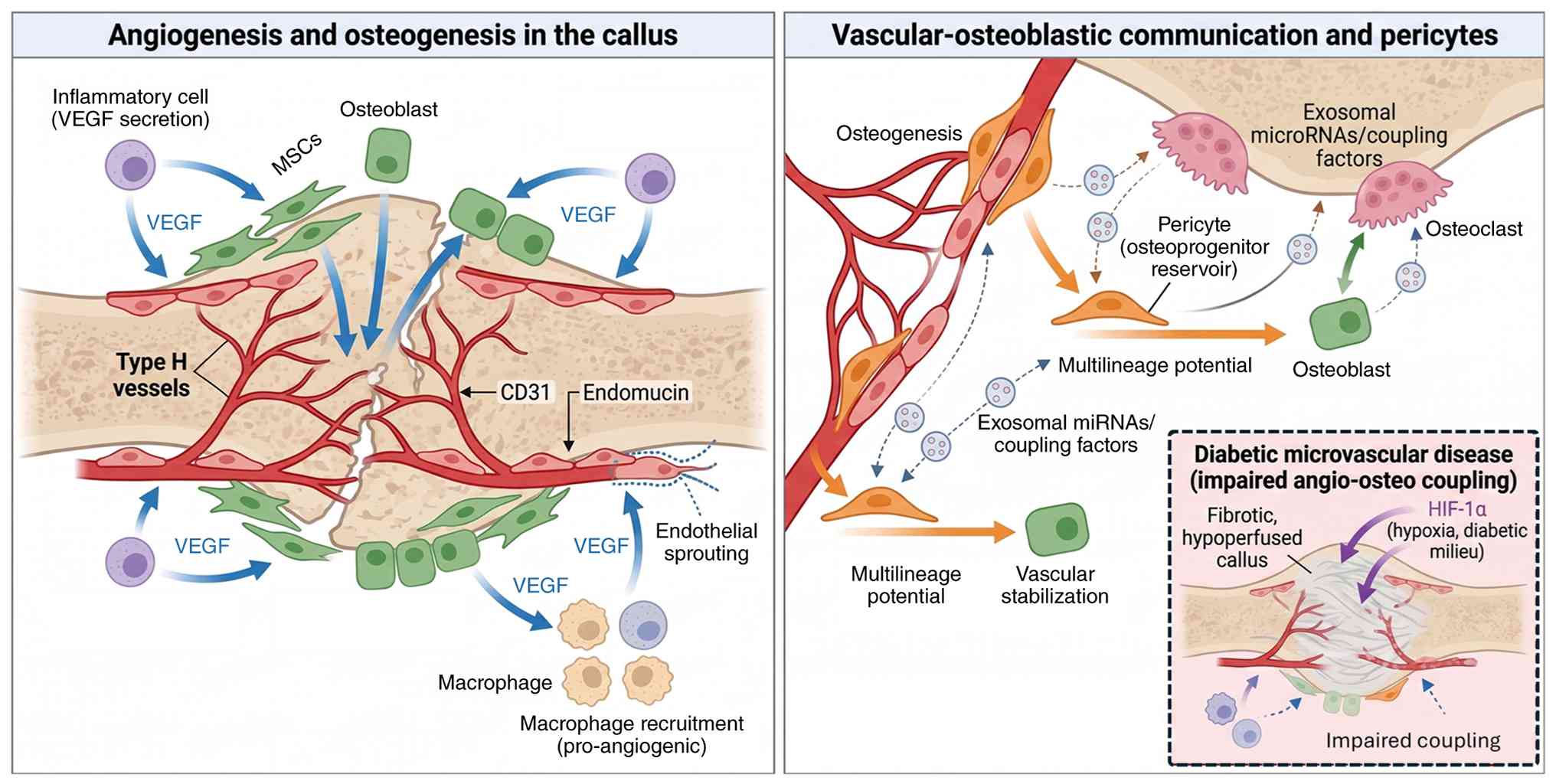
Within the callus, spatial organization of vessels
and bone-forming cells is non-random (179). VEGF produced by inflammatory
cells, MSCs, osteoblasts and hypertrophic chondrocytes promotes
endothelial proliferation and sprouting, while reciprocal signals
from endothelial cells enhance osteoblast and osteoclast activity
(188). Osteoblast-derived VEGF
plays key roles at multiple fracture healing stages, promoting
macrophage recruitment and angiogenic responses during
inflammation, coupling angiogenesis with osteogenesis in areas of
intramembranous ossification and stimulating blood vessels and
osteoclasts for cartilage resorption during endochondral
ossification (189). The
intimate bidirectional communication between endothelial cells and
bone cells involves angiocrine functions where endothelium
regulates osteoprogenitors, while immune cells recruited by the
vascular microenvironment modulate both angiogenesis and
osteogenesis (190).
Pericytes serve dual functions as vascular
stabilizers and osteoprogenitor reservoirs. Bone-specific
microvascular pericytes, tightly affiliated with capillaries,
exhibit multilineage potential and constitute the cellular reserve
for bone formation, remodeling and repair (191). Coupling factors and exosomal
microRNAs mediate crosstalk between osteoclasts, osteoblasts,
vessel-specific endothelial cells and perivascular pericytes,
playing central roles in angiogenesis-osteogenesis coupling
essential for bone remodeling (192). Perfusion geometry within the
callus determines nutrient delivery and waste removal, with
adequacy of vascular supply being critically dependent on
successful angiogenesis, as fracture healing can be prevented by
inhibiting angiogenesis (193).
In bone-origin repair applied beneath DFUs, the
interaction between angiogenesis and osteogenesis is a factor to be
taken into account. The hope is that a localized callus response
can augment perfusion in an otherwise ischemic, microangiopathic
environment. However, it is equally plausible that, in advanced
diabetic microvascular disease, the ability of bone to mount an
effective angiogenic response is constrained, leading to
under-perfused or fibrotic callus (194). The balance between these two
outcomes likely depends on the degree of vascular compromise and
the integrity of HIF-VEGF signaling in diabetic bone (Fig. 7) (34,195).
Diabetes mellitus exerts multifactorial effects on
bone regeneration, and these are expected to shape any callus
induced by bone-origin repair under a DFU (196,197). Hyperglycemia, AGE accumulation,
oxidative stress, low-grade inflammation and vascular damage
converge to impair osteoblast differentiation, increase osteoclast
activity and alter the function of osteocytes and MSCs (181,196,198). AGEs alter matrix mechanical
properties, disrupt molecular conformation, impair enzyme activity
and interfere with receptor functioning (199,200). Through interaction with RAGE,
these molecules trigger pro-inflammatory signaling and oxidative
stress that fundamentally derails normal callus kinetics (201). MSCs exposed to hyperglycemic or
AGE-rich environments show impaired proliferation and osteogenic
differentiation, increased adipogenic drift and enhanced
senescence, while osteoblasts exhibit reduced matrix production and
increased susceptibility to apoptosis (182,202).
On the immune side, diabetes alters macrophage
plasticity, prolonging pro-inflammatory phenotypes and delaying the
transition toward pro-reparative subsets (203). In bone, this can translate into
a prolonged inflammatory phase within the callus, with excessive
production of reactive oxygen species and pro-inflammatory
cytokines that interfere with osteoblast and chondrocyte function
(204). Aged and diabetic
macrophages show increased sensitivity to inflammatory signals, and
failure of M1-to-M2 repolarization leads to persistent
inflammation, increased osteoclast activation and decreased
osteoblast formation (168).
Proper macrophage polarization is essential for successful bone
healing, as M2 macrophages promote tissue regeneration while
prolonged M1 dominance inhibits repair (205). Microangiopathy compounds these
cellular defects by reducing perfusion to fracture sites, with
decreased blood supply increasing impaired healing incidence from
10 to 15 to 46% (206). The
diabetic microenvironment exhibits reduced VEGF responsiveness,
impaired endothelial function and diminished Type H vessel
abundance (207).
To facilitate future research design and clinical
trial endpoint selection, studies of callus biology in diabetic
bone repair should incorporate quantifiable measures across
structural, vascular, histologic and circulating domains (208-210). Structural evaluation may
include longitudinal CT or micro-CT to quantify callus volume and
mineral density (211,212), thereby characterizing the
three-dimensional composition and maturation of the regenerative
unit. Vascular assessment may include serial local perfusion
measurements using laser Doppler-based methods or angiographic
techniques to track angiogenic recovery (213). Where tissue-level analysis is
feasible, quantification of CD31hi/Emcnhi
type-H vessels may provide a useful marker of angiogenic-osteogenic
coupling (187,214-216). In parallel, serial measurement
of PDGF-BB and other candidate osteokines may help characterize
temporal circulating response patterns (214,217). At present, such circulating
profiles should be interpreted as exploratory biomarkers rather
than validated surrogate endpoints.
Over the past two decades, bone has been redefined
from a purely structural tissue to a multifunctional endocrine
organ. Osteoblasts, osteocytes and, to a lesser extent, osteoclasts
secrete bioactive factors, collectively termed osteokines, that
regulate energy metabolism, vascular biology and immune function
(218). In the context of DFUs,
a key distinction must be made between two levels of bone-derived
signaling: Systemic endocrine effects mediated by circulating
factors, and local paracrine or autocrine effects occurring within
the bone-wound microenvironment (15,219-221). While both may be relevant in
the diabetic state, their evidence levels and mechanistic
implications are not equivalent. Accordingly, the following
discussion distinguishes between osteokines that are more plausibly
involved as functional mediators of repair and those that may
currently be more effectively interpreted as associated
biomarkers.
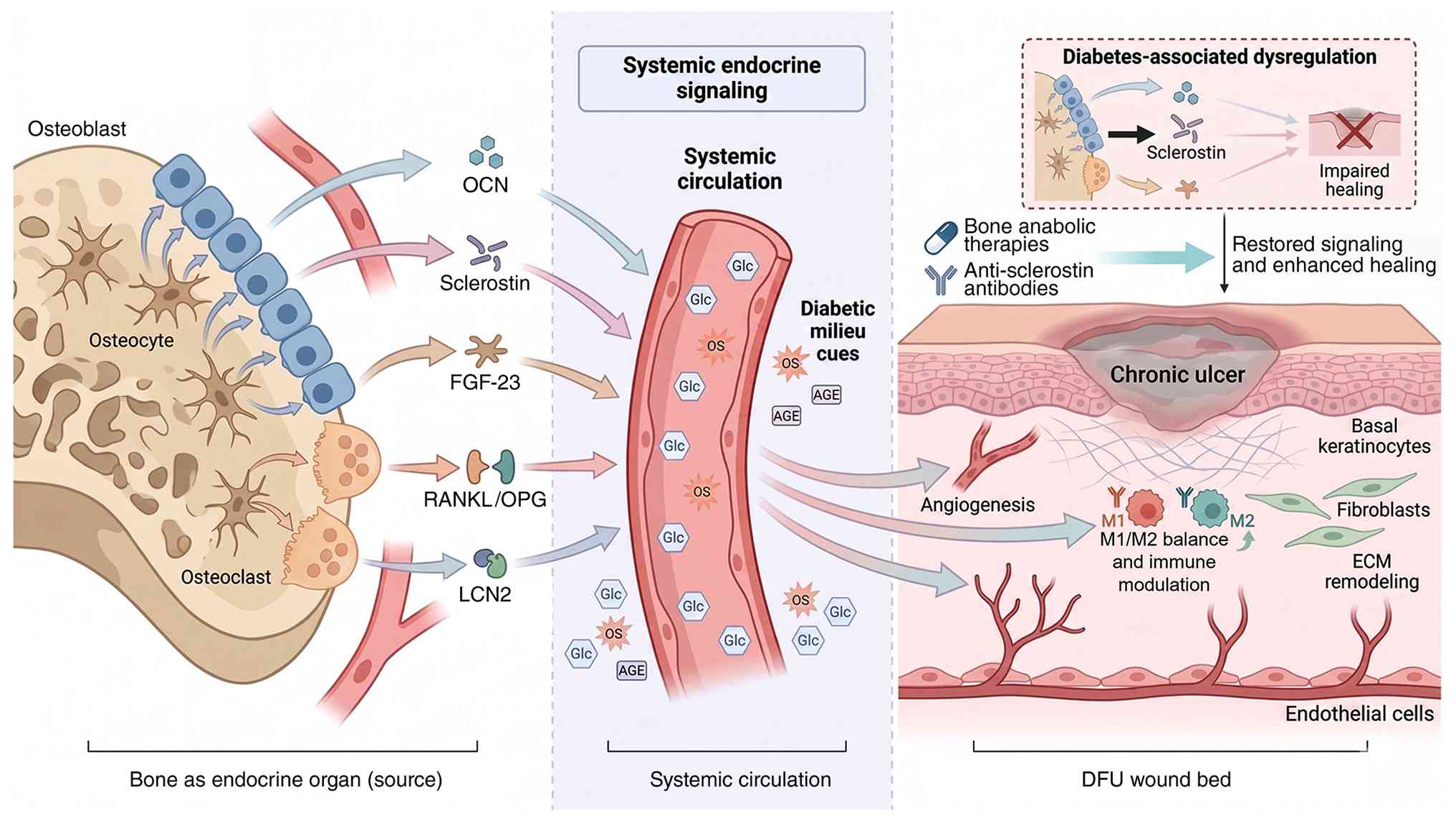
The recognition of bone as an endocrine organ has
fundamentally reshaped the understanding of skeletal biology beyond
its traditional structural and mineral storage functions (15,218). Bone cells, osteoblasts,
osteocytes and osteoclasts, secrete a constellation of bioactive
molecules termed 'osteokines' that exert systemic effects on
distant organs through endocrine, paracrine and autocrine
mechanisms (222). These
bone-derived factors represent key mediators associating skeletal
metabolism to wound healing, with particular relevance in the
diabetic context where both systems are profoundly dysregulated
(222).
OCN, the primary non-collagenous protein in the
bone matrix, exists in two major forms with distinct biological
associations: The carboxylated form, which facilitates
mineralization and the undercarboxylated (ucOCN) form (15), which has been implicated in
systemic metabolic regulation (223). While experimental murine models
indicate that ucOCN can potently stimulate insulin secretion and
peripheral sensitivity (19,224), its endocrine role in humans
remains a subject of ongoing debate (225-228). Human clinical data generally
show an inverse association between circulating OCN and glycemic
indices, such as HbA1c and fasting glucose, but these associations
are not uniform and often do not establish ucOCN as a definitive
causal mediator (223,226). This clinical discrepancy is
illustrated by reports of patients with type 2 diabetes exhibiting
lower circulating OCN levels than healthy controls (7.07±3.80
ng/mlvs. 20.41±13.50 ng/ml), alongside inverse associations with
glycemic markers (223).
Consequently, in the context of DFU pathogenesis, OCN is best
interpreted as a plausible metabolic mediator whose reduction in
diabetes may reflect altered bone-endocrine function, rather than a
fully validated driver of soft-tissue repair.
Sclerostin, encoded by the SOST gene and
predominantly secreted by osteocytes, is a potent inhibitor of
canonical Wnt/β-catenin signaling (15). By binding to LRP5/6 co-receptors
on osteoblasts, it suppresses bone formation, shifting the
remodeling balance toward an anti-anabolic state (229).
Beyond these local skeletal effects, circulating
sclerostin has been associated with metabolic dysfunction; however,
its role as a systemic endocrine mediator remains less clearly
defined than its established paracrine action in bone (230). While elevated sclerostin levels
in diabetes may contribute to a compromised bone environment
(231), direct evidence
associating sclerostin to DFU healing remains limited (232,233). Pharmacological inhibition of
sclerostin is under clinical investigation, with monoclonal
antibodies, including romosozumab, blosozumab, and BPS804
(setrusumab), being explored for their potent anabolic effects
(234-237).
FGF-23, produced by osteoblasts and osteocytes,
represents a bone-derived endocrine factor that regulates phosphate
metabolism primarily through renal actions (238). FGF-23 targets the kidney's
proximal tubule, the parathyroid gland and the cardiovascular
system; while its primary role is phosphate homeostasis, it has
also been implicated in inflammatory and vascular pathways relevant
to tissue repair (239,240). In diabetes, however,
interpretation of circulating FGF-23 is complicated by the frequent
coexistence of chronic kidney disease (CKD), making it difficult to
distinguish direct endocrine effects from CKD-related biomarker
elevation (15,241-244). Thus, FGF-23 is better
interpreted as a context-dependent systemic biomarker rather than
an established causal mediator of DFU repair.
By contrast, the RANKL/OPG axis constitutes a
fundamental regulatory system linking bone remodeling with immune
activation (245). The
RANKL/OPG ratio, regulated by osteoblasts and osteocytes,
influences immune cell subsets (for example, T cells and dendritic
cells) and endothelial cells, contributing to osteoimmune crosstalk
within the wound microenvironment. In diabetes, this ratio is
elevated, promoting excessive osteoclastogenesis and inflammatory
cytokine production (246-249). While RANKL-neutralizing
antibodies (for example, denosumab) represent potential strategies
to rebalance this signaling, their relevance to DFU remains
unproven (250,251). Therapeutic extrapolation must
remain cautious regarding potential risks, including increased
infection susceptibility, suppression of necessary bone remodeling
and impaired skeletal adaptation in the biomechanically complex
diabetic foot (252).
LCN2, secreted by osteoblasts, influences energy
metabolism by suppressing appetite in the brain and has been linked
to inflammatory processes (253). LCN2, can bind bacterial
siderophores and has been implicated in matrix degradation; subtly
altered LCN2 signaling might therefore affect the
protease-antiprotease balance and ECM turnover within chronic DFUs,
although this remains speculative.
LCN2 also known as neutrophil gelatinase-associated
lipocalin, is secreted by osteoblasts (253-255), and various immune cell types
(256,257), and has been implicated in
energy metabolism, innate immunity (256) and inflammatory signaling
(253,258). In the context of bone-derived
endocrine signaling, its interpretation remains highly
context-dependent. While LCN2 can bind bacterial siderophores
(259), and has been associated
with matrix remodeling (256,259), its specific causal relevance to
DFU repair is not yet established. In diabetes, current evidence
suggests that circulating LCN2 levels may reflect a systemic
dysmetabolic or inflammatory state rather than act as a primary
driver of wound healing (256,260), particularly because its
metabolic and immune effects are not consistently unidirectional.
Accordingly, LCN2 is best interpreted as a context-dependent
osteokine with biomarker relevance, while its direct therapeutic
modulation for DFU remains under investigation (254,256,261-263).
PDGF-BB, released locally following bone injury,
acts on fibroblasts, endothelial cells and pericytes (220,264). Among the osteokines discussed
here, PDGF-BB is more plausibly positioned as a direct paracrine
mediator than as an accompanying biomarker, because
bone-injury-triggered PDGF-BB signaling is mechanistically linked
to angiogenesis, fibroblast activation and improved diabetic wound
repair (265,266). PDGF-BB also influences
osteoclast precursor behavior, further supporting its role in
coordinating reparative responses across bone-associated tissues
(220,264). In the diabetic state, impaired
PDGF-BB release from bone may compromise this reparative influence
on distal soft tissues (220).
While topical recombinant PDGF-BB is an established clinical
therapy, strategies to leverage bone-derived release mechanisms or
targeted delivery platforms remain of interest and should be
regarded as investigational within the specific framework of
bone-origin repair (267).
To further illustrate the systemic roles of
bone-derived signals in diabetic wound healing, Table I summarizes the primary sources,
target tissues, functional effects, diabetes-associated alterations
and emerging therapeutic strategies for key osteokines implicated
in this endocrine network.
The diabetic milieu fundamentally disrupts bone
endocrine function, creating a pathological feedback loop wherein
skeletal dysfunction exacerbates systemic metabolic derangements
that further impair wound healing. Understanding these
disease-specific alterations and identifying therapeutic strategies
to restore beneficial bone-wound crosstalk represents a frontier in
diabetic wound management. The complex interaction between
bone-derived signals (osteokines) and diabetic wound healing plays
a key role in the pathophysiology of DFU (7,220). Osteoblasts, osteocytes and
osteoclasts secrete various osteokines, including OCN, sclerostin,
FGF-23, RANKL/OPG and LCN2, which exert systemic effects on
metabolism, angiogenesis and immune regulation (Fig. 8) (229,268).
Diabetes is associated with broad alterations in
osteokine secretion that may influence both systemic metabolism and
the local bone-wound microenvironment (223). Type 2 diabetic patients exhibit
reduced circulating OCN levels, specifically the undercarboxylated
form, showing substantial inverse associations with glycemic
control (223,269,270). Elevated circulating sclerostin,
increased FGF-23, frequently confounded by coexisting CKD,
dysregulated LCN2 profiles (260,271,272) and a shifted RANKL/OPG balance
(273) collectively suggest a
catabolic and pro-inflammatory endocrine/paracrine milieu. Although
the patterns imply that not only is the bone of the diabetic
patient structurally weakened, but it is also endocrinologically
abnormal, the relative contribution of each osteokine to the
failure of DFU repair remains unequal and, in some cases, needs
further mechanistic explanations.
Additional therapeutic considerations involve
attempts to modulate OCN biology, for instance, through vitamin K
supplementation to optimize carboxylation status, or interventions
aimed at reducing bone marrow adiposity (274). However, within the DFU context,
such strategies remain biologically plausible but clinically
unvalidated, particularly given the unresolved debate surrounding
the causal role of undercarboxylated OCN in human metabolic repair
(275). Furthermore, while the
emerging field of bone-targeted drug delivery, utilizing
bisphosphonates or bone-binding peptides, may offer opportunities
to localize osteoactive agents (276), its efficacy and potential
unintended consequences in the diabetic foot remain insufficiently
studied. Finally, the deliberate induction of controlled bone
remodeling to trigger osteokine release is a provocative concept
(220); its clinical
translation would require strict integration with established
standards of care, including infection control, vascular assessment
and mechanical off-loading (277).
Preclinical evidence suggests that controlled bone
injury can activate a bone-skin signaling response in diabetic
mice, with PDGF-BB identified as a key circulating mediator
associating tibial bone defects to accelerated foot wound healing
(220). In humans,
interventional studies of TTT in severe or recalcitrant DFUs have
reported improved healing, limb salvage, distal perfusion, and
small-vessel density, but these clinical data do not yet establish
that callus formation itself is the proven causal mechanism of
wound closure (132,278). These findings are reinforced by
a systematic review and meta-analysis of 7 studies involving 818
participants that found pooled healing and limb-salvage rates of
0.96 and 0.98, respectively, together with significant improvements
in ankle-brachial index and skin temperature (137). However, these clinical outcomes
do not yet establish that bone-injury-induced callus is itself the
proven causal mechanism of wound closure, and TTT must still be
interpreted alongside its reported risks, including fracture at the
transport site and pin-site infection.
The temporal cascade proceeds as PDGF-BB mediates
keratinocyte growth factor secretion from wound-site fibroblasts
via PDGFR signaling, thereby accelerating re-epithelialization
(220). Additional preclinical
evidence suggests that neurovascular signaling also contributes to
bone repair, as increased β2-adrenergic signaling has been
associated with enhanced callus neovascularization in fracture
healing models (279,280).
Several points of controversy and counterevidence
should be acknowledged to maintain a balanced perspective on the
bone-origin repair framework. First, the current human literature
regarding osteokine directionality, particularly for OCN,
sclerostin, FGF-23 and LCN2, remains heterogeneous (229,281,282). In several population studies,
these associations may reflect broader systemic metabolic or
inflammatory states rather than direct causal effects on wound
repair (283,284). Second, interpretation of FGF-23
in the diabetic population is substantially confounded by the high
prevalence of CKD, which can considerably alter circulating levels
and complicate mechanistic attribution to bone signaling alone
(285). Third, the clinical
utility of bone-targeted stimulation may be limited in severely
ischemic feet, where impaired macro- and microcirculation could
restrict both local callus formation and any downstream soft-tissue
benefit. Collectively, these considerations reinforce that the
bone-origin repair framework should presently be viewed as a
hypothesis-generating concept whose specific mediators, clinical
indications and therapeutic limits require prospective
validation.
The convergence of metabolic dysfunction,
neuropathy and vascular disease in diabetes creates profound
skeletal alterations (including cortical thinning, bone marrow
stromal cell exhaustion and impaired callus biology) that directly
compromise the bone-soft tissue unit and contribute to ulcer
chronicity. Beyond these structural deficits, the recognition of
bone as a dynamic endocrine organ secreting bioactive osteokines
has fundamentally reshaped the understanding of systemic factors
influencing wound repair.
The diabetic bone microenvironment is characterized
by dysregulated osteokine secretion patterns, with reduced levels
of beneficial factors such as OCN and elevated levels of inhibitory
molecules such as sclerostin, creating a catabolic and
pro-inflammatory milieu that impairs wound healing capacity both
locally and systemically. Hyperglycemia, AGE accumulation,
oxidative stress and chronic low-grade inflammation converge to
impair osteoblast function, alter macrophage polarization,
compromise angiogenesis and reduce the regenerative potential of
mesenchymal progenitors within bone marrow. These cellular and
molecular disruptions translate into prolonged healing times,
increased infection risk and elevated amputation rates in diabetic
patients with foot ulcers.
However, key caveats must be emphasized.
Bone-origin repair is not a universal solution for all DFUs.
Patient selection is paramount, active osteomyelitis, severe
structural instability, advanced Charcot neuroarthropathy and
critical ischemia represent absolute contraindications to
bone-targeted interventions. The concept should be understood as an
adjunctive strategy layered onto optimized conventional management
(revascularization, infection control, pressure off-loading and
meticulous debridement) rather than a replacement for foundational
care. The depth, spatial distribution and timing of bone
stimulation likely require patient-specific titration based on bone
quality, vascular status and ulcer characteristics. Furthermore,
the risk of complications, including fracture, infection spread,
ectopic ossification and over-lengthening, necessitates rigorous
clinical protocols and multidisciplinary expertise. Therapeutic
targeting of the bone-wound axis offers multiple avenues, including
anabolic bone agents, anti-sclerostin antibodies and optimization
of OCN bioactivity. Large-scale clinical trials are urgently needed
to validate safety and efficacy. The bone-wound axis represents a
frontier bridging osteoimmunology and regenerative medicine that
may unlock new limb salvage opportunities, though this promise
requires rigorous validation, careful patient selection and
multidisciplinary expertise.
Looking forward, precision medicine approaches that
phenotype individual patients' osteokine profiles and tailor
interventions accordingly may become feasible, offering
personalized strategies to harness skeletal endocrine function for
improved diabetic wound outcomes. Single-cell sequencing
technologies, advanced imaging modalities and biomarker discovery
platforms will be instrumental in identifying which patients are
most likely to benefit from bone-targeted approaches. Large-scale,
well-controlled clinical trials are urgently needed to validate the
safety and efficacy of bone-origin repair strategies, establish
standardized protocols and define optimal patient selection
criteria.
To focus future investigation, several testable
research questions emerge: i) Can controlled peri-ulcer bone
stimulation induce measurable local callus formation and improve
soft-tissue healing beyond optimized standard DFU care alone? ii)
Which ulcer phenotypes are most likely to benefit from this
strategy, particularly neuropathic-predominant vs. ischemic or
mixed presentations? iii) Which structural, vascular or circulating
biomarkers most reliably reflect biologically meaningful activation
of the bone-soft tissue unit? iv) Can candidate bone-derived
mediators such as PDGF-BB be prospectively associated with
downstream wound-healing outcomes in human DFU?
Not applicable.
SC and XC drafted the manuscript, edited and
revised the manuscript. XC designed the review. All authors read
and approved the final version of the manuscript. Data
authentication not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
No funding was received.
|
1
|
Bhowmik B, Siddiquee T, Moreira NC, Gupta
A, Saboo B, Khan AKA, Pathan S, Riley P and Chauhan AS: Global
assessment of insulin and oral hypoglycaemic agent accessibility
and affordability: A cross-sectional survey of international
diabetes federation member countries. Diabetes Metab Syndr.
20:1033782026. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Duncan BB, Magliano DJ and Boyko EJ: IDF
diabetes atlas 11th edition 2025: Global prevalence and projections
for 2050. Nephrol Dial Transplant. 41:7–9. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dawi J, Tumanyan K, Tomas K, Misakyan Y,
Gargaloyan A, Gonzalez E, Hammi M, Tomas S and Venketaraman V:
Diabetic foot ulcers: Pathophysiology, immune dysregulation, and
emerging therapeutic strategies. Biomedicines. 13:10762025.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McDermott K, Fang M, Boulton AJM, Selvin E
and Hicks CW: Etiology, epidemiology, and disparities in the burden
of diabetic foot ulcers. Diabetes Care. 46:209–221. 2023.
View Article : Google Scholar :
|
|
5
|
Sa BC, Maskan Bermudez N, Shimon SV and
Kirsner RS: Diabetic foot ulcers: A review of debridement
techniques. Surg Technol Int. 44:31–35. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Aditya C, Bukke SPN, Anitha K, Meeraraje
P, Goruntla N, Yadesa TM and Onohuean H: A comprehensive review on
diabetic foot ulcer addressing vascular insufficiency, impaired
immune response, and delayed wound healing mechanisms. Front
Pharmacol. 16:16220552025. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim J: The pathophysiology of diabetic
foot: A narrative review. J Yeungnam Med Sci. 40:328–334. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Parveen K, Hussain MA, Anwar S, Elagib HM
and Kausar MA: Comprehensive review on diabetic foot ulcers and
neuropathy: Treatment, prevention and management. World J Diabetes.
16:1003292025. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Soyoye DO, Abiodun OO, Ikem RT, Kolawole
BA and Akintomide AO: Diabetes and peripheral artery disease: A
review. World J Diabetes. 12:827–838. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Boulton AJM, Armstrong DG, Löndahl M,
Frykberg RG, Game FL, Edmonds ME, Orgill DP, Kramer K, Gurtner GC,
Januszyk M, et al: ADA Clinical Compendia Series. New
Evidence-Based Therapies for Complex Diabetic Foot Wounds. American
Diabetes Association. © 2022 by American Diabetes Association. All
rights reserved. None of the contents may be reproduced without the
written permission of the American Diabetes Association. Arlington,
VA: 2022
|
|
11
|
Raja JM, Maturana MA, Kayali S, Khouzam A
and Efeovbokhan N: Diabetic foot ulcer: A comprehensive review of
pathophysiology and management modalities. World J Clin Cases.
11:1684–1693. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu Y, Saiding Q, Zhou X, Wang J, Cui W and
Chen X: Electrospun fiber-based immune engineering in regenerative
medicine. Smart Med. 3:e202300342024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang N, Chen J, Chen Y, Chen L, Bao L and
Huang Z, Han X, Lu J, Cai Z, Cui W and Huang Z: Kneadable
dough-type hydrogel transforming from dynamic to rigid network to
repair irregular bone defects. Bioact Mater. 40:430–444.
2024.PubMed/NCBI
|
|
14
|
Zhuang P, Yang W, Chen Y, Zhang Y,
Leboucher C, Rosenholm JM and Zhang H: Biomaterials that passively
and actively target macrophages promote the regeneration of injured
tissues. Biomed Technol. 8:17–49. 2024. View Article : Google Scholar
|
|
15
|
Han Y, You X, Xing W, Zhang Z and Zou W:
Paracrine and endocrine actions of bone-the functions of secretory
proteins from osteoblasts, osteocytes, and osteoclasts. Bone Res.
6:162018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wawrzyniak A and Balawender K: Structural
and metabolic changes in bone. Animals (Basel). 12:19462022.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Streeten EA: Bone as a classic endocrine
organ: Interactions with non-bone tissues. Rev Endocr Metab Disord.
16:77–78. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Martiniakova M, Mondockova V, Biro R,
Kovacova V, Babikova M, Zemanova N, Ciernikova S and Omelka R: The
link between bone-derived factors osteocalcin, fibroblast growth
factor 23, sclerostin, lipocalin 2 and tumor bone metastasis. Front
Endocrinol (Lausanne). 14:11135472023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kanazawa I: Osteocalcin as a hormone
regulating glucose metabolism. World J Diabetes. 6:1345–1354. 2015.
View Article : Google Scholar :
|
|
20
|
Wei J, Hanna T, Suda N, Karsenty G and
Ducy P: Osteocalcin promotes β-cell proliferation during
development and adulthood through Gprc6a. Diabetes. 63:1021–1031.
2014. View Article : Google Scholar
|
|
21
|
Vasiliadis ES, Evangelopoulos DS, Kaspiris
A, Benetos IS, Vlachos C and Pneumaticos SG: The role of sclerostin
in bone diseases. J Clin Med. 11:8062022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gerosa L and Lombardi G: Bone-to-brain: A
round trip in the adaptation to mechanical stimuli. Front Physiol.
12:6238932021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hetta HF, Elsaghir A, Sijercic VC, Akhtar
MS, Gad SA, Moses A, Zeleke MS, Alanazi FE, Ahmed AK and Ramadan
YN: Mesenchymal stem cell therapy in diabetic foot ulcer: An
updated comprehensive review. Health Sci Rep. 7:e20362024.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rodriguez-Menocal L, Davis SC, Guzman W,
Gil J, Valdes J, Solis M, Higa A, Natesan S, Schulman CI, Christy
RJ and Badiavas EV: Model to inhibit contraction in third-degree
burns employing split-thickness skin graft and administered bone
marrow-derived stem cells. J Burn Care Res. 44:302–310. 2023.
View Article : Google Scholar
|
|
25
|
Zhu D, Chen F, Qiang H and Qi H: SPA
inhibits hBMSC osteogenic differentiation and M1 macrophage
polarization by suppressing SETD2 in acute suppurative
osteomyelitis. Sci Rep. 14:127282024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Voelker R: What are diabetic foot ulcers?
JAMA. 330:23142023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Armstrong DG, Tan TW, Boulton AJM and Bus
SA: Diabetic foot ulcers: A review. JAMA. 330:62–75. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu Y, Wang P, Li J, Chen L, Shu B, Wang
H, Liu H, Zhao S, Zhou J, Chen X and Xie J: Single-cell RNA
sequencing reveals the impaired epidermal differentiation and
pathological microenvironment in diabetic foot ulcer. Burns Trauma.
13:tkae0652025. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Monteiro-Soares M, Hamilton EJ, Russell
DA, Srisawasdi G, Boyko EJ, Mills JL, Jeffcoate W and Game F:
Guidelines on the classification of foot ulcers in people with
diabetes (IWGDF 2023 update). Diabetes Metab Res Rev. 40:e36482024.
View Article : Google Scholar
|
|
30
|
Huang F, Lu X, Yang Y, Yang Y, Li Y, Kuai
L, Li B, Dong H and Shi J: Microenvironment-based diabetic foot
ulcer nanomedicine. Adv Sci (Weinh). 10:e22033082023. View Article : Google Scholar :
|
|
31
|
Wang X, Yuan CX, Xu B and Yu Z: Diabetic
foot ulcers: Classification, risk factors and management. World J
Diabetes. 13:1049–1065. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Burgess JL, Wyant WA, Abdo Abujamra B,
Kirsner RS and Jozic I: Diabetic wound-healing science. Medicina
(Kaunas). 57:10722021. View Article : Google Scholar :
|
|
33
|
Banday MZ, Sameer AS and Nissar S:
Pathophysiology of diabetes: An overview. Avicenna J Med.
10:174–188. 2020. View Article : Google Scholar
|
|
34
|
Yang DR, Wang MY, Zhang CL and Wang Y:
Endothelial dysfunction in vascular complications of diabetes: A
comprehensive review of mechanisms and implications. Front
Endocrinol (Lausanne). 15:13592552024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dong H, Sun Y, Nie L, Cui A, Zhao P, Leung
WK and Wang Q: Metabolic memory: Mechanisms and diseases. Signal
Transduct Target Ther. 9:382024. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
González P, Lozano P, Ros G and Solano F:
Hyperglycemia and oxidative stress: An integral, updated and
critical overview of their metabolic interconnections. Int J Mol
Sci. 24:93522023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang Y, Zhang Z, Tu C, Chen X and He R:
Advanced glycation end products in disease development and
potential interventions. Antioxidants (Basel). 14:4922025.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou M, Zhang Y, Shi L, Li L, Zhang D,
Gong Z and Wu Q: Activation and modulation of the AGEs-RAGE axis:
Implications for inflammatory pathologies and therapeutic
interventions-A review. Pharmacol Res. 206:1072822024. View Article : Google Scholar
|
|
39
|
Wang B, Jiang T, Qi Y, Luo S, Xia Y, Lang
B, Zhang B and Zheng S: AGE-RAGE axis and cardiovascular diseases:
Pathophysiologic mechanisms and prospects for clinical
applications. Cardiovasc Drugs Ther. 39:1489–1506. 2025. View Article : Google Scholar :
|
|
40
|
Lin H, Yang Y, Wang X, Chung M, Zhang L,
Cai S, Pan X and Pan Y: Targeting the AGEs-RAGE axis: Pathogenic
mechanisms and therapeutic interventions in diabetic wound healing.
Front Med (Lausanne). 12:16676202025. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hudson BI and Lippman ME: Targeting RAGE
signaling in inflammatory disease. Annu Rev Med. 69:349–364. 2018.
View Article : Google Scholar
|
|
42
|
Dong H, Zhang Y, Huang Y and Deng H:
Pathophysiology of RAGE in inflammatory diseases. Front Immunol.
13:9314732022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Khalid M, Petroianu G and Adem A: Advanced
glycation end products and diabetes mellitus: Mechanisms and
perspectives. Biomolecules. 12:5422022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Riaz M, Iqbal MZ, Klar AS and Biedermann
T: Immunomodulatory mechanisms of chronic wound healing:
Translational and clinical relevance. MedComm (2020). 6:e703782025.
View Article : Google Scholar :
|
|
45
|
Cepas V, Collino M, Mayo JC and Sainz RM:
Redox signaling and advanced glycation endproducts (AGEs) in
diet-related diseases. Antioxidants (Basel). 9:1422020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Qin Y and Deng S: Inflammation, diabetic
foot and related treatments. Front Endocrinol (Lausanne).
16:16766212025. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wu XQ, Zhang DD, Wang YN, Tan YQ, Yu XY
and Zhao YY: AGE/RAGE in diabetic kidney disease and ageing kidney.
Free Radic Biol Med. 171:260–271. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang X, Dong T, Yao S, Lu S and Li W:
Application of transverse tibial bone transport and
microcirculation reconstruction in the treatment of diabetic foot
ulcer: A case report. Ann Palliat Med. 10:8358–8364. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu H, Yan XY, Li GQ, Wang BN, Wang D,
Zhang YH and Guo JL: Evaluation of wound temperature monitoring at
various anatomical sites in the management of patients with
diabetic foot undergoing microcirculation reconstruction. J Orthop
Surg Res. 19:7762024. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Van Dam PS, Cotter MA, Bravenboer B and
Cameron NE: Pathogenesis of diabetic neuropathy: focus on
neurovascular mechanisms. Eur J Pharmacol. 719:180–186. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wu B, Fu Z, Wang X, Zhou P, Yang Q, Jiang
Y and Zhu D: A narrative review of diabetic bone disease:
Characteristics, pathogenesis, and treatment. Front Endocrinol
(Lausanne). 13:10525922022. View Article : Google Scholar :
|
|
52
|
Araújo R, Páscoa R, Bernardino R and Gomes
PS: Impact of High glucose on bone collagenous matrix composition,
structure, and organization: An integrative analysis using an ex
vivo model. Cells. 14:1302025. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sharma P, Sharma RK and Gaur K:
Understanding the impact of diabetes on bone health: A clinical
review. Metabol Open. 24:1003302024. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gao Q, Jiang Y, Zhou D, Li G, Han Y, Yang
J, Xu K, Jing Y, Bai L, Geng Z, et al: Advanced glycation end
products mediate biomineralization disorder in diabetic bone
disease. Cell Rep Med. 5:1016942024. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Schwartz AV: TZDs and bone: A review of
the recent clinical evidence. PPAR Res. 2008:2978932008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Paschalis EP, Shane E, Lyritis G,
Skarantavos G, Mendelsohn R and Boskey AL: Bone fragility and
collagen cross-links. J Bone Miner Res. 19:2000–2004. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Saito M, Fujii K and Marumo K: Degree of
mineralization-related collagen crosslinking in the femoral neck
cancellous bone in cases of hip fracture and controls. Calcif
Tissue Int. 79:160–168. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Dubský M, Sojáková D, Fejfarová V and Jude
EB: Diabetic peripheral neuropathy: New diagnostics and treatment
perspectives. Drugs Aging. 43:29–48. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Deng H, Li B, Shen Q, Zhang C, Kuang L,
Chen R, Wang S, Ma Z and Li G: Mechanisms of diabetic foot
ulceration: A review. J Diabetes. 15:299–312. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Qin Q, Lee S, Patel N, Walden K,
Gomez-Salazar M, Levi B and James AW: Neurovascular coupling in
bone regeneration. Exp Mol Med. 54:1844–1849. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Hansen CS, Theilade S, Lajer M, Hansen TW
and Rossing P: Cardiovascular autonomic neuropathy and bone
metabolism in type 1 diabetes. Diabet Med. 35:1596–1604. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Rümenapf G, Abilmona N, Morbach S and Sigl
M: Peripheral arterial disease and the diabetic foot syndrome:
Neuropathy makes the difference! A narrative review. J Clin Med.
13:21412024. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Hu K and Olsen BR: The roles of vascular
endothelial growth factor in bone repair and regeneration. Bone.
91:30–38. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wu Z, Li W, Jiang K, Lin Z, Qian C, Wu M,
Xia Y, Li N, Zhang H, Xiao H, et al: Regulation of bone
homeostasis: Signaling pathways and therapeutic targets. MedComm
(2020). 5:e6572024. View Article : Google Scholar :
|
|
65
|
Luo M, Zhao Z and Yi J: Osteogenesis of
bone marrow mesenchymal stem cell in hyperglycemia. Front
Endocrinol (Lausanne). 14:11500682023. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Dama G, Du J, Zhu X, Liu Y and Lin J: Bone
marrow-derived mesenchymal stem cells: A promising therapeutic
option for the treatment of diabetic foot ulcers. Diabetes Res Clin
Pract. 195:1102012023. View Article : Google Scholar
|
|
67
|
Yu X, Liu P, Li Z and Zhang Z: Function
and mechanism of mesenchymal stem cells in the healing of diabetic
foot wounds. Front Endocrinol (Lausanne). 14:10993102023.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Shi Y, Yang X, Min J, Kong W, Hu X, Zhang
J and Chen L: Advancements in culture technology of adipose-derived
stromal/stem cells: Implications for diabetes and its
complications. Front Endocrinol (Lausanne). 15:13432552024.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Qadir A, Liang S, Wu Z, Chen Z, Hu L and
Qian A: Senile Osteoporosis: The involvement of differentiation and
senescence of bone marrow stromal cells. Int J Mol Sci. 21:3492020.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Li K, Hu S and Chen H: Cellular senescence
and other age-related mechanisms in skeletal diseases. Bone Res.
13:682025. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Wang YT, Zheng SY, Luo Y, Xiao WF, Huang C
and Li YS: Osteoimmunology and aging: Mechanisms, implications, and
therapeutic perspectives. Ageing Res Rev. 111:1028222025.
View Article : Google Scholar
|
|
72
|
Matsushita Y, Chu AKY, Ono W, Welch JD and
Ono N: Intercellular interactions of an adipogenic
CXCL12-expressing stromal cell subset in murine bone marrow. J Bone
Miner Res. 36:1145–1158. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Peng X, Zhou X, Yin Y, Luo B, Liu Y and
Yang C: Inflammatory microenvironment accelerates bone marrow
mesenchymal stem cell aging. Front Bioeng Biotechnol.
10:8703242022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhang W, Xie Y and Wang B: Senescence of
bone marrow mesenchymal stromal cells: A narrative review of
mechanisms, functional consequences, and rejuvenation strategies
for age-related disorders. Stem Cell Res Ther. 17:582025.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Liang F, Luo YF, Guo Z, Qian Q, Meng XB
and Mo ZH: MicroRNA-139-5p mediates BMSCs impairment in diabetes by
targeting HOXA9/c-Fos. FASEB J. 37:e226972023. View Article : Google Scholar
|
|
76
|
Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng
J, Li Y, Wang X and Zhao L: Inflammatory responses and
inflammation-associated diseases in organs. Oncotarget.
9:7204–7218. 2017. View Article : Google Scholar
|
|
77
|
Moiseeva V, Cisneros A, Cobos AC, Tarrega
AB, Oñate CS, Perdiguero E, Serrano AL and Muñoz-Cánoves P:
Context-dependent roles of cellular senescence in normal, aged, and
disease states. FEBS J. 290:1161–1185. 2023. View Article : Google Scholar
|
|
78
|
Schieber M and Chandel NS: ROS function in
redox signaling and oxidative stress. Curr Biol. 24:R453–R462.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Vujčić S, Kotur-Stevuljević J, Vekić J,
Perović-Blagojević I, Stefanović T, Ilić-Mijailović S, Koprivica
Uzelac B, Bosić S, Antonić T, Guzonjić A, et al: Oxidative stress
and inflammatory biomarkers in patients with diabetic foot.
Medicina (Kaunas). 58:18662022. View Article : Google Scholar
|
|
80
|
Zhang H, Yan Z, Zhu J, Li Z, Chen L, Zheng
W, Dai Z, Yang J, Yun X, Wang Y, et al: Extracellular
mitochondrial-derived vesicles affect the progression of diabetic
foot ulcer by regulating oxidative stress and mitochondrial
dysfunction. Adv Sci (Weinh). 12:e24075742025. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Li Y and Wang X: Chrysin attenuates high
glucose-induced BMSC dysfunction via the activation of the
PI3K/AKT/Nrf2 signaling pathway. Drug Des Devel Ther. 16:165–182.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Weinberg E, Maymon T and Weinreb M: AGEs
induce caspase-mediated apoptosis of rat BMSCs via TNFα production
and oxidative stress. J Mol Endocrinol. 52:67–76. 2014. View Article : Google Scholar
|
|
83
|
Rabbani PS, Soares MA, Hameedi SG, Kadle
RL, Mubasher A, Kowzun M and Ceradini DJ: Dysregulation of
Nrf2/Keap1 redox pathway in diabetes affects multipotency of
stromal cells. Diabetes. 68:141–155. 2019. View Article : Google Scholar
|
|
84
|
Huang X, Zhang Y, Qi B, Sun K, Liu N, Tang
B, Fang S, Zhu L and Wei X: HIF-1α: Its notable role in the
maintenance of oxygen, bone and iron homeostasis (Review). Int J
Mol Med. 50:1412022. View Article : Google Scholar
|
|
85
|
You J, Liu M, Li M, Zhai S, Quni S, Zhang
L, Liu X, Jia K, Zhang Y and Zhou Y: The role of HIF-1α in bone
regeneration: A new direction and challenge in bone tissue
engineering. Int J Mol Sci. 24:80292023. View Article : Google Scholar
|
|
86
|
Xing J, Ying Y, Mao C, Liu Y, Wang T, Zhao
Q, Zhang X, Yan F and Zhang H: Hypoxia induces senescence of bone
marrow mesenchymal stem cells via altered gut microbiota. Nat
Commun. 9:20202018. View Article : Google Scholar :
|
|
87
|
Zhang H, Fu ZS, Zhou Y, Wang SN, Ye SY,
Wang AN and Liu JT: Immunometabolic reprogramming in diabetic
osteomyelitis: From mechanisms to therapeutics. Front Cell Infect
Microbiol. 15:16063172025. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Ndosi M, Wright-Hughes A, Brown S,
Backhouse M, Lipsky BA, Bhogal M, Reynolds C, Vowden P, Jude EB,
Nixon J and Nelson EA: Prognosis of the infected diabetic foot
ulcer: A 12-month prospective observational study. Diabet Med.
35:78–88. 2018. View Article : Google Scholar :
|
|
89
|
Syauta D, Mulawardi Prihantono, Hendarto
J, Mariana N, Sulmiati, Kusumanegara J and Faruk M: Risk factors
affecting the degree of diabetic foot ulcers according to Wagner
classification in diabetic foot patients. Medicina Clínica
Práctica. 4:1002312021. View Article : Google Scholar
|
|
90
|
Wang Q, Liu C, An J, Liu J, Wang Y and Cai
Y: Mechanisms of microbial infection and wound healing in diabetic
foot ulcer: Pathogenicity in the inflammatory-proliferative phase,
chronicity, and treatment strategies. Front Endocrinol (Lausanne).
16:16579282025. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Bala Y, Zebaze R and Seeman E: Role of
cortical bone in bone fragility. Curr Opin Rheumatol. 27:406–413.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Iori G, Schneider J, Reisinger A, Heyer F,
Peralta L, Wyers C, Glüer CC, van den Bergh JP, Pahr D and Raum K:
Cortical thinning and accumulation of large cortical pores in the
tibia reflect local structural deterioration of the femoral neck.
Bone. 137:1154462020. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zebaze RM, Ghasem-Zadeh A, Bohte A,
Iuliano-Burns S, Mirams M, Price RI, Mackie EJ and Seeman E:
Intracortical remodelling and porosity in the distal radius and
post-mortem femurs of women: A cross-sectional study. Lancet.
375:1729–1736. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Cirovic A, Cirovic A, Djukic D, Djonic D,
Zivkovic V, Nikolic S, Djuric M and Milovanovic P:
Three-dimensional mapping of cortical porosity and thickness along
the superolateral femoral neck in older women. Sci Rep.
12:155442022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Pourebrahim R, Montoya RH, Alaniz Z,
Ostermann L, Lin PP, Liu B, Ayoub E, Burks JK and Andreeff M:
Mdm2/p53 levels in bone marrow mesenchymal stromal cells is
essential for maintaining the hematopoietic niche in response to
DNA damage. Res Sq. [Preprint] rs.3.rs-2544760. 2023.PubMed/NCBI
|
|
96
|
Yang X, Cheng J, Xiao J, Gu Z and Dong C:
Bone marrow mesenchymal stromal cells metabolic reprogramming in
systemic lupus erythematosus: Remodeling of bone marrow
microenvironment and regulation of immune cell fate. Front Immunol.
17:17252982026. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Leimkühler NB and Schneider RK:
Inflammatory bone marrow microenvironment. Hematology Am Soc
Hematol Educ Program. 2019:294–302. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Gomes AC, Saraiva M and Gomes MS: The bone
marrow hematopoietic niche and its adaptation to infection. Semin
Cell Dev Biol. 112:37–48. 2021. View Article : Google Scholar
|
|
99
|
Deng P, Yuan Q, Cheng Y, Li J, Liu Z, Liu
Y, Li Y, Su T, Wang J, Salvo ME, et al: Loss of KDM4B exacerbates
bone-fat imbalance and mesenchymal stromal cell exhaustion in
skeletal aging. Cell Stem Cell. 28:1057–1073.e7. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
McNally JN and Bruckner A: An unusual case
of proteus mirabilis-induced severe contiguous bacterial
osteomyelitis in an elderly nursing home resident: A case report.
Cureus. 16:e577102024.PubMed/NCBI
|
|
101
|
Winkler E, Schöni M, Krähenbühl N, Uçkay I
and Waibel FWA: Foot osteomyelitis location and rates of primary or
secondary major amputations in patients with diabetes. Foot Ankle
Int. 43:957–967. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Senneville É, Albalawi Z, van Asten SA,
Abbas ZG, Allison G, Aragón-Sánchez J, Embil JM, Lavery LA, Alhasan
M, Oz O, et al: IWGDF/IDSA guidelines on the diagnosis and
treatment of diabetes-related foot infections (IWGDF/IDSA 2023).
Diabetes Metab Res Rev. 40:e36872024. View Article : Google Scholar
|
|
103
|
Salvi M, Bonanni FR, Uccioli L, Bellizzi
E, Ruotolo V, Andreadi A, Bellia A, Lauro D and Meloni M: Heel
diabetic foot osteomyelitis: A current challenge in the clinical
practice. Int J Low Extrem Wounds. 153473462513761152025.Epub ahead
of print. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Anju VT, Busi S, Imchen M, Kumavath R,
Mohan MS, Salim SA, Subhaswaraj P and Dyavaiah M: Polymicrobial
infections and biofilms: Clinical significance and eradication
strategies. Antibiotics (Basel). 11:17312022. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Zambelli R, Santos AF, Moreira LR, Ribeiro
HM, Simões R, Magalhães JM, Constantino P, Salomão MC, Netto CC and
Leopoldino AO: Bacterial profile and antimicrobial resistance in
diabetic foot ulcer infections: A 10-year retrospective cohort
study. Braz J Infect Dis. 29:1045702025. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Masters EA, Trombetta RP, de Mesy Bentley
KL, Boyce BF, Gill AL, Gill SR, Nishitani K, Ishikawa M, Morita Y,
Ito H, et al: Evolving concepts in bone infection: redefining
'biofilm', 'acute vs. chronic osteomyelitis', 'the immune proteome'
and 'local antibiotic therapy'. Bone Res. 7:202019. View Article : Google Scholar
|
|
107
|
Villa F, Marchandin H, Lavigne J-P,
Schuldiner S, Cellier N, Sotto A and Loubet P: Anaerobes in
diabetic foot infections: Pathophysiology, epidemiology, virulence,
and management. Clin Microbiol Rev. 37:e00143232024. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Senneville EM, Lipsky BA, van Asten SAV
and Peters EJ: Diagnosing diabetic foot osteomyelitis. Diabetes
Metab Res Rev. 36(Suppl 1): e32502020. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Peterlin AAN, Jensen LK, Gleipner-Andersen
E and Gottlieb H: CLOSE-UP-a favourable protocol for limb-sparing
surgery of diabetic foot osteomyelitis. J Bone Jt Infect.
10:199–206. 2025. View Article : Google Scholar
|
|
110
|
Schöllmann H, Weichert V, Neidlein C,
Brinkmann N, Dudda M and Steinhausen E: Fiberfill©-A new bone
substitute for treatment of chronic osteomyelitis? J Clin Med.
15:12772026. View Article : Google Scholar
|
|
111
|
Pollak N, Janežič EG, Šink Ž and Ugwoke
CK: Crosstalk between skeletal muscle and proximal connective
tissues in lipid dysregulation in obesity and type 2 diabetes.
Metabolites. 15:5812025. View Article : Google Scholar :
|
|
112
|
Hasson M, Fernandes LM, Solomon H, Pepper
T, Huffman NL, Pucha SA, Bariteau JT, Kaiser JM and Patel JM:
Considering the cellular landscape in marrow stimulation techniques
for cartilage repair. Cells Tissues Organs. 213:523–537.
2024.PubMed/NCBI
|
|
113
|
Wang R, Gu S, Kim YH, Lee A, Lin H and
Jiang D: Diabetic wound repair: From mechanism to therapeutic
opportunities. MedComm (2020). 6:e704062025. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Mohsin F, Javaid S, Tariq M and Mustafa M:
Molecular immunological mechanisms of impaired wound healing in
diabetic foot ulcers (DFU), current therapeutic strategies and
future directions. Int Immunopharmacol. 139:1127132024. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Shanbhogue VV, Hansen S, Frost M,
Jørgensen NR, Hermann AP, Henriksen JE and Brixen K: Compromised
cortical bone compartment in type 2 diabetes mellitus patients with
microvascular disease. Eur J Endocrinol. 174:115–124. 2016.
View Article : Google Scholar
|
|
116
|
Cavanagh PR, Young MJ, Adams JE, Vickers
KL and Boulton AJ: Radiographic abnormalities in the feet of
patients with diabetic neuropathy. Diabetes Care. 17:201–209. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Argyropoulos M, Wynell-Mayow W, Johnson O,
Faroug R, Johal KS, Deol RS, Hakmi A and Mordecai S: Charcot
neuro-osteoarthropathy: A review of key concepts and an
evidence-based surgical management algorithm. Front Clin Diabetes
Healthc. 5:13443592024. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Altmann D, Waibel FWA, Forgo G, Grigorean
A, Lipsky BA, Uçkay I and Schöni M: Timing of revascularization and
parenteral antibiotic treatment associated with therapeutic
failures in ischemic diabetic foot infections. Antibiotics (Basel).
12:6852023. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Paulini MR, Leonardo Pitol D, Crivelaro do
Nascimento G, Buchaim DV, Rodrigues da Cunha M, Buchaim RL and
Mardegan Issa JP: Psychological stress modulates bone remodeling
pathways in normotensive and hypertensive rats: A cellular and
molecular approach. ACS Omega. 11:13558–13571. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Meloni M, Izzo V, Giurato L,
Lázaro-Martínez JL and Uccioli L: Prevalence, clinical aspects and
outcomes in a large cohort of persons with diabetic foot disease:
Comparison between neuropathic and ischemic ulcers. J Clin Med.
9:17802020. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Fitridge R, Chuter V, Mills J, Hinchliffe
R, Azuma N, Behrendt CA, Boyko EJ, Conte MS, Humphries M, Kirksey
L, et al: The intersocietal IWGDF, ESVS, SVS guidelines on
peripheral artery disease in people with diabetes and a foot ulcer.
Diabetes Metab Res Rev. 40:e36862024. View Article : Google Scholar
|
|
122
|
Dalle Carbonare L, Cominacini M, Trabetti
E, Bombieri C, Pessoa J, Romanelli MG and Valenti MT: The bone
microenvironment: New insights into the role of stem cells and cell
communication in bone regeneration. Stem Cell Res Ther. 16:1692025.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
He F, Lu T, Fang X, Feng S, Feng S, Tian
Y, Li Y, Zuo F, Deng X and Ye J: Novel extrusion-microdrilling
approach to fabricate calcium phosphate-based bioceramic scaffolds
enabling fast bone regeneration. ACS Appl Mater Interfaces.
12:32340–32351. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Schulze F, Lang A, Schoon J, Wassilew GI
and Reichert J: Scaffold guided bone regeneration for the treatment
of large segmental defects in long bones. Biomedicines. 11:3252023.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Zhu G, Zhang T, Chen M, Yao K, Huang X,
Zhang B, Li Y, Liu J, Wang Y and Zhao Z: Bone physiological
microenvironment and healing mechanism: Basis for future
bone-tissue engineering scaffolds. Bioact Mater. 6:4110–4140.
2021.PubMed/NCBI
|
|
126
|
Wang J, Wu Y, Li G, Zhou F, Wu X, Wang M,
Liu X, Tang H, Bai L, Geng Z, et al: Engineering large-scale
self-mineralizing bone organoids with bone matrix-inspired
hydroxyapatite hybrid bioinks. Adv Mater. 36:e23098752024.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Knight MN and Hankenson KD: Mesenchymal
stem cells in bone regeneration. Adv Wound Care (New Rochelle).
2:306–316. 2013. View Article : Google Scholar
|
|
128
|
Pajarinen J, Lin T, Gibon E, Kohno Y,
Maruyama M, Nathan K, Lu L, Yao Z and Goodman SB: Mesenchymal stem
cell-macrophage crosstalk and bone healing. Biomaterials.
196:80–89. 2019. View Article : Google Scholar :
|
|
129
|
Gomes JRCL, Vargas IA, Rodrigues AFA,
Gertz LC, Freitas MP, Miguens SAQ Jr, Ozkomur A and Hernandez PAG:
Micro-osteoperforation for enhancement of orthodontic movement: A
mechanical analysis using the finite element method. PLoS One.
19:e03087392024. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Brighton CT and Hunt RM: Early histologic
and ultrastructural changes in microvessels of periosteal callus. J
Orthop Trauma. 11:244–253. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Murao H, Yamamoto K, Matsuda S and Akiyama
H: Periosteal cells are a major source of soft callus in bone
fracture. J Bone Miner Metab. 31:390–398. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Chen Y, Kuang X, Zhou J, Zhen P, Zeng Z,
Lin Z, Gao W, He L, Ding Y, Liu G, et al: Proximal tibial cortex
transverse distraction facilitating healing and limb salvage in
severe and recalcitrant diabetic foot ulcers. Clin Orthop Relat
Res. 478:836–851. 2020. View Article : Google Scholar
|
|
133
|
Zhen PX, Su HJ, Yang SJ, Chen X, Lin ZM
and Liu SN: Comparison of clinical efficacy between tibial cortex
transverse transport and platelet-rich plasma treatment for severe
diabetic foot ulcers. Front Surg. 12:15079822025. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Zhao S, Guo F, Zang Y, Hu R, Yu X, Zhang
H, Xie T, Li X, Bai C, Shi H and Zhang D: The association of the
perioperative neutrophil-to-lymphocyte ratio with wound healing in
patients with Wagner grade 3 and 4 diabetic foot ulcers after
tibial cortex transverse transport surgery: A prospective
observational cohort study. Front Endocrinol (Lausanne).
15:14202322024. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Xu J, Lv K, Qiang K, Xia Z, Zhao J and Shi
N: Electrodiagnostic evidence of nerve regeneration in patients
with diabetic Charcot foot treated with proximal tibial cortex
transverse distraction. Jt Dis Relat Surg. 36:430–436. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Chen Y, Ding X, Zhu Y, Jia Z, Qi Y, Chen
M, Lu J, Kuang X, Zhou J, Su Y, et al: Effect of tibial cortex
transverse transport in patients with recalcitrant diabetic foot
ulcers: A prospective multicenter cohort study. J Orthop Translat.
36:194–204. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Hu XX, Xiu ZZ, Li GC, Zhang JY, Shu LJ,
Chen Z, Li H, Zou QF and Zhou Q: Effectiveness of transverse tibial
bone transport in treatment of diabetic foot ulcer: A systematic
review and meta-analysis. Front Endocrinol (Lausanne).
13:10953612023. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Yang Y, Li Y, Pan Q, Bai S, Wang H, Pan
XH, Ling KK and Li G: Tibial cortex transverse transport
accelerates wound healing via enhanced angiogenesis and
immunomodulation. Bone Joint Res. 11:189–199. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Giurato L, Meloni M, Izzo V and Uccioli L:
Osteomyelitis in diabetic foot: A comprehensive overview. World J
Diabetes. 8:135–142. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Dalla Paola L: Confronting a dramatic
situation: The charcot foot complicated by osteomyelitis. Int J Low
Extrem Wounds. 13:247–262. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Aragón-Sánchez J, Lázaro-Martínez JL,
Quintana-Marrero Y, Álvaro-Afonso FJ and Hernández-Herrero MJ:
Charcot neuroarthropathy triggered and complicated by
osteomyelitis. How limb salvage can be achieved. Diabet Med.
30:e229–e232. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Facchin A, Lemaire S, Toner LG, Argaw A
and Frenette J: When bone forms where it shouldn't: Heterotopic
ossification in muscle injury and disease. Int J Mol Sci.
26:75162025. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Meyers C, Lisiecki J, Miller S, Levin A,
Fayad L, Ding C, Sono T, McCarthy E, Levi B and James AW:
Heterotopic ossification: A comprehensive review. JBMR Plus.
3:e101722019. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Adejuyigbe B, Gharpure M, Wahle CF and
Kallini JR: Distraction osteogenesis: A comprehensive review. Appl
Biosci. 3:503–516. 2024. View Article : Google Scholar
|
|
145
|
Liu Z, Liu Q, Guo H, Liang J and Zhang Y:
Overview of physical and pharmacological therapy in enhancing bone
regeneration formation during distraction osteogenesis. Front Cell
Dev Biol. 10:8374302022. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Boulton AJ, Armstrong DG, Kirsner RS,
Attinger CE, Lavery LA, Lipsky BA, Mills JL and Steinberg JS:
Diagnosis and management of diabetic foot complications. American
Diabetes Association; Arlington, VA: 2018
|
|
147
|
Kim J, Nomkhondorj O, An CY, Choi YC and
Cho J: Management of diabetic foot ulcers: A narrative review. J
Yeungnam Med Sci. 40:335–342. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Schaper NC, van Netten JJ, Apelqvist J,
Bus SA, Fitridge R, Game F, Monteiro-Soares M and Senneville E;
IWGDF Editorial Board: Practical guidelines on the prevention and
management of diabetes-related foot disease (IWGDF 2023 update).
Diabetes Metab Res Rev. 40:e36572024. View Article : Google Scholar
|
|
149
|
Hingorani A, LaMuraglia GM, Henke P,
Meissner MH, Loretz L, Zinszer KM, Driver VR, Frykberg R, Carman
TL, Marston W, et al: The management of diabetic foot: A clinical
practice guideline by the Society for Vascular Surgery in
collaboration with the American Podiatric Medical Association and
the Society for Vascular Medicine. J Vasc Surg. 63(2 Suppl):
3S–21S. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Kota SK, Kota SK, Meher LK, Sahoo S,
Mohapatra S and Modi KD: Surgical revascularization techniques for
diabetic foot. J Cardiovasc Dis Res. 4:79–83. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Lei FR, Shen XF, Zhang C, Li XQ, Zhuang H
and Sang HF: Clinical efficacy of endovascular revascularization
combined with vacuum-assisted closure for the treatment of diabetic
foot. World J Diabetes. 15:1499–1508. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Huizing E, Schreve MA, Stuart JWC, de
Vries JP and Çağdaş Ü: Treatment of clinically uninfected diabetic
foot ulcers, with and without antibiotics. J Wound Care.
33:118–126. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Hinchliffe RJ, Forsythe RO, Apelqvist J,
Boyko EJ, Fitridge R, Hong JP, Katsanos K, Mills JL, Nikol S,
Reekers J, et al: Guidelines on diagnosis, prognosis, and
management of peripheral artery disease in patients with foot
ulcers and diabetes (IWGDF 2019 update). Diabetes Metab Res Rev.
36(Suppl 1): e32762020. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Arundel C, Mandefield L, Fairhurst C,
Baird K, Gkekas A, Saramago P and Chetter I; SWHSI-2 Trial
Investigators: Negative pressure wound therapy versus usual care in
patients with surgical wound healing by secondary intention in the
UK (SWHSI-2): An open-label, multicentre, parallel-group,
randomised controlled trial. Lancet. 405:1689–1699. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Tansley J, Collings R, Williams J and
Paton J: Off-loading and compression therapy strategies to treat
diabetic foot ulcers complicated by lower limb oedema: A scoping
review. J Foot Ankle Res. 16:562023. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Chang MC, Choo YJ, Park IS, Park MW and
Kim DH: Orthotic approach to prevention and management of diabetic
foot: A narrative review. World J Diabetes. 13:912–920. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Bahney CS, Zondervan RL, Allison P,
Theologis A, Ashley JW, Ahn J, Miclau T, Marcucio RS and Hankenson
KD: Cellular biology of fracture healing. J Orthop Res. 37:35–50.
2019. View Article : Google Scholar :
|
|
158
|
Wähnert D, Miersbach M, Colcuc C, Brianza
S, Vordemvenne T, Plecko M and Schwarz A: Promoting bone callus
formation by taking advantage of the time-dependent fracture gap
strain modulation. Front Surg. 11:13764412024. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Sheen JR, Mabrouk A and Garla VV: Fracture
healing overview. StatPearls [Internet]. StatPearls Publishing;
Treasure Island, FL: 2023
|
|
160
|
Ramirez GA, Manfredi AA and Maugeri N:
Misunderstandings between platelets and neutrophils build in
chronic inflammation. Front Immunol. 10:24912019. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Wang Y, Wang B, Liu D, Xu L, Hu X, Liu X,
Zheng H, Wang Z, Cheng M and Yan W: Osteoimmunology uncovered: How
macrophages and biomaterials revolutionize bone healing. Mater
Today Bio. 36:1026472025. View Article : Google Scholar
|
|
162
|
Camarena A, Kang L, Mirando AJ, Augustine
E, McMillian NS, Stinson NC, Agarwal SM, Becker ML, Hilton MJ and
Fernandez-Moure JS: Platelet-rich plasma enhances rib fracture
strength and callus formation in vivo. J Trauma Acute Care Surg.
97:884–890. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Han D, Liu W, Gong J, Ma Y and Sun Z:
Challenges and future perspectives in using mesenchymal stem cells
for efficient bone fracture healing. Front Bioeng Biotechnol.
13:15689142025. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Trompet D, Melis S, Chagin AS and Maes C:
Skeletal stem and progenitor cells in bone development and repair.
J Bone Miner Res. 39:633–654. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Liu L, Zhou N, Fu S, Wang L, Liu Y, Fu C,
Xu F, Guo W, Wu Y, Cheng J, et al: Endothelial cell-derived
exosomes trigger a positive feedback loop in
osteogenesis-angiogenesis coupling via up-regulating zinc finger
and BTB domain containing 16 in bone marrow mesenchymal stem cell.
J Nanobiotechnology. 22:7212024. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Qiu M, Li C, Cai Z, Li C, Yang K, Tulufu
N, Chen B, Cheng L, Zhuang C, Liu Z, et al: 3D biomimetic calcified
cartilaginous callus that induces type H vessels formation and
osteoclastogenesis. Adv Sci (Weinh). 10:e22070892023. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Wang H, Li Y, Li H, Yan X, Jiang Z, Feng
L, Hu W, Fan Y and Lin Sand Li G: T cell related osteoimmunology in
fracture healing: Potential targets for augmenting bone
regeneration. J Orthop Translat. 51:82–93. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Kushioka J, Chow SK, Toya M, Tsubosaka M,
Shen H, Gao Q, Li X, Zhang N and Goodman SB: Bone regeneration in
inflammation with aging and cell-based immunomodulatory therapy.
Inflamm Regen. 43:292023. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Halper J, Dolfi B, Ivanov S, Madel MB and
Blin-Wakkach C: Macrophages and osteoclasts: Similarity and
divergence between bone phagocytes. Front Immunol. 16:16838722025.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Sinder BP, Pettit AR and McCauley LK:
Macrophages: Their emerging roles in bone. J Bone Miner Res.
30:2140–2149. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Liao J, Wu T, Zhang Q, Shen P, Huang Z,
Wang J, Zhang P, Lin S, Pi J, Zhang N, et al: TGF-β/BMP signaling
in skeletal biology: Molecular mechanisms, regulatory networks, and
therapeutic implications in development, regeneration, and disease.
Bone Res. 14:62026. View Article : Google Scholar
|
|
172
|
Li S, Cai X, Guo J, Li X, Li W, Liu Y and
Qi M: Cell communication and relevant signaling pathways in
osteogenesis-angiogenesis coupling. Bone Res. 13:452025. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Wang L, Ruan M, Bu Q and Zhao C: Signaling
pathways driving MSC osteogenesis: Mechanisms, regulation, and
translational applications. Int J Mol Sci. 26:13112025. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Hadjiargyrou M, Lombardo F, Zhao S, Ahrens
W, Joo J, Ahn H, Jurman M, White DW and Rubin CT: Transcriptional
profiling of bone regeneration. Insight into the molecular
complexity of wound repair. J Biol Chem. 277:30177–30182. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Hadjiargyrou M, Kotsiopriftis M, Lauzier
D, Hamdy RC and Kloen P: Activation of Wnt signaling in human
fracture callus and nonunion tissues. Bone Rep. 22:1017802024.
View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Haffner-Luntzer M, Ragipoglu D, Ahmad M,
Schoppa A, Steppe L, Fischer V, Luther J, Yorgan T, Bockamp E,
Amling M, et al: Wnt1 boosts fracture healing by enhancing bone
formation in the fracture callus. J Bone Miner Res. 38:749–764.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Pelullo M, Zema S, Nardozza F, Checquolo
S, Screpanti I and Bellavia D: Wnt, Notch, and TGF-β pathways
impinge on hedgehog signaling complexity: An open window on cancer.
Front Genet. 10:7112019. View Article : Google Scholar
|
|
178
|
Chen W, Wu P, Yu F, Luo G, Qing L and Tang
J: HIF-1α regulates bone homeostasis and angiogenesis,
participating in the occurrence of bone metabolic diseases. Cells.
11:35522022. View Article : Google Scholar
|
|
179
|
Grosso A, Burger MG, Lunger A, Schaefer
DJ, Banfi A and Di Maggio N: It takes two to tango: Coupling of
angiogenesis and osteogenesis for bone regeneration. Front Bioeng
Biotechnol. 5:682017. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Ballhause TM, Jiang S, Baranowsky A,
Brandt S, Mertens PR, Frosch KH, Yorgan T and Keller J: Relevance
of notch signaling for bone metabolism and regeneration. Int J Mol
Sci. 22:13252021. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Li P, Alenazi KKK, Dally J, Woods EL,
Waddington RJ and Moseley R: Role of oxidative stress in impaired
type II diabetic bone repair: Scope for antioxidant therapy
intervention? Front Dent Med. 5:14640092024. View Article : Google Scholar
|
|
182
|
Sheng N, Xing F, Wang J, Zhang QY, Nie R,
Li-Ling J, Duan X and Xie HQ: Recent progress in bone-repair
strategies in diabetic conditions. Mater Today Bio. 23:1008352023.
View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Lin X, Patil S, Gao YG and Qian A: The
bone extracellular matrix in bone formation and regeneration. Front
Pharmacol. 11:7572020. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Ramasamy SK, Kusumbe AP, Wang L and Adams
RH: Endothelial Notch activity promotes angiogenesis and
osteogenesis in bone. Nature. 507:376–380. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Song S, Zhang G, Chen X, Zheng J, Liu X,
Wang Y, Chen Z, Wang Y, Song Y and Zhou Q: HIF-1α increases the
osteogenic capacity of ADSCs by coupling angiogenesis and
osteogenesis via the HIF-1α/VEGF/AKT/mTOR signaling pathway. J
Nanobiotechnology. 21:2572023. View Article : Google Scholar
|
|
186
|
Riddle RC, Khatri R, Schipani E and
Clemens TL: Role of hypoxia-inducible factor-1alpha in
angiogenic-osteogenic coupling. J Mol Med (Berl). 87:583–590. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Peng Y, Wu S, Li Y and Crane JL: Type H
blood vessels in bone modeling and remodeling. Theranostics.
10:426–436. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Cassuto J, Folestad A, Göthlin J, Malchau
H and Kärrholm J: VEGF-A, -C, -D, VEGFR1, -2, -3, PDGF-BB and FGF-2
join forces to induce vascular and lymphatic angiogenesis during
bone healing of hip implants. Bone Rep. 26:1018562025. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Hu K and Olsen BR: Osteoblast-derived VEGF
regulates osteoblast differentiation and bone formation during bone
repair. J Clin Invest. 126:509–526. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Perepletchikova D and Malashicheva A:
Communication between endothelial cells and osteoblasts in
regulation of bone homeostasis: Notch players. Stem Cell Res Ther.
16:562025. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Issabekova A, Kudaibergen G, Sekenova A,
Dairov A, Sarsenova M, Mukhlis S, Temirzhan A, Baidarbekov M,
Eskendirova S and Ogay V: The therapeutic potential of pericytes in
bone tissue regeneration. Biomedicines. 12:212023. View Article : Google Scholar
|
|
192
|
Zhu S, Yao F, Qiu H, Zhang G, Xu H and Xu
J: Coupling factors and exosomal packaging microRNAs involved in
the regulation of bone remodelling. Biol Rev Camb Philos Soc.
93:469–480. 2018. View Article : Google Scholar
|
|
193
|
Hankenson KD, Dishowitz M, Gray C and
Schenker M: Angiogenesis in bone regeneration. Injury. 42:556–561.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Lim JC, Ko KI, Mattos M, Fang M, Zhang C,
Feinberg D, Sindi H, Li S, Alblowi J, Kayal RA, et al: TNFα
contributes to diabetes impaired angiogenesis in fracture healing.
Bone. 99:26–38. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Qin Q, Liu Y, Yang Z, Aimaijiang M, Ma R,
Yang Y, Zhang Y and Zhou Y: Hypoxia-inducible factors signaling in
osteogenesis and skeletal repair. Int J Mol Sci. 23:112012022.
View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Souza ATP, Freitas GP, Lopes HB, Weffort
D, Adolpho LF, Gomes MPO, Oliveira FS, Almeida ALG, Beloti MM and
Rosa AL: Diabetes mellitus impairs the bone regeneration capacity
of mesenchymal stromal cell-based therapy. Arch Med Res.
56:1032342025. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Cai F and Liu Y, Liu K, Zhao R, Chen W,
Yusufu A and Liu Y: Diabetes mellitus impairs bone regeneration and
biomechanics. J Orthop Surg Res. 18:1692023. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Li Z, Yue M and Zhou Y: Advances in
material-based strategies for diabetic bone regeneration. Stem
Cells Transl Med. 13:243–254. 2024. View Article : Google Scholar :
|
|
199
|
Zgutka K, Tkacz M, Tomasiak P and
Tarnowski M: A role for advanced glycation end products in
molecular ageing. Int J Mol Sci. 24:98812023. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Twarda-Clapa A, Olczak A, Białkowska AM
and Koziołkiewicz M: Advanced glycation end-products (AGEs):
Formation, chemistry, classification, receptors, and diseases
related to AGEs. Cells. 11:13122022. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Guarneri F, Custurone P, Papaianni V and
Gangemi S: Involvement of RAGE and oxidative stress in inflammatory
and infectious skin diseases. Antioxidants (Basel). 10:822021.
View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Yin M, Zhang Y, Yu H and Li X: Role of
hyperglycemia in the senescence of mesenchymal stem cells. Front
Cell Dev Biol. 9:6654122021. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Shen Y, Zhang Y, Zhou Z, Wang J, Han D,
Sun J, Chen G, Tang Q, Sun W and Chen L: Dysfunction of macrophages
leads to diabetic bone regeneration deficiency. Front Immunol.
13:9904572022. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Sheppard AJ, Barfield AM, Barton S and
Dong Y: Understanding reactive oxygen species in bone regeneration:
A glance at potential therapeutics and bioengineering applications.
Front Bioeng Biotechnol. 10:8367642022. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Frade BB, Dias RB, Gemini Piperni S and
Bonfim DC: The role of macrophages in fracture healing: A narrative
review of the recent updates and therapeutic perspectives. Stem
Cell Investig. 10:42023. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Bahney CS, Hu DP, Miclau T III and
Marcucio RS: The multifaceted role of the vasculature in
endochondral fracture repair. Front Endocrinol (Lausanne). 6:42015.
View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Yang J and Liu Z: Mechanistic pathogenesis
of endothelial dysfunction in diabetic nephropathy and retinopathy.
Front Endocrinol (Lausanne). 13:8164002022. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Booth SL, Centi AJ and Gundberg C: Bone as
an endocrine organ relevant to diabetes. Curr Diab Rep. 14:5562014.
View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Figeac F, Tencerova M, Ali D, Andersen TL,
Appadoo DRC, Kerckhofs G, Ditzel N, Kowal JM, Rauch A and Kassem M:
Impaired bone fracture healing in type 2 diabetes is caused by
defective functions of skeletal progenitor cells. Stem Cells.
40:149–164. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Brown ML, Yukata K, Farnsworth CW, Chen
DG, Awad H, Hilton MJ, O'Keefe RJ, Xing L, Mooney RA and Zuscik MJ:
Delayed fracture healing and increased callus adiposity in a
C57BL/6J murine model of obesity-associated type 2 diabetes
mellitus. PLoS One. 9:e996562014. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Zondervan RL, Capobianco CA, Jenkins DC,
Reicha JD, Fredrick L, Lam C, Schmanski JT, Isenberg JS, Ahn J,
Marcucio RS and Hankenson KD: CD47 is required for mesenchymal
progenitor proliferation and fracture repair. Bone Res. 13:292025.
View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Wee H, Khajuria DK, Kamal F, Lewis GS and
Elbarbary RA: Assessment of bone fracture healing using
micro-computed tomography. J Vis Exp. 9: View Article : Google Scholar : 2022.
|
|
213
|
Xu C, Sellke FW and Abid MR: Assessments
of microvascular function in organ systems. Am J Physiol Heart Circ
Physiol. 322:H891–H905. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Xu T, Luo Y, Kong F, Lv B, Zhao S, Chen J,
Liu W, Cheng L, Zhou Z, Zhou Z, et al: GIT1 is critical for
formation of the CD31(hi)Emcn(hi) vessel subtype in coupling
osteogenesis with angiogenesis via modulating preosteoclasts
secretion of PDGF-BB. Bone. 122:218–230. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Zhang J, Pan J and Jing W: Motivating role
of type H vessels in bone regeneration. Cell Prolif. 53:e128742020.
View Article : Google Scholar :
|
|
216
|
Wang L, Zhou F, Zhang P, Wang H, Qu Z, Jia
P, Yao Z, Shen G, Li G, Zhao G, et al: Human type H vessels are a
sensitive biomarker of bone mass. Cell Death Dis. 8:e27602017.
View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Wang F, Ye Y, Zhang Z, Teng W, Sun H, Chai
X, Zhou X, Chen J, Mou H, Eloy Y, et al: PDGFR in PDGF-BB/PDGFR
signaling pathway does orchestrates osteogenesis in a temporal
manner. Research (Wash D C). 6:00862023.PubMed/NCBI
|
|
218
|
Guntur AR and Rosen CJ: Bone as an
endocrine organ. Endocr Pract. 18:758–762. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Wang J, Yang X, Zhou T, Ma H, Yuan X, Yan
S and Wang S: Microenvironment of diabetic foot ulcers:
Implications for healing and therapeutic strategies. J Res Med Sci.
30:192025. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Shen T, Dai K, Zhang S, Wang J and Liu C:
Injured bone-triggered osteokines secretion promotes diabetic wound
healing. Bone Res. 13:832025. View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Wang B, Zhao G, Zhang J, Chen W, Yang S
and Sun Y: Advances in stem cell therapy for diabetic foot ulcers.
Diabetes Metab Syndr Obes. 18:4021–4034. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Florencio-Silva R, Sasso GR, Sasso-Cerri
E, Simões MJ and Cerri PS: Biology of bone tissue: Structure,
function, and factors that influence bone cells. Biomed Res Int.
2015:4217462015. View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Tiwari R, Singh S, Bajpai M, Verma N and
Verma S: Impact of osteocalcin on glycemic regulation and insulin
sensitivity in type 2 diabetes mellitus patients. Cureus.
16:e716752024.PubMed/NCBI
|
|
224
|
Lei H, Liu J, Wang W, Yang X, Feng Z, Zang
P, Lu B and Shao J: Association between osteocalcin, a pivotal
marker of bone metabolism, and secretory function of islet beta
cells and alpha cells in Chinese patients with type 2 diabetes
mellitus: An observational study. Diabetol Metab Syndr. 14:1602022.
View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Lin X, Brennan-Speranza TC, Levinger I and
Yeap BB: Undercarboxylated osteocalcin: Experimental and human
evidence for a role in glucose homeostasis and muscle regulation of
insulin sensitivity. Nutrients. 10:8472018. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Zwakenberg SR, Gundberg CM, Spijkerman AM,
van der ADL, van der Schouw YT and Beulens JW: Osteocalcin is not
associated with the risk of type 2 diabetes: Findings from the
EPIC-NL Study. PLoS One. 10:e01386932015. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Booth SL, Centi A, Smith SR and Gundberg
C: The role of osteocalcin in human glucose metabolism: Marker or
mediator? Nat Rev Endocrinol. 9:43–55. 2013. View Article : Google Scholar
|
|
228
|
Hwang YC, Jee JH, Jeong IK, Ahn KJ, Chung
HY and Lee MK: Circulating osteocalcin level is not associated with
incident type 2 diabetes in middle-aged male subjects: Mean
8.4-year retrospective follow-up study. Diabetes Care.
35:1919–1924. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Wang JS, Mazur CM and Wein MN: Sclerostin
and osteocalcin: Candidate bone-produced hormones. Front Endocrinol
(Lausanne). 12:5841472021. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Holdsworth G, Roberts SJ and Ke HZ: Novel
actions of sclerostin on bone. J Mol Endocrinol. 62:R167–R185.
2019. View Article : Google Scholar
|
|
231
|
Wu X, Ai Y, He Y, Ma D, Li X, Huang X,
Cheng R and Wang B: Localized sclerostin accumulation in osteocyte
lacunar-canalicular system is associated with cortical bone
microstructural alterations and bone fragility in db/db male mice.
Front Cell Dev Biol. 13:15627642025. View Article : Google Scholar : PubMed/NCBI
|
|
232
|
Ke HZ, Richards WG, Li X and Ominsky MS:
Sclerostin and Dickkopf-1 as therapeutic targets in bone diseases.
Endocr Rev. 33:747–783. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Monaghan MG, Borah R, Thomsen C and Browne
S: Thou shall not heal: Overcoming the non-healing behaviour of
diabetic foot ulcers by engineering the inflammatory
microenvironment. Adv Drug Deliv Rev. 203:1151202023. View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Fulzele K, Lai F, Dedic C, Saini V, Uda Y,
Shi C, Tuck P, Aronson JL, Liu X, Spatz JM, et al:
Osteocyte-secreted wnt signaling inhibitor sclerostin contributes
to beige adipogenesis in peripheral fat depots. J Bone Miner Res.
32:373–384. 2017. View Article : Google Scholar :
|
|
235
|
Romero-Díaz C, Duarte-Montero D,
Gutiérrez-Romero SA and Mendivil CO: Diabetes and bone fragility.
Diabetes Ther. 12:71–86. 2021. View Article : Google Scholar :
|
|
236
|
Yu S, Li D, Zhang N, Ni S, Sun M, Wang L,
Xiao H, Liu D, Liu J, Yu Y, et al: Drug discovery of sclerostin
inhibitors. Acta Pharm Sin B. 12:2150–2170. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
237
|
Kocełak P, Puzianowska-Kuźnicka M,
Olszanecka-Glinianowicz M and Chudek J: Wnt signaling pathway and
sclerostin in the development of atherosclerosis and vascular
calcification. Adv Clin Exp Med. 33:519–532. 2024. View Article : Google Scholar
|
|
238
|
Erben RG: Physiological actions of
fibroblast growth factor-23. Front Endocrinol (Lausanne).
9:2672018. View Article : Google Scholar : PubMed/NCBI
|
|
239
|
Kurpas A, Supeł K, Idzikowska K and
Zielińska M: FGF23: A review of its role in mineral metabolism and
renal and cardiovascular disease. Dis Markers. 2021:88212922021.
View Article : Google Scholar : PubMed/NCBI
|
|
240
|
Bouma-de Krijger A and Vervloet MG:
Fibroblast growth factor 23: Are we ready to use it in clinical
practice? J Nephrol. 33:509–527. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
241
|
Kojima F, Uchida K, Ogawa T, Tanaka Y and
Nitta K: Plasma levels of fibroblast growth factor-23 and mineral
metabolism in diabetic and non-diabetic patients on chronic
hemodialysis. Int Urol Nephrol. 40:1067–1074. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
242
|
Tanaka H, Hamano T, Fujii N, Tomida K,
Matsui I, Mikami S, Nagasawa Y, Ito T, Moriyama T, Horio M, et al:
The impact of diabetes mellitus on vitamin D metabolism in
predialysis patients. Bone. 45:949–955. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
243
|
Ivey-Miranda JB, Stewart B, Cox ZL,
McCallum W, Maulion C, Gleason O, Meegan G, Amatruda JG,
Moreno-Villagomez J, Mahoney D, et al: FGF-23 (Fibroblast Growth
Factor-23) and cardiorenal interactions. Circ Heart Fail.
14:e0083852021. View Article : Google Scholar : PubMed/NCBI
|
|
244
|
van der Vaart A, Yeung SMH, van Dijk PR,
Bakker SJL and de Borst MH: Phosphate and fibroblast growth factor
23 in diabetes. Clin Sci (Lond). 135:1669–1687. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
245
|
De Leon-Oliva D, Barrena-Blázquez S,
Jiménez-Álvarez L, Fraile-Martinez O, García-Montero C,
López-González L, Torres-Carranza D, García-Puente LM, Carranza ST,
Álvarez-Mon MÁ, et al: The RANK-RANKL-OPG System: A multifaceted
regulator of homeostasis, immunity, and cancer. Medicina (Kaunas).
59:17522023. View Article : Google Scholar : PubMed/NCBI
|
|
246
|
Ma T, Zhang T, Peng C, Liu K, Xiong Y,
Chen K, Peng N, Wei Z, Kuang J and Ou L: Immune cells: the key
mediator between the gut microbiota and osteoporosis. Front
Immunol. 16:16800212025. View Article : Google Scholar : PubMed/NCBI
|
|
247
|
Zhang X, Sun Q, Xie X, Luo M, Zan J and
Cong Z: Epimedin B protects against bone loss and inflammation in
diabetic osteoporosis rats by regulating OPG/RANKL pathway. J
Orthop Surg Res. 20:4032025. View Article : Google Scholar : PubMed/NCBI
|
|
248
|
Walsh MC and Choi Y: Biology of the
RANKL-RANK-OPG System in immunity, bone, and beyond. Front Immunol.
5:5112014. View Article : Google Scholar : PubMed/NCBI
|
|
249
|
Chairatnathrongporn R, Tansriratanawong K,
Santiprabhob J, Boriboonhirunsarn C and Promsudthi A: Salivary gene
expression of RANK, RANKL, and OPG in type 1 diabetes mellitus and
periodontal disease patients. J Int Soc Prev Community Dent.
12:603–611. 2022. View Article : Google Scholar
|
|
250
|
Greco T, Mascio A, Comisi C, Polichetti C,
Caravelli S, Mosca M, Mondanelli N, Troiano E, Maccauro G and
Perisano C: RANKL-RANK-OPG pathway in charcot diabetic foot:
Pathophysiology and clinical-therapeutic implications. Int J Mol
Sci. 24:30142023. View Article : Google Scholar : PubMed/NCBI
|
|
251
|
Lu J, Hu D, Zhang Y, Ma C, Shen L and
Shuai B: Current comprehensive understanding of denosumab (the
RANKL neutralizing antibody) in the treatment of bone metastasis of
malignant tumors, including pharmacological mechanism and clinical
trials. Front Oncol. 13:11338282023. View Article : Google Scholar : PubMed/NCBI
|
|
252
|
Liu ZX, Huang MZ, Ding MC, Ma XP and Jiang
N: Therapeutic options and prognostic risk factors in diabetic foot
osteomyelitis: A narrative review. Int J Surg. 112:4799–4825. 2026.
View Article : Google Scholar
|
|
253
|
Narita K, Ohori F, Marahleh A, Ma J, Ren
J, Lin A, Fan Z, Murakami K and Kitaura H: Lipocalin-2 upregulation
in hypoxic murine osteocytes enhances RANKL-induced
osteoclastogenesis. Sci Rep. 16:45122026. View Article : Google Scholar : PubMed/NCBI
|
|
254
|
Mosialou I, Shikhel S, Luo N, Petropoulou
PI, Panitsas K, Bisikirska B, Rothman NJ, Tenta R, Cariou B, Wargny
M, et al: Lipocalin-2 counteracts metabolic dysregulation in
obesity and diabetes. J Exp Med. 217:e201912612020. View Article : Google Scholar : PubMed/NCBI
|
|
255
|
Mosialou I, Shikhel S, Liu JM, Maurizi A,
Luo N, He Z, Huang Y, Zong H, Friedman RA, Barasch J, et al:
MC4R-dependent suppression of appetite by bone-derived lipocalin 2.
Nature. 543:385–390. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
256
|
Jaberi SA, Cohen A, D'Souza C, Abdulrazzaq
YM, Ojha S, Bastaki S and Adeghate EA: Lipocalin-2: Structure,
function, distribution and role in metabolic disorders. Biomed
Pharmacother. 142:1120022021. View Article : Google Scholar : PubMed/NCBI
|
|
257
|
Krizanac M, Mass Sanchez PB, Weiskirchen R
and Schröder SK: Overview of the expression patterns and roles of
Lipocalin 2 in the reproductive system. Front Endocrinol
(Lausanne). 15:13656022024. View Article : Google Scholar : PubMed/NCBI
|
|
258
|
Tan Q, Zhang C, Rao X, Wan W, Lin W, Huang
S, Ying J, Lin Y and Hua F: The interaction of lipocalin-2 and
astrocytes in neuroinflammation: Mechanisms and therapeutic
application. Front Immunol. 15:13587192024. View Article : Google Scholar : PubMed/NCBI
|
|
259
|
Sólis-Suarez DL, Cifuentes-Mendiola SE,
González-Alva P, Rodríguez-Hernández AP, Martínez-Dávalos A,
Llamosas-Hernandez FE, Godínez-Victoria M and García-Hernández AL:
Lipocalin-2 as a fundamental protein in type 2 diabetes and
periodontitis in mice. J Periodontol. 96:369–382. 2025. View Article : Google Scholar :
|
|
260
|
Daoud MS, Alshareef FH, Alnaami AM, Amer
OE, Hussain SD and Al-Daghri NM: Prospective changes in lipocalin-2
and adipocytokines among adults with obesity. Sci Rep.
15:287942025. View Article : Google Scholar : PubMed/NCBI
|
|
261
|
Ernesto CS, Laura SD, Obed PI, David AR,
Eloy GJ and Lilia GA: LCN2 blockade mitigating metabolic
dysregulation and redefining appetite control in type 2 diabetes.
Metab Brain Dis. 40:972025. View Article : Google Scholar : PubMed/NCBI
|
|
262
|
Xie Z, Wang X, Luo X, Yan J, Zhang J, Sun
R, Luo A and Li S: Activated AMPK mitigates diabetes-related
cognitive dysfunction by inhibiting hippocampal ferroptosis.
Biochem Pharmacol. 207:1153742023. View Article : Google Scholar
|
|
263
|
Moon Y, Lee MH, Fujii N, Fujii T and Choi
JW: Lipocalin-2 levels, insulin resistance, and urinary albumin
excretion in type 2 diabetes subgroups. Clin Lab. 69:2023.
View Article : Google Scholar : PubMed/NCBI
|
|
264
|
Li DQ, Wan QL, Pathak JL and Li ZB:
Platelet-derived growth factor BB enhances osteoclast formation and
osteoclast precursor cell chemotaxis. J Bone Miner Metab.
35:355–365. 2017. View Article : Google Scholar
|
|
265
|
Chen Y, Jiang L, Lyu K, Lu J, Long L, Wang
X, Liu T and Li S: A promising candidate in tendon healing
events-PDGF-BB. Biomolecules. 12:15182022. View Article : Google Scholar : PubMed/NCBI
|
|
266
|
Xie H, Cui Z, Wang L, Xia Z, Hu Y, Xian L,
Li C, Xie L, Crane J, Wan M, et al: PDGF-BB secreted by
preosteoclasts induces angiogenesis during coupling with
osteogenesis. Nat Med. 20:1270–1278. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
267
|
Slade HB, Lynch SE and Dickerson JE Jr:
Safety of up to 140 daily applications of recombinant human
platelet-derived growth factor (rhPDGF-BB) onto skin wounds:
Unboxing the evidence. Wound Repair Regen. 33:e701082025.
View Article : Google Scholar : PubMed/NCBI
|
|
268
|
Li G, Qi M and Liang S: The role of
bone-derived factors in bone and muscle communication. Front
Endocrinol (Lausanne). 16:17021042025. View Article : Google Scholar : PubMed/NCBI
|
|
269
|
Alamri T, Alhumaydhi F and Wasti AZ:
Association between uncarboxylated osteocalcin, blood glucose, and
BMI among Saudi diabetic patients: An evaluation study. Eur Rev Med
Pharmacol Sci. 28:2005–2013. 2024.PubMed/NCBI
|
|
270
|
Bilotta FL, Arcidiacono B, Messineo S,
Greco M, Chiefari E, Britti D, Nakanishi T, Foti DP and Brunetti A:
Insulin and osteocalcin: Further evidence for a mutual cross-talk.
Endocrine. 59:622–632. 2018. View Article : Google Scholar :
|
|
271
|
Leanza G, Cannata F, Faraj M, Pedone C,
Viola V, Tramontana F, Pellegrini N, Vadalà G, Piccoli A, Strollo
R, et al: Bone canonical Wnt signaling is downregulated in type 2
diabetes and associates with higher advanced glycation end-products
(AGEs) content and reduced bone strength. Elife. 12:RP904372024.
View Article : Google Scholar : PubMed/NCBI
|
|
272
|
Lin X, Onda DA, Yang CH, Lewis JR,
Levinger I and Loh K: Roles of bone-derived hormones in type 2
diabetes and cardiovascular pathophysiology. Mol Metab.
40:1010402020. View Article : Google Scholar : PubMed/NCBI
|
|
273
|
Cipriani C, Colangelo L, Santori R,
Renella M, Mastrantonio M, Minisola S and Pepe J: The interplay
between bone and glucose metabolism. Front Endocrinol (Lausanne).
11:1222020. View Article : Google Scholar : PubMed/NCBI
|
|
274
|
Napoli N, Strollo R, Paladini A, Briganti
SI, Pozzilli P and Epstein S: The alliance of mesenchymal stem
cells, bone, and diabetes. Int J Endocrinol. 2014:6907832014.
View Article : Google Scholar : PubMed/NCBI
|
|
275
|
Xu Y, Shen L, Liu L, Zhang Z and Hu W:
Undercarboxylated osteocalcin and its associations with bone
mineral density, bone turnover markers, and prevalence of
osteopenia and osteoporosis in Chinese population: A
cross-sectional study. Front Endocrinol (Lausanne). 13:8439122022.
View Article : Google Scholar : PubMed/NCBI
|
|
276
|
Eller-Vainicher C, Cairoli E, Grassi G,
Grassi F, Catalano A, Merlotti D, Falchetti A, Gaudio A, Chiodini I
and Gennari L: Pathophysiology and management of type 2 diabetes
mellitus bone fragility. J Diabetes Res. 2020:76089642020.
View Article : Google Scholar : PubMed/NCBI
|
|
277
|
Gallagher KA, Mills JL, Armstrong DG,
Conte MS, Kirsner RS, Minc SD, Plutzky J, Southerland KW,
Tomic-Canic M; American Heart Association Council on Peripheral
Vascular Disease; et al: Current status and principles for the
treatment and prevention of diabetic foot ulcers in the
cardiovascular patient population: A scientific statement from the
American heart association. Circulation. 149:e232–e253. 2024.
View Article : Google Scholar :
|
|
278
|
Liu Y, Jiang C, Zhang X, Ma B, Ding Y, Jin
Y, Liu Y, Li L and Zhao C: Anterior superior iliac spine
distraction for severe and recalcitrant diabetic foot ulcers.
Injury. 54:778–783. 2023. View Article : Google Scholar
|
|
279
|
Jahn D, Knapstein PR, Otto E, Köhli P,
Sevecke J, Graef F, Graffmann C, Fuchs M, Jiang S, Rickert M, et
al: Increased β(2)-adrenergic signaling promotes fracture healing
through callus neovascularization in mice. Sci Transl Med.
16:eadk91292024. View Article : Google Scholar
|
|
280
|
Al-Karagholi MA, Kalatharan V, Fagerberg
PS and Amin FM: The vascular role of CGRP: A systematic review of
human studies. Front Neurol. 14:12047342023. View Article : Google Scholar : PubMed/NCBI
|
|
281
|
He T, Qin L, Chen S, Huo S, Li J, Zhang F,
Yi W, Mei Y and Xiao G: Bone-derived factors mediate crosstalk
between skeletal and extra-skeletal organs. Bone Res. 13:492025.
View Article : Google Scholar : PubMed/NCBI
|
|
282
|
Takashi Y and Kawanami D: The role of
bone-derived hormones in glucose metabolism, diabetic kidney
disease, and cardiovascular disorders. Int J Mol Sci. 23:23762022.
View Article : Google Scholar : PubMed/NCBI
|
|
283
|
Ferreira S, Gardy S, Linardatos J,
Churchward-Venne T, Josse A, Correa J and Gibbs J: Acute effects of
dietary whey protein supplementation after endurance exercise on
serum osteokine and inflammatory cytokine concentrations in
endurance runners. Appl Physiol Nutr Metab. 51:1–15. 2026.
View Article : Google Scholar : PubMed/NCBI
|
|
284
|
Yang Y, Ruan W, Li J, Dang R, An H, Zhao
W, Zhao Y, Xu L and Tan H: Evaluation of the efficacy of
retroperitoneoscopic debridement for lumbar tuberculosis: A
retrospective study and preliminary results. Int Orthop. Apr
23–2026.Epub ahead of print. View Article : Google Scholar :
|
|
285
|
Liu D, Yu S, Zhang Y, Li Q, Kang P, Wang
L, Han R, Cheng D, Chen A, Hou X, et al: Fibroblast growth factor
23 predicts incident diabetic kidney disease: A 4.6-year
prospective study. Diabetes Obes Metab. 27:2232–2241. 2025.
View Article : Google Scholar : PubMed/NCBI
|