Introduction
Cerebral small vessel disease (CSVD) is a complex
neurological condition characterized by compromised blood flow and
structural abnormalities in the small blood vessels of the brain
(1). The clinical manifestations
of CSVD include cognitive decline, stroke and severe neurological
deficit (2). The underlying
mechanisms that trigger CSVD are multifaceted, entailing an
intricate interplay among vascular conditions, inflammation and
neuronal integrity (3,4). The current treatments (antiplatelet
agents, statins and risk factor management) for CSVD often fail to
yield satisfactory outcomes, warranting the need for a
comprehensive understanding of the pathogenic mechanisms involved
to develop effective and safe treatments.
The high-temperature requirement serine peptidase A1
(HTRA1) belongs to the serine protease family, which is involved in
proteolysis and cell fate determination (5). It exhibits dual activity as a
chaperone and serine protease (6). HTRA1 plays crucial roles in
physiological processes, including extracellular matrix remodeling
by hydrolyzing fibronectin, decorin, aggrecan and elastin (7-9)
and regulating growth factors such as transforming growth factor-β
(TGF-β) (10,11). The expression levels of HTRA1
vary across tissues and under various conditions, notably in blood
vessels (12,13), which are involved in numerous
physiological processes. Recently, HTRA1 has been implicated in
age-associated macular degeneration, osteoarthritis, cancer, and
CSVD (14,15). Several heterozygous and
homozygous HTRA1 mutations cause CSVD (16,17). Heterozygous HTRA1
mutations cause cerebral autosomal dominant arteriopathy with
subcortical infarcts and leukoencephalopathy (CADASIL) type 2, also
known as heterozygous HTRA1 mutation carrier (mc) (18). Previous research has primarily
focused on case reports and potential pathogenic mechanisms
associated with mutation sites (19,20). Partial site mutations in
HTRA1 can lead to the downregulation of HTRA1 mRNA
and protein expression, resulting in reduced HTRA1 protease
activity (21-23). Decreased HTRA1 protease activity
is the primary pathogenic mechanism underlying HTRA1-associated
CSVD. However, the endothelial-specific mechanisms linking
HTRA1 mutations to CSVD phenotypes remain elusive and the
functional consequences of partial HTRA1 deficiency on
cerebrovascular homeostasis are underexplored (17).
Materials and methods
Subjects
Between January 2018 and December 2023, four
patients (three male, one female; age range, 44-54 years; median
age, 47.5 years) diagnosed as heterozygous HTRA1mcs and four
healthy controls (HCs; three male, one female; age range, 43-53
years; median age, 48 years) were recruited from the Department of
Neurology, First Hospital of Shanxi Medical University (Taiyuan,
China). The inclusion criteria were as follows: i) confirmation of
a heterozygous HTRA1 mutation by genetic testing; ii)
presence of typical CSVD-related neuroimaging features on brain
MRI; iii) age between 18 and 60 years; and iv) provision of written
informed consent. The exclusion criteria were as follows: i) other
hereditary or acquired causes of CSVD; ii) presence of severe
systemic diseases; iii) history of stroke not attributable to CSVD;
and iv) any contraindication to MRI. Inclusion criteria for HCs
were: i) absence of HTRA1 mutations confirmed by genetic
screening; ii) no clinical symptoms or neuroimaging features
suggestive of CSVD; iii) age and sex matched to the patient group;
and iv) provision of written informed consent. Exclusion criteria
for HCs were: i) any history of neurological disorders; ii)
significant systemic illnesses; or iii) any contraindications to
MRI; iv) any abnormal findings on brain MRI suggestive of CSVD or
other intracranial pathologies.
All patients exhibited typical CSVD imaging
features, with brain magnetic resonance imaging (MRI) lesions
corresponding to the clinical symptoms. The brain MRI scans were
performed using a 3.0-Tesla Siemens MAGNETOM Prisma scanner.
T2-weighted fluid-attenuated inversion recovery (FLAIR) sequences
were acquired with the following parameters: Repetition time, 9,000
msec, echo time, 94 msec, and inversion time, 2,500 msec. Clinical
and imaging data were assessed from the probands and their family
members. Cognitive function was evaluated using the Montreal
Cognitive Assessment (MoCA) and the Mini-Mental State Examination
(MMSE). The study was approved by the ethics committee of the First
Hospital of Shanxi Medical University (approval no. KYLL-2024-161),
and all participants provided written informed consent. Peripheral
blood samples were collected from patients for genetic analysis and
HCs.
Sanger sequencing
Genomic DNA was analyzed via Sanger sequencing of
DNA extracted from peripheral blood collected from the probands,
family members and HCs in anticoagulant EDTA-coated tubes. Genomic
DNA was extracted using the Ezup Column Blood Genomic DNA
Extraction kit (Sangon Biotech, cat. no. B518253) according to the
manufacturer's instructions. PCR was performed using Taq Plus DNA
Polymerase (Sangon Biotech, cat. no B600090). Thermocycling
conditions were as follows: Initial denaturation at 95°C for 5 min;
followed by 40 cycles of denaturation at 95°C for 30 sec, annealing
at 58°C for 30 sec, and elongation at 72°C for 30 sec; with a final
extension at 72°C for 10 min. The PCR products were detected by 1%
agarose gel electrophoresis. Following purification by gel
extraction, the products were sequenced on a 3730XL sequencer
(Applied Biosystems). Sanger sequencing and primer synthesis
(Table I) were performed by
Sangon Biotech Co., Ltd. The pathogenicity of the HTRA1
heterozygous mutations c.854C>T (p.P285L) and c.905G>A
(p.R302Q) was assessed using bioinformatics tools, including
Polyphen-2 (http://genetics.bwh.harvard.edu/pph2), Sorting
Intolerant From Tolerant (/sift.bii.a-star.edu.sg), Mutation Taster (https://www.mutationtaster.org), Combined
Annotation Dependent Depletion (https://cadd.gs.washington.edu/score), non-synonymous
single nucleotide polymorphism (nsSNP) Analyzer (https://snpanalyzer.uthsc.edu) and Mutation Assessor
(http://mutationassessor.org/r3).
According to the Ensembl database (https://www.ensembl.org), both mutations were
identified as single nucleotide polymorphisms, specifically
c.854C>T (rs587776446) and c.905G>A (rs2133449474). The
aforementioned tools predicted that both mutations were deleterious
and likely pathogenic, indicating a notable impact on HTRA1
function and potential involvement in associated phenotypic
mechanisms.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Primer | Sequence,
5'→3' | Product size,
bp |
|---|
| HTRA1 c.854
C>T | Forward:
TTGGTTTTCCATGATATCTGTGC | 275 |
| Reverse:
GTTGATGATGGCGTCGGTCT |
| HTRA1 c.905
G>A | Forward:
GTGGTCGCCATCGGAAGC | 384 |
| Reverse:
ATCACCCTAAAGCACCCAATAC |
RNA-seq
Total RNA was extracted using TRNzol Universal
Reagent (cat. no. DP424; TIANGEN Biotech Co., Ltd.) according to
the manufacturer's instructions. RNA integrity was assessed using
the Agilent 5400 Fragment Analyzer System (Agilent Technologies),
and the RNA integrity number (RIN) was determined for each sample.
Sequencing libraries were prepared and subjected to paired-end (PE)
sequencing with a read length of 150 bp (2×150 bp) on the Illumina
NovaSeq X Plus platform (Illumina, Inc.) using the NovaSeq X Series
25B Reagent Kit (300 cycles; cat. no. 20104706; Illumina, Inc.).
The final library loading concentration was measured by
quantitative PCR (qPCR) and was 4.03 nM for patient samples and
4.24 nM for normal controls. RNA-seq was performed by Novogene Co.,
Ltd. Raw reads were filtered to remove adapters and low-quality
sequences, aligned to the human genome (GRCh38: obtained from the
NCBI, assembly accession: GCF_000001405.26) using HISAT2
(daehwankimlab.github.io/hisat2/), and quantified using
feature counts. Differential expression analysis [DESeq2
(https://pubmed.ncbi.nlm.nih.gov/25516281),
|log2 FC|>1, FDR-adjusted P<0.05] was used to
identify significant genes, which were visualized using volcano or
heatmaps. Principal Component Analysis (PCA) was performed on the
normalized expression data of all detected genes using the prcomp
function in R software (version 4.3.1). Gene Ontology (GO,
http://geneontology.org/) and Kyoto Encyclopedia
of Genes and Genomes (KEGG; kegg.jp/) pathway enrichment analyses
were conducted using cluster Profiler (FDR <0.05) (version
4.19.6; https://doi.org/10.1089/omi.2011.0118).
ELISA
Anticoagulant-coated blood collection tubes were
used to collect 3-5 ml venous blood from the HTRA1mc and HC groups.
Following standing at 25°C for 30 min, the blood was centrifuged at
1,200 × g for 10 min at 4°C to obtain plasma, divided into
microcentrifuge tubes, and centrifuged at 3,200 × g for another 10
min at 4°C to remove platelets. The expression levels of HTRA1
(cat. no. HM11176) and NADPH oxidase 4 (NOX4; both Bioswamp Life
Science Lab; Wuhan Bienle Biotechnology Co., Ltd.; cat. no.
HM11179) were measured using ELISA kits, in accordance with the
manufacturer's instructions.
Construction of HtrA1 overexpression (OE)
and knockdown lentiviral vectors
The lentiviral vectors for OE included mCherry red
fluorescent LV8N (EF-1a/mCherry&Puro) and non-fluorescent LV6
(EF-1a/Puro) shuttle plasmid vectors. For knockdown, the short
hairpin (sh)HtrA1-RNA lentiviral vectors included GFP green
fluorescent LV3 (H1/GFP&Puro) and non-fluorescent LV2 (U6/Puro)
shuttle plasmid vectors (GenePharma). The coding and protein
sequences of mouse HtrA1 were obtained from the National
Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/). The HtrA1 gene
fragments were cloned into the NotI/NsiI sites of the
LV8N and LV6 shuttle plasmid vectors. Following verification via
Sanger sequencing, the HtrA1 gene OE plasmid was
constructed. The following shRNA sequences were incorporated into
the LV3 and LV2 shuttle plasmids for the mouse HtrA1 gene:
shRNA-HtrA1-mus-749, 5'-GGGTCAAGGTTGAGCTGAAGA-3';
shRNA-HtrA1-mus-885, 5'-GCTGAGACCTGGAGAATTTGT-3';
shRNA-HtrA1-mus-1079, 5'-GCGAGGTGATTGGGATTAACA-3'; and negative
control (NC), 5'-TTCTCCGAACGTGTCACGT-3'.
Recombinant viral plasmids for HtrA1 OE and
knockdown, along with three auxiliary packaging plasmids
(PG-P1-VSVG, PG-P2-REV and PG-P3-RRE) (GenePharma), were subjected
to high-purity non-toxic endotoxin extraction. Plasmid DNA was
extracted using a high-purity midi-prep kit (Shanghai GenePharma
Co.) and dissolved in sterile double-distilled water. The
concentration and purity of plasmid DNA were determined by UV
spectrophotometry, and only samples with an A260/A280 ratio between
1.8 and 2.0 were used for subsequent experiments. For lentivirus
production, the third-generation lentiviral packaging system was
used. 293T cells (GenePharma) were co-transfected with RNAi-Mate
reagent (GenePharma, cat. no. G04001) at 37°C. In total, 20
μg plasmid DNA was used, composed of the recombinant
HtrA1-OE or HtrA1-shRNA plasmid and three helper plasmids
(PG-P1-VSVG, PG-P2-REV, PG-P3-RRE) at a 1 : 1 : 1 : 1 mass ratio
(2.5 μg each). After 48 h, the lentiviral supernatant was
collected, filtered (0.45 μm), and concentrated. The final
lentivirus titer was determined to be ≥1×108 TU/ml
(GenePharma). bEnd.3 cells were infected at an MOI of 125 (Table II). The virus-containing medium
was removed after 24 h and replaced with fresh medium. Following a
48-h recovery, transduced cells were selected and maintained in
medium containing 5 μg/ml puromycin.
| Table IILentivirus vector titers. |
Table II
Lentivirus vector titers.
| Lentiviral
vector | Titer, TU/ml |
|---|
| LV8N-NC |
2.00×108 |
| LV8N-HtrA1 |
3.00×108 |
| LV3-NC |
5.00×108 |
|
LV3-HtrA1-Mus-749 |
1.00×108 |
| LV3-
HtrA1-Mus-885 |
1.00×108 |
| LV3-
HtrA1-Mus-1079 |
1.00×108 |
| LV6-NC |
1.29×108 |
| LV6-HtrA1 |
2.05×108 |
| LV2-NC |
4.00×108 |
| LV2-
HtrA1-Mus-1079 |
1.85×108 |
Culture and viral infection of bEnd.3
cells
The bEnd.3 cells (Procell Life Science) were
cultured in high-glucose DMEM (HyClone; Cytiva; cat. no.
SH30022.01) supplemented with 10% FBS (Wuhan Boster Biological
Technology, Ltd.; cat. no. PYG0001) at 37°C in a 5% CO2
atmosphere. Upon reaching ~50% confluence, the cells were exposed
to serum-free high-glucose DMEM along with lentiviral supernatant
overexpressing HtrA1 [multiplicity of infection=125]. After 24 h
incubation, the medium was replaced with complete medium containing
10% FBS. After 72 h, 5 μg/ml puromycin was used for
screening of HtrA1 OE and knockdown cells for 96 h. The groups were
as follows: Untransfected (UT)-OE; NC-OE (bEnd.3 cells infected
with LV8N-NC or LV6-NC lentiviral supernatant); HtrA1-OE (bEnd.3
cells infected with LV8N-HtrA1 or LV6-HtrA1 lentiviral
supernatant); sh-UT; sh-NC (bEnd.3 cells infected with LV3-NC or
LV2-NC lentiviral supernatant); and sh-HtrA1 (bEnd.3 cells infected
with LV3 or LV2 lentiviral supernatant). Cells in the sh-HtrA1
group were treated with 5 μM GLX351322 (MedChemExpress; cat.
no. HY-100111) for 24 h at 37°C to downregulate the expression of
NOX4.
Reverse transcription-quantitative (RT-q)
PCR
Total RNA was extracted from cells using TRIzol
reagent (Invitrogen, cat. no. 15596-026), followed by chloroform
and isopropanol precipitation. RNA concentration and purity were
assessed using a Take3 micro-detection plate (BioTek, RNA samples
were reverse transcribed into cDNA using the PrimeScript RT reagent
kit with gDNA Eraser according to the manufacturer's protocol
(Takara Bio, Inc.; cat. no. RR047A). The obtained cDNA was
amplified using a Roche Diagnostics LightCycler480II. The 20
μl reaction mixture comprised 10.0 μl TB Green Premix
Ex TaqII (Takara Bio, Inc.; cat. no. RR820A), 2.0 μl cDNA,
6.4 μl diethylpyrocarbonate water and 0.8 μl (10
μM) primer. Thermocycling conditions were as follows:
Initial denaturation at 95°C for 30 sec, followed by 40 cycles of
denaturation at 95°C for 5 sec, and annealing/extension at 60°C for
30 sec. GAPDH was used as an endogenous reference for the
normalization of mRNA expression. The HtrA1 mRNA levels were
determined using the 2−ΔΔCq method (24). The primer sequences (Sangon
Biotech Co., Ltd.) were as follows: HtrA1 forward,
5'-tgacggcgggcatctccttc-3' and reverse,
5'-tcttggtgacagctttccctttgg-3' and GAPDH forward,
5'-ccctggccaaggtcatccat-3' and reverse,
5'-tcacgccacagctttccaga-3'.
Western blotting
bEnd.3 cells and mouse brain tissue samples were
lysed in RIPA lysis buffer (cat. no. AR0102-100) containing
broad-spectrum protease inhibitors (both Wuhan Boster Biological
Technology Co.; cat. no. AR1182). Protein concentration was
determined using a BCA protein assay kit (Wuhan Boster Biological
Technology Co.; cat. no. AR0146) following the manufacturer's
instructions. Equal amounts (30 μg) of protein were
separated by 10% SDS-PAGE, followed by transfer onto a 0.22
μM polyvinylidene fluoride membrane. The membranes were
blocked at room temperature for 2 h with 5% (w/v) non-fat dry milk
in 0.1% TBST. After blocking for 2 h, the membranes were incubated
overnight with primary antibodies at 4°C as follows: Rabbit
anti-HtrA1 (1:250, Wuhan Boster Biological Technology Co.; cat. no.
A01801-1), anti-occludin (cat. no. A2601), anti-zona occludens
(ZO)1 (both 1:1,000, both ABclonal Biotech Co., Ltd.; A0659),
anti-claudin (CLDN)5 (1:250, BIOSS; cat. no. bs-10296R), anti-NOX4
(Wuhan Boster Biological Technology Co.; cat. no. BM4135),
anti-caspase3 (both 1:1,000, ABclonal Biotech Co., Ltd.; cat. no.
A2156), anti-GAPDH (1:5,000, Shanghai Abways Biotechnology Co.,
Ltd.; cat. no. AB0037) and anti-β-actin (1:5,000, ABclonal Biotech
Co., Ltd.; cat. no. AC038). After washing the membrane with TBST,
it was incubated with goat anti-rabbit IgG-HRP secondary antibody
(1:5,000; Wuhan Boster Biological Technology Co., Ltd.; cat. no.
BA1054) for 90 min at 25°C. The luminescent solution was prepared
using the ECL Luminescent Substrate kit (Wuhan Boster Biological
Technology Co., Ltd.; cat. no. AR1171) and the bands were
visualized using an imaging analysis system (Bio-Rad Laboratories,
Inc.; Chemidoc MP). The gray value of the target proteins was
analyzed using the ImageJ software (V1.8.0; National Institutes of
Health).
Cell Counting Kit-8 (CCK-8) assay
A total of 5×103 cells was seeded in a
96-well plate. After 12, 24, 48 and 72 h, cells were treated with
working solution containing 10 μl CCK-8 solution
(MedChemExpress; cat. no. HY-K0301) and 90 μl DMEM with 10%
FBS. After 2 h incubation in the dark, the absorbance was measured
at 450 nm using a multifunctional microplate reader (BioTek;
Agilent Technologies, Inc.; Synergy H1).
Real-time cellular analysis (RTCA)
RTCA was performed using the RTCA S16 system (ACEA
Biosciences, Inc.). A total of 50 μl DMEM with 10% FBS in
E-plate16 wells was used for determining the baseline. bEnd.3 cells
were seeded at a density of 5×103 cells/well in the
wells of E-plate16. Following 30 min incubation at 37°C, RTCA was
performed for 72 h, with impedance measured at 15-min
intervals.
Immunofluorescence staining of cells
A total of 5×103 bEnd.3 cells was seeded
in 24-well plates. The cells were fixed with 4% paraformaldehyde at
room temperature for 30 min. Following washing with PBS, 0.5%
Triton X-100 (Beyotime Biotechnology; cat. no. P0096-500 ml) was
added at room temperature for permeabilization of cell membrane.
Blocking was performed using 5% BSA (Bossis; cat. no. AR1006) for 2
h at 25°C. The cells were then incubated overnight at 4°C with the
primary antibodies as follows: Rabbit anti-CLDN5 (1:100, ABclonal
Biotech Co., Ltd.; A10207), anti-occludin (1:150, Bossis; cat. no.
bs-10011R), anti-ZO1 (1:150, ABclonal Biotech Co., Ltd.; cat. no.
A0659) and anti-NOX4 (1:100, Wuhan Boster Biological Technology Co.
Ltd.; BM4135). Cells were washed three times with PBS with 0.1%
Tween-20 (PBST) and incubated with Alexa Fluor 555-labeled donkey
(1:500, cat. no. A0453) and Alexa Fluor 488-labeled goat
anti-rabbit IgG (H+L; 1:200, both Beyotime Biotechnology; cat. no.
A0423) at 25°C for in the dark for 2 h. The cells were washed three
times with PBST for 15 min and imaged under a confocal microscope
(Leica GmbH). Fluorescence intensity was analyzed using ImageJ
software (V1.8.0; National Institutes of Health).
FITC-dextran permeability assay
A total of 2×105 cells were seeded in the
upper chamber of a 24-Transwell system (8 μm, Falcon;
Corning Life Sciences; cat. no. 353097) and incubated at 37°C in a
humidified 5% CO2 incubator for 24 h. The upper chamber
was filled with 200 μl DMEM (HyClone; Cytiva) with 10% FBS
(Boster, cat. no. PYG0001), whereas the lower chamber contained 600
μl PBS. Following 12 h incubation at 37°C, the medium from
the upper chamber was removed and the cells were washed three times
with PBS. A total of 100 μl 1 μg/ml FITC-dextran
(40,000 MW, Sigma-Aldrich; Merck KGaA; cat. no. SLCB8958) was added
to the upper chamber. The medium in the lower chamber was discarded
and replaced with 600 μl PBS. After the 1 h incubation at
37°C, the fluorescence intensity was measured using the SpectraMax
i3x detection system at excitation/emission (Ex/Em) wavelengths of
485/528 nm (Molecular Devices, LLC).
Reactive oxygen species (ROS) assay
bEnd.3 cells (1.5×105) were seeded in a
6-well plate and cultured for 24 h at 37°C. DCFH-DA
(MedChemExpress; cat. no. HY-D0940) staining was performed when the
cell density reached 80%. The culture medium was discarded and the
cells were washed three times with PBS. Subsequently, the cells
were incubated with 1 ml DCFH-DA working solution at a
concentration of 10 μM for 20 min at 37°C in the dark.
Images were captured under a fluorescence microscope (Ex/Em=488/525
nm) and the fluorescence intensity was analyzed using ImageJ
software (V1.8.0, National Institutes of Health).
Superoxide dismutase (SOD) and
malondialdehyde (MDA) assay
The cells were detached using 0.25% trypsin (Beijing
Solarbio Science & Technology Co., Ltd.; cat. no. T1300). The
cells were lysed using RIPA lysis buffer (Beijing Solarbio, cat.
no. R0010) containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton
X-100, 1% sodium deoxycholate, and 0.1% SDS. The protein
concentration was measured using the BCA method. The activity of
SOD and MDA were determined using the SOD (cat. no. A001-3) and MDA
Detection kits (both Nanjing Jiancheng Bioengineering Institute;
cat. no. A003-1-2), according to the manufacturer's
instructions.
Apoptosis detection
Apoptosis (early + late apoptotic cells) was
detected via flow cytometry using the Annexin V-FITC/propidium
iodide (PI) staining kit (Dalian Meilun Biology Technology Co.,
Ltd.; cat. no. MA0220) following the manufacturer's instructions.
Briefly, 2×105 bEnd.3 cells were seeded in 6-well plates
and cultured for 24 h at 37°C. Cells were detached using 0.25%
trypsin (Wuhan Boster Biological Technology Ltd.; cat. no. PYG0065)
and incubated with 5 μl Annexin V-FITC/PI staining solution
for 15 min at 25°C in the dark. Flow cytometry was performed using
a NovoCyte 3130 instrument (ACEA Biosciences, Inc.). Red and green
fluorescence signals were recorded and analyzed using the
NovoExpress software (Version 1.5.0, ACEA Biosciences, Inc.).
AAV/B130-shHtrA1 mouse model
All animal experiments were approved by the Animal
Welfare and Ethics Committee of the First Hospital of Shanxi
Medical University (approval no. DWYJ-2025-304, Taiyuan, China).
Male C57BL/6J mice (Spef Biotechnology Co., Ltd.; age, 6-8 weeks)
were housed in a specific pathogen-free animal facility at 25±2°C,
50-60% relative humidity and a 12/12-h light/dark cycle with free
access to food and water (n=24, weight, 20-25 g). Humane endpoints
were as follows: i) loss of more than 20 % of initial body weight
within 24 h, ii) inability to reach food or water, iii) persistent
hunched posture with ruffled fur, iv) moribund state and v)
respiratory distress. Animals reaching any of these endpoints were
immediately euthanized by cervical dislocation. None of the animals
reached the predefined humane endpoints during the study. The
endothelial cell-specific HtrA1-shRNA mouse model was created by
injecting AAV HBAAV2/BI30-m-HtrA1-shRNA-EGFP (Hanbio Biotechnology
Co., Ltd., cat. no. 80062053) into the tail vein of C57BL/6J mice.
Mice were divided into three groups (n=8): Vehicle (mice injected
with saline), AAV/B130-NC (mice injected with a virus control) and
AAV/B130-shHtrA1 group (mice injected with HtrA1-interfering AAV).
Anesthesia was induced with 5% isoflurane using a compact small
animal anesthesia machine (RWD Life Science Co., Ltd.), followed by
maintenance with 1.5-2.5% isoflurane. After 4 weeks, mice were
sacrificed by cervical dislocation and brain tissue was isolated.
To investigate the effects of oxidative stress on AAV/B130-shHtrA1
C57BL/6J mice were intraperitoneally administered GLX351322 (5
mg/kg/day) for 4 weeks.
Tissue immunofluorescence
Brain tissue was fixed in 4% paraformaldehyde in 0.1
M phosphate buffer (pH 7.4) for 24 h at 4°C. Tissue was sectioned
into 15 μm slices on a cryostat (Leica, RM2235). Antigen
retrieval was performed by heating the sections in EDTA antigen
retrieval solution (Zhongshan Jinqiao Biotechnology Co., Ltd.; cat.
no. ZLI-9069) at 95°C for 2 min in a water bath. After cooling to
room temperature, the sections were blocked with 5% BSA (Solarbio,
cat. no. SW3015) for 30 min. The sections were incubated with
primary antibodies overnight at 4°C and with the secondary
antibodies for 1 h at room temperature. The primary antibodies were
as follows: Mouse-anti-α-smooth muscle actin (SMA; Zenbio, Inc.;
cat. no. L29JLZC) and rabbit-anti-HtrA1 (both 1:50, Proteintech
Group, Inc.; cat. no. 00129131). The secondary antibodies were
Alexa Fluor 488-labeled goat anti-mouse (cat. no. A0428) and Alexa
Fluor 555-labeled donkey anti-rabbit IgG (H+L; both 1:500, both
Beyotime; cat. no. A0453). The nuclei were counterstained with DAPI
(Beijing Solarbio Science & Technology Co., Ltd.; C0065) for 10
min at 25°C. Fluorescent images were captured using a confocal
microscope (Leica GmbH).
Evans blue assay
Mice were intravenously injected with 0.5% Evans
blue (Absin, cat. no. Abs47002009) in saline at 3 ml/kg body weight
and euthanized 2 h after dye circulation. The weight of each
cerebral hemisphere was recorded and Evans blue extraction was
performed by incubating the brain tissue in acetone for 24 h at
25°C, followed by centrifugation at 12,000 × g for 30 min at 4°C.
Evans blue staining was quantified by measuring the absorbance at
620 nm using a spectrophotometer (Thermo Fisher Scientific,
Inc.).
Open-field test (OFT)
Each mouse underwent the OFT once to avoid
habituation or stress-associated behavioral changes. A plain
50×50×40 cm open-field arena was used to assess the locomotor
activity and anxiety-like behavior. Following 30 sec habituation,
the total distance traveled and time spent in the center arena
(16.7×16.7 cm) was recorded in a 5 min session. The frequencies of
rearing and defecation in the apparatus were recorded. At the end
of each experimental trial, the apparatus was thoroughly cleaned
and disinfected by spraying 75% ethyl alcohol to eliminate residual
olfactory cues. Mouse behavior was analyzed using VisuTrack
software (XR-VT version, Shanghai Xinruan Information Technology
Co., Ltd.).
Novel object recognition test (NORT)
Mice were placed in an open field with two identical
objects to familiarize them with their environment. Following 6 h,
the mice were returned to the same open field for testing. One of
the familiar objects was replaced with a novel object that had a
unique color and shape. In both phases of the experiment, each
mouse faced the wall of the apparatus and was allowed to explore
the objects freely for 5 min. Before each trial, the apparatus was
cleaned using 75% ethanol solution. Mouse behavior was recorded
using a video tracking system and analyzed using the VisuTrack
software. NOR memory was evaluated using the NOR index (NOI) as
follows: NOI=(time spent exploring the novel object)/(total
exploration time for both objects) ×100%.
Morris water maze (MWM) test
The MWM test was conducted in a white circular tank,
measuring 120 cm in diameter and 50 cm in height, filled with tap
water maintained at 22±3°C, and divided into four equal virtual
quadrants. Non-toxic white paint was added to ensure the water was
opaque. During the place navigation-training phase, which consisted
of four trials of 120 sec/day over 4 consecutive days, a circular
escape platform (diameter, 10 cm) was submerged 1 cm below the
water surface in a designated quadrant. Each mouse was released
from different starting positions, facing the tank wall and allowed
to locate the escape platform. Upon reaching the platform, the
mouse was allowed to stay on it for 3 sec. Latency to escape onto
the platform was recorded. If a mouse failed to locate the platform
within 120 sec, it was guided to the platform and allowed to remain
there for 30 sec. On day 5, a probe test was conducted, wherein the
mice swam freely without the platform for 120 sec. The proportion
of time spent in the target quadrant and the number of platform
crossings were recorded. Swimming paths were monitored and analyzed
using VisuTrack software.
Statistical analysis
Statistical analysis was performed using the
GraphPad Prism 9 (GraphPad Software Inc.; Dotmatics) and R (version
4.3.1; R Foundation for Statistical Computing) software. Data are
presented as the mean ± standard deviation of three independent
experiments. Comparisons were made using one-way ANOVA followed by
Tukey's, LSD t or Dunnett's post hoc test. Differentially expressed
genes were analyzed using the DESeq2 package. The Wald test was
used to analyze the differences between two groups, with the
filtering conditions set at |log2 FC|>1 and an
adjusted P-value <0.05. The significance of enrichment analysis
was assessed using a hypergeometric distribution test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Sanger sequencing, family pedigree,
clinical results and RNA-seq analysis for heterozygous
HTRA1mcs
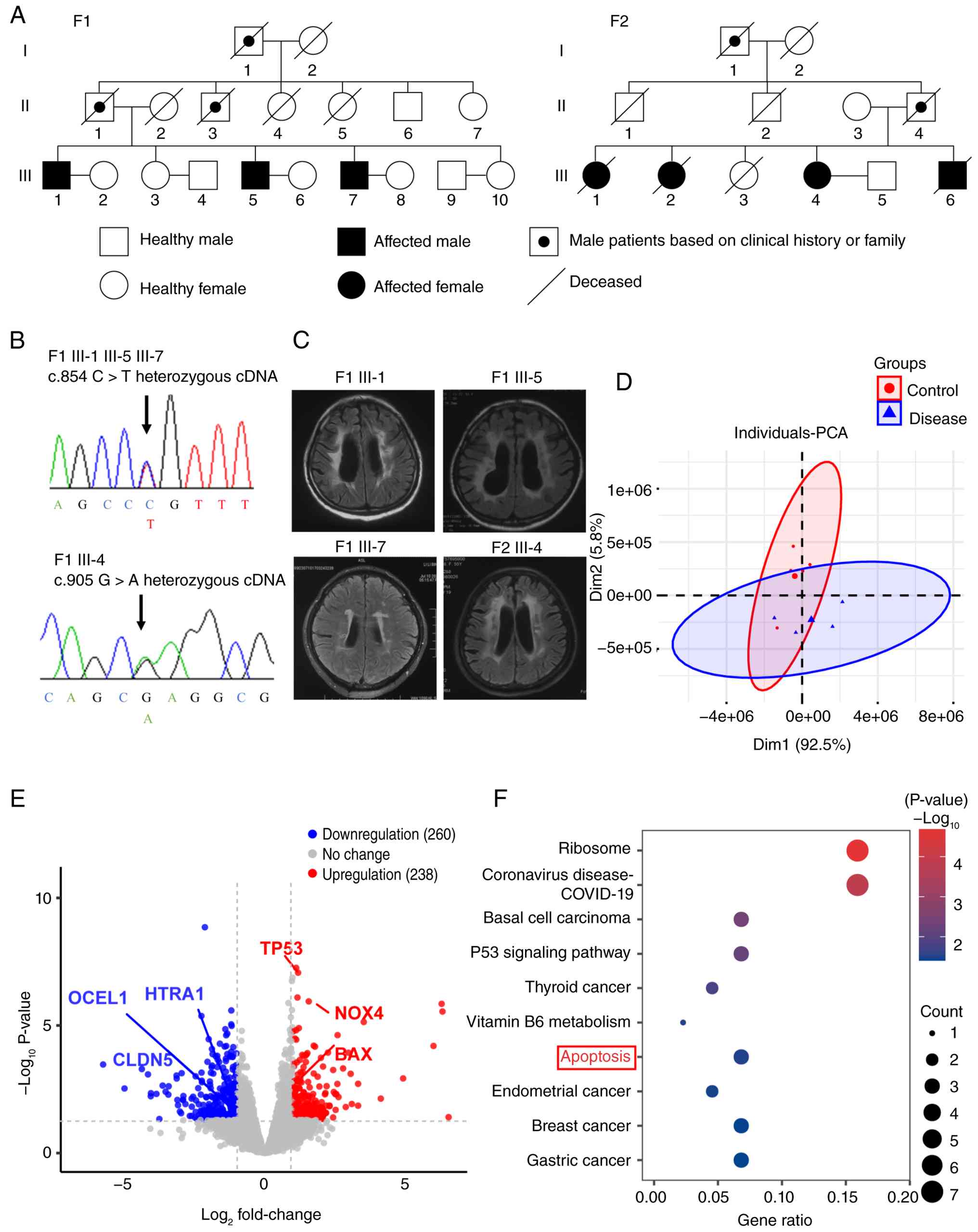
Sanger sequencing was performed to analyze the
carrier status of the pathogenic variants in two families (Fig. 1A and B). In family 1 (F1), the
proband (F1 III-5), his elder brother (F1 III-1) and younger
brother (F1 III-7) were found to carry a heterozygous HTRA1
mutation (c.854C>T, p.P285L). In F2, the proband (F2 III-1), two
sisters (F2 III-2 and -4) and a brother (F2 III-6) carried a
heterozygous HTRA1 mutation (c.905G>A, p.R302Q). The
proband (F2 III-1) and their sister (F2 III-2) and brother (F2
III-6) all died of cerebrovascular diseases.
The predicted pathogenicity of the HTRA1 c.854
C>T and c.905 G>A mutations is shown in Table III. The phenotypic
characteristics exhibited a notable degree of homogeneity, with the
clinical presentation, including subcortical ischemic events and
progressive cognitive decline, being characteristic of CSVD
(Table IV). The pedigrees of
families with heterozygous HTRA1 mutations are shown in
Fig. 1A.
| Table IIIPredicted pathogenicity of the HTRA1
c.854 C>T and c.905 G>A mutations. |
Table III
Predicted pathogenicity of the HTRA1
c.854 C>T and c.905 G>A mutations.
| Variant | Protein domain | ID SNP | Polyphen-2 | SIFT | Mutation
taster | CADD | nsSNP analyzer | Mutation
assessor |
|---|
| c.854 C>T
(p.P285L) | Serine
protease | rs587776446 | Probably
damaging | Damaging (score,
0.016) |
Disease-causing | Deleterious | Disease | Medium functional
impact |
| c.905 G>A
(p.R302Q) | Serine
protease | rs2133449474 | Probably
damaging | Damaging (score,
0.015) |
Disease-causing | Deleterious | Disease | Medium functional
impact |
| Table IVClinical features of heterozygous
high temperature requirement serine peptidase A1 mutation
carriers. |
Table IV
Clinical features of heterozygous
high temperature requirement serine peptidase A1 mutation
carriers.
| Characteristic | Patient
|
|---|
| F1 III-1 | F1 III-5 | F1 III-7 | F2 III-4 |
|---|
| Mutation | p.P285L | p.P285L | p.P285L | p.R302Q |
| Sex | Male | Male | Male | Female |
| Age, years | 49 | 46 | 44 | 54 |
| Age at first stroke
episode, years | 35 | 41 | 42 | 48 |
| Gait
disturbance | Present | Present | Absent | Present |
| Cognitive
decline | Present | Present | Present | Present |
| Spondylosis
deformans | Present | Present | Absent | Absent |
| Alopecia | Present | Present | Present | Absent |
| Hypertension | Absent | Absent | Present | Absent |
| Diabetes
mellitus | Absent | Absent | Present | Absent |
| Dyslipidemia | Absent | Absent | Absent | Absent |
| Pseudobulbar
palsy | Present | Present | Absent | Absent |
| Hyperreflexia of
limbs | Present | Present | Absent | Present |
| Babinski
reflex | Present | Present | Present | Present |
| Chronic heart
failure | Absent | Absent | Absent | Absent |
| Heavy alcohol
consumption | Absent | Absent | Absent | Absent |
| Smoking | Absent | Absent | Absent | Absent |
F1III-1 was a 49-year-old male patient who presented
with recurrent strokes and cognitive decline for 20 years. The
patient experienced a first stroke, manifesting as left-sided
weakness and dysphagia. The patient is bedridden because of limb
weakness, gait disturbance and pseudobulbar palsy. The patient had
a medical history of alopecia and spondylosis deformans. Brain MRI,
with fluid-attenuated inversion-recovery (FLAIR) sequences,
revealed multiple lacunar infarcts and confluent white matter
hyperintensity (WMH) in the periventricular regions (Fig. 1C). Cognitive evaluation at the
time of diagnosis demonstrated substantial impairment. The patient
achieved a Montreal Cognitive Assessment (MoCA) score of 13/30
(severe impairment; normal range ≥26) and a Mini-Mental State
Examination (MMSE) score of 16/30 (moderate impairment; normal
range ≥24).
F1III-5 was a 46-year-old male patient who presented
with right-sided weakness, dysarthria, history of cerebral
infarction, and cognitive decline. The patient had alopecia and
spondylosis deformans with lumbar disc surgery. Brain MRI with
FLAIR sequences revealed multiple lacunar infarcts and WMH in the
periventricular regions (Fig.
1C). Cognitive assessment revealed a notable decline (MoCA
score, 15; MMSE score, 18).
F1III-7 was a 44-year-old male patient who presented
with recurrent dizziness and gait unsteadiness. The patient
exhibited hypertension and diabetes. Brain MRI indicated multiple
lacunar infarcts and WMH (Fig.
1C). Cognitive assessment showed a mild decline (MoCA score,
25; MMSE score, 24).
F2III-4 was a 54-year-old female patient who
presented with recurrent dizziness, diplopia and impaired movement
of the right limbs, with a history spanning 8 years. Brain MRI
showed multiple subcortical lacunar infarcts and WMH (Fig. 1C), with cognitive assessment
indicating a mild decline (MoCA score, 23; MMSE score, 21).
PCA revealed significant differences between the
HTRA1mc and HC groups (Fig. 1D).
In total, 489 differentially expressed genes were identified, of
which 238 were up- and 260 were downregulated (Figs. 1E and 2A). In the HTRA1mc group, mRNA
expression of HTRA1, occludin-like protein 1 (OCEL1)
and CLDN5 was downregulated, whereas that of NOX4 and
Bcl-2 associated X (BAX) was upregulated (Fig. 1E). The KEGG enrichment analysis
showed significant enrichment in 'ribosome', 'P53 signaling
pathway', 'apoptosis', 'thyroid cancer', 'endometrial cancer',
'breast cancer', and gastric cancer (Fig. 1F). GO enrichment analysis
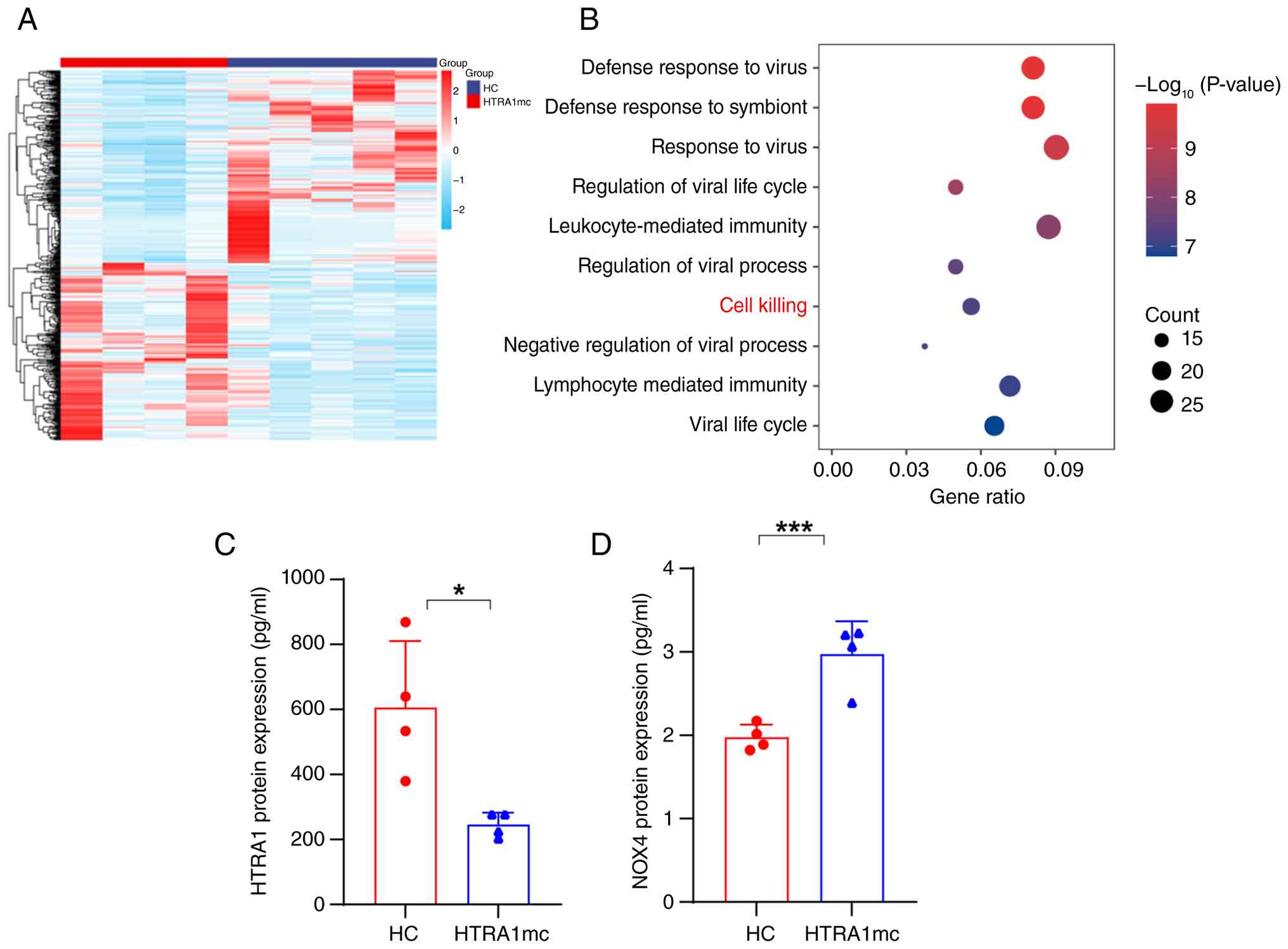
revealed significant enrichment in 'defense response to virus',
'leukocyte-mediated immunity', 'lymphocyte mediated immunity', and
'cell killing' (Fig. 2B). The
HTRA1mc group showed decreased plasma HTRA1 (Fig. 2C) and elevated NOX4 protein
(Fig. 2D) levels compared with
the HC group. These findings indicated that HTRA1 mutations
may increase oxidative stress and apoptosis, thereby contributing
to CSVD. Additionally, the downregulation of tight
junction-associated genes, such as OCEL1 and CLDN5,
may impair the blood-brain barrier (BBB) function.
Effect of HtrA1 OE on the viability and
oxidative stress of bEnd.3 cells
mCherry red fluorescence carried by lentivirus was
observed in both the NC-OE and HtrA1-OE groups (Fig. 3A). RT-qPCR and western blot
analyses showed that the HtrA1 mRNA (Fig. 3B) and protein (Fig. 3C) levels were significantly
higher in the HtrA1-OE group than in the NC-OE group. No
differences were observed between the UT-OE and NC-OE groups
(Fig. 3B and C). In the CCK-8
assay, cells in the HtrA1-OE group exhibited enhanced viability at
both 48 and 72 h compared with those in the NC-OE group (Fig. 3D). These findings were supported
by the RTCA results (Fig. 3E),
which suggested that HtrA1 OE improved the viability of bEnd.3
cells. DCFH-DA, SOD and MDA assays were performed to assess
oxidative stress. The HtrA1-OE group showed decreased NOX4
expression (Fig. 3C and F) and
reactive oxygen species (ROS) levels (Fig. 3G) compared with the NC-OE group,
whereas the SOD activity was higher and MDA levels were lower in
the HtrA1-OE group (Fig. 3H).
These results indicated that HtrA1 OE decreased oxidative stress
and exerted a protective effect on bEnd.3 cells.
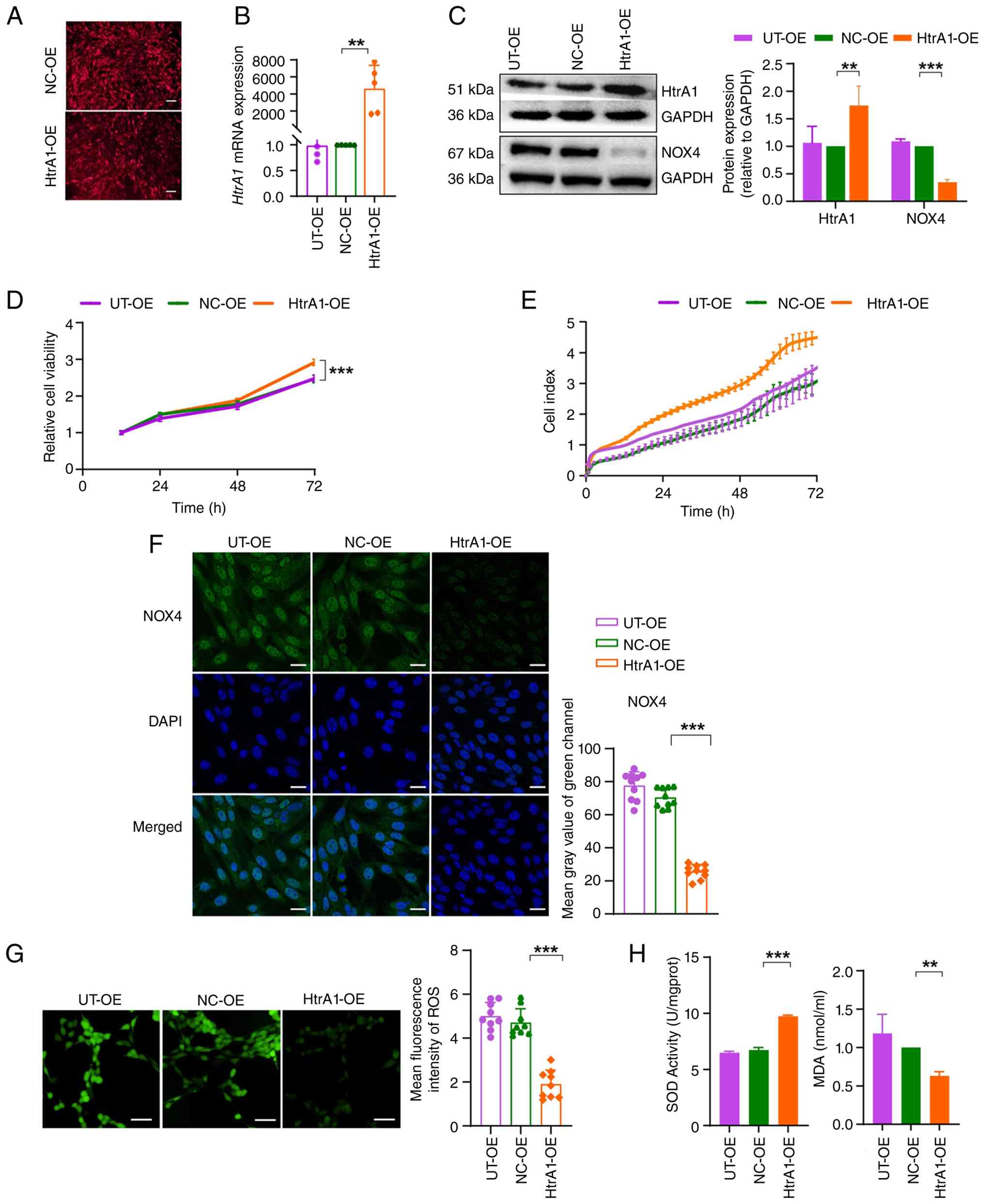 | Figure 3Effect of lentiviral vector-mediated
HtrA1 OE on bEnd.3 cell viability and oxidative stress. (A)
Infection with lentiviral supernatant induced OE of HtrA1 in bEnd.3
cells. Scale bar, 200 μm. (B) Reverse
transcription-quantitative PCR analysis of HtrA1 mRNA
expression in bEnd.3 cells. (C) Western blot analysis of HtrA1 and
NOX4 protein expression in bEnd.3 cells. (D) Cell viability
evaluated using the Cell Counting Kit-8 assay. (E) Real-time
monitoring of cell viability in each group using real-time cellular
analysis. (F) Immunofluorescence staining of NOX4 (green) and cell
nuclei (blue) in bEnd.3 cells. Scale bar, 25 μm. (G) DCFH-DA
analysis of intracellular ROS. Scale bar, 100 μm. (H)
Activity of SOD and concentration of MDA in bEnd.3 cells.
**P<0.01, ***P<0.001. HtrA1, high
temperature requirement serine peptidase A1; OE, overexpression;
SOD, superoxide dismutase; MDA, malondialdehyde; UT, untransfected;
NC, negative control; ROS, reactive oxygen species. |
Effect of HtrA1 OE on tight junctions,
permeability and apoptosis in bEnd.3 cells
HtrA1 OE in bEnd.3 cells significantly increased the
expression of tight junction proteins ZO1, occludin and CLDN5 in
the HtrA1-OE group compared with that in the NC-OE group (Fig. 4A). These results were
corroborated by immunofluorescence, with higher expression levels
of these proteins observed in the HtrA1-OE group (Fig. 4B and C). The FITC-dextran
experiment revealed decreased cell permeability in the HtrA1-OE
group compared with that in the NC-OE group (Fig. 4D), indicating strengthened tight
junctions. Western blotting revealed a significant decrease in
caspase3 expression in the HtrA1-OE group compared with that in the
NC-OE group (Fig. 4E). Flow
cytometry also indicated decreased apoptosis in the HtrA1-OE group
compared with the NC-OE group (Fig.
4F). These findings indicated that HtrA1 OE strengthened the
integrity of tight junctions and decreased apoptosis in bEnd.3
cells.
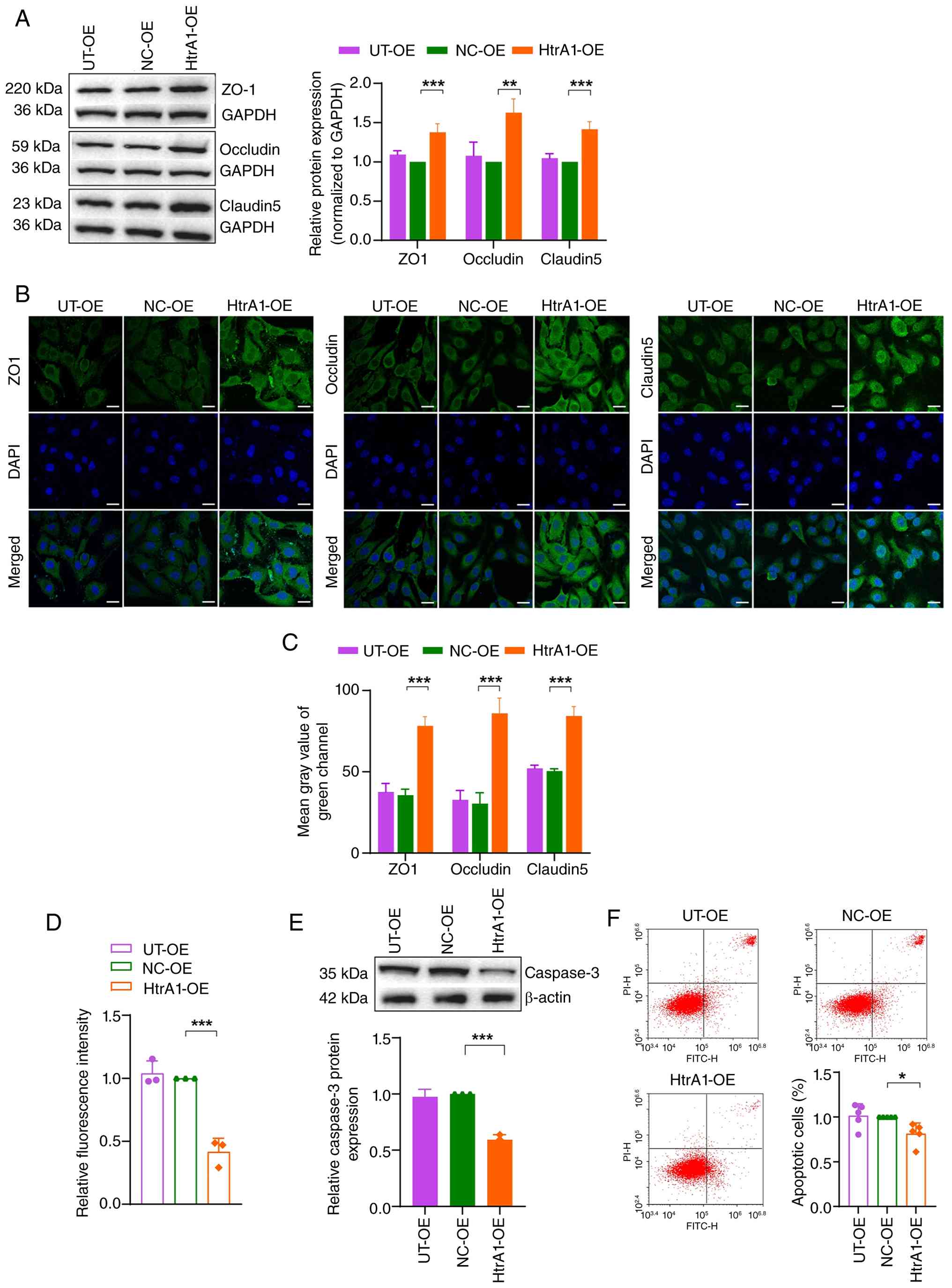 | Figure 4Effect of HtrA1 OE on tight
junctions, permeability and apoptosis in bEnd.3 cells. (A) Western
blot analysis of ZO1, occludin and claudin 5 proteins in bEnd.3
cells. (B) Immunofluorescence of ZO1, occludin, claudin 5 and
nuclei (blue) in bEnd.3 cells. Scale bar, 25 μm. (C) Mean
gray value of green channel of ZO1, occludin and claudin 5. (D)
Permeability of bEnd.3 cell monolayers was assessed using
FITC-dextran. (E) Western blot analysis of caspase3 protein in
bEnd.3 cells. (F) Flow cytometry analysis of bEnd.3 cell apoptosis.
*P<0.05, **P<0.01,
***P<0.001. HtrA1, high temperature requirement
serine peptidase A1; OE, overexpression; ZO1, zonula occludens
protein 1; UT, untransfected; NC, negative control. |
Effects of HtrA1 knockdown on bEnd.3 cell
viability and oxidative stress
Both the sh-NC and sh-HtrA1 groups showed green
fluorescence, confirming successful establishment of a stable HtrA1
knockdown cell line (Fig. 5A).
RT-qPCR and western blot analyses revealed significantly decreased
HtrA1 mRNA and protein levels in the shRNA-HtrA1-mus-1079
group compared with those in the sh-NC group (Fig. 5B and C). As the
shRNA-HtrA1-mus-1079 group showed the highest interference
efficiency, it was selected as the sh-HtrA1 group for subsequent
experiments. The CCK-8 assay showed decreased viability of cells in
the sh-HtrA1 group at 72 h compared with that in the sh-NC group
(Fig. 5D). Consistently, RTCA
(Fig. 5E) demonstrated decreased
cell viability following HtrA1 knockdown. The expression of NOX4
was increased in the sh-HtrA1 group compared with that in the sh-NC
group, as evidenced by western blot analysis (Fig. 5F) and immunofluorescence
(Fig. 5G). ROS levels were
higher in the sh-HtrA1 group compared with sh-NC group (Fig. 5H), whereas the SOD activity and
MDA levels were lower and higher, respectively (Fig. 5I). These findings indicated that
HtrA1 knockdown increased oxidative stress in bEnd.3 cells.
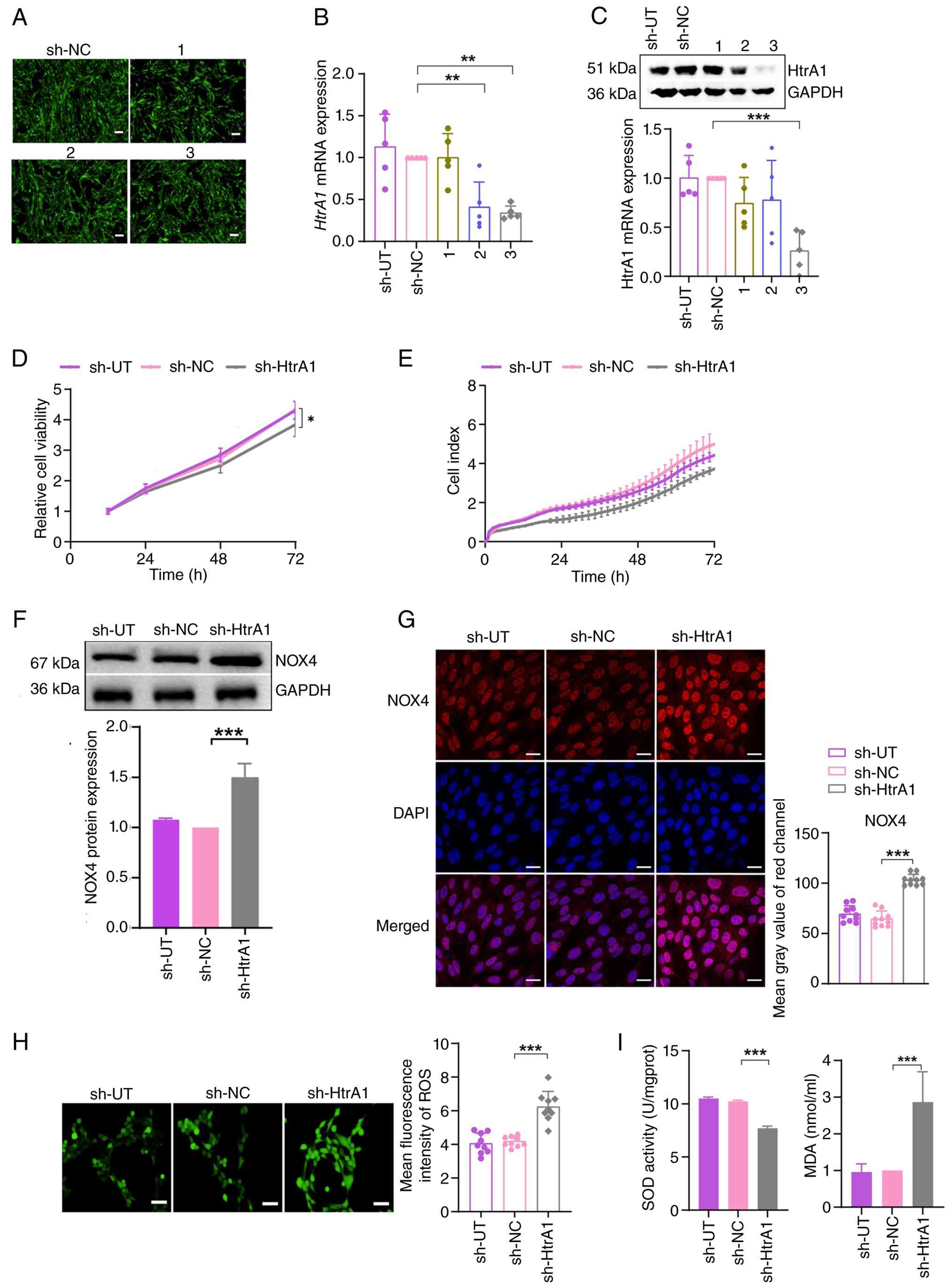 | Figure 5Effect of lentiviral vector-mediated
HtrA1 knockdown on bEnd.3 cell viability and oxidative stress. (A)
Infection with lentiviral supernatant induced knockdown of HtrA1 in
bEnd.3 cells. Scale bar, 200 μm. (B) Reverse
transcription-quantitative PCR analysis of HtrA1 mRNA expression.
(C) Western blot analysis of HtrA1 protein expression in bEnd.3
cells. (D) Cell viability was evaluated using Cell Counting Kit-8
assay. (E) Real-time monitoring of cell viability using real-time
cellular analysis. (F) Western blot analysis of NOX4 protein
expression in bEnd.3 cells. (G) Immunofluorescence analysis of NOX4
(red) and cell nuclei (blue) in bEnd.3 cells. Scale bar, 25
μm. (H) DCFH-DA staining showing intracellular ROS levels.
Scale bar, 100 μm. (I) Activity of SOD and concentration of
MDA in bEnd.3 cells. *P<0.05, **P<0.01,
***P<0.001. 1, shRNA-HtrA1-mus749; 2,
shRNA-HtrA1-mus885; 3, shRNA-HtrA1-mus1079; HtrA1, high temperature
requirement serine peptidase A1; ROS, reactive oxygen species; SOD,
superoxide dismutase; MDA, malondialdehyde; sh, short hairpin; UT,
untransfected; NC, negative control. |
Effect of HtrA1 knockdown on tight
junction, permeability and apoptosis in bEnd.3 cells
Following HtrA1 knockdown in bEnd.3 cells, western
blot analysis showed decreased expression of the tight junction
proteins ZO1, occludin and CLDN5 in the sh-HtrA1 group compared
with that in the sh-NC group (Fig.
6A). Confocal microscopy confirmed this reduction (Fig. 6B and C). The FITC-dextran assay
revealed increased permeability in the sh-HtrA1 group (Fig. 6D), indicating compromised tight
junction integrity and enhanced cell permeability following HtrA1
knockdown. Western blot analysis revealed elevated levels of
caspase3 in the sh-HtrA1 group compared with the sh-NC group
(Fig. 6E). Flow cytometry
confirmed increased apoptosis in the sh-HtrA1 group (Fig. 6F). These findings indicated that
HtrA1 knockdown increased apoptosis in bEnd.3 cells, emphasizing
the role of HtrA1 in regulating tight junctions and cell barrier
properties.
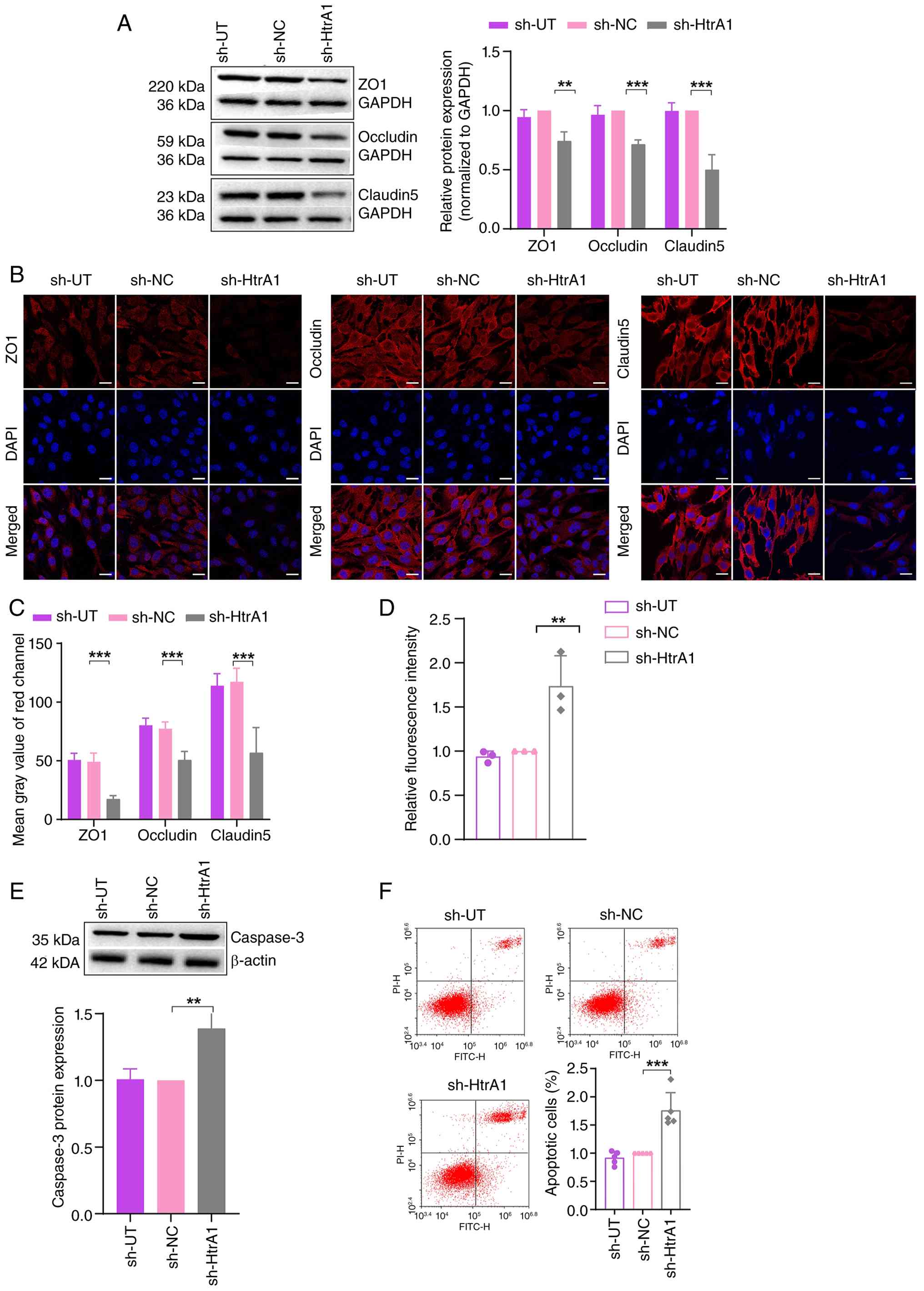 | Figure 6Effect of HtrA1 knockdown on tight
junctions, permeability and apoptosis in bEnd.3 cells. (A) Western
blot analysis of ZO1, occludin and claudin 5 proteins in bEnd.3
cells. (B) Immunofluorescence analysis of ZO1, occludin, claudin 5
and nuclei (blue) in bEnd.3 cells. Scale bar, 25 μm. (C)
Quantitative fluorescence analysis. (D) Permeability of bEnd.3 cell
monolayers was assessed using FITC-dextran. (E) Western blot
analysis of caspase3 proteins in bEnd.3 cells. (F) Flow cytometry
analysis of bEnd.3 cell apoptosis. **P<0.01,
***P<0.001. HtrA1, high temperature requirement
serine peptidase A1; ZO1, zonula occludens protein 1; sh, short
hairpin; UT, untransfected; NC, negative control. |
Interference with HtrA1 expression in
cerebral vascular endothelial cells of C57BL/6J mice
An endothelial cell-specific HtrA1 knockdown mouse
model was constructed by injecting AAV2/B130-TIE carrying a
specific endothelial promoter into the tail vein of C57BL/6J mice.
After 4 weeks, immunofluorescence staining of HtrA1 in mouse brain
slices showed notable HtrA1 expression in cerebrovascular
endothelial cells, colocalizing with the vascular marker α-SMA
(Fig. 7A). The green fluorescent
protein marker in the viral vector confirmed the successful
transfection of endothelial cells (Fig. 7B). RT-qPCR and western blotting
revealed significantly reduced HtrA1 mRNA (Fig. 7C) and protein levels (Fig. 7D) in the AAV/B130-shHtrA1 group,
indicating effective gene knockdown. Expression of tight junction
proteins ZO1, occludin and CLDN5 decreased, whereas that of NOX4
and caspase3 was increased in the AAV/B130-shHtrA1 compared with
that in the AAV/B130-NC group (Fig.
7D).
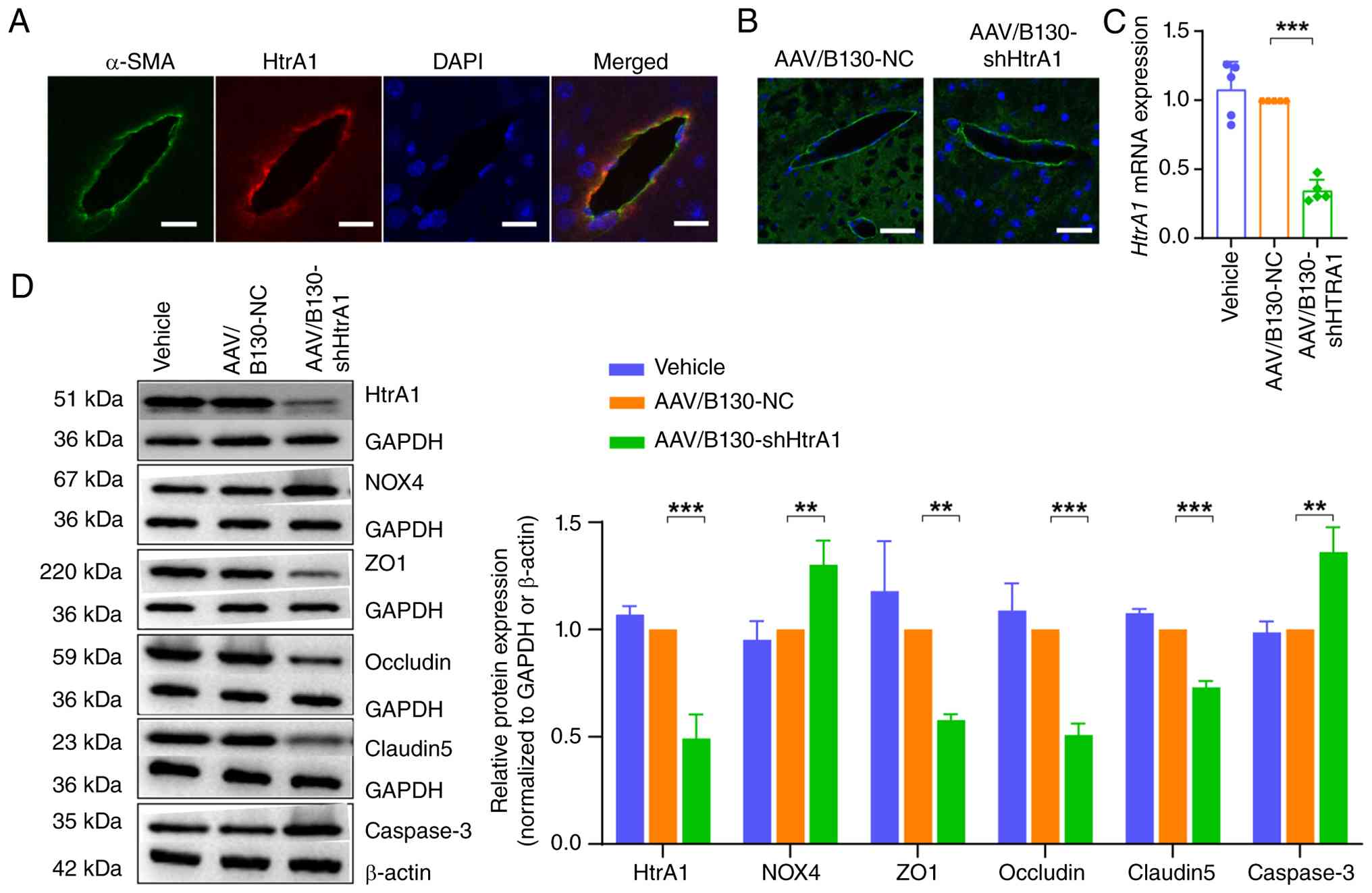 | Figure 7Interference with HtrA1 expression in
cerebral vascular endothelial cells of C57BL/6J mice. (A) Mouse
cerebral vascular endothelial cells showing HtrA1 expression
colocalized with the expression of vascular marker α-SMA. (B)
AAV2/B130-TIE was injected into the tail vein of C57BL/6J mice.
Scale bar, 25 μm. (C) Reverse transcription-quantitative PCR
analysis of HtrA1 mRNA expression in the brain tissue of C57BL/6J
mice. (D) Western blot analysis of HtrA1, NOX4, ZO1, occludin,
claudin 5 and caspase3 protein in the brain tissue of C57BL/6J
mice. **P<0.01, ***P<0.001. HtrA1, high
temperature requirement serine peptidase A1; SMA, smooth muscle
actin; AAV, adeno-associated virus; ZO1, zonula occludens protein
1; sh, short hairpin; NC, negative control. |
HtrA1 regulates tight junctions,
permeability and apoptosis of brain vascular endothelial cells via
the NOX4-dependent oxidative stress pathway
Functional rescue experiments using GLX351322 were
performed following HtrA1 knockdown in bEnd.3 cells. The sh-HtrA1 +
GLX351322 group exhibited a significant decrease in NOX4 expression
compared with the sh-NC group (Fig.
8A). The present study evaluated expression levels of tight
junction proteins, ZO1, occludin and CLDN5. Western blot analysis
did not indicate any significant differences between the sh-HtrA1 +
GLX351322 and sh-NC groups (Fig.
8A). In the FITC-dextran experiment, no significant increase in
permeability was observed for the sh-HtrA1 + GLX351322 compared
with the sh-NC group (Fig. 8B).
There was no significant difference in levels of caspase3 in the
sh-HtrA1 + GLX351322 compared with the sh-NC group (Fig. 8A). Moreover, flow cytometry
showed that the apoptosis rate in the sh-HtrA1 + GLX351322 group
was similar to that in the sh-NC group (Fig. 8C). These findings indicated that
inhibiting NOX4 expression enhanced the expression of tight
junction proteins, decreased cell permeability and mitigated
apoptosis. Western blot analysis (Fig. 8D) showed elevated protein
expression of NOX4 in the AAV/B130-shHtrA1 compared with the
AAV/B130-NC mice. GLX351322 treatment significantly decreased the
NOX4 levels, indicating effective inhibition. Additionally,
GLX351322 increased the expression of tight junction proteins ZO1,
occludin and CLDN5 and decreased the expression of caspase3
compared with that in the AAV/B130-shHtrA1 group. The Evans Blue
assay revealed decreased BBB permeability following GLX351322
injection (Fig. 8E).
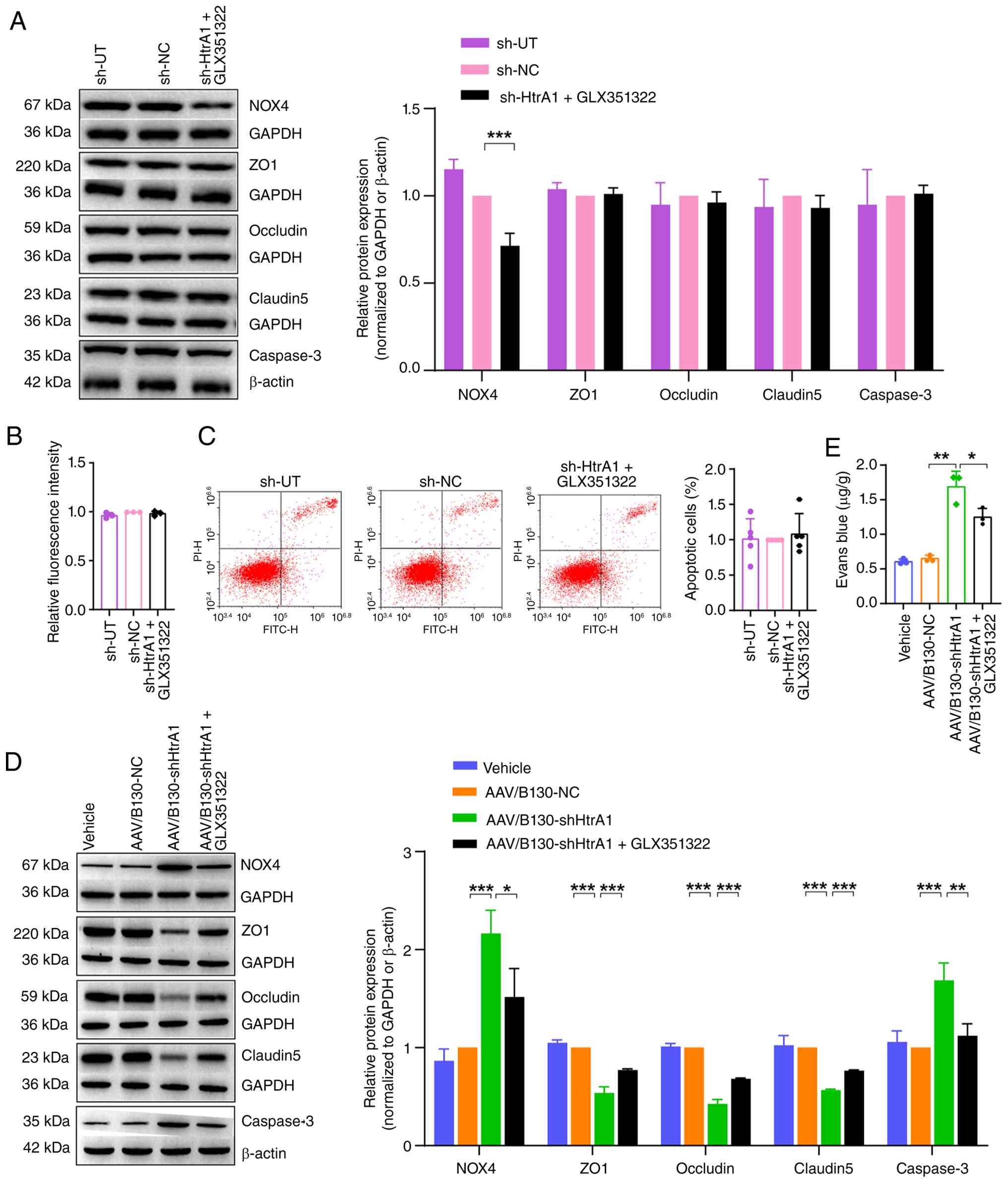 | Figure 8HtrA1 regulates tight junctions,
permeability and apoptosis of brain vascular endothelial cells via
the NOX4-dependent oxidative stress pathway. (A) Western blot
analysis of NOX4, ZO1, occludin, claudin 5 and caspase3 protein
expression in bEnd.3 cells. (B) Permeability of bEnd.3 cell
monolayers was assessed using FITC-dextran. (C) Flow cytometry
analysis of bEnd.3 cell apoptosis. (D) Western blot analysis of
NOX4, ZO1, occludin, claudin 5 and caspase3 in the mouse brain
tissue. (E) Blood-brain barrier permeability of the mouse brain
tissue was determined using Evans blue assay.
*P<0.05, **P<0.01,
***P<0.001. HtrA1, high temperature requirement
serine peptidase A1; ZO1, zonula occludens protein 1; sh, short
hairpin; UT, untransfected; NC, negative control. |
Intraperitoneal injection of GLX351322
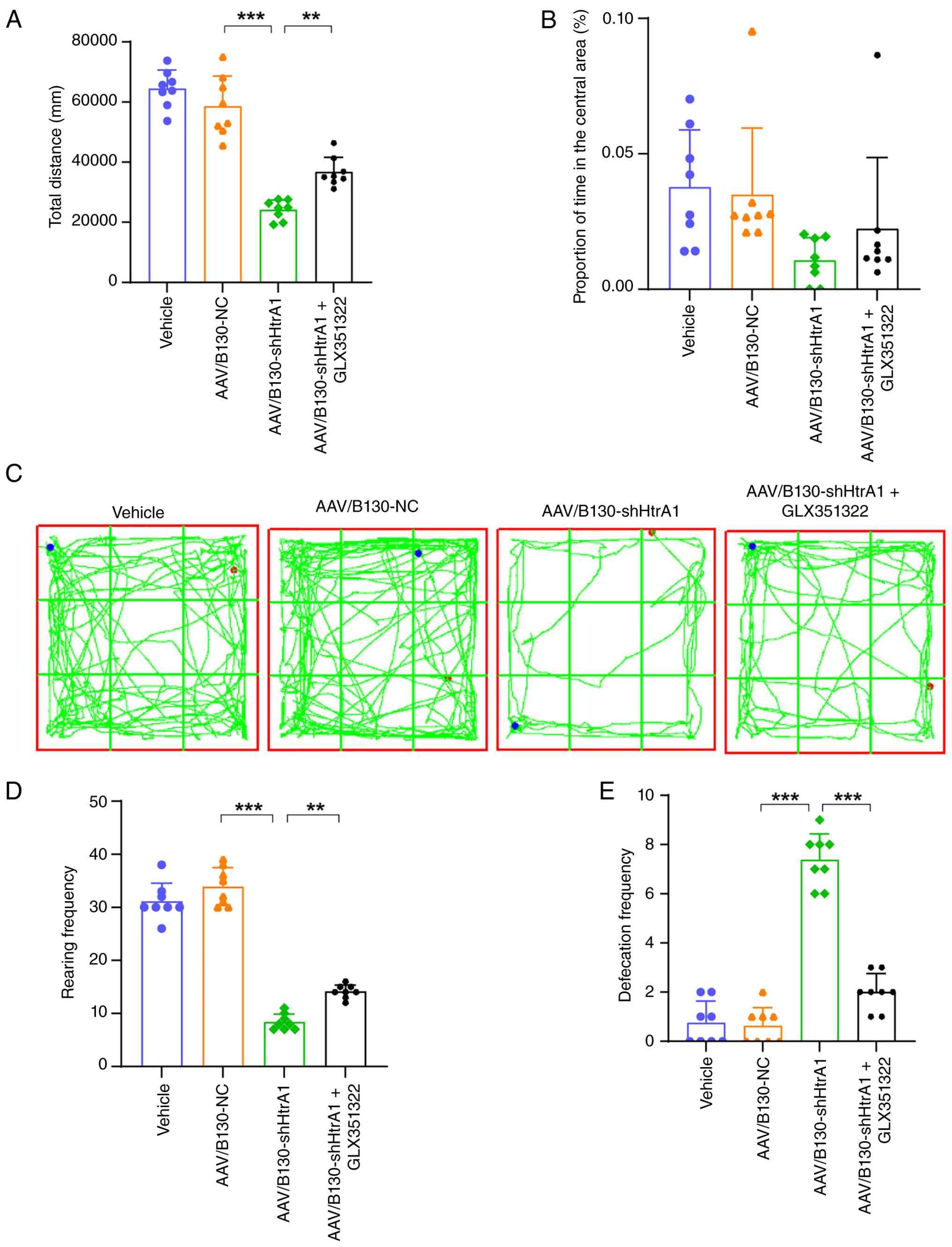
attenuates immobility and anxiety in AAV/B130-shHtrA1 mice
In the OFT, the AAV/B130-shHtrA1 mice exhibited
significantly decreased total distance traveled and decreased time
spent in the central region compared with the AAV/B130-NC mice
(Fig. 9A-C). However, GLX351322
significantly increased the total distance travelled and increased
time spent in the central region in the AAV/B130-shHtrA1 mice
(Fig. 9A-C). AAV/B130-shHtrA1
mice showed decreased rearing and increased defecation frequency
compared with the AAV/B130-NC mice (Fig. 9D and E). By contrast, the
AAV/B130-shHtrA1 + GLX351322 mice exhibited increased rearing and
decreased defecation frequency (Fig.
9D and E). These results indicated that GLX351322 effectively
reversed depressive immobility and anxiety in the AAV/B130-shHtrA1
mice.
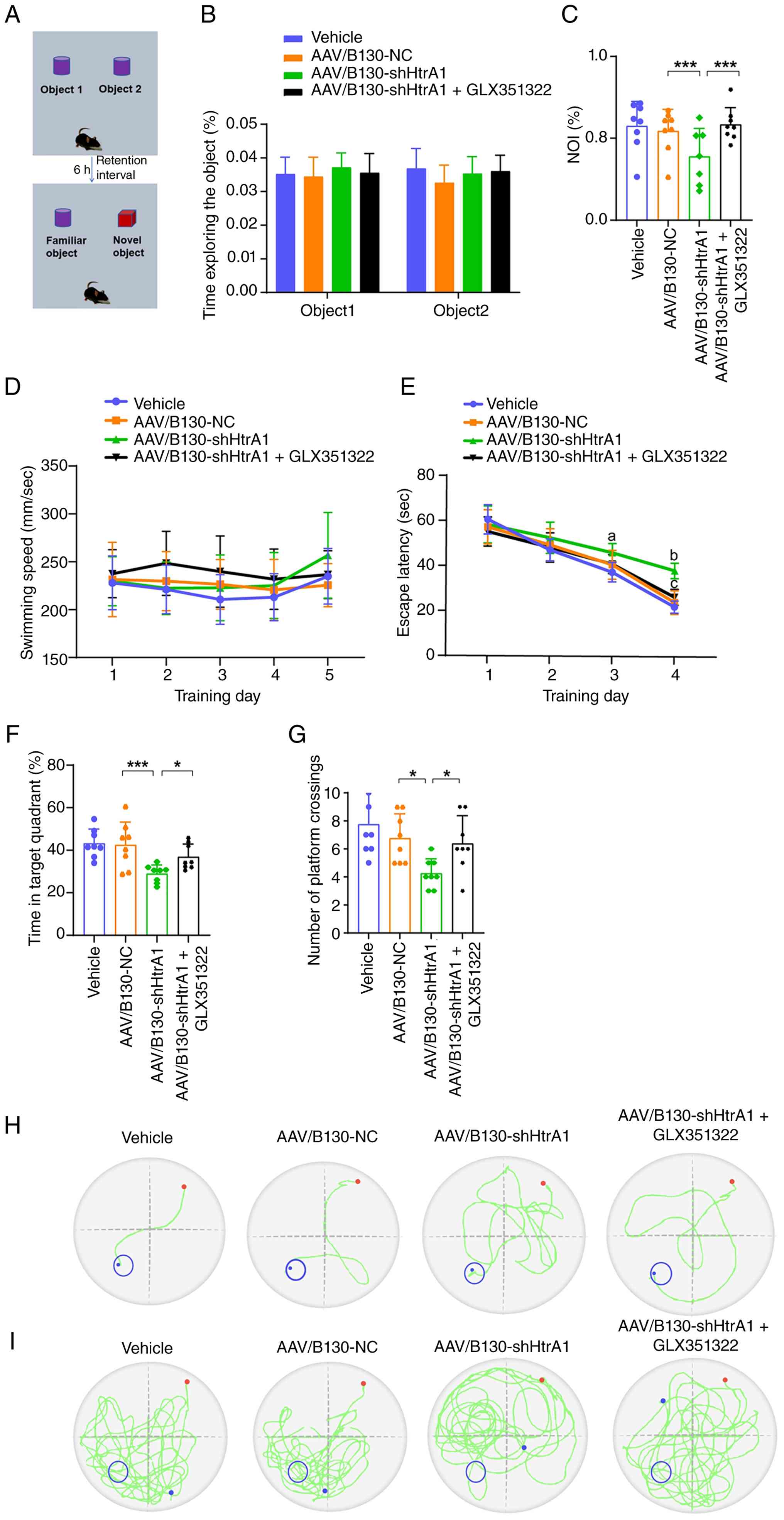
Intraperitoneal injection of GLX351322
ameliorates recognition and spatial memory deficit in
AAV/B130-shHtrA1 mice
In the NORT (Fig.
10A), no significant difference in the exploration time for two
identical objects during the familiarization period was observed
between groups (Fig. 10B).
During the test phase, the NOI in AAV/B130-shHtrA1 mice was
significantly lower than that in AAV/B130-NC mice, whereas
intraperitoneal injection of GLX351322 reversed this decline
(Fig. 10C). These results
indicated that the intraperitoneal injection of GLX351322 rescued
memory deficit in the AAV/B130-shHtrA1 mice. MWM test was used to
evaluate spatial learning and memory in mice. No difference in mean
swimming speed was observed between the groups during the entire
MWM test (Fig. 10D). The escape
latency of AAV/B130-shHtrA1 mice was markedly longer on training
days 3 and 4 than that of the AAV/B130-NC group, whereas the
intraperitoneal injection of GLX351322 significantly shortened the
escape latency of AAV/B130-shHtrA1 mice on day 4 during the place
navigation-training phase (Fig.
10E), indicating GLX351322 relieved the spatial learning
deficit in AAV/B130-shHtrA1 mice. In the probe test, the
AAV/B130-shHtrA1 mice demonstrated significantly decreased
percentage of swimming time in the target quadrant (Fig. 10F) and number of platform
crossings (Fig. 10G) compared
with the AAV/B130-NC mice, whereas these decreases were reversed in
AAV/B130-shHtrA1 + GLX351322 mice, indicating GLX351322
proficiently improved the spatial reference memory in
AAV/B130-shHtrA1 mice. Representative trajectories on training day
4 (Fig. 10H) and during the
probe test (Fig. 10I) are
shown.
Discussion
In F1, patients exhibited clinical features typical
of CSVD, consistent with clinical manifestations of HTRA1
mutations reported in the literature (25,26). Moreover, the symptoms in patients
from F2 also support the role of HTRA1 mutations in the
pathogenesis of CSVD (20). In
the present study, the heterozygous HTRA1 mutations were
predicted to be pathogenic. These findings are consistent with
previous studies indicating that HTRA1 heterozygous
mutations result in the loss of protein function, triggering
extracellular matrix accumulation, cerebrovascular and retinal
vascular degeneration and white matter lesions (27,28). RNA-seq results showed significant
differences in the expression of genes between heterozygous
HTRA1mcs and HCs, supporting the existing literature on the
crucial role of HTRA1 in cellular signaling and its regulation of
physiological functions (29,30). The present study found
significant changes in the gene expression of genes associated with
oxidative stress and apoptosis in HTRA1mcs compared with HCs. mRNA
levels of NOX4, a key oxidative stress marker, and
BAX, a key indicator of apoptosis, were upregulated in the
HTRA1mcs. Conversely, the expression of mRNAs encoding tight
junction proteins was significantly downregulated. These findings
indicated that a disruption in the integrity of the tight junctions
may contribute to the pathophysiology of CVSD. Functional analyses,
including KEGG pathway analysis, revealed significant enrichment in
'apoptosis'. Collectively, these data indicated that the
dysregulation of apoptosis signaling may serve a key role in the
disease process. This may be associated with the cerebral
microbleeds affected by heterozygous HTRA1 mutations
(31). These results suggest
heterozygous HTRA1 mutations not only affect HTRA1
expression but may also influence cell survival and function by
regulating downstream signaling pathways.
Previous studies have indicated that endothelial
dysfunction, oxidative stress and apoptosis are key factors
contributing to the development of CSVD (32-34). HTRA1 is expressed both intra- and
extracellularly and participates in various biological processes,
such as cell apoptosis, oxidative stress response (35-37) and maintenance of the BBB
(38). The pathological changes
observed in patients with cerebral autosomal recessive arteriopathy
with subcortical infarcts and leukoencephalopathy include fibrosis,
thinning of the adventitia of cerebral arterioles, loss of smooth
muscle cells and thickening of the intima (39). Loss of HTRA1 impairs the
maturation of smooth muscle cells and affects the function of
vascular smooth muscle (40).
Based on the moderate expression of HTRA1 in endothelial cells, it
was hypothesized that these cells serve a role in this process.
HtrA1 could enhance endothelial cell function by regulating
intracellular signaling pathways and promoting cell proliferation
and survival (41). However,
HTRA1 also inhibits the proliferation of esophageal squamous cell
carcinoma, ovarian and endometrial cancer, melanoma, neuroblastoma,
liver cancer, and thyroid cancer cells (42,43). Additionally, in polypoid
choroidal vasculopathy, HTRA1 OE decreases the proliferation of
chorioretinal and human umbilical vein endothelial cells (44). HTRA1 promotes skin cell viability
and proliferation, further confirming that it serves diverse roles
in different types of tissue (45). The BBB permeability is associated
with CSVD. Vascular endothelial cells and their tight junctions are
key elements of the BBB function and structure (46). Disruption of this barrier
involves increased paracellular permeability of endothelial cells,
primarily due to the degradation of junctional proteins, such as
ZO1, occludin and CLDN (46).
The decrease in tight junction proteins leads to increased gaps
between endothelial cells, making it easier for macromolecules and
harmful substances to penetrate the BBB, thereby increasing the
risk of neurological damage (47). The present study found elevated
expression of tight junction-associated proteins and decreased cell
permeability following HtrA1 OE, which further supported the
hypothesis that HtrA1 OE contributes to the maintenance of the
endothelial barrier integrity, which is key for preventing an
increase in vascular permeability and inflammatory response
(48). HtrA1 knockdown disrupts
the intercellular tight junctions, leading to a decrease in
endothelial barrier function, which may increase vascular
permeability and promote inflammatory responses (49). The present endothelial
cell-specific HtrA1 knockdown mouse model demonstrated
normal expression of HtrA1 contributes to the stabilization of BBB
structure and function. The present findings suggested that
HTRA1 mutations contributed to the development of CSVD by
affecting endothelial cell functionality and modulating the BBB
permeability. Impaired BBB function is hypothesized to be a common
pathological alteration in both CADASIL and HTRA1-associated CSVD
with heterozygous mutations (38).
HTRA1 promotes apoptosis in tumor cells (50,51) in a caspase-dependent or
caspase-independent manner, associated with the activation of
caspase3 and caspase7 (52).
HTRA1 OE triggers retinal epithelial cell apoptosis in age-related
macular degeneration (53), and
promotes fibroblast survival with antiapoptotic effects in a
porcine wound healing model (54). In summary, HtrA1 inhibited
apoptosis in vivo and in vitro. However, the effects
of HtrA1 are contradictory, depending on the specific cell type and
environmental context.
NOX4 is a key oxidase that serves a role in the
oxidative stress response of endothelial cells (55). High NOX4 expression causes
endothelial cell dysfunction, which leads to various cardiovascular
diseases (56-58). Therefore, HtrA1 may protect
cerebrovascular endothelial cells by inhibiting NOX4 expression,
thereby maintaining normal cell function. Moreover, the absence of
HtrA1 may result in increased sensitivity of endothelial cells to
oxidative stress, exacerbating cell damage and apoptosis (59,60). In summary, oxidative stress is
associated with the tight junctions between endothelial cells.
Elevated oxidative stress, characterized by increased catalase
expression and NADPH oxidase activity, leads to overall barrier
dysfunction. Oxidative stress increases the BBB permeability
(61), whereas NOX4 knockdown
restores the expression of tight junction proteins and preserves
the integrity of the endothelial cell barrier (62). Here, increased oxidative stress
from NOX4 decreased the expression of tight junction proteins and
increased the permeability of bEnd.3 cells, whereas inhibition of
NOX4 expression reversed this effect.
Oxidative stress is associated with endothelial
cell apoptosis and NOX4-dependent accumulation of ROS is a notable
cause of apoptosis in vascular endothelial cells (63). NOX4 induces apoptosis in brain
endothelial cells during inflammation-induced oxidative stress
(55,64). It also induces apoptosis in
arterial smooth muscle (65) and
human umbilical vein endothelial cells (66). NOX4 knockdown decreases the
production of ROS, caspase3 activity and the expression of Bcl-2
family members (67). Here,
HtrA1 knockdown increased the expression of NOX4 and the levels of
ROS and MDA and decreased SOD activity in bEnd.3 cells. Here, HtrA1
knockdown increased apoptosis in bEnd.3 cells, pointing to an
essential role of HtrA1 in preserving tight junctions and cell
barrier function. These results demonstrated the pathogenic
mechanism of CSVD associated with HTRA1 and provided a basis for
further investigations.
In the present study, downregulated HtrA1
expression in mouse cerebral vascular endothelial cells resulted in
increased anxiety-like behaviors, as well as notable decreases in
memory and spatial learning abilities. The present study showed
that HtrA1 was essential for regulating behavior in mice,
particularly via oxidative stress-related signaling pathways. NOX4
OE is associated with the development of depressive symptoms,
whereas its inhibition may improve depressive symptoms by
decreasing oxidative stress and inflammatory responses (68). In addition, the GLX351322
intervention significantly increased the anxiety-like behaviors in
mice in the OFT, suggesting that it may act by modulating the
neurotransmitter system (69).
As GLX351322 improved cognitive function in mice, HtrA1 may
influence behavior via the NOX4-mediated oxidative stress pathway.
These findings not only enhance understanding of the role of HtrA1
in hereditary CSVD but also provide a theoretical foundation for
developing future therapeutic strategies for this condition
(68,70,71). Consequently, targeting the NOX4
pathway may offer a promising therapeutic strategy for
counteracting the deleterious effects of HTRA1 mutations in
patients with CSVD, ultimately improving neurological outcomes.
The present study has several limitations. While
the present findings implicated the NOX4-mediated oxidative stress
pathway in HTRA1-associated CSVD, peripheral blood transcriptomics
serves primarily as a screening tool and does not constitute a
direct readout of cerebral endothelial events. The restricted
sample size (two families) and mutation spectrum (protease domain
variants p.P285L/p. R302Q) may limit generalizability. Expanding
cohorts to include mutations across functional domains may
strengthen genotype-phenotype associations. The AAV/B130-shHtrA1
model incompletely recapitulates the human heterozygous partial
loss-of-function effects. The shRNA knockdown model primarily
mimics a haploinsufficiency state. However, certain HTRA1
mutations may exert dominant-negative effects or alter protease
function in manners not captured by gene knockdown. In the future,
the use of knock-in animal models harboring specific
patient-derived mutations may elucidate these potential
mutation-specific effects. While the present study focused on
endothelial cells, future investigations examining other key
components of the neurovascular unit, such as pericytes, astrocytes
and microglia, may be essential to elucidate the pathophysiology of
CSVD. Although the present study identified HtrA1-NOX4
interactions, the precise regulatory mechanism remains unknown and
studies using coimmunoprecipitation and chromatin accessibility
assays are required.
Heterozygous HTRA1 mutations underlie CSVD
pathogenesis and mediate their effects via NOX4-mediated oxidative
stress, which disrupts endothelial homeostasis and BBB integrity.
By demonstrating the efficacy of NOX4 inhibition using GLX351322 in
rescuing cerebrovascular and cognitive deficit, the present study
provided preclinical evidence in support of the HTRA1-NOX4
interaction as a potential intervention target.
Availability of data and materials
The data generated in the present study may be
found in the Gene Expression Omnibus under accession number
GSE324041 or at the following URL: ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE324041.
Authors' contributions
SS, SX and CL conceived and designed the study. SS,
MS, WJ, WY and XL conducted the experiments. SS, WY, and JW
analyzed data. SS and JW confirm the authenticity of all the raw
data. SS drafted the manuscript. SX and CL revised the manuscript.
All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the ethics
committee of the First Hospital of Shanxi Medical University,
Taiyuan, China (approval No. KYLL-2024-161). All animal experiments
were approved by the Animal Welfare and Ethics Committee of the
First Hospital of Shanxi Medical University (approval no.
DWYJ-2025-304, Taiyuan, China). All participants provided written
informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
AAV
|
adeno-associated virus
|
|
BBB
|
blood-brain barrier
|
|
CADASIL
|
cerebral autosomal dominant
arteriopathy with subcortical infarcts and leukoencephalopathy
|
|
CCK-8
|
Cell Counting Kit-8
|
|
CLDN5
|
claudin 5
|
|
CSVD
|
cerebral small vessel disease
|
|
FLAIR
|
fluid-attenuated
inversion-recovery
|
|
GO
|
Gene Ontology
|
|
HC
|
healthy control
|
|
HTRA1mc
|
high-temperature requirement serine
peptidase A1 mutation carrier
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
MDA
|
malondialdehyde
|
|
MMSE
|
Mini-Mental State Examination
|
|
MoCA
|
Montreal Cognitive Assessment
|
|
MOI
|
multiplicity of infection
|
|
MWM
|
Morris water maze
|
|
NOI
|
novel object recognition index
|
|
NORT
|
novel object recognition test
|
|
NOX4
|
NADPH oxidase 4
|
|
OCEL1
|
occludin-like protein 1
|
|
OFT
|
open-field test
|
|
RT-q
|
reverse
transcription-quantitative
|
|
seq
|
sequencing
|
|
ROS
|
reactive oxygen species
|
|
RTCA
|
real-time cellular analysis
|
|
SOD
|
superoxide dismutase
|
|
TGF-β
|
transforming growth factor-β
|
|
WMH
|
white matter hyperintensity
|
Acknowledgements
Not applicable.
Funding
The present study was supported by Shanxi Basic Research Program
(grant nos. 202303021221222, 202403021212232, 202403021221350 and
202503021211261).
References
|
1
|
Dupré N, Drieu A and Joutel A:
Pathophysiology of cerebral small vessel disease: A journey through
recent discoveries. J Clin Invest. 134:e1728412024. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Salvadori E, Brambilla M, Maestri G,
Nicotra A, Cova I, Pomati S and Pantoni L: The clinical profile of
cerebral small vessel disease: Toward an evidence-based
identification of cognitive markers. Alzheimers Dement. 19:244–260.
2023. View Article : Google Scholar :
|
|
3
|
Chojdak-Łukasiewicz J, Dziadkowiak E,
Zimny A and Paradowski B: Cerebral small vessel disease: A review.
Adv Clin Exp Med. 30:349–356. 2021. View Article : Google Scholar
|
|
4
|
Gao Y, Li D, Lin J, Thomas AM, Miao J,
Chen D, Li S and Chu C: Cerebral small vessel disease: Pathological
mechanisms and potential therapeutic targets. Front Aging Neurosci.
14:9616612022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Clausen T, Kaiser M, Huber R and Ehrmann
M: HTRA proteases: Regulated proteolysis in protein quality
control. Nat Rev Mol Cell Biol. 12:152–162. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Krojer T, Garrido-Franco M, Huber R,
Ehrmann M and Clausen T: Crystal structure of DegP (HtrA) reveals a
new protease-chaperone machine. Nature. 416:455–459. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tossetta G, Fantone S, Licini C, Marzioni
D and Mattioli-Belmonte M: The multifaced role of HtrA1 in the
development of joint and skeletal disorders. Bone. 157:1163502022.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
An E, Sen S, Park SK, Gordish-Dressman H
and Hathout Y: Identification of novel substrates for the serine
protease HTRA1 in the human RPE secretome. Invest Ophthalmol Vis
Sci. 51:3379–3386. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tiaden AN, Breiden M, Mirsaidi A, Weber
FA, Bahrenberg G, Glanz S, Cinelli P, Ehrmann M and Richards PJ:
Human serine protease HTRA1 positively regulates osteogenesis of
human bone marrow-derived mesenchymal stem cells and mineralization
of differentiating bone-forming cells through the modulation of
extracellular matrix protein. Stem Cells. 30:2271–2282. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shiga A, Nozaki H, Yokoseki A, Nihonmatsu
M, Kawata H, Kato T, Koyama A, Arima K, Ikeda M, Katada S, et al:
Cerebral small-vessel disease protein HTRA1 controls the amount of
TGF-β1 via cleavage of proTGF-β1. Hum Mol Genet. 20:1800–1810.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Beaufort N, Scharrer E, Kremmer E, Lux V,
Ehrmann M, Huber R, Houlden H, Werring D, Haffner C and Dichgans M:
Cerebral small vessel disease-related protease HtrA1 processes
latent TGF-β binding protein 1 and facilitates TGF-β signaling.
Proc Natl Acad Sci USA. 111:16496–16501. 2014. View Article : Google Scholar
|
|
12
|
Hara K, Shiga A, Fukutake T, Nozaki H,
Miyashita A, Yokoseki A, Kawata H, Koyama A, Arima K, Takahashi T,
et al: Association of HTRA1 mutations and familial ischemic
cerebral small-vessel disease. N Engl J Med. 360:1729–1739. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
De Luca A, De Falco M, Severino A,
Campioni M, Santini D, Baldi F, Paggi MG and Baldi A: Distribution
of the serine protease HtrA1 in normal human tissues. J Histochem
Cytochem. 51:1279–1284. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yao Y and Li N: Effect of HtrA1
polymorphism on sensitivity to chemotherapy in patients with colon
cancer. Med Sci Monit. 26:e9219332020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tiaden AN and Richards PJ: The emerging
roles of HTRA1 in musculoskeletal disease. Am J Pathol.
182:1482–1488. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Uemura M, Nozaki H, Kato T, Koyama A,
Sakai N, Ando S, Kanazawa M, Hishikawa N, Nishimoto Y, Polavarapu
K, et al: HTRA1-related cerebral small vessel disease: A review of
the literature. Front Neurol. 11:5452020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu JY, Zhu YC, Zhou LX, Wei YP, Mao CH,
Cui LY, Peng B and Yao M: HTRA1-related autosomal dominant cerebral
small vessel disease. Chin Med J (Engl). 134:178–184. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Verdura E, Hervé D, Scharrer E, Amador
Mdel M, Guyant-Maréchal L, Philippi A, Corlobé A, Bergametti F,
Gazal S, Prieto-Morin C, et al: Heterozygous HTRA1 mutations are
associated with autosomal dominant cerebral small vessel disease.
Brain. 138:2347–2358. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu SY, Li HJ, Li S, Ren QQ, Liang JL and
Li CX: Heterozygous pathogenic and likely pathogenic symptomatic
HTRA1 variant carriers in cerebral small vessel disease. Int J Gen
Med. 16:1149–1162. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Qian E, Uemura M, Kobayashi H, Nakamura S,
Ozawa F, Yoshimatsu S, Ishikawa M, Onodera O, Morimoto S and Okano
H: A human induced pluripotent stem cell model from a patient with
hereditary cerebral small vessel disease carrying a heterozygous
R302Q mutation in HTRA1. Inflamm Regen. 43:232023. View Article : Google Scholar
|
|
21
|
Nozaki H, Kato T, Nihonmatsu M, Saito Y,
Mizuta I, Noda T, Koike R, Miyazaki K, Kaito M, Ito S, et al:
Distinct molecular mechanisms of HTRA1 mutants in manifesting
heterozygotes with CARASIL. Neurology. 86:1964–1974. 2016.
View Article : Google Scholar
|
|
22
|
Zellner A, Scharrer E, Arzberger T, Oka C,
Domenga-Denier V, Joutel A, Lichtenthaler SF, Müller SA, Dichgans M
and Haffner C: CADASIL brain vessels show a HTRA1 loss-of-function
profile. Acta Neuropathol. 136:111–125. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fasano A, Formichi P, Taglia I, Bianchi S,
Di Donato I, Battisti C, Federico A and Dotti MT: HTRA1 expression
profile and activity on TGF-β signaling in HTRA1 mutation carriers.
J Cell Physiol. 235:7120–7127. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
25
|
Xu SY, Li L, Sun WX, Shen JY and Li CX:
Case report: Hypnic headache responds to agomelatine-a potential
prophylactic treatment option. Front Neurol. 14:11793912023.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shang T, Pinho M, Ray D and Khera A: Two
unique mutations in HTRA1-related cerebral small vessel disease in
north America and Africa and literature review. J Stroke
Cerebrovasc Dis. 30:1060292021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Coste T, Hervé D, Neau JP, Jouvent E, Ba
F, Bergametti F, Lamy M, Cogez J, Derache N, Schneckenburger R, et
al: Heterozygous HTRA1 nonsense or frameshift mutations are
pathogenic. Brain. 144:2616–2624. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Poulsen ET, Nielsen NS, Scavenius C,
Mogensen EH, Risør MW, Runager K, Lukassen MV, Rasmussen CB,
Christiansen G, Richner M, et al: The serine protease HtrA1 cleaves
misfolded transforming growth factor β-induced protein (TGFBIp) and
induces amyloid formation. J Biol Chem. 294:11817–11828. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
May A, Su F, Dinh B, Ehlen R, Tran C,
Adivikolanu H and Shaw PX: Ongoing controversies and recent
insights of the ARMS2-HTRA1 locus in age-related macular
degeneration. Exp Eye Res. 210:1086052021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang Y, Shi C, Li Y, Yu W, Wei S, Fan Y,
Mao C, Yang Z, Yu L, Zhao Z, et al: Genetic study of cerebral small
vessel disease in Chinese Han population. Front Neurol.
13:8294382022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kobayashi Y, Kondo Y, Tazawa KI, Yamamoto
K, Yoshinaga T, Nakamura K and Sekijima Y: HTRA1-related cerebral
small-vessel disease causes cerebral microbleeds on the brainstem
surface. J Neurol Sci. 466:1232292024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li MT, Ke J, Guo SF, Wu Y, Bian YF, Shan
LL, Liu QY, Huo YJ, Guo C, Liu MY, et al: The protective effect of
quercetin on endothelial cells injured by hypoxia and
reoxygenation. Front Pharmacol. 12:7328742021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jiang S, Ma X, Chen Y, Gu B, Sun N and
Xiao H: Effects of ginkgo diterpene lactone on brain inflammation
and oxidative stress in rats with cognitive impairment of cerebral
small vessel disease. Am J Transl Res. 13:6382–6390.
2021.PubMed/NCBI
|
|
34
|
Lu YW, Hao RJ, Wei YY and Yu GR: The
protective effect of harpagoside on angiotensin II (Ang II)-induced
blood-brain barrier leakage in vitro. Phytother Res. 35:6241–6254.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Spugnini EP, Cardillo I, Fanciulli M,
Crispi S, Vincenzi B, Boccellino M, Quagliuolo L and Baldi A:
Electroporation as a strategy to promote HtrA1 gene uptake and
chemotherapy efficacy in a mouse model of mesothelioma. Front
Biosci (Elite Ed). 5:974–981. 2013.PubMed/NCBI
|
|
36
|
Tossetta G, Fantone S, Giannubilo SR,
Ciavattini A, Senzacqua M, Frontini A and Marzioni D: HTRA1 in
placental cell models: A possible role in preeclampsia. Curr Issues
Mol Biol. 45:3815–3828. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Clawson GA, Bui V, Xin P, Wang N and Pan
W: Intracellular localization of the tumor suppressor HtrA1/Prss11
and its association with HPV16 E6 and E7 proteins. J Cell Biochem.
105:81–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li Y, Ying Y, Yao T, Jia X, Liang H, Tang
W, Jia X, Song H, Shao X, Wang DJJ, et al: Decreased water exchange
rate across blood-brain barrier in hereditary cerebral small vessel
disease. Brain. 146:3079–3087. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fukutake T: Cerebral autosomal recessive
arteriopathy with subcortical infarcts and leukoencephalopathy
(CARASIL): From discovery to gene identification. J Stroke
Cerebrovasc Dis. 20:85–93. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ikawati M, Kawaichi M and Oka C: Loss of
HtrA1 serine protease induces synthetic modulation of aortic
vascular smooth muscle cells. PLoS One. 13:e01966282018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pan S, Liu M, Xu H, Chuan J and Yang Z:
Lipopolysaccharide activating NF-kB signaling by regulates HTRA1
expression in human retinal pigment epithelial cells. Molecules.
28:22362023. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xia J, Wang F, Wang L and Fan Q: Elevated
serine protease HtrA1 inhibits cell proliferation, reduces
invasion, and induces apoptosis in esophageal squamous cell
carcinoma by blocking the nuclear factor-κB signaling pathway.
Tumour Biol. 34:317–328. 2013. View Article : Google Scholar
|
|
43
|
Li Y, Yuan J, Rothzerg E, Wu X, Xu H, Zhu
S and Xu J: Molecular structure and the role of high-temperature
requirement protein 1 in skeletal disorders and cancers. Cell
Prolif. 53:e127462020. View Article : Google Scholar :
|
|
44
|
Jiang J, Huang L, Yu W, Wu X, Zhou P and
Li X: Overexpression of HTRA1 leads to down-regulation of
fibronectin and functional changes in RF/6A cells and HUVECs. PLoS
One. 7:e461152012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yamawaki S, Naitoh M, Kubota H, Aya R,
Katayama Y, Ishiko T, Tamura T, Yoshikawa K, Enoshiri T, Ikeda M
and Suzuki S: HtrA1 is specifically up-regulated in active keloid
lesions and stimulates keloid development. Int J Mol Sci.
19:12752018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ballabh P, Braun A and Nedergaard M: The
blood-brain barrier: An overview: Structure, regulation, and
clinical implications. Neurobiol Dis. 16:1–13. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Song D, Lee JY, Park EC, Choi NE, Nam HY,
Seo J and Lee J: Structure-activity relationship analysis of
activity-based probes targeting HTRA family of serine proteases.
Bioorg Med Chem Lett. 87:1292592023. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yamamoto Y, Kojima K, Taura D, Sone M,
Washida K, Egawa N, Kondo T, Minakawa EN, Tsukita K, Enami T, et
al: Human iPS cell-derived mural cells as an in vitro model of
hereditary cerebral small vessel disease. Mol Brain. 13:382020.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Guo F, Tao X, Wu Y, Dong D, Zhu Y, Shang D
and Xiang H: Carfilzomib relieves pancreatitis-initiated pancreatic
ductal adenocarcinoma by inhibiting high-temperature requirement
protein A1. Cell Death Discov. 10:582024. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chien J, Staub J, Hu SI, Erickson-Johnson
MR, Couch FJ, Smith DI, Crowl RM, Kaufmann SH and Shridhar V: A
candidate tumor suppressor HtrA1 is downregulated in ovarian
cancer. Oncogene. 23:1636–1644. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yu T, Chen CZ and Xing YQ: Inhibition of
cell proliferation, migration and apoptosis in blue-light
illuminated human retinal pigment epithelium cells by
down-regulation of HtrA1. Int J Ophthalmol. 10:524–529.
2017.PubMed/NCBI
|
|
52
|
Chien J, Aletti G, Baldi A, Catalano V,
Muretto P, Keeney GL, Kalli KR, Staub J, Ehrmann M, Cliby WA, et
al: Serine protease HtrA1 modulates chemotherapy-induced
cytotoxicity. J Clin Invest. 116:1994–2004. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chen Y, Xing X, Dai W, Tian L, Dong Z and
Yu S: Brain regions involved in fractional amplitude of
low-frequency fluctuation in cluster headache patients: A
resting-state functional MRI study. BMC Neurol. 22:3362022.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sabino F, Madzharova E and Auf dem Keller
U: Cell density-dependent proteolysis by HtrA1 induces
translocation of zyxin to the nucleus and increased cell survival.
Cell Death Dis. 11:6742020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Basuroy S, Bhattacharya S, Leffler CW and
Parfenova H: Nox4 NADPH oxidase mediates oxidative stress and
apoptosis caused by TNF-alpha in cerebral vascular endothelial
cells. Am J Physiol Cell Physiol. 296:C422–C432. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Davis CM, Lyon-Scott K, Varlamov EV, Zhang
WH and Alkayed NJ: Role of endothelial STAT3 in cerebrovascular
function and protection from ischemic brain injury. Int J Mol Sci.
23:121672022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kuppusamy M, Ottolini M and Sonkusare SK:
Role of TRP ion channels in cerebral circulation and neurovascular
communication. Neurosci Lett. 765:1362582021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Alves JV, da Costa RM, Awata WMC,
Bruder-Nascimento A, Singh S, Tostes RC and Bruder-Nascimento T:
NADPH oxidase 4-derived hydrogen peroxide counterbalances
testosterone-induced endothelial dysfunction and migration. Am J
Physiol Endocrinol Metab. 327:E1–E12. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Nivoit P, Mathivet T, Wu J, Salemkour Y,
Sankar DS, Baudrie V, Bourreau J, Guihot AL, Vessieres E, Lemitre
M, et al: Autophagy protein 5 controls flow-dependent endothelial
functions. Cell Mol Life Sci. 80:2102023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Storm J, Wu Y, Davies J, Moxon CA and
Craig AG: Testing the effect of PAR1 inhibitors on Plasmodium
falciparum-induced loss of endothelial cell barrier function.
Wellcome Open Res. 5:342020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Lochhead JJ, McCaffrey G, Quigley CE,
Finch J, DeMarco KM, Nametz N and Davis TP: Oxidative stress
increases blood-brain barrier permeability and induces alterations
in occludin during hypoxia-reoxygenation. J Cereb Blood Flow Metab.
30:1625–1636. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jiang J, Huang K, Xu S, Garcia JGN, Wang C
and Cai H: Targeting NOX4 alleviates sepsis-induced acute lung
injury via attenuation of redox-sensitive activation of
CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction. Redox
Biol. 36:1016382020. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ma Y and Li W, Yin Y and Li W: AST IV
inhibits H2O2-induced human umbilical vein
endothelial cell apoptosis by suppressing Nox4 expression through
the TGF-β1/Smad2 pathway. Int J Mol Med. 35:1667–1674. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Liu J, Chandaka GK, Zhang R and Parfenova
H: Acute antioxidant and cytoprotective effects of sulforaphane in
brain endothelial cells and astrocytes during inflammation and
excitotoxicity. Pharmacol Res Perspect. 8:e006302020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Pedruzzi E, Guichard C, Ollivier V, Driss
F, Fay M, Prunet C, Marie JC, Pouzet C, Samadi M, Elbim C, et al:
NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced
endoplasmic reticulum stress and apoptosis in human aortic smooth
muscle cells. Mol Cell Biol. 24:10703–10717. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Simon F and Fernández R: Early
lipopolysaccharide-induced reactive oxygen species production
evokes necrotic cell death in human umbilical vein endothelial
cells. J Hypertens. 27:1202–1216. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zhang Q, Liu X, Li N, Zhang J, Yang J and
Bu P: Sirtuin 3 deficiency aggravates contrast-induced acute kidney
injury. J Transl Med. 16:3132018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Liao J, Peng B, Huang G, Diao C, Qin Y,
Hong Y, Lin J, Lin Y, Jiang L, Tang N, et al: Inhibition of NOX4
with GLX351322 alleviates acute ocular hypertension-induced retinal
inflammation and injury by suppressing ROS mediated redox-sensitive
factors activation. Biomed Pharmacother. 165:1150522023. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Tao W, Yu L, Shu S, Liu Y, Zhuang Z, Xu S,
Bao X, Gu Y, Cai F, Song W, et al: miR-204-3p/Nox4 mediates memory
deficits in a mouse model of Alzheimer's disease. Mol Ther.
29:396–408. 2021. View Article : Google Scholar
|
|
70
|
Chen M, Yang S, Wu Y, Zhao Z, Zhai X and
Dong D: High temperature requirement A1 in cancer: Biomarker and
therapeutic target. Cancer Cell Int. 21:5132021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Anvari E, Wikström P, Walum E and Welsh N:
The novel NADPH oxidase 4 inhibitor GLX351322 counteracts glucose
intolerance in high-fat diet-treated C57BL/6 mice. Free Radic Res.
49:1308–1318. 2015. View Article : Google Scholar : PubMed/NCBI
|