Introduction
In recent years, with the increasing global burden
of obesity and metabolic syndrome, non-alcoholic fatty liver
disease (NAFLD) has become one of the most common chronic liver
diseases in adults. The global prevalence of NAFLD is estimated to
be around 29.8%, reaching as high as 30-40% in regions such as
Europe and North America (1,2).
The pathological spectrum of NAFLD ranges from simple hepatic
steatosis to non-alcoholic steatohepatitis (NASH), which is
characterized by inflammation and varying degrees of fibrosis. In
some patients, NASH can further progress to cirrhosis or even
hepatocellular carcinoma, significantly increasing the risk of both
liver-related and systemic complications. Epidemiological data show
that patients with NAFLD have a markedly increased incidence of
cardiovascular events and type 2 diabetes and approximately
1.5-6.5% of cases will progress to NASH within 10 years,
accompanied by worsening fibrosis and an exponentially increasing
risk of poor outcomes (3).
Moreover, NASH is closely associated with chronic kidney disease,
atherosclerosis and sleep apnea, suggesting that it is not merely
an isolated liver disorder, but a complex manifestation of systemic
multi-organ metabolic dysregulation, posing unprecedented
challenges for public health and clinical management.
The urea cycle, which is unique to mammalian
hepatocytes, is a central hub of nitrogen metabolism. It detoxifies
excess ammonia (NH3) by converting it into soluble urea,
thereby enabling its safe excretion and maintaining nitrogen
homeostasis. This cycle is accomplished through the cooperation of
mitochondrial and cytosolic compartments and involves six key
enzymes: N-acetyl-L-glutamate synthase (NAGS), carbamoyl phosphate
synthetase 1 (CPS1), ornithine transcarbamylase (OTC),
argininosuccinate synthase (ASS1), argininosuccinate lyase (ASL)
and cytosolic arginase 1 (ARG1). Among these, NAGS-produced
N-acetylglutamate is an essential allosteric activator of CPS1,
ensuring precise regulation of cycle flux. The expression and
activity of these enzymes are influenced by various physiological
hormones (such as glucagon and glucocorticoids) and nutritional
status and are further finely tuned by epigenetic mechanisms
including DNA methylation and histone modifications (4,5).
As well as detoxification, the urea cycle also plays important
roles in amino acid biosynthesis, one-carbon metabolism and its
crosstalk with the tricarboxylic acid (TCA) cycle (6). The tight coupling of its carbon
skeleton to the energy metabolic network highlights the critical
importance of coordinated nitrogen and carbon metabolism for
hepatic homeostasis (7).
A study has shown that urea cycle flux is markedly
impaired in the livers of NAFLD/NASH patients (8). Multiple transcriptomic and
proteomic analyses have revealed downregulation of key enzymes such
as CPS1, OTC and ASS1, accompanied by elevated intrahepatic free
ammonia concentrations and glutamine metabolic reprogramming.
During the development of NASH, hepatic CPS1 and OTC protein levels
and activities are found to decrease proportionally and these
changes can be partially reversed by refeeding with a normal diet,
suggesting a central role for epigenetic regulation in this process
(9). Meanwhile, loss or
dysfunction of ARG1 can exacerbate nitric oxide (NO) production
imbalance in hepatocytes, triggering excessive oxidative stress and
inflammatory cell apoptosis, which significantly promotes hepatic
fibrogenesis. Conversely, maintaining the metabolic integrity of
the arginine-citrulline-arginine cycle helps inhibit pathological
NO synthesis and mitigate hepatocyte inflammatory injury (10). Additionally, metabolomics studies
have identified characteristic alterations in urea cycle
intermediates and arginine metabolic branches (including nitric
oxide and polyamines) in the serum of NASH patients, underscoring a
bidirectional feedback relationship between nitrogen metabolic
imbalance and hepatic immunometabolic microenvironment remodeling
(11).
Materials and methods
The present study was a narrative review based
exclusively on published literature and no original data from our
group are included. Nevertheless, by integrating basic mechanistic
studies and clinical observations, it aimed to provide a coherent
framework linking urea-cycle impairment and arginine/NO metabolism
to NAFLD/NASH pathogenesis, to the development of tissue and
circulating biomarkers and to potential therapeutic strategies
targeting these pathways.
A narrative literature search was conducted using
the PubMed database (https://pubmed.ncbi.nlm.nih.gov/) from its inception
to October 31, 2025. The search combined Medical Subject Headings
and free-text terms related to non-alcoholic fatty liver disease
and nitrogen/arginine metabolism. Representative search strings
included combinations of the following terms: 'non-alcoholic fatty
liver disease' OR 'nonalcoholic steatohepatitis' OR NAFLD OR NASH;
and 'urea cycle' OR 'carbamoyl phosphate synthetase 1' OR CPS1 OR
'ornithine transcarbamylase' OR OTC OR 'argininosuccinate synthase'
OR ASS1 OR 'argininosuccinate lyase' OR ASL OR arginase OR ARG1 OR
ARG2 OR 'arginine metabolism' OR 'nitric oxide' OR iNOS OR 'hepatic
stellate cell*' OR 'Kupffer cell*' OR 'hepatic macrophage*' OR
'hyperammonemia' OR 'ammonia.' These terms were combined with
Boolean operators (AND/OR) to capture both mechanistic and clinical
studies relevant to urea-cycle impairment and arginine metabolism
in NAFLD/NASH. Eligible literature included peer-reviewed
English-language primary studies and selected reviews on
urea-cycle/arginine-NO/arginase biology in NAFLD/NASH (or related
metabolic liver injury/fibrogenesis), spanning mechanistic work in
cells/animals and translational evidence from human tissues,
cohorts, enzyme assays and multi-omics. The present study excluded
non-peer-reviewed or non-English articles, abstracts only, studies
focused mainly on inherited urea-cycle disorders without clear
NAFLD/NASH relevance and reports centered on non-metabolic liver
diseases.
Eligible publications were peer-reviewed articles in
English, including original basic research (cell culture, animal
models and human tissue studies), clinical observational studies,
interventional trials and high-quality narrative or systematic
reviews that provided mechanistic or translational insights into
urea-cycle and arginine metabolism in NAFLD/NASH. Exclusion
criteria comprised case reports or small case series without
mechanistic data; conference abstracts, editorials, or comments
lacking primary results; studies focusing exclusively on inborn
errors of the urea cycle or other liver diseases without clear
relevance to NAFLD/NASH; and articles not providing specific
information on urea-cycle enzymes, arginine/NO metabolism, or
related biomarkers/therapeutic targets. When multiple reports from
the same cohort or model were available, priority was given to the
most comprehensive or recent publication.
Fundamentals of the urea cycle and arginine
metabolism
Urea cycle
In mammals, the integration of nitrogen and carbon
metabolism forms a crucial network that maintains homeostasis, with
the urea cycle and L-arginine metabolism serving as central nodes.
The urea cycle efficiently converts toxic NH3, produced
by protein degradation and amino acid deamination, into non-toxic,
water-soluble urea for safe excretion and detoxification.
Meanwhile, arginine, a semi-essential amino acid, not only
participates in protein synthesis but also gives rise to multiple
functional molecules via various enzyme branches, including nitric
oxide NO, polyamines, creatine and citrulline. These form complex
metabolic pathways with increasingly recognized physiological and
pathological significance (7,12-14).
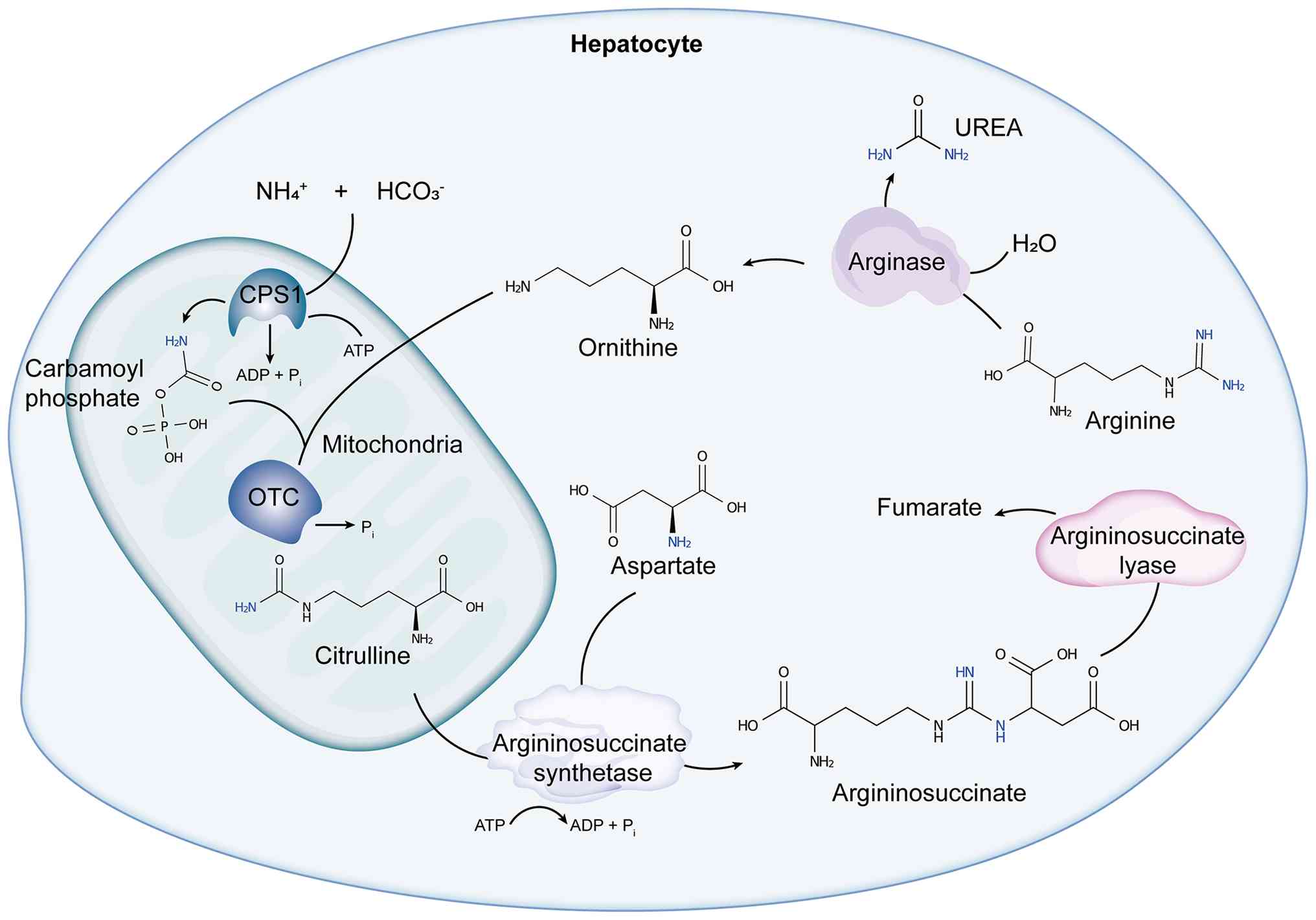
In hepatocytes, the urea cycle is divided into
mitochondrial and cytosolic phases, coordinated by six core enzymes
(15,16). It initiates in the mitochondrial
matrix, where NAGS catalyzes the formation of N-acetylglutamate
(NAG) from glutamate and acetyl-CoA. NAG serves as an essential
allosteric activator of CPS1, driving the formation of carbamoyl
phosphate from ammonia and bicarbonate in the presence of two ATP
molecules. OTC then combines carbamoyl phosphate and ornithine to
produce citrulline. After citrulline is transported to the cytosol,
ASS1 and ASL sequentially convert it into arginine and fumarate.
Finally, ARG1 hydrolyzes arginine into ornithine and urea, thus
completing the nitrogen metabolic cycle. This process consumes four
phosphate bonds (3 ATP → 2 ADP + AMP) per mole of urea produced and
is tightly coupled to the TCA cycle and other metabolic pathways to
maintain energy and material balance (7) (Fig.
1).
Regulation of the urea cycle
Efficient operation of the urea cycle in hepatocytes
relies on precise multi-level molecular regulation, including gene
transcription, epigenetic modifications and post-translational
modifications, ensuring that ammonia detoxification flux remains
synchronized with metabolic demands. NAGS, which catalyzes NAG
synthesis as the cycle's entry point, is tightly regulated at the
transcriptional level by various factors. For example,
phosphorylation of Sp1 and CREB in response to increased
intracellular cAMP enhances their binding to the NAGS promoter,
while liver-specific transcription factors hepatocyte nuclear
factor 1 and nuclear factor Y respond to glucagon and
glucocorticoid signals by synergizing with enhancer elements to
upregulate NAGS expression, adapting to changes in nitrogen
excretion under fasting or stress (17,18). Additionally, DNA methylation and
histone acetylation play key roles; methylation at CpG sites in the
NAGS promoter correlates negatively with its mRNA expression and
histone deacetylase 1-mediated deacetylation of H3K9ac represses
NAGS transcription (19-22).
As the rate-limiting enzyme of the urea cycle, CPS1
activity and expression depend on NAG allosteric activation and are
also finely regulated by post-translational modifications. In the
mitochondrial matrix, the NAD+-dependent deacetylase
SIRT5 can deacetylate CPS1 at K55 and K287, markedly enhancing its
catalytic efficiency; fasting or high-protein diets promote
mitochondrial NAD+ accumulation, increasing SIRT5
activity and accelerating ammonia detoxification. Sirt5-knockout
mice exhibit hyperammonemia and impaired urea production when
fasting, underscoring the core role of the SIRT5-CPS1 axis in
ammonia metabolism adaptation (23-26). Furthermore, SIRT5 also mediates
desuccinylation and demalonylation of CPS1, constructing a
multi-dimensional regulatory network responsive to energy state
fluctuations (24,26). Additionally, O-GlcNAc
glycosylation of CPS1 rises in hyperglycemia or aging, blocking NAG
binding and suppressing enzyme activity; caloric restriction or
low-carbohydrate diets reduce this modification and restore
function (23,27). Oxidative stress induces
peroxynitrite (ONOO−)-mediated tyrosine nitration of
CPS1, disrupting allosteric sites and further reducing activity, a
mechanism validated in drug-induced liver injury, implicating
protein nitration as a key node in ammonia metabolism imbalance
under oxidative/nitrosative stress (23,28). Phosphoproteomic studies also show
that CPS1 can be phosphorylated at serine/threonine sites by
kinases such as PKA and PKC, potentially altering mitochondrial
localization or protein interactions to couple energy and urea
cycle signaling (27,29).
The second enzyme in the cycle, OTC, is similarly
subject to multi-level regulation. Proteomics of Sirt5-knockout
mice reveals hyper-succinylation of OTC, indicating SIRT5's role in
maintaining OTC activity and ammonia flux (23). On the epigenetic level, DNA
hypermethylation of the OTC promoter in NAFLD/NASH patients is
closely associated with transcriptional downregulation, directly
impairing hepatocyte detoxification capacity and providing a
potential therapeutic target (30,31).
ASS1 catalyzes the critical step converting
citrulline to arginine and is regulated by cell proliferation and
stress signals. Tumor suppressor p53 can bind directly to the ASS1
promoter to induce transcription, thus protecting cell survival in
DNA damage or metabolic stress by suppressing excessive Akt pathway
activation; ASS1-deficient cells are more sensitive to radiotherapy
and chemotherapy, confirming its dual role as a p53 target gene
(32,33). Moreover, activation of the AMPK
pathway promotes ASS1 mRNA expression, enhancing the
citrulline-arginine cycle to meet the demand for arginine and
downstream NO production under energy deprivation (34,35).
ASL is not only key to arginine synthesis but also
influences downstream NO production, affecting vascular homeostasis
and immune responses. ASL transcription is jointly regulated by
hypoxia, insulin and glucocorticoid signals (13). In malnutrition models, glucose
and cAMP pathways suppress ASL mRNA levels, while glucocorticoids
upregulate ASL expression via GRE elements, partially mediated by
increased PGC-1α and FoxO, ensuring a continuous supply of urea
cycle intermediates for acute and chronic metabolic stress
adaptation (36-39).
Finally, ARG1 catalyzes the hydrolysis of arginine
to ornithine and urea, completing nitrogen excretion. Transcription
of ARG1 is directly regulated by the hepatic glucocorticoid
receptor (GR): In situ experiments and liver-specific GR
knockout models show that GR activity determines ARG1 mRNA and
protein expression. GR agonists (such as dexamethasone) robustly
induce ARG1 transcription, boosting urea synthesis and preventing
hyperammonemia; conversely, GR-deficient mice fail to induce ARG1
even with dexamethasone, resulting in ammonia metabolism disorders
and neuromuscular impairment (40). Additionally, caloric restriction
significantly upregulates ARG1, highlighting its adaptive role in
aging and nutrient limitation. Experiments in mice of different
ages show that fasting or caloric restriction elevates hepatic ARG1
mRNA and enzyme activity, delaying age-related ammonia metabolism
decline (41). In macrophage M2
polarization, IL-4 activates STAT6 and C/EBPβ, collaborating with
RAR/RXR heterodimers to recruit chromatin remodeling complexes and
the transcription elongation factor TFIIS, massively upregulating
ARG1 and promoting tissue repair and anti-inflammatory responses
(42-46). Moreover, hypoxia-inducible factor
HIF-1α has been shown to downregulate ARG1 in hyperuricemia-induced
liver injury, linking ARG1 to hepatic inflammation, oxidative
stress and apoptosis and highlighting its importance as a
regulatory target in metabolic and inflammatory diseases (47).
Arginine metabolism and its spatial
heterogeneity
Arginine is not only an intermediate of the urea
cycle but also a hub for multiple downstream metabolic pathways
(9). In the cytosol, ASS1 and
ASL form the citrulline-arginine cycle to regenerate arginine from
citrulline, which is indispensable for systemic arginine balance,
protein synthesis and NO production (48). Arginine metabolism branches
through various enzyme systems: Generation of NO and citrulline via
nitric oxide synthase (NOS), with NO serving as a signaling
molecule in hemodynamics, immune defense and cellular signaling;
polyamine production via ornithine decarboxylase (ODC), involved in
cell proliferation and differentiation; creatine biosynthesis via
arginine:glycine amidinotransferase, supporting energy supply for
muscle and nervous tissue; formation of methylated arginine
derivatives, such as asymmetric dimethylarginine (ADMA) and SDMA,
affecting endothelial function and cardiovascular risk assessment
(7). Overall, the complex
branches of arginine metabolism illustrate the intersection of
nitrogen metabolism with broad biological processes and its
physiological/pathological significance continues to expand with
ongoing research (7,14,49).
In the conversion of arginine to NO, three main NOS
isoforms, neuronal NOS (nNOS), inducible NOS (iNOS) and endothelial
NOS (eNOS), regulate NO production in a synergistic or antagonistic
manner (49-51). All NOS enzymes require NADPH,
molecular oxygen and tetrahydrobiopterin as cofactors, using an
electron transfer chain to oxidize arginine to Nω-hydroxyarginine
and subsequently to NO and citrulline (52,53). The isoforms differ in
calcium/calmodulin sensitivity, gene regulation, tissue
distribution and functional effects: nNOS is mainly found in the
nervous system (neurotransmission, plasticity), eNOS is restricted
to vascular endothelium (maintaining vascular tone and blood flow)
and iNOS is strongly induced during inflammation or infection,
producing high NO concentrations for antimicrobial and signaling
purposes (51,54,55).
Competing with the NOS pathway, arginase hydrolyzes
arginine to ornithine and urea (56). Mammals have two arginase
isoforms: Hepatic ARG1 (cytosolic, terminal step in the urea cycle)
and mitochondrial ARG2 (found in kidney, immune cells and other
tissues, involved in local ornithine and polyamine synthesis)
(57). Studies confirm that
arginase activity is essential not only for urea production but
also for competing with NOS for arginine, thereby regulating NO
levels and playing complex roles in immunity, fibrosis and
metabolic disease (58). For
example, upregulated Arg1 expression in activated hepatic stellate
cells (HSCs) promotes collagen synthesis and fibrosis progression,
while Arg1 inhibition alleviates fibrosis, indicating therapeutic
potential in tissue repair and pathology (59,60). Taken together, these enzymatic
branches highlight that arginine metabolism is not only
biochemically diversified but also poised to be differentially
engaged across tissues and cell types.
As well as this enzymatic complexity at the
whole-organ level, arginine metabolism is further shaped by
pronounced spatial organization within tissues, particularly in the
liver. Within the highly structured liver, urea cycle and arginine
metabolism activity exhibit significant spatial and functional
heterogeneity that operates at two interrelated levels: metabolic
zonation among hepatocytes and differences between parenchymal and
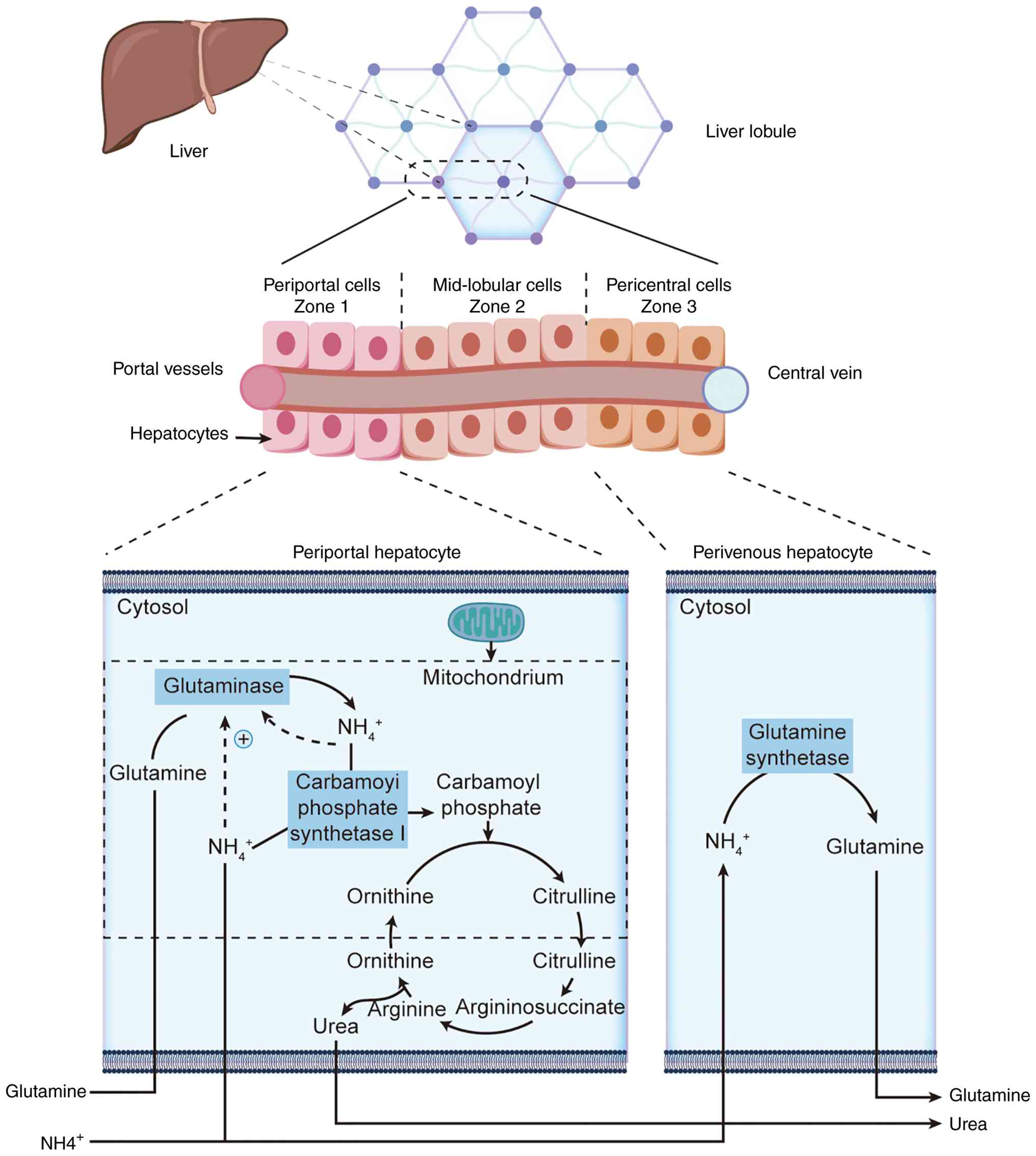
non-parenchymal cell types (61,62). The classical Couinaud
segmentation divides hepatocytes into periportal (zone 1), midzonal
(zone 2) and pericentral (zone 3) regions based on blood supply
gradients (63,64). In the periportal region, blood is
rich in oxygen and amino acids and urea cycle enzymes are highly
expressed to detoxify most ammonia and generate urea (65,66); minimal ammonia escaping to the
pericentral region is converted to glutamine by glutamine
synthetase at extremely low concentrations, ensuring blood ammonia
levels remain safe, demonstrating fine spatial division of labor
within hepatic lobules (67-70) (Fig. 2). Complementing this hepatocyte
zonation, non-parenchymal cells provide additional layers of
heterogeneity to hepatic nitrogen and immunometabolic cross-talk.
Kupffer cells, the most abundant resident macrophage population,
display strong phagocytic and secretory capacities and, depending
on their M1/M2 polarization state, express iNOS or Arg1, biasing
local NO or ornithine metabolism and influencing global
inflammation and repair; their plasticity and microenvironmental
dependence make them core players in NAFLD/NASH progression
(71-74). HSCs, located in the space of
Disse, store vitamin A in quiescence and upon activation
differentiate into myofibroblast-like cells with high Arg1
expression, promoting polyamine and collagen production to drive
fibrosis; hepatic sinusoidal endothelial cells utilize eNOS-derived
NO to regulate blood flow and the local microenvironment, which is
critical for oxygen gradient and metabolic zonation (55,60).
Metabolic reprogramming in hepatocytes
Under physiological homeostasis, hepatocytes
maintain systemic nitrogen balance and efficient ammonia
detoxification via a robust nitrogen metabolic network (75,76). Ammonia, primarily derived from
protein and amino acid catabolism, must be tightly regulated
intracellularly; otherwise, it can strongly inhibit mitochondrial
function and oxidative phosphorylation (77). In hepatocytes, the urea cycle
remains the only pathway in mammals capable of efficiently
converting ammonia into urea, with its flux sufficient to detoxify
tens of grams of ammonia daily and ensure blood ammonia
concentrations are kept within a safe range (<50 μmol/l)
(13,75).
Alterations of the urea cycle in
hepatocytes under diabetic conditions and their implications
Under high-fat and/or high-glucose conditions, the
urea cycle in hepatocytes often undergoes functional failure
(30) (Fig. 3). Animal and clinical studies
have demonstrated that in high-fat, high-cholesterol diet-induced
NASH rat models, the gene and protein expression of OTC and CPS1 is
markedly downregulated, accompanied by hypermethylation in their
promoter regions, decreased enzymatic activity and intrahepatic
ammonia accumulation. Restoration of a normal diet partially
reverses these epigenetic alterations, indicating a critical role
for DNA methylation in the plastic regulation of the urea cycle
under pathological states (30).
In human patients with NAFLD, liver tissue analyses from 20 cases
of steatosis and 15 cases of NASH also confirmed that mRNA levels
of CPS1, OTC, ASS1 and other urea cycle-related enzymes are
decreased by an average of 30-40%, with urea production capacity
significantly correlating with gene expression. This suggests that
lipid accumulation itself suppresses hepatic ammonia detoxification
via transcriptional and epigenetic mechanisms (8). In addition, in high-fructose,
high-fat-induced steatosis mouse hepatocytes, hypermethylation of
urea cycle enzyme gene promoters correlates with low expression,
further exacerbating the risk of cell injury under hyperammonemic
conditions (30). A further
epigenetic study indicated that the low expression of urea cycle
enzymes associated with NAFLD is not an isolated event but rather
part of a global remodeling of the hepatocellular DNA methylation
landscape (78). In a NASH-like
liver injury model induced in C57BL/6J and DBA/2J mice by a
lipogenic methyl-deficient diet, the liver exhibits not only marked
steatosis but also global and repetitive element DNA
hypomethylation, altered trimethylation of histone H3K9 and H3K27
and changes in H4K20 trimethylation, together with pronounced
alterations in the protein expression of the maintenance DNA
methyltransferase DNMT1 and the de novo methyltransferase
DNMT3A, suggesting that DNA methyltransferases themselves are
highly sensitive to nutritional and metabolic status during the
development of fatty liver injury (78). In human NAFLD liver biopsies,
methylation levels at specific CpG sites within the mitochondrial
MT ND6 gene are higher in patients with NASH than in those with
simple steatosis and the ratio of methylated to unmethylated DNA is
positively correlated with the NAFLD activity score and
histological severity, whereas MT ND6 mRNA and protein expression
are reduced; DNMT1 is markedly upregulated in NASH and subcellular
abnormalities such as disrupted mitochondrial cristae and increased
numbers of peroxisomes can be observed (79). Taken together with the global DNA
methylation and histone modification abnormalities described in the
mouse model, these findings suggest that as disease progresses from
simple steatosis to NASH, the liver undergoes methylation
reprogramming aligned with metabolic stress at both the nuclear and
mitochondrial genomic levels (78,79). These lines of evidence
collectively support the concept that NAFLD-related nutritional
imbalances, such as methyl donor deficiency and environmental
exposures such as reduced physical activity, can alter one-carbon
metabolism and DNMT expression or activity, thereby creating a
hepatic epigenetic milieu that is prone to aberrant CpG methylation
in the promoters of key metabolic genes and in turn provides a
permissive background for transcriptional repression of urea
cycle-related genes (78,79).
However, to the best of the authors' knowledge, no previous study
has directly analyzed whether epigenetic alterations at urea cycle
enzyme genes in NAFLD/NASH liver tissue directly drive their
reduced expression and the associated impairment of ammonia
detoxification. The most relevant to this possibility comes from a
epigenetic study of CPS1 (22),
the rate-limiting enzyme of the urea cycle. Analyses of human
hepatocellular carcinoma cell lines and tumor tissues have shown
that two CpG dinucleotides in the proximal CPS1 promoter and a
CpG-rich region within the first intron are hypermethylated,
coinciding with marked downregulation of CPS1 mRNA and loss of CPS1
protein in tumor specimens and HCC cells; treatment of HCC cells
with the demethylating agent 5-aza-2'-deoxycytidine reduces
methylation at these CpG sites, markedly restores CPS1
transcription and increases the activity of reporter constructs
containing the CPS1 promoter in parallel with CpG demethylation,
demonstrating that DNA methylation at specific CpG sites directly
mediates CPS1 transcriptional silencing in HCC (22). By measuring expression of the
liver-specific transcription factor HNF3β and the hepatocyte marker
gene AAT, this study also ruled out the possibility that CPS1
downregulation is mainly attributable to hepatocyte
dedifferentiation and a global collapse of liver-specific
transcriptional programs and in its discussion, drawing on prior
evidence, it emphasized that promoter CpG hypermethylation often
silences genes by blocking transcription factor access, thereby
providing a rationale for viewing CPS1 promoter CpG
hypermethylation as a potential mechanism regulating urea cycle
enzyme expression rather than definitive evidence in NAFLD/NASH
(22). Thus, integrating the
evidence for altered DNMT1 and DNMT3A expression in NAFLD animal
models, abnormal MT ND6 methylation in livers from patients with
NAFLD and CPS1 promoter and gene body CpG hypermethylation in HCC,
it can be hypothesized that under chronic nutritional imbalance,
metabolic overload and inflammatory stress, hepatocytes may undergo
DNMT-mediated CpG methylation reprogramming to target genes
encoding urea cycle enzymes, maintaining low expression of CPS1 and
other key enzymes and ultimately weakening hepatic ammonia
detoxification capacity; however, this interpretation remains
inferential and requires direct validation in NAFLD/NASH (22,78,79).
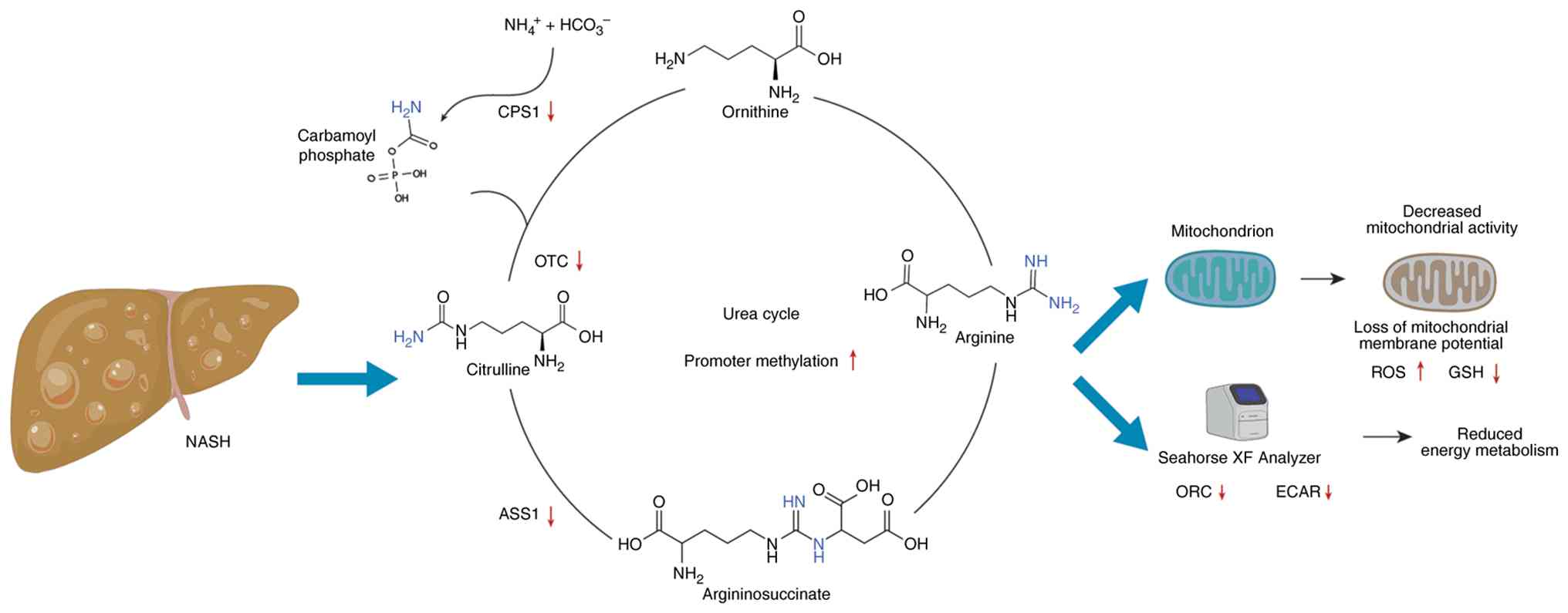 | Figure 3Epigenetic suppression of the
hepatocyte urea cycle under metabolic stress: in highfat,
highcholesterol diet-induced NASH rats, CpG hypermethylation of
CPS1 and OTC promoters leads to a marked drop in their mRNA/protein
levels, reduced enzymatic activity and intrahepatic ammonia
buildup-changes that are partially reversed upon return to a normal
diet. Likewise, human NAFLD (steatosis and NASH) liver samples show
a 30-40% decrease in CPS1, OTC and ASS1 transcripts correlating
with impaired urea production and highfructose, highfat mouse
hepatocytes display similar promoter hypermethylation and enzyme
downregulation. All figures were generated using Microsoft
PowerPoint (Microsoft Corporation). NASH, non-alcoholic
steatohepatitis; CPS1, carbamoyl phosphate synthetase 1; OTC,
ornithine transcarbamylase; NAFLD, non-alcoholic fatty liver
disease; ASS1, argininosuccinate synthase; ECAR, extracellular
acidification rate. |
Decreased urea cycle enzyme activity in the livers
of patients with NAFLD is positively correlated with intrahepatic
ammonia concentrations. Excess ammonia can induce HSCs activation
and fibrogenesis, providing a kinetic basis for the transition from
simple steatosis to inflammatory fibrosis (80). At the cellular level, in
vitro studies have revealed that when ammonia exceeds 100
μmol/l, it interacts with key enzymes of the mitochondrial
electron transport chain, inhibiting NADH:ubiquinone oxidoreductase
(complex I) and cytochrome c oxidase (complex IV), thereby
disrupting proton pump function in the respiratory chain (77). Experiments with isolated hepatic
mitochondria show that NH4+ concentrations of
1-20 mM significantly inhibit dehydrogenase activity, leading to
loss of mitochondrial membrane potential, opening of permeability
transition pores, excess ROS generation, lipid peroxidation,
glutathione (GSH) depletion and a precipitous drop in ATP
synthesis: Hallmarks of an energy crisis (81).
Further metabolic flux analysis shows that under
excess ammonia, glutamate dehydrogenase 2 reversibly aminates
α-ketoglutarate (α-KG), rapidly depleting TCA cycle α-KG, promoting
glutamate and glutamine synthesis and redirecting substrate flux to
non-oxidative branches: Ultimately impairing sustained TCA cycle
energy supply and metabolic homeostasis (77). This not only slows regeneration
of critical cycle intermediates, but also leads to diminished
proton-motive force in the mitochondrial matrix, hindering normal
oxidative phosphorylation. Meanwhile, ammonia exerts bidirectional
effects on glucose metabolism: Seahorse and metabolomics
experiments demonstrate that in cell models exposed to high
ammonia, both mitochondrial respiration (oxygen consumption rate)
and glycolytic function (extracellular acidification rate) sharply
decline, indicating that ammonia can rapidly suppress the two major
cellular energy pathways, further aggravating energy deficiency
(77).
Arginine-mediated nitric oxide cycle in
hepatocytes
As an intermediate of the urea cycle, arginine is
also the sole substrate for various NOS that generate NO. The
metabolic fate of arginine within hepatocytes directly determines
NO content and signal strength and the 'arginine-NO axis' finely
regulates mitochondrial function, TCA cycle activity and
glucose/lipid metabolism. Basal NO in hepatocytes primarily derives
from endogenous eNOS activity. This nanomolar-level NO activates
soluble guanylate cyclase (sGC) to catalyze GTP to cyclic GMP
(cGMP), subsequently activating cGMP-dependent protein kinase (PKG)
and downstream coactivator PGC-1α, thereby promoting mitochondrial
biogenesis and respiratory chain gene expression, increasing
mitochondrial density and oxidative phosphorylation capacity
(82,83). The sGC/cGMP pathway also inhibits
acetyl-CoA carboxylase (ACC), lowers malonyl-CoA, relieves
carnitine palmitoyltransferase-1 (CPT-1) inhibition, enhances fatty
acid transport into mitochondria and β-oxidation, ultimately
suppressing lipogenesis and boosting fatty acid utilization
(84).
Low NO exerts biphasic effects on the TCA cycle: It
mildly inhibits complex IV activity, transiently lowers membrane
potential, triggers ROS as signaling molecules and activates AMPK
and PGC-1α-mediated mitochondrial biogenesis. At moderate NO, the
pyruvate dehydrogenase (PDH) complex and mitochondrial aconitase
are reversibly inhibited, reducing pyruvate flux into the TCA cycle
and promoting glutamine-dependent anaplerosis, thereby sustaining
α-KG and ATP homeostasis (85).
When NO rises further, PDH and aconitase inhibition intensifies,
causing mitochondrial citrate accumulation that not only feeds back
to inhibit glycolytic enzymes (pyruvate kinase), but also
suppresses CPT-1, reducing fatty acid oxidation and shifting cell
metabolism toward glycolysis and glutamine pathways to adapt to
compromised oxidative phosphorylation (85).
In glucose metabolism, NO significantly inhibits
hepatic glycogen synthesis and gluconeogenesis: experiments show
that in primary rat hepatocytes treated with NO donor
S-nitroso-N-acetylcysteine, glycogen synthase activity and
gluconeogenic enzymes (pyruvate carboxylase, phosphoenolpyruvate
carboxykinase) are markedly reduced, with intracellular
glucose-6-phosphate and UDP-glucose accumulating; indicating that
NO suppresses glycogen synthase conversion to the active form and
limits glucose storage and gluconeogenic substrate availability
(86). eNOS-derived NO via
cGMP-PKG also downregulates G6Pase, reducing hepatic glucose output
and thereby synergistically modulating systemic glucose balance
(85).
In lipid metabolism, low NO via sGC/cGMP promotes
fatty acid oxidation, downregulates ACC, activates AMPK, increases
CPT-1 activity and enhances mitochondrial fatty acid uptake and
degradation (84,85). However, in inflammatory or
endotoxin-stimulated settings, iNOS becomes highly expressed in
hepatocytes, producing micromolar to millimolar NO that rapidly
reacts with superoxide to form ONOO−. ONOO−
causes nitration and oxidation of mitochondrial complexes I and III
and various metabolic enzymes (PDH, CPT-1), resulting in loss of
membrane potential, S-nitrosylation and persistent inhibition of
complex I, excessive ROS production, GSH depletion, energy crisis
and activation of apoptosis signals (87-90).
Thus, the competition between arginase and iNOS for
arginine in hepatocytes is pivotal: Arg2 knockout mice on a
high-fat diet exhibit worse hepatic lipid accumulation, upregulated
iNOS and exacerbated NO/ONOO−-mediated oxidative damage,
indicating that enhancing Arg2 activity can mitigate hepatic
inflammation and steatosis (91). Conversely, nonselective NOS
inhibitor l-NAME treatment in obese rats aggravates mitochondrial
dysfunction, triglyceride accumulation and proinflammatory cytokine
levels, further validating the bidirectional role of NO homeostasis
in hepatic metabolic adaptation (92).
In summary, the arginine-NO axis in hepatocytes
regulates mitochondrial biogenesis, TCA flux, glucose-lipid
metabolic balance and redox state in a concentration-dependent
manner. Its precise equilibrium is vital for energy homeostasis and
stress resilience. During NAFLD-to-NASH progression, inflammation
and/or DNA methylation-driven downregulation of urea cycle enzymes
and impaired Arg2 activity shift arginine metabolism toward iNOS,
exacerbating NO-ONOO−-induced mitochondrial and
metabolic injury.
Metabolic cross-talk between the hepatic
urea cycle and glucose-lipid metabolism in NAFLD/NASH
Under physiological conditions, amino acids enter
the tricarboxylic acid TCA cycle via transamination to generate
oxaloacetate and aspartate, the latter serving as a substrate for
the urea cycle. At the same time, fumarate generated by the urea
cycle can re-enter the TCA cycle, providing carbon backbones and
reducing equivalents for gluconeogenesis and fatty acid synthesis,
thereby tightly coupling nitrogen metabolism with hepatic glucose
and lipid metabolism (93). In
obesity-related NAFLD and NASH, hepatic fatty acid influx is
increased while fatty acid oxidation and very low-density
lipoprotein export are constrained, leading to excessive lipid
accumulation in hepatocytes and establishing a background of
metabolic imbalance together with insulin resistance and disordered
glucose metabolism. On this basis, expression and activity of key
urea cycle enzymes are downregulated, ureagenesis capacity is
diminished and hyperammonemia develops (8,30).
Transcriptomic and proteomic analyses of liver
tissue from patients with NAFLD show a coordinated downregulation
of multiple nitrogen-conversion pathways responsible for urea
production and ammonia clearance, whereas alternative pathways such
as glutamine synthetase are relatively preserved or even
upregulated. This indicates a redistribution of nitrogen flux away
from the urea cycle toward alternative ammonia-detoxifying routes,
a reprogramming pattern that correlates with intrahepatic fat
content and disease stage (8,30). Human stable isotope tracer and
metabolic flux studies further demonstrate that hepatic TCA cycle
and gluconeogenic fluxes are globally elevated in NAFLD and
positively correlated with intrahepatic triglyceride content,
suggesting that in fatty liver the mitochondrial carbon flux is
preferentially directed toward the TCA cycle and gluconeogenesis
(94). Against a background of
impaired urea cycle enzyme expression and function, this enhanced
carbon flux cannot be adequately matched to nitrogen metabolism,
favoring accumulation of ammonia and related metabolites in the
liver and systemically (8,30,80).
Glutamine is not only an alternative product for
ammonia detoxification but also a major energy and carbon source
for activated hepatic stellate cells. In NASH models and liver
tissue from NASH patients, glutamine metabolism and glutaminolysis
are enhanced, sustaining oxidative metabolism in activated stellate
cells and driving scar formation and matrix deposition, thereby
functionally linking suppression of the urea cycle and glutamine
metabolic reprogramming directly to the fibrotic phenotype
(80,93,95). In normal mice and isolated
perfused rat livers, acute glucagon stimulation rapidly enhances
amino acid clearance and urea production, reflecting physiological
hepatic glucagon sensitivity at the level of amino acid metabolism.
By contrast, in fatty liver models induced by high-fat diet or in
ob/ob obese mice, the same glucagon stimulus produces a markedly
attenuated increase in amino acid catabolism and ureagenesis,
manifesting as impaired amino acid clearance and blunted ureagenic
responses, consistent with a state of glucagon resistance primarily
affecting amino acid metabolism in fatty liver (5). In obese adult mice with hepatic
steatosis, intravenous glucagon elicits a relatively preserved
glycemic response, whereas the increments in amino acid clearance
and urea production are diminished, providing clinical evidence for
glucagon resistance predominantly at the level of amino acid
turnover. This selective resistance allows glucagon-induced
gluconeogenesis to be maintained while impairing the capacity of
the urea cycle to dispose of amino acids and excess nitrogen,
thereby aggravating hyperammonemia and amino acid overload
(96). Taken together, in
NAFLD/NASH, downregulation of urea cycle enzymes, redistribution of
nitrogen toward the glutamine pathway, increased TCA and
gluconeogenic fluxes and glucagon resistance specifically affecting
amino acid metabolism collectively form a self-reinforcing
metabolic circuit in which disordered nitrogen handling and
glucose-lipid metabolic reprogramming exacerbate each other and
drive the persistent progression of inflammation, fibrosis and
insulin resistance (30,80,95).
Interplay between the hepatic urea cycle
and one-carbon metabolism in NAFLD/NASH
In hepatocytes, nitrogen derived from amino acid
catabolism is primarily cleared via the urea cycle, whereas
one-carbon metabolism channels nutrients and metabolic substrates
such as serine into one-carbon units and reducing equivalents for
nucleotide synthesis, redox defense and the generation of methyl
donors; consequently, under conditions of nutrient excess and fatty
liver, nitrogen disposal capacity, methyl donor availability and
the epigenetic landscape tend to be perturbed in a coordinated
manner (97,98). One-carbon metabolism generates
S-adenosylmethionine via the methionine cycle and
S-adenosylmethionine functions as a universal methyl donor for DNA
and histone methylation, as well as for phosphatidylethanolamine
methylation by phosphatidylethanolamine N-methyltransferase to form
phosphatidylcholine; these reactions influence membrane
phospholipid composition, very-low-density lipoprotein assembly and
hepatic lipid export and are closely linked to the development and
progression of NAFLD (97,98). In a 52-week high-fat,
high-cholesterol diet-induced NAFLD mouse model, hepatic methionine
levels were depleted, whereas S-adenosylhomocysteine and
homocysteine concentrations were increased; at the same time,
serine was markedly depleted and glycine levels were reduced,
accompanied by downregulation of glycine N-methyltransferase and an
elevated phosphatidylcholine to phosphatidylethanolamine ratio,
indicating system-wide remodeling of the methionine cycle and
associated transmethylation and transsulfuration pathways (99). Among the genes examined in that
study, only the Hmgcr promoter exhibited hypermethylation, whereas
genes such as Fasn did not show similar changes, suggesting that
DNA methylation alterations may be targeted to specific loci rather
than occurring in a global, nonselective fashion (99). Methyl donor-deficient models
further demonstrate that insufficient one-carbon flux can elicit
NAFLD/NASH-like phenotypes accompanied by extensive epigenetic
reprogramming: male C57BL/6J and DBA/2J mice fed a lipogenic
methyl-deficient diet for 6-18 weeks developed liver injury
resembling human NASH, together with global and repetitive element
DNA hypomethylation, reduced DNMT1 expression, altered DNMT3A
expression and broad remodeling of histone modification profiles,
including changes in H3K9me3, H3K27me3 and H4K20me3 with
strain-dependent patterns (78).
In parallel, urea cycle impairment is a reproducible feature of
NAFLD/NASH: in Wistar rats fed a high-fat, high-cholesterol diet
for 10 months to induce NASH and subsequently switched to a normal
chow diet and in liver biopsies from patients with NAFLD or NASH,
hepatic OTC and CPS1 gene and protein expression and OTC enzymatic
activity were reduced, accompanied by elevated ammonia
concentrations and hypermethylation of OTC and CPS1 promoter
regions; in the rat model, these changes were at least partially
reversible upon dietary normalization, underscoring the plasticity
of the urea cycle in response to nutritional and metabolic cues
(30). Taken together, these
data support a model in which disturbed one-carbon metabolism, by
altering methyl donor supply and the network of methylation-related
enzymes, modulates the transcriptional regulation of key urea cycle
genes, while impaired urea cycle activity and consequent ammonia
accumulation feedback to exacerbate hepatocellular metabolic stress
and disrupt amino acid and one-carbon substrate homeostasis,
thereby establishing a feed-forward pathological circuit between
epigenetic remodeling and nitrogen disposal in NAFLD/NASH (30,97,100).
Metabolic reprogramming of the urea cycle
and arginine metabolism in HSCs
Against a background of hepatic injury and lipid
deposition, the transition of HSCs from quiescent to activated
states is accompanied by global remodeling of arginine metabolism
and related enzyme systems. This metabolic reprogramming not only
influences the proliferation, migration and matrix synthesis of
HSCs themselves, but also, through metabolic cross-talk with
hepatocytes, collectively drives fibrogenesis. The dynamic balance
between the urea cycle and NO cycle thus emerges as a central
regulatory hub in HSCs activation and the progression of NASH/NAFLD
(30,101,102).
In the quiescent state, HSCs reside in the
perisinusoidalyizho space of hepatic lobules, where their main
functions include storage of vitamin A and secretion of matrix
metalloproteinases. At this time, amino acid metabolism is low and
energy is mainly derived from lipid oxidation driven by
intracellular lipid droplets; the expression of ARG1/ARG2 is nearly
undetectable, iNOS is not expressed among NOS family members and
eNOS is mainly localized to microvascular endothelial cells,
providing limited NO to HSCs themselves (60,103). During this stage, arginine is
used mainly for basal metabolism, with little conversion to NO or
ornithine derivatives (104);
intracellular ornithine is primarily derived from urea cycle
intermediates secreted by hepatocytes or dietary amino acids.
Meanwhile, ornithine decarboxylase 1 activity is low and polyamine
synthesis is maintained at minimal levels, preventing uncontrolled
proliferation and excess matrix production (105). It is in this stable environment
that HSCs are highly sensitive to external injury signals such as
TGF-β and PDGF and upon stimulation rapidly rewire their metabolic
network, priming for activation.
Urea cycle remodeling in HSCs under
pathological conditions
When hepatic inflammation and apoptosis are
triggered by high-fat, high-glucose, or toxic insults, HSCs are
mobilized as principal fibrogenic effectors. Their morphology
shifts from stellate to flattened and α-smooth muscle actin
(α-SMA), type I collagen and fibronectin expression are markedly
upregulated (106,107). Accompanying this phenotypic
transformation, cellular energy metabolism switches from
mitochondrial lipid oxidation to a pattern reliant on both aerobic
glycolysis and glutamine metabolism, rapidly supplying ATP and
intermediate metabolites (108,109). In this process, ODC1 is
markedly upregulated in activated HSCs, catalyzing the
decarboxylation of ornithine to produce putrescine; subsequent
actions of spermidine synthase and spermine synthase convert
putrescine into spermidine and spermine, meeting high demands for
nucleic acid stability and protein modification during
proliferation and matrix synthesis (110,111) (Fig. 4).
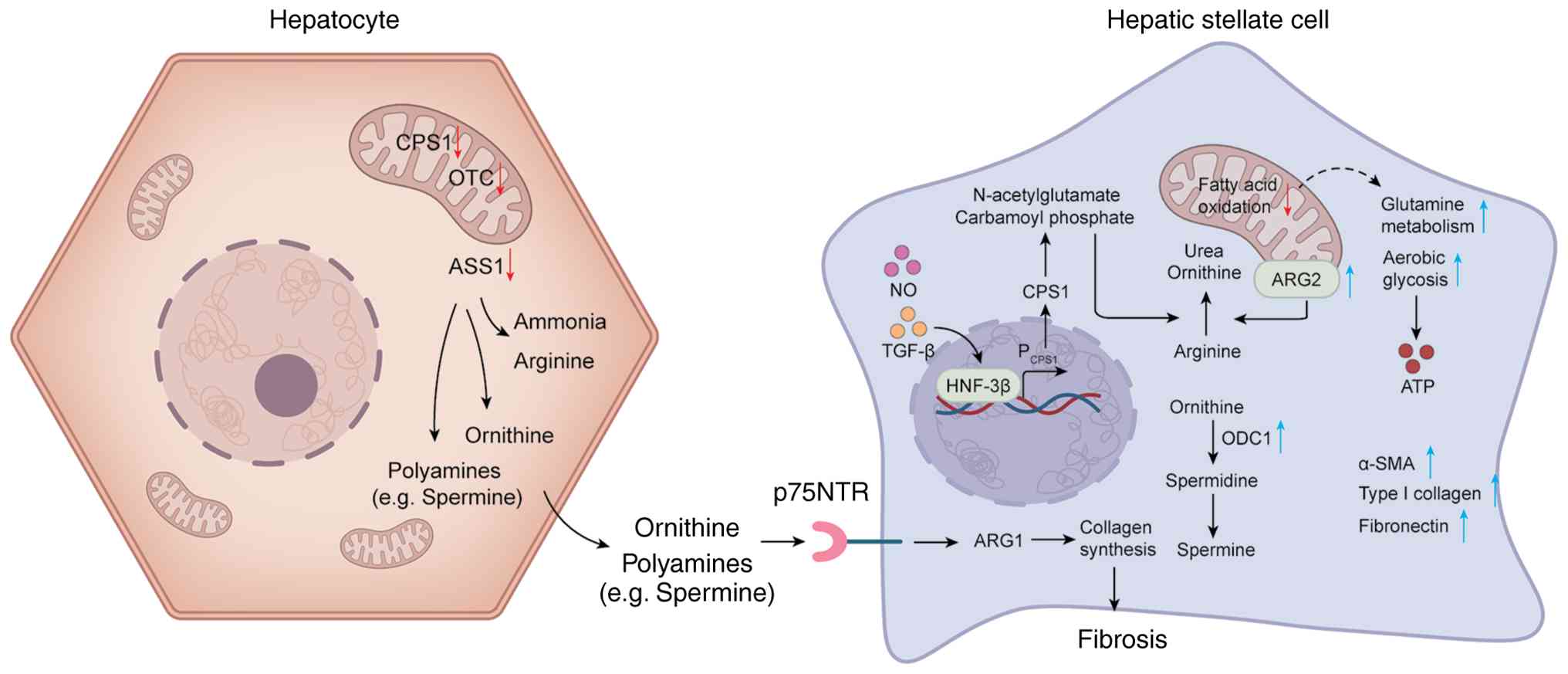 | Figure 4Metabolic reprogramming of hepatic
stellate cells and ornithine-polyamine flux during hepatic
inflammation and NAFLD/NASH. During hepatic inflammation, hepatic
stellate cells are mobilized, their morphology shifts from a
stellate to a flattened shape and the expression of α-SMA, type I
collagen and fibronectin is markedly upregulated. Cellular energy
metabolism is reprogrammed from mitochondrial fatty acid oxidation
toward a phenotype predominantly reliant on aerobic glycolysis and
glutamine metabolism, providing rapid ATP and biosynthetic
intermediates. In activated HSCs, ornithine ODC1 is strongly
upregulated, catalyzing the decarboxylation of ornithine and its
further conversion into the polyamines spermidine and spermine.
Mitochondrial ARG2 is upregulated and hydrolyzes arginine to
ornithine and urea. Regulation by NO and TGF-β increases the
affinity of the HNF-3β binding site within the CPS1 promoter,
maintaining CPS1 at a low-to-intermediate expression level, thereby
sustaining the production of carbamoyl phosphate and NAG and
providing essential intermediates for ARG2-driven ornithine
generation. In the pathological context of NAFLD/NASH, hepatic
expression of CPS1, OTC and ASS1 is reduced, leading to excessive
accumulation of ammonia and arginine. Hepatocyte-derived ornithine
and polyamines (such as spermine) can act on HSCs via receptors
including p75NTR and, through ARG1-mediated pathways, further
promote collagen synthesis, thereby establishing a vicious cycle of
local fibrogenesis. All figures were generated using Microsoft
PowerPoint (Microsoft Corporation). NAFLD, non-alcoholic fatty
liver disease; NASH, non-alcoholic steatohepatitis; α-SMA, α-smooth
muscle actin; HSCs, hepatic stellate cells; ODC1, ornithine
decarboxylase 1; ARG, cytosolic arginase; NO, nitric oxide; CPS1,
carbamoyl phosphate synthetase 1; OTC, ornithine transcarbamylase;
ASS1, argininosuccinate synthase. |
Simultaneously, ARG2 is significantly upregulated in
mitochondria, hydrolyzing arginine to ornithine and urea, thus
providing a constant substrate supply for ODC1; cytosolic ARG1
activity is also enhanced, supporting polyamine synthesis and
metabolic intermediate production (60). Notably, although classic urea
cycle enzymes such as CPS1 and OTC are generally downregulated in
activated HSCs, atypical modular reassembly occurs within the cell:
under the co-regulation of NO and TGF-β, the affinity of the HNF-3β
binding site in the CPS1 promoter increases, allowing CPS1 to be
maintained at low to moderate expression, continuously generating
carbamoyl phosphate and NAG and supplying essential intermediates
for ARG2-driven ornithine production and the polyamine pathway
(31,112-115). This atypical urea cycle
configuration enables HSCs to flexibly shift between limited urea
production and robust polyamine synthesis in response to
extracellular ammonia and synthetic demands, achieving fine-tuned
metabolic adaptation (112,114).
Under the pathological background of NASH/NAFLD, the
expression of key enzymes such as CPS1, OTC and ASS1 in hepatocytes
is significantly reduced due to high-fat, high-glucose and
inflammatory environments, leading to excessive accumulation of
ammonia and arginine in the periportal region (31). This surplus of ammonia and
arginine further upregulates ARG2 and ODC1 in HSCs, enhancing
ornithine and polyamine metabolism to meet the energy and matrix
synthesis demands of activation (116). Meanwhile, ornithine and
polyamines (such as spermine) released by hepatocytes can act on
HSCs via receptors like p75NTR and, through ARG1-mediated pathways,
further promote collagen synthesis, creating a vicious cycle of
localized fibrosis (60). In
advanced fibrotic stages, accumulated polyamines not only enhance
PARP and SMAD signaling in TGF-β-induced collagen gene
transcription and matrix crosslinking, but may also suppress
autophagy via excessive activation of mTOR, resulting in protein
aggregate accumulation and mitochondrial damage, pushing fibrotic
tissue toward irreversibility (116-119).
NO cycle remodeling in HSCs
In the quiescent state, HSCs express almost no iNOS
and respond only weakly to NO signals secreted by neighboring
Kupffer cells or hepatocytes; however, under inflammatory
stimulation (such as TNF-α, IL-1β, IL-6 and LPS), NOS2 gene
expression and iNOS activity in HSCs are rapidly induced, with both
mRNA and protein levels rising dramatically within hours, leading
to a surge in cellular NO production to micromolar levels (120-122). High concentrations of NO can
directly inhibit ODC1 activity, reducing polyamine synthesis and
through S-nitrosylation, post-translationally modify CPS1 and ARG2,
decreasing arginine consumption and channeling more substrate into
the NO pathway to amplify inflammatory signaling (120,123). NO reacts with superoxide to
form ONOO−, which nitrates mitochondrial complexes
I/III, the PDH complex and CPT-1, leading to loss of mitochondrial
membrane potential, massive ROS generation, ATP depletion and
consequently HSC apoptosis or progression to a pro-fibrotic
phenotype (120,124).
By contrast, low to moderate NO signals can
reversibly inhibit oxidative phosphorylation and activate the
AMPK/PGC-1α pathway, inducing compensatory aerobic glycolysis and
TCA cycle remodeling, ensuring abundant ATP and NADPH for matrix
synthesis (125,126). This biphasic effect makes NO a
key determinant in HSC activation: even minor changes in
concentration can create a tipping point between fibrosis
progression and spontaneous resolution (125,126). In early activation, low NO
mainly acts through cGMP/PKG-mediated AMPK phosphorylation to boost
aerobic glycolysis and TCA cycle activity in HSCs, supporting
proliferation and matrix generation; at moderate NO, reversible
inhibition of PDH and aconitase shifts metabolism toward glutamine
dependence and lactate fermentation, producing more matrix
precursors and antioxidant NADPH (127,128).
However, sustained iNOS upregulation and extensive
ONOO− formation can cause complete mitochondrial
failure: irreversible loss of membrane potential, activation of
mitochondria-mediated apoptosis, disruption of local cell renewal,
microenvironmental imbalance and accelerated fibrotic scar
expansion (129). Multiple
in vitro and animal studies of NASH have shown that iNOS and
ARG2 mRNA levels are significantly higher in activated HSCs than in
quiescent states, reflecting the simultaneous amplification of urea
detoxification and NO generation, driving HSCs into a state of high
metabolic stress and promoting progression to irreversible fibrosis
(120). Furthermore, high
levels of NO produced by HSCs can diffuse into neighboring
hepatocytes, where S-nitrosylation inhibits CPS1 and ARG1, further
impairing the urea cycle, causing increased arginine and ammonia
leakage and establishing a positive feedback loop between HSCs and
hepatocytes that exacerbates NASH/NAFLD-related fibrosis (28).
Hepatic macrophages
Alterations of the urea cycle and NO
pathways in macrophages
In recent years, hepatic macrophages, including
resident Kupffer cells and infiltrating monocyte-derived
macrophages (MoMFs), have garnered wide attention for their roles
in NAFLD and its progressive form, NASH. In pathological contexts,
hepatic macrophages contribute to inflammatory responses, matrix
remodeling and fibrogenesis by secreting inflammatory mediators,
phagocytosing apoptotic cells and regulating intercellular
metabolic signaling, thus exerting multilayered control over the
hepatic microenvironment (130-133). In NAFLD/NASH, metabolic
stressors such as lipid accumulation and insulin resistance drive
metabolic reprogramming of hepatic macrophages, inducing dynamic
switching between pro-inflammatory (M1) and
anti-inflammatory/repair (M2) polarization states, which mediate
divergent functional outcomes at different disease stages (134).
In the early inflammatory phase, Kupffer cells and
infiltrating macrophages preferentially polarize toward the M1
phenotype, characterized metabolically by a reliance on glycolysis
for ATP production and regulatory metabolite generation and by
robust upregulation of iNOS. Through iNOS, L-arginine is converted
to large amounts of NO, amplifying pro-inflammatory signaling and
inducing high expression of cytokines such as TNF-α, IL-1β and
IL-6, which further exacerbate oxidative stress and apoptosis in
hepatocytes and propagate inflammatory infiltration throughout
hepatic tissue (30,135). Evidence shows that NO not only
directly damages mitochondrial function and increases ROS, but also
transiently suppresses TGF-β1 signaling in HSCs, briefly delaying
collagen matrix secretion. However, excessive NO production
intensifies hepatocyte toxicity, causing more severe liver injury
and accelerating inflammatory deterioration (31,136). Under M1 polarization,
macrophage arginine flux is directed predominantly toward the iNOS
pathway, with relatively suppressed Arg1 expression, maximizing NO
production and amplifying the local pro-inflammatory milieu
(137). IL-1β further acts on
hepatocytes via its receptor, on the one hand upregulating
lipogenic genes and promoting triglyceride accumulation and on the
other hand impairing insulin signal transduction, thereby
increasing the susceptibility of hepatocytes to lipotoxic and
inflammatory insults (138,139). Meanwhile, TNF-α secreted by
Kupffer cells can activate the hepatocellular TNFR-JNK axis,
inducing changes in mitochondrial permeability and cytochrome
c release; in dietary steatohepatitis mouse models,
pharmacological or genetic blockade of TNF-α markedly attenuates
hepatocyte apoptosis and hepatic inflammation (140). A study has further shown that
Kupffer cell-derived TNF-α can induce hepatocytes to secrete the
chemokine C-X-C motif chemokine ligand 1, thereby driving massive
neutrophil recruitment and amplifying necrotic liver injury, which
establishes a TNF-α-CXCL1-mediated amplification loop between
macrophages and hepatocytes (141).
Conversely, in M2-polarized hepatic macrophages,
Arg1 drives the urea cycle, converting L-arginine into urea and
L-ornithine. This reaction helps eliminate excess nitrogen through
urea excretion while providing ornithine for proline synthesis, a
step that is essential for matrix remodeling and tissue repair
(142). M2 macrophages also
downregulate iNOS, suppress NO generation and secrete
anti-inflammatory mediators, attenuating excessive immune responses
and stabilizing tissue during late-stage fibrosis (143,144). This Arg1 driven metabolic shift
not only curbs NO overproduction, but also provides macrophages
with energy and precursors for extracellular matrix and collagen
synthesis, thus accelerating fibrogenesis and tissue remodeling
(145). At the level of
metabolic crosstalk between macrophages and hepatocytes,
alternatively activated Kupffer cells secrete anti-inflammatory
cytokines such as IL-10, which act via the hepatocellular IL-10
receptor to suppress excessive STAT3 phosphorylation and
acute-phase protein expression, thereby limiting triglyceride
accumulation and impairment of insulin signaling (146). In high-fat diet-fed mice,
depletion of Kupffer cells using clodronate liposomes or global
knockout of IL-10 both lead to a marked reduction in hepatic IL-10,
enhanced STAT3 signaling and increased intrahepatic triglyceride
levels, indicating that Kupffer cell-derived IL-10 plays a pivotal
role in restraining hepatocyte lipotoxicity (146). In obesity-related models,
chimeric mice reconstituted with PPARδ-deficient bone marrow cells
exhibit downregulation of hepatic fatty acid oxidation genes and
pronounced steatosis; in follow-up conditioned culture experiments,
supernatants from PPARδ-deficient macrophages added to primary
hepatocytes reduce the expression of oxidative genes such as Cpt1a
and Acox1, lower mitochondrial oxygen consumption and exacerbate
triglyceride accumulation, demonstrating that M2-associated
metabolic mediators can directly remodel hepatocyte lipid
metabolism via paracrine mechanisms (147). In the early phase of high-fat
feeding, free fatty acids released from hepatocytes, particularly
palmitate, activate Kupffer cells, inducing robust TNF-α secretion
that acts on hepatocyte TNFR1 to drive citrate accumulation,
increased de novo lipogenesis and enhanced fatty acid
uptake; blockade of TNF-α signaling markedly reduces hepatocellular
lipid droplet burden, suggesting that metabolites and cytokines
together establish a bidirectional crosstalk loop between
macrophages and hepatocytes (148). Taken together, these
bidirectional cytokine- and metabolite-driven interactions are not
only central to lipotoxic and inflammatory amplification, but also
provide a mechanistic context in which hepatocellular metabolic
stress, mitochondrial dysfunction and altered arginine handling may
converge to impair urea cycle function in NAFLD/NASH.
In NAFLD/NASH patients, global hepatic urea cycle
activity is clearly impaired. Studies show that mRNA and protein
expression of key enzymes such as CPS1 and OTC are markedly reduced
in NASH tissues, severely compromising the capacity for urea
synthesis in both hepatocytes and hepatic macrophages. This
contributes to hyperammonemia and worsens liver injury (8,31). The degree of CPS1 downregulation
(as the rate-limiting urea cycle enzyme) correlates positively with
the loss of urea synthetic capacity in NASH patients, while
declining OTC expression is associated with greater fibrosis and
worse clinical outcomes (8).
Thus, functional impairment of the urea cycle spans the full
spectrum of NASH, underpinning hyperammonemia and hepatocellular
damage and requiring compensatory ammonia clearance via macrophage
Arg1 dependent mechanisms.
Despite global urea cycle impairment, Arg1
expression in hepatic macrophages is often upregulated across
various research models. This apparent paradox may reflect a
compensatory hepatic strategy: Enhancing local urea cycling in
macrophages to mitigate ammonia toxicity. However, when Arg1
activity becomes excessive, arginine is quickly depleted, limiting
the substrate for iNOS, lowering NO production and weakening the
early pro-inflammatory removal of cytokines and apoptotic cells,
which ultimately slows lesion resolution and tissue repair
(135,149). At the same time, abundant
ornithine generated by Arg1 is utilized by HSCs for proline and
collagen precursor synthesis, accelerating matrix deposition and
fibrosis progression; this is supported by evidence from both NASH
animal models and clinical liver tissue (144,149). Hence, precise regulation of
Arg1 activity is critical for balancing hepatic inflammation
clearance vs. fibrogenic progression in NAFLD/NASH.
Mechanistic studies further demonstrate that Arg1
knockout or inhibition in macrophages restores iNOS-mediated NO
production, thereby enhancing the early pro-inflammatory clearance
capacity and reducing intrahepatic inflammation, oxidative stress
and lipid peroxidation in NASH mouse models. Nonetheless, this
situation can perpetuate mitochondrial dysfunction, boost
superoxide generation and amplify oxidative stress, which
collectively heighten hepatocellular necrosis and apoptosis and
accelerate fibrogenesis, underscoring the intricate role Arg1 plays
in regulating inflammation (31,149). Conversely, in late-stage NASH,
excessive Arg1 activity may acutely clear ammonia via urea
production and protect hepatocytes, but surplus ornithine synthesis
further drives HSC activation and collagen deposition, thereby
worsening fibrosis, increasing hyperammonemia and potentially
predisposing to hepatic encephalopathy and other complications
(144,149). Therefore, the delicate balance
between Arg1 and iNOS activities in hepatic macrophages ultimately
determines whether the liver manifests inflammatory or fibrotic
phenotypes during NAFLD/NASH progression.
At the microenvironmental level, excess free fatty
acids activate TLR4 signaling, upregulate iNOS and promote M1
polarization, while pro-inflammatory cytokines further reinforce
the M1 phenotype. Conversely, IL-4, IL-13 and PPARγ/δ agonists
enhance Arg1, driving M2 polarization, so hepatic macrophages
display remarkable phenotypic and metabolic plasticity in response
to complex microenvironmental cues (31,143). Additionally, negative
regulators such as galectin-12 inhibit Arg1 in macrophages;
galectin-12 deficiency promotes M2 polarization in Kupffer cells
with elevated Arg1 and TGF-β1, enhancing HSC activation and
collagen deposition, indicating a key regulatory role for
galectin-12 in maintaining M1/M2 metabolic balance (150). Various transcription factors
altered in NASH also directly regulate urea cycle gene
transcription (CPS1, OTC and ASS1), while mTOR signaling influences
downstream effectors (S6K and 4EBP1) to control macrophage
metabolic fate, indirectly affecting Arg1 and iNOS levels. For
example, mTORC1 hyperactivation stabilizes HIF-1α, promoting iNOS
and M1 polarization, whereas mTOR inhibition favors M2 polarization
and Arg1 transcription, substantially shaping hepatic macrophage
phenotype and function (114).
At the epigenetic level, histone acetylation and DNA methylation
changes in early NASH also affect expression of key arginine
metabolic enzymes, further complexifying the regulatory landscape
of macrophage metabolic reprogramming.
Single cell transcriptomic analyses demonstrate
pronounced heterogeneity among hepatic macrophages in NASH.
Inflammation related subsets often show contrasting levels of Arg1
and iNOS, cells with abundant Arg1 cluster within fibrotic regions,
whereas those with higher iNOS drive the early inflammatory
response (150-152). For example, in NASH mouse
models, 15-20% of MoMFs differentiate into
Arg1hi/Retnlahi M2-like cells under certain
inflammatory stimuli, whereas about 30% of Kupffer cells display
high iNOS, TNF and IL-1β, indicative of strong M1 features. Such
spatial and temporal dynamics underscore the distinct contributions
of macrophage subpopulations to fibrosis progression and
inflammatory spread (153,154).
Inflammatory state changes mediated by
urea and NO cycle alterations
At the level of intercellular interactions,
M2-polarized macrophages secrete large amounts of L-ornithine and
its proline derivatives, providing precursors for collagen
synthesis by HSCs. Additionally, various metabolites (such as urea)
can modulate TGF-β1/Smad signaling in HSCs in a
concentration-dependent manner during fibrosis: low urea
concentrations promote HSC survival and collagen synthesis, whereas
high urea may induce HSC apoptosis, potentially facilitating matrix
remodeling and fibrosis regression at late stages (144,149). On the cytokine level, NO
secreted by M1 macrophages can directly inhibit TGF-β1 signaling in
HSCs, briefly suppressing collagen deposition; however, excess NO
becomes cytotoxic to hepatocytes, disrupting mitochondrial membrane
potential, inducing apoptosis and releasing DAMPs
(damage-associated molecular patterns) that recruit and activate
further macrophages, fueling a vicious cycle and ultimately
accelerating fibrosis (31,136).
As well as metabolic interplay with HSCs, hepatic
macrophages also secrete chemokines and cytokines that tightly
interact with hepatocytes. M1-derived TNF-α and IL-1β can induce
hepatocyte MCP-1 expression, enhancing monocyte recruitment and
establishing a pro-inflammatory circuit; conversely, M2 macrophage
IL-10 and unsaturated fatty acid-binding proteins, via PI3K/Akt
signaling, suppress hepatocyte apoptosis and promote proliferation
and repair, conferring both protective and pro-fibrotic actions
during pathology and recovery (114,144). HSCs, in turn, not only respond
to M2-derived ornithine and collagen precursors, but can secrete
IL-33, CXCL12 and other factors that induce macrophage CD163 and
MERTK, further promoting M2 polarization and creating complex
positive-feedback networks that stabilize and amplify the fibrotic
microenvironment (155).
Clinical biomarkers and therapeutic
targets
Biomarker value of urea cycle enzymes and
arginine metabolites in serum and liver tissue
Liver tissue assessment
At the tissue level, consistent with the central
role of CPS1 and other urea-cycle enzymes in hepatic ammonia
detoxification aforementioned, multiple investigations using
gene-expression profiling, proteomics and enzyme-activity assays
have documented sharp reductions in these rate-limiting enzymes in
NAFLD and NASH. One hepatic mRNA-sequencing study comparing 20
non-diabetic patients with NAFLD (eight with simple steatosis and
12 with NASH) to healthy controls reported a roughly 3.5-fold
decrease in CPS1 transcripts (P<0.0001); this decline closely
paralleled diminished urea-synthetic capacity (P=0.03) (75). Given that CPS1 catalyzes the
entry step of the urea cycle, its downregulation provides a direct
mechanistic explanation for impaired ammonia detoxification and the
tight correlation between CPS1 expression and urea production
supports CPS1 as a mechanistically grounded tissue biomarker of
urea-cycle failure in NAFLD/NASH. The same analysis and other
studies observed significant downregulation of OTC, ASS1 and ASL,
whereas GS expression rose by more than 1.5-fold and showed a
negative correlation with CPS1 (P=0.004) (8). This enzyme-expression pattern
mirrors the shift from urea-based detoxification toward glutamine
trapping of excess ammonia described in experimental models,
indicating that quantitative measurement of CPS1, OTC, ASS1, ASL
and GS in liver tissue reflects not only the severity of metabolic
remodeling but also the specific route by which ammonia is handled.
Indeed, human liver specimen analyses demonstrate that lower CPS1,
OTC, ASS1 and ASL expression is strongly associated with higher
serum ALT and AST concentrations and more advanced fibrosis,
indicating that these enzymes can serve as pathophysiologically
informed tissue biomarkers for staging NAFLD/NASH and assessing
hepatic injury (8,31).
Complementing transcriptomic data, studies
utilizing western blotting, immunohistochemistry and in
vitro enzyme assays in a rat NASH model and liver biopsies from
35 human fatty liver patients further confirmed that CPS1 and OTC
protein levels and activities were markedly reduced (30). Importantly, promoter
hypermethylation of CPS1 and OTC, triggered by a high-fat,
high-cholesterol diet, was tightly associated with the loss of
enzyme expression, whereas switching back to a normal diet partly
restored both methylation status and enzyme levels (30). These findings link a specific
epigenetic mechanism (promoter hypermethylation) to measurable loss
of CPS1/OTC function and underscore that tissue CPS1/OTC expression
simultaneously captures an actionable regulatory lesion and a
potential therapeutic target.
Mechanistically, suppression of urea-cycle flux
leads to intrahepatic and systemic ammonia accumulation, which in
turn drives mitochondrial dysfunction and HSC activation as
outlined in sections Fundamentals of the urea cycle and arginine
metabolism and Metabolic reprogramming in hepatocytes.
Clinically, this is reflected by a study showing that peripheral
blood ammonia levels in NASH patients are significantly higher than
in healthy controls (P<0.01) and intrahepatic ammonia content is
positively correlated with fibrosis stage (r=0.62; P<0.001)
(30). Thus, hyperammonemia is
not only an effector of fibrogenesis but also serves as a
circulating biomarker that integrates upstream defects in
CPS1/OTC/ASS1/ASL into a clinically accessible readout of NAFLD
severity.
Taken together, these data indicate that
downregulation of CPS1, OTC, ASS1 and related enzymes and the
resulting hyperammonemia, are not only central drivers of
mitochondrial dysfunction and fibrogenesis but also provide
mechanistic justification for using urea-cycle enzyme expression
and systemic ammonia levels as biomarkers to stage NAFLD/NASH.
Blood-based detection
In serum and plasma, measurement of ammonia and
arginine metabolites not only reflects hepatic urea cycle
impairment but also reveals systemic metabolic disturbances.
Research showed that blood-ammonia concentrations are significantly
higher in NASH patients (P<0.01). Circulating ammonia also
closely mirrors intrahepatic ammonia content and fibrosis stage
(r=0.62; P<0.001), supporting blood ammonia as a non-invasive
biochemical marker of urea-cycle impairment and fibrosis
progression (30).
For arginine derivatives, one of the clinical study
measured plasma ADMA in 70 patients with NAFLD (53 NASH; 17 NAFLD)
and 12 healthy controls, finding that ADMA was significantly higher
in the NAFLD group (0.81±0.25 vs. 0.48±0.24 μmol/l; P=0.005)
and that this elevation was independent of insulin resistance or
body composition, suggesting ADMA as a sensitive early warning
marker for endothelial dysfunction and cardiovascular risk in NAFLD
(156).
A Chinese cohort study reported that serum
homocitrulline was significantly elevated in patients with NAFLD,
serving as a diagnostic metabolic biomarker (157). A plasma metabolomics study of
non-diabetic patients with NAFLD and NASH, found significantly
decreased homoglutathione and glutamyl-dipeptide, with distinctive
fluctuations in citrulline, ornithine, arginine and other urea
cycle substrates and derivatives, along with widespread
dysregulation of branched-chain amino acids, glutamate and
pyruvate. These metabolic fingerprints reflect global amino acid
metabolic imbalance and tight linkage to disease progression,
providing data to support multi-marker diagnostic models (158).
Genetic testing
At the genetic level, studies of polymorphisms
provide biomarkers for NAFLD/NASH susceptibility and risk
stratification. Large-scale association and clinical phenotype
analyses have identified that the A allele of the CPS1 gene SNP
rs1047891 confers partial protection against diet-induced urea
cycle impairment and is associated with a significantly reduced
risk of NAFLD progression, indicating this SNP as a genetic marker
for early risk screening (31).
Potential therapeutic strategies
targeting the urea cycle
In hepatocytes, the urea cycle is the principal
pathway for detoxifying ammonia (8,31); efficient arginine-citrulline
cycling not only maintains nitrogen homeostasis but also promotes
mitochondrial biogenesis and fatty-acid oxidation through nanomolar
nitric-oxide production (159,160). In late-stage disease or under
high-fat/high-glucose conditions, downregulation of key urea-cycle
enzymes and substrate depletion trigger hyper-ammonemia and
energy-metabolic stress (8,31). By contrast, activated HSCs and M2
macrophages preferentially channel arginine into ornithine and
polyamine synthesis to fuel proliferation, extracellular-matrix
production and fibrogenesis (60,75), whereas pro-inflammatory M1
macrophages, via iNOS, generate excessive NO and peroxynitrite,
amplifying oxidative and inflammatory injury (161,162).
Given this cell- and stage-specific metabolic
reprogramming, the urea and NO cycles emerge as regulatable
therapeutic nodes. Maintaining adequate substrate levels and
balanced enzyme activity in hepatocytes, while continuously
tracking blood ammonia, urea and NO derivatives, can restore or
enhance physiological urea synthesis and beneficial NO signaling,
yet prevent excessive substrate diversion into pro-fibrotic or
pro-inflammatory pathways (163,164).
Accordingly, successful NASH therapies must
precisely modulate arginine availability and urea-cycle
homeostasis, demanding high target specificity plus ongoing
monitoring of hepatic function, nitrogen metabolism and fibrosis
markers, with dose adjustments based on efficacy and tolerance. As
metabolic wiring varies across cell types and disease stages, these
pathways remain versatile drug targets: Dynamically optimizing
arginine or citrulline supplementation, with real-time read-outs of
blood ammonia, urea and NO derivatives, can enhance ammonia
detoxification and hepatoprotective NO signaling while limiting
flux into polyamine or excess NO production (163,164). Finally, integrating single-cell
transcriptomics and epigenomics allows patient stratification
through deep molecular profiling (165), whereas multimodal serum and
metabolomics monitoring throughout treatment helps gauge hepatic
architecture, fibrosis progression and metabolic performance,
achieving a balanced profile of efficacy and safety.
Although multiple drugs targeting the urea
(Table I) (163,166-170) and NO cycles (Table II) (168-176) have been approved or are in
clinical trials for other diseases, their application in NASH/NAFLD
remains to be further validated and refined for targeted
therapy.
 | Table IApproved and investigational drugs
targeting the urea cycle. |
Table I
Approved and investigational drugs
targeting the urea cycle.
| Authors, year | Drug |
Mechanism/target | Drug type | Development
stage | Indications | (Refs.) |
|---|
| Sugiyama et
al, 2020 | Carglumic acid
(Carbaglu) | CPS1 activator
(N-carbamoyl-L-glutamate) | Small molecule | Approved | NAGS deficiency;
organic acidemia-related hyperammonemia | (166) |
| Butterworth et
al, 2019 and Conte et al, 2022 |
L-ornithine-L-aspartate (LOLA) | Supplement
ornithine/aspartate to enhance urea cycle | Small
molecule/amino-acid complex | Clinical (Phase
II/III; approved in multiple countries) | Hepatic
encephalopathy; NAFLD/NASH (improvement in liver enzymes,
triglycerides and liver/spleen CT ratio) | (163,167) |
| Naing et al,
2024 | CB-1158
(INCB001158) | Arginase 1
inhibitor | Small molecule | Phase I/II
clinical | Metastatic solid
tumors (tumor-microenvironment immunomodulation) | (168) |
| Fenwick et
al, 2024 | BCT-100 | PEGylated
recombinant human Arginase 1 (arginase) | Biologic
(enzyme-replacement therapy) | Phase I/II
clinical | Arginine-dependent
adult and pediatric tumors | (169) |
| Russo et al,
2024 | Pegzilarginase | PEGylated human
Arginase 1 | Biologic
(enzyme-replacement therapy) | Phase III
clinical | Arginase 1
deficiency (ARG1-D) | (170) |
| Table IIApproved and investigational drugs
targeting the NO-sGC-cGMP signaling pathway. |
Table II
Approved and investigational drugs
targeting the NO-sGC-cGMP signaling pathway.
| Authors, year | Drug |
Mechanism/target | Drug type | Development
stage | Indications | (Refs.) |
|---|
| Lian et al,
2017 | Riociguat
(Adempas) | sGC stimulator | Small molecule | Approved | PAH; CTEPH | (171) |
| Trujillo et
al, 2023 | Vericiguat
(Verquvo) | sGC stimulator | Small molecule | Approved | HFrEF; reduces risk
of heart-failure-related death and hospitalization | (172) |
| Hanrahan et
al, 2020 and Schwartzkopf et al, 2022 | Praliciguat
(IW-1973) | sGC stimulator | Small molecule | Phase II
clinical | Type 2 diabetes
with hypertension; diabetic kidney disease and other metabolic
disorders | (173,174) |
| Hingorany et
al, 2011 | Cinaciguat (BAY
58-2667) | sGC activator | Small molecule | Phase II
clinical | Acute decompensated
heart failure; symptomatic heart failure | (175) |
| Gawrys et
al, 2025 | Mosliciguat (BAY
1237592) | sGC activator | Small molecule | Clinical
(inhaled) | Pulmonary arterial
hypertension (inhalation therapy) | (176) |
| Sawabe et
al, 2019 | TY-55002 | Short-acting sGC
activator | Small molecule | Preclinical | Acute decompensated
heart failure | (177) |
| Bhogal et
al, 2019 | Sildenafil
(Revatio) | PDE5 inhibitor
(enhances NO-cGMP signaling) | Small molecule | Approved | PAH | (178) |
| Rosenzweig et
al, 2010 | Tadalafil
(Adcirca/Cialis) | PDE5 inhibitor
(enhances NO-cGMP signaling) | Small molecule | Approved | PAH; ED; BPH | (179) |
In children with CPS1 deficiency, carglumic acid
administered in combination with other ammonia-lowering measures
can maintain relatively low blood ammonia and glutamine levels and
improve tolerance to dietary protein intake, suggesting that
supplementation with a cofactor mimetic related to CPS1 activation
may enhance urea synthesis in inherited urea cycle disorders with
residual enzymatic activity (166). By contrast,
L-ornithine-L-aspartate does not directly activate a specific
rate-limiting enzyme, but primarily supports ammonia detoxification
and related metabolic conversion by providing ornithine and
aspartate. Available clinical summaries indicate that oral
L-ornithine-L-aspartate in patients with NAFLD/NASH is associated
with improvements in liver enzymes, triglycerides and imaging
parameters. As well as ammonia reduction, its potential benefits
may also involve increased synthesis of glutamine, arginine and
glutathione, as well as antioxidant, anti-inflammatory and
microcirculatory effects (163,177). INCB001158 has shown measurable
pharmacodynamic activity and an overall acceptable tolerability
profile in advanced or metastatic solid tumors; however, its
clinical responses have not clearly exceeded the background
response rates of the respective tumor types, suggesting that
increasing arginine availability alone may be insufficient to
produce durable clinical benefit (168). Unlike ARG1 inhibition, BCT-100
and pegzilarginase represent strategies based on systemic arginine
depletion and enzyme replacement therapy, respectively. BCT-100 has
achieved sustained depletion of circulating arginine in children
and adolescents with relapsed or refractory tumors, supporting the
clinical feasibility of systemic metabolic remodeling mediated by
exogenous arginase (169).
Pegzilarginase, in turn, has demonstrated efficacy in reducing
plasma arginine levels and improving functional outcomes in
patients with ARG1 deficiency, indicating that enzyme replacement
therapy can confer benefit through correction of circulating
metabolic abnormalities (170).
In NAFLD and NASH, dysregulated arginine metabolism
is reflected not only by altered utilization of urea cycle
substrates and impaired ammonia clearance, but also by
redistribution toward the NO-producing branch. Accordingly, the
NO-sGC-cGMP pathway may be regarded as a druggable downstream node
of the arginine metabolic network. Riociguat and vericiguat are
both sGC stimulators that directly stimulate soluble guanylate
cyclase and enhance its responsiveness to endogenous NO, thereby
increasing cGMP production; they have been approved for diseases
such as pulmonary arterial hypertension, chronic thromboembolic
pulmonary hypertension and heart failure with reduced ejection
fraction (171,172). Praliciguat is likewise an sGC
stimulator and has entered clinical development for metabolic
disorders. It has been evaluated in a randomized placebo-controlled
trial in patients with type 2 diabetes and hypertension and has
also shown metabolic effects in diet-induced obese mice, supporting
its translational relevance at the interface between arginine-NO
signaling and metabolic homeostasis (173,174). By contrast, cinaciguat,
mosliciguat and TY-55002 are sGC activators. Rather than enhancing
the coupling between endogenous NO and native sGC, these agents
activate oxidized or heme-free forms of sGC. At present, cinaciguat
has mainly been investigated in acute decompensated heart failure,
mosliciguat is under clinical development as an inhaled agent and
TY-55002 remains at the preclinical stage. Collectively, these
agents suggest that even when arginine flux is diverted toward the
NO branch, downstream signaling can still be pharmacologically
activated without directly supplementing NO itself (175-177). Sildenafil and tadalafil
represent another level of intervention. As PDE5 inhibitors, they
prolong the duration of existing NO-cGMP signaling by inhibiting
cGMP degradation. Their pharmacological significance therefore lies
in preserving the second-messenger effects already generated by the
arginine-NO branch, rather than directly promoting the upstream
urea cycle or arginine resynthesis (178,179).
Conclusion and future perspectives
NAFLD, particularly its advanced form, NASH, has
emerged as a leading cause of chronic liver disease, significantly
increasing the global burden of metabolic disorders (80). This comprehensive review
highlighted the pivotal role of the urea cycle and arginine
metabolism dysregulation in the pathogenesis and progression of
NAFLD/NASH. Liver hepatocytes exhibit a profound impairment in key
urea cycle enzymes, including CPS1, OTC, ASS1 and ASL (30). This enzymatic downregulation is
closely associated with epigenetic modifications such as DNA
hypermethylation and histone modifications, which result in
decreased urea synthesis and elevated intrahepatic ammonia levels
(22). The accumulation of
ammonia not only exacerbates hepatic injury by disrupting
mitochondrial oxidative phosphorylation and inducing oxidative
stress but also promotes HSC activation and fibrosis progression
(81).
Importantly, the arginine-NO axis emerges as a
critical metabolic switch that governs hepatic metabolic
homeostasis (7). In early
stages, physiological levels of NO generated by eNOS enhance
mitochondrial biogenesis, oxidative phosphorylation and fatty acid
oxidation, providing a protective metabolic phenotype (82). However, sustained inflammation in
NASH results in increased iNOS expression in Kupffer cells and
hepatic infiltrating monocyte-derived macrophages, elevating NO and
reactive nitrogen species production to pathological levels
(134). This pathologic NO
elevation leads to extensive mitochondrial damage, metabolic
dysfunction and intensified hepatic inflammation and apoptosis
(85). Moreover, activated HSCs
reprogram their arginine metabolism, favoring polyamine synthesis
pathways via arginase, which drives fibrogenesis through enhanced
collagen deposition and extracellular matrix remodeling (60). Concurrently, the interplay
between macrophages and HSCs modulates local metabolic fluxes,
exacerbating hepatic fibrosis through intricate feedback loops
involving NO and polyamine metabolism (102).
A clinical study highlights the potential of urea
cycle metabolites and arginine derivatives as biomarkers for
diagnosing and monitoring NAFLD/NASH, particularly blood ammonia
levels, plasma ADMA and CPS1 genetic variants, which correlate with
hepatic dysfunction and cardiovascular risk (156). Advances in single-cell
transcriptomics and metabolomics have further clarified hepatic
cellular heterogeneity, emphasizing the therapeutic value of
precisely targeting distinct metabolic pathways (153). Thus, strategies aiming to
restore urea cycle functionality and rebalance arginine-NO
metabolism (such as supplementation with citrulline or arginine,
modulating enzyme activities through epigenetic and
post-translational regulation and selective inhibition of harmful
pathways like iNOS-driven NO overproduction or excessive polyamine
synthesis) represent promising therapeutic approaches (164). Successful implementation of
these therapies necessitates rigorous clinical trials, detailed
biochemical monitoring and molecular profiling to achieve optimal
efficacy and personalized intervention, potentially revolutionizing
NAFLD/NASH management by mitigating liver injury, fibrosis
progression and systemic metabolic dysfunction.
Availability of data and materials
Not applicable.
Authors' contributions
BZ, CW, PL and ZQ contributed to writing and
editing of the manuscript. RQ, SC and HN revised the manuscript.
Data authentication is not applicable. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present review was funded by a project supported by Henan
provincial Medical Science and Technology Research Project (grant
no. LHGJ20240441).
References
|
1
|
Eric KK, Blaise YL, Leopold KK, Niaore SB,
Innocent MK, Arnaud AY and Michel K: Madelung's deformity: A case
report and review of the literature. Pan Afr Med J. 23:1372016.
|
|
2
|
Davydov DM, Lobanov AV, Morozov SG,
Gribova IE and Murashev AN: Neurodevelopment and
phenotype-modulating functions of S100B protein: A pilot study.
Physiol Behav. 140:188–196. 2015. View Article : Google Scholar
|
|
3
|
Wang D, Liu Q, Xiao D, Guo T, Ma Y, Duan
K, Wang J, Lu X, Feng B and Weng J: Microparticle entrapment for
drug release from porous-surfaced bone implants. J Microencapsul.
32:443–449. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Haenecour P, Floss C, Jose J, Amari S,
Lodders K, Jadhav M, Wang A and Gyngard F: Coordinated analysis of
two graphite grains from the Co3.0 Lap 031117 meteorite: First
identification of a co nova graphite and a presolar iron sulfide
subgrain. Astrophys J. 825:882016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Winther-Sørensen M, Galsgaard KD, Santos
A, Trammell SAJ, Sulek K, Kuhre RE, Pedersen J, Andersen DB,
Hassing AS, Dall M, et al: Glucagon acutely regulates hepatic amino
acid catabolism and the effect may be disturbed by steatosis. Mol
Metab. 42:1010802020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Erdal R, Birsoy K and Unlu G: Amino acid
metabolism in liver mitochondria: From homeostasis to disease.
Metabolites. 15:4462025. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fung TS, Ryu KW and Thompson CB: Arginine:
At the crossroads of nitrogen metabolism. EMBO J. 44:1275–1293.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Eriksen PL, Vilstrup H, Rigbolt K, Suppli
MP, Sørensen M, Heebøll S, Veidal SS, Knop FK and Thomsen KL:
Non-alcoholic fatty liver disease alters expression of genes
governing hepatic nitrogen conversion. Liver Int. 39:2094–2101.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Reece TB and Cleveland JC Jr: Noteworthy
literature in cardiac surgery 2015. Semin Cardiothorac Vasc Anesth.
20:34–39. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang J, He WS, Wang C, Yan YG, Ouyang ZH,
Xue JB, Li XL and Wang WJ: Application of vascularized fibular
graft for reconstruction and stabilization of multilevel cervical
tuberculosis: A case report. Medicine (Baltimore). 97:e93822018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lysaght J: The 'obesity paradox' in action
with cancer immunotherapy. Nat Rev Endocrinol. 15:132–133. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tang K, Zhang H, Deng J, Wang D, Liu S, Lu
S, Cui Q, Chen C, Liu J, Yang Z, et al: Ammonia detoxification
promotes CD8(+) T cell memory development by urea and citrulline
cycles. Nat Immunol. 24:162–173. 2023. View Article : Google Scholar
|
|
13
|
Haberle J, Boddaert N, Burlina A,
Chakrapani A, Dixon M, Huemer M, Karall D, Martinelli D, Crespo PS,
Santer R, et al: Suggested guidelines for the diagnosis and
management of urea cycle disorders. Orphanet J Rare Dis. 7:322012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Morris SM Jr: Arginine metabolism
revisited. J Nutr. 146:2579S–2586S. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Makris G, Veit L, Rüfenacht V, Klassa S,
Zürcher N, Matsumoto S, Poms M and Häberle J: Expression and
function of the urea cycle in widely-used hepatic cellular models.
J Inherit Metab Dis. 47:1228–1238. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Morris SM Jr: Regulation of enzymes of the
urea cycle and arginine metabolism. Annu Rev Nutr. 22:87–105. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Caldovic L, Ahn J, Jandricovic J, Balick
VM, Brayer M, Chansky PA, Dawson T, Edwards AC, Felsen SE, Ismat K,
et al: Datamining approaches for examining the low prevalence of
N-acetylglutamate synthase deficiency and understanding
transcriptional regulation of urea cycle genes. J Inherit Metab
Dis. 47:1175–1193. 2024. View Article : Google Scholar :
|
|
18
|
Heibel SK, Lopez GY, Panglao M, Sodha S,
Mariño-Ramírez L, Tuchman M and Caldovic L: Transcriptional
regulation of N-acetylglutamate synthase. PLoS One. 7:e295272012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brulport A, Vaiman D, Bou-Maroun E,
Chagnon MC and Corre LL: Hepatic transcriptome and DNA methylation
patterns following perinatal and chronic BPS exposure in male mice.
BMC Genomics. 21:8812020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ko S, Russell JO, Tian J, Gao C, Kobayashi
M, Feng R, Yuan X, Shao C, Ding H, Poddar M, et al: Hdac1 regulates
differentiation of bipotent liver progenitor cells during
regeneration via Sox9b and Cdk8. Gastroenterology. 156:187–202.e14.
2019. View Article : Google Scholar
|
|
21
|
Hernandez Altemir F, Hernandez Montero S
and Moros Pena M: Combitube SA through submental route. A technical
innovation. J Craniomaxillofac Surg. 31:257–259. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu H, Dong H, Robertson K and Liu C: DNA
methylation suppresses expression of the urea cycle enzyme
carbamoyl phosphate synthetase 1 (CPS1) in human hepatocellular
carcinoma. Am J Pathol. 178:652–661. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Soria LR, Makris G, D'Alessio AM, De
Angelis A, Boffa I, Pravata VM, Rüfenacht V, Attanasio S, Nusco E,
Arena P, et al: O-GlcNAcylation enhances CPS1 catalytic efficiency
for ammonia and promotes ureagenesis. Nat Commun. 13:52122022.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yuan T, Kumar S, Skinner ME, Victor-Joseph
R, Abuaita M, Keijer J, Zhang J, Kunkel TJ, Liu Y, Petrunak EM, et
al: Human SIRT5 variants with reduced stability and activity do not
cause neuropathology in mice. iScience. 27:1099912024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nakagawa T, Lomb DJ, Haigis MC and
Guarente L: SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and
regulates the urea cycle. Cell. 137:560–570. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Park J, Chen Y, Tishkoff DX, Peng C, Tan
M, Dai L, Xie Z, Zhang Y, Zwaans BM, Skinner ME, et al:
SIRT5-mediated lysine desuccinylation impacts diverse metabolic
pathways. Mol Cell. 50:919–930. 2013. View Article : Google Scholar
|
|
27
|
Wu J, Liu J, Lapenta K, Desrouleaux R, Li
MD and Yang X: Regulation of the urea cycle by CPS1 O-GlcNAcylation
in response to dietary restriction and aging. J Mol Cell Biol.
14:mjac0162022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Takakusa H, Mohar I, Kavanagh TJ, Kelly
EJ, Kaspera R and Nelson SD: Protein tyrosine nitration of
mitochondrial carbamoyl phosphate synthetase 1 and its functional
consequences. Biochem Biophys Res Commun. 420:54–60. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bak S, Leon IR, Jensen ON and Hojlund K:
Tissue specific phosphorylation of mitochondrial proteins isolated
from rat liver, heart muscle and skeletal muscle. J Proteome Res.
12:4327–4339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
De Chiara F, Heeboll S, Marrone G,
Montoliu C, Hamilton-Dutoit S, Ferrandez A, Andreola F, Rombouts K,
Grønbæk H, Felipo V, et al: Urea cycle dysregulation in
non-alcoholic fatty liver disease. J Hepatol. 69:905–915. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gallego-Duran R, Ampuero J, Pastor-Ramirez
H, Álvarez-Amor L, Del Campo JA, Maya-Miles D, Montero-Vallejo R,
Cárdenas-García A, Pareja MJ, Gato-Zambrano S, et al: Liver injury
in non-alcoholic fatty liver disease is associated with urea cycle
enzyme dysregulation. Sci Rep. 12:34182022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Miyamoto T, Lo PHY, Saichi N, Ueda K,
Hirata M, Tanikawa C and Matsuda K: Argininosuccinate synthase 1 is
an intrinsic Akt repressor transactivated by p53. Sci Adv.
3:e16032042017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lim LQJ, Adler L, Hajaj E, Soria LR, Perry
RB, Darzi N, Brody R, Furth N, Lichtenstein M, Bab-Dinitz E, et al:
ASS1 metabolically contributes to the nuclear and cytosolic
p53-mediated DNA damage response. Nat Metab. 6:1294–1309. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Boycott C, Beetch M, Lubecka-Gajewska K,
Ramsey BS, Torregrosa-Allen S, Elzey BD, Cox A, Lanman NA, de Conti
A, Li M, et al: AMPK-Dependent epigenetic regulation of metabolism
mediates the anti-cancer action of pterostilbene in hepatocellular
carcinoma. Mol Nutr Food Res. 69:e702172025. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mun GI, Kim IS, Lee BH and Boo YC:
Endothelial argininosuccinate synthetase 1 regulates nitric oxide
production and monocyte adhesion under static and laminar shear
stress conditions. J Biol Chem. 286:2536–2542. 2011. View Article : Google Scholar
|
|
36
|
Sun N and Zhao X: Argininosuccinate
synthase 1, arginine deprivation therapy and cancer management.
Front Pharmacol. 13:9355532022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Renouf S, Buquet C, Fairand A, Benamar M
and Husson A: Changes in levels of argininosuccinate lyase mRNA
during induction by glucagon and cyclic AMP in cultured foetal-rat
hepatocytes. Biochem J. 291(Pt 2): 609–613. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Latasa MU, Carretero MV, Garcia-Trevijano
ER, Torres L, Mato JM and Avila MA: Identification of
argininosuccinate lyase as a hypoxia-responsive gene in rat
hepatocytes. J Hepatol. 33:709–715. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Husson A, Renouf S, Fairand A, Buquet C,
Benamar M and Vaillant R: Expression of argininosuccinate lyase
mRNA in foetal hepatocytes. Regulation by glucocorticoids and
insulin. Eur J Biochem. 192:677–681. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Okun JG, Conway S, Schmidt KV, Schumacher
J, Wang X, de Guia R, Zota A, Klement J, Seibert O, Peters A, et
al: Molecular regulation of urea cycle function by the liver
glucocorticoid receptor. Mol Metab. 4:732–740. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Majaw T and Sharma R: Arginase I
expression is upregulated by dietary restriction in the liver of
mice as a function of age. Mol Cell Biochem. 407:1–7. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xu Y, Liu J, Wang J, Wang J, Lan P and
Wang T: USP25 stabilizes STAT6 to promote IL-4-induced macrophage
M2 polarization and fibrosis. Int J Biol Sci. 21:475–489. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pinos I, Yu J, Pilli N, Kane MA and
Amengual J: Functional characterization of interleukin 4 and
retinoic acid signaling crosstalk during alternative macrophage
activation. Biochim Biophys Acta Mol Cell Biol Lipids.
1868:1592912023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lee B, Wu CY, Lin YW, Park SW and Wei LN:
Synergistic activation of Arg1 gene by retinoic acid and IL-4
involves chromatin remodeling for transcription initiation and
elongation coupling. Nucleic Acids Res. 44:7568–7579. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gray MJ, Poljakovic M, Kepka-Lenhart D and
Morris SM Jr: Induction of arginase I transcription by IL-4
requires a composite DNA response element for STAT6 and C/EBPbeta.
Gene. 353:98–106. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sheldon KE, Shandilya H, Kepka-Lenhart D,
Poljakovic M, Ghosh A and Morris SM Jr: Shaping the murine
macrophage phenotype: IL-4 and cyclic AMP synergistically activate
the arginase I promoter. J Immunol. 191:2290–2298. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Huang L, He X, Peng W, He X, Xu B, Xu H,
Wang Y, Xu W, Chen W, Wang S, et al: Hyperuricemia induces liver
injury by upregulating HIF-1alpha and inhibiting arginine
biosynthesis pathway in mouse liver and human L02 hepatocytes.
Biochem Biophys Res Commun. 617(Pt 2): 55–61. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xu M, Guo Y, Wang M, Luo X, Shen X, Li Z,
Wang L and Guo W: L-arginine homeostasis governs adult neural stem
cell activation by modulating energy metabolism in vivo. EMBO J.
42:e1126472023. View Article : Google Scholar
|
|
49
|
Wu G and Morris SM Jr: Arginine
metabolism: nitric oxide and beyond. Biochem J. 336(Pt 1): 1–17.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Farahani A, Farahani A, Kashfi K and
Ghasemi A: Inducible nitric oxide synthase (iNOS): More than an
inducible enzyme? Rethinking the classification of NOS isoforms.
Pharmacol Res. 216:1077812025. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Forstermann U and Sessa WC: Nitric oxide
synthases: Regulation and function. Eur Heart J. 33:829–837.
837a–837d. 2012. View Article : Google Scholar
|
|
52
|
Król M and Kepinska M: Human nitric oxide
synthase-its functions, polymorphisms and inhibitors in the context
of inflammation, diabetes and cardiovascular diseases. Int J Mol
Sci. 22:562020. View Article : Google Scholar
|
|
53
|
Rochette L, Lorin J, Zeller M, Guilland
JC, Lorgis L, Cottin Y and Vergely C: Nitric oxide synthase
inhibition and oxidative stress in cardiovascular diseases:
Possible therapeutic targets? Pharmacol Ther. 140:239–257. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Dong J, Li D, Kang L, Luo C and Wang J:
Insights into human eNOS, nNOS and iNOS structures and medicinal
indications from statistical analyses of their interactions with
bound compounds. Biophys Rep. 9:159–175. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Stuehr DJ: Mammalian nitric oxide
synthases. Biochim Biophys Acta. 1411:217–230. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Caldwell RW, Rodriguez PC, Toque HA,
Narayanan SP and Caldwell RB: Arginase: A multifaceted enzyme
important in health and disease. Physiol Rev. 98:641–665. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ren Y, Li Z, Li W, Fan X, Han F, Huang Y,
Yu Y, Qian L and Xiong Y: Arginase: Biological and therapeutic
implications in diabetes mellitus and its complications. Oxid Med
Cell Longev. 2022:24194122022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
West EE, Merle NS, Kaminski MM, Palacios
G, Kumar D, Wang L, Bibby JA, Overdahl K, Jarmusch AK, Freeley S,
et al: Loss of CD4(+) T cell-intrinsic arginase 1 accelerates Th1
response kinetics and reduces lung pathology during influenza
infection. Immunity. 56:2036–2053 e12. 2023. View Article : Google Scholar
|
|
59
|
Heuser SK, Li J, Pudewell S, LoBue A, Li Z
and Cortese-Krott MM: Biochemistry, pharmacology and in vivo
function of arginases. Pharmacol Rev. 77:1000152025. View Article : Google Scholar
|
|
60
|
Zhang M, Wu Z, Salas SS, Aguilar MM,
Trillos-Almanza MC, Buist-Homan M and Moshage H: Arginase 1
expression is increased during hepatic stellate cell activation and
facilitates collagen synthesis. J Cell Biochem. 124:808–817. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Paolini E, Longo M, Meroni M and
Dongiovanni P: Hepatic zonation in MASLD: Old question, new
challenge in the era of spatial omics. Int J Mol Sci. 26:107012025.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Haussinger D, Lamers WH and Moorman AF:
Hepatocyte heterogeneity in the metabolism of amino acids and
ammonia. Enzyme. 46:72–93. 1992.PubMed/NCBI
|
|
63
|
Cunningham RP and Porat-Shliom N: Liver
zonation-revisiting old questions with new technologies. Front
Physiol. 12:7329292021. View Article : Google Scholar
|
|
64
|
Lamers WH, Hilberts A, Furt E, Smith J,
Jonges GN, van Noorden CJ, Janzen JW, Charles R and Moorman AF:
Hepatic enzymic zonation: A reevaluation of the concept of the
liver acinus. Hepatology. 10:72–76. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yasaka TM, Kim CK, Meadows V and Monga SP:
Zonation, zonation, zonation: The real estate of the liver. Annu
Rev Pathol. 21:185–212. 2026. View Article : Google Scholar :
|
|
66
|
Brosnan ME and Brosnan JT: Hepatic
glutamate metabolism: A tale of 2 hepatocytes. Am J Clin Nutr.
90:857S–861S. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Frieg B, Görg B, Gohlke H and Häussinger
D: Glutamine synthetase as a central element in hepatic glutamine
and ammonia metabolism: Novel aspects. Biol Chem. 402:1063–1072.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Villar VH, Allega MF, Deshmukh R,
Ackermann T, Nakasone MA, Vande Voorde J, Drake TM, Oetjen J, Bloom
A, Nixon C, et al: Hepatic glutamine synthetase controls
N(5)-methylglutamine in homeostasis and cancer. Nat Chem Biol.
19:292–300. 2023. View Article : Google Scholar :
|
|
69
|
Gebhardt R and Matz-Soja M: Liver
zonation: Novel aspects of its regulation and its impact on
homeostasis. World J Gastroenterol. 20:8491–8504. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Gebhardt R, Baldysiak-Figiel A, Krugel V,
Ueberham E and Gaunitz F: Hepatocellular expression of glutamine
synthetase: An indicator of morphogen actions as master regulators
of zonation in adult liver. Prog Histochem Cytochem. 41:201–266.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Li W and He F: Infusion of kupffer cells
expanded in vitro ameliorated liver fibrosis in a murine model of
liver injury. Cell Transplant. 30:96368972110040902021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Chen J, Deng X, Liu Y, Tan Q, Huang G, Che
Q, Guo J and Su Z: Kupffer cells in non-alcoholic fatty liver
disease: Friend or foe? Int J Biol Sci. 16:2367–2378. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Dixon LJ, Barnes M, Tang H, Pritchard MT
and Nagy LE: Kupffer cells in the liver. Compr Physiol. 3:785–797.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Rath M, Muller I, Kropf P, Closs EI and
Munder M: Metabolism via arginase or nitric oxide synthase: Two
competing arginine pathways in macrophages. Front Immunol.
5:5322014. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Vilstrup H, Eriksen PL, Kjærgaard K,
Sørensen M, Thomsen KL and Ott P: Down the road towards hepatic
encephalopathy. Urea synthesis-the liver workhorse of nitrogen
metabolism. Metab Brain Dis. 40:492024. View Article : Google Scholar
|
|
76
|
Simpson KL, MacLeod EL, Kakajiwala A,
Gropman AL and Ah Mew N: Urea Cycle Disorders Overview.
GeneReviews® [Internet]. Adam MP, Feldman J, Mirzaa GM, Pagon RA,
Wallace SE and Amemiya A: University of Washington; Seattle, WA:
1993
|
|
77
|
Drews L, Zimmermann M, Westhoff P,
Brilhaus D, Poss RE, Bergmann L, Wiek C, Brenneisen P, Piekorz RP,
Mettler-Altmann T, et al: Ammonia inhibits energy metabolism in
astrocytes in a rapid and glutamate dehydrogenase 2-dependent
manner. Dis Model Mech. 13:dmm0471342020. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Pogribny IP, Tryndyak VP, Bagnyukova TV,
Melnyk S, Montgomery B, Ross SA, Latendresse JR, Rusyn I and Beland
FA: Hepatic epigenetic phenotype predetermines individual
susceptibility to hepatic steatosis in mice fed a lipogenic
methyl-deficient diet. J Hepatol. 51:176–186. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Pirola CJ, Gianotti TF, Burgueño AL,
Rey-Funes M, Loidl CF, Mallardi P, Martino JS, Castaño GO and
Sookoian S: Epigenetic modification of liver mitochondrial DNA is
associated with histological severity of nonalcoholic fatty liver
disease. Gut. 62:1356–1363. 2013. View Article : Google Scholar
|
|
80
|
Thomsen KL, Eriksen PL, Kerbert AJ, De
Chiara F, Jalan R and Vilstrup H: Role of ammonia in NAFLD: An
unusual suspect. JHEP Rep. 5:1007802023. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Niknahad H, Jamshidzadeh A, Heidari R,
Zarei M and Ommati MM: Ammonia-induced mitochondrial dysfunction
and energy metabolism disturbances in isolated brain and liver
mitochondria and the effect of taurine administration: Relevance to
hepatic encephalopathy treatment. Clin Exp Hepatol. 3:141–151.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Nisoli E and Carruba MO: Nitric oxide and
mitochondrial biogenesis. J Cell Sci. 119(Pt 14): 2855–2862. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Seth RK, Das S, Dattaroy D,
Chandrashekaran V, Alhasson F, Michelotti G, Nagarkatti M,
Nagarkatti P, Diehl AM, Bell PD, et al: TRPV4 activation of
endothelial nitric oxide synthase resists nonalcoholic fatty liver
disease by blocking CYP2E1-mediated redox toxicity. Free Radic Biol
Med. 102:260–273. 2017. View Article : Google Scholar
|
|
84
|
Garcia-Villafranca J, Guillen A and Castro
J: Involvement of nitric oxide/cyclic GMP signaling pathway in the
regulation of fatty acid metabolism in rat hepatocytes. Biochem
Pharmacol. 65:807–812. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Pappas G, Wilkinson ML and Gow AJ: Nitric
oxide regulation of cellular metabolism: Adaptive tuning of
cellular energy. Nitric Oxide. 131:8–17. 2023. View Article : Google Scholar
|
|
86
|
Sprangers F, Sauerwein HP, Romijn JA, van
Woerkom GM and Meijer AJ: Nitric oxide inhibits glycogen synthesis
in isolated rat hepatocytes. Biochem J. 330(Pt 2): 1045–1049. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Cruz-Ojeda P, Navarro-Villarán E,
Fuertes-Agudo M, Mata A, López-Lluch G, Navas P, Cadenas S, Casado
M and Muntané J: Peroxynitrite is involved in the mitochondrial
dysfunction induced by Sorafenib in liver cancer cells. Free Radic
Biol Med. 229:251–263. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Babarasul MH, Bapir R, Rahman DHK,
Fakhralddin SS, Kakamad FH, Tahir SH, Ali RM, Abdalla BA, Mohammed
SH and Hussein DM: Synchronous ipsilateral papillary renal cell
carcinoma and urothelial carcinoma: A case report. Oncol Lett.
25:2212023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Riobo NA, Clementi E, Melani M, Boveris A,
Cadenas E, Moncada S and Poderoso JJ: Nitric oxide inhibits
mitochondrial NADH:ubiquinone reductase activity through
peroxynitrite formation. Biochem J. 359(Pt 1): 139–145. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Clementi E, Brown GC, Feelisch M and
Moncada S: Persistent inhibition of cell respiration by nitric
oxide: Crucial role of S-nitrosylation of mitochondrial complex I
and protective action of glutathione. Proc Natl Acad Sci USA.
95:7631–7636. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Navarro LA, Wree A, Povero D, Berk MP,
Eguchi A, Ghosh S, Papouchado BG, Erzurum SC and Feldstein AE:
Arginase 2 deficiency results in spontaneous steatohepatitis: A
novel link between innate immune activation and hepatic de novo
lipogenesis. J Hepatol. 62:412–420. 2015. View Article : Google Scholar
|
|
92
|
Sheldon RD, Padilla J, Jenkins NT,
Laughlin MH and Rector RS: Chronic NOS inhibition accelerates NAFLD
progression in an obese rat model. Am J Physiol Gastrointest Liver
Physiol. 308:G540–G549. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Delgado TC, de Las Heras J and
Martínez-Chantar ML: Understanding gut-liver axis nitrogen
metabolism in Fatty liver disease. Front Endocrinol (Lausanne).
13:10581012022. View Article : Google Scholar :
|
|
94
|
Sunny NE, Parks EJ, Browning JD and
Burgess SC: Excessive hepatic mitochondrial TCA cycle and
gluconeogenesis in humans with nonalcoholic fatty liver disease.
Cell Metab. 14:804–810. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Du K, Chitneni SK, Suzuki A, Wang Y, Henao
R, Hyun J, Premont RT, Naggie S, Moylan CA, Bashir MR, et al:
Increased glutaminolysis marks active scarring in nonalcoholic
steatohepatitis progression. Cell Mol Gastroenterol Hepatol.
10:1–21. 2020. View Article : Google Scholar :
|
|
96
|
Suppli MP, Bagger JI, Lund A, Demant M,
van Hall G, Strandberg C, Kønig MJ, Rigbolt K, Langhoff JL, Wewer
Albrechtsen NJ, et al: Glucagon resistance at the level of amino
acid turnover in obese subjects with hepatic steatosis. Diabetes.
69:1090–1099. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ducker GS and Rabinowitz JD: One-carbon
metabolism in health and disease. Cell Metab. 25:27–42. 2017.
View Article : Google Scholar
|
|
98
|
Radziejewska A, Muzsik A, Milagro FI,
Martínez JA and Chmurzynska A: One-carbon metabolism and
nonalcoholic fatty liver disease: The crosstalk between nutrients,
microbiota and genetics. Lifestyle Genom. 13:53–63. 2020.
View Article : Google Scholar
|
|
99
|
Pacana T, Cazanave S, Verdianelli A, Patel
V, Min HK, Mirshahi F, Quinlivan E and Sanyal AJ: Dysregulated
hepatic methionine metabolism drives homocysteine elevation in
diet-induced nonalcoholic fatty liver disease. PLoS One.
10:e01368222015. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Sun C, Fan JG and Qiao L: Potential
epigenetic mechanism in non-alcoholic fatty liver disease. Int J
Mol Sci. 16:5161–5179. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Trivedi P, Wang S and Friedman SL: The
power of plasticity-metabolic regulation of hepatic stellate cells.
Cell Metab. 33:242–257. 2021. View Article : Google Scholar
|
|
102
|
Yang F, Li H, Li Y, Hao Y, Wang C, Jia P,
Chen X, Ma S and Xiao Z: Crosstalk between hepatic stellate cells
and surrounding cells in hepatic fibrosis. Int Immunopharmacol.
99:1080512021. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Han YP: Matrix metalloproteinases, the
pros and cons, in liver fibrosis. J Gastroenterol Hepatol. 21(Suppl
3): S88–S91. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Pudewell S, Lissy J, Nakhaeizadeh H, Taha
MS, Akbarzadeh M, Rezaei Adariani S, Nakhaei-Rad S, Li J, Kordes C,
Häussinger D, et al: Physical Interaction between Embryonic Stem
Cell-Expressed Ras (ERas) and Arginase-1 in quiescent hepatic
stellate cells. Cells. 11:5082022. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Tsuchida T and Friedman SL: Mechanisms of
hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol.
14:397–411. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Povero D, Panera N, Eguchi A, Johnson CD,
Papouchado BG, de Araujo Horcel L, Pinatel EM, Alisi A, Nobili V
and Feldstein AE: Lipid-induced hepatocyte-derived extracellular
vesicles regulate hepatic stellate cell via microRNAs targeting
PPAR-gamma. Cell Mol Gastroenterol Hepatol. 1:646–663 e4. 2015.
View Article : Google Scholar
|
|
107
|
Higashi T, Friedman SL and Hoshida Y:
Hepatic stellate cells as key target in liver fibrosis. Adv Drug
Deliv Rev. 121:27–42. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Yin X, Peng J, Gu L, Liu Y, Li X, Wu J, Xu
B, Zhuge Y and Zhang F: Targeting glutamine metabolism in hepatic
stellate cells alleviates liver fibrosis. Cell Death Dis.
13:9552022. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Chen Y, Choi SS, Michelotti GA, Chan IS,
Swiderska-Syn M, Karaca GF, Xie G, Moylan CA, Garibaldi F, Premont
R, et al: Hedgehog controls hepatic stellate cell fate by
regulating metabolism. Gastroenterology. 143:1319–1329 e11. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Nunez-Sanchez MA, Martinez-Sanchez MA,
Sierra-Cruz M, Lambertos A, Rico-Chazarra S, Oliva-Bolarín A,
Balaguer-Román A, Yuste JE, Martínez CM, Mika A, et al: Increased
hepatic putrescine levels as a new potential factor related to the
progression of metabolic dysfunction-associated steatotic liver
disease. J Pathol. 264:101–111. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Shi B, Wang W, Ye M, Liang M, Yu Z, Zhang
Y, Liu Z, Liang X, Ao J, Xu F, et al: Spermidine suppresses the
activation of hepatic stellate cells to cure liver fibrosis through
autophagy activator MAP1S. Liver Int. 43:1307–1319. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Huang R, Cui H, Yahya Ali Alshami MA, Fu
C, Jiang W, Cai M, Zhou S, Zhu X and Hu C: LOX-1 rewires glutamine
ammonia metabolism to drive liver fibrosis. Mol Metab.
96:1021322025. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Tang Z, Lin B, Li W, Li X, Liu F and Zhu
X: Y-box binding protein 1 promotes chromatin accessibility to
aggravate liver fibrosis. Cell Signal. 109:1107502023. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Rojas A, Garcia-Lozano MR, Gil-Gomez A,
Romero-Gomez M and Ampuero J: Glutaminolysis-ammonia-urea cycle
axis, non-alcoholic fatty liver disease progression and development
of novel therapies. J Clin Transl Hepatol. 10:356–362. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Chen Z, Tang N, Wang X and Chen Y: The
activity of the carbamoyl phosphate synthase 1 promoter in human
liver-derived cells is dependent on hepatocyte nuclear factor
3-beta. J Cell Mol Med. 21:2036–2045. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Jalan R, De Chiara F, Balasubramaniyan V,
Andreola F, Khetan V, Malago M, Pinzani M, Mookerjee RP and
Rombouts K: Ammonia produces pathological changes in human hepatic
stellate cells and is a target for therapy of portal hypertension.
J Hepatol. 64:823–833. 2016. View Article : Google Scholar
|
|
117
|
Zhang H, Alsaleh G, Feltham J, Sun Y,
Napolitano G, Riffelmacher T, Charles P, Frau L, Hublitz P, Yu Z,
et al: Polyamines control eIF5A hypusination, TFEB translation and
autophagy to reverse B cell senescence. Mol Cell. 76:110–125 e9.
2019. View Article : Google Scholar
|
|
118
|
Okumura S, Teratani T, Fujimoto Y, Zhao X,
Tsuruyama T, Masano Y, Kasahara N, Iida T, Yagi S, Uemura T, et al:
Oral administration of polyamines ameliorates liver
ischemia/reperfusion injury and promotes liver regeneration in
rats. Liver Transpl. 22:1231–1244. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Akinyele O and Wallace HM: Understanding
the polyamine and mTOR pathway interaction in breast cancer cell
growth. Med Sci (Basel). 10:512022.PubMed/NCBI
|
|
120
|
Iwakiri Y and Kim MY: Nitric oxide in
liver diseases. Trends Pharmacol Sci. 36:524–536. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Ozturk Akcora B, Vassilios Gabriel A,
Ortiz-Perez A and Bansal R: Pharmacological inhibition of STAT3
pathway ameliorates acute liver injury in vivo via inactivation of
inflammatory macrophages and hepatic stellate cells. FASEB Bioadv.
2:77–89. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Okina Y, Sato-Matsubara M, Kido Y,
Urushima H, Daikoku A, Kadono C, Nakagama Y, Nitahara Y, Hoang TH,
Thuy LTT, et al: Nitric oxide derived from cytoglobin-deficient
hepatic stellate cells causes suppression of cytochrome c oxidase
activity in hepatocytes. Antioxid Redox Signal. 38:463–479. 2023.
View Article : Google Scholar :
|
|
123
|
Bauer PM, Fukuto JM, Buga GM, Pegg AE and
Ignarro LJ: Nitric oxide inhibits ornithine decarboxylase by
S-nitrosylation. Biochem Biophys Res Commun. 262:355–358. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Ren XY, Li YN, Qi JS and Niu T:
Peroxynitrite-induced protein nitration contributes to liver
mitochondrial damage in diabetic rats. J Diabetes Complications.
22:357–364. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Mueller BJ, Roberts MD, Mobley CB, Judd RL
and Kavazis AN: Nitric oxide in exercise physiology: Past and
present perspectives. Front Physiol. 15:15049782025. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Lira VA, Brown DL, Lira AK, Kavazis AN,
Soltow QA, Zeanah EH and Criswell DS: Nitric oxide and AMPK
cooperatively regulate PGC-1 in skeletal muscle cells. J Physiol.
588(Pt 18): 3551–3566. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Rockey DC and Chung JJ: Inducible nitric
oxide synthase in rat hepatic lipocytes and the effect of nitric
oxide on lipocyte contractility. J Clin Invest. 95:1199–1206. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Palmieri EM, Gonzalez-Cotto M, Baseler WA,
Davies LC, Ghesquière B, Maio N, Rice CM, Rouault TA, Cassel T,
Higashi RM, et al: Nitric oxide orchestrates metabolic rewiring in
M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase.
Nat Commun. 11:6982020. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Blas-Garcia A and Apostolova N: Novel
therapeutic approaches to liver fibrosis based on targeting
oxidative stress. Antioxidants (Basel). 12:15672023. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Gao H, Jin Z, Bandyopadhyay G, Cunha E,
Rocha K, Liu X, Zhao H, Zhang D, Jouihan H, Pourshahian S, et al:
MiR-690 treatment causes decreased fibrosis and steatosis and
restores specific Kupffer cell functions in NASH. Cell Metab.
34:978–990.e4. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Blériot C, Barreby E, Dunsmore G, Ballaire
R, Chakarov S, Ficht X, De Simone G, Andreata F, Fumagalli V, Guo
W, et al: A subset of Kupffer cells regulates metabolism through
the expression of CD36. Immunity. 54:2101–2116.e6. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Baffy G: Kupffer cells in non-alcoholic
fatty liver disease: The emerging view. J Hepatol. 51:212–223.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Ye D, Li FY, Lam KS, Li H, Jia W, Wang Y,
Man K, Lo CM, Li X and Xu A: Toll-like receptor-4 mediates
obesity-induced non-alcoholic steatohepatitis through activation of
X-box binding protein-1 in mice. Gut. 61:1058–1067. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Kazankov K, Jorgensen SMD, Thomsen KL,
Møller HJ, Vilstrup H, George J, Schuppan D and Grønbæk H: The role
of macrophages in nonalcoholic fatty liver disease and nonalcoholic
steatohepatitis. Nat Rev Gastroenterol Hepatol. 16:145–159. 2019.
View Article : Google Scholar
|
|
135
|
Yin Y, Wang Q, Qi M, Zhang C, Li Z and
Zhang W: Ghrelin ameliorates nonalcoholic steatohepatitis induced
by chronic low-grade inflammation via blockade of Kupffer cell M1
polarization. J Cell Physiol. 236:5121–5133. 2021. View Article : Google Scholar
|
|
136
|
Zhang W and Lang R: Macrophage metabolism
in nonalcoholic fatty liver disease. Front Immunol. 14:12575962023.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Liu Y, Xu R, Gu H, Zhang E, Qu J, Cao W,
Huang X, Yan H, He J and Cai Z: Metabolic reprogramming in
macrophage responses. Biomark Res. 9:12021. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Miura K, Kodama Y, Inokuchi S, Schnabl B,
Aoyama T, Ohnishi H, Olefsky JM, Brenner DA and Seki E: Toll-like
receptor 9 promotes steatohepatitis by induction of
interleukin-1beta in mice. Gastroenterology. 139:323–334.e7. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Nov O, Kohl A, Lewis EC, Bashan N, Dvir I,
Ben-Shlomo S, Fishman S, Wueest S, Konrad D and Rudich A:
Interleukin-1beta may mediate insulin resistance in liver-derived
cells in response to adipocyte inflammation. Endocrinology.
151:4247–4256. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Zhang W, Kudo H, Kawai K, Fujisaka S, Usui
I, Sugiyama T, Tsukada K, Chen N and Takahara T: Tumor necrosis
factor-alpha accelerates apoptosis of steatotic hepatocytes from a
murine model of non-alcoholic fatty liver disease. Biochem Biophys
Res Commun. 391:1731–1736. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Su L, Li N, Tang H, Lou Z, Chong X, Zhang
C, Su J and Dong X: Kupffer cell-derived TNF-α promotes hepatocytes
to produce CXCL1 and mobilize neutrophils in response to necrotic
cells. Cell Death Dis. 9:3232018. View Article : Google Scholar
|
|
142
|
Viola A, Munari F, Sanchez-Rodriguez R,
Scolaro T and Castegna A: The metabolic signature of macrophage
responses. Front Immunol. 10:14622019. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Scheja L and Kluwe J: Arginine and NASH-do
macrophages deliver the first hit? J Hepatol. 62:260–261. 2015.
View Article : Google Scholar
|
|
144
|
Tada Y, Kasai K, Makiuchi N, Igarashi N,
Kani K, Takano S, Honda H, Yanagibashi T, Watanabe Y,
Usui-Kawanishi F, et al: Roles of macrophages in advanced liver
fibrosis, identified using a newly established mouse model of
diet-induced non-alcoholic steatohepatitis. Int J Mol Sci.
23:132512022. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Yadav P, Ortega JG, Tamaki W, Chien C,
Chang KC, Biswas N, Pan S, Nilsson J, Yin X, Bhattacharyya A, et
al: Macrophage-fibroblast crosstalk drives Arg1-dependent lung
fibrosis via ornithine loading. J Clin Invest. 135:e1887342025.
View Article : Google Scholar
|
|
146
|
Clementi AH, Gaudy AM, van Rooijen N,
Pierce RH and Mooney RA: Loss of Kupffer cells in diet-induced
obesity is associated with increased hepatic steatosis, STAT3
signaling and further decreases in insulin signaling. Biochim
Biophys Acta. 1792:1062–1072. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Odegaard JI, Ricardo-Gonzalez RR, Red
Eagle A, Vats D, Morel CR, Goforth MH, Subramanian V, Mukundan L,
Ferrante AW and Chawla A: Alternative M2 activation of Kupffer
cells by PPARdelta ameliorates obesity-induced insulin resistance.
Cell Metab. 7:496–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Jentsch HFR, Dietrich M and Eick S:
Non-surgical periodontal therapy with adjunctive
amoxicillin/metronidazole or metronidazole when no aggregatibacter
actinomycetemcomitans is detected-A Randomized clinical trial.
Antibiotics (Basel). 9:6862020. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Liu C, Rajapakse AG, Riedo E, Fellay B,
Bernhard MC, Montani JP, Yang Z and Ming XF: Targeting arginase-II
protects mice from high-fat-diet-induced hepatic steatosis through
suppression of macrophage inflammation. Sci Rep. 6:204052016.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Lee JL, Wang YC, Hsu YA, Chen CS, Weng RC,
Lu YP, Chuang CY and Wan L: Galectin-12 modulates Kupffer cell
polarization to alter the progression of nonalcoholic fatty liver
disease. Glycobiology. 33:673–682. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Xiong X, Kuang H, Ansari S, Liu T, Gong J,
Wang S, Zhao XY, Ji Y, Li C, Guo L, et al: Landscape of
intercellular crosstalk in healthy and NASH liver revealed by
single-cell secretome gene analysis. Mol Cell. 75:644–660 e5. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Ramachandran P, Dobie R, Wilson-Kanamori
JR, Dora EF, Henderson BEP, Luu NT, Portman JR, Matchett KP, Brice
M, Marwick JA, et al: Resolving the fibrotic niche of human liver
cirrhosis at single-cell level. Nature. 575:512–518. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Zhang Y, Li J, Li H, Jiang J, Guo C, Zhou
C, Zhou Z and Ming Y: Single-cell RNA sequencing to dissect the
immunological network of liver fibrosis in Schistosoma
japonicum-infected mice. Front Immunol. 13:9808722022. View Article : Google Scholar
|
|
154
|
Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y,
Haseeb M, Tanaka KE and Czaja MJ: Impaired macrophage autophagy
increases the immune response in obese mice by promoting
proinflammatory macrophage polarization. Autophagy. 11:271–284.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Pelletier K, Cote G, Madsen K, Chen S, Kim
SJ, Chan CT, Mattsson J, Pasic I and Kitchlu A: Chronic kidney
disease, survival and graft-versus-host-disease-free/relapse-free
survival in recipients of allogeneic hematopoietic stem cell
transplant. Clin Kidney J. 15:1583–1592. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Boga S, Alkim H, Koksal AR, Bayram M,
Ozguven MB, Ergun M, Neijmann ST, Ozgon G and Alkim C: Increased
plasma levels of asymmetric dimethylarginine in nonalcoholic fatty
liver disease: Relation with insulin resistance, inflammation and
liver histology. J Investig Med. 63:871–877. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Yang Y, Huang Z, Yang Z, Qi Y, Shi H, Zhou
Y, Wang F and Yang M: Serum metabolomic profiling reveals an
increase in homocitrulline in Chinese patients with nonalcoholic
fatty liver disease: A retrospective study. PeerJ. 9:e113462021.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Kalhan SC, Guo L, Edmison J, Dasarathy S,
McCullough AJ, Hanson RW and Milburn M: Plasma metabolomic profile
in nonalcoholic fatty liver disease. Metabolism. 60:404–413. 2011.
View Article : Google Scholar
|
|
159
|
Kim SK, Joe Y, Zheng M, Kim HJ, Yu JK, Cho
GJ, Chang KC, Kim HK, Han J, Ryter SW and Chung HT: Resveratrol
induces hepatic mitochondrial biogenesis through the sequential
activation of nitric oxide and carbon monoxide production. Antioxid
Redox Signal. 20:2589–2605. 2014. View Article : Google Scholar
|
|
160
|
Dai Z, Wu Z, Yang Y, Wang J, Satterfield
MC, Meininger CJ, Bazer FW and Wu G: Nitric oxide and energy
metabolism in mammals. Biofactors. 39:383–391. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Kingery JR, Hamid T, Lewis RK, Ismahil MA,
Bansal SS, Rokosh G, Townes TM, Ildstad ST, Jones SP and Prabhu SD:
Leukocyte iNOS is required for inflammation and pathological
remodeling in ischemic heart failure. Basic Res Cardiol.
112:192017. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Jia L, Wang Y, Jin C, Ma Y, Wang Y, Song
L, Shen J, Xie Y and Xiang M: Macrophage heme oxygenase-1 modulates
peroxynitrite-mediated vascular injury and exacerbates abdominal
aortic aneurysm development. Am J Physiol Cell Physiol.
328:C1808–C1821. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Butterworth RF and Canbay A:
Hepatoprotection by L-ornithine L-aspartate in non-alcoholic fatty
liver disease. Dig Dis. 37:63–68. 2019. View Article : Google Scholar :
|
|
164
|
Jegatheesan P, Beutheu S, Ventura G,
Nubret E, Sarfati G, Bergheim I and De Bandt JP: Citrulline and
nonessential amino acids prevent fructose-induced nonalcoholic
fatty liver disease in rats. J Nutr. 145:2273–2279. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Steixner-Kumar AA, Santacruz D, Geiger T,
Rust W, Böttner D, Krenkel O, Bahrami E, Okafo G, Barth TFE, Haenle
M, et al: Single-cell landscape of peripheral immune cells in
MASLD/MASH. Hepatol Commun. 9:e06432025. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Sugiyama Y, Shimura M, Ogawa-Tominaga M,
Ebihara T, Kinouchi Y, Isozaki K, Matsuhashi T, Tajika M, Fushimi
T, Ichimoto K, et al: Therapeutic effect of N-carbamylglutamate in
CPS1 deficiency. Mol Genet Metab Rep. 24:1006222020.PubMed/NCBI
|
|
167
|
Conte C and Muscaritoli M: Editorial:
Muscle mass and function in COVID-19. Front Nutr. 9:10127422022.
View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Naing A, Papadopoulos KP, Pishvaian MJ,
Rahma O, Hanna GJ, Garralda E, Saavedra O, Gogov S, Kallender H,
Cheng L, et al: First-in-human phase 1 study of the arginase
inhibitor INCB001158 alone or combined with pembrolizumab in
patients with advanced or metastatic solid tumours. BMJ Oncol.
3:e0002492024. View Article : Google Scholar
|
|
169
|
Fenwick N, Weston R, Wheatley K, Hodgson
J, Marshall L, Elliott M, Makin G, Ng A, Brennan B, Lowis S, et al:
PARC: A phase I/II study evaluating the safety and activity of
pegylated recombinant human arginase BCT-100 in relapsed/refractory
cancers of children and young adults. Front Oncol. 14:12965762024.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Russo RS, Gasperini S, Bubb G, Neuman L,
Sloan LS, Diaz GA and Enns GM; PEACE Investigators: Efficacy and
safety of pegzilarginase in arginase 1 deficiency (PEACE): A phase
3, randomized, double-blind, placebo-controlled, multi-centre
trial. EClinicalMedicine. 68:1024052024. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Lian TY, Jiang X and Jing ZC: Riociguat: A
soluble guanylate cyclase stimulator for the treatment of pulmonary
hypertension. Drug Des Devel Ther. 11:1195–1207. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Trujillo ME, Ayalasomayajula S, Blaustein
RO and Gheyas F: Vericiguat, a novel sGC stimulator: Mechanism of
action, clinical and translational science. Clin Transl Sci.
16:2458–2466. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Hanrahan JP, Seferovic JP, Wakefield JD,
Wilson PJ, Chickering JG, Jung J, Carlson KE, Zimmer DP, Frelinger
AL III, Michelson AD, et al: An exploratory, randomised,
placebo-controlled, 14 day trial of the soluble guanylate cyclase
stimulator praliciguat in participants with type 2 diabetes and
hypertension. Diabetologia. 63:733–743. 2020. View Article : Google Scholar :
|
|
174
|
Schwartzkopf CD, Hadcock JR, Liu G,
Germano P, Roux J, Shea CM, Buys ES and Jones JE: Beneficial
metabolic effects of praliciguat, a soluble guanylate cyclase
stimulator, in a mouse diet-induced obesity model. Front Pharmacol.
13:8520802022. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Hingorany S and Frishman WH: Soluble
guanylate cyclase activation with cinaciguat: A new approach to the
treatment of decompensated heart failure. Cardiol Rev. 19:23–29.
2011. View Article : Google Scholar
|
|
176
|
Gawrys O, Kala P, Sadowski J, Melenovsky
V, Sandner P and Cervenka L: Soluble guanylyl cyclase stimulators
and activators: Promising drugs for the treatment of hypertension?
Eur J Pharmacol. 987:1771752025. View Article : Google Scholar
|
|
177
|
Sawabe T, Chiba T, Kobayashi A, Nagasaka
K, Aihara K and Takaya A: A novel soluble guanylate cyclase
activator with reduced risk of hypotension by short-acting
vasodilation. Pharmacol Res Perspect. 7:e004632019. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Bhogal S, Khraisha O, Al Madani M, Treece
J, Baumrucker SJ and Paul TK: Sildenafil for pulmonary arterial
hypertension. Am J Ther. 26:e520–e526. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Rosenzweig EB: Tadalafil for the treatment
of pulmonary arterial hypertension. Expert Opin Pharmacother.
11:127–132. 2010. View Article : Google Scholar
|