Introduction
Immune cells continuously adapt to dynamic nutrient
availability, tissue oxygenation gradients and inflammatory
signals, yet they must simultaneously preserve long-term
homeostasis to execute rapid effector programs(1,2).
This adaptive capacity is governed by immunometabolism, a process
in which innate and adaptive responses are intrinsically coupled to
coordinated shifts in glycolysis, oxidative phosphorylation, lipid
and amino acid metabolism that shape effector function, memory
formation, exhaustion and tolerance in immune cells (3). In this regard, mitochondria are
increasingly appreciated as key integrators, transitioning from a
canonical role in bioenergetics and metabolism to one that
encompasses multifunctional immune signaling. Specifically,
mitochondrial effectors modulate pattern recognition receptor
signaling, inflammasome activation, type I interferon responses and
programmed cell death, thereby linking metabolic reprogramming
directly to immune function (4).
Accordingly, mitochondrial dysfunction is increasingly viewed as a
primary driver of immune dysregulation across infection, cancer,
autoimmunity and chronic inflammation rather than merely as a
secondary consequence of inflammation (5,6).
Recent studies have revealed that mitochondrial
function is shaped not only by metabolic flux, but also by the
spatial organization of mitochondrial organelle interfaces, a
mitochondria-centric interaction network composed of two primary
regulatory modalities: Membrane contact sites (MCSs) and vesicular
trafficking pathways, including mitochondrial-derived vesicles
(MDVs) and mitochondrial extracellular vesicles (mitoEVs).
MCSs are defined as regions where two organelle
membranes are closely apposed without fusion (7). These contacts facilitate the rapid
and spatially restricted exchange of ions, metabolites, lipids and
redox signals, serving as platforms for the assembly and
coordination of signaling complexes, and thereby regulating the
magnitude and duration of downstream responses (8,9).
Mitochondria establish such contacts with the endoplasmic reticulum
(ER) (10,11), lysosomes (12,13), peroxisomes (14), the Golgi apparatus (15), lipid droplets (LDs) (16,17), the nucleus (18,19) and the plasma membrane (20,21). In parallel with contact-based
crosstalk, mitochondria generate MDVs that selectively transport
oxidized proteins, lipids and nucleic acids to
endolysosomal/lysosomal compartments (22,23), phagosomes (24) and peroxisomal compartments
(25,26). Furthermore, mitoEVs distribute
mitochondrial cargo to neighboring cells when lysosomal function is
impaired (27,28) or when intercellular vesicle
exchange remodels the recipient cell organelle network (29-31). Collectively, membrane
contact-mediated crosstalk and MDV trafficking establish a
mitochondria-centric interaction network that enables rapid,
spatially restricted exchange of essential molecular cargo and
signaling mediators both within the cell and across intercellular
boundaries. This spatial layer of regulation is particularly
critical in immune cells, where it coordinates organelle function
with immune response-driven metabolic rewiring and signaling,
ultimately influencing effector polarization, cytokine secretion,
antimicrobial and antiviral responses and cell fate decisions
(32-34). Mitochondria-centered organelle
crosstalk is dynamically remodeled during immune cell activation
and chronic stress, which determines whether stress signals are
buffered, resolved, or amplified into sustained inflammation
(9-35). When dysregulated, it can skew
immune signaling and promote immunopathology across diverse
diseases, including infection, sepsis, cancer, autoimmunity and
chronic inflammatory disorders (36). Accordingly, coordinated control
of mitochondrial organelle communication through MCSs and selective
vesicle trafficking is emerging as a key determinant of immune cell
fate and a tractable therapeutic target in immunopathology.
In the present review, the spatial organization of
immune regulation was explored focusing on mitochondria-centered
coordination in immunometabolism. MCSs and vesicle-mediated
communication were characterized, elucidating how interfaces
between mitochondria and other organelles coordinate signaling and
metabolic circuits that tune innate and adaptive immune states,
with an emphasis on lipid transfer, Ca2+ handling,
mitophagy, redox control and epigenetic imprinting. Additionally,
shared and context specific patterns of interfacial rewiring were
summarized, and it was considered how these structural shifts
contribute to immunopathology, including infectious, septic,
neoplastic and autoimmune disorders. Crucially, emerging
therapeutic strategies that target interfacial dynamics or vesicle
trafficking were highlighted, including small molecule agents,
nanobiotechnology and cellular therapies that may advance our
understanding of how inter-organelle coordination shapes immune
cell states in health and disease, thereby expediting the clinical
realization of precision immunotherapies. Finally, the precision
and limitations of emerging methodologies were evaluated to ensure
objective validation of these nanoscale interactions, bridging the
gap between fundamental mechanistic discovery and the development
of next-generation interface-targeted diagnostic and therapeutic
clinical applications.
Structural organization of the mitochondrial
interface network
The mitochondrial interface network coordinates
cellular processes through structured interactions with multiple
organelles, mediated by nanometer-scale MCSs, multi-organelle
junctions and vesicle-mediated exchange pathways. By imposing
proximity and molecular selectivity, the interface network supports
controlled metabolite transfer, lipid trafficking, ion flux and
stress sensing, thereby coupling organelle state to immune
signaling and effector programs. In the present review, the
structural basis of mitochondrial inter-organelle communication was
described, its core functional roles discussed, and it was
highlighted how alterations in these interfaces shape immune cell
behavior in diverse conditions.
ER-mitochondria contacts in immune
signaling
ER-mitochondria contact sites (ERMCS) function as
highly adaptable immuno-metabolic signaling platforms, integrating
calcium flux, lipid exchange, stress sensing and antiviral
signaling to fine-tune immune cell fate across developmental,
inflammatory and pathological contexts (Fig. 1). ERMCS, often termed
mitochondria-associated membranes (MAMs), are specialized
microdomains structurally defined by core tethering complexes. This
apposition is primarily stabilized by mitofusin 2 (MFN2) and the
vesicle-associated membrane protein-associated protein B
(VAPB)-protein tyrosine phosphatase-interacting protein 51
(PTPIP51) complex (10,37,38). Beyond physical anchoring, MAMs
form dynamic functional hubs dedicated to localized lipid handling
and ion flux. For lipid transfer, proteins such as
oxysterol-binding protein-related proteins 5 and 8 (ORP5/8)
facilitate non-vesicular phosphatidylserine flux by coupling to the
mitochondrial MIB/MICOS architecture, which is essential for
maintaining cristae organization, mitochondrial morphology and
respiratory competence (39,40). Concurrently, calcium transfer is
structurally embedded into ERMCS via the inositol
1,4,5-trisphosphate receptor (IP3R)-glucose-regulated protein 75
(GRP75)-voltage-dependent anion channel (VDAC) axis, which funnels
ER-released Ca2+ across the outer mitochondrial membrane
(41,42). Subsequent entry into the
mitochondrial matrix is tightly gated by the mitochondrial calcium
uniporter complex and its regulators (EMRE and MICU1/2) (43,44). Functionally, this privileged
Ca2+ transport route directly fuels mitochondrial
oxidative metabolism and ATP production, while preventing calcium
overload-induced cell death. Ultimately, this localized structural
and metabolic network, continuously modulated by molecular
chaperones (for example, Sig-1R and DJ-1) and stress sensors (for
example, IRE1α and PERK), directly couples cellular energetic
states to downstream immune effector pathways (45-47).
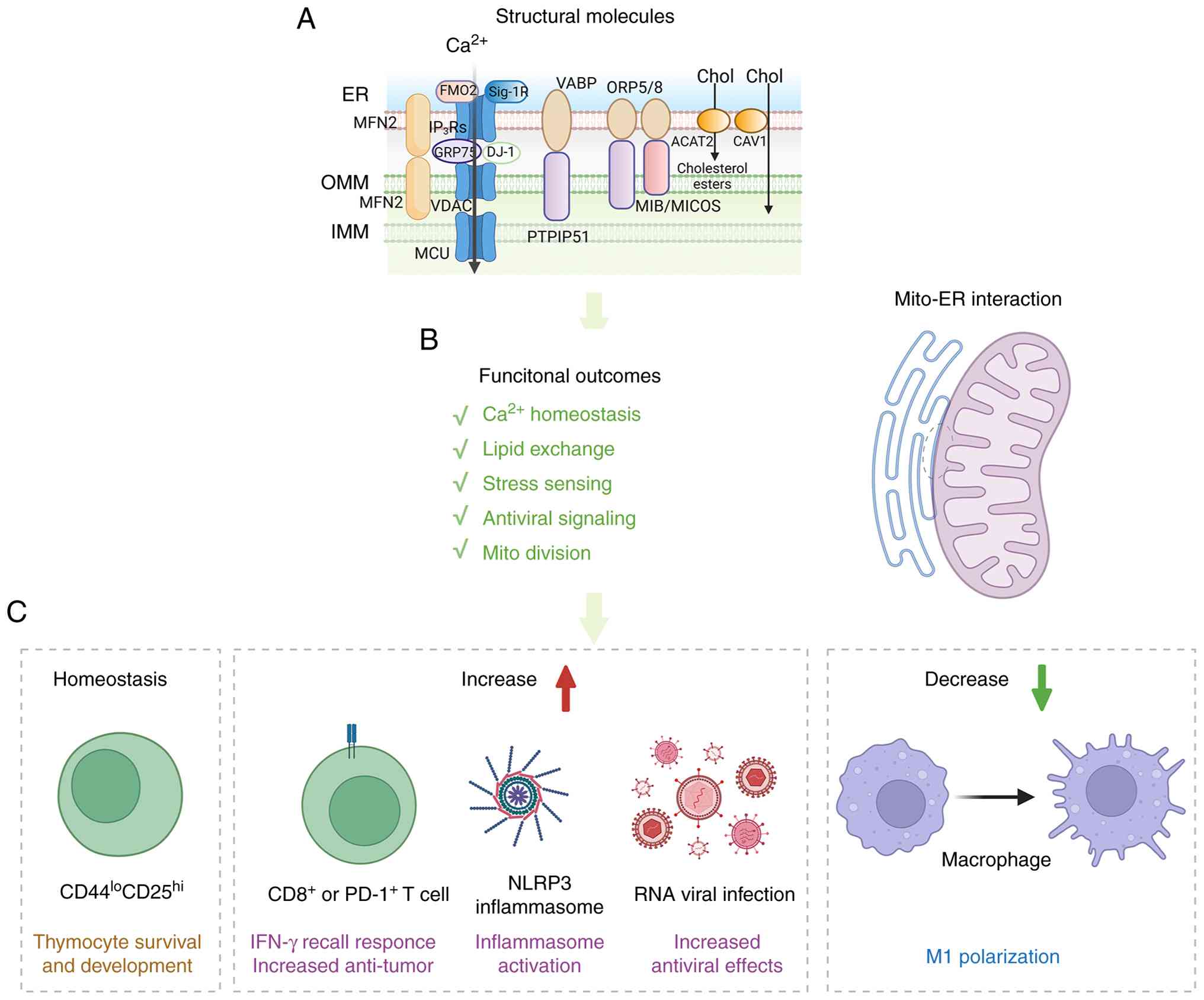 | Figure 1ER-mitochondria contacts as
immuno-metabolic signaling hubs in immune cells. (A) Schematic
illustration of the structural organization of ER-mitochondria
contact sites, highlighting representative tethering and signaling
molecules involved in Ca2+ transfer, lipid exchange and
stress sensing. (B) Major functional outcomes of ER-mitochondria
coupling, including Ca2+ homeostasis, lipid exchange,
stress sensing, antiviral signaling and mitochondrial division. (C)
Representative immunological consequences of ER-mitochondria
interface remodeling. Under homeostatic conditions, ER-mitochondria
contacts support thymocyte survival and development. Increased or
reinforced coupling promotes IFN-γ recall responses and antitumor
activity in CD8+ or PD-1+ T cells, enhances
NLRP3 inflammasome activation and antiviral signaling, whereas
reduced coupling is associated with diminished M1 polarization in
macrophages. ER, endoplasmic reticulum; OMM, outer mitochondrial
membrane; IMM, inner mitochondrial membrane; MFN2, mitofusin 2;
FMO2, flavin-containing monooxygenase 2; Sig-1R, sigma-1 receptor;
IP3R, inositol 1,4,5-trisphosphate receptor; GRP75,
glucose-regulated protein 75; VDAC, voltage-dependent anion
channel; MCU, mitochondrial calcium uniporter; DJ-1, protein DJ-1;
VAPB, vesicle-associated membrane protein-associated protein B;
PTPIP51, protein tyrosine phosphatase-interacting protein 51;
ORP5/8, oxysterol-binding protein-related proteins 5 and 8; MIB,
mitochondrial intermembrane space bridging; MICOS, mitochondrial
contact site and cristae organizing system; ACAT2, acetyl-CoA
acetyltransferase 2; CAV1, caveolin-1; NLRP3, NOD-, LRR- and pyrin
domain-containing protein 3. |
In immune cells, this ER-mitochondrial coupling is
not merely a housekeeping function but a tunable platform that
translates microenvironmental stress into distinct immuno-metabolic
programs. Proximity ligation assays (PLAs) reveal that during early
thymocyte development (DN3 stage, defined by CD44lo
CD25hi), the assembly of the IP3R-GRP75-VDAC
Ca2+ transfer axis at ER-mitochondria contacts is
essential. Loss of the chaperone GRP75 disrupts this axis,
triggering mitochondrial stress, aberrant mitochondrial DNA (mtDNA)
release and type I interferon signaling that ultimately impairs
thymocyte survival and development (48). In mature populations, MAMs serve
as spatially restricted signaling hubs, and the functional
heterogeneity of ER-mitochondria contacts depends on the specific
immune cell subset and its distinct metabolic demands. For
instance, transmission electron microscopy (TEM) and PLAs reveal
that the abundance of ER-mitochondria contacts is increased in
effector memory CD8+ T cells (CD62L−
CD45RA− populations). These contact sites spatially
organize the mTORC2-AKT-GSK3β signaling pathway, which recruits
hexokinase I to mitochondrial VDAC, accelerating the substrate flux
and respiration required for rapid IFN-γ production during the
recall response of memory CD8+ T cells. Genetic ablation
of the mTORC2 structural component Rictor, as well as
pharmacological inhibition targeting the mTOR/AKT axis, profoundly
abrogates the recall effector functions (49). Furthermore, in the metabolically
hostile tumor microenvironment (TME), TEM and interaction assays
reveal that strengthening ER-mitochondria coupling via MFN2-SERCA2
interactions in tumor-infiltrating CD8+ T cells
(PD-1+) prevents mitochondrial Ca2+ overload
and preserves metabolic fitness. T cell specific Mfn2
conditional knockout models disrupt these contacts and precipitate
metabolic collapse, whereas MFN2 overexpression restored organelle
tethering, preserved T cell metabolic fitness, and significantly
augments antitumor efficacy, highlighting MAM integrity as a
determinant of immunotherapy efficacy (50).
In innate immunity, ER-mitochondria contact dynamics
are intricately linked to macrophage polarization and inflammatory
thresholds. Confocal microscopy (CM) and TEM demonstrate that
classically activated pro-inflammatory macrophage (LPS +
IFN-γ-induced) actively orchestrate a distinct spatial uncoupling
between the ER and mitochondria, thereby enforcing the glycolytic
metabolic switch requisite for robust inflammatory effector
functions. By contrast, IL-4+ IL-13-induced
anti-inflammatory macrophages do not exhibit altered
ER-mitochondria interactions (51). Specific MAM tethering components
also act as critical inflammatory switches. Subcellular
fractionation and CM reveal that the ER-resident tether ORMDL3
physically anchors mitochondrial dynamics regulating protein Fis-1
in human macrophages (THP-1), facilitating targeted calcium flux
and concentrated reactive oxygen species (ROS) generation that
drive classical NLRP3 inflammasome activation and IL-1β release.
Small interfering RNA (siRNA)-mediated ORMDL3 knockdown dismantles
inter-organelle contacts and completely abrogates inflammasome
assembly (52). In
disease-associated macrophages (CD14+ monocytes
differentiated), calcium transfer through MAM sites and GSK3β
inactivation drive mitochondrial hyperactivation and amplify
macrophage inflammatory responses in diseases such as rheumatoid
arthritis and coronary artery disease (53). Beyond metabolic tuning,
ER-mitochondria interfaces function as essential signaling
scaffolds during host defense. CM identifies that MAMs as the
principal platform on which MAVS is spatially anchored to RIG-I
during RNA virus infection. This compartmentalization orchestrates
downstream antiviral signaling to drive interferon responses,
making MAMs a prime membrane-proximal target for viral antagonism
(34). Collectively, even subtle
shifts in contact abundance, spacing, or composition can markedly
alter calcium transfer, metabolic flux and inflammatory signaling,
underscoring the sensitivity of immune responses to the precise
organization of ER-mitochondria coupling.
Mitochondria-lysosome contacts and immune
quality control
Rather than serving primarily as a signaling
scaffolds, mitochondria-lysosome interfaces act as adaptive
buffering modules that calibrate organelle turnover, degradative
capacity and inflammatory stress in response to immune challenge
(54) (Fig. 2). Mitochondria-lysosome contacts
are governed by a Ras-related protein Rab7A (Rab7) centered
tethering cycle, in which GTP-bound lysosomal Rab7 promotes contact
formation, whereas mitochondrial fission 1 protein (FIS1) recruits
TBC1D15 (TBC1 domain family member 15, a Rab7 GTPase-activating
protein) to drive Rab7 GTP hydrolysis and contact release, thereby
creating a reversible interface that spatially coordinates the two
organelle networks and biases sites of mitochondrial remodeling
(55). During this process,
mitochondria, the ER and lysosomes are spatially integrated into a
multidimensional crosstalk network. Super-resolution and live-cell
imaging further show that ER tubules physically constrict
mitochondria to mark the division sites, which subsequently
recruits lysosomes in a process driven by RAB7 GTP hydrolysis
(56) and spatially regulated by
ORP1L-mediated PI(4)P signaling
at the ER-lysosome-mitochondrion interface (57). Beyond direct tethering,
mitochondria communicate with the endolysosomal system via MDVs.
Through PINK1/Parkin-induced and syntaxin 17-mediated pathways,
MDVs selectively route oxidized cargo to lysosomes, providing a
steady-state quality control route independent of canonical
mitophagy (23,58).
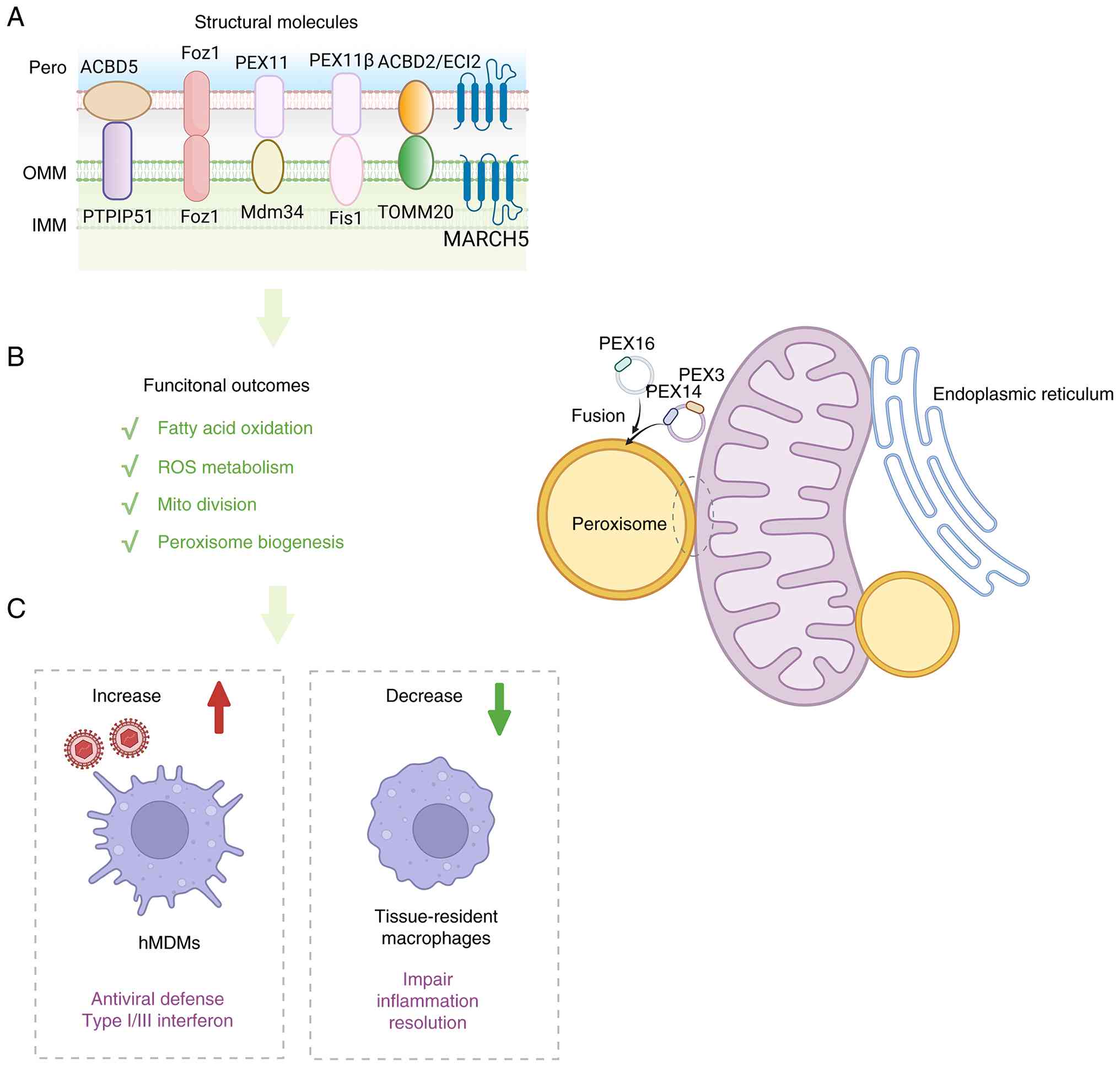
 | Figure 2Mitochondria-peroxisome crosstalk
coordinates lipid metabolism, redox buffering and antiviral
defense. (A) Schematic representation of structural molecules at
mitochondria-peroxisome interfaces, which collectively contribute
to tethering, peroxisome biogenesis, organelle division and
metabolite exchange. (B) Functional outputs of
mitochondria-peroxisome communication, including fatty acid
oxidation, ROS metabolism, mitochondrial division and peroxisome
biogenesis. (C) Representative immunological consequences of
mitochondria-peroxisome coupling. Increased coordination between
the two organelles enhances antiviral defense and type I/III
interferon responses in hMDMs, whereas impaired peroxisomal support
in tissue-resident macrophages is associated with defective
inflammation resolution. ACBD5, acyl-CoA-binding domain-containing
protein 5; PTPIP51, protein tyrosine phosphatase-interacting
protein 51; Fzo1, fuzzy onions 1; PEX, peroxin; Mdm34,
mitochondrial distribution and morphology protein 34; Fis1,
mitochondrial fission 1 protein; ACBD2, acyl-CoA-binding
domain-containing protein 2; ECI2, enoyl-CoA delta isomerase 2;
TOMM20, translocase of outer mitochondrial membrane 20; MARCH5,
membrane-associated ring-CH-type finger 5; ROS, reactive oxygen
species; hMDMs, human monocyte-derived macrophages; IFN,
interferon. |
On the mitochondrial side, these contacts mark
active sites of mitochondrial fission. Super-resolution imaging
shows that lysosomes, often alongside the ER, serve as active
organizers of mitochondrial division (55). Functionally, these contacts act
as fission-competent geometries that can tune broader cell
behaviors, such as migration and proliferation (59). Additionally, these interfaces
serve as local ion exchange sites. Proximity mapping reveals that
lysosomal Ca2+ efflux (for example, via TRPML1) is
routed to mitochondria, tuning mitochondrial Ca2+
dynamics and linking inter-organelle contact remodeling to stress
adaptation programs (58). On
the lysosomal side, mitochondrial activity constrains lysosomal
competence through energetics, spatial organization and
transcriptional adaptation. For instance, mitochondrial metabolic
dysfunction can elevate lysosomal pH and impair autophagic flux
(60). Furthermore, contact
tethering dynamics, including mitochondrial Mid51/Fis1
organization, actively shape lysosomal motility, positioning and
network remodeling (61).
Transcriptionally, mitochondrial damage and redox states are
effectively transduced into lysosomal signaling. Elevated
mitochondrial ROS and AMPK activation trigger TFEB nuclear
translocation, driving a wave of lysosomal biogenesis and
increasing autophagic capacity during stress (62,63). Collectively, these pathways
establish mitochondria-lysosome interfaces as vital regulatory
checkpoints coordinating lysosomal degradative function and
positioning with mitochondrial energetic and redox outputs. This
tight coupling is particularly consequential in immune cells, which
must synchronize endolysosomal processing with rapid metabolic
reprogramming to maintain effective immune signaling.
Disruption of mitochondria-lysosome can jointly
impair metabolic fitness and signaling integration, reshaping
T-cell activation, fate decisions and dysfunction across diverse
diseases (64). In
CD4+ T lymphocytes, Tfam loss or pharmacological
inhibition of mitochondrial respiration severely impairs lysosomal
degradation capacity and calcium mobilization. This physical and
metabolic uncoupling disrupts endolysosomal trafficking and
triggers a compensatory but aberrant activation of the
transcription factor EB (TFEB) network and synergy causally
subverts T cell differentiation towards pro-inflammatory Th1 and
Th17 subsets. NAD+ restoration rescues lysosomal
function, and failure of these mitochondria to lysosome
NAD+ transfer promotes pro-inflammatory CD4+
T cell skewing and worsens inflammation in vivo (65). Advanced spatial transcriptomics
and dynamic live-cell proximity assays demonstrated that the intact
spatial signaling between mitochondria and lysosomes is
indispensable for orchestrating regulatory T cells (Tregs)
metabolic plasticity. Treg cell-specific mitochondria Opa1
deletion or lysosome Flcn deletion phenocopies and this
interplay reprograms Treg cell differentiation and functions with
direct consequences for immune tolerance (66). In THP-1 cells, TFAM functions
unconventionally as a dedicated autophagy receptor when mtDNA leaks
into the cytosol through physically bridges the interaction between
the mislocated cytosolic mtDNA and the LC3 protein residing on the
autophagosomal membrane. Targeted Tfam knockdown and global
knockout completely abrogated this receptor-mediated physical
capture. The misplaced mtDNA pathologically hyperactivated the
cGAS-STING innate immune sensing pathway, culminating in an
explosive downstream cascade of type I interferons and sterile
inflammation (67). Furthermore,
mitochondria-lysosome coupling also shapes inflammasome biology and
antimicrobial defense. For example, in bone marrow-derived
macrophage (BMDM), high-resolution confocal fluorescence microscopy
with LC-MS/MS-targeted metabolomics reveals that upon stimulation
by pathogens such as Salmonella typhimurium, the lysosomal
biogenesis factor (TFEB) is activated and translocates to the
nucleus to upregulate the transcription of aconitate decarboxylase
1 (Irg1/Acod1), thereby driving the massive synthesis of itaconate
within mitochondria. The mitochondria-derived itaconate precisely
target the Salmonella-containing vacuole to suppressing the
survival of proliferative Salmonella in macrophage. Both
Tfeb, Irg1 and Rab32-deficient models confirmed that targeted
blockade at any steps of this spatial delivery pathway disrupts the
spatial transport of itaconate inhibiting the bactericidal capacity
of macrophages (68). Moreover,
infection-induced metabolic stress activates AMPK, which
subsequently promotes the spatial translocation of TFEB from the
cytoplasm or lysosomal surface to the nucleus, synergistically
regulating lysosomal biogenesis and mitochondrial function,
resulting the accelerated clearance of damaged organelles, ultimate
enhancement of the immune clearance capacity against intracellular
pathogens, and the fine-tuning of programmed cell death to prevent
inflammatory storms (32).
Overall, mitochondria-lysosome contacts extend far beyond basic
organelle quality control to serve as essential signaling hubs in
immunity. By coordinating the clearance of inflammatory triggers
(for example, damaged mtDNA) and tuning metabolic flux, these
interfaces directly dictate immune cell fate, support robust
antimicrobial defenses, and prevent aberrant inflammation, which
translating cellular quality control directly into systemic
immunological homeostasis.
Mitochondria-peroxisome crosstalk in
antiviral immunity
In mammalian cells, de novo peroxisome
formation can proceed through mitochondria-derived pre-peroxisomal
vesicles in which PEX3 and PEX14 first target the mitochondrial
outer membrane and are released into vesicular intermediates that
later fuse with ER-derived PEX16 carriers (69) (Fig. 3). The outer mitochondrial
membrane E3 ubiquitin ligase MARCH5 is essential for this selective
biogenic route, specifically controlling the formation of
PEX3-positive pre-peroxisomes to maintain organelle abundance and
functionality (70,71). Once formed, peroxisomes engage
mitochondria through functional contact sites (PerMit) that act as
bidirectional hubs coupling lipid flux, redox buffering and
organelle homeostasis. These contacts rely on specific tethers,
such as the ACBD5-PTPIP51 complex, which facilitates the transfer
of mitochondria-produced ROS into the peroxisome for antioxidant
buffering under oxidative stress (72), and ACBD2/ECI2, which supports
lipid/cholesterol trafficking (73). Additionally, mitochondrial fusion
proteins (such as Fzo1 in yeast and MFN1/MFN2 in mammals) promote
spatial co-clustering to enhance metabolic coordination, such as
transferring peroxisomal citrate to support the mitochondrial TCA
cycle (74,75). Productive coupling also depends
on spatial distribution and shared remodeling mechanisms.
Peroxisome-targeted MIRO1 variants rewire organelle motility and
positioning to optimize functional engagement in regions of high
metabolic demand (76).
Furthermore, mitochondria and peroxisomes utilize a shared, modular
division machinery toolkit including DRP1, FIS1, MFF and PEX11β
that enable synchronized tuning of organelle morphology under
metabolic stress (77-80), with components such as ERMES
Mdm34 further stabilizing these functional contacts (81). Methodological advances,
particularly Split-TurboID proximity labeling, now enable precise,
high-resolution proteomic mapping of these dynamic PerMit
interfaces (82). Peroxisomes
perform the initial steps of very long chain fatty acid (FA)
β-oxidation, generating shortened lipid intermediates and
acyl-carnitines that are subsequently exported for further
oxidation in mitochondria (83).
Conversely, disruption of peroxisome biogenesis (for example, PEX3
or PEX5 loss) directly feeds back onto mitochondrial dynamics,
inducing DRP1-dependent fragmentation and sensitizing cells to
apoptosis, highlighting peroxisomes as active modulators of
mitochondrial stress responses (84).
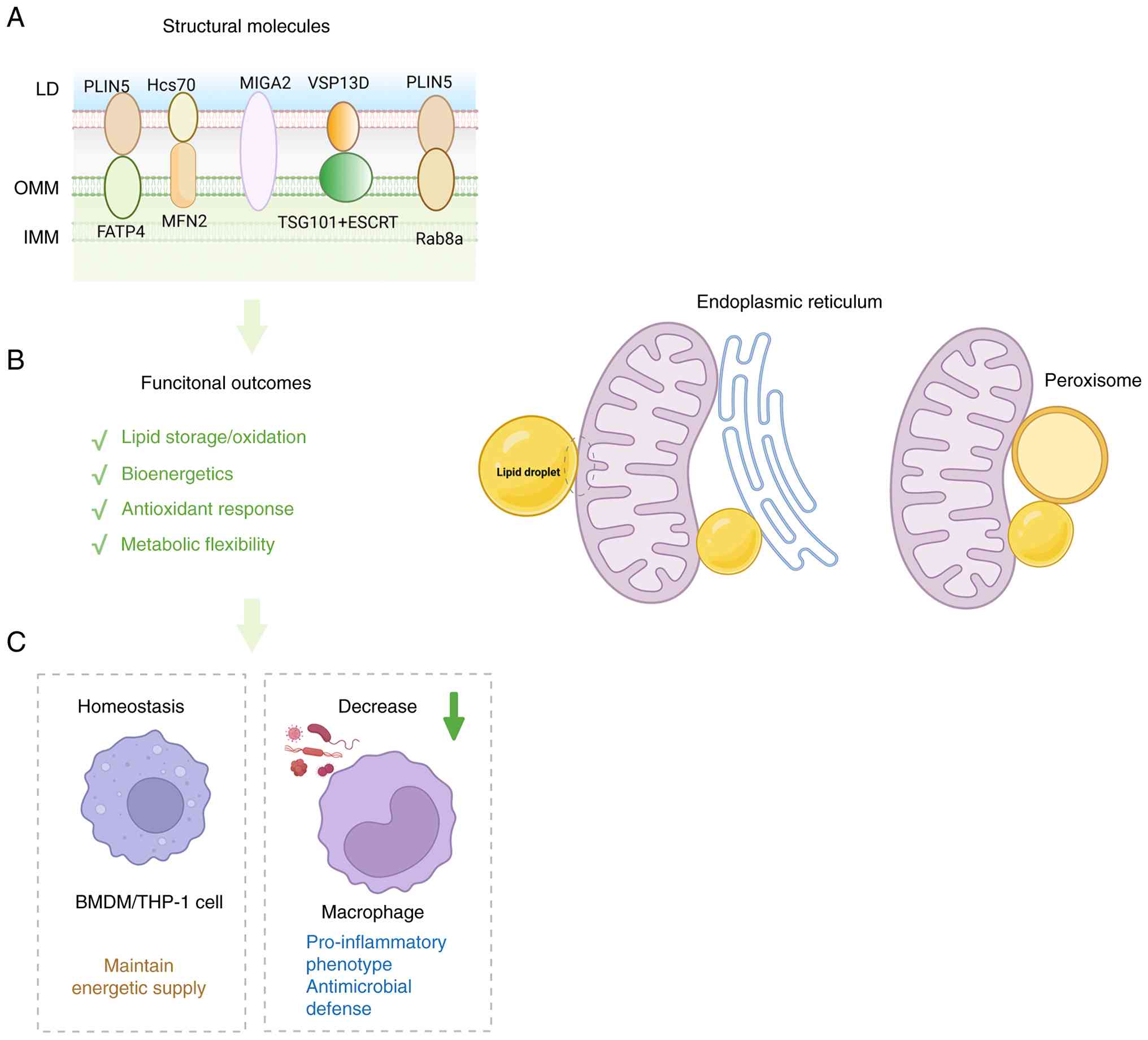
 | Figure 3Mitochondria-LD contacts support
metabolic flexibility and inflammatory rewiring. (A) Schematic
illustration of the structural organization of mitochondria-LD
contact sites, showing representative tethering and regulatory
molecules. (B) Major functional outcomes of mitochondria-LD
coupling, including lipid storage/oxidation, bioenergetics,
antioxidant response and metabolic flexibility. (C) Representative
immune consequences of remodeling at this interface. Under
homeostatic conditions, mitochondria-LD coupling helps maintain
energetic supply in BMDM/THP-1 cells. By contrast, reduced coupling
in macrophages limits mitochondrial fatty acid oxidation and is
associated with a pro-inflammatory phenotype and enhanced
antimicrobial defense. LD, lipid droplet; PLIN5, perilipin 5;
Hsc70, heat shock cognate 70; FATP4, fatty acid transport protein
4; MIGA2, mitoguardin 2; VSP13D, vacuolar protein
sorting-associated protein 13D; TSG101, tumor susceptibility gene
101; ESCRT, endosomal sorting complexes required for transport;
Rab8a, Ras-related protein Rab-8A; BMDM, bone marrow-derived
macrophage. |
In immune metabolic condition, peroxisomes and
mitochondria form an interdependent metabolic redox unit, in which
coordinated FA handling and ROS control. In immune
microenvironment, this coupling may tune inflammatory signaling
thresholds by linking lipid remodeling and oxidative stress
management to mitochondrial fitness, thereby shaping how cells
respond to infectious or inflammatory stress (85). The Mitochondrial Antiviral
Signaling protein (MAVS) localizes not exclusively to mitochondria
but also to peroxisomes using high-resolution confocal
immunofluorescence microscopy and rigorous subcellular
fractionation. This dual localization dictates a bifurcated
immunological outcome: Peroxisomal MAVS (pexMAVS) orchestrates a
rapid, transient, and Type I interferon-independent induction of
interferon-stimulated genes (ISGs, such as Viperin), whereas
mitochondrial MAVS (mitoMAVS) coordinates a delayed, stable Type I
interferon (IFN-α/β) response. MAVS-deficient (Mavs−/−)
genetic background and performed complementation assays by
overexpressing MAVS variants engineered to selectively target
either the peroxisome (PEX11β-MAVS) or the mitochondrion
(mito-MAVS) indicates that peroxisomes and mitochondria act
sequentially and cooperatively as innate immune signaling platforms
during the host's antiviral defense (86). In human monocyte-derived
macrophages, both peroxisomal and mitochondrial MAVS are capable of
activating the classic RLR-dependent Type I and III interferon
pathways. However, the hepatitis C virus NS3/4A protease localizes
to both organelles but exhibits distinct cleavage efficiencies,
effectively dismantling MAVS at these spatial hubs to truncate the
innate immune response, and the causality was established through
MAVS knockout followed by the lentiviral overexpression of
organelle-restricted MAVS constructs (MAVS-pex and MAVS-mito)
(87).
More broadly, after severe respiratory viral
infection, including SARS-CoV-2 infection, tissue-resident alveolar
macrophages undergo marked peroxisomal remodeling. Peroxisomes
support macrophage-specific lipid metabolism, including ether lipid
synthesis and very-long-chain fatty acid processing, and thereby
help preserve mitochondrial metabolic fitness under inflammatory
stress. When this peroxisomal support is impaired, alveolar
macrophages show reduced mitochondrial respiratory capacity,
increased mitochondrial stress and aberrant inflammatory
activation. Conditional Pex5 deletion in CD11c+ lung
myeloid cells, together with complementary macrophage-lineage
models, impaired inflammation resolution after viral clearance and
disrupted alveolar epithelial repair. Mechanistically,
Pex5-deficient alveolar macrophages exhibited reduced ether lipid
production, mitochondrial dysfunction, increased inflammasome
activation and excessive IL-1β release, which promoted dysplastic
KRT8high transitional epithelial progenitor
accumulation, defective alveolar regeneration and chronic
post-viral lung pathology (88).
Collectively, peroxisome-mitochondria coordination is viewed
through shared lipid metabolism and ROS control, and through
organelle-localized signaling modules (for example, MAVS) that
together shape antimicrobial and antiviral effects.
Mitochondria-LD crosstalk in
immuno-metabolic fueling
Unlike other mitochondrial interfaces that primarily
organize signaling or degradative buffering, mitochondria-LD
contacts are uniquely positioned to govern how immune cells
partition lipids between storage, oxidation and inflammatory
mediator production (Fig. 4).
Mitochondria-LD contacts constitute specialized membrane sites
coupling neutral lipid storage with oxidative bioenergetic
membranes (89). These
interactions range from transient 'kiss-and-run' contacts
supporting rapid lipid flux to stable anchors (90), which supports high-demand
processes such as thermogenesis in brown adipocytes (91). Mitochondria bound to LDs
(peridroplet mitochondria) exhibit distinct proteomes and
bioenergetics that preferentially supply ATP for LD expansion while
exhibiting reduced FA oxidation (FAO) and fusion/fission dynamics
(92). At the molecular level,
diverse tethering modules assemble these interfaces. Core scaffolds
include LD-localized PLIN5, which couples with FATP4 (93) or mitochondrial Rab8a during
energy stress (94) and
MFN2-Hsc70 in cardiomyocytes (17). Moreover, systems-level spectral
imaging and PLAs reveals the spatial coupling among the ER,
mitochondria, and LDs constitutes a tri-organelle architectures via
ESYT-VAPB complexes (16), while
factors such as MIGA2 and the VPS13D-ESCRT axis integrate spatial
tethering with lipid transfer and membrane remodeling capabilities
(95,96) to efficiently execute downstream
metabolic programs. However, this tri-organelle crosstalk only
reported in non-immune cells (for example, adipocytes and
fibroblasts), thus future investigations are required to determine
whether the dynamic assembly of the ER-Mito-LD axis play a
biophysical switch governing immune cell metabolic fate.
Functionally, LD-mitochondria contacts coordinate lipid flux and
stress buffering to dictate organelle fate. On the mitochondrial
side, peridroplet mitochondria structurally partition activities to
preferentially support processes such as thermogenic fuel delivery
in brown adipocytes (92).
During nutrient and oxidative stress, these contacts execute
critical protective functions. For instance, DGAT1-dependent LD
biogenesis buffers excess FAs to prevent acylcarnitine accumulation
and mitochondrial lipotoxicity (97), while LD-centered antioxidant
responses mitigate lipid peroxidation to preserve mitochondrial
fitness (98,99). Efficient FA transfer for
β-oxidation and metabolic adaptation is dynamically driven by
specific tethers under stress, including PLIN5-FATP4 (93) and AMPK-regulated Rab8a-PLIN5 in
starved muscle cells (94), as
well as MFN2-Hsc70 and PLIN5-mediated coupling in lipid-loaded
oxidative tissues (100).
Furthermore, tripartite ER-LD-mitochondria junctions act as
essential hubs for sustained FAO and lipotoxic resistance (16). On the LD side, these interfaces
actively regulate lipid retention and mobilization. For instance,
PLIN5 restrains basal lipolysis while permitting stimulus-dependent
mobilization (101), and the
VPS13D-TSG101-ESCRT pathway dynamically remodels the LD surface to
enhance transfer efficiency during starvation (96).
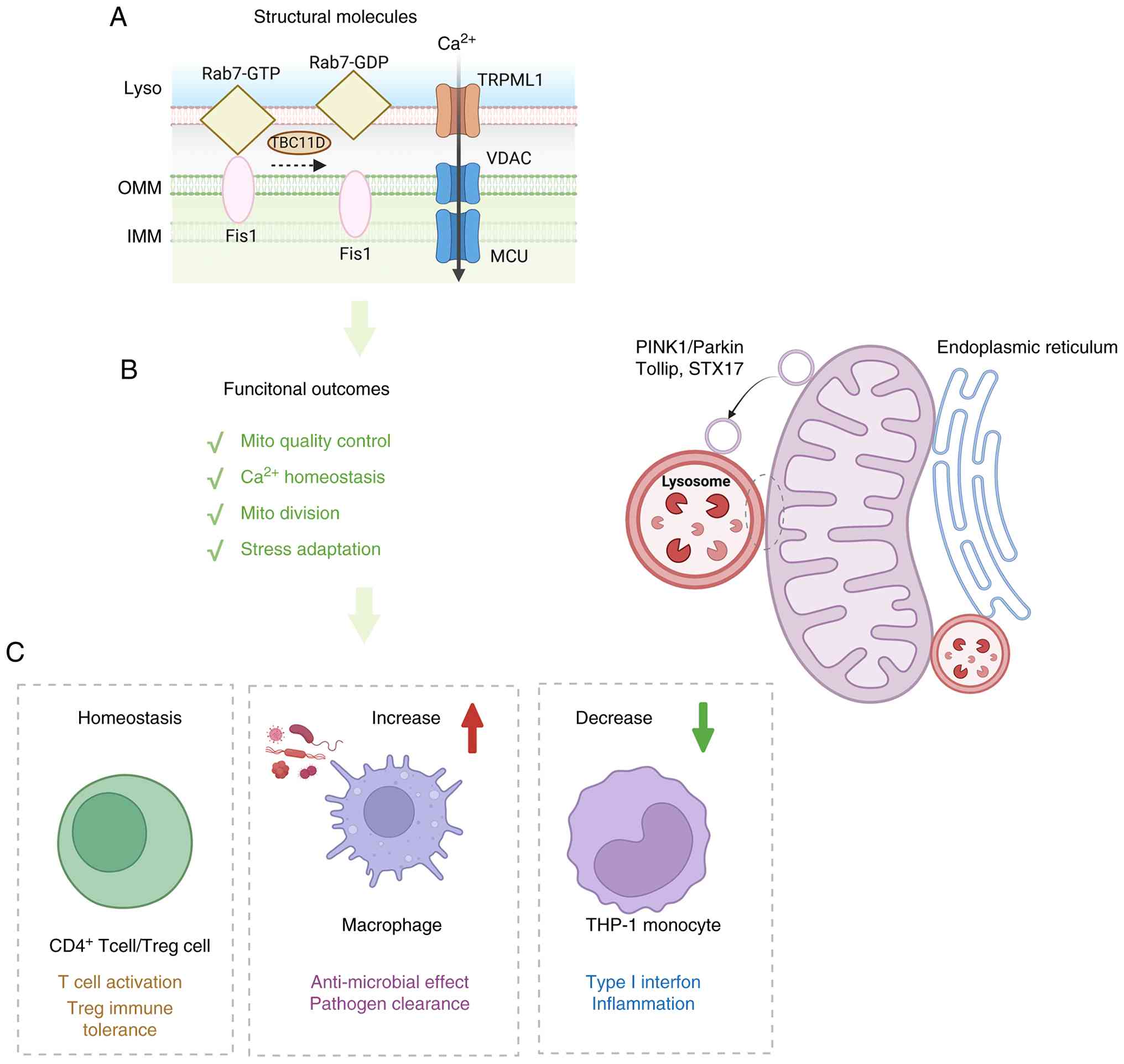
 | Figure 4Mitochondria-lysosome interfaces
integrate mitochondrial quality control with immune adaptation. (A)
Schematic illustration of the molecular architecture of
mitochondria-lysosome contacts. (B) Major functional outcomes of
mitochondria-lysosome crosstalk, including mitochondrial quality
control, Ca2+ homeostasis, mitochondrial division and
stress adaptation. Mitochondrial cargo is also delivered to
lysosomes through vesicular trafficking pathways associated with
PINK1/Parkin, Tollip and STX17. (C) Representative immunological
consequences of remodeling at this interface. Under homeostatic
conditions, mitochondria-lysosome communication supports T-cell
activation and Treg immune tolerance. Increased signaling through
this axis promotes macrophage antimicrobial activity and pathogen
clearance, whereas reduced coordination in THP-1 monocytes is
associated with altered type I interferon signaling and
inflammatory responses. Rab7, Ras-related protein Rab7; GTP,
guanosine triphosphate; GDP, guanosine diphosphate; TBC1D15, TBC1
domain family member 15; TRPML1, transient receptor potential
mucolipin 1; PINK1, PTEN-induced kinase 1; Parkin, parkin RBR E3
ubiquitin protein ligase; Tollip, Toll interacting protein; STX17,
syntaxin 17; Treg, regulatory T cell. |
In immune cells, the physical coupling between LDs
and mitochondria acts as a critical metabolic checkpoint that
directly dictates inflammatory signaling and immune cell
polarization. Rather than functioning merely as passive lipid
storage depots, LDs serve as active innate immune hubs that
concentrate lipid metabolic enzymes (such as cyclooxygenases and
lipoxygenases) to drive the robust synthesis of potent
pro-inflammatory lipid mediators, including eicosanoids and
prostaglandins, which intertwined with classic lipid-mediated
inflammatory signaling pathways (102). Utilizing high-resolution
live-cell confocal microscopy, advanced subcellular fractionation
and organelle-targeted proteomics, LDs undergo profound spatial and
functional reprogramming to serve as active innate immune hubs in
both murine BMDM and THP-1 cells. In resting macrophages, LDs
maintain extensive physical contacts with mitochondria to supply
FAs for OXPHOS. However, upon pathogen sensing (LPS-induced), this
deliberate physical uncoupling restricts mitochondrial FA
β-oxidation, thereby forcing a metabolic shift away from oxidative
phosphorylation and toward aerobic glycolysis. By preventing the
mitochondria from consuming these lipids for ATP production, the
immune cell effectively preserves arachidonic acid and other
essential FAs within LDs to fuel the massive production of
inflammatory mediators. Furthermore, this uncoupling alters
mitochondrial ROS production, which further amplifies antimicrobial
signaling, increases LD pathogen engagement, and contributes to
cellular autonomous defense. Pharmacological inhibitors to ablate
LD formation, as well as genetic knockdown models targeting key LD
coat proteins (for example, Plin2) rescued intracellular bacterial
survival and unequivocally establishing the obligate role of
LD-mitochondria spatial uncoupling in host defense (103). During human cytomegalovirus
infection, the interferon inducible enzyme viperin is relocalized
to mitochondria, where it perturbs mitochondrial FA β-oxidation by
engaging mitochondrial metabolic machinery. This mitochondrial
rewiring promotes lipogenic flux and LD accumulation, which in turn
supports efficient production of infectious virions, indicating
that a mitochondria-centered switch can drive LD biogenesis and
lipid supply during antiviral stress that is exploited by HCMV for
replication (104). Overall,
the dynamic remodeling of LD-mitochondria contacts is a fundamental
driver of immune cell polarization, dictating the balance between
lipid-driven inflammatory mediator synthesis, antimicrobial defense
and immune resolution. Although emerging evidence supports a role
for LD-mitochondria coupling or uncoupling in shaping inflammatory
lipid mediator availability and antiviral effector programs, direct
mechanistic studies of the LD-mitochondria axis in immune cells
remain relatively limited, making this an important direction for
future investigation.
Mitochondria-nucleus signaling and
epigenetic imprinting
Mitochondria and the nucleus maintain continuous,
bidirectional communication driven by defined spatial organization
rather than acting as independent compartments. The central
structural basis for this coupling is the regulated positioning of
nucleus-associated mitochondria (NAM) at the perinuclear region.
This extreme spatial proximity enables the direct, highly localized
channeling of retrograde signals such as mtROS and TCA cycle
intermediates directly into the nucleoplasm without cytosolic
dilution, profoundly influencing chromatin remodeling and
transcriptomic reprogramming (105) (Fig. 5). During mitochondrial
dysfunction, a pro-survival mitochondrial retrograde response is
facilitated by direct contact sites between mitochondria and the
nucleus that create localized communication microdomains. These
promote nuclear stabilization of pro-survival transcriptional
effects, and the translocator protein-dependent control of
mitochondrial quality as being required for this
mitochondria-nucleus coupling, positioning physical proximity as an
enabling step for adaptive nuclear reprogramming (19). Upon specific mitochondrial inner
membrane stress (such as that induced by mitochondrial uncouplers
or misfolded proteins), the resident mitochondrial protease OMA1 is
activated and subsequently cleaves the mitochondria-localized
protein DELE1. The cleaved form of DELE1 physically translocates
from the mitochondria to the cytosol to binds and activates the
eIF2α kinase HRI and attenuation of global cellular translation
alongside the selective nuclear expression of cytoprotective
factors (such as ATF4) to restore mitochondrial homeostasis.
Conversely, upon sensing an elevated AMP/ATP ratio, the cellular
energy sensor AMPK directly phosphorylates the transcriptional
coactivator PGC-1α at specific residues and enables the spatial
translocation and transactivation of PGC-1α within the nucleus to
induce the mitochondrial biogenesis. The loss-of-function
interventions (for example, pharmacological AMPK inhibitor and
Pgc-1α knockdown) completely abrogated the transcription of
mitochondrial genes in response to energetic stress (106). Similarly, activated mTOR
promotes the interaction between the transcription factor Yin Yang
1 (YY1) and the coactivator PGC-1α, promoting the transcription of
critical mitochondrial genes to match nutrient availability.
Pharmacological inhibition using Rapamycin (a specific mTOR
inhibitor) and siRNA-mediated specific knockdown of YY1 decoupled
mTOR signaling from PGC-1α transactivation, resulting in severe
mitochondrial respiratory defects and diminished oxidative function
(107). Collectively, spatial
coupling via perinuclear NAM contacts, integrated with these
stress-responsive signaling modules, constitutes a vital structural
platform for bidirectional mitonuclear coordination.
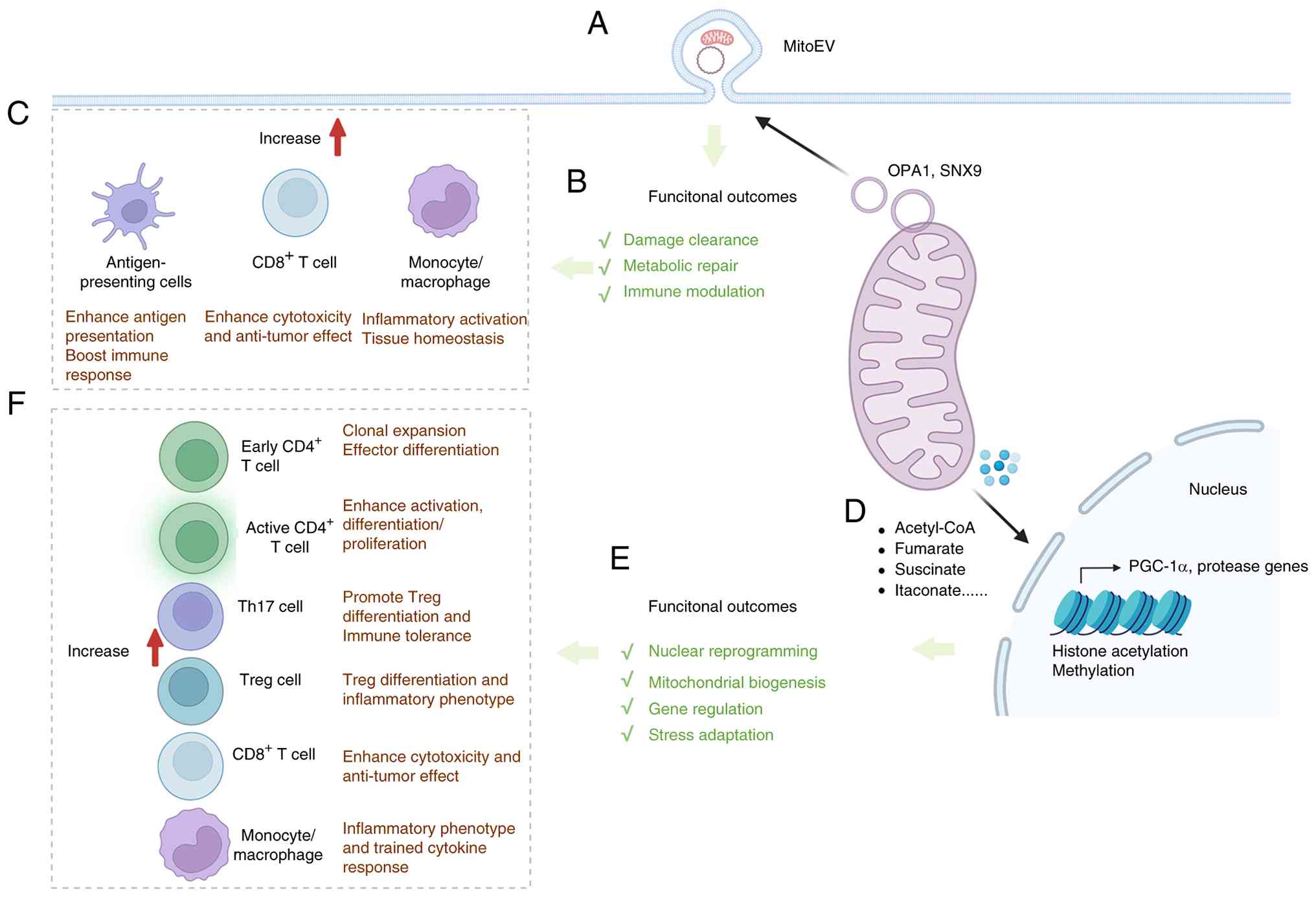 | Figure 5Vesicle-mediated mitochondrial
communication and mitochondria-nucleus signaling extend the
mitochondrial interface network in immunity. (A) Schematic
illustration of the release of mitochondrial-derived vesicles and
mitoEVs as an extracellular route of mitochondrial communication
and cargo export. (B) Major functional outcomes of mitoEV release,
including damage clearance, metabolic repair and immune modulation.
OPA1- and SNX9-related pathways are shown as representative
regulators of mitochondrial cargo sorting and extracellular vesicle
biogenesis. (C) Representative immunological consequences of
mitoEV-mediated signaling in recipient cells, including enhanced
antigen presentation by antigen-presenting cells, increased
cytotoxicity and antitumor effects in CD8+ T cells, and
inflammatory activation or tissue-homeostatic responses in
monocyte/macrophage populations. (D) Schematic illustration of
mitochondria-nucleus communication mediated by the transfer of
metabolites, which regulate chromatin remodeling and
transcriptional programs. (E) Major functional outcomes of
mitochondria-nucleus signaling, including nuclear reprogramming,
mitochondrial biogenesis, gene regulation and stress adaptation.
(F) Representative immune consequences of mitochondria-nucleus
communication, including clonal expansion and effector
differentiation of early and active CD4+ T cells,
altered Th17/Treg balance, enhanced CD8+ T-cell
cytotoxicity and antitumor activity, and inflammatory reprogramming
in monocytes/macrophages. OPA1, optic atrophy 1; SNX9, sorting
nexin 9; PGC-1α, peroxisome proliferator-activated receptor gamma
coactivator 1-alpha; Th17, T helper 17; Treg, regulatory T cell;
mitoEVs, mitochondria-containing extracellular vesicles. |
Moreover, mitochondrial metabolites orchestrate
nuclear gene expression by providing essential substrates for
chromatin modifications, competitively inhibiting epigenetic
enzymes, and acting as direct signaling intermediates to stabilize
transcription factor complexes. For instance, classic mitochondrial
metabolic enzymes, specifically the branched-chain ketoacid
dehydrogenase (BCKDHA) and pyruvate dehydrogenase (PDC) complexes,
physically translocate to the nucleus and anchor to the mediator
transcriptional coactivator complex, resulting de novo
production of acetyl-CoA directly at the chromatin, bypassing the
need for mitochondria to nucleus metabolite diffusion. This
targeted metabolite production directly fuels localized histone
acetylation. Targeted knockdown BCKDHA and PDC or using small
molecule inhibitors abolished site-specific histone acetylation and
disrupted normal cell state transitions (108). Under hypoxic conditions,
mitochondrial metabolism is rewired, specifically altering the
spatial efflux of citrate and acetyl-CoA from the mitochondria into
the cytosolic and nuclear compartments, altering the global
landscape of histone acetylation. Exogenous supplementation of
acetate or inhibit ATP-citrate lyase (ACLY) reversed the
hypoxia-induced epigenetic changes (109). Conversely, the accumulation of
specific mitochondrial metabolites exerts regulatory control by
competitively inhibiting chromatin-modifying enzymes (110). For example, in cells harboring
mutations in the mitochondrial TCA enzymes fumarate hydratase (FH)
or succinate dehydrogenase, there is a massive spatial accumulation
of fumarate and succinate within the mitochondria, which
subsequently spill over into the cytosol and nucleus. These
accumulated metabolites act as competitive inhibitors of nuclear
α-KG-dependent dioxygenases, including multiple histone
demethylases and TET family DNA demethylases and resulting
fundamentally alteration the cellular transcriptomic landscape
(111,112). When severe mitochondrial
dysfunction triggers the spatial activation of the Integrated
Stress Response (ISR) (often via the OMA1-DELE1-HRI axis), it
strongly upregulates the transcription factor CHOP. CHOP acts as a
highly specific transcriptional tuner to represses the overarching
expression of the ISR master regulator ATF4, effectively scaling
down the stress response to prevent cytotoxic hyper-activation and
allow cellular viability adaptation (113). Thus, mitochondria-nucleus
communication couples metabolic state to chromatin and
transcriptional regulation while enabling the nucleus to
continuously recalibrate mitochondrial function, together
supporting sustained cellular adaptation beyond the initiating
cue.
In immune cells, mitonuclear signaling can lock in
durable activation states, lineage choices and inflammatory
thresholds. Upon classic T cell receptor (TCR) and CD28
co-stimulation, early CD4+ T cell (CD4+
CD62L+ CD44−) undergo a massive program of
mitochondrial biogenesis and proteome remodeling to specifically
upregulate mitochondrial enzymes involved in one-carbon (1C)
metabolism (such as SHMT2) to generate formate from serine. This
formate then physically effluxes from the mitochondria into the
cytosol and nucleus, providing the carbon units necessary for de
novo purine synthesis and epigenetic methylation, which is key
for successful clonal expansion and effector differentiation of T
cells. Mitochondrial translation inhibitor and tfam genetic
deficiency lead to a profound collapse in 1C metabolism and
completely arresting T cell activation and proliferation in
vitro and in vivo (114). In pathogenic Th17 cells
(CD4+ IL-17A+ subsets), MTHFD2 restrains
inappropriate transcription factor FoxP3 induction, whereas MTHFD2
deficiency favors Treg differentiation (CD4+
FoxP3+), positioning this axis as a tunable lever for
anti-inflammatory immunotherapy target (115). In parallel, upon
CD4+ T cells activation, the PDH complex, a canonical
mitochondrial enzyme, undergoes a notable nuclear translocation.
This spatial repositioning allows for the localized, on-site
generation of acetyl-CoA directly at the chromatin, which acts as a
rate-limiting step to fuel H3K27ac-mediated histone acetylation,
promoting T cell activation, differentiation and proliferation,
driven by the classic TCR signaling pathway. In PDH-deficiency T
cells, acetyl-CoA generation and subsequent histone modification
was impaired with limited T cell activation (116). In Treg cells (CD4+
Poxp3+), elevated levels of the mitochondrial TCA cycle
metabolite α-ketoglutarate (αKG) induce a profound metabolic
rewiring including oxidative metabolism and altered lipid
homeostasis through increasing DNA demethylation, which actively
restricting Treg differentiation and shifting the balance toward an
inflammatory phenotype. Cell permeable αKG analogs alongside
pharmacological inhibitors of mitochondrial complex II (TTFA) and
diacylglycerol acyltransferase (DGAT1/2 inhibitors) effectively
rescued Treg differentiation (117). In CD8+ cytotoxic T
lymphocytes (CTLs), mitochondria-derived glutarate exerts its
effects by translocating to the nucleocytosolic space, where it
functions as a competitive inhibitor of nuclear αKG-dependent
dioxygenases (such as KDM5 histone demethylases and TET enzymes).
This profoundly enhanced the CD8+ T cell cytotoxicity,
persistence and effector function, significantly boosting antitumor
immunity within the classic cytotoxic T cell activation network.
Direct in vivo dietary supplementation of glutarate, as well
as the genetic knockout and pharmacological inhibition of the
glutarate-producing enzyme DHTKD1 heighten their antitumor efficacy
(118). During inflammatory
activation of macrophage (murine BMDMs and human PBMCs),
macrophages highly upregulate the mitochondrial enzyme IRG1,
catalyzing the production of itaconate to translocation from the
mitochondria directly into the nucleus. Within the nucleoplasm,
itaconate functions as a direct, competitive inhibitor of
Ten-eleven translocation (TET) DNA dioxygenases and inhibit the DNA
demethylation at specific loci to dampens the overarching
transcription of secondary inflammatory genes, serving as a
negative feedback loop for classic NF-κB inflammatory signaling
pathways. Genetic knockout mouse model lacking the
itaconate-producing enzyme (Irg1−/−), along with
treatment using the cell-permeable derivative octyl-itaconate
ablation of mitochondrial itaconate production unleashed unbridled
nuclear TET activity and exacerbated inflammation (119).
In trained immunity, primary human CD14+
monocytes exposed with β-glucan (activating the classic Dectin-1
trained immunity pathway) triggers a sustained upregulation of
mitochondrial glutaminolysis, causing the specific accumulation of
the TCA cycle intermediate fumarate inside the mitochondria.
Fumarate subsequently spatially translocates into the
nucleocytosolic compartment, where it acts as a competitive
inhibitor of nuclear KDM5 family histone demethylases to stable
maintaining the activation of epigenetic marks (H3K4me3) at
pro-inflammatory gene promoters. Consequently, these cells mount an
enhanced, trained cytokine response upon subsequent heterologous
infections. Blocking mitochondrial glutamine utilization (treated
with glutaminolysis inhibitor BPTES) erased the trained epigenetic
phenotype, whereas exogenous fumarate supplementation casually
induced trained immunity independently of receptor stimulation
(120). Consistently,
mevalonate acting as a key mediator via insulin-like growth
factor-1 receptor (IGF-1R) -mTOR signaling and downstream histone
modifications, establishing a long-lived heightened responsiveness
of innate immune cells, directly connecting metabolic state to
persistent functional rewiring (121). Under stress condition,
herpesviruses induce mtDNA stress is required to fully license
interferon stimulated gene expression and robust type I interferon
responses (122). TFAM
functions as a selective autophagy receptor through an
LC3-interacting region, enabling delivery of cytosolic mtDNA-TFAM
complexes into the autolysosomal system. By clearing leaked mtDNA,
this lysosome-dependent pathway restrains cGAS-STING inflammatory
signaling amplification triggered by mitochondrial damage, whereas
failure to clear leaked mtDNA can sustain inflammatory signaling
(67,123). Together, these studies
underscore mitochondria-nucleus communication as a key axis in
immune cell regulation, whereby metabolic flux and mitochondrial
genome integrity are coupled to nuclear transcriptional and
epigenetic programs.
Vesicle-mediated mitochondrial
communication in immunity
MDVs (~70-150 nm) and mitoEVs are crucial carriers
mediating selective intracellular organelle communication and
extracellular cell-to-cell signaling (36,124) (Fig. 5). The biogenesis, cargo selection
and routing of these vesicles are orchestrated by a comprehensive
network of regulatory molecules. Initial membrane tubulation and
scission are driven by MIRO1, MIRO2 and DRP1, coordinated with
mitochondrial fission adaptors MID49, MID51 and MFF (125). Following budding, vesicle fate
is determined by specific sorting mechanisms. The PINK1-Parkin
axis, along with fusion factors such as Syntaxin 17 and the
endosomal adaptor Tollip, routes damaged mitochondrial material
toward late endosomal and lysosomal pathways (58,123). Alternatively, OPA1 and SNX9
regulate the loading of mitochondrial cargo into extracellular
vesicle routes (126), while a
distinct pathway involving the E3 ligase MARCH5 and PEX3 generates
pre-peroxisomal carriers (70).
By packaging functional macromolecular complexes (for example, ATP
synthase) rather than random fragments, MDV biogenesis serves as an
early, highly selective remodeling step responsive to stress
(124). These vesicular
pathways exert profound functional impacts on mitochondrial fitness
and partner organelles through degradative, metabolic replenishment
and signal efflux routes. First, the degradative MDV-to-lysosome
route provides rapid quality control. It selectively clears damaged
components independently of bulk mitophagy and acts as an essential
compensatory disposal pathway when LC3-dependent autophagy is
compromised, thereby preserving mitochondrial homeostasis (127). Second, for metabolic
replenishment, specific MDVs support de novo peroxisome
biogenesis. By routing defined mitochondrial components through an
ER-dependent relay, this pathway tightly couples mitochondrial
vesiculation to cellular lipid metabolic capacity and peroxisome
abundance (25). Beyond
intracellular circuits, mitochondrial components can be loaded into
multivesicular bodies and released as mitoEVs (27). This signal efflux enables
intercellular metabolic repair by transferring functional mtDNA and
proteins to support respiration in recipient cells and serves as an
alternative degradative clearance route during lysosomal impairment
(128). Crucially, the sorting
mechanisms governing MDV/mitoEV fate impose strict regulations on
what mitochondrial material exits the cell. This selectively limits
the release of potentially pro-inflammatory mitochondrial cargo
[for example, damage-associated molecular patterns (DAMPs)],
directly shaping intercellular communication. In the tissue
microenvironment, secreted mitoEVs are captured by professional
phagocytes, linking the routing of mitochondrial material directly
to phagosomal processing. Ultimately, this dynamic vesicular
continuum, balancing internal lysosomal turnover with external
vesicle release, determines the final disposal site of
mitochondrial antigens and profoundly modulates local inflammatory
states and immune responses under stress (27,126).
In immunity, MDVs and mitoEVs constitute an
additional mechanism of mitochondrial quality control that
intersects with antigen presentation, innate immune sensing, and
tissue level immuno-metabolic crosstalk (36,124). In professional antigen
presenting immune cells, mitochondrial proteins can be routed for
MHC class I presentation, and this pathway is restrained by the
Parkinson's-linked PINK1-Parkin quality control axis.
Mechanistically, PINK1-Parkin suppresses MDV dependent delivery of
mitochondrial cargo into the endolysosomal system for antigen
loading, thereby limiting mitochondrial antigen presentation
(MitAP) under inflammatory stimuli, thereby coupling organelle
surveillance decisions to adaptive immune monitoring (129). In an infection model, loss of
PINK1 is associated with rewired early immune responses and altered
inflammatory programs, where peripheral myeloid cells emerge as the
earliest highly dysregulated subset, subsequently triggering an
aberrant CD8+ cytotoxic T cell response. This is
consistent with mitochondrial quality control acting as an upstream
constraint on immunogenic mitochondrial cargoes which shape
downstream innate and adaptive immune programs. Although that
study's focus was not limited to vesicle ultrastructure, it links a
mitochondrial quality control defect to detectable alterations in
host immune response dynamics during infection (130). Beyond MitAP, infection can
trigger remodeling of MAMs and outer membrane shedding. For
example, during infection-associated mitochondrial import stress,
mitochondria can generate large outer membrane positive structures
(SPOTs) consistent with a vesiculation remodeling response that
redistributes outer membrane components. Functionally, this
stress-induced remodeling influences infection consequences,
suggesting how mitochondrial membrane remodeling can be hijacked
during in host pathogen interactions with downstream consequences
for inflammatory damage and cellular defense capacity (131). Under stress condition, EV
loading is selectively regulated to avoid exporting oxidized
mitochondrial proteins that function as pro-inflammatory DAMPs.
Mechanistically, OPA1 and SNX9 promote MDVs' trafficking
mitochondrial proteins into EVs, whereas Parkin redirects damaged
mitochondrial content toward lysosomal disposal, collectively
shaping whether mitochondrial signals are immunologically silent or
inflammation enhancing (126).
Conversely, when mitochondrial and nuclear DNA are packaged into
EVs, transfer of EV-associated dsDNA to macrophages can engage the
cGAS-STING pathway and amplify inflammatory programs in Crohn's
disease relevant inflammatory condition, providing a direct
mechanistic link between mitoEV cargo and innate immune activation
(132). Under inflammasome
related pyroptotic stress, caspase-1 and gasdermin D promote mtDNA
escape into the cytosol, then drive intraluminal vesicle formation
that enables mtDNA loading into intraluminal vesicles (ILVs) and
release via exosomes. Extracellular mtDNA containing exosomes
trigger excessive inflammation that induces Behçet's syndrome such
as pathology, directly connecting inflammatory cell death to
mitoEV-mediated propagation of sterile inflammation (133). At the tissue level,
mitochondrial export pathways can interface with professional
phagocytes to support immunologically controlled organelle
disposal. In the healthy myocardium, resident macrophages actively
take up cardiomyocyte-derived components including mitochondria,
supporting tissue homeostasis through phagocytic disposal of
expelled mitochondrial cargo (134). Conversely, thermogenically
stressed brown adipocytes eject oxidatively damaged mitochondrial
cargos within EVs, and resident macrophages remove these mitoEVs to
prevent extracellular accumulation to preserve thermogenic
competence. When macrophage clearance is compromised, mitoEV excess
accumulation is associated with impaired adaptive thermogenesis,
linking immune phagocyte function to mitochondrial quality control
at the tissue level (135). In
addition, when lysosomal function is compromised, cells can reroute
mitochondrial components into EVs generated through late endosome
or MVB pathways, creating an extracellular offloading route for
mitochondrial components. In vivo, these mitochondria
containing EVs can be captured by phagocytes and ultimately
processed in recipient cell lysosomes, positioning mitoEV release
as compensatory organelle quality control with immunological
consequences (27).
Collectively, these findings support that vesicle-mediated
mitochondrial trafficking via MDVs and mitoEVs coordinates cellular
intrinsic mitochondrial quality control with immune recognition and
intercellular inflammatory signaling and indicate that
context-dependent imbalances between immunologically silent
disposal and inflammatory mitochondrial export can remodel tissue
immune state and influence susceptibility to
inflammation-associated disease states.
Rewiring mitochondrial interfaces in immune
dysregulation-associated diseases
Immune dysregulation across diverse diseases is
increasingly recognized that mitochondria function as integrated
signaling and metabolic hubs within inter-organelle communication
landscapes rather than as autonomous bioenergetic units.
Mitochondrial MCSs and vesicle-mediated trafficking routes
coordinate metabolite flux, calcium handling, and redox control
with innate immune sensing and antigen processing, thereby allowing
host defense interface modules to, when chronically activated or
aberrantly dysregulated, sustain maladaptive inflammatory responses
and tissue injury in infection and sepsis (123,136), cancer (137,138), and autoimmune or chronic
inflammatory disorders (139,140). In the present review, the
shared and disease specific patterns of mitochondrial interface
rewiring were summarized and were linked to mechanism-targeted
immunomodulatory strategies (Fig.
6).
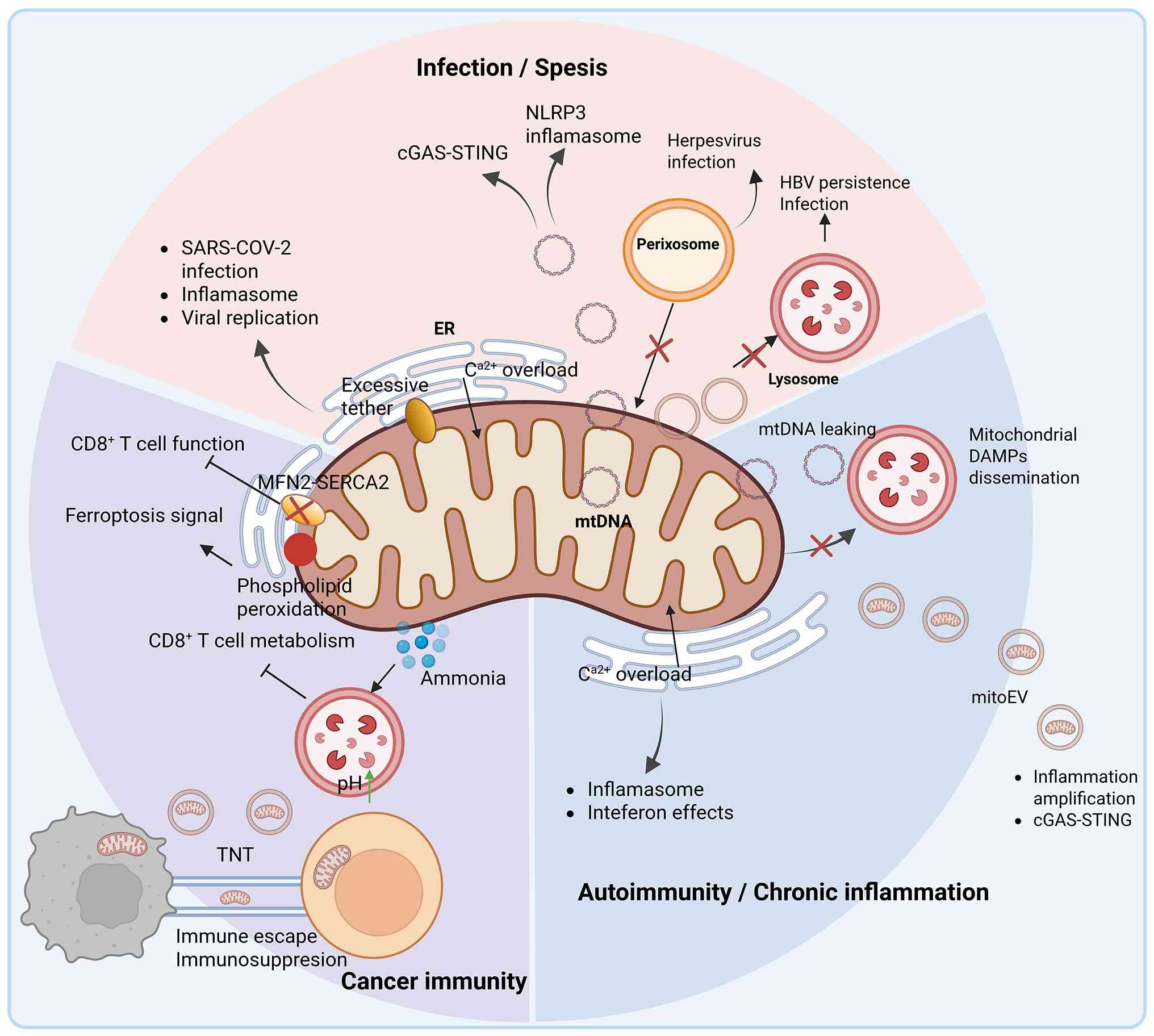 | Figure 6Maladaptive rewiring of mitochondrial
interfaces in immunopathology. Mitochondrial organelle interfaces
and trafficking routes are remodeled during disease, shifting from
homeostatic regulation to pathogenic signaling. In infection and
sepsis, pathogens (such as SARS-CoV-2) induce aberrant
ER-mitochondria tethering and Ca2+ overload, triggering
NLRP3 inflammasome activation and viral replication. Mitochondrial
stress leads to mtDNA leakage and cGAS-STING activation, while
peroxisomes contribute to antiviral signaling. Additionally,
inhibition of lysosomal function (for example, during HBV
persistence) prevents the clearance of damaged organelles and viral
components. In autoimmunity and chronic inflammation, sustained ER
stress and Ca2+ overload promote the release of
mitochondrial DAMPs and oxidized mtDNA. Defective lysosome
clearance amplifies sterile inflammation via inflammasome and
interferon pathways, while mitoEVs disseminate inflammatory signals
systemically. In cancer immunity, the tumor microenvironment
disrupts the MFN2-SERCA2 axis at ER-mitochondria contacts,
impairing the metabolic fitness of CD8+ T cell.
Furthermore, interface-associated phospholipid peroxidation
promotes ferroptosis signaling, while lysosome accumulation of
ammonia inhibits T cell metabolism. Intercellular transfer of
mitochondria via TNTs further contributes to immunosuppression and
tumor immune escape. NLRP3, NOD-, LRR- and pyrin domain-containing
protein 3; mtDNA, mitochondrial DNA; cGAS, cyclic GMP-AMP synthase;
STING, stimulator of interferon genes; HBV, hepatitis B virus;
mitoEV, mitochondria-containing extracellular vesicle; SERCA2,
sarcoplasmic/endoplasmic reticulum Ca2+-ATPase 2; DAMPS,
damage-associated molecular patterns; ER, endoplasmic reticulum;
TNTs, tunneling nanotubes. |
Infection and sepsis
Mitochondria shape host defense in infection and
sepsis through interface circuits couple organelle organization to
innate signaling and damage handling. During viral infection,
pathogens can rewire ER-mitochondria contact topology rather than
altering mitochondrial metabolism alone. In human cytomegalovirus
infection, stabilized ER encapsulation structures around
mitochondria become a dominant contact phenotype that supports
viral production, while premature strengthening of ER-mitochondria
tethering can activate STING linked antiviral programs, revealing
that contact remodeling can tune the balance between viral
replication and immunity (4,141). During SARS-CoV-2 infection,
viral structural proteins perturb host calcium homeostasis and
remodel ERMCS, linking altered contact architecture to
mitochondrial dysfunction and downstream cell stress programs that
can influence inflammatory injury in infected tissues (142). SARS-CoV-2 also engages
mitochondrial quality control to dampen antiviral signaling. Open
reading frame 10 (ORF10) suppresses type I interferon programs by
promoting MAVS degradation through mitophagy, thereby connecting a
virus driven shift in mitochondrial turnover to impaired innate
antiviral response (143).
Within infected cells, ERMCS provide spatial control points for
inflammasome priming. Enhanced ER-mitochondria coupling can
increase localized Ca2+ delivery to mitochondria and
promote permeability transition-associated release of oxidized
mtDNA fragments that support NLRP3 activation, positioning
Ca2+ hotspots at the interface as a proximal switch for
caspase 1-dependent cytokine activation (144). Crucially, genetic knockout or
pharmacological blockade of the mitochondrial permeability
transition pore and VDAC completely abolishes the cytosolic escape
of oxidized mtDNA, confirming the physical opening of these
interface channels is the strict molecular trigger that precedes,
and directly causes, subsequent NLRP3 inflammasome assembly and
pyroptosis. ER stress can further tighten ER-mitochondria
communication and augment Ca2+ dependent NLRP3 responses
together with remodeling of MAM-associated factors and
mitochondrial dynamics, reinforcing that inflammatory state is
regulated at contact sites (33).
Antiviral signaling is also spatially organized
across mitochondria and peroxisomes, as MAVS signals from both
membranes and peroxisomes can function as antiviral signaling sites
(86). In herpesvirus infection,
mitochondria to cytosol communication via mtDNA stress enhance the
innate antiviral signaling. Mitochondrial genome instability
promotes mtDNA release that engages cGAS-STING and elevates
interferon-stimulated gene expression, strengthening antiviral
defense, but prolonged activation provides a route to excessive
inflammatory signaling that can contribute to immunopathology
during infection (122).
Consistently, viperin can engage the peroxisome biogenesis factor
PEX19 and has been proposed to recruit peroxisomes to MAVS
signaling hubs, supporting an inter-organelle view for interferon
induction (145). In addition,
mitochondria-lysosome coupling and vesicle-mediated trafficking
constrain how mitochondrial damage is processed during infection.
Mitophagy and MDVs can route selected mitochondrial cargo to
multivesicular bodies and lysosomes for degradation, limiting
persistent mtROS producing substructures and reducing uncontrolled
DAMP release (125,127). When lysosomal function is
compromised, mitochondrial cargo can be diverted toward
extracellular disposal, including the secretion of mitoEVs under
lysosome inhibited conditions, shifting the balance from
degradation toward export with potential immunological consequences
(27). In HBV infection,
mitochondria-lysosome coupling emerges as a decisive antiviral
control level. Suppression of mitochondrial respiratory chain
activity weakens lysosomal acidification through combined redox and
energetic mechanisms, which reduces autophagic degradation of viral
material and thereby favors HBV persistence and disease
progression, whereas restoration of respiratory chain support
reinstates lysosomal function and promotes viral clearance
(146). Specifically,
pharmacological inhibition of the mitochondrial respiratory chain
using Rotenone or Antimycin A directly triggers defective lysosomal
acidification and halts autophagic flux, resulting in delayed HBV
clearance; conversely, metabolic restoration of respiratory chain
function fully reverses these lysosomal deficits, establishing
mitochondria-lysosome energetic crosstalk as the upstream causal
determinant of viral clearance. Metabolic stress can also intersect
with these routes, because fumarate-driven mitochondrial stress
promotes an MDV pathway that enables mtDNA release and cGAS-STING
linked inflammation (147).
Crucially, while conditional deletion of FH triggers early mtDNA
cytosolic release, targeted genetic depletion of Sorting Nexin 9
(SNX9) completely abolishes the generation of these MDVs and
prevents the subsequent STING-dependent inflammatory response. This
mechanistically proves that SNX9-dependent vesicular trafficking is
the indispensable sequence bridging metabolic stress to innate
immunity (147). Together,
these observations support that infection reshapes mitochondria-ER,
mitochondria-lysosome, mitochondria-peroxisome and vesicle mediated
mitochondrial routes to balance microbial clearance against host
damage.
In sepsis, the same interface circuits are adaptive
during early infection can become persistently engaged and augment
immunopathology. Circulating mtDNA can activate STING and
simultaneously disrupt lysosomal acidification and autophagic
clearance, creating a feed forward circuit that sustains
inflammatory injury in sepsis associated lung injury models
(148). Targeted administration
of the STING-specific small molecule inhibitor C-176, or genetic
knockdown of STING, explicitly blocks mtDNA-induced microglial
polarization, rescues autophagic dysfunction, and significantly
alleviates sepsis-associated inflammatory injury, confirming STING
as the essential causal trigger in this pathological cascade.
Maladaptive interface remodeling also involves the direct
dysregulation of mitochondria-lysosome contacts; specifically,
reduced TBC1D15-dependent contact control aggravates inflammatory
lung injury while restoring TBC1D15 function can improve mitophagy
flux and tissue outcomes (149). Knockdown or conditional
deletion of the Rab7 TBC1D15 disrupts these contacts, impairs
autophagic clearance, and thereby exacerbates sepsis-induced acute
lung injury, whereas restoration of TBC1D15 reverses this
pathogenic cascade (149).
Beyond intracellular contacts, vesicle-mediated mitochondrial
transfer can propagate damage signals across immune cells, because
pyroptotic macrophage-derived microvesicles can deliver
mitochondria to neutrophils and enhance NET formation and
coagulopathy, and microvesicle-transferred mitochondria have been
linked to cGAS-STING activation and inflammatory reprogramming in
sepsis (150). Specifically,
genetic knockout of Gasdermin D (GSDMD−/−) or treatment
with the GSDMD inhibitor Disulfiram strictly prevents the packaging
of GSDMD-N-expressing mitochondria into microvesicles, subsequently
abolishing microvesicle-induced NETosis and protecting against
sepsis-induced acute injury. This clarifies that GSDMD-mediated
pore formation is the upstream causal event dictating intercellular
mitochondrial transfer. Collectively, these studies indicate that
mitochondria-centered contact sites and vesicle-based trafficking
act as tunable targets during infection but can become maladaptive
drivers of sustained inflammation and organ injury in sepsis.
Cancer immunity
The TME enforces chronic bioenergetic and catabolic
stress on infiltrating immune cells, and accumulating evidence
indicates that mitochondria-centered coupling and trafficking
translate this stress into antitumor immune phenotypes. Hypoxic and
lactate-enriched niches reprogram solute transport and carbon
utilization in tumor infiltrating exhausted T cells, thereby
constraining mitochondrial metabolic limits and effector capacity,
which provides metabolic conditions in which organelle coordination
becomes rate limiting for immune effector function (151). At the interface,
ER-mitochondria coupling provides direct evidence that interface
architecture can determine intratumoral T cell effector capacity.
In TME, ERMCS act as prime hotspots for phospholipid peroxidation
that initiates and propagates ferroptotic signaling, indicating
that contact site remodeling can tune ferroptosis sensitivity and
thereby reshape immune pressure within the tumor ecosystem
(152). In CD8+ T
cells, an MFN2-SERCA2 axis supports ER-mitochondria calcium
homeostasis and mitochondrial metabolic competence, and disruption
of this module compromises effector function and antitumor
immunity, while reinforcement improves therapeutic efficacy,
positioning ER-mitochondria interfaces as regulatory hubs that
connect chronic stimulation to durable dysfunction (50). Deletion of MFN2 explicitly in T
cells (Cd4-Cre mediated deletion in vivo) disrupts the
SERCA2 interaction, proving that the structural loss of this
specific ER-mitochondria tether is the direct upstream trigger for
intracellular Ca2+ dysregulation. This timeline of
events, initial contact loss, followed by calcium and metabolic
collapse, definitively shows that interfacial disruption actively
drives CD8+ T cell exhaustion in the TME, rather than
being a secondary byproduct of the exhausted state.
Mitochondria-lysosome coupling is also poised to shape immune cell
persistence by specifying whether stressed mitochondrial regions
are repaired, routed to lysosomal degradation, or rerouted toward
inflammatory signals. In effector CD8+ T cells, ammonia
produced by mitochondrial metabolism is routed into lysosomes, and
excessive accumulation elevates lysosomal pH, triggers
reflux-associated mitochondrial injury, and blocks effective
clearance, thereby weakening antitumor immunity and adoptive T cell
therapy responses (153).
Mitochondria-lysosome coupling as a core immuno-metabolic module in
T cells, where coordinated lysosomes with mitochondrial
bioenergetics and redox to shape autophagy, trafficking, and fate
decisions, highlighting contact site biology as a still unresolved
determinant of exhaustion progression and therapy responsiveness
(64).
Vesicle-mediated mitochondrial communication
provides an additional aspect that allowing tumors to distribute
mitochondrial signals and even mitochondria to reshape immunity at
a distance. Senescent or stressed tumor cells can release mtDNA in
EVs that are taken up by polymorphonuclear myeloid-derived
suppressor cells, thereby activating cGAS-STING together with an ER
stress pathway involving protein kinase R-like ER kinase (PERK) to
reinforce an immunosuppressive program in the TME (154). In the TME, cancer cells can
transfer dysfunctional mitochondria carrying mutant mtDNA into T
cells, and these imported mitochondria persist rather than being
efficiently cleared, driving metabolic disruption and functional
decline that favors immune escape (138). Consistently, tumor cells can
acquire mitochondria from immune cells through physical nanotubes,
metabolically empowering cancer cells while depleting mitochondrial
capacity in immune cells and weakening their antitumor activity
(155). Conversely, bone marrow
stromal cells form intercellular nanotubes that deliver
mitochondria into CD8+ T cells, thereby restoring
respiratory capacity, improving resistance to exhaustion, and
increasing antitumor efficacy across preclinical models (156). Collectively, these studies
indicate that tumors reshape ER-mitochondria contact architecture
and lysosome linked quality control within immune cells while
deploying mitoEV cargo transfer and direct mitochondrial exchange
to impose spatially organized, mitochondria-centered
immunosuppression across the TME.
Autoimmunity and chronic
inflammation
Mitochondria organelle interfaces provide a core
checkpoint that determines whether mitochondrial stress is resolved
or converted into persistent sterile inflammation in autoimmunity
and chronic inflammatory disease. When nucleoid turnover and
endolysosomal disposal are compromised, mtDNA and inner
membrane-associated immunostimulatory species accumulate in
aberrant location and chronically activate cytosolic DNA sensing
and inflammasome pathways, including cGAS-STING and NLRP3 pathway,
thereby biasing myeloid and stromal compartments toward type I
interferon and IL-1 family responses (144,157,158). Under homeostatic conditions,
mtDNA stress is buffered by routing into autophagy and lysosome
pathways, and TFAM-dependent sequestration of cytoplasmic mtDNA and
nucleoid material restricts inappropriate DNA sensing and
inflammatory signaling (67).
When Tfam is conditionally ablated (using Cre-loxP conditional
knockout models) in specific immune or neural cell lineages, the
immediate molecular trigger is the failure of mitochondria to
deliver mtDNA to autophagosomes. This localized interface failure
rigorously precedes the subsequent pathological accumulation of
cytosolic mtDNA and the downstream hyperactivation of the
cGAS-STING pathway, proving that the disruption of TFAM-mediated
organelle crosstalk is the primary initiator of sterile autoimmune
pathology.
Mitochondrial function also feeds back on the
degradative pathway of this circuit, because mitochondrial
respiratory defects can impair lysosome competence and
endolysosomal trafficking, linking primary mitochondrial
dysfunction to reduced clearance capacity and exaggerated
inflammatory programs (65). In
parallel, chronic ER stress and sustained ER-mitochondria calcium
coupling at contact sites can increase mitochondrial permeability
and facilitate escape of oxidized mitochondrial danger signals,
reinforcing inflammasome and interferon circuits (33,144). Vesicle-based routing further
determines whether organelle stress remains local or becomes
systemically pro-inflammatory, as MDVs execute selective cargo
sorting that favors lysosomal disposal of damaged mitochondrial
material while limiting extracellular dissemination of
mitochondrial DAMPs, and this selection is reinforced by Parkin
dependent MDV trafficking mechanisms that require endosomal
adaptors such as Tollip (159).
More generally, mitochondrial components can be packaged into EVs
in a regulated way that shapes cargo oxidation state and
inflammatory potential, supporting the idea that vesicle selection
is an active checkpoint that separates protective disposal from
DAMP dissemination (126). In
inflammatory condition, mitochondria to lysosome signaling controls
Treg cell state stability which determine whether tissue
inflammation resolves or persists. Mitochondrial stress-driven
signaling engages AMPK- and TFEB-dependent lysosomal programs,
shifting Tregs away from highly oxidative suppressive states and
reducing effective tissue immune suppression, which can permit
sustained inflammation and thereby worsen disease-associated immune
dysregulation (32). In systemic
lupus erythematosus, platelet activation drives release of
extracellular mitochondria and mtDNA, providing circulating
mitochondrial antigens and nucleic acids that can be taken up by
immune cells and linked to interferon-biased programs (160). Collectively,
mitochondria-lysosome and mitochondria-ER contact site signaling,
together with MDV-based cargo selection, define an integrated
inter-organelle network that gates mitochondrial danger signals and
provides a mechanistic foundation for sustained autoimmunity and
chronic inflammation.
Therapeutic targeting of mitochondrial
interfaces
Small molecule and biologic targeting
contact sites and mitochondrial traffic
Therapeutic manipulation of mitochondria-organelle
interfaces aims to reroute inflammatory signals flow at nanoscale
contact sites and vesicular trafficking checkpoints rather than
globally suppressing mitochondrial metabolism. In this context,
small molecules currently represent the most tractable therapeutic
modality, whereas interface-selective biologic strategies remain
largely experimental (161). A
pharmacologically accessible target is the sigma 1 receptor
enriched at ER-mitochondria contacts, which constrains IRE1α RNase
activity during stress. In murine models of systemic inflammation
and sepsis, pharmacologic sigma-1 receptor agonism with fluvoxamine
suppressed IRE1-dependent cytokine programs and improved survival,
supporting an interface-enriched molecular pathway as a druggable
immunomodulatory target (162).
In LPS or fecal-induced inflammatory murine models, fluvoxamine
dampened cytokine productions in ex vivo LPS-stimulated
human whole-blood leukocytes (162). More direct MERC-directed
screening has also yielded compound 24, first identified in HCT116
cells as a MERC enhancer, and subsequently shown to restore
defective cholesterol trafficking, increased MERCs, enhanced LD
formation and restoration of mitochondrial cristae architecture in
an amyotrophic lateral sclerosis patient-derived cell model
(163). In parallel, several
clinically used or bioactive compounds appear to remodel MERC
function in a context-dependent manner rather than by directly
targeting tether proteins. For example, metformin normalized
excessive sarcoplasmic reticulum (SR)/ER-mitochondria coupling in
ventricular cardiomyocytes. This drug decreased myocardial MICU1
expression, mitochondrial Ca2+ content and improved
complex I-driven respiration (164). Sulforaphane was reported to
counteract MAM disruption and exaggerated glucose production in
palmitate-treated mouse hepatocytes and diabetic animal models
(165). Luteolin, identified in
a neuronal cell-based high-throughput screen and validated in
primary neurons, increased mitochondria-ER contacts, mitochondrial
Ca2+ and Ca2+-dependent PDC activity without
changing mitochondrial mass or structure (166). In neuronal cells, urolithin A
treatment reduced mitochondrial Ca2+ influx, mtROS
accumulation, and cognitive impairment in high glucose and in
streptozotocin-induced diabetic mice (167). Oxidative stress can promote
efflux of mtDNA fragments through pores associated with VDAC
oligomerization at the mitochondrial outer membrane, thereby
potentiating cytosolic DNA sensing and downstream inflammatory
responses. Pharmacologic inhibition of VDAC oligomerization with
VBIT-4 reduced mtDNA release and attenuated interferon linked
pathology in a lupus model, providing proof of principle that
restricting mitochondrial danger signals escape can indirectly
dampen downstream organelle coupled inflammatory signaling,
including cGAS-dependent STING signaling (168). The translational attractiveness
of this strategy lies in its action at a proximal checkpoint
upstream of several inflammatory sensing pathways, although this
advantage is tempered by a relatively narrow safety window, because
higher concentrations of VBIT-4 can suppress mitochondrial
respiration, perturb membrane potential and reduce cell viability
(169). Pharmacologic
manipulation of MDV routing remains at an early stage, yet recent
mechanistic work has begun to identify druggable intervention
targets. Parkin, acting in concert with the endosomal adaptor
Tollip, facilitates the incorporation of MDVs into the endosomal
system for subsequent lysosomal delivery independently of canonical
mitophagy, supporting the concept that vesicle-mediated
mitochondrial quality control can be selectively reinforced to
constrain inflammatory mitochondrial cargo (159). In this context, inhibition of
the mitochondrial deubiquitylase USP30 represents a practical
chemical lever because it amplifies PINK1-Parkin-dependent
ubiquitin signaling positioned upstream of multiple mitochondrial
disposal routes, including vesicle mediated trafficking and
lysosome directed clearance, thereby providing a plausible way to
shift mitochondrial traffic toward protective containment when the
pathway is intact or only partially compromised (170). Compared with VDAC blockade,
USP30 inhibition benefits from enzymatic tractability and
measurable pharmacodynamic readouts, although its efficacy is still
likely to depend on residual integrity of PINK1/Parkin-associated
quality control pathways in the target cells. Translational
development will require precise characterization of therapeutic
windows, given that interface components regulate essential calcium
and lipid exchange. Excessive enhancement of ER-mitochondria
calcium coupling may increase susceptibility to permeability
transition, whereas excessive attenuation can impair antimicrobial
signaling and lymphocyte activation, indicating that efficacy and
clinical acceptability are likely to depend on cell type, disease
stage and dosing regimen (162). These drugs are translationally
attractive because it combines repurposable agents with established
human pharmacology, yet their further clinical translation remains
limited by several issues. The field lacks robust biomarkers,
target engagement assays, and delivery strategies capable of
demonstrating selective MERC remodeling in immune disease relevant
conditions. Moreover, the relevance of these agents to
immune-related disease remains incompletely defined, as most have
yet to be validated in bona fide immune cells or in established
models of immune-mediated pathology. Future translational studies
should therefore focus on demonstrating target engagement in
relevant immune cell subsets.
For mitochondria-lysosome communication, the
current small-molecule landscape is somewhat more mechanistically
focused, although it remains at an early stage. In
palmitate-injured human endothelial cells and renal tubular
epithelial cells, TRPML1 agonists ML-SA5 and ML-SA8 reduced
mitochondrial ROS, indicating that pharmacologic activation of
lysosomal Ca2+ signaling can stabilize mitochondrial
homeostasis through the lysosome-mitochondria axis (171). Importantly, this pathway also
has emerging relevance to immune biology, because ML-SA5 suppresses
IL-1β secretion in bone marrow-derived macrophages and RAW264.7
cells exposed to LPS, zymosan A, or resiquimod, thereby linking
lysosomal signaling directly to inflammatory control in macrophages
(172). Currently available
pharmacology for mitochondria-LD and mitochondria-peroxisome
interfaces is rare and is dominated mainly by indirect metabolic
remodeling rather than direct tether targeting. At the
mitochondria-LD interface, atorvastatin promoted PKA-dependent
phosphorylation of PLIN5 in livers of high-fat diet-fed mice and in
isolated hepatocytes, thereby enhancing lipolysis and reducing
hepatic triglyceride accumulation (173), whereas glycycoumarin induced
the PLIN5-SIRT1 axis in vitro and in vivo and
attenuated palmitate-induced hepatocyte lipo-apoptosis and
inflammatory responses (174).
Because PLIN5 is a key organizer of LD-mitochondria coupling, these
observations are therapeutically relevant, but they also indicate
that existing compounds mainly act through metabolic regulators of
the interface rather than through dedicated contact site ligands.
At the mitochondria-peroxisome interface, the best developed small
molecules are PPAR agonists. Fenofibrate enhanced the expression of
peroxisomal genes and proteins involved in peroxisomal biogenesis
and function in livers of high-fat diet-fed mice, while also
improving FAO programs linked to mitochondrial metabolism (175). Bezafibrate was explored in
VLCAD-deficient patient fibroblasts as a metabolic reprogrammer for
mitochondrial FAO disorders (176), although subsequent work in
stressed VLCAD-deficient cells showed that bezafibrate-driven PPAR
activation could also disturb mitochondrial redox bioenergetics and
reduce cell viability (177).
Several drugs (for example, fenofibrate and bezafibrate) already
have systemic pharmacology, oral dosing paradigms, or early
clinical safety information, which lowers the barrier to
repurposing; however, their limitations are especially relevant for
immune-oriented interpretation. Accordingly, for immune-related
diseases these compounds are considered enabling metabolic
modulators whose relevance to organelle interface immunobiology
remains to be established directly.
Interface-targeted delivery and
nanotechnologies
Following from pharmacologic manipulation of
interface proteins, targeted delivery and nanotechnology provide a
targeting strategy that localizes bioactivity at nanoscale
interfaces and MDV pathways, thereby improving efficacy while
limiting system wide perturbation of organelle homeostasis. A
practical design principle is dual or sequential subcellular
targeting, in which a mitochondria-enriched motif is combined with
a second motif that biases accumulation at a partner compartment
such as the ER, increasing the probability that payloads reside
near contact zones where calcium, redox and lipid exchange are
integrated. In ER-mitochondria co-targeting strategies, the
molecular delivery platform DDY enables simultaneous mitochondrial
and ER localization with glutathione (GSH) responsive intracellular
parent drug release. Cancer cells exhibit significantly higher GSH
concentrations than normal cells; DDY safely and selectively
releases the repurposed antihistamine desloratadine directly at
mitochondria-ER interfaces. This precision delivery induces
profound ferroptosis in xenograft and metastasis tumor models while
avoiding systemic toxicity, highlighting a translational pathway to
reactivate approved drugs lacking innate anticancer activity. An
additional interface-directed strategy is mitochondria-ER click
systems built on the benzothiophene-fused azacyclononyne BT9N. By
conjugating an ER-directing sulfonamide moiety with mitochondria
maintaining thiophene and thiourea groups, BT9N relies on
mitochondrial membrane potential independent targeting to undergo
direct intra-organelle click reactions with azides. This modularity
enables organelle resolved functionalization proximal to
mitochondria-ER interfaces, providing a contact proximate
perturbation strategy that avoids bulk organelle disruption
(178). Interface-resolved
profiling further enables rational anchor selection, specifically,
proximity-dependent biotinylation and split proximity labeling at
LD-mitochondria contact sites identified a multimeric
ESYT1-ESYT2-VAPB/VAPC architecture localized at LD-mitochondria-ER
junctions that supports FA transfer and links tether composition to
lipotoxic stress control with implications for metabolic disease
biology (16). Photoactivatable
THTTPy-PTSA enables sequential ER-mitochondria targeting to rewire
ER-mitochondria coupling and enhance immunogenic cell death coupled
antitumor immunity in vivo (179). Mechanistically, THTTPy-PTSA
employs a p-toluene-sulfonamide (PTSA) group for initial ER
localization. Upon light irradiation, its tetrahydropyridine group
undergoes photo-oxidative dehydrogenation into a
mitochondria-targeting pyridinium moiety, driving a spatial
translocation that triggers cascade-amplified ER stress. This
targeted perturbation overcomes ER-mitochondria mediated immune
escape by releasing abundant DAMPs to induce widespread ICD
(179). Similarly, the
nanocomposite (Cu2Se-CaO2)@LA evokes mitochondria-associated ER
stress and engages PERK-mediated eIF2α phosphorylation to drive ICD
with dendritic cell maturation and cytotoxic T cell recruitment
(180). Triggered by second
near-infrared (NIR-II) irradiation, this
H2O2-self-supplying platform integrates
photothermal and chemodynamic therapies (PTT/CDT) to induce severe
Ca2+ overload and ROS formation. This sequence
selectively damages the mitochondria and ER networks, exerting
potent in vivo tumor suppression with high biosafety
(180). Additional
ER-mitochondria dual targeting phototherapeutic implementations
include the near infrared photosensitizer TCy5-I-3F that
synchronizes ER stress with mitochondrial dysfunction to inhibit
tumor growth and enhance immune activation under irradiation (ER
and mitochondrial double-targeted NIR photosensitizer
synergistically promote tumor cell death). Additionally, the
colorectal cancer nanoplatform (NP) HA/pCe6-A to NPs that
suppresses cancer stemness-linked resistance while enhancing
immunotherapy, consistent with the principle that
interface-synchronized ER stress and mitochondrial damage can be
therapeutically beneficial (181). By utilizing a hyaluronic acid
coating to actively target CD44 receptors on cancer stem-like cells
via endocytosis, this platform co-delivers the ER-targeting
photosensitizer pCe6 and the mitochondrial inhibitor atovaquone.
This dual organelle disruption effectively reprograms the
immunosuppressive TME by promoting apoptosis and ROS production,
achieving superior synergistic tumor inhibition compared with
conventional monotherapies (181).
Beyond ER coupling, mitochondria-lysosome
co-targeting nanostrategies are emerging through dual localization
photosensitizer or nanoparticle designs to modulate autophagy flux
and mitochondria-lysosome crosstalk. For example, a biomimetic
nanoplatform utilizing a hollow MnO2 core loaded with
tetrandrine and a mitochondria-targeting photosensitizer (Ce6-Apt)
initiates a triple-killing mechanism. Under near-infrared
irradiation, Ce6-Apt inflicts mitochondrial photodynamic damage to
activate mitophagy, while tetrandrine induces lysosomal
alkalinization to block the mitophagic flux and simultaneously
triggers excessive macropinocytosis (methuosis). This synergistic
spatiotemporal control completely severs the compensatory energy
supply of tumor cells, offering a precision strategy to overcome
metabolic drug resistance (182). Vesicle focused engineering
provides an interface informed delivery level because
mitochondria-lysosome quality control decisions can be switched
toward extracellular secretion when lysosomal function is impaired,
as lysosomal inhibition increases the secretion of mitochondria
within large EVs generated in multivesicular bodies and released
independently of autophagy, establishing a degradation vs. export
switch that is directly relevant to MDVs or mitoEVs (27), while regulated cargo selection
limits dissemination of oxidized mitochondrial DAMP proteins into
EVs, indicating that vesicle loading is an active checkpoint that
separates protective disposal from pro-inflammatory release
(126). Enabling EV platforms
that enhance active cargo loading and endosomal escape can be
adapted to interface-targeted delivery, when coupled to validated
dual organelle targeting signals. Examples include TOP-EVs for
intracellular protein delivery (183) and multimodal engineered EVs
that combine mini-intein mediated cargo loading with VSV-G driven
endosomal escape (184). By
contrast, strategies such as IDEA-mediated cGAS delivery mainly
demonstrate intracellular pathway engagement, with limited evidence
for preferential enrichment at organelle contact microdomains
(185). Overall, current
interface-directed delivery is best developed for ER-mitochondria
coupling and for mitochondria linked endolysosomal quality control
and offloading routing, whereas other mitochondria organelle
interfaces remain comparatively limited, underscoring the need for
future interface resolved profiling guided ligand design and
validation.
Despite these innovative designs, the clinical
translation of nanoparticle-based delivery systems is severely
hindered by in vivo bottlenecks, including poor solid tissue
penetration, rapid clearance by the mononuclear phagocyte system,
and variable immunological responses and translational uncertainty
in humans (186). The leap from
murine xenograft models to the complex human immune ecosystem
remains a formidable challenge. Alternatively, vesicle-focused
engineering (such as EVs) provides an interface-informed delivery
platform with superior biocompatibility (126). Strategies such as TOP-EVs
(183) and mini-intein-mediated
EVs (184) enhance precise
cargo loading. However, scaling up engineered EVs for clinical
grade manufacturing with consistent batch-to-batch uniformity
remains a core industrial obstacle. Future translational efforts
must prioritize simplifying these complex delivery platforms into
scalable, immune-cell-directed formulations that maintain high
interface specificity with low systemic exposure.
Engineering interfaces for adoptive cell therapy.
Distinct from systemic in vivo nanomedicine, targeted
modulation of mitochondrial interfaces during the cellular
manufacturing phase provides a highly translatable strategy to
imprint metabolic resilience and sustain long-term antitumor
efficacy in adoptive cell therapies. Sustained efficacy of
adoptively transferred tumor reactive CD8+ T cells relies on
mitochondrial fitness integrated across organelle interfaces and
mitochondria nucleus renewal programs under chronic stimulation. In
solid tumors, persistent antigen exposure and microenvironmental
stress drive impaired ADP coupled oxidative phosphorylation,
oxidative damage, reduced mitophagy and accumulation of
dysfunctional mitochondria, which promote terminal exhaustion and
limit persistence (187). One
engineering strategy strengthens mitochondria-ER coupling through
the MFN2-SERCA2 axis, in which MFN2 stabilizes contact-dependent
calcium exchange and sustains metabolic fitness in tumor
infiltrating CD8+ T cells to improve effector function
and antitumor immunity in solid tumor, providing a strategy for
reinforcing contact points during adoptive manufacturing or genetic
engineering (50). Mitochondria
to lysosome quality control is a second leverage targetable step
because repeated antigen stimulation drives progressive
accumulation of damaged mitochondria that cannot be compensated by
late intervention. In metabolically engineered CAR-T cells,
combined semaglutide and urolithin A treatment increased autophagy
through mTOR inhibition and enhanced mitophagy through ATG4B,
thereby improving persistence, sustaining long term antitumor
activity, and reducing cytokine release syndrome liability in
preclinical tumor models (188). Related interventions further
support this axis, as succinate preconditioning promoted
Bcl-2/adenovirus E1B 19 kDa-interacting protein 3 (BNIP3) mediated
mitophagy and preserved stem like CD8+ T programs that
improved antitumor immunity and adoptive efficacy in vivo
(189), and pharmacologic
inhibition of the mitochondrial deubiquitylase USP30 restored
mitophagy and mitochondrial fitness under chronic stimulation while
rejuvenating effector function, highlighting a druggable target
upstream of lysosome-directed mitochondrial disposal routes
(190). In parallel,
mitochondria-nucleus coupling can be engineered to secure
mitochondrial renewal capacity during sustained tumor challenge. In
CAR-T cells, expression of an inhibitory resistant PGC-1α isoform
reinforced mitochondrial biogenesis programs, stabilized oxidative
fitness, and supported durable antitumor potency, providing a
transcriptional module that cooperates with contact switch
reinforcement and mitochondria to lysosome throughput by sustaining
mitochondrial replacement over time (191). Collectively, adoptive cell
therapy can be potentiated by coordinated manipulation of
mitochondria-ER contact dependent calcium handling,
mitochondria-lysosome quality control throughput, and
mitochondria-nucleus biogenesis programs, while the limited
availability of selective tools for other mitochondria organelle
interfaces remains a major opportunity for future identification
and validation. Compared with the systemic administration of small
molecules or nanoparticles, manipulating mitochondrial interfaces
ex vivo during adoptive cell therapy manufacturing bypasses
numerous of the aforementioned systemic delivery and safety
bottlenecks, offering exceptional practical translation value. By
confining interface modulation to the ex vivo expansion
phase, researchers can hardwire metabolic resilience into
engineered immune cells, maximizing therapeutic efficacy while
might be eliminating the risk of systemic pleiotropic toxicity to
the patient's native tissues. However, the broader clinical
translation of all interface-targeted strategies is universally
hindered by shared preclinical and diagnostic bottlenecks. The
cornerstone of advancing these targeted therapies into clinical
trials is rigorous safety evaluation, yet current preclinical
testing models represent a profound bottleneck. Murine models often
fail to faithfully recapitulate the complex immuno-metabolic
architectures of human patients (192). Establishing a reliable safety
evaluation path requires a paradigm shift: Bypassing exclusive
reliance on simple animal models in favor of human immune-competent
organoids and patient-derived immune cells (193). Furthermore, across all
therapeutic modalities, a critical limitation in early-phase
clinical trials is the lack of validated, non-invasive biomarkers
that specifically report interface dysfunction, rather than generic
mitochondrial stress. Therefore, translating these therapies
requires the concurrent development of quantitative spatial
proteomics and localized in vivo dynamic sensors (194,195) to continuously monitor
off-target interface toxicities and ensure precise pharmacological
dosing.
Technical methodologies for probing
mitochondrial interfaces: Applications, limitations and potential
biases
The rigorous translation of mitochondrial interface
biology relies heavily on the precision of the methodologies used
to detect and quantify these nanoscale interactions. Because
organelle contacts are highly dynamic, transient and structurally
complex, a major bottleneck in the field is that no single
technique can simultaneously capture high-resolution architecture,
rapid temporal changes, and the comprehensive proteomic landscape.
To meet the rigorous standards of modern immunometabolism research
and avoid overinterpreting experimental findings, it is essential
to critically evaluate the specific operational principles,
applicable scopes, advantages and inherent biological biases of
current probing technologies developed over the past decade.
Fluorescence and super-resolution
microscopy
The traditional optical diffraction limit of ~200
nm has historically hindered the precise characterization of MCSs,
as the physical cleft between mitochondria and partner organelles
typically ranges from 10 to 30 nm (196). Over the past decade, the advent
of advanced fluorescence and super-resolution microscopy has
bridged this gap, allowing researchers to visualize these
interfaces. Crucially, the accuracy of these optical techniques
relies on the selection of robust, validated biomolecular markers.
For tracking mitochondria-ER contacts, commonly utilized markers
include the IP3R-GRP75-VDAC1 complex, MFN2, and the VAPB-PTPIP51
tethering axis, often visualized alongside bulk organelle markers
such as TOMM20 for mitochondria and SEC61β or calreticulin for the
ER (197). When visualizing
mitochondria-lysosome crosstalk, researchers frequently co-stain
TOMM20 with late endosomal or lysosomal markers such as Rab7 or
LAMP1 (55), whereas
mitochondria-LD interfaces are effectively tracked using PLIN2/5
paired with mitochondrial tracking dyes (198). By leveraging these specific
markers, researchers can balance spatial resolution, temporal
dynamics and cell viability across a progressive toolkit of
microscopy techniques.
Standard live-cell confocal microscopy, and its
advanced iterations mathematically push the resolution limit down
to ~120-140 nm by utilizing concentric detector arrays to reassign
photon detection (199). This
technique is broadly utilized for live-cell tracking of bulk
inter-organelle network changes, such as macroscopic
mitochondria-lysosome kissing events during autophagy marked by
TOMM20 and LAMP1 overlap (55),
or broad ER-mitochondria co-localization in microglia (200). Its core advantage lies in its
high accessibility, deep tissue penetration and low phototoxicity,
allowing for continuous, multi-hour tracking of organelle networks
without inducing artificial metabolic stress. However, its primary
limitation is the inability to physically resolve the nanoscale MCS
gap. Due to the overlap of point-spread functions, two organelles
separated too close will appear merged under diffraction-limited
imaging, leading to an overestimation of actual contact surface
areas and an increase false-positive interpretations of tethering
(201). To achieve true
super-resolution while maintaining live-cell compatibility,
structured illumination microscopy (SIM) projects patterned grid
light onto the sample, mathematically reconstructing the resulting
moiré interference fringes to double the spatial resolution to ~100
nm in three dimensions (202).
Multispectral SIM has been heavily applied to map highly dynamic,
multi-organelle networks, including the spatiotemporal rewiring of
ER-mitochondria (203). SIM
represents the optimal compromise between spatial resolution and
temporal dynamics. Its low illumination intensity minimizes
photobleaching and phototoxicity, making it the preferred modality
for tracking rapid, real-time 'kiss-and-run' interface dynamics in
delicate, highly photosensitive primary cells such as T cells.
Despite this dynamic superiority, a 100-nm resolution is still
mathematically insufficient to visualize the actual 10-30 nm
physical cleft, confirming close apposition but fundamentally
failing to identify the ultrastructural architecture of the tethers
(202). Pushing spatial
resolution further, stimulated emission depletion (STED) nanoscopy
utilizes a conventional excitation laser paired with a donut-shaped
depletion laser, forcing peripheral fluorophores back to the ground
state via stimulated emission and driving resolution down to 30-50
nm (204). STED is specifically
utilized to unmask intricate sub-organellar architecture,
physically distinguishing the opposing membranes of multi-organelle
and mitochondria interaction by resolving specific markers or
identifying cristae topology (205). While STED provides true
nanoscale resolution capable of resolving the actual contact gap in
living or minimally fixed cells, the steep cost of this resolution
is phototoxicity (206). The
ultra-high-intensity depletion laser rapidly photobleaches
conventional dyes and induces massive oxidative stress. This
severely restricts the duration of time-lapse imaging and
necessitates the use of highly specialized, photostable synthetic
probes, strictly limiting its broad application in unperturbed
primary immune cells. Representing the pinnacle of optical spatial
resolution, single-molecule localization microscopy techniques such
as STORM and PALM rely on the stochastic photoswitching of
individual fluorophores, generating a composite image with
near-molecular resolution of 10-20 nm (207). STORM and related
single-molecule localization microscopy approaches are well suited
to resolving the nanoscale organization and spatial topology of
proteins at inter-organelle interfaces. STORM achieves the highest
spatial resolution among all optical technologies, effectively
matching the structural precision of electron microscopy while
simultaneously providing specific molecular identification of the
tethering components. However, the fundamental trade-off is the
absolute loss of temporal dynamics, the need to acquire thousands
of frames greatly prolongs acquisition time and increases
phototoxicity, therfore STORM is still most often applied to
chemically fixed cells and remains poorly suited to capturing rapid
organelle crosstalk dynamics (207). Ultimately, for future clinical
applications, such as evaluating the metabolic resilience of
engineered CAR-T cells, relying on a single optical modality is
insufficient. Researchers must employ a tiered approach, utilizing
SIM for live-cell tracking of multi-organelle network markers,
followed by STORM or STED to validate the precise molecular
assembly of tethers at the nanoscale. For future clinical
application, however, a significant limitation is that primary
immune cells are notoriously sensitive to phototoxicity (208), which can artificially induce
metabolic stress or mitophagy during prolonged observation.
Furthermore, these methods rely heavily on the exogenous expression
of fluorescent fusion proteins. The overexpression of these
organelle-targeted tags can inadvertently alter membrane lipid
curvature or artificially stabilize otherwise transient contacts,
leading to false-positive interpretations of interface frequency
and duration. Therefore, while super-resolution imaging excels in
mapping temporal dynamics, its objectivity is often compromised by
tag-induced structural artifacts and light-induced metabolic
stress.
Genetically encoded contact sensors
To translate visual co-localization observed in
traditional microscopy into quantifiable physical proximity, the
last decade has seen the rapid evolution of genetically encoded
contact sensors. These systems bridge the gap between static
imaging and functional biochemistry by converting the physical
narrowing of the inter-organelle cleft into a definitive,
measurable signal. The first major breakthrough in this domain was
the development of split-fluorescence contact sensors, most notably
the split-GFP-based contact site sensor (SPLICS). The core
principle of SPLICS relies on splitting a beta-barrel fluorescent
protein into two non-fluorescent fragments, which are targeted to
the outer membranes of opposing organelles; fluorescence is only
reconstituted when the organelles are within a strictly defined
physical gap. Originally engineered to quantify ER-mitochondria
contacts (209), the SPLICS
architecture has since been adapted into an extensive toolkit.
Researchers have subsequently extended split-fluorescence
approaches to visualize a broader repertoire of inter-organelle
contacts, including mitochondria-associated interfaces with
lysosomes and peroxisomes (210). The paramount advantage of
split-GFP sensors is their ability to provide highly specific,
tunable and high-throughput readouts of contact events without
requiring complex super-resolution equipment. However, the most
critical limitation of early split-protein systems is the
irreversible tethering artifact. Because the functional readout
relies on the high-affinity physical reconstitution of the protein
fragments, the sensor itself acts as an artificial molecular
staple, trapping the organelles together and masking the natural
disassembly of the contact site.
To overcome this irreversible tethering trap and
capture the true transient nature of mitochondrial crosstalk, the
field progressively transitioned toward reversible genetically
encoded sensors. These advanced tools utilize low-affinity
fragments that dynamically associate and dissociate in strict
accordance with physiological membrane distancing. For example,
reversible proximity sensors such as MERLIN have expanded the
field's ability to quantify dynamic mitochondria-ER interactions in
living cells without enforcing irreversible tethering (211). More recently, this reversible
paradigm has been extended to map mitochondria-LD contacts. By
utilizing a reversible, fluorogen-activated bimolecular
complementation toolkit (FABCON) based on splitFAST, researchers
can quantitatively track the dynamic association and dissociation
of mito-LD foci during rapid shifts in lipid biogenesis and
nutrient deprivation, thereby bypassing the irreversible tethering
trap of traditional GFP sensors (212). Despite these massive
technological leaps, critical gaps remain in the biosensor
repertoire, particularly concerning the mitochondria-nucleus axis
and vesicular transport pathways. Currently, the field severely
lacks dedicated and widely validated genetically encoded proximity
sensors for the mitochondria-nucleus interface. Furthermore, while
genetically encoded sensors excel at mapping stable membrane
appositions, they are fundamentally ill-equipped to monitor MDVs or
mitoEVs. Because MDVs are highly transient trafficking
intermediates that bud from the mitochondria to deliver specialized
cargo to lysosomes or peroxisomes, attempting to tag them with
standard membrane-anchored split-proteins fails to capture their
dynamic budding, target fusion and release. Consequently, precisely
quantifying MDV and mitoEV dynamics still relies almost entirely on
pH-sensitive fluorescent cargo trackers or biochemical
fractionation, highlighting a major technical limitation and a
critical opportunity for future bioengineering. Ultimately,
translating the profound insights gained from these cellular
sensors into clinical practice will require identifying
non-invasive, systemic biomarkers that accurately reflect the
interface dynamics first unmasked by these genetic tools.
Proximity-dependent biotinylation and
spatial proteomics
The translation of mitochondrial interface biology
relies on the precision of spatial proteomics to map the dynamic
inter organelle contactome in living cells. Traditional biochemical
fractionation protocols frequently fail to preserve weak or
transient tethers (213). This
prompted the development of proximity dependent biotinylation
methodologies. The core principle of these techniques involves
genetically fusing a promiscuous enzyme to a known interface
resident protein. Upon activation, the enzyme covalently tags
neighboring proteins within a defined nanometer radius of 10 to 20
nm with biotin, enabling subsequent affinity purification and mass
spectrometry identification. The earliest iteration, BioID,
utilized a mutant Escherichia coli biotin ligase, but its
slow kinetics limited the study of acute metabolic shifts. To
capture rapid interface dynamics, the field advanced toward APEX2,
an engineered ascorbate peroxidase capable of biotinylating targets
within 1 min upon the addition of hydrogen peroxide and biotin
phenol (214). TurboID, an
evolved biotin ligase, offers a reduced cytotoxicity associated
with APEX2-mediated oxidative stress, improving the temporal
resolution of spatial proteomics (215).
Researchers leverage these enzymes to decode
tethering architectures across the mitochondrial network. For
mitochondria-ER contacts, standard single enzyme targeting
occasionally resulted in high cytosolic background. To resolve
this, researchers developed split TurboID, a system where the
enzyme is divided into two inactive fragments respectively targeted
to the outer mitochondrial membrane and the ER. Enzymatic activity
is reconstituted upon the direct physical apposition of the
organelles, allowing for the enrichment of over 100 endogenous
interface proteins in mammalian cells (82). This targeted enzyme logic has
been expanded to mitochondria-LD interactions. Utilizing spatially
restricted proximity proteomics targeted explicitly to LD and
mitochondrial outer membranes in hepatic cell models revealed a
multimeric architecture. This approach identified extended
synaptotagmins and VAPB at LD-mitochondria tri-junctions that
facilitate FA transfer during lipotoxic stress (16). For mitochondria-peroxisome
proximity proteomics, a dual-purification APEX2-based system was
developed to isolate mitochondrial subpopulations adjacent to
peroxisomes in U2OS cells, enabling interface-enriched proteomic
mapping distinct from the bulk mitochondrial network (216). Spatial proteomics is
constrained by inherent biases, particularly concerning vesicular
transport such as MDVs or mitoEVs. While researchers use APEX2 or
TurboID targeted to the mitochondrial matrix or outer membrane to
catalog internal cargo packaged into EVs prior to release,
capturing the transient membrane budding interface of a vesicle
remains difficult. The primary limitation of proximity labeling
tools is the bystander effect. The 10 to 20 nm labeling radius is a
chemical gradient, not a defined boundary. Abundant cytosolic
proteins or unrelated organelles rapidly diffusing past the
targeted interface during the labeling window can be inadvertently
biotinylated and identified as contact residents. In the context of
dynamic vesicle budding, this bystander noise obscures the specific
vesiculation machinery. To translate these high throughput datasets
into clinical targets, researchers are advised to cross reference
spatial proteomes with orthogonal structural validation and
functional screens.
Ultrastructural imaging: EM and
Cryo-ET
Establishing the definitive physical architecture
and absolute membrane distances of inter organelle contact sites
necessitates the sub-nanometer resolution of ultrastructural
imaging. Conventional electron microscopy utilizes heavy metal
staining and electron scattering to map cellular architecture at
nanometer resolution. Researchers apply conventional TEM to
quantify the physical gap between mitochondria and other
organelles. For example, researchers utilized electron microscopy
alongside structured illumination to identify dynamic
mitochondria-lysosome contact sites, demonstrating that these
specific interfaces regulate mitochondrial fission events via
localized GTPase activity (55).
The primary advantage of conventional TEM is its high spatial
resolution capable of distinguishing individual lipid bilayers.
However, a notable limitation is its reliance on chemical fixation
and dehydration. These processing steps introduce osmotic stress
and membrane blebbing, potentially shrinking or expanding the
perceived contact gap and creating morphological biases (217). Furthermore, conventional
electron microscopy provides a static snapshot, lacking the
temporal resolution to capture dynamic tethering events.
To overcome the limitations of two-dimensional
cross sections, the field advanced toward three-dimensional volume
electron microscopy techniques such as focused ion beam scanning
electron microscopy. This technique sequentially mills the sample
surface with gallium ions and images the newly exposed face,
allowing computational reconstruction of large cellular volumes
(218). Focused ion beam
scanning electron microscopy is widely applied to map the complex
three-dimensional architecture of multi-organelle junctions.
Researchers utilize this technique to reconstruct holistic cellular
maps encompassing the ER, mitochondria, LDs, lysosomes,
peroxisomes, and the nucleus, quantifying how mitochondria
intricately associate with LDs to facilitate efficient FA transfer
during metabolic shifts (219).
The core advantage of focused ion beam scanning electron microscopy
is its ability to contextualize organelle interactions within broad
cellular volumes, revealing extensive contact networks rather than
isolated focal points. Despite this spatial advantage, the
technique remains destructive and relies on heavy metal staining,
which limits specific molecular identification of the tethering
proteins involved.
Representing the pinnacle of ultrastructural
preservation, cryo-electron tomography captures samples in
vitrified ice at cryogenic temperatures. This approach preserves
biological structures in near native states without chemical
fixatives, followed by tilt series imaging to reconstruct
high-resolution three-dimensional tomograms. Cryo-electron
tomography is increasingly applied to map the native
three-dimensional architecture of MCSs, including
mitochondria-associated interfaces, under near-native conditions
(220). Additionally,
researchers utilize electron tomography to observe the biogenesis
of mitochondrial-derived compartments, demonstrating that these
vesicular structures emerge directly from mitochondrial tubules at
specific ER contact sites to sequester distinct cargo (221). The defining advantage of
cryo-electron tomography is the ability to visualize molecular
tethers and membrane curvature in an unperturbed physiological
context. However, the technique faces notable limitations. The
imaging depth is severely restricted to thin cellular regions,
often requiring complex cryogenic focused ion beam milling prior to
imaging (222). This technical
complexity reduces throughput and limits sample sizes. Looking
toward future clinical applications, validating therapeutic
interventions targeting mitochondrial interfaces relies on an
integrated structural approach. Researchers are encouraged to
combine the high throughput spatial mapping of volume electron
microscopy with the native structural preservation of cryo-electron
tomography. This complementary strategy helps mitigate the
individual biases of each imaging modality, ensuring that newly
identified interface regulators or mitochondria-derived vesicular
pathways represent genuine physiological targets rather than sample
preparation artifacts.
Conclusions and perspectives
The mitochondria-centered organelle networking
functions as a unifying integrative model for immune regulation, in
which spatially organized interfaces and mitochondrial trafficking
routes enforce a critical level of governance of immunometabolism
and inflammatory signaling (223,224). It integrates contact-dependent
signaling with vesicle-mediated redistribution of mitochondrial
cargo to explain how mitochondria partner organelle coupling
coordinates Ca2+ flux, lipid exchange, redox control,
mitochondrial quality control, and epigenetic adaptation to shape
innate and adaptive immune states (225-227). It is proposed that interface
architecture, tether composition, organelle positioning and
mitochondrial vesicle flux collectively determine whether
activation driven stress is limited and resolved or propagated into
persistent immune dysfunction. By integrating shared and context
specific interface remodeling across infection and sepsis, cancer,
autoimmune and chronic inflammatory disorders, it links organelle
network failure to endolysosomal routing defects, nucleic acid
sensing, and maladaptive inflammatory program. Finally, therapeutic
relevance was dissected, emphasizing that progress will depend on
interface resolved profiling, rigorous validation in human immune
systems, and development of selective tools capable of perturbing
defined contact sites or mitochondrial trafficking checkpoints
(105,228).
Several limitations hamper mechanistic inference
and clinical translation. Organelle contacts are transient and
heterogeneous at the nanoscale yet are frequently assessed by
qualitative co-localization or indirect reporters that fail to
discriminate contact identity, lifetime, or functional effect
(229,230). Moreover, commonly used tagging
and imaging strategies can perturb native architectures and
introduce technical confounders. These issues are exacerbated in
complex tissues, where proximity labeling and spatial proteomics
remain challenged by background labeling, context-dependent
effective labeling distance, and limited in vivo
deployability, collectively restricting causal interpretation in
human immune pathophysiology (82,231). Clinical translation will
require stringent target prioritization and validation in human
relevant systems. Numerous interface components are pleiotropic and
embedded within essential calcium and lipid homeostasis, which can
narrow therapeutic windows and impose strong dependence on cell
type, disease stage and tissue context. In addition, biomarkers
that report interface dysfunction remain less developed than global
mitochondrial stress signatures.
An important limitation of current models of
mitochondrial interface regulation is that these structures are
often discussed as relatively static, although available evidence
indicates that their organization and function are modified by
physiological and pathological context. The clearest evidence
currently concerns ERMCS, also termed MAMs, which regulate
Ca2+ transfer, phospholipid metabolism, inflammasome
activation, antiviral responses and ER stress-related signaling in
diverse cell types, including innate immune cells (226). Sex-related regulation should be
interpreted cautiously. Available studies support that sex
hormones, particularly estrogen, modulate mitochondrial
bioenergetics, redox signaling and fusion-fission regulators
including MFN1, MFN2, OPA1 and DRP1. However, most of these data
come from cardiovascular and renal systems, and direct evidence for
sex-dependent remodeling of mitochondrial interfaces in immune
cells remains limited (232,233). Aging provides more direct
evidence that interface states are not fixed. ITPR2-dependent
ER-to-mitochondria Ca2+ transfer has been shown to
promote senescence, whereas Itpr2 deficiency reduces
ER-mitochondria contacts and is associated with attenuated aging
phenotypes in mice, supporting a role for contact-dependent
Ca2+ signaling in age-related dysfunction (234). Consistent with the view that
remodeling is context dependent, therapy-induced senescence has
been reported to increase MERCS' contact surface while reducing
ER-mitochondria Ca2+ flux because of decreased IP3R
expression and reduced IP3R1-VDAC1 interaction (235). A recent review further conclude
that MAMs undergo morphological and functional alterations during
aging and senescence (236).
The tissue and TME represents another important regulatory layer.
Current evidence indicates that hypoxia, oxidative stress, nutrient
limitation and metabolite accumulation reprogram mitochondrial
metabolism in infiltrating immune cells within the TME, whereas
direct quantitative evidence for remodeling of specific
mitochondrial contact sites in these cells remains limited
(237). In parallel, aging and
diet further reshape the metabolic architecture of the tumor-immune
microenvironment, thereby changing the conditions under which
mitochondrial signaling operates in immune cells (238). Taken together, the available
literature supports physiological plasticity of mitochondrial
interfaces, but in immune cells this conclusion is currently best
substantiated for ER-mitochondria contacts, while direct evidence
for other interface classes remains insufficient. Progress is
therefore likely to depend on integrated validation strategy that
combine immune competent organoids and patient derived immune cells
with minimally perturbative contact reporters, quantitative sensors
for local calcium, lipid flux and redox dynamics, and orthogonal
spatial proteomics to connect nanoscale organization with
stratification features and therapeutic response (239,240). Future work should enable a
transition from correlative evidence to actionable intervention by
expanding chemical and delivery platform that enrich pharmacology
at defined contact sites or mitochondria derived trafficking
checkpoints, and by integrated these capabilities into adoptive
cell manufacturing strategies designed to preserve interface states
required for persistence and function under chronic stimulation.
Limited druggability across several mitochondria organelle
interfaces should be interpreted as a tool gap rather than evidence
limited physiological importance and supports systematic target
discovery and validation across additional contact systems and
trafficking routes that remain underexplored in immunological
conditions.
Availability of data and materials
Not applicable.
Authors' contributions
YW, HY and LX conceived the overall structure of
the manuscript and drafted the article. LY provided technical and
material support and contributed to manuscript preparation. YZ and
QY provided critical revisions and editorial comments. All authors
read and approved the final version of the manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Medical Research Project
of Chengdu Municipal Health Commission (grant no. 2022416).
References
|
1
|
Bacigalupa ZA, Landis MD and Rathmell JC:
Nutrient inputs and social metabolic control of T cell fate. Cell
Metab. 36:10–20. 2024. View Article : Google Scholar
|
|
2
|
Kao KC, Vilbois S, Tsai CH and Ho PC:
Metabolic communication in the tumour-immune microenvironment. Nat
Cell Biol. 24:1574–1583. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
O'Neill LA and Pearce EJ: Immunometabolism
governs dendritic cell and macrophage function. J Exp Med.
213:15–23. 2016. View Article : Google Scholar :
|
|
4
|
Marchi S, Guilbaud E, Tait SWG, Yamazaki T
and Galluzzi L: Mitochondrial control of inflammation. Nat Rev
Immunol. 23:159–173. 2023. View Article : Google Scholar
|
|
5
|
Murphy MP and O'Neill L: A break in
mitochondrial endosymbiosis as a basis for inflammatory diseases.
Nature. 626:271–279. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li H, Jin W, Liu J, Zhou Y, Shan X, Zhang
Y, Kou Y, Deng C, Jin C, Kuang J, et al: Optimizing mitochondria
function in immune cells: implications for cancer immunotherapy.
Trends Cancer. 11:1170–1184. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Calì T, Bayer EM, Eden ER, Hajnóczky G,
Kornmann B, Lackner L, Liou J, Reinisch K, Rhee HW, Rizzuto R, et
al: Key challenges and recommendations for defining organelle
membrane contact sites. Nat Rev Mol Cell Biol. 26:776–796. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lackner LL: The expanding and unexpected
functions of mitochondria contact sites. Trends Cell Biol.
29:580–590. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen B, Lyssiotis CA and Shah YM:
Mitochondria-organelle crosstalk in establishing compartmentalized
metabolic homeostasis. Mol Cell. 85:1487–1508. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Larrañaga-SanMiguel A, Bengoa-Vergniory N
and Flores-Romero H: Crosstalk between mitochondria-ER contact
sites and the apoptotic machinery as a novel health meter. Trends
Cell Biol. 35:33–45. 2025. View Article : Google Scholar
|
|
11
|
Chu Q, Wang J, Du Y, Zhou T, Shi A, Xiong
J, Ji WK and Deng L: Oligomeric CHMP7 mediates three-way ER
junctions and ER-mitochondria interactions. Cell Death Differ.
30:94–110. 2023. View Article : Google Scholar
|
|
12
|
Cisneros J, Belton TB, Shum GC, Molakal CG
and Wong YC: Mitochondria-lysosome contact site dynamics and
misregulation in neurodegenerative diseases. Trends Neurosci.
45:312–322. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Giamogante F, Barazzuol L, Maiorca F,
Poggio E, Esposito A, Masato A, Napolitano G, Vagnoni A, Calì T and
Brini M: A SPLICS reporter reveals [Formula: see text]-synuclein
regulation of lysosome-mitochondria contacts which affects TFEB
nuclear translocation. Nat Commun. 15:15162024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sargsyan Y and Thoms S: Staying in healthy
contact: How peroxisomes interact with other cell organelles.
Trends Mol Med. 26:201–214. 2020. View Article : Google Scholar
|
|
15
|
Dolman NJ, Gerasimenko JV, Gerasimenko OV,
Voronina SG, Petersen OH and Tepikin AV: Stable Golgi-mitochondria
complexes and formation of Golgi Ca(2+) gradients in pancreatic
acinar cells. J Biol Chem. 280:15794–15799. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bezawork-Geleta A, Devereux CJ, Keenan SN,
Lou J, Cho E, Nie S, De Souza DP, Narayana VK, Siddall NA,
Rodrigues CHM, et al: Proximity proteomics reveals a mechanism of
fatty acid transfer at lipid droplet-mitochondria-endoplasmic
reticulum contact sites. Nat Commun. 16:21352025. View Article : Google Scholar
|
|
17
|
Hu L, Tang D, Qi B, Guo D, Wang Y, Geng J,
Zhang X, Song L, Chang P, Chen W, et al: Mfn2/Hsc70 complex
mediates the formation of mitochondria-lipid droplets membrane
contact and regulates myocardial lipid metabolism. Adv Sci (Weinh).
11:e23077492024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zervopoulos SD, Boukouris AE, Saleme B,
Haromy A, Tejay S, Sutendra G and Michelakis ED: MFN2-driven
mitochondria-to-nucleus tethering allows a non-canonical nuclear
entry pathway of the mitochondrial pyruvate dehydrogenase complex.
Mol Cell. 82:1066–1077.e7. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Desai R, East DA, Hardy L, Faccenda D,
Rigon M, Crosby J, Alvarez MS, Singh A, Mainenti M, Hussey LK, et
al: Mitochondria form contact sites with the nucleus to couple
prosurvival retrograde response. Sci Adv. 6:eabc99552020.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Montes de Oca Balderas P:
Mitochondria-plasma membrane interactions and communication. J Biol
Chem. 297:1011642021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Won J, Choi Y, Yun Y and Lee HH:
Biochemical characterization of the Num1-Mdm36 complex at the
mitochondria-plasma membrane contact site. Mol Cells. 44:207–213.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Soubannier V, McLelland GL, Zunino R,
Braschi E, Rippstein P, Fon EA and McBride HM: A vesicular
transport pathway shuttles cargo from mitochondria to lysosomes.
Curr Biol. 22:135–141. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
McLelland GL, Lee SA, McBride HM and Fon
EA: Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial
vesicles to the endolysosomal system. J Cell Biol. 214:275–291.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Abuaita BH, Schultz TL and O'Riordan MX:
Mitochondria-derived vesicles deliver antimicrobial reactive oxygen
species to control phagosome-localized staphylococcus aureus. Cell
Host Microbe. 24:625–636.e5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Neuspiel M, Schauss AC, Braschi E, Zunino
R, Rippstein P, Rachubinski RA, Andrade-Navarro MA and McBride HM:
Cargo-selected transport from the mitochondria to peroxisomes is
mediated by vesicular carriers. Curr Biol. 18:102–108. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Braschi E, Goyon V, Zunino R, Mohanty A,
Xu L and McBride HM: Vps35 mediates vesicle transport between the
mitochondria and peroxisomes. Curr Biol. 20:1310–1315. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liang W, Sagar S, Ravindran R, Najor RH,
Quiles JM, Chi L, Diao RY, Woodall BP, Leon LJ, Zumaya E, et al:
Mitochondria are secreted in extracellular vesicles when lysosomal
function is impaired. Nat Commun. 14:50312023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gong Y, Zhou Y, Feng L and Zhao Y: The
roles of secretory autophagy in mitochondria release via
extracellular vesicles: Waste disposal and food delivery? Front
Cell Dev Biol. 12:13928102024. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Iorio R, Petricca S, Di Emidio G, Falone S
and Tatone C: Mitochondrial Extracellular Vesicles (mitoEVs):
Emerging mediators of cell-to-cell communication in health, aging
and age-related diseases. Ageing Res Rev. 101:1025222024.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Crewe C, Funcke JB, Li S, Joffin N,
Gliniak CM, Ghaben AL, An YA, Sadek HA, Gordillo R, Akgul Y, et al:
Extracellular vesicle-based interorgan transport of mitochondria
from energetically stressed adipocytes. Cell Metab.
33:1853–1868.e11. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kou L, Wang Y, Li J, Zou W, Jin Z, Yin S,
Chi X, Sun Y, Wu J, Wang T and Xia Y:
Mitochondria-lysosome-extracellular vesicles axis and
nanotheranostics in neurodegenerative diseases. Exp Neurol.
376:1147572024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
He Q, Huang T, Chen Z, Sha Z and Wu H:
Mitochondria-lysosome crosstalk in microbial infections. Sci China
Life Sci. 68:3587–3599. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pereira AC, De Pascale J, Resende R,
Cardoso S, Ferreira I, Neves BM, Carrascal MA, Zuzarte M, Madeira
N, Morais S, et al: ER-mitochondria communication is involved in
NLRP3 inflammasome activation under stress conditions in the innate
immune system. Cell Mol Life Sci. 79:2132022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Horner SM, Liu HM, Park HS, Briley J and
Gale M Jr: Mitochondrial-associated endoplasmic reticulum membranes
(MAM) form innate immune synapses and are targeted by hepatitis C
virus. Proc Natl Acad Sci USA. 108:14590–14595. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Voeltz GK, Sawyer EM, Hajnóczky G and
Prinz WA: Making the connection: How membrane contact sites have
changed our view of organelle biology. Cell. 187:257–270. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
König T and McBride HM:
Mitochondrial-derived vesicles in metabolism, disease, and aging.
Cell Metab. 36:21–35. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Elwakiel A, Mathew A and Isermann B: The
role of endoplasmic reticulum-mitochondria-associated membranes in
diabetic kidney disease. Cardiovasc Res. 119:2875–2883. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gordaliza-Alaguero I,
Sànchez-Fernàndez-de-Landa P, Radivojevikj D, Villarreal L,
Arauz-Garofalo G, Gay M, Martinez-Vicente M, Seco J,
Martín-Malpartida P, Vilaseca M, et al: Endogenous interactomes of
MFN1 and MFN2 provide novel insights into interorganelle
communication and autophagy. Autophagy. 21:957–978. 2025.
View Article : Google Scholar :
|
|
39
|
Galmes R, Houcine A, van Vliet AR,
Agostinis P, Jackson CL and Giordano F: ORP5/ORP8 localize to
endoplasmic reticulum-mitochondria contacts and are involved in
mitochondrial function. EMBO Rep. 17:800–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Monteiro-Cardoso VF, Rochin L, Arora A,
Houcine A, Jääskeläinen E, Kivelä AM, Sauvanet C, Le Bars R, Marien
E, Dehairs J, et al: ORP5/8 and MIB/MICOS link ER-mitochondria and
intra-mitochondrial contacts for non-vesicular transport of
phosphatidylserine. Cell Rep. 40:1113642022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rahhali K, Di Lisa F and Kaludercic N:
IP(3) receptor trafficking at the ER-mitochondria contacts impacts
on mitochondrial Ca(2+) homeostasis and metabolism. Cell Calcium.
110:1027002023. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Szabadkai G, Bianchi K, Várnai P, De
Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T and Rizzuto
R: Chaperone-mediated coupling of endoplasmic reticulum and
mitochondrial Ca2+ channels. J Cell Biol. 175:901–911. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hirabayashi Y, Kwon SK, Paek H, Pernice
WM, Paul MA, Lee J, Erfani P, Raczkowski A, Petrey DS, Pon LA and
Polleux F: ER-mitochondria tethering by PDZD8 regulates Ca(2+)
dynamics in mammalian neurons. Science. 358:623–630. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fan M, Zhang J, Tsai CW, Orlando BJ,
Rodriguez M, Xu Y, Liao M, Tsai MF and Feng L: Structure and
mechanism of the mitochondrial Ca(2+) uniporter holocomplex.
Nature. 582:129–133. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu Y, Ma X, Fujioka H, Liu J, Chen S and
Zhu X: DJ-1 regulates the integrity and function of ER-mitochondria
association through interaction with IP3R3-Grp75-VDAC1. Proc Natl
Acad Sci USA. 116:25322–25328. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hayashi T and Su TP: Sigma-1 receptor
chaperones at the ER-mitochondrion interface regulate Ca(2+)
signaling and cell survival. Cell. 131:596–610. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kumar V and Maity S: ER stress-sensor
proteins and ER-Mitochondrial crosstalk-signaling beyond (ER)
stress response. Biomolecules. 11:1732021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhao F, Cui Z, Wang P, Zhao Z, Zhu K, Bai
Y, Jin X, Wang L and Lu L: GRP75-dependent mitochondria-ER contacts
ensure cell survival during early mouse thymocyte development. Dev
Cell. 59:2643–2658.e7. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bantug GR, Fischer M, Grählert J, Balmer
ML, Unterstab G, Develioglu L, Steiner R, Zhang L, Costa ASH,
Gubser PM, et al: Mitochondria-endoplasmic reticulum contact sites
function as immunometabolic hubs that orchestrate the rapid recall
response of memory CD8(+) T cells. Immunity. 48:542–555.e6. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yang JF, Xing X, Luo L, Zhou XW, Feng JX,
Huang KB, Liu H, Jin S, Liu YN, Zhang SH, et al: Mitochondria-ER
contact mediated by MFN2-SERCA2 interaction supports CD8(+) T cell
metabolic fitness and function in tumors. Sci Immunol.
8:eabq24242023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Assis LHP, Dorighello GG and Oliveira HCF:
Pro-inflammatory polarization of macrophages is associated with
reduced endoplasmic reticulum-mitochondria interaction. Biochem
Biophys Res Commun. 606:61–67. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sharma J, Khan S, Singh NC, Sahu S, Raj D,
Prakash S, Bandyopadhyay P, Sarkar K, Bhosale V, Chandra T, et al:
ORMDL3 regulates NLRP3 inflammasome activation by maintaining
ER-mitochondria contacts in human macrophages and dictates
ulcerative colitis patient outcome. J Biol Chem. 300:1071202024.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zeisbrich M, Yanes RE, Zhang H, Watanabe
R, Li Y, Brosig L, Hong J, Wallis BB, Giacomini JC, Assimes TL, et
al: Hypermetabolic macrophages in rheumatoid arthritis and coronary
artery disease due to glycogen synthase kinase 3b inactivation. Ann
Rheum Dis. 77:1053–1062. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wong YC, Kim S, Peng W and Krainc D:
Regulation and function of mitochondria-lysosome membrane contact
sites in cellular homeostasis. Trends Cell Biol. 29:500–513. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wong YC, Ysselstein D and Krainc D:
Mitochondria-lysosome contacts regulate mitochondrial fission via
RAB7 GTP hydrolysis. Nature. 554:382–386. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Leisten ED, Woods AC and Wong YC:
Super-resolution microscopy: Insights into mitochondria-lysosome
crosstalk in health and disease. J Cell Biol. 222:e2023050322023.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Boutry M and Kim PK: ORP1L mediated PI(4)P
signaling at ER-lysosome-mitochondrion three-way contact
contributes to mitochondrial division. Nat Commun. 12:53542021.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
McLelland GL, Soubannier V, Chen CX,
McBride HM and Fon EA: Parkin and PINK1 function in a vesicular
trafficking pathway regulating mitochondrial quality control. EMBO
J. 33:282–295. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Qiu K, Zou W, Fang H, Hao M, Mehta K, Tian
Z, Guan JL, Zhang K, Huang T and Diao J: Light-activated
mitochondrial fission through optogenetic control of
mitochondria-lysosome contacts. Nat Commun. 13:43032022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhang YY, Lv ZD, Wang M, Wang JX, Limbu
SM, Qiao F, Chen LQ, Zhang ML, Luo Y and Du ZY: Mitochondrial fatty
acid oxidation dysfunction impairs autophagic flux through ATP
deficiency-caused lysosomal pH abnormity. J Nutr Biochem.
143:1099432025. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wong YC, Kim S, Cisneros J, Molakal CG,
Song P, Lubbe SJ and Krainc D: Mid51/Fis1 mitochondrial
oligomerization complex drives lysosomal untethering and network
dynamics. J Cell Biol. 2022.221:e2022061402022. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Malik N, Ferreira BI, Hollstein PE, Curtis
SD, Trefts E, Weiser Novak S, Yu J, Gilson R, Hellberg K, Fang L,
et al: Induction of lysosomal and mitochondrial biogenesis by AMPK
phosphorylation of FNIP1. Science. 380:eabj55592023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhang X, Cheng X, Yu L, Yang J, Calvo R,
Patnaik S, Hu X, Gao Q, Yang M, Lawas M, et al: MCOLN1 is a ROS
sensor in lysosomes that regulates autophagy. Nat Commun.
7:121092016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Norton EG, Chapman NM and Chi H:
Mitochondria and lysosomes in T cell immunometabolism. Trends
Immunol. 46:635–651. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Baixauli F, Acín-Pérez R,
Villarroya-Beltrí C, Mazzeo C, Nuñez-Andrade N, Gabandé-Rodriguez
E, Ledesma MD, Blázquez A, Martin MA, Falcón-Pérez JM, et al:
Mitochondrial respiration controls lysosomal function during
inflammatory T cell responses. Cell Metab. 22:485–498. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Saravia J, Chapman NM, Sun Y, Meng X,
Risch I, Li W, Guy C, Shi H, Hu H, Dhungana Y, et al: Mitochondrial
and lysosomal signaling orchestrates heterogeneous metabolic states
of regulatory T cells. Sci Immunol. 10:eads94562025. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Liu H, Zhen C, Xie J, Luo Z, Zeng L, Zhao
G, Lu S, Zhuang H, Fan H, Li X, et al: TFAM is an autophagy
receptor that limits inflammation by binding to cytoplasmic
mitochondrial DNA. Nat Cell Biol. 26:878–891. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Schuster EM, Epple MW, Glaser KM, Mihlan
M, Lucht K, Zimmermann JA, Bremser A, Polyzou A, Obier N,
Cabezas-Wallscheid N, et al: TFEB induces mitochondrial itaconate
synthesis to suppress bacterial growth in macrophages. Nat Metab.
4:856–866. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Sugiura A, Mattie S, Prudent J and McBride
HM: Newly born peroxisomes are a hybrid of mitochondrial and
ER-derived pre-peroxisomes. Nature. 542:251–254. 2027. View Article : Google Scholar
|
|
70
|
Zheng J, Chen J, Cao Z, Wu K, Wang J, Guo
Y and Zhuang M: Ubiquitin ligase MARCH5 controls the formation of
mitochondria-derived pre-peroxisomes. Dev Cell. 60:30–39.e3. 2025.
View Article : Google Scholar
|
|
71
|
Verhoeven N, Oshima Y, Cartier E, Bippes
CC, Neutzner A, Boyman L and Karbowski M: Outer mitochondrial
membrane E3 Ub ligase MARCH5 controls de novo peroxisome
biogenesis. Dev Cell. 60:40–50.e5. 2025. View Article : Google Scholar
|
|
72
|
DiGiovanni LF, Khroud PK, Carmichael RE,
Schrader TA, Gill SK, Germain K, Jomphe RY, Wiesinger C, Boutry M,
Kamoshita M, et al: ROS transfer at peroxisome-mitochondria contact
regulates mitochondrial redox. Science. 389:157–162. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Fan J, Li X, Issop L, Culty M and
Papadopoulos V: ACBD2/ECI2-mediated peroxisome-mitochondria
interactions in leydig cell steroid biosynthesis. Mol Endocrinol.
30:763–782. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Alsayyah C, Singh MK, Morcillo-Parra MA,
Cavellini L, Shai N, Schmitt C, Schuldiner M, Zalckvar E, Mallet A,
Belgareh-Touzé N, et al: Mitofusin-mediated contacts between
mitochondria and peroxisomes regulate mitochondrial fusion. PLoS
Biol. 22:e30026022024. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Huo Y, Sun W, Shi T, Gao S and Zhuang M:
The MFN1 and MFN2 mitofusins promote clustering between
mitochondria and peroxisomes. Commun Biol. 5:4232022. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Okumoto K, Ono T, Toyama R, Shimomura A,
Nagata A and Fujiki Y: New splicing variants of mitochondrial Rho
GTPase-1 (Miro1) transport peroxisomes. J Cell Biol. 217:619–633.
2018. View Article : Google Scholar :
|
|
77
|
Koch A, Thiemann M, Grabenbauer M, Yoon Y,
McNiven MA and Schrader M: Dynamin-like protein 1 is involved in
peroxisomal fission. J Biol Chem. 278:8597–8605. 2003. View Article : Google Scholar
|
|
78
|
Koch A, Yoon Y, Bonekamp NA, McNiven MA
and Schrader M: A role for Fis1 in both mitochondrial and
peroxisomal fission in mammalian cells. Mol Biol Cell.
16:5077–5086. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Otera H, Wang C, Cleland MM, Setoguchi K,
Yokota S, Youle RJ and Mihara K: Mff is an essential factor for
mitochondrial recruitment of Drp1 during mitochondrial fission in
mammalian cells. J Cell Biol. 191:1141–1158. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Schrader TA, Carmichael RE, Islinger M,
Costello JL, Hacker C, Bonekamp NA, Weishaupt JH, Andersen PM and
Schrader M: PEX11β and FIS1 cooperate in peroxisome division
independently of mitochondrial fission factor. J Cell Sci.
135:jcs2599242022. View Article : Google Scholar
|
|
81
|
Court DA, Khetoo S, Shuvo SR, Reitmeier SD
and Hausner G: In silico analysis of coevolution among ERMES
proteins, Pex11, and Lam6. Can J Microbiol. 63:984–997. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Cho KF, Branon TC, Rajeev S, Svinkina T,
Udeshi ND, Thoudam T, Kwak C, Rhee HW, Lee IK, Carr SA and Ting AY:
Split-TurboID enables contact-dependent proximity labeling in
cells. Proc Natl Acad Sci USA. 117:12143–12154. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Houten SM, Wanders RJA and Ranea-Robles P:
Metabolic interactions between peroxisomes and mitochondria with a
special focus on acylcarnitine metabolism. Biochim Biophys Acta Mol
Basis Dis. 1866:1657202020. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Tanaka H, Okazaki T, Aoyama S, Yokota M,
Koike M, Okada Y, Fujiki Y and Gotoh Y: Peroxisomes control
mitochondrial dynamics and the mitochondrion-dependent apoptosis
pathway. J Cell Sci. 132:jcs2247662019. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Fransen M, Lismont C and Walton P: The
peroxisome-mitochondria connection: How and why? Int J Mol Sci.
18:11262017. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Dixit E, Boulant S, Zhang Y, Lee AS,
Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, et
al: Peroxisomes are signaling platforms for antiviral innate
immunity. Cell. 141:668–681. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Bender S, Reuter A, Eberle F, Einhorn E,
Binder M and Bartenschlager R: Activation of type I and III
interferon response by mitochondrial and peroxisomal MAVS and
inhibition by hepatitis C Virus. PLoS Pathog. 11:e10052642015.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Wei X, Qian W, Narasimhan H, Chan T, Liu
X, Arish M, Young S, Li C, Cheon IS, Yu Q, et al: Macrophage
peroxisomes guide alveolar regeneration and limit SARS-CoV-2 tissue
sequelae. Science. 387:eadq25092025. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Olzmann JA and Carvalho P: Dynamics and
functions of lipid droplets. Nat Rev Mol Cell Biol. 20:137–155.
2019. View Article : Google Scholar
|
|
90
|
Cui L and Liu P: Two types of contact
between lipid droplets and mitochondria. Front Cell Dev Biol.
8:6183222020. View Article : Google Scholar :
|
|
91
|
Cui L, Mirza AH, Zhang S, Liang B and Liu
P: Lipid droplets and mitochondria are anchored during brown
adipocyte differentiation. Protein Cell. 10:921–926. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Benador IY, Veliova M, Mahdaviani K,
Petcherski A, Wikstrom JD, Assali EA, Acín-Pérez R, Shum M,
Oliveira MF, Cinti S, et al: Mitochondria bound to lipid droplets
have unique bioenergetics, composition, and dynamics that support
lipid droplet expansion. Cell Metab. 27:869–885.e6. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Miner GE, So CM, Edwards W, Ragusa JV,
Wine JT, Wong Gutierrez D, Airola MV, Herring LE, Coleman RA, Klett
EL and Cohen S: PLIN5 interacts with FATP4 at membrane contact
sites to promote lipid droplet-to-mitochondria fatty acid
transport. Dev Cell. 58:1250–1265.e6. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Ouyang Q, Chen Q, Ke S, Ding L, Yang X,
Rong P, Feng W, Cao Y, Wang Q, Li M, et al: Rab8a as a
mitochondrial receptor for lipid droplets in skeletal muscle. Dev
Cell. 58:289–305.e6. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Freyre C, Rauher PC, Ejsing CS and Klemm
RW: MIGA2 links mitochondria, the ER, and lipid droplets and
promotes de novo lipogenesis in adipocytes. Mol Cell.
76:811–825.e14. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Wang J, Fang N, Xiong J, Du Y, Cao Y and
Ji WK: An ESCRT-dependent step in fatty acid transfer from lipid
droplets to mitochondria through VPS13D-TSG101 interactions. Nat
Commun. 12:12522021. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Nguyen TB, Louie SM, Daniele JR, Tran Q,
Dillin A, Zoncu R, Nomura DK and Olzmann JA: DGAT1-Dependent lipid
droplet biogenesis protects mitochondrial function during
starvation-induced autophagy. Dev Cell. 42:9–21.e5. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Li J, Zhang Y, Peng J, Yang W, Pan Y, Xu
Y, Yao J, Lin S, Li Y, Gu X, et al: A regulator of amino acid
sensing links lipid peroxidation and lipid droplet-dependent
antioxidant response. Mol Cell. 85:3225–3240.e10. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Mathiowetz AJ and Olzmann JA: Lipid
droplets and cellular lipid flux. Nat Cell Biol. 26:331–345. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Kien B, Kolleritsch S, Kunowska N, Heier
C, Chalhoub G, Tilp A, Wolinski H, Stelzl U and Haemmerle G: Lipid
droplet-mitochondria coupling via perilipin 5 augments respiratory
capacity but is dispensable for FA oxidation. J Lipid Res.
63:1001722022. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Wang H, Sreenivasan U, Hu H, Saladino A,
Polster BM, Lund LM, Gong DW, Stanley WC and Sztalryd C: Perilipin
5, a lipid droplet-associated protein, provides physical and
metabolic linkage to mitochondria. J Lipid Res. 52:2159–2168. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Melo RC and Weller PF: Lipid droplets in
leukocytes: Organelles linked to inflammatory responses. Exp Cell
Res. 340:193–197. 2016. View Article : Google Scholar
|
|
103
|
Bosch M, Sánchez-Álvarez M, Fajardo A,
Kapetanovic R, Steiner B, Dutra F, Moreira L, López JA, Campo R,
Marí M, et al: Mammalian lipid droplets are innate immune hubs
integrating cell metabolism and host defense. Science.
370:eaay80852020. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Seo JY and Cresswell P: Viperin regulates
cellular lipid metabolism during human cytomegalovirus infection.
PLoS Pathog. 9:e10034972013. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Campanella M and Kannan B: Mitochondrial
sites of contact with the nucleus. J Cell Biol. 223:e2023050102024.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Jäger S, Handschin C, St-Pierre J and
Spiegelman BM: AMP-activated protein kinase (AMPK) action in
skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl
Acad Sci USA. 104:12017–12022. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Cunningham JT, Rodgers JT, Arlow DH,
Vazquez F, Mootha VK and Puigserver P: mTOR controls mitochondrial
oxidative function through a YY1-PGC-1alpha transcriptional
complex. Nature. 450:736–740. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Russo M, Gualdrini F, Vallelonga V,
Prosperini E, Noberini R, Pedretti S, Borriero C, Di Chiaro P,
Polletti S, Imperato G, et al: Acetyl-CoA production by
Mediator-bound 2-ketoacid dehydrogenases boosts de novo histone
acetylation and is regulated by nitric oxide. Mol Cell.
84:967–980.e10. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Pouikli A, Maleszewska M, Parekh S, Yang
M, Nikopoulou C, Bonfiglio JJ, Mylonas C, Sandoval T, Schumacher
AL, Hinze Y, et al: Hypoxia promotes osteogenesis by facilitating
acetyl-CoA-mediated mitochondrial-nuclear communication. EMBO J.
41:e1112392022. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Reid MA, Dai Z and Locasale JW: The impact
of cellular metabolism on chromatin dynamics and epigenetics. Nat
Cell Biol. 19:1298–1306. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H,
Liu L, Liu Y, Yang C, Xu Y, et al: Inhibition of α-KG-dependent
histone and DNA demethylases by fumarate and succinate that are
accumulated in mutations of FH and SDH tumor suppressors. Genes
Dev. 26:1326–1338. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Baksh SC and Finley LWS: Metabolic
coordination of cell fate by α-Ketoglutarate-dependent
dioxygenases. Trends Cell Biol. 31:24–36. 2021. View Article : Google Scholar
|
|
113
|
Kaspar S, Oertlin C, Szczepanowska K,
Kukat A, Senft K, Lucas C, Brodesser S, Hatzoglou M, Larsson O,
Topisirovic I and Trifunovic A: Adaptation to mitochondrial stress
requires CHOP-directed tuning of ISR. Sci Adv. 7:eabf09712021.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Ron-Harel N, Santos D, Ghergurovich JM,
Sage PT, Reddy A, Lovitch SB, Dephoure N, Satterstrom FK, Sheffer
M, Spinelli JB, et al: Mitochondrial biogenesis and proteome
remodeling promote one-carbon metabolism for T cell activation.
Cell Metab. 24:104–117. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Sugiura A, Andrejeva G, Voss K, Heintzman
DR, Xu X, Madden MZ, Ye X, Beier KL, Chowdhury NU, Wolf MM, et al:
MTHFD2 is a metabolic checkpoint controlling effector and
regulatory T cell fate and function. Immunity. 55:65–81.e9. 2022.
View Article : Google Scholar
|
|
116
|
Mocholi E, Russo L, Gopal K, Ramstead AG,
Hochrein SM, Vos HR, Geeven G, Adegoke AO, Hoekstra A, van Es RM,
et al: Pyruvate metabolism controls chromatin remodeling during
CD4(+) T cell activation. Cell Rep. 42:1125832023. View Article : Google Scholar
|
|
117
|
Matias MI, Yong CS, Foroushani A,
Goldsmith C, Mongellaz C, Sezgin E, Levental KR, Talebi A, Perrault
J, Rivière A, et al: Regulatory T cell differentiation is
controlled by αKG-induced alterations in mitochondrial metabolism
and lipid homeostasis. Cell Rep. 37:1099112021. View Article : Google Scholar
|
|
118
|
Minogue E, Cunha PP, Wadsworth BJ, Grice
GL, Sah-Teli SK, Hughes R, Bargiela D, Quaranta A, Zurita J,
Antrobus R, et al: Glutarate regulates T cell metabolism and
anti-tumour immunity. Nat Metab. 5:1747–1764. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Chen LL, Morcelle C, Cheng ZL, Chen X, Xu
Y, Gao Y, Song J, Li Z, Smith MD, Shi M, et al: Itaconate inhibits
TET DNA dioxygenases to dampen inflammatory responses. Nat Cell
Biol. 24:353–363. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Arts RJ, Novakovic B, Ter Horst R,
Carvalho A, Bekkering S, Lachmandas E, Rodrigues F, Silvestre R,
Cheng SC, Wang SY, et al: Glutaminolysis and fumarate accumulation
integrate immunometabolic and epigenetic programs in trained
immunity. Cell Metab. 24:807–819. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Bekkering S, Novakovic B, Ter Horst R,
Carvalho A, Bekkering S, Lachmandas E, Rodrigues F, Silvestre R,
Cheng SC, Wang SY, et al: Metabolic induction of trained immunity
through the mevalonate pathway. Cell. 172:135–146.e9. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
West AP, Khoury-Hanold W, Staron M, Tal
MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff
DA, et al: Mitochondrial DNA stress primes the antiviral innate
immune response. Nature. 520:553–557. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Hu MM and Shu HB: Mitochondrial
DNA-triggered innate immune response: Mechanisms and diseases. Cell
Mol Immunol. 20:1403–1412. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Zhou X, Liu S, Lu Y, Wan M, Cheng J and
Liu J: MitoEVs: A new player in multiple disease pathology and
treatment. J Extracell Vesicles. 12:e123202023. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
König T, Nolte H, Aaltonen MJ, Tatsuta T,
Krols M, Stroh T, Langer T and McBride HM: MIROs and DRP1 drive
mitochondrial-derived vesicle biogenesis and promote quality
control. Nat Cell Biol. 23:1271–1286. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Todkar K, Chikhi L, Desjardins V,
El-Mortada F, Pépin G and Germain M: Selective packaging of
mitochondrial proteins into extracellular vesicles prevents the
release of mitochondrial DAMPs. Nat Commun. 12:19712021. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Towers CG, Wodetzki DK, Thorburn J, Smith
KR, Caino MC and Thorburn A: Mitochondrial-derived vesicles
compensate for loss of LC3-mediated mitophagy. Dev Cell.
56:2029–2042.e5. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Lou P, Zhou X, Zhang Y, Xie Y, Wang Y,
Wang C, Liu S, Wan M, Lu Y and Liu J: Harnessing tissue-derived
mitochondria-rich extracellular vesicles (Ti-mitoEVs) to boost
mitochondrial biogenesis for regenerative medicine. Sci Adv.
11:eadt13182025. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Matheoud D, Sugiura A, Bellemare-Pelletier
A, Laplante A, Rondeau C, Chemali M, Fazel A, Bergeron JJ, Trudeau
LE, Burelle Y, et al: Parkinson's disease-related proteins PINK1
and parkin repress mitochondrial antigen presentation. Cell.
166:314–327. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Recinto SJ, Kazanova A, Liu L, Cordeiro B,
Premachandran S, Bessaiah H, Allot A, Afanasiev E, Mukherjee S, Pei
J, et al: PINK1 deficiency rewires early immune responses in a
mouse model of Parkinson's disease triggered by intestinal
infection. NPJ Parkinsons Dis. 11:1332025. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Li X, Straub J, Medeiros TC, Mehra C, den
Brave F, Peker E, Atanassov I, Stillger K, Michaelis JB, Burbridge
E, et al: Mitochondria shed their outer membrane in response to
infection-induced stress. Science. 375:eabi43432022. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Zhao F, Zheng T, Gong W, Wu J, Xie H, Li
W, Zhang R, Liu P, Liu J, Wu X, et al: Extracellular vesicles
package dsDNA to aggravate Crohn's disease by activating the STING
pathway. Cell Death Dis. 12:8152021. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Konaka H, Kato Y, Hirano T, Tsujimoto K,
Park J, Koba T, Aoki W, Matsuzaki Y, Taki M, Koyama S, et al:
Secretion of mitochondrial DNA via exosomes promotes inflammation
in Behçet's syndrome. EMBO J. 42:e1125732023. View Article : Google Scholar
|
|
134
|
Nicolás-Ávila JA, Lechuga-Vieco AV,
Esteban-Martínez L, Sánchez-Díaz M, Díaz-García E, Santiago DJ,
Rubio-Ponce A, Li JL, Balachander A, Quintana JA, et al: A network
of macrophages supports mitochondrial homeostasis in the heart.
Cell. 183:94–109.e23. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Rosina M, Ceci V, Turchi R, Chuan L,
Borcherding N, Sciarretta F, Sánchez-Díaz M, Tortolici F, Karlinsey
K, Chiurchiù V, et al: Ejection of damaged mitochondria and their
removal by macrophages ensure efficient thermogenesis in brown
adipose tissue. Cell Metab. 34:533–548.e12. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Hu D, Sheeja Prabhakaran H, Zhang YY, Luo
G, He W and Liou YC: Mitochondrial dysfunction in sepsis:
mechanisms and therapeutic perspectives. Crit Care. 28:2922024.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Scharping NE, Menk AV, Moreci RS,
Whetstone RD, Dadey RE, Watkins SC, Ferris RL and Delgoffe GM: The
tumor microenvironment represses T cell mitochondrial biogenesis to
drive intratumoral T cell metabolic insufficiency and dysfunction.
Immunity. 45:374–388. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Ikeda H, Kawase K, Nishi T, Watanabe T,
Takenaga K, Inozume T, Ishino T, Aki S, Lin J, Kawashima S, et al:
Immune evasion through mitochondrial transfer in the tumour
microenvironment. Nature. 638:225–236. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Lood C, Blanco LP, Purmalek MM,
Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA,
Elkon KB and Kaplan MJ: Neutrophil extracellular traps enriched in
oxidized mitochondrial DNA are interferogenic and contribute to
lupus-like disease. Nat Med. 22:146–153. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Rodero MP, Tesser A, Bartok E, Rice GI,
Della Mina E, Depp M, Beitz B, Bondet V, Cagnard N, Duffy D, et al:
Type I interferon-mediated autoinflammation due to DNase II
deficiency. Nat Commun. 8:21762017. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Cook KC, Tsopurashvili E, Needham JM,
Thompson SR and Cristea IM: Restructured membrane contacts rewire
organelles for human cytomegalovirus infection. Nat Commun.
13:47202022. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Poggio E, Vallese F, Hartel AJW,
Morgenstern TJ, Kanner SA, Rauh O, Giamogante F, Barazzuol L,
Shepard KL, Colecraft HM, et al: Perturbation of the host cell
Ca(2+) homeostasis and ER-mitochondria contact sites by the
SARS-CoV-2 structural proteins E and M. Cell Death Dis. 14:2972023.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Li X, Hou P, Ma W, Wang X, Wang H, Yu Z,
Chang H, Wang T, Jin S, Wang X, et al: SARS-CoV-2 ORF10 suppresses
the antiviral innate immune response by degrading MAVS through
mitophagy. Cell Mol Immunol. 19:67–78. 2022. View Article : Google Scholar
|
|
144
|
Xian H, Watari K, Sanchez-Lopez E,
Offenberger J, Onyuru J, Sampath H, Ying W, Hoffman HM, Shadel GS
and Karin M: Oxidized DNA fragments exit mitochondria via mPTP- and
VDAC-dependent channels to activate NLRP3 inflammasome and
interferon signaling. Immunity. 55:1370–1385.e8. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Khantisitthiporn O, Shue B, Eyre NS, Nash
CW, Turnbull L, Whitchurch CB, Van der Hoek KH, Helbig KJ and Beard
MR: Viperin interacts with PEX19 to mediate peroxisomal
augmentation of the innate antiviral response. Life Sci Alliance.
4:e2020009152021. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Wei Z, Yan Y, Zhao L, Li C, Qi J, Chen D,
Huang X, Li M, Xiao Z, Lou G, et al: Mitochondrial respiratory
chain regulates HBV clearance through dual modulation of lysosomal
acidification. Emerg Microbes Infect. 14:25630792025. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Zecchini V, Paupe V, Herranz-Montoya I,
Janssen J, Wortel IMN, Morris JL, Ferguson A, Chowdury SR,
Segarra-Mondejar M, Costa ASH, et al: Fumarate induces vesicular
release of mtDNA to drive innate immunity. Nature. 615:499–506.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Liu Q, Wu J, Zhang X, Li X, Wu X, Zhao Y
and Ren J: Circulating mitochondrial DNA-triggered autophagy
dysfunction via STING underlies sepsis-related acute lung injury.
Cell Death Dis. 12:6732021. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Han H, Zhang Y, Huang E, Zhou S, Huang Z,
Qin K and Du X: The role of TBC1D15 in sepsis-induced acute lung
injury: Regulation of mitochondrial homeostasis and mitophagy. Int
J Biol Macromol. 293:1392892025. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Kuang L, Wu Y, Shu J, Yang J, Zhou H and
Huang X: Pyroptotic macrophage-derived microvesicles accelerate
formation of neutrophil extracellular traps via GSDMD-N-expressing
mitochondrial transfer during sepsis. Int J Biol Sci. 20:733–750.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Peralta RM, Xie B, Lontos K, Nieves-Rosado
H, Spahr K, Joshi S, Ford BR, Quann K, Frisch AT, Dean V, et al:
Dysfunction of exhausted T cells is enforced by MCT11-mediated
lactate metabolism. Nat Immunol. 25:2297–2307. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Sassano ML, Tyurina YY, Diokmetzidou A,
Vervoort E, Tyurin VA, More S, La Rovere R, Giordano F, Bultynck G,
Pavie B, et al: Endoplasmic reticulum-mitochondria contacts are
prime hotspots of phospholipid peroxidation driving ferroptosis.
Nat Cell Biol. 27:902–917. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Zhang H, Liu J, Yuan W, Zhang Q, Luo X, Li
Y, Peng Y, Feng J, Liu X, Chen J, et al: Ammonia-induced lysosomal
and mitochondrial damage causes cell death of effector CD8(+) T
cells. Nat Cell Biol. 26:1892–1902. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Lai P, Liu L, Bancaro N, Troiani M, Calì
B, Li Y, Chen J, Singh PK, Arzola RA, Attanasio G, et al:
Mitochondrial DNA released by senescent tumor cells enhances
PMN-MDSC-driven immunosuppression through the cGAS-STING pathway.
Immunity. 58:811–825.e7. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Saha T, Dash C, Jayabalan R, Khiste S,
Kulkarni A, Kurmi K, Mondal J, Majumder PK, Bardia A, Jang HL and
Sengupta S: Intercellular nanotubes mediate mitochondrial
trafficking between cancer and immune cells. Nat Nanotechnol.
17:98–106. 2022. View Article : Google Scholar
|
|
156
|
Baldwin JG, Heuser-Loy C, Saha T, Schelker
RC, Slavkovic-Lukic D, Strieder N, Hernandez-Lopez I, Rana N,
Barden M, Mastrogiovanni F, et al: Intercellular nanotube-mediated
mitochondrial transfer enhances T cell metabolic fitness and
antitumor efficacy. Cell. 187:6614–6630.e21. 2024. View Article : Google Scholar
|
|
157
|
Yan C, Liu X, Xu H and Wang L: Cytoplasmic
mtDNA clearance suppresses inflammatory immune responses. Trends
Cell Biol. 34:897–900. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Jiménez-Loygorri JI, Villarejo-Zori B,
Viedma-Poyatos Á, Zapata-Muñoz J, Benítez-Fernández R, Frutos-Lisón
MD, Tomás-Barberán FA, Espín JC, Area-Gómez E, Gomez-Duran A and
Boya P: Mitophagy curtails cytosolic mtDNA-dependent activation of
cGAS/STING inflammation during aging. Nat Commun. 15:8302024.
View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Ryan TA, Phillips EO, Collier CL, Jb
Robinson A, Routledge D, Wood RE, Assar EA and Tumbarello DA:
Tollip coordinates Parkin-dependent trafficking of
mitochondrial-derived vesicles. EMBO J. 39:e1025392020. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Melki I, Allaeys I, Tessandier N, Lévesque
T, Cloutier N, Laroche A, Vernoux N, Becker Y, Benk-Fortin H,
Zufferey A, et al: Platelets release mitochondrial antigens in
systemic lupus erythematosus. Sci Transl Med. 13:eaav59282021.
View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Ko M, You J, Cho E and Kwon HJ: Small
molecule modulation of organelle membrane contact sites. Trends
Pharmacol Sci. 47:490–503. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Rosen DA, Seki SM, Fernández-Castañeda A,
Beiter RM, Eccles JD, Woodfolk JA and Gaultier A: Modulation of the
sigma-1 receptor-IRE1 pathway is beneficial in preclinical models
of inflammation and sepsis. Sci Transl Med. 11:eaau52662019.
View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Etxebeste-Mitxeltorena M, Flores-Romero H,
Ramos-Inza S, Masiá E, Nenchova M, Montesinos J, Martinez-Gonzalez
L, Porras G, Orzáez M, Vicent MJ, et al: Modulation of
mitochondria-endoplasmic reticulum contacts (MERCs) by small
molecules as a new strategy for restoring lipid metabolism in an
amyotrophic lateral sclerosis model. J Med Chem. 68:1179–1194.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Angebault C, Panel M, Lacôte M, Rieusset
J, Lacampagne A and Fauconnier J: Metformin reverses the enhanced
myocardial SR/ER-Mitochondria interaction and impaired complex
I-Driven respiration in dystrophin-deficient mice. Front Cell Dev
Biol. 8:6094932021. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Tubbs E, Axelsson AS, Vial G, Wollheim CB,
Rieusset J and Rosengren AH: Sulforaphane improves disrupted
ER-mitochondria interactions and suppresses exaggerated hepatic
glucose production. Mol Cell Endocrinol. 461:205–214. 2018.
View Article : Google Scholar
|
|
166
|
Naia L, Pinho CM, Dentoni G, Liu J, Leal
NS, Ferreira DMS, Schreiner B, Filadi R, Fão L, Connolly NMC, et
al: Neuronal cell-based high-throughput screen for enhancers of
mitochondrial function reveals luteolin as a modulator of
mitochondria-endoplasmic reticulum coupling. BMC Biol. 19:572021.
View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Lee HJ, Jung YH, Choi GE, Kim JS, Chae CW,
Lim JR, Kim SY, Yoon JH, Cho JH, Lee SJ and Han HJ: Urolithin A
suppresses high glucose-induced neuronal amyloidogenesis by
modulating TGM2-dependent ER-mitochondria contacts and calcium
homeostasis. Cell Death Differ. 28:184–202. 2021. View Article : Google Scholar
|
|
168
|
Kim J, Gupta R, Blanco LP, Yang S,
Shteinfer-Kuzmine A, Wang K, Zhu J, Yoon HE, Wang X, Kerkhofs M, et
al: VDAC oligomers form mitochondrial pores to release mtDNA
fragments and promote lupus-like disease. Science. 366:1531–1536.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Belosludtsev KN, Ilzorkina AI, Matveeva
LA, Chulkov AV, Semenova AA, Dubinin MV and Belosludtseva NV:
Effect of VBIT-4 on the functional activity of isolated
mitochondria and cell viability. Biochim Biophys Acta Biomembr.
1866:1843292024. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Rusilowicz-Jones EV, Jardine J, Kallinos
A, Pinto-Fernandez A, Guenther F, Giurrandino M, Barone FG,
McCarron K, Burke CJ, Murad A, et al: USP30 sets a trigger
threshold for PINK1-PARKIN amplification of mitochondrial
ubiquitylation. Life Sci Alliance. 3:e2020007682020. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Feng X, Cai W, Li Q, Zhao L, Meng Y and Xu
H: Activation of lysosomal Ca2+ channels mitigates mitochondrial
damage and oxidative stress. J Cell Biol. 224:e2024031042025.
View Article : Google Scholar :
|
|
172
|
Xing Y, Wang MM, Zhang F, Xin T, Wang X,
Chen R, Sui Z, Dong Y, Xu D, Qian X, et al: Lysosomes finely
control macrophage inflammatory function via regulating the release
of lysosomal Fe(2+) through TRPML1 channel. Nat Commun. 16:9852025.
View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Gao X, Nan Y, Zhao Y, Yuan Y, Ren B, Sun
C, Cao K, Yu M, Feng X and Ye J: Atorvastatin reduces lipid
accumulation in the liver by activating protein kinase A-mediated
phosphorylation of perilipin 5. Biochim Biophys Acta Mol Cell Biol
Lipids. 1862:1512–1519. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Zhang E, Yin S, Zhao C, Fan L and Hu H:
Involvement of activation of PLIN5-Sirt1 axis in protective effect
of glycycoumarin on hepatic lipotoxicity. Biochem Biophys Res
Commun. 528:7–13. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Jiang S, Uddin MJ, Yu X, Piao L, Dorotea
D, Oh GT and Ha H: Peroxisomal fitness: A potential protective
mechanism of fenofibrate against high fat diet-induced
non-alcoholic fatty liver disease in mice. Diabetes Metab J.
46:829–842. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Yamaguchi S, Li H, Purevsuren J, Yamada K,
Furui M, Takahashi T, Mushimoto Y, Kobayashi H, Hasegawa Y,
Taketani T, et al: Bezafibrate can be a new treatment option for
mitochondrial fatty acid oxidation disorders: Evaluation by in
vitro probe acylcarnitine assay. Mol Genet Metab. 107:87–91. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Lund M, Andersen KG, Heaton R, Hargreaves
IP, Gregersen N and Olsen RKJ: Bezafibrate activation of PPAR
drives disturbances in mitochondrial redox bioenergetics and
decreases the viability of cells from patients with VLCAD
deficiency. Biochim Biophys Acta Mol Basis Dis. 1867:1661002021.
View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Vidyakina AA, Silonov SA, Ivanov AY,
Shpakova EA, Kim MD, Podolskaya EP, Gladchuk AS, Balova IA, Bräse S
and Danilkina NA: Dual organelle targeting for intra-organelle
click: mitochondria and endoplasmic reticulum-directed
benzothiophene-fused cycloalkyne probes. J Mater Chem B.
13:10525–10535. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Wang X, Qian J, Yang Z, Song Y, Pan W, Ye
Y, Qin X, Yan X, Huang X, Wang X, et al: Photodynamic modulation of
endoplasmic reticulum and mitochondria network boosted cancer
immunotherapy. Adv Mater. 36:e23109642024. View Article : Google Scholar
|
|
180
|
Feng X, Lin T, Chen D, Li Z, Yang Q, Tian
H, Xiao Y, Lin M, Liang M, Guo W, et al: Mitochondria-associated ER
stress evokes immunogenic cell death through the ROS-PERK-eIF2α
pathway under PTT/CDT combined therapy. Acta Biomater. 160:211–224.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Wang Y, Cheng W, Chen Y, Tian H, Huang C
and Li K: Endoplasmic reticulum-mitochondria dual-targeting
nanoplatform suppresses cancer stemness and enhances colorectal
cancer immunotherapy. J Control Release. 387:1142302025. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Xu X, Huang Q, Liu Y, Liu J, Yang D, Song
Y and Han X: Smart nanoparticles disrupting energy supply through
triple mechanisms to kill tumors via dual disruption of
mitochondria and lysosomes. Adv Sci (Weinh). 13:e173732026.
View Article : Google Scholar :
|
|
183
|
Ilahibaks NF, Ardisasmita AI, Xie S,
Gunnarsson A, Brealey J, Vader P, de Jong OG, de Jager S, Dekker N,
Peacock B, et al: TOP-EVs: Technology of Protein delivery through
Extracellular Vesicles is a versatile platform for intracellular
protein delivery. J Control Release. 355:579–592. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Liang X, Gupta D, Xie J, Van Wonterghem E,
Van Hoecke L, Hean J, Niu Z, Ghaeidamini M, Wiklander OPB, Zheng W,
et al: Engineering of extracellular vesicles for efficient
intracellular delivery of multimodal therapeutics including genome
editors. Nat Commun. 16:40282025. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Ma D, Xie A, Lv J, Min X, Zhang X, Zhou Q,
Gao D, Wang E, Gao L, Cheng L and Liu S: Engineered extracellular
vesicles enable high-efficient delivery of intracellular
therapeutic proteins. Protein Cell. 15:724–743. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Mitchell MJ, Billingsley MM, Haley RM,
Wechsler ME, Peppas NA and Langer R: Engineering precision
nanoparticles for drug delivery. Nat Rev Drug Discov. 20:101–124.
2021. View Article : Google Scholar
|
|
187
|
Yu YR, Imrichova H, Wang H, Chao T, Xiao
Z, Gao M, Rincon-Restrepo M, Franco F, Genolet R, Cheng WC, et al:
Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell
exhaustion. Nat Immunol. 21:1540–1551. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Akhtar A, Shakir M, Ansari MS, Divya,
Faizan MI, Chauhan V, Singh A, Alam R, Azmi I, Sharma S, et al:
Bioengineering the metabolic network of CAR T cells with GLP-1 and
Urolithin A increases persistence and long-term anti-tumor
activity. Cell Rep Med. 6:1020212025. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Ma K, Cheng H, Wang L, Xiao H, Liu F, Yang
Y, Xiao Z, Tang K, Li S, Wang G, et al: Succinate preserves CD8(+)
T cell fitness to augment antitumor immunity. Immunity.
58:2505–2523.e8. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Zhang R, Gao F, Li J, Jin J, Chen K,
Chaudhuri S, Liao Z, Xiao T, Xu Y, Wen H, et al: USP30 inhibition
augments mitophagy to prevent T cell exhaustion. Sci Adv.
11:eadv69022025. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Lontos K, Wang Y, Joshi SK, Frisch AT,
Watson MJ, Kumar A, Menk AV, Wang Y, Cumberland R, Lohmueller J, et
al: Metabolic reprogramming via an engineered PGC-1α improves human
chimeric antigen receptor T-cell therapy against solid tumors. J
Immunother Cancer. 11:e0065222023. View Article : Google Scholar
|
|
192
|
Bar-Ephraim YE, Kretzschmar K and Clevers
H: Organoids in immunological research. Nat Rev Immunol.
20:279–293. 2020. View Article : Google Scholar
|
|
193
|
Suhito IR, Sunil C and Tay A: Engineering
human immune organoids for translational immunology. Bioact Mater.
44:164–183. 2025.
|
|
194
|
Hung V, Lam SS, Udeshi ND, Svinkina T,
Guzman G, Mootha VK, Carr SA and Ting AY: Proteomic mapping of
cytosol-facing outer mitochondrial and ER membranes in living human
cells by proximity biotinylation. Elife. 6:e244632017. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Zhu Y, Akkaya KC, Ruta J, Yokoyama N, Wang
C, Ruwolt M, Lima DB, Lehmann M and Liu F: Cross-link assisted
spatial proteomics to map sub-organelle proteomes and membrane
protein topologies. Nat Commun. 15:32902024. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Scorrano L, De Matteis MA, Emr S, Giordano
F, Hajnóczky G, Kornmann B, Lackner LL, Levine TP, Pellegrini L,
Reinisch K, et al: Coming together to define membrane contact
sites. Nat Commun. 10:12872019. View Article : Google Scholar
|
|
197
|
Csordás G, Weaver D and Hajnóczky G:
Endoplasmic reticulum-mitochondrial contactology: Structure and
Signaling Functions. Trends Cell Biol. 28:523–540. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Gemmink A, Daemen S, Kuijpers HJH, Schaart
G, Duimel H, López-Iglesias C, van Zandvoort MAMJ, Knoops K and
Hesselink MKC: Super-resolution microscopy localizes perilipin 5 at
lipid droplet-mitochondria interaction sites and at lipid droplets
juxtaposing to perilipin 2. Biochim Biophys Acta Mol Cell Biol
Lipids. 1863:1423–1432. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
York AG, Chandris P, Nogare DD, Head J,
Wawrzusin P, Fischer RS, Chitnis A and Shroff H: Instant
super-resolution imaging in live cells and embryos via analog image
processing. Nat Methods. 10:1122–1126. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Zhang J, Shen SY, Zhai MY, Shen ZQ, Li W,
Liang LF, Yin SY, Han QQ, Li B, Zhang YQ and Yu J: Augmented
microglial endoplasmic reticulum-mitochondria contacts mediate
depression-like behavior in mice induced by chronic social defeat
stress. Nat Commun. 15:51992024. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Benhammouda S, Vishwakarma A, Gatti P and
Germain M: Mitochondria endoplasmic reticulum contact sites
(MERCs): Proximity ligation assay as a tool to study organelle
interaction. Front Cell Dev Biol. 9:7899592021. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Gustafsson MG, Shao L, Carlton PM, Wang
CJ, Golubovskaya IN, Cande WZ, Agard DA and Sedat JW:
Three-dimensional resolution doubling in wide-field fluorescence
microscopy by structured illumination. Biophys J. 94:4957–4970.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Valm AM, Cohen S, Legant WR, Melunis J,
Hershberg U, Wait E, Cohen AR, Davidson MW, Betzig E and
Lippincott-Schwartz J: Applying systems-level spectral imaging and
analysis to reveal the organelle interactome. Nature. 546:162–167.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Wegel E, Göhler A, Lagerholm BC, Wainman
A, Uphoff S, Kaufmann R and Dobbie IM: Imaging cellular structures
in super-resolution with SIM, STED and Localisation Microscopy: A
practical comparison. Sci Rep. 6:272902016. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Liu T, Stephan T, Chen P, Keller-Findeisen
J, Chen J, Riedel D, Yang Z, Jakobs S and Chen Z: Multi-color
live-cell STED nanoscopy of mitochondria with a gentle inner
membrane stain. Proc Natl Acad Sci USA. 119:e22157991192022.
View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Yang X, Yang Z, Wu Z, He Y, Shan C, Chai
P, Ma C, Tian M, Teng J, Jin D, et al: Mitochondrial dynamics
quantitatively revealed by STED nanoscopy with an enhanced
squaraine variant probe. Nat Commun. 11:36992020. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Lelek M, Gyparaki MT, Beliu G, Schueder F,
Griffié J, Manley S, Jungmann R, Sauer M, Lakadamyali M and Zimmer
C: Single-molecule localization microscopy. Nat Rev Methods
Primers. 1:392021. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Gómez-de-Mariscal E, Del Rosario M,
Pylvänäinen JW, Jacquemet G and Henriques R: Harnessing artificial
intelligence to reduce phototoxicity in live imaging. J Cell Sci.
137:jcs2615452024. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Cieri D, Vicario M, Giacomello M, Vallese
F, Filadi R, Wagner T, Pozzan T, Pizzo P, Scorrano L, Brini M and
Calì T: SPLICS: A split green fluorescent protein-based contact
site sensor for narrow and wide heterotypic organelle
juxtaposition. Cell Death Differ. 25:1131–1145. 2018. View Article : Google Scholar :
|
|
210
|
Tashiro S, Kakimoto Y, Shinmyo M, Fujimoto
S and Tamura Y: Improved Split-GFP systems for visualizing
organelle contact sites in yeast and human cells. Front Cell Dev
Biol. 8:5713882020. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Hertlein V, Flores-Romero H, Das KK,
Fischer S, Heunemann M, Calleja-Felipe M, Knafo S, Hipp K, Harter
K, Fitzgerald JC and García-Sáez AJ: MERLIN: A novel BRET-based
proximity biosensor for studying mitochondria-ER contact sites.
Life Sci Alliance. 3:e2019006002020. View Article : Google Scholar
|
|
212
|
Li X, Gamuyao R, Wu ML, Cho WJ, King SV,
Petersen RA, Stabley DR, Lindow C, Climer LK, Shirinifard A, et al:
A fluorogenic complementation tool kit for interrogating lipid
droplet-organelle interaction. J Cell Biol. 223:e2023111262024.
View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Moreira CMDN, Kelemen CD, Obado SO,
Zahedifard F, Zhang N, Holetz FB, Gauglitz L, Dallagiovanna B,
Field MC, Kramer S and Zoltner M: Impact of inherent biases built
into proteomic techniques: Proximity labeling and affinity capture
compared. J Biol Chem. 299:1027262023. View Article : Google Scholar
|
|
214
|
Hung V, Udeshi ND, Lam SS, Loh KH, Cox KJ,
Pedram K, Carr SA and Ting AY: Spatially resolved proteomic mapping
in living cells with the engineered peroxidase APEX2. Nat Protoc.
11:456–475. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Branon TC, Bosch JA, Sanchez AD, Udeshi
ND, Svinkina T, Carr SA, Feldman JL, Perrimon N and Ting AY:
Efficient proximity labeling in living cells and organisms with
TurboID. Nat Biotechnol. 36:880–887. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Cunningham CN, Van Vranken JG, Larios J,
Heyden K, Gygi SP and Rutter J: A dual-purification system to
isolate mitochondrial subpopulations. J Cell Sci.
138:jcs2636932025. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Giamogante F, Barazzuol L, Brini M and
Calì T: ER-Mitochondria contact sites reporters: Strengths and
weaknesses of the available approaches. Int J Mol Sci. 21:81572020.
View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Kizilyaprak C, Daraspe J and Humbel BM:
Focused ion beam scanning electron microscopy in biology. J
Microsc. 254:109–114. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Lee RG, Rudler DL, Raven SA, Peng L,
Chopin A, Moh ESX, McCubbin T, Siira SJ, Fagan SV, DeBono NJ, et
al: Quantitative subcellular reconstruction reveals a lipid
mediated inter-organelle biogenesis network. Nat Cell Biol.
26:57–71. 2024. View Article : Google Scholar
|
|
220
|
Ching C, Maufront J, di Cicco A, Lévy D
and Dezi M: Cool-contacts: Cryo-electron microscopy of membrane
contact sites and their components. Contact (Thousand Oaks).
7:251525642412313642024.PubMed/NCBI
|
|
221
|
Wilson ZN, West M, English AM, Odorizzi G
and Hughes AL: Mitochondrial-derived compartments are multilamellar
domains that encase membrane cargo and cytosol. J Cell Biol.
223:e2023070352024. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Villa E, Schaffer M, Plitzko JM and
Baumeister W: Opening windows into the cell: focused-ion-beam
milling for cryo-electron tomography. Curr Opin Struct Biol.
23:771–777. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Xu K, Saaoud F, Shao Y, Lu Y, Yang Q,
Jiang X, Wang H and Yang X: A new paradigm in intracellular
immunology: Mitochondria emerging as leading immune organelles.
Redox Biol. 76:1033312024. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Marques E, Kramer R and Ryan DG:
Multifaceted mitochondria in innate immunity. NPJ Metab Health Dis.
2:62024. View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Barazzuol L, Giamogante F and Calì T:
Mitochondria associated membranes (MAMs): Architecture and
physiopathological role. Cell Calcium. 94:1023432021. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Namgaladze D, Khodzhaeva V and Brüne B:
ER-Mitochondria communication in cells of the innate immune system.
Cells. 8:10882019. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
He Q, Qu M, Shen T, Su J, Xu Y, Xu C,
Barkat MQ, Cai J, Zhu H, Zeng LH and Wu X: Control of
mitochondria-associated endoplasmic reticulum membranes by protein
S-palmitoylation: Novel therapeutic targets for neurodegenerative
diseases. Ageing Res Rev. 87:1019202023. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Jeltema D, Abbott K and Yan N: STING
trafficking as a new dimension of immune signaling. J Exp Med.
220:e202209902023. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Obara CJ, Nixon-Abell J, Moore AS, Riccio
F, Hoffman DP, Shtengel G, Xu CS, Schaefer K, Pasolli HA, Masson
JB, et al: Motion of VAPB molecules reveals ER-mitochondria contact
site subdomains. Nature. 626:169–176. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Calì T and Brini M: Quantification of
organelle contact sites by split-GFP-based contact site sensors
(SPLICS) in living cells. Nat Protoc. 16:5287–5308. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Cho KF, Branon TC, Udeshi ND, Myers SA,
Carr SA and Ting AY: Proximity labeling in mammalian cells with
TurboID and split-TurboID. Nat Protoc. 15:3971–3999. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
232
|
Shulha AS, Shyshenko V, Schibalski RS,
Jones AC, Faulkner JL, Stadler K and Ilatovskaya DV: An update on
the role of sex hormones in the function of the cardiorenal
mitochondria. Biochem Soc Trans. 52:2307–2319. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Lynch S, Boyett JE, Smith MR and
Giordano-Mooga S: Sex hormone regulation of proteins modulating
mitochondrial metabolism, dynamics and inter-organellar cross talk
in cardiovascular disease. Front Cell Dev Biol. 8:6105162021.
View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Ziegler DV, Vindrieux D, Goehrig D, Jaber
S, Collin G, Griveau A, Wiel C, Bendridi N, Djebali S, Farfariello
V, et al: Calcium channel ITPR2 and mitochondria-ER contacts
promote cellular senescence and aging. Nat Commun. 12:7202021.
View Article : Google Scholar : PubMed/NCBI
|
|
235
|
Puebla-Huerta A, Huerta H,
Quezada-Gutierez C, Morgado-Cáceres P, Casanova-Canelo C, Niño SA,
Linsambarth S, Díaz-Rivera O, López-Domínguez JA, Rodríguez-López
S, et al: Calcium (Ca(2+)) fluxes at mitochondria-ER contact sites
(MERCS) are a new target of senolysis in therapy-induced senescence
(TIS). NPJ Aging. 11:112025. View Article : Google Scholar : PubMed/NCBI
|
|
236
|
Wang Z, Du X, Yu S, Yan X and Xin Y:
Mitochondria-associated membranes in aging and senescence. Aging
Dis. 16:2250–2272. 2024.PubMed/NCBI
|
|
237
|
Ahn M, Ali A and Seo JH: Mitochondrial
regulation in the tumor microenvironment: targeting mitochondria
for immunotherapy. Front Immunol. 15:14538862024. View Article : Google Scholar : PubMed/NCBI
|
|
238
|
Plata-Gómez AB and Ho P: Age- and
diet-instructed metabolic rewiring of the tumor-immune
microenvironment. J Exp Med. 222:e202411022025. View Article : Google Scholar : PubMed/NCBI
|
|
239
|
Polak R, Zhang ET and Kuo CJ: Cancer
organoids 2.0: Modelling the complexity of the tumour immune
microenvironment. Nat Rev Cancer. 24:523–539. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
240
|
Grönholm M, Feodoroff M, Antignani G,
Martins B, Hamdan F and Cerullo V: Patient-derived organoids for
precision cancer immunotherapy. Cancer Res. 81:3149–3155. 2021.
View Article : Google Scholar : PubMed/NCBI
|














