Introduction
In recent years, the role of organelles in the
pathogenesis of human diseases has emerged as a central focus of
scientific research. Cell survival and function depend on the
maintenance of organelle homeostasis. However, organelles do not
function in isolation; instead, they establish a highly
interconnected and collaborative intracellular network.
Inter-organellar communication primarily occurs via two mechanisms:
Vesicular transport and the formation of specialized membrane
contact sites. These contact sites enable direct material exchange
and signal transduction between organelles, thereby facilitating
the synergistic or antagonistic regulation of cellular processes
(1-4). Advances in imaging, particularly
electron microscopy and subcellular fluorescence techniques, have
been crucial for identifying and characterizing these membrane
contact sites. Membrane contact sites are defined as stable yet
dynamic regions where the membranes of two distinct organelles, or
an organelle and the plasma membrane (PM), come into close contact
without fusing. At these junctions, the membranes are consistently
separated by a well-defined gap of ~10-30 nanometers (5). Among the diverse inter-organellar
connections, the physical and functional interface between
mitochondria and the endoplasmic reticulum (ER), known as
mitochondria-ER contact sites (MERCs), is the most extensively
studied and characterized.
MERCs are ubiquitous in eukaryotic cells. Rather
than acting as static bridges, they are dynamic assemblies that are
remodeled in response to cellular signals and can cover ~10-15% of
the mitochondrial membrane surface area (6-9).
These specialized domains function as concentrated macromolecular
hubs accommodating hundreds of proteins, including tethering
molecules, structural scaffolds, ion channels and transporters,
lipid-modifying enzymes and numerous signaling proteins. The
molecular composition of MERCs varies across different tissue types
and species, reflecting the adaptation to specialized cellular
functions. Over the past decade, extensive research has
substantially enhanced our understanding of the interaction between
MERCs and core cellular activities. It is now clear that MERCs are
crucial for numerous fundamental physiological processes, including
the regulation of calcium (Ca2+) (10,11) and zinc (Zn2+)
signaling (12-14), the control of mitochondrial
dynamics (fission, fusion and motility) (15,16), the modulation of inflammatory
responses (17), the maintenance
of redox balance (18), the
synthesis and transfer of lipids (19), the management of ER stress
(20,21), the execution of autophagy and
mitophagy (22-24), the initiation of apoptosis
(25,26) and the regulation of cellular
senescence (27,28). Therefore, the structural
integrity and functional plasticity of MERCs are of critical
importance for cellular adaptation, survival and overall
homeostasis.
Despite substantial progress, a crucial challenge
persists in understanding how MERCs hierarchically integrate these
diverse functions. Ca2+ flux, lipid transfer and redox
signaling at MERCs are not isolated activities; instead, they are
interdependent processes that are coordinately regulated to
determine cellular outcomes. For instance, ER-to-mitochondria
Ca2+ transfer efficiency is modulated by the local lipid
composition of the contact-site membrane, which in turn is
influenced by MERC-resident lipid transfer proteins (19,29). Concurrently, the redox
environment at MERCs, governed by oxidoreductases such as Ero1α and
TMX1, directly regulates the activity of Ca2+ channels
and pumps. As a result, it calibrates the Ca2+ signal
that drives mitochondrial metabolism and, under pathological
conditions, apoptosis (30-33). The present review aims to move
beyond listing individual MERC functions by synthesizing how these
activities are integrated into a coherent signaling network.
Given the central role of MERCs in cellular
physiology, it is unsurprising that dysregulation of MERC
architecture or function is increasingly linked with the
pathogenesis of a wide range of human diseases. Abnormalities in
MERC composition, abundance, or activity have been implicated in
neurodegenerative disorders (for example, Alzheimer's, Parkinson's
and amyotrophic lateral sclerosis), metabolic diseases [for
example, diabetes and non-alcoholic fatty liver disease (NAFLD)],
cardiovascular diseases, cancer and various orthopedic conditions
(34-39). These dysfunctions often manifest
as disrupted ion homeostasis, bioenergetic failure, excessive
oxidative stress, and impaired organelle quality control,
ultimately resulting in cellular dysfunction and tissue
pathology.
A comprehensive and in-depth understanding of MERCs
is therefore essential for the advancement of fundamental cell
biology and for the development of precise therapeutic strategies
aimed at modulating MERC homeostasis in disease scenarios. The
present review provides a comprehensive and integrated overview of
the current knowledge about MERCs. The organizational principles
and molecular composition that define these contact sites were
systematically summarized, the regulatory mechanisms that govern
their dynamics were investigated, elaborating on their diverse
roles in mediating essential cellular functions, and MERC
alterations in disease were described. Finally, the emerging
perspectives and future directions for the targeted therapeutic
modulation of MERCs were discussed, highlighting the considerable
potential and significant challenges involved in translating this
knowledge into clinical applications.
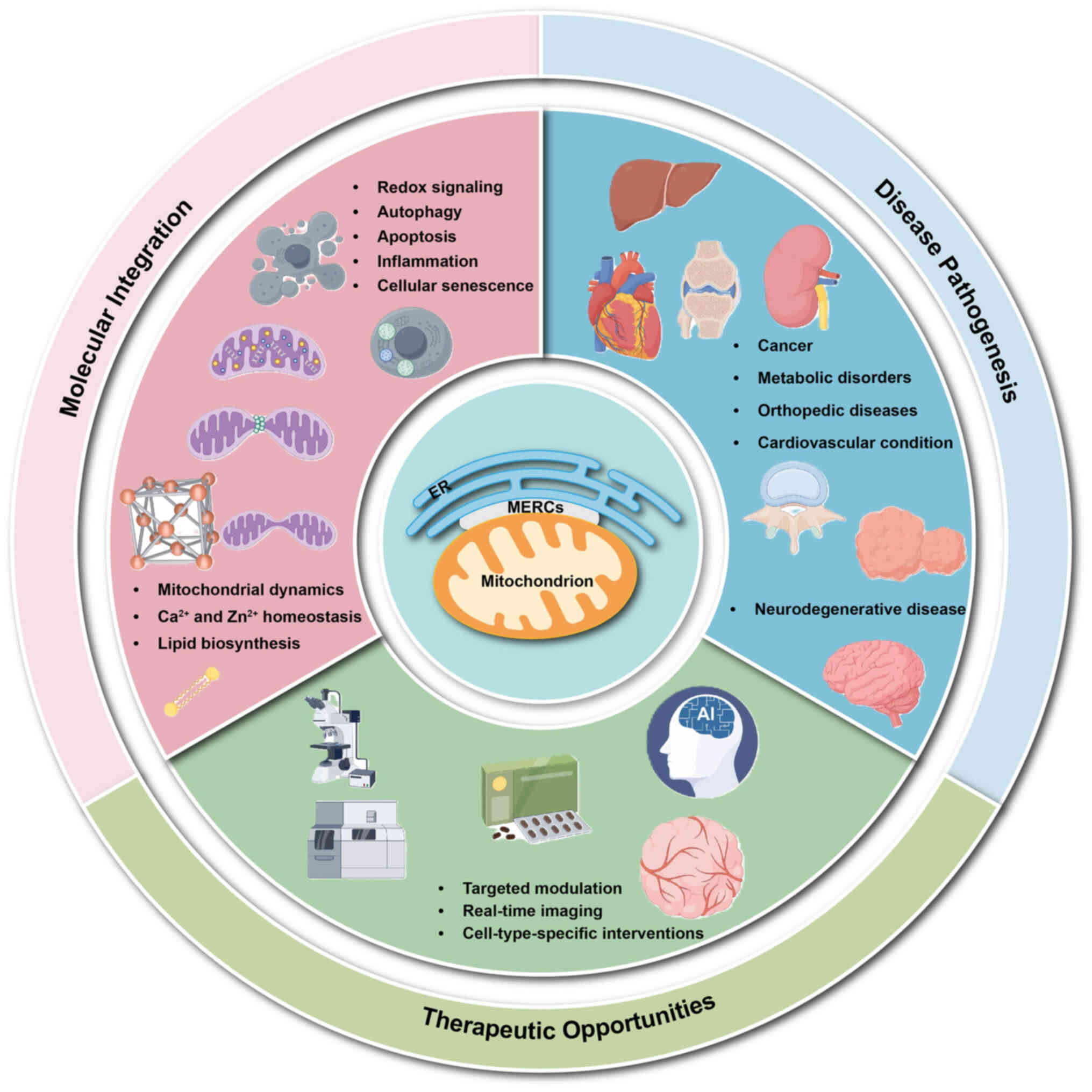
To provide a unifying framework for the diverse
functions and pathological roles discussed in the present review, a
three-tier model of MERC-mediated signal integration were proposed.
In the first tier (Sensing), MERCs detect and transduce rapid ionic
signals, mainly Ca2+ and Zn2+, together with
local redox changes that convey information regarding cellular
metabolic status and stress. In the second tier (Processing), these
signals are decoded by MERC-resident protein complexes that
coordinate mitochondrial dynamics (fission, fusion and mitophagy),
lipid biosynthesis and transfer and autophagic flux. In the third
tier (Execution), the integrated output is translated into cell
fate decisions, such as metabolic adaptation, apoptosis, senescence
and inflam- matory responses, thereby influencing tissue
homeostasis or pathology. This hierarchical model, used throughout
the review, provides a conceptual framework for comprehending how
dysfunction at any level of MERC signaling can propagate and
contribute to disease.
A recurring pattern in the literature, and a
unifying theme for the present review, is the dualistic nature of
MERC function in disease. Evidence from multiple systems
demonstrates that the relationship between MERC abundance or
activity and cellular health is non-linear. In pathological
contexts, both decreased and increased ER-mitochondria coupling
have been observed, with the direction of the change depending on
the specific molecular insult, cell type and disease stage.
Insufficient coupling impairs the essential inter-organellar
transfer of Ca2+ and lipids, leading to bioenergetic
failure and defective organelle quality control (39-42). Conversely, excessive coupling can
result in mitochondrial Ca2+ overload, elevated
oxidative stress and apoptotic cell death (27,43-45). This context-dependent duality
provides a framework for understanding why MERC phenotypes differ
across diseases. For example, contact sites are diminished in
Charcot-Marie-Tooth disease type 2A and amyotrophic lateral
sclerosis, whereas they are increased in certain cancers and
diabetic cardiomyopathy (DCM) (42,45,46). Throughout the current review,
this dualistic pattern was highlighted and its implications for
therapeutic strategies aimed at restoring MERC homeostasis were
discussed (Fig. 1).
Organization and evaluation of MERCs
MERCs were first detected by electron microscopy in
the 1950s (47,48) and were later biochemically
identified in the 1990s (49).
In 2006, electron tomography revealed that subdomains of the outer
mitochondrial membrane (OMM) and the ER are connected by tethers
that vary in size and shape (50). Because electron microscopy
provides resolution that matches the scale of these nanoscale
junctions, it remains the only method capable of direct
visualization. Three-dimensional (3D) reconstructions generated
through electron tomography provide the most detailed and
comprehensive views of ER-mitochondria interactions (50,51). Whole-cell, three-dimensional maps
of ER-mitochondria contacts have since been generated using focused
ion beam scanning electron microscopy (FIB-SEM) in neurons at 4 nm
resolution (52), serial
tilt-angle tomography in yeast cells (51), and, more recently, soft X-ray
tomography in lymphoblastoid cells with a 50 nm resolution
(53). However, data acquisition
and reconstruction processes associated with these 3D techniques
remain highly time-consuming, limiting their widespread use for
generating statistically robust organelle geometry datasets.
Several groups have analyzed ER-mitochondria
interaction parameters using transmission electron microscopy (TEM)
to enable statistical comparisons, yet variability in measurement
methodologies has made cross-study comparisons difficult. The main
quantitative parameters include the length of the contact interface
and the width of the gap between the ER and OMM. Some studies have
measured contact lengths normalized to mitochondrial perimeter
(54), whereas others have
quantified the number or frequency of ER-mitochondria contacts
(55), typically normalized per
mitochondrion. However, TEM is labor-intensive and captures only
static snapshots of cellular ultrastructure. More importantly, TEM
detects physical proximity rather than functional MERCs; an ER
tubule may surround a mitochondrion without forming active MERCs,
as close apposition alone does not confirm molecular tethering or
biological activity. Immuno-gold labeling in EM has been used to
detect specific proteins localized at ER-mitochondria contact
sites.
Among various microscopy-based methods, scanning
electron microscopy (SEM) has been proposed as a tool for examining
ER-mitochondria interactions across large specimen volumes.
Conventional SEM uses low-energy electrons to generate
surface-specific images (52,56), whereas backscattered electrons
can capture signals from the first few nanometers beneath the
surface, offering high Z-resolution. Over the past two decades,
advanced techniques have markedly improved the quality, efficiency
and resolution of full 3D image reconstruction. These approaches
use an automated ultramicrotome within the SEM chamber for serial
block-face imaging (57) or a
focused ion or plasma beam (FIB-SEM) (58,59) to sequentially remove thin layers
from a hardened sample. Both methods can be automated to acquire
hundreds of serial images. Increasing evidence indicates that
immunogold labeling can be combined with SEM to detect resident
proteins at the ER-mitochondria interface (60). Despite these technological
advances, generating comprehensive 3D reconstructions remains
challenging due to the substantial computational power and time
required to process large datasets.
Confocal microscopy is another approach for
assessing the juxtaposition of mitochondria and the ER, using
organelle-specific tracker probes (61) or fluorescent proteins (FPs)
(62) targeted to these
compartments. The degree of colocalization between the ER and
mitochondria is typically quantified using Manders' colocalization
coefficient in ImageJ (62).
However, mitochondrial membrane potential (MMP) influences the
accumulation of MitoTracker dyes thereby affecting organelle
labeling efficiency. Therefore, mitochondria-targeted genetically
encoded FPs are strongly recommended, as they remain stable under
varying mitochondrial conditions, including fluctuations in
membrane potential and redox state (63). Morphological changes in
organelles can also be misleading and complicate the interpretation
of fluorescence imaging data (64). Most critically, the physical
thickness of MERCs is far below the resolution limit of
conventional confocal microscopes (~150-300 nm). Consequently, this
method measures organelle proximity rather than the formation of
bona fide MERCs.
To address these limitations, confocal imaging has
been used to monitor dynamic changes in MERCs under normal and
stress conditions using a split-GFP-based reporter system (65). The split-GFP system consists of
two non-fluorescent fragments of a superfolder GFP variant: one
containing β-strands 1-10 and the other comprising β-strand 11,
which can spontaneously reassemble into a functional β-barrel
structure capable of emitting fluorescence (66,67). Fluorescence is restored only when
the two fragments are brought into close proximity, allowing each
moiety to be targeted to one of the adjacent membranes of interest.
Reconstituted split-GFP signals appear as distinct puncta localized
between various organelle pairs, including the ER and mitochondria,
vacuoles, peroxisomes and lipid droplets. These findings suggest
that individual organelles simultaneously establish discrete
contact sites with specific membrane domains of their partners
(68,69). The split-GFP system therefore
holds promise as a tool for identifying regulatory proteins and
previously unknown tethering factors at inter-organellar
interfaces. Advanced optical microscopy techniques with improved
resolution are also available. Super-resolution fluorescence
microscopy (SRM) offers enhanced spatial and temporal resolution,
enabling more detailed investigation of ER-mitochondria contact
site dynamics and fine structural architecture (70,71). Structured illumination microscopy
(SIM), a type of SRM suitable for rapid live-cell imaging, has been
used to visualize the kinetics and subcellular organization of
ER-mitochondria interactions (72,73). Nevertheless, SIM remains costly
and technically demanding, limiting its widespread application in
characterizing MERCs. It also requires specialized instrumentation
and specialized technical expertise.
Several methods detect sites of organelle contact
without providing information on their structural architecture.
Fluorescence resonance energy transfer (FRET) represents another
promising approach for studying contact site dynamics, as it is
highly sensitive to intermembrane distance. This technique detects
energy transfer between two fluorophores targeted to specific
organelles. These fluorophores are engineered with
rapamycin-inducible dimerization domains (74); upon addition of rapamycin, they
dimerize and generate a maximal FRET signal. Although initially
developed to visualize ER-mitochondria juxtaposition (75), this method could theoretically be
adapted to assess any potential contact site by attaching
appropriate targeting sequences to each fluorescent protein
independently. A major limitation of FRET, however, is the
requirement for equimolar expression of the two probe components,
which can hinder reproducibility and sensitivity.
The proximity ligation assay (PLA) can be tailored
to investigate proteins localized at two-membrane interfaces. It
enables highly specific detection of proximal protein-protein
interactions in cells or tissues (76,77). The method uses proximity probes
consisting of oligonucleotide-conjugated antibodies directed
against two target proteins. When these proteins are in close
proximity, the bound antibodies facilitate the formation of a
circular DNA strand. This circular DNA, covalently linked to the
antibody-antigen complex, serves as a template for rolling-circle
amplification. The amplified product is then detected via
hybridization with fluorescently labeled complementary
oligonucleotides. Notably, PLA can function as a molecular ruler
for estimating distances between epitopes by adjusting the length
of the oligonucleotides on the proximity probes and the size of the
protein-binding agents. Söderberg et al (78) estimated that the maximum
detectable distance, accounting for the sizes of both antibodies
and the connecting oligonucleotides, is ~30 nm. Longer
oligonucleotides can extend this range when needed, whereas shorter
DNA linkers combined with more compact binding agents can reduce
the detection distance to just over 10 nm, thereby enhancing
spatial resolution (78). A key
limitation of PLA, however, is that signal intensity depends on the
expression levels of the target proteins, which may vary across
samples. Additionally, background signals can arise because the PLA
partners are not always exclusively localized at organelle contact
sites. Finally, because sample fixation is required prior to
analysis, PLA provides only a static snapshot of cellular
conditions, raising the possibility of artifacts and limiting
insights into dynamic processes.
Subcellular fractionation using sucrose gradient
centrifugation is a widely used method for studying the biochemical
properties of ER-mitochondria interface sites. When ER-mitochondria
interactions were first observed in liver mitochondrial
preparations isolated via sucrose-based fractionation in the late
1950s, they were initially attributed to contamination (79). Since then, extensive efforts have
been devoted to refining protocols for enriching
mitochondrial-associated membranes (MAMs) during cell or tissue
fractionation. Throughout the present review, two related but
non-interchangeable terms were clearly distinguished. 'MERCs'
denote the physical, nanoscale membrane domains where the two
organelles closely appose each other and engage in functional
crosstalk. 'MAMs' specifically refer to the biochemical membrane
fraction that copurifies with mitochondria during subcellular
fractionation and is enriched in MERC-resident proteins. MAMs are
therefore an experimental preparation that captures MERC
components. However, not all MAM proteins are exclusively localized
to MERCs, and not all MERC functions can be studied using MAM
preparations alone. MAMs represent a distinct membrane fraction
that remains biochemically associated with mitochondria and have
played a key role in establishing the functional and physical link
between the ER and mitochondria (80-82).
To separate MAMs from pure mitochondria, Wieckowski
et al (83) established a
comprehensive protocol starting with HeLa cells or rat liver
tissue. Crude mitochondria containing both MAMs and intact
mitochondria are first isolated. Subsequently, differential
high-speed centrifugations in solutions of varying mannitol and
Percoll concentrations enable the separation of MAM and pure
mitochondrial fractions (83).
Rigorous controls are required to validate successful MAM
isolation. Specifically, western blot analysis of
well-characterized marker proteins should be performed to confirm
effective fractionation. Voltage-dependent anion channel (VDAC),
fatty acid-CoA ligase 4 (FACL4) and inositol 1,4,5-trisphosphate
receptor 3 (ITPR3) are commonly recognized as enriched MAM-resident
proteins. To assess potential cross-contamination, markers specific
to pure mitochondria or ER should also be examined: Cytochrome c
and NADH dehydrogenase 1 alpha subcomplex subunit 9 indicate
mitochondrial purity, whereas calnexin and calreticulin serve as ER
markers and help rule out ER contamination in non-MAM fractions.
Finally, contamination from other organelles, such as lysosomes,
Golgi apparatus, peroxisomes, nucleus and PM, must be excluded.
Given that proteomic analysis can identify novel MAM-localized
proteins following purification, subcellular fractionation remains
one of the most widely employed approaches for investigating
ER-mitochondria tethering and may uncover new molecular components
at these contact sites (84). It
is important to note, however, that the prolonged purification
process may alter protein composition through post-translational
modifications (for example, phosphorylation) or disruption of
protein complexes (for example, dimerization). Additionally,
residual contamination from non-target membranes may compromise the
specificity of isolated MAM proteins. Findings derived from this
method should therefore be validated using complementary
techniques.
Several approaches have been used to identify novel
proteins localized at MERCs or involved in their formation and
function. A genetic screen in yeast for mutations suppressible by
expression of a synthetic tether revealed the ER-mitochondrial
encounter structure (85),
whereas a functional screen based on the role of ER-mitochondria
contacts in lipid transfer identified the ER membrane protein
complex (EMC) (86). The
VDAC1-GRP75-ITPR and VAPB-PTPIP51 interactions were initially
discovered through yeast two-hybrid screening of putative tethering
components. Numerous studies have reported mass spectrometry (MS)
analysis of ER-mitochondria interactions using MAM fractions
isolated via gradient centrifugation from mouse brain and liver
(87), as well as from
virus-infected cells (88,89). Importantly, resident proteins at
ER-mitochondria interfaces have been successfully identified using
an engineered ascorbate peroxidase (APEX) (90,91). Unlike horseradish peroxidase,
which is commonly used but inactive under reducing conditions, APEX
remains functional when expressed in the cytoplasm, mitochondria
and other reducing environments. Upon treatment of live cells with
H2O2 and biotin-phenol for 1 min, APEX
catalyzes the one-electron oxidation of biotin-phenol, generating a
highly reactive, short-lived biotin-phenoxyl radical that
covalently labels proximal endogenous proteins. Biotinylated
proteins can then be efficiently enriched using streptavidin-based
pull-down assays. Lam et al (90) demonstrated that introducing a
single A134P mutation enhances the stability and sensitivity of
APEX. Cho et al (91)
showed that this technique can be coupled with MS to identify
previously unknown proteins residing at the ER-mitochondria
interface. By targeting the APEX probe to the outer membranes of
distinct organelles, researchers have mapped the proteomic
landscape associated with specific inter-organellar contact sites
(92). Furthermore, APEX2 has
been split into two inactive fragments that reassemble into an
active peroxidase when brought into close proximity. These
fragments are fused to rapamycin-inducible dimerization domains,
requiring rapamycin addition to induce reconstitution of enzymatic
activity (93). Similar to
FRET-based systems, the applicability of this split-APEX system is
limited by its dependence on rapamycin.
In summary, research on MERCs has advanced from the
initial ultrastructural observations to a well-developed,
multimethodological domain. The techniques can be broadly
classified into two categories: Those that reveal structural
morphology (for example, various EM modalities and super-resolution
microscopy) and those that detect functional interaction or
molecular composition (for example, biochemical fractionation,
split-GFP, PLA, FRET and APEX-based proximity labeling). Each
method has distinct advantages and inherent limitations. These
limitations range from the high spatial resolution but static and
labor-intensive nature of EM to the dynamic and functional readouts
of fluorescent probes, which frequently sacrifice direct structural
visualization for physiological relevance. Looking ahead, several
key challenges and opportunities shape the future direction of MERC
research. First, there is an urgent need to integrate complementary
techniques, by correlating high-resolution 3D structural data from
advanced EM with dynamic, functional readouts from live-cell
imaging and molecular mapping, to establish a comprehensive
understanding of MERCs. Second, developing more accessible,
high-throughput and automated methods for 3D EM analysis will be
critical for generating statistically reliable datasets on
organelle connectivity and MERC architecture across various
cellular states. Third, future tools should minimize perturbation,
as numerous current probes rely on overexpression or chemical
induction, which may modify native MERC physiology. Fourth,
improving the spatial and temporal resolution of live-cell imaging
to the nanometer scale, perhaps through next-generation
super-resolution techniques or novel biosensors, will be essential
for capturing the rapid dynamics of tether formation, disassembly
and functional regulation. Finally, expanding the molecular toolbox
to systematically discover and validate new tethering components
and regulatory mechanisms, perhaps through in-situ cryo-EM
or refined proximity proteomics in native environments, will
enhance our understanding of how MERCs integrate cellular
signaling, lipid metabolism and organelle homeostasis in health and
disease.
A crucial consideration when interpreting the MERC
literature is that no single method can unambiguously define a
'true' functional contact site. Each technique captures a distinct
aspect of the ER-mitochondria association, and findings from
different approaches are not always concordant. Electron
microscopy, whether TEM or FIB-SEM, remains the gold standard for
resolving nanoscale architecture, yet it detects physical proximity
rather than functional coupling; an ER tubule passing within 30 nm
of a mitochondrion may or may not represent a genuine
signaling-competent MERC (50,52). Proximity-based assays such as PLA
and split-GFP report on protein-protein interactions or membrane
apposition, but they depend on the expression levels of the
targeted proteins and may detect transient or nonfunctional
encounters (65,76). Functional readouts, such as
measurements of ER-to-mitochondria Ca2+ transfer or
lipid exchange, offer evidence of active communication. By
themselves, however, they cannot determine whether this
communication occurs through stable contact sites or via diffusible
intermediates. Biochemical MAM isolation provides proteomic
information but is prone to post-lysis artifacts and contamination
from other membrane compartments (82). These methodological disparities
are a key source of apparent discrepancies in the literature. For
instance, a study using TEM-based contact length quantification
might report a decrease in MERCs under a specific condition,
whereas a FRET-based assay could detect unchanged or even increased
functional coupling. Future studies should, therefore, adopt
orthogonal and complementary approaches. Ideally, these approaches
should integrate high-resolution structural imaging with dynamic
functional readouts to draw reliable conclusions about MERC status
in both health and disease.
Composition of MERCs
A core set of proteins mediates the tethering of
mitochondria to the ER in mammalian cells. These proteins either
form direct physical linkages or regulate the assembly and
stability of tethering complexes at MERCs (Fig. 2). Similar to other PM-anchoring
proteins such as junctophilins, STIM1 and extended synaptotagmins,
an intermembrane tether can consist of a single protein containing
two distinct membrane-binding domains. ATPase family AAA
domain-containing protein 3 (ATAD3) is currently the only known
natural candidate that functions as a single-protein linker between
the ER and mitochondria (94).
While certain proteins (for example, ITPR and FATE1/EMD) are
associated with relatively wide intermembrane gaps, numerous
tethers serve to narrow the distance between the ER and OMM.
Notably, FATE1/EMD has been shown to inhibit functions requiring
close membrane apposition (95).
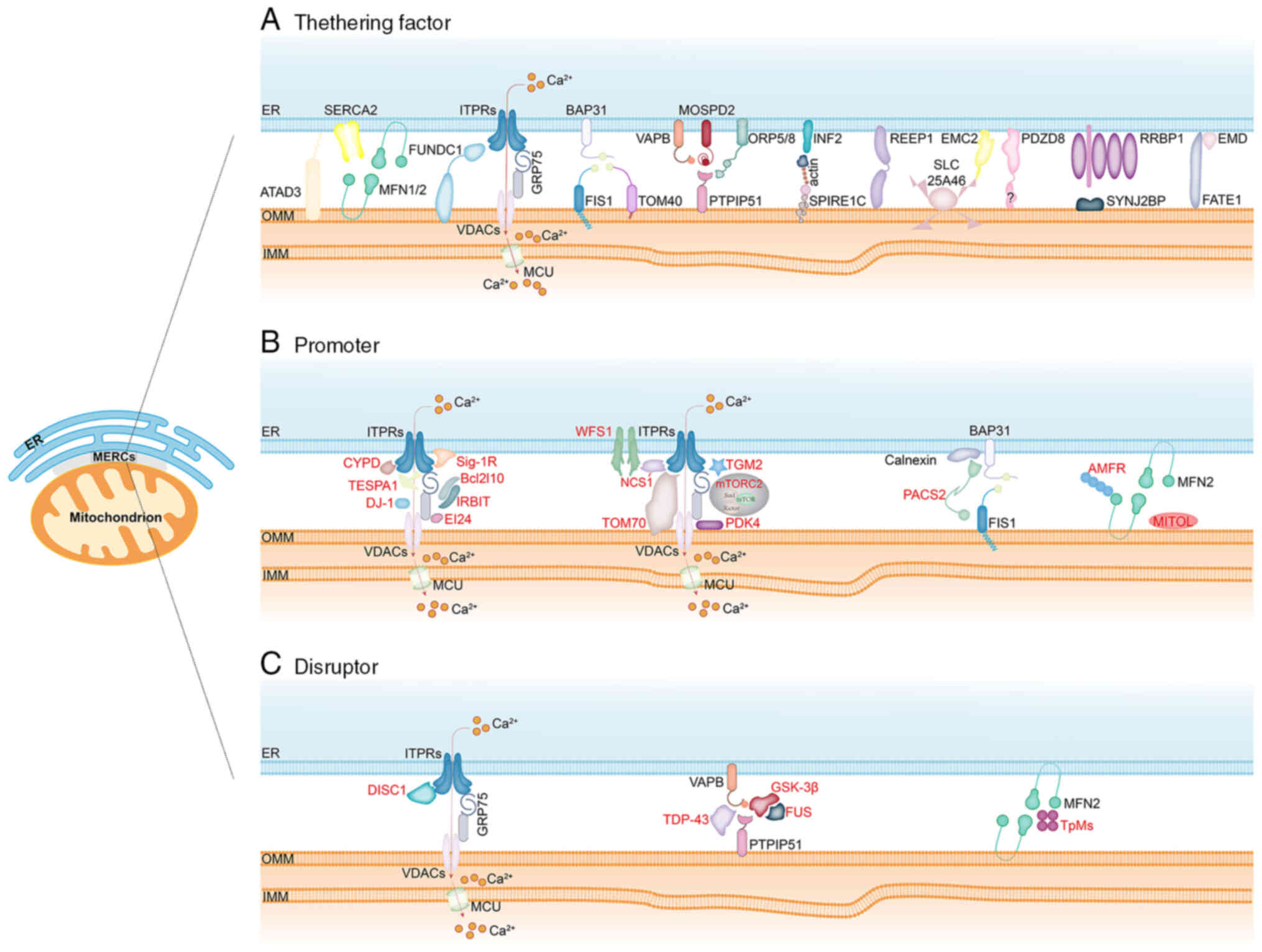 | Figure 2Composition of MERCs. MERCs are
specialized membrane domains formed by the close apposition of the
endoplasmic reticulum membrane and the OMM, serving as physical
bridges between the two organelles. These structures are organized
through: (A) Tethering factors, including ATAD3, MFN2-MFN1/2,
MFN2-SERCA2, ITPRs-GRP75-VDACs, FUNDC1-ITPRs, BAP31-FIS1,
BAP31-TOM40, VAPB-PTPIP51, MOSPD2-PTPIP51, ORP5/8-PTPIP51,
INF2-actin-SPIRE1C, REEP1, EMC2-SLC25A46, PDZD8, RRBP1-SYNJ2BP and
FATE1-EMD; (B) promoters, such as CYPD, TESPA1, DJ-1, Sig-1R,
IRBIT/AHCYL1, EI24, WFS1-NCS1, TOM70, TGM2, mTORC2, PDK4, PACS2,
AMFR and MITOL/MARCH5; and (C) disruptors, including DISC1, TDP-43,
FUS, GSK3β and Trichoplein/TpMs. MERCs, mitochondria-endoplasmic
reticulum contact sites; OMM, outer mitochondrial membrane; IMM,
inner mitochondrial membrane; ATAD3, ATPase family AAA
Domain-containing protein 3; MFN1/2, mitofusin 1/2; SERCA2,
sarco-endoplasmic reticulum calcium Ca2+ ATPase 2;
ITPRs, inositol 1,4,5-trisphosphate receptors; GRP75, glucose
regulate protein 75; VDACs, voltage-dependent anion channels;
FUNDC1, the FUN14 domain containing 1; FIS1, mitochondrial fission
1 protein; TOM40, translocase of OMM 40; BAP31, B-cell
receptor-associated protein 31; VAPB, vesicle-associated
membrane-protein-associated protein B; PTPIP51, protein tyrosine
phosphatase-interacting protein 51; MOSPD2, motile sperm
domain-containing protein 2; ORP5/ORP8, oxysterol-binding protein
related-protein 5 and 8; INF2, inverted formin 2; SPIRE1C, spire
type actin nucleation factor 1; REEP1, receptor accessory protein
1; PDZD8, PDZ domain containing 8; SLC25A46, solute carrier family
25 member 46; SYNJ2BP, synaptojanin 2 binding protein; RRBP1,
ribosome binding protein 1; FATE1, fetal and adult testis expressed
1; EMD, Emerin; CYPD, cyclophilin D; TESPA1, thymocyte-expressed,
positive selection-associated gene 1; Sig-1R, Sigma-1 receptor;
EI24, etoposide-induced protein 2.4; WFS1, Wolfram syndrome 1;
NCS1, neuronal calcium sensor 1; TOM70, translocase of the outer
membrane 70; TGM2, transglutaminase type 2; mTORC2, mechanistic
target of rapamycin kinase 2; PDK4, pyruvate dehydrogenase kinases
4; PACS2, phosphofurin acidic cluster sorting protein 2; MITOL,
mitochondrial ubiquitin ligase; AMFR, autocrine motility factor
receptor; DISC1, disrupted-in-schizophrenia 1; TDP-43, TAR
DNA-Binding Protein-43; GSK3β, glycogen synthase kinase-3β. |
Well-characterized MERC-resident proteins include
the following: (i) Tethering factors: ATAD3 (94,96), MFN2-MFN1/2 (mitofusin 1/2)
(62), MFN2-sarco-ER calcium
Ca2+ ATPase 2 (SERCA2) (39), GRP75-ITPRs(1, 2, and 3)-VDACs
(82), mitochondrial fission 1
protein (FIS1)/translocase of OMM 40 (TOM40)-B-cell
receptor-associated protein 31 (BAP31) (81,97), VAPB-PTPIP51 (46,98), PDZ domain containing 8 (PDZD8)
(99), inverted formin 2
(INF2)-spire type actin nucleation factor 1 (SPIRE1C) (16,100), oxysterol-binding protein
related-protein 5 and 8 (ORP5/ORP8)-PTPIP51 (29), receptor accessory protein 1
(REEP1) (101), synaptojanin 2
binding protein (SYNJ2BP)-ribosome binding protein 1 (RRBP1)
(92), motile sperm
domain-containing protein 2 (MOSPD2)-PTPIP51 (102), the FUN14 domain containing 1
(FUNDC1)-ITPRs (103), fetal
and adult testis expressed 1 (FATE1)-Emerin (EMD) (95), EMC2-solute carrier family 25
member 46 (SLC25A46) (40); (ii)
Promoters: phosphofurin acidic cluster sorting protein 2 (PACS2)
(25), cyclophilin D (CYPD)
(104), Sigma-1 receptor
(Sig-1R) (105),
thymocyte-expressed, positive selection-associated gene 1 (TESPA1)
(106), autocrine motility
factor receptor (AMFR) (107-109), Caveolin 1 (CAV1) (110), dynamin-related protein 1 (DRP1)
(111,112), IRBIT/adenosylhomocysteinase
like 1 (AHCYL1) (113),
membrane associated ring-ch-type finger 5 (MARCH5)/mitochondrial
ubiquitin ligase (MITOL) (114), mitochondrial Rho GTPase 1
(MIRO1) (115,116), mechanistic target of rapamycin
kinase 2 (mTORC2) (117),
reticulon 1C (RTN-1C) (118),
translocase of the outer membrane 70 (TOM70) (119), transglutaminase type 2 (TGM2)
(120), Wolfram syndrome 1
(WFS1) (121), pyruvate
dehydrogenase kinases 4 (PDK4) (122), etoposide-induced protein 2.4
(EI24) (123,124), DJ-1 (125), DsbA-L (35), Lonp1 (41); (iii) Disruptors:
disrupted-in-schizophrenia 1 (DISC1) (126,127), FUS (128), glycogen synthase kinase-3β
(GSK3β) (129,130), TAR DNA-Binding Protein-43
(TDP-43) (98), Trichoplein/TpMs
(131).
Notably, the precise role of MFN2 in MERCs remains a
topic of active research and debate. Although early studies
recognized MFN2 as a crucial tether that physically connects the
two organelles (62), later work
suggested that MFN2 might instead regulate the inter-organellar
distance. Paradoxically, its ablation can increase ER-mitochondria
coupling in specific contexts (54). These inconsistent findings likely
arise from cell-type-specific compensatory mechanisms and the
pleiotropic functions of MFN2 in mitochondrial fusion, underscoring
the need for careful interpretation of MERC phenotypes in genetic
models. The functional roles of the MERC-resident proteins
discussed in the present review are summarized in Table I.
 | Table ISummary of the functional roles of
MERCs-resident proteins. |
Table I
Summary of the functional roles of
MERCs-resident proteins.
| Authors, year | Protein | Mitochondria-ER
contact role | Relevant
interactions | Relevant
functions | (Refs.) |
|---|
| Issop et al,
2015 | ATAD3 | Tethering
factor | Interacts with
DRP1 | Facilitates optimal
cholesterol transfer from the endoplasmic reticulum to mitochondria
for steroidogenesis. | (96) |
| Yang et al,
2023; de Brito et al, 2008 | MFN2 | Tethering
factor | Interacts with
MFN1/2, SERCA2 | Mediates
ER-mitochondria tethering, a prerequisite for efficient
mitochondrial Ca2+ uptake. Enhances SERCA2-mediated
Ca2+ reuptake into the ER, thereby preventing excessive
mitochondrial Ca2+ accumulation and cell death. | (39,62) |
| Yang et al,
2023 | SERCA2 | Tethering
factor | Interacts with
MFN2 | Enhances
SERCA2-mediated Ca2+ reuptake into the ER, thereby
preventing excessive mitochondrial Ca2+ accumulation and
cell death. | (39) |
| Rizzuto et
al, 1998 | ITPRs | Tethering
factor | Form complex with
GRP75 and VDACs; Interacts with FUNDC1 | Participates in the
regulation of ER Ca2+ release. | (10) |
| GRP75 | Tethering
factor | Form complex with
ITPRs and VDACs | Participates in the
regulation of ER Ca2+ release. | |
| VDACs | Tethering
factor | Form complex with
ITPRs and GRP75 | Participates in the
regulation of ER Ca2+ release. | |
| Wu et al,
2017 | FUNDC1 | Tethering
factor | Interacts with
ITPRs | Binds ITPR2 to
modulate Ca2+ release from the ER into the cytosol and
mitochondria. | (103) |
| Iwasawa et
al, 2011; Namba et al, 2019 | BAP31 | Tethering
factor | Interacts with
FIS1, and TOM40 | The FIS1-BAP31
complex serves as a platform for apoptosis signaling. The
BAP31-TOM40 complex regulates oxygen consumption and mitochondrial
complex I activity. | (81,97) |
| Iwasawa et
al, 2011 | FIS1 | Tethering
factor | Interacts with
BAP31 | The FIS1-BAP31
complex serves as a platform for apoptosis signaling. | (81) |
| Namba et al,
2019 | TOM40 | Tethering
factor | Interacts with
BAP31 | The BAP31-TOM40
complex regulates oxygen consumption and mitochondrial complex I
activity. | (97) |
| De Vos et
al, 2012 | VAPB | Tethering
factor | Interacts with
PTPIP51 | VAPBP56S enhances
Ca2+ uptake and modifies PTPIP51 binding. | (80) |
| De Vos et
al, 2012; Di Mattia et al, 2018; Galmes et al,
2016 | PTPIP51 | Tethering
factor | Interacts with
VAPB, MOSPD2, and OPR5/8 | VAPB-P56S enhances
mitochondrial Ca2+ uptake and alters PTPIP51 binding
affinity. Tethers the ER and other organelles. ORP5 and ORP8
require a functional lipid-binding/transfer ORD domain to interact
with mitochondrial PTPIP51. | (29,80,102) |
| Di Mattia et
al, 2018 | MOSPD2 | Tethering
factor | Interacts with
PTPIP51 | Tethers the ER and
other organelles. | (102) |
| Galmes et
al, 2016 | OPR5/8 | Tethering
factor | Interacts with
PTPIP51 | ORP5 and ORP8
require a functional lipid-binding/transfer ORD domain to interact
with mitochondrial PTPIP51. | (29) |
| Manor et al,
2015 | INF2 | Tethering
factor | Interacts with
SPIRE1C | Spire1C promotes
actin assembly on mitochondrial surfaces through interaction with
INF2. | (100) |
| SPIRE1C | Tethering
factor | Interacts with
INF2 | Spire1C promotes
actin assembly on mitochondrial surfaces through interaction with
INF2. | |
| Lim et al,
2015 | REEP1 | Tethering
factor | | REEP1 facilitates
ER-mitochondria interactions. | (101) |
| Hirabayashi et
al, 2017 | PDZD8 | Tethering
factor | | PDZD8 tethers
mitochondria to the ER, regulating dendritic Ca2+
dynamics. | (99) |
| Hung et al,
2017 | RRBP1 | Tethering
factor | Interacts with
SYNJ2BP | A companion of
SYNJ2BP for ERM binding was discovered as RRBP1
byimmunoprecipitationmass spectrometry. | (92) |
| SYNJ2BP | Tethering
factor | Interacts with
INF2 | RRBP1 was
identified as a binding partner of SYNJ2BP in ER-mitochondria
membrane (ERM) contact sites via immunoprecipitation-mass
spectrometry. | |
| Dong et al,
2024 | EMC2 | Tethering
factor | Interacts with
SLC25A46 | The
EMC2-SLC25A46-Mic19 axis represents a pathway regulating
ER-mitochondria interactions. | (40) |
| SLC25A46 | Tethering
factor | Interacts with
EMC2 | One route
controlling ER-mitochondria interactions is the EMC2-SLC25A46-Mic19
axis. | |
| Doghman-Bouguerra
et al, 2016 | FATE1 | Tethering
factor | Interacts with
EMD | FATE1-mediated
uncoupling of ER-mitochondria may protect against apoptosis. | (95) |
| EMD | Tethering
factor | Interacts with
FATE1 | FATE1-mediated
uncoupling of ER-mitochondria may protect against apoptosis. | |
| Paillard et
al, 2013 | CYPD | Promoter | Interacts with
VDAC1/GRP75/ITPR1 complex | Modulates
mitochondrial Ca2+ overload through the
CYPD/VDAC1/GRP75/ITPR1 complex. | (104) |
| Matsuzaki et
al, 2013 | TESPA1 | Promoter | Interacts with
VDAC1/GRP75/ITPR3 complex | TESPA1 regulates
ITPR3-mediated Ca2+ release and mitochondrial
Ca2+ uptake by directly interacting with GRP75, but not
VDAC1. | (106) |
| Liu et al,
2019 | DJ-1 | Promoter | Interacts with
VDAC1/GRP75/ITPR3 complex | Contributes to the
regulation of Ca2+ crosstalk and structural integrity
between the ER and mitochondria. | (125) |
| Hayashi et
al, 2007 | Sig-1R | Promoter | Interacts with
VDAC/GRP75/ITPR3 complex | Plays a role in
controlling cell viability and inter-organellar ER-mitochondrial
Ca2+ signaling. | (105) |
| Bonneaue et
al, 2016 | IRBIT | Promoter | Interacts with
VDAC/GRP75/ITPR complex | Regulates Bcl2l10
activity and ER-mitochondria coupling. | (113) |
| Yuan et al,
2020 | EI24 | Promoter | Interacts with
VDACs/GRP75/ITPRs complex | Promotes
ER-to-mitochondria Ca2+ flux and enhances DNA
damage-induced apoptosis by interacting with VDAC2 to facilitate
ER-mitochondria contact formation. | (123,124) |
| Angebault et
al, 2018 | WFS1 | Promoter | Interacts with
VDAC1/GRP75/ITPR1 complex | Forms a complex
with NCS1 and ITPR1 to activate ER-mitochondria Ca2+
crosstalk, thereby stimulating mitochondrial respiratory chain
activity and tricarboxylic acid (TCA) cycle function. | (121) |
| Filadi et
al, 2018 | TOM70 | Promoter | Interacts with
VDAC1/GRP75/ITPR3 complex | Interacts with
ITPR3 to regulate mitochondrial respiration, influencing autophagy,
proliferation, and cellular bioenergetics. | (119) |
| D'Eletto et
al, 2018 | TGM2 | Promoter | Interacts with
VDAC1/GRP75/ITPR3 complex | Interacts with
GRP75 to modulate Ca2+ exchange between the ER and
mitochondria. | (120) |
| Betz et al,
2013 | mTORC2 | Promoter | Interacts with
VDAC1/GRP75/ITPR2 complex | Regulates
mitochondrial function and MERC integrity by Akt-mediated
phosphorylation of MERC-associated ITPR2. | (117) |
| Thoudam et
al, 2019 | PDK4 | Promoter | Interacts with
VDAC1/GRP75/ITPR1 complex | Stabilizes the
ITPR1-GRP75-VDAC1 complex, which plays a role in regulating ER
stress, mitochondrial dysfunction, and mitochondrial
Ca2+ accumulation. | (122) |
| Simmen et
al, 2005 | PACS2 | Promoter | Interacts with
BAP31/FIS1 complex | Contributes to the
control of apoptosis, ER homeostasis, and Ca2+ exchange
between the ER and mitochondria. | (25) |
| Wang et al,
2015 | AMFR | Promoter | Interacts with
MFN2 | Maintains MERC
integrity. | (107) |
| Sugiura et
al, 2013 | MITOL | Promoter | Interacts with
MFN2 | MITOL is required
for GTP-dependent MFN2 oligomerization. | (114) |
| Sala-Vila et
al, 2016 | CAV1 | Promoter | | CAV1 is an
essential component of hepatic MERCs. | (110) |
| Prudent et
al, 2015 | DRP1 | Promoter | | SUMOylated DRP1
functionally stabilizes ER-mitochondrial interactions. | (111) |
| Macaskill et
al, 2009 | MIRO1 | Promoter | | Links KIF5 motor
proteins to mitochondria. | (116) |
| Reali et al,
2015 | RTN-1C | Promoter | | Regulates lipid
exchange between the ER and mitochondria and maintains
intracellular Ca2+ homeostasis by modulating
mitochondrial morphology and function. | (118) |
| Yang et al,
2019 | DsbA-L | Promoter | | Preserves MERC
integrity to exert antiapoptotic effects. | (35) |
| Li et al,
2023 | Lonp1 | Promoter | | Controls the
unfolded protein response in the ER, mitochondrial fission, and
MERC stability. | (41) |
| Park et al,
2017 | DISC1 | Disruptor | Interacts with
ITPR1 | Participates in the
regulation of Ca2+ dynamics between mitochondria and the
ER. | (127) |
| Stoica et
al, 2014 | TDP-43 | Disruptor | Interacts with
VAPB-PTPIP51 complex | TDP-43,
pathologically linked to frontotemporal dementia and amyotrophic
lateral sclerosis, disrupts ER-mitochondria contacts, impairs the
VAPB-PTPIP51 interaction, and perturbs cellular Ca2+
homeostasis. | (98) |
| Stoica et
al, 2016 | FUS | Disruptor | Interacts with
VAPB-PTPIP51 complex | FUS disrupts
ER-mitochondria connections and the VAPB-PTPIP51 interaction,
leading to altered Ca2+ uptake. | (128) |
| Stoica et
al, 2014 | GSK3β | Disruptor | Interacts with
VAPB-PTPIP51 complex | Regulates the
VAPB-PTPIP51 interaction. | (98) |
| Cerqua et
al, 2010 | TpMs | Disruptor | Interacts with
MFN2 | Induces apoptosis
inhibition, mitochondrial fragmentation, and loss of ER
anchoring. | (131) |
In summary, the composition of MERCs is highly
complex and dynamic, involving a sophisticated network of proteins
that can be functionally classified as tethers, promoters, or
disruptors. This comprehensive list emphasizes that MERCs are not
formed by a single, universal mechanism but are organized through a
modular and likely cell-type-specific array of molecular complexes.
These components jointly regulate the physical distance, stability
and functional output of the contact sites. Beyond simple
structural bridging, MERC-resident proteins integrate core cellular
processes such as calcium signaling, lipid transfer, apoptosis
regulation, mitochondrial dynamics and metabolic adaptation,
establishing MERCs as central signaling hubs.
Important physiological roles of MERCs
MERCs participate in multiple key physiological
processes, including Ca2+ homeostasis, Zn2+
homeostasis, ER stress responses, redox signaling, autophagy,
mitochondrial dynamics, apoptosis, inflammation, lipid metabolism
and cellular senescence (Figs 3,
4, 5). These diverse functions can be
categorized into three interconnected tiers. The first tier
consists of ion signaling, specifically Ca2+ and
Zn2+ flux, which functions as the most rapid and direct
form of inter-organellar communication at MERCs. The second tier
includes organelle quality control mechanisms, such as
mitochondrial dynamics (fission, fusion and mitophagy) and
autophagy, which are regulated by MERC-localized ion signals and
lipid intermediates. The third tier involves cell fate decisions,
namely apoptosis, inflammation and cellular senescence, which
represent long-term outcomes triggered when MERC-regulated stress
signals surpass homeostatic thresholds. These three tiers are
underpinned by the fundamental activities of lipid
biosynthesis/transfer and redox signaling, which both modulate and
are modulated by the ion flux and quality control mechanisms.
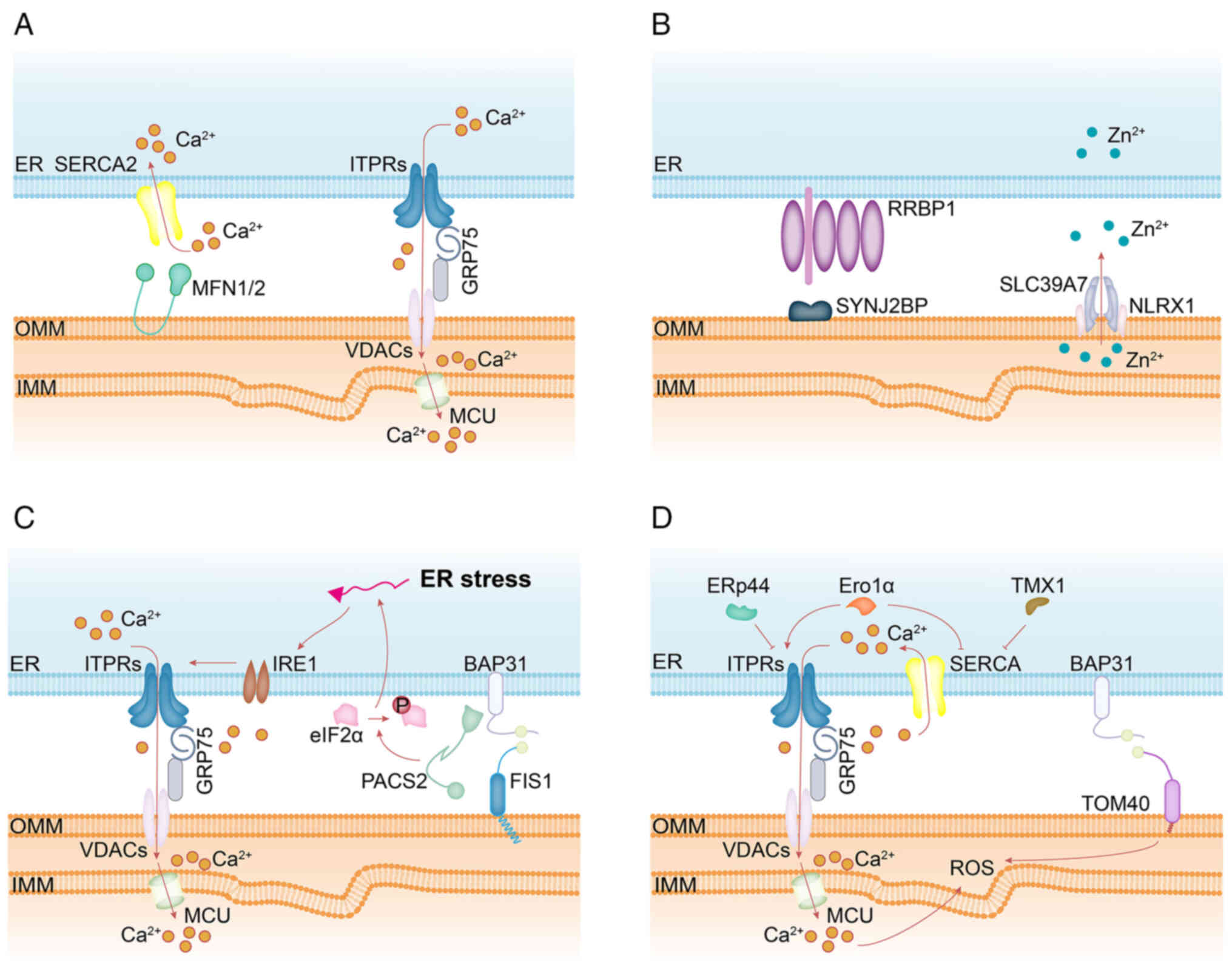 | Figure 3Schematic summary of MERC-associated
proteins involved in: (A) Ca2+ homeostasis-calcium is
transferred from the ER to the outer mitochondrial membrane via the
ITPR-GRP75-VDAC complex, promoting mitochondrial activity; MFN2
facilitates ER Ca2+ reuptake through SERCA2 at MERCs.
(B) Zn2+ homeostasis-SYNJ2BP interacts with RRBP1 to
support MERC integrity and promotes the interaction between NLRX1
and SLC39A7, thereby regulating mitochondrial Zn2+
dynamics. (C) ER stress-the ER stress sensor IRE1, localized at
MERCs, enhances mitochondrial Ca2+ uptake by activating
the ITPR-VDAC axis; depletion of PACS2 leads to a transient
increase in phosphorylated eIF2α, indicating induction of ER
stress. (D) redox signaling control-MERC-localized ROS are
predicted to trigger ITPR activation and SERCA inactivation,
amplifying Ca2+ transfer; ERp44 and Ero1α modulate ER
Ca2+ release through regulation of the ITPR-VDAC axis;
TMX1 and Ero1α inhibit SERCA activity, reducing cytosolic
Ca2+ clearance; loss of BAP31 suppresses mitochondrial
oxidative phosphorylation, although whether this depends on its
interaction with TOM40 remains unclear due to incomplete
understanding of BAP31's targeting mechanism to MERCs. ER,
endoplasmic reticulum; MERCs, mitochondria-ER contact sites; MFN2,
mitofusin 2; SERCA2, sarco-endoplasmic reticulum calcium
Ca2+ ATPase 2; ITPRs, inositol 1,4,5-trisphosphate
receptors; GRP75, glucose regulate protein 75; VDACs,
voltage-dependent anion channels; TOM40, translocase of outer
mitochondrial membrane 40; BAP31, B-cell receptor-associated
protein 31; SYNJ2BP, synaptojanin 2 binding protein; RRBP1,
ribosome binding protein 1; PACS2, phosphofurin acidic cluster
sorting protein 2; IRE1, inositol-requiring enzyme 1; ERp44, ER
protein 44; Ero1α, ER oxidoreductase 1 alpha; TMX1, thioredoxin
related transmembrane protein 1; SLC39A7, solute carrier family 39
member 7; NLRX1, NLR family member X1; ROS, reactive oxygen
species. |
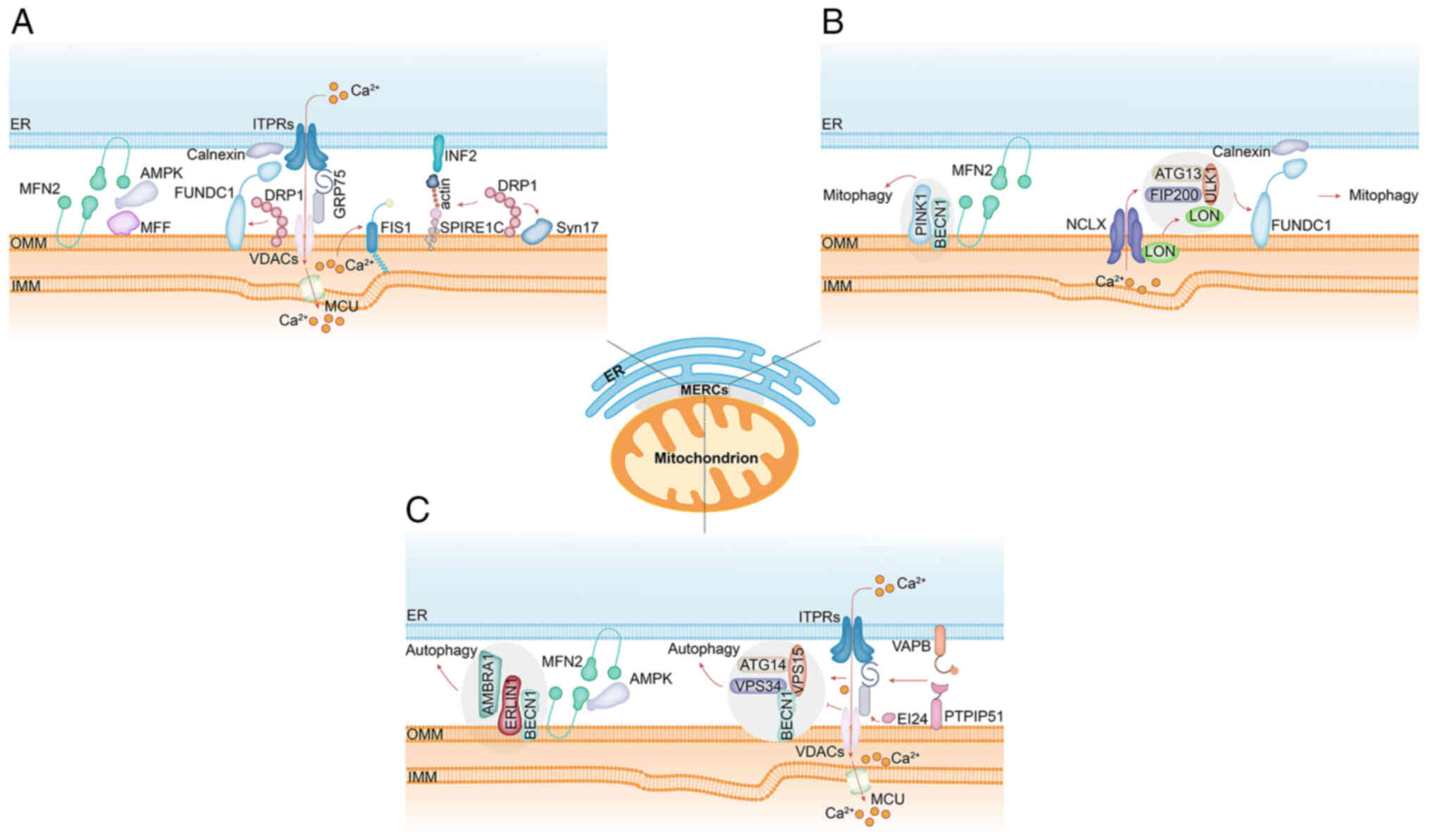 | Figure 4Schematic summary of MERC-associated
proteins involved in: (A) Mitochondrial fission-under energy
stress, the interaction between MFN2 and AMPK is reduced; this may
release active AMPK to phosphorylate MFF, thereby regulating
mitochondrial division. FUNDC1 accumulates at MERCs through
interaction with the ER-resident protein calnexin and acts as a
novel mitochondrial receptor for DRP1 to initiate fission under
hypoxic conditions. Spire1C promotes actin assembly on
mitochondrial surfaces by binding INF2; disruption of its actin- or
formin-binding activities impairs mitochondrial constriction and
division. MERCs contain raft-like microdomains enriched with
syntaxin 17 (Syn17), which regulates DRP1 localization and activity
to promote mitochondrial fission. (B) Mitophagy-during autophagy,
PINK1 and Beclin 1 (BECN1) relocalize to MERCs, facilitating
autophagosome initiation site formation and enhancing
ER-mitochondrial interface functionality. Under hypoxic conditions,
stress-induced mitochondrial protease Lon accumulates at MERCs and
functions as a molecular chaperone by stabilizing the FUNDC1-ULK1
complex in an NCLX-dependent manner, thereby triggering mitophagy.
(C) Autophagy-upon energy stress, substantial levels of AMPK
translocate from the cytosol to mitochondria and MERCs, where it
directly interacts with MFN2 to promote mitochondrial fission. At
MERC raft-like microdomains, ERLIN1 interacts with AMBRA1 and
recruits it to the BECN1 complex, a critical step for autophagosome
formation. Core components of the autophagy initiation
complex-including BECN1, ATG14, VPS15 and VPS34-localize to MERCs.
Overexpression of VAPB or PTPIP51 inhibits autophagosome
biogenesis. The C-terminal domain of EI24 is essential for both
MERC integrity and autophagic flux, and it interacts with the
ITPR-VDAC axis. ER, endoplasmic reticulum; MERCs, mitochondria-ER
contact sites; MFN2, mitofusin 2; MFF, mitochondrial fission
factor; ITPRs, inositol 1,4,5-trisphosphate receptors; VDACs,
voltage-dependent anion channels; FUNDC1, the FUN14 domain
containing 1; VAPB, vesicle-associated membrane-protein-associated
protein B; PTPIP51, protein tyrosine phosphatase-interacting
protein 51; INF2, inverted formin 2; SPIRE1C, spire type actin
nucleation factor 1; EI24, etoposide-induced protein 2.4; DRP1,
dynamin-related protein 1; PINK1, PTEN-induced putative kinase 1;
BECN1, Beclin 1; ULK1, unc-51 like autophagy activating kinase 1;
ATG14, autophagy related 14; VPS34, vacuolar protein sorting 34;
VPS15, vacuolar protein sorting 15; ERLIN1, ER lipid raft
associated 1; AMBRA1, autophagy and beclin 1 regulator 1. |
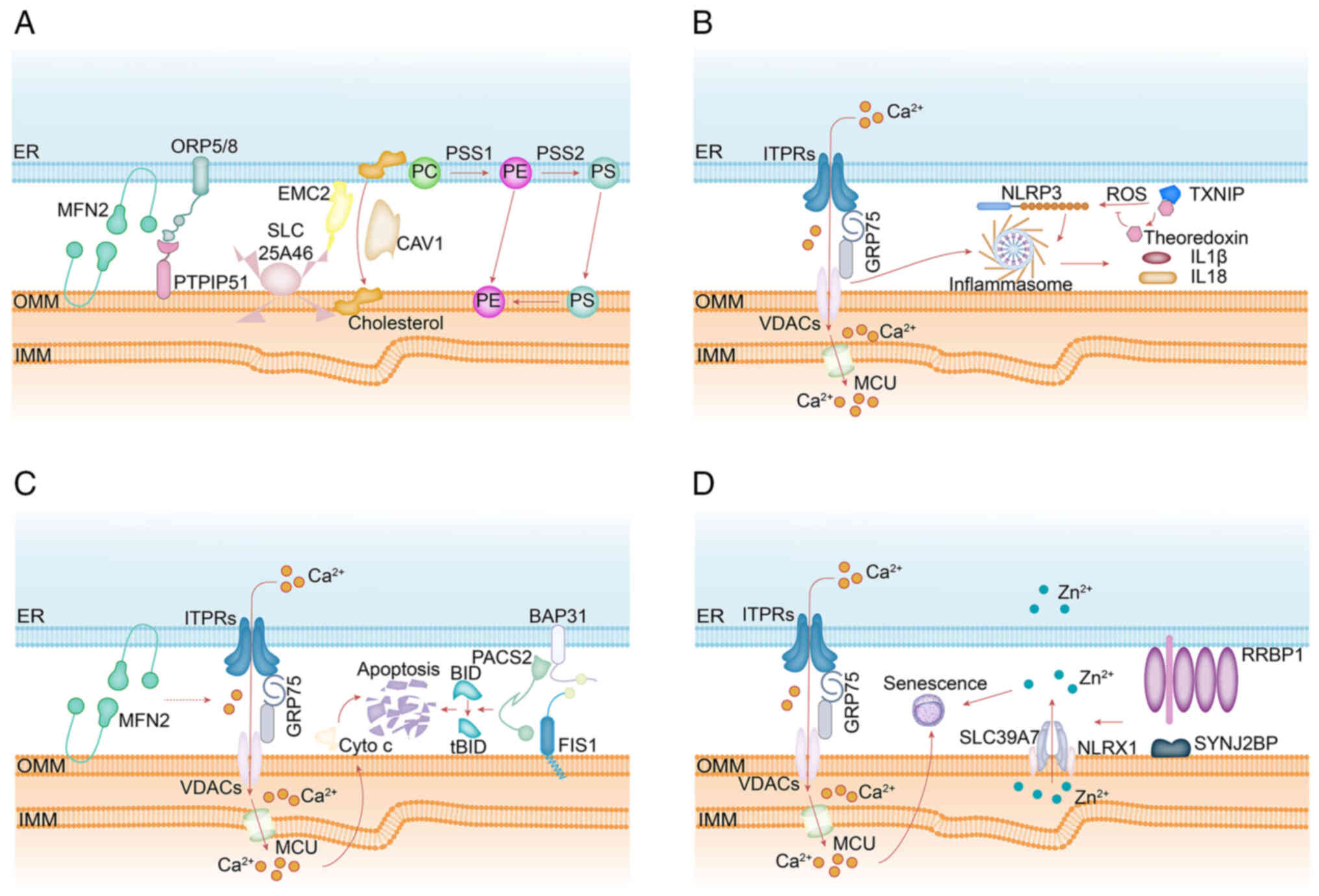 | Figure 5Schematic summary of MERC-associated
proteins involved in: (A) Lipid metabolism-ORP5 and ORP8, ER
membrane proteins, together with PTPIP51 localized on the
mitochondrial membrane, mediate PS transport to mitochondria for PE
synthesis. CAV1 regulates cholesterol transfer from the ER to
mitochondria to support lipid biosynthesis. PSS1 catalyzes the
conversion of PC to PS, while PSS2 facilitates the reverse
conversion of PE to PS. The SLC25A46-EMC2 axis may regulate
disruptions in mitochondrial phospholipid metabolism, including
reduced cardiolipin synthesis. MFN2 specifically binds PS and
mediates its transport into mitochondria. (B) Inflammation-NLRP3
localizes to the ER membrane in its inactive state; upon
activation, both NLRP3 and its adaptor protein ASC redistribute to
the ER-mitochondria interface. Inhibition of VDAC disrupts
mitochondrial function and suppresses inflammasome activation.
ROS-induced dissociation of TXNIP from thioredoxin promotes
formation of TXNIP-NLRP3 complexes, which translocate to MERCs and
facilitate inflammasome assembly and activation. (C)
Apoptosis-PACS2 facilitates the translocation of Bid to
mitochondria, leading to cytochrome c release and generation of
tBid, thereby initiating the intrinsic apoptotic cascade and
resulting in cell death. Following hypoxic injury, elevated levels
of ITPR2-enriched at MERCs-cause mitochondrial Ca2+
overload, which triggers photoreceptor apoptosis. (D) Cellular
senescence-calcium transfer to mitochondria via the ITPR-VDAC
complex induces premature senescence. ER, endoplasmic reticulum;
MERCs, mitochondria-ER contact sites; PS, phosphatidylserine; PE,
phosphatidylethanolamine; CAV-1, caveolin-1; PC,
phosphatidylcholine; tBid, truncated Bid; SYNJ2BP, a
senescence-promoting factor, interacts with RRBP1 to maintain
mitochondrial Zn2+ homeostasis and enhance the
NLRX1-SLC39A7 interaction. MFN2, mitofusin 2; SLC25A46, solute
carrier family 25 member 46; ITPRs, inositol 1,4,5-trisphosphate
receptors; GRP75, glucose regulate protein 75; VDACs,
voltage-dependent anion channels; PTPIP51, protein tyrosine
phosphatase-interacting protein 51; ORP5/ORP8, oxysterol-binding
protein related-protein 5 and 8; SYNJ2BP, synaptojanin 2 binding
protein; RRBP1, ribosome binding protein 1; PACS2, phosphofurin
acidic cluster sorting protein 2; PSS1, phosphatidylserine
synthase1; NLRP3, NOD-like receptor family, pyrin domain-containing
protein 3, TXNIP, tioredoxin-interacting protein; OMM, outer
mitochondrial membrane; IMM, inner mitochondrial membrane. |
MERCs and Ca2+ homeostasis
As a key intracellular messenger, Ca2+
regulates cellular functions such as gene expression, muscle
contraction and cell proliferation (132). Numerous proteins localized at
MERCs modulate Ca2+ dynamics. Among these, the inositol
1,4,5-trisphosphate receptor (ITPR) is a major ER-resident
Ca2+ release channel. It mediates the release of
concentrated Ca2+ from the ER lumen into the cytosol.
Upon binding of IP3 to ITPR, the channel is activated,
triggering pore opening and enabling Ca2+ efflux from
the ER into the cytoplasm (133). Additionally, VDAC1, located on
the OMM, has been shown to regulate mitochondrial Ca2+
uptake (134,135). Through GRP75, ITPRs interact
with VDAC1 to form the ITPR-GRP75-VDAC1 complex, which plays a
central role in mediating Ca2+ exchange between the ER
and mitochondria and reflects the coordinated molecular mechanisms
operating within MERCs (136).
After passage through VDAC1 in the OMM, Ca2+ enters the
mitochondrial matrix via the mitochondrial calcium uniporter (MCU)
at the inner mitochondrial membrane (IMM), a process regulated by
additional auxiliary factors (137). Through activation of ITPR-VDAC1
channels at specialized MERCs, the ER can directly stimulate
mitochondrial enzymatic activity (138). Alternatively, the ER indirectly
influences mitochondrial function by modulating cytoplasmic
Ca2+ levels, thereby affecting MMP via SERCA pumps on
the ER membrane (139).
According to Yang et al (39), the MFN2-SERCA2 interaction exerts
dual regulatory effects on inter-organellar Ca2+
transfer. On the one hand, the physical proximity facilitated by
this interaction enhances ER-mitochondria coupling and promotes
mitochondrial Ca2+ influx. On the other hand, MFN2
supports SERCA2-mediated Ca2+ reuptake into the ER at
contact sites, potentially preventing excessive mitochondrial
Ca2+ accumulation and thereby preserving both
mitochondrial Ca2+ homeostasis and cell survival
(39).
By facilitating the formation of specialized
microdomains characterized by high Ca2+ flux efficiency,
MERCs enable the rapid and precise transfer of Ca2+ from
the ER lumen directly to the mitochondrial matrix via coordinated
protein complexes such as ITPR-GRP75-VDAC and the downstream MCU.
This direct coupling supports key mitochondrial metabolic processes
but is also precisely regulated by interactions such as
MFN2-SERCA2, which can adjust the transfer efficiency to avoid
cytotoxic Ca2+ overload. The structural and functional
integrity of MERCs is therefore essential for maintaining calcium
homeostasis, directly connecting organelle communication to
fundamental cellular processes and survival. The highly organized
spatial arrangement of these Ca2+ handling complexes
defines MERCs as privileged signaling microdomains. Importantly,
this Ca2+ flux does not function in isolation; it serves
as the central mediator of MERC communication, interpreted in a
context-specific manner. The amplitude, frequency and spatial
localization of mitochondrial Ca2+ transients are
decoded by Ca2+-sensitive dehydrogenases in the TCA
cycle to align ATP production with cellular demand. Meanwhile,
sustained increases in matrix Ca2+ reduce the threshold
for permeability transition pore opening, directly connecting
metabolic control to cell fate decisions (140,141).
MERCs and Zn2+ homeostasis
Like Ca2+, Zn2+ acts as a
second messenger, and its precise spatiotemporal control is crucial
for cellular function. Intriguingly, numerous of the same MERC
platforms that coordinate Ca2+ transfer also participate
in Zn2+ regulation. Zinc, an essential trace element
with diverse physiological functions, plays a key role in
regulating various cellular signaling pathways, including
Zn2+ signaling (142). Previous studies have shown that
intracellular free Zn2+, similar to elevated cytosolic
Ca2+, acts as a signaling ion by modulating second
messenger metabolism, protein activation and phosphorylation, and
signal transduction processes (143,144). Cellular Zn2+ levels
must be tightly regulated, as both deficiency and excess can impair
cell function.
Two families of Zn2+ transporters mediate
Zn2+ flux across cellular and organellar membranes: Zrt-
and Irt-like proteins (ZIPs/SLC39A) facilitate Zn2+
efflux from intracellular stores into the cytosol (145), whereas Zn2+
transporters (ZnTs/SLC30A) promote Zn2+ sequestration
into organelles or extrusion from the cell. ZIP7 (solute carrier
family 39 member 7, SLC39A7), one of the best-characterized ZIP
transporters, was initially identified in the early secretory
pathway, particularly within the ER and Golgi apparatus, where it
regulates Zn2+ mobilization between the cytosol and
intracellular vesicles. Recent evidence suggests that SLC39A7 may
also redistribute to or localize within mitochondria (13). Pathological dysregulation of
SLC39A7 leads to mitochondrial Zn2+ accumulation and
impaired mitophagy (146),
thereby exacerbating mitochondrial oxidative stress. Song et
al (147) showed that NLR
family member X1 (NLRX1) is required to maintain SLC39A7
localization in mitochondria. Upon activation of NLRX1 using the
agonist NX-13, they observed reduced association between SLC39A7
and NLRX1, along with diminished mitochondrial targeting of
SLC39A7. NLRX1 transitions into an active conformation to promote
mitophagy. Thus, NLRX1-mediated retention of SLC39A7 in
mitochondria is critical for preserving local MMP, and its loss at
sites of mitochondrial damage suggests a role in localized
depolarization without affecting global membrane potential
(147). This finding highlights
how fine-tuned mitochondrial adaptations, such as transient
fluctuations or 'flickering' of membrane potential, are
physiologically regulated (148,149).
Another group of Zn2+ transporters,
including SLC30A9, SLC25A25 and SLC30A5, also contributes to
MERC-dependent regulation of mitochondrial Zn2+
distribution. Their colocalization at MERCs facilitates efficient
Zn2+ transfer from the ER to mitochondria. Moreover,
certain Zn2+ transporters exhibit dynamic relocalization
among organelles, enabling spatiotemporal control of
Zn2+ concentrations. In aged cardiomyocytes, altered
expression of Zn2+ transporters in mitochondria
(increased SLC30A7 and SLC30A8, no change in SLC39A7 and SLC39A8)
and ER (increased SLC39A7, decreased SLC30A7, no change in SLC39A8
and SLC30A8) correlates with oxidative stress-induced cardiac
dysfunction (13). Recent
findings indicate that SYNJ2BP enhances the interaction between
NLRX1 and SLC39A7, thereby supporting mitochondrial Zn2+
homeostasis. As a structural component of MERCs through its binding
to RRBP1, SYNJ2BP plays a key role in maintaining contact site
integrity. Loss of SYNJ2BP expression or disruption of MERC
architecture not only reduces formation of the SLC39A7-NLRX1
complex but also prevents mitochondrial localization of SLC39A7
(14). Similar to calcium,
Zn2+ functions as a crucial signaling ion, and its
accurate compartmentalization is essential for cellular well-being.
These findings indicate that MERCs act as strategic platforms that
promote this regulation by localizing key Zn2+
transporters, such as SLC39A7/ZIP7, and their regulatory partners,
including NLRX1 and SYNJ2BP. The formation of dynamic protein
complexes at the contact interface, as exemplified by the
SYNJ2BP-RRBP1 tether that enables the SLC39A7-NLRX1 interaction,
supports efficient Zn2+ transfer and buffering between
the ER and mitochondria. This localized control is important for
maintaining MMP, alleviating oxidative stress, and supporting
processes such as mitophagy. Dysregulation of these MERC-centered
Zn2+ regulatory mechanisms, as observed in aging
cardiomyocytes, is directly associated with mitochondrial
dysfunction and tissue pathology. Collectively, the evidence
indicates that MERCs are not merely passive bridges but active,
integrative hubs that coordinate Zn2+ signaling to
preserve mitochondrial quality and cellular resilience. Notably,
disruption of MERC integrity, which impairs Zn2+
homeostasis, can simultaneously trigger ER stress, since the
protein-folding environment of the ER is highly sensitive to
alterations in both ion flux and MERC architecture. This
bidirectional relationship between MERC status and ER stress is
discussed in the following section.
MERCs and ER stress
When ER homeostasis is disrupted, misfolded or
unfolded proteins accumulate, leading to ER stress and activation
of the unfolded protein response (UPR) (150). The adaptive phase of the UPR
aims to restore cellular homeostasis by promoting ER-associated
degradation of misfolded proteins and protecting cells from damage;
however, prolonged ER stress can ultimately trigger cellular
dysfunction and apoptosis (151). Although the protective effect
of enhanced MERC formation on ER stress depends on timing, a
previous study showed that tunicamycin-induced ER stress increases
MERC formation in liver and muscle tissues (20). Specifically, during the early
stages of ER stress, transient MERC expansion enhances
Ca2+ transfer from the ER to the mitochondrial matrix,
thereby boosting mitochondrial bioenergetics and ATP production.
This increased energy supply supports efficient ER protein folding
and facilitates the clearance of misfolded proteins, contributing
to cellular resilience against ER stress. By contrast, in obese
hepatocytes, chronic enrichment of MERCs results in sustained
elevation of mitochondrial matrix Ca2+ levels,
eventually leading to Ca2+ overload, mitochondrial
dysfunction, and impaired oxidative phosphorylation (OXPHOS)
(20).
The ER stress sensor inositol-requiring enzyme 1
(IRE1), enriched at MERCs, plays a key role in regulating
mitochondrial Ca2+ uptake by mediating the splicing of
X-box binding protein 1 (XBP1) (152). XBP1 is generally considered
cytoprotective and, through transcriptional regulation, modulates
various cellular processes in a context- and cell-type-specific
manner (153). Notably, MERCs
are not merely downstream effectors of ER stress; conversely, MERC
dysfunction can itself induce ER stress by impairing
inter-organellar Ca2+ signaling (25). For instance, depletion of
phosphofurin acidic cluster sorting protein 2 (PACS2) leads to a
transient increase in phosphorylated eIF2α, followed by restoration
of ER homeostasis and normal protein synthesis rates after two days
(25). MERCs are dynamically
regulated by the UPR and, in turn, actively help determine cellular
fate during ER stress. In the initial, adaptive stage, a transient
increase in MERC formation promotes beneficial Ca2+
transfer to mitochondria, enhancing ATP production to support
protein folding and restore ER homeostasis. In chronic pathological
conditions, however, sustained MERC expansion disrupts this
delicate equilibrium, resulting in mitochondrial Ca2+
overload, dysfunction and worsening of stress. Key molecular
factors, such as the MERC-localized sensor IRE1 and its downstream
effector XBP1, are crucial for this crosstalk. Importantly, the
relationship is cyclic: MERC dysfunction can itself trigger ER
stress, as demonstrated by PACS2 depletion, creating a harmful
feedback loop. MERCs thus serve as critical decision points, where
their functional state can either resolve or propagate ER stress,
highlighting the therapeutic potential of targeting MERC dynamics
to interrupt this cycle in diseases characterized by chronic ER
stress.
MERCs and redox signaling control
A key downstream consequence of ER stress is
increased generation of reactive oxygen species (ROS), especially
at the interface between the ER and mitochondria. As described
below, MERCs function as crucial platforms for redox communication
between these two organelles. Moreover, the ROS generated during ER
stress can further regulate MERC function, establishing
bidirectional regulatory loops. ROS are normally generated at
physiological levels necessary for maintaining cellular
homeostasis. Excessive ROS production, however, can lead to
oxidative damage to DNA, lipids, and proteins (154). Under basal conditions,
mitochondria, peroxisomes, and the ER maintain low ROS levels due
to intrinsic antioxidant defense mechanisms. However, perturbations
in redox balance, particularly within the mitochondrial respiratory
chain-can significantly enhance ROS generation and trigger
mitochondrial-derived oxidative stress (155,156). The amount of ROS produced by
organelles varies depending on oxygen tension, tissue type and cell
type. Among intracellular compartments, the ER is estimated to be
the largest contributor of ROS to the cytosol, accounting for ~60%
of total cellular ROS, with mitochondria and peroxisomes each
contributing ~20%. This estimation is based on both the relative
permeability to ROS and the absolute rates of ROS production across
these three organelles (157).
Given that both the ER and mitochondria function as
major intracellular redox hubs, redox-mediated communication
between them is highly plausible. It has been proposed that
MERC-localized ROS can activate inositol 1,4,5-trisphosphate
receptors (ITPRs) (30) and
inhibit sarco/ER Ca2+-ATPase (SERCA) (31), thereby establishing a
feed-forward loop that amplifies Ca2+ transfer between
the ER and mitochondria. In reducing environments, ER protein 44
(ERp44) binds to ITPR1 to prevent ER Ca2+ release,
serving a protective role (32).
By contrast, certain oxidoreductases promote ER-mitochondrial
Ca2+ crosstalk. Thioredoxin-related transmembrane
protein 1 (TMX1) and ER oxidoreductase 1 alpha (Ero1α) are two
well-characterized examples. Ero1α enhances ER Ca2+
release by modifying ITPRs and potentially competing with ERp44
(33), while simultaneously
reducing mitochondrial Ca2+ uptake (158). Conversely, TMX1 decreases
cytosolic Ca2+ clearance by inactivating SERCA (159). Glutathione peroxidase 8 (GPx8)
has a more complex role: Although it inactivates SERCA, it also
reduces ER Ca2+ stores, resulting in diminished
ER-mitochondria Ca2+ flux (160). BAP31, a key calnexin-binding
partner localized at MERCs, interacts with the mitochondrial outer
membrane translocase TOM40. Additionally, BAP31 associates with
NDUFS4, a core subunit of mitochondrial complex I (ubiquinone
oxidoreductase), which is part of the MERC-associated proteome
(97). Loss of BAP31 suppresses
mitochondrial OXPHOS; however, whether this effect depends on its
interaction with TOM40 remains unclear, as the mechanism underlying
BAP31 targeting to MERCs has not yet been fully elucidated. These
oxidoreductases, once considered solely components of the ER
protein folding machinery, are now recognized to cooperate in
forming a redoxosome at MERCs, where they are predominantly
localized (159,160). The redoxosome is a multimeric,
multi-organellar protein complex hypothesized to facilitate or
regulate the formation and function of MERCs in a redox-sensitive
manner. As the two primary intracellular sources and targets of
ROS, the ER and mitochondria employ MERCs to promote essential
redox communication. This inter-organellar crosstalk has two key
features: First, ROS localized at MERCs can enhance
inter-organellar calcium signaling through the modulation of ITPRs
and sarco/ER calcium-ATPase (SERCA), establishing a potential
positive feedback loop. Second, MERCs contain a set of specialized
oxidoreductases, such as ERp44, TMX1, Ero1α and GPx8, which jointly
regulate calcium flux in response to the local redox environment,
fulfilling protective or regulatory functions. The emerging concept
of a 'redoxosome', a multimeric protein complex at MERCs,
encompasses this coordinated activity, defining these contact sites
as highly sophisticated redox-sensitive centers. Moreover, the
interaction between MERC proteins, such as BAP31, and mitochondrial
complex I subunits directly links contact site integrity to the
regulation of OXPHOS. Therefore, MERCs are not merely passive
bystanders but active integrators that convert redox fluctuations
into precisely calibrated functional outputs, safeguarding cellular
homeostasis and highlighting how their malfunction could propagate
oxidative stress in diseases.
MERCs and mitochondrial dynamics
The redox environment at MERCs not only modulates
ion flux but also directly affects mitochondrial morphology. As
described below, the sites of mitochondrial fission are physically
determined by contacts between the ER and mitochondria, and local
redox conditions can regulate the recruitment and activity of the
fission machinery. Mitochondria are double-membrane-bound
organelles that regulate cellular metabolism and cell function.
Mitochondrial dynamics include mitophagy, motility, fusion and
fission (161). Mitochondrial
fission is a multistep process by which one mitochondrion divides
into two daughter mitochondria (162). This division predominantly
occurs at sites where the OMM undergoes constriction, driven by
actin polymerization or physical contact with the ER. During this
process, several OMM adaptor proteins-including mitochondrial
fission 1 protein (FIS1), mitochondrial fission factor (MFF),
mitochondrial dynamics proteins MiD49 and MiD51-recruit the
cytosolic GTPase dynamin-related protein 1 (Drp1) to the
mitochondrial surface (163,164). Subsequently, the
mitochondrial-specific phospholipid cardiolipin (CL) activates
highly oligomerized DRP1, promoting the formation of large helical
structures and enhancing GTPase activity at fission sites.
Nucleotide-induced allosteric regulation enables DRP1 to
self-assemble, undergo conformational changes, and disassemble,
thereby wrapping around the mitochondrion to mediate membrane
scission.
Notably, recent studies have shown that MERCs form
prior to Drp1 recruitment and mark the initial sites of
mitochondrial constriction. This finding indicates that MERCs
actively participate in the early stages of mitochondrial fission
by defining division sites. Drp1 is then recruited and assembled at
these pre-constricted locations (15). Further research has demonstrated
that the initial mitochondrial constriction and division can be
initiated by actin polymerization at MERCs, which is triggered by
ER-localized inverted formin 2 (INF2) (16,165). On the one hand, actin
polymerization induces OMM constriction by narrowing the
mitochondrial tubule to a diameter compatible with DRP1
oligomerization. On the other hand, spire type actin nucleation
factor 1C (Spire1C) localizes to mitochondria and establishes
direct links between the ER and the actin cytoskeleton via
mitochondrial anchoring. Spire1C promotes actin assembly on
mitochondrial surfaces through interaction with INF2. Disruption of
Spire1C's actin- or formin-binding activities reduces mitochondrial
division and constriction (100). In addition to actin, several
other proteins regulate DRP1 activity at MERCs. Syntaxin 17
(Syn17), a SNARE protein localized to raft-like microdomains within
MERCs, controls the localization and function of DRP1 in
nutrient-replete conditions, thereby stimulating mitochondrial
fission. This regulatory mechanism depends on the C-terminal
hydrophobic hairpin domain of Syn17 (166). Another MERC-associated protein,
FUNDC1, regulates hypoxia-induced mitochondrial dynamics. By
interacting with the ER-resident chaperone calnexin, FUNDC1
accumulates at MERCs and acts as a novel mitochondrial receptor for
DRP1, initiating fission under hypoxic conditions (167). Additional studies revealed that
ablation of FUNDC1 reduces intracellular Ca2+ levels,
leading to decreased binding of the cAMP response element-binding
protein (CREB) to the FIS1 promoter, which in turn suppresses
mitochondrial fission and FIS1 expression (103). Hu et al (168) found that while a subset of
AMP-activated protein kinase (AMPK) binds to MFN2 under basal
conditions, this interaction decreases during energy stress.
Intriguingly, the released active AMPK may phosphorylate MFF,
thereby regulating mitochondrial fission (168). Collectively, MERCs play a
crucial role in both the initiation and completion of mitochondrial
fission by determining division sites and facilitating fission
through multiple coordinated molecular pathways.
Mitochondrial fusion is a multistep process
involving several key proteins. First, dynamin-related GTPases such
as mitofusin 1 and 2 (MFN1/2) on the OMM, FAM73a/FAM73b, and optic
atrophy protein 1 (OPA1) on the IMM are activated. GTP hydrolysis
then drives OMM fusion, followed by IMM fusion, and finally the
mixing of intramitochondrial contents (169-171). Mitochondrial fusion contributes
to the maintenance of mitochondrial homogeneity and functional
stability by diluting mutant mtDNA and damaged proteins through
mixing with healthy matrix components. Genetic knockout of MFN1
and/or MFN2 disrupts mitochondrial architecture, leading to severe
cellular defects including fragmented and smaller mitochondria,
reduced MMP, impaired respiratory activity and diminished ATP
production. These abnormalities ultimately inhibit cell
proliferation (172). The human
ubiquitin ligase MITOL regulates MERC formation and selectively
ubiquitinates the mitochondrial pool of MFN2, but not the
ER-localized MFN2, thereby enhancing its fusogenic activity.
Sugiura et al (114)
identified lysine 192 (K192) in the GTPase domain of MFN2 as the
primary ubiquitination site targeted by MITOL. While both MFN1 and
MFN2 mediate OMM fusion, only MFN1 is required for OPA1-dependent
IMM fusion (173). This finding
suggests intermembrane communication during fusion events and
supports the possibility of direct interaction between MFN1 and
OPA1, a hypothesis that has become increasingly plausible given
recent insights into the topology and conformational dynamics of
MFN proteins (174). Notably,
MERCs appear to mark sites of mitochondrial fusion, providing
compelling evidence for their involvement in facilitating fusion,
although the underlying molecular mechanisms remain largely
undefined.
Mitophagy is essential for maintaining both
mitochondrial quantity and quality. It is an evolutionarily
conserved process that selectively eliminates damaged or excess
mitochondria through autophagy (175). Mitophagy involves two key
stages: First, double-membrane autophagosomes recognize and
selectively engulf impaired mitochondrial segments; second, the
fusion of autophagosomes with lysosomes forms autolysosomes, where
hydrolytic enzymes degrade the engulfed mitochondria. Two critical
regulators of mitophagy, PTEN-induced putative kinase 1 (PINK1) and
Beclin 1 (BECN1), relocalize to MERCs during autophagy, promoting
the formation of autophagosome initiation sites and enhancing
ER-mitochondrial interface functionality. Following mitophagy
induction, Parkin RBR E3 ubiquitin protein ligase (PARK2) is also
upregulated at MERCs. However, PINK1 silencing reduces BECN1
enrichment at MERCs regardless of PARK2 status, indicating a
PARK2-independent role for PINK1 in regulating mitophagy (22). The adaptor protein FUNDC1 has
been shown to play a crucial role in hypoxia-induced mitophagy by
enabling the recruitment of LC3 to the OMM. FUNDC1 accumulates at
MERCs through interaction with calnexin and subsequently binds DRP1
to promote mitochondrial fission. The fragmented mitochondria then
recruit the unc-51-like autophagy-activating kinase 1 (ULK1)
complex to initiate mitophagy (167,176). Recent findings using colon and
oral cancer cells have demonstrated that under hypoxic conditions,
the stress-induced mitochondrial protease Lon accumulates at MERCs
and acts as a molecular chaperone by stabilizing the FUNDC1-ULK1
complex in a manner dependent on the mitochondrial
Na+/Ca2+ exchanger (NCLX), thereby triggering
mitophagy (177). Rather than
acting as passive bystanders, MERCs actively organize these events
by acting as privileged platforms for the spatiotemporal assembly
of molecular machinery. They determine the initial sites for
mitochondrial division by recruiting and coordinating proteins such
as INF2, Spire1C and actin polymers to induce constriction before
DRP1-mediated scission. Simultaneously, through proteins such as
MFN2 and its regulator MITOL, MERCs promote and demarcate sites of
mitochondrial fusion, ensuring content mixing and functional
homogenization. Moreover, MERCs are crucial hubs for initiating
quality control through mitophagy, where proteins such as PINK1,
FUNDC1 and Lon protease converge to connect mitochondrial stress
sensing, fission and autophagosome formation. These processes are
highly interconnected; for example, fission promoted at MERCs often
provides substrates for subsequent mitophagy. MERCs thus emerge as
central decision-making nodes that physically and functionally
integrate the biogenesis, remodeling and turnover of mitochondria,
directly linking organelle dynamics to cellular energy status,
stress adaptation, and overall health.
MERCs and autophagy
Autophagy is a fundamental, evolutionarily
conserved cellular degradation process that eliminates various
macromolecules and subcellular components, including proteins,
lipids, nucleic acids and organelles. This lysosome-dependent
mechanism has attracted considerable attention for its critical
function in maintaining cellular homeostasis under both
physiological and pathological conditions (178). Autophagy contributes to
numerous cellular responses and functions, extending well beyond
basic clearance to include roles in metabolism, immunity and stress
adaptation. Previous studies have revealed that MERCs play a key
role in autophagosome biogenesis. Notably, core components of the
autophagy initiation complex, including Beclin 1 (BECN1),
autophagy-related protein 14 (ATG14) and vacuolar protein sorting
34 (VPS34), localize to MERCs (179,180). The two main modes of autophagy
induction are starvation-induced autophagy and rapamycin-inducible
autophagy, which occurs through inhibition of the mammalian target
of rapamycin (mTOR). Intriguingly, overexpression of VAPB or
PTPIP51 impairs rapamycin-inducible autophagy, whereas
starvation-induced autophagy remains unaffected (180). Subsequent research has linked
VAPB and PTPIP51 to ER-mitochondrial Ca2+ exchange,
clarifying their regulatory roles in autophagy. Specifically,
blockade of ITPR-mediated Ca2+ transfer from the ER to
mitochondria abolishes the inhibitory effect of VAPB and PTPIP51
overexpression on autophagy (180). By contrast, autophagy is
suppressed in primary pancreatic β cells lacking EI24. It has been
shown that EI24 interacts with VDAC1, ITPR, and the chaperone GRP75
on the OMM. Further mechanistic studies reveal that EI24 is
enriched at the ER-mitochondria interface and that both autophagic
flux and MERC integrity depend on the C-terminal domain of EI24
(123). Consistent with these
findings, energy stress induces mitochondrial fission and
autophagy, but more notably increases MERC formation, a process
mediated by AMP-activated protein kinase (AMPK). Under energy
stress, substantial levels of AMPK translocate from the cytosol to
mitochondria and MERCs, promoting mitochondrial fission. Notably,
AMPK directly interacts with MFN2 (168). More recently, Manganelli et
al (181) demonstrated that
knocking down ERLIN1 disrupts its interaction with autophagy and
beclin 1 regulator 1 (AMBRA1), preventing recruitment of AMBRA1 to
the BECN1 complex at raft-like microdomains within MERCs. This
interaction is essential for autophagosome formation during
nutrient starvation. Moreover, the structural integrity of key
proteins such as MFN2 is required for this association to occur
(181). The localization of
core autophagy machinery components, such as the BECN1-ATG14-VPS34
complex, to MERCs emphasizes these contact sites as preferential
locations for autophagosome formation. The regulatory mechanism is
multi-faceted, incorporating signals from nutrient status, energy
stress and inter-organellar communication. For example, the
VAPB-PTPIP51 tether regulates autophagy by influencing
ER-to-mitochondria Ca2+ signaling. Meanwhile, proteins
such as EI24 and AMPK directly associate MERC integrity and
mitochondrial dynamics with autophagic flux. Notably, the data
indicate that specific protein complexes at specialized MERC
subdomains, such as the ERLIN1-AMBRA1 interaction at raft-like
microdomains, are crucial for recruiting and activating the
autophagy machinery. Collectively, this evidence demonstrates that
MERCs are not simply passive structural bridges but rather dynamic
signaling hubs that physically and functionally integrate cellular
stress perception, organelle remodeling, and the autophagic
degradation pathway, thereby playing a key role in cellular
homeostasis and adaptation.
MERCs and lipid metabolism
A growing body of evidence indicates that the
regulation of phospholipid biosynthesis and intermembrane lipid
trafficking is one of the primary functions of MERCs (19). MERCs establish an aqueous
microenvironment between the ER and mitochondria that facilitates
the bidirectional, non-vesicular transport of lipids (182). Although most mitochondrial
phospholipids and their precursors are initially synthesized in the
ER, certain phospholipids, such as CL and phosphatidylethanolamine
(PE), are essential for mitochondrial respiratory function and can
be further processed or generated within mitochondria (183). Phosphatidylserine synthase
(PSS), localized at MERCs (184), catalyzes the synthesis of
phosphatidylserine (PS), a key precursor for PE production. It has
been previously shown that downregulation of PSS1 and PSS2 leads to
reduced PS levels, which alters the steady-state distribution of PS
in the ER and redirects lipid metabolism toward triacylglycerol and
diacylglycerol synthesis, ultimately promoting intracellular lipid
accumulation (185). MFN2
specifically binds PS and mediates its transfer from the ER to
mitochondria. Knockout of MFN2 results in decreased PSS1 expression
and impaired PS translocation, disrupting phospholipid synthesis
and inducing ER stress. Moreover, while PSS1 overexpression
enhances PS synthesis, it fails to restore PE production (36). Similarly, oxysterol-binding
protein-related proteins ORP5 and ORP8 localize to MERCs and
interact with OMM protein PTPIP51, the mitochondrial contact site
and cristae organizing system complex, and the mitochondrial
intermembrane bridging complex, thereby mediating PS transfer from
the ER to mitochondria. Depletion of ORP5 or ORP8 leads to defects
in mitochondrial respiratory function and abnormal mitochondrial
morphology (29,186). Cholesterol is likely
transported into mitochondria via specific MERC subdomains.
Caveolin-1 (CAV1), a MERC-associated protein, has been shown to
regulate cholesterol trafficking from the ER to mitochondria
(110). Inhibition of CAV1 is
associated with reduced physical extension and structural integrity
of MERCs, as well as abnormal intracellular accumulation of free
cholesterol (187). Recent
findings highlight the role of the Mic19-SLC25A46-EMC2 axis in
regulating ER-mitochondria interactions. Deficiency in Mic19
disrupts mitochondrial phospholipid metabolism, including
diminished CL biosynthesis, which compromises mitochondrial
membrane architecture and ultimately impairs fatty acid metabolism
(40). MERCs establish a
specialized microenvironment that enables the efficient transfer of
lipid precursors, such as PS, from their synthesis site in the ER
to mitochondria for subsequent processing into essential
phospholipids, including PE and CL. This coordinated transfer is
mediated by specific tethering and lipid-transfer proteins located
at MERCs, including MFN2, ORP5/ORP8 (which interact with PTPIP51),
and the enzyme PSS. Disruption of these components undermines lipid
homeostasis, resulting in defective mitochondrial respiration,
abnormal morphology and ER stress. Moreover, proteins such as CAV1
and the Mic19-SLC25A46-EMC2 axis highlight the role of MERCs in
cholesterol trafficking and the broader regulation of membrane
architecture and lipid metabolism. Collectively, the evidence
indicates that MERCs are not merely structural bridges but active
metabolic platforms; their integrity is crucial for maintaining
lipid balance, and their dysfunction directly contributes to
lipotoxicity and associated pathologies.
MERCs and inflammation
Upon infection or cellular stress, inflammatory
cytokines such as interleukin-1β (IL-1β) and interleukin-18 (IL-18)
are processed to their mature forms and activate the immune system.
This maturation process is mediated by inflammasomes, cytosolic
molecular platforms that assemble in response to danger signals.
Inflammasomes are classified into four main subfamilies based on
their sensor proteins: NLR family CARD domain-containing 4 (NLRC4),
NOD-like receptor family pyrin domain-containing 3 (NLRP3),
NOD-like receptor family pyrin domain-containing 1 (NLRP1), and
absent in melanoma 2 (AIM2) (188). NLRP3 is known to reside at the
ER membrane in its inactive state; upon activation, both NLRP3 and
its adaptor protein ASC relocalize to the ER-mitochondria
interface. To date, the NLRP3 inflammasome is the only inflammasome
complex shown to associate with MERCs (17). Notably, unlike other NLR family
members, NLRP3 can recognize not only pathogen-associated molecular
patterns but also danger-associated molecular patterns released by
damaged cells, including mitochondrial DNA (mtDNA), perturbed
Ca2+ signaling, and mitochondrial ROS (mtROS) (189).
Suppression of the VDAC)disrupts mitochondrial
function, leading to reduced ROS production and impaired
inflammasome activation (17).
In response to oxidative stress, the NLRP3 inflammasome
translocates to MERCs via the ROS sensor thioredoxin-interacting
protein (TXNIP). ROS inducers such as uric acid crystals trigger
the dissociation of TXNIP from thioredoxin and promote the
formation of TXNIP-NLRP3 complexes, which then translocate to MERCs
and facilitate inflammasome assembly and activation (190,191). MERCs function as a strategic
platform at which danger signals, such as mtROS, released mtDNA and
disrupted calcium signaling, converge to initiate the assembly and
activation of the inflammasome complex. The recruitment of NLRP3
and its adaptor ASC to the ER-mitochondria interface, facilitated
by the ROS-sensitive protein TXNIP, underscores MERCs as a
specialized domain for integrating metabolic and oxidative stress
signals into immune activation. Moreover, the reliance on
functional mitochondrial channels such as VDAC demonstrates how
MERC integrity and mitochondrial health directly govern
inflammatory output. Collectively, this indicates that MERCs are
not only structural connections but also dynamic signaling hubs
that enhance innate immune responses; their dysregulation may
contribute to pathological inflammation. The identified gap in
understanding how TXNIP or inflammasome activation mutually
modifies MERC composition points to an important area for future
research on the bidirectional communication between organelle
contact sites and inflammatory pathways.
MERCs and apoptosis
Apoptosis is a form of programmed cell death
executed by caspases, leading to the ordered disassembly of
cellular structures (192). In
earlier studies, Yang et al (35) demonstrated that overexpression of
disulfide bond A oxidoreductase-like protein (DsbA-L) in HK-2 cells
preserved MERC integrity and reduced apoptosis induced by high
glucose. The upregulation of FATE-1, a MERC-uncoupling protein,
partially reversed these protective effects, suggesting that
maintenance of MERC integrity exerts an antiapoptotic role
(35). PACS2, a multifunctional
sorting protein, also contributes to MERC formation and
inter-organellar communication. PACS2 specifically links
ER-mitochondria crosstalk to mitochondria-dependent apoptosis. In
response to apoptotic stimuli, PACS2 facilitates the translocation
of Bid to mitochondria, thereby initiating the intrinsic apoptotic
cascade through cytochrome c release and generation of truncated
Bid (tBid) (25).
As described in Section 'MERCs and Ca2+
homeostasis', the ITPR-GRP75-VDAC axis mediates tightly regulated
Ca2+ transfer from the ER to mitochondria at MERCs
(141). Under physiological
conditions, this Ca2+ flux supports oxidative metabolism
by activating Ca2+-dependent TCA cycle enzymes (193). However, under pathological
stress, this same machinery can drive excessive mitochondrial
Ca2+ accumulation, which sensitizes the permeability
transition pore (mPTP) to opening, resulting in the release of
proapoptotic factors including cytochrome c (140). Li et al (43) showed that MERCs are significantly
enhanced in photoreceptors following hypoxia induction, both in
vivo and in vitro (43). This increase coincided with
activation of mitochondrial apoptosis and disruption of
mitochondrial homeostasis. Following hypoxic injury, elevated
levels of ITPR2, enriched at MERCs, led to mitochondrial
Ca2+ overload, ultimately causing photoreceptor cell
death. Notably, ITPR2 knockdown reduced MERC formation and
consequently attenuated mitochondrial Ca2+ accumulation
during hypoxia, not only restoring mitochondrial morphology but
also rescuing mitochondrial function in photoreceptors (43). Another study revealed that
sevoflurane-induced MERC dysfunction is associated with MFN2
downregulation, and that enhanced ER-mitochondria coupling promotes
mitochondrial Ca2+ overload, leading to mitochondrial
permeability transition pore (mPTP) opening and neuronal apoptosis
(44).
MERCs function as a central hub where pro-survival
and pro-death signals converge, ultimately determining cell fate.
First, MERC integrity has an antiapoptotic effect, as evidenced by
proteins such as DsbA-L. Maintaining contact site stability
safeguards cells against apoptotic stimuli. Second, MERCs are
essential platforms for initiating and amplifying apoptotic
signaling. This proapoptotic function is mainly mediated through
precise regulation of mitochondrial Ca2+ uptake via
MERC-localized channels, for example, the ITPR-GRP75-VDAC complex.
Under stress conditions, heightened ER-mitochondria coupling can
result in pathogenic mitochondrial Ca2+ overload, which
triggers mPTP opening and cytochrome c release. Moreover, MERCs
facilitate the recruitment and activation of key apoptotic factors,
such as PACS2-dependent translocation of Bid to mitochondria. MERCs
thus act as a dynamic signaling switch: Their proper formation and
function are essential for cellular health, yet their
dysregulation, whether excessive coupling or disruption, can
actively drive the cell toward apoptosis. Nevertheless, the precise
molecular mechanisms by which individual MERC components govern the
critical steps of cytochrome c release and mPTP opening remain
incompletely understood, highlighting a crucial area for future
research to clarify how MERC architecture determines the commitment
to cell death.
MERCs and cellular senescence
Whereas the aforementioned MERC-mediated
Ca2+ overload acts as an acute trigger for apoptosis,
lower-intensity or chronic MERC dysfunction can, by contrast,
promote cellular senescence, a state of permanent proliferative
arrest with distinct functional consequences. Cellular senescence
can be triggered by various stressors and is characterized by a
sustained arrest in cell proliferation accompanied by the
development of a senescence-associated secretory phenotype, which
includes proinflammatory cytokines, profibrotic factors and matrix
metalloproteinases (194).
Calcium plays a crucial role in regulating numerous cellular and
molecular processes, including secretion, autophagy, migration,
proliferation and cell death (195). More recent evidence has shown
that calcium signaling regulates cellular senescence and influences
its progression (196,197).
Consistent with the central role of MERC-mediated
Ca2+ signaling aformentioned, evidence demonstrates that
calcium transfer from the ER to mitochondria via ITPR channels at
MERCs is a key driver of cellular senescence. In normal human
cells, this ITPR-dependent mitochondrial calcium accumulation
induces senescence (197,198). ITPR2 knockout (KO) mice exhibit
delayed aging phenotypes, including extended lifespan, improved
metabolic stress resistance, reduced immuno-senescence, and
decreased hepatic steatosis and fibrosis. Notably, both in
vivo and in vitro ablation of ITPR2 reduces the number
of mitochondria-ER contacts, and artificially enforced
ER-mitochondria tethering accelerates aging (27). Additionally, other studies have
shown that cyanidin 3-O-arabinoside suppresses p38-mediated VDAC1
expression, a pathway involved in MERC formation and calcium flux
through VDAC1-ITPR1 complexes. Furthermore,
dihydrotestosterone-induced MERC formation was found to increase
senescence in dermal papilla cells (199). A link between MERC-associated
genes and cellular senescence in osteoarthritis (OA) has been also
proposed by the authors. Two MERC-related genes, PTPN1 and ITPR1,
show positive correlations with senescence-associated genes
(MAP2K1, ABL1, PTEN and MAPK14) (200).
In addition to calcium regulation, intracellular
free Zn2+ has been shown to play a significant role in
cellular senescence. Recent research revealed that SYNJ2BP promotes
the NLRX1-SLC39A7 interaction and maintains mitochondrial
Zn2+ homeostasis. SYNJ2BP is essential for MERC
structural organization through its interaction with RRBP1. Loss of
SYNJ2BP expression or disruption of MERC structure not only reduces
formation of the SLC39A7-NLRX1 complex but also prevents
mitochondrial localization of SLC39A7 (14). By integrating published MERC
proteomic data with transcriptomic profiles from nucleus pulposus
cells in a tert-butyl hydroperoxide-induced degenerative model, it
was further demonstrated that SYNJ2BP downregulation is a key
pathological feature of intervertebral disc degeneration and
nucleus pulposus cell senescence, strongly associated with MERC
disruption (14). MERCs
therefore emerge as crucial signaling nodes. The dysregulation of
inter-organellar ion homeostasis, specifically calcium and zinc,
directly drives the aging process. Excessive calcium transfer
through the ITPR-VDAC axis at MERCs has been established as a key
factor in senescence, as evidenced by the significant antiaging
phenotypes observed in ITPR2 knockout models. Conversely, the
integrity of MERCs is essential for maintaining mitochondrial zinc
homeostasis via complexes such as SYNJ2BP-RRBP1 and SLC39A7-NLRX1.
Disruption of this structure accelerates senescence-associated
pathologies, such as intervertebral disc degeneration. These
findings collectively indicate that MERCs are not passive
bystanders but active regulators of cellular aging. Their enhanced,
diminished, or dysfunctional state can either induce or alleviate
senescence. The association between MERC-associated genes and
senescence markers in OA further emphasizes their broad
significance in age-related diseases. Ultimately, the evidence
presents MERCs as dynamic therapeutic targets. Modulating their
structure and function could provide a novel approach to intervene
in the fundamental processes of aging and senescence-related
disorders.
Collectively, the aforementioned physiological
functions exemplify the three-tier integration model of MERCs. Ion
fluxes and redox changes act as rapid sensing signals (Tier 1),
which are processed by organelle quality control and metabolic
mechanisms (Tier 2), ultimately determining cell fate outcomes
(Tier 3). In the following section, it is examined how
disease-associated perturbations at each of these tiers contribute
to the development of diseases.
Pathophysiology of MERCs
Before discussing individual diseases, it is useful
to define the core functional deficits arising from MERC
dysfunction and differentiate them from secondary pathological
consequences. The primary disruptions due to altered MERC biology
can be categorized into three interrelated groups: (i) Dysregulated
ion signaling, specifically abnormal ER-to-mitochondria
Ca2+ and Zn2+ transfer; (ii) Bioenergetic
failure, caused by impaired mitochondrial Ca2+-dependent
metabolism and lipid homeostasis; and (iii) defective organelle
quality control, including disrupted mitochondrial dynamics
(fission, fusion and mitophagy) and autophagy. These core defects,
in turn, trigger secondary pathological processes (such as
excessive oxidative stress, chronic inflammation and cellular
senescence) that exacerbate tissue injury and contribute to disease
progression. In the disease-specific discussions below, it is
indicated, whenever possible, which of these core deficits are the
primary drivers and which represent the downstream consequences of
MERC dysfunction.
The dual nature of MERC dysfunction offers a
conceptual framework for interpreting the diverse alterations
observed across diseases. As shown in the following subsections,
pathologies as distinct as CMT2A and DCM can both be associated
with MERC dysfunction. However, one is marked by a loss of
contacts, whereas the other entails excessive coupling (42,45). This pattern emphasizes the
context-specific nature of MERC biology and warns against
therapeutic approaches that aim to uniformly augment or diminish
MERC abundance without considering the specific disease context. It
is increasingly evident that alterations in MERC plasticity and
composition significantly contribute to the pathogenesis of certain
diseases, as supported by a growing body of evidence emphasizing
the critical role of MERCs in maintaining cellular homeostasis and
regulating cell fate (201)
(Table II). It is crucial to
note that the interpretation of altered MERC parameters in disease
contexts is complicated by several factors. First, the structural
and functional definition of MERCs lacks uniformity across studies.
Electron microscopy-based measurements of inter-organellar
distance, biochemical MAM isolation and PLA-based detection of
protein proximity capture different aspects of ER-mitochondria
association and may lead to non-concordant results. Second, MERC
composition and abundance are highly cell-type-specific. This means
that pathological changes observed in one tissue may not be
applicable to others. Third, the dynamic nature of MERCs means that
their abundance and coupling efficiency can vary over the course of
disease progression. For example, an early compensatory increase in
ER-mitochondria Ca2+ transfer may be followed by a
maladaptive loss of contact site integrity in later stages.
Therefore, the following disease-specific discussions aim to
distinguish, whenever possible, between causal mechanistic evidence
obtained from genetic or pharmacological rescue experiments and
correlative observations from patient samples or descriptive
models.
| Table IIRoles of MERCs in diseases. |
Table II
Roles of MERCs in diseases.
| Authors, year | Diseases | MERCs resident
protein | Principal
function | (Refs.) |
|---|
| Bernard-Marissal
et al, 2019 | CMT | MFN2↓ | Increases ER
stress. Alters Ca2+ homeostasis | (42) |
| Cartes-Saavedra
et al, 2021 | ADOA | ITPRs↑ | Alters
Ca2+ homeostasis | (206) |
| Stoica et
al, 2014; Stoica et al, 2016; Gomez-Suaga et al,
2022 | ALS/FTD | VAPB-PTPIP51↓ | Alters
Ca2+ homeostasis. Decreases mitochondrial ATP
production | (98,128,208) |
| Paillusson et
al, 2017 | PD | VAPB↓ | Alters
Ca2+ homeostasis. Decreases mitochondrial ATP
production | (212) |
| Lau et al,
2020 | AD | VAPB-PTPIP51↓ | NR | (218) |
| Paillusson et
al, 2017 | | Sig-1R↓ | Alters
Ca2+ homeostasis. Promotes mitochondrial
dysfunction | (212) |
| Cherubini et
al, 2020 | HD | MFN2↓ | NR | (224) |
| ITPR3↓ | NR | |
| GRP75↓ | NR | |
| DRP1↑ | Alters
Ca2+ homeostasis. Increase ROS production | |
| Yang et al,
2019 | DKD | DsbA-L↓ | Increases ER
stress. Increases apoptosis | (35) |
| Xue et al,
2021 | | PACS2↓ | Promotes
mitochondrial dysfunction | (229) |
| Chen et al,
2024 | | MFN2-VDR↓ | Inhibits
mitophagy | (230) |
| Li et al,
2023 | | DRP1↑ | Promotes
mitochondrial fission | (231) |
| Xie et al,
2022 | | VDAC1-RTN1A↑ | Increases
apoptosis. Activates inflammasome pathways | (232) |
| Xu et al,
2022 | NAFLD | Cds2↓ | Reduces lipid
metabolism | (237) |
| Dong et al,
2024 | |
EMC2-SLC25A46-Mic19↓ | Reduces lipid
metabolism. Reduces mitochondrial unfolded protein stress
response | (40) |
| Wu et al,
2017 | HF | FUNDC1↓ | Alters
Ca2+ homeostasis. Inhibits mitochondrial fission | (103) |
| LI et al,
2023 | | LonP1↓ | Promotes
mitochondrial fission. Activates ER unfolded protein stress
response | (41) |
| Wu et al,
2019; Salin Raj et al, 2023 | DCM | FUNDC1↑ PACS2↑
VDAC1-ITPR2↑ | Alters
Ca2+ homeostasis. Promotes mitochondrial fission.
Increases apoptosis | (45,245) |
| Li et al,
2024 | OA | PTPN1↑ | Promotes cellular
senescence | (201) |
| ITPR1↑ | Promotes cellular
senescence | |
| Song et al,
2024 | IDD | SYNJ2BP-RRBP1↓ | Alters
Zn2+ homeostasis. Promotes cellular senescence | (14) |
| Hernández-Alvarez
et al, 2019 | Cancer | MFN2↓ | Reduces lipid
metabolism. Increases ER stress | (36) |
| Wu et al,
2016; Chai et al, 2021 | | FUNDC1↑ | Promotes
mitochondrial fission | (167,250) |
| Hu et al,
2021 | | MFF↑ | Promotes
mitochondrial fission | (168) |
| Gomez-Suaga et
al, 2017 | | VAPB-PTPIP51↑ | Inhibits autophagy.
Alters Ca2+ homeostasis | (180) |
| Ponneri
Babuharisankar et al, 2023 | | FUNDC1-ULK1↑ | Promotes
autophagy | (177) |
| Raturi et
al, 2016 | | SERCA2b↑ | Alters
Ca2+ homeostasis | (159) |
| Zheng et al,
2018 | | EI24-VDAC2↑ | Alters
Ca2+ homeostasis. Increases apoptosis | (124) |
| Li et al,
2022 | | GRP75-VDAC1↑ | Alters
Ca2+ homeostasis | (252) |
Genetic disorders
Charcot-Marie-Tooth disease (CMT) is usually
inherited in an autosomal dominant manner, although X-linked and
autosomal recessive forms also exist (202). The most common genetic
mutations associated with CMT involve gap junction protein beta 1
(GJB1), peripheral myelin protein 22 (PMP22), myelin protein zero
(MPZ), and MFN2, which have become key targets in research efforts
aimed at developing disease-modifying therapies (202). The hallmark of CMT is
progressive degeneration of peripheral nerve axons and myelin
sheaths, leading to reduced nerve conduction velocity (203). Based on clinical and
electrophysiological features, four main subtypes of CMT can be
distinguished (203). CMT1 is
characterized by demyelination and is inherited in an autosomal
dominant pattern. CMT2 is an axonal form that may be inherited in
either a dominant or recessive pattern. CMTX, which is
predominantly X-linked, is associated with intermediate nerve
conduction velocities, although recessive and autosomal dominant
intermediate forms have also been reported. CMT4 exhibits a
demyelinating phenotype but follows an autosomal recessive
inheritance pattern.
Because dominant mutations in the MFN2 gene
predominantly impair mitochondrial fusion and motility, CMT2A is
the most common subtype among patients with CMT2 (204). It has been recently
demonstrated that expression of MFN2R94Q induces distal axonal
degeneration even in the absence of overt neuronal death (42). In primary neurons, motor neurons
from a CMT2A animal model, and fibroblasts derived from CMT2A
patients, the mutant protein reduces the extent of ER-mitochondria
contacts. These alterations occur concurrently with ER stress,
dysregulation of calcium homeostasis, and abnormalities in
mitochondrial morphology and axonal transport. Notably,
pharmacological interventions that alleviate ER stress or enhance
ER-mitochondria crosstalk restore mitochondrial morphology and
prevent axonal degeneration. These findings establish MERC
dysfunction as a key pathogenic mechanism through which mutant MFN2
drives CMT2A pathology (42).
Autosomal dominant optic atrophy (ADOA) leads to
degeneration of retinal neurons, resulting in optic nerve atrophy
and progressive visual impairment (205). Studies have shown that ADOA is
associated with mutations in two genes (OPA1 and OPA3) and three
established loci (OPA4, OPA5 and OPA8), which encode proteins
localized to the IMM. Additionally, other genetic loci (OPA2, OPA6
and OPA7) may be linked to X-linked or recessive forms of optic
atrophy (205). Previous
research has demonstrated that human fibroblasts derived from
patients with ADOA lacking functional OPA1 exhibit enhanced
ER-mitochondria coupling and a leftward shift in cytoplasmic
Ca2+ dependence, indicating more efficient
ER-to-mitochondria calcium transfer compared with wild-type cells
(206).
The evidence indicates that mutations in genes
encoding MERC-associated proteins, such as MFN2 in CMT2A and OPA1
in ADOA, cause distinct but significant alterations in MERC
structure and function. In CMT2A, the MFN2R94Q mutation decreases
ER-mitochondria contacts, initiating a series of events including
ER stress, calcium dyshomeostasis and mitochondrial dysfunction,
which ultimately leads to axonal degeneration. In ADOA, OPA1
deficiency results in increased ER-mitochondria coupling and more
efficient calcium transfer, which may paradoxically contribute to
retinal neuron pathology. These examples demonstrate that MERCs
represent a crucial functional interface where genetic lesions
converge to disrupt cellular homeostasis, and that the pathological
outcome, whether due to reduced or excessive connectivity, depends
on the specific molecular lesion and cellular context. Importantly,
the discovery that pharmacological restoration of ER-mitochondria
communication can reverse phenotypes in CMT2A models underscores
the therapeutic potential of targeting MERC dynamics to alter
disease progression. Thus, CMT2A and ADOA exemplify the two
contrasting poles of MERC dysfunction: In CMT2A, a primary loss of
ER-mitochondria contacts leads to axonal degeneration, whereas in
ADOA, pathological hyper-coupling and excessive Ca2+
transfer result in retinal neuron loss. These opposite deviations
from the normal MERC state emphasize how the functional
requirements at these contact sites are cell-type-specific.
Neurodegenerative diseases
Amyotrophic lateral sclerosis and frontotemporal
dementia (ALS/FTD) share overlapping clinical, pathological and
genetic features, establishing them as closely linked
neurodegenerative disorders. Clinical manifestations in patients
with FTD often resemble those observed in ALS, and vice versa
(207). A key pathological
hallmark shared by both conditions is the abnormal aggregation of
TDP-43, which plays a critical role in disease progression and is
associated with neuronal dysfunction (207). Stoica et al (98) demonstrated that MERCs are
significantly reduced in the presence of both wild-type and mutant
TDP-43, a protein implicated in familial ALS. Notably, this
reduction occurs without changes in the expression levels of MFN2,
PTPIP51, or VAPB. Specifically, VAPB levels were markedly decreased
in ALS tissues compared with controls, whereas no significant
alterations were detected in PTPIP51, ITPR3, or VDAC1. Although
TDP-43 does not directly bind to VAPB or PTPIP51 in vitro or
in vivo, it disrupts their interaction (98). The binding between VAPB and
PTPIP51 is diminished upon GSK3β activation, but enhanced by GSK3β
inhibitors, which promote MERC formation (98,128). Furthermore, overexpression of
both wild-type and four distinct mutant forms of TDP-43 was shown
to activate GSK3β (98).
Similarly, both wild-type and mutant FUS impair VAPB-PTPIP51
tethering and ER-mitochondria connectivity. These disruptions
adversely affect mitochondrial ATP synthesis and calcium
homeostasis. Additional studies indicate that neither wild-type nor
mutant FUS directly interacts with VAPB or PTPIP51; instead, their
effect is mediated through GSK3β activation. Treatment with the
GSK3β inhibitor AR-A014418 mitigates FUS-induced defects in
ER-mitochondrial coupling, restoring Ca2+ signaling and
mitochondrial ATP production (128).
Disruption of the VAPB-PTPIP51 tether has also been
observed in neurons derived from induced pluripotent stem (iPS)
cells of patients carrying the C9orf72 mutation, as well as in
transgenic mice expressing the ALS/FTD-associated mutant C9orf72.
All three C9orf72 dipeptide repeat proteins (DPRs) significantly
reduce VAPB-PTPIP51 and ITPR1-VDAC1 interactions, thereby impairing
ITPR-mediated Ca2+ transfer from the ER to mitochondria
(208), in contrast to control
samples. However, the absence of direct binding between DPRs and
either VAPB or PTPIP51 suggests an indirect mechanism, supporting
further evidence that DPR-induced damage to the VAPB-PTPIP51
complex is facilitated by GSK3β activation (208). It is crucial to note that
although the disruption of VAPB-PTPIP51 tethering by TDP-43, FUS
and C9orf72 DPRs has been repeatedly verified across multiple model
systems, the causal chain that links this disruption to specific
neurodegenerative endpoints in vivo remains an area under
active investigation. The correlative evidence from postmortem
human tissue and iPSC-derived neurons is robust, yet conclusive
proof that restoring this single tether is sufficient to halt
disease progression in animal models has not been established
(46,128,208). In this context, the disruption
of Ca2+ homeostasis and ATP production caused by
defective VAPB-PTPIP51 tethering can be regarded as a core
MERC-driven pathogenic mechanism. Meanwhile, the downstream
activation of GSK3β and the subsequent protein aggregation are
secondary events that amplify the initial insult.
Parkinson's disease (PD) is a neurodegenerative
disorder characterized by the death or degeneration of
dopamine-producing neurons in the substantia nigra (SN), a brain
region critical for motor control. Since dopaminergic neurons
regulate movement and coordination, patients with PD commonly
exhibit symptoms such as bradykinesia, tremors and muscle rigidity
(209). Two primary
pathophysiological hallmarks of PD are the abnormal accumulation of
α-synuclein (α-Syn) protein, leading to the formation of Lewy
bodies, and the dysfunction of dopaminergic neurons in the SN.
These pathological changes disrupt neural circuitry, ultimately
giving rise to the clinical manifestations of PD. As the disease
progresses, misfolded α-Syn aggregation, dysregulated dopamine
metabolism, oxidative stress and neuronal loss form a
self-reinforcing cycle (210).
Guardia-Laguarta et al (211) demonstrated that in both human
and mouse brain tissues, wild-type α-Syn localizes predominantly to
MERCs, rather than to mitochondria alone. Notably, pathogenic point
mutations in α-Syn reduce MERC formation. In contrast to cells
expressing wild-type α-Syn, mutant α-Syn-expressing cells exhibit
weakened ER-mitochondria associations, impaired MERC function, and
increased mitochondrial fragmentation (211). Another study focused on the
interaction between α-Syn and VAPB, showing that overexpression of
both wild-type and familial mutant α-Syn disrupts the VAPB-PTPIP51
tether, thereby loosening ER-mitochondria contacts (212). This disruption is also observed
in neurons derived from iPSCs of patients with PD with triplication
of the α-Syn locus. α-Syn-mediated weakening of ER-mitochondria
coupling impairs inter-organellar Ca2+ exchange and
compromises mitochondrial ATP production (212).
Alzheimer's disease (AD) is the leading cause of
cognitive impairment in a significant proportion of older adults.
The primary pathogenic features of AD include the degeneration of
neuronal connections and cells, along with the abnormal
accumulation of misfolded proteins, particularly intracellular
neurofibrillary tangles composed of hyperphosphorylated tau protein
and extracellular β-amyloid (Aβ) plaques (213). Familial forms of AD are
associated with mutations in presenilin-1 (PSEN1), presenilin-2
(PSEN2) and the amyloid precursor protein (APP). APP processing
generates Aβ, the pathological peptide that accumulates in AD
brains. The γ-secretase complex, which cleaves APP to produce Aβ,
contains presenilins as catalytic subunits (214). Both presenilins and APP
localize to MERCs, where they participate in APP processing and Aβ
generation (215,216). Mutations in these proteins, as
well as Aβ accumulation itself, disrupt MERC functions, including
mitochondrial biogenesis, ATP production, ER stress responses,
Ca2+ signaling, lipid metabolism, autophagy,
inflammation and synaptic activity (217). Lau et al (218) investigated the
neuropathological impact of AD across brain regions by analyzing
postmortem brain tissues from control subjects and AD patients.
Their findings indicate that the cerebellum is relatively spared in
AD, whereas the temporal cortex is highly vulnerable. In late-stage
AD, VAPB and PTPIP51 protein expression remained unchanged in the
cerebellum but was significantly reduced in the cerebral cortex.
Further analysis revealed that the VAPB-PTPIP51 tethering complex
was disrupted in pyramidal neurons from individuals at Braak stages
III-IV, whereas Purkinje cells retained intact ER-mitochondria
contacts. To determine whether VAPB loss correlates with reduced
PTPIP51 levels in the temporal cortex, neuron-specific
enolase-normalized VAPB and PTPIP51 signals were analyzed in
individual samples. The results demonstrated a strong positive
correlation between the two proteins (218). The chaperone protein sigma-1
receptor (Sig-1R) is predominantly localized at MERCs and performs
multiple critical cellular functions. Deletion of Sig-1R leads to
diminished ER-mitochondria coupling, altered mitochondrial
dynamics, disrupted Ca2+ homeostasis, and increased ER
stress (219). It has been
recently shown that N,N-dimethyltryptamine (DMT) restores neuronal
ER-mitochondria crosstalk by activating Sig-1R, a mechanism linked
to DMT's potential anti-AD effects. DMT enhances TCA cycle
activity, alleviates mitochondrial dysfunction in AD models, and
modulates ER-mitochondria contacts and mitochondrial calcium uptake
in both in vitro and in vivo systems (220).
Huntington's disease (HD) is an autosomal dominant,
progressive and fatal neurological disorder characterized by motor
dysfunction, including limb tremors, and cognitive decline
(221). Histopathological
examination of HD patient brains has revealed neurodegeneration in
multiple brain regions, particularly the striatum, subthalamic
nucleus, hypothalamus, caudate nucleus, and putamen. HD is caused
by an expanded CAG trinucleotide repeat in exon 1 of the huntingtin
(HTT) gene, which encodes a polyglutamine tract and leads to the
production of a mutant protein with toxic gain-of-function
properties (222). Selective
degeneration of the striatum in HD is associated with
well-established pathological features, including disruption of
Ca2+ homeostasis and mitochondrial dysfunction (223). A significant reduction in the
levels of the chaperone GRP75 and the ER-anchored ITPR3 in the
striatum of both patients with HD and two distinct HD animal
models, but not in the cortex or hippocampus, has been previously
demonstrated (224). MFN2
expression was found to be decreased specifically in the putamen of
patients with HD. The association between reduced MERC-associated
protein levels and the onset of behavioral abnormalities in HD
animal models suggests that impaired ER-mitochondria connectivity
contributes to striatal vulnerability, rather than being merely a
secondary consequence. Moreover, reductions in MERC proteins occur
during early to mid-stages of the disease, indicating that these
changes actively participate in HD progression rather than simply
reflecting late-stage neuronal damage. Disruptions in
ER-mitochondria interface proteins have also been implicated in
other neurodegenerative diseases, where they can either enhance or
diminish ER-mitochondria juxtaposition (224). Excessive mitochondrial fission
(overfission) has been observed in HD models, and treatment of R6/1
mutant huntingtin striatal neurons with the Drp1 inhibitor Mdivi-1
restored ER-mitochondria contacts and Ca2+ transfer,
thereby ameliorating Ca2+ homeostasis defects (224).
Despite having distinct etiologies, a common theme
emerges: The specific disruption of key tethering complexes, most
notably the VAPB-PTPIP51 interaction, impairs inter-organellar
communication. This disruption is instigated by disease-specific
proteins (for example, TDP-43, FUS, α-synuclein, Aβ, mutant
huntingtin), often via indirect pathways involving kinases such as
GSK3β. The subsequent loss of MERC integrity results in a
characteristic triad of defects: Dysregulated Ca2+
signaling, impaired mitochondrial bioenergetics (decreased ATP
production), and heightened cellular stress. Significantly, MERC
alterations frequently occur at an early stage of disease
progression, contributing to neuronal vulnerability rather than
merely being a late-stage consequence. The evidence indicates a
vicious cycle in which protein aggregates disrupt MERCs, and
dysfunctional MERCs in turn promote further proteostasis failure
and metabolic deficiency. Collectively, these findings firmly
establish MERCs as crucial hubs, the dysfunction of which is
integral to neurodegenerative pathogenesis, thus positioning the
restoration of ER-mitochondria connectivity as a promising
therapeutic strategy for multiple disorders.
Metabolic diseases
Diabetic kidney disease (DKD) is characterized by
early alterations in glomerular filtration rate and increased
urinary excretion of proteins, ions and other solutes. Advanced
stages of DKD are marked by extracellular matrix (ECM) protein
accumulation, interstitial fibrosis, cellular senescence, and
ultimately renal cell loss and organ failure (225). The pathogenesis of DKD involves
multiple mechanisms, including hemodynamic and metabolic
disturbances that trigger proinflammatory and profibrotic signaling
pathways in the kidney, leading to progressive histopathological
changes (226). Both human
patients and experimental murine models of DKD have been shown to
exhibit compromised MERC integrity. In a study by Yang et al
(227), the structural
integrity of MERCs was examined in renal biopsies from individuals
at various stages of DKD. Electron microscopy revealed a reduced
number of MERCs in DKD samples compared with controls.
Additionally, the expression levels of key MERC components,
including the tethering protein MFN2, the structural regulator
PACS2, and disulfide-bond A oxidoreductase-like protein (DsbA-L), a
MERC-localized enzyme, were found to be downregulated in DKD
biopsies. Furthermore, co-immunoprecipitation analysis showed
decreased interaction between the ER-resident ITPR1 and the
mitochondrial VDAC1, two MERC components critical for calcium
transfer, in the tubular compartment of DKD biopsies relative to
controls (227). MERC integrity
was negatively correlated with markers of kidney injury and
fibrosis in DKD (227). A more
recent study reported diminished expression of MFN2, a core MERC
protein, in kidney biopsies from patients with DKD. MFN2 was nearly
undetectable in DKD tissues, whereas it was robustly expressed in
the glomerular compartment of control samples (228). DsbA-L-deficient mice exhibited
elevated markers of ER stress and an exacerbated DKD phenotype,
whereas overexpression of DsbA-L in renal tubular cells conferred
protection against high glucose-induced apoptosis, ER stress and
MERC disruption (35).
Similarly, diabetic mice lacking PACS2, a structural component of
MERCs, displayed worsened kidney function and increased
proteinuria. By contrast, in vivo overexpression of PACS2
via intravenous delivery of an adenoviral vector, as well as its
overexpression in cultured renal tubular cells (HK-2), preserved
MERC integrity and protected against glucose-induced cellular
dysfunction (229). Chen et
al (230) demonstrated a
significant downregulation of vitamin D receptor (VDR), PINK1,
Parkin, FUNDC1, LC3II, ATG5, MFN2 and MFN1 in renal tubular cells
of diabetic rats. In streptozotocin-induced diabetic rats,
calcitriol treatment attenuated tubulointerstitial fibrosis and
reduced urinary albumin and serum creatinine levels. Moreover, VDR
agonists restored MERC integrity, alleviated mitophagy impairment,
and suppressed mitochondrial fission and ROS production (230).
Other studies suggest a more nuanced role for MERCs
in DKD progression through dynamic changes in their abundance or
composition. For instance, upregulation of A-kinase anchoring
protein 1 (AKAP1) at MERCs explains the increased MERC enrichment
observed in podocytes from biopsies of patients with DKD and in
podocytes exposed to high glucose in vitro. AKAP1 promotes
mitochondrial fission by enhancing DRP1 phosphorylation and
translocation. Pharmacological inhibition of DRP1 mitigates
podocyte dysfunction and excessive mitochondrial fission induced by
AKAP1 overexpression (231).
Tubule-specific overexpression of RTN1A exacerbated DKD in diabetic
mice, as evidenced by increased tubular damage, tubulointerstitial
fibrosis and impaired renal function. However, RTN1A overexpression
did not aggravate diabetes-induced albuminuria or glomerular
injury. Notably, RTN1A overexpression amplified ER stress and
mitochondrial dysfunction in diabetic tubular epithelial cells by
disrupting MERCs. ER-localized RTN1A interacted with both VDAC1 and
mitochondrial hexokinase-1 (HK1), interfering with their
association. Dissociation of VDAC1 from HK1 triggered apoptotic and
inflammasome activation pathways, contributing to TEC damage and
loss (232). The literature on
MERC alterations in DKD emphasizes the importance of distinguishing
correlative relationships from causal ones. Although the decreased
expression of MERC components, such as MFN2 and PACS2, is
associated with the disease severity in patient biopsies, the
functional rescue experiments, in which the overexpression of
DsbA-L or PACS2 mitigates renal injury in diabetic models, provide
more convincing evidence for a causal role of MERC dysfunction in
DKD pathogenesis (35,227,229). Taken together, the evidence
from DKD models indicates that the loss of MERC integrity
represents an early, primary event that initiates a cascade of
secondary pathologies, such as ER stress, mitochondrial fission and
inflammasome activation. These secondary pathologies collectively
lead to tubular injury and fibrosis.
NAFLD is a collective term encompassing a spectrum
of liver conditions, including hepatic steatosis and non-alcoholic
steatohepatitis (NASH), which may progress to cirrhosis and
hepatocellular carcinoma. A defining feature of NAFLD232 is
steatosis, defined as the accumulation of fat in >5% of
hepatocytes (233).
Contributing factors to NAFLD include diabetes, insulin resistance,
metabolic syndrome and genetic variants in TM6SF2 (transmembrane 6
superfamily member 2) and PNPLA3 (patatin-like phospholipase
domain-containing protein 3) (234,235). In individuals with NASH,
mitochondrial dysfunction is frequently observed and plays a
critical role in the progression from simple steatosis to NASH
(236). Liver biopsies from
patients with NASH show reduced MFN2 expression. Similarly, mouse
models of steatosis or NASH exhibit decreased Mfn2 levels, and
re-expression of Mfn2 in a NASH model ameliorates disease
pathology. Mice with liver-specific ablation of Mfn2 develop
fibrosis, hepatocellular malignancy, lipid accumulation and
inflammation. MFN2 has been shown to bind PS and facilitate its
transport into specific membrane domains, thereby promoting PS
transfer to mitochondria and subsequent mitochondrial PE synthesis.
Consequently, loss of hepatic Mfn2 impairs phospholipid
biosynthesis and PS trafficking, leading to ER stress, development
of a NASH-like phenotype, and increased susceptibility to liver
cancer (36). Recently, Xu et
al (237) demonstrated that
Cds2 expression is downregulated in both diet-induced and
genetically engineered NAFLD animal models. Hepatic deficiency of
Cds2 results in steatosis, inflammation and fibrosis. Loss of Cds2
alters the MERCs proteome, potentially affecting both the structure
and function of MERCs. The ER-mitochondria PS transfer, mediated by
EMC subunits and MFN2, is significantly impaired in Cds2-deficient
livers due to substantial downregulation of these proteins
(237). Additionally, MFN2
serves as a physical tether between mitochondria and the ER.
Reduced PS transport to mitochondria likely results from diminished
MFN2 protein levels and increased distance between the ER and
mitochondria in the livers of Cds2f/f; AlbCre mice (237). Mic19, a core component of the
mitochondrial contact site and cristae organizing system (MICOS)
complex, was shown by Dong and colleagues to regulate
ER-mitochondria interactions via the EMC2-SLC25A46-Mic19 axis.
Hepatocytes from mice with liver-specific Mic19 knockout (LKO)
exhibit impaired mitochondrial fatty acid β-oxidation and disrupted
lipid metabolism, which can spontaneously lead to NASH and liver
fibrosis. LKO also leads to reduced ER-mitochondrial contacts,
disordered mitochondrial lipid metabolism, cristae disorganization,
and activation of the mitochondrial UPR. By contrast, re-expression
of Mic19 in Mic19 LKO hepatocytes prevents the development of liver
disease. Furthermore, overexpression of Mic19 attenuates methionine
and choline-deficient diet-induced fatty liver disease (40).
Dysfunction of MERCs represents a central and
active pathogenic mechanism in metabolic diseases, particularly DKD
and NAFLD. A common characteristic in both conditions is the
downregulation or impaired function of core tethering and
regulatory proteins, such as MFN2, PACS2 and DsbA-L, which results
in a loss of MERC integrity. This structural disruption triggers a
series of functional deficits. In DKD, it impairs Ca2+
signaling through the ITPR1-VDAC1 axis, exacerbates ER stress, and
promotes renal cell apoptosis and fibrosis. In NAFLD, it disrupts
the crucial transfer of phospholipids such as PS, leading to
hepatic lipid accumulation, ER stress, inflammation, and the
progression to steatohepatitis. Significantly, the evidence
indicates a vicious cycle in which metabolic insults (for example,
hyperglycemia and lipotoxicity) damage MERCs, and dysfunctional
MERCs in turn intensify metabolic dysfunction and cellular injury.
Interventions that restore MERC structure or function, either by
overexpressing tethering components or by using agents such as VDR
agonists, exhibit protective effects in disease models,
highlighting the therapeutic potential of targeting these organelle
contact sites. MERCs are therefore identified as critical
integrators of cellular metabolism, and their deterioration is a
key factor in the pathogenesis and progression of complex metabolic
disorders.
Cardiovascular diseases
Heart failure (HF) is a clinical syndrome
characterized by the heart's inability to pump sufficient oxygen
and nutrients to meet the body's demands, leading to impaired
physical function and commonly presenting with fatigue, edema and
dyspnea (238). Mitochondrial
function and Ca2+ homeostasis are well-established
contributors to ventricular remodeling and the progression of HF
(239,240). Cardiac contraction and
rhythmicity require substantial energy, which is primarily
generated through mitochondrial OXPHOS (241). Additionally, optimal
Ca2+ levels necessary for excitation-contraction
coupling are regulated by the sarcoplasmic reticulum (SR), a
specialized membrane system enriched with Ca2+-ATPases.
Disruptions in Ca2+ homeostasis can lead to aberrant
energy metabolism and ER stress, both of which contribute to the
development of HF (242). MERCs
have been shown to play critical roles in cellular physiology,
including regulation of mitochondrial dynamics and Ca2+
signaling (43,231).
Patients with HF exhibit significantly reduced
expression of FUNDC1 and fewer SR-mitochondria contacts compared
with healthy individuals. The diminished FUNDC1 levels at MERCs
result in impaired ubiquitin-dependent inhibition of ITPR2
degradation, thereby compromising SR-to-mitochondria
Ca2+ transfer. This disruption ultimately affects
cardiac function through dysregulation of the CREB/Fis1 pathway.
Furthermore, FUNDC1-deficient animals display both diastolic and
systolic dysfunction (103).
LonP1, a protease localized at MERCs, plays a key regulatory role.
Depletion of LonP1 leads to a significant reduction in MERC
formation and causes mitochondrial fragmentation. Moreover, loss of
LonP1 in mouse cardiomyocytes disrupts mitochondrial fusion and
MERC integrity and activates the ER UPR. Consequently,
cardiomyocyte-specific deletion of LonP1 induces abnormal metabolic
reprogramming and pathological cardiac remodeling (41).
DCM is a distinct form of cardiomyopathy that
occurs independently of coronary artery disease or hypertension. In
individuals with diabetes, DCM is strongly associated with an
increased incidence of HF and elevated mortality rates (243). The pathophysiological hallmarks
of DCM include myocardial interstitial fibrosis, cardiomyocyte
hypertrophy, necrosis and apoptosis. Its development involves
multiple complex mechanisms, including insulin resistance,
disturbances in cardiac energy metabolism, oxidative stress,
inflammatory responses, Ca2+ imbalance, impaired
autophagy, and other contributing factors (244). A direct link between MERCs and
the onset of DCM has been recently suggested (45). Compared with cardiac tissues from
non-diabetic donors, those from diabetic individuals exhibit
significantly higher levels of FUNDC1. Cardiomyocyte-specific
deletion of Fundc1 abolished diabetes-induced MERC formation,
prevented mitochondrial Ca2+ overload, mitochondrial
fragmentation and apoptosis, and was associated with improved
mitochondrial functional capacity and cardiac performance (45). Based on these findings, several
research groups have investigated the effects of high glucose
exposure and demonstrated that such conditions reduce mitochondrial
biogenesis, fusion and OXPHOS while promoting MERC formation
through upregulation of PACS2, ITPR2, FUNDC1 and VDAC1. These
observations highlight the critical roles of mitochondria-driven
apoptotic pathways, such as the SMAC-HTRA2-ARTS-XIAP cascade, in
the molecular pathogenesis of DCM. Additionally, diabetic rats
exhibited signs of cardiomyopathy, including elevated levels of
troponin, TNNI3K and heart mass index. Both in vitro and
in vivo, ferulic acid effectively suppressed
hyperglycemia-induced alterations in MERCs and ameliorated the
associated cellular abnormalities (245).
The evidence indicates that MERC dysfunction is a
common factor, albeit presenting differently in various disease
contexts. In HF, a decline in MERC integrity, mediated by reduced
levels of proteins such as FUNDC1 and LonP1, disrupts the accurate
calcium signaling between the SR and mitochondria, impedes
mitochondrial fusion, and initiates maladaptive stress responses.
Collectively, these effects compromise cardiac energetics and
contractile function. Conversely, in DCM, a pathological elevation
in MERC formation, driven by the upregulation of proteins such as
FUNDC1, ITPR2 and PACS2, leads to excessive mitochondrial calcium
influx, resulting in Ca2+ overload, oxidative stress,
and the activation of apoptotic pathways. These contrasting
dysregulations, whether it is diminished or excessive
ER-mitochondria coupling, both culminate in the same adverse
outcomes: Disrupted mitochondrial bioenergetics, impaired calcium
homeostasis, and ultimately, cardiomyocyte death and cardiac
remodeling. The protective effects observed following genetic or
pharmacological modulation of MERC components emphasize that
restoring the physiological balance of these contact sites is a
feasible therapeutic strategy. Therefore, MERCs emerge as dynamic
and crucial regulators of cardiac health, where their precise
structural and functional adjustment is essential to prevent the
progression from metabolic stress to overt HF. The contrast between
HF and DCM clearly illustrates the dualistic nature of MERC
dysfunction in cardiac disease. In HF, the reduced levels of FUNDC1
and LonP1 result in diminished MERC formation and impaired
Ca2+ transfer. By contrast, in DCM, the upregulation of
FUNDC1, PACS2 and ITPR2 leads to excessive MERC coupling and
mitochondrial Ca2+ overload. Both of these extreme
situations ultimately compromise cardiac function, which reinforces
the concept that MERC integrity must be maintained within a
physiological range appropriate to the cell type and metabolic
context.
Orthopedic diseases
OA is a whole-joint disease characterized by
structural alterations in articular cartilage, subchondral bone,
ligaments, joint capsule, synovial membrane and periarticular
muscles (246). OA has a
complex etiology involving mechanical, inflammatory and metabolic
factors that ultimately lead to structural failure of the synovial
joint. Rather than being a passive degenerative condition or a
so-called 'wear-and-tear' disease as it is often described, OA
represents an active and dynamic process resulting from an
imbalance between tissue repair and degradation in the joint
(247). Research on the
involvement of MERCs in OA remains limited; however, the authors'
most recent work suggests a potential role for MERCs in OA
pathogenesis (200). The
present findings revealed a strong association between genes linked
to cellular senescence in OA and hub genes related to MERCs.
Further evidence from in vitro experiments demonstrated
positive correlations between OA-related senescence genes (PTEN,
ABL1, MAPK14 and MAP2K1) and MERCs-associated genes (PTPN1 and
ITPR1) (200).
Intervertebral disc degeneration (IDD) and its
associated pathological changes are the leading causes of low back
pain and neck pain, two of the most prevalent medical conditions
contributing to increased years lived with disability and
heightened demand for rehabilitation services (248). Accumulating evidence indicates
that disrupted mitochondrial function exacerbates key pathological
processes during IDD progression, including ECM degradation,
inflammatory responses, cellular senescence and cell death
(249). Previous studies have
demonstrated that MERCs facilitate the transfer of SLC39A7 from the
ER to mitochondria, thereby regulating intracellular
Zn2+ homeostasis (12,13). Furthermore, proteomic analyses
have revealed that NLRX1 localizes to both the ER membrane and the
OMM, suggesting a potential role in MERC formation and
stabilization (92). A recent
study found that SYNJ2BP promotes the interaction between NLRX1 and
SLC39A7 and is essential for maintaining mitochondrial
Zn2+ homeostasis. SYNJ2BP contributes to MERC structural
organization through its interaction with RRBP1. In addition to
reducing the formation of the SLC39A7-NLRX1 complex, loss of
SYNJ2BP expression and disruption of MERC architecture impair
mitochondrial localization of SLC39A7. Moreover, SYNJ2BP
downregulation was identified as a key pathological feature of
nucleus pulposus cell senescence and IDD progression, strongly
associated with MERC disruption (14).
Although research is still in its nascent stages,
preliminary evidence firmly indicates that MERC dysfunction
contributes to disease progression via distinct yet converging
pathways. In OA, a correlative association has been established
between MERC-associated genes (PTPN1 and ITPR1) and key factors
driving cellular senescence, suggesting that disrupted
inter-organellar communication may expedite the senescent phenotype
that is central to joint degradation. In IDD, a more well-defined
molecular mechanism is presented. Specifically, the downregulation
of the MERC component SYNJ2BP disrupts contact site architecture,
impedes formation of the NLRX1-SLC39A7 complex, and consequently
leads to dysregulation of mitochondrial zinc homeostasis. This
dysregulation promotes senescence of nucleus pulposus cells,
degradation of the ECM, and ultimately disc failure. Collectively,
these findings establish MERCs as crucial, yet relatively
under-investigated, cellular hubs in orthopedic health. The
impairment of their structural integrity or functional capacity
appears to intensify core degenerative processes, specifically, the
impairment of ion homeostasis (Zn2+) and the
acceleration of cellular senescence, which underlie both OA and
IDD. This highlights a common theme across various diseases: MERC
dysfunction disrupts basic cellular housekeeping, resulting in
tissue-specific pathology. In the future, these insights present a
promising new direction for research, indicating that therapeutic
strategies designed to preserve or restore MERC function may
potentially alleviate the progression of degenerative orthopedic
conditions by targeting their fundamental cellular origins.
Cancer
Increasing evidence indicates that MERCs play
essential roles in tumor initiation and progression. Key
intracellular mechanisms supported by MERCs, such as lipid
exchange, mitochondrial fission, autophagy, Ca2+
signaling and ROS regulation, are critically involved in cancer
cell survival, susceptibility to cell death, invasion and
metastasis (1). Galmes et
al (29) demonstrated that
in HeLa cells, the lipid-binding/transfer domains of ORP5 and ORP8,
oxysterol-binding protein-related proteins, target ER-mitochondria
contact sites and interact with the mitochondrial outer membrane
protein PTPIP51, thereby modulating mitochondrial morphology and
function. Additionally, hepatic deficiency of MFN2 has been shown
to impair phospholipid biosynthesis and ER-mitochondrial PS
transfer, ultimately leading to ER stress, development of a
NASH-like phenotype, and hepatocellular carcinogenesis (36). Multiple studies have linked
MERC-associated proteins to the regulation of tumor growth through
the mitochondrial fission pathway. For example, Wu et al
(167) reported that under
hypoxic conditions in HeLa cells, the ER membrane protein calnexin
interacts with FUNDC1, an OMM protein localized at MERCs, promoting
its accumulation at ER-mitochondria interfaces. This interaction
facilitates the recruitment of Drp1 to MERCs, initiating
mitochondrial fission. Thus, FUNDC1 is specifically required for
mitochondrial division in cervical cancer cells under hypoxic
stress (167). Moreover,
ubiquitin-specific protease 19 (USP19), an ER-resident
deubiquitinase, enhances Drp1 homopolymerization by binding to and
deubiquitinating FUNDC1 at ER-mitochondria contact sites in hypoxic
HeLa cells. USP19 also modulates Drp1's hydrolytic enzyme activity
and GTP binding capacity, thereby mediating mitochondrial fission
(250).
Notably, MERC-mediated regulation of autophagy also
contributes to cancer development. In a study by Hu et al
(168), acute energy stress
induces translocation of the cellular energy sensor AMPK from the
cytosol to MERCs in human cervical carcinoma HeLa cells. There,
AMPK directly interacts with the MERC tethering protein MFN2 and
plays a critical role in energy stress-induced autophagy.
Remarkably, following prolonged stress, a significant fraction of
AMPK switches to bind and activate the MFF, thereby coordinating
mitochondrial fission (168).
Furthermore, Gomez-Suaga et al (180) showed that the ER protein VAPB
and the mitochondrial protein PTPIP51 form tethering structures at
MERCs, enhancing ER-mitochondria connectivity and facilitating
ER-mitochondrial Ca2+ exchange, which suppresses
autophagosome formation in HeLa cells (180). Ponneri Babuharisankar et
al (177) further
demonstrated that under hypoxia, the stress-induced mitochondrial
chaperone Lon accumulates at ER-mitochondria contact sites in colon
and oral cancer cells. Lon stabilizes the FUNDC1-ULK1 complex in a
manner dependent on the mitochondrial NCLX, thereby promoting
mitophagy and supporting cancer cell survival and progression
(177). The fate of cancer
cells is also influenced by oncogenes and tumor suppressors that
disrupt Ca2+ homeostasis through complex interactions at
ER-mitochondria contact sites, directly altering Ca2+
flux from the ER to mitochondria within the context of
MERC-mediated Ca2+ signaling (251). TMX1 inhibits SERCA2b, a
Ca2+ pump enriched at ER-mitochondria contact sites,
thereby reducing ER-mitochondrial Ca2+ transfer in HeLa
cervical cancer and A375P melanoma cells (159). Additionally, Zheng et al
(124) showed that tubular ER
undergoes extensive expansion in response to DNA damage, a process
dependent on p53-mediated transcriptional activation of ER-shaping
proteins REEP1, REEP2 and EI24. Subsequently, EI24 interacts with
VDAC2 to promote ER-mitochondria contact formation, increasing
Ca2+ transfer from the ER to mitochondria and promoting
DNA damage-induced apoptosis (124). Li et al (252) also found that
cisplatin-resistant ovarian cancer (OC) cells exhibit elevated
levels of GRP75 and VDAC1, proteins known to stabilize MERCs. These
cells display resistance to cisplatin by preventing proapoptotic
ROS accumulation and mitigating cisplatin-induced mitochondrial
dysfunction (252). The
evidence indicates that MERCs are not passive structures; rather,
they are actively remodeled within tumors to support cell survival,
proliferation and adaptation to stress. Through specific tethering
complexes, such as ORP5/ORP8-PTPIP51 for lipid transfer and
VAPB-PTPIP51 for calcium exchange, MERCs regulate fundamental
processes that contribute to tumorigenesis. They enable cells to
survive under hypoxic conditions by promoting fission through the
FUNDC1-Drp1 axis and modulate the energy stress response by
recruiting AMPK to regulate autophagy. Moreover, MERCs play a
crucial role in determining cell fate in response to therapy. They
can mediate proapoptotic calcium transfer in response to DNA damage
(for example, via the EI24-VDAC2 axis), yet they can also be
exploited to promote therapy resistance, as observed in
cisplatin-resistant OC, where upregulated GRP75-VDAC1 stabilizes
MERCs to inhibit apoptosis. This dual functionality highlights the
context-dependent nature of MERC function in cancer, positioning
them as dynamic therapeutic targets whose modulation may disrupt
the adaptive mechanisms that enable tumors to grow and resist
treatment.
Strategies for targeting MERCs
Building upon the comprehensive overview of MERC
biology and pathology presented in the present review, the
development of therapeutic strategies targeting these contact sites
represents both a compelling opportunity and a formidable
challenge. Current approaches, although in their infancy, can be
broadly categorized into three levels of intervention: Direct
modulation of the contact-site machinery, regulation of individual
MERC-associated proteins, and indirect modulation through upstream
metabolic or signaling pathways. Direct strategies are aimed at
precisely manipulating the physical tethering or functional output
at the interface. The Drp1 inhibitor Mdivi-1 (253), which restores MERC integrity
and calcium homeostasis in HD models (224), serves as an example of this by
targeting a key executor of MERC-defined mitochondrial fission.
Similarly, the utilization of a cell-penetrating peptide to
displace Hexokinase 2 from MERCs, thereby inducing lethal
Ca2+ overload specifically in cancer cells (254,255), demonstrates the potential of
precisely disrupting pro-survival microdomains. These examples
highlight the therapeutic potential of molecules that can stabilize
beneficial contacts (for example, in neurodegeneration) or
destabilize pathological ones (for example, in cancer or diabetic
complications). The second level involves modulating the expression
or post-translational modification of core tethering components,
such as genetically or pharmacologically targeting MFN2, PACS2, or
cyclophilin D (104). While
effective in specific experimental settings, such as protecting
cardiomyocytes from ischemia-reperfusion injury, this approach is
severely restricted by the pleiotropic functions of these proteins
outside of MERCs. As demonstrated by the systemic metabolic
disruptions in liver-specific Mfn2 knockout mice (256) or the hepatic insulin resistance
in CypD knockout animals (257), global manipulation often
results in unacceptable on-target, off-site effects. This
underscores a fundamental challenge: The majority of MERC
regulators are not confined solely to the contact site but are
essential components of more extensive cellular networks that
govern fusion, calcium signaling and metabolism. Consequently, the
future of MERC-targeted therapy depends on overcoming this
limitation through the attainment of context-specificity, which
involves specifically influencing the protein's function at the
ER-mitochondria interface, and functional-selectivity, which
entails modulating only its tethering-related activity without
interfering with its other functions.
The translation of MERC-targeted strategies into
clinical applications encounters several fundamental obstacles.
First, the majority of MERC-resident proteins are not solely
localized at contact sites but also carry out essential functions
in other parts of the cell. For instance, MFN2 is essential for
mitochondrial fusion in all cell types, and its deficiency in
liver-specific knockout mice results in severe metabolic disruption
and hepatocarcinoma (36,256).
Likewise, cyclophilin D, while regulating MERC stability, serves as
a crucial regulator of the mitochondrial permeability transition
pore throughout the body (104,257). Pharmacological modulation of
these pleiotropic proteins will inevitably result in on-target but
off-site effects, which may overshadow the therapeutic benefits.
Second, the molecular composition and functional output of MERCs
are highly specific to cell types. A MERC-stabilizing intervention
aimed at restoring neuronal calcium homeostasis in AD could, in
theory, exacerbate cardiomyocyte pathology in HF by promoting
excessive mitochondrial calcium loading, as indicated by the
pathological MERC expansion observed in DCM (45,245). Third, the dynamic nature of
MERCs determines that their abundance, inter-membrane distance, and
protein composition continuously adapt to the cellular metabolic
status. This plasticity undermines the concept of a simple
'inhibitor' or 'activator' of MERCs. Instead, therapeutic
interventions may need to achieve context-dependent normalization
of MERC function. Strategies relying on cell-type-specific delivery
systems, such as ligand-functionalized nanoparticles or viral
vectors with tissue-specific promoters, may ultimately address the
specificity challenge. Nevertheless, such approaches are still in
the early pre-clinical stage and demand extensive validation in
physiologically relevant human models, including patient-derived
organoids and organ-on-chip platforms.
To actualize this paradigm shift, future research
should pivot towards addressing several fundamental biological
questions and developing next-generation technologies. First, a
significant knowledge gap pertains to the molecular code for MERC
targeting. Specifically, do conserved structural motifs or
lipid-binding domains universally direct proteins to this
interface, or is targeting accomplished through unique,
protein-specific combinatorial signals? Deciphering this 'MERC zip
code' via comparative proteomics of contact-site subdomains and
structural biology is of utmost importance. Such knowledge would
facilitate the rational design of targeted therapeutics, including
small molecules that allosterically modulate a tether only when it
is engaged at the contact site, or biologics conjugated to
MERC-homing peptides. Second, there exists an urgent requirement
for advanced tools that enable real-time, high-resolution in
vivo observation and manipulation of MERCs. The development of
more robust, minimally perturbative dynamic reporters beyond
split-GFP, perhaps based on novel biosensors for local lipid flux,
nanometer-scale distance changes, or specific enzymatic activities
at MERCs, will be vital for screening and validating candidate
drugs. Third, nanotechnology-based delivery systems present a
promising approach to surmount specificity challenges. Designing
liposomes or nanoparticles functionalized with ligands for
MERC-enriched surface proteins could enable site-specific delivery
of therapeutic cargoes. For example, gene-editing tools could be
used to correct mutant MFN2 exclusively in affected neurons in
CMT2A, or small interfering RNA could be employed to knock down
pathological FUNDC1 overexpression specifically in diabetic
cardiomyocytes. Finally, considering the dynamic and
cell-type-specific characteristics of MERCs identified in diverse
diseases, therapeutic strategies need to be highly customized. This
requires transitioning from conventional cell lines to conducting
tests in more physiologically relevant models, such as
patient-derived organoids or organ-on-a-chip systems that maintain
tissue-specific MERC architecture. In summary, the future direction
involves moving from a simplistic, protein-focused pharmacopeia to
a refined, interface-focused toolkit. The ultimate objective is to
develop 'MERC stabilizers' and 'MERC normalizers' that can
accurately adjust the distance, duration and functional output of
these crucial communication hubs, thus restoring cellular
homeostasis in a broad spectrum of diseases marked by their
dysfunction, ranging from neurodegeneration and cardiomyopathy to
cancer and metabolic syndrome. The three-tier model of MERC
signaling, namely sensing, processing and execution, further
suggests that therapeutic interventions can be strategically
targeted at a specific tier according to the disease context. For
example, normalizing abnormal ion sensing might be adequate in
early-stage diseases, whereas interventions targeting downstream
execution pathways may be necessary once organelle damage has been
established.
Prospects and remarks
Over the past several decades, scientific interest
has significantly shifted towards comprehending intracellular
communication through close-range, non-vesicular organelle
interactions. Among these, the contact sites between the ER and
mitochondria, two organelles essential for cellular homeostasis,
have emerged as prominent, multifunctional signaling hubs. As
elaborated in the present review, MERCs are not simply static
bridges but dynamic, protein-rich platforms that integrate and
regulate an astonishing variety of cellular processes, including
Ca2+ and Zn2+ flux, lipid biosynthesis and
transfer, redox balance, mitochondrial dynamics (fission, fusion
and mitophagy), autophagy, apoptosis, inflammation and senescence.
Current evidence clearly indicates that the exact structure and
function of these nanoscale domains are often altered during the
pathogenesis of a broad range of diseases, spanning
neurodegeneration and metabolic disorders to cancer and
cardiovascular conditions. MERC dysfunction is therefore now widely
recognized as a central pathogenic mechanism, rendering targeted
modulation of these interfaces a compelling yet complex therapeutic
frontier.
However, the translation of this knowledge into
effective clinical strategies is hindered by several fundamental
challenges that delineate the future trajectory of the field.
First, the intrinsic dynamic and heterogeneous characteristics of
MERCs pose a substantial obstacle (258,259). Their composition, abundance and
inter-membrane distance are not static but are precisely regulated
by cellular metabolic status, stress signals and tissue context
(260). This plasticity renders
broad, non-specific pharmacological manipulation perilous. A more
viable approach involves initially deciphering the endogenous,
homeostatic mechanisms that regulate MERC assembly and disassembly.
Progress in tool development is crucial in this regard. Emerging
techniques, including split-BioID for in situ proteomic
mapping of contact sites (261,262), next-generation
split-fluorescent or bioluminescent reporters for real-time
monitoring of MERC dynamics in live cells and organisms, and
high-throughput CRISPR screens targeting MERC morphology, will be
essential for uncovering these regulatory networks. Second, the
functional impact of MERCs is highly tissue- and context-specific,
which necessitates the adoption of a precision medicine approach.
In neurons, MERCs play a crucial role in supplying Ca2+
and ATP to maintain synaptic activity and plasticity. In
cardiomyocytes, they regulate the excitation-contraction coupling;
however, their over-activation may lead to Ca2+ overload
and apoptosis. In hepatocytes, they are essential for lipid
metabolism, and any disruption to them can result in steatosis.
Therefore, a therapeutic intervention aimed at stabilizing MERCs to
benefit neurons in AD might inadvertently aggravate the pathology
in DCM by facilitating pathological calcium transfer. Future
research should prioritize the creation of detailed 'MERC atlases'
that define their distinct molecular compositions and functional
outputs across various cell types and disease states to guide the
development of cell-selective modulators. Third, MERCs demonstrate
a crucial dualism, in which their activity needs to be sustained
within a narrow physiological range. Optimal coupling improves
mitochondrial bioenergetics and promotes survival. By contrast,
inadequate or excessive coupling can lead to pathology, either
through bioenergetic failure or via apoptotic Ca2+
overload, excessive fission and impaired quality control (39-41,229,232). This Yin-Yang characteristic
emphasizes that therapeutic strategies cannot simply aim to
'increase' or 'decrease' MERCs across the board. Instead, the
objective must be to' 'normalize' or 'rescue' their function,
restoring homeostasis regardless of whether the initial defect is
hyper-or hypo-connectivity. This necessitates drugs that can detect
and rectify the underlying functional deficit, rather than merely
addressing the structural parameter. Fourthly, the potential of
MERCs to act as sources of novel biomarkers remains largely
unexploited, yet it confronts both technical and conceptual
obstacles. Although the circulating levels of MERC-associated
proteins (such as GRP78 in diabetes) (263) exhibit promising prospects,
their lack of specificity represents a significant limitation,
given that these proteins play diverse roles within cells (264). More sophisticated strategies
are required. These may encompass PLA in circulating immune cells
to detect specific tethering interactions, advanced lipidomic
profiling of plasma to uncover signatures of disrupted MERC lipid
transfer, or the isolation and proteomic analysis of MERC-enriched
fractions from accessible tissues. The advancement of non-invasive
imaging techniques capable of assessing MERC integrity in
vivo persists as a distant yet potentially transformative
objective.
In conclusion, the research on MERCs has advanced
from descriptive ultrastructural analysis to an understanding of
their role as key integrators in cellular life-and-death decisions.
Their malfunction represents a common pathological node across
various diseases. The future research path requires a
multidisciplinary approach: (i) Fundamental discovery: Employing
cutting-edge tools in spatial proteomics, super-resolution imaging,
and organelle-specific gene editing to elucidate the regulatory
mechanisms of MERCs. (ii) Contextual precision: Systematically
mapping the biological characteristics of MERCs across various
tissues and disease stages to enable targeted interventions. (iii)
Therapeutic innovation: Transcending conventional targets to
develop a novel category of 'interface therapeutics'-small
molecules, peptides, or nanotechnologies that can precisely
modulate the MERC interactome. (iv) Biomarker translation:
Developing specific, MERC-centered diagnostic markers for early
disease detection and patient stratification. By adopting this
comprehensive framework, future research will not only enhance the
understanding of these remarkable organelles but also open up new
possibilities for diagnosing and treating some of the most
widespread and challenging human diseases. The ultimate objective
is to harness the potential of inter-organellar communication,
shifting the therapeutic paradigm from targeting individual
pathways to restoring the balance of the cellular network.
Availability of data and materials
Not applicable.
Authors' contributions
HML, YL, WW and XC wrote the manuscript, and
prepared the figures and tables. WW and CT conceived and revised
the manuscript. All authors read and approved the final version of
the manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Central Zhejiang Science
and Technology Innovation Corridor Joint Fund of Zhejiang
Provincial Natural Science Foundation of China (grant no.
LJHSQY26H060003) and the Key R&D Program of Zhejiang (grant no.
2026C02A1194).
References
|
1
|
Prinz WA, Toulmay A and Balla T: The
functional universe of membrane contact sites. Nat Rev Mol Cell
Biol. 21:7–24. 2020. View Article : Google Scholar
|
|
2
|
Scorrano L, De Matteis MA, Emr S, Giordano
F, Hajnóczky G, Kornmann B, Lackner LL, Levine TP, Pellegrini L,
Reinisch K, et al: Coming together to define membrane contact
sites. Nat Commun. 10:12872019. View Article : Google Scholar
|
|
3
|
Csordás G, Weaver D and Hajnóczky G:
Endoplasmic reticulum-mitochondrial contactology: Structure and
signaling functions. Trends Cell Biol. 28:523–540. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Helle SC, Kanfer G, Kolar K, Lang A,
Michel AH and Kornmann B: Organization and function of membrane
contact sites. Biochim Biophys Acta. 1833:2526–2541. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rowland AA and Voeltz GK: Endoplasmic
reticulum-mitochondria contacts: function of the junction. Nat Rev
Mol Cell Biol. 13:607–625. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Copeland DE and Dalton AJ: An association
between mitochondria and the endoplasmic reticulum in cells of the
pseudobranch gland of a teleost. J Biophys Biochem Cytol.
5:393–296. 1959. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Montisano DF, Cascarano J, Pickett CB and
James TW: Association between mitochondria and rough endoplasmic
reticulum in rat liver. Anat Rec. 203:441–450. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pickett CB, Montisano D, Eisner D and
Cascarano J: The physical association between rat liver
mitochondria and rough endoplasmic reticulum. I. Isolation,
electron microscopic examination and sedimentation equilibrium
centrifugation analyses of rough endoplasmic
reticulum-mitochondrial complexes. Exp Cell Res. 128:343–352. 1980.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou X, Feng M, Li Z, Gan T and Zhang Y,
Ma X, Wu R, Xie Y, Song F, Yang G and Zhang Y: ER-Mitochondria
Tethering and Calcium Flux: A Core Mechanism for Biomineralization.
FASEB J. 40:e717512026. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rizzuto R, Pinton P, Carrington W, Fay FS,
Fogarty KE, Lifshitz LM, Tuft RA and Pozzan T: Close contacts with
the endoplasmic reticulum as determinants of mitochondrial Ca2+
responses. Science. 280:1763–1766. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Csordás G, Thomas AP and Hajnóczky G:
Quasi-synaptic calcium signal transmission between endoplasmic
reticulum and mitochondria. EMBO J. 18:96–108. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tuncay E, Bitirim CV, Olgar Y, Durak A,
Rutter GA and Turan B: Zn(2+)-transporters ZIP7 and ZnT7 play
important role in progression of cardiac dysfunction via affecting
sarco(endo) plasmic reticulum-mitochondria coupling in
hyperglycemic cardiomyocytes. Mitochondrion. 44:41–52. 2019.
View Article : Google Scholar
|
|
13
|
Olgar Y, Tuncay E and Turan B:
Mitochondria-targeting antioxidant provides cardioprotection
through regulation of cytosolic and mitochondrial Zn(2+) levels
with re-distribution of Zn(2+)-transporters in aged rat
cardiomyocytes. Int J Mol Sci. 20:37832019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Song Y, Geng W, Zhu D, Liang H, Du Z, Tong
B, Wang K, Li S, Gao Y, Feng X, et al: SYNJ2BP ameliorates
intervertebral disc degeneration by facilitating
mitochondria-associated endoplasmic reticulum membrane formation
and mitochondrial Zn(2+) homeostasis. Free Radic Biol Med.
212:220–233. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Friedman JR, Lackner LL, West M,
DiBenedetto JR, Nunnari J and Voeltz GK: ER tubules mark sites of
mitochondrial division. Science. 334:358–362. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Korobova F, Ramabhadran V and Higgs HN: An
actin-dependent step in mitochondrial fission mediated by the
ER-associated formin INF2. Science. 339:464–746. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou R, Yazdi AS, Menu P and Tschopp J: A
role for mitochondria in NLRP3 inflammasome activation. Nature.
469:221–225. 2011. View Article : Google Scholar
|
|
18
|
Booth DM, Enyedi B, Geiszt M, Várnai P and
Hajnóczky G: Redox Nanodomains Are Induced by and Control Calcium
Signaling at the ER-Mitochondrial Interface. Mol Cell. 63:240–248.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vance JE: MAM (mitochondria-associated
membranes) in mammalian cells: lipids and beyond. Biochim Biophys
Acta. 1841:595–609. 2014. View Article : Google Scholar
|
|
20
|
Arruda AP, Pers BM, Parlakgül G, Güney E,
Inouye K and Hotamisligil GS: Chronic enrichment of hepatic
endoplasmic reticulum-mitochondria contact leads to mitochondrial
dysfunction in obesity. Nat Med. 20:1427–1435. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Casas-Martinez JC, Xia Q, Li P,
Borja-Gonzalez M, Miranda-Vizuete A, McDermott E, Dockery P,
Quinlan LR, Goljanek-Whysall K, Samali A and McDonagh B: Adaptive
ER stress promotes mitochondrial remodelling and longevity through
PERK-dependent MERCS assembly. Cell Death Differ. 33:732–747. 2026.
View Article : Google Scholar :
|
|
22
|
Gelmetti V, De Rosa P, Torosantucci L,
Marini ES, Romagnoli A, Di Rienzo M, Arena G, Vignone D, Fimia GM
and Valente EM: PINK1 and BECN1 relocalize at
mitochondria-associated membranes during mitophagy and promote
ER-mitochondria tethering and autophagosome formation. Autophagy.
13:654–669. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hamasaki M, Furuta N, Matsuda A, Nezu A,
Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et
al: Autophagosomes form at ER-mitochondria contact sites. Nature.
495:389–393. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang JY and Yang WY: Bit-by-bit autophagic
removal of parkin-labelled mitochondria. Nat Commun. 4:24282013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Simmen T, Aslan JE, Blagoveshchenskaya AD,
Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM and
Thomas G: PACS-2 controls endoplasmic reticulum-mitochondria
communication and Bid-mediated apoptosis. EMBO J. 24:717–729. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Giorgi C, Wieckowski MR, Pandolfi PP and
Pinton P: Mitochondria associated membranes (MAMs) as critical hubs
for apoptosis. Commun Integr Biol. 4:334–335. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ziegler DV, Vindrieux D, Goehrig D, Jaber
S, Collin G, Griveau A, Wiel C, Bendridi N, Djebali S, Farfariello
V, et al: Calcium channel ITPR2 and mitochondria-ER contacts
promote cellular senescence and aging. Nat Commun. 12:7202021.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Puebla-Huerta A, Huerta H,
Quezada-Gutierez C, Morgado-Caceres P, Casanova-Canelo C, Nino SA,
Linsambarth S, Diaz-Rivera O, Lopez-Dominguez JA, Rodriguez-Lopez
S, et al: Calcium (Ca(2+)) fluxes at mitochondria-ER contact sites
(MERCS) are a new target of senolysis in therapy-induced senescence
(TIS). NPJ Aging. 11:112025. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Galmes R, Houcine A, van Vliet AR,
Agostinis P, Jackson CL and Giordano F: ORP5/ORP8 localize to
endoplasmic reticulum-mitochondria contacts and are involved in
mitochondrial function. EMBO Rep. 17:800–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lock JT, Sinkins WG and Schilling WP:
Protein S-glutathionylation enhances Ca2+-induced Ca2+ release via
the IP3 receptor in cultured aortic endothelial cells. J Physiol.
590:3431–3447. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li JH, Yue W, Huang Z, Chen ZQ, Zhan Q,
Ren FB, Liu JY and Fu SB: Calcium overload induces C6 rat glioma
cell apoptosis in sonodynamic therapy. Int J Radiat Biol.
87:1061–1066. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Higo T, Hattori M, Nakamura T, Natsume T,
Michikawa T and Mikoshiba K: Subtype-specific and ER lumenal
environment-dependent regulation of inositol 1,4,5-trisphosphate
receptor type 1 by ERp44. Cell. 120:85–98. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li G, Mongillo M, Chin KT, Harding H, Ron
D, Marks AR and Tabas I: Role of ERO1-alpha-mediated stimulation of
inositol 1,4,5-triphosphate receptor activity in endoplasmic
reticulum stress-induced apoptosis. J Cell Biol. 186:783–792. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li C, Li L, Yang M, Yang J, Zhao C, Han Y,
Zhao H, Jiang N, Wei L, Xiao Y, et al: PACS-2 ameliorates tubular
injury by facilitating endoplasmic reticulum-mitochondria contact
and mitophagy in diabetic nephropathy. Diabetes. 71:1034–1050.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang M, Zhao L, Gao P, Zhu X, Han Y, Chen
X, Li L, Xiao Y, Wei L, Li C, et al: DsbA-L ameliorates high
glucose induced tubular damage through maintaining MAM integrity.
EBioMedicine. 43:607–619. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hernández-Alvarez MI, Sebastián D, Vives
S, Ivanova S, Bartoccioni P, Kakimoto P, Plana N, Veiga SR,
Hernández V, Vasconcelos N, et al: Deficient endoplasmic
reticulum-mitochondrial phosphatidylserine transfer causes liver
disease. Cell. 177:881–895.e17. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rieusset J, Fauconnier J, Paillard M,
Belaidi E, Tubbs E, Chauvin MA, Durand A, Bravard A, Teixeira G,
Bartosch B, et al: Disruption of calcium transfer from ER to
mitochondria links alterations of mitochondria-associated ER
membrane integrity to hepatic insulin resistance. Diabetologia.
59:614–623. 2016. View Article : Google Scholar
|
|
38
|
Shuwen H, Yinhang W, Jing Z, Qiang Y,
Yizhen J, Quan Q, Yin J, Jiang L and Xi Y: Cholesterol induction in
CD8(+) T cell exhaustion in colorectal cancer via the regulation of
endoplasmic reticulum-mitochondria contact sites. Cancer Immunol
Immunother. 72:4441–4456. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang JF, Xing X, Luo L, Zhou XW, Feng JX,
Huang KB, Liu H, Jin S, Liu YN, Zhang SH, et al: Mitochondria-ER
contact mediated by MFN2-SERCA2 interaction supports CD8(+) T cell
metabolic fitness and function in tumors. Sci Immunol.
8:eabq24242023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dong J, Chen L, Ye F, Tang J, Liu B, Lin
J, Zhou PH, Lu B, Wu M, Lu JH, et al: Mic19 depletion impairs
endoplasmic reticulum-mitochondrial contacts and mitochondrial
lipid metabolism and triggers liver disease. Nat Commun.
15:1682024. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li Y, Huang D, Jia L, Shangguan F, Gong S,
Lan L, Song Z, Xu J, Yan C, Chen T, et al: LonP1 links
Mitochondria-ER interaction to regulate heart function. Research
(Wash D C). 6:01752023.PubMed/NCBI
|
|
42
|
Bernard-Marissal N, van Hameren G, Juneja
M, Pellegrino C, Louhivuori L, Bartesaghi L, Rochat C, El Mansour
O, Médard JJ, Croisier M, et al: Altered interplay between
endoplasmic reticulum and mitochondria in Charcot-Marie-Tooth type
2A neuropathy. Proc Natl Acad Sci USA. 116:2328–2337. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xu L, Xu Y, Jiang Y, Jiang J, Chen S, Sun
D, Li S, Wei F and Zhu H: IP3R2 regulates apoptosis by Ca2+
transfer through mitochondria-ER contacts in hypoxic photoreceptor
injury. Exp Eye Res. 245:1099652024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhu R, Liu L, Mao T, Wang X, Li Y, Li T,
Lv S, Zeng S, Fu N, Li N, et al: Mfn2 regulates mitochondria and
mitochondria-associated endoplasmic reticulum membrane function in
neurodegeneration induced by repeated sevoflurane exposure. Exp
Neurol. 377:1148072024. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wu S, Lu Q, Ding Y, Wu Y, Qiu Y, Wang P,
Mao X, Huang K, Xie Z and Zou MH: Hyperglycemia-driven inhibition
of AMP-Activated protein kinase α2 induces diabetic cardiomyopathy
by promoting mitochondria-associated endoplasmic reticulum
membranes in vivo. Circulation. 139:1913–1936. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Fueller J, Egorov MV, Walther KA, Sabet O,
Mallah J, Grabenbauer M and Kinkhabwala A: Subcellular partitioning
of protein tyrosine phosphatase 1B to the endoplasmic reticulum and
mitochondria depends sensitively on the composition of its tail
anchor. PLoS One. 10:e01394292015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bernhard W and Rouiller C: Close
topographical relationship between mitochondria and ergastoplasm of
liver cells in a definite phase of cellular activity. J Biophys
Biochem Cytol. 2(4 Suppl): S73–S78. 1956. View Article : Google Scholar
|
|
48
|
Bernhard W, Haguenau F, Gautier A and
Oberling C: Submicroscopical structure of cytoplasmic basophils in
the liver, pancreas and salivary gland; study of ultrafine slices
by electron microscope. Z Zellforsch Mikrosk Anat. 37:281–300.
1952.In Undetermined Language. View Article : Google Scholar
|
|
49
|
Vance JE: Phospholipid synthesis in a
membrane fraction associated with mitochondria. J Biol Chem.
265:7248–7256. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Csordás G, Renken C, Várnai P, Walter L,
Weaver D, Buttle KF, Balla T, Mannella CA and Hajnóczky G:
Structural and functional features and significance of the physical
linkage between ER and mitochondria. J Cell Biol. 174:915–921.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Murley A, Lackner LL, Osman C, West M,
Voeltz GK, Walter P and Nunnari J: ER-associated mitochondrial
division links the distribution of mitochondria and mitochondrial
DNA in yeast. Elife. 2:e004222013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wu Y, Whiteus C, Xu CS, Hayworth KJ,
Weinberg RJ, Hess HF and De Camilli P: Contacts between the
endoplasmic reticulum and other membranes in neurons. Proc Natl
Acad Sci USA. 114:E4859–E4867. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Elgass KD, Smith EA, LeGros MA, Larabell
CA and Ryan MT: Analysis of ER-mitochondria contacts using
correlative fluorescence microscopy and soft X-ray tomography of
mammalian cells. J Cell Sci. 128:2795–2804. 2015.PubMed/NCBI
|
|
54
|
Filadi R, Greotti E, Turacchio G, Luini A,
Pozzan T and Pizzo P: Mitofusin 2 ablation increases endoplasmic
reticulum-mitochondria coupling. Proc Natl Acad Sci USA.
112:E2174–E2181. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhao YG, Chen Y, Miao G, Zhao H, Qu W, Li
D, Wang Z, Liu N, Li L, Chen S, et al: The ER-Localized
transmembrane protein EPG-3/VMP1 Regulates SERCA Activity to
Control ER-Isolation membrane contacts for autophagosome formation.
Mol Cell. 67:974–989.e6. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kremer A, Lippens S, Bartunkova S,
Asselbergh B, Blanpain C, Fendrych M, Goossens A, Holt M, Janssens
S, Krols M, et al: Developing 3D SEM in a broad biological context.
J Microsc. 259:80–96. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Denk W and Horstmann H: Serial block-face
scanning electron microscopy to reconstruct three-dimensional
tissue nanostructure. PLoS Biol. 2:e3292004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bushby AJ, P'Ng KM, Young RD, Pinali C,
Knupp C and Quantock AJ: Imaging three-dimensional tissue
architectures by focused ion beam scanning electron microscopy. Nat
Protoc. 6:845–858. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Obara CJ, Nixon-Abell J, Moore AS, Riccio
F, Hoffman DP, Shtengel G, Xu CS, Schaefer K, Pasolli HA, Masson
JB, et al: Motion of VAPB molecules reveals ER-mitochondria contact
site subdomains. Nature. 626:169–176. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Goldberg MW and Fiserova J: Immunogold
labelling for scanning electron microscopy. Methods Mol Biol.
657:297–313. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ma JH, Shen S, Wang JJ, He Z, Poon A, Li
J, Qu J and Zhang SX: Comparative proteomic analysis of the
mitochondria-associated ER membrane (MAM) in a long-term type 2
diabetic rodent model. Sci Rep. 7:20622017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
de Brito OM and Scorrano L: Mitofusin 2
tethers endoplasmic reticulum to mitochondria. Nature. 456:605–610.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Brooks C, Wei Q, Cho SG and Dong Z:
Regulation of mitochondrial dynamics in acute kidney injury in cell
culture and rodent models. J Clin Invest. 119:1275–1285. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Doghman-Bouguerra M and Lalli E:
ER-mitochondria interactions: Both strength and weakness within
cancer cells. Biochim Biophys Acta Mol Cell Res. 1866:650–662.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yang Z, Zhao X, Xu J, Shang W and Tong C:
A novel fluorescent reporter detects plastic remodeling of
mitochondria-ER contact sites. J Cell Sci. 131:jcs2086862018.
View Article : Google Scholar
|
|
66
|
Magliery TJ, Wilson CG, Pan W, Mishler D,
Ghosh I, Hamilton AD and Regan L: Detecting protein-protein
interactions with a green fluorescent protein fragment reassembly
trap: scope and mechanism. J Am Chem Soc. 127:146–157. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Cabantous S, Terwilliger TC and Waldo GS:
Protein tagging and detection with engineered self-assembling
fragments of green fluorescent protein. Nat Biotechnol. 23:102–107.
2005. View Article : Google Scholar
|
|
68
|
Cieri D, Vicario M, Giacomello M, Vallese
F, Filadi R, Wagner T, Pozzan T, Pizzo P, Scorrano L, Brini M and
Calì T: SPLICS: A split green fluorescent protein-based contact
site sensor for narrow and wide heterotypic organelle
juxtaposition. Cell Death Differ. 25:1131–1145. 2018. View Article : Google Scholar :
|
|
69
|
Kakimoto Y, Tashiro S, Kojima R, Morozumi
Y, Endo T and Tamura Y: Visualizing multiple inter-organelle
contact sites using the organelle-targeted split-GFP system. Sci
Rep. 8:61752018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Shim SH, Xia C, Zhong G, Babcock HP,
Vaughan JC, Huang B, Wang X, Xu C, Bi GQ and Zhuang X:
Super-resolution fluorescence imaging of organelles in live cells
with photoswitchable membrane probes. Proc Natl Acad Sci USA.
109:13978–13983. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Modi S, López-Doménech G, Halff EF,
Covill-Cooke C, Ivankovic D, Melandri D, Arancibia-Cárcamo IL,
Burden JJ, Lowe AR and Kittler JT: Miro clusters regulate
ER-mitochondria contact sites and link cristae organization to the
mitochondrial transport machinery. Nat Commun. 10:43992019.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhanghao K, Chen X, Liu W, Li M, Liu Y,
Wang Y, Luo S, Wang X, Shan C, Xie H, et al: Super-resolution
imaging of fluorescent dipoles via polarized structured
illumination microscopy. Nat Commun. 10:46942019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Guo Y, Li D, Zhang S, Yang Y, Liu JJ, Wang
X, Liu C, Milkie DE, Moore RP, Tulu US, et al: Visualizing
intracellular organelle and cytoskeletal interactions at nanoscale
resolution on millisecond timescales. Cell. 175:1430–1442.e17.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Inoue T, Heo WD, Grimley JS, Wandless TJ
and Meyer T: An inducible translocation strategy to rapidly
activate and inhibit small GTPase signaling pathways. Nat Methods.
2:415–418. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Csordás G, Várnai P, Golenár T, Roy S,
Purkins G, Schneider TG, Balla T and Hajnóczky G: Imaging
interorganelle contacts and local calcium dynamics at the
ER-mitochondrial interface. Mol Cell. 39:121–132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Fredriksson S, Gullberg M, Jarvius J,
Olsson C, Pietras K, Gústafsdóttir SM, Ostman A and Landegren U:
Protein detection using proximity-dependent DNA ligation assays.
Nat Biotechnol. 20:473–477. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Gullberg M, Gústafsdóttir SM, Schallmeiner
E, Jarvius J, Bjarnegård M, Betsholtz C, Landegren U and
Fredriksson S: Cytokine detection by antibody-based proximity
ligation. Proc Natl Acad Sci USA. 101:8420–8424. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Söderberg O, Gullberg M, Jarvius M,
Ridderstråle K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P,
Bahram F, Larsson LG and Landegren U: Direct observation of
individual endogenous protein complexes in situ by proximity
ligation. Nat Methods. 3:995–1000. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Lever JD and Chappell JB: Mitochondria
isolated from rat brown adipose tissue and liver. J Biophys Biochem
Cytol. 4:287–290. 1958. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
De Vos KJ, Mórotz GM, Stoica R, Tudor EL,
Lau KF, Ackerley S, Warley A, Shaw CE and Miller CC: VAPB interacts
with the mitochondrial protein PTPIP51 to regulate calcium
homeostasis. Hum Mol Genet. 21:1299–1311. 2012. View Article : Google Scholar :
|
|
81
|
Iwasawa R, Mahul-Mellier AL, Datler C,
Pazarentzos E and Grimm S: Fis1 and Bap31 bridge the
mitochondria-ER interface to establish a platform for apoptosis
induction. EMBO J. 30:556–568. 2011. View Article : Google Scholar
|
|
82
|
Szabadkai G, Bianchi K, Várnai P, De
Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T and Rizzuto
R: Chaperone-mediated coupling of endoplasmic reticulum and
mitochondrial Ca2+ channels. J Cell Biol. 175:901–911. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Wieckowski MR, Giorgi C, Lebiedzinska M,
Duszynski J and Pinton P: Isolation of mitochondria-associated
membranes and mitochondria from animal tissues and cells. Nat
Protoc. 4:1582–1590. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
David C, Koch J, Oeljeklaus S, Laernsack
A, Melchior S, Wiese S, Schummer A, Erdmann R, Warscheid B and
Brocard C: A combined approach of quantitative interaction
proteomics and live-cell imaging reveals a regulatory role for
endoplasmic reticulum (ER) reticulon homology proteins in
peroxisome biogenesis. Mol Cell Proteomics. 12:2408–2425. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Kornmann B, Currie E, Collins SR,
Schuldiner M, Nunnari J, Weissman JS and Walter P: An
ER-mitochondria tethering complex revealed by a synthetic biology
screen. Science. 325:477–481. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Lahiri S, Chao JT, Tavassoli S, Wong AK,
Choudhary V, Young BP, Loewen CJ and Prinz WA: A conserved
endoplasmic reticulum membrane protein complex (EMC) facilitates
phospholipid transfer from the ER to mitochondria. PLoS Biol.
12:e10019692014. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Poston CN, Krishnan SC and Bazemore-Walker
CR: In-depth proteomic analysis of mammalian
mitochondria-associated membranes (MAM). J Proteomics. 79:219–230.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Zhang A, Williamson CD, Wong DS, Bullough
MD, Brown KJ, Hathout Y and Colberg-Poley AM: Quantitative
proteomic analyses of human cytomegalovirus-induced restructuring
of endoplasmic reticulum-mitochondrial contacts at late times of
infection. Mol Cell Proteomics. 10:M111.0099362011. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Horner SM, Wilkins C, Badil S,
Iskarpatyoti J and Gale M Jr: Proteomic analysis of
mitochondrial-associated ER membranes (MAM) during RNA virus
infection reveals dynamic changes in protein and organelle
trafficking. PLoS One. 10:e01179632015. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Lam SS, Martell JD, Kamer KJ, Deerinck TJ,
Ellisman MH, Mootha VK and Ting AY: Directed evolution of APEX2 for
electron microscopy and proximity labeling. Nat Methods. 12:51–54.
2015. View Article : Google Scholar :
|
|
91
|
Cho IT, Adelmant G, Lim Y, Marto JA, Cho G
and Golden JA: Ascorbate peroxidase proximity labeling coupled with
biochemical fractionation identifies promoters of endoplasmic
reticulum-mitochondrial contacts. J Biol Chem. 292:16382–16392.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Hung V, Lam SS, Udeshi ND, Svinkina T,
Guzman G, Mootha VK, Carr SA and Ting AY: Proteomic mapping of
cytosol-facing outer mitochondrial and ER membranes in living human
cells by proximity biotinylation. Elife. 6:e244632017. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Han Y, Branon TC, Martell JD, Boassa D,
Shechner D, Ellisman MH and Ting A: Directed evolution of split
APEX2 peroxidase. ACS Chem Biol. 14:619–635. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Baudier J: ATAD3 proteins: Brokers of a
mitochondria-endoplasmic reticulum connection in mammalian cells.
Biol Rev Camb Philos Soc. 93:827–844. 2018. View Article : Google Scholar
|
|
95
|
Doghman-Bouguerra M, Granatiero V, Sbiera
S, Sbiera I, Lacas-Gervais S, Brau F, Fassnacht M, Rizzuto R and
Lalli E: FATE1 antagonizes calcium- and drug-induced apoptosis by
uncoupling ER and mitochondria. EMBO Rep. 17:1264–1280. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Issop L, Fan J, Lee S, Rone MB, Basu K,
Mui J and Papadopoulos V: Mitochondria-associated membrane
formation in hormone-stimulated Leydig cell steroidogenesis: Role
of ATAD3. Endocrinology. 156:334–345. 2015. View Article : Google Scholar
|
|
97
|
Namba T: BAP31 regulates mitochondrial
function via interaction with Tom40 within ER-mitochondria contact
sites. Sci Adv. 5:eaaw13862019. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Stoica R, De Vos KJ, Paillusson S, Mueller
S, Sancho RM, Lau KF, Vizcay-Barrena G, Lin WL, Xu YF, Lewis J, et
al: ER-mitochondria associations are regulated by the VAPB-PTPIP51
interaction and are disrupted by ALS/FTD-associated TDP-43. Nat
Commun. 5:39962014. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Hirabayashi Y, Kwon SK, Paek H, Pernice
WM, Paul MA, Lee J, Erfani P, Raczkowski A, Petrey DS, Pon LA and
Polleux F: ER-mitochondria tethering by PDZD8 regulates Ca(2+)
dynamics in mammalian neurons. Science. 358:623–630. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Manor U, Bartholomew S, Golani G,
Christenson E, Kozlov M, Higgs H, Spudich J and Lippincott-Schwartz
J: A mitochondria-anchored isoform of the actin-nucleating spire
protein regulates mitochondrial division. Elife. 4:e088282015.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Lim Y, Cho IT, Schoel LJ, Cho G and Golden
JA: Hereditary spastic paraplegia-linked REEP1 modulates
endoplasmic reticulum/mitochondria contacts. Ann Neurol.
78:679–696. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Di Mattia T, Wilhelm LP, Ikhlef S,
Wendling C, Spehner D, Nominé Y, Giordano F, Mathelin C, Drin G,
Tomasetto C and Alpy F: Identification of MOSPD2, a novel scaffold
for endoplasmic reticulum membrane contact sites. EMBO Rep.
19:e454532018. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X,
Huang K, Xie Z and Zou MH: Binding of FUN14 Domain Containing 1
With Inositol 1,4,5-Trisphosphate receptor in
mitochondria-associated endoplasmic reticulum membranes maintains
mitochondrial dynamics and function in hearts in vivo. Circulation.
136:2248–2266. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Paillard M, Tubbs E, Thiebaut PA, Gomez L,
Fauconnier J, Da Silva CC, Teixeira G, Mewton N, Belaidi E, Durand
A, et al: Depressing mitochondria-reticulum interactions protects
cardiomyocytes from lethal hypoxia-reoxygenation injury.
Circulation. 128:1555–1565. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Hayashi T and Su TP: Sigma-1 receptor
chaperones at the ER-mitochondrion interface regulate Ca(2+)
signaling and cell survival. Cell. 131:596–610. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Matsuzaki H, Fujimoto T, Tanaka M and
Shirasawa S: Tespa1 is a novel component of mitochondria-associated
endoplasmic reticulum membranes and affects mitochondrial calcium
flux. Biochem Biophys Res Commun. 433:322–326. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Wang PT, Garcin PO, Fu M, Masoudi M,
St-Pierre P, Panté N and Nabi IR: Distinct mechanisms controlling
rough and smooth endoplasmic reticulum contacts with mitochondria.
J Cell Sci. 128:2759–2765. 2015.PubMed/NCBI
|
|
108
|
Goetz JG, Genty H, St-Pierre P, Dang T,
Joshi B, Sauvé R, Vogl W and Nabi IR: Reversible interactions
between smooth domains of the endoplasmic reticulum and
mitochondria are regulated by physiological cytosolic Ca2+ levels.
J Cell Sci. 120(Pt 20): 3553–3564. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Wang HJ, Guay G, Pogan L, Sauvé R and Nabi
IR: Calcium regulates the association between mitochondria and a
smooth subdomain of the endoplasmic reticulum. J Cell Biol.
150:1489–1498. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Sala-Vila A, Navarro-Lérida I,
Sánchez-Alvarez M, Bosch M, Calvo C, López JA, Calvo E, Ferguson C,
Giacomello M, Serafini A, et al: Interplay between hepatic
mitochondria-associated membranes, lipid metabolism and caveolin-1
in mice. Sci Rep. 6:273512016. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Prudent J, Zunino R, Sugiura A, Mattie S,
Shore GC and McBride HM: MAPL SUMOylation of Drp1 stabilizes an
ER/Mitochondrial platform required for cell death. Mol Cell.
59:941–955. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Sesaki H, Adachi Y, Kageyama Y, Itoh K and
Iijima M: In vivo functions of Drp1: Lessons learned from yeast
genetics and mouse knockouts. Biochim Biophys Acta. 1842:1179–1185.
2014. View Article : Google Scholar
|
|
113
|
Bonneau B, Ando H, Kawaai K, Hirose M,
Takahashi-Iwanaga H and Mikoshiba K: IRBIT controls apoptosis by
interacting with the Bcl-2 homolog, Bcl2l10, and by promoting
ER-mitochondria contact. Elife. 5:e198962016. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Sugiura A, Nagashima S, Tokuyama T, Amo T,
Matsuki Y, Ishido S, Kudo Y, McBride HM, Fukuda T, Matsushita N, et
al: MITOL regulates endoplasmic reticulum-mitochondria contacts via
Mitofusin2. Mol Cell. 51:20–34. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Yi M, Weaver D and Hajnóczky G: Control of
mitochondrial motility and distribution by the calcium signal: A
homeostatic circuit. J Cell Biol. 167:661–672. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Macaskill AF, Rinholm JE, Twelvetrees AE,
Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D
and Kittler JT: Miro1 is a calcium sensor for glutamate
receptor-dependent localization of mitochondria at synapses.
Neuron. 61:541–555. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Betz C, Stracka D, Prescianotto-Baschong
C, Frieden M, Demaurex N and Hall MN: Feature Article: mTOR complex
2-Akt signaling at mitochondria-associated endoplasmic reticulum
membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad
Sci USA. 110:12526–12534. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Reali V, Mehdawy B, Nardacci R, Filomeni
G, Risuglia A, Rossin F, Antonioli M, Marsella C, Fimia GM,
Piacentini M and Di Sano F: Reticulon protein-1C is a key component
of MAMs. Biochim Biophys Acta. 1853:733–745. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Filadi R, Leal NS, Schreiner B, Rossi A,
Dentoni G, Pinho CM, Wiehager B, Cieri D, Calì T, Pizzo P and
Ankarcrona M: TOM70 Sustains Cell Bioenergetics by Promoting
IP3R3-Mediated ER to Mitochondria Ca(2+) Transfer. Curr Biol.
28:369–382.e6. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
D'Eletto M, Rossin F, Occhigrossi L,
Farrace MG, Faccenda D, Desai R, Marchi S, Refolo G, Falasca L,
Antonioli M, et al: Transglutaminase type 2 regulates
ER-mitochondria contact sites by interacting with GRP75. Cell Rep.
25:3573–3581.e4. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Angebault C, Fauconnier J, Patergnani S,
Rieusset J, Danese A, Affortit CA, Jagodzinska J, Mégy C, Quiles M,
Cazevieille C, et al: ER-mitochondria cross-talk is regulated by
the Ca(2+) sensor NCS1 and is impaired in Wolfram syndrome. Sci
Signal. 11:eaaq13802018. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Thoudam T, Ha CM, Leem J, Chanda D, Park
JS, Kim HJ, Jeon JH, Choi YK, Liangpunsakul S, Huh YH, et al: PDK4
Augments ER-Mitochondria Contact to Dampen Skeletal Muscle Insulin
Signaling During Obesity. Diabetes. 68:571–586. 2019. View Article : Google Scholar
|
|
123
|
Yuan L, Liu Q, Wang Z, Hou J and Xu P:
EI24 tethers endoplasmic reticulum and mitochondria to regulate
autophagy flux. Cell Mol Life Sci. 77:1591–1606. 2020. View Article : Google Scholar
|
|
124
|
Zheng P, Chen Q, Tian X, Qian N, Chai P,
Liu B, Hu J, Blackstone C, Zhu D, Teng J and Chen J: DNA damage
triggers tubular endoplasmic reticulum extension to promote
apoptosis by facilitating ER-mitochondria signaling. Cell Res.
28:833–854. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Liu Y, Ma X, Fujioka H, Liu J, Chen S and
Zhu X: DJ-1 regulates the integrity and function of ER-mitochondria
association through interaction with IP3R3-Grp75-VDAC1. Proc Natl
Acad Sci USA. 116:25322–25328. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Norkett R, Modi S, Birsa N, Atkin TA,
Ivankovic D, Pathania M, Trossbach SV, Korth C, Hirst WD and
Kittler JT: DISC1-dependent regulation of mitochondrial dynamics
controls the morphogenesis of complex neuronal dendrites. J Biol
Chem. 291:613–629. 2016. View Article : Google Scholar :
|
|
127
|
Park SJ, Lee SB, Suh Y, Kim SJ, Lee N,
Hong JH, Park C, Woo Y, Ishizuka K, Kim JH, et al: DISC1 modulates
neuronal stress responses by gate-keeping ER-mitochondria Ca(2+)
transfer through the MAM. Cell Rep. 21:2748–2759. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Stoica R, Paillusson S, Gomez-Suaga P,
Mitchell JC, Lau DH, Gray EH, Sancho RM, Vizcay-Barrena G, De Vos
KJ, Shaw CE, et al: ALS/FTD-associated FUS activates GSK-3β to
disrupt the VAPB-PTPIP51 interaction and ER-mitochondria
associations. EMBO Rep. 17:1326–1342. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Cosson P, Marchetti A, Ravazzola M and
Orci L: Mitofusin-2 independent juxtaposition of endoplasmic
reticulum and mitochondria: an ultrastructural study. PLoS One.
7:e462932012. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Lynes EM, Bui M, Yap MC, Benson MD,
Schneider B, Ellgaard L, Berthiaume LG and Simmen T: Palmitoylated
TMX and calnexin target to the mitochondria-associated membrane.
EMBO J. 31:457–470. 2012. View Article : Google Scholar
|
|
131
|
Cerqua C, Anesti V, Pyakurel A, Liu D,
Naon D, Wiche G, Baffa R, Dimmer KS and Scorrano L:
Trichoplein/mitostatin regulates endoplasmic reticulum-mitochondria
juxtaposition. EMBO Rep. 11:854–860. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Maltan L, Weiß S, Najjar H, Leopold M,
Lindinger S, Höglinger C, Höbarth L, Sallinger M, Grabmayr H,
Berlansky S, et al: Photocrosslinking-induced CRAC channel-like
Orai1 activation independent of STIM1. Nat Commun. 14:12862023.
View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Gross PA, Rodstein M, LaMontagne JR,
Kaslow RA, Saah AJ, Wallenstein S, Neufeld R, Denning C, Gaerlan P
and Quinnan GV: Epidemiology of acute respiratory illness during an
influenza outbreak in a nursing home. A prospective study. Arch
Intern Med. 148:559–561. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Gincel D, Zaid H and Shoshan-Barmatz V:
Calcium binding and translocation by the voltage-dependent anion
channel: A possible regulatory mechanism in mitochondrial function.
Biochem J. 358(Pt 1): 147–155. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Shoshan-Barmatz V and Golan M:
Mitochondrial VDAC1: Function in cell life and death and a target
for cancer therapy. Curr Med Chem. 19:714–735. 2012. View Article : Google Scholar
|
|
136
|
Benade J, Sher L, De Klerk S, Deshpande G,
Bester D, Marnewick JL, Sieck G, Laher I and Essop MF: The impact
of sugar-sweetened beverage consumption on the liver: A
proteomics-based analysis. Antioxidants (Basel). 9:5692020.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Belosludtsev KN, Dubinin MV, Belosludtseva
NV and Mironova GD: Mitochondrial Ca2+ transport: mechanisms,
molecular structures, and role in cells. Biochemistry (Mosc).
84:593–607. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Várnai P, Balla A, Hunyady L and Balla T:
Targeted expression of the inositol 1,4,5-triphosphate receptor
(IP3R) ligand-binding domain releases Ca2+ via endogenous IP3R
channels. Proc Natl Acad Sci USA. 102:7859–7864. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Tan Y, Mui D, Toan S, Zhu P, Li R and Zhou
H: SERCA overexpression improves mitochondrial quality control and
attenuates cardiac microvascular ischemia-reperfusion injury. Mol
Ther Nucleic Acids. 22:696–707. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Giorgi C, Baldassari F, Bononi A, Bonora
M, De Marchi E, Marchi S, Missiroli S, Patergnani S, Rimessi A,
Suski JM, et al: Mitochondrial Ca(2+) and apoptosis. Cell Calcium.
52:36–43. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Murphy E and Steenbergen C: Regulation of
mitochondrial Ca(2+) uptake. Annu Rev Physiol. 83:107–126. 2021.
View Article : Google Scholar
|
|
142
|
Stefanidou M, Maravelias C, Dona A and
Spiliopoulou C: Zinc: A multipurpose trace element. Arch Toxicol.
80:1–9. 2006. View Article : Google Scholar
|
|
143
|
Xu Z and Zhou J: Zinc and myocardial
ischemia/reperfusion injury. Biometals. 26:863–878. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Myers SA: Zinc transporters and zinc
signaling: new insights into their role in type 2 diabetes. Int J
Endocrinol. 2015:1675032015. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Kambe T, Tsuji T, Hashimoto A and Itsumura
N: The physiological, biochemical, and molecular roles of zinc
transporters in zinc homeostasis and metabolism. Physiol Rev.
95:749–784. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Zhang H, Yang N, He H, Chai J, Cheng X,
Zhao H, Zhou D, Teng T, Kong X, Yang Q and Xu Z: The zinc
transporter ZIP7 (Slc39a7) controls myocardial reperfusion injury
by regulating mitophagy. Basic Res Cardiol. 116:542021. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Song Y, Liang H, Li G, Ma L, Zhu D, Zhang
W, Tong B, Li S, Gao Y, Wu X, et al: The NLRX1-SLC39A7 complex
orchestrates mitochondrial dynamics and mitophagy to rejuvenate
intervertebral disc by modulating mitochondrial Zn(2+) trafficking.
Autophagy. 20:809–829. 2024. View Article : Google Scholar :
|
|
148
|
Burman JL, Pickles S, Wang C, Sekine S,
Vargas JNS, Zhang Z, Youle AM, Nezich CL, Wu X, Hammer JA and Youle
RJ: Mitochondrial fission facilitates the selective mitophagy of
protein aggregates. J Cell Biol. 216:3231–3247. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Murata D, Yamada T, Tokuyama T, Arai K,
Quirós PM, López-Otín C, Iijima M and Sesaki H: Mitochondrial
Safeguard: a stress response that offsets extreme fusion and
protects respiratory function via flickering-induced Oma1
activation. EMBO J. 39:e1050742020. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Kaufman RJ: Orchestrating the unfolded
protein response in health and disease. J Clin Invest.
110:1389–1398. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Fan Y, Lee K, Wang N and He JC: The role
of endoplasmic reticulum stress in diabetic nephropathy. Curr Diab
Rep. 17:172017. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Carreras-Sureda A, Jaña F, Urra H, Durand
S, Mortenson DE, Sagredo A, Bustos G, Hazari Y, Ramos-Fernández E,
Sassano ML, et al: Non-canonical function of IRE1α determines
mitochondria-associated endoplasmic reticulum composition to
control calcium transfer and bioenergetics. Nat Cell Biol.
21:755–767. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Acosta-Alvear D, Zhou Y, Blais A, Tsikitis
M, Lents NH, Arias C, Lennon CJ, Kluger Y and Dynlacht BD: XBP1
controls diverse cell type- and condition-specific transcriptional
regulatory networks. Mol Cell. 27:53–66. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Djordjević VB: Free radicals in cell
biology. Int Rev Cytol. 237:57–89. 2004. View Article : Google Scholar
|
|
155
|
Kowaltowski AJ, de Souza-Pinto NC,
Castilho RF and Vercesi AE: Mitochondria and reactive oxygen
species. Free Radic Biol Med. 47:333–343. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Bak DW and Weerapana E: Cysteine-mediated
redox signalling in the mitochondria. Mol Biosyst. 11:678–697.
2015. View Article : Google Scholar
|
|
157
|
Yoboue ED, Sitia R and Simmen T: Redox
crosstalk at endoplasmic reticulum (ER) membrane contact sites
(MCS) uses toxic waste to deliver messages. Cell Death Dis.
9:3312018. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Anelli T, Bergamelli L, Margittai E,
Rimessi A, Fagioli C, Malgaroli A, Pinton P, Ripamonti M, Rizzuto R
and Sitia R: Ero1α regulates Ca(2+) fluxes at the endoplasmic
reticulum-mitochondria interface (MAM). Antioxid Redox Signal.
16:1077–1087. 2012. View Article : Google Scholar
|
|
159
|
Raturi A, Gutiérrez T, Ortiz-Sandoval C,
Ruangkittisakul A, Herrera-Cruz MS, Rockley JP, Gesson K, Ourdev D,
Lou PH, Lucchinetti E, et al: TMX1 determines cancer cell
metabolism as a thiol-based modulator of ER-mitochondria Ca2+ flux.
J Cell Biol. 214:433–444. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Yoboue ED, Rimessi A, Anelli T, Pinton P
and Sitia R: Regulation of calcium fluxes by GPX8, a Type-II
transmembrane peroxidase enriched at the mitochondria-associated
endoplasmic reticulum membrane. Antioxid Redox Signal. 27:583–595.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Che Y, Chen Y, Wang Z, Zheng S, Xing K,
Yuan S and Zhong X: The combination of rhodosin and MMF prolongs
cardiac allograft survival by inhibiting DC maturation by promoting
mitochondrial fusion. Oxid Med Cell Longev. 2022:72603052022.
View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Al Ojaimi M, Salah A and El-Hattab AW:
Mitochondrial fission and fusion: Molecular mechanisms, biological
functions, and related disorders. Membranes (Basel). 12:8932022.
View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Otera H, Ishihara N and Mihara K: New
insights into the function and regulation of mitochondrial fission.
Biochim Biophys Acta. 1833:1256–1268. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Fonseca TB, Sánchez-Guerrero Á, Milosevic
I and Raimundo N: Mitochondrial fission requires DRP1 but not
dynamins. Nature. 570:E34–E42. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Chakrabarti R, Ji WK, Stan RV, de Juan
Sanz J, Ryan TA and Higgs HN: INF2-mediated actin polymerization at
the ER stimulates mitochondrial calcium uptake, inner membrane
constriction, and division. J Cell Biol. 217:251–268. 2018.
View Article : Google Scholar :
|
|
166
|
Arasaki K, Shimizu H, Mogari H, Nishida N,
Hirota N, Furuno A, Kudo Y, Baba M, Baba N, Cheng J, et al: A role
for the ancient SNARE syntaxin 17 in regulating mitochondrial
division. Dev Cell. 32:304–317. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Wu W, Lin C, Wu K, Jiang L, Wang X, Li W,
Zhuang H, Zhang X, Chen H, Li S, et al: FUNDC1 regulates
mitochondrial dynamics at the ER-mitochondrial contact site under
hypoxic conditions. EMBO J. 35:1368–1384. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Hu Y, Chen H, Zhang L, Lin X, Li X, Zhuang
H, Fan H, Meng T, He Z, Huang H, et al: The AMPK-MFN2 axis
regulates MAM dynamics and autophagy induced by energy stresses.
Autophagy. 17:1142–1156. 2021. View Article : Google Scholar :
|
|
169
|
van der Bliek AM, Shen Q and Kawajiri S:
Mechanisms of mitochondrial fission and fusion. Cold Spring Harb
Perspect Biol. 5:a0110722013. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Chan DC: Mitochondrial fusion and fission
in mammals. Annu Rev Cell Dev Biol. 22:79–99. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Gao Z, Li Y, Wang F, Huang T, Fan K, Zhang
Y, Zhong J, Cao Q, Chao T, Jia J, et al: Mitochondrial dynamics
controls anti-tumour innate immunity by regulating CHIP-IRF1 axis
stability. Nat Commun. 8:18052017. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Pyakurel A, Savoia C, Hess D and Scorrano
L: Extracellular regulated kinase phosphorylates mitofusin 1 to
control mitochondrial morphology and apoptosis. Mol Cell.
58:244–254. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Cipolat S, Martins de Brito O, Dal Zilio B
and Scorrano L: OPA1 requires mitofusin 1 to promote mitochondrial
fusion. Proc Natl Acad Sci USA. 101:15927–15932. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Mattie S, Riemer J, Wideman JG and McBride
HM: A new mitofusin topology places the redox-regulated C terminus
in the mitochondrial intermembrane space. J Cell Biol. 217:507–515.
2018. View Article : Google Scholar :
|
|
175
|
Palikaras K, Lionaki E and Tavernarakis N:
Mechanisms of mitophagy in cellular homeostasis, physiology and
pathology. Nat Cell Biol. 20:1013–1022. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Wu W, Tian W, Hu Z, Chen G, Huang L, Li W,
Zhang X, Xue P, Zhou C, Liu L, et al: ULK1 translocates to
mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO
Rep. 15:566–575. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Ponneri Babuharisankar A, Kuo CL, Chou HY,
Tangeda V, Fan CC, Chen CH, Kao YH and Lee AY: Mitochondrial
Lon-induced mitophagy benefits hypoxic resistance via
Ca(2+)-dependent FUNDC1 phosphorylation at the ER-mitochondria
interface. Cell Death Dis. 14:1992023. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Miller DR and Thorburn A: Autophagy and
organelle homeostasis in cancer. Dev Cell 2021. 56:906–918.
2021.
|
|
179
|
Hamasaki M, Shibutani ST and Yoshimori T:
Up-to-date membrane biogenesis in the autophagosome formation. Curr
Opin Cell Biol. 25:455–460. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Gomez-Suaga P, Paillusson S, Stoica R,
Noble W, Hanger DP and Miller CCJ: The ER-mitochondria tethering
complex VAPB-PTPIP51 regulates autophagy. Curr Biol. 27:371–385.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Manganelli V, Matarrese P, Antonioli M,
Gambardella L, Vescovo T, Gretzmeier C, Longo A, Capozzi A,
Recalchi S, Riitano G, et al: Raft-like lipid microdomains drive
autophagy initiation via AMBRA1-ERLIN1 molecular association within
MAMs. Autophagy. 17:2528–2548. 2021. View Article : Google Scholar :
|
|
182
|
Rusiñol AE, Cui Z, Chen MH and Vance JE: A
unique mitochondria-associated membrane fraction from rat liver has
a high capacity for lipid synthesis and contains pre-Golgi
secretory proteins including nascent lipoproteins. J Biol Chem.
269:27494–27502. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Horvath SE and Daum G: Lipids of
mitochondria. Prog Lipid Res. 52:590–614. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Stone SJ and Vance JE: Phosphatidylserine
synthase-1 and -2 are localized to mitochondria-associated
membranes. J Biol Chem. 275:34534–34540. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Yang X, Liang J, Ding L, Li X, Lam SM,
Shui G, Ding M and Huang X: Phosphatidylserine synthase regulates
cellular homeostasis through distinct metabolic mechanisms. PLoS
Genet. 15:e10085482019. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Monteiro-Cardoso VF, Rochin L, Arora A,
Houcine A, Jääskeläinen E, Kivelä AM, Sauvanet C, Le Bars R, Marien
E, Dehairs J, et al: ORP5/8 and MIB/MICOS link ER-mitochondria and
intra-mitochondrial contacts for non-vesicular transport of
phosphatidylserine. Cell Rep. 40:1113642022. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Fu Y, Hoang A, Escher G, Parton RG,
Krozowski Z and Sviridov D: Expression of caveolin-1 enhances
cholesterol efflux in hepatic cells. J Biol Chem. 279:14140–14146.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Schroder K and Tschopp J: The
inflammasomes. Cell. 140:821–832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Rathinam VA, Vanaja SK and Fitzgerald KA:
Regulation of inflammasome signaling. Nat Immunol. 13:333–342.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Saxena G, Chen J and Shalev A:
Intracellular shuttling and mitochondrial function of
thioredoxin-interacting protein. J Biol Chem. 285:3997–4005. 2010.
View Article : Google Scholar
|
|
191
|
Zhou R, Tardivel A, Thorens B, Choi I and
Tschopp J: Thioredoxin-interacting protein links oxidative stress
to inflammasome activation. Nat Immunol. 11:136–140. 2010.
View Article : Google Scholar
|
|
192
|
Savitskaya MA and Onishchenko GE:
Mechanisms of apoptosis. Biochemistry (Mosc). 80:1393–1405. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Kuo IY, Brill AL, Lemos FO, Jiang JY,
Falcone JL, Kimmerling EP, Cai Y, Dong K, Kaplan DL, Wallace DP, et
al: Polycystin 2 regulates mitochondrial Ca(2+) signaling,
bioenergetics, and dynamics through mitofusin 2. Sci Signal.
12:eaat73972019. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Gorgoulis V, Adams PD, Alimonti A, Bennett
DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K,
Ferbeyre G, et al: Cellular senescence: Defining a path forward.
Cell. 179:813–827. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Berridge MJ, Lipp P and Bootman MD: The
versatility and universality of calcium signalling. Nat Rev Mol
Cell Biol. 1:11–21. 2000. View Article : Google Scholar
|
|
196
|
Farfariello V, Iamshanova O, Germain E,
Fliniaux I and Prevarskaya N: Calcium homeostasis in cancer: A
focus on senescence. Biochim Biophys Acta. 1853:1974–1979. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Martin N and Bernard D: Calcium signaling
and cellular senescence. Cell Calcium. 70:16–23. 2018. View Article : Google Scholar
|
|
198
|
Ma X, Warnier M, Raynard C, Ferrand M,
Kirsh O, Defossez PA, Martin N and Bernard D: The nuclear receptor
RXRA controls cellular senescence by regulating calcium signaling.
Aging Cell. 17:e128312018. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Jung YH, Chae CW, Choi GE, Shin HC, Lim
JR, Chang HS, Park J, Cho JH, Park MR, Lee HJ and Han HJ: Cyanidin
3-O-arabinoside suppresses DHT-induced dermal papilla cell
senescence by modulating p38-dependent ER-mitochondria contacts. J
Biomed Sci. 29:172022. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Li HM, Wang C, Liu Q, Tong Z, Song B, Wei
W and Teng C: Correlation between mitochondria-associated
endoplasmic reticulum membrane-related genes and cellular
senescence-related genes in osteoarthritis. ACS Omega.
9:19169–19181. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Mao H, Chen W, Chen L and Li L: Potential
role of mitochondria-associated endoplasmic reticulum membrane
proteins in diseases. Biochem Pharmacol. 199:1150112022. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Morena J, Gupta A and Hoyle JC:
Charcot-marie-tooth: From molecules to therapy. Int J Mol Sci.
20:34192019. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Saporta AS, Sottile SL, Miller LJ, Feely
SM, Siskind CE and Shy ME: Charcot-Marie-Tooth disease subtypes and
genetic testing strategies. Ann Neurol. 69:22–33. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Fridman V, Bundy B, Reilly MM, Pareyson D,
Bacon C, Burns J, Day J, Feely S, Finkel RS, Grider T, et al: CMT
subtypes and disease burden in patients enrolled in the inherited
neuropathies consortium natural history study: A cross-sectional
analysis. J Neurol Neurosurg Psychiatry. 86:873–878. 2015.
View Article : Google Scholar :
|
|
205
|
Lenaers G, Hamel C, Delettre C,
Amati-Bonneau P, Procaccio V, Bonneau D, Reynier P and Milea D:
Dominant optic atrophy. Orphanet J Rare Dis. 7:462012. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Cartes-Saavedra B, Macuada J, Lagos D,
Arancibia D, Andrés ME, Yu-Wai-Man P, Hajnóczky G and Eisner V:
OPA1 modulates mitochondrial Ca(2+) uptake through ER-mitochondria
coupling. Front Cell Dev Biol. 9:7741082022. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Verde F, Otto M and Silani V:
neurofilament light chain as biomarker for amyotrophic lateral
sclerosis and frontotemporal dementia. Front Neurosci.
15:6791992021. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Gomez-Suaga P, Mórotz GM, Markovinovic A,
Martín-Guerrero SM, Preza E, Arias N, Mayl K, Aabdien A, Gesheva V,
Nishimura A, et al: Disruption of ER-mitochondria tethering and
signalling in C9orf72-associated amyotrophic lateral sclerosis and
frontotemporal dementia. Aging Cell. 21:e135492022. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Cabreira V and Massano J: Parkinson's
disease: Clinical review and update. Acta Med Port. 32:661–670.
2019.In Portuguese. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Bidesi NSR, Vang Andersen I, Windhorst AD,
Shalgunov V and Herth MM: The role of neuroimaging in Parkinson's
disease. J Neurochem. 159:660–689. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Guardia-Laguarta C, Area-Gomez E, Rüb C,
Liu Y, Magrané J, Becker D, Voos W, Schon EA and Przedborski S:
α-Synuclein is localized to mitochondria-associated ER membranes. J
Neurosci. 34:249–259. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Paillusson S, Gomez-Suaga P, Stoica R,
Little D, Gissen P, Devine MJ, Noble W, Hanger DP and Miller CCJ:
α-Synuclein binds to the ER-mitochondria tethering protein VAPB to
disrupt Ca(2+) homeostasis and mitochondrial ATP production. Acta
Neuropathol. 134:129–149. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Castellani RJ, Rolston RK and Smith MA:
Alzheimer disease. Dis Mon. 56:484–546. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Schellenberg GD and Montine TJ: The
genetics and neuropathology of Alzheimer's disease. Acta
Neuropathol. 124:305–323. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Del Prete D, Suski JM, Oulès B, Debayle D,
Gay AS, Lacas-Gervais S, Bussiere R, Bauer C, Pinton P,
Paterlini-Bréchot P, et al: Localization and processing of the
Amyloid-β protein precursor in mitochondria-associated membranes. J
Alzheimers Dis. 55:1549–1570. 2017. View Article : Google Scholar
|
|
216
|
Pera M, Larrea D, Guardia-Laguarta C,
Montesinos J, Velasco KR, Agrawal RR, Xu Y, Chan RB, Di Paolo G,
Mehler MF, et al: Increased localization of APP-C99 in
mitochondria-associated ER membranes causes mitochondrial
dysfunction in Alzheimer disease. EMBO J. 36:3356–3371. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Area-Gomez E, de Groof A, Bonilla E,
Montesinos J, Tanji K, Boldogh I, Pon L and Schon EA: A key role
for MAM in mediating mitochondrial dysfunction in Alzheimer
disease. Cell Death Dis. 9:3352018. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Lau DHW, Paillusson S, Hartopp N, Rupawala
H, Mórotz GM, Gomez-Suaga P, Greig J, Troakes C, Noble W and Miller
CCJ: Disruption of endoplasmic reticulum-mitochondria tethering
proteins in post-mortem Alzheimer's disease brain. Neurobiol Dis
2020. 143:1050202020. View Article : Google Scholar
|
|
219
|
Bernard-Marissal N, Médard JJ, Azzedine H
and Chrast R: Dysfunction in endoplasmic reticulum-mitochondria
crosstalk underlies SIGMAR1 loss of function mediated motor neuron
degeneration. Brain. 138(Pt 4): 875–890. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Cheng D, Lei ZG, Chu K, Lam OJH, Chiang CY
and Zhang ZJ: N, N-Dimethyltryptamine, a natural hallucinogen,
ameliorates Alzheimer's disease by restoring neuronal Sigma-1
receptor-mediated endoplasmic reticulum-mitochondria crosstalk.
Alzheimers Res Ther. 16:952024. View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Bates GP: History of genetic disease: The
molecular genetics of Huntington disease-a history. Nat Rev Genet.
6:766–773. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Reddy PH: Increased mitochondrial fission
and neuronal dysfunction in Huntington's disease: Implications for
molecular inhibitors of excessive mitochondrial fission. Drug
Discov Today. 19:951–955. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Browne SE: Mitochondria and Huntington's
disease pathogenesis: Insight from genetic and chemical models. Ann
N Y Acad Sci. 1147:358–382. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Cherubini M, Lopez-Molina L and Gines S:
Mitochondrial fission in Huntington's disease mouse striatum
disrupts ER-mitochondria contacts leading to disturbances in Ca(2+)
efflux and Reactive Oxygen Species (ROS) homeostasis. Neurobiol
Dis. 136:1047412020. View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Anders HJ, Huber TB, Isermann B and
Schiffer M: CKD in diabetes: Diabetic kidney disease versus
nondiabetic kidney disease. Nat Rev Nephrol. 14:361–377. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Reidy K, Kang HM, Hostetter T and Susztak
K: Molecular mechanisms of diabetic kidney disease. J Clin Invest.
124:2333–2340. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Yang M, Han Y, Luo S, Xiong X, Zhu X, Zhao
H, Jiang N, Xiao Y, Wei L, Li C, et al: MAMs protect against
ectopic fat deposition and lipid-related kidney damage in DN
patients. Front Endocrinol (Lausanne). 12:6095802021. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Tang X, Yuan Y, Liang X and Jiang X:
Mitofusin2 expression is associated with podocyte injury in IgA
nephropathy. Eur J Med Res. 28:1422023. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Xue M, Fang T, Sun H, Cheng Y, Li T, Xu C,
Tang C, Liu X, Sun B and Chen L: PACS-2 attenuates diabetic kidney
disease via the enhancement of mitochondria-associated endoplasmic
reticulum membrane formation. Cell Death Dis. 12:11072021.
View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Chen H, Zhang H, Li AM, Liu YT, Liu Y,
Zhang W, Yang C, Song N, Zhan M and Yang S: VDR regulates
mitochondrial function as a protective mechanism against renal
tubular cell injury in diabetic rats. Redox Biol. 70:1030622024.
View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Li X, Yang Q, Liu S, Song S and Wang C:
Mitochondria-associated endoplasmic reticulum membranes promote
mitochondrial fission through AKAP1-Drp1 pathway in podocytes under
high glucose conditions. Exp Cell Res. 424:1135122023. View Article : Google Scholar : PubMed/NCBI
|
|
232
|
Xie Y, E J, Cai H, Zhong F, Xiao W, Gordon
RE, Wang L, Zheng YL, Zhang A, Lee K and He JC: Reticulon-1A
mediates diabetic kidney disease progression through endoplasmic
reticulum-mitochondrial contacts in tubular epithelial cells.
Kidney Int. 102:293–306. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Sanyal AJ, Brunt EM, Kleiner DE, Kowdley
KV, Chalasani N, Lavine JE, Ratziu V and McCullough A: Endpoints
and clinical trial design for nonalcoholic steatohepatitis.
Hepatology. 54:344–353. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Tarantino G and Finelli C: What about
non-alcoholic fatty liver disease as a new criterion to define
metabolic syndrome? World J Gastroenterol. 19:3375–3384. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
235
|
Romeo S, Kozlitina J, Xing C, Pertsemlidis
A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC and Hobbs HH:
Genetic variation in PNPLA3 confers susceptibility to nonalcoholic
fatty liver disease. Nat Genet. 40:1461–1465. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
236
|
Pérez-Carreras M, Del Hoyo P, Martín MA,
Rubio JC, Martín A, Castellano G, Colina F, Arenas J and
Solis-Herruzo JA: Defective hepatic mitochondrial respiratory chain
in patients with nonalcoholic steatohepatitis. Hepatology.
38:999–1007. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
237
|
Xu J, Chen S, Wang W, Man Lam S, Xu Y,
Zhang S, Pan H, Liang J, Huang X, Wang Y, et al: Hepatic
CDP-diacylglycerol synthase 2 deficiency causes mitochondrial
dysfunction and promotes rapid progression of NASH and fibrosis.
Sci Bull (Beijing). 67:299–314. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
238
|
Alpert CM, Smith MA, Hummel SL and Hummel
EK: Symptom burden in heart failure: Assessment, impact on
outcomes, and management. Heart Fail Rev. 22:25–39. 2017.
View Article : Google Scholar
|
|
239
|
Brown DA, Perry JB, Allen ME, Sabbah HN,
Stauffer BL, Shaikh SR, Cleland JG, Colucci WS, Butler J, Voors AA,
et al: Expert consensus document: Mitochondrial function as a
therapeutic target in heart failure. Nat Rev Cardiol. 14:238–250.
2017. View Article : Google Scholar :
|
|
240
|
Shi X, Zhang Y, Chen R, Gong Y, Zhang M,
Guan R, Rotstein OD, Liu X and Wen XY: ndufa7 plays a critical role
in cardiac hypertrophy. J Cell Mol Med. 24:13151–13162. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
241
|
Kimball TH and Vondriska TM: Metabolism,
epigenetics, and causal inference in heart failure. Trends
Endocrinol Metab. 31:181–191. 2020. View Article : Google Scholar
|
|
242
|
Ren J, Bi Y, Sowers JR, Hetz C and Zhang
Y: Endoplasmic reticulum stress and unfolded protein response in
cardiovascular diseases. Nat Rev Cardiol. 18:499–521. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
243
|
Liu P, Zhang Z, Chen H and Chen Q:
Pyroptosis: Mechanisms and links with diabetic cardiomyopathy.
Ageing Res Rev. 94:1021822024. View Article : Google Scholar : PubMed/NCBI
|
|
244
|
Ritchie RH and Abel ED: Basic mechanisms
of diabetic heart disease. Circ Res. 126:1501–1525. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
245
|
Salin Raj P, Nair A, Preetha Rani MR,
Rajankutty K, Ranjith S and Raghu KG: Ferulic acid attenuates high
glucose-induced MAM alterations via PACS2/IP3R2/FUNDC1/VDAC1
pathway activating proapoptotic proteins and ameliorates
cardiomyopathy in diabetic rats. Int J Cardiol. 372:101–109. 2023.
View Article : Google Scholar
|
|
246
|
Brandt KD, Radin EL, Dieppe PA and van de
Putte L: Yet more evidence that osteoarthritis is not a cartilage
disease. Ann Rheum Dis. 65:1261–1264. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
247
|
Fu K, Robbins SR and McDougall JJ:
Osteoarthritis: The genesis of pain. Rheumatology (Oxf).
57(suppl_4): iv43–iv50. 2018. View Article : Google Scholar
|
|
248
|
GBD 2016 Disease, Injury Incidence and
Prevalence Collaborators: Global, regional, and national incidence,
prevalence, and years lived with disability for 328 diseases and
injuries for 195 countries, 1990-2016: A systematic analysis for
the Global Burden of Disease Study 2016. Lancet. 390:1211–1259.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
249
|
Madhu V, Hernandez-Meadows M, Boneski PK,
Qiu Y, Guntur AR, Kurland IJ, Barve RA and Risbud MV: The mitophagy
receptor BNIP3 is critical for the regulation of metabolic
homeostasis and mitochondrial function in the nucleus pulposus
cells of the intervertebral disc. Autophagy. 19:1821–1843. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
250
|
Chai P, Cheng Y, Hou C, Yin L, Zhang D, Hu
Y, Chen Q, Zheng P, Teng J and Chen J: USP19 promotes
hypoxia-induced mitochondrial division via FUNDC1 at
ER-mitochondria contact sites. J Cell Biol. 220:e2020100062021.
View Article : Google Scholar : PubMed/NCBI
|
|
251
|
Ivanova H, Kerkhofs M, La Rovere RM and
Bultynck G: Endoplasmic reticulum-mitochondrial Ca(2+) fluxes
underlying cancer cell survival. Front Oncol. 7:702017. View Article : Google Scholar : PubMed/NCBI
|
|
252
|
Li J, Qi F, Su H, Zhang C, Zhang Q, Chen
Y, Chen P, Su L, Chen Y, Yang Y, et al: GRP75-faciliated
mitochondria-associated ER Membrane (MAM) integrity controls
cisplatin-resistance in ovarian cancer patients. Int J Biol Sci.
18:2914–2931. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
253
|
Cassidy-Stone A, Chipuk JE, Ingerman E,
Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR
and Nunnari J: Chemical inhibition of the mitochondrial division
dynamin reveals its role in Bax/Bak-dependent mitochondrial outer
membrane permeabilization. Dev Cell. 14:193–204. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
254
|
Ciscato F, Masgras I, Gori A, Fantuz M,
Bergamaschi G, Komarov D, La Spina M, Ghasemi-Firouzabadi S, Pizzi
M, Dei Tos AP, et al: The use of hexokinase 2-displacing peptides
as an anti-neoplastic approach for malignant peripheral nerve
sheath tumors. Cells. 13:11622024. View Article : Google Scholar : PubMed/NCBI
|
|
255
|
Ciscato F, Filadi R, Masgras I, Pizzi M,
Marin O, Damiano N, Pizzo P, Gori A, Frezzato F, Chiara F, et al:
Hexokinase 2 displacement from mitochondria-associated membranes
prompts Ca(2+) -dependent death of cancer cells. EMBO Rep.
21:e491172020. View Article : Google Scholar : PubMed/NCBI
|
|
256
|
Stone SJ, Levin MC, Zhou P, Han J, Walther
TC and Farese RV Jr: The endoplasmic reticulum enzyme DGAT2 is
found in mitochondria-associated membranes and has a mitochondrial
targeting signal that promotes its association with mitochondria. J
Biol Chem. 284:5352–5361. 2009. View Article : Google Scholar
|
|
257
|
Sebastián D, Hernández-Alvarez MI, Segalés
J, Sorianello E, Muñoz JP, Sala D, Waget A, Liesa M, Paz JC,
Gopalacharyulu P, et al: Mitofusin 2 (Mfn2) links mitochondrial and
endoplasmic reticulum function with insulin signaling and is
essential for normal glucose homeostasis. Proc Natl Acad Sci USA.
109:5523–5528. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
258
|
Bravo-Sagua R, Torrealba N, Paredes F,
Morales PE, Pennanen C, López-Crisosto C, Troncoso R, Criollo A,
Chiong M, Hill JA, et al: Organelle communication: Signaling
crossroads between homeostasis and disease. Int J Biochem Cell
Biol. 50:55–59. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
259
|
Li YE, Sowers JR, Hetz C and Ren J: Cell
death regulation by MAMs: From molecular mechanisms to therapeutic
implications in cardiovascular diseases. Cell Death Dis.
13:5042022. View Article : Google Scholar : PubMed/NCBI
|
|
260
|
Tubbs E and Rieusset J: Metabolic
signaling functions of ER-mitochondria contact sites: Role in
metabolic diseases. J Mol Endocrinol. 58:R87–R106. 2017. View Article : Google Scholar
|
|
261
|
Vallese F, Catoni C, Cieri D, Barazzuol L,
Ramirez O, Calore V, Bonora M, Giamogante F, Pinton P, Brini M and
Calì T: An expanded palette of improved SPLICS reporters detects
multiple organelle contacts in vitro and in vivo. Nat Commun.
11:60692020. View Article : Google Scholar : PubMed/NCBI
|
|
262
|
Calì T and Brini M: Quantification of
organelle contact sites by split-GFP-based contact site sensors
(SPLICS) in living cells. Nat Protoc. 16:5287–5308. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
263
|
Prasad M, Pawlak KJ, Burak WE, Perry EE,
Marshall B, Whittal RM and Bose HS: Mitochondrial metabolic
regulation by GRP78. Sci Adv. 3:e16020382017. View Article : Google Scholar : PubMed/NCBI
|
|
264
|
Nourbakhsh M, Sharifi R, Heydari N,
Nourbakhsh M, Ezzati-Mobasser S and Zarrinnahad H: Circulating TRB3
and GRP78 levels in type 2 diabetes patients: Crosstalk between
glucose homeostasis and endoplasmic reticulum stress. J Endocrinol
Invest. 45:649–655. 2022. View Article : Google Scholar
|