Introduction
Sepsis-induced cardiomyopathy (SIC) is an acute and
reversible myocardial dysfunction caused by sepsis. It is
clinically characterized by acute systolic and/or diastolic
impairment of either one or both ventricles, which is not
attributable to coronary artery disease (1). SIC is a relatively common
complication of sepsis. Of all patients with sepsis, ~50% exhibit
some degree of left or right ventricular dysfunction (2). Patients with SIC have a 2-3 times
higher mortality rate than septic patients without cardiac
dysfunction (3). Currently, no
evidence-based guidelines, effective prevention strategies or
specific therapies exist for SIC, representing a major unmet need.
Urgent development of novel treatment strategies is key to
increasing survival and improving prognoses for these patients.
Sepsis involves multiple pathogenic pathways,
including dysregulated inflammatory mediators, mitochondrial
dysfunction, oxidative stress and calcium handling abnormalities
(4). Bioenergetic and metabolic
derangements serve a central role in the development of SIC
(5). As the primary energy
source of the heart, mitochondria generate the vast majority of ATP
required to sustain myocardial contraction (6). Increasing evidence indicates that
severe sepsis induces both structural and functional impairments in
cardiac mitochondria, resulting in ATP depletion and cardiomyocyte
apoptosis (7-10). Thus, mitochondrial-targeted
therapy may represent a promising therapeutic strategy for SIC.
AMP-activated protein kinase (AMPK), a cellular energy sensor,
regulates diverse physiological processes in the cardiovascular
system (11). AMPK pathway
inhibition was observed in SIC (12,13). In addition to metabolic
regulation, AMPK activation also regulates mitochondrial biogenesis
and dynamics (fusion-fission) by activating peroxisome
proliferator-activated receptor γ co-activator-1α (PGC-1α), thereby
enhancing mitochondrial function (11). Adiponectin receptor 1 (AdipoR1)
is established as an upstream activator in the AMPK signaling
pathway. Inhibition of the AdipoR1 signaling pathway may lead to
myocardial mitochondrial dysfunction and uncoupling (14). This evidence suggests that the
AdipoR1/AMPK pathway may serve as a pivotal mediator for promoting
mitochondrial function in SIC.
Myonectin, also known as C1q tumor necrosis
factor-related protein 15 (CTRP15) or erythroferrone, serves a role
in regulating glucose, fatty acid and iron metabolism (15,16). To date, studies on its role in
cardiovascular disease remain limited. Otaka et al (17) demonstrated that myonectin
suppressed inflammatory responses and apoptosis by activating the
sphingosine-1-phosphate (S1P)/cAMP/Akt pathway, ameliorating
myocardial ischemia-reperfusion (I/R) injury in mice. Another study
elucidated that myonectin inhibited cardiac fibrosis through
activation of the insulin receptor (IR)/insulin receptor
substrate-1 (IRS-1)/Akt signaling pathway (18). These findings suggest that
myonectin has a cardioprotective effect. Furthermore, Ozaki et
al (19) found that
myonectin promoted mitochondrial function in skeletal muscle by
activating the AMPK/PGC-1α pathway. Whether myonectin can exert
cardioprotective effects in SIC by modulating mitochondrial
function remains to be elucidated. To address this gap, we
hypothesize that myonectin ameliorates SIC by enhancing
mitochondrial function through promoting mitochondrial biogenesis
and regulating mitochondrial dynamics via the AdipoR1/AMPK pathway.
The present study aimed to investigate the protective potential of
myonectin against SIC and to elucidate the mechanisms involved.
Materials and methods
Animals and treatments
Male C57BL/6J mice (6-8 weeks old; 20-22 g) were
purchased from Jilin Qianhe Model Biotechnology Co., Ltd. During
the study period, mice were maintained under specific pathogen-free
(SPF) conditions with controlled temperature (20-22°C) and humidity
(45-50%), under a 12-h light/12-h dark cycle. Mice had free access
to food and water. All animal experiments were approved by the
Institutional Ethics Committee of the School of Basic Medical
Sciences, Jilin University (Jilin, China; approval nos. 2025-597
and 2026-491). All procedures were conducted in accordance with the
institution's guidelines for the care and use of laboratory
animals. To eliminate potential sex-related variability and
hormonal fluctuations associated with the estrous cycle, only male
mice were included in the present study.
A total of 24 mice were randomly divided into four
groups (n=6 per group): Control, control+myonectin,
lipopolysaccharide (LPS) and LPS+myonectin groups. Mice in the LPS
group received an intraperitoneal injection of LPS (from E.
coli O111:B4; L2630; MilliporeSigma) at 10 mg/kg to establish a
SIC model (20,21). The LPS+myonectin group received a
tail vein injection of recombinant mouse myonectin (rMyonectin;
cat.no. 00393-06-100; Aviscera Bioscience, Inc.) at 200 ng/g, a
dose based on previous literature (17), 2 h before LPS administration. The
control group received equal volumes of PBS via tail vein and
intraperitoneal injections, separately. The control+myonectin group
received rMyonectin at 200 ng/g via tail vein injection, followed
by an equal volume of PBS via intraperitoneal injection 2 h later.
Cardiac function was assessed by echocardiography 24 h after LPS
injection. Subsequently, mice remained deeply anesthetized with an
overdose of 5% isoflurane. After the disappearance of corneal
reflexes and pedal withdrawal reflexes, which indicated a deep
unconscious state, blood was collected via intracardiac puncture,
and heart tissues were harvested as an adjunctive means to confirm
death. This procedure ensured euthanasia while the animals were
unconscious. The total experimental duration was 26 h. Humane
endpoints were predefined as meeting any one of the following
conditions: Severe respiratory distress evidenced by open-mouth
breathing, a decline in body weight >20% relative to baseline,
hypothermia (<32°C), lateral recumbency or loss of
responsiveness to external stimuli.
For the cecal ligation and puncture (CLP) model, a
total of 24 mice were randomly divided into four groups (n=6 per
group): Sham, sham+myonectin, CLP and CLP+myonectin groups. The
experimental procedure for CLP was based on previously published
literature (22,23). Mice were anesthetized with
isoflurane (4% for induction and 2% for maintenance). Following
disinfection of the lower abdomen with 75% ethanol, a 1.5 cm
midline longitudinal incision was made to expose the cecum. The
distal three-quarters of the cecum was ligated. A total of two
holes were then punctured in the ligated segment using a 22-gauge
needle, and a small amount of fecal content was gently expressed.
The cecum was subsequently returned to the peritoneal cavity, and
the abdominal incision was closed in layers with sutures.
Immediately after surgery, 1 ml sterile saline was administered
intraperitoneally to compensate for fluid loss. Sham-operated mice
were subjected to the identical surgical procedure except that CLP
was omitted. No antibiotics were administered during the
experimental period. The sham+myonectin and the CLP+myonectin
groups were pretreated with rMyonectin (200 ng/g) via tail vein
injection 2 h prior to the procedure. Cardiac function was assessed
by echocardiography 24 h after surgery. The total experimental
duration was 26 h. The procedures for anesthesia, euthanasia and
blood and tissue collection, as well as the humane endpoints, were
identical to those described for the LPS model.
To determine the survival rate, separate experiments
were conducted. For the LPS model, mice were randomly divided into
four groups (n=10 per group): Control, control+myonectin, LPS and
LPS+myonectin groups. For the CLP model, mice were randomly divided
into four groups (n=10 per group): Sham, sham+myonectin, CLP and
CLP+myonectin groups. Mice were pretreated via tail vein injection
with either rMyonectin (200 ng/g) or PBS 2 h prior to an
intraperitoneal dose of LPS (15 mg/kg) or CLP surgery. Survival was
monitored every 12 h for 3 days as described previously (24,25).
A total of 128 mice were used in the present study,
of which 123 were euthanized, while 5 were found dead (attributed
to SIC-mediated cardiac emergencies). Of the 123 mice that were
euthanized, 24 were euthanized upon reaching humane endpoints, and
the remaining 99 were euthanized at the scheduled end of the
study.
Echocardiographic assessment
Echocardiography was performed under isoflurane
anesthesia (4% for induction and 2% for maintenance) in mice from
each group using a GE Vivid E95 ultrasound system (GE Healthcare
Technologies, Inc.) to non-invasively evaluate changes in cardiac
structure and function. M-mode images were acquired from the left
ventricular long-axis view, and left ventricular parameters
including left ventricular ejection fraction (LVEF), left
ventricular fractional shortening (LVFS), left ventricular internal
diameter at end-systole (LVIDs), left ventricular internal diameter
at end-diastole (LVIDd) and heart rate (HR) were measured. The
echocardiographer was blinded to the treatment groups.
Histological analysis
Cardiac tissues were fixed in 4% paraformaldehyde at
4°C for 48 h, embedded in paraffin and sectioned at 4-μm
thickness. Sections were deparaffinized with dimethylbenzene and
rehydrated through a graded ethanol series (100, 95, 80 and 70%).
Subsequently, parallel sections were processed for hematoxylin and
eosin (H&E) staining and terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) assay. The stained sections were
observed and imaged under the microscope (OLYMPUS BX53), and the
images were analyzed using ImageJ software (National Institutes of
Health; v1.54f). The apoptotic index was calculated as the
integrated density of TUNEL-positive cells divided by the total
DAPI-stained nuclear area.
ATP assessment
The relative levels of ATP were determined using the
ATP assay kit (cat. no. S0027; Beyotime Biotechnology) according to
the manufacturer's instructions. Samples (20 mg cardiac tissue or
cells from one well of a 6-well plate) were homogenized with ATP
lysis buffer (200 μl). Cell lysates or tissue homogenates
were centrifuged at 12,000 × g for 5 min at 4°C, and the
supernatants were collected and kept on ice. ATP working solution
(100 μl) was added to each well of 96-well plates and
allowed to equilibrate at room temperature for 3-5 min.
Subsequently, 20 μl sample or standard solution was added to
each well and mixed thoroughly. The values, relative light unit
(RLU), were measured using the multimode microplate reader
(CLARIOSTAR, BMG Labtech GmbH).
Enzyme-linked immunosorbent assay
(ELISA)
Serum was obtained by centrifugation (1,000 × g; 15
min; 4°C) of blood samples after allowing them to clot at room
temperature for 2 h. The levels of cardiac troponin I (cTn-I) and
brain natriuretic peptide (BNP) in the serum were measured using
the commercial ELISA kits (cat. nos. CSB-E08421m and CSB-E07971m;
Cusabio Technology, LLC) according to the manufacturer's
instructions.
Transmission electron microscopy
(TEM)
Mitochondrial ultrastructure was observed using TEM.
Heart tissue was initially fixed in 3% glutaraldehyde overnight at
4°C, post-fixed in 1% osmium tetroxide for 2 h at 4°C and rinsed
with PBS. Following dehydration through a graded ethanol series,
the samples were embedded in Epon resin. Ultrathin sections (50 nm)
were cut using a diamond knife and mounted on copper grids. The
sections were then stained with uranyl acetate for 15 min at room
temperature and lead citrate for 10 min at room temperature. Images
were acquired using the TEM (Hitachi H600 Electron Microscope,
Hitachi, Ltd.) at magnifications of ×3,000, ×7,000 and ×20,000 to
assess overall morphology and detailed structures.
Isolation and culture of neonatal mouse
cardiomyocytes (NMCMs)
The isolation and culture of NMCMs were performed
based on a previously described method (26). Neonatal C57BL/6J mice (1-3 days
old) were derived from an in-house SPF breeding colony established
by mating male and female mice purchased from Jilin Qianhe Model
Biotechnology Co., Ltd. Following anesthesia induction with 2%
isoflurane (on a thermostatic heating pad at 37°C for ~2 min) and
confirmation of loss of consciousness and reflexes, mice were
euthanized by rapid decapitation with sterile scissors, and hearts
were aseptically harvested, promptly chilled, washed, and minced in
cold Ca2+- and Mg2+-free Hank's Balanced Salt
Solution (cat. no. 14175095; Gibco; Thermo Fisher Scientific,
Inc.). The tissue fragments were then digested in a solution of 1
mg/ml trypsin (cat. no. T4799; MilliporeSigma) at 4°C for 7 h. The
trypsin solution was then removed. The tissue fragments were
subjected to sequential digestions with 0.8 mg/ml Collagenase Type
II (cat. no. C6885; MilliporeSigma) in a 37°C water bath for 5 min
per cycle, until no visible tissue fragments remained. The released
cell suspension from each digestion was collected and kept on ice.
The cell suspension was centrifuged at 200 × g for 5 min at 4°C.
The pellet was resuspended in Dulbecco's modified Eagle medium/F12
Ham medium (DMEM/F12; cat. no. C11330500BT; Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% Fetal Bovine Serum (FBS;
cat. no. A5256701; Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin solution. The cell suspension was filtered
through a cell strainer and cultured for 90 min under standard
culture conditions (37°C, 5% CO2) to remove fibroblasts.
Cardiomyocytes were then collected, plated onto culture dishes
pre-coated with 0.01% poly-L-lysine (cat. no. ST508; Beyotime
Biotechnology) and cultured until a synchronous, spontaneous
beating rhythm was observed, after which they were ready for
subsequent treatments. NMCMs were pretreated for 2 h with
rMyonectin or with the AMPK inhibitor Compound C (CC; 10 μM;
cat. no. HY-13418A; MedChemExpress) (27,28), followed by stimulation with 10
μg/ml LPS for 24 h (29).
Small interfering (si)RNA
interference
The AdipoR1-targeting siRNA was designed and
synthesized by GenCefe Biotech and had the following sequences:
Sense strand, 5'-GGCUCUUCCACACUGUCUTT-3'; antisense strand,
5'-UAGACAGUGUGGAAGAGCCTT-3'. The negative control siRNA sequences
were: Sense strand, 5'-UUCUCCGAACGUGUCACGUTT-3'; antisense strand,
5'-ACGUGACACGUUCGGAGAATT-3'. Cells were transfected with 150 pmol
siRNA per well in a 6-well plate using 7.5 μl
GP-transfect-Mate reagent (cat. no. G04008; Shanghai GenePharma
Co., Ltd.). After 6 h incubation at 37°C, the medium was replaced
with fresh culture medium, and the cells were further cultured. The
levels of mRNA and protein were assessed at 24 and 48 h,
respectively.
Cell counting kit-8 (CCK-8) assay
Cell viability of NMCMs was assessed using the CCK-8
assay (cat. no. BA00208; Bioss) according to the manufacturer's
instructions. Absorbance was measured at 450 nm.
Intracellular lactate dehydrogenase (LDH)
activity assay
The activity of intracellular LDH was determined
with the commercial assay kit (cat. no. BC0685; Beijing Solarbio
Science & Technology Co., Ltd.) per the manufacturer's
instructions. Cells were lysed in the provided extraction buffer
and sonicated on ice. After centrifugation of the lysates (8,000 ×
g; 10 min; 4°C), the absorbance of the supernatant was measured at
450 nm. The protein concentration of the supernatant was determined
using a BCA Protein Assay Kit (cat. no. C05-02001; Bioss) to
normalize the LDH activity. Finally, the specific activity of LDH
was calculated and expressed as units per milligram of protein.
Mitochondrial membrane potential (MMP)
assessment
MMP was evaluated using a JC-1 assay kit (cat. no.
C2003S; Beyotime Biotechnology) following the manufacturer's
instructions. NMCMs were plated on glass-bottom confocal culture
dishes and stained with 1 μM JC-1 probes for 20 min at 37°C
in the dark. After washing twice with PBS, fluorescent images were
immediately acquired using the confocal microscope (Nikon
Corporation). The fluorescent signals of JC-1 aggregates (red) and
monomers (green) were recorded. The ratio of red-to-green
fluorescence intensity was quantified using ImageJ software
(National Institutes of Health, v1.54f) to evaluate MMP.
Mitochondrial respiratory chain complex I
and III activity assay
The activities of mitochondrial respiratory chain
complexes I and III were assessed using the Cell Mitochondrial
Complex I (NADH-CoQ Reductase) Activity Assay Kit (cat. no.
E-BC-K834-M; Wuhan Elabscience Biotechnology Co., Ltd.) and the
Cell Mitochondrial Complex III (Coenzyme Q-Cytochrome C Reductase)
Activity Assay Kit (cat. no. E-BC-K836-M; Wuhan Elabscience
Biotechnology Co., Ltd.), respectively. Cardiomyocytes were
harvested, resuspended in the provided extraction solution and
processed according to the manufacturer's instructions for
mitochondrial respiratory chain complex extraction. The absorbance
was measured at 340 nm (for complex I) and 550 nm (for complex
III).
Measurement of oxygen consumption rate
(OCR)
After treatment according to the experimental
groups, cells were cultured for 24 h. OCR was evaluated using the
Enhanced Oxygen Consumption Rate Fluorometric Assay Kit (cat. no.
E-BC-F070; Wuhan Elabscience Biotechnology Co., Ltd.). The
microplate was placed in the multifunctional microplate reader
(Synergy H1, BioTek; Agilent Technologies, Inc.) set at 37°C in
dynamic reading mode. The excitation wavelength was set at 405 nm,
the emission wavelength at 675 nm, and measurements were taken
every 2 min for 100 min. Finally, the fluorescence values were
plotted against time, and OCR was represented by the slopes of the
curves.
Flow cytometry
Cell apoptosis was analyzed using flow cytometry.
Following harvest by trypsinization, cells were stained with
Annexin V-FITC and propidium iodide (PI), and then immediately
analyzed by a DxFLEX flow cytometer (Beckman Coulter, Inc.). The
data were analyzed using FlowJo software (v10.8.1; BD Biosciences)
to quantify the percentage of apoptotic cells in each group.
Western blot
Proteins were extracted from cardiac tissue and
NMCMs using RIPA lysis buffer (cat. no. PC101; Shanghai Epizyme
Biopharmaceutical Technology Co., Ltd.) supplemented with protease
and phosphatase inhibitors. The protein concentration was
quantified with the BCA Protein Assay Kit (cat. no. C05-02001;
Bioss). Equal amounts of proteins (20 μg per lane) were
denatured in SDS-PAGE loading buffer (cat. no. P1040; Beijing
Solarbio Science & Technology Co., Ltd.) at 100°C for 5 min,
separated by 8-12% SDS-PAGE and transferred to PVDF membranes. The
membranes were blocked with 5% skim milk for 1 h at room
temperature and then incubated overnight at 4°C with the following
primary antibodies: Bax (1:1,000; cat. no. ab32503; Abcam), Bcl-2
(1:1,000; cat. no. A19693; ABclonal Biotech Co., Ltd.), cleaved
caspase-3 (1:600; cat. no. WL02117; Wanleibio Co., Ltd.), caspase-3
(1:500; cat. no. T40044; Abmart Pharmaceutical Technology Co.,
Ltd.), PGC-1α (1:2,000; cat. no. ab313559; Abcam), nuclear
respiratory factor 1 (NRF1; 1:1,000; cat. no. A3252; ABclonal
Biotech Co., Ltd.), mitochondrial transcription factor A (TFAM;
1:1,000; cat. no. 15218; Cell Signaling Technology, Inc.),
mitofusin 2 (Mfn2; 1:3,000; cat. no. ab124773; Abcam), optic
atrophy 1 (OPA1; 1:1,000; cat. no. ab157457; Abcam), phosphorylated
(p)-dynamin-related protein 1 (Drp1; Ser616; 1:1,000; cat. no.
ab314755; Abcam), Drp1 (1:4,000; cat. no. ab184247; Abcam), p-AMPK
(1:1,000; cat. no. ab133448; Abcam), AMPK (1:1,000; cat. no.
ab207442; Abcam), AdipoR1 (1:4,000; cat. no. bs-0610R; Bioss),
β-actin (1:8,000; cat. no. P30002; Abmart Pharmaceutical Technology
Co., Ltd.) and β-tubulin (1:10,000; cat. no. A12289; ABclonal
Biotech Co., Ltd.). After washing, the membranes were incubated
with HRP-conjugated Goat Anti-Rabbit secondary antibody (1:10,000;
cat. no. ab6721; Abcam) for 1 h at room temperature. After
incubation with an enhanced chemiluminescence (ECL) substrate, the
bands were visualized using MiniChemi 610 System (SinSage
Technology, Co., Ltd.) and SageCapture™ software. Band intensities
were quantified using ImageJ software (National Institutes of
Health, v1.54f) and normalized to β-actin or β-tubulin.
Reverse transcription-quantitative PCR
(RT-qPCR) analysis
Total RNA was extracted using the Fast Total RNA
Extraction Kit (cat. no. RP1201; BioTeke Corporation). cDNA was
synthesized from the extracted RNA with the ToloScript All-in-One
RT EasyMix for qPCR (cat. no. 22107-01; Tolo Biotech Co., Ltd.)
according to the manufacturer's instructions. The primers were
synthesized by Comate Bioscience. The primer sequences used in the
present study were as follows: AdipoR1: Forward
5'-GAGAAGATGGAGGAGTTCGTGTA-3', reverse
5'-AGCAGGTAGTCGTTGTCTTTCAG-3'; β-actin: Forward
5'-GTACTCTGTGTGGATCGGTGG-3', reverse 5'-GCAGCTCAGTAACAGTCCG-3'.
Following the mixture of cDNA, primers, and the 2X Q3 SYBR qPCR
Master Mix (cat. no. 22204; Tolo Biotech Co., Ltd.), qPCR was
performed using the Real-Time PCR system (ABI 7500 DX). The
thermocycling protocol was as follows: Initial denaturation at 95°C
for 30 sec, followed by 40 cycles of 95°C for 10 sec and 60°C for
30 sec. A melt curve analysis was performed to verify amplification
specificity. Relative gene expression was quantified via the
2−ΔΔCq method (30),
where β-actin served as internal control.
Statistical analysis
Data from three or more independent experiments are
presented as the mean±SEM. Statistical analyses were performed
using GraphPad Prism software (version 10.0; Dotmatics).
Comparisons among multiple groups were performed using one-way
ANOVA followed by Tukey's multiple comparison test. Survival was
assessed using the Kaplan-Meier method, and differences between
groups were compared with the Gehan-Breslow-Wilcoxon test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
rMyonectin improves cardiac function,
attenuates myocardial injury and inhibits apoptosis in SIC
mice
Following pretreatment with rMyonectin via tail vein
injection, SIC was induced in mice by intraperitoneal injection of
LPS after a 2-h interval. Cardiac function was evaluated by
echocardiography 24 h post-induction. Subsequently, blood samples
and heart tissue were collected under anesthesia for analysis
(Fig. 1A). The results showed
that rMyonectin administration significantly improved the survival
rate of mice challenged with LPS (Fig. 1B). Echocardiographic assessment
revealed that LPS-challenged mice displayed significant impairments
in cardiac systolic function compared with controls, characterized
by reduced LVEF and LVFS, alongside increased LVIDs (Fig. 1C-F). Consistent with these
functional deficits, serum levels of BNP and cTn-I were markedly
elevated in LPS-challenged mice (Fig. 1G and H), confirming the
successful induction of SIC. Notably, pretreatment with rMyonectin
effectively attenuated these pathophysiological alterations,
improving systolic performance and reducing myocardial injury
(Fig. 1C-H). Histopathological
analysis revealed that myocardial fibers in the control group were
orderly arranged with preserved interstitial integrity. rMyonectin
treatment alone induced no structural alterations. By contrast, LPS
challenge induced distinct pathological changes, including
cardiomyocyte disarray, interstitial edema and vacuolar
degeneration. Importantly, rMyonectin pretreatment significantly
attenuated these LPS-induced structural abnormalities (Fig. 1I). Furthermore, rMyonectin
significantly attenuated LPS-induced cardiomyocyte apoptosis, as
shown by a reduced apoptosis index (Fig. 1J and K), downregulated cleaved
caspase-3 and Bax, and upregulated Bcl-2 in myocardial tissue
(Fig. 1L).
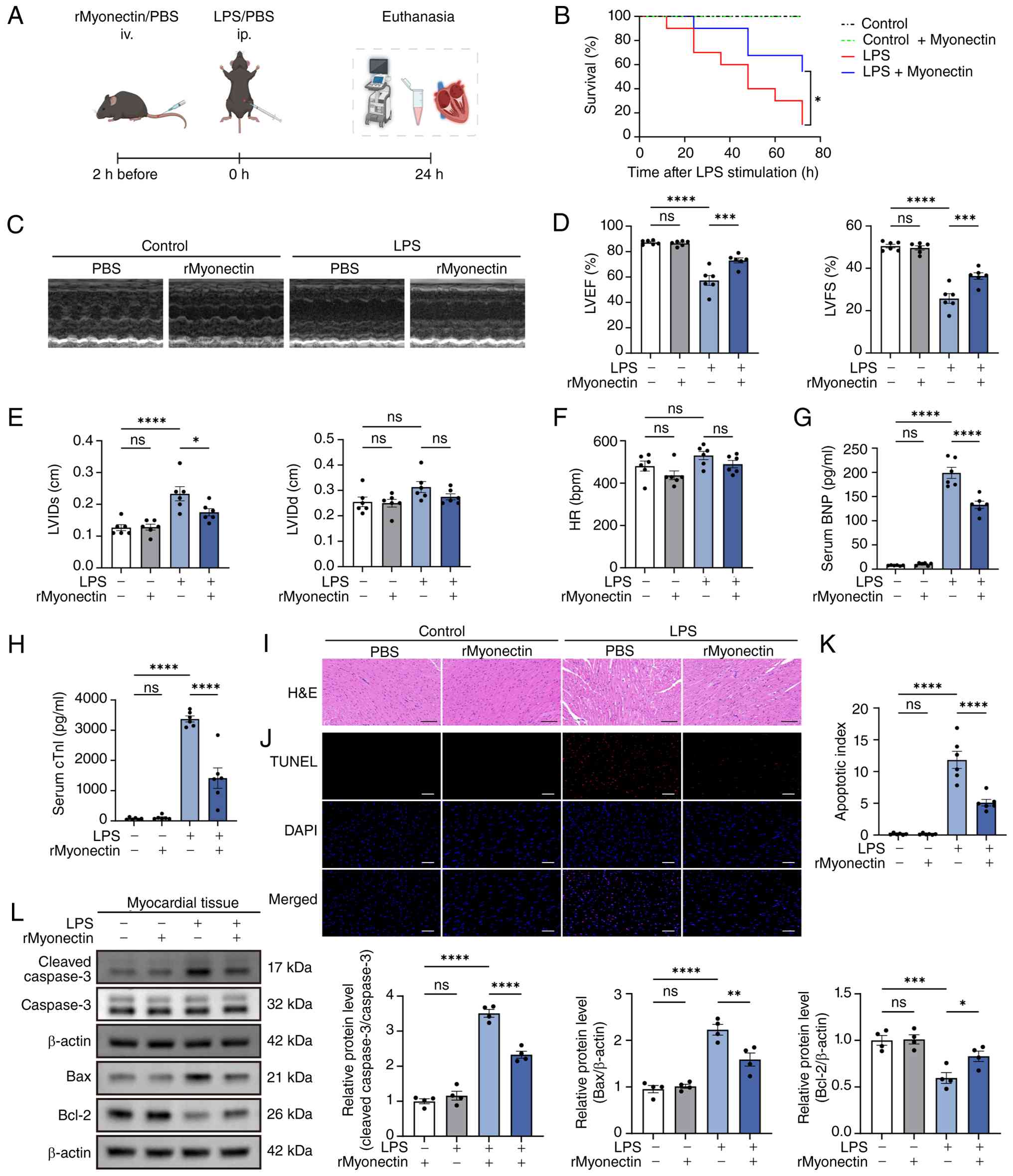 | Figure 1rMyonectin improves cardiac function,
attenuates myocardial injury and inhibits apoptosis in LPS-induced
sepsis-induced cardiomyopathy mice. (A) Schematic diagram of the
timeline for LPS and rMyonectin administration and subsequent
cardiac evaluation. (B) Survival curves. (C) Representative images
from echocardiography. (D) LVEF and LVFS. (E) LVIDs and LVIDd. (F)
HR. The serum levels of (G) BNP and (H) cTn-I. (I) H&E staining
of heart sections. Scale bar, 100 μm. (J) TUNEL assay of
heart sections. Scale bar, 50 μm. (K) Quantification of
apoptosis in TUNEL-stained sections. (L) Western blot analysis and
semi-quantification of cleaved caspase-3, caspase-3, Bax and Bcl-2
protein expression in myocardial tissue. The data are presented as
mean±SEM. *P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. The schematic
diagram was created using BioRender.com. rMyonectin, recombinant myonectin;
LVEF, left ventricular ejection fraction; LVFS, left ventricular
fractional shortening; LVIDs, left ventricular internal diameter at
end-systole; LVIDd, left ventricular internal diameter at
end-diastole; HR, heart rate; BNP, brain natriuretic peptide;
cTn-I, cardiac troponin I; TUNEL, terminal deoxynucleotidyl
transferase dUTP nick end labeling; LPS, lipopolysaccharide;
H&E, hematoxylin and eosin. |
Moreover, a CLP-induced model was established in
C57BL/6J mice to further evaluate the effects of rMyonectin on SIC
(Fig. 2A). The results showed
that rMyonectin significantly improved the survival rate (Fig. 2B), ameliorated cardiac
dysfunction (Fig. 2C-G) and
myocardial injury (Fig. 2H and
I), and inhibited cardiomyocyte apoptosis (Fig. 2J-L) in CLP-induced SIC mice.
These results demonstrate that rMyonectin protects against SIC
irrespective of the sepsis model, whether endotoxemic or
polymicrobial.
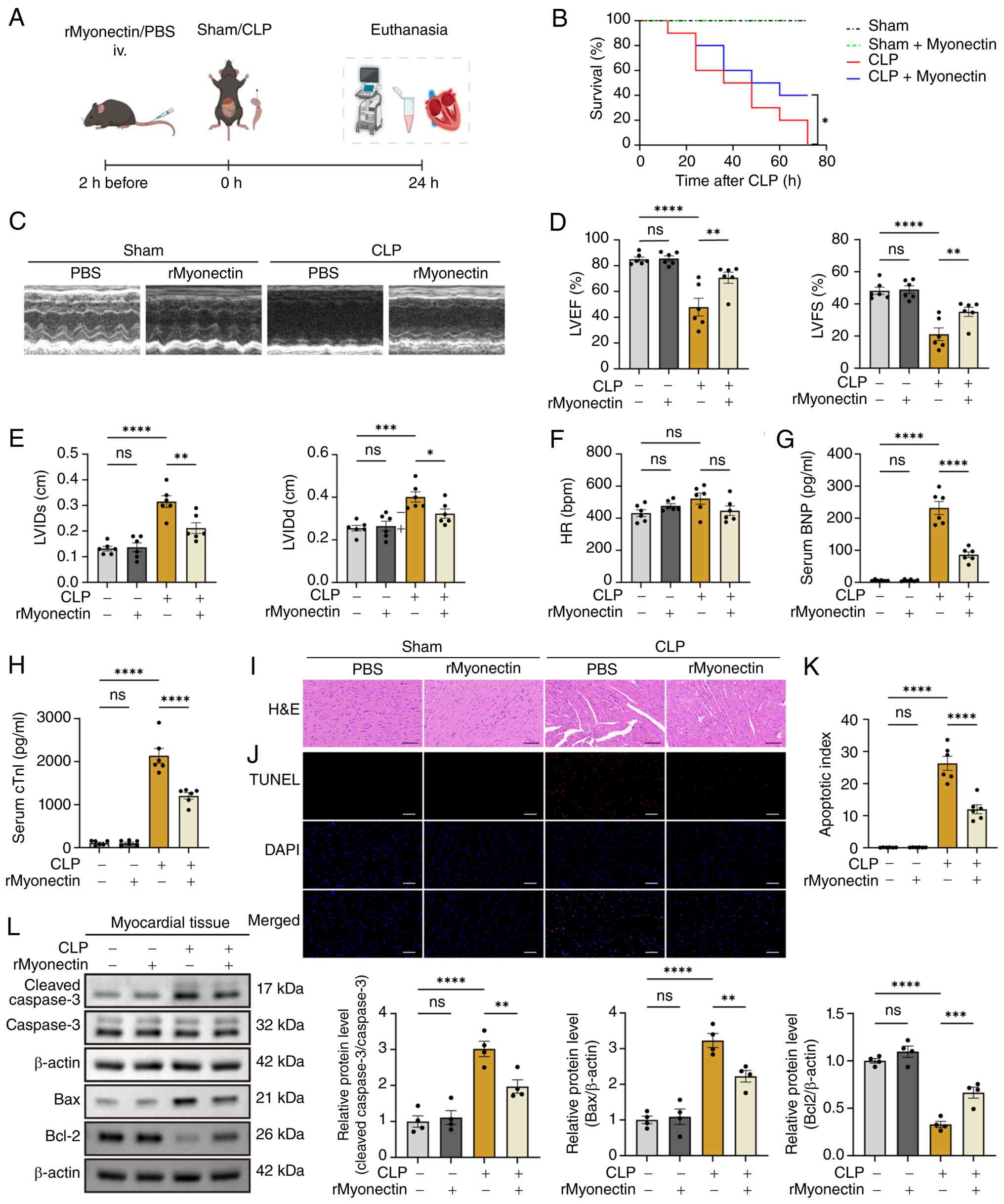 | Figure 2rMyonectin improves cardiac function,
attenuates myocardial injury and inhibits apoptosis in CLP-induced
sepsis-induced cardiomyopathy mice. (A) Schematic diagram of the
timeline for CLP surgery and rMyonectin administration, and
subsequent cardiac evaluation. (B) Survival curves. (C)
Representative images from echocardiography. (D) LVEF and LVFS. (E)
LVIDs and LVIDd. (F) HR. The serum levels of (G) BNP and (H) cTn-I.
(I) H&E staining of heart sections. Scale bar, 100 μm.
(J) TUNEL assay of heart sections. Scale bar, 50 μm. (K)
Quantification of apoptosis in TUNEL-stained sections. (L) Western
blot analysis and semi-quantification of cleaved caspase-3,
caspase-3, Bax, and Bcl-2 protein expression in myocardial tissue.
The data are presented as mean±SEM. *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001. The schematic diagram was created using
BioRender.com. rMyonectin, recombinant
myonectin; CLP, cecal ligation and puncture; LVEF, left ventricular
ejection fraction; LVFS, left ventricular fractional shortening;
LVIDs, left ventricular internal diameter at end-systole; LVIDd,
left ventricular internal diameter at end-diastole; HR, heart rate;
BNP, brain natriuretic peptide; cTn-I, cardiac troponin I; TUNEL,
terminal deoxynucleotidyl transferase dUTP nick end labeling;
H&E, hematoxylin and eosin. |
rMyonectin protects cardiac mitochondria
in SIC mice by promoting mitochondrial biogenesis and maintaining
mitochondrial dynamics
The present study revealed that rMyonectin
attenuated the LPS-induced decrease in myocardial ATP levels
(Fig. 3A). This indicated a
beneficial role for myonectin in cardiac energy metabolism during
SIC. Due to the central role of mitochondria in cardiac energetics,
their morphology was assessed using TEM. Results revealed that LPS
challenge induced mitochondrial shortening, swelling, cristae
disruption and increased vacuolation. These structural defects were
mitigated by rMyonectin pretreatment (Fig. 3B). At the molecular level,
relative to LPS administration alone, pretreatment with rMyonectin
upregulated the expression of PGC-1α, NRF1, TFAM, OPA1 and Mfn2
while reducing the p-Drp1(Ser616)/Drp1 ratio, thereby promoting
mitochondrial biogenesis and restoring the balance of mitochondrial
fusion-fission dynamics (Fig.
3C). Consistently, in CLP-induced SIC mice, rMyonectin
pretreatment also attenuated myocardial ATP depletion (Fig. 4A), improved mitochondrial
ultrastructure (Fig. 4B),
promoted mitochondrial biogenesis and restored the balance of
mitochondrial fusion-fission dynamics (Fig. 4C).
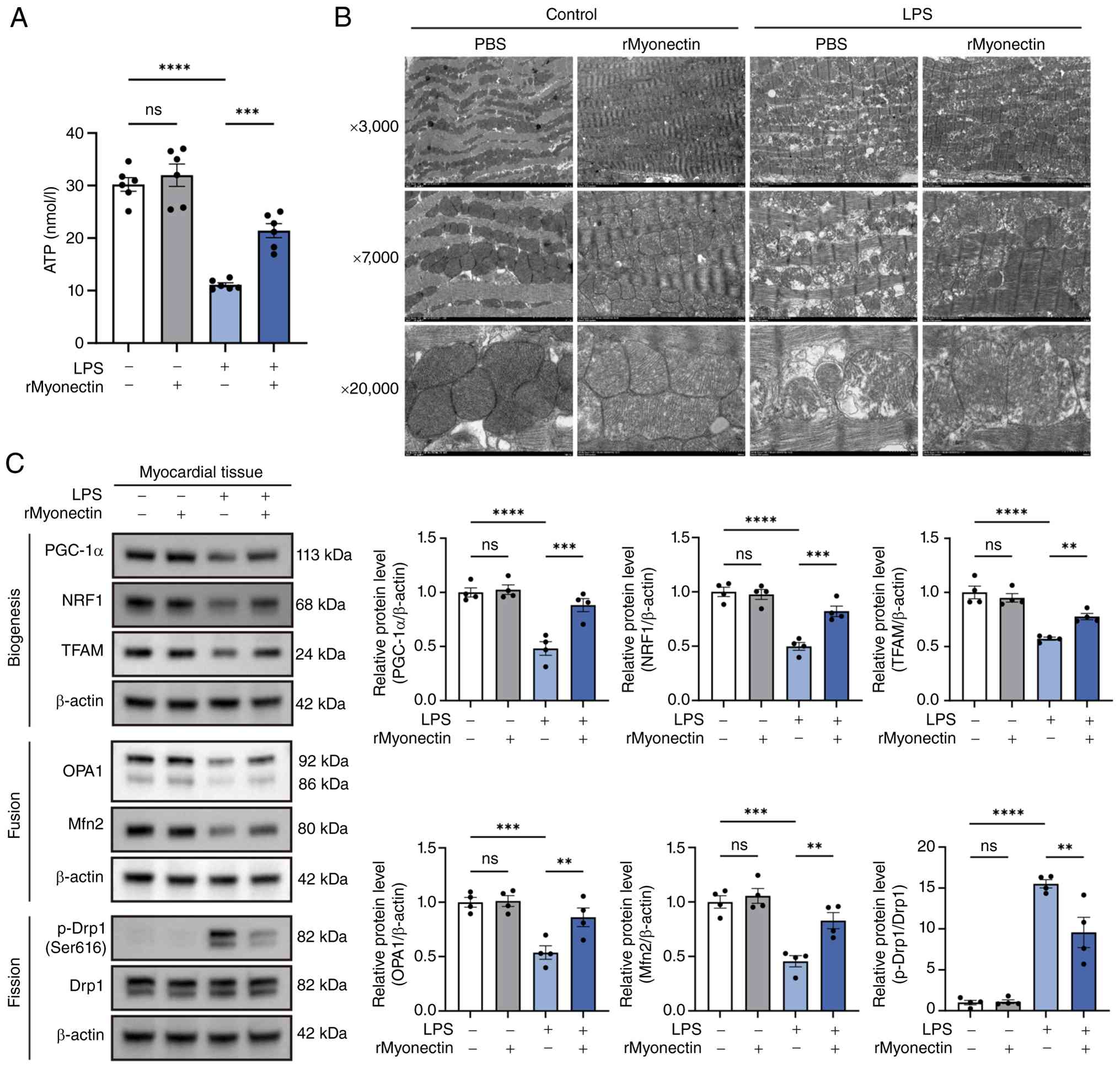 | Figure 3rMyonectin protects cardiac
mitochondria in LPS-induced sepsis-induced cardiomyopathy mice by
promoting mitochondrial biogenesis and maintaining mitochondrial
dynamics. (A) The ATP content in myocardial tissue. (B)
Mitochondrial ultrastructure of myocardial tissue was observed by
TEM. Magnification, ×3,000, ×7,000 and ×20,000. (C) Western blot
analysis and semi-quantification of PGC-1α, NRF1, TFAM, OPA1, Mfn2,
p-Drp1 at Ser616 and Drp1 in myocardial tissue. The data are
presented as mean±SEM. **P<0.01,
***P<0.001, ****P<0.0001. rMyonectin,
recombinant myonectin; TEM, transmission electron microscopy;
PGC-1α, peroxisome proliferator-activated receptor γ co-activator-1
α; NRF1, nuclear respiratory factor 1; TFAM, mitochondrial
transcription factor A; Mfn2, mitofusin 2; OPA1, optic atrophy 1;
Drp1, dynamin-related protein 1; LPS, lipopolysaccharide; p-,
phosphorylated. |
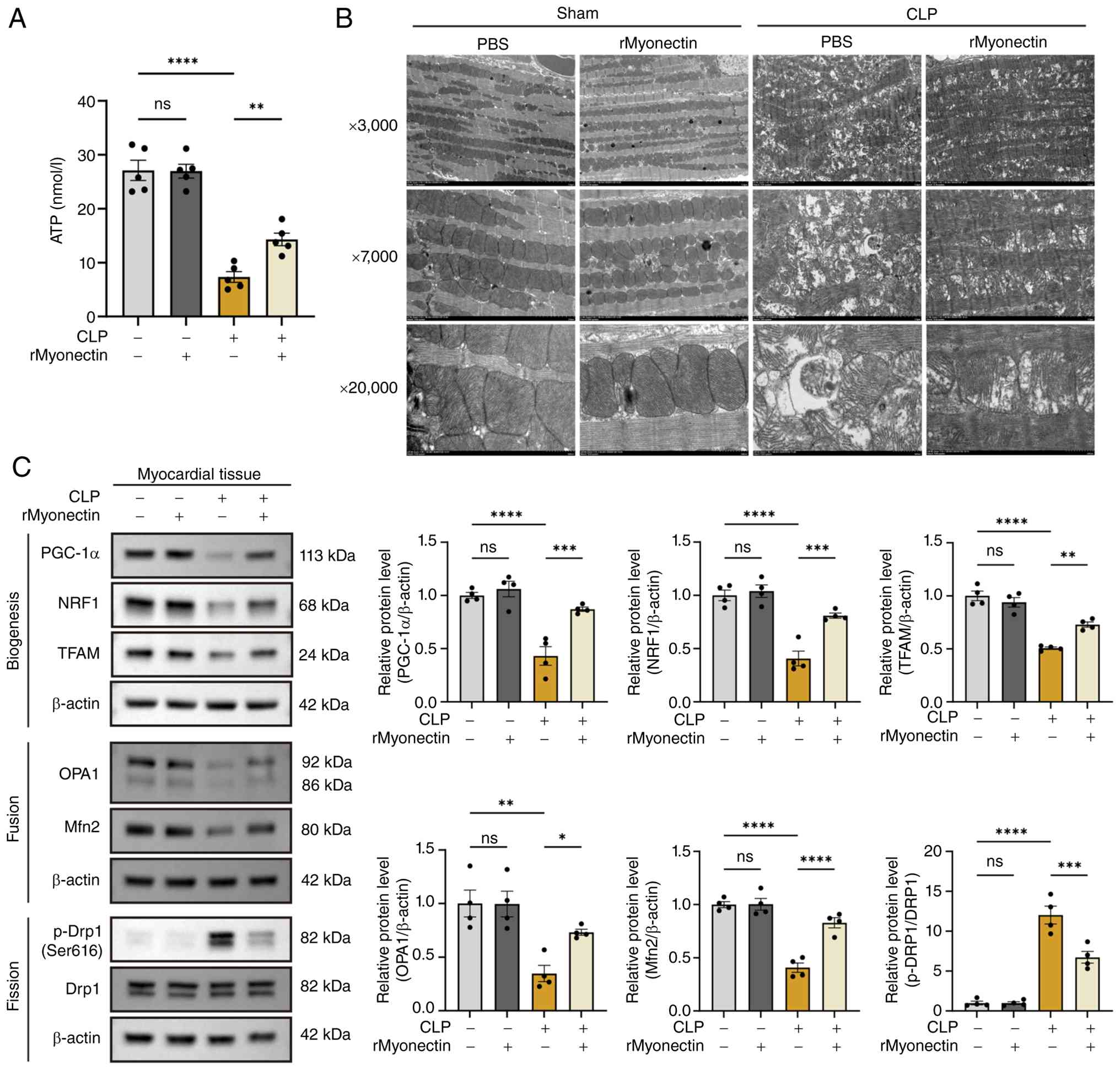 | Figure 4rMyonectin protects cardiac
mitochondria in CLP-induced sepsis-induced cardiomyopathy mice by
promoting mitochondrial biogenesis and maintaining mitochondrial
dynamics. (A) The ATP content in myocardial tissue. (B)
Mitochondrial ultrastructure of myocardial tissue was observed by
TEM. Magnification, ×3,000, ×7,000 and ×20,000. (C) Western blot
analysis and semi-quantification of PGC-1α, NRF1, TFAM, OPA1, Mfn2,
p-Drp1 at Ser616 and Drp1 in myocardial tissue. The data are
presented as mean±SEM. *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001. rMyonectin, recombinant myonectin; CLP,
cecal ligation and puncture; TEM, transmission electron microscopy;
PGC-1α, peroxisome proliferator-activated receptor γ co-activator-1
α; NRF1, nuclear respiratory factor 1; TFAM, mitochondrial
transcription factor A; Mfn2, mitofusin 2; OPA1, optic atrophy 1;
Drp1, dynamin-related protein 1; p-, phosphorylated. |
rMyonectin attenuates LPS-induced
cardiomyocyte injury and inhibits apoptosis
NMCMs were isolated from 1-3-day-old C57BL/6J mice
and treated with LPS to establish an in vitro model of SIC
(Fig. 5A). To evaluate the
cellular safety of rMyonectin, NMCMs were treated with increasing
concentrations of rMyonectin (1, 2, 5 and 8 μg/ml). The
CCK-8 assay showed that rMyonectin at these concentrations had no
significant effect on cell viability (Fig. 5B). To assess its protective
effect, NMCMs were pretreated with the same concentrations of
rMyonectin prior to LPS challenge. Analysis revealed that
rMyonectin reduced the activity of LPS-induced intracellular LDH.
Notably, LDH activity was significantly reduced at a concentration
of 5 μg/ml, but no further reduction was observed at 8
μg/ml (Fig. 5C).
Therefore, 5 μg/ml rMyonectin was selected for subsequent
experiments. At this optimal concentration, rMyonectin pretreatment
reduced LPS-induced apoptosis (Fig.
5D and E) and downregulated cleaved caspase-3 and Bax, while
upregulating Bcl-2 expression (Fig.
5F). Taken together, these data indicate that rMyonectin
attenuates LPS-induced cardiomyocyte injury and inhibits
apoptosis.
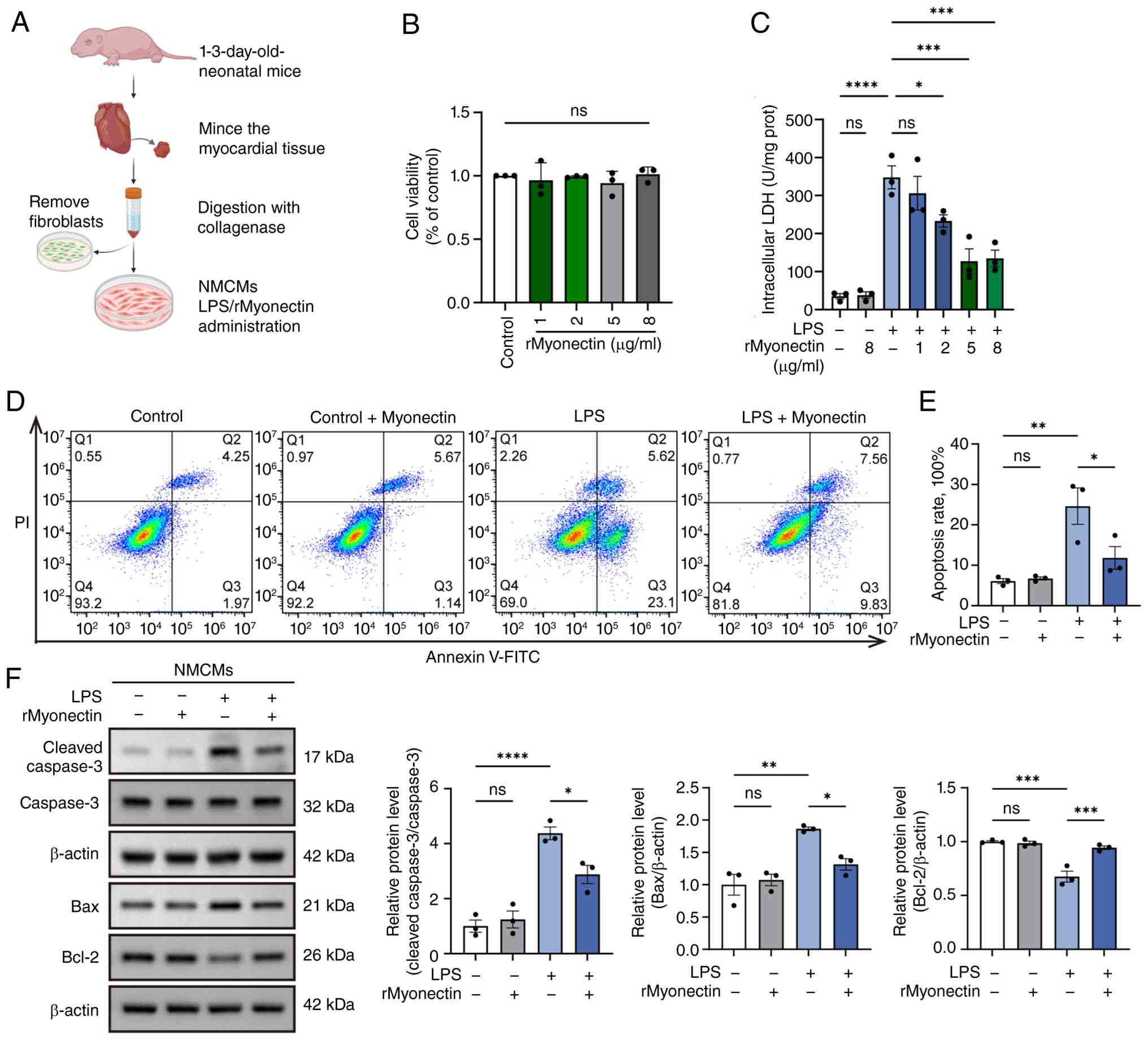 | Figure 5rMyonectin attenuates LPS-induced
cardiomyocyte injury and inhibits apoptosis. (A) Schematic diagram
showing the isolation and culture of NMCMs. NMCMs were pretreated
for 2 h with rMyonectin, followed by stimulation with LPS for 24 h.
(B) Cell viability of NMCMs was determined by Cell Counting Kit-8
assay after 24-h treatment with increasing concentrations of
rMyonectin (1, 2, 5, 8 μg/ml). (C) Intracellular LDH
activity in NMCMs pretreated with rMyonectin (1, 2, 5, 8
μg/ml) prior to LPS stimulation. (D) Apoptosis was assessed
by flow cytometry after double labeling with Annexin V-FITC and PI.
(E) The percentage of apoptotic cells detected using flow
cytometry. (F) Western blot analysis and semi-quantification of
cleaved caspase-3, caspase-3, Bax and Bcl-2 protein expression in
NMCMs. The data are presented as mean±SEM. *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001. The schematic diagram was created using
BioRender.com. rMyonectin, recombinant
myonectin; NMCMs, neonatal mouse cardiomyocytes; LDH, lactate
dehydrogenase; LPS, lipopolysaccharide; PI, propidium iodide. |
rMyonectin ameliorates LPS-induced
mitochondrial dysfunction in cardiomyocytes
In cardiomyocytes, rMyonectin reversed the
LPS-induced decrease in ATP levels (Fig. 6A), ameliorated the reductions in
OCR (Fig. 6B) and in the
activities of mitochondrial respiratory chain complexes I and III
(Fig. 6C), and stabilized MMP
(Fig. 6D). Consistent with the
in vivo observations, LPS challenge downregulated PGC-1α,
NRF1, TFAM, OPA1 and Mfn2 while increasing the p-Drp1(Ser616)/Drp1
ratio, all of which were reversed by rMyonectin (Fig. 6E). These results indicate that
rMyonectin ameliorates LPS-induced mitochondrial dysfunction, as
evidenced by enhanced mitochondrial respiratory function,
stabilized membrane potential, and restored mitochondrial dynamics
and biogenesis.
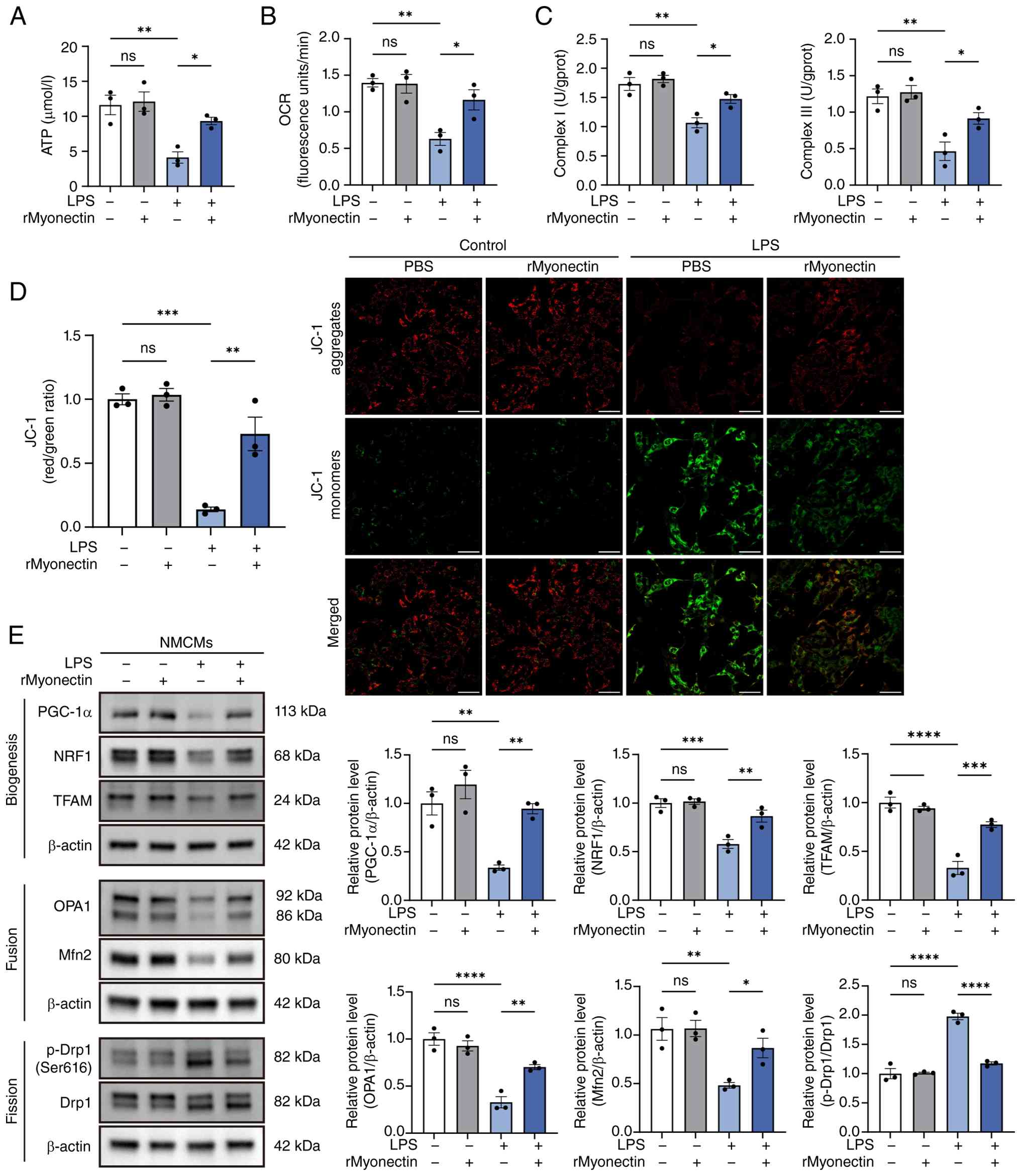 | Figure 6rMyonectin ameliorates LPS-induced
mitochondrial dysfunction in cardiomyocytes. (A) The ATP content in
NMCMs. (B) Relative OCR. (C) Detection of the activities of
mitochondrial respiratory chain complexes I and III. (D) Analysis
of MMP using JC-1 staining. Scale bar, 50 μm. (E) Western
blot analysis and semi-quantification of PGC-1α, NRF1, TFAM, OPA1,
Mfn2, p-Drp1 at Ser616, and Drp1 in NMCMs. The data are presented
as mean±SEM. *P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. rMyonectin,
recombinant myonectin; NMCMs, neonatal mouse cardiomyocytes; OCR,
oxygen consumption rate; MMP, mitochondrial membrane potential;
PGC-1α, peroxisome proliferator-activated receptor γ co-activator-1
α; NRF1, nuclear respiratory factor 1; TFAM, mitochondrial
transcription factor A; Mfn2, mitofusin 2; OPA1, optic atrophy 1;
Drp1, dynamin-related protein 1; LPS, lipopolysaccharide; p-,
phosphorylated. |
Protective effect of rMyonectin against
LPS-induced apoptosis and mitochondrial dysfunction in
cardiomyocytes is mediated by AMPK activation
Due to the key role of AMPK in energy metabolism,
the present study first assessed AMPK activation. Analysis revealed
that rMyonectin effectively attenuated the LPS-induced reduction in
AMPK phosphorylation in both myocardial tissue and NMCMs (Fig. 7A and B). To determine whether
AMPK activation mediates the protective effects of rMyonectin, the
present study inhibited AMPK in cardiomyocytes using CC in addition
to rMyonectin pretreatment (Fig.
7C). CC abolished the protective effects of rMyonectin against
cardiomyocyte injury (Fig. 7D)
and apoptosis (Fig. 7E-G).
Furthermore, CC attenuated the ability of rMyonectin to elevate
intracellular ATP levels (Fig.
8A), to increase OCR (Fig.
8B) and activities of mitochondrial respiratory chain complexes
I and III (Fig. 8C), and to
preserve MMP (Fig. 8D). Western
blot analysis further demonstrated that the rMyonectin-induced
upregulation of PGC-1α, NRF1, TFAM, OPA1 and Mfn2, as well as the
downregulation of the p-Drp1(Ser616)/Drp1 ratio, was abolished upon
AMPK inhibition (Fig. 8E). These
results demonstrate that the anti-apoptotic and
mitochondrial-protective effects of rMyonectin are mediated by AMPK
activation.
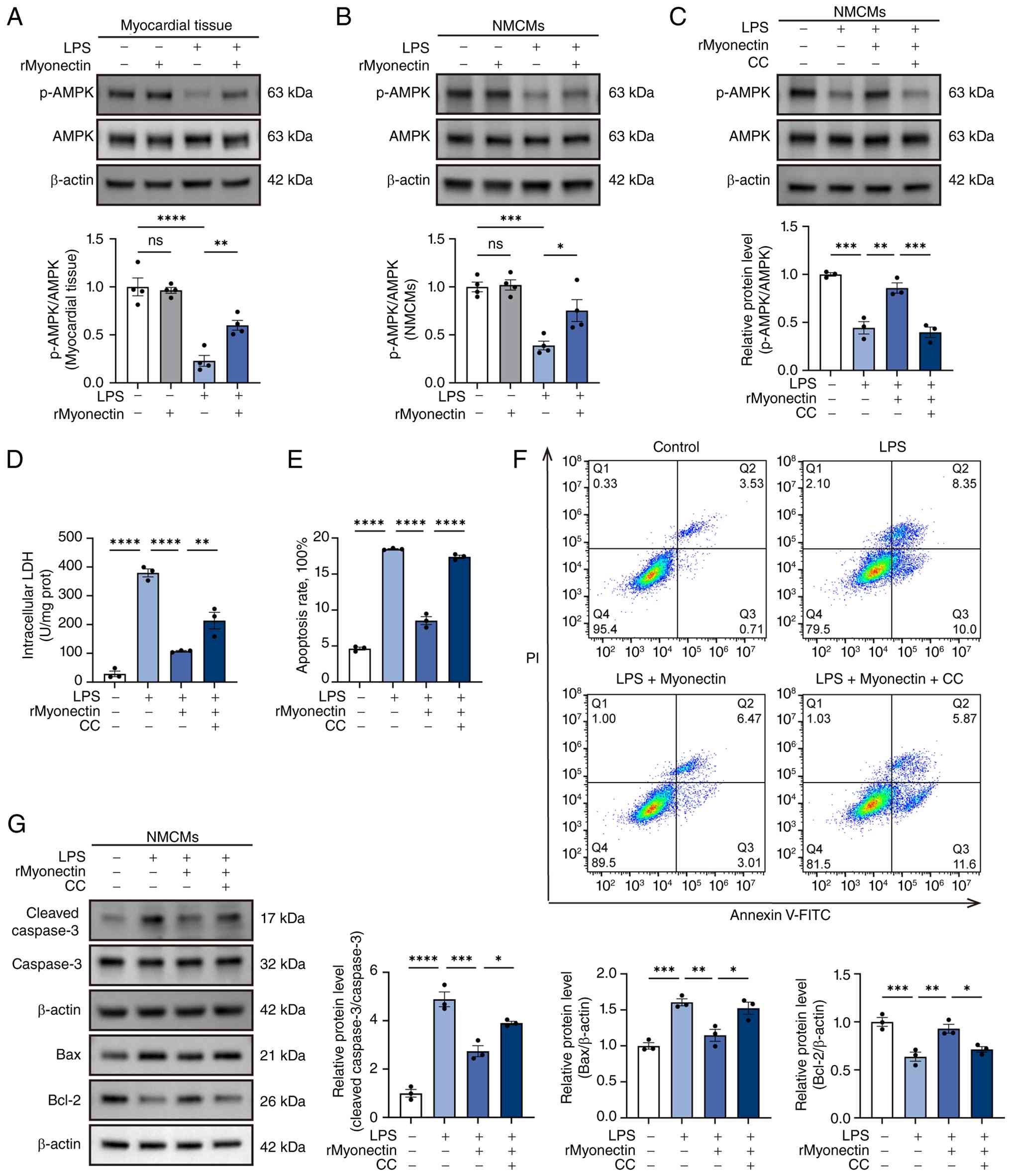 | Figure 7Protective effect of rMyonectin
against LPS-induced apoptosis in cardiomyocytes is mediated by AMPK
activation. (A) Western blot analysis and semi-quantification of
p-AMPK and AMPK in myocardial tissue. (B) Western blot analysis and
semi-quantification of p-AMPK and AMPK in NMCMs. (C) Expression of
p-AMPK and AMPK in NMCMs after CC treatment. (D) Intracellular LDH
activity in NMCMs. (E) The percentage of apoptotic cells detected
using flow cytometry. (F) Apoptosis was assessed using flow
cytometry after double labeling with Annexin V-FITC and PI. (G)
Western blot analysis and semi-quantification of cleaved caspase-3,
caspase-3, Bax and Bcl-2 protein expression in NMCMs. The data are
presented as mean±SEM. *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001. rMyonectin, recombinant myonectin;
NMCMs, neonatal mouse cardiomyocytes; AMPK, AMP-activated protein
kinase; CC, Compound C; LDH, lactate dehydrogenase; LPS,
lipopolysaccharide; PI, propidium iodide; p-, phosphorylated. |
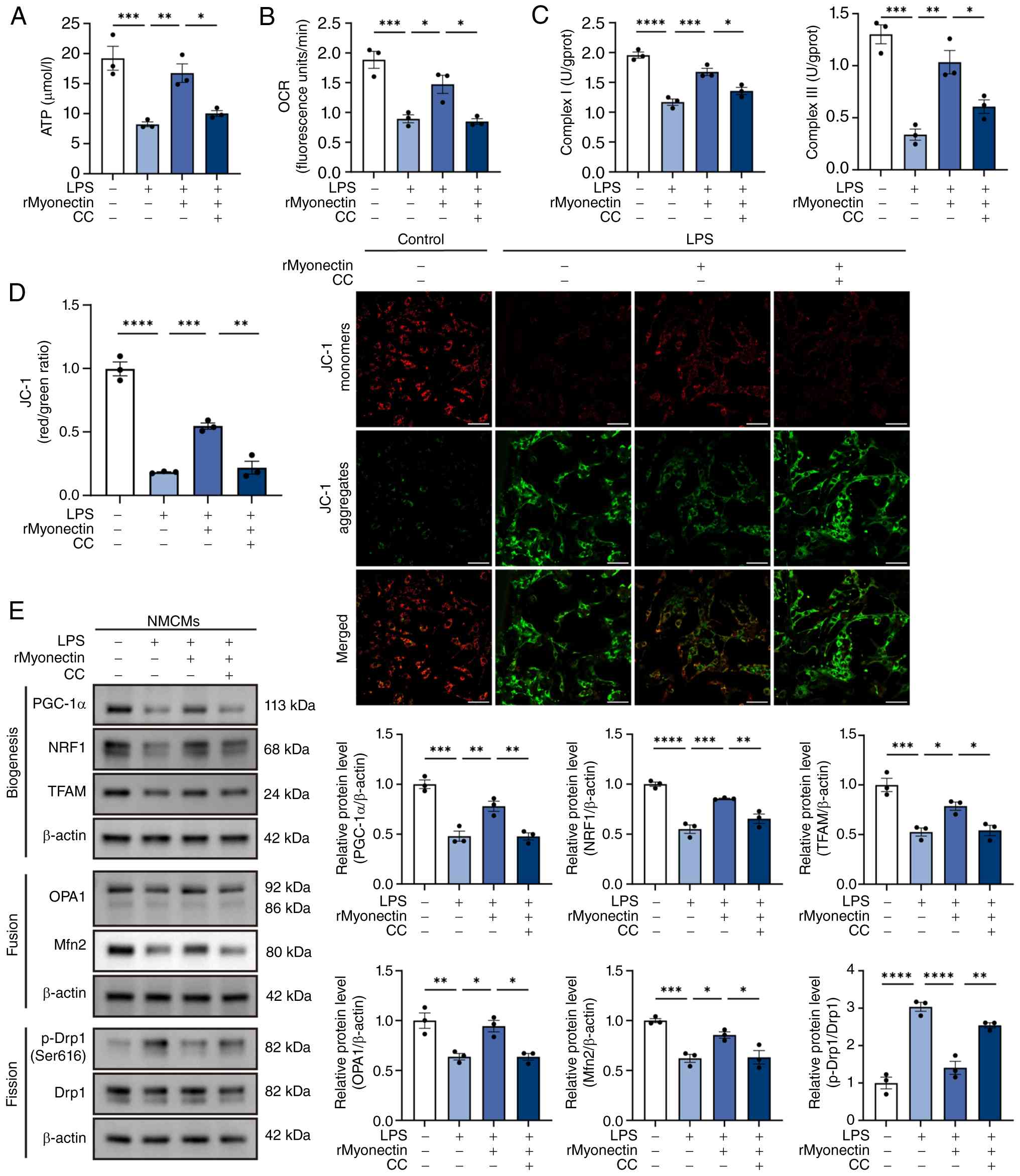 | Figure 8Protective effect of rMyonectin
against LPS-induced mitochondrial dysfunction in cardiomyocytes is
mediated by AMPK activation. (A) The ATP content in NMCMs. (B)
Relative OCR. (C) Detection of the activities of mitochondrial
respiratory chain complexes I and III. (D) Analysis of MMP using
JC-1 staining. Scale bar, 50 μm. (E) Western blot analysis
and semi-quantification of PGC-1α, NRF1, TFAM, OPA1, Mfn2, p-Drp1
at Ser616, and Drp1 in NMCMs. The data are presented as mean±SEM.
*P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. rMyonectin,
recombinant myonectin; NMCMs, neonatal mouse cardiomyocytes; OCR,
oxygen consumption rate; MMP, mitochondrial membrane potential;
PGC-1α, peroxisome proliferator-activated receptor γ co-activator-1
α; NRF1, nuclear respiratory factor 1; TFAM, mitochondrial
transcription factor A; Mfn2, mitofusin 2; OPA1, optic atrophy 1;
Drp1, dynamin-related protein 1; LPS, lipopolysaccharide; p-,
phosphorylated. |
AdipoR1 knockdown abolishes the
protective effect of rMyonectin against LPS-induced apoptosis and
mitochondrial dysfunction in cardiomyocytes
Myonectin is a paralog of adiponectin, sharing
similar structural and functional characteristics. Adiponectin
mediates its cardioprotective effects primarily through activating
the adiponectin receptors. Notably, AdipoR1 has been established as
an upstream activator in the AMPK signaling pathway (31,32). To determine whether myonectin
protected cardiomyocytes through the AdipoR1/AMPK pathway, the
present study measured AdipoR1 protein levels in myocardial tissue
and NMCMs. The results indicated that rMyonectin treatment reversed
the LPS-induced downregulation of AdipoR1 expression in these
models (Fig. 9A and B).
Following siRNA-mediated knockdown of AdipoR1 in NMCMs (with
transfection efficiency validated in Figs. S1 and S2), AMPK phosphorylation
was reduced (Fig. 9C and D), and
the protective effects of rMyonectin were abolished. Specifically,
AdipoR1 knockdown reversed the protective effects of rMyonectin
against cardiomyocyte injury (Fig.
9E) and apoptosis (Fig.
9F-H). Moreover, it eliminated the rMyonectin-induced
enhancements in intracellular ATP levels (Fig. 10A), OCR (Fig. 10B), the activities of
mitochondrial respiratory chain complexes I and III (Fig. 10C), as well as the restoration
of MMP (Fig. 10D). Western blot
analysis further demonstrated that the rMyonectin-induced
upregulation of PGC-1α, NRF1, TFAM, OPA1 and Mfn2, as well as the
downregulation of the p-Drp1(Ser616)/Drp1 ratio, was abolished by
AdipoR1 knockdown (Fig. 10E).
Collectively, these results demonstrate that rMyonectin protects
cardiomyocytes from LPS-induced injury via the AdipoR1/AMPK
pathway.
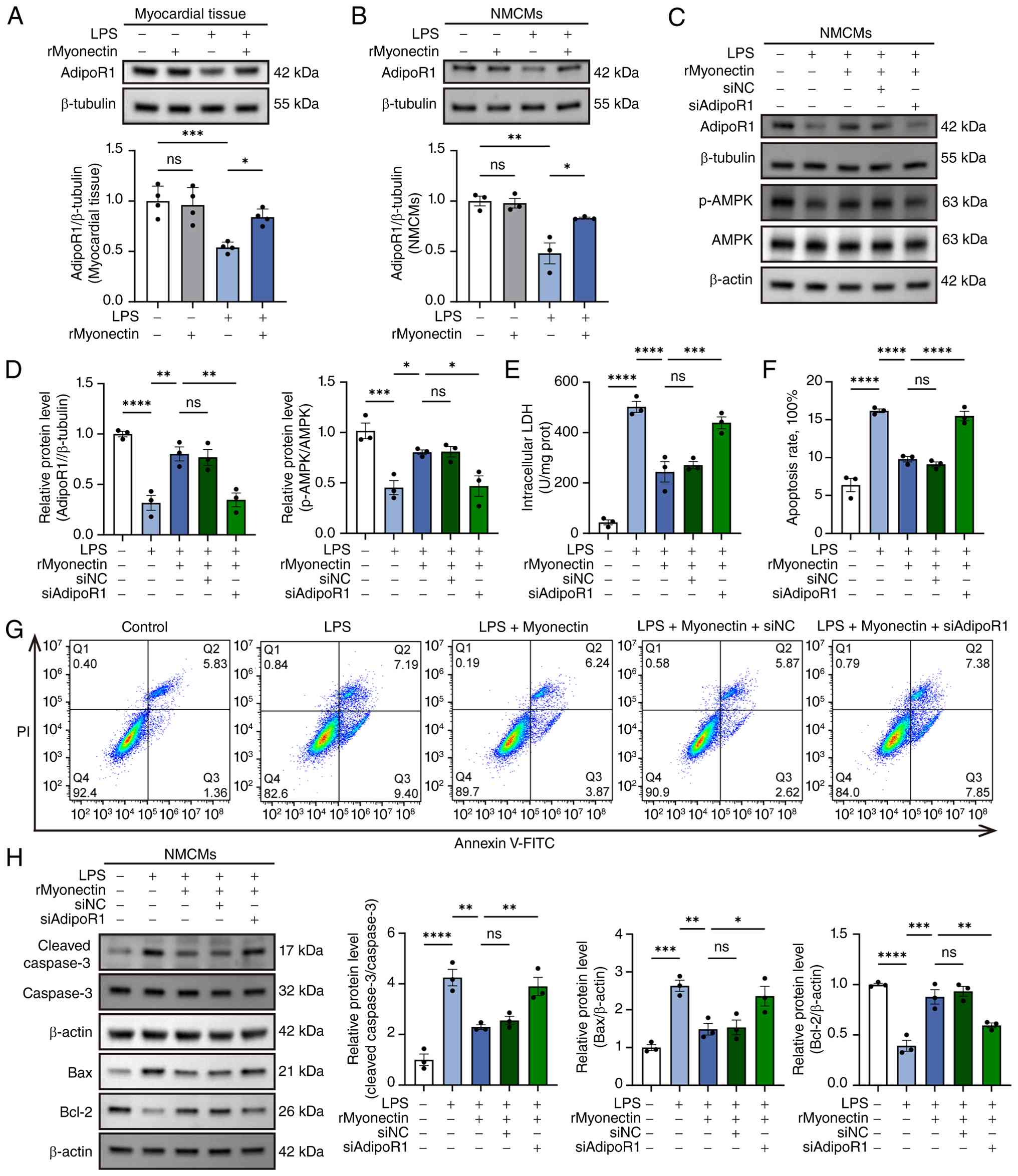 | Figure 9AdipoR1 knockdown abolishes the
protective effect of rMyonectin against LPS-induced apoptosis in
cardiomyocytes. (A) Western blot analysis and semi-quantification
of AdipoR1 in myocardial tissue. (B) Western blot analysis and
semi-quantification of AdipoR1 in NMCMs. (C) Representative western
blots showing the expression of AdipoR1, p-AMPK and AMPK in NMCMs
following AdipoR1 knockdown. (D) Semi-quantification of AdipoR1,
p-AMPK and AMPK protein levels in NMCMs following AdipoR1
knockdown. (E) Intracellular LDH activity in NMCMs. (F) The
percentage of apoptotic cells detected using flow cytometry. (G)
Apoptosis was assessed using flow cytometry after double labeling
with Annexin V-FITC and PI. (H) Western blot analysis and
semi-quantification of cleaved caspase-3, caspase-3, Bax, and Bcl-2
protein expression in NMCMs. The data are presented as mean±SEM.
*P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. rMyonectin,
recombinant myonectin; NMCMs, neonatal mouse cardiomyocytes;
AdipoR1, adiponectin receptor 1; AMPK, AMP-activated protein
kinase; LDH, lactate dehydrogenase; si, small interfering RNA; NC,
negative control; siAdipoR1, siRNA targeting AdipoR1; LPS,
lipopolysaccharide; p-, phosphorylated; PI, propidium iodide. |
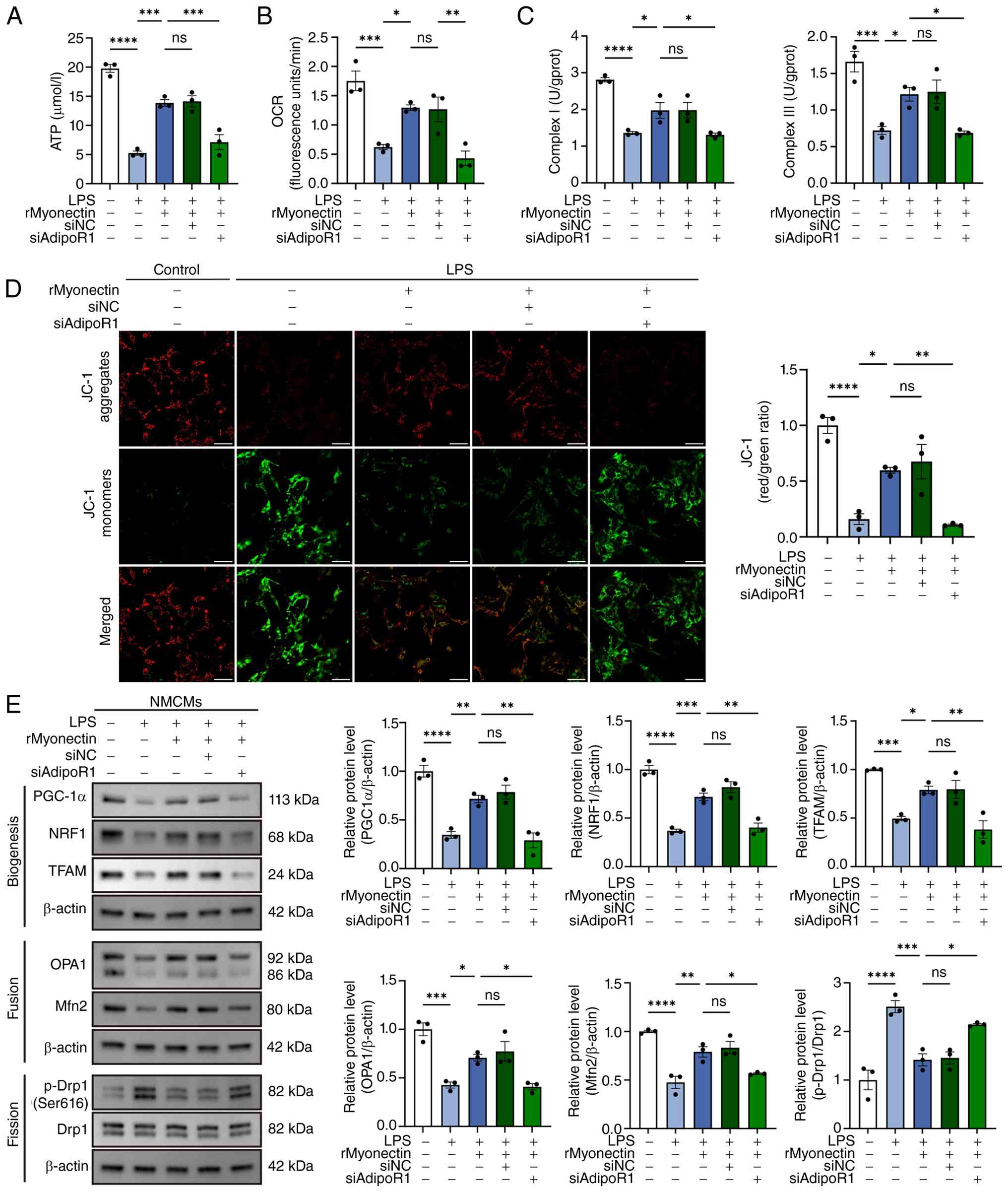 | Figure 10AdipoR1 knockdown abolishes the
protective effect of rMyonectin against LPS-induced mitochondrial
dysfunction in cardiomyocytes. (A) The ATP content in NMCMs. (B)
Relative OCR. (C) Detection of the activities of mitochondrial
respiratory chain complexes I and III. (D) Analysis of MMP using
JC-1 staining. Scale bar, 50 μm. (E) Western blot analysis
and semi-quantification of PGC-1α, NRF1, TFAM, OPA1, Mfn2, p-Drp1
at Ser616 and Drp1 in NMCMs. The data are presented as mean±SEM.
*P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. rMyonectin,
recombinant myonectin; NMCMs, neonatal mouse cardiomyocytes;
AdipoR1, adiponectin receptor 1; AMPK, AMP-activated protein
kinase; OCR, oxygen consumption rate; MMP, mitochondrial membrane
potential; PGC-1α, peroxisome proliferator-activated receptor γ
co-activator-1 α; NRF1, nuclear respiratory factor 1; TFAM,
mitochondrial transcription factor A; Mfn2, mitofusin 2; OPA1,
optic atrophy 1; Drp1, dynamin-related protein 1; si, small
interfering RNA; NC, negative control; siAdipoR1, siRNA targeting
AdipoR1; LPS, lipopolysaccharide; p-, phosphorylated. |
Discussion
To the best of our knowledge, the present study
elucidated, for the first time, the protective role of rMyonectin
in SIC by using both LPS- and CLP-induced in vivo models,
together with LPS-stimulated cardiomyocytes for in vitro
validation. Mechanistically, rMyonectin exerted its
cardioprotective effects by activating the AdipoR1/AMPK pathway,
which in turn ameliorated mitochondrial dysfunction (as evidenced
by enhanced mitochondrial biogenesis, restored homeostasis of
mitochondrial dynamics, increased OCR, elevated activities of
mitochondrial respiratory chain complexes I and III and enhanced
ATP production) and suppressed cardiomyocyte apoptosis (Fig. 11). Collectively, these findings
suggest that rMyonectin holds promise as a protective candidate for
SIC.
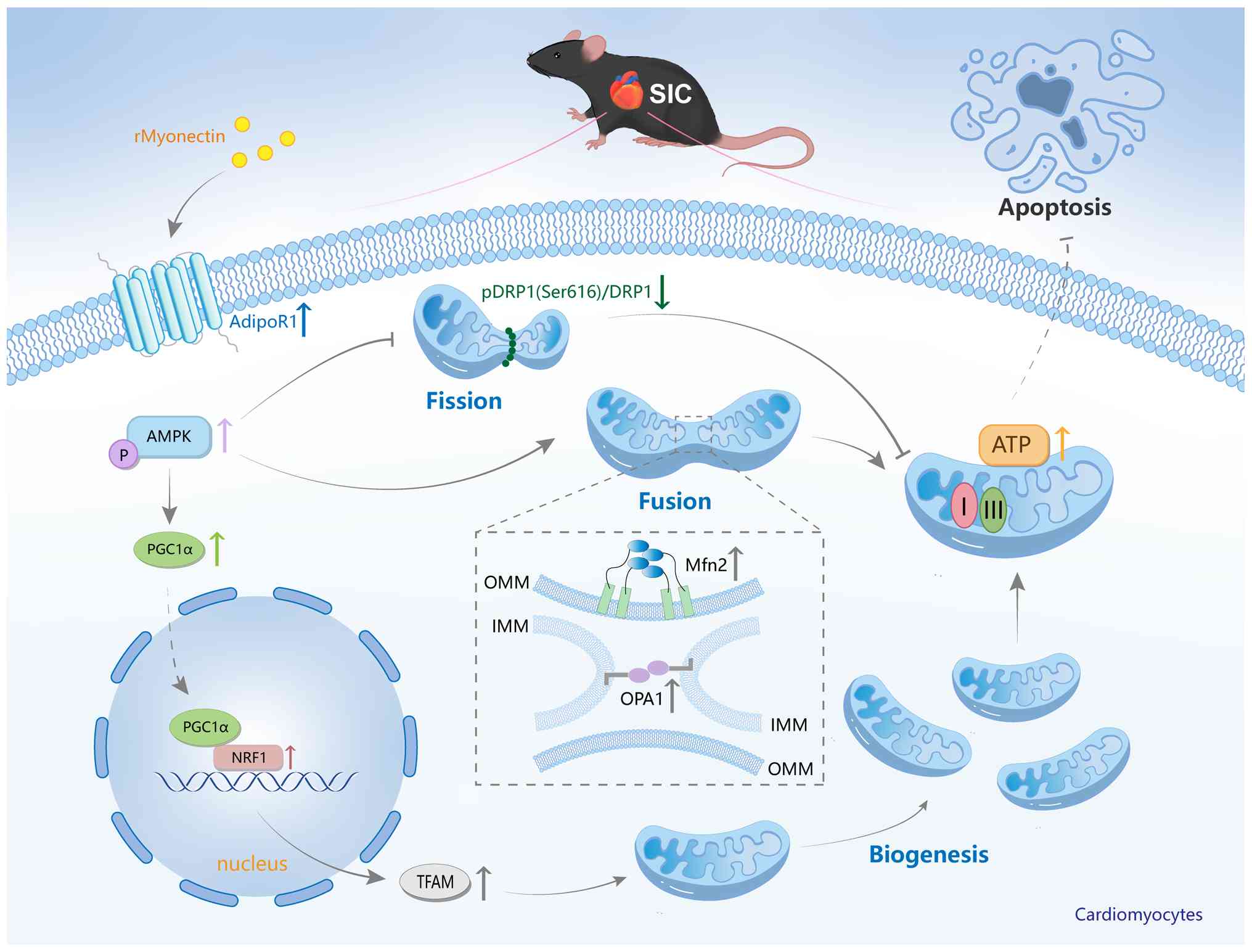 | Figure 11Molecular mechanism by which
rMyonectin ameliorates SIC. rMyonectin ameliorates SIC by
alleviating mitochondrial dysfunction and inhibiting cardiomyocyte
apoptosis via activation of the AdipoR1/AMPK pathway. rMyonectin,
recombinant myonectin; SIC, sepsis-induced cardiomyopathy; OMM,
outer mitochondrial membranes; IMM, inner mitochondrial membranes;
AdipoR1, adiponectin receptor 1; AMPK, AMP-activated protein
kinase; PGC-1α, peroxisome proliferator-activated receptor γ
co-activator-1 α; NRF1, nuclear respiratory factor 1; TFAM,
mitochondrial transcription factor A; Mfn2, mitofusin 2; OPA1,
optic atrophy 1; Drp1, dynamin-related protein 1; I, mitochondrial
respiratory chain complex I; III, mitochondrial respiratory chain
complex III; p-, phosphorylated. |
Myonectin, also known as CTRP15, is a member of the
CTRP family. The CTRPs are paralogs of adiponectin and serve
diverse roles in metabolism and cardiovascular physiology. The
majority of CTRPs, with the exception of CTRP4, share a common
structure composed of four distinct domains: A signal peptide at
the N terminus, a short variable region, a collagenous domain and a
C-terminal globular domain that is homologous to C1q. Despite this
common architectural framework, individual CTRPs exhibit distinct
tissue specific expression profiles and regulate a wide array of
physiological functions (31).
The heart not only expresses several members of the CTRPs but also
serves as an important target organ for their actions (33). Given this, their roles in cardiac
pathologies such as SIC are of substantial interest. Accumulating
evidence has elucidated the roles and underlying mechanisms of
CTRPs in SIC. For instance, Wei et al (34) revealed that CTRP3 expression was
upregulated in cardiac tissue upon LPS stimulation. CTRP3 protected
against sepsis-induced cardiac dysfunction by activating the AMPKα
pathway, thereby inhibiting inflammatory responses and apoptosis.
Similarly, CTRP1 has been shown to exert a protective effect
against LPS-induced cardiac injury through a sirtuin 1-dependent
mechanism that attenuates oxidative stress and inflammation
(25). CTRP9 also protects
against LPS-induced acute myocardial injury by inhibiting the
inflammatory response via an AdipoR1/AMPK-dependent pathway
(35).
Myonectin is a myokine primarily synthesized and
secreted by skeletal muscle and is also expressed in cardiac and
smooth muscle. Exercise stimulates the upregulation of myonectin in
skeletal muscle and increases its circulating concentration
(36). To date, its precise
functions and mechanisms in cardiovascular diseases remain poorly
characterized. Otaka et al (17) systematically elucidated the role
of myonectin in myocardial I/R injury using a multifaceted
experimental approach that included generating myonectin-knockout
mice, performing AAV-mediated knockdown of myonectin in skeletal
muscle, and administering rMyonectin intravenously. They found that
skeletal muscle-derived myonectin protected against myocardial I/R
injury in mice by suppressing the inflammatory response in
macrophages and apoptosis in cardiomyocytes through the
S1P/cAMP/Akt signaling pathway. Zhao et al (18) demonstrated that myonectin
exhibited markedly higher expression in cardiomyocytes compared
with cardiac fibroblasts. The study further revealed that myonectin
attenuated myocardial fibrosis by promoting the phosphorylation of
the IR and IRS-1, thereby activating the PI3K/Akt signaling
pathway. These findings establish myonectin as a functionally
relevant factor in cardiovascular pathophysiology. To investigate
the role of myonectin in SIC, the present study employed two
complementary murine models (intraperitoneal LPS challenge and CLP)
together with LPS-challenged NMCMs for in vitro validation,
with rMyonectin administered accordingly. The results consistently
demonstrated that rMyonectin effectively ameliorated cardiac
dysfunction in vivo, and reduced myocardial injury as well
as suppressed cardiomyocyte apoptosis in both in vivo and
in vitro settings, collectively validating its
cardioprotective effect.
To investigate the potential mechanisms of myonectin
in the heart, the present study drew an analogy based on its known
functions in skeletal muscle. Existing literature indicated that in
skeletal tissue, myonectin activated the AMPK/PGC-1α signaling
pathway, promoted mitochondrial biogenesis, and thus ameliorated
mitochondrial dysfunction (19).
Mitochondrial dysfunction serves as an important pathogenic
mechanism underlying SIC. This dysfunction is characterized by
structural abnormalities, oxidative stress, altered mitochondrial
permeability, mitochondrial uncoupling and dysregulation of
mitochondrial quality control systems along with disrupted calcium
homeostasis (37,38). Mitochondrial quality control is
maintained through three interconnected processes: Dynamics (fusion
and fission), biogenesis and mitophagy (39). Under physiological conditions, a
continuous cycle of fusion and fission sustains mitochondrial
structural integrity and functional homeostasis. Fission and fusion
are indispensable for a range of key cellular functions, including
ATP production, mitochondrial DNA (mtDNA) distribution,
mitochondrial respiratory activity, cell survival, apoptosis and
calcium signaling (40). Fusion
of the outer and inner mitochondrial membranes is primarily
mediated by mitofusins (Mfn1/2) and OPA1, respectively.
Mitochondrial fusion enables the dynamic repair of reversibly
damaged portions of mitochondria, forming functional elongated
organelles (41).
Mitochondrial fission is a process whereby Drp1
triggers constriction of the mitochondrial membrane, resulting in
the division of a single mitochondrion into two separate organelles
(40). Phosphorylation of Drp1
at serine 616 is an activating modification that drives its
recruitment to the outer mitochondrial membranes, leading to
mitochondrial fragmentation (42). A homeostatic balance between
mitochondrial fusion and fission is essential for maintaining
mitochondrial network integrity and ensuring proper cellular
physiological function. In septic mice, myocardial Drp1 activation
and Mfn2 downregulation promote a pathological shift toward
mitochondrial fission, leading to an imbalance in mitochondrial
dynamics and, ultimately, to mitochondrial dysfunction and
apoptosis (8,37). Mitochondrial biogenesis can be
defined as the growth and division of pre-existing mitochondria
(43), which requires
coordinated mtDNA replication and expression, import of
nuclear-encoded proteins, and phospholipid transport for membrane
assembly (44). PGC-1α is the
primary regulator of mitochondrial biogenesis. It co-activates
NRF1, which in turn activates the promoter of TFAM. Through this
transcriptional cascade, PGC-1α mediates the expression of
mitochondrial proteins encoded by both nuclear DNA and mtDNA,
promoting mitochondrial biogenesis, regulating the intracellular
mitochondrial content, increasing mitochondrial ATP production and
coordinating cellular energy metabolism (45). The findings of the present study
indicate that rMyonectin protects against SIC by activating the
AMPK pathway, which upregulates PGC-1α, NRF1, TFAM, OPA1 and Mfn2,
while downregulating p-Drp1(Ser616)/Drp1, thereby restoring the
myocardial mitochondrial fission-fusion balance and promoting
mitochondrial biogenesis. The present study provides the first
evidence that myonectin regulates mitochondrial dynamics.
How myonectin activates AMPK to alleviate
mitochondrial dysfunction warrants further investigation. Notably,
the specific receptors for adiponectin, AdipoR1 and AdipoR2,
mediate AMPK and PPARα activation, respectively (32). Koentges et al (14) showed that loss of AdipoR1 induced
cardiac mitochondrial dysfunction and uncoupling in mice, a
phenotype not observed with AdipoR2 loss. Given that myonectin is a
paralog of adiponectin, the two proteins share structural
similarities (31). This led to
the investigation of whether myonectin acts through a similar
receptor-mediated pathway. Having demonstrated that myonectin
activated the AMPK pathway, we hypothesized that AdipoR1 mediated
the protective effects of myonectin in SIC. The present data
confirmed this hypothesis, as AdipoR1 knockdown abolished the
protective effects of rMyonectin on mitochondrial dysfunction and
cardiomyocyte apoptosis in NMCMs.
Several limitations of the present study should be
acknowledged. First, rMyonectin was administered systemically via
tail vein injection. This non-targeted delivery resulted in its
broad distribution across multiple tissues and organs, which
confounded the interpretation of its cardiac-specific effects in
vivo. Consequently, the cardiac specificity of myonectin in SIC
remains to be elucidated in future studies. Second, the present
study employed a pretreatment protocol, which is a classic design
in mechanistic research to elucidate the protective effects and
signaling pathways of rMyonectin. However, this prophylactic
administration protocol is not fully aligned with real-world
clinical practice, and the present study did not evaluate the
efficacy of therapeutic administration. Future studies are
warranted to further assess the therapeutic efficacy of rMyonectin
when administered after the onset of sepsis, in order to determine
its potential for clinical translation. Finally, the in vivo
experiments in the present study were conducted exclusively in male
mice, a long-standing convention in cardiovascular basic research.
The primary rationale for this approach was to avoid potential
interference of the female estrous cycle with cardiac contractile
function. Although a study has suggested that under conventional
group-housing conditions the estrous cycle in female mice may be
irregular, thereby reducing such interference to some extent
(46), the influence of the
estrous cycle on cardiac function cannot be completely excluded.
Nevertheless, the exclusive use of male mice in this design
inevitably introduces a sex-specific bias, limiting the
generalizability across sexes. Future studies should repeat the key
experiments in female mice to assess the impact of sex on the
results.
In conclusion, the present study demonstrates that
rMyonectin protects against SIC by activating AdipoR1/AMPK
signaling, thereby restoring mitochondrial function and suppressing
apoptosis to alleviate cardiac injury and dysfunction. These
findings position rMyonectin as a novel and promising protective
agent for SIC.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
PL contributed to study design, data analysis and
manuscript drafting. RC, HZ, HL and LL contributed to study design
and conducted the initial data interpretation. BD and PY
contributed to study design, critical review and manuscript
revision. All authors reviewed the manuscript critically. BD and PY
confirm the authenticity of all the raw data. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
All animal procedures were approved by the
Institutional Ethics Committee of the School of Basic Medical
Sciences, Jilin University (approval nos. 2025-597 and
2026-491).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
We are grateful to the following colleagues from the
Scientific Research Center of China-Japan Union Hospital of Jilin
University for their guidance and support with flow cytometry: Dr
Yucheng Zhang, Ms. Yingnan Liu (lab technician), Ms. Huiying Lv
(lab technician) and Ms. Puyu Gao (lab technician).
Funding
The present study was supported by the National Natural Science
Foundation of China (grant nos. 82470327 and 82470400) and the
Project of Jilin Provincial Department of Finance (grant no.
2022SCZ40).
References
|
1
|
Aissaoui N, Boissier F, Chew M, Singer M
and Vignon P: Sepsis-induced cardiomyopathy. Eur Heart J.
46:3339–3353. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lanspa MJ, Cirulis MM, Wiley BM, Olsen TD,
Wilson EL, Beesley SJ, Brown SM, Hirshberg EL and Grissom CK: Right
ventricular dysfunction in early sepsis and septic shock. Chest.
159:1055–1063. 2021. View Article : Google Scholar
|
|
3
|
Kuroshima T, Kawaguchi S and Okada M:
Current perspectives of mitochondria in sepsis-induced
cardiomyopathy. Int J Mol Sci. 25:47102024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu YC, Yu MM, Shou ST and Chai YF:
Sepsis-induced cardiomyopathy: Mechanisms and treatments. Front
Immunol. 8:10212017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stanzani G, Duchen MR and Singer M: The
role of mitochondria in sepsis-induced cardiomyopathy. Biochim
Biophys Acta Mol Basis Dis. 1865:759–773. 2019. View Article : Google Scholar
|
|
6
|
Lopaschuk GD, Karwi QG, Tian R, Wende AR
and Abel ED: Cardiac energy metabolism in heart failure. Circ Res.
128:1487–1513. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Suliman HB, Welty-Wolf KE, Carraway M,
Tatro L and Piantadosi CA: Lipopolysaccharide induces oxidative
cardiac mitochondrial damage and biogenesis. Cardiovasc Res.
64:279–288. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen XS, Cui JR, Meng XL, Wang SH, Wei W,
Gao YL, Shou ST, Liu YC and Chai YF: Angiotensin-(1-7) ameliorates
sepsis-induced cardiomyopathy by alleviating inflammatory response
and mitochondrial damage through the NF-κB and MAPK pathways. J
Transl Med. 21:22023. View Article : Google Scholar
|
|
9
|
Zhang S, Xu Y, Zhu J, Ma J, Niu Q and Wang
X: Carbon monoxide attenuates LPS-induced myocardial dysfunction in
rats by regulating the mitochondrial dynamic equilibrium. Eur J
Pharmacol. 889:1737262020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen XS, Wang SH, Liu CY, Gao YL, Meng XL,
Wei W, Shou ST, Liu YC and Chai YF: Losartan attenuates
sepsis-induced cardiomyopathy by regulating macrophage polarization
via TLR4-mediated NF-κB and MAPK signaling. Pharmacol Res.
185:1064732022. View Article : Google Scholar
|
|
11
|
Kim TT and Dyck JR: Is AMPK the savior of
the failing heart? Trends Endocrinol Metab. 26:40–48. 2015.
View Article : Google Scholar
|
|
12
|
Wan S, Cui Z, Wu L, Zhang F, Liu T, Hu J,
Tian J, Yu B, Liu F, Kou J and Li F: Ginsenoside Rd promotes
omentin secretion in adipose through TBK1-AMPK to improve
mitochondrial biogenesis via WNT5A/Ca2+ pathways in
heart failure. Redox Biol. 60:1026102023. View Article : Google Scholar
|
|
13
|
Jin Z, Li X, Liu H, He T, Jiang W, Peng L,
Wu X, Chen M, Fan Y, Lu Z, et al: MEGF9 prevents
lipopolysaccharide-induced cardiac dysfunction through activating
AMPK pathway. Redox Rep. 30:24352522025. View Article : Google Scholar :
|
|
14
|
Koentges C, König A, Pfeil K, Hölscher ME,
Schnick T, Wende AR, Schrepper A, Cimolai MC, Kersting S, Hoffmann
MM, et al: Myocardial mitochondrial dysfunction in mice lacking
adiponectin receptor 1. Basic Res Cardiol. 110:372015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Petro JL, Gallo-Villegas J and Calderón
JC: Myonectin and metabolic health: A systematic review. Front
Endocrinol (Lausanne). 16:15571422025. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Srole DN and Ganz T: Erythroferrone
structure, function, and physiology: Iron homeostasis and beyond. J
Cell Physiol. 236:4888–4901. 2021. View Article : Google Scholar
|
|
17
|
Otaka N, Shibata R, Ohashi K, Uemura Y,
Kambara T, Enomoto T, Ogawa H, Ito M, Kawanishi H, Maruyama S, et
al: Myonectin is an exercise-induced myokine that protects the
heart from ischemia-reperfusion injury. Circ Res. 123:1326–1338.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao Q, Zhang CL, Xiang RL, Wu LL and Li
L: CTRP15 derived from cardiac myocytes attenuates TGFβ1-induced
fibrotic response in cardiac fibroblasts. Cardiovasc Drugs Ther.
34:591–604. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ozaki Y, Ohashi K, Otaka N, Kawanishi H,
Takikawa T, Fang L, Takahara K, Tatsumi M, Ishihama S, Takefuji M,
et al: Myonectin protects against skeletal muscle dysfunction in
male mice through activation of AMPK/PGC1α pathway. Nat Commun.
14:46752023. View Article : Google Scholar
|
|
20
|
Saiyang X, Qingqing W, Man X, Chen L, Min
Z, Yun X, Wenke S, Haiming W, Xiaofeng Z, Si C, et al: Activation
of Toll-like receptor 7 provides cardioprotection in septic
cardiomyopathy-induced systolic dysfunction. Clin Transl Med.
11:e2662021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ni L, Lin B, Shen M, Li C, Hu L, Fu F,
Chen L, Yang J and Shi D: PKM2 deficiency exacerbates gram-negative
sepsis-induced cardiomyopathy via disrupting cardiac calcium
homeostasis. Cell Death Discov. 8:4962022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fang Y, Yang B, Zhu D, Zhang X, Jiang X,
Wang H, Zhang L, Yu Y, Yang Y, Yuan M, et al: Anemoside B4
ameliorates septic cardiomyopathy by activating SRC-mediated
PI3K/AKT/mTOR pathway to alleviate mitochondrial dysfunction and
cardiomyocyte senescence. Int Immunopharmacol. 171:1161082026.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang B, Li T, Wang Z, Zhu Y, Niu K, Hu S,
Lin Z, Zheng X, Jin X and Shen C: Ruxolitinib-based senomorphic
therapy mitigates cardiomyocyte senescence in septic cardiomyopathy
by inhibiting the JAK2/STAT3 signaling pathway. Int J Biol Sci.
20:4314–4340. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wan TT, Li Y, Li JX, Xiao X, Liu L, Li HH
and Guo SB: ACE2 activation alleviates sepsis-induced
cardiomyopathy by promoting MasR-Sirt1-mediated mitochondrial
biogenesis. Arch Biochem Biophys. 752:1098552024. View Article : Google Scholar
|
|
25
|
Jiang W, Li W, Hu X, Hu R, Li B and Lan L:
CTRP1 prevents sepsis-induced cardiomyopathy via Sirt1-dependent
pathways. Free Radic Biol Med. 152:810–820. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ravi V, Jain A, Taneja A, Chatterjee K and
Sundaresan NR: Isolation and culture of neonatal murine primary
cardiomyocytes. Curr Protoc. 1:e1962021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang H, Wang L, Hu F, Wang P, Xie Y, Li F
and Guo B: Neuregulin-4 attenuates diabetic cardiomyopathy by
regulating autophagy via the AMPK/mTOR signalling pathway.
Cardiovasc Diabetol. 21:2052022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang F, Qin Y, Wang Y, Meng S, Xian H, Che
H, Lv J, Li Y, Yu Y, Bai Y and Wang L: Metformin inhibits the NLRP3
inflammasome via AMPK/mTOR-dependent effects in diabetic
cardiomyopathy. Int J Biol Sci. 15:1010–1019. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang J, Pu X, Zhuang H, Guo Z, Wang M,
Yang H, Li C and Chang X: Astragaloside IV alleviates septic
myocardial injury through DUSP1-Prohibitin 2 mediated mitochondrial
quality control and ER-autophagy. J Adv Res. 75:561–580. 2025.
View Article : Google Scholar
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−delta delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
31
|
Schäffler A and Buechler C: CTRP family:
Linking immunity to metabolism. Trends Endocrinol Metab.
23:194–204. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nguyen TMD: Adiponectin: Role in
physiology and pathophysiology. Int J Prev Med. 11:1362020.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schanbacher C, Hermanns HM, Lorenz K,
Wajant H and Lang I: Complement 1q/tumor necrosis factor-related
proteins (CTRPs): Structure, receptors and signaling. Biomedicines.
11:5592023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wei WY, Ma ZG, Zhang N, Xu SC, Yuan YP,
Zeng XF and Tang QZ: Overexpression of CTRP3 protects against
sepsis-induced myocardial dysfunction in mice. Mol Cell Endocrinol.
476:27–36. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kambara T, Shibata R, Ohashi K, Matsuo K,
Hiramatsu-Ito M, Enomoto T, Yuasa D, Ito M, Hayakawa S, Ogawa H, et
al: C1q/tumor necrosis factor-related protein 9 protects against
acute myocardial injury through an adiponectin receptor
I-AMPK-dependent mechanism. Mol Cell Biol. 35:2173–2185. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Seldin MM, Peterson JM, Byerly MS, Wei Z
and Wong GW: Myonectin (CTRP15), a novel myokine that links
skeletal muscle to systemic lipid homeostasis. J Biol Chem.
287:11968–11980. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lin Y, Xu Y and Zhang Z: Sepsis-induced
myocardial dysfunction (SIMD): The pathophysiological mechanisms
and therapeutic strategies targeting mitochondria. Inflammation.
43:1184–1200. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sato R, Sanfilippo F, Lanspa M, Duggal A
and Dugar S: Sepsis-induced cardiomyopathy: Mechanism, prevalence,
assessment, prognosis, and management. Chest. 168:1383–1394. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu BH, Xu CZ, Liu Y, Lu ZL, Fu TL, Li GR,
Deng Y, Luo GQ, Ding S, Li N and Geng Q: Mitochondrial quality
control in human health and disease. Mil Med Res.
11:322024.PubMed/NCBI
|
|
40
|
Jin JY, Wei XX, Zhi XL, Wang XH and Meng
D: Drp1-dependent mitochondrial fission in cardiovascular disease.
Acta Pharmacol Sin. 42:655–664. 2021. View Article : Google Scholar
|
|
41
|
Forte M, Schirone L, Ameri P, Basso C,
Catalucci D, Modica J, Chimenti C, Crotti L, Frati G, Rubattu S, et
al: The role of mitochondrial dynamics in cardiovascular diseases.
Br J Pharmacol. 178:2060–2076. 2021. View Article : Google Scholar
|
|
42
|
Serasinghe MN and Chipuk JE: Mitochondrial
fission in human diseases. Handb Exp Pharmacol. 240:159–188. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jornayvaz FR and Shulman GI: Regulation of
mitochondrial biogenesis. Essays Biochem. 47:69–84. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu L, Li Y, Chen G and Chen Q: Crosstalk
between mitochondrial biogenesis and mitophagy to maintain
mitochondrial homeostasis. J Biomed Sci. 30:862023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Murphy MP and Hartley RC: Mitochondria as
a therapeutic target for common pathologies. Nat Rev Drug Discov.
17:865–886. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
MacDonald JK, Pyle WG, Reitz CJ and
Howlett SE: Cardiac contraction, calcium transients, and
myofilament calcium sensitivity fluctuate with the estrous cycle in
young adult female mice. Am J Physiol Heart Circ Physiol.
306:H938–H953. 2014. View Article : Google Scholar : PubMed/NCBI
|














