Introduction
Fracture healing is a complex biological process
that requires precise spatiotemporal coordination among the
nervous, vascular and skeletal systems (1,2).
With the accelerated aging of the global population, fractures have
emerged as a major and growing public health challenge (3). The global burden is substantial,
with 178 million new cases reported in 2019 alone, a 33.4% increase
since 1990 (3). This significant
incidence highlights the urgent clinical need to understand the
fundamental biology of fracture repair, particularly when this
intricate process fails (3).
The successful healing of these fractures is often
critically impaired by underlying conditions, with osteoporosis
being a predominant risk factor that compromises bone quality
(4,5). The repair process relies on a
sophisticated crosstalk between systems. Specifically, the nervous
system modulates bone metabolism through neuropeptides, such as
calcitonin gene-related peptide (CGRP). In parallel, a specialized
capillary subtype, known as type H vessels (the 'H' denotes high
expression of both CD31 and endomucin), couples angiogenesis with
osteogenesis via angiocrine factors, such as Wnt (wingless-related
integration site) ligands (6-8).
However, the molecular mechanisms that spatiotemporally integrate
these multisystemic cues remain incompletely elucidated.
Initially recognized as a neuronal guidance cue and
synaptogenesis regulator, neuropilin-1 (NRP1) has since been
identified as a pleiotropic co-receptor for a wide spectrum of
ligands, including vascular endothelial growth factor (VEGF),
semaphorin 3A (SEMA3A) and TGF-β (9-12). Its potential role is further
supported by research demonstrating that NRP1 mediates SEMA3A
signaling to enhance osteogenesis and accelerate fracture repair
(13). Emerging evidence
currently suggests that NRP1 lies at the crossroads of
nerve-vessel-bone crosstalk, functioning as a potential signaling
node that coordinates the distribution of nerves, vascular
patterning, and osteogenic differentiation during skeletal
regeneration (14-18).
In the context of impaired fracture healing, as
commonly observed in osteoporosis, NRP1 signaling is frequently
disrupted, potentially leading to dysregulated bone remodeling
(19). This is associated with
poor repair outcomes, including dysregulated arterial capillary
formation, abnormal axonal orientation and delayed callus
mineralization (14,18,20). The present review summarizes the
multifaceted functions of NRP1 in orchestrating the intercellular
communication essential for bone repair. It discusses the
preliminary translational potential of targeting NRP1 signaling as
a candidate therapeutic strategy to recalibrate the impaired
healing microenvironment and restore bone homeostasis.
Structure and distribution of NRP1
Structure of NRP1
NRP1 is a conserved transmembrane glycoprotein of
~130 kDa that is widely expressed across vertebrate species. It
exhibits a modular three-domain structure consisting of a large
extracellular domain, a single transmembrane helix and a short
cytoplasmic tail (21,22).
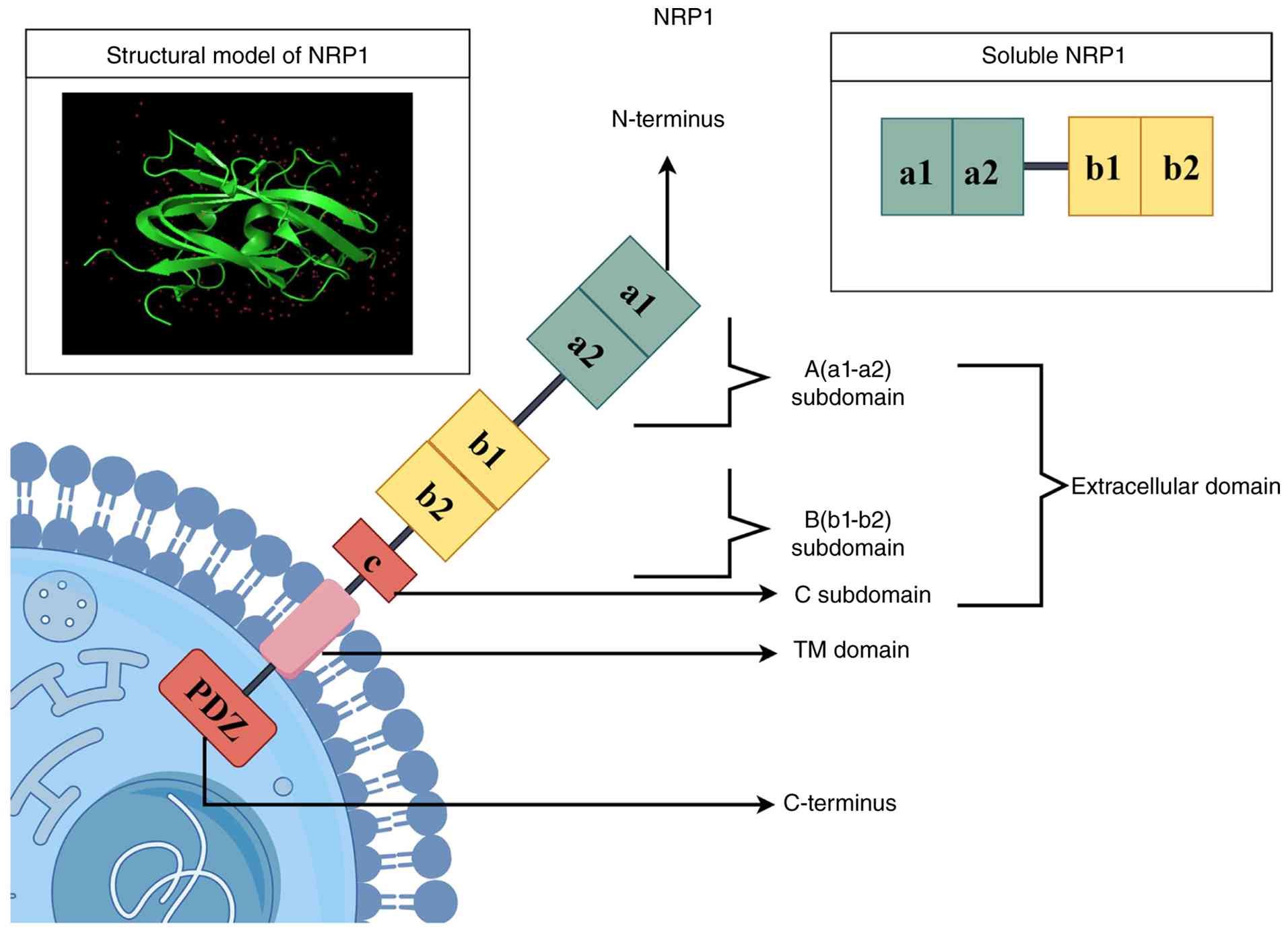
Extracellular domain
The extracellular domain of NRP1 consists of five
subdomains arranged sequentially from the N-terminus to the
C-terminus, designated as a1, a2, b1, b2 and c domains, and this
region serves as the core ligand-binding domain of NRP1 (23,24). Among these, the a1/a2 domains are
mainly responsible for binding to SEMA3 family molecules (25). The b1/b2 domains are the primary
binding sites for VEGF and exhibit a strong preference for the
VEGFA165 isoform. This interaction provides a key basis for
VEGF-mediated endothelial cell migration (26). In addition to the aforementioned
classical interactions, the b1/b2 domains can also mediate the
binding to C-end rule (CendR) peptides (27-29). The C domain mediates
homodimerization of NRP1, a key step in the assembly of functional
receptor complexes (30), as
illustrated in Fig. 1.
Transmembrane domain
The hydrophobic transmembrane domain of NRP1
contains a GxxxG motif that promotes its homodimerization (31). NRP1 stabilizes its
oligomerization state through the synergistic effect of the
extracellular c domain and the GxxxG motif, providing the necessary
structural framework for the formation of functional complexes with
receptors such as VEGF receptor 2 (VEGFR2) and platelet-derived
growth factor receptor β (PDGFRβ) (22,31-35), as illustrated in Fig. 1.
Cytoplasmic domain
The cytoplasmic domain of NRP1 is a short tail
structure with no kinase activity; yet, it undertakes indispensable
biological functions. A postsynaptic density protein-95/discs
large/zonula occludens-1 (PDZ)-binding motif known as the SEA
tripeptide sequence (Ser-Glu-Ala, the three C-terminal residues of
the cytoplasmic tail) exists at its C-terminus (21,36). This motif can specifically bind
to intracellular adaptor proteins containing PDZ domains, thereby
regulating cell adhesion and cytoskeletal rearrangement. In
addition, NRP1 acts as a coreceptor to enhance VEGFR2
phosphorylation, thereby amplifying downstream p38 MAPK and ERK1/2
signaling (37,38). Moreover, NRP1 can maintain its
own stability through a Rab11A-dependent recycling pathway and
undergo post-translational modifications, such as glycosaminoglycan
and glycosylation modifications (39,40) (Fig. 1)
Soluble NRP1 (sNRP1)
In addition to transmembrane NRP1, a soluble isoform
of NRP1, sNRP1, also exists. sNRP1 is generated by alternative
splicing or proteolytic cleavage and contains only the
extracellular a and b domains (41,42). As a decoy receptor, sNRP1 can
competitively bind to free VEGFA165 and prevent it from binding to
the NRP1/VEGFR2 complex on the cell surface (41), as illustrated in Fig. 1.
Regional and cell-specific distribution
of NRP1 in bone tissue
NRP1 exhibits regional and cell-specific
distribution in bone tissue, which is highly consistent with its
functions in osteogenesis, bone repair and neurovascular coupling
(17,18,43). NRP1 is localized in bone tissue
regions with active osteogenesis and bone repair, such as the
metaphyses of long bones and trabecular bone margins. These regions
are rich in osteoblasts and bone marrow endothelial cells, and this
specific localization provides an anatomical basis for
NRP1-mediated bone formation and bone repair signaling pathways
(18,43). At the cellular level, NRP1 is
expressed in osteoblasts, osteoclast precursors, chondrocytes,
intraosseous vascular endothelial cells and sensory nerve endings
in bone tissue. This expression pattern has been confirmed by
relevant studies in both mouse and human bone tissues, further
supporting its potential involvement in mediating bone metabolism
and repair-related signaling pathways (6,14,17,44).
NRP1 regulates the functions of key vascular
cells and structural homeostasis
NRP1 regulates the functions of key
vascular cells
Precapillary arterioles and capillaries, composed of
vascular endothelial cells and accompanying pericytes, are key
vascular structures responsible for nutrient transport and
metabolic exchange in bone tissue (6). Research on non-skeletal systems has
confirmed that an abnormal NRP1 expression leads to vascular
structural disorganization and impaired maturation (45).
Endothelial cells
Existing evidence suggests that NRP1 may regulate
endothelial cell function and play a potential role in angiogenesis
during bone regeneration. In vivo research using a rabbit
ischemic bone defect regeneration model confirmed that the local
sustained release of NRP1 via a 3D-printed magnesium alloy
composite scaffold significantly promoted neovascularization in the
defect area (46). In
vitro experiments have further clarified its molecular
mechanism: As a co-receptor of VEGFR2, NRP1 forms a stable ternary
complex with VEGFA and VEGFR2 on the surface of endothelial cells,
thereby enhancing signal transduction efficiency and activating the
phosphatidylinositol 3-kinase-protein kinase B (PI3K-AKT) pathway.
This ultimately upregulates the expression of VEGFA, fibroblast
growth factor 2 and the nuclear receptor Nr4a1 in endothelial
cells, thereby regulating endothelial cell migration and
proliferation (46), as
demonstrated in Fig. 2.
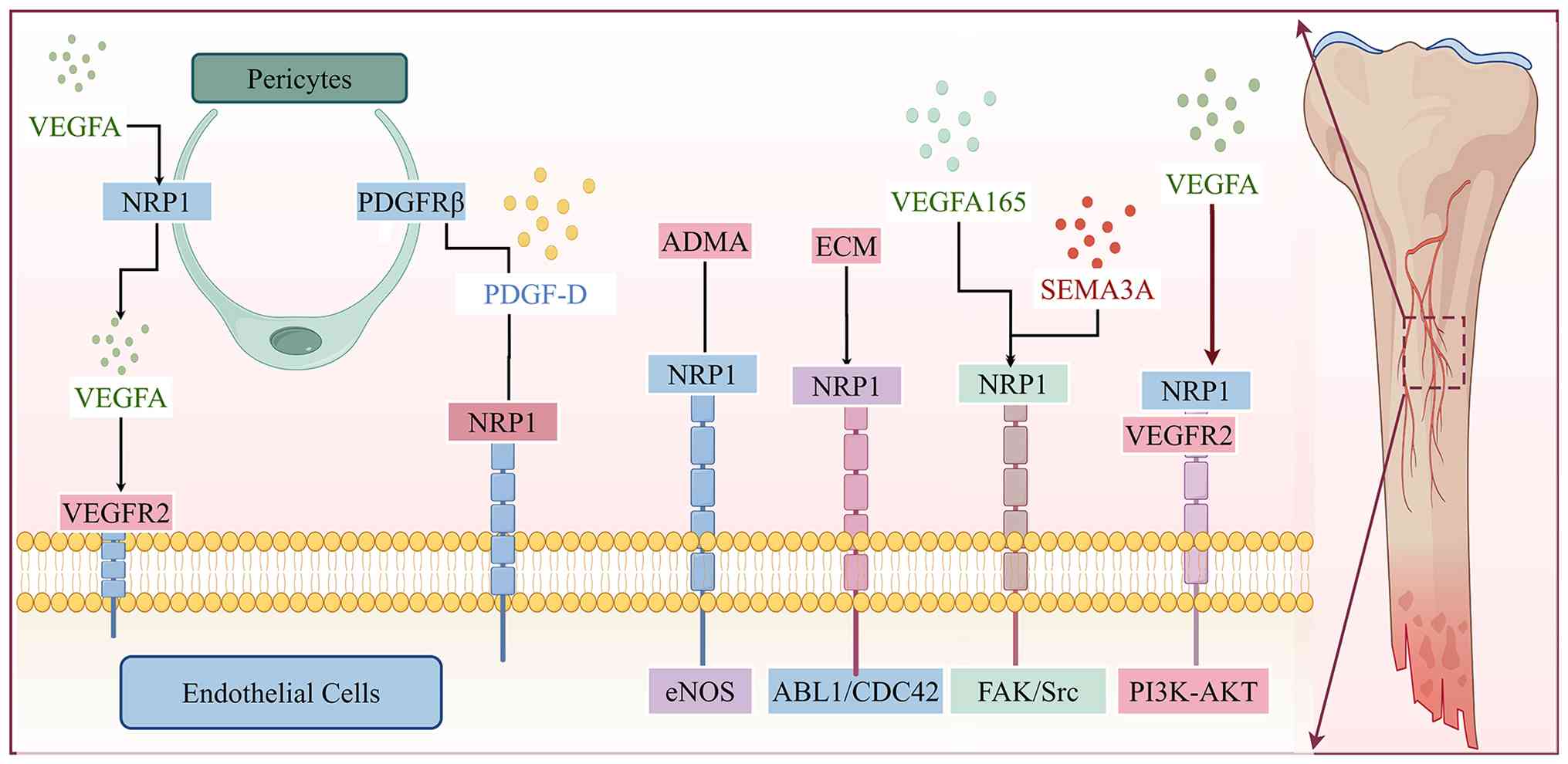 | Figure 2Schematic illustration of the
molecular mechanisms by which NRP1, as a multiligand co-receptor,
regulates angiogenesis and vascular homeostasis maintenance. At the
endothelial cell level, VEGFA forms a ternary complex with NRP1 and
VEGFR2 to activate the PI3K-AKT pathway, promoting
neovascularization in bone defect areas; this mechanism has been
directly validated in bone regeneration models. The extracellular
matrix and VEGFA165/SEMA3A mediate the ABL1/CDC42 and FAK/Src
pathways, respectively, via NRP1 to regulate vascular sprouting and
branching. NRP1 also maintains endothelial cell survival and
barrier function through the eNOS pathway. In terms of
endothelial-pericyte crosstalk, pericyte NRP1 can capture VEGFA and
present it to endothelial VEGFR2 to form a transcellular complex.
At the same time, PDGF-D simultaneously binds to endothelial NRP1
and pericyte PDGFRβ to form a ternary complex, jointly regulating
vascular permeability and pericyte coverage. NRP1, neuropilin-1;
VEGF, vascular endothelial growth factor; SEMA3A, semaphorin 3A;
eNOS, extracellular nitric oxide synthase; PDGF, platelet-derived
growth factor receptor. |
Although the macroscopic pro-angiogenic effect has
been confirmed in bone regeneration, the in-depth analysis of the
multidimensional mechanistic network of NRP1 in endothelial cells
still requires drawing on studies in non-skeletal systems. The
following mechanisms, including ligand competition, directional
vascular sprouting and the maintenance of endothelial barrier
homeostasis, have not yet been directly verified in bone
regeneration models, but can provide a potential reference
framework for the study of bone repair mechanisms.
During the initiation phase of angiogenesis, NRP1
regulates the directional sprouting of blood vessels. In
vitro experiments have confirmed that SEMA3A selectively
inhibits the VEGFA165-induced phosphorylation of focal adhesion
kinase and Src in endothelial cells via NRP1, thereby blocking the
downstream angiogenic signals of VEGF (47). In vivo angiogenesis models
have further confirmed that VEGFA165 and SEMA3A exert functional
antagonism through NRP1, jointly regulating the intensity and scope
of vascular sprouting (47).
This ligand-competition mechanism was first directly verified by
Miao et al (48) through
biochemical and functional experiments: On the surface of
NRP1-expressing cells, SEMA3A and VEGFA165 share overlapping
binding sites in the b1/b2 domains of NRP1 and can competitively
inhibit each other's binding to NRP1 (48). The aforementioned studies on
non-skeletal systems suggest that NRP1 itself does not preset a
pro-angiogenic or anti-angiogenic direction, and its functional
output may depend on the relative concentrations and temporal
expression of the two ligands in the microenvironment, rather than
being linearly determined by a single signaling pathway. The
applicability of this regulatory mode in angiogenesis during
fracture healing remains to be verified. Clinical studies on
rheumatoid arthritis have provided pathological evidence for this
ligand competition mechanism. Clinical studies have found that
synovial vascular density is significantly higher in patients with
rheumatoid arthritis with VEGFA165-positive expression than in
those with a negative expression (49). Subsequent research has
demonstrated that Sema3A expression in the synovial lining cells of
joints in patients with rheumatoid arthritis is significantly
decreased. When Sema3A expression is insufficient, the dominant
occupancy of NRP1 on the surface of synovial endothelial cells by
VEGFA enhances pro-angiogenic signals and drives the formation of
pathological pannus (50), as
illustrated in Fig. 2.
Vascular sprouting is a crucial step in blood vessel
formation. During vascular sprouting, NRP1 is highly expressed in
endothelial tip cells. It mediates the activation of the Abelson
murine leukemia viral oncogene homolog 1/cell division cycle 42
(ABL1/CDC42) pathway by the extracellular matrix, thereby promoting
actin remodeling and filopodia formation, which are necessary for
the directional migration of tip cells and normal vascular
branching (51), as demonstrated
in Fig. 2. Moreover, in
vitro experiments using human umbilical artery endothelial
cells have shown that NRP1 specifically binds to the endocytic
adaptor protein GAIP-interacting protein C terminus 1 via the
conserved SEA motif at the C-terminus of its cytoplasmic domain. It
selectively mediates the endocytosis of the active conformation of
integrin α5β1 at the cell membrane into Rab5-positive early
endosomes, followed by their subsequent recycling to adhesion
sites, ultimately enhancing endothelial cell adhesion to
fibronectin (37).
Following blood vessel formation, NRP1 also plays a
critical role in endothelial cell survival and in maintaining
endothelial barrier homeostasis. In a previous study, in
vitro functional experiments using human umbilical vein
endothelial cells (HUVECs) and in vivo analyses using
endothelial-specific NRP1 knockout mice confirmed that NRP1
maintains dimethylarginine dimethylaminohydrolase 1 (DDAH1)
expression in endothelial cells via a post-transcriptional
regulatory mechanism (52).
DDAH1 has been identified as the key enzyme responsible for
degrading asymmetric dimethylarginine (ADMA) in vivo, an
endogenous inhibitor of nitric oxide (NO) synthase (NOS). By
eliminating intracellular ADMA, it relieves its inhibitory effect
on NOS and restores NO bioavailability in endothelial cells
(53). A previous study using a
mouse hindlimb ischemia model further validated the endothelial
protective effects of NRP1 (54). NRP1 carried by mesenchymal stem
cell-derived extracellular vesicles was shown to act on endothelial
cells in ischemic tissues, significantly promoting nitric oxide
production and inhibiting endothelial cell apoptosis by activating
the endothelial NOS pathway, thereby exerting endothelial
protection (54) (Fig. 2).
Furthermore, research using an in vitro HUVEC
model confirmed that endothelial NRP1 is a component of adherens
junctions. It stabilizes endothelial adherens junctions by
promoting the binding of vascular endothelial cadherin
(VE-cadherin) to p120 catenin, thereby maintaining the integrity
and homeostasis of the endothelial barrier (55). In addition to the aforementioned
regulatory mechanisms, NRP1 also modulates endothelial barrier
function through a mechanical force-dependent pathway. Studies have
demonstrated that the synthetic NRP1-specific agonist RCαβ
activates the NRP1-MET proto-oncogene, hepatocyte growth factor
receptor (NRP1-MET) signaling axis, which in turn upregulates Ras
homolog family member A (RhoA) activity, promotes the assembly of
actin stress fibers in endothelial cells, and increases cell
stiffness. Furthermore, this NRP1-mediated signaling drives the
redistribution of VE-cadherin from continuous intercellular
junctions to punctate focal adherens junctions, recruits vinculin
to enhance intercellular mechanical force transmission, and
ultimately increases endothelial permeability (56,57). It should be noted that this
effect is caused by the activation of the MET signaling axis by the
synthetic NRP1-specific agonist RCαβ. It is not contradictory to
the aforementioned function of NRP1 in maintaining barrier
homeostasis under physiological conditions, but rather reflects the
dependence of the downstream functional output of NRP1 on ligand
type.
Taken together, the prominent effect of NRP1 as a
co-receptor of VEGFR2 in driving endothelial angiogenesis has been
directly verified in a rabbit ischemic bone defect regeneration
model (46). However, its
multidimensional regulatory mechanisms in upstream ligand
competition, directional vascular sprouting, and the maintenance of
endothelial barrier homeostasis are currently mainly derived from
studies in non-skeletal systems, which provide a conceptual
framework for deciphering the temporal regulatory patterns of NRP1
on blood vessels during fracture healing (33,58-60).
Pericytes
Historically, the role of NRP1 in the vascular
network has been centered on endothelial cells; however,
accumulating evidence indicates that it also exerts multi-faceted
regulatory functions in pericytes. High-resolution confocal imaging
has confirmed that NRP1 is expressed on pericytes covering the
vascular wall (61), providing a
foundation for subsequent functional studies. It should be noted
that all regulatory mechanisms of NRP1 in pericytes identified to
date are derived from studies in non-skeletal systems. These
findings provide potential research directions for understanding
impaired vascular maturation during bone repair. Yet, there is no
direct evidence from fracture models supporting its role in
intraosseous pericytes, which remains a critical gap in this
field.
Research on non-skeletal systems has shown that NRP1
is involved throughout the pericyte life cycle, from
differentiation to the execution of physiological functions. An
in vitro tumor microenvironment model first confirmed that
PDGF-BB secreted by tumor cells directly interacts with NRP1 on the
surface of mouse embryonic mesenchymal stem cells, significantly
upregulating the expression of pericyte-specific markers, such as
α-smooth muscle actin and desmin, and driving the
transdifferentiation of mesenchymal stem cells into a pericyte
phenotype (58). After pericytes
mature, NRP1 becomes a crucial foundation for maintaining their
functions. Research using pericyte-specific gene knockout has
confirmed that neuron-glial antigen 2/PDGFRβ-positive pericytes in
the kidneys of adult mice highly express NRP1 (59). Although NRP1 deficiency does not
affect the number of surviving pericytes, it disrupts basement
membrane protein homeostasis, impairs glomerular basement membrane
integrity, and leads to microscopic hematuria and glomerular
hyperfiltration (59).
Beyond its intracellular regulation within single
cells, NRP1 plays unique roles in communications between pericytes
and endothelial cells. Pericytes can capture extracellular VEGFA in
a paracrine manner via NRP1 and present it to VEGFR2 on endothelial
cells, forming a transcellular complex. This complex
transcellularly regulates the endothelial VEGFR2 pathway and
vascular permeability homeostasis by delaying VEGFR2 endocytosis
and enhancing the Y949-Src-VE-cadherin signaling axis (60). Muhl et al (33) further confirmed that NRP1 can
mediate transcellular signaling between endothelial cells and
pericytes. PDGF-D simultaneously binds to NRP1 on endothelial cells
and PDGFRβ on adjacent pericytes to form a ternary complex. This
complex accumulates at cell contact protrusions, thereby forming a
local signaling hub. This unique signaling mode enables PDGF-D to
maintain vascular pericyte coverage and to mediate
contact-dependent pericyte migration, ultimately indirectly
regulating vascular homeostasis (33), as illustrated in Fig. 2.
In addition, the function of NRP1 in pericytes is
not linearly determined by a single signaling pathway, but rather
exhibits a high degree of context dependence. A previous study on
skin microvessels in gene knockout mouse models demonstrated that
the regulatory effect of NRP1 on vascular permeability depends on
the expression ratio of NRP1 in pericytes and endothelial cells
(60). When NRP1 is highly
expressed in pericytes, endothelial NRP1/VEGFR2
cis-complexes maintain moderate permeability; if endothelial
NRP1 is deficient, pericyte NRP1 forms trans-complexes with
endothelial VEGFR2, significantly enhancing permeability. When NRP1
expression is low in pericytes, vascular permeability is always
dominated by endothelial NRP1/VEGFR2 cis-complexes (60). This further confirms that the
function of NRP1 depends on its cellular expression pattern rather
than on linear regulation by a single signaling pathway.
Taken together, the regulatory functions of NRP1 in
pericyte differentiation, transcellular communication and vascular
permeability have been preliminarily verified in non-skeletal
systems such as the kidney and skin. There is currently no direct
evidence from fracture models supporting the aforementioned roles
of NRP1 in intraosseous pericytes, which remains an urgent gap in
the field of vascular maturation during bone repair. The functional
regulation of intraosseous pericytes may follow similar principles,
but the expression dynamics and functions of NRP1 in pericytes
during fracture healing still require direct verification.
NRP1 and type H vessels
Type H vessels are a subtype of capillaries
characterized by a high expression of cluster of differentiation 31
(CD31) and endomucin. They form a network through vertically
arranged vascular columns connected by vascular loops or arches.
They are mainly distributed in osteogenically active regions such
as the metaphyses of long bones and the endosteum. They are
considered the key structural carriers of bone-vessel coupling
(6,62,63).
Studies on bone development models have demonstrated
that VEGF is a key regulatory factor of skeletal angiogenesis and
participates in the angiogenesis-osteogenesis coupling process
mediated by type H vessels (6,64,65). Further studies on bone
homeostasis have indicated that osteoclast precursors can directly
promote the growth of type H vessels by secreting PDGF-BB (66-68). These findings have established
that type H vessels depend on VEGF and PDGF signaling.
In the field of systemic vasculature, NRP1 has been
identified as a co-receptor for both the VEGF and PDGF signaling
axes. It is involved in endothelial-pericyte crosstalk and in
vascular maturation and stabilization (33). This dual co-receptor property
biochemically demonstrate that NRP1 possesses the molecular basis
to simultaneously integrate VEGF and PDGF signals, suggesting that
it may participate in the regulation of intraosseous type H
vessels. However, there is currently no direct research evidence
regarding the expression and function of NRP1 in type H vessels of
bone tissue in bone repair models. Its regulatory role in type H
vessels remains an important hypothesis to be verified in the field
of bone-vessel coupling. Future studies can first detect the
expression and distribution of NRP1 in intraosseous type H vessels,
and then use fracture models with endothelial cell- or
pericyte-specific NRP1 knockout to examine its function in the
formation and maintenance of type H vessels. Further studies are
required to confirm that NRP1 regulates type H vessels, as this may
provide a new perspective on the coupling mechanism between
intraosseous angiogenesis and osteogenesis.
As summarized above, there are critical differences
in the sufficiency of research evidence supporting the distinct
mechanisms of NRP1 in the regulation of bone tissue vasculature.
Among these, the role of NRP1 as a co-receptor of VEGFR2 in
promoting neovascularization in bone defect areas has been fully
validated through animal experiments. It represents the most
clearly established core function to date. By contrast, the
functions of NRP1 in guiding the direction of angiogenic sprouting,
mediating signal communication between pericytes and endothelial
cells and regulating the formation of type H vessels in bone have
not yet been directly investigated in fracture models. Existing
studies on non-skeletal systems suggest that the intensity and
extent of angiogenic sprouting are co-regulated by the relative
concentrations of VEGFA165 and SEMA3A ligands in the local
microenvironment; however, whether this regulatory principle
applies to the unique microenvironment of fracture healing remains
to be definitively determined by experimental evidence.
Interaction of NRP1 with two angiogenic
factors
The pro-angiogenic function of NRP1 is highly
dependent on its specific interactions with the two major signaling
molecule families, VEGF and PDGF. At present, the elucidation of
the relevant mechanisms is mainly derived from studies on
non-skeletal systems, and direct validation in the fracture
microenvironment remains very limited (6,69-71).
Among the VEGF family, VEGFR1 and VEGFR2 are key
receptors that regulate endothelial cell function and are also the
main targets for interaction with NRP1 (72,73).
The pro-angiogenic effect of the NRP1-VEGFR2
signaling axis has been directly verified in a rabbit ischemic bone
defect model through the synergistic action of 3D-printed magnesium
alloy scaffolds (46). This
model also confirmed that Mg2+ can further enhance the
signal transduction efficiency of the NRP1-VEGFR2 ternary complex
on the surface of endothelial cells by upregulating VEGFA
expression in bone marrow mesenchymal stem cells (BMSCs) (46).
However, the majority of the additional potential
molecular mechanisms underlying the interaction between NRP1 and
VEGF are derived primarily from in vitro studies conducted
on non-bone tissues (74-76).
In vitro studies on receptor kinetics and endothelial cell
function have biochemically elucidated the interaction patterns
between NRP1 and members of the VEGFR family on endothelial cell
surfaces (74). In the
ligand-free resting state, VEGFR1 and NRP1 form a constitutive
stable complex. This complex regulates the intracellular
trafficking and membrane distribution homeostasis of both
receptors, and it is a consensus in the field that it can buffer
aberrant VEGFR2 activation under basal conditions (74). Upon VEGFA binding, the formation
of stable complexes between NRP1 and membrane-localized VEGFR2 is
significantly enhanced. This process increases the phosphorylation
and activation of VEGFR2 and its downstream ERK signaling, thereby
synergistically amplifying VEGFA-mediated endothelial cell signal
transduction and angiogenic sprouting (75,76). In addition to binding membrane
receptors, NRP1 on endothelial cells can bind soluble VEGFR1,
thereby finely regulating endothelial cell adhesion and migration
(10).
Notably, similar amplification mechanisms have also
been identified in other pathological models. In tumor and liver
fibrosis models, VEGFA165 can enhance the stability of NRP1/VEGFR2
complexes on the surface of vascular endothelial cells and liver
sinusoidal endothelial cells, respectively, amplify downstream
intracellular signaling pathways, including ERK and PI3K-Akt, and
drive pathological angiogenesis (77,78). On the other hand, the abnormal
inhibition of this signaling axis will also lead to severe vascular
network defects. For example, in a previous study, in a mouse model
of neonatal exposure to diethylstilbestrol, the downregulation of
VEGFR2 and its co-receptor NRP1 can directly cause uterine
pathological angiogenic dysfunction and abnormal vascular
maturation (79).
In the aforementioned bone defect, tumor and liver
fibrosis models, endothelial NRP1 amplifies downstream signaling by
stabilizing the VEGFA/VEGFR2 complex, and this core biochemical
mechanism is highly conserved across models. However, the vascular
phenotypes driven by this mechanism differ: It manifests as ordered
reparative angiogenesis in bone defects, but as disordered
pathological angiogenesis in tumors. This discrepancy suggests that
the final functional output of the NRP1-VEGFR2 signaling axis is
not linearly determined by a single mechanism, but may depend on
the intensity and duration of signals in the local
microenvironment. Specifically, the temporal expression of VEGFA in
bone defects may contribute to the formation of a reparative
vascular network, whereas persistent ligand stimulation in tumors
may drive abnormal vascular proliferation. At present, the effect
of this axis in mediating reparative angiogenesis in bone defects
has been directly verified. Still, its functional switching
mechanism under different pathological microenvironments remains to
be further clarified in the context of fracture.
In addition to the VEGF signaling axis, the PDGF
signaling axis plays an important role in mediating intercellular
communication, pericyte recruitment, and maintenance of blood
vessel homeostasis. Although all direct experimental evidence for
the regulation of this signaling axis by NRP1 currently comes from
non-skeletal systems, these findings provide a potential
theoretical framework for elucidating the mechanisms of vascular
maturation disorders during bone repair.
At the level of cellular behavior regulation, NRP1
is an important auxiliary molecule for mesenchymal stem cells
(MSCs) to respond to PDGF signals. It is an essential condition for
the complete phosphorylation of PDGFRα homodimers induced by
PDGF-AA. It can partially enhance the activation level of
PDGF-BB-mediated PDGFRβ homodimers, thereby regulating the
migration and proliferation of MSCs (71). In addition, NRP1/PDGF also plays
a crucial role in intercellular communications. NRP1 can function
as a specific coreceptor for PDGF-D, mediating trans-signaling
between endothelial cells and pericytes to maintain pericyte
coverage and, indirectly, regulate vascular homeostasis (33).
Taken together, NRP1 provides the molecular basis
for regulating the entire angiogenesis process by integrating the
VEGF and PDGF signaling axes. However, the spatiotemporal
expression patterns of VEGF and PDGF and the modes of intercellular
communication in the bone repair microenvironment are fundamentally
different from those in existing non-skeletal system studies.
Therefore, whether NRP1 can replicate these regulatory patterns in
the in situ bone defect area to achieve orderly coordination
of angiogenesis and maturation remains a key gap in this field. A
summary of the strength and classification of evidence for
NRP1-mediated bone angiogenesis regulation is presented in Table I.
 | Table IStrength and classification of
evidence for NRP1-mediated bone angiogenesis regulation. |
Table I
Strength and classification of
evidence for NRP1-mediated bone angiogenesis regulation.
| Evidence level | Model type | Cell type | Regulatory
mechanism | Functional
effect |
|---|
| Bone regeneration
model | In vitro
bone cell model + in vivo bone repair model | Endothelial
cells | NRP1 acts as a
VEGFR2 coreceptor, forming a VEGFA-NRP1-VEGFR2 temary complex
andactivating the PI3K-AKT pathway | Promotes
endothelial migration and proliferation, accelerating angiogenesis
in bone defect areas |
| Non-skeletal
system | In vitro
non-bone cell model + In vivo non-skeletal model +clinical
research | Endothelial
cells | SEMA3A
competitively binds to NRP1 with VEGF165, inhibiting FAK/Src
phosphorylation | Antagonistically
regulates vascular sprouting; drives pathological angiogenesis in
rheumatoid arthritis |
| Non-skeletal
system | In vitro
non-bone cell model | Endothelial tip
cells | NRPl mediates
ECMinduced activation of the ABL1/CDC42 pathway | Promotes
directional migration of tip cells and vascular branching |
| Non-skeletal
system | In vitro
non-bone cell model | Endothelial
cells | NRP1 binds to
GIPCl, mediating the endocytic recycling of integrin α5β1 | Enhances
endothelial cell adhesion to fibronectin |
| Non-skeletal
system | In vitro
non-bone cell model + in vivo non-skeletal model | Endothelial
cells | NRPl maintains
DDAH1 expression and activates the eNOS pathway | Inhibits
endothelial cell apoptosis |
| Non-skeletal
system | In vitro
non-bone cell model | Endothelial
cells | NRPl promotes the
binding of VE-cadherin to pl20 catenin | Stabilizes
endothelial adherens junctions and maintains barrier integrity |
| Non-skeletal
system | In vitro
non-bone cell model | Endothelial
cells | RCαβ activates the
NRP1-MET-RhoA signaling axis | Increases
endothelial cell permeability |
| Non-skeletal
system | In vitro
non-bone cell model | Mesenchymal stem
cells/Pericytes | PDGF-BB directly
binds to NRPl on the surface of mesenchymal stem cells | Drives the
differentiation of mesenchymal stem cells into pericytes |
| Non-skeletal
system | In vivo
non-skeletal model | Pericytes | NRP1 deficiency
disrupts basement membrane protein homeostasis | Impairs the
integrity of the glomerular basement membrane |
| Non-skeletal
system | In vitro
non-bone cell model | Pericytes +
Endothelial cells | Pericyte NRP1
captures VEGFA and presents it to endothelial VEGFR2, forming a
transcellular complex | Regulates the
homeostasis of vascular permeability |
| Non-skeletal
system | In vitro
non-bone cell model | Pericytes +
Endothelial cells | PDGF-D binds to
endothelial NRP1 and pericyte PDGFR β to form a temary complex | Maintains vascular
pericyte coverage and regulates vascular homeostasis |
| Non-skeletal
system | In vivo
non-skeletal model | Pericytes +
Endothelial cells | Vascular
permeability depends on the ratio of NRP1 expression between
pericytes and endothelial cells | Bidirectionally
regulates vascular permeability |
| Non-skeletal
system | In vitro
non-bone cell model | Endothelial
cells | VEGF-A enhances the
stability of the NRP1-VEGFR2 complex and amplifies VEGFR2-Erk
signaling | Promotes vascular
sprouting |
| Non-skeletal
system | In vitro
non-bone cell model | Endothelial
cells | NRP1 binds to
soluble VEGFR1 | Finely regulates
endothelial cell adhesion and migration |
| Non-skeletal
system | In vivo
non-skeletal model | Endothelial
cells | VEGF-A165 enhances
the stability of the NRP1/VEGFR2 complex, amplifying downstream
signaling | Drives pathological
angiogenesis |
| Non-skeletal
system | In vivo
non-skeletal model | Endothelial
cells | Downregulation of
VEGFR2 and NRP1 expression | Leads to impaired
uterine angiogenesis and abnormal vascular maturation |
Functions of NRP1 in regulating key cells
involved in bone repair
Fracture repair relies on the synergistic action of
osteoblasts, osteoclasts, chondrocytes and BMSCs. NRP1 is
considered to participate in the signaling regulation of this
cellular network potentially and to affect the proliferation,
differentiation and maintenance of homeostasis of the
aforementioned cells through direct and indirect pathways (17,44,80,81).
Osteoblasts
Osteoblasts are the primary executors of new bone
formation, and existing studies suggest that NRP1 may regulate
osteogenic differentiation and bone regeneration (17,82). Early in vivo research
demonstrated that in the developing bones of mice and chick
embryos, NRP1 was localized to osteoblasts migrating along blood
vessels in the metaphyses, to osteoblasts at the edge of trabeculae
in the marrow cavity, and to endothelial cells in bone tissue,
which also widely expressed NRP1 (43). This expression pattern indicates
that NRP1 may participate in skeletal development through dual
pathways: Regulating osteoblast function and mediating angiogenesis
(43).
In vitro experiments on BMSCs have shown that
SEMA3A binds to NRP1 on the cell surface, promoting osteogenic
differentiation, while inhibiting adipogenic differentiation by
activating the canonical Wnt/β-catenin signaling pathway. This
effect was further verified in a mouse cortical bone defect model,
where the local application of recombinant SEMA3A significantly
increased the proportion of osteoblasts in the defect area and
accelerated bone regeneration and repair (17) (Fig. 3).
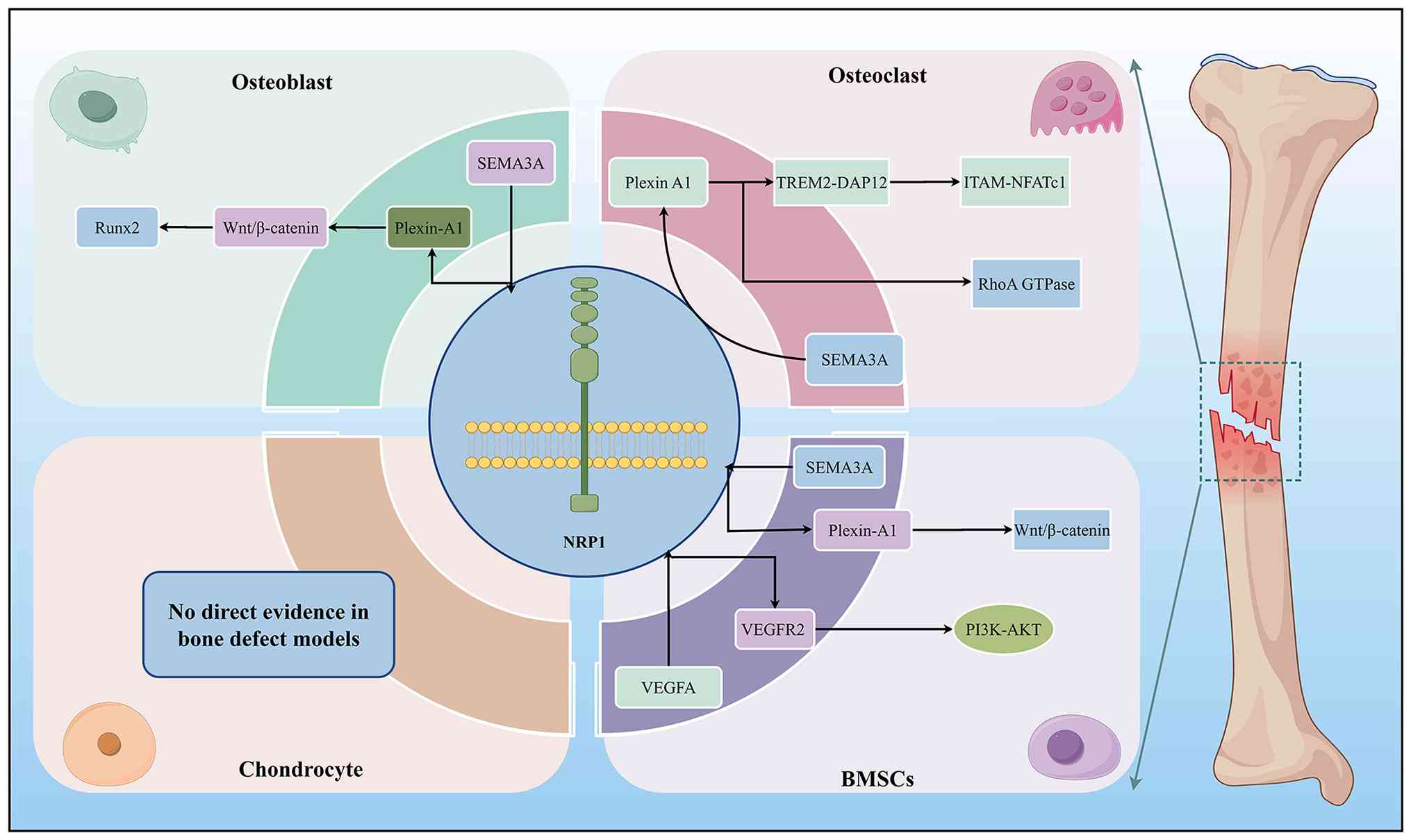 | Figure 3Schematic illustration of the
molecular mechanisms by which NRP1, as a core signaling node,
regulates the functions of osteoblasts, osteoclasts, BMSCs and
chondrocytes during bone repair. At the osteoblast level, SEMA3A
binds to the NRP1-Plexin-A1 receptor complex, activates the
Wnt/β-catenin pathway, and upregulates Runx2 expression, promoting
osteogenic differentiation and inhibiting adipogenic
differentiation. At the osteoclast level, SEMA3A simultaneously
blocks two independent downstream pathways through the
NRP1-Plexin-A1 complex: it inhibits the TREM2-DAP12-ITAM-NFATc1
signaling axis to reduce osteoclast differentiation and maturation.
It suppresses RhoA GTPase activity to block the directional
migration of osteoclast precursors, ultimately inhibiting bone
resorption. At the BMSC level, the SEMA3A-NRP1-Plexin-A1 pathway
activates Wnt/β-catenin signaling to promote osteogenic
differentiation; VEGFA forms a ternary complex with NRP1 and
VEGFR2, activating the PI3K-AKT pathway to promote BMSC migration.
All the above regulatory mechanisms in osteoblasts, osteoclasts,
and BMSCs have been directly validated in bone repair models. There
is currently no direct experimental evidence in bone repair models
for NRP1-mediated functional regulation in chondrocytes. NRP1,
neuropilin-1; BMSCs, bone marrow mesenchymal stem cells; SEMA3A,
semaphorin 3A; VEGFA, vascular endothelial growth factor A; VEGFR2,
VEGF receptor 2; Runx, runt-related transcription factor 2; TREM2,
triggering receptor expressed on myeloid cells 2; DAP12, DNAX
activation protein of 12 kDa; ITAM, immunoreceptor tyrosine-based
activation motif; NFATc1, nuclear factor of activated T cells
cytoplasmic 1. |
In addition to the canonical pathway mediated by
SEMA3A, NRP1 can also regulate the osteogenic process by
interacting with the collagen receptor discoidin domain receptor 2
(DDR2). In vitro preosteoblast models have demonstrated that
NRP1 overexpression can significantly upregulate the mRNA and
protein levels of osteogenic markers, enhance alkaline phosphatase
activity and promote mineralized nodule formation. The underlying
mechanism is that NRP1 co-localizes with DDR2 on the cell membrane
and prolongs its half-life, thereby amplifying the downstream
ERK1/2-Runt-related transcription factor 2 (Runx2) signaling
cascade (82,83).
The aforementioned osteogenic regulatory mechanisms,
which are of utmost significance under physiological conditions,
are equally important under pathological conditions. NRP1 also
exerts a protective effect in counteracting metabolic osteogenic
inhibition. In vitro and in vivo research using
diabetic pathological models has demonstrated that supplementation
with SEMA3A can restore osteoblast activity via NRP1 and increase
bone mineral density and bone mass (20). In addition, in in vitro
osteogenic models of glucocorticoid-induced inhibition, the NRP1
pathway can antagonize glucocorticoid inhibition of osteoblasts
(84).
Osteoclasts
Osteoclasts are the key functional cells responsible
for bone resorption in the skeletal system, and their excessive
activation is the central cause of pathological bone loss (85,86).
In a previous study using a mouse cortical bone
defect model, tje local application of recombinant SEMA3A
significantly reduced the proportion of osteoclasts in the defect
area and inhibited bone resorption activity (17). In vitro experiments using
primary bone marrow mononuclear cells further elucidated the
molecular mechanism underlying this effect. Upon binding to NRP1 on
the cell surface, SEMA3A was shown to reverse the receptor
activator of nuclear factor κB ligand (RANKL)-induced
downregulation of NRP1 expression. It promoted the formation of a
stable complex between NRP1 and Plexin-A1 (17). This complex competitively blocked
the interaction between Plexin-A1 and the triggering receptor
expressed on myeloid cells 2-DNAX activation protein of 12 kDa
(TREM2-DAP12) signaling complex, thereby inhibiting downstream
immunoreceptor tyrosine-based activation motif (ITAM) signaling and
nuclear factor of activated T-cells cytoplasmic 1 (NFATc1)
transcriptional activation. Moreover, the SEMA3A-NRP1-Plexin-A1
pathway also inhibited RhoA GTPase activity and blocks the
directional migration of osteoclast precursors to bone resorption
sites. The synergistic action of these two independent pathways
ultimately inhibits the maturation, differentiation and
bone-resorptive function of osteoclasts (17), as illustrated in Fig. 3.
In addition to the canonical SEMA3A-mediated pathway
that has been verified in bone defect models, the negative
regulatory effect of NRP1 on osteoclasts has been further supported
by in vitro research in other non-fracture models. At the
functional level, small molecules such as wedelolactone have been
confirmed to inhibit osteoclast differentiation via the
NRP1-Plexin-A1 pathway (87).
sNRP1 secreted by osteocytes can also directly inhibit osteoclast
differentiation through a paracrine pathway (88). The research results from the
aforementioned non-fracture models collectively suggest that NRP1
is a potential negative regulator of osteoclast differentiation and
function and may play an important role in maintaining bone
homeostasis. However, the specific role of this negative regulatory
effect in the physiological bone remodeling process during fracture
healing remains to be verified.
Chondrocytes
Chondrocytes are the key components that maintain
the structural integrity of the callus and play a critical role in
fracture healing. To date, to the best of our knowledge, there are
no direct in vivo studies on the regulation of chondrocyte
function by NRP1 during fracture healing.
In terms of non-fracture acute mechanical stress
defense, a previous in vitro study using a high-magnitude
cyclic tensile strain model of ATDC5 chondrocytes suggested that
excessive mechanical stress downregulated endogenous SEMA3A
expression in chondrocytes, while, as a compensation, it
upregulated its co-receptor NRP1 and signaling receptor Plexin-A1
on the chondrocyte surface (89). The exogenous supplementation of
SEMA3A was found to bind to the aforementioned upregulated receptor
complex, inhibit the excessive activation of the AKT, ERK and NF-κB
pathways, and reduce the production of pro-inflammatory cytokines
and matrix-degrading enzymes, thereby protecting the chondrocyte
extracellular matrix (89). The
results from this in vitro acute mechanical stress model
indicate that the SEMA3A-NRP1 signaling axis exerts a
chondroprotective effect under acute mechanical stress, providing a
reference framework for understanding the mechanism by which early
fracture cartilaginous callus resists abnormal stress. However,
given the differences in phenotype and mechanical microenvironment
between the ATDC5 cell line and in vivo cartilaginous callus
chondrocytes, the actual role of this mechanism in fracture healing
remains to be directly verified in vivo.
By sharp contrast, NRP1 exerts an opposite
regulatory effect in chronic inflammatory microenvironments of
cartilage degeneration, such as osteoarthritis. SEMA3A and NRP1
mRNA are synchronously upregulated in knee chondrocytes from
patients with osteoarthritis, and their expression levels are
closely associated with the progression of cartilage degeneration
(44,80). Further in vitro studies
using inflammatory chondrocyte models have demonstrated that
interleukin (IL)-1β and TNF-α coordinately upregulate SEMA3A and
NRP1 expression in chondrocytes via the CCAAT/enhancer-binding
protein β transcriptional pathway. Overactivated SEMA3A-NRP1
signaling inhibits the PI3K-AKT pro-survival pathway in an
NRP1-dependent manner, driving chondrocytes to shift toward a
catabolic phenotype and increasing apoptosis (80). Moreover, this signaling axis
specifically upregulates matrix metalloproteinase 13, a key enzyme
in cartilage matrix degradation and accelerates type II collagen
degradation (44). In
vitro intervention studies have further confirmed this
pro-degenerative effect: exosomal miR-485-3p derived from synovial
mesenchymal stem cells can target and silence NRP1 mRNA in
chondrocytes, correct the abnormality of the PI3K-AKT pathway,
reduce chondrocyte apoptosis, and restore the expression of
anabolic markers (90).
The research results from the aforementioned in
vitro acute mechanical stress model and chronic inflammatory
model of osteoarthritis reveal a microenvironment-dependent
bidirectional regulatory pattern of chondrocytes in non-fracture
scenarios: NRP1 exerts a chondroprotective effect under acute
mechanical stress, whereas it may drive cartilage degeneration in a
chronic inflammatory microenvironment. Since both effects are
derived from non-fracture models, they cannot be directly
extrapolated to the cartilaginous callus during fracture healing.
Therefore, targeted interventions against NRP1 may carry the risk
of opposite effects, and this functional uncertainty is a key issue
that urgently needs to be addressed in research on NRP1 in bone
repair.
BMSCs
In an ischemic bone defect model, exogenous NRP1
loaded on 3D-printed magnesium alloy scaffolds can form a
synergistic regulatory axis with sustained-release magnesium ions
and exert effects through two independent pathways: In addition to
activating the endothelial NRP1-VEGFR2-PI3K-AKT pathway to promote
angiogenesis as described above, it can also significantly enhance
the migration ability of BMSCs through the same signaling axis
(46). Beyond VEGF signaling,
NRP1 can also function as a functional co-receptor for SEMA3A to
regulate the differentiation fate of BMSCs through a completely
distinct downstream pathway. Hayashi et al (17) confirmed that this signaling axis
exerts dual bone-protective effects, promoting osteogenic
differentiation. It inhibits adipogenic differentiation in BMSCs
and suppresses the differentiation and function of osteoclast
precursors. The promoting effect on bone regeneration was further
verified in a mouse cortical bone defect model (17) (Fig. 3).
In addition to the regulation of angiogenesis and
osteogenic differentiation, which have been verified in the
aforementioned bone defect models, the majority of the other
functions of NRP1 in BMSCs and their upstream regulatory mechanisms
are derived from studies outside the fracture-healing system. At
the molecular mechanism level of osteogenic differentiation, Shi
et al (13) further
refined the SEMA3A-NRP1-Wnt pathway through in vitro
experiments using mouse BMSCs. They found that activated
Wnt/β-catenin can, in turn, transcriptionally upregulate NRP1
expression, forming a 'SEMA3A-NRP1-Wnt/β-catenin-NRP1' positive
feedback loop that continuously amplifies the osteogenic effect
(13). At the level of
maintaining aging-related functions, research using a mouse model
of aging-related bone loss demonstrated that pulsed electromagnetic
fields can induce sensory nerves to secrete SEMA3A. After binding
to NRP1 on the surface of LepR+ BMSCs, SEMA3A restores
their osteogenic potential and antagonizes cellular senescence
(91).
In addition, NRP1 expression is also finely
regulated by the non-coding RNA network. In models of
periodontitis, miR-148a can target and silence NRP1 in periodontal
ligament stem cells, thereby inhibiting their osteogenic
differentiation (92). In models
of osteoporosis, long non-coding RNA (lncRNA) MALAT1 indirectly
upregulates NRP1 expression in BMSCs by competitively sponging
miR-320a, thereby promoting their osteogenic differentiation
(93).
Collectively, only two effects of NRP1 in BMSCs,
namely vascular migration and osteogenic differentiation, have been
directly verified in bone defect models. The remaining regulatory
mechanisms, including the Wnt-NRP1 positive feedback loop, the
anti-aging effect of neurogenic SEMA3A, and the
post-transcriptional regulation by non-coding RNAs, are all derived
from in vitro cell experiments or non-bone injury systems,
such as periodontitis and osteoporosis, and their mode of action in
fracture healing remains unclear. The strength and classification
of evidence for the NRP1 regulation of key bone repair cell
functions is summarized in Table
II.
| Table IIStrength and classification of
evidence for NRP1 regulation of key bone repair cell functions. |
Table II
Strength and classification of
evidence for NRP1 regulation of key bone repair cell functions.
| Evidence | Model type | Cell type | Regulatory
mechanism | Functional
effect |
|---|
| Bone repair
model | In vitro
bone cell model + in vivo bone repair model | Osteoblasts | SEMA3A-NRP1
activates Wnt/β-catenin pathway | Promotes
osteogenesis, inhibits adipogenesis, and accelerates bone
regeneration |
| Bone repair
model | In vitro
bone cell model + in vivo bone repair model | Osteoclasts |
SEMA3A-NRP1-Plexin-A1 blocks
TREM2-DAP12-NFATc1 pathway | Inhibits osteoclast
differentiation and bone resorption |
| Bone repair
model | In vitro
bone cell model + in vivo bone repair model | Osteoclasts |
SEMA3A-NRP1-Plexin-A1 inhibits RhoA GTPase
activity | Inhibits the
directional migration of osteoclast precursors |
| Bone repair
model | In vitro
bone cell model + in vivo bone repair model | BMSCs | NRP1 synergizes
with magnesium ions to amplify the VEGFA-VEGFR2-PI3K-AKT
pathway | Promotes BMSC
migration and drives angiogenesis |
| Bone repair
model | In vitro
bone cell model + in vivo bone repair model | BMSCs | SEMA3A-NRP1
activates Wnt/β-catenin pathway | Promotes BMSC
osteogenesis, inhibits adipogenesis, and provides dual bone
protection |
| Bone biology
field | In vitro
bone cell model | Osteoblasts | NRP1 extends DDR2
half-life and amplifies the ERK1/2-Runx2 pathway | Enhances osteogenic
differentiation and mineralization |
| Bone biology
field | In vitro
bone cell experiment + in vivo non-bone-repair model | Osteoblasts | SEMA3A mediates
signaling through NRP1 | Restores osteogenic
activity and antagonizes metabolic osteogenic inhibition |
| Bone biology
field | In vitro
bone cell model | Osteoblasts | Activation of
NRP1-related signaling pathways | Antagonizes
glucocorticoid-induced osteogenic inhibition |
| Bone biology
field | In vitro
bone cell model | Osteoclasts | Wedelolactone acts
through the NRP1-PlexinA1 pathway | Inhibits osteoclast
differentiation |
| Bone biology
field | In vitro
bone cell model | Osteoclasts | Soluble NRP1 exerts
a paracrine effect on osteoclast precursors | Directly inhibits
osteoclast differentiation |
| Bone biology
field | In vitro
bone cell model | Chondrocytes | Exogenous SEMA3A
binds to NRP1/Plexin-A1, inhibiting AKT/ERK/NF-κB pathways | Reduces matrix
degradation and protects cartilage matrix |
| Bone biology
field | In vitro
bone cell model + Clinical research | Chondrocytes | IL-1β/TNF-α
upregulates SEMA3A/NRP1, inhibits PI3K/AKT, and upregulates
MMP13 | Promotes
chondrocyte apoptosis and matrix degradation, driving cartilage
degeneration |
| Bone biology
field | In vitro
bone cell model | Chondrocytes | miR-485-3p targets
and silences NRP1 in chondrocytes | Reduces chondrocyte
apoptosis |
| Bone biology
field | In vitro
bone cell model | BMSCs | Forms a
SEMA3A-NRP1-Wnt/β-catenin-NRP1 positive feedback loop | Continuously
amplifies the osteogenic effect |
| Bone biology
field | In vitro
bone cell model | BMSCs | Neurogenic SEMA3A
binds to NRP1 on the surface of LepR+ BMSCs | Restores BMSC
osteogenic potential and antagonizes cellular senescence |
| Bone biology
field | In vitro
bone cell model | BMSCs | miR-148a targets
and silences NRP1 in periodontal ligament stem cells (PDLSCs) | Inhibits osteogenic
differentiation of periodontal ligament stem cells |
| Bone biology
field | In vitro
bone cell model | BMSCs | lncRNA MALAT1
sponges miR-320a, indirectly upregulating NRP1 | Promotes BMSC
osteogenic differentiation |
NRP1-mediated neuro-bone crosstalk in bone
tissue
The mammalian skeleton is a highly innervated,
dynamic organ. The nervous system not only mediates skeletal
nociception, but also serves as a key regulator of skeletal
development, injury repair and homeostasis maintenance (94-97). Of note, the majority of current
studies on the role of NRP1 in the aforementioned neuro-bone
regulation are derived from neural development models and bone
homeostasis maintenance models, and no direct in vivo
functional verification has been obtained in the context of
fracture healing, at least to the best of our knowledge.
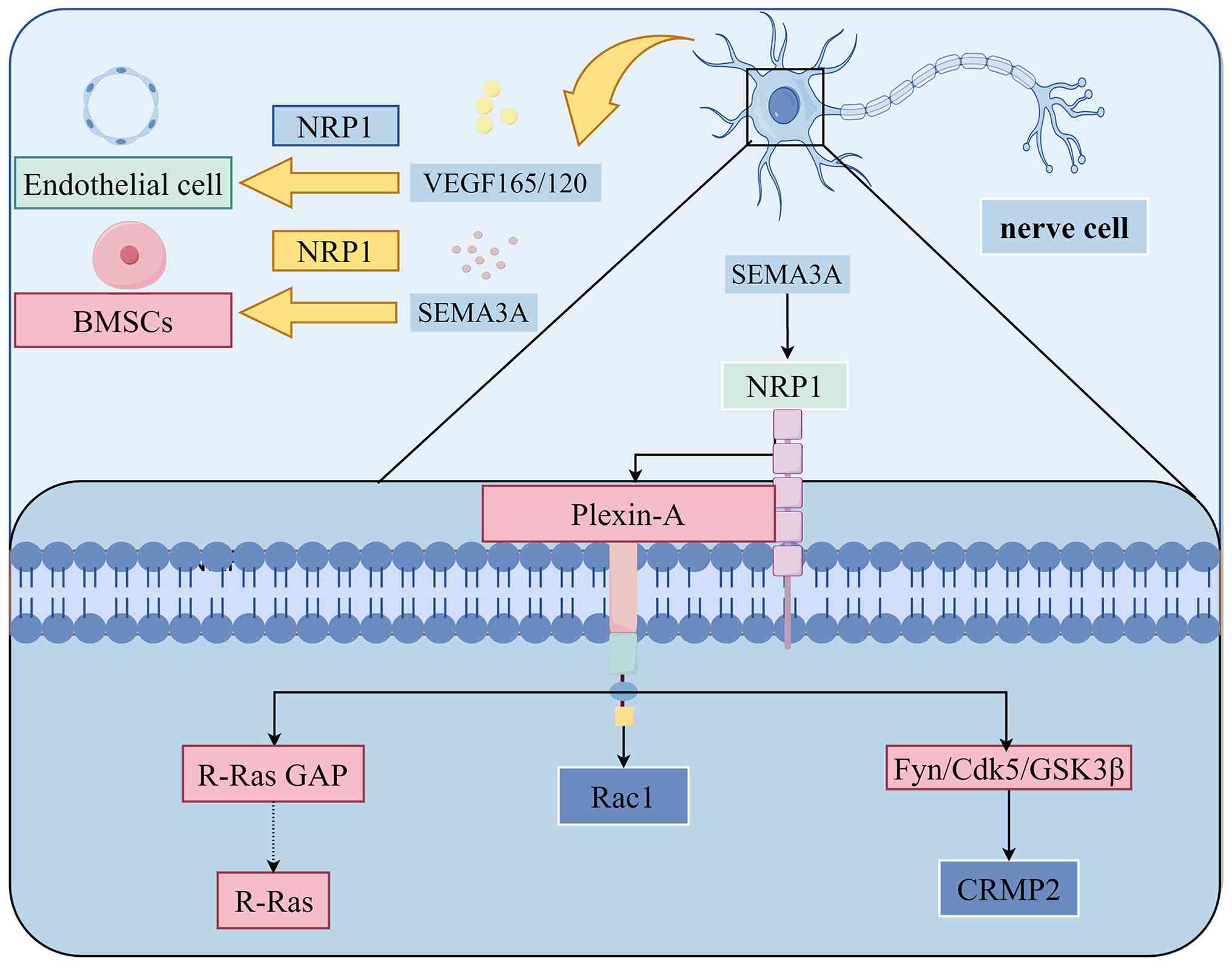
It has been elucidated in the field of neurobiology
that SEMA3A can induce growth cone collapse via its primary
receptor, NRP1, forcing axons to avoid SEMA3A-enriched regions,
which constitutes the core molecular basis for its regulation of
precise neural projection (16,98-100). The underlying regulatory
mechanism has been clarified in in vitro axon guidance
models: NRP1 must form a functional receptor complex with Plexin-A,
and the intracellular domain of Plexin-A transduces downstream
signals (99). This complex
induces repulsive responses mainly through three synergistic
pathways: i) It activates Rac1 to drive growth cone plasma membrane
endocytosis and actin cytoskeleton rearrangement, directly inducing
growth cone collapse; ii) it phosphorylates collapsin response
mediator protein 2 through the Fyn-Cdk5-GSK3β kinase cascade,
inhibiting microtubule assembly to block axon extension; third, it
inhibits integrin-mediated cell adhesion via the R-Ras GAP activity
of Plexin-A, synergistically enhancing the repulsive effect
(99) as illustrated in Fig. 4.
Studies on classical bone development and bone
homeostasis have confirmed that neuronal paracrine SEMA3A is a key
functional isoform that maintains bone homeostasis under
physiological conditions, and that its effects are mediated through
NRP1, a key receptor (14,98). The study by Fukuda et al
(14), first elucidated the
central role of NRP1 in this regulation. Neuron-derived SEMA3A
specifically regulates the projection and innervation density of
CGRP-positive sensory nerves by binding to the NRP1-Plexin-A4
complex on intraosseous sensory nerve endings, thereby maintaining
normal intraosseous innervation and indirectly regulating bone
homeostasis (14,101). CGRP-positive sensory nerves
have been further confirmed to participate in the regulation of
bone repair by secreting CGRP (102).
In addition to the indirect regulation of
innervation, a recent study (13) on bone homeostasis have further
revealed the direct regulatory effect of neurogenic SEMA3A on BMSCs
via NRP1. Specifically, in that study, in a mouse tibial
denervation model, the loss of sensory innervation led to the
synchronous downregulation of SEMA3A and NRP1 in bone, accompanied
by the impaired osteogenic capacity of BMSCs. In vitro
co-culture experiments further confirmed that neurogenic SEMA3A
secreted by dorsal root ganglia directly promoted the osteogenic
differentiation of BMSCs through the SEMA3A-NRP1-Wnt/β-catenin
pathway elucidated above (13).
Previous research on disuse osteoporosis in rats further validated
this mechanism in vivo. Following sciatic nerve transection,
the expression of Sema3A and NRP1 in bone tissue was synchronously
downregulated, which inhibited the activity of Wnt/β-catenin
signaling, resulting in decreased osteogenic activity and abnormal
osteoclast activation, and subsequently led to bone loss and
destruction of bone microstructure (103) (Fig. 4).
In addition to directly regulating the function of
BMSCs, nerves may indirectly regulate skeletal homeostasis by
guiding vascular differentiation and spatial distribution. Research
has confirmed that peripheral nerves serve as a template for the
differentiation and spatial distribution of cutaneous arteries
(104). Sensory neurons and
Schwann cells can secrete VEGF164/120, induce endothelial cells to
express arterial-specific markers, such as NRP1 and ephrinB2, and
guide blood vessels to grow along nerve branches. The loss of
sensory nerves or Schwann cells prevents cutaneous small blood
vessels from inducing the expression of arterial markers, such as
NRP1, resulting in significantly impaired arterial differentiation
(104). Given that bone tissue
is also densely populated with sensory nerve fibers, this
'neuro-vascular' template mechanism may also apply to the spatial
configuration of the intraosseous vascular network. Still, its
actual role in bone tissue and fracture healing remains to be
experimentally verified (Fig.
4). The strength and classification of evidence for
NRP1-mediated neuro-osseous crosstalk is summarized in Table III.
| Table IIIStrength and classification of
evidence for NRP1-mediated neuro-osseous crosstalk. |
Table III
Strength and classification of
evidence for NRP1-mediated neuro-osseous crosstalk.
| Evidence level | Model type | Tissue and cell
type | Regulatory
mechanism | Functional
effect |
|---|
| Bone biology
field | In vivo
non-bone-repair model | Intraosseous
sensory nerve terminals | Neuronal SEMA3A
binds to intraosseous sensory nerve NRP1-Plexin-A4 | Regulates the
projection density of intraosseous sensory nerves, indirectly
maintaining bone homeostasis |
| Bone biology
field | In vivo bone
cell model + in vivo non-bone-repair model | BMSCs | Sensory nerve
SEMA3A acts on BMSCs through the Wnt/β-catenin/NRP1 positive
feedback loop | Promotes BMSC
osteogenic differentiation |
| Non-skeletal
system | In vivo
non-bone cell model + in vivo non-skeletal model | Neuronal axonal
growth cones |
SEMA3A-NRP1-Plexin-A activates Rac1,
driving plasma membrane endocytosis and actin rearrangement | Directly induces
growth cone collapse |
| Non-skeletal
system | In vivo
non-bone cell model + in vivo non-skeletal model | Neuronal axonal
growth cones |
SEMA3A-NRP1-Plexin-A phosphorylates CRMP2
through Fyn-dk5-GSK3β | Inhibits
microtubule assembly and blocks axon extension |
| Non-skeletal
system | In vivo
non-bone cell model + in vivo non-skeletal model | Neuronal axonal
growth cones |
SEMA3A-NRP1-Plexin-A inhibits integrin
adhesion through R-Ras GAP activity | Enhances the axonal
repulsion effect and regulates precise neural projection |
| Non-skeletal
system | In vivo
non-skeletal model | Sensory nerves | Sensory
neurons/Schwann cells secrete VEGF164/120, inducing endothelial
expression of arterial markers such as NRP1 | Guides blood
vessels to grow along nerves, regulating the differentiation and
distribution of skin arteries |
Dysregulated expression of NRP1 in
pathological states of bone tissue
Systemic pathological conditions directly
associated with impaired fracture healing
Diabetes mellitus and osteoporosis included in this
section are both clinically recognized independent high-risk
factors for delayed fracture union and nonunion, and belong to
systemic pathological conditions directly related to impaired
fracture healing. Existing experimental evidence suggests that the
dysregulated expression and function of NRP1, driven by systemic
pathological states, may be a potential mechanism mediating the
imbalance in osteoclast-osteoblast homeostasis and poor fracture
healing. However, the causal association between the dysregulated
expression of NRP1 in the aforementioned pathological states and
delayed fracture union has not yet been established.
Diabetes mellitus
Diabetes mellitus is an independent high risk
factor for delayed fracture union and nonunion. Its core
pathological mechanisms involve the accumulation of advanced
glycation end products induced by hyperglycemia, the excessive
production of reactive oxygen species, amplified chronic
inflammation and abnormal insulin signaling. These factors
collectively lead to increased apoptosis and inhibited osteoblast
differentiation, excessive osteoclast activation, and impaired
angiogenesis and innervation (105). Recent studies (106,20) have shown that the SEMA3A/NRP1
signaling axis may be a potential regulator of the aforementioned
pathological processes.
Qiao et al (106) found that, in a rat model of
type 2 diabetes mellitus, endogenous SEMA3A expression in BMSCs was
significantly decreased, and exogenous supplementation with
recombinant SEMA3A upregulated osteogenesis-related gene expression
and restored the mineralization capacity of the cells (106). Subsequent the in-depth study
focusing on osteoblasts by Zhang et al (20) further elucidated the molecular
mechanisms underlying osteogenic inhibition in a high glucose
environment. Specifically, in vitro experiments on
osteoblasts by Zhang et al (20) confirmed that high glucose can
downregulate the expression of SEMA3A and NRP1 in osteoblasts in a
concentration-dependent manner, and inhibit the expression of core
osteogenic markers, such as ALP and Runx2, as well as the formation
of mineralized nodules. Exogenous supplementation with SEMA3A can
significantly reverse the high-glucose-induced inhibition of
osteogenic differentiation via the SEMA3A-NRP1-Wnt/β-catenin
pathway. In vivo experiments using mice with
streptozotocin-induced diabetes further verified that the
downregulation of the SEMA3A/NRP1 axis was accompanied by bone
loss, destruction of trabecular bone microstructure, and imbalance
of bone turnover homeostasis, and exogenous SEMA3A intervention
effectively ameliorated the abnormalities in bone mineral density,
bone microstructure, and bone metabolic markers in mice (20).
Collectively, the inhibition of the SEMA3A/NRP1
signaling axis by a high-glucose environment is one of the
potential regulatory mechanisms of diabetic bone loss, and
restoring its activity can improve osteogenic function and bone
structure in mice with diabetes. However, whether this mechanism
mediates the impaired healing of fractures complicated by diabetes
mellitus and whether targeted intervention can promote the healing
of diabetic fractures remains to be further verified.
Osteoporosis
A previous study demonstrated that, in the bone
tissue of ovariectomized rats with osteoporosis, the expression of
lncRNA MALAT1 was significantly downregulated, while the expression
of miR-320a was abnormally elevated (93). Under physiological conditions,
MALAT1 in BMSCs sequestered miR-320a via the competing endogenous
RNA (ceRNA) mechanism, relieving its inhibitory effect on the
SEMA3A-NRP1-Wnt/β-catenin pathway. This regulatory balance was
shown to be disrupted in osteoporosis, leading to the excessive
activation of miR-320a, which ultimately impeded the osteogenic
differentiation of BMSCs and resulted in insufficient bone
formation (93).
In osteoclastogenesis, miR-148a in osteoclast
precursor cells is a key positive regulator of osteoclast
differentiation, and its expression is significantly upregulated in
the serum of patients with postmenopausal osteoporosis and in the
bone tissue of ovariectomized mice with osteoporosis. miR-148a can
directly target and bind to the 3' untranslated region of NRP1 and
inhibit its protein expression, thereby relieving the negative
regulation of NRP1 on the NFATc1/c-Fos signaling axis, further
excessively promoting the differentiation and maturation of
osteoclasts and their bone resorption function, and ultimately
exacerbating estrogen deficiency-induced bone loss (19).
In addition, increased osteocyte apoptosis is
another pathological feature of postmenopausal and age-related
osteoporosis, and NRP1 on the surface of mature osteocytes plays a
key regulatory role in this process (107). Under physiological conditions,
estrogen upregulates SEMA3A expression by inhibiting miR-497/195.
After binding to NRP1 on the surface of osteocytes, SEMA3A
activates the mature osteocyte-specific soluble guanylate
cyclase-cyclic guanosine monophosphate-protein kinase G pathway and
inhibits osteocyte apoptosis. Estrogen deficiency leads to the
inactivation of this protective axis, increased osteocyte apoptosis
and aggravated bone loss (107).
All the aforementioned regulatory associations are
based on simple models of osteoporosis, revealing the role of NRP1
in regulating bone homeostasis through multicellular pathways.
However, to the best of our knowledge, there is currently no
evidence to indicate that it exerts the same effect in the complex
scenario of osteoporosis complicated with fracture, and whether it
mediates impaired fracture healing in such patients remains to be
verified.
Other bone metabolic disorders with
mechanistic reference significance
The following pathological states differ
significantly from fracture nonunion in terms of pathogenesis and
pathological nature. Relevant studies cannot be used directly as
evidence of impaired fracture healing; however, they can provide a
reference for understanding the functional complexity of NRP1 in
regulating bone metabolism.
Infectious osteolysis
In Porphyromonas gingivalis
lipopolysaccharide (P-LPS)-induced infectious osteolysis, the
Toll-like receptor 4 pathway on the surface of osteoclast
precursors and osteoblast precursors is specifically activated by
P-LPS. This pathway can significantly downregulate the expression
of the SEMA3A/NRP1 axis in both cell types. This regulatory
imbalance disrupts bone homeostasis in a bidirectional manner. It
not only promotes the differentiation and the activation of
osteoclasts and their bone-resorptive function, but also inhibits
the proliferation, differentiation and mineralization of
osteoblasts. These changes eventually induce progressive bone loss.
Exogenous supplementation with recombinant SEMA3A can
simultaneously reverse the excessive osteoclast activation and
osteogenic dysfunction described above and significantly alleviate
P-LPS-mediated osteolysis (108). It should be clearly stated that
this infection model is characterized by chronic, persistent
inflammation-driven pathological bone destruction, which differs
fundamentally from the temporally controlled physiological bone
remodeling process during fracture healing. Its core reference
value lies in suggesting that NRP1 may play a bone-protective role
in inflammation-driven bone loss. This also provides a potential
research direction for the subsequent exploration of the molecular
mechanism of delayed fracture healing under infectious
conditions.
Genetic association between NRP1 gene
variants and bone metabolic homeostasis
At the genetic level, research on the Chinese Han
population has demonstrated that mutations in the NRP1 rs2070296
and rs180868035 loci are significantly associated with increased
osteoporosis risk and decreased bone mineral density. Among these,
the missense mutation NRP1 rs180868035 can directly disrupt SEMA3A
binding to NRP1 and disrupt bone metabolic homeostasis by
inhibiting osteoblast proliferation and differentiation and
promoting osteoclast activation (109). This genetic finding provides
population-level evidence of the role of NRP1 in maintaining bone
homeostasis. However, the association between these findings and
fracture healing remains speculative, and there is currently no
research data available (to the best of our knowledge) on the
association between these gene loci and fracture-healing
phenotypes. Therefore, this finding can only provide
population-level reference for the study of bone repair mechanisms,
and cannot be directly used as evidence for the association between
NRP1 dysregulation and impaired fracture healing.
Periprosthetic osteolysis (PPO) and
the microenvironment-dependent functional difference
phenomenon
Clinical specimens and in vitro experiments
have shown that multinucleated osteoclast-like cells phagocytosing
wear particles in PPO lesions exhibit a high expression of NRP1 and
its ligand SEMA3A (110).
During the process of wear particle-induced osteoclast
differentiation, the expression of NRP1 is continuously upregulated
with cell maturation, and wear particles can further amplify this
effect (110). In the
aforementioned pathological environment of PPO, the highly
expressed NRP1 fails to effectively inhibit osteoclast bone
resorption, leading instead to persistent pathological bone
destruction. This result constitutes a clear functional difference
from the aforementioned conclusion that NRP1 negatively regulates
osteoclast differentiation in bone homeostasis models (110).
Combined with the known characteristics of NRP1
signaling pathways, the aforementioned phenomenon may be mediated
by dual mechanisms. First, the chronic foreign-body inflammatory
environment may specifically downregulate Plexin-A1 expression in
osteoclast precursor cells. By contrast, the osteoclast inhibitory
effect of SEMA3A depends on the functional complex formed by NRP1
and Plexin-A1 (17). Therefore,
the high expression of NRP1 alone may not effectively transduce
inhibitory signals. Second, a large amount of VEGFA165 and PDGF-BB
produced under the stimulation of wear particles can competitively
occupy the b1/b2 binding sites of NRP1 (48), which not only blocks the binding
of SEMA3A, but also may turn to activate pro-osteoclast signals.
All the aforementioned mechanisms are reasonable speculations based
on existing evidence and remain to be directly verified by
subsequent experiments.
It can be seen that the dissociation between NRP1
expression and function in PPO is not a mere experimental
discrepancy, but directly reflects the high
microenvironment-dependent nature of NRP1 function. Whether it
ultimately exerts a bone-protective or bone-destructive effect may
be jointly determined by the microenvironmental signal background,
such as the type of local inflammation, the co-receptor expression
profile and the abundance of competitive ligands, rather than a
simple linear correlation with its expression level. This is
precisely a key scientific issue worthy of in-depth exploration in
the current field of NRP1 in bone metabolism. The strength and
classification of evidence for NRP1 dysregulation in pathological
bone states is summarized in Table
IV.
| Table IVStrength and classification of
evidence for NRP1 dysregulation in pathological bone states. |
Table IV
Strength and classification of
evidence for NRP1 dysregulation in pathological bone states.
| Pathological state
classification | Pathology type | Evidence level | Model type | Molecular
mechanism | Degree of
association with fracture healing |
|---|
| Systemic
pathological states directly related to bone repair impairment | Diabetes | Bone biology
field | In vitro
bone cell experiment + in vivo non-bone-repair model | High glucose
inhibits the SEMA3A/NRP1-Wnt/β-catenin axis, suppressing
osteogenesis and promoting osteoclastogenesis | High-risk factor
for fracture |
| Systemic
pathological states directly related to bone repair impairment | Osteoporosis | Bone biology
field | In vitro
bone cell experiment + in vivo non-bone-repair model | BMSCs: miR-320a
upregulation inhibits NRP1/β-catenin, impeding osteogenesis;
osteoclast precursors: miR-148a upregulation silences NRP1,
promoting osteoclastogenesis; osteocytes: Estrogen deficiency
inactivates the SEMA3A/NRP1 pathway, increasing osteocyte
apoptosis | High-risk factor
for fracture |
| Bone metabolic
disorders with mechanistic reference significance | Infectious
osteolysis | Bone biology
field | In vitro
bone cell experiment + in vivo non-bone-repair model | P-LPS activates the
TLR4 pathway, downregulating the SEMA3A/NRP1 axis in
osteoblasts/osteoclasts, bidirectionally disrupting bone
homeostasis | Different
pathological nature, provides mechanistic reference only |
| Bone metabolic
disorders with mechanistic reference significance | NRP1 gene
mutation | Bone biology
field | In vitro
bone cell experiment + clinical samples | Mutations like
rs180868035 disrupt SEMA3A-NRP1 binding, inhibiting osteogenesis
and promoting osteoclastogenesis | Different
pathological nature, provides mechanistic reference only |
| Bone metabolic
disorders with mechanistic reference significance | Periprosthetic
osteolysis (PPO) | Bone biology
field | In vitro
bone cell experiment + clinical samples | Wear particles
induce high expression of NRP1 in osteoclast-like cells, which lose
their osteoclast-inhibitory function and paradoxically promote bone
destruction | Different
pathological nature, provides mechanistic reference only |
Translational prospects and challenges of
NRP1 as a therapeutic target for bone repair
NRP1 is involved in the interactive regulation of
nerves, blood vessels and bone, and has potential mediating value
in maintaining bone homeostasis and repairing injuries. Given the
widespread expression of NRP1 across multiple systemic tissues,
systemic intervention may entail substantial off-target risks and
an uncertain safety profile; thus, it is critical to explore
effective and precise intervention strategies (111-113).
Intervention strategies validated in bone
regeneration-related models
Targeted activation strategy of the
SEMA3A-NRP1 signaling axis
As the canonical ligand of NRP1, the targeted
activation strategy of SEMA3A has been directly validated in mouse
cortical bone defect models, which simultaneously achieve dual
bone-protective effects by inhibiting bone resorption and promoting
bone formation (17).
Mechanistically, its osteoclast-inhibitory effect has been
clarified through the dual mechanisms described above. Moreover, it
can promote osteogenic differentiation and block the adipogenic
transformation of MSCs via the FARP2-Rac1-Wnt/β-catenin pathway,
and all effects are strictly dependent on the Sema-binding domain
of NRP1 (17). Both SEMA3A
knockout mice and NRP1-SEMA3A-binding site mutant knock-in mice
exhibit severe bone mass reduction phenotypes, genetically
confirming that this axis is a key regulatory pathway for
maintaining bone homeostasis. The local or systemic administration
of recombinant SEMA3A can significantly accelerate the repair of
mouse cortical bone defects and reverse postmenopausal bone loss,
providing direct in vivo evidence for the application of
NRP1 agonist-based interventions in bone regeneration (17).
However, the study by Hayashi et al
(17) still has certain
limitations. The core experiments are all based on mouse models.
Although the effects have been preliminarily verified in
human-derived cells, there is a lack of long-term efficacy and
safety data from large animals. Recombinant SEMA3A has a short
half-life and poor bone targeting, and systemic administration may
cause off-target effects. In addition, the study only evaluated the
short-term administration effects and lacked long-term safety
monitoring data.
Local delivery of NRP1 based on
functionalized scaffolds
To address the bottlenecks of the widespread
systemic expression of NRP1, leading to off-target effects and the
easy degradation of recombinant proteins, the local delivery system
of liposome-encapsulated NRP1 loaded onto 3D-printed magnesium
alloy scaffolds has been fully validated in ischemic bone defect
models. It is currently one of the research strategies with
preliminary exploratory value in bone regeneration. This system
achieves stable, sustained release of NRP1 via a composite coating
and synergizes with the osteogenic effect of magnesium ions to
enhance local angiogenesis via the NRP1-VEGFR2-PI3K-AKT pathway,
ultimately realizing synergistic repair of the
angiogenesis-osteogenesis coupling (46). This strategy can improve the
local bioavailability of NRP1 without obvious systemic adverse
reactions, effectively avoiding the off-target risks of systemic
interventions, and has potential translational value in clinical
scenarios such as ischemic bone injury and fracture nonunion.
However, the study described above (46) still has certain limitations. The
animal experiments are based solely on rabbit ischemic bone defect
models and lack long-term efficacy and safety verification in large
animals. In addition, this strategy conducted only a 12-week
short-term safety monitoring period, and the long-term systemic
safety and metabolic effects of degradation products remain to be
investigated.
Intervention strategies with biological
rationality but not yet validated in fracture models
The following intervention strategies have all been
mechanistically validated in non-fracture systems, and their
efficacy in bone repair remains to be confirmed in vivo.
Targeted strategy of the miR-148a-NRP1
axis
The inhibition of osteogenic differentiation by the
inflammatory microenvironment is a potential pathological mechanism
underlying impaired repair of inflammatory bone defects. In a model
of LPS-induced osteogenic dysfunction of periodontal ligament stem
cells, it was found that the upregulated expression of miR-148a
negatively regulates NRP1 expression by directly targeting the 3'
untranslated region of NRP1 Mrna (19), thereby impairing the osteogenic
capacity of cells. The inhibition of miR-148a or the overexpression
of NRP1 can significantly reverse this inflammation-mediated
osteogenic dysfunction, whereas the knockdown of NRP1 can
completely abrogate the osteogenic-promoting effect of miR-148a
inhibitors (92). The negative
regulatory relationship between miR-148a and NRP1 has also been
verified in clinical periodontitis tissues and in derived stem
cells (92). It should be
clarified that although the miR-148a-NRP1 axis exhibits potential
to regulate osteogenic differentiation in the context of
periodontitis, the evidence presented above is derived solely from
in vitro cell models. Whether this axis exerts the same
efficacy in the complex in vivo environment of fracture
healing remains to be directly verified.
Based on findings in periodontitis models, the
miR-148a-NRP1 axis may be a potential target for intervening in
osteogenic dysfunction in inflammatory microenvironments. Given
that inflammation is also a key inducer of impaired fracture
healing, this strategy has only theoretical potential application
in inflammatory fracture-healing disorders. Currently, it lacks
in vivo intervention evidence from fracture animal
models.
Regulatory potential of natural small
molecules
Previous studies have confirmed that natural
products, such as flavonoids, saponins and polyphenols, can promote
angiogenesis and bone repair (114-117). Recent studies on luteolin and
naringin have found that NRP1 and its mediated signaling pathways
are also potential molecular targets for these naturally active
ingredients to regulate bone homeostasis (84,103).
In vitro experiments using primary
osteoblasts have shown that luteolin can significantly upregulate
mRNA and the protein expression of the SEMA3A/NRP1/Plexin-A1
pathway in glucocorticoid-injured rat primary calvarial
osteoblasts, promote osteoblast proliferation and inhibit abnormal
apoptosis (84). In a previous
study, naringin was shown to exert a potent in vivo
bone-protective effect in a rat model of disuse osteoporosis
(103). It upregulated NRP1
expression in bone tissue in a dose-dependent manner,
simultaneously promoting bone formation and inhibiting bone
resorption through the SEMA3A-NRP1-Wnt/β-catenin pathway, and
effectively maintaining bone mass and bone microstructure (103).
The aforementioned studies collectively indicate
that the SEMA3A-NRP1 axis is a potential molecular target for
traditional Chinese medicine active ingredients to regulate bone
metabolism. Luteolin provides a new intervention direction for
glucocorticoid-induced osteoblast dysfunction, while naringin
further verifies the feasibility of targeting this pathway to
prevent and treat bone metabolic disorders in vivo. However,
it should be noted that luteolin has undergone only preliminary
mechanistic exploration in vitro in primary osteoblasts and
lacks in vivo verification in glucocorticoid-induced
osteoporosis and fracture-healing animal models. Naringin has also
not been studied in the context of fracture repair. The clinical
translational value and optimal administration regimens of both
still require further in-depth exploration.
Exploratory strategies with insufficient
evidence
sNRP1 can be stably detected in serum and has been
confirmed as an independent biomarker for a poor prognosis in
various malignant tumors, including breast cancer, ovarian cancer
and hepatocellular carcinoma, in cancer research. Its high
expression is significantly associated with a shorter patient
survival, and the association between its level and the efficacy of
anti-angiogenic therapy has been reported (118-122). It should be
strictly clarified that all the aforementioned evidence is derived
entirely from the field of oncology, and there are currently (to
the best of our knowledge) no in vitro or in vivo
studies available on fracture healing that directly examine the
association between sNRP1 and the healing process. In addition,
malignant tumors are a chronic, continuously progressive and
uncontrollable process of pathological angiogenesis. By contrast,
angiogenesis in fracture healing is acute, self-limiting and
precisely regulated by physiological repair programs. Whether sNRP1
has similar clinical significance across these two distinct
biological backgrounds remains an open question. Given the
established regulatory functions of NRP1 in bone tissue injury and
repair, and the application of sNRP1 as a biomarker in other
diseases, exploring its association with fracture-healing
phenotypes has only pure theoretical value. Positioning it as a
potential biomarker for fracture healing still lacks clinical or
preclinical data to support this claim.
Opportunities in translational research
and existing barriers Translational opportunities
NRP1 may serve as a potential nexus of neural,
vascular and skeletal signals, providing a unique opportunity to
develop pleiotropic bone repair strategies.
Multi-pathway regulatory effect. Unlike
traditional bone repair therapies that target a single cytokine,
the modulation of NRP1 can simultaneously engage multiple important
pathways, including VEGF-mediated angiogenesis, SEMA3A-mediated
osteogenic differentiation and osteoclast inhibition (17,46). In addition, based on the
regulatory roles of NRP1 in the projection of intraosseous sensory
nerves and in promoting BMSC osteogenic differentiation via
neurogenic SEMA3A, it theoretically has the potential to integrate
neuro-osseous coupling repair. Still, to date, to the best of our
knowledge, no direct evidence has been obtained in fracture models
(13,14). If this multi-pathway synergistic
effect is fully confirmed, it is expected to yield new insight into
the clinical problem of the limited efficacy of single-target
therapies.
The rise of smart biomaterials.
Technological breakthroughs in smart biomaterials have enabled
novel, targeted and precise delivery solutions for NRP1 and the
exertion of its functions. Recent research has confirmed that the
local delivery system based on 3D-printed biodegradable magnesium
alloy scaffolds can achieve spatiotemporally synergistic controlled
release of NRP1 and Mg2+, providing the first verifiable
technical solution to the problem of systemic off-target effects of
NRP1 (46). This strategy
provides potential research and translational ideas worth exploring
to address the clinically intractable ischemic osteogenic disorder
and promote bone defect repair.
Existing barriers
Despite its promising prospects, translating NRP1
research from the laboratory to clinical application remains
challenging, particularly due to biological complexity and clinical
safety concerns.
Spatial specificity and systemic
off-target risks
NRP1 is widely expressed across multiple tissues,
and its function is highly microenvironment-dependent. This
characteristic indicates that systemic intervention is highly
likely to simultaneously disrupt the normal physiological functions
of NRP1 across the cardiovascular, nervous, and immune systems,
raising fundamental questions about the feasibility of systemic
therapeutic modulation (112).
This characteristic also directly indicates that the systemic
therapeutic regulation viewpoint proposed in this paper remains
impractical at present. How to achieve precise intervention at the
fracture site, while avoiding the disruption of the physiological
functions of NRP1 in the cardiovascular, nervous and immune systems
is a critical safety challenge that needs to be overcome for the
clinical translation of its targeting strategies. Although the
current local delivery systems have shown no systemic adverse
reactions in the short term, their safety in the context of
long-term intervention is completely unknown.
General lack of large animal models
and clinical evidence
Currently, the functional evidence for NRP1 in bone
repair is primarily based on mouse models and in vitro
systems. Due to critical differences between the human skeletal
system and rodents in metabolic rate, mechanical loading patterns,
and healing time course, the therapeutic efficacy of NRP1 remains
unverified in standardized large-animal models and human clinical
trials. This gap is not a skippable developmental step, but a
critical link that must be completed in the logical chain of target
validation.
Incomplete elucidation of the
spatiotemporal and context-dependent nature of the mechanism of
action
As stated multiple times in the present review, the
function of NRP1 is not fixed. NRP1 undergoes bidirectional
functional switching across different pathological
microenvironments, and the threshold for switching between its
protective and destructive effects has not yet been elucidated,
thereby preventing the determination of the time window and
direction of targeted intervention. For example, in the chronic
foreign-body inflammatory environment of periprosthetic osteolysis,
NRP1 exhibits an abnormally high expression, but loses its proper
bone-protective function, leading instead to progressive bone
destruction (110). This
bidirectional regulatory capacity means that the optimal
intervention window for NRP1 remains unclear during the dynamic
process of fracture healing, as the microenvironment is constantly
changing. Until these basic parameters are elucidated, NRP1
intervention strategies will lack a fundamental basis.
Summary and prospects
The present review article systematically
summarizes the research progress on the regulatory mechanisms of
NRP1-mediated bone repair. The functions that have been directly
validated in bone defect models to date include: NRP1 functioning
as a co-receptor of VEGFR2 to drive endothelial angiogenesis,
enhancing the migration and paracrine function of BMSCs via the
NRP1-VEGFR2 axis, and synchronously promoting osteogenesis and
inhibiting osteoclastogenesis through the SEMA3A axis (17,46).
However, significant gaps remain in the regulatory
network of NRP1 in bone repair. At the cellular and tissue levels,
the regulation of NRP1 in pericytes, type H vessels and
chondrocytes is derived from non-skeletal systems, and its role in
fracture healing remains unclear. At the signaling network level,
the function of the NRP1-PDGF axis in vascular maturation and the
regulatory mechanisms of neuro-osseous crosstalk have not been
directly verified in fracture models. At the pathological and
translational levels, the causal association between NRP1
dysregulation and impaired healing has not been established, and
multiple intervention strategies have not yet been translated from
bone homeostasis models to fracture models.
In bone defect models that have been directly
validated, the effects mediated by NRP1, including promoting
osteogenesis, inhibiting osteoclastogenesis and regulating the
osteogenic differentiation of BMSCs, mainly depend on the binding
of SEMA3A. However, the regulatory mode of competitive binding
between VEGFA165 and SEMA3A during the initiation of vascular
sprouting is currently mainly derived from studies on non-skeletal
systems, suggesting that its functional outcome may be determined
by the local concentration ratio of the two ligands. Furthermore,
the function of NRP1 exhibits cell-type specificity: It has been
confirmed in bone defect models that NRP1 can drive angiogenesis in
endothelial cells; studies in non-skeletal systems further indicate
that it can also guide vascular sprouting and maintain barrier
homeostasis in endothelial cells, and regulate vascular
permeability and maturation in pericytes. It can promote
differentiation and mineralization, counteract metabolic osteogenic
inhibition in osteoblasts, inhibit maturation, differentiation and
bone resorption in osteoclast precursor cells, enhance migration
and paracrine function, and regulate the balance between osteogenic
and adipogenic differentiation in bone marrow mesenchymal stem
cells.
In addition, NRP1 activation exerets significant
context-dependent bidirectional effects in chondrocytes and
osteoclasts. In chondrocytes, under acute mechanical stress, NRP1
can inhibit excessive activation of the AKT, ERK and NF-κB
pathways, thereby protecting the cartilage matrix. By contrast,
chronic inflammation in osteoarthritis can inhibit the PI3K-AKT
pro-survival pathway, driving chondrocyte apoptosis and matrix
degradation. In the osteoclast lineage, under physiological
conditions, NRP1 inhibits bone resorption through SEMA3A signaling;
in diabetes and osteoporosis, the downregulated expression of NRP1
is accompanied by enhanced bone resorption; while in periprosthetic
osteolysis, a paradoxical phenomenon of abnormally high NRP1
expression coexisting with bone destruction is observed. This
functional switch from protection to destruction suggests that the
pros and cons of NRP1 activation are not determined solely by the
pathway itself, but are jointly shaped by the local
microenvironment's inflammatory state and the cellular context.
It is precisely this high
microenvironment-dependent nature of function that constitutes the
core challenge of current NRP1 research and also points the way for
future research. First, elucidating the molecular switches that
govern the directional conversion of NRP1 function, particularly
the inflammatory threshold and signaling nodes at which it shifts
from protection to destruction, will directly determine the
direction of targeting strategies. Second, there is an urgent need
to systematically validate the regulatory functions of NRP1 in
pericytes, type H vessels, chondrocytes and neuro-osseous crosstalk
in fracture models. At the technical level, although the existing
local delivery strategy based on 3D-printed magnesium alloy
scaffolds has shown preliminary efficacy, transitioning from
sustained release to precise regulation within a specific time
window remains a barrier to clinical applicability. In the longer
term, NRP1 may become a promising research target for bone repair,
and its spatiotemporal regulation at specific stages of fracture
healing is a key area for interdisciplinary collaboration among
materials science, bone biology and pharmacology.
Core scientific issues and future
research directions
Based on current research on NRP1 in bone repair, a
series of key scientific issues remain to be elucidated, and future
research can be prioritized and pursued sequentially based on
evidence. The most prominent problem in current research is the
severe lack of evidence for key mechanisms. The role of NRP1 in
type H vessels, intraosseous pericytes, and fracture callus
formation and remodeling remains poorly verified in fracture
models. The molecular switches underlying its ligand dependence and
cell specificity have also not been clearly elucidated. Moreover,
the mechanisms underlying the bidirectional effects of NRP1 across
different pathological scenarios remain unclear. This molecule is
downregulated in osteoporosis and diabetes, but is abnormally
upregulated in periprosthetic osteolysis. The key threshold and
core signaling mechanism underlying its functional shift from bone
protection to bone destruction remain unclear, which makes it
difficult to determine the direction of targeted interventions. At
the translational research level, most existing intervention
strategies have been validated only in small-animal bone defect
models, lacking support from clinically relevant composite
pathological models such as diabetes and aging. Furthermore, the
optimal spatiotemporal window for local delivery of NRP1 has not
yet been established; thus, it is difficult to support the
development of precise and effective targeted interventions.
Future research is required to prioritize
addressing the core shortcomings outlined above, such as the
following: First, to systematically validate the regulatory effects
of NRP1 on type H vessels, pericytes and chondrocytes in standard
fracture healing models, elucidate the molecular mechanism of its
bidirectional functional conversion, and consolidate the core
theoretical foundation. Second, to construct composite animal
models of diabetes and osteoporosis complicated by fractures,
clarify the direct causal association between dysregulated NRP1
expression and delayed fracture union or nonunion, and fill a key
evidence gap in the pathological mechanisms underlying these
conditions. At the translational application level, focus should be
on developing local delivery systems for NRP1 with spatiotemporally
controlled release, reducing systemic off-target risks, while
ensuring repair efficacy, and improving the safety and clinical
applicability of targeted interventions.
Availability of data and materials
Not applicable.
Authors' contributions
KZ was involved in the conceptualization of the
study, in the writing of the original draft of the manuscript, and
in literature curation. TL was involved in the critical literature
analysis and synthesis, visualization, and in the writing reviewing
and editing of the manuscript. HL was involved in the validation of
data from the literature, in the writing of the original draft of
the manuscript, and in the writing reviewing and editing of the
manuscript. HT was involved in literature curation and validation,
and in the writing reviewing and editing of the manuscript. ZZ was
involved in literature curation, provision of academic resources
(institutional database subscriptions, scientific visualization
software licenses) and in the writing reviewing and editing of the
manuscript. ZY was involved in the formal analysis, validation of
data from the literature, and in the writing reviewing and editing
of the manuscript. JW, XYan, HH and XYao were involved in
literature curation and validation, and in the writing reviewing
and editing of the manuscript. ZL was involved in literature
curation, in critical literature analysis and synthesis, and in the
writing reviewing and editing of the manuscript. SS was involved in
formal analysis, validation of data from the literature, and in the
writing reviewing and editing of the manuscript. XW provided
resources, and was involved in the validation of data from the
literature, in the writing reviewing and editing of the manuscript.
FLY was involved in funding acquisition, study supervision and
correspondence. XC was involved in the conceptualization of the
study, in study supervision and correspondence. This work is
presented as a narrative review. All cited literature and original
materials for figures and tables have been thoroughly reviewed by
KZ and XC, who jointly confirm the accuracy and objectivity of the
narrative synthesis presented in this review. All authors have read
and approved the final manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, AI tools were
used to improve the readability and language of the manuscript or
to generate images, and subsequently, the authors revised and
edited the content produced by the AI tools as necessary, taking
full responsibility for the ultimate content of the present
manuscript.
Acknowledgments
Not applicable.
Funding
The present study was supported by the following funding
programs: The National Natural Science Foundation of China (funding
institution: National Natural Science Foundation Committee of
China, grant nos. 82372412 and 82572767); the Jiangsu Provincial
Social Development Program (funding institution: Department of
Science and Technology of Jiangsu Province, grant no. BE2022701);
the Jiangsu Provincial Traditional Chinese Medicine Science and
Technology Development Program (funding institution: Jiangsu
Administration of Traditional Chinese Medicine, grant no.
MS2023069); the Jiangsu Provincial Traditional Chinese Medicine
Leading Talents Training Program (funding institution: Jiangsu
Administration of Traditional Chinese Medicine, grant no. SLJ0322);
Wuxi Municipal Health Commission Scientific Research Program
(funding institution: Wuxi Municipal Health Commission, grant no.
Z202405); and the Wuxi Municipal Health Commission Major Scientific
Research Program (funding institution: Wuxi Municipal Health
Commission, grant no. Z202322).
References
|
1
|
Du H, Zhang W, Liu Z and Liu W: LncRNA
GABARAPL2 promotes non-union long bone fractures by suppressing
bone mesenchymal stromal cell osteogenesis. J Orthop Surg Res.
20:7412025. View Article : Google Scholar
|
|
2
|
Horenberg AL, Ren Y, Zeng EZ, Rindone AN,
Pathak AP and Grayson WL: 3D imaging reveals changes in the
neurovascular architecture of the murine calvarium with aging. Bone
Res. 13:242025. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
GBD 2019 Fracture Collaborators: Global,
regional, and national burden of bone fractures in 204 countries
and territories, 1990-2019: A systematic analysis from the global
burden of disease study 2019. Lancet Healthy Longev. 2:e580–e592.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zura R, Xiong Z, Einhorn T, Watson JT,
Ostrum RF, Prayson MJ, Della Rocca GJ, Mehta S, McKinley T, Wang Z
and Steen RG: Epidemiology of fracture nonunion in 18 human bones.
JAMA Surg. 151:e1627752016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Siris ES, Miller PD, Barrett-Connor E,
Faulkner KG, Wehren LE, Abbott TA, Berger ML, Santora AC and
Sherwood LM: Identification and fracture outcomes of undiagnosed
low bone mineral density in postmenopausal women: Results from the
national osteoporosis risk assessment. JAMA. 286:2815–2822. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kusumbe AP, Ramasamy SK and Adams RH:
Coupling of angiogenesis and osteogenesis by a specific vessel
subtype in bone. Nature. 507:323–328. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Augustin HG and Koh GY: Organotypic
vasculature: From descriptive heterogeneity to functional
pathophysiology. Science. 357:eaal23792017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hamiti Y, Liu K, Wang S, Yang X, Kadier X
and Yusufu A: CGRP-dependent molecular signaling drives bone marrow
stem cell osteogenesis in distraction osteogenesis. Front Bioeng
Biotechnol. 13:16414762025. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Telley L, Cadilhac C, Cioni JM, Saywell V,
Jahannault-Talignani C, Huettl RE, Sarrailh-Faivre C, Dayer A,
Huber AB and Ango F: Dual function of NRP1 in axon guidance and
subcellular target recognition in cerebellum. Neuron. 91:1276–1291.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Colotti G, Failla CM, Lacal PM, Ungarelli
M, Ruffini F, Di Micco P, Orecchia A and Morea V: Neuropilin-1 is
required for endothelial cell adhesion to soluble vascular
endothelial growth factor receptor 1. FEBS J. 289:183–198. 2022.
View Article : Google Scholar :
|
|
11
|
Lee PS, Gao N, Dike M, Shkilnyy O, Me R,
Zhang Y and Yu FX: Opposing effects of neuropilin-1 and -2 on
sensory nerve regeneration in wounded corneas: Role of Sema3C in
ameliorating diabetic neurotrophic keratopathy. Diabetes.
68:807–818. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jeon HM, Shin YJ, Lee J, Chang N, Woo DH,
Lee WJ, Nguyen D, Kang W, Cho HJ, Yang H, et al: The semaphorin
3A/neuropilin-1 pathway promotes clonogenic growth of glioblastoma
via activation of TGF-β signaling. JCI Insight. 8:e1670492023.
View Article : Google Scholar
|
|
13
|
Shi J, Zhang B, Wu Z, Zhang Y, Gupta A,
Wang X, Wang J, Pan L, Xiao M, Zhang S and Wang L: Peripheral
nerve-derived Sema3A promotes osteogenic differentiation of
mesenchymal stem cells through the wnt/β-catenin/Nrp1 positive
feedback loop. J Cell Mol Med. 28:e182012024. View Article : Google Scholar
|
|
14
|
Fukuda T, Takeda S, Xu R, Ochi H, Sunamura
S, Sato T, Shibata S, Yoshida Y, Gu Z, Kimura A, et al: Sema3A
regulates bone-mass accrual through sensory innervations. Nature.
497:490–493. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kitsukawa T, Shimizu M, Sanbo M, Hirata T,
Taniguchi M, Bekku Y, Yagi T and Fujisawa H: Neuropilin-semaphorin
III/D-mediated chemorepulsive signals play a crucial role in
peripheral nerve projection in mice. Neuron. 19:995–1005. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gomez C, Burt-Pichat B, Mallein-Gerin F,
Merle B, Delmas PD, Skerry TM, Vico L, Malaval L and Chenu C:
Expression of semaphorin-3A and its receptors in endochondral
ossification: potential role in skeletal development and
innervation. Dev Dyn. 234:393–403. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hayashi M, Nakashima T, Taniguchi M,
Kodama T, Kumanogoh A and Takayanagi H: Osteoprotection by
semaphorin 3A. Nature. 485:69–74. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Termini CM, Pang A, Fang T, Roos M, Chang
VY, Zhang Y, Setiawan NJ, Signaevskaia L, Li M, Kim MM, et al:
Neuropilin 1 regulates bone marrow vascular regeneration and
hematopoietic reconstitution. Nat Commun. 12:69902021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pan B, Zheng L, Liu S, Fang J, Lou C, Hu
X, Ye L, Lai H, Gao J, Zhang Y, et al: MiR-148a deletion protects
from bone loss in physiological and estrogen-deficient mice by
targeting NRP1. Cell Death Discov. 8:4702022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang L, Zheng L, Li C, Wang Z, Li S and
Xu L: Sema3a as a novel therapeutic option for high
glucose-suppressed osteogenic differentiation in diabetic
osteopathy. Front Endocrinol (Lausanne). 10:5622019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
He Z and Tessier-Lavigne M: Neuropilin is
a receptor for the axonal chemorepellent semaphorin III. Cell.
90:739–751. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Soker S, Takashima S, Miao HQ, Neufeld G
and Klagsbrun M: Neuropilin-1 is expressed by endothelial and tumor
cells as an isoform-specific receptor for vascular endothelial
growth factor. Cell. 92:735–745. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Appleton BA, Wu P, Maloney J, Yin J, Liang
WC, Stawicki S, Mortara K, Bowman KK, Elliott JM, Desmarais W, et
al: Structural studies of neuropilin/antibody complexes provide
insights into semaphorin and VEGF binding. EMBO J. 26:4902–4912.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ikeda M, Hosoda Y, Hirose S, Okada Y and
Ikeda E: Expression of vascular endothelial growth factor isoforms
and their receptors flt-1, KDR, and neuropilin-1 in synovial
tissues of rheumatoid arthritis. J Pathol. 191:426–433. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gu C, Limberg BJ, Whitaker GB, Perman B,
Leahy DJ, Rosenbaum JS, Ginty DD and Kolodkin AL: Characterization
of neuropilin-1 structural features that confer binding to
semaphorin 3A and vascular endothelial growth factor 165. J Biol
Chem. 277:18069–18076. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mamluk R, Gechtman Z, Kutcher ME, Gasiunas
N, Gallagher J and Klagsbrun M: Neuropilin-1 binds vascular
endothelial growth factor 165, placenta growth factor-2, and
heparin via its b1b2 domain. J Biol Chem. 277:24818–24825. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Baek DS, Kim JH, Kim YJ and Kim YS:
Immunoglobulin fc-fused peptide without C-terminal arg or lys
residue augments neuropilin-1-dependent tumor vascular
permeability. Mol Pharm. 15:394–402. 2018. View Article : Google Scholar
|
|
28
|
Teesalu T, Sugahara KN, Kotamraju VR and
Ruoslahti E: C-end rule peptides mediate neuropilin-1-dependent
cell, vascular, and tissue penetration. Proc Natl Acad Sci USA.
106:16157–16162. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vander Kooi CW, Jusino MA, Perman B, Neau
DB, Bellamy HD and Leahy DJ: Structural basis for ligand and
heparin binding to neuropilin B domains. Proc Natl Acad Sci USA.
104:6152–6157. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Renzi MJ, Feiner L, Koppel AM and Raper
JA: A dominant negative receptor for specific secreted semaphorins
is generated by deleting an extracellular domain from neuropilin-1.
The Journal of Neuroscience: J Neurosci. 19:7870–7880. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Roth L, Nasarre C, Dirrig-Grosch S, Aunis
D, Crémel G, Hubert P and Bagnard D: Transmembrane domain
interactions control biological functions of neuropilin-1. Mol Biol
Cell. 19:646–654. 2008. View Article : Google Scholar :
|
|
32
|
Cao S, Yaqoob U, Das A, Shergill U,
Jagavelu K, Huebert RC, Routray C, Abdelmoneim S, Vasdev M, Leof E,
et al: Neuropilin-1 promotes cirrhosis of the rodent and human
liver by enhancing PDGF/TGF-beta signaling in hepatic stellate
cells. J Clin Invest. 120:2379–2394. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Muhl L, Folestad EB, Gladh H, Wang Y,
Moessinger C, Jakobsson L and Eriksson U: Neuropilin 1 binds PDGF-D
and is a co-receptor in PDGF-D-PDGFRβ signaling. J Cell Sci.
130:1365–1378. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Parker MW, Xu P, Li X and Vander Kooi CW:
Structural basis for selective vascular endothelial growth factor-a
(VEGF-a) binding to neuropilin-1. J Biol Chem. 287:11082–11089.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rossignol M, Gagnon ML and Klagsbrun M:
Genomic organization of human neuropilin-1 and neuropilin-2 genes:
Identification and distribution of splice variants and soluble
isoforms. Genomics. 70:211–222. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cai H and Reed RR: Cloning and
characterization of neuropilin-1-interacting protein: A
PSD-95/dlg/ZO-1 domain-containing protein that interacts with the
cytoplasmic domain of neuropilin-1. J Neurosci. 19:6519–6527. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Valdembri D, Caswell PT, Anderson KI,
Schwarz JP, König I, Astanina E, Caccavari F, Norman JC, Humphries
MJ, Bussolino F and Serini G: Neuropilin-1/GIPC1 signaling
regulates alpha-5beta1 integrin traffic and function in endothelial
cells. PLoS Biol. 7:e252009. View Article : Google Scholar
|
|
38
|
Kawamura H, Li X, Goishi K, van Meeteren
LA, Jakobsson L, Cébe-Suarez S, Shimizu A, Edholm D, Ballmer-Hofer
K, Kjellén L, et al: Neuropilin-1 in regulation of VEGF-induced
activation of p38MAPK and endothelial cell organization. Blood.
112:3638–3649. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ballmer-Hofer K, Andersson AE, Ratcliffe
LE and Berger P: Neuropilin-1 promotes VEGFR-2 trafficking through
Rab11 vesicles thereby specifying signal output. Blood.
118:816–826. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shintani Y, Takashima S, Asano Y, Kato H,
Liao Y, Yamazaki S, Tsukamoto O, Seguchi O, Yamamoto H, Fukushima
T, et al: Glycosaminoglycan modification of neuropilin-1 modulates
VEGFR2 signaling. EMBO J. 25:3045–3055. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gagnon ML, Bielenberg DR, Gechtman Z, Miao
HQ, Takashima S, Soker S and Klagsbrun M: Identification of a
natural soluble neuropilin-1 that binds vascular endothelial growth
factor: In vivo expression and antitumor activity. Proc Natl Acad
Sci USA. 97:2573–2578. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mehta V, Fields L, Evans IM, Yamaji M,
Pellet-Many C, Jones T, Mahmoud M and Zachary I: VEGF (vascular
endothelial growth factor) induces NRP1 (neuropilin-1) cleavage via
ADAMs (a disintegrin and metalloproteinase) 9 and 10 to generate
novel carboxy-terminal NRP1 fragments that regulate angiogenic
signaling. Arterioscler Thromb Vasc Biol. 38:1845–1858. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Harper J, Gerstenfeld LC and Klagsbrun M:
Neuropilin-1 expression in osteogenic cells: Down-regulation during
differentiation of osteoblasts into osteocytes. J Cell Biochem.
81:82–92. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Stöckl S, Reichart J, Zborilova M,
Johnstone B and Grässel S: Semaphorin 3A-neuropilin-1 signaling
modulates MMP13 expression in human osteoarthritic chondrocytes.
Int J Mol Sci. 23:141802022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Vempati P, Popel AS and Mac Gabhann F:
Extracellular regulation of VEGF: Isoforms, proteolysis, and
vascular patterning. Cytokine Growth Factor Rev. 25:1–19. 2014.
View Article : Google Scholar
|
|
46
|
Pei Z, Xu H, Zhao P, Wen Y, Zhang Z, Huang
L, Wang M, Peng B, Wen L, Wen P and Sun F: Reconstructing the
ischemic osteogenic microenvironment through hierarchical scaffolds
orchestrating Mg2+ signaling and neuropilin-1-mediated
angiogenesis. Bioact Mater. 62:738–758. 2026.PubMed/NCBI
|
|
47
|
Acevedo LM, Barillas S, Weis SM, Göthert
JR and Cheresh DA: Semaphorin 3A suppresses VEGF-mediated
angiogenesis yet acts as a vascular permeability factor. Blood.
111:2674–2680. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Miao HQ, Soker S, Feiner L, Alonso JL,
Raper JA and Klagsbrun M: Neuropilin-1 mediates
collapsin-1/semaphorin III inhibition of endothelial cell motility:
Functional competition of collapsin-1 and vascular endothelial
growth factor-165. J Cell Biol. 146:233–242. 1999.PubMed/NCBI
|
|
49
|
Ouyang X, Ma Q, Zhou C, Tang J, Li M, Qing
J, Lei X, Huang D, Liu H and Zhang G: Natural bioactive products in
the regulation of bone metabolism and regeneration. Front
Pharmacol. 16:16832792025. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xu R: Semaphorin 3A: A new player in bone
remodeling. Cell Adh Migr. 8:5–10. 2014. View Article : Google Scholar
|
|
51
|
Fantin A, Lampropoulou A, Gestri G,
Raimondi C, Senatore V, Zachary I and Ruhrberg C: NRP1 regulates
CDC42 activation to promote filopodia formation in endothelial tip
cells. Cell Rep. 11:1577–1590. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang Y, Wang E, Zhang Y, Madamsetty VS, Ji
B, Radisky DC, Grande JP, Misra S and Mukhopadhyay D: Neuropilin-1
maintains dimethylarginine dimethylaminohydrolase 1 expression in
endothelial cells, and contributes to protection from angiotensin
II-induced hypertension. FASEB J. 33:494–500. 2019. View Article : Google Scholar
|
|
53
|
Hu X, Atzler D, Xu X, Zhang P, Guo H, Lu
Z, Fassett J, Schwedhelm E, Böger RH, Bache RJ and Chen Y:
Dimethylarginine dimethylaminohydrolase-1 is the critical enzyme
for degrading the cardiovascular risk factor asymmetrical
dimethylarginine. Arterioscler Thromb Vasc Biol. 31:1540–1546.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Łabędź-Masłowska A, Vergori L,
Kędracka-Krok S, Karnas E, Bobis-Wozowicz S, Sekuła-Stryjewska M,
Sarna M, Andriantsitohaina R and Zuba-Surma EK: Mesenchymal stem
cell-derived extracellular vesicles exert pro-angiogenic and
pro-lymphangiogenic effects in ischemic tissues by transferring
various microRNAs and proteins including ITGa5 and NRP1. J
Nanobiotechnology. 22:602024. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bosseboeuf E, Chikh A, Chaker AB, Mitchell
TP, Vignaraja D, Rajendrakumar R, Khambata RS, Nightingale TD,
Mason JC, Randi AM, et al: Neuropilin-1 interacts with VE-cadherin
and TGFBR2 to stabilize adherens junctions and prevent activation
of endothelium under flow. Sci Signal. 16:eabo48632023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Rezaei M, Martins Cavaco AC, Seebach J,
Niland S, Zimmermann J, Hanschmann EM, Hallmann R, Schillers H and
Eble JA: Signals of the neuropilin-1-MET axis and cues of
mechanical force exertion converge to elicit inflammatory
activation in coherent endothelial cells. J Immunol. 202:1559–1572.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Niland S and Eble JA: Neuropilins in the
context of tumor vasculature. Int J Mol Sci. 20:6392019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Dhar K, Dhar G, Majumder M, Haque I, Mehta
S, Van Veldhuizen PJ, Banerjee SK and Banerjee S: Tumor
cell-derived PDGF-B potentiates mouse mesenchymal stem
cells-pericytes transition and recruitment through an interaction
with NRP-1. Mol Cancer. 9:2092010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wnuk M, Anderegg MA, Graber WA, Buergy R,
Fuster DG and Djonov V: Neuropilin1 regulates glomerular function
and basement membrane composition through pericytes in the mouse
kidney. Kidney Int. 91:868–879. 2017. View Article : Google Scholar
|
|
60
|
Pal S, Su Y, Nwadozi E, Claesson-Welsh L
and Richards M: Neuropilin-1 controls vascular permeability through
juxtacrine regulation of endothelial adherens junctions.
Angiogenesis. 28:72024. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Jurisic G, Maby-El Hajjami H, Karaman S,
Ochsenbein AM, Alitalo A, Siddiqui SS, Ochoa Pereira C, Petrova TV
and Detmar M: An unexpected role of semaphorin3a-neuropilin-1
signaling in lymphatic vessel maturation and valve formation. Circ
Res. 111:426–436. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ruan Z, Yin H, Wan TF, Lin ZR, Zhao SS,
Long HT, Long C, Li ZH, Liu YQ, Luo H, et al: Metformin accelerates
bone fracture healing by promoting type H vessel formation through
inhibition of YAP1/TAZ expression. Bone Res. 11:452023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Liu X, Zhang P, Gu Y, Guo Q and Liu Y:
Type H vessels: Functions in bone development and diseases. Front
Cell Dev Biol. 11:12365452023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Peng Y, Wu S, Li Y and Crane JL: Type H
blood vessels in bone modeling and remodeling. Theranostics.
10:426–436. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yu Y, Dai K, Gao Z, Tang W, Shen T, Yuan
Y, Wang J and Liu C: Sulfated polysaccharide directs therapeutic
angiogenesis via endogenous VEGF secretion of macrophages. Sci Adv.
7:eabd82172021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Xie H, Cui Z, Wang L, Xia Z, Hu Y, Xian L,
Li C, Xie L, Crane J, Wan M, et al: PDGF-BB secreted by
preosteoclasts induces angiogenesis during coupling with
osteogenesis. Nat Med. 20:1270–1278. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zhang J, Pan J and Jing W: Motivating role
of type H vessels in bone regeneration. Cell Prolif. 53:e128742020.
View Article : Google Scholar :
|
|
68
|
Gao B, Deng R, Chai Y, Chen H, Hu B, Wang
X, Zhu S, Cao Y, Ni S, Wan M, et al: Macrophage-lineage TRAP+ cells
recruit periosteum-derived cells for periosteal osteogenesis and
regeneration. J Clin Invest. 129:2578–2594. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Gerber HP, Vu TH, Ryan AM, Kowalski J,
Werb Z and Ferrara N: VEGF couples hypertrophic cartilage
remodeling, ossification and angiogenesis during endochondral bone
formation. Nat Med. 5:623–628. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wang Y, Wan C, Deng L, Liu X, Cao X,
Gilbert SR, Bouxsein ML, Faugere MC, Guldberg RE, Gerstenfeld LC,
et al: The hypoxia-inducible factor alpha pathway couples
angiogenesis to osteogenesis during skeletal development. J Clin
Invest. 117:1616–1626. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Ball SG, Bayley C, Shuttleworth CA and
Kielty CM: Neuropilin-1 regulates platelet-derived growth factor
receptor signalling in mesenchymal stem cells. Biochem J.
427:29–40. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ferrara N, Gerber HP and LeCouter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Olsson AK, Dimberg A, Kreuger J and
Claesson-Welsh L: VEGF receptor signalling in control of vascular
function. Nat Rev Mol Cell Biol. 7:359–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Sarabipour S, Kinghorn K, Quigley KM,
Kovacs-Kasa A, Annex BH, Bautch VL and Mac Gabhann F: Trafficking
dynamics of VEGFR1, VEGFR2, and NRP1 in human endothelial cells.
PLoS Comput Biol. 20:e10117982024. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Sharma S, Ehrlich M, Zhang M, Blobe GC and
Henis YI: NRP1 interacts with endoglin and VEGFR2 to modulate VEGF
signaling and endothelial cell sprouting. Commun Biol. 7:1122024.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Whitaker GB, Limberg BJ and Rosenbaum JS:
Vascular endothelial growth factor receptor-2 and neuropilin-1 form
a receptor complex that is responsible for the differential
signaling potency of VEGF(165) and VEGF(121). J Biol Chem.
276:25520–25531. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Smith GT, Radin DP and Tsirka SE: From
protein-protein interactions to immune modulation: therapeutic
prospects of targeting neuropilin-1 in high-grade glioma. Front
Immunol. 13:9586202022. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Noah AA, El-Mezayen NS, El-Ganainy SO,
Darwish IE and Afify EA: Reversal of fibrosis and portal
hypertension by empagliflozin treatment of CCl4-induced liver
fibrosis: Emphasis on gal-1/NRP-1/TGF-β and gal-1/NRP-1/VEGFR2
pathways. Eur J Pharmacol. 959:1760662023. View Article : Google Scholar
|
|
79
|
Yamashita S, Kudo A, Kawakami H and Okada
Y: Mechanisms of angiogenic suppression in uteri exposed to
diethylstilbestrol neonatally in the mouse. Biol Reprod.
88:1162013. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Sun J, Wei X, Wang Z, Liu Y, Lu J, Lu Y,
Cui M, Zhang X and Li F: Inflammatory milieu cultivated Sema3A
signaling promotes chondrocyte apoptosis in knee osteoarthritis. J
Cell Biochem. 119:2891–2899. 2018. View Article : Google Scholar
|
|
81
|
Wu K, Huang D and Huang X: The effects of
semaphorin 3A in bone and cartilage metabolism: Fundamental
mechanism and clinical potential. Front Cell Dev Biol.
11:13211512023. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zhang Y, Su J, Teng Y, Zhang J, Wang J, Li
K, Yao L and Li X: Nrp1, a neuronal regulator, enhances
DDR2-ERK-Runx2 cascade in osteoblast differentiation via
suppression of DDR2 degradation. Cell Physiol Biochem. 36:75–84.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhang Y, Su J, Wu S, Teng Y, Yin Z, Guo Y,
Li J, Li K, Yao L and Li X: DDR2 (discoidin domain receptor 2)
suppresses osteoclastogenesis and is a potential therapeutic target
in osteoporosis. Sci Signal. 8:ra312015. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Zheng L: Luteolin stimulates proliferation
and inhibits late differentiation of primary rat calvarial
osteoblast induced by high-dose dexamethasone via
Sema3A/NRP1/pleixin A1. Curr Pharm Biotechnol. 22:1538–1545. 2021.
View Article : Google Scholar
|
|
85
|
Feng X and Teitelbaum SL: Osteoclasts: New
insights. Bone Res. 1:11–26. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Einhorn TA and Gerstenfeld LC: Fracture
healing: mechanisms and interventions. Nat Rev Rheumatol. 11:45–54.
2015. View Article : Google Scholar :
|
|
87
|
Liu YQ, Han XF, Bo JX and Ma HP:
Wedelolactone enhances osteoblastogenesis but inhibits
osteoclastogenesis through Sema3A/NRP1/PlexinA1 pathway. Front
Pharmacol. 7:3752016. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Azab E, Chandler KB, Uda Y, Sun N, Hussein
A, Shuwaikan R, Lu V, Costello CE, McComb ME and Divieti Pajevic P:
Osteocytes control myeloid cell proliferation and differentiation
through gsα-dependent and -independent mechanisms. FASEB J.
34:10191–10211. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Sumi C, Hirose N, Yanoshita M, Takano M,
Nishiyama S, Okamoto Y, Asakawa Y and Tanimoto K: Semaphorin 3A
inhibits inflammation in chondrocytes under excessive mechanical
stress. Mediators Inflamm. 2018:57036512018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Qiu M, Xie Y, Tan G, Wang X, Huang P and
Hong L: Synovial mesenchymal stem cell-derived exosomal miR-485-3p
relieves cartilage damage in osteoarthritis by targeting the
NRP1-mediated PI3K/akt pathway: Exosomal miR-485-3p relieves
cartilage damage. Heliyon. 10:e240422024. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Wang T, Liang Z, Wang C, Chen J, Ma Y,
Chen S, Zhou D and Hong Z: Pulsed electromagnetic fields mediate
sensory nerve regulation for bone formation in aging models. Nat
Commun. 16:82232025. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Bao L, Zhang X, Xu Y, Wang M, Song Y, Gu
Y, Zheng Y, Xiao J, Wang Y, Zhou Q, et al: Dysfunction of
MiR-148a-NRP1 functional axis suppresses osteogenic differentiation
of periodontal ligament stem cells under inflammatory
microenvironment. Cell Reprogram. 21:314–322. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Su C, Wang H, Xu L, Zhang Y and Li Y:
MALAT1/miR-320a in bone marrow mesenchymal stem cells function may
shed light on mechanisms underlying osteoporosis. Arch Med Sci.
18:1638–1649. 2021.PubMed/NCBI
Tomlinson RE, Li Z, Zhang Q, Goh BC, Li Z,
Thorek DLJ, Rajbhandari L, Brushart TM, Minichiello L, Zhou F, et
al: NGF-TrkA signaling by sensory nerves coordinates the
vascularization and ossification of developing endochondral bone.
Cell Rep. 16:2723–2735. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Li Z, Meyers CA, Chang L, Lee S, Li Z,
Tomlinson R, Hoke A, Clemens TL and James AW: Fracture repair
requires TrkA signaling by skeletal sensory nerves. J Clin Invest.
129:5137–5150. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Qin Q, Lee S, Patel N, Walden K,
Gomez-Salazar M, Levi B and James AW: Neurovascular coupling in
bone regeneration. Exp Mol Med. 54:1844–1849. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Ma YX, Jiao K, Wan QQ, Li J, Liu MY, Zhang
ZB, Qin W, Wang KY, Wang YZ, Tay FR and Niu LN: Silicified collagen
scaffold induces semaphorin 3A secretion by sensory nerves to
improve in-situ bone regeneration. Bioact Mater. 9:475–490.
2022.
|
|
97
|
Li Z, Hao J, Duan X, Wu N, Zhou Z, Yang F,
Li J, Zhao Z and Huang S: The role of semaphorin 3A in bone
remodeling. Front Cell Neurosci. 11:402017.PubMed/NCBI
|
|
98
|
Negishi M, Oinuma I and Katoh H: Plexins:
Axon guidance and signal transduction. Cell Mol Life Sci.
62:1363–1371. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Gu C, Rodriguez ER, Reimert DV, Shu T,
Fritzsch B, Richards LJ, Kolodkin AL and Ginty DD: Neuropilin-1
conveys semaphorin and VEGF signaling during neural and
cardiovascular development. Dev Cell. 5:45–57. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Liu YH, Liang KH, Zhang W, Rao GW and
Zheng Q: Small molecule inhibitors of the co-receptor neuropilin-1.
Drug Discov Today. 30:1045232025. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Wee NKY, Novak S, Ghosh D, Root SH,
Dickerson IM and Kalajzic I: Inhibition of CGRP signaling impairs
fracture healing in mice. J Orthop Res. 41:1228–1239. 2023.
View Article : Google Scholar
|
|
102
|
Ma X, Lv J, Sun X, Ma J, Xing G, Wang Y,
Sun L, Wang J, Li F, Li Y and Zhao Z: Naringin ameliorates bone
loss induced by sciatic neurectomy and increases semaphorin 3A
expression in denervated bone. Sci Rep. 6:245622016. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Mukouyama Y suke, Shin D, Britsch S,
Taniguchi M and Anderson DJ: Sensory nerves determine the pattern
of arterial differentiation and blood vessel branching in the skin.
Cell. 109:693–705. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Jiao H, Xiao E and Graves DT: Diabetes and
its effect on bone and fracture healing. Curr Osteoporos Rep.
13:327–335. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Qiao Q, Xu X, Song Y, Song S, Zhu W and Li
F: Semaphorin 3A promotes osteogenic differentiation of BMSC from
type 2 diabetes mellitus rats. J Mol Histol. 49:369–376. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Hayashi M, Nakashima T, Yoshimura N,
Okamoto K, Tanaka S and Takayanagi H: Autoregulation of osteocyte
Sema3A orchestrates estrogen action and counteracts bone aging.
Cell Metab. 29:627–637.e5. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Feng J, Fu D, Phan S, Hu X and Xing Q: P.
Gingivalis LPS drives osteoclastogenesis and impairs osteoblast
differentiation by suppressing Sema3A/Nrp1 via TLR-4. Int Dent J.
75:1039412025. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Zhou HL, Wei MH, Di DS, Zhang RY, Zhang
JL, Yuan TT, Liu Q, Zhou TT, Huang Q and Wang Q: Association
between SEMA3A signaling pathway genes and BMD/OP risk: An
epidemiological and experimental study. Front Endocrinol
(Lausanne). 13:10144312022. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Saad S, Dharmapatni AASSK, Crotti TN,
Cantley MD, Algate K, Findlay DM, Atkins GJ and Haynes DR:
Semaphorin-3a, neuropilin-1 and plexin-A1 in prosthetic-particle
induced bone loss. Acta Biomater. 30:311–318. 2016. View Article : Google Scholar
|
|
110
|
Pellet-Many C, Frankel P, Jia H and
Zachary I: Neuropilins: Structure, function and role in disease.
Biochem J. 411:211–226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Chuckran CA, Liu C, Bruno TC, Workman CJ
and Vignali DA: Neuropilin-1: A checkpoint target with unique
implications for cancer immunology and immunotherapy. J Immunother
Cancer. 8:e0009672020. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Liu LN, Li XM, Ye DQ and Pan HF: Emerging
role of semaphorin-3A in autoimmune diseases. Inflammopharmacology.
26:655–665. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Gao ZR, Feng YZ, Zhao YQ, Zhao J, Zhou YH,
Ye Q, Chen Y, Tan L, Zhang SH, Feng Y, et al: Traditional Chinese
medicine promotes bone regeneration in bone tissue engineering.
Chin Med. 17:862022. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Ramesh P, Jagadeesan R, Sekaran S,
Dhanasekaran A and Vimalraj S: Flavonoids: Classification,
function, and molecular mechanisms involved in bone remodelling.
Front Endocrinol (Lausanne). 12:7796382021. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Li Y, Li L, Li X, Luo B, Ye Q, Wang H,
Yang L, Zhu X, Han L, Zhang R, et al: A mechanistic review of
chinese medicine polyphenols on bone formation and resorption.
Front Pharmacol. 13:10175382022. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Zhang Y, Xu L, Xu H, Zhang Y, Wang L,
Jiang S, Sun Y and Yu Y: Plant-derived food bioactives in bone
health: from multi-targeted roles to molecular mechanisms in
osteoporosis. Front Nutr. 12:17300532026. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Rachner TD, Kasimir-Bauer S, Goebel A,
Erdmann K, Hoffmann O, Rauner M, Hofbauer LC, Kimmig R and Bittner
AK: Soluble neuropilin-1 is an independent marker of poor prognosis
in early breast cancer. J Cancer Res Clin Oncol. 147:2233–2238.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Fernández-Palanca P, Payo-Serafín T,
Fondevila F, Méndez-Blanco C, San-Miguel B, Romero MR, Tuñón MJ,
Marin JJG, González-Gallego J and Mauriz JL: Neuropilin-1 as a
potential biomarker of prognosis and invasive-related parameters in
liver and colorectal cancer: A systematic review and meta-analysis
of human studies. Cancers(Basel). 14:34552022.PubMed/NCBI
|
|
119
|
Klotz DM, Kuhlmann JD, Link T, Goeckenjan
M, Hofbauer LC, Göbel A, Rachner TD and Wimberger P: Clinical
impact of soluble neuropilin-1 in ovarian cancer patients and its
associa- tion with its circulating ligands of the HGF/c-MET axis.
Front Oncol. 12:9748852022. View Article : Google Scholar
|
|
120
|
Mäenpää N, Tiainen L, Hämäläinen M,
Luukkaala T, Tanner M, Lahdenperä O, Vihinen P, Karihtala P,
Kellokumpu-Lehtinen PL, Moilanen E and Jukkola A: Neuropilin-1 and
placental growth factor as prognostic factors in metastatic breast
cancer. BMC Cancer. 24:3312024. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Rodrigues EM, Giovanini AF, Ribas CAPM,
Malafaia O, Roesler R and Isolan GR: The nervous system development
regulator neuropilin-1 as a potential prognostic marker and
therapeutic target in brain cancer. Cancers(Basel).
15:49222023.PubMed/NCBI
|