Introduction
Within the spectrum of populations at high risk for
sepsis, patients with cancer constitute a particularly susceptible
subgroup that is distinguished by substantially unfavorable
clinical outcomes. Contemporary multinational, multicenter
epidemiological investigations have uniformly revealed that these
patients account for >20% of all sepsis-related hospital
admissions and have a markedly increased risk of all-cause
mortality (1,2). Historically, clinical research has
ascribed the increased sepsis predisposition in patients with
cancer to factors that induce acquired immunodeficiency, including
neutropenia, chemotherapy-mediated compromise of mucosal integrity
within the gastrointestinal and respiratory epithelia and
immunosuppressive antineoplastic interventions (3,4).
Undoubtedly, these conventional risk factors represent the most
well-recognized and clinically actionable drivers of sepsis in
patients with cancer and serve as the primary targets for infection
prevention and control in clinical practice (3,4).
However, established risk factors explain only a fraction of
malignancy-associated sepsis cases. Notably, some patients with
advanced cancer develop refractory, life-threatening infections
that rapidly progress to sepsis or septic shock despite a lack of
severe neutropenia or evident mucosal barrier damage (4), suggesting that the
pathophysiological mechanisms are partially independent of
conventional risk factors.
In contemporary oncology, chronic stress responses
in the tumor microenvironment critically link malignant progression
to systemic host dysfunction (5). Among these stress responses,
endoplasmic reticulum (ER) stress and its master regulator
glucose-regulated protein 78 (GRP78/BiP/HSPA5), a core heat shock
protein 70 family member, have attracted significant attention
because of their pleiotropic functions (6,7).
As the primary ER molecular chaperone, GRP78 orchestrates the
unfolded protein response (UPR) to maintain proteostasis, modulate
calcium-dependent protein folding and regulate apoptotic signaling
under physiological and stress conditions (8). A growing body of basic and
translational research implicates dysregulated GRP78 expression in
increased infection susceptibility and accelerated sepsis
progression among patients with cancer. First, robust experimental
evidence has indicated that the tumor microenvironment induces
sustained, chronic ER stress, leading to the marked upregulation of
GRP78 expression in neoplastic cells, along with aberrant membrane
translocation and extracellular secretion that increase the
systemic levels of secreted GRP78 (sGRP78) (9). Second, cell surface-localized GRP78
acts as a key entry receptor or cofactor for diverse viral and
fungal pathogens while also modulating host immune responses to
bacterial components, thereby increasing infection susceptibility
(10,11). Preliminary clinical studies
further confirmed positive correlations between elevated sGRP78
concentrations, the severity of multiorgan dysfunction and
short-term mortality in sepsis patients, supporting its potential
utility as a prognostic biomarker (12,13).
While preclinical studies have robustly demonstrated
GRP78-mediated pathogen invasion and immunomodulation and studies
of non-cancer sepsis cohorts link elevated circulating levels of
GRP78 to mortality, no prospective or retrospective study has
specifically examined the use of GRP78 as a predictor of the sepsis
risk, severity, or outcome in an oncology population. The present
review focused specifically on GRP78 dysregulation induced by the
tumor microenvironment. It systematically elucidated the
pathological pathways through which GRP78 affects sepsis
susceptibility in patients with cancer via a dual mechanism:
Aberrant GRP78 expression not only facilitates pathogen invasion
but also dysregulates inflammatory responses and promotes
immunosuppression. Finally, GRP78-targeted therapeutic strategies
are explored. The aberrant expression of GRP78 and its functional
implications in the tumor microenvironment are shown in Fig. 1.
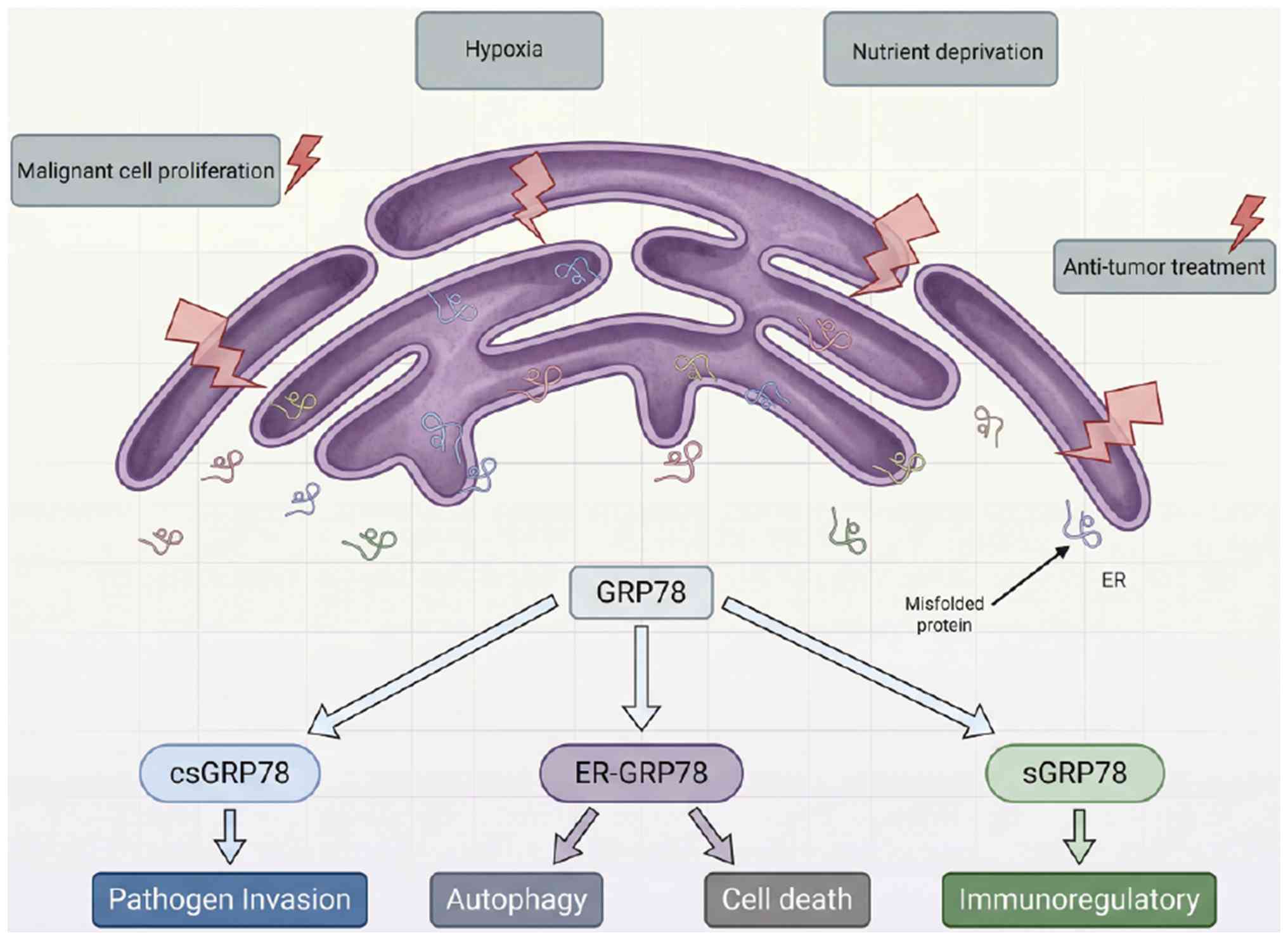 | Figure 1GRP78 dysregulation connects the
tumor microenvironment and sepsis susceptibility. Tumor-derived
GRP78 drives sepsis pathogenesis through dual mechanisms. i)
Pathogen invasion: csGRP78 acts as a critical coreceptor,
facilitating the entry of specific viruses (such as SARS-CoV-2,
dengue virus and Japanese encephalitis virus) and Mucorales fungi
(such as Rhizopus spp.), thereby increasing infection
efficiency. For bacterial pathogens, the predominant cause of
clinical sepsis, direct evidence for csGRP78 as an adhesion or
invasion receptor is currently limited to Mycoplasma
hyopneumoniae (a nonhuman pathogen). In bacterial infections,
the predominant cause of clinical sepsis, csGRP78 does not function
as a canonical adhesion receptor for major human pathogens.
Instead, GRP78 contributes to the progression of bacterial sepsis
through the indirect regulation of host inflammatory responses,
phagocytic function and epithelial barrier integrity. ii)
Immunosuppression: sGRP78 functions as a potent immunosuppressive
cytokine, triggering a 'cytokine storm' in early sepsis followed by
profound immune paralysis in late sepsis. This immunosuppressive
milieu, combined with direct pathogen invasion, compromises host
defense mechanisms, leading to uncontrolled infection, vascular
leakage, multiorgan dysfunction and ultimately septic shock. The
figure was constructed using the Figdraw 2.0 tool (https://www.figdraw.com/#/), with official
authorization obtained by the authors (authorization no.
PPPIU1c449). GRP78, glucose-regulated protein 78; cs, cell-surface;
s, secreted. |
GRP78: Beyond its role as an ER
chaperone
GRP78, an ER-resident molecular chaperone, undergoes
a subcellular redistribution under stress conditions, resulting in
the formation of three functional states: endoplasmic reticulum
GRP78 (ER-GRP78), sGRP78 and cell-surface GRP78 (csGRP78) (6,14,15). The dynamic interconversion among
these three states constitutes a crucial mechanism by which tumor
cells adapt to microenvironmental stress. Fig. 2 depicts the three functional
states of GRP78, the central roles of GRP78 in maintaining ER
homeostasis under physiological conditions and the GRP78-mediated
regulatory mechanism that orchestrates the activation of the
unfolded protein response activation to restore ER homeostasis
during pathological stress.
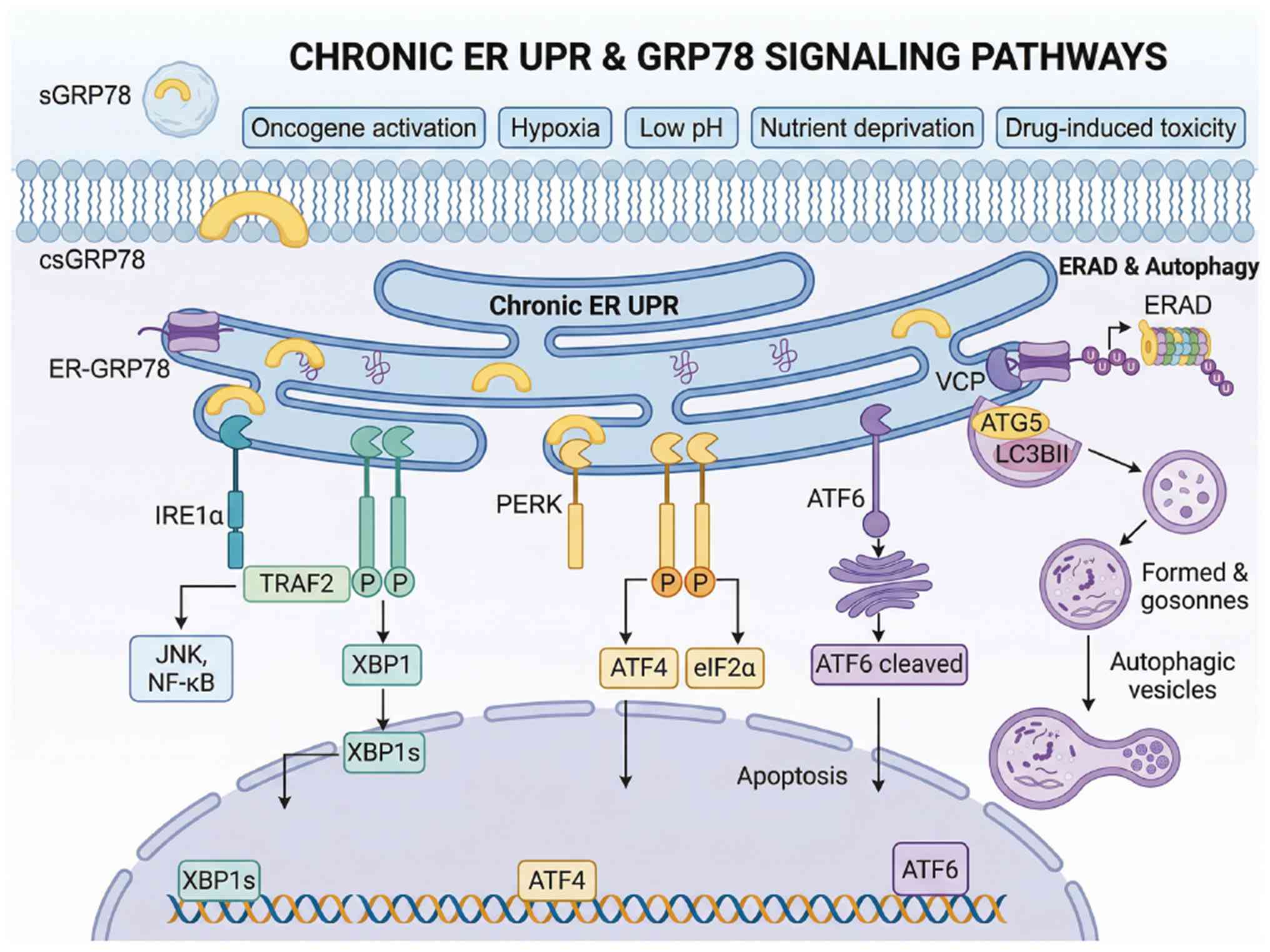 | Figure 2Schematic of GRP78-mediated
maintenance of ER homeostasis and the UPR regulatory feedback loop
under physiological and stress conditions. GRP78 is expressed in
three well-characterized functional states: the ER-resident form
(ER-GRP78) with intrinsic protein-folding chaperone activity, the
cell surface-localized form (csGRP78) that acts as a transmembrane
signaling receptor and the soluble extracellular form (sGRP78) that
acts as a secreted intercellular signaling factor. Under resting
conditions, ER-resident GRP78 maintains cellular homeostasis
through three key functions: preserving ER proteostasis through its
molecular chaperone activity; indirectly inhibiting apoptosis
through the suppression of CHOP and caspase activation; and
negatively regulating the UPR by binding to and repressing three
core ER stress sensors (PERK, IRE1 and ATF6). Upon exposure to
pathological stress, unfolded/misfolded proteins that accumulate in
the ER compete for binding to GRP78, leading to the differential
activation of three interconnected UPR branches: The
PERK-eIF2α-ATF4, ATF6 cleavage-nuclear translocation and IRE1-sXBP1
pathways. The ATF6 and IRE1-XBP1 pathways primarily converge to
upregulate GRP78 transcription, whereas PERK signaling contributes
indirectly, resulting in the formation of a negative feedback loop
to restore ER homeostasis. The figure was constructed using the
Figdraw 2.0 tool (https://www.figdraw.com/#/), with official
authorization obtained by the authors (authorization no.
RPWUW59df5). GRP78, glucose-regulated protein 78; ER, endoplasmic
reticulum; ER-GRP78, endoplasmic reticulum-resident GRP78; csGRP78,
cell-surface GRP78; sGRP78, secreted GRP78; UPR, unfolded protein
response; PERK, protein kinase R-like ER kinase; IRE1,
inositol-requiring enzyme 1; ATF6, activating transcription factor
6; CHOP, C/EBP homologous protein; eIF2α, eukaryotic initiation
factor 2 alpha; ATF4, activating transcription factor 4; sXBP1,
spliced X-box binding protein 1; XBP1, X-box binding protein 1. |
ER-GRP78: The canonical guardian of
proteostasis in the endoplasmic reticulum
ER-GRP78 represents the canonical functional state
of GRP78, which is predominantly localized within the ER lumen
(16). Under physiological
conditions, ER-GRP78 is maintained within the endoplasmic reticulum
through the binding of its C-terminal KDEL (Lys-Asp-Glu-Leu)
sequence to KDEL receptors (KDELRs) (17). As the central regulator of the
UPR, ER-GRP78 binds to and inhibits transmembrane stress sensors,
including protein kinase R-like ER kinase (PERK),
inositol-requiring enzyme 1 alpha (IRE1α) and activating
transcription factor 6 (ATF6), thereby maintaining these proteins
in an inactive conformation (18,19). When unfolded or misfolded
proteins accumulate within the ER lumen, GRP78 dissociates from
these sensors to bind to the aberrant proteins, initiating the UPR
signaling cascade. This response restores proteostasis through
three coordinated mechanisms: Inhibition of global protein
synthesis, upregulation of molecular chaperone expression and
enhancement of ER-associated degradation. Under normal
physiological conditions, the vast majority of GRP78 exists as
ER-GRP78, serving as the cornerstone of cellular protein quality
control.
sGRP78: A systemic messenger in
intercellular communication
GRP78 can be secreted extracellularly as sGRP78 and
is detected in various body fluids, including blood, urine and
cerebrospinal fluid (15). Its
secretion occurs through both canonical (ER-Golgi apparatus
pathway) and noncanonical (direct release via exosomes or vesicles)
mechanisms (20). In the tumor
microenvironment, exosomes secreted by GRP78-induced M2 macrophages
can transfer GRP78 to colorectal cancer cells, promoting cancer
stem cell properties and chemotherapeutic resistance (15,20). Clinical investigations revealed
significantly elevated serum sGRP78 levels in lung patients with
cancer, with 80.2% sensitivity and 68.9% specificity for an
early-stage lung cancer diagnosis and an area under the curve of
0.788, outperforming the conventional biomarker carcinoembryonic
antigen (14). sGRP78 levels are
positively correlated with the tumor burden, disease stage and a
poor prognosis; notably, non-small cell lung cancer (NSCLC)
patients with high sGRP78 expression experience significantly
shorter overall survival (14).
csGRP78: Stress-driven cell surface
translocation
In response to tumor microenvironmental stressors
(hypoxia, nutrient deprivation and therapeutic stress) or ER stress
induction, GRP78 translocates to the cell surface to form csGRP78
through the following four primary mechanisms.
First, the ER retention and recycling system is
saturated. Under pathological stress conditions, sustained GRP78
overexpression exceeds the retrograde transport capacity of KDEL
receptors (KDELRs) (16).
Consequently, substantial quantities of unretrieved GRP78 enter the
anterograde secretory pathway via COPII-coated vesicles and are
ultimately trafficked to the plasma membrane through post-Golgi
secretory vesicles (21).
Second, KDELRs are dysfunctional. Pathological
conditions induce the aberrant activation of the tyrosine-protein
kinase SRC, which mediates the phosphorylation of ASAP1 and the
accumulation of Arf1-GTP, leading to the dispersion of KDEL
receptors from the Golgi complex (17,22). This process downregulates their
retrograde transport activity, impairing the effective retrieval of
escaped GRP78 and facilitating its entry into secretory pathways
(22).
Third, vesicular trafficking and secretory
pathway-mediated mechanisms are involved. GRP78 is trafficked
through two routes: One subset follows the canonical secretory
pathway and is transported from the ER to the Golgi apparatus via
COPII vesicles and subsequently to the plasma membrane through
secretory vesicles (22). A
second subset utilizes exosome-mediated pathways, potentially
bypassing Golgi apparatus processing through alternative routes
(23).
Fourth, plasma membrane anchoring occurs. GRP78 is
trafficked to the plasma membrane and stably anchored to the outer
membrane leaflet through interactions with cell surface
glycoproteins, phosphatidylserine and other lipid moieties,
resulting in the formation of functional csGRP78 (23). Following membrane anchoring,
csGRP78 undergoes specific conformational rearrangements that
expose its substrate-binding domain to the extracellular milieu,
significantly increasing ligand accessibility (24). This structural adaptation confers
the capacity to specifically bind extracellular ligands, pathogen
structural proteins and cell surface receptors (17).
Notably, the expression of csGRP78 is markedly
increased on the surface of multiple solid tumor cells but is
minimal on cells in normal tissues, establishing it as an ideal
target for tumor-specific therapeutic interventions.
Tumor-driven ER stress and GRP78
dysregulation in sepsis susceptibility
Aberrant expression patterns of GRP78
across different tumor types
Tumorigenesis and progression are accompanied by
persistent ER stress driven by malignant cell proliferation,
hypoxia, nutrient deprivation and antitumor treatment, which are
the core drivers of aberrant GRP78 overexpression, subcellular
redistribution (especially membrane translocation) and
extracellular release (6,25).
Unlike total intracellular GRP78, which acts primarily as a
chaperone to regulate tumor progression and therapeutic resistance,
csGRP78 and sGRP78 represent plausible molecular mediators that may
link tumor pathology to increased sepsis susceptibility in patients
with cancer, but these findings require direct clinical
validation.
Notably, most existing studies have focused on the
correlations between GRP78 expression and the tumor stage,
chemotherapy resistance and overall survival, while direct clinical
evidence verifying the causal association between GRP78
dysregulation and sepsis incidence/prognosis in patients with
cancer remains scarce. Table I
systematically summarized the changes in GRP78 expression directly
related to the infection/sepsis risk across common human
malignancies, with a focus on membrane expression and secretion
features that are closely associated with sepsis pathogenesis
(15,26-61).
 | Table IGRP78 dysregulation in human
malignancies: Expression features, underlying mechanisms and
implications for sepsis susceptibility. |
Table I
GRP78 dysregulation in human
malignancies: Expression features, underlying mechanisms and
implications for sepsis susceptibility.
| Tumor category | Tumor type | GRP78 expression
features | Core regulatory
mechanisms | Implications for
sepsis susceptibility | (Refs.) |
|---|
| Hematologic
malignancies | AML | High surface
expression; high in leukemia stem cells | ER stress and UPR
activation | Increases
post-chemotherapy sepsis risk | (26) |
| ALL | High in high-risk
pediatric tumors; co-expressed with CXCR4 | ER stress and
migration pathway activation | Amplifies
immunosuppression and infection risk | (27) |
| CLL | High surface
expression; elevated in progressive stage | HIF-1α/BAG3
synergistic regulation | Induces immune
escape and inhibits anti-infective immunity | (28,29) |
| MM | No total
difference; elevated in relapsed bone marrow plasma | ER stress from high
secretory state | Mediates
immunosuppression and infection risk | (30) |
| DLBCL | Abnormally high
surface expression | ER stress and
TGF-β1 synergistic regulation | Promotes
immunosuppression and post-chemotherapy infection | (31) |
| Central nervous
system tumors | GBM | High expression;
correlates with malignancy/ recurrence; poor prognosis | Hypoxia-driven ER
stress and UPR activation | Increases CNS
infection and sepsis-associated encephalopathy | (32,33) |
| Pediatric brain
tumors | Heterogeneous;
highest in ependymomas | ER stress and
oncogene regulation | Increases CNS
infection and systemic immunosuppression | (34) |
| Thoracic and breast
tumors | NSCLC | Upregulated; higher
in advanced stages; chemo-resistant | Ubiquitination
imbalance, hypoxia/ROS-induced UPR, O-glycosylation | Promotes pulmonary
infection progression to sepsis | (35-37) |
| Breast cancer | Upregulated; higher
in TNBC | TDP-43 activation,
ATF6α-AKT pathway, ER stress translocation | Increases
post-chemotherapy sepsis risk | (38-40) |
| ESCC/ EA | High in 71.7% ESCC;
higher in early-stage EA | Hypoxia-induced ER
stress and lncRNA-activated MAPK | Impairs
gastrointestinal barrier; increases infection risk | (41-44) |
| Gastrointestinal
cancers | Gastric cancer | Overexpressed;
linked to stage and poor prognosis | m5C stabilization,
hypoxia-MAPK, ER stress-PKM2 feedback | Increases
intra-abdominal infection and sepsis | (45-47) |
| CRC | Overexpressed;
higher in KRAS-mutant tumors | MARylation-UPR,
uPA-uPAR translocation | Promotes microbiota
translocation and infection | (48,49) |
| PDAC | High expression;
poor prognosis, chemo-resistant | Sp1 activation, ER
stress-UPR | Increases
biliary/intra-abdominal infection and sepsis | (50-52) |
| HCC | Overexpressed;
linked to invasion, metastasis, drug resistance | CYP2E1-ATF6,
targeted therapy-induced IRE1α | Impairs intestinal
barrier; increases sepsis risk in cirrhosis | (15,53) |
| Genitourinary
cancers | Prostate
cancer | Upregulated;
elevated after ADT | UPR/AKT/mTOR
activation, ADT-induced ER stress | Increases
post-treatment sepsis risk | (54-56) |
| RCC | Upregulated in
tumors and serum | VHL deletion-HIF-α,
hypoxia-ER stress | Increases urinary
tract infection and sepsis | (57) |
| Bladder cancer | Upregulated; linked
to progression and poor prognosis | VEGFA/VEGFR2/PI3K/
AKT activation | Promotes urinary
tract infection progression to sepsis | (58,59) |
| UTUC | Controversial
expression and clinical significance | Tumor progression
pathway-driven | Sepsis risk
association unclear | (60,61) |
Core mechanisms driving GRP78 membrane
translocation and extracellular release in tumors
Chronic, persistent ER stress in the tumor
microenvironment not only upregulates total GRP78 expression but
also, more importantly, specifically increases its membrane
translocation and extracellular release (6,8).
These two processes represent the primary sources of persistently
elevated csGRP78 and sGRP78 levels in patients with cancer,
resulting in the formation of an immunosuppressive microenvironment
that may contribute to systemic immune dysfunction (62,63). Compared with physiological GRP78
trafficking in normal cells, tumor cells and antitumor treatments
increase GRP78 membrane translocation and secretion through
multiple nonredundant mechanisms, including SRC-mediated KDELR
dispersion and DNAJC3-dependent endosomal transport, with
significant heterogeneity across tumor types and treatment
modalities (17).
Tumor-intrinsic pathological processes
drive constitutive GRP78 dysregulation
The most potent constitutive induction of GRP78
membrane translocation and secretion occurs in highly secretory
tumors and tumors with sustained hypoxia/ER stress (26,53). Among these tumors, hematologic
malignancies and highly hypoxic solid tumors show the most
significant baseline GRP78 membrane localization and secretion
(53,64). The core mechanisms include two
aspects, which are described below.
Persistent inactivation of the ER
retention-retrieval system: Oncogenic activation of SRC kinase in
tumor cells disrupts KDEL receptor-mediated retrograde transport by
inducing KDELR dispersion from the Golgi apparatus, while the ER
stress-induced upregulation of GRP78 promotes its translocation to
the cell surface via multiple mechanisms, including the suppression
of Golgi-to-ER retrieval (65).
Hyperactivation of noncanonical secretory pathways:
Under hypoxic and nutrient-deprived conditions, tumor cells widely
utilize exosome-mediated unconventional secretion to release GRP78
into the extracellular space (66). This noncanonical pathway operates
alongside classical secretion mechanisms, with ER-stressed tumor
cells showing enhanced packaging of GRP78 into exosomes that bypass
traditional ER-Golgi quality control checkpoints. This effect is
most prominent in GBM, PDAC and other hypoxic solid tumors and is
the main source of circulating GRP78 in patients with advanced
malignancies (23,53).
Antitumor therapies mediate the secondary
amplification of GRP78 dysregulation
Antitumor treatments are the most important
exogenous inducers of GRP78 membrane translocation and
extracellular release and are the core drivers of the sharp
increase in the sepsis risk in patients with cancer during
treatment. Among these treatments, myelosuppressive chemotherapy,
multitarget tyrosine kinase inhibitors (TKIs) and androgen
deprivation therapy (ADT) have the strongest induction effects.
Chemotherapy: Platinum-based chemotherapy and
anthracycline-based chemotherapy significantly exacerbate ERS in
tumor cells, upregulate GRP78 expression and simultaneously promote
its membrane translocation and exosomal secretion (67,68). This process forms a vicious cycle
of 'treatment-induced stress-GRP78
dysregulation-immunosuppression-infection susceptibility', which is
the core mechanism of postchemotherapy sepsis in patients with
hematologic malignancies and solid tumors. Paradoxically, while
chemotherapy aims to eliminate tumor cells, it inadvertently
amplifies GRP78-mediated immunosuppression, creating a transient
'window of vulnerability' where the host defense against pathogens
is severely compromised.
Targeted therapy: Sorafenib activates the
IRE1α-mediated unfolded protein response pathway, promoting GRP78
membrane translocation and extracellular release (53). The sGRP78 level measured in
patients after targeted therapy can be higher than that at
baseline, which is closely related to the increased infection risk
during treatment (53).
EGFR-TKIs may disrupt ER homeostasis and are associated with an
increased infection risk (69).
Hormone therapy: ADT may upregulate GRP78
expression; however, direct clinical evidence linking ADT-induced
GRP78 dysregulation to sepsis risk in prostate patients with cancer
is currently lacking (55,56).
From membrane surface exposure to pathogen
invasion
Pathogen infection represents the core pathogenic
foundation of sepsis development and progression and spans the
entire disease course (70).
Current research has progressively established that csGRP78 serves
as a critical alternative receptor or coreceptor for pathogen
invasion (71), providing a
hypothetical pathological mechanism that may underlie the
progression of infection to sepsis in patients with cancer, which
requires prospective clinical validation. In patients with cancer,
csGRP78 detected at sites of pathogen invasion may originate from
two distinct cellular compartments with different
pathophysiological implications: i) The first is tumor-derived
csGRP78. Malignant cells constitutively overexpress csGRP78 because
of chronic ER stress driven by hypoxia, nutrient deprivation and
oncogenic signaling (6,8). ii) Host-derived csGRP78 in
nonmalignant host cells, including mucosal epithelial cells,
vascular endothelial cells, alveolar epithelial cells and innate
immune cells (macrophages and dendritic cells), is obtained from
the de novo upregulation of csGRP78 expression in response
to infection-induced ER stress, hypoxia and inflammatory cytokines
(72). The relative
contributions of tumor-derived vs. host-derived csGRP78 to pathogen
invasion likely vary by pathogen tropism, the anatomical site of
infection and cancer type. Table
II summarizes the probable cellular sources of csGRP78 for each
major pathogen class discussed in the present review (73-98).
| Table IIProbable cellular sources of csGRP78
mediating pathogen invasion in cancer-associated sepsis. |
Table II
Probable cellular sources of csGRP78
mediating pathogen invasion in cancer-associated sepsis.
| Pathogen class | Pathogen | Primary target cell
type | Predominant csGRP78
Source | Evidence basis |
|---|
| RNA viruses | SARS-CoV-2 | Alveolar epithelial
cells, monocytes | Host-derived
(VeroE6-ACE2, THP-1) | Carlos et al
(73) used VeroE6-ACE2 cells for
mechanistic studies; Han et al (74) used THP-1 monocytes and primary
CD14+ monocytes for clinical translational validation |
| DENV | Hepatocytes | Cancer cell
line-derived (HepG2, Huh-7) | Alhoot et al
(75) and Jindadamrongwech et
al (76) used HepG2 cells;
Chumchanchira et al (77)
used Huh-7 cells. All cell lines are derived from hepatocellular
carcinoma |
| ZIKV | Lung epithelial
cells | Cancer cell
line-derived (A549) | Khongwichit et
al (78) explicitly used
A549 cells for all experiments, including infection,
co-immunoprecipitation, immunofluorescence, flow cytometry,
antibody inhibition and siRNA knockdown |
| JEV | Lung epithelial
cells, kidney cells | Cancer cell
line-derived (Neuro2a, 293T, Huh-7, BHK-21) | Nain et al
(79) used Neuro2a and Huh-7
cells; Wu et al (80)
confirmed that the ER stress response protein GRP78 is co-opted by
JEV |
| CVA9 | Kidney cells,
rhabdomyosarcoma cells, cervical carcinoma cells | Host-derived
(GMK) | Triantafilou et
al (81) primarily used GMK
cells for infection experiments |
| IAV | Lung epithelial
cells | Cancer cell
line-derived (A549) | Rashid et al
(82) and Feng et al
(83) both used A549 cells. No
evidence from primary respiratory epithelial cells or macrophages
is available |
| HIV-1 | T lymphocytes,
macrophages | Host-derived
(BSC-1) | Earl et al
(84) used BSC-1 cells for gp160
studies; Elshemey et al (85) provided computational evidence.
Direct binding of csGRP78 in primary cells has not been
confirmed |
| EBOV/ MARV | Endothelial cells,
monocytes, macrophages | Cancer cell
line-derived (primary human GBM cells, A549) | Booth et al
(86) used multiple tumor cell
lines for in vitro mechanistic studies, not primary
endothelial cells or monocytes |
| CHIKV | Epithelial cells,
fibroblasts, endothelial cells | Host-derived (THP-1
cells) | Gupta et al
(87) did not explicitly detect
direct GRP78 binding; evidence is limited to UPR pathway
modulation |
| VEEV | Neural cells,
endothelial cells, macrophages | Host-derived (Vero,
293T) | Barrera et
al (88) confirmed the
interaction between VEEV E2 protein and GRP78 via
co-immunoprecipitation and colocalization |
| MV | Human airway
epithelial cells | Cancer cell
line-derived (HEp-2) | Bolt et al
(89) confirmed that measles
virus infection upregulates GRP78 expression |
| DNA viruses | HBV | Hepatocytes | Cancer cell
line-derived (Huh-6, HepG2, HepAd38) | Suwanmanee et
al (90) suggested that
GRP78 interacts with the preS2 domain via its ATPase domain |
| Fungi | Rhizopus
spp./ Mucorales | Nasal epithelial
cells, vascular endothelial cells | Mixed (CCL-30,
A549) | Gebremariam et
al (91) suggested that
endothelial cell GRP78 binds to the fungal CotH3 protein; Alqarihi
et al (92) identified
the specific interaction between nasal epithelial cell GRP78 and
fungal CotH3 |
| P.
jirovecii | Alveolar epithelial
cells (type II pneumocytes) | Host-derived (rat
primary alveolar & airway epithelial cells) | Kottom et al
(93) suggested that GRP78
mediates organism attachment |
| Bacteria | M.
hyopneumoniae | Respiratory
epithelial cells | Host-derived (293T,
PTEC) | Pan et al
(94) suggested that Mhp271
interacts with host GRP78 |
| S.
pneumoniae | Alveolar
macrophages | Host-derived
(BMCs) | Cho et al
(95) did not directly detect
GRP78; the study focused on the NLRP3 inflammasome |
| P.
aeruginosa | Bronchial
epithelial cells | Host-derived
(16HBE) | van 't Wout et
al (96) used the 16HBE
human bronchial epithelial cell line. No direct GRP78 binding has
been confirmed |
| M.
tuberculosis | Macrophages | Host-derived
(BMDMs) | Lim et al
(97) suggested that
tuberculosis infection induces ER stress-mediated apoptosis. No
direct GRP78 binding has been confirmed |
| H.
pylori | Gastric epithelial
cells | Cancer cell
line-derived (AZ-521) | Akazawa et
al (98) suggested that VacA
toxin induces ER stress contributing to apoptosis. No direct GRP78
binding has been confirmed |
GRP78 as a critical cofactor for viral
invasion
Accumulating evidence from in vitro and in
vivo studies confirms that csGRP78 acts as a bona fide
functional receptor or critical coreceptor for a broad spectrum of
pathogenic viruses (73,74,79). It mediates viral invasion via two
core mechanisms: Acting as a primary receptor to directly drive
viral adhesion and endocytosis or as an attachment cofactor to
increase viral binding affinity to cognate receptors and modulate
post-invasion intracellular trafficking, thereby markedly
increasing infection efficiency (74). This pro-infective role has been
validated across multiple viral families (including
Flaviviridae, Coronaviridae, Orthomyxoviridae
and Enteroviridae), positioning GRP78 as a promising
broad-spectrum antiviral target (21).
For Flaviviridae members, including dengue
virus, Zika virus and Japanese encephalitis virus, GRP78 serves as
a key host factor via direct interactions with viral envelope
proteins and silencing/knockdown of GRP78 consistently inhibits
viral invasion, replication and progeny production (75-80). csGRP78 also acts as a critical
coreceptor for Coxsackievirus A9 (Enteroviridae), an
essential host factor for influenza A virus
(Orthomyxoviridae) and a key cooperative receptor for severe
acute respiratory syndrome coronavirus 2 (SARS-CoV-2,
Coronaviridae) (81-83). For SARS-CoV-2, in particular,
GRP78 directly binds the spike protein receptor-binding domain and
forms a functional complex with ACE2 to mediate viral entry, with
clinical data linking elevated sGRP78 levels to COVID-19 infection
susceptibility (21,73).
The receptor/coreceptor function of csGRP78 in
driving viral infection has also been confirmed for retroviruses,
filoviruses and other pathogenic viruses (84,86,89,99). Collectively, these findings
establish csGRP78 as a core host factor that mediates viral
invasion, which underpins the progression to severe disease and
sepsis secondary to viral infection (Table III) (73-90,99-104).
| Table IIICore roles and underlying mechanisms
of GRP78 in infection by diverse pathogenic viruses. |
Table III
Core roles and underlying mechanisms
of GRP78 in infection by diverse pathogenic viruses.
| Pathogen | Viral family | Primary role | Key mechanism | (Refs.) |
|---|
| SARS-CoV-2 |
Coronaviridae | Key auxiliary
factor for viral entry/ ACE2-independent entry receptor | csGRP78 binds spike
protein RBD; forms functional complex with ACE2 to mediate viral
entry | (73,74,99) |
| DENV |
Flaviviridae | Key host factor for
viral entry and replication | csGRP78 is core
component of viral receptor complex to mediate entry; infection
upregulates GRP78 to promote replication | (75-77) |
| ZIKV |
Flaviviridae | Essential host
factor for viral entry and replication | csGRP78 binds
envelope E protein to mediate entry; intracellular GRP78 stabilises
viral proteins and regulates replication/progeny production | (78,100,101) |
| JEV |
Flaviviridae | Key host factor for
viral entry and replication | csGRP78 binds
envelope protein domain III to mediate entry; intracellular GRP78
regulates viral RNA replication, protein synthesis and progeny
release | (79,80,102) |
| CVA9 |
Picornaviridae | Critical
co-receptor for viral entry | csGRP78 cooperates
with integrin αvβ3 for initial adhesion; forms complex with MHC-I
to mediate virus-receptor complex endocytosis | (81) |
| IAV |
Orthomyxoviridae | Essential host
factor for viral entry and replication | csGRP78 binds
hemagglutinin to mediate entry; intracellular GRP78 modulates
replication via ER stress pathway regulation | (82,83,103) |
| HIV-1 |
Retroviridae | Potential auxiliary
factor for viral entry/key regulator of viral envelope
maturation | csGRP78 may mediate
viral adhesion/entry; intracellular GRP78 acts as chaperone for
envelope protein folding/maturation | (84,85) |
| EBOV/ MARV |
Filoviridae | Essential host
factor for viral replication | Regulates viral
glycoprotein folding/maturation; modulates host viral receptor
expression to enhance infection/ replication | (86) |
| Alphavirus
spp. (CHIKV, VEEV) |
Togaviridae | Key host factor for
viral infection and replication | csGRP78 binds E2
protein to regulate entry; infection activates UPR to upregulate
GRP78, further promoting replication | (87,88) |
| MV |
Paramyxoviridae | Key chaperone for
viral glycoprotein maturation | Intracellular GRP78
acts as molecular chaperone for viral glycoprotein folding,
maturation and trafficking | (89) |
| HBV |
Hepadnaviridae | Auxiliary factor
for viral entry/ regulator of antigen secretion | csGRP78 facilitates
viral adhesion/entry; intracellular GRP78 regulates viral antigen
folding and secretion | (90,104) |
Roles of GRP78 in fungal adhesion and
invasion
csGRP78 serves as a critical receptor for host cell
invasion by certain opportunistic pathogenic fungi, with the most
comprehensive mechanistic studies focusing on Mucorales fungi,
which represent important pathogens that cause invasive fungal
infections that progress to sepsis in immunocompromised hosts.
Mucormycosis is a lethal invasive fungal infection
caused by Mucorales fungi such as Rhizopus, predominantly
affecting individuals with diabetic ketoacidosis (DKA),
hematological malignancies, or those undergoing chemotherapy or
immunosuppressive therapy, that results in immunocompromised states
(92). Mechanistic studies have
confirmed that under pathological conditions such as DKA, csGRP78
expression is significantly upregulated in host nasal epithelial
cells and vascular endothelial cells. csGRP78 directly and
specifically binds to the CotH3 protein on the outer wall of
Mucorales fungal spores, mediating spore adhesion to and invasion
of host epithelial cells and vascular endothelial cells, thereby
leading to vascular invasion and tissue necrosis. This process
represents the core mechanism underlying the specific infection of
immunocompromised hosts by Mucorales fungi (91,105).
Research on other common pathogenic fungi indicates
has not produced definitive evidence that currently confirms that
Candida albicans, Aspergillus fumigatus,
Cryptococcus neoformans, or Histoplasma capsulatum
directly utilize host csGRP78 as an invasion receptor. Only studies
on Pneumocystis jirovecii suggest that it may bind to GRP78
on the surface of lung epithelial cells, with csGRP78 potentially
serving as a putative receptor for host cell adhesion, although the
relevant mechanisms require further validation (93). A summary of the differential
roles of GRP78 in the invasion of various pathogenic fungi is
presented in Table IV (91-93,105-110).
| Table IVCore roles and underlying mechanisms
of GRP78 in pathogenic fungal infection. |
Table IV
Core roles and underlying mechanisms
of GRP78 in pathogenic fungal infection.
| Pathogen | Fungal
taxonomy | Functional
category | Primary role | Key mechanism | (Refs.) |
|---|
| Mucorales
fungi | Order
Mucorales | Host GRP78 core
function | Essential host
functional receptor for fungal invasion | Host csGRP78
directly binds to fungal CotH3 protein to mediate spore adhesion to
and invasion of host epithelial and vascular endothelial cells | (91,92,105) |
| Pneumocystis
jirovecii | Family
Pneumocystidaceae | Host GRP78 core
function | Potential host
receptor for fungal adhesion | Host csGRP78
mediates fungal adhesion to host lung epithelial cells | (93) |
| Candida
albicans | Family
Saccharomycetaceae | Other host
regulatory function | Host cytoprotective
factor against fungal-induced cell damage | Sustained
expression of host GRP78 attenuates fungal-induced epithelial cell
death | (106) |
| Aspergillus
fumigatus | Family
Aspergillaceae | Other host
regulatory function | Regulator of host
inflammatory and stress responses | Host GRP78
participates in the regulation of fungal-induced ER stress and
inflammatory signaling in host cells | (107,108) |
| Cryptococcus
neoformans | Family
Cryptococcaceae | Fungal-encoded
GRP78 homolog function | Essential molecule
for fungal pathogenicity | Fungal-encoded
Kar2/BiP (GRP78 ortholog) regulates the virulence of the
pathogen | (109) |
| Histoplasma
capsulatum | Family
Ajellomycetaceae | Host response
marker | Biomarker of host
stress response to fungal infection | Fungal infection
induces significant upregulation of host GRP78 expression | (110) |
Role of GRP78 in bacterial
infections
Compared with viruses and fungi, the current
evidence regarding the direct role of csGRP78 in mediating
bacterial adhesion and invasion remains relatively limited. The
existing research focuses primarily on bacterial infection-induced
ER stress and changes in GRP78 expression in host cells, with only
a few pathogens confirmed to directly interact with csGRP78 to
promote the infection process (94); meanwhile, the indirect regulatory
role of GRP78 in bacterial sepsis, which is equally important, has
been underemphasized.
The membrane protein Mhp271 of Mycoplasma
hyopneumoniae can directly and specifically bind to the NBD of
host cell csGRP78, promoting mycoplasmal adhesion to and invasion
of host cells, which represents an important mechanism in its
infection process (94).
Although Mycoplasma hyopneumoniae is a rare cause of severe
sepsis in humans, this interaction provides a proof-of-concept that
csGRP78 can function as a bacterial receptor. Research on other
common sepsis-causing pathogens has shown that infections with
Staphylococcus aureus, Escherichia coli,
Pseudomonas aeruginosa, Salmonella, Mycobacterium
tuberculosis, Helicobacter pylori and Streptococcus
pneumoniae can induce ER stress in host cells and upregulate
GRP78 expression (95-98,111-113). However, no definitive direct
evidence currently confirms that these bacteria utilize host
csGRP78 as an adhesion or invasion receptor.
Notably, the contribution of GRP78 to bacterial
sepsis is not dependent on its role as a direct receptor; instead,
its indirect regulatory effects on host inflammatory responses,
phagocytic function and barrier integrity constitute a temporally
ordered, functionally interdependent pathogenic sequence that
drives bacterial sepsis progression. It is hypothesized that GRP78
functions as a systems-level coordinator of the failure of the host
response through the following integrated sequence.
Phase I: Barrier disruption and bacterial
translocation. Bacterial infection-induced ER stress contributes to
the downregulation of tight junction proteins in intestinal
epithelial cells, disrupting barrier function and creating an
anatomical portal for bacterial translocation (111,112). Escherichia coli-derived
LPS aggravates ER stress and ER stress-mediated apoptosis in ER
stress-responsive IPEC-J2 cells; the crosstalk between nuclear
GRP78 and p53 triggers this LPS-induced increase in apoptosis
(112). Salmonella
infection activates the IRE1α-UPR signaling axis via curli
proteins, upregulating GRP78 expression (113). In patients with cancer,
chemotherapy and radiotherapy pre-compromise mucosal integrity,
while tumor-derived sGRP78 may further impair barrier repair by
suppressing local immune surveillance, increasing the width of the
anatomical portal and decreasing its defense.
Phase II: Innate immune dysregulation and delayed
clearance. As bacteria translocate, host macrophages upregulate
GRP78 in response to infection-induced ER stress. However, this
upregulation paradoxically impairs rather than enhances
antimicrobial function. Streptococcus pneumoniae infection
induces the significant upregulation of GRP78 expression in aged
lungs, which is associated with decreased NLRP3 inflammasome
activation and impaired bacterial clearance by senescent
macrophages (95). Additionally,
ER stress-mediated GRP78 overexpression can trigger macrophage
death through the PERK/ATF4/CHOP pathway, further reducing the
phagocytic capacity (114).
Phase III: Inflammatory amplification and tissue
injury. When the bacterial burden exceeds the blunted clearance
capacity of Phase II, sustained ER stress drives the dissociation
of GRP78 from UPR sensors, activating the NF-κB, MAPK and AP-1
signaling pathways and triggering excessive proinflammatory
cytokine secretion (18,115). In the context of persistent ER
stress, CHOP activation downstream of the PERK-eIF2α axis induces
apoptosis, exacerbating tissue damage and systemic inflammation
(114). Staphylococcus
aureus infection activates the GRP78-EIF2α-ATF4 pathway and ER
stress-autophagy axis, leading to host cell apoptosis (111). In patients with cancer,
chemotherapy-induced tumor cell death releases damage-associated
molecular patterns, which may synergize with bacterial
pathogen-associated molecular patterns to amplify the inflammatory
cytokine storm beyond the threshold observed in non-cancer
sepsis.
Phase IV: Endothelial dysfunction and multiorgan
failure. The sustained inflammatory milieu drives the translocation
of GRP78 to endothelial cell surfaces, where csGRP78-mediated
signaling disrupts vascular integrity. GRP78-induced
O-GlcNAcylation of VE-cadherin compromises endothelial junctions,
whereas the activation of the PERK/IRE1α pathway promotes NF-κB
binding to the iNOS promoter, driving excessive nitric oxide
production that induces vasodilation, hypotension and tissue
hypoperfusion (114,116,117). These vascular and barrier
effects culminate in the hemodynamic collapse and multiorgan
dysfunction characteristic of septic shock.
Fundamental mechanistic distinction: Thus, unlike in
viral and fungal infections, where csGRP78 functions as a canonical
adhesion or coreceptor, csGRP78 does not appear to serve as a
primary bacterial receptor for major pathogens causing sepsis in
humans. Instead, GRP78 modulates host inflammatory responses,
phagocytic function and epithelial barrier integrity through an
indirect, temporally ordered, four-phase mechanism that coordinates
the failure of the host response from the initial bacterial
encounter to terminal organ dysfunction. This systems-level role
represents a fundamental mechanistic distinction across pathogen
classes.
A summary of the roles of GRP78 in different
bacterial infections is presented in Table V (94-98,111-113,118-124), which highlights that the
indirect regulatory effects of GRP78 on host immunity and barrier
function are far more extensive than its direct role as a bacterial
receptor and that these indirect effects are crucial for the
progression of common bacterial infections to sepsis.
| Table VCore roles and underlying mechanisms
of GRP78 in bacterial and mycoplasma infection. |
Table V
Core roles and underlying mechanisms
of GRP78 in bacterial and mycoplasma infection.
| Pathogen | Bacterial
taxonomy | Functional
category | Primary role of
GRP78 | Key mechanism | (Refs.) |
|---|
| Mycoplasma
hyopneumoniae | Order
Mycoplasmatales, Family Mycoplasmataceae | Direct receptor for
pathogen adhesion and invasion | Key interacting
molecule mediating mycoplasma adhesion and invasion | Host csGRP78
directly binds to mycoplasmal Mhp271 protein via its NBD to
facilitate pathogen adhesion and host cell invasion | (94) |
| Clostridium
butyricum | Order
Eubacteriales, Family Clostridiaceae | Therapeutic target
and immune regulatory function | Regulatory target
of tumor immune microenvironment | Binds to host
csGRP78 to inhibit pro-inflammatory signaling and improve the
efficacy of tumor immunotherapy | (118) |
| Chlamydia
pneumoniae/ Chlamydia psittaci | Order
Chlamydiales, Family Chlamydiaceae | Host stress and
autophagy regulatory function | Regulator of host
autophagy and stress response | Pathogen infection
activates the ER stress pathway to upregulate GRP78 expression,
which further modulates host autophagy | (119,120) |
| Streptococcus
pneumoniae | Order
Lactobacillales, Family Streptococcaceae | Host immune
regulatory function | Regulator of
macrophage immune function | Modulates
inflammasome activation in senescent macrophages during
infection | (95) |
| Staphylococcus
aureus | Order
Bacillales, Family Staphylococcaceae | Host cell death and
inflammatory regulatory function | Regulator of host
cell death and inflammatory response | Activates the
GRP78-EIF2α-ATF4 pathway and ER stress-autophagy signaling axis;
mediates ferroptosis and apoptosis of host cells via downstream
CHOP activation, exacerbating tissue damage | (111,121) |
| Pseudomonas
aeruginosa | Order
Pseudomonadales, Family Pseudomonadaceae | Host stress
response regulatory function | Regulator of host
stress response | Pathogen infection
activates the host UPR pathway to upregulate GRP78 expression | (96) |
| Escherichia
coli | Order
Enterobacterales, Family Enterobacteriaceae | Host epithelial
apoptosis and inflammatory regulatory function | Regulator of
intestinal epithelial apoptosis and inflammation | Nuclear GRP78-p53
signaling axis mediates LPS-induced intestinal epithelial cell
apoptosis | (112,122) |
| Salmonella
spp. | Order
Enterobacterales, Family Enterobacteriaceae | Host stress
response regulatory function | Activator of host
UPR pathway | Bacterial curli
protein activates the IRE1α-UPR signaling axis to upregulate GRP78
expression | (113) |
| Mycobacterium
tuberculosis | Order
Mycobacteriales, Family Mycobacteriaceae | Host cell death and
autophagy regulatory function | Regulator of
macrophage apoptosis and autophagy | Modulates apoptosis
and autophagy of infected macrophages via ER stress signaling | (97,123) |
| Helicobacter
pylori | Order
Campylobacterales, Family Helicobacteraceae | Host cell apoptosis
and carcinogenesis regulatory function | Regulator of host
cell apoptosis and carcinogenesis | Modulates ER
stress-mediated cell apoptosis and intracellular ROS levels during
infection | (98,124) |
In summary, the existing research has fully
confirmed that csGRP78 serves as a critical receptor or coreceptor
for host cell invasion by multiple viruses and Mucorales fungi. Its
abnormal overexpression can directly increase host susceptibility
to these pathogens, providing the core pathological foundation for
the subsequent progression of infection to sepsis. Although direct
evidence that csGRP78 acts as a bacterial receptor is currently
limited, the role of GRP78 in the progression of bacterial sepsis,
the primary cause of clinical sepsis, through its indirect
regulation of inflammatory responses, phagocytic function and
barrier integrity is indispensable.
Immunoregulatory role of GRP78 in
sepsis
Based on preclinical evidence, in addition to its
proposed role in mediating pathogen invasion, the inflammatory and
immunoregulatory functions of GRP78 are hypothesized to be
critically governed by two determining factors: Cellular origin
(tumor-derived vs. host-derived) and the temporal phase of sepsis
(early systemic inflammatory response syndrome vs. late
immunosuppressive phase) (115,125). Specifically, tumor-derived
GRP78, which predominantly exists as sGRP78, establishes a baseline
immunosuppressive state that increases the vulnerability of
patients with cancer to infectious complications (115). By contrast, host-derived GRP78,
encompassing both csGRP78 and intracellular pools, exhibits
stage-specific bidirectional immunomodulatory effects: it amplifies
proinflammatory responses during early sepsis but subsequently
drives immune exhaustion and paralysis in the late
immunosuppressive phase (125).
These ontologically and temporally distinct mechanisms collectively
establish GRP78 dysregulation as a pivotal link between cancer
pathophysiology and sepsis progression. Table VI comprehensively summarizes the
regulatory effects of GRP78 on key inflammatory cytokines during
immune responses (116,126-142), with explicit stratification by
the cellular origin of GRP78 and the temporal phase of sepsis.
| Table VIRegulatory roles and core mechanisms
of GRP78 in inflammatory immune mediators. |
Table VI
Regulatory roles and core mechanisms
of GRP78 in inflammatory immune mediators.
| Major mediator
category | Inflammatory
mediator | GRP78 origin | Core role of
GRP78 | Key regulatory
mechanism | (Refs.) |
|---|
| Pro-inflammatory
cytokines | TNF-α | Host-derived
(DC) | Negative
regulation | sGRP78 inhibits the
TLR4 signaling pathway to suppress TNF-α production | (126) |
| IL-1β | Host-derived
(pSGC) | Bidirectional
regulation | Modulates the
maturation and activation of the NLRP3 inflammasome | (127) |
| IL-6 | Host-derived
(DC) | Negative
regulation | sGRP78 inhibits the
TLR4 signaling pathway to suppress IL-6 production | (126) |
| IFN-γ | Host-derived
(PBMCs) | Positive
regulation | IFN-γ levels
positively correlate with the frequency of GRP78-positive clones in
PBMCs | (128) |
| IL-12 | Host-derived
(PBMCs) | Negative
regulation | Drives M2
macrophage polarization and inhibits IL-12 secretion | (129) |
| IL-18 | Host-derived
(pSGC) | Positive
regulation | Regulates the NLRP3
inflammasome to promote IL-18 maturation and release | (127) |
| IL-17A | Host-derived
(HaCaT) | Negative
regulation | Exerts targeted
inhibition on the CCR6/IL-17A signaling axis | (130) |
| IL-2 | Host-derived (BM
cells) | Negative
regulation | Maintains immune
cell homeostasis and regulates compensatory IL-2 secretion | (131) |
| Anti-inflammatory
mediators | IL-10 | Host-derived
(PBMCs) | Negative
regulation | Promotes M2
macrophage polarization and upregulates IL-10 expression in
macrophages | (129) |
| TGF-β1 | Tumor-derived
(DLD1, HCT-116, SW480) | Positive
regulation | Promotes TGF-β1
expression and secretion and activates the Smad2/3 signaling
pathway | (132) |
| IL-4 | Host-derived
(PBMCs) | Positive
regulation | Forms a positive
feedback loop with M2 macrophage polarization | (129) |
| IL-13 | Host-derived
(BMDMs) | Negative
regulation | Inhibits IL-13
signaling transduction | (133) |
| Chemokines | IL-8/CXCL8 | Host-derived
(HPAECs) | Positive
regulation | Promotes IL-8
transcriptional expression via the ER stress pathway | (134) |
| MCP-1/CCL2 | Host-derived (human
monocytes) | Positive
regulation | CCL2 levels
positively correlate with the frequency of CD45+sGRP78+cells | (135) |
| MIP-1α/β | Host-derived
(BMDMs, AMs) | Positive
regulation | Promotes chemokine
expression via the NF-κB/IRE1 signaling pathway | (136,137) |
| Vasoactive and
endothelial-related mediators | NO | Tumor-derived (RAW
264.7) | Positive
regulation | Regulates iNOS
expression and NO synthesis via the ER stress pathway | (138) |
| ICAM-1/ VCAM-1 | Host-derived
(hAECs) | Positive
regulation | csGRP78 activates
the NF-κB pathway to upregulate adhesion molecules | (116) |
| Other key
inflammatory mediators | ROS | Host-derived (NRVM,
H9c2, rat renal cells) | Bidirectional
regulation of oxidative stress | Bidirectionally
modulates ER stress and antioxidant signaling pathways | (139-141) |
| MMPs | Tumor-derived
(SMMC7721, HepG2) | Positive
regulation | Promotes
MMP-2/MMP-9 transcriptional activation and regulates ECM
degradation | (142) |
Regulation of proinflammatory cytokines:
Origin- and stage-dependent bidirectional control
The regulation of proinflammatory cytokines by GRP78
is the core of its stage-dependent effects, with distinct roles for
tumor-derived and host-derived GRP78.
Tumor-derived sGRP78: Baseline
immunosuppression (pre-infection stage)
Tumor cells secrete large amounts of sGRP78 into the
systemic circulation, which predominantly exerts
anti-inflammatory/immunosuppressive effects to establish a baseline
state of immune hyporesponsiveness in patients with cancer
(62,115); this key factor increases the
susceptibility of these patients to bacterial/viral infection.
Specifically, sGRP78 binds to CD14 on macrophages/neutrophils,
promotes TLR4 endocytic degradation and inhibits TLR4 dimerization,
thereby suppressing LPS-induced TNF-α and IL-6 production (126). Qin et al (126) showed that 40 µg/ml
sGRP78 almost fully abolished TNF-α release in bone marrow-derived
dendritic cells, mimicking the effect of TLR4 knockout; this
phenomenon is particularly prominent in patients with cancer and
with high circulating sGRP78 levels, reducing their ability to
mount an effective early inflammatory response against invading
pathogens. This tumor-induced baseline immune suppression creates a
'predisposed' state: Even in the absence of traditional risk
factors such as neutropenia, patients with cancer with high sGRP78
levels are more likely to develop persistent infections that
progress to sepsis.
Host-derived GRP78: Stage-dependent
bidirectional regulation (post-infection stage)
Upon infection, host cells (immune cells, epithelial
cells and endothelial cells) upregulate GRP78 expression and induce
its subcellular redistribution, with effects varying significantly
between early and late sepsis.
Early sepsis: Intracellular GRP78 and csGRP78
(host-derived) predominantly exert proinflammatory effects to
eliminate pathogens. Intracellular GRP78 activates the PERK pathway
to promote TNF-α secretion, whereas csGRP78 activates the
NF-κB/MAPK pathway to upregulate IL-6 expression; these responses
are necessary for pathogen clearance, but their excessive
activation can amplify the inflammatory cytokine storm (143). For example, in a sepsis-related
acute kidney injury (AKI) model, LPS-induced ER stress upregulated
intracellular GRP78 and TNF-α levels and inhibiting the GRP78/PERK
axis reduced renal TNF-α levels and tissue injury (143). With respect to IL-1β,
intracellular GRP78 initially activates the NLRP3 inflammasome to
promote IL-1β maturation, enhancing the early inflammatory response
against pathogens (144).
However, when an infection persists and ER stress
remains unresolved, secondary remodeling of GRP78 expression and
subcellular localization occur in host cells (62,115). The functional of GRP78
subsequently shifts from 'driving effective inflammation to clear
pathogens' to 'excessively suppressing immune responses to mitigate
tissue damage,' ultimately precipitating the development of immune
paralysis (62,125).
Late sepsis: Sustained ER stress and excessive
sGRP78 accumulation lead to the functional exhaustion of GRP78,
shifting its role to anti-inflammatory/immunosuppressive effects.
In vitro studies using human coronary artery endothelial
cells demonstrate that chronic ER stress promotes GRP78 secretion
(145). The GRP78 released into
the conditioned medium subsequently attenuates ER stress and
endothelial inflammation (145).
GRP78 also orchestrates Th1/Th17-related cytokine
networks in a stage-dependent manner. In the pre-infection stage,
tumor-derived sGRP78 suppresses the expression of IL-12 (an M1
macrophage marker) and promotes M2 polarization, impairing Th1
anti-infective immunity and increasing the risk of sepsis (129). In early sepsis, host-derived
GRP78 is positively correlated with IFN-γ secretion, enhancing
Th1-mediated pathogen clearance (128); it also activates the p38 MAPK
pathway to upregulate IL-18, driving M1 microglial polarization and
exacerbating inflammatory injury (127). In late sepsis, sGRP78
negatively regulates IL-17 by targeting the CCR6/IL-17A axis in γδ
T cells, mitigating tissue damage but further weakening
anti-infective immunity (130).
Additionally, GRP78 indirectly regulates IL-2 by maintaining immune
homeostasis: hematopoietic cell-specific GRP78 knockout leads to
hematopoietic stem cell apoptosis, reduced numbers of lymphocytes
and a compensatory increase in IL-2 levels, disrupting sepsis
resolution (131).
Regulation of anti-inflammatory
mediators: Amplifying immune paralysis in late sepsis
The regulation of anti-inflammatory mediators by
GRP78 primarily serves to amplify immune suppression in late
sepsis, with consistent effects observed for tumor-derived and
host-derived GRP78.
IL-10: A key mediator of GRP78-induced
immune paralysis
GRP78 promotes IL-10 production in a cell
type-specific manner, predominantly by driving M2 macrophage
polarization, a core mechanism of immunosuppression in late sepsis.
In macrophages, both tumor-derived sGRP78 and host-derived csGRP78
upregulate IL-10 expression: GRP78 overexpression significantly
increases IL-10 release, while GRP78 knockdown markedly suppresses
it (129). This effect is
particularly pronounced in late sepsis, in which sustained GRP78
activation drives extensive M2 polarization, further inhibiting
proinflammatory responses and impairing pathogen clearance. By
contrast, GRP78-induced tolerogenic dendritic cells exhibit an
immature phenotype but show no significant alteration in IL-10
production (146); this
exception reflects the cell type specificity of the
anti-inflammatory effects of GRP78.
TGF-β and IL-4/IL-13: Synergistic
amplification of immunosuppression
The role of GRP78 in regulating TGF-β levels in
patients with sepsis remains unclear, but existing oncological
research provides indirect evidence that GRP78 overexpression in
colon cancer cells promotes TGF-β1 secretion and activates the
Smad2/3 pathway to regulate tissue remodeling (132). In late sepsis, this pathway may
contribute to multiorgan fibrotic injury, although direct
validation in sepsis models is needed.
With respect to the IL-4/IL-13 axis (key inducers of
M2 polarization), GRP78 forms a positive feedback loop with these
cytokines to amplify immune suppression: IL-4/IL-13 upregulates
GRP78 expression in macrophages (129,147) and GRP78 further promotes M2
polarization via the JAK/STAT pathway, increasing anti-inflammatory
signaling (129). Notably,
serum IL-13 levels are elevated fourfold in macrophage-specific
GRP78-deficient mice, suggesting a negative feedback mechanism by
which GRP78 regulates IL-13 secretion (133). This feedback mechanism may
prevent excessive immunosuppression in early sepsis but fails in
late sepsis because of sustained GRP78 overactivation. The current
evidence is primarily derived from tumor-associated macrophage
models and the precise role of this axis in sepsis requires further
validation (129).
Regulation of chemokines: Stage-dependent
modulation of inflammatory cell infiltration
The regulation of chemokines (IL-8, MCP-1 and
MIP-1α/β) by GRP78 is closely linked to sepsis-induced tissue
inflammation, with the effects varying by the sepsis stage.
Early sepsis: Intracellular GRP78 (host-derived)
acts as a positive regulator of IL-8 and MCP-1 levels, promoting
endothelial inflammatory responses and leukocyte infiltration to
facilitate pathogen clearance. In an LPS-induced acute lung injury
model, the cleavage of intracellular GRP78 suppressed IL-8
expression and pulmonary inflammation (134) and Apelin-36 reduced LPS-induced
MCP-1 release by inhibiting the GRP78/ASK1/JNK pathway (148), confirming the proinflammatory
role of GRP78 in early sepsis.
Late sepsis: Excessive sGRP78 (tumor and
host-derived) inhibits chemokine production, reducing leukocyte
recruitment to infection sites and impairing pathogen clearance.
For example, the GRP78-specific monoclonal antibody N88 reduces
CCL3/CCL4 (MIP-1α/β homologues) expression in LPS-stimulated
macrophages (136), whereas the
inhibition of the IRE1/XBP-1 pathway (downstream of GRP78) reduces
MIP-1α/β production and alleviated acute lung injury (137); these effects reflect the shift
of GRP78 function towards immunosuppression in late sepsis.
This stage-specific switch in chemokine regulation
by GRP78 underscores its central role in balancing inflammatory
cell infiltration: promoting pathogen clearance in early sepsis but
contributing to immune evasion in late sepsis.
Regulation of vasoactive and endothelial
function-related inflammatory mediators
GRP78 modulates vasoactive mediators and endothelial
inflammation in a stage-dependent manner, contributing to sepsis
progression.
Early sepsis: Host-derived csGRP78 and intracellular
GRP78 drive vascular dysfunction and inflammatory amplification.
Intracellular GRP78 activates the PERK/IRE1α pathway, promoting
NF-κB binding to the iNOS promoter and excessive NO production to
induce vasodilation, hypotension and tissue hypoperfusion (138). In endothelial cells, csGRP78
binds anti-GRP78 autoantibodies, activating NF-κB to upregulate
ICAM-1/VCAM-1 and promoting leukocyte adhesion and transendothelial
migration (116). These effects
are necessary for local inflammation but, when they are excessive,
contribute to systemic endothelial dysfunction.
Late sepsis: Sustained GRP78 activation exacerbates
endothelial barrier disruption and multiorgan dysfunction.
Excessive NO production (mediated by GRP78/iNOS) leads to
irreversible vascular hyporesponsiveness, whereas persistent
endothelial activation (via csGRP78) disrupts barrier integrity,
which are key features of septic shock (70,149).
In patients with cancer, tumor-derived sGRP78
further exacerbates vascular dysfunction by disrupting endothelial
barrier integrity, increasing the likelihood of developing septic
shock compared with patients with non-cancer sepsis. Specifically,
sGRP78 promotes angiogenesis and endothelial cell proliferation
under physiological conditions (150,151); however, during sustained ER
stress in individuals with sepsis, the cell surface translocation
of GRP78 induces the O-GlcNAcylation of VE-cadherin and disrupts
endothelial junctions, leading to increased vascular permeability
and barrier dysfunction (117).
This paradoxical shift from proangiogenic to barrier-disruptive
functions contributes to the hemodynamic instability characteristic
of septic shock in patients with cancer.
Regulation of other key inflammatory
mediators
The regulation of ROS and MMPs by GRP78 also
follows a stage-dependent pattern.
ROS regulation: In early sepsis, intracellular
GRP78 (host-derived) maintains redox homeostasis by suppressing ER
stress and increasing the activity of Nrf2/HO-1 antioxidant axis
(139,140), mitigating oxidative damage. In
late sepsis, persistent ER stress induces GRP78 dysfunction,
disrupting oxidative-antioxidant equilibrium and exacerbating
cellular injury and decreased GRP78 expression is correlated with
the exacerbation of oxidative stress in individuals with
sepsis-induced AKI (141).
MMP regulation: In early sepsis, GRP78 upregulates
MMP-2/9/14 expression via the FAK-Src-JNK-c-Jun pathway, promoting
extracellular matrix degradation and leukocyte infiltration
(142). In late sepsis,
excessive MMP activation (mediated by GRP78) contributes to tissue
remodeling and multiorgan fibrosis; PI3K-γ inhibition attenuates
GRP78-related ER stress and MMP-9 expression, improving pulmonary
pathology in individuals with sepsis-associated acute lung injury
(152).
Therapeutic approaches for targeting
GRP78
GRP78-targeted inhibitors can be divided into seven
categories based on their structural properties and mechanisms of
action (Table VII) (88,153-181): i) Natural products, ii)
synthetic products, iii) modified bacterial toxins, iv) metal-based
drugs, v) monoclonal antibodies, vi) peptide agents and vii)
nucleic acid drugs. Accumulating evidence has indicated that the
expression and function of GRP78 can be regulated at multiple
levels, including transcriptional regulation, post-transcriptional
regulation, translational regulation and post-translational
regulation, providing multiple actionable targets for the
development of GRP78-targeted interventions.
| Table VIIRepresentative GRP78-targeted agents:
Mechanisms and dual therapeutic values in cancer and sepsis
intervention. |
Table VII
Representative GRP78-targeted agents:
Mechanisms and dual therapeutic values in cancer and sepsis
intervention.
| Drug category | Representative
compounds | Precise mechanism
of action | (Refs.) |
|---|
| Synthetic small
molecules | HA15, CXL146 | Target GRP78 ATPase
domain; inhibit chaperone activity | (88,153,154) |
| AR12
(OSU-03012) | Activate PERK/ATF4
pathway; downregulate GRP78 at transcriptional level | (155) |
| Rosuvastatin,
Simvastatin | Inhibit GRP78
expression at transcriptional level | (156) |
| Sevoflurane | Suppress
GRP78-mediated ER stress | (157) |
| Ribociclib,
Piperine | Negatively regulate
GRP78 transcription | (158) |
| Monoclonal
antibodies | SAM-6 | Specifically bind
to O-glycosylated epitope of tumor-specific csGRP78 | (159) |
| N88 | Target GRP78
NBD | (160) |
| C38, C107,
anti-CDT | Target GRP78
C-terminal SBD | (161,162) |
| Peptide agents | Pep42 | Specifically bind
to csGRP78; mediate targeted endocytosis for drug delivery | (163) |
| M4 peptide | Target ER-localized
GRP78 via C/F helices | (164) |
| Bag-1 peptide,
GIRLRG peptide, BC71 | Target GRP78
ATPase/SBD domain; inhibit chaperone activity | (165-167) |
| Kringle 5 | Inhibit pERK1/2
signaling; downregulate GRP78 expression | (168) |
| Natural
products | EGCG, honokiol | High-affinity
binding to GRP78 ATPase domain; inhibit chaperone activity | (169) |
| Genistein | Antagonize NF-Y/CBF
binding; repress GRP78 transcription | (170) |
| Triptolide | Induce chronic ERS;
downregulate GRP78 expression | (171) |
| Plumbagin, DATS,
Isoliquiritigenin | Inhibit GRP78
activity or downregulate its expression | (172-174) |
| Metal-based
drugs | NKP-1339
(IT-139) | Inhibit
HSPA5 promoter activity; downregulate GRP78 (Phase II
clinical trial) | (175) |
| Ruthenium (II)
complex 1 | Induce ROS
production; trigger GRP78 conformational changes | (176) |
| Modified bacterial
toxins | SubAB5 | Site-specific
cleavage of ER-localized GRP78; abrogate its chaperone
function | (177) |
| VCD, Prunustatin A,
JBIR-04, JBIR-05 | Block GRP78
transcription | (178-180) |
| Nucleic acid
drugs | miR-181a | Directly target
HSPA5 3′-UTR; inhibit GRP78 translation | (181) |
However, a critical 'double-edged sword' dilemma
exists when these strategies are applied to cancer-associated
sepsis. First, systemic GRP78 inhibition disrupts ER proteostasis
in host immune and epithelial cells. GRP78 is essential for
maintaining the unfolded protein response in neutrophils,
macrophages and intestinal epithelial cells under infectious
stress; global suppression of this activity impairs cell viability
and exacerbates barrier dysfunction (131,182). Second, nonselective inhibition
abolishes the stage-dependent protective role of host-derived
GRP78. In early sepsis, intracellular GRP78 attenuates
CHOP-mediated apoptosis and preserves the phagocytic capacity,
whereas in late sepsis, persistent GRP78 activation paradoxically
limits NLRP3 overactivation to prevent excessive tissue injury
(114,125,127). Third, in vivo studies of
sepsis have not been conducted in tumor-bearing animals. The
existing preclinical efficacy data are derived exclusively from
tumor xenograft models without concurrent infection, suggesting
that the net effect of GRP78 inhibition on sepsis outcomes in
patients with cancer is entirely hypothetical (88,153-181). Thus, the feasibility of
GRP78-targeted therapy hinges on resolving two core questions:
'When to intervene?' and 'how to deliver the drug precisely?'.
Transcriptional and post-transcriptional
regulation of GRP78
The transcriptional activation of the
GRP78-encoding gene HSPA5 is primarily mediated by the ERSE in its
promoter region, where ERSE-binding transcription factors such as
NF-Y, YY1, general TFII-I and ATF6 play key roles (183), with most transcriptional
regulators acting by interfering with the binding of these factors
to the HSPA5 promoter. For instance, genistein represses GRP78
transcription by antagonizing NF-Y/CBF binding to ERSE (170). In addition to this canonical
ERSE-dependent pathway, ERSE-independent regulatory axes also serve
as potential targets for GRP78 transcriptional intervention. Under
pathological ER stress, ATF4 activates the HSPA5 promoter by
forming a complex with ATF1 and CREB1, but in bortezomib-resistant
osteosarcoma cells, ATF4 and its activators can repress GRP78
transcription via alternative mechanisms, reflecting
context-dependent bidirectional regulation (158) and agents such as AR12, Kringle
5 (K5), piperine and ribociclib target this axis through distinct
pathways (155,158,168).
With respect to post-transcriptional regulation,
nucleic acid drugs are the most well-developed targeting agents and
an in vitro study has confirmed that miR-181a directly
targets the 3′-untranslated region (3′-UTR) of the GRP78
mRNA to inhibit GRP78 translation (181). Notably, transcriptional and
post-transcriptional inhibitors non-selectively suppress GRP78
expression in both tumor cells and host immune/epithelial cells,
posing significant feasibility risks in treating sepsis. For
example, GRP78 is essential for maintaining cellular homeostasis in
normal tissues; a global reduction in the level of GRP78 of 50% in
whole body or specific tissues potently suppresses tumorigenesis,
but requires a careful evaluation of potential side effects on
normal organs (182). In the
context of sepsis, where host cells rely on GRP78 to manage ER
stress and maintain barrier function, non-selective inhibition may
exacerbate tissue damage and impair immune cell viability (115). This finding is particularly
concerning given that GRP78 levels are correlated with disease
severity in sepsis patients and that both insufficient and
excessive ER stress responses can be detrimental (6). Thus, these non-selective strategies
are not suitable for direct application in patients with cancer
with active sepsis unless they are combined with tumor-specific
delivery systems.
Translational and post-translational
regulation of GRP78
The biological function of GRP78 can be modulated
at the translational and post-translational levels primarily
through direct targeting of the GRP78 protein, including blocking
its functional domains, inducing conformational changes, promoting
protein cleavage or degradation and inhibiting its subcellular
translocation. csGRP78 represents the most feasible target for
balancing antitumor and anti-infective effects, as it is
selectively overexpressed on the membranes of malignant and
stressed epithelial cells but minimally expressed on normal
quiescent cells (17,115). This selectivity arises because
normal cells retain GRP78 within the ER through KDEL-mediated
retrieval, whereas cancer cells and virally infected cells display
significantly higher levels of csGRP78 because of constitutive ER
stress and impaired retrograde transport (17,115). This selectivity allows the
targeted inhibition of tumor-associated csGRP78 without
significantly impairing host GRP78 function.
Monoclonal antibodies and peptides serve as the
primary targeting modalities, with monoclonal antibodies, including
SAM-6, C38, C107, anti-CDT and N88, effectively blocking csGRP78
function (159-162). Importantly, these antibodies
predominantly target the tumor cell marker csGRP78, inhibiting
tumor progression while avoiding the direct suppression of host
immune cells to reduce the risk of exacerbating sepsis-related
immunosuppression. For example, the GRP78-neutralizing human
monoclonal antibody hMAb159 specifically blocks csGRP78-mediated
pathogen invasion without affecting the chaperone function of
intracellular GRP78 in host cells (73), making it a promising candidate
for treating cancer-associated sepsis.
While monoclonal antibodies represent a
well-established class of csGRP78-targeting agents, peptide-based
therapeutics have also been widely developed for this purpose.
Pep42, a 13-amino acid cyclic peptide, specifically binds csGRP78
on tumor cells and mediates its lysosomal internalization, serving
as a tumor-targeted drug delivery carrier (163,184), whereas other peptides exert
antitumor effects by targeting different csGRP78 or intracellular
GRP78 domains (164-167); however, the effects of
intracellular GRP78 inhibitors require careful consideration in
treating sepsis: Inhibiting intracellular GRP78 in host cells may
disrupt ER homeostasis and exacerbate inflammatory dysregulation,
particularly in early sepsis, in which host-derived intracellular
GRP78 is critical for controlling the inflammatory cytokine
storm.
There are two major classes of inhibitors that
target intracellular GRP78 that have been developed: Natural
products and synthetic small molecules. Natural products and
synthetic small molecules are the main inhibitors targeting
intracellular GRP78 domains, with honokiol showing promise as a
treatment for intracranial diseases because of its ability to cross
the blood-brain barrier (153,154,156,157,169,171-174). In addition to inhibitors
targeting specific domains, two alternative strategies have been
explored to disrupt GRP78 function: inducing protein cleavage and
triggering conformational changes. Modified bacterial toxins (such
as SubAB) induce site-specific GRP78 cleavage to inactivate it
(177), whereas metal-based
drugs modulate GRP78 via conformational changes or promoter
inhibition, with NKP-1339 entering clinical trials as a treatment
for solid tumors (175,176). However, these intracellular
inhibitors lack tumor selectivity and may impair host cell function
in individuals with sepsis; their clinical application requires
tumor-specific delivery systems to reduce off-target toxicity.
Balancing antitumor and anti-infective
effects: Timing and precision delivery in patients with
cancer-associated sepsis
GRP78 is proposed as a pivotal molecular nexus that
may connect tumor progression with increased susceptibility to
sepsis in patients with cancer. Targeting GRP78 presents a
distinctive dual therapeutic potential within this population, but
its clinical feasibility depends on addressing the 'double-edged
sword' effect through stage-specific intervention and
origin-specific delivery.
Stage-specific intervention: When to
target GRP78
The role of GRP78 in sepsis has evolved dynamically
and tailored intervention strategies based on the sepsis stage are
needed. For instance, prophylactic administration of a
csGRP78-neutralizing antibody such as hMAb159 could be evaluated in
high-risk patients with cancer prior to chemotherapy-induced
neutropenia, with breakthrough infection incidence and dynamic
sGRP78 kinetics as primary endpoints, although no such trial has
yet been conducted or registered.
Pre-infection stage: Tumor-derived sGRP78 is the
primary driver of baseline immunosuppression (62). Targeting tumor-derived csGRP78 or
sGRP78 can reduce immunosuppression and the sepsis risk without
affecting host GRP78 function; this is the most feasible window for
a GRP78-targeted intervention (62). For example, pretreatment with
SAM-6 in high-risk patients with cancer has the potential to reduce
sGRP78 levels and enhance anti-infective immunity (159).
Early sepsis: Host-derived GRP78 plays a critical
role in maintaining cellular homeostasis and ER stress responses
during infection (17). In the
context of sepsis, GRP78 expression is significantly increased in
host immune and epithelial cells, serving as a protective mechanism
to manage ER stress and prevent cell death (13). While the proinflammatory role of
host GRP78 in pathogen clearance is complex and context dependent,
its cytoprotective function in managing ER stress and preventing
apoptosis is well established (115). Non-selective inhibition of
GRP78 at this stage could exacerbate tissue damage by disrupting ER
homeostasis and promoting apoptosis via CHOP activation (185). Thus, intervention strategies
should prioritize the selective targeting of tumor-derived GRP78
while preserving host GRP78 function to maintain cellular viability
during infection (62).
Late sepsis: The sustained increase in sGRP78
levels contributes significantly to systemic immunosuppression
(125). Therapeutic strategies
targeting circulating sGRP78, such as neutralization with
monoclonal antibodies or blockade of its interactions with immune
receptors, represent promising approaches to reverse
immunosuppression and restore the pathogen clearance capacity while
avoiding the risk of exacerbating the initial inflammatory storm
(62). However, careful
consideration is needed, as the excessive inhibition of
host-derived GRP78 may compromise cellular homeostasis and lead to
tissue damage (186).
Therefore, achieving an optimal therapeutic balance between
alleviating immunosuppression and preserving essential host cell
functions remains critical for an effective clinical
intervention.
Origin-specific precision delivery:
How to target GRP78
Precision delivery strategies are essential to
selectively target tumor-derived GRP78 while protecting
host-derived GRP78, thus resolving the 'double-edged sword'
dilemma.
Tumor-specific targeting carriers: Peptides,
antibody-drug conjugates and tumor-specific nanoparticles can
deliver GRP78 inhibitors exclusively to tumor cells. For example, a
Pep42-conjugated GRP78 siRNA specifically silences GRP78 in tumor
cells without affecting host immune cells, achieving antitumor
effects while preserving host anti-infective immunity (163,184).
csGRP78-selective inhibitors: Monoclonal antibodies
(such as hMAb159 and SAM-6) that specifically bind to csGRP78 on
tumor cells can block tumor progression and pathogen invasion
without interfering with the chaperone function of intracellular
GRP78 in host cells, making them ideal treatments for
cancer-associated sepsis (73,159).
Context-dependent combination therapy: While GRP78
inhibition shows promise in enhancing pathogen clearance in viral
infections (187), its
combination with antibiotics in treating bacterial sepsis requires
further investigation. The inhibition of ER stress combined with
conventional sepsis treatments may improve outcomes by modulating
immune homeostasis (188).
Proposed priorities for the clinical
development of GRP78-targeted therapy for cancer-associated
sepsis
The aforementioned mechanistic analysis and
stage-origin framework (Balancing antitumor and anti-infective
effects: Timing and precision delivery in patients with
cancer-associated sepsis) can be translated into a concrete,
prioritized clinical development roadmap. Three criteria guide the
ranking: i) Tumor selectivity (agents must spare host GRP78 to
avoid exacerbating the infection); (ii) mechanistic clarity (agents
with validated target engagement in human systems are preferred
over preclinical leads) and (iii) regulatory feasibility (agents
with an existing IND/IMPD status or prior oncology phase I data are
prioritized for repurposing as sepsis treatments if their safety
profile permits acute administration).
Pre-infection prophylaxis (highest
priority)
The pre-infection window offers the most favorable
risk-benefit ratio because tumor-derived csGRP78/sGRP78 can be
suppressed without compromising host ER stress responses. The
humanized monoclonal antibody hMAb159 is the lead candidate: It
depletes cell surface GRP78 and reduces ACE2 expression in
epithelial cells, blocking SARS-CoV-2 entry in vitro
(73). However, the following
critical caveats apply: hMAb159 has not been tested in
tumor-bearing models and its epithelial cell target profile may not
translate to tumor-specific csGRP78 blockade. SAM-6 is a secondary
candidate; this fully human IgM antibody targets an O-glycosylated
tumor-specific variant of GRP78 with a molecular weight of 82 kDa,
an epitope specific for malignant cells (159). Its fully human backbone reduces
the immunogenic risk, but the IgM isotype may limit tissue
penetration and pharmacokinetics.
Early sepsis (moderate priority,
restricted indication)
Early sepsis is the most dangerous window for
intervention because host-derived GRP78 is cytoprotective.
Tumor-specific delivery systems may facilitate the selective
suppression of treatment-induced amplification of GRP78 expression
in the tumor. Pep42-drug conjugates represent a proof-of-concept:
The cyclic 13-mer peptide specifically binds tumor csGRP78,
undergoes clathrin-mediated endocytosis and traffics to lysosomes
for drug release (163). In
addition to the use of cytotoxic payloads, the Pep42 platform could
theoretically be extended to nucleic acid delivery; however, while
Pep42 has demonstrated tumor-specific delivery of cytotoxic drugs
(doxorubicin and paclitaxel) in xenograft models, siRNA conjugation
has not been reported to date (184). A phase I/II trial would require
biopsy confirmation of csGRP78 expression prior to enrolment, with
real-time pharmacodynamic monitoring of circulating sGRP78.
Late sepsis (moderate priority,
immunomodulatory focus: Mechanism re-evaluation required)
In late sepsis, sustained sGRP78 accumulation
drives immune paralysis. The N88 monoclonal antibody targets the
NH2-terminal domain of GRP78 and inhibits NFκB
activation and proinflammatory cytokine expression in
LPS-stimulated bone marrow-derived macrophages (136,160). However, the anti-inflammatory
activity of N88 presents a paradox for sepsis therapy: While late
sepsis is characterized by immunosuppression, N88 further dampens
innate immunity via the LRP1/NMDA-R pathway (136).
Agents deprioritized for sepsis
indications
Transcriptional inhibitors (genistein, ribociclib
and AR12) and global translational inhibitors (miR-181a) are
deprioritized due to the risk of nonselective suppression of host
GRP78 activity (86,158,170). Inhibitors targeting the
intracellular domain (HA15, CXL146 and honokiol) are deprioritized
for systemic administration but retained for CNS-specific delivery
given their blood-brain barrier penetrance (88,154,169).
Current limitations and future
directions
Three fundamental constraints limit the conclusions
of the present review and the field at large. First, direct
clinical evidence is lacking. No study has investigated the link
between GRP78 dysregulation and sepsis susceptibility in patients
with cancer. Published clinical data on the role of GRP78 in
oncology populations address exclusively tumor progression and
cancer-specific survival, not infectious outcomes. Furthermore,
while sGRP78 expression is related to a poor prognosis in patients
with non-cancer sepsis, these findings cannot be extrapolated to
patients with cancer-associated sepsis, in which tumor-derived
sGRP78 expression creates a unique pathological microenvironment.
Second, tumor-bearing sepsis models do not exist. All preclinical
efficacy data for GRP78-targeted agents are derived either from
tumor xenograft models or from non-tumor sepsis models (such as
LPS-induced lung injury and cecal ligation and puncture models)
evaluated in isolation. No study has combined an analysis of the
tumor burden with polymicrobial sepsis. Consequently, whether GRP78
inhibition prolongs or shortens survival in this context remains
unknown. The proposed 'double-edged sword', simultaneous tumor
suppression and impaired host defenses, cannot be rigorously
assessed without such models. Third, a biomarker threshold has not
been defined. The absence of an established sGRP78 cut-off value
for sepsis risk stratification or therapeutic monitoring further
hinders clinical translation.
Future research should prioritize addressing this
gap by launching large-scale, multicenter prospective cohort
studies in high-risk patients with cancer to clarify the
correlation between baseline sGRP78 levels, dynamic changes during
infection and sepsis outcomes, along with nested case-control
studies to explore causal relationships. Complementing this
approach, efforts should focus on developing tumor-specific
delivery systems, establishing stage-specific intervention
protocols and validating efficacy in tumor-bearing preclinical
sepsis models. In summary, the translation of GRP78 into a viable
diagnostic biomarker and therapeutic target for cancer-associated
sepsis hinges on filling the critical gap in research on sGRP78 and
sepsis outcomes in cancer-specific clinical cohorts.
Availability of data and materials
Not applicable.
Authors' contributions
HR was responsible for literature collection and
the initial draft of the manuscript. MPZ, LJZ and SYL contributed
to the analysis and interpretation of experimental data related to
vascular smooth muscle cell function in sepsis; they were
additionally responsible for image rendering, table processing and
the scientific validation of graphical data presentation and
critically revised the manuscript for content accuracy. SSL
contributed to revising the manuscript critically for intellectual
content and approved the final version for publication. Data
authentication is not applicable. All authors reviewed the
manuscript critically for intellectual content and read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
GRP78
|
glucose-regulated protein 78
|
|
HSPA5
|
GRP78-encoding gene
|
|
UPR
|
unfolded protein response
|
|
ER
|
endoplasmic reticulum
|
|
BiP
|
immunoglobulin heavy-chain binding
protein
|
Acknowledgments
The authors acknowledge the use of Figdraw 2.0
(http://www.figdraw.com) for generating the
figures presented in this work.
Funding
The present study was supported by the China Postdoctoral
Science Foundation (grant no. 2025M772200).
References
|
1
|
Hensley MK, Donnelly JP, Carlton EF and
Prescott HC: Epidemiology and outcomes of cancer-related versus
non-cancer-related sepsis hospitalizations. Crit Care Med.
47:1310–1316. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Husby M, Frydenberg H, Myklebust TA, Skei
NV, Solligård E, Thune I, Gustad LT and Furberg AS: Cancer patients
with sepsis: Prognostic insights from a population-based cohort
study in norway. Cancer Epidemiol Biomarkers Prev. 35:264–275.
2026. View Article : Google Scholar :
|
|
3
|
MacPhail A, Dendle C, Slavin M, Weinkove
R, Bailey M, Pilcher D and McQuilten Z: Neutropenic sepsis in the
intensive care unit: Differences in clinical profile and outcomes
according to the cause of neutropenia. Open Forum Infect Dis.
11:ofae2892024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nates JL, Pène F, Darmon M, Mokart D,
Castro P, David S, Povoa P, Russell L, Nielsen ND, Gorecki GP, et
al: Septic shock in the immunocompromised cancer patient: A
narrative review. Crit Care. 28:2852024. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tian W, Liu Y, Cao C, Zeng Y, Pan Y, Liu
X, Peng Y and Wu F: Chronic stress: Impacts on tumor
microenvironment and implications for anti-cancer treatments. Front
Cell Dev Biol. 9:7770182021. View Article : Google Scholar :
|
|
6
|
Amaresan R and Gopal U: Cell surface
GRP78: A potential mechanism of therapeutic resistant tumors.
Cancer Cell Int. 23:1002023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
UniProt Consortium: UniProt: The universal
protein knowledge-base in 2021. Nucleic Acids Res. 49(D1):
D480–D489. 2021. View Article : Google Scholar
|
|
8
|
Peng C, Wang J, Wang S, Zhao Y, Wang H,
Wang Y, Ma Y and Yang J: Endoplasmic reticulum stress: Triggers
microenvironmental regulation and drives tumor evolution. Cancer
Med. 14:e706842025. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Guo W, Wang M, Yang Z, Liu D, Ma B, Zhao
Y, Chen Y and Hu Y: Recent advances in small molecule and peptide
inhibitors of glucose-regulated protein 78 for cancer therapy. Eur
J Med Chem. 261:1157922023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shin J, Toyoda S, Fukuhara A and Shimomura
I: GRP78, a novel host factor for SARS-CoV-2: The emerging roles in
COVID-19 related to metabolic risk factors. Biomedicines.
10:19952022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Han X, Du J, Li W, Yang S, Sun H, Wang G
and Cong H: Zika virus and host protein interactions for
understanding molecular mechanisms of pathogenesis and therapeutic
development. Front Cell Infect Microbiol. 16:17544652026.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Doerflinger M, Reljic B, Menassa J, Nedeva
C, Jose I, Faou P, Mackiewicz L, Mansell A, Pellegrini M, Hotchkiss
R and Puthalakath H: Circulating BiP/Grp78 is a novel prognostic
marker for sepsis-mediated immune cell death. FEBS J.
288:1809–1821. 2021. View Article : Google Scholar
|
|
13
|
Li F, Lin Q, Shen L, Zhang Z, Wang P,
Zhang S, Xing Q, Xia Z, Zhao Z, Zhang Y and Zhu B: The diagnostic
value of endoplasmic reticulum stress-related specific proteins
GRP78 and CHOP in patients with sepsis: A diagnostic cohort study.
Ann Transl Med. 10:4702022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang F, Li X, Zhao N, Duan L and Chen Y:
Circulating GRP78 acts as a biomarker in the early diagnosis of
lung cancer. Int J Clin Exp Pathol. 11:5223–5231. 2018.PubMed/NCBI
|
|
15
|
Chen L, Zheng H, Yu X, Liu L, Li H, Zhu H,
Zhang Z, Lei P and Shen G: Tumor-Secreted GRP78 promotes the
establishment of a pre-metastatic niche in the liver
microenvironment. Front Immunol. 11:5844582020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cela I, Dufrusine B, Rossi C, Luini A, De
Laurenzi V, Federici L and Sallese M: KDEL receptors:
Pathophysiological functions, therapeutic options, and
biotechnological opportunities. Biomedicines. 10:12342022.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee AS: Stress-induced translocation of
the endoplasmic reticulum chaperone GRP78/BiP and its impact on
human disease and therapy. Proc Natl Acad Sci USA.
122:e24122461222025. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Luo Q, Gao X, Meng P, Qi X, Wang W, Li S
and Duan L: Endoplasmic reticulum stress in non-small cell lung
cancer: A review of therapeutic agents, mechanistic insights, and
implications for therapy. Front Cell Dev Biol. 13:16930232025.
View Article : Google Scholar :
|
|
19
|
Read A and Schröder M: The unfolded
protein response: An overview. Biology (Basel).
10:3842021.PubMed/NCBI
|
|
20
|
Tian J, Zhang L, La X, Fan X and Li Z:
MiR-769-5p of macrophage exosomes induced by GRP78 promotes
stemness and chemoresistance in colorectal cancer. Cell Death Dis.
16:1562025. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ibrahim IM, Abdelmalek DH and Elfiky AA:
GRP78: A cell's response to stress. Life Sci. 226:156–163. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tsai YL, Ha DP, Zhao H, Carlos AJ, Wei S,
Pun TK, Wu K, Zandi E, Kelly K and Lee AS: Endoplasmic reticulum
stress activates SRC, relocating chaperones to the cell surface
where GRP78/CD109 blocks TGF-β signaling. Proc Natl Acad Sci USA.
115:E4245–E4254. 2018. View Article : Google Scholar
|
|
23
|
Van Krieken R, Tsai YL, Carlos AJ, Ha DP
and Lee AS: ER residential chaperone GRP78 unconventionally
relocalizes to the cell surface via endosomal transport. Cell Mol
Life Sci. 78:5179–5195. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Elshemey WM, Ibrahim IM and Elfiky AA:
Understanding the association of cell-surface proteins (ACE2 and
GRP78) facilitating pathogen recognition: A computational approach.
J Recept Signal Transduct Res. 45:182–188. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen X and Cubillos-Ruiz JR: Endoplasmic
reticulum stress signals in the tumour and its microenvironment.
Nat Rev Cancer. 21:71–88. 2021. View Article : Google Scholar
|
|
26
|
Staquicini DI, D'Angelo S, Ferrara F,
Karjalainen K, Sharma G, Smith TL, Tarleton CA, Jaalouk DE,
Kuniyasu A, Baze WB, et al: Therapeutic targeting of
membrane-associated GRP78 in leukemia and lymphoma: Preclinical
efficacy in vitro and formal toxicity study of BMTP-78 in rodents
and primates. Pharmacogenomics J. 18:436–443. 2018. View Article : Google Scholar
|
|
27
|
Angeles-Floriano T, Rivera-Torruco G,
García-Maldonado P, Juárez E, Gonzalez Y, Parra-Ortega I,
Vilchis-Ordoñez A, Lopez-Martinez B, Arriaga-Pizano L, Orozco-Ruíz
D, et al: Cell surface expression of GRP78 and CXCR4 is associated
with childhood high-risk acute lymphoblastic leukemia at
diagnostics. Sci Rep. 12:23222022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ijabi R, Roozehdar P, Afrisham R,
Moradi-Sardareh H, Kaviani S, Ijabi J and Sahebkar A: Association
of GRP78, HIF-1α and BAG3 expression with the severity of chronic
lymphocytic leukemia. Anticancer Agents Med Chem. 20:429–436. 2020.
View Article : Google Scholar
|
|
29
|
Huergo-Zapico L, Gonzalez-Rodriguez AP,
Contesti J, Gonzalez E, López-Soto A, Fernandez-Guizan A,
Acebes-Huerta A, de Los Toyos JR, Lopez-Larrea C, Groh V, et al:
Expression of ERp5 and GRP78 on the membrane of chronic lymphocytic
leukemia cells: Association with soluble MICA shedding. Cancer
Immunol Immunother. 61:1201–1210. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Steiner N, Borjan B, Hajek R, Jöhrer K,
Göbel G, Willenbacher W, Kern J, Gunsilius E and Untergasser G:
Expression and release of glucose-regulated protein-78 (GRP78) in
multiple myeloma. Oncotarget. 8:56243–56254. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nitta H, Takizawa H, Mitsumori T,
Iizuka-Honma H, Araki Y, Fujishiro M, Tomita S, Kishikawa S,
Hashizume A, Sawada T, et al: Possible new histological prognostic
index for large B-Cell lymphoma. J Clin Med. 12:63242023.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee HK, Xiang C, Cazacu S, Finniss S,
Kazimirsky G, Lemke N, Lehman NL, Rempel SA, Mikkelsen T and Brodie
C: GRP78 is overexpressed in glioblastomas and regulates glioma
cell growth and apoptosis. Neuro Oncol. 10:236–243. 2008.
View Article : Google Scholar
|
|
33
|
Wen X and Chen X and Chen X: Increased
expression of GRP78 correlates with adverse outcome in recurrent
glioblastoma multiforme patients. Turk Neurosurg. 30:11–16.
2020.
|
|
34
|
Ibanez J, Hebbar N, Thanekar U, Yi Z,
Houke H, Ward M, Nevitt C, Tian L, Mack SC, Sheppard H, et al:
GRP78-CAR T cell effector function against solid and brain tumors
is controlled by GRP78 expression on T cells. Cell Rep Med.
4:1012972023. View Article : Google Scholar
|
|
35
|
Zhang P, Li C, Li H, Yuan L, Dai H, Peng
Z, Deng Z, Chang Z, Cui CP and Zhang L: Ubiquitin ligase CHIP
regulates OTUD3 stability and suppresses tumour metastasis in lung
cancer. Cell Death Differ. 27:3177–3195. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li X, Hu F, Lu T, Wu S, Ma G, Lin Y and
Zhang H: Endoplasmic reticulum stress in non-small cell lung
cancer. Am J Cancer Res. 15:1829–1851. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Song J, Liu W, Wang J, Hao J, Wang Y, You
X, Du X, Zhou Y, Ben J, Zhang X, et al: GALNT6 promotes invasion
and metastasis of human lung adenocarcinoma cells through
O-glycosylating chaperone protein GRP78. Cell Death Dis.
11:3522020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang J, Xu H and Tang L: Targeting
TDP-43-activated GRP78/endoplasmic reticulum stress axis suppresses
triple-negative breast cancer progression. Biochem Pharmacol.
242(Pt 2): 1173312025. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang H, Yang X, Deng L, Zhou X, Tao J, Wu
Z and Chen H: ATF6α inhibits ΔNp63α expression to promote breast
cancer metastasis by the GRP78-AKT1-FOXO3a signaling. Cell Death
Dis. 16:2892025. View Article : Google Scholar
|
|
40
|
Yerushalmi R, Raiter A, Nalbandyan K and
Hardy B: Cell surface GRP78: A potential marker of good prognosis
and response to chemotherapy in breast cancer. Oncol Lett.
10:2149–2155. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ren P, Chen C, Yue J, Zhang J and Yu Z:
High expression of glucose-regulated protein 78 (GRP78) is
associated with metastasis and poor prognosis in patients with
esophageal squamous cell carcinoma. Onco Targets Ther. 10:617–625.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Langer R, Feith M, Siewert JR, Wester HJ
and Hoefler H: Expression and clinical significance of glucose
regulated proteins GRP78 (BiP) and GRP94 (GP96) in human
adenocarcinomas of the esophagus. BMC Cancer. 8:702008. View Article : Google Scholar :
|
|
43
|
Lin R, Ma M, Han B, Zheng Y, Wang Y and
Zhou Y: Esophageal cancer stem cells reduce hypoxia-induced
apoptosis by inhibiting the GRP78-perk-eIF2α-ATF4-CHOP pathway in
vitro. J Gastrointest Oncol. 14:1669–1693. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu Q, Xie Z, Li W, Li M and Qi Y:
Aging-associated mechanisms and metabolic vulnerabilities in
esophageal carcinoma: an integrative review. Front Cell Dev Biol.
13:17148242025. View Article : Google Scholar :
|
|
45
|
Zheng B, Zhao W, Jin Y, Sun W, Li Y, Zhang
Q, Li Y, Wang J, Chen D, Ren K, et al: LncRNA SLC16A1-AS1
cooperates with NSUN2 to stabilize GRP78 mRNA via m5C modification
in gastric cancer. iScience. 29:1146142026. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang Y, Wang JH, Zhang XL, Wang XL and
Yang L: Endoplasmic reticulum chaperone glucose-regulated protein
78 in gastric cancer: An emerging biomarker. Oncol Lett.
15:6087–6093. 2018.PubMed/NCBI
|
|
47
|
Zhou B, Lu D, Wang A, Cui J, Zhang L, Li
J, Fan L, Wei W, Liu J and Sun G: Endoplasmic reticulum stress
promotes sorafenib resistance via miR-188-5p/hnRNPA2B1-mediated
upregulation of PKM2 in hepatocellular carcinoma. Mol Ther Nucleic
Acids. 26:1051–1065. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang S, Chen X, Gong Q, Huang J, Tang Y,
Xiao M, Li M, Li Q and Wang Y: The critical role of GRP78/BiP
MARylation in ER stress of KRAS-mutant colorectal cancer. JCI
Insight. 11:e1828092026. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Li Z, Zhang L, Zhao Y, Li H, Xiao H, Fu R,
Zhao C, Wu H and Li Z: Cell-surface GRP78 facilitates colorectal
cancer cell migration and invasion. Int J Biochem Cell Biol.
45:987–994. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ha DP, Tsai YL and Lee AS: Suppression of
ER-stress induction of GRP78 as an anti-neoplastic mechanism of the
cardiac glycoside Lanatoside C in pancreatic cancer: Lanatoside C
suppresses GRP78 stress induction. Neoplasia. 23:1213–1226. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Niu Z, Wang M, Zhou L, Yao L, Liao Q and
Zhao Y: Elevated GRP78 expression is associated with poor prognosis
in patients with pancreatic cancer. Sci Rep. 5:160672015.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Dauer P, Sharma NS, Gupta VK, Nomura A,
Dudeja V, Saluja A and Banerjee S: GRP78-mediated antioxidant
response and ABC transporter activity confers chemoresistance to
pancreatic cancer cells. Mol Oncol. 12:1498–1512. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Li S, Oh GH, Hong JA, Choi S, Kim M, Kwon
H, Ko SK, Park SJ, Kim HK, Choi HJ and Song JJ: Combined
downregulation of TGF-β1 and GRP78 is responsible for overcoming
acquired sorafenib resistance, which is initiated by rewiring the
cell surface CD44-GRP78-IGF-1R signaling circuit. Cancer Gene Ther.
32:884–898. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yang M, Weng K, Guo Y, Huang L, Chen J and
Lu H: GRP78 promotes bone metastasis of prostate cancer by
regulating bone microenvironment through Sonic hedgehog signaling.
Mol Carcinog. 63:494–509. 2024. View Article : Google Scholar
|
|
55
|
Enriquez C, Cancila V, Ferri R, Sulsenti
R, Fischetti I, Milani M, Ostano P, Gregnanin I, Mello-Grand M,
Berrino E, et al: Castration-induced downregulation of SPARC in
stromal cells drives neuroendocrine differentiation of prostate
cancer. Cancer Res. 81:4257–4274. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fu X, Liu J, Liu D, Zhou Y, Guo Y, Wang Z,
Yang S, He W, Chen P, Wang X, et al: Glucose-regulated protein 78
modulates cell growth, epithelial-mesenchymal transition, and
oxidative stress in the hyperplastic prostate. Cell Death Dis.
13:782022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kumar M, Garg H, Gupta N, Sharma A,
Kaushal S, Kumar R and Dinda AK: Glucose-regulated protein 78
(GRP78) in renal cell carcinoma: A novel biomarker for predicting
tumor behavior. Heliyon. 7:e073002021. View Article : Google Scholar
|
|
58
|
Wang Y, Wang J, Liu Y, Wang X and Ren M:
Multidimensional pan-cancer analysis of HSPA5 and its validation in
the prognostic value of bladder cancer. Heliyon. 10:e271842024.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wang Q, Ke S, Liu Z, Shao H, He M and Guo
J: HSPA5 promotes the proliferation, metastasis and regulates
ferroptosis of bladder cancer. Int J Mol Sci. 24:51442023.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Park CH, Choi MS, Ha JY, Kim BH, Park CH
and Kim CI: Effect of overexpression of glucose-regulated protein
78 and bcl-2 on recurrence and survival in patients with ureter
tumors. Korean J Urol. 54:671–676. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Uematsu K, Ogata S, Nakanishi K, Hiroi S,
Tominaga S, Aida S and Kawai T: Glucose-regulated protein 78
expression in urothelial carcinoma of the upper urinary tract. BJU
Int. 106:873–878. 2010. View Article : Google Scholar
|
|
62
|
Wu Z, Zheng P, Xiao Y, Wang Q, Pan X, Zhou
X, Lv Y, Xiao J, He Y, Huang T and Lei P: Targeted degradation of
sGRP78 alleviates the immunosuppressive tumor microenvironment. Adv
Sci (Weinh). 12:e099212025. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Alvarez-Elizondo MB, Raiter A, Yerushalmi
R and Weihs D: Chemotherapy-induced cell-surface GRP78 expression
as a prognostic marker for invasiveness of metastatic
triple-negative breast cancer. Ann Biomed Eng. 53:881–890. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wei S, Fan X, Li X, Zhou W, Zhang Z, Dai
S, Lv H, Liu Y, Shan B, Zhao L, et al: Hypoxia induced Lnc191
upregulation dictates the progression of esophageal squamous cell
carcinoma by activating GRP78/ERK pathway. Adv Sci (Weinh).
12:e24066742025. View Article : Google Scholar :
|
|
65
|
Dufrusine B, Cela I, Gramegna Tota C,
Palumbo M and Sallese M: Signalling pathways and cellular functions
of KDEL receptors: Implications in cancer biology. Cell Mol Life
Sci. 82:2992025. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Tsurusawa N, Iha K, Sato A, Tsai HY,
Sonoda H, Watabe S, Yoshimura T, Wu DC, Lin MW and Ito E:
Ultrasensitive detection of GRP78 in exosomes and observation of
migration and proliferation of cancer cells by application of
GRP78-Containing exosomes. Cancers (Basel). 14:38872022. View Article : Google Scholar :
|
|
67
|
Ou Y, Zhang K, Shuai Q, Wang C, Hu H, Cao
L, Qi C, Guo M, Li Z, Shi J, et al: USP51/GRP78/ABCB1 axis confers
chemoresistance through decreasing doxorubicin accumulation in
triple-negative breast cancer cells. Acta Pharm Sin B.
15:2593–2611. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Raiter A, Lipovetsky J, Hyman L, Mugami S,
Ben-Zur T and Yerushalmi R: Chemotherapy controls metastasis
through stimulatory effects on GRP78 and Its transcription factor
CREB3L1. Front Oncol. 10:15002020. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Ma Q, Gu LY, Ren YY, Zeng LL, Gong T and
Zhong DS: Increased risk of severe infections in cancer patients
treated with vascular endothelial growth factor receptor tyrosine
kinase inhibitors: A meta-analysis. Onco Targets Ther. 8:2361–2374.
2015.PubMed/NCBI
|
|
70
|
Ruan H, Zhang Q, Zhang YP, Li SS and Ran
X: Unraveling the role of HIF-1α in sepsis: From pathophysiology to
potential therapeutics-a narrative review. Crit Care. 28:1002024.
View Article : Google Scholar
|
|
71
|
Gonzalez-Gronow M, Gopal U, Austin RC and
Pizzo SV: Glucose-regulated protein (GRP78) is an important cell
surface receptor for viral invasion, cancers, and neurological
disorders. IUBMB Life. 73:843–854. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Akinyemi AO, Simpson KE, Oyelere SF, Nur
M, Ngule CM, Owoyemi BCD, Ayarick VA, Oyelami FF, Obaleye O, Esoe
DP, et al: Unveiling the dark side of glucose-regulated protein 78
(GRP78) in cancers and other human pathology: A systematic review.
Mol Med. 29:1122023. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Carlos AJ, Ha DP, Yeh DW, Van Krieken R,
Tseng CC, Zhang P, Gill P, Machida K and Lee AS: The chaperone
GRP78 is a host auxiliary factor for SARS-CoV-2 and GRP78 depleting
antibody blocks viral entry and infection. J Biol Chem.
296:1007592021. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Han B, Lv Y, Moser D, Zhou X, Woehrle T,
Han L, Osterman A, Rudelius M, Choukér A and Lei P:
ACE2-independent SARS-CoV-2 virus entry through cell surface GRP78
on monocytes-evidence from a translational clinical and
experimental approach. EBioMedicine. 98:1048692023. View Article : Google Scholar
|
|
75
|
Alhoot MA, Wang SM and Sekaran SD: RNA
interference mediated inhibition of dengue virus multiplication and
entry in HepG2 cells. PLoS One. 7:e340602012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Jindadamrongwech S, Thepparit C and Smith
DR: Identification of GRP 78 (BiP) as a liver cell expressed
receptor element for dengue virus serotype 2. Arch Virol.
149:915–927. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Chumchanchira C, Sornjai W, Roytrakul S,
Lithanatudom P and Smith DR: Increased expression of chaperone
proteins in response to DENV 2 infection of Huh-7 liver cells. PLoS
One. 20:e03297832025. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Khongwichit S, Sornjai W, Jitobaom K,
Greenwood M, Greenwood MP, Hitakarun A, Wikan N, Murphy D and Smith
DR: A functional interaction between GRP78 and Zika virus E
protein. Sci Rep. 11:3932021. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Nain M, Mukherjee S, Karmakar SP, Paton
AW, Paton JC, Abdin MZ, Basu A, Kalia M and Vrati S: GRP78 is an
important host factor for Japanese encephalitis virus entry and
replication in mammalian cells. J Virol. 91:e02274–16. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Wu YP, Chang CM, Hung CY, Tsai MC,
Schuyler SC and Wang RY: Japanese encephalitis virus co-opts the
ER-stress response protein GRP78 for viral infectivity. Virol J.
8:1282011. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Triantafilou K, Fradelizi D, Wilson K and
Triantafilou M: GRP78, a coreceptor for coxsackievirus A9,
interacts with major histocompatibility complex class I molecules
which mediate virus internalization. J Virol. 76:633–643. 2002.
View Article : Google Scholar
|
|
82
|
Rashid MU, Yasmin T and Coombs KM: HSPA5,
a host cellular heat-shock protein required for influenza a virus
replication. Int J Mol Sci. 26:109982025. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Feng X, Ning M, Chen B, Li X, Sun H, Pu J,
Liu J, Wang N and Huang Y: Comparative IP-MS reveals HSPA5 and
HSPA8 interacting with hemagglutinin protein to promote the
replication of influenza A virus. Pathogens. 14:5352025. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Earl PL, Moss B and Doms RW: Folding,
interaction with GRP78-BiP, assembly, and transport of the human
immunodeficiency virus type 1 envelope protein. J Virol.
65:2047–2055. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Elshemey WM, Mostafa HIA and Elfiky AA:
Computational characterization of HIV envelope interactions with
cellular GRP78 as a potential entry mechanism. Cell Stress
Chaperones. 30:1001282025. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Booth L, Roberts JL, Ecroyd H, Tritsch SR,
Bavari S, Reid SP, Proniuk S, Zukiwski A, Jacob A, Sepúlveda CS, et
al: AR-12 inhibits multiple chaperones concomitant with stimulating
autophagosome formation collectively preventing virus replication.
J Cell Physiol. 231:2286–2302. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Gupta S, Mishra KP, Kumar B, Singh SB and
Ganju L: Andrographolide mitigates unfolded protein response
pathway and apoptosis involved in chikungunya virus infection. Comb
Chem High Throughput Screen. 4:849–859. 2021. View Article : Google Scholar
|
|
88
|
Barrera MD, Callahan V, Akhrymuk I, Bhalla
N, Zhou W, Campbell C, Narayanan A and Kehn-Hall K: Proteomic
discovery of VEEV E2-Host partner interactions identifies GRP78
inhibitor HA15 as a potential therapeutic for alphavirus
infections. Pathogens. 10:2832021. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Bolt G: The measles virus (MV)
glycoproteins interact with cellular chaperones in the endoplasmic
reticulum and MV infection upregulates chaperone expression. Arch
Virol. 146:2055–2068. 2001. View Article : Google Scholar
|
|
90
|
Suwanmanee Y, Wada M and Ueda K:
Functional roles of GRP78 in hepatitis B virus infectivity and
antigen secretion. Microbiol Immunol. 65:189–203. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Gebremariam T, Liu M, Luo G, Bruno V, Phan
QT, Waring AJ, Edwards JE Jr, Filler SG, Yeaman MR and Ibrahim AS:
CotH3 mediates fungal invasion of host cells during mucormycosis. J
Clin Invest. 124:237–250. 2014. View Article : Google Scholar :
|
|
92
|
Alqarihi A, Gebremariam T, Gu Y,
Swidergall M, Alkhazraji S, Soliman SSM, Bruno VM, Edwards JE Jr,
Filler SG, Uppuluri P and Ibrahim AS: GRP78 and integrins play
different roles in host cell invasion during mucormycosis. mBio.
11:e01087–20. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Kottom TJ, Hebrink DM and Limper AH:
Binding of Pneumocystis carinii to the lung epithelial cell
receptor HSPA5 (GRP78). J Med Microbiol. 67:1772–1777. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Pan Q, Xu Q, Liu T, Zhang Y and Xin J:
Mycoplasma hyopneumoniae membrane protein Mhp271 interacts with
host UPR protein GRP78 to facilitate infection. Mol Microbiol.
118:208–222. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Cho SJ, Rooney K, Choi AMK and
Stout-Delgado HW: NLRP3 inflammasome activation in aged macrophages
is diminished during Streptococcus pneumoniae infection. Am J
Physiol Lung Cell Mol Physiol. 314:L372–L387. 2018. View Article : Google Scholar
|
|
96
|
van 't Wout EF, van Schadewijk A, van
Boxtel R, Dalton LE, Clarke HJ, Tommassen J, Marciniak SJ and
Hiemstra PS: Virulence factors of pseudomonas aeruginosa induce
both the unfolded protein and integrated stress responses in airway
epithelial cells. PLoS Pathog. 11:e10049462015. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Lim YJ, Choi JA, Lee JH, Choi CH, Kim HJ
and Song CH: Mycobacterium tuberculosis 38-kDa antigen induces
endoplasmic reticulum stress-mediated apoptosis via toll-like
receptor 2/4. Apoptosis. 20:358–370. 2015. View Article : Google Scholar
|
|
98
|
Akazawa Y, Isomoto H, Matsushima K, Kanda
T, Minami H, Yamaghchi N, Taura N, Shiozawa K, Ohnita K, Takeshima
F, et al: Endoplasmic reticulum stress contributes to Helicobacter
pylori VacA-induced apoptosis. PLoS One. 8:e823222013. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Sabirli R, Koseler A, Goren T, Turkcuer I
and Kurt O: High GRP78 levels in Covid-19 infection: A case-control
study. Life Sci. 265:1187812021. View Article : Google Scholar
|
|
100
|
Royle J, Ramírez-Santana C, Akpunarlieva
S, Donald CL, Gestuveo RJ, Anaya JM, Merits A, Burchmore R, Kohl A
and Varjak M: Glucose-regulated protein 78 interacts with zika
virus envelope protein and contributes to a productive infection.
Viruses. 12:5242020. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Chen J, Chen Z, Liu M, Qiu T, Feng D, Zhao
C, Zhang S, Zhang X and Xu J: Placental alkaline phosphatase
promotes zika virus replication by stabilizing viral proteins
through BIP. mBio. 11:e01716–20. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Yadav P, Chakraborty P, Jha NK, Dewanjee
S, Jha AK, Panda SP, Mishra PC, Dey A and Jha SK: Molecular
mechanism and role of japanese encephalitis virus infection in
central nervous system-mediated diseases. Viruses. 14:26862022.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Mazel-Sanchez B, Iwaszkiewicz J, Bonifacio
JPP, Silva F, Niu C, Strohmeier S, Eletto D, Krammer F, Tan G,
Zoete V, et al: Influenza A viruses balance ER stress with host
protein synthesis shutoff. Proc Natl Acad Sci USA.
118:e20246811182021. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Liu M, Yuan M, Ma Y, Wang J, Cheng X, Shi
Y, Shang J, He M, Bai L, Du L and Tang H: Wild-Type and
rtA181T/sW172* mutant strains of hepatitis B virus drive
hepatocarcinogenesis via distinct GRP78-Mediated ER stress
pathways. J Med Virol. 97:e701512025. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Szebenyi C, Gu Y, Gebremariam T, Kocsubé
S, Kiss-Vetráb S, Jáger O, Patai R, Spisák K, Sinka R, Binder U, et
al: cotH genes are necessary for normal spore formation and
virulence in mucor lusitanicus. mBio. 14:e03386222023. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Shang T, Yu Q, Ren T, Wang XT, Zhu H, Gao
JM, Pan G, Gao X, Zhu Y, Feng Y and Li MC: Xuebijing injection
maintains GRP78 expression to prevent candida albicans-induced
epithelial death in the kidney. Front Pharmacol. 10:14162019.
View Article : Google Scholar
|
|
107
|
Wang S, Gong L, Mo Y, Zhang J, Jiang Z,
Tian Z and Shao C: Resveratrol attenuates inflammation and
apoptosis through alleviating endoplasmic reticulum stress via
Akt/mTOR pathway in fungus-induced allergic airways inflammation.
Int Immunopharmacol. 103:1084892022. View Article : Google Scholar
|
|
108
|
Zhou H, Zhang J, Wang R, Huang J, Xin C
and Song Z: The unfolded protein response is a potential
therapeutic target in pathogenic fungi. FEBS J. 292:5008–5025.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Jung KW, Kang HA and Bahn YS: Essential
roles of the Kar2/BiP molecular chaperone downstream of the UPR
pathway in Cryptococcus neoformans. PLoS One. 8:e589562013.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
English BC, Van Prooyen N, Örd T, Örd T
and Sil A: The transcription factor CHOP, an effector of the
integrated stress response, is required for host sensitivity to the
fungal intracellular pathogen Histoplasma capsulatum. PLoS Pathog.
13:e10065892017. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Yi Y, Gao K, Zhang R, Lin P, Wang A and
Jin Y: Staphylococcus aureus induces goat endometrial epithelial
cells apoptosis via the autophagy and endoplasmic reticulum stress
pathway. Animals (Basel). 12:7112022. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Jiang Q, Liu G, Chen J, Yao K and Yin Y:
Crosstalk between nuclear glucose-regulated protein 78 and tumor
protein 53 contributes to the lipopolysaccharide aggravated
apoptosis of endoplasmic reticulum stress-responsive porcine
intestinal epithelial cells. Cell Physiol Biochem. 48:2441–2455.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Grando K, Bessho S, Harrell K, Kyrylchuk
K, Pantoja AM, Olubajo S, Albicoro FJ, Klein-Szanto A and Tükel Ç:
Bacterial amyloid curli activates the host unfolded protein
response via IRE1α in the presence of HLA-B27. Gut Microbes.
16:23928772024. View Article : Google Scholar
|
|
114
|
Sun J, Mai K and Ai Q: Effects of GRP78 on
endoplasmic reticulum stress and inflammatory response in
macrophages of large yellow croaker (Larimichthys crocea). Int J
Mol Sci. 24:58552023. View Article : Google Scholar :
|
|
115
|
Li T and Fu J, Cheng J, Elfiky AA, Wei C
and Fu J: New progresses on cell surface protein HSPA5/BiP/GRP78 in
cancers and COVID-19. Front Immunol. 14:11666802023. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Crane ED, Al-Hashimi AA, Chen J, Lynn EG,
Won KD, Lhoták Š, Naeim M, Platko K, Lebeau P, Byun JH, et al:
Anti-GRP78 autoantibodies induce endothelial cell activation and
accelerate the development of atherosclerotic lesions. JCI Insight.
3:e993632018. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Lenin R, Nagy PG, Jha KA and Gangaraju R:
GRP78 translocation to the cell surface and O-GlcNAcylation of
VE-Cadherin contribute to ER stress-mediated endothelial
permeability. Sci Rep. 9:107832019. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Xie M, Yuan K, Zhang Y, Zhang Y, Zhang R,
Gao J, Wei W, Jiang L, Li T, Ding Y, et al: Tumor-resident
probiotic Clostridium butyricum improves aPD-1 efficacy in
colorectal cancer models by inhibiting IL-6-mediated
immunosuppression. Cancer Cell. 43:1885–1901.e10. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Chen L, Huang Q, Bai Q, Tong T, Zhou Y, Li
Z, Xiao C and Chen L: Chlamydia psittaci induces autophagy in human
bronchial epithelial cells via PERK and IRE1α, but Not ATF6
pathway. Infect Immun. 90:e00079222022. View Article : Google Scholar
|
|
120
|
Walenna NF, Kurihara Y, Chou B, Ishii K,
Soejima T and Hiromatsu K: Chlamydia pneumoniae infection-induced
endoplasmic reticulum stress causes fatty acid-binding protein 4
secretion in murine adipocytes. J Biol Chem. 295:2713–2723. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Bao L, Zhao Y, Duan S, Wu K, Shan R, Liu
Y, Yang Y, Chen Q, Song C and Li W: Ferroptosis is involved in
Staphylococcus aureus-induced mastitis through autophagy activation
by endoplasmic reticulum stress. Int Immunopharmacol.
140:1128182024. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Jiang Q, Chen S, Ren W, Liu G, Yao K, Wu G
and Yin Y: Escherichia coli aggravates endoplasmic reticulum stress
and triggers CHOP-dependent apoptosis in weaned pigs. Amino Acids.
49:2073–2082. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Liang S, Wang F, Bao C, Han J, Guo Y, Liu
F and Zhang Y: BAG2 ameliorates endoplasmic reticulum
stress-induced cell apoptosis in Mycobacterium
tuberculosis-infected macrophages through selective autophagy.
Autophagy. 16:1453–1467. 2020. View Article : Google Scholar :
|
|
124
|
Dixit P, Suratkal SS, Kokate SB,
Chakraborty D, Poirah I, Samal S, Rout N, Singh SP, Sarkar A and
Bhattacharyya A: Siah2-GRP78 interaction regulates ROS and provides
a proliferative advantage to Helicobacter pylori-infected gastric
epithelial cancer cells. Cell Mol Life Sci. 79:4142022. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Wu Z, Xu Z, Zhou X, Li H, Zhao L, Lv Y,
Guo Y, Shen G, He Y and Lei P: sGRP78 enhances selective autophagy
of monomeric TLR4 to regulate myeloid cell death. Cell Death Dis.
13:5872022. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Qin K, Ma S, Li H, Wu M, Sun Y, Fu M, Guo
Z, Zhu H, Gong F, Lei P and Shen G: GRP78 Impairs production of
lipopolysaccharide-induced cytokines by interaction with CD14.
Front Immunol. 8:5792017. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
He J, Xu M, Chen Y and Wu S: Grp78
regulates NLRP3 inflammasome and participates in Sjogren's
syndrome. Int Immunopharmacol. 140:1128152024. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Raiter A, Lipovetzki J, Lubin I and
Yerushalmi R: GRP78 expression in peripheral blood mononuclear
cells is a new predictive marker for the benefit of taxanes in
breast cancer neoadjuvant treatment. BMC Cancer. 20:3332020.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Zhang H, Wang SQ, Hang L, Zhang CF, Wang
L, Duan CJ, Cheng YD, Wu DK and Chen R: GRP78 facilitates M2
macrophage polarization and tumour progression. Cell Mol Life Sci.
78:7709–7732. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Zhao L, Li J, Jiang B, Yang J, Lan J, Li
D, Wen J, Xia Y, Nie W, Wang Z, et al: GRP78 downregulation in
keratinocytes promotes skin inflammation through the recruitment
and activation of CCR6(+) IL-17A-Producing γδ T cells. J Invest
Dermatol. 144:1557–1567.e11. 2024. View Article : Google Scholar
|
|
131
|
Wey S, Luo B and Lee AS: Acute inducible
ablation of GRP78 reveals its role in hematopoietic stem cell
survival, lymphogenesis and regulation of stress signaling. PLoS
One. 7:e390472012. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Zhang L, Li Z, Fan Y, Li H, Li Z and Li Y:
Overexpressed GRP78 affects EMT and cell-matrix adhesion via
autocrine TGF-β/Smad2/3 signaling. Int J Biochem Cell Biol.
64:202–211. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Kim JH, Lee E, Friedline RH, Suk S, Jung
DY, Dagdeviren S, Hu X, Inashima K, Noh HL, Kwon JY, et al:
Endoplasmic reticulum chaperone GRP78 regulates macrophage function
and insulin resistance in diet-induced obesity. FASEB J.
32:2292–2304. 2018. View Article : Google Scholar
|
|
134
|
Leonard A, Grose V, Paton AW, Paton JC,
Yule DI, Rahman A and Fazal F: Selective inactivation of
intracellular BiP/GRP78 attenuates endothelial inflammation and
permeability in acute lung injury. Sci Rep. 9:20962019. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Angeles-Floriano T, Sanjuan-Méndez A,
Rivera-Torruco G, Parra-Ortega I, Lopez-Martinez B, Martinez-Castro
J, Marin-Santiago S, Alcántara-Hernández C, Martínez-Martínez A,
Márquez-González H, et al: Leukocyte surface expression of the
endoplasmic reticulum chaperone GRP78 is increased in severe
COVID-19. J Leukoc Biol. 113:1–10. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Gunner CB, Azmoon P, Mantuano E, Das L,
Zampieri C, Pizzo SV and Gonias SL: An antibody that targets
cell-surface glucose-regulated protein-78 inhibits expression of
inflammatory cytokines and plasminogen activator inhibitors by
macrophages. J Cell Biochem. 124:743–752. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Zhao Y, Jiang Y, Chen L, Zheng X, Zhu J,
Song X, Shi J, Li Y and He W: Inhibition of the endoplasmic
reticulum (ER) stress-associated IRE-1/XBP-1 pathway alleviates
acute lung injury via modulation of macrophage activation. J Thorac
Dis. 12:284–295. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Ho HJ, Huang DY, Ho FM, Lee LT and Lin WW:
Inhibition of lipopolysaccharide-induced inducible nitric oxide
synthase expression by endoplasmic reticulum stress. Cell Signal.
24:2166–2178. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Bi X, Zhang G, Wang X, Nguyen C, May HI,
Li X, Al-Hashimi AA, Austin RC, Gillette TG, Fu G, et al:
Endoplasmic reticulum chaperone GRP78 protects heart from
ischemia/reperfusion injury through Akt activation. Circ Res.
122:1545–1554. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Ji H, Xiao F, Li S, Wei R, Yu F and Xu J:
GRP78 effectively protect hypoxia/reperfusion-induced myocardial
apoptosis via promotion of the Nrf2/HO-1 signaling pathway. J Cell
Physiol. 236:1228–1236. 2021. View Article : Google Scholar
|
|
141
|
Karaboğa İ, Okuyan HM, Doğan S, Ayçiçek Ş
and Çakıroğlu H: Ebselen alleviates sepsis-induced acute kidney
injury by regulating endoplasmic reticulum stress, apoptosis, and
oxidative stress. Vet Med Sci. 11:e703182025. View Article : Google Scholar
|
|
142
|
Li H, Song H, Luo J, Liang J, Zhao S and
Su R: Knockdown of glucose-regulated protein 78 decreases the
invasion, metalloproteinase expression and ECM degradation in
hepatocellular carcinoma cells. J Exp Clin Cancer Res. 31:392012.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Chen Y, Wei W, Fu J, Zhang T, Zhao J and
Ma T: Forsythiaside A ameliorates sepsis-induced acute kidney
injury via anti-inflammation and antiapoptotic effects by
regulating endoplasmic reticulum stress. BMC Complement Med Ther.
23:352023. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Jia YJ, Xiong S, Yao M, Wei Y and He Y:
HMGB1 inhibition blocks ferroptosis and oxidative stress to
ameliorate sepsis-induced acute lung injury by activating the Nrf2
pathway. Kaohsiung J Med Sci. 40:710–721. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Repges E, Al Zaidi M, Jansen F, Zimmer S,
Tiyerlili V and Aksoy A: The endoplasmic reticulum (ER) chaperone
GRP78 is secreted during ER Stress and alleviates endothelial cell
inflammation. Europ Heart J. 43:ehad655.32902022. View Article : Google Scholar
|
|
146
|
Yang M, Zhang F, Qin K, Wu M, Li H, Zhu H,
Ning Q, Lei P and Shen G: Glucose-regulated protein 78-induced
myeloid antigen-presenting cells maintained tolerogenic signature
upon LPS stimulation. Front Immunol. 7:5522016. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Wang Z and Wang Z: The role of macrophages
polarization in sepsis-induced acute lung injury. Front Immunol.
14:12094382023. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Long Y, Zhao Y, Ma X, Zeng Y, Hu T, Wu W,
Deng C, Hu J and Shen Y: Endoplasmic reticulum stress contributed
to inflammatory bowel disease by activating p38 MAPK pathway. Eur J
Histochem. 66:86–103. 2022. View Article : Google Scholar
|
|
149
|
Liu SY, Ruan H and Li SS: HIF-1α: A bridge
connecting sepsis and acute respiratory distress syndrome. Eur J
Med Res. 30:8272025. View Article : Google Scholar
|
|
150
|
Ni M, Zhang Y and Lee AS: Beyond the
endoplasmic reticulum: atypical GRP78 in cell viability, signalling
and therapeutic targeting. Biochem J. 434:181–188. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Sato A and Ito E: GRP78: A multifaceted
role in cancer progression and infectious disease transmission. J
Cell Immunol. 7:9–13. 2025. View Article : Google Scholar
|
|
152
|
Han J, Zhang X, Lau JK, Fu K, Lau HC, Xu
W, Chu ES, Lan H and Yu J: Bone marrow-derived macrophage
contributes to fibrosing steatohepatitis through activating hepatic
stellate cells. J Pathol. 248:488–500. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Cerezo M, Lehraiki A, Millet A, Rouaud F,
Plaisant M, Jaune E, Botton T, Ronco C, Abbe P, Amdouni H, et al:
Compounds triggering ER stress exert anti-melanoma effects and
overcome BRAF inhibitor resistance. Cancer Cell. 29:805–819. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Bian T, Tagmount A, Vulpe C, Vijendra KC
and Xing C: CXL146, a novel 4H-Chromene derivative, targets GRP78
to selectively eliminate multidrug-resistant cancer cells. Mol
Pharmacol. 97:402–408. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Booth L, Roberts JL, Cash DR, Tavallai S,
Jean S, Fidanza A, Cruz-Luna T, Siembiba P, Cycon KA, Cornelissen
CN and Dent P: GRP78/BiP/HSPA5/Dna K is a universal therapeutic
target for human disease. J Cell Physiol. 230:1661–1676. 2015.
View Article : Google Scholar :
|
|
156
|
Saleh DO, Mansour DF and Mostafa RE:
Rosuvastatin and simvastatin attenuate cisplatin-induced
cardiotoxicity via disruption of endoplasmic reticulum
stress-mediated apoptotic death in rats: Targeting ER-Chaperone
GRP78 and Calpain-1 pathways. Toxicol Rep. 7:1178–1186. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Liu D, Jin X, Zhang C and Shang Y:
Sevoflurane relieves hepatic ischemia-reperfusion injury by
inhibiting the expression of Grp78. Biosci Rep. 38:BSR201805492018.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Luo J, Xia Y, Yin Y, Luo J, Liu M, Zhang
H, Zhang C, Zhao Y, Yang L and Kong L: ATF4 destabilizes RET
through nonclassical GRP78 inhibition to enhance chemosensitivity
to bortezomib in human osteosarcoma. Theranostics. 9:6334–6353.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Rauschert N, Brändlein S, Holzinger E,
Hensel F, Müller-Hermelink HK and Vollmers HP: A new tumor-specific
variant of GRP78 as target for antibody-based therapy. Lab Invest.
88:375–386. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
de Ridder GG, Ray R and Pizzo SV: A murine
monoclonal antibody directed against the carboxyl-terminal domain
of GRP78 suppresses melanoma growth in mice. Melanoma Res.
22:225–235. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Misra UK and Pizzo SV: Ligation of cell
surface GRP78 with antibody directed against the COOH-terminal
domain of GRP78 suppresses Ras/MAPK and PI 3-kinase/AKT signaling
while promoting caspase activation in human prostate cancer cells.
Cancer Biol Ther. 9:142–152. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Gopal U, Mowery Y, Young K and Pizzo SV:
Targeting cell surface GRP78 enhances pancreatic cancer
radiosensitivity through YAP/TAZ protein signaling. J Biol Chem.
294:13939–13952. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Yoneda Y, Steiniger SC, Capková K, Mee JM,
Liu Y, Kaufmann GF and Janda KD: A cell-penetrating peptidic GRP78
ligand for tumor cell-specific prodrug therapy. Bioorg Med Chem
Lett. 18:1632–1636. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Gupta P, Walter MR, Su ZZ, Lebedeva IV,
Emdad L, Randolph A, Valerie K, Sarkar D and Fisher PB: BiP/GRP78
is an intracellular target for MDA-7/IL-24 induction of
cancer-specific apoptosis. Cancer Res. 66:8182–8191. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Kao C, Chandna R, Ghode A, Dsouza C, Chen
M, Larsson A, Lim SH, Wang M, Cao Z, Zhu Y, et al: Proapoptotic
cyclic peptide BC71 targets cell-surface GRP78 and functions as an
anticancer therapeutic in mice. EBioMedicine. 33:22–32. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Maddalo D, Neeb A, Jehle K, Schmitz K,
Muhle-Goll C, Shatkina L, Walther TV, Bruchmann A, Gopal SM, Wenzel
W, et al: A peptidic unconjugated GRP78/BiP ligand modulates the
unfolded protein response and induces prostate cancer cell death.
PLoS One. 7:e456902012. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Kapoor V, Dadey DY, Nguyen K, Wildman SA,
Hoye K, Khudanyan A, Bandara N, Rogers BE, Thotala D and Hallahan
DE: Tumor-specific binding of radiolabeled PEGylated GIRLRG
peptide: A novel agent for targeting cancers. J Nucl Med.
57:1991–1997. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Fang S, Hong H, Li L, He D, Xu Z, Zuo S,
Han J, Wu Q, Dai Z, Cai W, et al: Plasminogen kringle 5 suppresses
gastric cancer via regulating HIF-1α and GRP78. Cell Death Dis.
8:e31442017. View Article : Google Scholar
|
|
169
|
Martin S, Lamb HK, Brady C, Lefkove B,
Bonner MY, Thompson P, Lovat PE, Arbiser JL, Hawkins AR and Redfern
CP: Inducing apoptosis of cancer cells using small-molecule plant
compounds that bind to GRP78. Br J Cancer. 109:433–443. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Zhou Y and Lee AS: Mechanism for the
suppression of the mammalian stress response by genistein, an
anticancer phytoestrogen from soy. J Natl Cancer Inst. 90:381–388.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Mujumdar N, Banerjee S, Chen Z, Sangwan V,
Chugh R, Dudeja V, Yamamoto M, Vickers SM and Saluja AK: Triptolide
activates unfolded protein response leading to chronic ER stress in
pancreatic cancer cells. Am J Physiol Gastrointest Liver Physiol.
306:G1011–G1020. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Kawiak A, Domachowska A, Jaworska A and
Lojkowska E: Plumbagin sensitizes breast cancer cells to
tamoxifen-induced cell death through GRP78 inhibition and Bik
upregulation. Sci Rep. 7:437812017. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Zhang Y, Xie WP, Zhang YK, Chen YQ, Wang
DL, Li G and Guan DH: Experimental study of inhibitory effects of
diallyl trisulfide on the growth of human osteosarcoma Saos-2 cells
by downregulating expression of glucose-regulated protein 78. Onco
Targets Ther. 11:271–277. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Hu FW, Yu CC, Hsieh PL, Liao YW, Lu MY and
Chu PM: Targeting oral cancer stemness and chemoresistance by
isoliquiritigenin-mediated GRP78 regulation. Oncotarget.
8:93912–93923. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Bakewell SJ, Rangel DF, Ha DP, Sethuraman
J, Crouse R, Hadley E, Costich TL, Zhou X, Nichols P and Lee AS:
Suppression of stress induction of the 78-kilodalton glucose
regulated protein (GRP78) in cancer by IT-139, an anti-tumor
ruthenium small molecule inhibitor. Oncotarget. 9:29698–29714.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Purushothaman B, Arumugam P, Ju H, Kulsi
G, Samson AAS and Song JM: Novel ruthenium(II) triazine complex
[Ru(bdpta) (tpy)](2+) co-targeting drug resistant GRP78 and
subcellular organelles in cancer stem cells. Eur J Med Chem.
156:747–759. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Paton AW, Beddoe T, Thorpe CM, Whisstock
JC, Wilce MC, Rossjohn J, Talbot UM and Paton JC: AB5 subtilase
cytotoxin inactivates the endoplasmic reticulum chaperone BiP.
Nature. 443:548–552. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Zhao P, Ueda JY, Kozone I, Chijiwa S,
Takagi M, Kudo F, Nishiyama M, Shin-ya K and Kuzuyama T: New
glycosylated derivatives of versipelostatin, the GRP78/Bip
molecular chaperone down-regulator, from Streptomyces versipellis
4083-SVS6. Org Biomol Chem. 7:1454–1460. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Izumikawa M, Ueda JY, Chijiwa S, Takagi M
and Shin-ya K: Novel GRP78 molecular chaperone expression
down-regulators JBIR-04 and -05 isolated from Streptomyces
violaceoniger. J Antibiot (Tokyo). 60:640–644. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Thomas S, Sharma N, Gonzalez R, Pao PW,
Hofman FM, Chen TC, Louie SG, Pirrung MC and Schönthal AH:
Repositioning of Verrucosidin, a purported inhibitor of chaperone
protein GRP78, as an inhibitor of mitochondrial electron transport
chain complex I. PLoS One. 8:e656952013. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Luo C and Qiu J: miR-181a inhibits
cervical cancer development via downregulating GRP78. Oncol Res.
25:1341–1348. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Ji C, Kaplowitz N, Lau MY, Kao E, Petrovic
LM and Lee AS: Liver-specific loss of glucose-regulated protein 78
perturbs the unfolded protein response and exacerbates a spectrum
of liver diseases in mice. Hepatology. 54:229–239. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Tung J, Huang L, George G, Harding HP, Ron
D and Ordonez A: A genome-wide CRISPR/Cas9 screen identifies
calreticulin as a selective repressor of ATF6α. Elife.
13:RP969792024. View Article : Google Scholar
|
|
184
|
Liu Y, Steiniger SC, Kim Y, Kaufmann GF,
Felding-Habermann B and Janda KD: Mechanistic studies of a peptidic
GRP78 ligand for cancer cell-specific drug delivery. Mol Pharm.
4:435–447. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Luo J, Xia Y, Luo J, Li J, Zhang C, Zhang
H, Ma T, Yang L and Kong L: GRP78 inhibition enhances ATF4-induced
cell death by the deubiquitination and stabilization of CHOP in
human osteosarcoma. Cancer Lett. 410:112–123. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Tscheschner H, Meinhardt E, Schlegel P,
Jungmann A, Lehmann LH, Müller OJ, Most P, Katus HA and Raake PW:
CaMKII activation participates in doxorubicin cardiotoxicity and is
attenuated by moderate GRP78 overexpression. PLoS One.
14:e02159922019. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Ha DP, Shin WJ, Hernandez JC, Neamati N,
Dubeau L, Machida K and Lee AS: GRP78 inhibitor YUM70 suppresses
SARS-CoV-2 viral entry, spike protein production and ameliorates
lung damage. Viruses. 15:11182023. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Cheng W, Zhang J, Li D, Xie Y, Lei X, Wang
H and Cui N: The endoplasmic reticulum stress status of CD4+ T
lymphocytes and its association with mTOR-mediated
autophagic-lysosomal disorder in elderly sepsis patients. Front
Immunol. 16:16480752025. View Article : Google Scholar : PubMed/NCBI
|














