Osteoarthritis (OA) is a chronic degenerative joint
disorder characterized primarily by progressive articular cartilage
degeneration, often accompanied by synovial inflammation,
osteophyte formation and subchondral bone sclerosis (1). Its major clinical manifestations
include chronic joint pain, morning stiffness and progressive
decline in joint function, all of which markedly impair the quality
of life of patients (2). With
the aging of the population, the rising prevalence of obesity and
the increasing incidence of joint injury, the global burden of OA
continues to increase. In 2019, the number of individuals affected
by OA was >500 million worldwide (3), and this figure is expected to
increase further in the coming decades (4,5).
OA not only causes substantial functional disability, but also
imposes a considerable socioeconomic burden (6). Traditionally, OA has been regarded
as a disease driven mainly by mechanical wear and tear (7,8).
However, accumulating evidence currently indicates that OA is a
whole-joint disorder involving multiple tissues, including
cartilage, synovium and subchondral bone, and is closely associated
with metabolic dysregulation (9,10).
Previous research on OA has focused mainly on local
pathological processes within the joint, such as cartilage matrix
degradation (11), the crosstalk
between cartilage and subchondral bone (12) and local inflammatory responses
(13). Nevertheless, increasing
evidence suggests that the initiation and progression of OA are not
driven solely by local factors, but rather reflect a complex
process involving disrupted homeostasis across multiple systems,
including metabolic (14),
immune (15), neural (16), skeletal and intestinal systems
(17). In this context, the
interplay between systemic metabolic status and the joint
microenvironment has attracted increasing attention (18). A large body of experimental and
clinical evidence indicates that endocrine hormones and metabolic
products can influence joint tissue homeostasis through the
circulatory system and regulate key pathological processes,
including chondrocyte metabolism, synovial inflammatory responses
and subchondral bone remodeling, thereby contributing to the onset
and progression of OA (19-25). Based on these observations, the
present review adopted the 'endocrine-skeletal axis' as a
conceptual framework and highlights the bidirectional regulatory
association between the endocrine system and the osteoarticular
system. By integrating hormonal signaling, metabolic status and
inflammatory responses, this axis mediates the dynamic interplay
between systemic metabolism and local joint homeostasis, providing
a critical perspective for understanding the systemic pathogenesis
of OA. A schematic illustration of the systemic endocrine-metabolic
network driving OA is presented in Fig. 1.
The present review aimed to move beyond the
conventional single hormone-centered research paradigm, and to
systematically summarize the key regulatory roles of hormonal,
metabolic and inflammatory networks involved in the pathogenesis
and progression of OA from an integrated perspective. Particular
emphasis is placed on the interactions between endocrine signaling
and metabolic pathways, as well as their influence on major
pathological phenotypes, such as cartilage degradation,
dysregulated bone remodeling and synovial inflammation. In
addition, the present review outlines recent advances in relevant
preclinical and clinical studies, and discusses potential
therapeutic strategies targeting the endocrine-metabolic
interaction network, with the goal of providing a theoretical basis
for the precision prevention and treatment of OA.
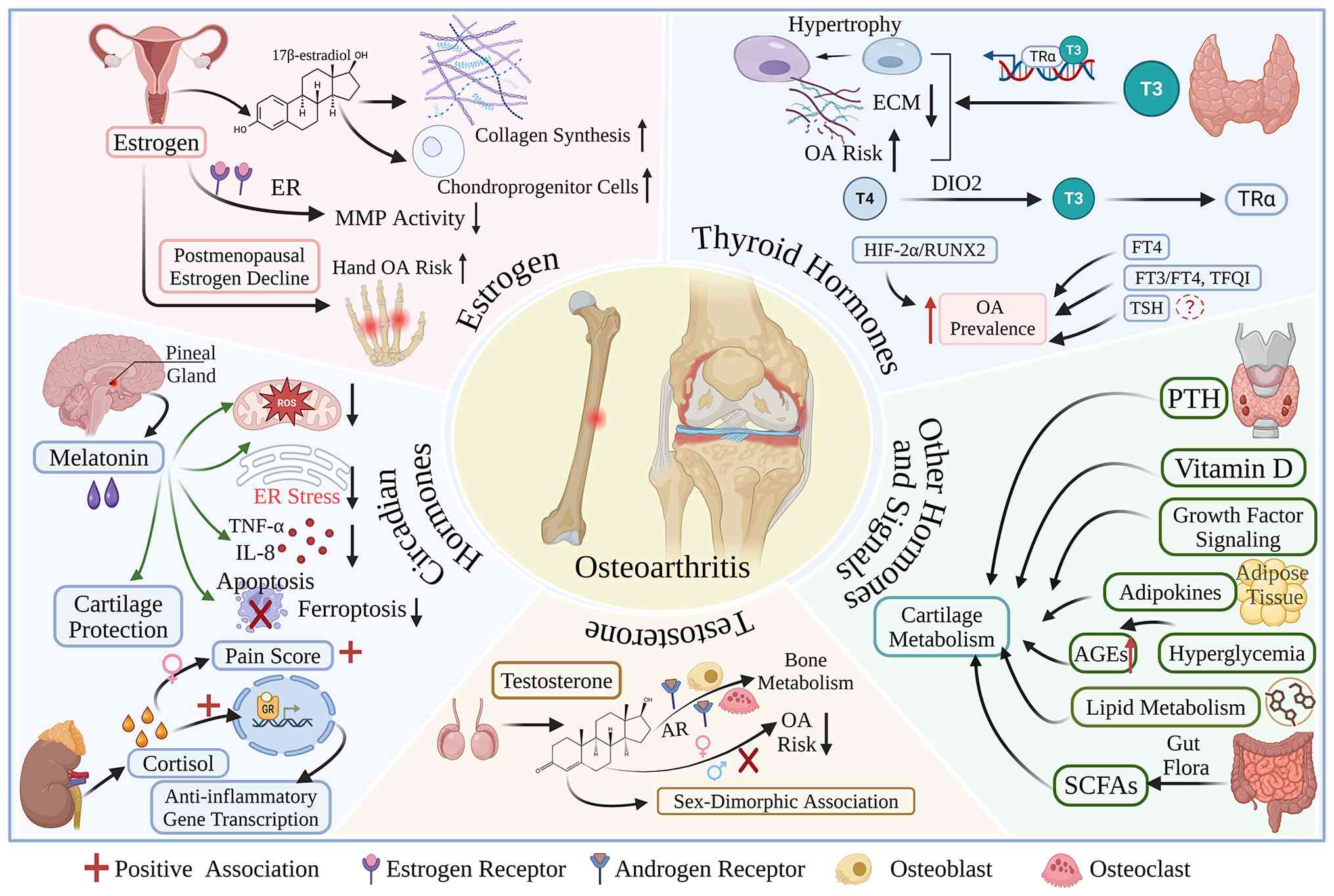
Evidence suggests that estrogen plays a crucial
regulatory role in maintaining the homeostasis of articular
cartilage and subchondral bone, and its deficiency can
significantly increase the risk of developing OA in postmenopausal
women, while accelerating disease progression (26-28). Within the physiological
concentration range, 17β-estradiol promotes the chondrogenic
potential of cartilage progenitor cells and chondrocytes and
enhances the synthesis of key cartilage matrix components,
including type II collagen, thereby preserving the normal phenotype
of cartilage tissue (29). In
addition, estrogen can inhibit matrix degradation mediated by
matrix metalloproteinases (MMPs) through estrogen receptor-mediated
signaling, thereby reducing cartilage matrix loss and preserving
cartilage structural integrity (30).
The expression of estrogen receptor (ER)α and β has
been detected in human articular chondrocytes. Although the overall
expression levels of these receptors do not differ markedly between
normal cartilage and cartilage affected by OA, receptor expression
patterns and hormonal sensitivity may exhibit sex-related
differences (31,32). Studies have indicated that the
decline in estrogen levels after menopause is associated with an
increased risk and greater severity of knee and hand OA (33). At the clinical level, previous
studies have demonstrated that estrogen replacement therapy may, to
a certain extent, alleviate structural degeneration and improve
joint symptoms in postmenopausal OA (34-36). Moreover, estrogen levels are
negatively associated with the volume of knee effusion-synovitis,
and this association appears to be more evident in women than in
men, suggesting that estrogen may contribute to sex-related
differences in the pathogenesis of OA (37,38).
Testosterone is a critical sex hormone for
maintaining bone metabolic homeostasis. It regulates the functions
of osteoblasts and osteoclasts through androgen receptor signaling
and thereby participates in the dynamic balance between bone
formation and bone resorption (39,40). At present, the association
between testosterone and OA remains somewhat controversial. This
inconsistency may partly reflect differences in the testosterone
indicators examined, including total, free and bioavailable
testosterone, as well as age-related hormonal status, sex-specific
responses, the site of OA and study design. Genetic analyses
suggest that bioavailable testosterone levels may have a causal
association with the risk of developing OA (41), whereas epidemiological research
has reported that low testosterone levels are associated with an
increased risk of developing OA (42). These findings suggest that
differences in study populations and OA subtypes may influence the
observed associations. Therefore, testosterone-related associations
should be interpreted in a site- and population-specific
manner.
As regards sex differences, some studies have found
that endogenous testosterone levels are negatively associated with
the risk of developing knee OA in middle-aged and older women,
whereas no similar association has been observed in men (43,44). In addition, testosterone levels
may also be associated with the severity of pain and functional
impairment in patients with OA (45). For example, in patients
undergoing total knee arthroplasty, higher total testosterone
levels have been shown to be associated with improved
post-operative pain relief and functional recovery (46). Hormonal balances may also affect
the risk of developing OA. Evidence suggests that the
testosterone-to-estradiol ratio is negatively associated with risk
of developing OA, and this association appears to be more
pronounced in women (47).
Furthermore, chondrocytes exhibit sex-specific responses to
androgens and estrogens, indicating that sex hormone signaling may
contribute to sex-specific pathological processes in OA (48).
At the level of clinical intervention, the safety of
testosterone replacement therapy has been preliminarily evaluated
in certain orthopedic populations. For example, as previously
demonstrated, in patients undergoing reverse shoulder arthroplasty,
short-term preoperative testosterone replacement therapy did not
significantly increase the risk of revision, infection, or fracture
(49). Existing research also
suggests that testosterone replacement therapy may influence a
range of OA-related orthopedic outcomes, including the incidence of
joint replacement, post-operative recovery and bone healing;
however, its overall benefits and risks warrant further
investigation (50).
Thyroid hormones, with triiodothyronine (T3) as the
principal active form, participate in chondrocyte differentiation,
endochondral ossification and adult bone remodeling through thyroid
hormone receptor α and the deiodinase system. Dysfunction of this
axis is closely associated with the onset and progression of OA
(51). Mechanistic analyses have
demonstrated that T3 can induce chondrocyte hypertrophic
differentiation and promote the expression of ossification-related
genes, thereby accelerating cartilage matrix degradation and
driving OA progression (52).
Genetic studies have further suggested a possible causal
association between thyroid dysfunction and the risk of developing
OA. For example, genetic susceptibility to thyrotoxicosis has been
associated with an increased risk of developing knee OA, whereas no
similar association has been observed for hip OA (53). In population-based studies,
elevated free thyroxine (FT4) levels have been associated with an
increased prevalence, greater disease severity and a higher risk of
progression of knee OA. This association appears to be more
pronounced in individuals with obesity or those exposed to high
mechanical loading, whereas the association between
thyroid-stimulating hormone and OA remains inconclusive (54). In addition, several indicators
reflecting thyroid hormone sensitivity, including the free
triiodothyronine (FT3)/FT4 ratio and the thyroid feedback
quantile-based index (TFQI), have also been reported to be
associated with OA prevalence, with the TFQI exhibiting stronger
predictive performance in certain populations (55). Notably, a local thyroid hormone
regulatory network has been identified in the synovial tissue of
patients with OA. The inflammatory cytokine, tumor necrosis
factor-α (TNF-α) can regulate the expression of deiodinases and
thyroid hormone receptors, suggesting potential crosstalk between
inflammation and thyroid hormone signaling (56).
The deiodinase family, including types I, II and III
deiodinase (DIO1, DIO2 and DIO3, respectively), constitutes a key
enzymatic system that regulates the local activation and
inactivation of thyroid hormones and plays an important role in OA
pathogenesis (57). Among these,
DIO2 has been regarded as a potential susceptibility gene for OA,
and its polymorphisms have been associated with disease risk
(58). It has been suggested
that, in certain genetic backgrounds, upregulated DIO2 expression
may promote cartilage matrix degradation and cartilage
mineralization through pathways, such as hypoxia-inducible
factor-2α (HIF-2α) and runt-related transcription factor 2, thereby
accelerating the progression of OA, whereas the inhibition of
related deiodinase activity may help preserve cartilage homeostasis
(59). In addition, the D2
enzyme encoded by DIO2 is responsible for converting thyroxine into
T3, and its dysfunction may disrupt local thyroid hormone
homeostasis and increase the risk of developing OA (60). Clinical studies further suggest
that, in individuals at a high risk of developing knee OA,
levothyroxine treatment may be associated with a reduced quadriceps
muscle mass, and this decline in muscle mass may further alter
joint loading and promote the development of OA (61).
Melatonin is a key hormone involved in maintaining
circadian homeostasis, and abnormal melatonin secretion has been
implicated in the onset and progression of OA (62,63). A growing body of evidence
indicates that melatonin exerts multiple protective effects against
OA, including antioxidant, anti-inflammatory and metabolic
regulatory actions. Mechanistic studies suggest that melatonin can
inhibit chondrocyte apoptosis and ferroptosis by regulating
mitochondrial function and endoplasmic reticulum homeostasis, while
also reducing the release of pro-inflammatory cytokines, such as
TNF-α and interleukin (IL)-8 and suppressing the activity of MMPs,
thereby preserving cartilage matrix homeostasis (64-68). At the molecular level, melatonin
can regulate oxidative stress and inflammatory responses through
melatonin receptor 1 and downstream intracellular signaling
pathways, thereby attenuating OA-related cartilage injury (69-71). In addition, some clinical studies
suggest that melatonin may have analgesic potential, as its use has
been found to be associated with reduced subsequent analgesic
requirements and a lower risk of joint replacement (72). Current basic and animal studies
generally support the potential protective role of melatonin in OA
(73-76), and further suggest that it may
serve as a candidate agent for OA prevention or treatment (77). However, the available evidence is
still derived mainly from experimental studies, and large-scale
clinical investigations remain limited. Its precise efficacy,
optimal dosage and target populations therefore require further
clarification (78).
In addition to melatonin, hormones associated with
the hypothalamic-pituitary-adrenal axis may also participate in the
pathological process of OA. It has been demonstrated that the
association between cortisol and OA is markedly heterogeneous and
may be influenced by factors, such as measurement methods, sex and
population characteristics (79). In population-based studies, pain
severity in women with OA has been reported to be positively
associated with cortisol levels (80). By contrast, in patients with knee
OA, serum cortisol levels have also been reported to be negatively
associated with knee pain scores, and to be associated with the
levels of multiple inflammatory mediators (81). Moreover, some studies have found
that patients with knee OA exhibit a blunted cortisol awakening
response and reduced morning cortisol levels after waking. This
circadian alteration is associated with greater pain interference
and a lower pain threshold, and may also exhibit sex-related
differences (82). At the
molecular level, cortisol may exert anti-inflammatory effects by
regulating glucocorticoid receptor-related transcriptional
activity, thereby contributing to the modulation of inflammatory
responses in OA (83). As
cortisol secretion displays pronounced circadian rhythmicity, it
may exert distinct biological effects across different
concentrations and time phases. For example, high cortisol levels
may promote inflammatory responses, whereas moderate levels may
help maintain the balance between stress adaptation and
inflammation (63).
Parathyroid hormone (PTH) plays a central role in
the regulation of bone metabolism and its association with OA
appears to be complex. Genetic studies suggest that elevated serum
levels of PTH may have a potential causal association with a
reduced risk of developing both hip and knee OA, with the
association appearing to be more pronounced in knee OA (84,85). Experimental studies have also
shown that PTH (1-34) can attenuate cartilage
degeneration and improve subchondral bone microarchitecture in
animal models of OA, thereby helping to maintain joint tissue
homeostasis (86,87). Mechanistic evidence further
suggests that PTH may exert protective effects by regulating
chondrocyte proliferation, matrix synthesis and inflammatory
responses (88). However, some
studies have proposed that PTH may also promote the progression of
OA by altering local bone metabolism and the joint microenvironment
(89), indicating that its
effects may be context-dependent and closely related to the mode of
administration and disease stage.
Vitamin D also plays an important role in the
regulation of bone and cartilage metabolism, although its value in
the prevention and treatment of OA remains controversial.
Epidemiological studies have shown that vitamin D deficiency is
common in patients with knee OA and is associated with multisite
joint pain and greater disease severity (90). At the molecular level, vitamin D
may contribute to the maintenance of cartilage homeostasis by
regulating chondrocyte autophagy and metabolic pathways (91). However, multiple clinical studies
have demonstrated that vitamin D supplementation has limited
effects on OA-related pain and functional improvement, and has not
demonstrated clear protective benefits against structural changes,
such as cartilage volume loss or joint space narrowing (92,93). Therefore, although vitamin D
deficiency may be associated with the onset of OA, consistent
evidence is still lacking as to whether vitamin D supplementation
can delay disease progression (94).
In addition to the hormones described above, several
growth factor-related signaling pathways may also contribute to the
onset and progression of OA. For example, growth hormone and its
downstream mediator insulin-like growth factor-1 are involved in
the maintenance of joint homeostasis through the regulation of bone
metabolism and chondrocyte function, and abnormal levels of these
factors may affect bone microarchitecture and increase the risk of
developing OA (95). In
addition, growth hormone-releasing hormone has been reported to
promote chondrocyte proliferation and enhance extracellular matrix
(ECM) synthesis, thereby exerting protective effects against
cartilage injury (96). The
transforming growth factor-β (TGF-β) family plays an essential role
in cartilage development and tissue repair, and its biological
effects may be concentration-dependent. Low levels appear to
support cartilage homeostasis, whereas aberrant activation may
aggravate joint structural damage (97,98). Vascular endothelial growth factor
(VEGF) is closely associated with OA-related angiogenesis and pain
regulation, and its abnormal expression may promote cartilage
degeneration and contribute to OA pain development (99).
Metabolic OA is increasingly recognized as a key
clinical subtype of OA, and its onset and progression are closely
associated with metabolic syndrome and related metabolic
disturbances. This subtype is typically driven by the combined
effects of obesity, insulin resistance, dyslipidemia and chronic
low-grade inflammation. Through the synergistic interaction between
systemic metabolic abnormalities and alterations in the local joint
microenvironment, these factors ultimately lead to structural joint
degeneration and functional impairment (100).
From a clinical perspective, metabolic OA is most
commonly observed in individuals aged 45 to 65 years, frequently
affects the knee and hand joints, and is often characterized by
more pronounced inflammatory responses and pain symptoms. Patients
with this subtype commonly present with metabolic comorbidities,
such as obesity, diabetes and cardiovascular disease, suggesting
that the development of OA is not determined solely by local joint
factors, but is also closely linked to systemic metabolic status
(101). Epidemiological
research has demonstrated that, in middle-aged women, the severity
of metabolic syndrome is significantly associated with the
structural progression of knee OA over subsequent years, including
osteophyte formation, bone marrow lesions and cartilage defects.
Among these factors, indicators, such as waist circumference, blood
glucose levels and high-density lipoprotein (HDL) cholesterol may
serve as critical predictors (102).
In recent years, it has become increasingly evident
that metabolic abnormalities can disrupt joint tissue homeostasis
through multiple mechanisms, including systemic inflammation,
immune dysregulation and altered metabolic signaling pathways.
Examples include gut microbiota imbalances, changes in immune cell
polarization and the dysregulation of lipid metabolism-related
signaling pathways (102-105). These mechanisms provide a key
theoretical basis for understanding the development and progression
of metabolic OA, and may also provide potential directions for
future biomarker discovery and the development of targeted
intervention strategies.
Obesity is one of the most critical modifiable risk
factors for OA. Its impact on OA arises not only from the increased
mechanical loading associated with excess body weight, but also
from the metabolic inflammation mediated by a wide range of
adipokines secreted by adipose tissue (106,107). Adipose tissue dysfunction can
lead to dysregulated adipokine secretion and trigger chronic
low-grade inflammation, thereby affecting cartilage metabolism,
synovial inflammation and subchondral bone remodeling. This process
is considered a major pathological basis of obesity-related OA
(107,108). Among these adipokines, leptin
is one of the most extensively studied. Its levels are
significantly elevated in patients with obesity and OA, and it can
promote the expression of pro-inflammatory mediators and MMPs
through the activation of signaling pathways, such as the
JAK/signal transducer and activator of transcription (STAT) pathway
and nuclear factor κB (NF-κB) signaling, while simultaneously
suppressing cartilage matrix synthesis. In this manner, leptin
accelerates cartilage degradation and contributes to synovitis and
abnormal bone remodeling (21,23). Epidemiological research has
further demonstrated that serum leptin levels are positively
associated with the risk of developing hand and knee OA, and may
partly explain the link between obesity and OA. This effect appears
to be more evident in women and in individuals with knee OA
(109).
In addition to leptin, several other adipokines are
involved in the pathological process of obesity-related OA. For
example, adiponectin may exert bidirectional and context-dependent
effects in OA. On the one hand, it participates in metabolic
regulation through pathways, such as AMP-activated protein kinase
(AMPK). On the other hand, it may promote inflammatory responses
and influence chondrocyte apoptosis and autophagy (110,111). Adipokines, such as visfatin and
resistin are upregulated in obesity and can aggravate joint tissue
injury by promoting the expression of inflammatory mediators and
matrix-degrading enzymes. Their levels have also been associated
with OA severity (107,112). Notably, adipokines are involved
not only in structural joint degeneration, but also in the
regulation of OA pain through peripheral inflammation and
mechanisms related to neural sensitization (108). Taken together, adipose tissue
in OA is not merely a source of excessive mechanical loading, but
also an important endocrine organ, and the adipokines it secretes
play a central role in linking systemic metabolic abnormalities
with local joint inflammation.
Glucotoxicity and insulin resistance are
increasingly recognized as key pathological links underlying the
comorbidity of type 2 diabetes mellitus (T2DM) and OA. Through the
interplay between metabolic dysregulation and inflammatory
responses, these processes jointly promote joint degeneration.
Chronic hyperglycemia can disrupt the homeostasis of the joint
microenvironment through multiple mechanisms. For example, high
glucose levels can enhance glycolysis in synovial macrophages and
promote lactate accumulation, thereby inducing CD11b lactylation
and impairing macrophage efferocytosis, which in turn aggravates
synovial inflammation. In this process, the acetyltransferase
CREB-binding protein may serve as a critical mediator, and targeted
CREB-binding protein knockdown has been shown to partially delay
the progression of hyperglycemia-associated OA (113). At the same time, hyperglycemia
can promote the accumulation of advanced glycation end products in
fibroblast-like synoviocytes (FLSs) through the HIF-1α/glucose
transporter 1 (GLUT1) pathway and induce endoplasmic reticulum
stress, further increasing the expression of inflammatory
mediators, such as TNF-α and IL-6, as well as matrix-degrading
enzymes including MMPs and a disintegrin and metalloproteinase with
thrombospondin motifs (ADAMTS). These changes suppress cartilage
matrix synthesis and accelerate matrix degradation (114). In addition, high-glucose
conditions can induce oxidative stress and exacerbate joint tissue
injury, further supporting glucotoxicity as a key mechanistic link
between metabolic abnormalities and joint inflammation (19).
Epidemiological studies also support a critical role
for insulin resistance in the development of OA. Multiple
population-based studies have shown that insulin resistance-related
indicators, including the triglyceride-glucose index, its derived
indices and the metabolic score for insulin resistance, are all
significantly associated with the risk of developing OA. These
findings suggest that insulin resistance may serve as a key
metabolic marker linking metabolic syndrome to the onset of OA, and
may provide a potential basis for early screening and risk
assessment (115-118). At the molecular level, abnormal
insulin signaling can amplify inflammatory responses and suppress
autophagy through pathways, such as phosphoinositide 3-kinase
(PI3K)/protein kinase B (Akt)/mechanistic target of rapamycin
(mTOR)/NF-κB, thereby promoting the secretion of inflammatory
mediators and chemokines by FLSs and upregulating
cartilage-degrading molecules such as MMP-9 and MMP-13, which
ultimately aggravates synovial inflammation and cartilage damage
(119). Notably, OA and T2DM
share a common metabolic background characterized by nutrient
excess, chronic inflammation, hyperglycemia and mitochondrial
dysfunction. Among the key regulatory nodes involved, AMPK has been
highlighted as a central integrator linking energy metabolism,
inflammatory signaling, and cellular stress responses across these
pathological processes (120).
At the clinical level, T2DM and poor glycemic
control have been reported to be significantly associated with an
increased risk of symptomatic knee OA, independently of traditional
risk factors such as age and body mass index, suggesting that
glycemic management may represent a key strategy for preventing
adverse OA-related outcomes (121). In addition, several widely used
glucose-lowering agents, such as metformin and glucagon-like
peptide-1 receptor agonists, are considered to exert potential
protective effects against OA by improving metabolic inflammation,
regulating energy metabolism and suppressing cartilage degradation.
However, their precise therapeutic value still requires further
confirmation in clinical studies (122).
The association between dyslipidemia and OA remains
somewhat heterogeneous. Epidemiological research suggests that
dyslipidemia is associated with an increased risk of developing OA,
although the findings are not fully consistent across different
study designs (123). For
example, some studies have reported that dyslipidemia is associated
with a higher risk of developing hand OA, with the association for
elevated levels of triglycerides appearing to be relatively
consistent, whereas the evidence linking low-density lipoprotein
(LDL) and HDL levels to hand OA remains inconclusive (124). At the same time, several
studies have not identified a clear association between
dyslipidemia and knee OA (125). In addition, lipid-related
indices may also be associated with OA pain phenotypes. For
instance, HDL cholesterol may exert a protective effect, whereas
elevated triglycerides may increase risk (126). Overall, dyslipidemia may
contribute to the association between obesity and OA by promoting
systemic inflammatory responses, but its causal role and clinical
significance still require further clarification (123,127).
To more directly synthesize the inconsistent
epidemiological findings, current evidence suggests that elevated
levels of triglycerides exhibit relatively consistent positive
associations with OA, particularly in studies on hand OA and
OA-related pain phenotypes (124,126). By contrast, although HDL
cholesterol may be associated with certain OA-related pain
phenotypes, its association with structural OA remains inconsistent
(124,126), whereas LDL cholesterol and
total cholesterol have not exhibited a uniform association with OA
across studies (123,124). The association between
dyslipidemia and OA also appears to be joint-site specific, being
more evident for hand OA than for knee OA, for which some studies
have reported no clear association (124,125). This heterogeneity may partly
reflect differences in the joint site examined, the lipid
parameters assessed, outcome definitions and study design,
including cross-sectional, case-control and cohort approaches
(123-126).
Beyond circulating lipid levels, the local
dysregulation of lipid metabolism within the joint is increasingly
regarded as a key mechanism underlying the onset and progression of
OA. Disrupted cholesterol homeostasis in chondrocytes can impair
ECM homeostasis through several mechanisms. For example, defective
cholesterol efflux or abnormal cholesterol metabolic pathways can
activate signaling cascades, such as Ras/Raf/MEK/ERK, thereby
promoting cartilage matrix degradation (128,129). In addition, abnormal fatty acid
metabolism is also involved in the pathogenesis of OA. In
obesity-related OA, enhanced fatty acid oxidation (FAO) in
chondrocytes can aggravate abnormalities in cartilage matrix
metabolism through metabolic reprogramming and epigenetic
regulation (130). Different
types of fatty acids exert distinct effects on OA. Among these, n-3
polyunsaturated fatty acids, such as eicosapentaenoic acid and
docosahexaenoic acid, may exert protective effects through pathways
including peroxisome proliferator-activated receptor γ and NF-κB,
whereas n-6 fatty acids may promote inflammatory responses and
accelerate cartilage degradation (131). Moreover, lipid peroxidation is
considered a critical mechanism of cartilage injury. For example,
lipid peroxidation mediated by acyl-CoA synthetase long-chain
family member 4 can induce ferroptosis and amplify joint
inflammatory responses (132).
Taken together, abnormal lipid metabolism not only affects systemic
inflammatory status, but also disrupts cartilage homeostasis
through local metabolic reprogramming.
Oxidized LDL (oxLDL) is considered a key molecular
link between lipid metabolic abnormalities and OA. Upon binding to
lectin-like oxidized low-density lipoprotein receptor-1, oxLDL can
induce oxidative stress and inflammatory responses, thereby
promoting cartilage degeneration and osteophyte formation (133,134). In chondrocytes, oxLDL can
suppress transcription factor EB activity through the activation of
the ERK1/2 and mTOR pathways, leading to impaired autophagy and
lysosomal dysfunction and ultimately inducing cell death (135). In synovial tissue, oxLDL can
activate macrophages and fibroblasts, further aggravating synovial
inflammation (136,137). Based on these mechanisms, the
modulation of lipid metabolism or the inhibition of related
signaling pathways has been considered to hold therapeutic
potential. For example, statins or agents targeting the mTOR
pathway have been reported in some studies to possibly attenuate
the progression of OA, although their clinical efficacy warrants
further validation (135).
Gut microbiota dysbiosis is increasingly regarded as
a crucial initiating factor that drives the dysfunction of the
gut-joint axis and contributes to the onset and progression of OA.
Studies have shown that the composition of the gut microbiota is
significantly altered in patients with OA, as reflected by an
increased abundance of potentially pathogenic bacteria, such as
Actinomycetaceae and Bilophila, together with a
reduction in the numbers of beneficial microbes, including
Roseburia and Bifidobacterium. These changes are
accompanied by abnormalities in amino acid, carbohydrate and
lipid-related metabolic pathways (138-140). Alterations in microbial
composition not only impair intestinal metabolic function, but can
also promote systemic inflammation by disrupting the intestinal
barrier. For example, microbial dysbiosis can increase zonulin
levels and downregulate the expression of tight junction proteins,
such as zonula occludens-1 and occludin, thereby increasing
intestinal permeability. At the same time, it can induce the
release of inflammatory mediators including TNF-α and IFN-γ, and
weaken the structural integrity of the mucus barrier (138,139,141). Under these conditions,
bacterial metabolites, such as lipopolysaccharide (LPS) and other
inflammatory mediators can enter the circulation and trigger
low-grade inflammatory responses, which are considered an important
mechanism linking gut microbiota abnormalities to OA-related
inflammation.
Short-chain fatty acids (SCFAs) produced by the gut
microbiota are considered key molecular mediators regulating the
gut-joint axis. Among these, acetate, propionate and butyrate
account for the vast majority of total intestinal SCFAs, and can
regulate immune responses and intestinal barrier function through
mechanisms, such as the activation of G protein-coupled receptors
(GPRs), including GPR41, GPR43 and GPR109A, as well as the
inhibition of histone deacetylases. Through these actions, SCFAs
help maintain the Treg/Th17 balance and regulate macrophage
polarization (142-146). Among all SCFAs, butyrate
appears to exert particularly prominent anti-inflammatory effects.
It can inhibit the NF-κB and mitogen-activated protein kinase
(MAPK) signaling pathways through GPR43, reduce the expression of
inflammatory and matrix-degrading molecules, such as IL-1β, TNF-α
and MMP13, and improve chondrocyte autophagy, as well as type II
collagen expression (147-149). Clinical research further
suggests that butyrate supplementation may, to a certain extent,
improve pain and functional scores in patients with knee OA
(150). In addition,
interventions, such as high-fiber diets, probiotic or prebiotic
supplementation and fecal microbiota transplantation may improve
the intestinal metabolic environment by increasing SCFA levels,
thereby providing potential avenues for the prevention and
treatment of OA (144). A
summary of key endocrine and metabolic mediators involved in the
pathogenesis of OA is presented in Table I.
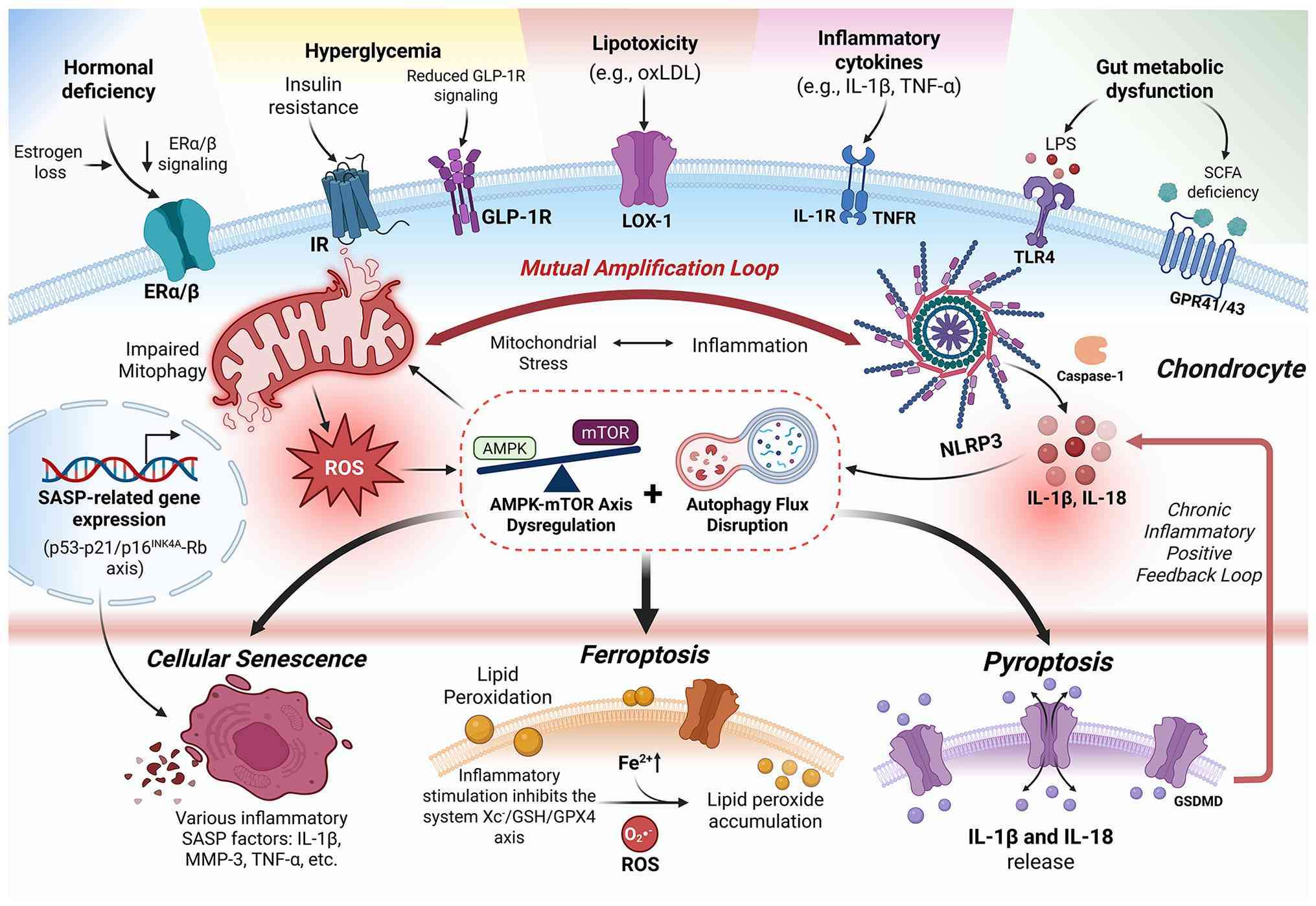
In OA, metabolic abnormalities can function as
upstream drivers of low-grade inflammation. A central feature of
this process is the imbalance between ECM synthesis and degradation
in cartilage (152,153). Immunometabolic reprogramming
further amplifies this imbalance. For example, pyruvate kinase
M2-mediated glycolytic activation and microRNA-576-5p deficiency
can induce chondrocyte stress and promote the release of
damage-associated molecular patterns (DAMPs), including adenosine
triphosphate (ATP), high mobility group box 1 and S100A8/A9
(152-155). These DAMPs activate pattern
recognition receptors, such as Toll-like receptor 4 and NLR family
pyrin domain-containing 3 (NLRP3), leading to the activation of
NF-κB, MAPK and inflammasome signaling. This response increases MMP
expression, chondrocyte apoptosis and the release of IL-1β, thereby
sustaining low-grade inflammation within the joint (152-156). Thus, immunometabolic
reprogramming establishes an inflammation-related positive feedback
loop that contributes to the progression of chronic OA. Enhanced
glycolysis and reactive oxygen species (ROS) accumulation are
central metabolic nodes involved in this process (157). The link between immunometabolic
stress and endocrine-metabolic imbalance with chondrocyte
degeneration in OA is illustrated in Fig. 2.
Within the OA microenvironment, multiple stimuli,
including inflammatory cytokines, hyperglycemia and mechanical
injury, can induce metabolic reprogramming in synovial cells and
chondrocytes, and establish an inflammation-amplifying network
centered on glycolysis. For example, under IL-1β stimulation,
exosomes released by inflammatory FLSs can be taken up by
macrophages and enhance HIF1A activity, thereby driving the
expression of key glycolytic enzymes such as GLUT1 and hexokinase 2
and increasing glycolytic flux in macrophages (158). At the same time, chondrocytes
exposed to IL-1β activate aerobic glycolysis through the NF-κB
pathway and upregulate lactate dehydrogenase A expression,
exhibiting metabolic features reminiscent of the Warburg effect
(159). In addition,
hyperglycemia can directly promote glycolysis in synovial
macrophages and enhance lactate production, thereby further
amplifying metabolic dysregulation (113). Taken together, these findings
indicate that glycolytic reprogramming is a critical link
connecting inflammatory stimulation with immunometabolic
imbalance.
Enhanced glycolysis leads to the accumulation of
metabolic byproducts, such as lactate and ROS, which further
amplify inflammatory responses through multiple mechanisms. For
example, lactate can upregulate nicotinamide adenine dinucleotide
phosphate oxidase 4 (NOX4) and promote ROS generation through
HCAR1/PI3K/Akt signaling, while ROS in turn activates the NLRP3
inflammasome and induces the release of inflammatory mediators
(159-162). Moreover, increased glycolysis
in macrophages promotes the secretion of IL-1β and TNF-α, which
further stimulates FLSs to release more inflammation-related
exosomes, thereby forming a sustained inflammatory amplification
loop (158). ROS and associated
inflammatory signaling can also promote MMP expression and
aggravate cartilage damage, leading to the release of additional
DAMPs and further reinforcing metabolic abnormalities. This
positive feedback network enables inflammation to persist beyond
the initial trigger and ultimately drives the progression of OA
(159). Therefore, targeting
key glycolytic nodes, such as HIF1A and lactate dehydrogenase A, or
ROS-related pathways, including NOX4 and NLRP3, may represent
promising strategies for intervening in immunometabolic
inflammation in OA.
The inflammatory microenvironment, including IL-1β
and TNF-α, together with metabolic stressors, such as mechanical
loading, obesity and aging, can synergistically induce disturbances
in chondrocyte energy metabolism and are considered key triggers of
mitochondrial dysfunction in OA (163-165). These stimuli can disrupt
cellular energy regulatory networks and affect the AMPK-mTOR
signaling axis, thereby altering the levels of autophagy and
mitophagy. As a key sensor of cellular energy homeostasis, AMPK is
activated through phosphorylation at the Thr172 site of its α
subunit and promotes autophagy initiation by inhibiting mTOR
activity, thereby helping to maintain energy balance (166-168). However, under OA-related
metabolic stress, this regulatory axis is often impaired. For
example, insufficient β-hydroxybutyrate (βOHB) can weaken
HCAR2-mediated AMPK activation and suppress PTEN-induced putative
kinase 1 (PINK1)/Parkin-dependent mitophagy, thereby impairing the
clearance of damaged mitochondria (167). By contrast, excessive
enhancement of FAO may disrupt cartilage matrix homeostasis by
suppressing AMPK activity and promoting SRY-box transcription
factor 9 (SOX9) degradation (130). Taken together, these findings
suggest that the AMPK-mTOR axis serves as a key regulatory node
linking metabolic stress to mitochondrial quality control.
Impaired autophagy and mitophagy directly disrupt
mitochondrial quality control and promote the accumulation of
oxidative stress. When mitophagy is insufficient, damaged
mitochondria progressively accumulate within cells, manifesting as
cristae disruption, loss of membrane potential and reduced ATP
production, together with excessive generation of ROS (168). Metabolic regulators, such as
α-ketoglutarate (α-KG) and sirtuin (SIRT)4 are also involved in
this process, and alterations in their expression can aggravate
mitochondrial fragmentation and promote ROS production (169,170). Furthermore, ROS not only
damages mitochondrial DNA and respiratory chain complexes, but also
reinforces the vicious cycle of mitochondrial dysfunction, ROS
accumulation and autophagic imbalance through feedback regulation
of the AMPK-mTOR pathway (171).
This energy crisis and oxidative stress ultimately
translate into metabolic imbalance of the cartilage ECM. For
example, ROS can activate inflammatory signaling and upregulate the
expression of MMP13 and ADAMTS5, while suppressing the synthesis of
collagen type II alpha 1 chain (Col2a1) and aggrecan, thereby
promoting cartilage degeneration (169). In addition, enhanced FAO may
further inhibit the expression of ECM synthesis-related genes
through epigenetic reprogramming (130). Therefore, modulation of the
AMPK-mTOR signaling axis or restoration of mitochondrial metabolic
homeostasis, such as by targeting FAO or supplementing metabolites,
including βOHB and α-KG, may represent promising strategies for
alleviating metabolic abnormalities and cartilage injury in OA
(130,167).
The progression of OA is closely associated with the
aberrant activation of PCD in chondrocytes. Accumulating evidence
indicates that multiple forms of PCD are involved in OA, including
apoptosis, pyroptosis, ferroptosis, necroptosis, autophagy,
cuproptosis and PANoptosis (172-174). Through complex molecular
networks, these cell death modalities collectively influence
chondrocyte survival and function, and contribute to inflammatory
amplification and cartilage matrix destruction. Among these,
pyroptosis and ferroptosis have attracted particular attention in
recent years due to their close links to inflammatory responses and
oxidative stress, as well as their potential therapeutic relevance.
To clarify these overlapping pathways, Table II summarizes their major
triggers, molecular mediators, OA-related consequences, current
evidence status and provides supporting references.
Pyroptosis is a form of PCD accompanied by a robust
inflammatory response and can contribute to OA-related cartilage
injury through both canonical, namely caspase-1-dependent, and
non-canonical, namely caspase-4/5/11-dependent, pathways (175). In the canonical pathway,
stimuli such as IL-1β, TNF-α, LPS, ATP, ROS and mechanical stress
can activate NF-κB or MAPK signaling, thereby inducing the
expression of NLRP3 inflammasome-related components and promoting
inflammasome assembly, followed by the recruitment and activation
of caspase-1. Activated caspase-1 cleaves gasdermin D (GSDMD),
resulting in the formation of membrane pores, while also promoting
the maturation and release of IL-1β and IL-18, thereby amplifying
inflammatory responses (176).
The non-canonical pathway is initiated by the direct activation of
caspase-4/5 in humans or caspase-11 in mice by cytosolic LPS, and
similarly induces pyroptosis through GSDMD cleavage (177). Both pathways ultimately
converge on GSDMD-mediated membrane rupture and inflammatory
cytokine release, which further upregulate the expression of
matrix-degrading enzymes such as MMP1, MMP3, MMP13 and ADAMTS4/5,
thereby suppressing cartilage matrix synthesis and aggravating
synovitis and cartilage degeneration (178).
Ferroptosis is a form of PCD driven by iron overload
and lipid peroxidation, with the core regulatory axis centered on
system Xc−/glutathione (GSH)/glutathione peroxidase 4
(GPX4) (179). System
Xc− is composed of solute carrier family (SLC)7 member
11 (SLC7A11) and SLC3A2, and maintains GSH synthesis through
cystine-glutamate exchange, whereas GSH serves as an essential
cofactor for GPX4 to reduce lipid peroxides and limit oxidative
injury (180). In the OA
microenvironment, inflammatory stimulation or mechanical stress can
suppress SLC7A11 expression and reduce GPX4 activity, thereby
leading to the gradual accumulation of lipid peroxides (181). At the same time, dysregulated
iron metabolism can further amplify this process. For example,
transferrin receptor 1-mediated iron uptake, ferritin degradation,
and reduced iron efflux can all increase intracellular free
Fe2+ levels and promote ROS generation through the
Fenton reaction (182). Lipid
metabolic reprogramming also provides key substrates for
ferroptosis. For instance, acyl-CoA synthetase long-chain family
member 4 and lysophosphatidylcholine acyltransferase 3 promote the
formation of polyunsaturated fatty acid-phosphatidylethanolamine,
which can subsequently undergo peroxidation under the catalysis of
lipoxygenases, thereby disrupting membrane stability (183). Once activated, ferroptosis not
only induces chondrocyte injury, but also promotes MMP13 expression
and suppresses type II collagen synthesis, thereby further
aggravating cartilage matrix imbalance and driving OA progression
(184).
Cellular senescence and the SASP it mediates are
increasingly recognized as key drivers of OA progression, and their
regulation involves multiple interconnected pathways related to
cell cycle control, inflammatory signaling and metabolic stress
(191,192). In general, the
p53-p21/p16INK4A-Rb axis is primarily involved in the
initiation of senescence, whereas the mTOR pathway mainly regulates
SASP at the translational level, and NF-κB contributes to the
transcriptional activation of SASP. In addition, signaling
pathways, such as IL-6-STAT3, ROS-MAPK and autophagy-GATA4 also
participate in shaping the SASP regulatory network (193,194).
mTOR signaling also plays a key role in the
regulation of cellular senescence and SASP in OA. It has been
demonstrated that the selective inhibition of mTOR complex 1 can
activate Akt signaling through negative feedback and enhance
autophagic flux, thereby attenuating IL-1β-induced cellular
senescence and reducing the secretion of matrix-degrading SASP
factors (198). By contrast,
lactate accumulation in the OA microenvironment can promote p53 and
p21 expression through the arginase 2-mTOR/ribosomal protein S6
kinase β-1/eukaryotic translation initiation factor 4B signaling
cascade and induce G1/S arrest in synovial cells, while also
enhancing the secretion of multiple inflammatory SASP factors and
aggravating synovial inflammation (199). In addition, fibroblast growth
factor 21 can inhibit mTOR phosphorylation through SIRT1 and
promote transcription factor EB-mediated autophagy activation,
thereby reducing the expression of p16INK4A and p21 and
alleviating chondrocyte senescence, while also suppressing SASP
secretion and promoting cartilage matrix synthesis (200). Taken together, these findings
highlight the critical role of the mTOR-autophagy axis in
regulating senescence and the SASP network in OA.
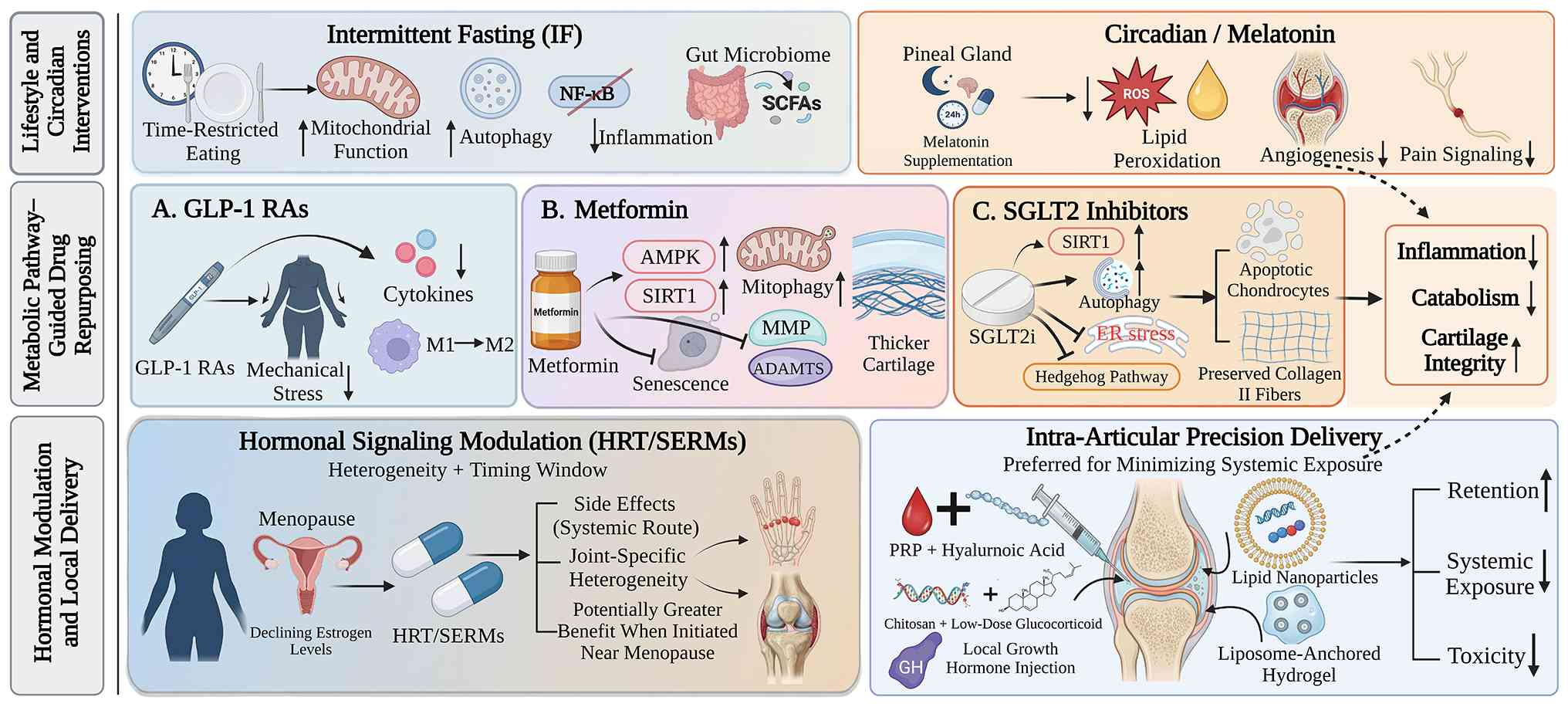
The efficacy and risks of hormone replacement
therapy (HRT) and selective estrogen receptor modulators (SERMs) in
OA exhibit substantial heterogeneity, and their effects are
influenced by factors such as the affected joint site, timing of
intervention, and route of administration. Current evidence
suggests that systemic HRT is associated with an increased risk of
the onset of knee OA and joint replacement (201). By contrast, combined
estrogen-progestin HRT initiated during the early perimenopausal
period may reduce the risk of developing hand OA, although this
potential benefit appears to be largely confined to the treatment
period, and the risk may rise again following discontinuation
(202). A previous
meta-analysis of animal models indicated that estrogen therapy can
upregulate type II collagen expression and reduce the levels of
cartilage degradation markers, including C-terminal telopeptide of
type II collagen and cartilage oligomeric matrix protein, thereby
ameliorating cartilage degeneration (36). SERMs may also exert protective
effects by suppressing cartilage turnover (36). However, clinical studies have
demonstrated that HRT does not confer clear advantages in improving
hand OA-related function, while long-term systemic use may increase
the risk of developing cardiovascular disease and hormone-related
malignancies, which limits its broader application in the treatment
of OA (203,204). Overall, the available clinical
and observational evidence for HRT in OA remains inconsistent and
appears to be influenced by joint site, timing of initiation,
formulation and treatment duration, whereas the evidence supporting
SERMs is still derived mainly from preclinical models.
The therapeutic window is considered a critical
determinant of hormonal intervention strategies. It has been
demonstrated that estrogen therapy initiated during the early
postmenopausal stage can significantly improve cartilage structure
and attenuate joint degeneration, with effects that are clearly
superior to those achieved with late intervention (36). In population-based studies, the
efficacy of HRT also appears to be closely related to formulation
type and duration of use. For example, oral formulations have
exhibited some advantage in reducing the risk of developing hand
OA, whereas patients receiving treatment for ≥5 years tend to
exhibit an increased risk of developing knee OA (202). These findings suggest that the
potential benefits of hormonal therapy in OA depend on precise
population stratification and appropriate timing of
intervention.
Compared with systemic administration,
intra-articular local delivery strategies can increase local drug
concentrations, while minimizing systemic exposure and are
therefore emerging as an important technological direction in the
treatment of OA. In recent years, the application of nanodelivery
systems and hydrogel-based materials has markedly improved
intra-articular retention and the controlled release of therapeutic
agents. For example, lipid nanoparticles can enhance drug
accumulation within the joint cavity and improve the stability and
cellular uptake efficiency of mRNA-based therapeutics (205). Liposome-anchored hydrogels can
achieve sustained release and prolong in vivo activity to
~22 days, while exerting synergistic anti-inflammatory and
pro-chondrogenic effects without evident organ toxicity (206). In addition, combined injection
of platelet-rich plasma and hyaluronic acid has shown more durable
pain relief and functional improvement than either treatment alone
(207), whereas chitosan
combined with low-dose glucocorticoids or local growth hormone
injection has also demonstrated some potential for cartilage
protection and repair (208,209). Overall, local delivery systems
may improve intra-articular retention and reduce systemic exposure.
However, the maturity of evidence varies across platforms.
Platelet-rich plasma combined with HA has been evaluated in
clinical studies, whereas lipid nanoparticles, liposome-anchored
hydrogels, and other advanced delivery systems remain largely
preclinical or early translational platforms that require further
clinical validation (205-209).
The potential benefits of glucagon-like peptide-1
receptor agonists (GLP-1 RAs) in OA extend beyond weight reduction
and may also involve direct joint-protective effects through
multiple weight-independent mechanisms. It has been demonstrated
that GLP-1R is expressed in both human knee chondrocytes and
synovial tissue (210),
providing a molecular basis for local actions. In terms of
weight-dependent effects, GLP-1 RAs promote substantial weight loss
by suppressing appetite and delaying gastric emptying, thereby
reducing mechanical stress on weight-bearing joints and attenuating
obesity-related systemic inflammation (211). In terms of weight-independent
mechanisms, these agents can suppress NF-κB signaling and reduce
the release of pro-inflammatory mediators, such as TNF-α and IL-1β,
while also promoting the polarization of macrophages from the M1 to
the M2 phenotype and downregulating the expression of
cartilage-degrading enzymes, such as MMP-3 and MMP-13 (210-212). Clinically, the STEP-9 trial
revealed that after 68 weeks of treatment with semaglutide at 2.4
mg weekly, patients with obesity and OA achieved an additional
14.1-point improvement in the Western Ontario and McMaster
Universities Osteoarthritis Index (WOMAC) pain score compared with
the patients treated with the placebo (213). In addition, a cohort study
suggested that the long-term use of GLP-1 RA was associated with a
slower knee cartilage loss and a lower risk of joint replacement
(214). Clinically, the STEP-9
trial, reported by Bliddal et al (213), demonstrated that the weekly
administration of semaglutide at 2.4 mg for 68 weeks produced an
additional 14.1-point improvement in the Western Ontario and
McMaster Universities Osteoarthritis Index pain score compared with
placebo in individuals with obesity and knee osteoarthritis.
Metformin, as a key activator of the AMPK signaling
pathway, exerts multitarget metabolic regulatory effects and is
considered to have substantial repurposing potential in metabolic
OA. Available evidence indicates that its protective effects depend
primarily on activation of the AMPKα1 isoform (215). In experimental models,
metformin can inhibit chondrocyte senescence by regulating the
inducible nitric oxide synthase/peroxynitrite/p53 signaling axis,
while also promoting chondrocyte proliferation through the
downregulation of microRNA-34a and the release of its inhibitory
effect on SIRT1 (216,217). In addition, metformin can
activate PINK1/Parkin-mediated mitophagy and promote the clearance
of damaged mitochondria, while suppressing the expression of
matrix-degrading enzymes from the MMP and ADAMTS families, thereby
attenuating cartilage degeneration (218,219). In animal models, metformin has
been shown to increase articular cartilage thickness and reduce
cartilage damage, with more pronounced protective effects in
obesity-related or high-fat diet-associated OA models (215,219). Clinical studies further suggest
that the long-term use of metformin is associated with a lower rate
of knee cartilage volume loss and may further reduce the risk of
joint replacement when used in combination with cyclooxygenase-2
inhibitors (220,221). Taken together, preclinical
evidence and observational cohort studies support the potential
disease-modifying effects of metformin in metabolic OA; however,
randomized OA-specific clinical trials are still required to
confirm its therapeutic efficacy.
The potential value of sodium-glucose cotransporter
2 (SGLT2) inhibitors in OA intervention lies mainly in their
anti-inflammatory and oxidative stress-modulating effects. Studies
have shown that agents such as dapagliflozin can activate SIRT1
signaling and inhibit protein kinase R-like endoplasmic reticulum
kinase/eukaryotic translation initiation factor 2α/C/EBP homologous
protein-mediated endoplasmic reticulum stress, thereby reducing
chondrocyte apoptosis and producing a chondroprotective phenotype
characterized by increased type II collagen expression and
decreased MMP13 and ADAMTS5 levels (222). In addition, these drugs can
regulate the balance between autophagy and apoptosis through the
activation of the AMPK pathway, upregulate autophagy-related
proteins, such as Beclin 1 and ULK1, and suppress the expression of
key Hedgehog pathway molecules, including sonic hedgehog and
glioma-associated oncogene homolog 1 (223). In vivo studies have
further indicated that SGLT2 inhibitors can reduce the serum levels
of IL-1β, IL-6 and cartilage oligomeric matrix protein in animal
models of OA, and improve joint space narrowing and osteophyte
formation, without obvious toxicity within the reported dose range
(222,223). Moreover, their combined use
with methotrexate may further enhance anti-inflammatory and
joint-protective effects, providing a novel potential therapeutic
strategy for patients with OA accompanied by metabolic
abnormalities (223). On the
whole, the majority of current evidence for SGLT2 inhibitors in OA
is derived from chondrocyte studies and animal models, and
prospective clinical validation in patients with OA is still
lacking.
Melatonin, a crucial endogenous hormone that
regulates circadian rhythm, has exhibited multidimensional
protective potential in OA intervention. It has been demonstrated
that melatonin can activate the PI3K/Akt-ERK-miR-185a signaling
axis through the melatonin receptor 1, thereby suppressing synovial
inflammation and angiogenesis and reducing the release of TNF-α,
IL-8 and VEGF (66). In
addition, melatonin can inhibit chondrocyte ferroptosis through the
regulation of the NOX4/GRP78/GPX4 axis, reduce the accumulation of
ROS and lipid peroxidation, and improve mitochondrial function
(65). In terms of cartilage
homeostasis, melatonin can regulate matrix metabolism through the
SIRT1/NF-κB and TGF-β1/Smad2 pathway, promote the expression of
Col2a1 and aggrecan, and suppress MMP activity (224). At the same time, melatonin can
downregulate pain-related neuro mediators, such as nerve growth
factor and calcitonin gene-related peptide, and has been shown to
be associated with a reduced risk of joint replacement in patients
with OA (72). In recent years,
multiple delivery systems, such as melatonin-loaded poly
(lactic-co-glycolic acid) nanoparticles functionalized with a
collagen-binding peptide (MT@PLGA-COLBP), have been developed to
improve the in vivo bioavailability of melatonin, and animal
studies have demonstrated favorable long-term tolerability without
evident organ toxicity (73,75). Therefore, although melatonin has
exhibited consistent protective effects in experimental OA models
and limited observational evidence in humans, its disease-modifying
efficacy, optimal dosing, and target population remain to be
established in prospective clinical trials.
Intermittent fasting is regarded as a potential
non-pharmacological intervention through its effects on metabolic
remodeling and inflammatory regulation. Intermittent fasting can
induce a shift in metabolic substrate utilization and activate the
AMPK/SIRT1 signaling axis, thereby improving mitochondrial function
and alleviating insulin resistance (225). At the same time, intermittent
fasting can reduce the release of pro-inflammatory mediators, such
as TNF-α and IL-1β by suppressing NF-κB signaling, and can promote
the clearance of damaged mitochondria through the activation of
autophagy (225). Recent
research further suggests that intermittent fasting can inhibit
osteocyte-derived neuropeptide Y-mediated pro-inflammatory
macrophage polarization and osteoclastogenesis, thereby weakening
the pathological amplification process linking inflammation, bone
destruction and cartilage injury (226). In addition, intermittent
fasting may lower systemic inflammation through the gut-joint axis
by reshaping gut microbial composition and promoting the production
of SCFAs (227). In animal
models and preliminary clinical studies, intermittent fasting has
been reported to preserve cartilage structural integrity, suppress
osteophyte formation, and improve pain and motor function. These
metabolic and joint-protective effects may be further enhanced when
intermittent fasting is combined with a high-protein diet (225,226). However, current evidence for
intermittent fasting in OA is still based mainly on animal models
and early clinical observations, and long-term human studies are
warranted to determine its efficacy, safety, adherence and
applicability across different OA phenotypes.
OA exhibits marked clinical and biological
heterogeneity, which is also a key reason why conventional
empirical treatments often produce variable outcomes.
Classification based solely on the affected joint site or imaging
findings often fails to capture the dominant pathogenic mechanisms
underlying the disease (228).
In recent years, increasing evidence has indicated that systemic
metabolic dysregulation is a critical driver of OA, and that
substantial differences in metabolic and endocrine phenotypes exist
among patients. However, these factors have rarely been
incorporated into traditional therapeutic decision-making
frameworks, resulting in a lack of mechanism-oriented interventions
(229). As the research
paradigm of OA has gradually expanded from that of a local joint
disorder to a systemic disease associated with disrupted whole-body
metabolic homeostasis (14), the
development of an endocrine-metabolic phenotyping framework may
provide a basis for mechanism-oriented stratification and future
therapeutic exploration.
From this perspective, patients with OA may be
provisionally grouped according to dominant endocrine-metabolic
features into two candidate phenotypic patterns, namely a hormone
deficiency-dominant pattern and a metabolic syndrome-dominant
pattern. The hormone deficiency-dominant pattern is primarily
associated with abnormalities in endocrine regulation involving sex
hormones, thyroid hormones, vitamin D, or PTH. Its pathological
features are more inclined toward chondrocyte senescence,
dysregulated subchondral bone remodeling, and impaired cartilage
matrix synthesis, and it is commonly observed in postmenopausal
women or individuals with endocrine disorders (230-232). By contrast, the metabolic
syndrome-dominant pattern is driven mainly by systemic metabolic
disturbances such as obesity, insulin resistance and dyslipidemia,
with key pathological processes including chronic low-grade
inflammation, abnormal adipokine signaling and oxidative stress
(233-235). For example, signaling through
the receptor for advanced glycation end products and related
inflammatory responses can promote cartilage degradation, while
metabolic abnormalities mediated by senescent immune cells may
further aggravate damage to both the joint and musculoskeletal
systems (236,237). In clinical practice, some
patients may simultaneously exhibit hormone deficiency and
metabolic abnormalities, thereby forming a mixed
endocrine-metabolic pattern that requires more refined
stratification and management strategies. This framework should be
regarded as a hypothesis-generating model rather than a validated
clinical taxonomy.
With the development of multi-omics technologies
and artificial intelligence (AI), molecular feature-based
mechanism-oriented phenotyping and stratified therapeutic
exploration may become increasingly feasible. Integrative
multi-omics analyses can identify potential key molecules and
regulatory networks through datasets such as transcriptomics and
immunomics, including candidate targets, such as Jun proto-oncogene
and VEGFA, as well as molecular biomarkers related to autophagy or
inflammation, such as V-Erb-B2 avian erythroblastic leukemia viral
oncogene homolog 2, thereby supporting the identification of
candidate pathological patterns (238,239). At the same time, AI
technologies can be used to automatically assess structural joint
changes and assist therapeutic decision-making. For example,
imaging analysis tools and clinical decision support systems may
help predict disease progression or treatment response (240). In addition, federated learning
provides a novel technical pathway for multicenter data integration
and may improve model generalizability while preserving data
privacy (241). Overall, the
synergistic application of multi-omics and AI provides an essential
technical foundation for mechanism-oriented OA phenotyping,
stratified intervention design and treatment response assessment. A
summary of the therapeutic strategies targeting the
endocrine-metabolic axis in OA is illustrated in Fig. 3 and presented in Table III.
In recent years, the conceptual framework of OA has
gradually shifted from that of a local degenerative disorder driven
primarily by mechanical wear to that of a whole-joint disease
jointly driven by local tissue injury and systemic
endocrine-metabolic disequilibrium (7,242,243). As summarized in the present
review, hormonal dysregulation, metabolic syndrome-related
abnormalities, and immunometabolic stress do not act in isolation.
Instead, they are tightly interconnected through key pathological
processes including inflammatory amplification, disordered energy
metabolism, mitochondrial dysfunction, PCD, and cellular
senescence, and together they ultimately drive cartilage
degeneration, synovial inflammation, and dysregulated subchondral
bone remodeling (183,244,245). This evolving perspective not
only broadens the systemic pathological landscape of OA, but also
highlights its substantial biological heterogeneity. In particular,
these provisional phenotypic patterns based on hormonal status and
metabolic features, namely the hormone deficiency-dominant pattern
and the metabolic syndrome-dominant pattern, may help explain, at
the mechanistic level, the differences in disease progression,
clinical phenotype, and therapeutic response observed among
patients (230,246,247).
Although increasing evidence supports the role of
the endocrine-metabolic axis in OA, several critical bottlenecks
still hinder its clinical translation. First, the majority of
existing models focus on a single mechanical insult, a single
hormonal abnormality, or a single metabolic factor, and therefore
remain insufficient to fully recapitulate the complex disease
course of human OA, which is characterized by long-term
accumulation and dynamic interaction of multiple pathogenic
factors. Second, current clinical studies still provide an
inadequate characterization of patient heterogeneity. This is
particularly evident in the limited integration of evidence
regarding sex differences, menopausal status, the synergistic
effects of obesity and insulin resistance, and the identification
of dominant mechanisms across candidate phenotypic patterns
(21,127,248). In addition, endocrine-metabolic
interventions, including GLP-1 receptor agonists, metformin,
melatonin and intermittent fasting, have shown encouraging effects
in preclinical studies, observational analyses or early clinical
settings (213-215). Intra-articular local delivery
systems, such as lipid nanoparticle- and hydrogel-based platforms,
may also enhance joint retention and reduce systemic exposure
(205-207). However, there remains a
substantial gap between mechanistic association and confirmed
clinical benefit, suggesting that future OA research should move
beyond descriptive correlations and establish mechanistic
frameworks that are testable, stratifiable and translatable.
Despite these advances, several limitations of the
current available evidence should be acknowledged. The
translational evidence for endocrine-metabolic interventions
remains uneven. For example, melatonin has been shown to exert
protective effects in experimental OA models and limited
observational evidence in humans; however, long-term prospective
human data are still insufficient, and its optimal dosage,
treatment duration and target populations remain unclear (72-78). Similarly, evidence for hormonal
interventions, including HRT and SERMs, remains heterogeneous and
appears to be influenced by joint site, menopausal timing,
formulation, treatment duration and long-term safety
considerations, including cardiovascular and hormone-related risks
(201-204). In addition, although
multi-omics profiling, imaging analysis, and AI-based prediction
models have identified promising molecular signatures and
decision-support tools, few clinical studies have prospectively
integrated endocrine indicators, metabolic status, imaging
phenotypes, and multi-omics data to validate mechanism-based OA
stratification (238-241,249-251). Finally, the hormone
deficiency-dominant and metabolic syndrome-dominant patterns
discussed in the present review should still be regarded as
provisional phenotypic patterns rather than validated clinical
categories. Future studies are required to define operational
thresholds, predictive biomarkers, and treatment-response
indicators before these patterns can be used to guide clinical
decision-making (230,246,247).
Future OA research may no longer focus on
identifying a single intervention target applicable to all
patients, but rather on building an integrated classification
framework based on hormonal status, metabolic characteristics and
patterns of immunometabolic stress. On this basis, multi-omics
technologies may further identify key molecular networks and
biomarkers for different candidate phenotypic patterns (249,250), while AI, machine learning and
clinical decision support tools may improve the feasibility of
patient stratification, therapeutic response prediction, and
dynamic monitoring (240,251). Overall, understanding OA
through the lens of endocrine-metabolic interaction and advancing
mechanism-oriented intervention through stratified classification
may represent a key direction for future OA research and clinical
management. It may also provide a useful reference for
translational studies of other metabolism-related osteoarticular
diseases.
Not applicable.
XC was involved in the conceptualization of the
review, supervision, writing, reviewing and editing of the
manuscript, and project administration. RY, HZ and QX were involved
in the literature search, evidence extraction and organization,
interpretation of data from the literature, and in the writing of
the original draft of the manuscript. YX and YL were involved in
the literature search, evidence organization, figure preparation,
and in the writing, reviewing and editing of the manuscript. All
authors have read and approved the final manuscript. Data
authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present review was supported by the Nanchang University
Second Affiliated Hospital In-Hospital Project (grant no.
2023efyC02), the Nanchang University Medical Interdisciplinary
Innovation Fund Project (grant no. 2024JC002), the Clinical
Research Project of the Second Affiliated Hospital of Nanchang
University (grant no. 2024efyA01), the National Natural Science
Foundation of China Incubation Project (grant no. 2023YNFY12009),
the National College Students' Innovative Entrepreneurial Training
Plan Program (grant no. 202410403067), the Innovation and
Entrepreneurship Training Program for College Students in Jiangxi
Province (grant no. S202410403035), and the Doctoral Start-up Fund
(grant no. B3477).
|
1
|
Zhu C, Zhang L, Ding X, Wu W and Zou J:
Non-coding RNAs as regulators of autophagy in chondrocytes:
Mechanisms and implications for osteoarthritis. Ageing Res Rev.
99:1024042024. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tang S, Zhang C, Oo WM, Fu K, Risberg MA,
Bierma-Zeinstra SM, Neogi T, Atukorala I, Malfait AM, Ding C and
Hunter DJ: Osteoarthritis. Nat Rev Dis Primers. 11:102025.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cao F, Xu Z, Li XX, Fu ZY, Han RY, Zhang
JL, Wang P, Hou S and Pan HF: Trends and cross-country inequalities
in the global burden of osteoarthritis, 1990-2019: A
population-based study. Ageing Res Rev. 99:1023822024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Courties A, Kouki I, Soliman N, Mathieu S
and Sellam J: Osteoarthritis year in review 2024: Epidemiology and
therapy. Osteoarthritis Cartilage. 32:1397–1404. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Turkiewicz A, Petersson IF, Björk J,
Hawker G, Dahlberg LE, Lohmander LS and Englund M: Current and
future impact of osteoarthritis on health care: A population-based
study with projections to year 2032. Osteoarthritis Cartilage.
22:1826–1832. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Leifer VP, Katz JN and Losina E: The
burden of OA-health services and economics. Osteoarthritis
Cartilage. 30:10–16. 2022. View Article : Google Scholar
|
|
7
|
Katz JN, Arant KR and Loeser RF: Diagnosis
and treatment of Hip and knee osteoarthritis: A review. JAMA.
325:568–578. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kloppenburg M, Namane M and Cicuttini F:
Osteoarthritis. Lancet. 405:71–85. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wieland HA, Michaelis M, Kirschbaum BJ and
Rudolphi KA: Osteoarthritis-an untreatable disease? Nat Rev Drug
Discov. 4:331–344. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou R, Fu W, Vasylyev D, Waxman SG and
Liu CJ: Ion channels in osteoarthritis: Emerging roles and
potential targets. Nat Rev Rheumatol. 20:545–564. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pinals RS: Mechanisms of joint
destruction, pain and disability in osteoarthritis. Drugs. 52(Suppl
3): S14–S20. 1996. View Article : Google Scholar
|
|
12
|
Goldring SR and Goldring MB: Changes in
the osteochondral unit during osteoarthritis: Structure, function
and cartilage-bone crosstalk. Nat Rev Rheumatol. 12:632–644. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Knights AJ, Redding SJ and Maerz T:
Inflammation in osteoarthritis: The latest progress and ongoing
challenges. Curr Opin Rheumatol. 35:128–134. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mobasheri A, Rayman MP, Gualillo O, Sellam
J, van der Kraan P and Fearon U: The role of metabolism in the
pathogenesis of osteoarthritis. Nat Rev Rheumatol. 13:302–311.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang J, Xin Y, Dong Z, Li S and Yang G:
The interplay between the immune microenvironment and bone aging:
From molecular mechanisms to therapeutic interventions. Exp
Gerontol. 212:1129742025. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tang Y, Wang Z, Cao J and Tu Y: Bone-brain
crosstalk in osteoarthritis: Pathophysiology and interventions.
Trends Mol Med. 31:281–295. 2025. View Article : Google Scholar
|
|
17
|
Longo UG, Lalli A, Bandini B, de Sire R,
Angeletti S, Lustig S, Ammendolia A, Budhiparama NC and de Sire A:
Role of the gut microbiota in osteoarthritis, rheumatoid arthritis,
and spondylarthritis: An update on the gut-joint axis. Int J Mol
Sci. 25:32422024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Han Z, Wang K, Ding S and Zhang M:
Cross-talk of inflammation and cellular senescence: A new insight
into the occurrence and progression of osteoarthritis. Bone Res.
12:692024. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Veronese N, Cooper C, Reginster JY,
Hochberg M, Branco J, Bruyère O, Chapurlat R, Al-Daghri N, Dennison
E, Herrero-Beaumont G, et al: Type 2 diabetes mellitus and
osteoarthritis. Semin Arthritis Rheum. 49:9–19. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Scotece M and Mobasheri A: Leptin in
osteoarthritis: Focus on articular cartilage and chondrocytes. Life
Sci. 140:75–78. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Collins KH, Lenz KL, Pollitt EN, Ferguson
D, Hutson I, Springer LE, Oestreich AK, Tang R, Choi YR, Meyer GA,
et al: Adipose tissue is a critical regulator of osteoarthritis.
Proc Natl Acad Sci USA. 118:e20210961182021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xie C and Chen Q: Adipokines: New
therapeutic target for osteoarthritis? Curr Rheumatol Rep.
21:712019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ait Eldjoudi D, Cordero Barreal A,
Gonzalez-Rodríguez M, Ruiz-Fernández C, Farrag Y, Farrag M, Lago F,
Capuozzo M, Gonzalez-Gay MA, Mera Varela A, et al: Leptin in
osteoarthritis and rheumatoid arthritis: Player or bystander? Int J
Mol Sci. 23:28592022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gulati M, Dursun E, Vincent K and Watt FE:
The influence of sex hormones on musculoskeletal pain and
osteoarthritis. Lancet Rheumatol. 5:e225–e238. 2023. View Article : Google Scholar
|
|
25
|
Lan Y, Zheng Q, Li M, Chen J, Huang D and
Lin L: Associations between surrogate insulin resistance indexes
and osteoarthritis: NHANES 2003-2016. Sci Rep. 15:15782025.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sniekers YH, Weinans H, van Osch GJ and
van Leeuwen JP: Oestrogen is important for maintenance of cartilage
and subchondral bone in a murine model of knee osteoarthritis.
Arthritis Res Ther. 12:R1822010. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Roman-Blas JA, Castañeda S, Largo R and
Herrero-Beaumont G: Osteoarthritis associated with estrogen
deficiency. Arthritis Res Ther. 11:2412009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ootake T, Ishii T, Sueishi K, Watanabe A,
Ishizuka Y, Amano K, Nagao M, Nishimura K and Nishii Y: Effects of
mechanical stress and deficiency of dihydrotestosterone or
17β-estradiol on temporomandibular joint osteoarthritis in mice.
Osteoarthritis Cartilage. 29:1575–1589. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gilmer G, Iijima H, Hettinger ZR, Jackson
N, Bergmann J, Bean AC, Shahshahan N, Creed E, Kopchak R, Wang K,
et al: Menopause-induced 17β-estradiol and progesterone loss
increases senescence markers, matrix disassembly and degeneration
in mouse cartilage. Nat Aging. 5:65–86. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hu Z, Chen L, Zhao J, Zhang W, Jin Z, Sun
Y, Li Z, Chang B, Shen P and Yang Y: Lipoxin A4
ameliorates knee osteoarthritis progression in rats by antagonizing
ferroptosis through activation of the ESR2/LPAR3/Nrf2 axis in
synovial fibroblast-like synoviocytes. Redox Biol. 73:1031432024.
View Article : Google Scholar
|
|
31
|
Ushiyama T, Ueyama H, Inoue K, Ohkubo I
and Hukuda S: Expression of genes for estrogen receptors alpha and
beta in human articular chondrocytes. Osteoarthritis Cartilage.
7:560–566. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kinney RC, Schwartz Z, Week K, Lotz MK and
Boyan BD: Human articular chondrocytes exhibit sexual dimorphism in
their responses to 17beta-estradiol. Osteoarthritis Cartilage.
13:330–337. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Srikanth VK, Fryer JL, Zhai G, Winzenberg
TM, Hosmer D and Jones G: A meta-analysis of sex differences
prevalence, incidence and severity of osteoarthritis.
Osteoarthritis Cartilage. 13:769–781. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ham KD, Loeser RF, Lindgren BR and Carlson
CS: Effects of long-term estrogen replacement therapy on
osteoarthritis severity in cynomolgus monkeys. Arthritis Rheum.
46:1956–1964. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yasuoka T, Nakashima M, Okuda T and
Tatematsu N: Effect of estrogen replacement on temporomandibular
joint remodeling in ovariectomized rats. J Oral Maxillofac Surg.
58:189–197. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gilmer G, Bean AC, Iijima H, Jackson N,
Thurston RC and Ambrosio F: Uncovering the 'riddle of femininity'
in osteoarthritis: A systematic review and meta-analysis of
menopausal animal models and mathematical modeling of estrogen
treatment. Osteoarthritis Cartilage. 31:447–457. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jin X, Wang BH, Wang X, Antony B, Zhu Z,
Han W, Cicuttini F, Wluka AE, Winzenberg T, Blizzard L, et al:
Associations between endogenous sex hormones and MRI structural
changes in patients with symptomatic knee osteoarthritis.
Osteoarthritis Cartilage. 25:1100–1106. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xu H, Xiao W, Ding C, Zou J, Zhou D, Wang
J, Ding L, Jin C, Sun L and Li Y: Global burden of osteoarthritis
among postmenopausal women in 204 countries and territories: A
systematic analysis for the global burden of disease study 2021.
BMJ Glob Health. 10:e0171982025. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Almeida M, Laurent MR, Dubois V, Claessens
F, O'Brien CA, Bouillon R, Vanderschueren D and Manolagas SC:
Estrogens and androgens in skeletal physiology and pathophysiology.
Physiol Rev. 97:135–187. 2017. View Article : Google Scholar
|
|
40
|
Khosla S and Monroe DG: Regulation of bone
metabolism by sex steroids. Cold Spring Harb Perspect Med.
8:a0312112018. View Article : Google Scholar :
|
|
41
|
Jiang Z, Yao X, Yang Y, Tang F, Ma W, Yao
X and Lan W: The causal impact of bioavailable testosterone levels
on osteoarthritis: A bidirectional Mendelian randomized study. BMC
Musculoskelet Disord. 26:3872025. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ma N and Gao F: Correlation between low
testosterone levels and the risk of osteoarthritis: A
cross-sectional analysis of NHANES data (2011-2016). BMC
Musculoskelet Disord. 26:232025. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Szilagyi IA, Schiphof D, Chaker L, Boer
CG, Aribas E, Kavousi M, Ikram MA, Bierma-Zeinstra SMA and van
Meurs JBJ: Associations between testosterone and knee and hand
osteoarthritis among males and females from the general population.
Osteoarthritis Cartilage. 33:1237–1245. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang J, Yin J, Zhang X, Wang J, Xing X, Tu
J and Cai G: The association between endogenous sex hormones and
knee osteoarthritis in women: A population-based cohort study.
Osteoarthritis Cartilage. 33:1229–1236. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Masoud O, Morris L, Al-Hamdani M,
Al-Haidose A and Abdallah AM: Association between clinical
laboratory indicators and WOMAC scores in Qatar Biobank
participants: The impact of testosterone and fibrinogen on pain,
stiffness, and functional limitation. Scand J Pain. 25:2025.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Freystaetter G, Fischer K, Orav EJ, Egli
A, Theiler R, Münzer T, Felson DT and Bischoff-Ferrari HA: Total
serum testosterone and western ontario and mcmaster universities
osteoarthritis index pain and function among older men and women
with severe knee osteoarthritis. Arthritis Care Res (Hoboken).
72:1511–1518. 2020. View Article : Google Scholar :
|
|
47
|
Tian X and Zhang B: The association
between sex hormones and prevalence of OA in US adults. Front Med
(Lausanne). 11:14252102024. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Pan Q, O'Connor MI, Coutts RD, Hyzy SL,
Olivares-Navarrete R, Schwartz Z and Boyan BD: Characterization of
osteoarthritic human knees indicates potential sex differences.
Biol Sex Differ. 7:272016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Parmar RP, Cronen A, Hui C, Stickels M,
Lederman E and Shah A: Testosterone replacement therapy is not
associated with greater revision rates in reverse total shoulder
arthroplasty. J Clin Med. 14:13412025. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cohn RM, Ganz MP and Scuderi GR:
Testosterone replacement therapy in orthopaedic surgery. J Am Acad
Orthop Surg. 32:331–338. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bassett JH and Williams GR: Role of
thyroid hormones in skeletal development and bone maintenance.
Endocr Rev. 37:135–187. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Li C, Tu Y, Rong R, Zhang Z, Chen W, Long
L, Zhang Y, Wang C, Pan B, Wu X, et al: Association of thyroid
hormone with osteoarthritis: From mendelian randomization and RNA
sequencing analysis. J Orthop Surg Res. 19:4292024. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
He Z, Gong Z, Jiao S, Xiong W, Hao X, Cui
J and Zhang J: Genetic predisposition to thyrotoxicosis and onset
of knee osteoarthritis. Front Endocrinol (Lausanne).
15:13640272024. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Xu Y, Szilagyi IA, Boer CG,
Sedaghati-Khayat B, Visser WE, van Meurs JB and Chaker L:
Association between thyroid function and osteoarthritis: A
population-based cohort study. Osteoarthritis Cartilage.
33:888–896. 2025. View Article : Google Scholar
|
|
55
|
Chen S, Sun X, Zhou G, Jin J and Li Z:
Association between sensitivity to thyroid hormone indices and the
risk of osteoarthritis: An NHANES study. Eur J Med Res. 27:1142022.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Pörings AS, Lowin T, Dufner B, Grifka J
and Straub RH: A thyroid hormone network exists in synovial
fibroblasts of rheumatoid arthritis and osteoarthritis patients.
Sci Rep. 9:132352019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Luongo C, Dentice M and Salvatore D:
Deiodinases and their intricate role in thyroid hormone
homeostasis. Nat Rev Endocrinol. 15:479–488. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Deng Y, Han Y, Gao S, Dong W and Yu Y: The
physiological functions and polymorphisms of type II deiodinase.
Endocrinol Metab (Seoul). 38:190–202. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Bomer N, den Hollander W, Ramos YF, Bos
SD, van der Breggen R, Lakenberg N, Pepers BA, van Eeden AE,
Darvishan A, Tobi EW, et al: Underlying molecular mechanisms of
DIO2 susceptibility in symptomatic osteoarthritis. Ann Rheum Dis.
74:1571–1579. 2015. View Article : Google Scholar :
|
|
60
|
Dayan CM and Panicker V: Novel insights
into thyroid hormones from the study of common genetic variation.
Nat Rev Endocrinol. 5:211–218. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Mohajer B, Moradi K, Guermazi A, Mammen
JSR, Hunter DJ, Roemer FW and Demehri S: Levothyroxine use and
longitudinal changes in thigh muscles in at-risk participants for
knee osteoarthritis: Preliminary analysis from osteoarthritis
initiative cohort. Arthritis Res Ther. 25:582023. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jahanban-Esfahlan R, Mehrzadi S, Reiter
RJ, Seidi K, Majidinia M, Baghi HB, Khatami N, Yousefi B and
Sadeghpour A: Melatonin in regulation of inflammatory pathways in
rheumatoid arthritis and osteoarthritis: Involvement of circadian
clock genes. Br J Pharmacol. 175:3230–3238. 2018. View Article : Google Scholar
|
|
63
|
Hossain FM and Hong Y, Jin Y, Choi J and
Hong Y: Physiological and pathological role of circadian hormones
in osteoarthritis: Dose-dependent or time-dependent? J Clin Med.
8:14152019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Hosseinzadeh A, Kamrava SK, Joghataei MT,
Darabi R, Shakeri-Zadeh A, Shahriari M, Reiter RJ, Ghaznavi H and
Mehrzadi S: Apoptosis signaling pathways in osteoarthritis and
possible protective role of melatonin. J Pineal Res. 61:411–425.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wang Q, Qi B, Shi S, Jiang W, Li D, Jiang
X and Yi C: Melatonin alleviates osteoarthritis by regulating NADPH
oxidase 4-induced ferroptosis and mitigating mitochondrial
dysfunction. J Pineal Res. 76:e129922024. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Liu SC, Tsai CH, Wang YH, Su CM, Wu HC,
Fong YC, Yang SF and Tang CH: Melatonin abolished proinflammatory
factor expression and antagonized osteoarthritis progression in
vivo. Cell Death Dis. 13:2152022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zhao Z, Bi B, Cheng G, Zhao Y, Wu H, Zheng
M and Cao Z: Melatonin ameliorates osteoarthritis rat cartilage
injury by inhibiting matrix metalloproteinases and JAK2/STAT3
signaling pathway. Inflammopharmacology. 31:359–368. 2023.
View Article : Google Scholar
|
|
68
|
Ke C, Li H, Yang D, Ying H, Zhu H, Wang J,
Xu J and Wang L: Melatonin attenuates the progression of
osteoarthritis in rats by inhibiting inflammation and related
oxidative stress on the surface of knee cartilage. Orthop Surg.
14:2230–2237. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhou X, Zhang Y, Hou M, Liu H, Yang H,
Chen X, Liu T, He F and Zhu X: Melatonin prevents cartilage
degradation in early-stage osteoarthritis through activation of
miR-146a/NRF2/HO-1 axis. J Bone Miner Res. 37:1056–1072. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Guan H, Cao R, Zhang J, Liu C, Duan X,
Zhao Y, Xing F, Kong N, Li Y, Wu Z, et al: Melatonin attenuates
synovial hyperplasia, inflammation, and fibrosis and postpones
osteoarthritis progression. FASEB J. 40:e712952026. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Li S, Si H, Xu J, Liu Y and Shen B: The
therapeutic effect and mechanism of melatonin on osteoarthritis:
From the perspective of non-coding RNAs. Front Genet.
13:9689192022. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Li H, Zhou B, Wu J, Zhang Y, Zhang W,
Doherty M, Deng X, Wang N, Xie D, Wang Y, et al: Melatonin is a
potential novel analgesic agent for osteoarthritis: Evidence from
cohort studies in humans and preclinical research in rats. J Pineal
Res. 76:e129452024. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Liang H, Yan Y, Sun W, Ma X, Su Z, Liu Z,
Chen Y and Yu B: Preparation of melatonin-loaded nanoparticles with
targeting and sustained release function and their application in
osteoarthritis. Int J Mol Sci. 24:87402023. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Xiong Z, Peng G, Deng J, Liu M, Ning X,
Zhuang Y, Yang H and Sun H: Therapeutic targets and potential
delivery systems of melatonin in osteoarthritis. Front Immunol.
15:13319342024. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Paulino Silva KM, de Sousa FL, Alves ACB,
Rocha PA, da Costa HNAF, Ferreira WR, Reis TS, de Oliveira TKB,
Cabral Batista SR, Lapa Neto CJC, et al: Chondroprotective effect
of melatonin and strontium ranelate in animal model of
osteoarthritis. Heliyon. 7:e067602021. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhang Y, Liu T, Yang H, He F and Zhu X:
Melatonin: A novel candidate for the treatment of osteoarthritis.
Ageing Res Rev. 78:1016352022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Lu KH, Lu PW, Lu EW, Tang CH, Su SC, Lin
CW and Yang SF: The potential remedy of melatonin on
osteoarthritis. J Pineal Res. 71:e127622021. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Bagherifard A, Hosseinzadeh A, Koosha F,
Sheibani M, Karimi-Behnagh A, Reiter RJ and Mehrzadi S: Melatonin
and bone-related diseases: An updated mechanistic overview of
current evidence and future prospects. Osteoporos Int.
34:1677–1701. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Villafañe JH, Pedersini P, Bertozzi L,
Drago L, Fernandez-Carnero J, Bishop MD and Berjano P: Exploring
the relationship between chronic pain and cortisol levels in
subjects with osteoarthritis: Results from a systematic review of
the literature. Osteoarthritis Cartilage. 28:572–580. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Carlesso LC, Sturgeon JA and Zautra AJ:
Exploring the relationship between disease-related pain and
cortisol levels in women with osteoarthritis. Osteoarthritis
Cartilage. 24:2048–2054. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Mehta O, Vijay A, Gohir SA, Kelly T, Zhang
W, Doherty M, Walsh DA, Aithal G and Valdes AM: Serum metabolome
analysis identified amino-acid metabolism associated with pain in
people with symptomatic knee osteoarthritis-a cross-sectional
study. J Pain. 24:1251–1261. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Paschali M, Lazaridou A, Paschalis T,
Moradian JR, Sadora J, Vilsmark ES and Edwards RR: Individual
variation in diurnal cortisol in patients with knee osteoarthritis:
Clinical correlates. Int J Psychophysiol. 167:1–6. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Galuh S, Faught E, Klaassen I, Koorneef
LL, Brinks J, van Dijk EHC, Elewaut D, Schlingemann RO, Schaaf MJM,
Boon CJF and Meijer OC: The glucocorticoid receptor is affected by
its target ZBTB16 in a dissociated manner. J Endocrinol.
266:e2402832025. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Huang G, Zhong Y, Li W, Liao W and Wu P:
Causal relationship between parathyroid hormone and the risk of
osteoarthritis: A mendelian randomization study. Front Genet.
12:6869392021. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Qu Z, Yang F, Yan Y, Huang J, Zhao J, Hong
J, Li S, Jiang G, Wang W and Yan S: A Mendelian randomization study
on the role of serum parathyroid hormone and 25-hydroxyvitamin D in
osteoarthritis. Osteoarthritis Cartilage. 29:1282–1290. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Zhang H, Bei M, Zheng Z, Liu N, Cao X,
Xiao Y, Lian Q, Wang Y, Hou X and Tian F: Parathyroid hormone
(1-34) attenuates cartilage degradation and preserves subchondral
bone micro-architecture in rats with patella
baja-induced-patellofemoral joint osteoarthritis. Calcif Tissue
Int. 111:87–95. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Shao LT, Gou Y, Fang JK, Hu YP, Lian QQ,
Yang Z, Zhang YY, Wang YD, Tian FM and Zhang L: The protective
effects of parathyroid hormone (1-34) on cartilage and subchondral
bone through down-regulating JAK2/STAT3 and WNT5A/ROR2 in a
collagenase-induced osteoarthritis mouse model. Orthop Surg.
13:1662–1672. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Shao L, Ding L, Li W, Zhang C, Xia Y, Zeng
M, Ye Z and Deng DYB: Let-7a-5p derived from parathyroid hormone
(1-34)-preconditioned BMSCs exosomes delays the progression of
osteoarthritis by promoting chondrocyte proliferation and
migration. Stem Cell Res Ther. 16:2992025. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Zhang M, Hu W, Cai C, Wu Y, Li J and Dong
S: Advanced application of stimuli-responsive drug delivery system
for inflammatory arthritis treatment. Mater Today Bio.
14:1002232022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Swailem K, Sadhan M, Al-Mashramah GA and
Saghir MA: Association between vitamin D deficiency, inflammatory
markers, and knee osteoarthritis: A retrospective study. J Orthop
Surg Res. 20:7942025. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Saengsiwaritt W, Ngamtipakon P and
Udomsinprasert W: Vitamin D and autophagy in knee osteoarthritis: A
review. Int Immunopharmacol. 123:1107122023. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Chevalley T, Brandi ML, Cashman KD,
Cavalier E, Harvey NC, Maggi S, Cooper C, Al-Daghri N, Bock O,
Bruyère O, et al: Role of vitamin D supplementation in the
management of musculoskeletal diseases: Update from an European
society of clinical and economical aspects of osteoporosis,
osteoarthritis and musculoskeletal diseases (ESCEO) working group.
Aging Clin Exp Res. 34:2603–2623. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Kuswanto W and Baker MC: Repurposing drugs
for the treatment of osteoarthritis. Osteoarthritis Cartilage.
32:886–895. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Szulc M, Świątkowska-Stodulska R,
Pawłowska E and Derwich M: Vitamin D3 metabolism and its
role in temporomandibular joint osteoarthritis and autoimmune
thyroid diseases. Int J Mol Sci. 24:40802023. View Article : Google Scholar
|
|
95
|
Yuen KCJ: Growth hormone and bone:
Preclinical and clinical perspectives. Endocr Pract. 31:1197–1206.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Li Z and Li Y: Effect of growth hormone
releasing hormone on chondrocytes of osteoarthritis. Korean J
Intern Med. 37:222–229. 2022. View Article : Google Scholar :
|
|
97
|
Yoo KH, Thapa N, Chwae YJ, Yoon SH, Kim
BJ, Lee JO, Jang YN and Kim J: Transforming growth factor-β family
and stem cell-derived exosome therapeutic treatment in
osteoarthritis (review). Int J Mol Med. 49:622022. View Article : Google Scholar
|
|
98
|
Blaney Davidson EN, van der Kraan PM and
van den Berg WB: TGF-beta and osteoarthritis. Osteoarthritis
Cartilage. 15:597–604. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Ma K, Singh G, Wang J, O-Sullivan I,
Votta-Velis G, Bruce B, Anbazhagan AN, van Wijnen AJ and Im HJ:
Targeting vascular endothelial growth factor receptors as a
therapeutic strategy for osteoarthritis and associated pain. Int J
Biol Sci. 19:675–690. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Sampath SJP, Venkatesan V, Ghosh S and
Kotikalapudi N: Obesity, metabolic syndrome, and osteoarthritis-an
updated review. Curr Obes Rep. 12:308–331. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
huo Q, Yang W, Chen J and Wang Y:
Metabolic syndrome meets osteoarthritis. Nat Rev Rheumatol.
8:729–737. 2012. View Article : Google Scholar
|
|
102
|
Jansen NEJ, Molendijk E, Schiphof D, van
Meurs JBJ, Oei EHG, van Middelkoop M and Bierma-Zeinstra SMA:
Metabolic syndrome and the progression of knee osteoarthritis on
MRI. Osteoarthritis Cartilage. 31:647–655. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Jiménez-Muro M, Soriano-Romaní L, Mora G,
Ricciardelli D and Nieto JA: The microbiota-metabolic syndrome axis
as a promoter of metabolic osteoarthritis. Life Sci.
329:1219442023. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Warmink K, Vinod P, Korthagen NM, Weinans
H and Rios JL: Macrophage-driven inflammation in metabolic
osteoarthritis: Implications for biomarker and therapy development.
Int J Mol Sci. 24:61122023. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Zhang C, Mao Y, Xin Y, Li H, Cai M, Lin Y,
Zhang X, Huang Y, Chen Y, Huang Z, et al: Fatty acid binding
protein 4 induces osteogenesis and angiogenesis as pathogenesis of
metabolic osteoarthritis. Mol Med. 32:92025. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Kim D, Ansari MM, Ghosh M, Heo Y, Choi KC
and Son YO: Implications of obesity-mediated cellular dysfunction
and adipocytokine signaling pathways in the pathogenesis of
osteoarthritis. Mol Aspects Med. 103:1013612025. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
De Roover A, Escribano-Núñez A, Monteagudo
S and Lories R: Fundamentals of osteoarthritis: Inflammatory
mediators in osteoarthritis. Osteoarthritis Cartilage.
31:1303–1311. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Binvignat M, Sellam J, Berenbaum F and
Felson DT: The role of obesity and adipose tissue dysfunction in
osteoarthritis pain. Nat Rev Rheumatol. 20:565–584. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Kroon FPB, Veenbrink AI, de Mutsert R,
Visser AW, van Dijk KW, le Cessie S, Rosendaal FR and Kloppenburg
M: The role of leptin and adiponectin as mediators in the
relationship between adiposity and hand and knee osteoarthritis.
Osteoarthritis Cartilage. 27:1761–1767. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Ilia I, Nitusca D and Marian C:
Adiponectin in osteoarthritis: Pathophysiology, relationship with
obesity and presumptive diagnostic biomarker potential. Diagnostics
(Basel). 12:4552022. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Feng X, Xiao J and Bai L: Role of
adiponectin in osteoarthritis. Front Cell Dev Biol. 10:9927642022.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Franco-Trepat E, Guillán-Fresco M,
Alonso-Pérez A, Jorge-Mora A, Francisco V, Gualillo O and Gómez R:
Visfatin connection: Present and future in osteoarthritis and
osteoporosis. J Clin Med. 8:11782019. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Zhou H, Xiao Y, Xue X, Liu J, Xu H, Wang
J, Zhou J, Zuo Q, Liang W, Song H, et al: Hyperglycemia exacerbates
osteoarthritis by impairing macrophage efferocytosis through
modulation of CD11b lactylation. Nat Commun. 17:7242025. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Li Q, Wen Y, Wang L, Chen B, Chen J, Wang
H and Chen L: Hyperglycemia-induced accumulation of advanced
glycosylation end products in fibroblast-like synoviocytes promotes
knee osteoarthritis. Exp Mol Med. 53:1735–1747. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Selvi NMK, Nandhini S, Sakthivadivel V,
Lokesh S, Srinivasan AR and Sumathi S: Association of
triglyceride-glucose index (TyG index) with HbA1c and insulin
resistance in type 2 diabetes mellitus. Maedica (Bucur).
16:375–381. 2021.PubMed/NCBI
|
|
116
|
Huang J, Rozi R, Ma J, Fu B, Lu Z, Liu J
and Ding Y: Association between higher triglyceride glucose index
and increased risk of osteoarthritis: Data from NHANES 2015-2020.
BMC Public Health. 24:7582024. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Zhang S, Jiang Z, Liao H, Bian H, Zhou J,
Wang H and Jiang T: Association between obesity indices, insulin
resistance markers, and osteoarthritis in middle-aged and elderly
Chinese adults. Front Nutr. 12:16274212025. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Weng Y, Yang H, Li L, Lv S and Chen G:
Association between the metabolic score for insulin resistance and
osteoarthritis prevalence: A cross-sectional population-based
study. Medicine (Baltimore). 104:e448502025. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Qiao L, Li Y and Sun S: Insulin
exacerbates inflammation in fibroblast-like synoviocytes.
Inflammation. 43:916–936. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Tchetina EV, Markova GA and Sharapova EP:
Insulin resistance in osteoarthritis: Similar mechanisms to type 2
diabetes mellitus. J Nutr Metab. 2020:41438022020. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Zheng J, Huang X, Huang J, Meng B, Li F,
Liu H, Chen L, Zhou R, Zou M and Wu X: Association of diabetes
mellitus status and hyperglycemia with symptomatic knee
osteoarthritis. Arthritis Care Res (Hoboken). 75:509–518. 2023.
View Article : Google Scholar
|
|
122
|
Halabitska I, Babinets L, Oksenych V and
Kamyshnyi O: Diabetes and osteoarthritis: Exploring the
interactions and therapeutic implications of insulin, metformin,
and GLP-1-based interventions. Biomedicines. 12:16302024.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Xiong J, Long J, Chen X, Li Y and Song H:
Dyslipidemia might be associated with an increased risk of
osteoarthritis. Biomed Res Int. 2020:31052482020. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Hsueh TY and Chen YP: The association
between dyslipidemia and hand osteoarthritis: A systematic review
and meta-analysis. Clin Rheumatol. 44:1439–1448. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Xie Y, Zhou W, Zhong Z, Zhao Z, Yu H,
Huang Y and Zhang P: Metabolic syndrome, hypertension, and
hyperglycemia were positively associated with knee osteoarthritis,
while dyslipidemia showed no association with knee osteoarthritis.
Clin Rheumatol. 40:711–724. 2021. View Article : Google Scholar
|
|
126
|
Chen X, Liu J and Wang G: Dyslipidemia in
osteoarthritis: A study combining bibliometric analysis and
retrospective data mining. Medicine (Baltimore). 104:e422302025.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Yan L, Ge H, Xu Q, Jiang D, Shen A, Yang
M, Zheng Y and Cao Y: Dyslipidemia induced inflammation mediated
the association between obesity and Osteoarthritis: A
population-based study. BMC Public Health. 24:31552024. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Cao C, Shi Y, Zhang X, Li Q, Zhang J, Zhao
F, Meng Q, Dai W, Liu Z, Yan W, et al: Cholesterol-induced LRP3
downregulation promotes cartilage degeneration in osteoarthritis by
targeting Syndecan-4. Nat Commun. 13:71392022. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Lee G, Yang J, Kim SJ, Tran TT, Lee SY,
Park KH, Kwon SH, Chung KH, Koh JT, Huh YH, et al: Enhancement of
intracellular cholesterol efflux in chondrocytes leading to
alleviation of osteoarthritis progression. Arthritis Rheumatol.
77:151–162. 2025. View Article : Google Scholar :
|
|
130
|
Mei Z, Yilamu K, Ni W, Shen P, Pan N, Chen
H, Su Y, Guo L, Sun Q, Li Z, et al: Chondrocyte fatty acid
oxidation drives osteoarthritis via SOX9 degradation and epigenetic
regulation. Nat Commun. 16:48922025. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Abshirini M, Ilesanmi-Oyelere BL and
Kruger MC: Potential modulatory mechanisms of action by long-chain
polyunsaturated fatty acids on bone cell and chondrocyte
metabolism. Prog Lipid Res. 83:1011132021. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Zou Z, Hu W, Kang F, Xu Z, Li Y, Zhang J,
Li J, Zhang Y and Dong S: Interplay between lipid dysregulation and
ferroptosis in chondrocytes and the targeted therapy effect of
metformin on osteoarthritis. J Adv Res. 69:515–529. 2025.
View Article : Google Scholar :
|
|
133
|
Hashimoto K and Akagi M: The role of
oxidation of low-density lipids in pathogenesis of osteoarthritis:
A narrative review. J Int Med Res. 48:3000605209316092020.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Hashimoto K, Mori S, Oda Y, Nakano A,
Sawamura T and Akagi M: Lectin-like oxidized low density
lipoprotein receptor 1-deficient mice show resistance to
instability-induced osteoarthritis. Scand J Rheumatol. 45:412–422.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Lee JS, Kim YH, Jhun J, Na HS, Um IG, Choi
JW, Woo JS, Kim SH, Shetty AA, Kim SJ and Cho ML: Oxidized LDL
accelerates cartilage destruction and inflammatory chondrocyte
death in osteoarthritis by disrupting the TFEB-regulated
autophagy-lysosome pathway. Immune Netw. 24:e152024. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Shen P, Zhu Y, Zhu L, Weng F, Li X and Xu
Y: Oxidized low density lipoprotein facilitates tumor necrosis
factor-α mediated chondrocyte death via autophagy pathway. Mol Med
Rep. 16:9449–9456. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
de Munter W, van der Kraan PM, van den
Berg WB and van Lent PL: High systemic levels of low-density
lipoprotein cholesterol: Fuel to the flames in inflammatory
osteoarthritis? Rheumatology (Oxford). 55:16–24. 2016. View Article : Google Scholar
|
|
138
|
You Y, Xiang T, Yang C, Xiao S, Tang Y,
Luo G, Ling Z, Luo F and Chen Y: Interactions between the gut
microbiota and immune cell dynamics: Novel insights into the
gut-bone axis. Gut Microbes. 17:25454172025. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Fu L, Zhang P, Wang Y and Liu X:
Microbiota-bone axis in ageing-related bone diseases. Front
Endocrinol (Lausanne). 15:14143502024. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Wei J, Zhang C, Zhang Y, Zhang W, Doherty
M, Yang T, Zhai G, Obotiba AD, Lyu H, Zeng C and Lei G: Association
between gut microbiota and symptomatic hand osteoarthritis: Data
from the Xiangya osteoarthritis study. Arthritis Rheumatol.
73:1656–1662. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Guido G, Ausenda G, Iascone V and Chisari
E: Gut permeability and osteoarthritis, towards a mechanistic
understanding of the pathogenesis: A systematic review. Ann Med.
53:2380–2390. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Koh A, De Vadder F, Kovatcheva-Datchary P
and Bäckhed F: From dietary fiber to host physiology: short-chain
fatty acids as key bacterial metabolites. Cell. 165:1332–1345.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Hu J, Lin S, Zheng B and Cheung PCK:
Short-chain fatty acids in control of energy metabolism. Crit Rev
Food Sci Nutr. 58:1243–1249. 2018. View Article : Google Scholar
|
|
144
|
Han J, Meng X, Kong H, Li X, Chen P and
Zhang XA: Links between short-chain fatty acids and osteoarthritis
from pathology to clinic via gut-joint axis. Stem Cell Res Ther.
16:2512025. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Mann ER, Lam YK and Uhlig HH: Short-chain
fatty acids: Linking diet, the microbiome and immunity. Nat Rev
Immunol. 24:577–595. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Postler TS and Ghosh S: Understanding the
holobiont: how microbial metabolites affect human health and shape
the immune system. Cell Metab. 26:110–130. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Pirozzi C, Francisco V, Guida FD, Gómez R,
Lago F, Pino J, Meli R and Gualillo O: Butyrate modulates
inflammation in chondrocytes via GPR43 receptor. Cell Physiol
Biochem. 51:228–243. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Cho KH, Na HS, Jhun J, Woo JS, Lee AR, Lee
SY, Lee JS, Um IG, Kim SJ, Park SH and Cho ML: Lactobacillus (LA-1)
and butyrate inhibit osteoarthritis by controlling autophagy and
inflammatory cell death of chondrocytes. Front Immunol.
13:9305112022. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Korsten SGPJ, Hartog M, Berends AJ,
Koenders MI, Popa CD, Vromans H, Garssen J, van de Ende CHM,
Vermeiden JPW and Willemsen LEM: A sustained-release butyrate
tablet suppresses ex vivo T helper cell activation of
osteoarthritis patients in a double-blind placebo-controlled
randomized trial. Nutrients. 16:33842024. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Karim A, Khan HA, Ahmad F and Qaisar R:
Butyrate (short-chain fatty acid) alleviates
lipopolysaccharide-binding proteins and improves physical function
in knee osteoarthritis patients. Int J Biol Macromol.
307:1420172025. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Hartley A, Sanderson E, Paternoster L,
Teumer A, Kaplan RC, Tobias JH and Gregson CL: Mendelian
randomization provides evidence for a causal effect of higher serum
IGF-1 concentration on risk of hip and knee osteoarthritis.
Rheumatology (Oxford). 60:1676–1686. 2021. View Article : Google Scholar :
|
|
152
|
Shu Z, Miao X, Tang T, Zhan P, Zeng L and
Jiang Y: The GSK-3β/ β-catenin signaling pathway is involved in
HMGB1-induced chondrocyte apoptosis and cartilage matrix
degradation. Int J Mol Med. 45:769–778. 2020.PubMed/NCBI
|
|
153
|
van Lent PLEM, Blom AB, Schelbergen RFP,
Slöetjes A, Lafeber FPJG, Lems WF, Cats H, Vogl T, Roth J and van
den Berg WB: Active involvement of alarmins S100A8 and S100A9 in
the regulation of synovial activation and joint destruction during
mouse and human osteoarthritis. Arthritis Rheum. 64:1466–1476.
2012. View Article : Google Scholar
|
|
154
|
Zhang B, Chen H, Ouyang J, Xie Y and Chen
L, Tan Q, Du X, Su N, Ni Z and Chen L: SQSTM1-dependent autophagic
degradation of PKM2 inhibits the production of mature IL1B/IL-1β
and contributes to LIPUS-mediated anti-inflammatory effect.
Autophagy. 16:1262–1278. 2020. View Article : Google Scholar :
|
|
155
|
Weng M, Yang Z and Feng X: Serum
miR-576-5p as a novel diagnostic biomarker and therapeutic target
for osteoarthritis via targeting KLF10-mediated chondrocyte
dysfunction. BMC Musculoskelet Disord. 26:10882025. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Terkawi MA, Ebata T, Yokota S, Takahashi
D, Endo T, Matsumae G, Shimizu T, Kadoya K and Iwasaki N: Low-grade
inflammation in the pathogenesis of osteoarthritis: Cellular and
molecular mechanisms and strategies for future therapeutic
intervention. Biomedicines. 10:11092022. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Shi J and Du G: Metabolic reprogramming of
glycolysis favors cartilage progenitor cells rejuvenation. Joint
Bone Spine. 91:1056342024. View Article : Google Scholar
|
|
158
|
Liu B, Xian Y, Chen X, Shi Y, Dong J, Yang
L, An X, Shen T, Wu W, Ma Y, et al: Inflammatory fibroblast-like
synoviocyte-derived exosomes aggravate osteoarthritis via enhancing
macrophage glycolysis. Adv Sci (Weinh). 11:e23073382024. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Arra M, Swarnkar G, Ke K, Otero JE, Ying
J, Duan X, Maruyama T, Rai MF, O'Keefe RJ, Mbalaviele G, et al:
LDHA-mediated ROS generation in chondrocytes is a potential
therapeutic target for osteoarthritis. Nat Commun. 11:34272020.
View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Lan W, Chen X, Yu H, Ruan J, Kang J, Nie
X, Cao Y, Tang S and Ding C: UGDH lactylation aggravates
osteoarthritis by suppressing glycosaminoglycan synthesis and
orchestrating nucleocytoplasmic transport to activate MAPK
signaling. Adv Sci (Weinh). 12:e24137092025. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Huang YF, Wang G, Ding L, Bai ZR, Leng Y,
Tian JW, Zhang JZ, Li YQ, Ahmad, Qin YH, et al: Lactate-upregulated
NADPH-dependent NOX4 expression via HCAR1/PI3K pathway contributes
to ROS-induced osteoarthritis chondrocyte damage. Redox Biol.
67:1028672023. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Liu X, Li Y, Zhao J, Hu Z, Fang W, Ke J,
Li W and Long X: Pyroptosis of chondrocytes activated by synovial
inflammation accelerates TMJ osteoarthritis cartilage degeneration
via ROS/NLRP3 signaling. Int Immunopharmacol. 124:1107812023.
View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Liu D, Cai ZJ, Yang YT, Lu WH, Pan LY,
Xiao WF and Li YS: Mitochondrial quality control in cartilage
damage and osteoarthritis: New insights and potential therapeutic
targets. Osteoarthritis Cartilage. 30:395–405. 2022. View Article : Google Scholar
|
|
164
|
Shen T, Alvarez-Garcia O, Li Y, Olmer M
and Lotz MK: Suppression of Sestrins in aging and osteoarthritic
cartilage: Dysfunction of an important stress defense mechanism.
Osteoarthritis Cartilage. 25:287–296. 2017. View Article : Google Scholar
|
|
165
|
Wang B and Zhang H, Fan Z, Shi H, Huang F
and Zhang H: KLF5 aggravates osteoarthritis progression by
inhibiting chondrocyte autophagy via transcriptional activation of
PLK2. Sci Rep. 15:389632025. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Bohensky J, Leshinsky S, Srinivas V and
Shapiro IM: Chondrocyte autophagy is stimulated by HIF-1 dependent
AMPK activation and mTOR suppression. Pediatr Nephrol. 25:633–642.
2010. View Article : Google Scholar
|
|
167
|
Zhuang H, Ren X, Zhang Y, Li H and Zhou P:
β-Hydroxybutyrate enhances chondrocyte mitophagy and reduces
cartilage degeneration in osteoarthritis via the
HCAR2/AMPK/PINK1/Parkin pathway. Aging Cell. 23:e142942024.
View Article : Google Scholar
|
|
168
|
Lin X, Liu H, Qiao L, Deng H, Bao M, Yang
Z, He Y, Xiang R, He H and Han J: Chondrocyte autophagy mediated by
T-2 toxin via AKT/TSC/Rheb/mTOR signaling pathway and protective
effect of CSA-SeNP. Osteoarthritis Cartilage. 32:1283–1294. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Liu L, Zhang W, Liu T, Tan Y, Chen C, Zhao
J, Geng H and Ma C: The physiological metabolite α-ketoglutarate
ameliorates osteoarthritis by regulating mitophagy and oxidative
stress. Redox Biol. 62:1026632023. View Article : Google Scholar
|
|
170
|
Lin S, Wu B, Hu X and Lu H: Sirtuin 4
(Sirt4) downregulation contributes to chondrocyte senescence and
osteoarthritis via mediating mitochondrial dysfunction. Int J Biol
Sci. 20:1256–1278. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Chen Y, Wu YY, Si HB, Lu YR and Shen B:
Mechanistic insights into AMPK-SIRT3 positive feedback
loop-mediated chondrocyte mitochondrial quality control in
osteoarthritis pathogenesis. Pharmacol Res. 166:1054972021.
View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Li H, Chen P, Cheng W, Zhang Z, Yang J,
Zhang H, Jin F, Kan L, Chen L and Wang H: Programmed cell death in
osteoarthritis. Apoptosis. 31:32026. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Chen B, Wang L, Xie D and Wang Y:
Exploration and breakthrough in the mode of chondrocyte death-A
potential new mechanism for osteoarthritis. Biomed Pharmacother.
170:1159902024. View Article : Google Scholar
|
|
174
|
Guan M, Yu Q, Zhou G, Wang Y, Yu J, Yang W
and Li Z: Mechanisms of chondrocyte cell death in osteoarthritis:
Implications for disease progression and treatment. J Orthop Surg
Res. 19:5502024. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Li X, Ji L, Men X, Chen X, Zhi M, He S and
Chen S: Pyroptosis in bone loss. Apoptosis. 28:293–312. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Lou C, Fang Y, Mei Y, Hu W, Sun L, Jin C,
Chen H and Zheng W: Cucurbitacin B attenuates osteoarthritis
development by inhibiting NLRP3 inflammasome activation and
pyroptosis through activating Nrf2/HO-1 pathway. Phytother Res.
38:3352–3369. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Ebata T, Terkawi MA, Kitahara K, Yokota S,
Shiota J, Nishida Y, Matsumae G, Alhasan H, Hamasaki M, Hontani K,
et al: Noncanonical pyroptosis triggered by macrophage-derived
extracellular vesicles in chondrocytes leading to cartilage
catabolism in osteoarthritis. Arthritis Rheumatol. 75:1358–1369.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Tang H, Gong X, Dai J, Gu J, Dong Z, Xu Y,
Hu Z, Zhao C, Deng J and Dong S: The IRF1/GBP5 axis promotes
osteoarthritis progression by activating chondrocyte pyroptosis. J
Orthop Translat. 44:47–59. 2023. View Article : Google Scholar
|
|
179
|
Ru Q, Li Y, Xie W, Ding Y, Chen L, Xu G,
Wu Y and Wang F: Fighting age-related orthopedic diseases: Focusing
on ferroptosis. Bone Res. 11:122023. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Miao Y, Chen Y, Xue F, Liu K, Zhu B, Gao
J, Yin J, Zhang C and Li G: Contribution of ferroptosis and GPX4's
dual functions to osteoarthritis progression. EBioMedicine.
76:1038472022. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Wang S, Li W, Zhang P, Wang Z, Ma X, Liu
C, Vasilev K, Zhang L, Zhou X, Liu L, et al: Mechanical overloading
induces GPX4-regulated chondrocyte ferroptosis in osteoarthritis
via Piezo1 channel facilitated calcium influx. J Adv Res. 41:63–75.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Sun K, Hou L, Guo Z, Wang G, Guo J, Xu J,
Zhang X and Guo F: JNK-JUN-NCOA4 axis contributes to chondrocyte
ferroptosis and aggravates osteoarthritis via ferritinophagy. Free
Radic Biol Med. 200:87–101. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Xia S, Li L, Shi Z, Sun N and He Y:
Ferroptosis in osteoarthritis: Metabolic reprogramming,
immunometabolic crosstalk, and targeted intervention strategies.
Front Immunol. 16:16046522025. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Chen BY, Pathak JL, Lin HY, Guo WQ, Chen
WJ, Luo G, Wang LJ, Sun XF, Ding Y, Li J, et al: Inflammation
triggers chondrocyte ferroptosis in TMJOA via HIF-1α/TFRC. J Dent
Res. 103:712–722. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Stolberg-Stolberg J, Sambale M, Hansen U,
Raschke ASM, Bertrand J, Pap T and Sherwood J: Cartilage trauma
induces necroptotic chondrocyte death and expulsion of cellular
contents. Int J Mol Sci. 21:42042020. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Khaleque MA, Kim JH, Tanvir MAH, Park JB
and Kim YY: Significance of necroptosis in cartilage degeneration.
Biomolecules. 14:11922024. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Tsvetkov P, Coy S, Petrova B, Dreishpoon
M, Verma A, Abdusamad M, Rossen J, Joesch-Cohen L, Humeidi R,
Spangler RD, et al: Copper induces cell death by targeting
lipoylated TCA cycle proteins. Science. 375:1254–1261. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Chang B, Hu Z, Chen L, Jin Z and Yang Y:
Development and validation of cuproptosis-related genes in
synovitis during osteoarthritis progress. Front Immunol.
14:10905962023. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Jiang Z, Li J, Shi M, Li Y, Sun N, Xi K,
Li X, Zhao D, Leng X, Zhou Z and Dong H: Hypoxia, cuproptosis, and
osteoarthritis: Unraveling the molecular crosstalk. Redox Biol.
85:1037572025. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Zhou D, Luo Y, Li F, Liu T, Mei Y, Li F,
Hou X, Fu Z and Liu Z: Exploring the mechanisms of PANoptosis in
osteoarthritis and the therapeutic potential of andrographolide
through bioinformatics and single-cell analysis. Biol Direct.
20:412025. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Loeser RF, Collins JA and Diekman BO:
Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol.
12:412–420. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Coryell PR, Diekman BO and Loeser RF:
Mechanisms and therapeutic implications of cellular senescence in
osteoarthritis. Nat Rev Rheumatol. 17:47–57. 2021. View Article : Google Scholar
|
|
193
|
Chen X, Gong W, Shao X, Shi T, Zhang L,
Dong J, Shi Y, Shen S, Qin J, Jiang Q and Guo B: METTL3-mediated
m6A modification of ATG7 regulates autophagy-GATA4 axis
to promote cellular senescence and osteoarthritis progression. Ann
Rheum Dis. 81:87–99. 2022. View Article : Google Scholar
|
|
194
|
Roh K, Noh J, Kim Y, Jang Y, Kim J, Choi
H, Lee Y, Ji M, Kang D, Kim MS, et al: Lysosomal control of
senescence and inflammation through cholesterol partitioning. Nat
Metab. 5:398–413. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Faust HJ, Zhang H, Han J, Wolf MT, Jeon
OH, Sadtler K, Peña AN, Chung L, Maestas DR Jr, Tam AJ, et al:
IL-17 and immunologically induced senescence regulate response to
injury in osteoarthritis. J Clin Invest. 130:5493–5507. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Jeon OH, Kim C, Laberge RM, Demaria M,
Rathod S, Vasserot AP, Chung JW, Kim DH, Poon Y, David N, et al:
Local clearance of senescent cells attenuates the development of
post-traumatic osteoarthritis and creates a pro-regenerative
environment. Nat Med. 23:775–781. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Diekman BO, Sessions GA, Collins JA,
Knecht AK, Strum SL, Mitin NK, Carlson CS, Loeser RF and Sharpless
NE: Expression of p16INK4a is a biomarker of chondrocyte
aging but does not cause osteoarthritis. Aging Cell. 17:e127712018.
View Article : Google Scholar
|
|
198
|
Kakiuchi Y, Yurube T, Kakutani K, Takada
T, Ito M, Takeoka Y, Kanda Y, Miyazaki S, Kuroda R and Nishida K:
Pharmacological inhibition of mTORC1 but not mTORC2 protects
against human disc cellular apoptosis, senescence, and
extracellular matrix catabolism through Akt and autophagy
induction. Osteoarthritis Cartilage. 27:965–976. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Huang Y, Yue S, Yan Z, Liu Y, Qiao J,
Zhang M, Dong Y and Zheng J: Lactate-upregulated ARG2 expression
induces cellular senescence in fibroblast-like synoviocytes of
osteoarthritis via activating the mTOR/S6K1 signaling pathway. Int
Immunopharmacol. 142:1130712024. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Lu H, Jia C, Wu D, Jin H, Lin Z, Pan J, Li
X and Wang W: Fibroblast growth factor 21 (FGF21) alleviates
senescence, apoptosis, and extracellular matrix degradation in
osteoarthritis via the SIRT1-mTOR signaling pathway. Cell Death
Dis. 12:8652021. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Hou WY, Zhu CY, Gu YF, Zhu L and Zhou ZX:
Association of hormone replacement therapy and the risk of knee
osteoarthritis: A meta-analysis. Medicine (Baltimore).
101:e324662022. View Article : Google Scholar :
|
|
202
|
Burkard T, Rauch M, Spoendlin J,
Prieto-Alhambra D, Jick SS and Meier CR: Risk of hand
osteoarthritis in new users of hormone replacement therapy: A
nested case-control analysis. Maturitas. 132:17–23. 2020.
View Article : Google Scholar
|
|
203
|
Williams JAE, Chester-Jones M, Minns Lowe
C, Goff MV, Francis A, Brewer G, Marian I, Morris SL, Warwick D,
Eldridge L, et al: Hormone replacement therapy (conjugated
oestrogens plus bazedoxifene) for post-menopausal women with
symptomatic hand osteoarthritis: Primary report from the HOPE-e
randomised, placebo-controlled, feasibility study. Lancet
Rheumatol. 4:e725–e737. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Pang H, Chen S, Klyne DM, Harrich D, Ding
W, Yang S and Han FY: Low back pain and osteoarthritis pain: A
perspective of estrogen. Bone Res. 11:422023. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Sun M, Ma B, Pan Z, Zhao Y, Tian L, Fan Y,
Kong W, Wang J, Xu B, Ao Y, et al: Targeted therapy of
osteoarthritis via intra-articular delivery of
lipid-nanoparticle-encapsulated recombinant human FGF18 mRNA. Adv
Healthc Mater. 13:e24008042024. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Li G, Liu S, Chen Y, Zhao J, Xu H, Weng J,
Yu F, Xiong A, Udduttula A, Wang D, et al: An injectable
liposome-anchored teriparatide incorporated gallic acid-grafted
gelatin hydrogel for osteoarthritis treatment. Nat Commun.
14:31592023. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Gupta N, Khatri K, Lakhani A, Dahuja A,
Randhawa A, Bansal V and Bansal K: Long-term effectiveness of
intra-articular injectables in patients with knee osteoarthritis: A
systematic review and Bayesian network meta-analysis. J Orthop Surg
Res. 20:2272025. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Liu J, Huang X, Lv T, Cao L and Lu L:
Intra-articular injection of chitosan combined with low-dose
glucocorticoid for the treatment of knee osteoarthritis in early
and middle stages. Medicine (Baltimore). 103:e399242024. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Ok SM, Kim JH, Kim JS, Jeong EG, Park YM,
Jeon HM, Heo JY, Ahn YW, Yu SN, Park HR, et al: Local injection of
growth hormone for temporomandibular joint osteoarthritis. Yonsei
Med J. 61:331–340. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Meurot C, Martin C, Sudre L, Breton J,
Bougault C, Rattenbach R, Bismuth K, Jacques C and Berenbaum F:
Liraglutide, a glucagon-like peptide 1 receptor agonist, exerts
analgesic, anti-inflammatory and anti-degradative actions in
osteoarthritis. Sci Rep. 12:15672022. View Article : Google Scholar
|
|
211
|
Karacabeyli D and Lacaille D:
Glucagon-like peptide-1 receptor agonists in arthritis: Current
insights and future directions. Nat Rev Rheumatol. 21:671–683.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Ryan M, Megyeri S, Nuffer W and Trujillo
JM: The potential role of GLP-1 receptor agonists in
osteoarthritis. Pharmacotherapy. 45:177–186. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Bliddal H, Bays H, Czernichow S, Uddén
Hemmingsson J, Hjelmesæth J, Hoffmann Morville T, Koroleva A, Skov
Neergaard J, Vélez Sánchez P, Wharton S, et al: Once-weekly
semaglutide in persons with obesity and knee osteoarthritis. N Engl
J Med. 391:1573–1583. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Zhu H, Zhou L, Wang Q, Cai Q, Yang F, Jin
H, Chen Y, Song Y and Zhang C: Glucagon-like peptide-1 receptor
agonists as a disease-modifying therapy for knee osteoarthritis
mediated by weight loss: Findings from the Shanghai Osteoarthritis
Cohort. Ann Rheum Dis. 82:1218–1226. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Li J, Zhang B, Liu WX, Lu K, Pan H, Wang
T, Oh CD, Yi D, Huang J, Zhao L, et al: Metformin limits
osteoarthritis development and progression through activation of
AMPK signalling. Ann Rheum Dis. 79:635–645. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Hou J, Lin Y, Zhu C, Chen Y, Lin R, Lin H,
Liu D, Guan D, Yu B, Wang J, et al: Zwitterion-lubricated hydrogel
microspheres encapsulated with metformin ameliorate age-associated
osteoarthritis. Adv Sci (Weinh). 11:e24024772024. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Yan S, Dong W, Li Z, Wei J, Han T, Wang J
and Lin F: Metformin regulates chondrocyte senescence and
proliferation through microRNA-34a/SIRT1 pathway in osteoarthritis.
J Orthop Surg Res. 18:1982023. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Wang C, Yang Y, Zhang Y, Liu J, Yao Z and
Zhang C: Protective effects of metformin against osteoarthritis
through upregulation of SIRT3-mediated PINK1/Parkin-dependent
mitophagy in primary chondrocytes. Biosci Trends. 12:605–612. 2019.
View Article : Google Scholar
|
|
219
|
Li D, Ruan G, Zhang Y, Zhao Y, Zhu Z, Ou
Q, Huang H, Chen J, Han W, Tang S, et al: Metformin attenuates
osteoarthritis by targeting chondrocytes, synovial macrophages and
adipocytes. Rheumatology (Oxford). 62:1652–1661. 2023. View Article : Google Scholar
|
|
220
|
Lambova SN: Pleiotropic effects of
metformin in osteoarthritis. Life (Basel). 13:4372023.PubMed/NCBI
|
|
221
|
Li BZ, Wei HX, Li SZ, Yang YZ, Zhang AR
and Guo HZ: Update on metformin for osteoarthritis treatment.
Inflammopharmacology. 33:5963–5975. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Liu Z, Huang J, Wang X, Deng S, Zhou J,
Gong Z, Li X, Wang Y, Yang J and Hu Y: Dapagliflozin suppress
endoplasmic reticulum stress mediated apoptosis of chondrocytes by
activating Sirt1. Chem Biol Interact. 384:1107242023. View Article : Google Scholar : PubMed/NCBI
|
|
223
|
El-Demerdash AA, Darwish SF, El-Derany MO
and El-Demerdash E: Dapagliflozin targets the crosstalk between
apoptosis, autophagy, and Hedgehog signaling pathways through AMPK
activation in the adjuvant-induced arthritic rat model.
Inflammopharmacology. 33:3157–3176. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Zhao M, Song X, Chen H, Ma T, Tang J, Wang
X, Yu Y, Lv L, Jia L and Gao L: Melatonin prevents chondrocyte
matrix degradation in rats with experimentally induced
osteoarthritis by inhibiting nuclear factor-κB via SIRT1.
Nutrients. 14:39662022. View Article : Google Scholar
|
|
225
|
Park S and Shin BK: Intermittent fasting
with a high-protein diet mitigated osteoarthritis symptoms by
increasing lean body mass and reducing inflammation in
osteoarthritic rats with Alzheimer's disease-like dementia. Br J
Nutr. 127:55–67. 2022. View Article : Google Scholar
|
|
226
|
Qian YX, Rao SS, Tan YJ, Wang Z, Yin H,
Wan TF, He ZH, Wang X, Hong CG, Zeng HJ, et al: Intermittent
fasting targets osteocyte neuropeptide Y to relieve osteoarthritis.
Adv Sci (Weinh). 11:e24001962024. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Sun N, Zhao Y, Wang J, Zhang A and He Y:
Intermittent fasting in osteoarthritis: From mechanistic insights
to therapeutic potential. Front Nutr. 12:16048722025. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Sharma L: Osteoarthritis of the knee. N
Engl J Med. 384:51–59. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Conaghan PG, Cook AD, Hamilton JA and Tak
PP: Therapeutic options for targeting inflammatory osteoarthritis
pain. Nat Rev Rheumatol. 15:355–363. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Lu Y, Yin Y, Kats ER, Sun J, Liu S and Lin
H: Estrogen receptor-α loss accelerates cartilage degradation
through CLEC3B-mediated chondrocyte hypertrophy and inflammation.
Osteoarthritis Cartilage. 33:1443–1453. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Jin WJ, Jiang SD, Jiang LS and Dai LY:
Differential responsiveness to 17β-estradiol of mesenchymal stem
cells from postmenopausal women between osteoporosis and
osteoarthritis. Osteoporos Int. 23:2469–2478. 2012. View Article : Google Scholar
|
|
232
|
Montemor CN, Fernandes MTP, Marquez AS,
Bignardi PR, Poli RC, Gâmbaro GA, da Silva RA, Ngomo S and
Fernandes KBP: Impact of reduced vitamin D levels on pain,
function, and severity in knee or hip osteoarthritis. Nutrients.
17:4472025. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Batushansky A, Zhu S, Komaravolu RK, South
S, Mehta-D'souza P and Griffin TM: Fundamentals of OA. An
initiative of osteoarthritis and cartilage. Obesity and metabolic
factors in OA. Osteoarthritis Cartilage. 30:501–515. 2022.
View Article : Google Scholar
|
|
234
|
Rogers-Soeder TS, Lane NE, Walimbe M,
Schwartz AV, Tolstykh I, Felson DT, Lewis CE, Segal NA and Nevitt
MC; Multicenter Osteoarthritis (MOST) Study Group: Association of
diabetes mellitus and biomarkers of abnormal glucose metabolism
with incident radiographic knee osteoarthritis. Arthritis Care Res
(Hoboken). 72:98–106. 2020. View Article : Google Scholar
|
|
235
|
Man TM, Ma Y, Zhao YG, He QS, Li GS and Wu
XF: Machine learning-driven insights into lipid metabolism and
inflammatory pathways in knee osteoarthritis. Front Nutr.
12:15520472025. View Article : Google Scholar : PubMed/NCBI
|
|
236
|
Awan UN, Waraich RS, Nangrejo R, Noor SS,
Siddiqui IA and Ikram K: RAGE signalling contributes to oxidative
stress and inflammation in knee osteoarthritis patients with
metabolic syndrome. Clin Exp Rheumatol. 42:2258–2264.
2024.PubMed/NCBI
|
|
237
|
Xiang W, Zhang T, Li B, Li S, Zhang B,
Fang S, Chen L, Gong Y, Huang B, Feng D, et al: Senescent
macrophages induce ferroptosis in skeletal muscle and accelerate
osteoarthritis-related muscle atrophy. Nat Aging. 5:1295–1316.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
238
|
Gao Q, Yao D, Yin Z, Yu G, Shi B and Wang
J: Comprehensive multi-omics approach reveals potential therapeutic
targets and agents for osteoarthritis. Postgrad Med J. 101:464–474.
2025. View Article : Google Scholar
|
|
239
|
Cai Y, Yu D, Meng J, Qiu Y, Liu J and Yao
J: Multi-omics analysis and validation of autophagy-related
diagnostic biomarker in osteoarthritis. Ann Med. 57:25480452025.
View Article : Google Scholar : PubMed/NCBI
|
|
240
|
Jayakumar P, Moore MG, Furlough KA, Uhler
LM, Andrawis JP, Koenig KM, Aksan N, Rathouz PJ and Bozic KJ:
Comparison of an artificial intelligence-enabled patient decision
aid vs educational material on decision quality, shared
decision-making, patient experience, and functional outcomes in
adults with knee osteoarthritis: A randomized clinical trial. JAMA
Netw Open. 4:e20371072021. View Article : Google Scholar : PubMed/NCBI
|
|
241
|
Saxer F, Jansen G, Bierma-Zeinstra SMA,
Holzhauer B, Demanse D, Melnick J, Vukadinovic Greetham D, Rall T,
Mesenbrink P and Schieker M: Why is there no treatment for
osteoarthritis-opportunity for AI based big data analytics to
advance the field. Osteoarthritis Cartilage. 34:657–666. 2026.
View Article : Google Scholar
|
|
242
|
Collins KH, Lenz KL, Welhaven HD, Ely E,
Springer LE, Paradi S, Tang R, Braxton L, Akk A, Yan H, et al:
Adipose-derived leptin and complement factor D mediate
osteoarthritis severity and pain. Sci Adv. 11:eadt59152025.
View Article : Google Scholar : PubMed/NCBI
|
|
243
|
Vincent TL: OA synovial fluid: Biological
insights into a whole-joint disease. Osteoarthritis Cartilage.
30:765–766. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
244
|
Tomura T, Ishii T, Kasahara N and Nishii
Y: Dihydrotestosterone and 17β-estradiol modulate TMJ
osteoarthritis development and reveal sex-specific differences in
pathogenesis. Sci Rep. 15:187402025. View Article : Google Scholar
|
|
245
|
Zhang S, Wang D, Zhao J, Zhao H, Xie P,
Zheng L, Sheng P, Yuan J, Xia B, Wei F and Zhang Z: Metabolic
syndrome increases osteoarthritis risk: Findings from the UK
Biobank prospective cohort study. BMC Public Health. 24:2332024.
View Article : Google Scholar : PubMed/NCBI
|
|
246
|
Walrabenstein W, Wagenaar CA, van de Put
M, van der Leeden M, Gerritsen M, Twisk JWR, van der Esch M, van
Middendorp H, Weijs PJM, Roorda LD and van Schaardenburg D: A
multidisciplinary lifestyle program for metabolic
syndrome-associated osteoarthritis: the 'Plants for Joints'
randomized controlled trial. Osteoarthritis Cartilage.
31:1491–1500. 2025. View Article : Google Scholar
|
|
247
|
Pragasam SSJ and Venkatesan V: Metabolic
syndrome predisposes to osteoarthritis: Lessons from model system.
Cartilage. 13(1 Suppl): 1598S–1609S. 2021. View Article : Google Scholar
|
|
248
|
Hernandez PA, Bradford JC, Brahmachary P,
Ulman S, Robinson JL, June RK and Cucchiarini M: Unraveling
sex-specific risks of knee osteoarthritis before menopause: Do sex
differences start early in life? Osteoarthritis Cartilage.
32:1032–1044. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
249
|
Fan Y, Bian X, Meng X, Li L, Fu L, Zhang
Y, Wang L, Zhang Y, Gao D, Guo X, et al: Unveiling inflammatory and
prehypertrophic cell populations as key contributors to knee
cartilage degeneration in osteoarthritis using multi-omics data
integration. Ann Rheum Dis. 83:926–944. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
250
|
Wu Y, Liu J, Yu W, Wang X, Li J and Zeng
W: Single-cell transcriptome and multi-omics integration reveal
ferroptosis-driven immune microenvironment remodeling in knee
osteoarthritis. Front Immunol. 16:16083782025. View Article : Google Scholar : PubMed/NCBI
|
|
251
|
Castagno S, Birch M, van der Schaar M and
McCaskie A: Predicting rapid progression in knee osteoarthritis: A
novel and interpretable automated machine learning approach, with
specific focus on young patients and early disease. Ann Rheum Dis.
84:124–135. 2025. View Article : Google Scholar : PubMed/NCBI
|