Introduction
Primary effusion lymphoma (PEL) is a subtype of
non-Hodgkin’s B-cell lymphoma, which is mostly found in patients
with AIDS, but is also sometimes found in immunosuppressed patients
such as those who have undergone organ transplantation (1,2). PEL
usually presents as a lymphomatous effusion in body cavities and is
caused by human herpes virus 8 (HHV-8), also known as Kaposi’s
sarcoma-associated herpes virus (KSHV) (2). HHV-8 infects endothelial and
B-lymphoid cells and is responsible for the development of Kaposi’s
sarcoma and PEL. Despite the improved therapeutic outcome in
AIDS-related lymphomas after the introduction of highly active
antiretroviral therapy (HAART), PEL generally still has an
extremely aggressive clinical course and the prognosis in patients
with PEL is poor with a median survival of 6.2 months (3). Optimal treatment for PEL has not yet
been established and development of novel therapeutic agents is
needed.
A number of signaling pathways, including NF-κB,
JAK/STAT and phosphoinositide 3-kinase (PI3-K) pathways, are
constitutively activated and play critical roles in the survival
and growth of PEL cells. HHV-8 encodes a virus Fas-associated death
domain-like interleukin-1β-converting enzyme (FLICE) inhibitory
protein (vFLIP) that has the ability to activate the NF-κB pathway
(4,5). vFLIP has been shown to bind to the
IKK complex to induce constitutive kinase activation, and as a
result, PEL cells have high NF-κB pathway activity (6,7). In
fact, it has been shown that inhibition of NF-κB induces apoptosis
in PEL cells (8,9). These studies suggest that
vFLIP-mediated NF-κB activation is essential for the survival of
PEL cells and that this pathway can be a target of therapy for
PEL.
Diethyldithiocarbamate (DDTC), a member of the
dithiocarbamate family and metabolite of disulfiram (10,11),
is a potent copper-chelating compound. Some of the known biological
activities of DDTC are the inhibition of the proteasome and the
induction of apoptosis in cancer cells (12). DDTC has also been used for the
treatment of alcoholism, metal poisoning and HIV (13–16).
Because dithiocarbamates are well-known inhibitors of NF-κB
(17), we explored the possibility
that DDTC could be a therapeutic agent for PEL.
In this study, we investigated the inhibitory
effects of DDTC on the growth of PEL cell lines in vitro and
in vivo. DDTC inhibited cell growth and induced apoptosis in
HHV-8-infected PEL cells. Our findings suggest that DDTC is a
promising agent for the treatment of PEL.
Materials and methods
Cell culture
HHV-8-infected human PEL cell lines, BCBL-1
(obtained through the AIDS Research and Reference Reagent Program,
Division of AIDS, NIAID, NIH) (18), BC-1 and BC-3 (purchased from the
American Type Culture Collection, Manassas, VA), and
HHV-8-uninfected human cell lines, K562 (obtained from RIKEN Cell
Bank, Tsukuba, Japan), were maintained in RPMI-1640 supplemented
with 10% heat inactivated fetal calf serum, penicillin (100 U/ml)
and streptomycin (100 μg/ml) in a humidified incubator at 37°C and
5% CO2.
Animal studies
Balb/c Rag-2-deficient (Rag-2−/−) mice
were crossed with Balb/c Jak-3-deficient (Jak-3−/−) mice
to establish Balb/c Rag-2/Jak-3 double-deficient
(Rag-2−/−Jak3−/−) mice as we have described
recently (19). The mice were
housed and monitored in a vivarium in compliance with the
guidelines of the animal facility center of Kumamoto University.
All experiments were performed according to the protocols approved
by the Animal Welfare Committee of Kumamoto University
(#A19–115).
Reagents and antibodies
DDTC was purchased from Sigma-Aldrich Co. (St.
Louis, MO, USA). Antibody against ubiquitinated protein (clone FK2)
was purchased from BIOMOL; antibody against cleaved caspase-3 was
purchased from Cell Signaling Technologies; antibodies against p65
(Sc-8008), actin (Sc-1616), γ-tubulin (Sc-7396) were purchased from
Santa Cruz Biotechnology.
Tetrazolium dye methylthiotetrazole (MTT)
assay
The antiproliferative effects of DDTC against
HHV-8-infected and -uninfected cell lines were measured by the MTT
method. Cells were incubated in triplicate in a 96-well
microculture plate in the presence of different concentrations of
DDTC in a final volume of 0.1 ml for 24 h at 37°C. Subsequently,
MTT (0.5 mg/ml final concentration) was added to each well. After 3
h of additional incubation, 100 μl of a solution of 0.04 N HCl in
isopropanol were added to dissolve the crystal. The absorption
values at 595 nm were determined with an automatic enzyme-linked
immnosorbent assay (ELISA) plate reader (Multiskan, Thermo
Electron, Vantaa, Finland). Values are normalized to the untreated
samples.
Annexin V assay
Apoptosis was measured by dual-labeling with the
Annexin V-FITC Apoptosis Detection kit I (BD Biosciences
Pharmingen). Briefly, after treatment with DDTC, cells were
harvested, washed and then incubated with Annexin V-FITC and
propidium iodide (PI) for 20 min in the dark, before being analyzed
on an LSR II cytometer.
Cell cycle analysis
Cells were washed with phosphate-buffered saline
(PBS) and fixed with 70% ethanol. Fixed cells were washed by
centrifugation in FACS washing buffer (0.5% NaN3, 2% FBS
in PBS) and stained with PI (10 μg/ml in PBS) for 30 min in the
dark. Samples were analyzed on a LSR II cytometer after nylon mesh
filtration.
Cell lysis and immunoblotting
Cells were lysed with lysis buffer (25 mM HEPES, 10
mM Na4P2O7·10H2O, 100
mM NaF, 5 mM EDTA, 2 mM Na3VO4, 1% Triton
X-100). For nuclear extraction, cells were washed and resuspended
in 150 μl of cold buffer containing 10 mM HEPES-KOH (pH 7.9), 10 mM
KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol and 0.5 mM
phenylmethylsulfonyl fluoride (PMSF). The cells were then allowed
to swell on ice for 15 min, after which 10 μl of 10% Nonidet P-40
solution was added, and the samples were vigorously vortexed for 10
sec. The homogenate was centrifuged at 15,000 × g for 1 min at 4°C.
The nuclear pellet was resuspended in 50 μl of ice-cold buffer
containing 20 mM HEPES-KOH (pH 7.9), 0.4 M NaCl, 1 mM EDTA, 1 mM
EGTA, 1 mM dithiothreitol and 1 mM PMSF, the tube was vigorously
vortexed for 15 min at 4°C. Then the nuclear extract was
centrifuged at 20,000 × g for 5 min at 4°C and the clear
supernatant was collected.
Real-time RT-PCR analysis
Total RNA was isolated from cell lines using TRIzol
(Invitrogen, Carlsbad, CA). Quantitative real-time RT-PCR analysis
of IL-6 was carried out with Prime Script RT reagent kit and SYBR
Premix Ex Taq II (Takara Bio Inc., Ohtsu, Japan) according to the
manufacturer’s instructions. PCR amplifications were performed
using iQ5 thermal cycler (Bio-Rad Laboratories, Inc., Hercules, CA)
with the following amplification conditions: 95°C for 3 min, for 40
cycles at 95°C for 10 sec, at 55°C for 30 sec. The Ct values for
each gene amplification were normalized by subtracting the Ct value
calculated for β-actin (internal control). The normalized gene
expression values were expressed as the relative quantity of
gene-specific mRNA compared with control mRNA (fold induction). The
oligonucleo-tide primers used in this study are as shown below.
IL-6-Fw: 5′-GCACTGGCAGAAAACAACCT; IL-6-Rv: 5′-CAGGGGT
GGTTATTGCATCT; β-actin-Fw: 5′-GCTAT C CAGGCTGTG; β-actin-Rv:
5′-TGTCACGCACGATTTCC.
Statistical analysis
Data are presented as mean ± SE. Significance of the
difference between groups was assessed with one-way ANOVA. A
P<0.05 was considered statistically significant.
Results
Effects of DDTC on immunodeficient mice
inoculated with BC-3 cells
To evaluate the HHV-8-associated PEL response to
DDTC treatment, we established a mouse model of PEL. Balb/c
Rag-2−/−Jak3−/− mice were inoculated
intraperitoneally with BC-3 cells, an HHV-8-infected PEL cell line.
DDTC (2.5 or 100 mg/kg) or PBS alone was administered
intraperitoneally on day 21 after cell inoculation and 5 days per
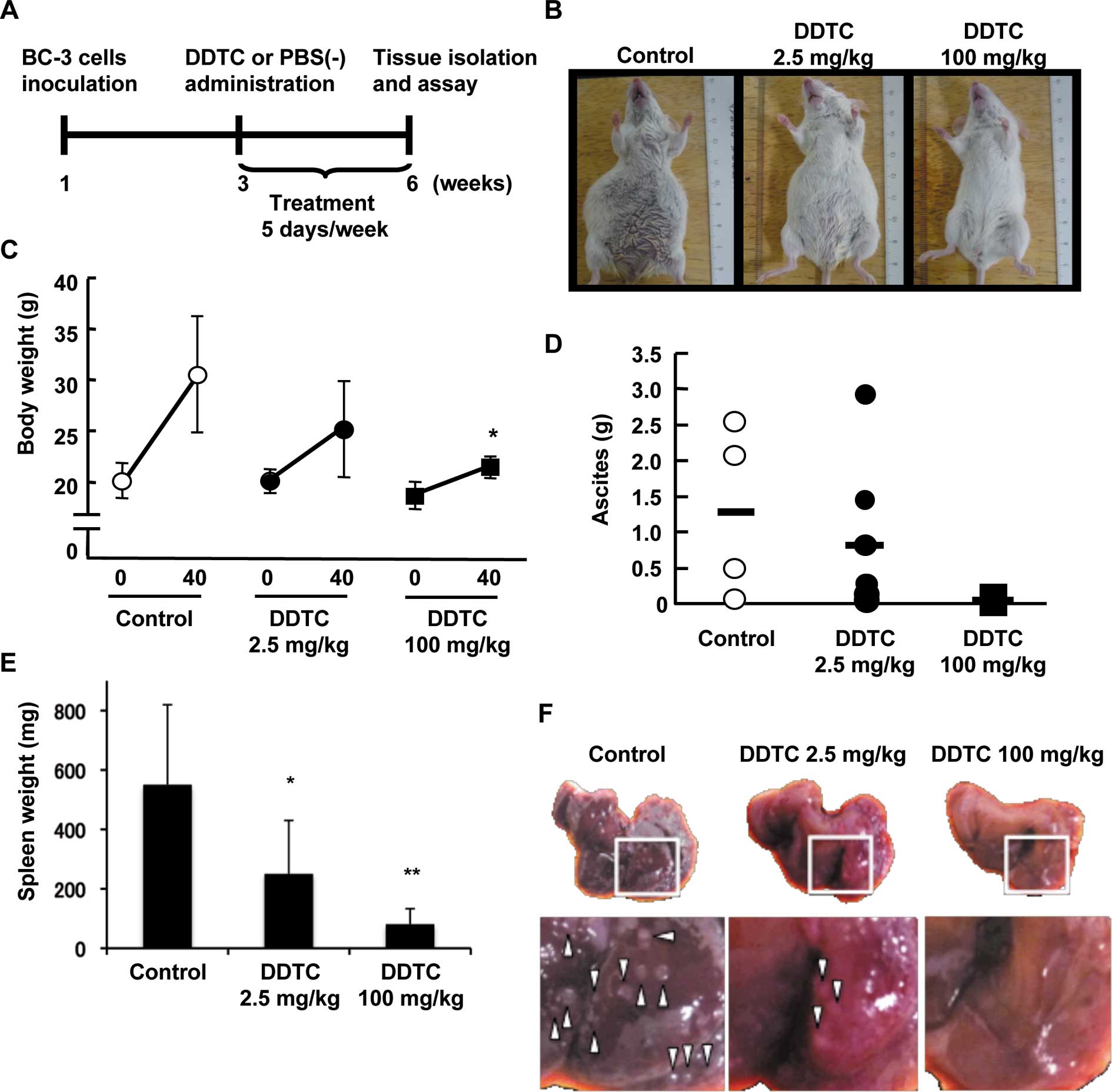
week thereafter for 3 weeks (Fig.
1A). It has been reported that treatment with this dose of DDTC
has no toxicity in mice (20).
BC-3 cells produced massive ascites in peritoneal
body cavity with evident abdominal distention, while the abdominal
distention was not observed in mice that received injection of DDTC
(Fig. 1B). The increase of body
weight in BC-3-inoculated mice was significantly reduced by DDTC
treatment in a dose-dependent manner (Fig. 1C) consistent with the difference in
appearance. The alleviation of abdominal distention and decrease in
body weight implied that DDTC treatment suppressed the production
of ascites, therefore we examined the effect of DDTC on the amount
of ascites in BC-3-inoculated mice. Although no significant
decrease in the weight of ascites was observed, there was a
tendency of ascites to be reduced in DDTC-treated mice in a
dose-dependent manner (Fig. 1D).
Previously, it has been shown that HHV-8-encoded IL-6 induces
splenomegaly (21). Consistent
with this report, splenomegaly was induced in mice inoculated with
BC-3 cells (Fig. 1E; control).
Treatment with DDTC, however, dose-dependently suppressed the
development of splenomegaly (Fig.
1E). Moreover, hepatic tumorigenesis was prevented in
DDTC-treated mice, but not in PBS-treated control mice (Fig. 1F). These results indicate that DDTC
treatment significantly inhibits the growth of PEL cells in
vivo.
DDTC has proteasome inhibitory activity
in HHV-8-infected and -uninfected cells
We next determined how DDTC controls the progression
of PEL. Because DDTC was shown to have proteasome inhibitory
activity and induces apoptosis in prostate cancer cells (12), we investigated whether DDTC
inhibits the proteasome activity in HHV-8-infected and -uninfected
cells. To examine the accumulation of ubiquitinated proteins,
HHV-8-infected BC-3 cells and HHV-8-uninfected K562 cells were
treated for 12 or 24 h with 3 μM DDTC, an appropriate concentration
for leukemia cells as determined previously (22), and cell lysates were analyzed using
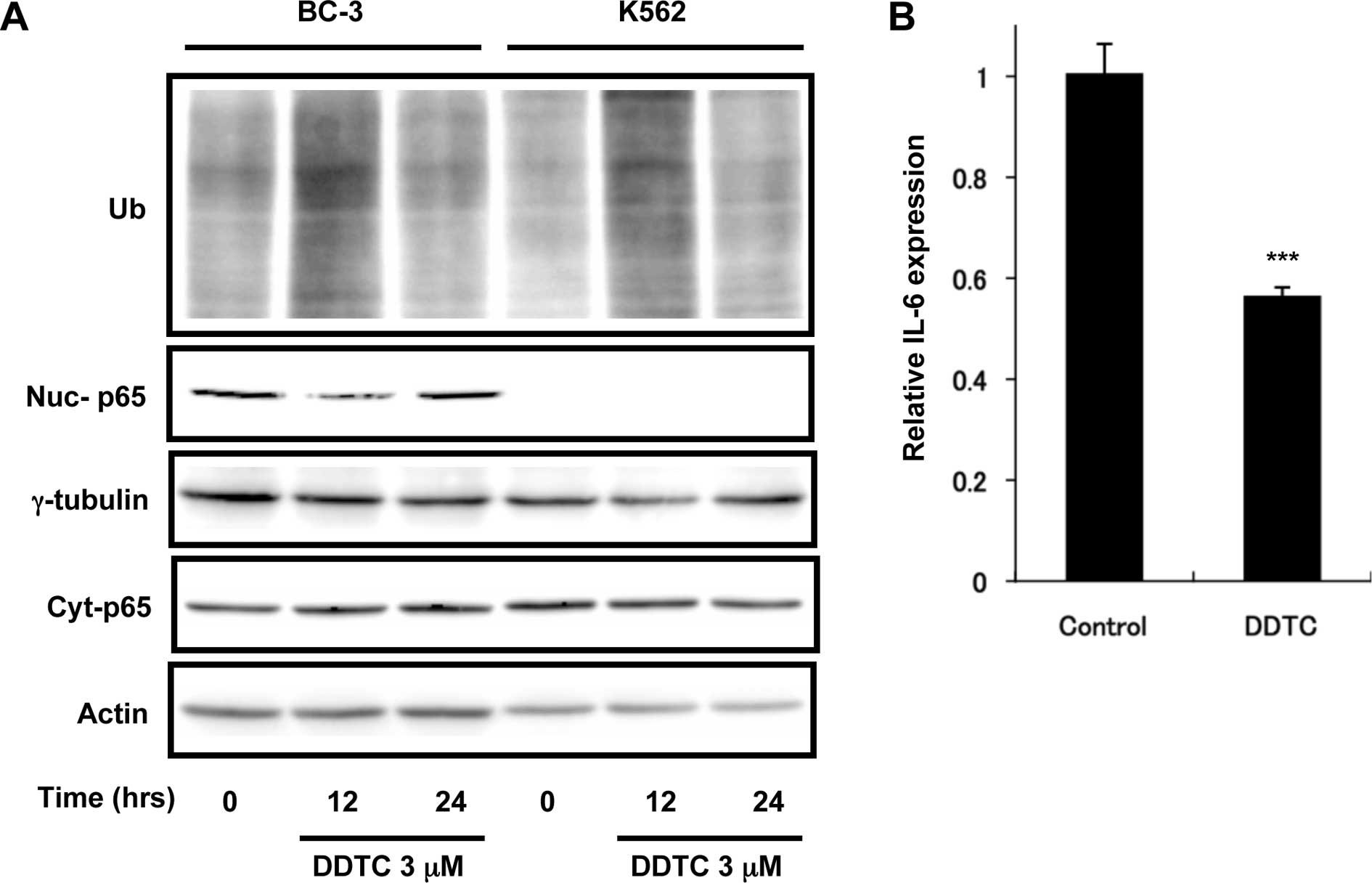
anti-ubiquitinated protein antibody. As shown in Fig. 2A, the accumulation of ubiquitinated
proteins was observed in both BC-3 and K562 cells after treatment
with DDTC for 12 h, but not for 24 h. This result suggests that
DDTC has proteasome inhibitory activity irrespective of HHV-8
infection.
DDTC suppresses the NF-κB pathway in
HHV-8-infected PEL cells
DDTC is known to suppress the activation of NF-κB
pathway (17), thus we next
examined whether DDTC influences the NF-κB pathway in
HHV-8-infected and -uninfected cells. To determine the localization
and expression of p65, a subtype of NF-κB, BC-3 and K562 cells were
treated with DDTC for 12 or 24 h and the nuclear extracts were
subjected to immunoblotting with anti-p65 antibody. Nuclear p65
expression (Nuc-p65) was detected in control BC-3 cells, but this
was reduced in DDTC-treated BC-3 cells at 12 h of treatment and
returned to basal level at 24 h of treatment (Fig. 2A; Nuc-p65). The level of cytosolic
p65 (Cyt-p65) was not remarkably altered (Fig. 2A). On the other hand, nuclear p65
was undetectable in K562 cells. Moreover, we examined by
quantitative RT-PCR the mRNA expression of the growth factor IL-6
in BC-3 cells. IL-6 is one of the target genes of NF-κB and
accelerates the cell growth in HHV-8-infected PEL cells (23). As expected, DDTC treatment for 12 h
decreased the IL-6 mRNA expression (Fig. 2B), indicating that DDTC inhibited
the NF-κB signaling pathway.
DDTC induces caspase-3 dependent
apoptosis in HHV-8 infected PEL cells
It has been demonstrated that inhibition of NF-κB
induces the apoptosis of HHV-8-infected PEL cells (9,24).
To determine whether inhibition of NF-κB by DDTC treatment was
attributed to cell cycle arrest or apoptosis in HHV-8-infected
cells, BC-3 and K562 cells were left untreated or treated with DDTC
for 48 h. Cells were fixed and cell cycle fraction was determined
by flow cytometry. As shown in Fig.
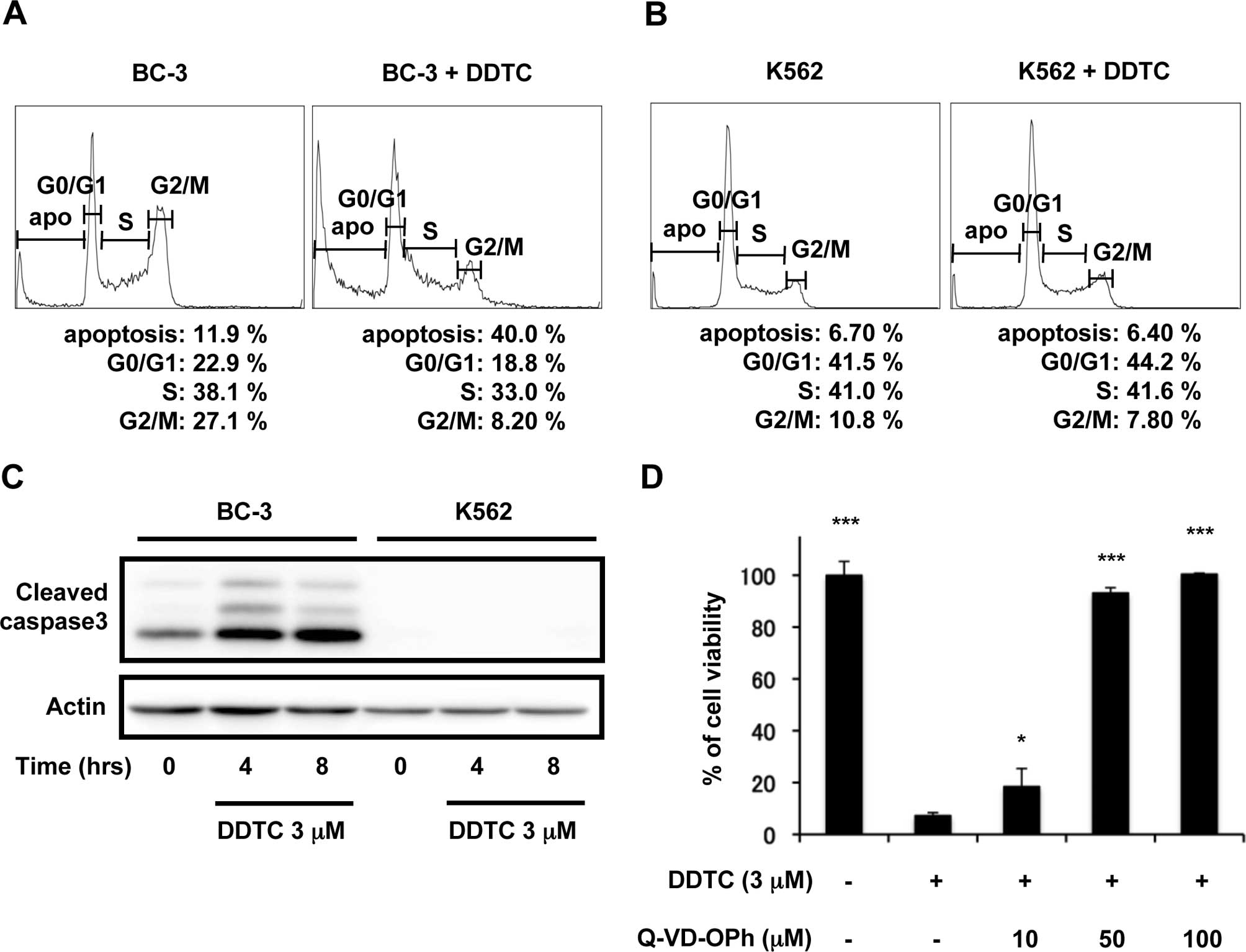
3A, DDTC treatment increased the population of sub-G1 cells
from 12 to 40% in BC-3 cells. This increase in sub-G1 population
was accompanied by reduction in G0/G1, S and G2 phases, an
indication that these cells were undergoing apoptosis (25). On the other hand, there was no
increase in the sub-G1 population after DDTC treatment in K562
cells (Fig. 3B). Since caspases
are important mediators of apoptosis, we investigated whether DDTC
treatment activates caspase-3. Caspase-3 is the main executioner
protease and its activation marks a point-of-no-return in the
complicated cascade of apoptosis induction (26). Cells were treated with DDTC, and
cell lysates were immunoblotted with anti-cleaved caspase-3
antibody. As shown in Fig. 3C,
DDTC treatment resulted in caspase-3 cleavage in BC-3 cells, but
did not activate caspase-3 in K562 cells. Furthermore, pretreatment
of BC-3 cells with various concentrations of Q-VD-OPh, a
pan-caspase inhibitor, abrogated the cell death induced by DDTC
treatment in a dose-dependent manner (Fig. 3D). These results suggest that DDTC
treatment selectively induces apoptosis in BC-3 cells in
caspase-3-dependent pathway.
DDTC causes a dose-dependent inhibition
of proliferation and induces apoptosis in various HHV-8-infected
PEL cell lines
Next, we determined whether DDTC treatment leads to
cell death in other HHV-8-infected cell lines. Three HHV-8-infected
cell lines (BCBL-1, BC-1 and BC-3) and an HHV-8-uninfected cell
line (K562) were cultured in the presence of 0.1, 0.3, 1 and 3 μM
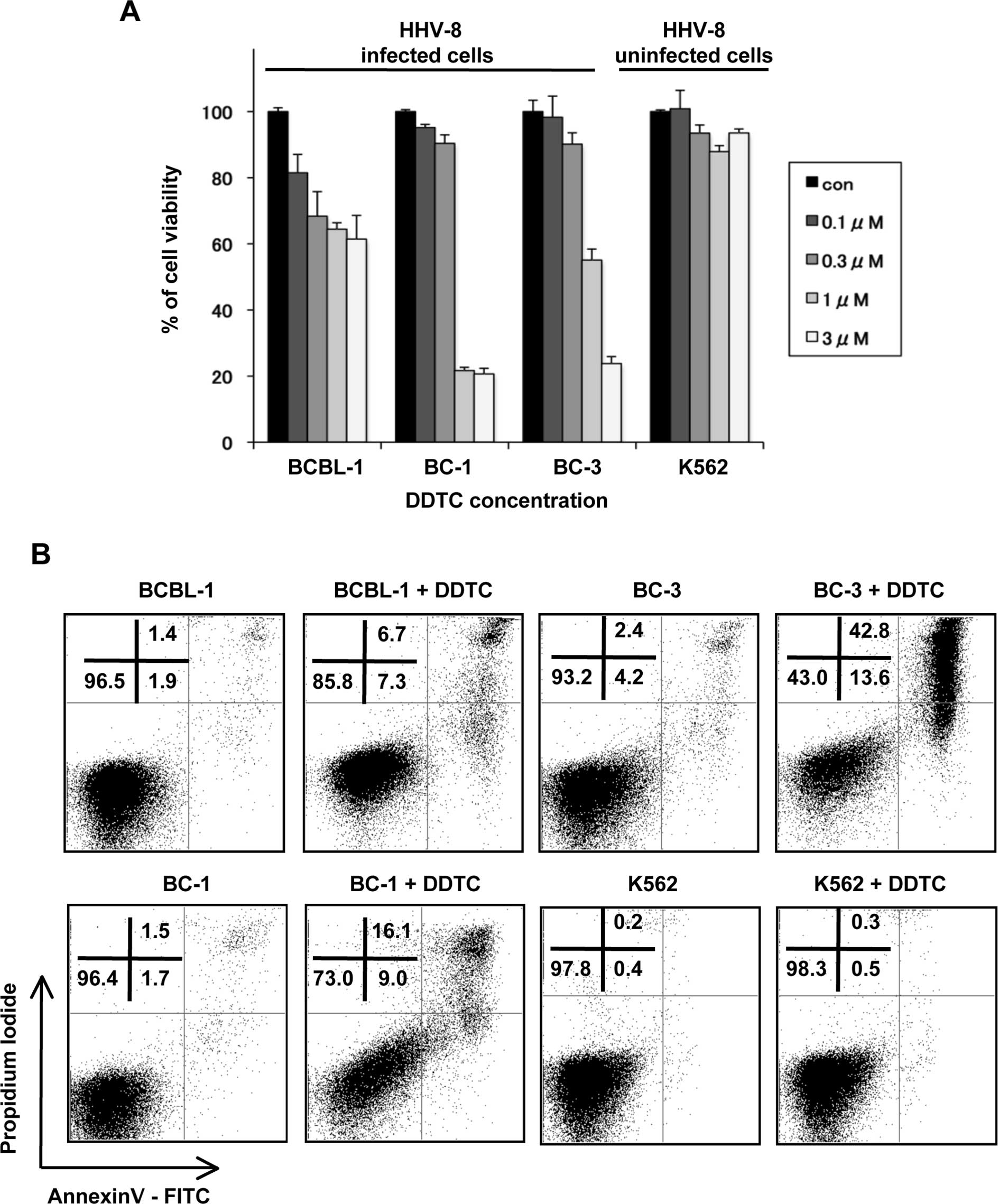
DDTC for 24 h, and MTT assay was performed. DDTC treatment
decreased the viable cells in a dose-dependent manner in
HHV-8-infected PEL cells (Fig.
4A). In contrast, treatment with DDTC did not affect the cell
viability in K562 cells. Moreover, we carried out Annexin V binding
assay followed by flow cytometry. Annexin V and propidium iodide
(PI) dual staining allows separation of cells at early phases of
apoptosis (Annexin V-positive, PI-negative) from those at the later
stages of cell death (Annexin V-positive and PI-positive). DDTC
treatment increased the population of early and late phases of
apoptosis in BCBL-1, BC-1 and BC-3 cells, whereas it did not induce
cell death in K562 cells (Fig.
4B). These results indicated that DDTC treatment induced
apoptosis in multiple HHV-8-infected PEL cell lines thereby
suppressing the growth of PEL cells.
Discussion
In this study we showed for the first time that DDTC
is a possible PEL therapeutic agent. DDTC suppressed the
constitutively activated NF-κB pathway and induced
caspase-3-dependent apoptosis in several PEL cell lines. DDTC also
suppressed the growth of PEL cells in vivo. The reported use
of DDTC or disulfiram as inhibitor of the canonical NF-κB pathway
is due to the ability of DDTC to block the release and degradation
of IκB by abolishing its phosphorylation, thereby preventing the
nuclear translocation of NF-κB subunit p65 (17). It has been demonstrated that DDTC
has proteasome-inhibitory and apoptotic functions (12,27),
and indeed we observed that DDTC increased the accumulation of
ubiquitinated proteins in BC-3 cells as well as in K562 cells
(Fig. 2A). However, apoptosis was
highly induced only in HHV-8-infected (BC-3) cells but not in K562
cells (Fig. 3A and B).
HHV-8-infected PEL cells are known to possess a constitutively
active NF-κB pathway, and its survival and proliferation are
largely dependent on this pathway (8,9).
Consistent with these reports, our results revealed that BC-3 cells
have a high level of p65 protein expression in the nucleus that was
reduced by DDTC treatment for 12 h. On the other hand, p65 protein
was not detected in the nucleus of K562 (Fig. 2A), in agreement with the report
that NF-κB is not constitutively expressed in K562 cells but could
be stimulated by ionizing radiation (28). Thus, DDTC could suppress the
abnormally activated NF-κB pathway in BC-3 cells and trigger
apoptosis in these cells. These findings imply that DDTC may
selectively cause apoptosis in NF-κB-dependent proliferative cells.
Hence it is possible that DDTC induces apoptosis in not only PEL
cell lines but also in other lymphoma cells that has abnormally
activated NF-κB pathway. Indeed, we detected by MTT assay that DDTC
treatment decreased the viability of Raji cells, a Burkitt lymphoma
cell line (data not shown), in which NF-κB pathway is
constitutively activated (29). In
agreement with this finding, a recent report showed that treatment
with pharmacological inhibitors of NF-κB pathway, including DDTC,
reduced the growth of medulloblastoma in vivo (30). It is also interesting to note the
study of Kanno et al, wherein they showed that human
leukemia cell lines NALM-6 and HL-60, which have constitutively
activated NF-κB signaling (31,32),
are sensitive to DDTC treatment while K562 cell line was mostly
unaffected by DDTC (22). The lack
of apoptosis, despite the proteasomal inhibition, observed in
DDTC-treated K562 cells (Figs. 2A
and 3B) would suggest that
proteasomal inhibition mediated by DDTC does not necessarily lead
to cell death. Rather, the induction of apoptotic pathway is also
contingent to the molecular constitution of the cells. This concept
may be relevant when determining and assessing the potential
therapeutic effects of DDTC on other cancer cells especially
because accumulated studies on DDTC demonstrating its function as
ubiquitin-proteasome inhibitor has fostered interest on DDTC as a
possible anti-tumor agent (reviewed in refs. 27,33,34).
DDTC is a metabolite of disulfiram, which can be
administrated orally and has few adverse effects (10). The dosage of DDTC used in this
study (2.5 mg/kg, 100 mg/kg) has no toxicity in mice (20). The human equivalent dose of 100
mg/kg in mice is 300 mg/m2 according to the standard
conversion table. This dose is less than 400–600 mg/m2,
the maximally tolerated dose of DDTC in human (35). Because PEL, unlike most
non-Hodgkin’s lymphomas, is relatively resistant to standard
cytotoxic chemotherapy, and virtually all PEL patients succumb to
the disease (36), finding a
viable therapeutic drug is imperative. Although further
investigations are required to determine the most effective dosage
in vivo, disulfiram could be a new therapeutic agent for PEL
with few side effects.
Acknowledgements
This study is supported by grants from the Ministry
of Education, Science, Sports and Culture (MEXT) of Japan, and the
Global COE Program (Cell Fate Regulation Research and Education
Unit), MEXT, Japan.
References
|
1
|
Chen YB, Rahemtullah A and Hochberg E:
Primary effusion lymphoma. Oncologist. 12:569–576. 2007.
|
|
2
|
Nador RG, Cesarman E, Chadburn A, Dawson
DB, Ansari MQ, Sald J and Knowles DM: Primary effusion lymphoma: a
distinct clinicopathologic entity associated with the Kaposi’s
sarcoma-associated herpes virus. Blood. 88:645–656. 1996.
|
|
3
|
Boulanger E, Gerard L, Gabarre J, Molina
JM, Rapp C, Abino JF, Cadranel J, Chevret S and Oksenhendler E:
Prognostic factors and outcome of human herpesvirus 8-associated
primary effusion lymphoma in patients with AIDS. J Clin Oncol.
23:4372–4380. 2005.
|
|
4
|
An J, Sun Y, Sun R and Rettig MB: Kaposi’s
sarcoma-associated herpesvirus encoded vFLIP induces cellular IL-6
expression: the role of the NF-kappaB and JNK/AP1 pathways.
Oncogene. 22:3371–3385. 2003.
|
|
5
|
Guasparri I, Keller SA and Cesarman E:
KSHV vFLIP is essential for the survival of infected lymphoma
cells. J Exp Med. 199:993–1003. 2004.
|
|
6
|
Field N, Low W, Daniels M, Howell S,
Daviet L, Boshoff C and Collins M: KSHV vFLIP binds to IKK-gamma to
activate IKK. J Cell Sci. 116:3721–3728. 2003.
|
|
7
|
Liu L, Eby MT, Rathore N, Sinha SK, Kumar
A and Chaudhary PM: The human herpes virus 8-encoded viral FLICE
inhibitory protein physically associates with and persistently
activates the Ikappa B kinase complex. J Biol Chem.
277:13745–13751. 2002.
|
|
8
|
Keller SA, Hernandez-Hopkins D, Vider J,
Ponomarev V, Hyjek E, Schattner EJ and Cesarman E: NF-kappaB is
essential for the progression of KSHV- and EBV-infected lymphomas
in vivo. Blood. 107:3295–3302. 2006.
|
|
9
|
Keller SA, Schattner EJ and Cesarman E:
Inhibition of NF-kappaB induces apoptosis of KSHV-infected primary
effusion lymphoma cells. Blood. 96:2537–2542. 2000.
|
|
10
|
Suh JJ, Pettinati HM, Kampman KM and
O’Brien CP: The status of disulfiram: a half of a century later. J
Clin Psychopharmacol. 26:290–302. 2006.
|
|
11
|
Sunderman FW Sr: The extended therapeutic
role of dithiocarb (sodium diethyldithiocarbamate) from nickel
poisoning to AIDS. Ann Clin Lab Sci. 22:245–248. 1992.
|
|
12
|
Pang H, Chen D, Cui QC and Dou QP: Sodium
diethyldithiocarbamate, an AIDS progression inhibitor and a
copper-binding compound, has proteasome-inhibitory and
apoptosis-inducing activities in cancer cells. Int J Mol Med.
19:809–816. 2007.
|
|
13
|
Brewer C: Long-term, high-dose disulfiram
in the treatment of alcohol abuse. Br J Psychiatry. 163:687–689.
1993.
|
|
14
|
Hersh EM, Brewton G, Abrams D, Bartlett J,
Galpin J, Gill P, Gorter R, Gottlieb M, Jonikas JJ, Landesman S, et
al: Ditiocarb sodium (diethyldithiocarbamate) therapy in patients
with symptomatic HIV infection and AIDS. A randomized,
double-blind, placebo-controlled, multicenter study. JAMA.
265:1538–1544. 1991.
|
|
15
|
Reisinger EC, Kern P, Ernst M, Bock P,
Flad HD and Dietrich M: Inhibition of HIV progression by
dithiocarb. German DTC Study Group. Lancet. 335:679–682. 1990.
|
|
16
|
Shinobu LA, Jones SG and Jones MM:
Mobilization of aged cadmium deposits by dithiocarbamates. Arch
Toxicol. 54:235–242. 1983.
|
|
17
|
Cvek B and Dvorak Z: Targeting of nuclear
factor-kappaB and proteasome by dithiocarbamate complexes with
metals. Curr Pharm Des. 13:3155–3167. 2007.
|
|
18
|
Renne R, Zhong W, Herndier B, McGrath M,
Abbey N, Kedes D and Ganem D: Lytic growth of Kaposi’s
sarcoma-associated herpesvirus (human herpesvirus 8) in culture.
Nat Med. 2:342–346. 1996.
|
|
19
|
Ono A, Hattori S, Kariya R, Iwanaga S,
Taura M, Harada H, Suzu S and Okada S: Comparative study of human
hematopoietic cell engraftment into BALB/c and C57BL/6 strain of
rag-2/jak3 double-deficient mice. J Biomed Biotechnol.
2011.539748:
|
|
20
|
Hersh EM, Funk CY, Petersen EA, Ryschon KL
and Mosier DE: Dose response and timing effects in the therapy of
the LP-BM5 murine retrovirus-induced lymphoproliferative
immunodeficiency disease with diethyldithiocarbamate. Int J
Immunopharmacol. 15:137–143. 1993.
|
|
21
|
Aoki Y, Jaffe ES, Chang Y, Jones K,
Teruya-Feldstein J, Moore PS and Tosato G: Angiogenesis and
hematopoiesis induced by Kaposi’s sarcoma-associated
herpesvirus-encoded interleukin-6. Blood. 93:4034–4043. 1999.
|
|
22
|
Kanno S, Matsukawa E, Miura A, Shouji A,
Asou K and Ishikawa M: Diethyldithiocarbamate-induced cytotoxicity
and apoptosis in leukemia cell lines. Biol Pharm Bull. 26:964–968.
2003.
|
|
23
|
Asou H, Said JW, Yang R, Munker R, Park
DJ, Kamada N and Koeffler HP: Mechanisms of growth control of
Kaposi’s sarcoma-associated herpes virus-associated primary
effusion lymphoma cells. Blood. 91:2475–2481. 1998.
|
|
24
|
Takahashi-Makise N, Suzu S, Hiyoshi M,
Ohsugi T, Katano H, Umezawa K and Okada S: Biscoclaurine alkaloid
cepharanthine inhibits the growth of primary effusion lymphoma in
vitro and in vivo and induces apoptosis via suppression of the
NF-kappaB pathway. Int J Cancer. 125:1464–1472. 2009.
|
|
25
|
Zhang C, Hazarika P, Ni X, Weidner DA and
Duvic M: Induction of apoptosis by bexarotene in cutaneous T-cell
lymphoma cells: relevance to mechanism of therapeutic action. Clin
Cancer Res. 8:1234–1240. 2002.
|
|
26
|
Porter AG and Janicke RU: Emerging roles
of caspase-3 in apoptosis. Cell Death Differ. 6:99–104. 1999.
|
|
27
|
Sauna ZE, Shukla S and Ambudkar SV:
Disulfiram, an old drug with new potential therapeutic uses for
human cancers and fungal infections. Mol Biosyst. 1:127–134.
2005.
|
|
28
|
Cataldi A, Rapino M, Centurione L,
Sabatini N, Grifone G, Garaci F and Rana R: NF-kappaB activation
plays an antiapoptotic role in human leukemic K562 cells exposed to
ionizing radiation. J Cell Biochem. 89:956–963. 2003.
|
|
29
|
Herrero JA, Mathew P and Paya CV: LMP-1
activates NF-kappa B by targeting the inhibitory molecule I kappa B
alpha. J Virol. 69:2168–2174. 1995.
|
|
30
|
Spiller SE, Logsdon NJ, Deckard LA and
Sontheimer H: Inhibition of nuclear factor kappa-B signaling
reduces growth in medulloblastoma in vivo. BMC Cancer.
11:1362011.
|
|
31
|
Safa M, Zand H, Mousavizadeh K, Kazemi A,
Bakhshayesh M and Hayat P: Elevation of cyclic AMP causes an
imbalance between NF-kappaB and p53 in NALM-6 cells treated by
doxorubicin. FEBS Lett. 584:3492–3498. 2010.
|
|
32
|
Han SS, Kim K, Hahm ER, Park CH, Kimler
BF, Lee SJ, Lee SH, Kim WS, Jung CW, Park K, Kim J, Yoon SS, Lee JH
and Park S: Arsenic trioxide represses constitutive activation of
NF-kappaB and COX-2 expression in human acute myeloid leukemia,
HL-60. J Cell Biochem. 94:695–707. 2005.
|
|
33
|
Cvek B and Dvorak Z: The value of
proteasome inhibition in cancer. Can the old drug, disulfiram, have
a bright new future as a novel proteasome inhibitor? Drug Discov
Today. 13:716–722. 2008.
|
|
34
|
Kona FR, Buac D and Burger AM: Disulfiram,
and disulfiram derivatives as novel potential anticancer drugs
targeting the ubiquitin proteasome system in both preclinical and
clinical studies. Curr Cancer Drug Targets. 11:338–346. 2011.
|
|
35
|
Kaplan CS, Petersen EA, Yocum D and Hersh
EM: A randomized, controlled dose response study of intravenous
sodium diethyldithiocarbamate in patients with advanced human
immunodeficiency virus infection. Life Sci. 45:iii–ix. 1989.
|
|
36
|
Komanduri KV, Luce JA, McGrath MS,
Herndier BG and Ng VL: The natural history and molecular
heterogeneity of HIV-associated primary malignant lymphomatous
effusions. J Acquir Immune Defic Syndr Hum Retrovirol. 13:215–226.
1996.
|