Introduction
Prostate cancer is the most frequently diagnosed
malignancy and second leading cause of cancer death in males both
in the United States and United Kingdom. A unique characteristic of
these tumors is that they are exquisitely dependent on androgen for
development, growth and survival (1–3).
Hence, in addition to their normal physiological roles in the
growth and development of male sex organs, androgens play an
equally critical function in the abnormal growth of prostate
cancer. In both contexts, these effects are mediated through
activation of the androgen receptor (AR) signaling pathway
(4). Upon diagnosis patients are
typically subjected to androgen ablation therapy involving either
surgical or chemical castration. This latter process is achieved
through the use of selective antiandrogen agents, such as
gonadotropin-releasing hormone (GnRH) agonists or second generation
AR inhibitors like bicalutamide (Casodex). Unfortunately, while
blockade of AR is initially effective in achieving clinically
relevant remissions, most patients relapse and progress with
castration-resistant disease within 12–24 months (5). Previously considered to be
androgen-independent, it is now emerging that these recurrent
prostate cancers may still rely on AR signaling for growth and
survival. The mechanisms by which these more aggressive tumors
retain AR activity and AR-mediated gene expression are still
unclear, although it has been hypothesized that the development of
intratumoral steroidogenesis may contribute to castration-resistant
tumor growth (6). First-line
treatment for castration-resistant prostate cancer typically
consists of docetaxel in combination with prednisone or
estramustine (7). However, no
consensus exists regarding the best approach following docetaxel
failure (8). Second-line
approaches including hormone therapy, taxane or immunotherapy often
fail to dramatically impact patient survival. A unifying feature of
advanced or metastatic disease therefore is that patient prognosis
is dismal as therapeutic options become more limited.
The AR receptor pathway mediates the transcriptional
regulation of multiple genes in prostate cancer, including those
that promote tumor cell survival and proliferation. In addition,
the ability of AR to crosstalk with other key growth factor
signaling pathways in prostate cancer has been established
(9). In particular, recent studies
have identified several mechanisms for regulation of AR by the
mammalian target of rapamycin (mTOR) signaling cascade and vice
versa (10–12). mTOR is a serine/threonine kinase
downstream of PI3K/AKT that acts as a checkpoint for both cellular
nutritional status and cell cycle control (13–15).
Of note, dysregulation of the PI3K/AKT/mTOR pathway has been
implicated in the malignant transformation accompanying prostate
cancer progression (16).
Moreover, loss-of-function of the tumor suppressor gene PTEN, which
results in constitutive activation of AKT and upregulation of mTOR
activity, has been implicated in the etiology of numerous human
cancers including more than 50% of advanced prostate tumors
(17–19). Further, it has been demonstrated
that tumors that harbor deletions or defects in PTEN are, in
general, hypersensitive to inhibition of mTOR (20–22).
This provides a clear biological rationale for the blockade of mTOR
activity as a potential therapeutic point of intervention for
prostate cancer.
Ridaforolimus (AP23573, MK-8669), a non-prodrug
analog of rapamycin, is a potent and selective inhibitor of mTOR
(23) currently under clinical
investigation as a targeted cancer. Interestingly, ridaforolimus
has shown promising single-agent activity in a phase II trial of
advanced, progressive endometrial cancer (24). Similar to prostate cancer, this
tumor type is also characterized by a high incidence of functional
inactivation of PTEN (25–27). In preclinical models of endometrial
cancer, we previously demonstrated an association between PTEN loss
and ridaforolimus sensitivity (28). A number of reports have also
suggested targeting mTOR in prostate cancer (29–31).
Here we investigated both the effects of mTOR inhibition alone, as
well as in combination with AR blockade, in models of prostate
cancer. We show that simultaneous treatment with ridaforolimus and
bicalutamide results in synergistic antiproliferative effects in
vitro and in vivo. These findings support the potential
therapeutic value of dual inhibition of the AR and mTOR signaling
pathways as a valid approach for the treatment of patients with
this disease.
Materials and methods
Cell lines and reagents
All cell lines used in this study were obtained from
the American Type Culture Collection with the exception of the C4-2
line, which was a kind gift from Dr George Thalmann (University of
Bern, Switzerland). Cells were maintained and cultured according to
standard techniques at 37°C in 5% (v/v) CO2 using culture medium
recommended by the supplier. Ridaforolimus (AP23573; MK-8669) was
synthesized at ARIAD Pharmaceuticals and prepared in ethanol to a 1
mM stock concentration. For in vitro cellular assays
ridaforolimus was diluted in the optimal medium. For in vivo
experiments, ridaforolimus was diluted in a vehicle of 4% ethanol,
5% Tween 80, and 5% propylene glycol. Bicalutamide (Casodex) was
purchased from Zheijang Esun Chemical Co., Ltd. (China).
In vitro proliferation assays
Proliferation assays were performed as previously
described (23). Briefly,
exponentially growing cell lines were plated into two 96-well
plates and incubated overnight at 37°C. Twenty-four hours later one
plate was aspirated and stored at −80°C and the other treated with
10-fold serial concentrations of ridaforolimus (1000 nM to 0.0001
nM) or vehicle (ethanol). Following 72 h culture at 37°C, the
plates were assessed simultaneously for cell growth using the
CyQUANT Cell Proliferation Assay kit (Invitrogen). The parameters
measured were: Doubling time (DT) = [0.301*(72)/LOG(Day4/Day1)];
Doublings = 72/DT; and Cell growth rate (%) = Doublings
ridaforolimus /Doublings vehicle *100. The maximal inhibitory
effect (Imax) measure was used to determine relative
sensitivity of each cell line. Imax = 100 – cell growth
rate (%) at the dose whereby maximum inhibition is observed.
Compound combination proliferation assays were performed similarly
except cell growth was determined as the change in cell number
between vehicle control and compound treated cells after 72 h in
culture. The average (± SD) of n ≥3 individual experiments are
reported.
Median effect analysis
Cells were seeded as described for the in
vitro proliferation assay and combination treatments of
ridaforolimus and bicalutamide were performed with fixed 1:1 ratios
of concentrations that induced half the maximal effect (i.e.
EC50 values) for each drug. Two-fold serial dilutions
above and below the EC50 values were added to the cell
cultures for 72 h. The nature of the ridaforolimus-bicalutamide
combination interaction was evaluated using the combination index
(CI) method of Chou and Talalay (32) and values were generated using
Median Effect analysis (CalcuSyn Software; Biosoft).
Anchorage-independent cell proliferation
analysis
C4-2 cells were immobilized in 6-well dishes in
culture medium containing 0.3% agarose. The soft agar layer
containing the cells was then overlaid with a liquid media layer
containing one of the following: 0.5 nM ridaforolimus, 10 μM
bicalutamide, 0.5 nM ridaforolimus + 10 μM bicalutamide, or
media alone (no treatment). The cells were cultured at 37°C for 2
weeks, with the liquid media layer being replaced with fresh
media/drug treatment every three to four days. Colonies were then
counted using a Universal Hood II Molecular Imager®
ChemiDoc™XRS System and Quantity One® SW 1-D Analysis
software (Bio-Rad Laboratories, Inc.). The relative colony
formation for each treatment group was then calculated as a
percentage of the untreated group using the formula: (# colonies
treatment group/ # colonies untreated group)*100. The p-value was
calculated using the Student’s t-test.
Flow cytometric analysis
C4-2 cells cultured in 10 cm dishes were treated for
24 h with one of the following: 50 nM ridaforolimus, 50 μM
bicalutamide, 50 nM ridaforolimus + 50 μM bicalutamide, or
vehicle control. The cells were then harvested and fixed with 70%
ethanol / 30% PBS overnight at 4°C. Fixed cells were washed and
then sequentially incubated with 50 μg/ml RNase A (37°C for
30 min) and 20 μg/ml propidium iodide (room temperature for
30 min in the dark). DNA content was analyzed using a FACSort flow
cytometer and CellQuest V3.1 software (Becton-Dickinson and
Company). The percentage of cells in each phase of the cell cycle
was then estimated from the FL2-A channel data using ModFit LT for
Mac V2.0 software (Verity Software House, Inc.). The p-value was
calculated using the Student’s t-test.
Biomarker expression and in vitro
pharmacodynamics
Cell lines were harvested during exponential growth
phase and immunoblotted for markers of the AR and AKT/mTOR
pathways. Lysates were loaded left to right according to decreasing
level of ridaforolimus sensitivity determined by Imax.
For pharmacodynamic analysis, cells were treated with 0.5 nM
ridaforolimus and/or 10 μM bicalutamide then harvested and
assessed by immunoblotting for PTEN, AKT, p-AKT
(Ser473), S6, p-S6 (Ser235/236), p-4E-BP1
(Ser65/Thr70) and VDAC (loading control)
expression (Cell Signaling Technology). PTEN status of cell lines
was determined using the Sanger Cancer Genome Project mutation
database (http://www.sanger.ac.uk/genetics/CGP) and confirmed by
immunoblotting.
PSA ELISA
Cells were treated for 72 h with single agent or
combination compound treatment as described for the in vitro
proliferation assay, supernatants were harvested, and PSA
Quantikine Immunoassay was performed according to the manufacturers
protocols (R&D Systems).
Subcutaneous tumor model
Prostate cancer xenografts were established by the
subcutaneous implantation of C4-2 cells (5×106 cells +
matrigel) at the right flank area of six to eight week old male
nude mice (nu/nu strain) (Charles River Laboratories; Wilmington,
MA). For analysis of efficacy, when the average tumor volume
reached approximately 200 mm3 mice were administered the
indicated dose of ridaforolimus i.p. (n=10 mice/condition) daily
for 5 days followed by a 2 day break, or bicalutamide p.o. daily,
or the combination. Three cycles of dosing were completed (21
days). Mean tumor volume volumes were calculated for each treatment
group by caliper measurements using the following formula: tumor
volume = (length × width2)/2. Blood was harvested from
mice on days 0, 7, 14 and 21 for PSA ELISA. Differences between the
multiple treatment groups were analyzed by one-way ANOVA test.
Ethical treatment of animals
All of the animal experiments were conducted in
strict accordance with the National Institute of Health Guide for
the Care and Use of Laboratory Animals. The protocol was approved
by the Institutional Animal Care and Use Committees, ARIAD
Pharmaceuticals, Inc. (Protocol Number: 08-01). All efforts were
made to minimize suffering.
Results
Loss of PTEN and elevated AKT/mTOR
activity are associated with ridaforolimus sensitivity in prostate
cell lines
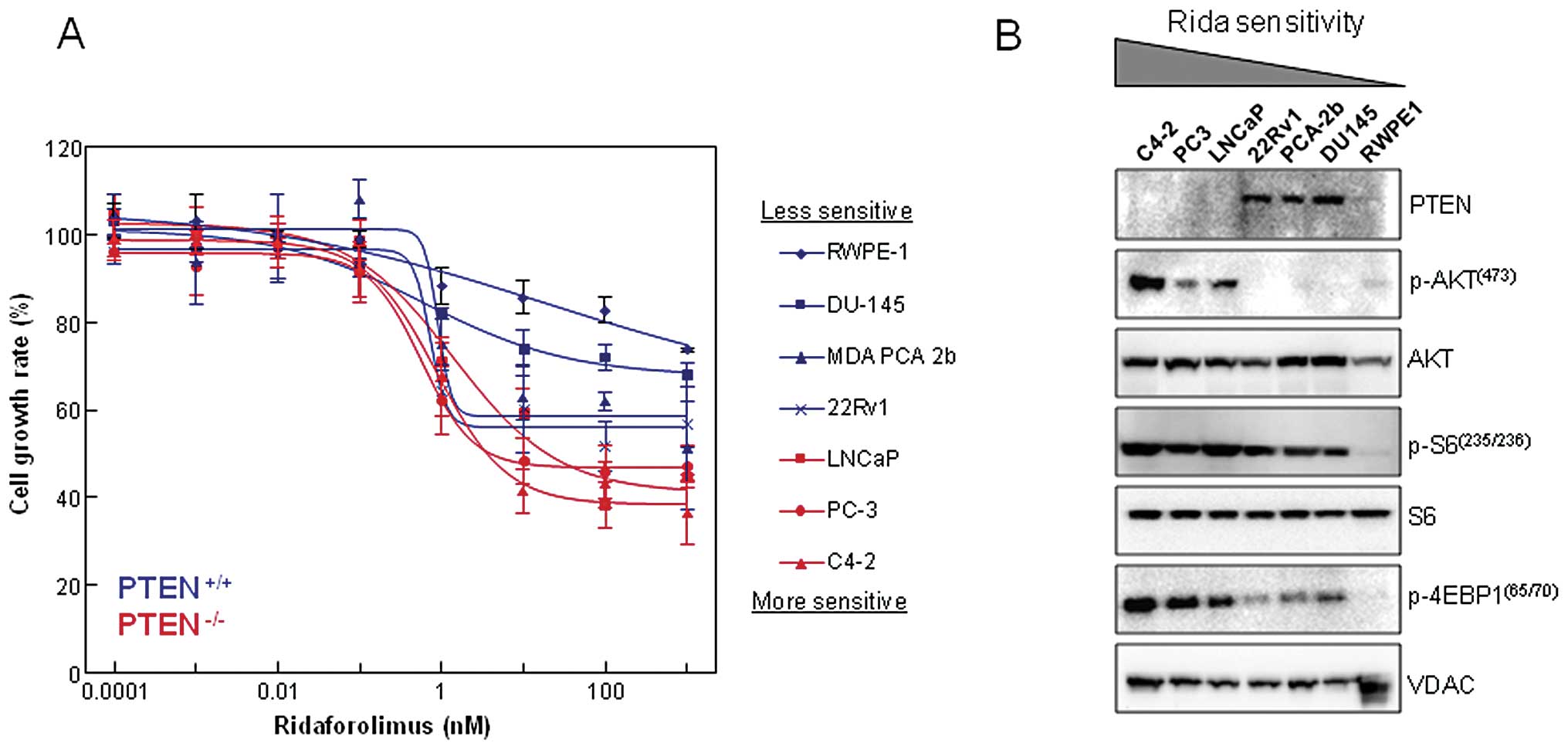
We examined the effect that single agent
ridaforolimus treatment had on cell proliferation using a series of
prostate-derived cell lines (Fig.
1A). Exposure to nanomolar concentrations of ridaforolimus
reduced cellular proliferation in each of the 6 prostate cancer
cell lines (DU-145, MDA PCA 2b, 22Rv1, LNCaP, PC-3 and C4-2) with
maximal inhibition (Imax) ranging from 20–60%. In
contrast, ridaforolimus was least effective in inhibiting the cell
growth rate of the immortalized normal prostatic epithelial line
RWPE-1. Notably, cellular PTEN status was associated with drug
sensitivity, as the PTEN-null cancer cell lines showed the greatest
degree of inhibition. Loss of PTEN typically leads to constitutive
activation of downstream components of the PI3K pathway, including
the AKT and mTOR kinases. As confirmation, we examined the
expression levels and activation state of this signaling cascade
and compared that to ridaforolimus sensitivity in our panel of
prostate lines (Fig. 1B). As
expected, there was a concomitant increase in phospho-AKT
(Ser473) levels observed in the PTEN−/− cell
lines (LNCaP, PC-3, C4-2). These same three lines also demonstrated
hyperactivation of the mTOR pathway, as evidenced by elevated
phosphorylation of the key downstream targets ribosomal protein S6
and 4E-BP1 (Fig. 1B). Together,
these data indicate that PTEN loss and aberrant mTOR signaling are
intrinsic cellular properties associated with ridaforolimus
sensitivity in prostate cancer lines.
Simultaneous blockade of both AR and mTOR
pathways in cancer, but not normal prostate lines, results in
synergistic growth inhibition
Loss of PTEN and consequent elevation of AKT
activity can promote both mTOR as well as AR-dependent
proliferation (33). Further, it
has been suggested that one function of AR in PTEN-deficient
prostate cancer cells is to promote the pathologic activation of
mTOR (11), providing a potential
mechanistic link between these two pathways in prostate cancer.
Based on these considerations, we evaluated the combinatorial
effects of ridaforolimus and the antiandrogen bicalutamide in
inhibiting the growth of prostate cell lines. To examine this, the
normal prostate PTEN wild-type line, RWPE-1, and two
PTEN−/− tumor lines, LNCaP and C4-2, were treated in
vitro with a combination of ridaforolimus and bicalutamide.
LNCaP is a well characterized, androgen-dependent cell line that
represents the early stages of prostate cancer progression. The
metastatically derived subline of LNCaP, C4-2, has been
traditionally considered androgen-independent. Indeed, we have
found that C4-2 can grow in the absence of androgens in
vitro and in castrated mice, although proliferation in both
model systems is substantially reduced (data not shown). However,
they do respond to androgen and are sensitive to bicalutamide in an
androgen-rich environment (see below). Therefore, C4-2 cells are a
more progressed form of prostate cancer compared with LNCaP, but
they are not fully androgen-independent. Combinatorial activity was
determined using the Median Effect method to establish whether the
combinations exhibited antagonistic, additive or synergistic
activity (Fig. 2A). RWPE-1 cells
displayed only a simple additive effect when treated with these two
agents together. In stark contrast, the combination was found to be
strongly synergistic in both prostate cancer cell lines. The
combinatorial benefit was further demonstrated by significant
inhibition of anchorage-independent growth in the C4-2 cell line
(Fig. 2B). These findings suggest
the potential for combining therapy in prostate cancer patients
with inhibitors of both AR and mTOR pathways.
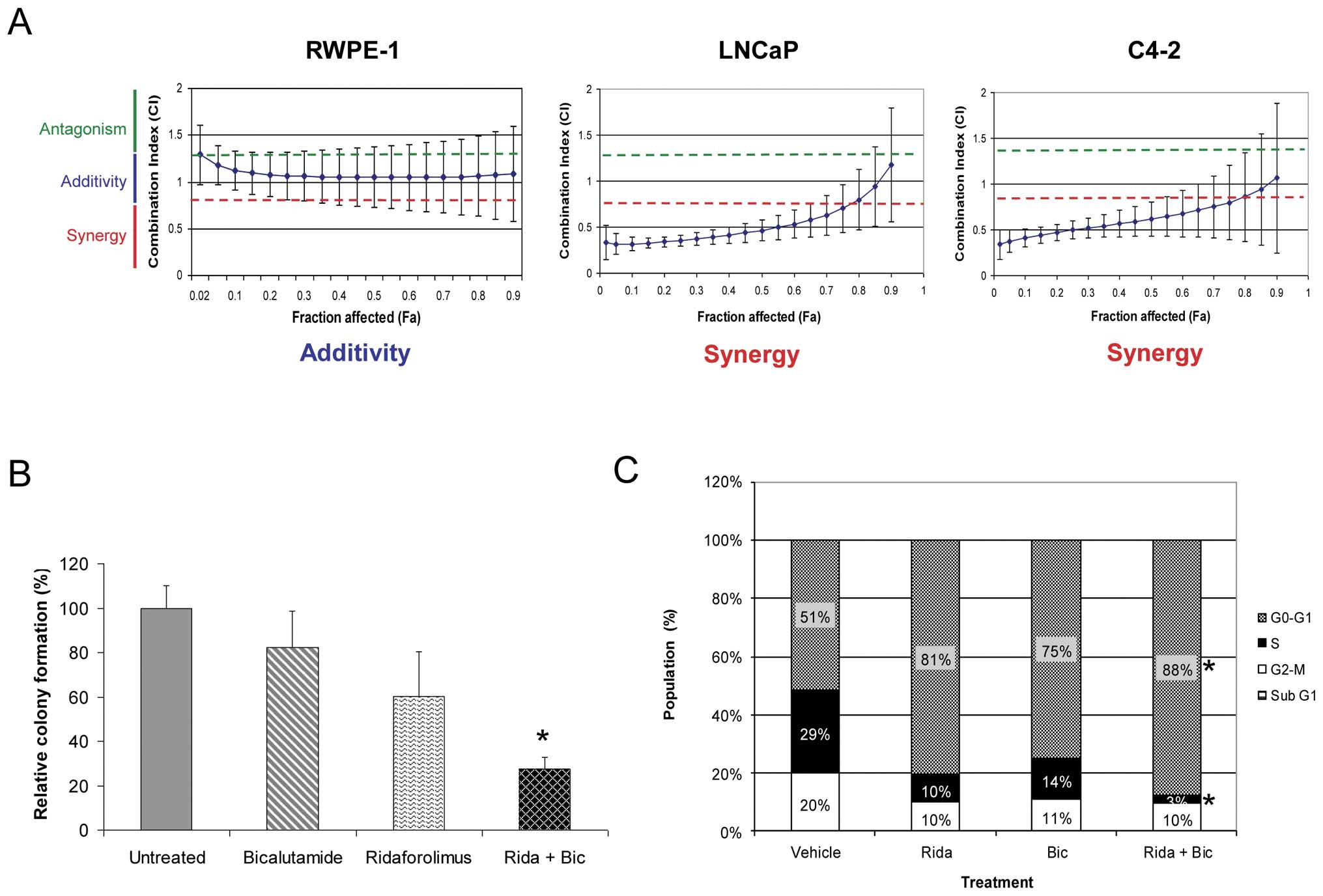 | Figure 2Simultaneous AR and mTOR pathway
blockade results in synergistic growth inhibition in prostate
cancer lines. (A) RWPE-1, LNCaP and C4-2 cell lines were treated
with increasing concentrations of ridaforolimus, bicalutamide, or
both, and the effects on proliferation determined. The Combination
Index (CI) was calculated using Median Effect analysis. Strict
criteria were applied to drug interaction analysis, where synergy
was defined as CI <0.75, additivity as >0.75 CI <1.25, and
antagonism as CI >1.25. Data expressed as mean CI (± SD),
determined for a range of drug concentrations and a fractional
effect (Fa) of 0.2 to 0.8 over the complete dosing range. (B) Soft
agar clonogenic assay to determine the effects of the ridaforolimus
and bicalutamide combination on anchorage-independent growth of
C4-2 prostate cancer cells. C4-2 cells were treated with medium
alone, bicalutamide (10 μM), ridaforolimus (0.5 nM) or the
combination (Rida + Bic) for 2 weeks. The percentage colony
formation (compared to untreated controls) are presented as means ±
SD for duplicate experiments. *p-value ≤0.01 compared
with vehicle treated cells. (C) Ridaforolimus and bicalutamide in
combination induce cell cycle arrest in prostate cancer cells. C4-2
prostate cancer cells were treated with vehicle, ridaforolimus
(Rida; 50 nM), bicalutamide (Bic; 50 μM) or the combination
(Rida + Bic) for 24 h. Cells were harvested, stained with propidium
iodide and analyzed by flow cytometry to determine DNA content. The
percentage of cells in G1, S or G2/M phase was calculated from FL-2
histograms using ModFit Lt software. *p-value ≤0.05
compared with single agents or vehicle treated cells. |
Ridaforolimus and bicalutamide
combination treatment promotes cell cycle arrest in prostate cancer
cells
Ridaforolimus exerts its antiproliferative effects
on cancer cells through primarily cytostatic, not cytotoxic,
activities (23). We thus
investigated the mode of action of the ridaforolimusbicalutamide
combination on cell cycle progression and survival of C4-2 cells.
Single agent treatment alone led to a decrease in both S and G2/M
phases with a concomitant accumulation in the G1 phase of the cell
cycle (Fig. 2C). Consistent with
the enhanced growth inhibition of these cells shown in Fig. 2A, the effect of combination
treatment was more pronounced, resulting in an almost complete G1
arrest in this cell line. Indeed, both the increase in the
proportion of cells in G1 phase and reduction in S phase were
significantly different from either vehicle controls or each
individual agent alone. No increase in the sub-G1 fraction was
observed with any treatment indicating no significant pro-apoptotic
activity. This result was confirmed by additional studies which
failed to detect an increase in other apoptotic markers including
cleaved PARP and caspase-3 (data not shown).
PSA levels mirror cell growth of
ridaforolimus-bicalutamide treated prostate cancer lines
mTOR blockade can result in increased AR
transcriptional activity and consequent PSA expression, independent
of effects on cell growth (12).
This finding has important clinical implications, as plasma PSA
levels in prostate cancer patients are used as a measure of tumor
growth and disease progression. As shown in Fig. 3A (left panel), the immortalized
RWPE-1 cell line did not exhibit elevation of AR or PSA expression,
as expected. Bicalutamide treatment resulted in significant
suppression of PSA levels and moderate decrease of AR expression in
both LNCaP and C4-2 prostate cancer lines. In contrast, addition of
ridaforolimus to the androgen-dependent LNCaP line resulted in an
increase in PSA expression and a similar response was observed in
the C4-2 cells. However, in both cases, the combination of
ridaforolimus and bicalutamide abrogated this effect. Indeed,
combination treatment resulted in the temporal inhibition of both
PSA and AR expression (Fig. 3A,
right panel) thus, bicalutamide not only inhibited basal levels of
AR transcriptional activity, but was sufficient to block the
ridaforolimus-induced stimulation of PSA. Ridaforolimus alone and
in combination promoted a modest increase of p-AKT levels in both
cancer lines at the 24 h time point, however, this effect was not
sustained with the combination over the longer 72 h time course. As
expected, single agent ridaforolimus reduced phospho-S6 levels in
the tumor lines. Interestingly, the addition of bicalutamide also
potentiated this effect, providing further evidence of pathway
crosstalk in these cancer cells.
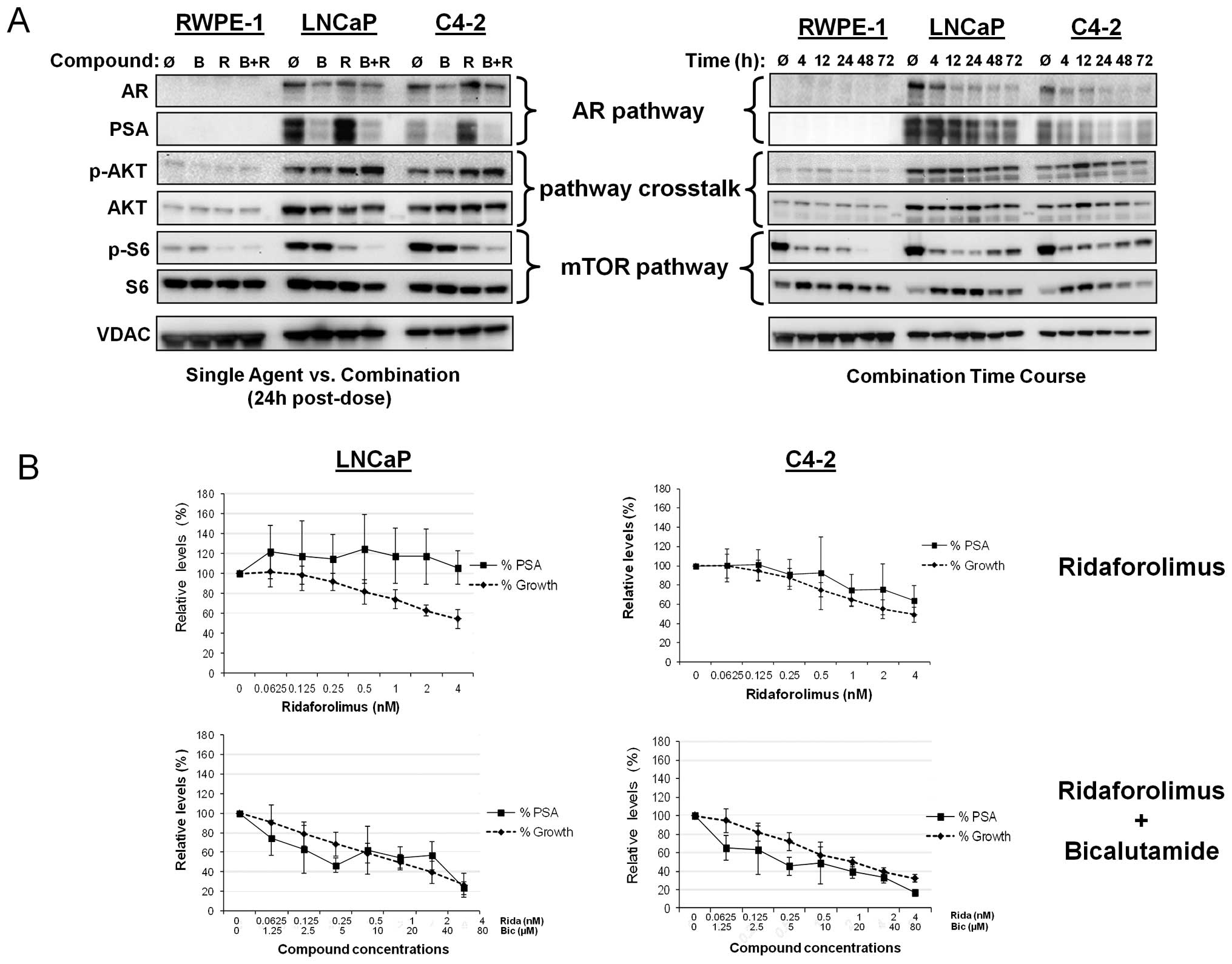 | Figure 3Combination of ridaforolimus plus
bicalutamide inhibits AR and mTOR signaling; PSA levels mirror cell
growth in combination-treated prostate cell lines. (A) In the left
panel RWPE-1, LNCaP and C4-2 cells were treated for 24 h with
vehicle alone (Ǿ), 10 μM bicalutamide (B), 0.5 nM
ridaforolimus (R), or the combination (B+R). In the right panel,
RWPE-1, LNCaP and C4-2 cells were treated with the combination of
ridaforolimus and bicalutamide for up to 3 days with lysates
harvested at the indicated times. Cellular extracts were
immunoblotted for AR, PSA, phospho-S6 (Ser235/236),
ribosomal protein S6 and VDAC (as a control). (B) LNCaP and C4-2
cells were treated with ridaforolimus as a single agent at the
indicated concentrations (upper panels), or with the
ridaforolimus/bicalutamide combination at the range of
concentrations indicated (lower panels) for 3 days. PSA levels in
the supernatant were determined by ELISA and relative secretion
presented as the ratio of compound- versus vehicle-treated cells.
The effects on proliferation were evaluated and cell growth shown
as a percentage of vehicle controls. Data presented as means of ≥3
individual experiments ± SD. |
Having determined that mTOR inhibition by
ridaforolimus leads to increased PSA expression, we next examined
whether this effect related to changes in cell growth. To
investigate this, cell lines were treated with either ridaforolimus
alone or bicalutamide and the relative levels of cellular
proliferation and PSA secretion compared (Fig. 3B). As expected, RWPE-1 cells, which
lack AR signaling and exhibit low endogenous mTOR activity, did not
secrete PSA (data not shown). In LNCaP cells, despite a
dose-dependent decrease in cell proliferation following
ridaforolimus treatment, the levels of secreted PSA did not change.
Combination treatment, however, did result in a parallel effect on
both PSA secretion and cell growth (Fig. 3B). In C4-2 cells, a decrease in
cell growth with ridaforolimus treatment was accompanied by a
slight decrease in PSA levels at the highest doses tested. Similar
to the LNCaP line, a marked reduction in PSA levels mirrored growth
inhibition after addition of the ridaforolimus-bicalutamide
combination. Taken together, these data support the utility of PSA
secretion as a readout of cell proliferation in combination treated
cancer lines in vitro.
Ridaforolimus plus bicalutamide inhibits
prostate tumor growth and reduces plasma PSA level in vivo
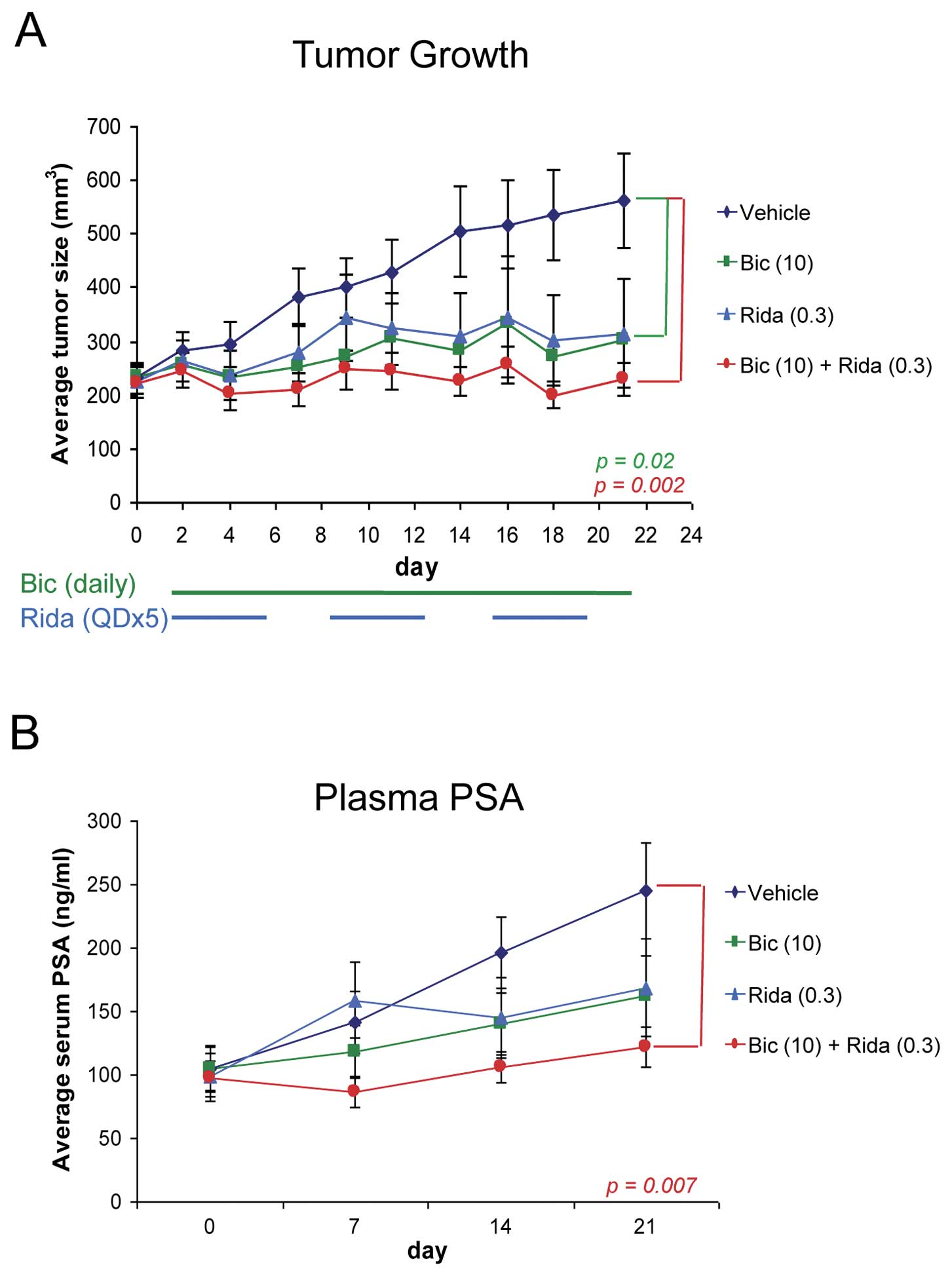
Finally, we evaluated the combinatorial effect of
ridaforolimus and bicalutamide on prostate tumor growth in the C4-2
xenograft model. We used intact nude mice for this study because
although C4-2 cells are able to grow tumors in castrated mice, we
found that the growth is not robust enough to use for evaluation of
efficacy (data not shown). Daily dosing of bicalutamide and a 5
days per week schedule for ridaforolimus were used as this
recapitulates the dosing regimens being explored in clinical
studies. Single agent ridaforolimus and bicalutamide reduced tumor
growth by 73% and 79%, respectively, at defined submaximal doses
Fig. 4A). Consistent with the
earlier in vitro observations, the
ridaforolimus-bicalutamide combination exhibited improved and
potent antitumor activity, almost completely abrogating tumor
growth (TGI = 98%). The combination was also well tolerated, as
evidenced by no significant changes in body weight over the course
of treatment (data not shown). Importantly, plasma PSA levels were
again tightly linked to tumor growth in the combination-treated
mice (Fig. 4B), suggesting that
PSA may be an accurate and relevant marker of tumor growth in
patients undergoing combination therapy.
Discussion
A number of reports have implicated mTOR signaling
as a prominent factor during prostate cancer progression (16,29–31).
This can be explained in part by functional loss of PTEN and
concomitant activation of the mTOR pathway which is predicted to
result in hypersensitivity to mTOR inhibitors (20–22).
Consistent with this, we found mTOR signaling to be elevated in
PTEN−/− prostate cancer lines relative to
PTEN+/+ lines, and that PTEN−/− lines exhibit
greater sensitivity to ridaforolimus in vitro. This suggests
PTEN status may predict for sensitivity to ridaforolimus in this
tumor type. Several lines of evidence link PTEN inactivation to
disease progression and risk of recurrence in prostate cancer
(34–38). Moreover in engineered mouse models,
loss-of-function of PTEN leads to high grade PIN and/or carcinoma
(39,40) and can amplify the tumor-promoting
effects of other oncogenes including p53 and p27 (41,42).
Taken together, our results identify a potential molecular
predictor of response to ridaforolimus treatment in prostate
cancer, and also support the possible therapeutic utility of mTOR
blockade for treating this disease.
Targeting AR through androgen ablation therapy is
the mainstay of prostate cancer treatment. However, these cancers
often progress and as a result, treatment options become limited.
While often termed ‘androgen-independent’, recent work has shown
that the majority of these tumors continue to rely on AR signaling
for growth and survival, and several mechanisms have been
postulated for reactivation of AR in the castrate environment
(3). Overexpression of AR through
genomic amplification, as well as mutations that allow activation
by reduced androgen levels or by other endogenous steroids, has
been observed in recurrent tumors. These cellular alterations are
effective in sensitizing the AR pathway since hormone ablation does
not completely eliminate serum androgens, with around 10% of
baseline testosterone levels still present in castrated men
resulting from peripheral conversion in the adrenal glands
(43). More recently, ongoing
steroidogenesis within prostate tumors and maintenance of
intratumoral androgens has been identified as a possible means for
these cancers to circumvent low levels of circulating hormones
(6). In addition, crosstalk with
other growth factor signaling pathways can both stabilize AR and
enhance its transcriptional activity. Compensatory regulation
between the AR and mTOR pathways has emerged as a key mechanism in
the pathogenesis of prostate cancer (10–12).
This provides a clear rationale for the investigation of
combinatorial strategies using inhibitors of both of these
signaling pathways.
We show that simultaneous blockade of both pathways
using a combination of ridaforolimus with the antiandrogen
bicalutamide resulted in synergistic antiproliferative effects in
prostate cancer cell lines, but not in an immortalized normal
prostate epithelial line. In normal prostatic epithelia, basal
levels of mTOR and AR signaling are each low, as observed in the
RWPE-1 cell line. As such, normal tissues would be expected to be
relatively unresponsive to treatment with this drug combination.
However, in transformed cells, the sustained amplification of both
pathways (e.g. through functional PTEN loss) leads to a
proliferative circuit and enhanced crosstalk between the two
signaling cascades. In this instance, effective blockade of both
signaling pathways results in enhanced inhibition of tumor cell
proliferation, thus accounting for the strong synergy displayed by
ridaforolimus-bicalutamide treatment. This model is supported by
recent studies that demonstrate dual AR/mTOR inhibition using
antiandrogens with rapamycin or its analogs (12,33,44).
In considering mechanism, both androgen deprivation and mTOR
inhibition are known to cause cell cycle arrest (45,46).
Our data suggest that it is an enhancement of this cytostatic
mechanism that accounts for the augmented effects on growth
inhibition. In contrast, Wang et al(12) have reported that
bicalutamide-rapamycin treatment can induce apoptosis in prostate
cancer cell lines, although it is not clear whether compound
differences, cell lines or duration of treatment account for this
discrepancy.
An important finding of this study is that the
antiproliferative effects observed following combination treatment
in vitro translated to potent antitumor activity in
vivo using a C4-2 xenograft model. This cell line, a derivative
of LNCaP, has previously been described as androgen-independent
(47,48) and has served as a model for
studying growth inhibition in advanced disease. While we have found
that C4-2 can grow in the absence of androgens in vitro and
in castrated mice, our data reveal that these cells do respond to
androgen signaling as shown by their sensitivity to bicalutamide
treatment in an androgen-rich environment (Fig. 4A). This is supported by previous
findings (49) that the endogenous
activity of AR in this line exhibits both androgen-inducibility and
ligand-independent elements. Although not examined here, additional
mechanisms such as intratumoral production of androgens may
potentially contribute to the anti-androgen responsiveness of these
cells in vivo. Single agent blockade of either the AR or
mTOR pathway showed comparable levels of tumor response; however
the combination treatment regimen (based on the clinical dosing
schedules for each) resulted in virtually complete inhibition of
tumor growth. Consistent with our observations, it was recently
reported that the addition of rapamycin enhanced the efficacy of
antiandrogen treatment in a PTEN-null, androgen-dependent
transgenic mouse model of prostate cancer (33).
A finding of particular significance was our
observation that combination treatment resulted in parallel effects
on tumor cell growth and plasma PSA levels in the xenograft model.
As single agents, ridaforolimus and bicalutamide exerted opposing
effects on AR transcriptional activity in vitro. In
accordance with other reports of mTOR inhibition (12,44),
ridaforolimus treatment of the prostate tumor lines increased
expression of PSA, despite its inhibitory effects on cell growth.
However, the addition of bicalutamide was sufficient to block the
ridaforolimus-induced stimulation of PSA, thereby restoring the
correlation between growth inhibition and secreted PSA levels both
in vitro and in vivo. This finding has important
clinical implications, as serum PSA levels are used to track tumor
burden in prostate cancer patients (50,51).
Therefore, the antitumor activity and concomitant reduction of
serum PSA exhibited by the combination treatment suggests that PSA
would provide a relevant clinical marker of tumor growth in
patients treated with this regimen.
In summary, we have shown that the mTOR inhibitor
ridaforolimus exhibits robust antiproliferative activity in
preclinical models of prostate cancer, alone and in combination
with an antiandrogen. The degree of sensitivity is associated with
the activation state of the AKT pathway. The addition of
bicalutamide results in potent antiproliferative and antitumor
activity and represents a potential effective combination strategy
for the treatment of prostate cancers. Notably, serum PSA levels
reflect the tumor inhibition seen in murine models using this
treatment regimen. Taken together, these observations provide
strong preclinical support for the exploration of this combination
as a novel therapeutic approach in prostate cancer patients,
particularly those with loss of the tumor suppressor PTEN.
Abbreviations:
|
mTOR
|
mammalian target of rapamycin
|
|
AR
|
androgen receptor
|
|
PTEN
|
phosphatase and tensin homolog
|
|
PI3K/AKT
|
phosphoinositide 3-kinase/protein
kinase B
|
|
PSA
|
prostate specific antigen
|
|
PIN
|
prostate intraepithelial neoplasia
|
References
|
1
|
Balk SP and Knudsen KE: AR, the cell
cycle, and prostate cancer. Nucl Recept Signal.
6:E0012008.PubMed/NCBI
|
|
2
|
Lee JT, Lehmann BD, Terrian DM, et al:
Targeting prostate cancer based on signal transduction and cell
cycle pathways. Cell Cycle. 7:1745–1762. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen Y, Sawyers CL and Scher HI: Targeting
the androgen receptor pathway in prostate cancer. Curr Opin
Pharmacol. 8:440–448. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li J and Al-Azzawi F: Mechanism of
androgen receptor action. Maturitas. 63:142–148. 2009. View Article : Google Scholar
|
|
5
|
Arnold JT and Isaacs JT: Mechanisms
involved in the progression of androgen-independent prostate
cancers: it is not only the cancer cell’s fault. Endocr Relat
Cancer. 9:61–73. 2002.
|
|
6
|
Montgomery RB, Mostaghel EA, Vessella R,
et al: Maintenance of intratumoral androgens in metastatic prostate
cancer: a mechanism for castration-resistant tumor growth. Cancer
Res. 68:4447–4454. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
NCCN Clinical Practice Guidelines in
Oncology. Prostate Cancer Journal V.3. 2010.
|
|
8
|
Tan WW: Novel agents and targets in
managing patients with metastatic prostate cancer. Cancer Control.
13:194–198. 2006.PubMed/NCBI
|
|
9
|
Zhu ML and Kyprianou N: Androgen receptor
and growth factor signaling cross-talk in prostate cancer cells.
Endocr Relat Cancer. 15:841–849. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cinar B, De Benedetti A and Freeman MR:
Post-transcriptional regulation of the androgen receptor by
Mammalian target of rapamycin. Cancer Res. 65:2547–2553. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu Y, Chen SY, Ross KN and Balk SP:
Androgens induce prostate cancer cell proliferation through
mammalian target of rapamycin activation and post-transcriptional
increases in cyclin D proteins. Cancer Res. 66:7783–7792. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y, Mikhailova M, Bose S, Pan CX,
deVere White RW and Ghosh PM: Regulation of androgen receptor
transcriptional activity by rapamycin in prostate cancer cell
proliferation and survival. Oncogene. 27:7106–7117. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hay N and Sonenberg N: Upstream and
downstream of mTOR. Genes Dev. 18:1926–1945. 2004. View Article : Google Scholar
|
|
14
|
Bjornsti MA and Houghton PJ: The TOR
pathway: a target for cancer therapy. Nat Rev Cancer. 4:335–348.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Guertin DA and Sabatini DM: An expanding
role for mTOR in cancer. Trends Mol Med. 11:353–361. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kremer CL, Klein RR, Mendelson J, et al:
Expression of mTOR signaling pathway markers in prostate cancer
progression. Prostate. 66:1203–1212. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Suzuki H, Freije D, Nusskern DR, et al:
Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple
metastatic prostate cancer tissues. Cancer Res. 58:204–209.
1998.PubMed/NCBI
|
|
18
|
Wang SI, Parsons R and Ittmann M:
Homozygous deletion of the PTEN tumor suppressor gene in a subset
of prostate adenocarcinomas. Clin Cancer Res. 4:811–815.
1998.PubMed/NCBI
|
|
19
|
McCall P, Witton CJ, Grimsley S, Nielsen
KV and Edwards J: Is PTEN loss associated with clinical outcome
measures in human prostate cancer? Br J Cancer. 99:1296–1301. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Neshat MS, Mellinghoff IK, Tran C, et al:
Enhanced sensitivity of PTEN-deficient tumors to inhibition of
FRAP/mTOR. Proc Natl Acad Sci USA. 98:10314–10319. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mills GB, Lu Y and Kohn EC: Linking
molecular therapeutics to molecular diagnostics: inhibition of the
FRAP/RAFT/TOR component of the PI3K pathway preferentially blocks
PTEN mutant cells in vitro and in vivo. Proc Natl Acad Sci USA.
98:10031–10033. 2001. View Article : Google Scholar
|
|
22
|
Podsypanina K, Lee RT, Politis C, et al:
An inhibitor of mTOR reduces neoplasia and normalizes p70/S6 kinase
activity in PTEN+/− mice. Proc Natl Acad Sci USA.
98:10320–10325. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rivera VM, Squillace RM, Miller D, et al:
Ridaforolimus (AP23573, MK-8669), a potent mTOR inhibitor, has
broad antitumor activity and can be optimally administered using
intermittent dosing regimens. Mol Cancer Ther. 10:1059–1071. 2011.
View Article : Google Scholar
|
|
24
|
Colombo N, McMeekin S, Schwartz P, et al:
A phase II trial of the mTOR inhibitor AP23573 as a single agent in
advanced endometrial cancer. J Clin Oncol. 25(18S): 55162007.
|
|
25
|
Mutter GL: Pten, a protean tumor
suppressor. Am J Pathol. 158:1895–1898. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mutter GL, Lin MC, Fitzgerald JT, et al:
Altered PTEN expression as a diagnostic marker for the earliest
endometrial precancers. J Natl Cancer Inst. 92:924–930. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chow LM and Baker SJ: PTEN function in
normal and neoplastic growth. Cancer Lett. 241:184–196. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Squillace RM, Miller D, Cookson M, et al:
Antitumor activity of ridaforolimus and potential cell cycle
determinants of sensitivity in sarcoma and endometrial cancer
models. Mol Cancer Ther. 10:1959–1968. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Majumder PK, Febbo PG, Bikoff R, et al:
mTOR inhibition reverses Akt-dependent prostate intraepithelial
neoplasia through regulation of apoptotic and HIF-1-dependent
pathways. Nat Med. 10:594–601. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mousses S, Wagner U, Chen Y, et al:
Failure of hormone therapy in prostate cancer involves systematic
restoration of androgen responsive genes and activation of
rapamycin sensitive signaling. Oncogene. 20:6718–6723. 2001.
View Article : Google Scholar
|
|
31
|
Gao N, Zhang Z, Jiang BH and Shi X: Role
of PI3K/AKT/mTOR signaling in the cell cycle progression of human
prostate cancer. Biochem Biophys Res Commun. 310:1124–1132. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: the combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang W, Zhu J, Efferson CL, et al:
Inhibition of tumor growth progression by antiandrogens and mTOR
inhibitor in a Pten-deficient mouse model of prostate cancer.
Cancer Res. 69:7466–7472. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gray IC, Phillips SM, Lee SJ, Neoptolemos
JP, Weissenbach J and Spurr NK: Loss of the chromosomal region
10q23-25 in prostate cancer. Cancer Res. 55:4800–4803.
1995.PubMed/NCBI
|
|
35
|
Komiya A, Suzuki H, Ueda T, et al: Allelic
losses at loci on chromosome 10 are associated with metastasis and
progression of human prostate cancer. Genes Chromosomes Cancer.
17:245–253. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gray IC, Stewart LM, Phillips SM, et al:
Mutation and expression analysis of the putative prostate
tumour-suppressor gene PTEN. Br J Cancer. 78:1296–1300. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
McMenamin ME, Soung P, Perera S, Kaplan I,
Loda M and Sellers WR: Loss of PTEN expression in paraffin-embedded
primary prostate cancer correlates with high Gleason score and
advanced stage. Cancer Res. 59:4291–4296. 1999.PubMed/NCBI
|
|
38
|
Burton JL, Oakley N and Anderson JB:
Recent advances in the histopathology and molecular biology of
prostate cancer. BJU Int. 85:87–94. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang S, Gao J, Lei Q, et al:
Prostate-specific deletion of the murine Pten tumor suppressor gene
leads to metastatic prostate cancer. Cancer Cell. 4:209–221. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gao H, Ouyang X, Banach-Petrosky WA, Shen
MM and Abate-Shen C: Emergence of androgen independence at early
stages of prostate cancer progression in Nkx3.1; Pten mice. Cancer
Res. 66:7929–7933. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen Z, Trotman LC, Shaffer D, et al:
Crucial role of p53-dependent cellular senescence in suppression of
Pten-deficient tumorigenesis. Nature. 436:725–730. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Di Cristofano A, De Acetis M, Koff A,
Cordon-Cardo C and Pandolfi PP: Pten and p27KIP1 cooperate in
prostate cancer tumor suppression in the mouse. Nat Genet.
27:222–224. 2001.PubMed/NCBI
|
|
43
|
Hellerstedt BA and Pienta KJ: The current
state of hormonal therapy for prostate cancer. CA Cancer J Clin.
52:154–179. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Schayowitz A, Sabnis G, Njar VC and Brodie
AM: Synergistic effect of a novel antiandrogen, VN/124-1, and
signal transduction inhibitors in prostate cancer progression to
hormone independence in vitro. Mol Cancer Ther. 7:121–132. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Murillo H, Huang H, Schmidt LJ, Smith DI
and Tindall DJ: Role of PI3K signaling in survival and progression
of LNCaP prostate cancer cells to the androgen refractory state.
Endocrinology. 142:4795–4805. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chan S: Targeting the mammalian target of
rapamycin (mTOR): a new approach to treating cancer. Br J Cancer.
91:1420–1424. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Thalmann GN, Anezinis PE, Chang SM, et al:
Androgen-independent cancer progression and bone metastasis in the
LNCaP model of human prostate cancer. Cancer Res. 54:2577–2581.
1994.PubMed/NCBI
|
|
48
|
Thalmann GN, Sikes RA, Wu TT, et al: LNCaP
progression model of human prostate cancer: androgen-independence
and osseous metastasis. Prostate. 44:91–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Dehm SM and Tindall DJ: Ligand-independent
androgen receptor activity is activation function-2-independent and
resistant to antiandrogens in androgen refractory prostate cancer
cells. J Biol Chem. 281:27882–27893. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nash AF and Melezinek I: The role of
prostate specific antigen measurement in the detection and
management of prostate cancer. Endocr Relat Cancer. 7:37–51. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ryan CJ, Smith A, Lal P, et al: Persistent
prostate-specific antigen expression after neoadjuvant androgen
depletion: an early predictor of relapse or incomplete androgen
suppression. Urology. 68:834–839. 2006. View Article : Google Scholar
|